
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Pharma Eurasia 2026, supported by Ministry of Health, Republic of Uzbekistan!

![]()
![]()
![]()
Uzbekistan ![]() calling…
calling…
![]() Excited to announce Pharma Eurasia 2026, supported by the Pharmaceutical Industry Development Agency and the Ministry of Health, Republic of Uzbekistan!
Excited to announce Pharma Eurasia 2026, supported by the Pharmaceutical Industry Development Agency and the Ministry of Health, Republic of Uzbekistan! ![]() Join 400+ exhibitors and 7000+ industry visitors from across the globe from 20–22 May 2026 at the Tashkent Pharma Park, Uzbekistan
Join 400+ exhibitors and 7000+ industry visitors from across the globe from 20–22 May 2026 at the Tashkent Pharma Park, Uzbekistan ![]() – your gateway to pharma opportunities in the CIS & Eurasia region.
– your gateway to pharma opportunities in the CIS & Eurasia region. ![]()
![]()
![]() Contact: +91-9526974225 (M. Harikrishnan)
Contact: +91-9526974225 (M. Harikrishnan) ![]()
![]() Visit: www.pharmedxworld.com
Visit: www.pharmedxworld.com
About Pharma Eurasia
Pharma Eurasia is more than just an exhibition — it’s your gateway to the most dynamic pharmaceutical markets in Central Asia, CIS, and Eurasia. Taking place on 20–22 May 2026 in Tashkent, Uzbekistan, the event unites global manufacturers, exporters, regional distributors, and investors — bringing the entire pharma ecosystem under one roof.
Here, industry leaders connect, collaborate, and explore opportunities to shape the future of pharmaceuticals in one of the world’s fastest-growing hubs.
Why Tashkent
Position your business at the heart of Central Asia’s trade crossroads. Tashkent offers unmatched connectivity to CIS, Eurasian, Middle Eastern, and South Asian markets — supported by government incentives, zero-duty pharma zones, and a rapidly growing demand for medicines and medical technology.
Join us in Tashkent, Uzbekistan, from 20–22 May 2026, and unlock the gateway to new partnerships, expanding networks, and a market ready for your products.
SPSR Excellence Award 2025 – SPSR Global Pharmacist Excellence Awards 2025

Honoured to get SPSR Excellence Award 2025 – SPSR Global Pharmacist Excellence Awards 2025
From the Society of Pharmaceutical Sciences and Research (SPSR)!
In recognition of your excellent work and valuable contributions in the field of Pharmaceutical Sciences and allied disciplines, it gives us great pleasure to confer upon you the:
SPSR Excellence Award 2025 – SPSR Global Pharmacist Excellence Awards 2025
This prestigious award will be conferred during the:
101st SPSR International Webinar & SPSR World Pharmacists Day Excellence Awards 2025
Theme: “Think Health, Think Pharmacist”
Event Details
Date: Thursday, 25th September 2025
Time: 8:00 PM – 10:00 PM IST
Mode: Online (YouTube Live)
YouTube Link: https://youtube.com/live/BcUPHOGweIA?feature=share
The SPSR World Pharmacist Day Excellence Awards are instituted to acknowledge individuals who have demonstrated outstanding dedication, innovation, and impact in advancing pharmacy, healthcare, and allied sciences.
We extend our heartfelt congratulations and look forward to honoring your achievements on this prestigious occasion.
Warm regards,
Mrs. Monika Sabharwal
Founder and National Secretary
Society of Pharmaceutical Sciences and Research (SPSR)
Website – www.spsrpharma.org
Email – secretary@spsrpharma.org
Follow us – https://in.linkedin.com/company/spsr2010
SPSR WhatsApp Channel – https://whatsapp.com/channel/0029Va8rRBpDZ4LjgD2QQV0P
Brimarafenib
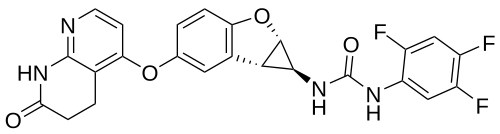
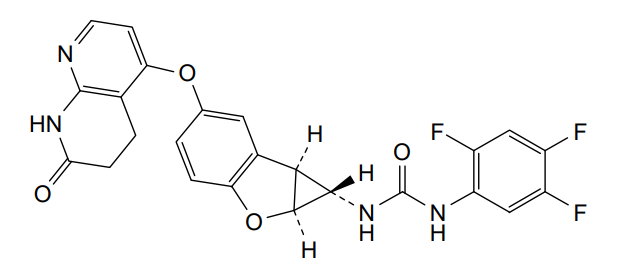
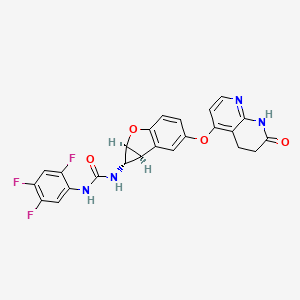
Brimarafenib
CAS 1643326-82-2
MF C24H17F3N4O4 MW482.4 g/mol
N-{(1S,1aS,6bS)-5-[(7-oxo-5,6,7,8-tetrahydro-1,8-naphthyridin-4-yl)oxy]-1a,6b-dihydro-1H-cyclopropa[b]benzofuran-1-yl}-N′-(2,4,5-trifluorophenyl)urea
rapidly accelerated fibrosarcoma (Raf) kinase inhibitor,
- 1-((1S,1aS,6bS)-5-((7-oxo-6,8-dihydro-5H-1,8-naphthyridin-4-yl)oxy)-1a,6b-dihydro-1H-cyclopropa(b)(1)benzofuran-1-yl)-3-(2,4,5-trifluorophenyl)urea
- 1-[(1S,1aS,6bS)-5-[(7-oxo-6,8-dihydro-5H-1,8-naphthyridin-4-yl)oxy]-1a,6b-dihydro-1H-cyclopropa[b][1]benzofuran-1-yl]-3-(2,4,5-trifluorophenyl)urea
Antineoplastic, MapKure, LLC, SpringWorks Therapeutics, BeiGene, BGB-3245, BGB 3245, GXS33OY2CB
Brimarafenib is an investigational new drug that is being evaluated for the treatment of cancer. It targets the proto-oncogene BRAF with activating mutations BRAF mutations (such as V600E), non-V600 BRAF mutations, and RAF fusions.[1][2]
It is being developed by MapKure, LLC, a joint venture between SpringWorks Therapeutics and BeiGene.[1]
Brimarafenib is an orally available inhibitor of both monomer and dimer forms of activating mutations of the serine/threonine-protein kinase BRAF (B-raf) protein, including V600 BRAF mutations, non-V600 BRAF mutations, and RAF fusions, with potential antineoplastic activity. Upon administration, brimarafenib targets and binds to both monomeric and dimeric forms of activating BRAF mutations and fusions. This may result in the inhibition of BRAF-mediated signaling and inhibit proliferation in tumor cells expressing BRAF mutations and fusions. BRAF belongs to the RAF family of serine/threonine protein kinases and plays a role in regulating the mitogen-activated protein kinase (MAPK)/ extracellular signal-regulated kinase (ERK) signaling pathway, which is often dysregulated in human cancers and plays a key role in tumor cell proliferation and survival. BRAF mutations and fusions have been identified in a number of solid tumors and are drivers of cancer growth.
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014206343&_cid=P22-MG0802-32937-1
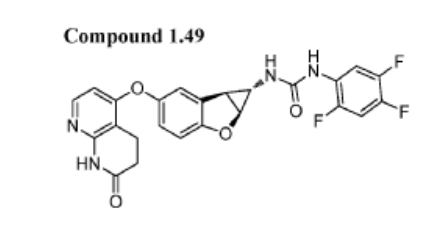
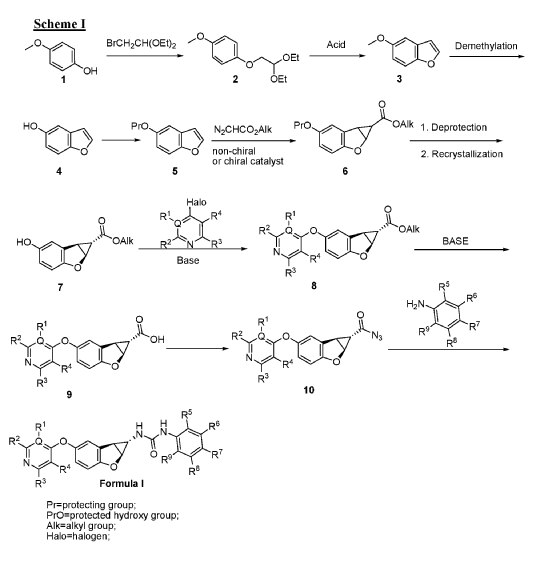

PAT
Fused tricyclic urea compounds as raf kinase and/or raf kinase dimer inhibitors
Publication Number: WO-2014206343-A1
Priority Date: 2013-06-28
- Fused tricyclic urea compounds as raf kinase and/or raf kinase dimer inhibitorsPublication Number: US-2016368914-A1Priority Date: 2013-06-28
- Fused tricyclic urea compounds as raf kinase and/or raf kinase dimer inhibitorsPublication Number: US-2017233391-A1Priority Date: 2013-06-28
- Fused tricyclic urea compounds as raf kinase and/or raf kinase dimer inhibitorsPublication Number: US-2019144446-A1Priority Date: 2013-06-28
- Fused tricyclic urea compounds as Raf kinase and/or Raf kinase dimer inhibitorsPublication Number: US-9670203-B2Priority Date: 2013-06-28Grant Date: 2017-06-06
- Fused tricyclic urea compounds as raf kinase and/or raf kinase dimer inhibitorsPublication Number: US-9920055-B2Priority Date: 2013-06-28Grant Date: 2018-03-20



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
| Clinical data | |
|---|---|
| Other names | BGB-3245 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1643326-82-2 |
| PubChem CID | 117807031 |
| IUPHAR/BPS | 13203 |
| ChemSpider | 129144353 |
| UNII | GXS33OY2CB |
| Chemical and physical data | |
| Formula | C24H17F3N4O4 |
| Molar mass | 482.419 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Brimarafenib”.
- Tellenbach FL, Seiler LL, Johnson M, Rehrauer H, Schukla P, Martinez-Gomez J, et al. “Combination of the Novel Raf Dimer Inhibitor Brimarafenib with the Mek Inhibitor Mirdametinib is Effective Against Nras Mutant Melanoma”. SSRN: 4934723. doi:10.2139/ssrn.4934723.
///////Brimarafenib, Antineoplastic, MapKure, LLC, SpringWorks Therapeutics, BeiGene, BGB-3245, BGB 3245, GXS33OY2CB
Brezivaptan
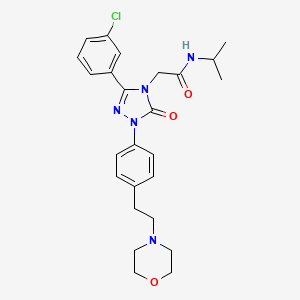
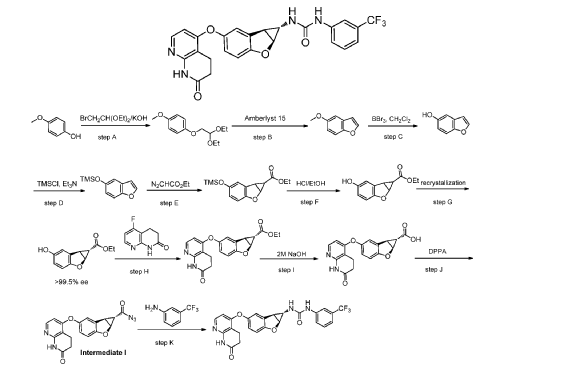
Brezivaptan
CAS 1370444-22-6
ANC-501, THY-1773, TS-121, 575OB1CKN0
MF C25H30ClN5O3 MW 484.0 g/mol
2-[3-(3-chlorophenyl)-1-{4-[2-(morpholin-4-yl)ethyl]phenyl}-5-oxo-1,5-dihydro-4H-1,2,4-triazol-4-yl]-N-(propan-2-yl)acetamide
2-[3-(3-chlorophenyl)-1-[4-(2-morpholin-4-ylethyl)phenyl]-5-oxo-1,2,4-triazol-4-yl]-N-propan-2-ylacetamide
vasopressin receptor antagonist
- ANC-501 in the Treatment of Adults With Major Depressive DisorderCTID: NCT05439603Phase: Phase 2Status: CompletedDate: 2024-12-31
- A Study to Evaluate the Safety and Efficacy of TS-121 as an Adjunctive Treatment for Major Depressive DisorderCTID: NCT03093025Phase: Phase 2Status: TerminatedDate: 2020-07-14
- Exploratory Study Using Positron Emission Tomography With TS-121 and [11C]TASP0410699 in Healthy Adult Male SubjectsCTID: NCT02448212Phase: Phase 1Status: CompletedDate: 2017-02-14
Brezivaptan[1] (developmental code names ANC-501, THY-1773, TS-121) is an orally active, selective vasopressin V1B receptor antagonist which is under development by Taisho Pharmaceutical for the adjunctive treatment of major depressive disorder.[2][3][4] As of November 2022, it is in phase II clinical trials for this indication.[2][3][5]
ANC-501 is under investigation in clinical trial NCT05439603 (ANC-501 in the Treatment of Adults With Major Depressive Disorder).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US90328697&_cid=P11-MFYT6K-98384-1
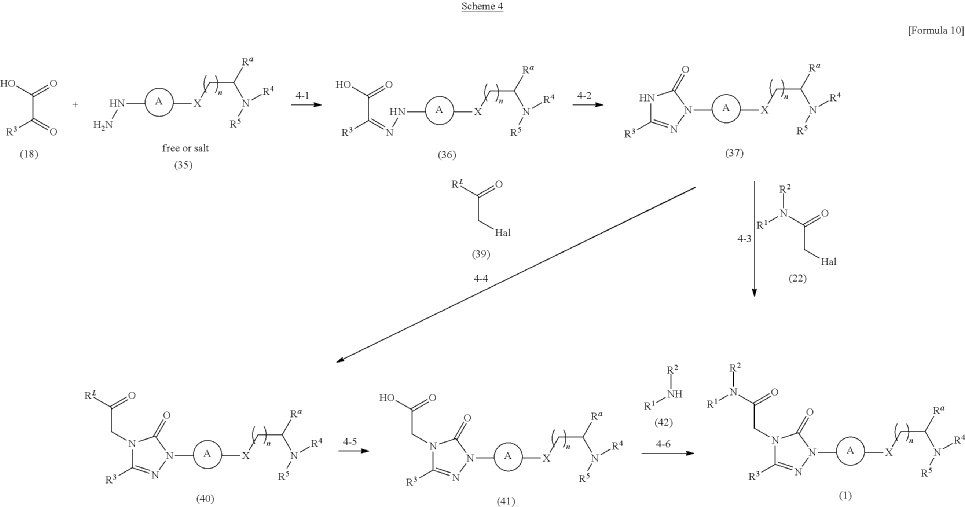
Synthesis of Example Aa-1
2-[3-(3-Chlorophenyl)-1-{4-[2-(morpholin-4-yl)ethyl]phenyl}-5-oxo-1,5-dihydro-4H-1,2,4-triazol-4-yl]-N-(propan-2-yl)acetamide
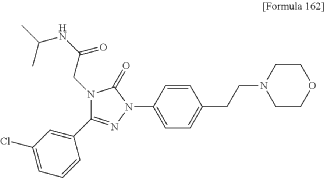
| A mixture of the compound (100 mg) prepared in Reference Example P-I1, morpholine (0.03 mL), N,N-diisopropylethylamine (0.35 mL), and MeCN (3.00 mL) was stirred at an outside temperature of 80° C. overnight. After cooling, the solvent was distilled off under reduced pressure. The residue was purified by column chromatography (SNAP Cartridge HP-Sil: 10 g, mobile phase: CHCl 3/MeOH=98/2 to 85/15 (v/v); and SNAP Cartridge KP-NH: 28 g, mobile phase: n-hexane/CHCl 3=80/20 to 0/100 (v/v)) and preparative thin-layer chromatography (PTLC) (1.0 mm silica gel 60F 254 plate, mobile phase: EtOAc/MeOH=95/5 (v/v)). The resulting crude product was washed with a solvent mixture of EtOAc and n-hexane (EtOAc/n-hexane=1/4 (v/v)) with stirring to yield the title compound (70 mg, colorless solid). |
PAT
- 1,2,4-triazolone derivativePublication Number: NZ-608729-APriority Date: 2010-10-01
- 1, 2, 4-triazolone derivativePublication Number: US-2013197217-A1Priority Date: 2010-10-01
- 1, 2, 4-triazolone derivative and use thereof as an antagonist on the arginine-vasopressin 1B receptorPublication Number: US-9193695-B2Priority Date: 2010-10-01Grant Date: 2015-11-24
- 1,2,4-triazolone derivative, substance and pharmaceutical compositionPublication Number: BR-112013007389-B1Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: EP-2623499-A1Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: EP-2623499-B1Priority Date: 2010-10-01Grant Date: 2015-04-22
- DERIVAT 1,2,4-TRIAZOLONAPublication Number: HR-P20150462-T1Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: HU-E025729-T2Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: IL-225091-APriority Date: 2010-10-01
- Methods of treating depression with 1,2,4-triazolone derivativesPublication Number: WO-2023235785-A1Priority Date: 2022-06-01
- 1,2,4-triazolone derivativePublication Number: AU-2011308403-A1Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: AU-2011308403-B2Priority Date: 2010-10-01Grant Date: 2014-08-21
- 1,2,4-Triazolone DerivativesPublication Number: CN-103119028-APriority Date: 2010-10-01
- 1,2,4-Triazolone DerivativesPublication Number: CN-103119028-BPriority Date: 2010-10-01Grant Date: 2016-05-25
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- PubChem. “Brezivaptan”. pubchem.ncbi.nlm.nih.gov. Retrieved 2024-08-15.
- “TS 121 -“. AdisInsight. Springer Nature Switzerland AG.
- “New Drug Pipeline – Taisho Pharmaceutical Holdings”.
- Kamiya M, Sabia HD, Marella J, Fava M, Nemeroff CB, Umeuchi H, Iijima M, Chaki S, Nishino I (September 2020). “Efficacy and safety of TS-121, a novel vasopressin V1B receptor antagonist, as adjunctive treatment for patients with major depressive disorder: A randomized, double-blind, placebo-controlled study”. Journal of Psychiatric Research. 128: 43–51. doi:10.1016/j.jpsychires.2020.05.017. PMID 32521250. S2CID 219587135.
- Inatani S, Mizuno-Yasuhira A, Kamiya M, Nishino I, Sabia HD, Endo H (May 2021). “Prediction of a clinically effective dose of THY1773, a novel V1B receptor antagonist, based on preclinical data”. Biopharmaceutics & Drug Disposition. 42 (5): 204–217. doi:10.1002/bdd.2273. PMC 8252455. PMID 33734452.
External links
- Clinical trial number NCT03093025 for “A Study to Evaluate the Safety and Efficacy of TS-121 as an Adjunctive Treatment for Major Depressive Disorder” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Other names | TS-121; TS121; TS-1211; TS1211; THY1773; THY-1773; ANC-501; ANC501 |
| Routes of administration | By mouth |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1370444-22-6 |
| PubChem CID | 56952080 |
| DrugBank | DB18907 |
| ChemSpider | 129325033 |
| UNII | 575OB1CKN0 |
| ChEMBL | ChEMBL5314910 |
| Chemical and physical data | |
| Formula | C25H30ClN5O3 |
| Molar mass | 484.00 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////////Brezivaptan, ANC-501, THY-1773, TS-121, ANC 501, THY 1773, TS 121, 575OB1CKN0
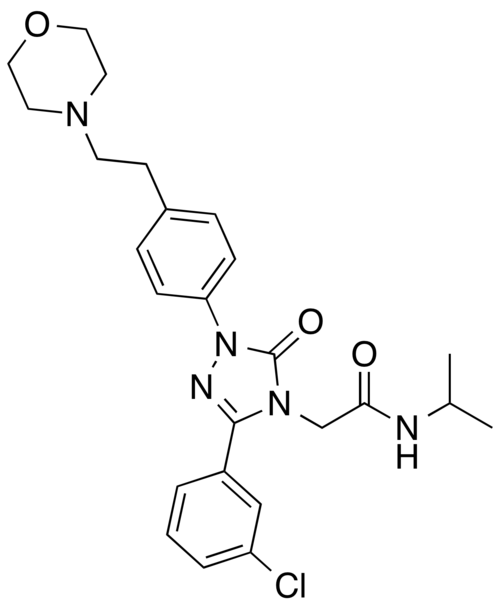
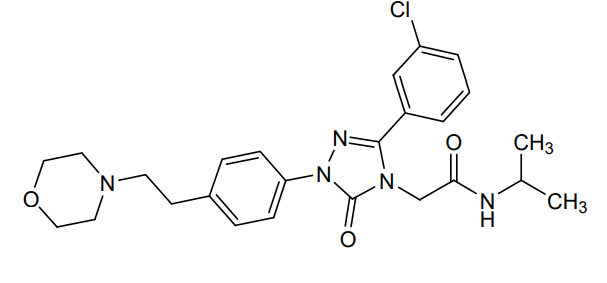
Atumelnant
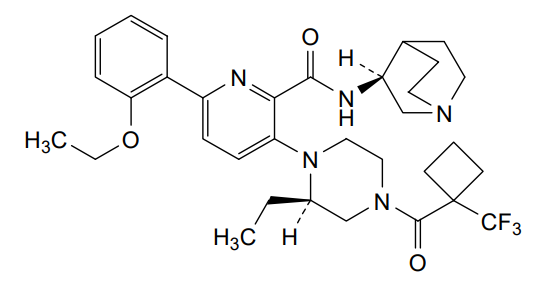
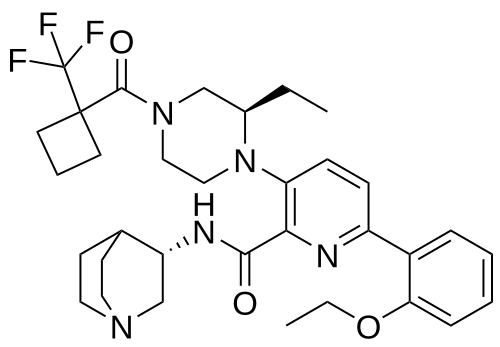
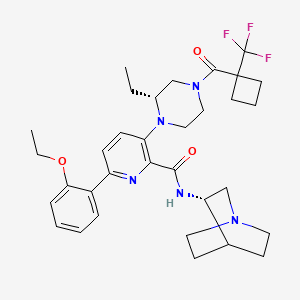
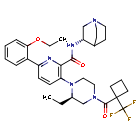
Atumelnant
CAS 2392970-97-5
MF C33H42F3N5O3 MW 613.7 g/mol
CRN04894, NR57FH6U1N
CRINETICS PHARMA, Orphan Drug Status, Congenital adrenal hyperplasia
N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide
N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-{(2R)-2-ethyl-4-[1-(trifluoromethyl) cyclobutane-1-carbonyl]piperazin-1-yl}pyridine-2-carboxamide
Adrenocorticotropic hormone receptor antagonist
- OriginatorCrinetics Pharmaceuticals
- ClassAmides; Antineoplastics; Antisecretories; Benzene derivatives; Cyclobutanes; Ethers; Fluorocarbons; Ketones; Piperazines; Pyridines; Quinuclidines; Small molecules
- Mechanism of ActionMelanocortin type 2 receptor antagonists
- Orphan Drug StatusYes – Congenital adrenal hyperplasia
- Phase IICongenital adrenal hyperplasia; Cushing syndrome
- No development reportedEctopic ACTH syndrome
- 21 Aug 2025Atumelnant receives Orphan Drug status for Congenital adrenal hyperplasia in the US
- 07 Aug 2025Crinetics pharmaceuticals plans phase II/III clinical trial for Cushing’s disease in 1H 2026
- 08 May 2025Crinetics Pharmaceuticals plans the phase III CALM-CAH trial for Congenital adrenal hyperplasia (In adults) (PO), in the second half of 2025
Atumelnant (INNTooltip International Nonproprietary Name; developmental code name CRN04894) is an investigational new drug developed by Crinetics Pharmaceuticals for the treatment of adrenocorticotropic hormone (ACTH)-dependent endocrine disorders.[1] It is a selective antagonist of the melanocortin type 2 receptor (MC2R), also known as the ACTH receptor, which is primarily expressed in the adrenal glands.[1][2] The drug is orally active.[1] Atumelnant is being evaluated to treat conditions such as congenital adrenal hyperplasia (CAH) and ACTH-dependent Cushing’s syndrome caused for example by pituitary adenomas.[3]
Atumelnant is an orally bioavailable nonpeptide antagonist of the adrenocorticotropic hormone (ACTH) receptor (ACTHR; melanocortin receptor 2; MC2R), with potential steroid hormone production inhibitory activity. Upon oral administration, atumelnant competes with ACTH for receptor binding to MC2R in the adrenal cortex and inhibits ACTH signaling. This may inhibit the synthesis and secretion of steroid hormones. MC2R, a member of the melanocortin receptor subfamily of type 1 G protein-coupled receptors, plays a key role in adrenal steroidogenesis.
PAPER
Discovery of CRN04894: A Novel Potent Selective MC2R Antagonist
Publication Name: ACS Medicinal Chemistry Letters
Publication Date: 2024-03-19, PMCID: PMC11017392, PMID: 38628803
DOI: 10.1021/acsmedchemlett.3c00514
PATENTS
- Melanocortin subtype-2 receptor antagonists and uses thereofPublication Number: IL-279152-B2Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2024300920-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor antagonists and uses thereofPublication Number: IL-279152-B1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: JP-2024009837-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: KR-102695210-B1Priority Date: 2018-06-05Grant Date: 2024-08-13
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2024109866-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: CN-112533904-BPriority Date: 2018-06-05Grant Date: 2024-10-29
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10981894-B2Priority Date: 2018-06-05Grant Date: 2021-04-20
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2021002254-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2021238164-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-11566015-B2Priority Date: 2018-06-05Grant Date: 2023-01-31
- Melanocortin subtype-2 receptor (MC2R) antagonists and their usesPublication Number: JP-7359783-B2Priority Date: 2018-06-05Grant Date: 2023-10-11
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2020216415-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10766877-B2Priority Date: 2018-06-05Grant Date: 2020-09-08
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: CN-112533904-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: EP-3802500-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: KR-20210005995-APriority Date: 2018-06-05
- Melanocortin subtype-2 receptor (MC2R) antagonists for the treatment of diseasePublication Number: CN-117043146-APriority Date: 2021-03-19
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10562884-B2Priority Date: 2018-06-05Grant Date: 2020-02-18
- Melanocortin subtype-2 receptor (MC2R) antagonists and uses thereofPublication Number: US-10604507-B2Priority Date: 2018-06-05Grant Date: 2020-03-31
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2019367481-A1Priority Date: 2018-06-05
- Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereofPublication Number: US-2020010452-A1Priority Date: 2018-06-05
Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: US-2022313691-A1Priority Date: 2021-03-19 - Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: WO-2022197798-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: TW-202302108-APriority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: AU-2022240609-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of diseasePublication Number: EP-4308553-A1Priority Date: 2021-03-19
- Melanocortin subtype-2 receptor (mc2r) antagonist for the treatment of acth-dependent cushing’s syndromePublication Number: WO-2024211343-A1Priority Date: 2023-04-05
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: TW-202430167-APriority Date: 2022-12-16
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: US-2024208963-A1Priority Date: 2022-12-16
- Crystalline melanocortin subtype-2 receptor (mc2r) antagonistPublication Number: WO-2024130091-A1Priority Date: 2022-12-16
- Treatment of congenital adrenal hyperplasia and polycystic ovary syndromePublication Number: WO-2023163945-A1Priority Date: 2022-02-2
SYN
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US278278493&_cid=P22-MFXDN2-76849-1
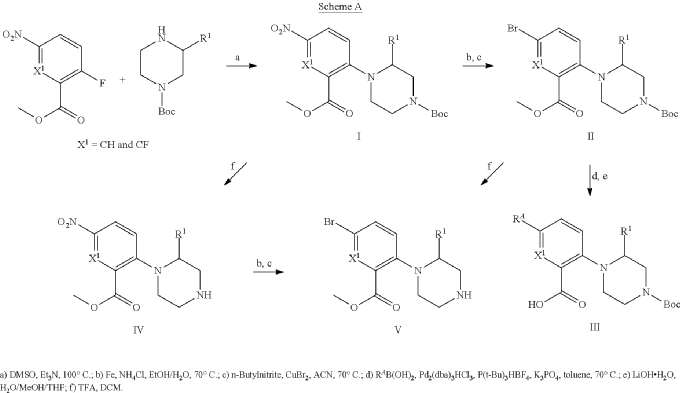
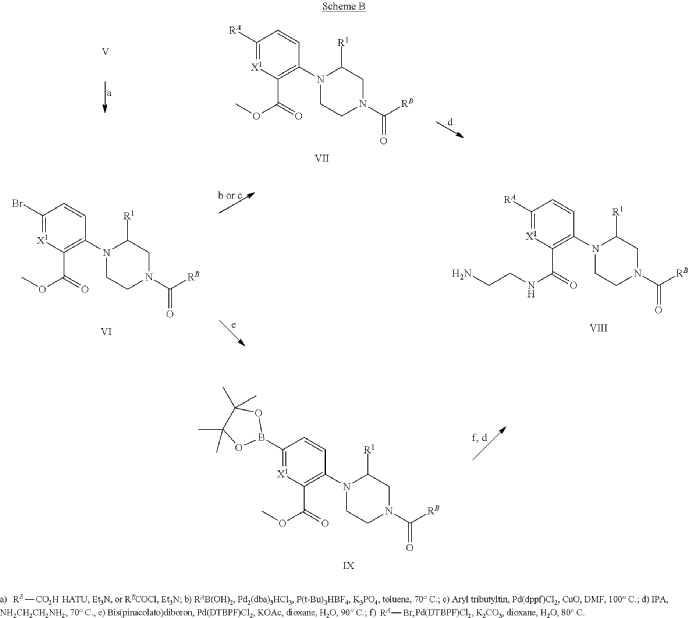
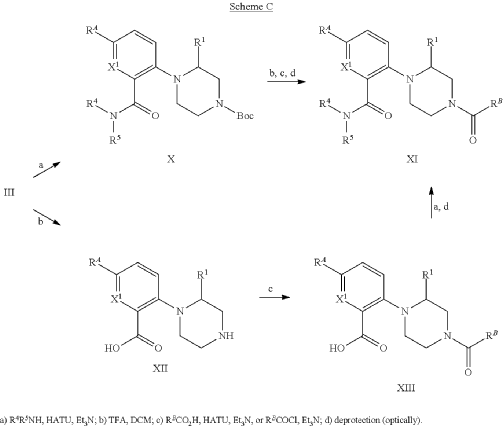
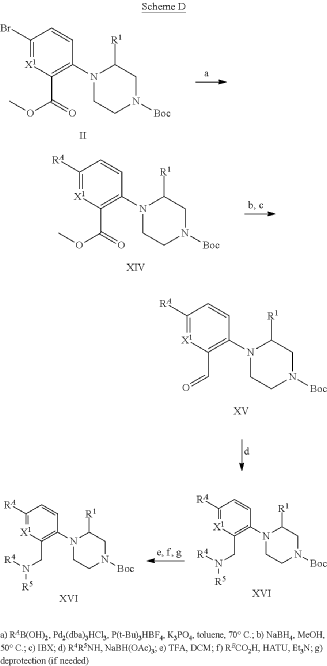
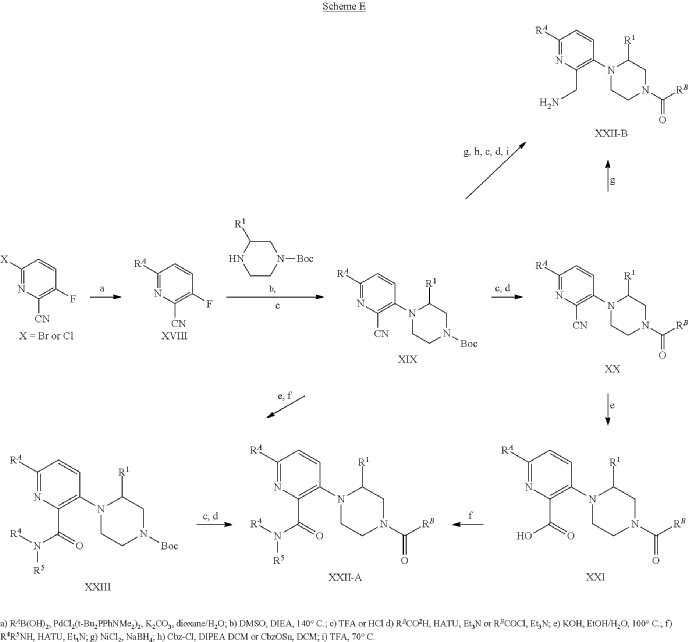
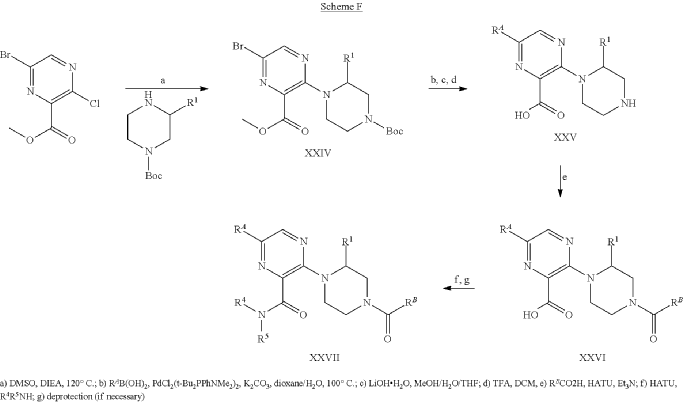
Example 31: N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide (Compound 1-410)
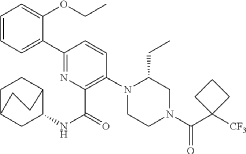
Step 31-1, Preparation of 6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxylic acid
Step 31-2, Preparation of N-[(3S)-1-azabicyclo[2.2.2]octan-3-yl]-6-(2-ethoxyphenyl)-3-[(2R)-2-ethyl-4-[1-(trifluoromethyl)cyclobutanecarbonyl]piperazin-1-yl]pyridine-2-carboxamide
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Crinetics Pharmaceuticals”. AdisInsight. 21 January 2025. Retrieved 25 February 2025.
- “Atumelnant (CRN04894)”. crinetics.com. 14 August 2020.
- Varlamov EV, Gheorghiu ML, Fleseriu M (December 2024). “Pharmacological management of pituitary adenomas – what is new on the horizon?”. Expert Opinion on Pharmacotherapy. 26 (2): 119–125. doi:10.1080/14656566.2024.2446625. PMID 39718553.
| Clinical data | |
|---|---|
| Other names | CRN04894 |
| Routes of administration | Oral[1] |
| Drug class | Melanocortin MC2 receptor antagonist[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2392970-97-5 |
| PubChem CID | 146361282 |
| IUPHAR/BPS | 13339 |
| ChemSpider | 129750231 |
| UNII | NR57FH6U1N |
| KEGG | D13102 |
| Chemical and physical data | |
| Formula | C33H42F3N5O3 |
| Molar mass | 613.726 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Atumelnant, CRN04894, CRN 04894, NR57FH6U1N, CRINETICS PHARMA, Orphan Drug Status, Congenital adrenal hyperplasia, PHASE 3
Ateganosine
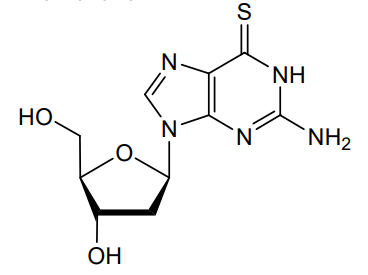
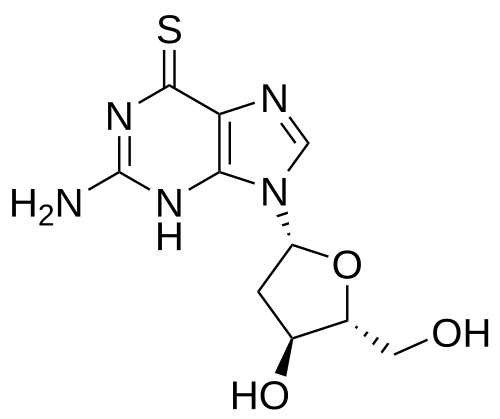
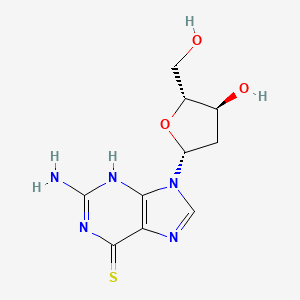
Ateganosine
CAS 789-61-7
MF C10H13N5O3S MW 283.31 g/mol
2′-deoxy-6-thioguanosine
nucleoside analogue, antineoplastic
- 6-THIO-2′-DEOXYGUANOSINE
- 2′-Deoxythioguanosine
- TGdR
- Thioguanine deoxyriboside
- KR0RFB46DF
- NSC-71261
Ateganosine is a telomerase inhibitor[1] and apoptosis inducer currently under investigation for the treatment of various cancers, including non-small cell lung cancer (NSCLC).[2]
Beta-Thioguanine Deoxyriboside is a thiopurine nucleoside derivative with antineoplastic activity. After conversion to the triphosphate, beta-thioguanine deoxyriboside is incorporated into DNA, resulting in inhibition of DNA replication. This agent is cytotoxic against leukemia cell lines and has demonstrated some activity against leukemia cells in vivo. Beta-thioguanine deoxyriboside demonstrates antineoplastic activity against 6-thioguanine-resistant tumor cells. (NCI04)
- THIO Sequenced With Cemiplimab in Advanced NSCLCCTID: NCT05208944Phase: Phase 2Status: RecruitingDate: 2025-05-31
- A Phase III Study With THIO + Cemiplimab vs Chemotherapy as 3rd Line Treatment in Advanced/Metastatic NSCLCCTID: NCT06908304Phase: Phase 3Status: Not yet recruitingDate: 2025-04-08
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Eglenen-Polat B, Kowash RR, Huang HC, Siteni S, Zhu M, Chen K, et al. (January 2024). “A telomere-targeting drug depletes cancer initiating cells and promotes anti-tumor immunity in small cell lung cancer”. Nature Communications. 15 (1) 672. Bibcode:2024NatCo..15..672E. doi:10.1038/s41467-024-44861-8. PMC 10803750. PMID 38253555.
- “Ateganosine”. PatSnap.
| Clinical data | |
|---|---|
| Other names | 2′-Deoxythioguanosine |
| Identifiers | |
| IUPAC name | |
| CAS Number | 789-61-7 |
| PubChem CID | 3000603 |
| DrugBank | DB18117 |
| ChemSpider | 2272164 |
| UNII | KR0RFB46DF |
| KEGG | D13071 |
| ChEMBL | ChEMBL3250476 |
| CompTox Dashboard (EPA) | DTXSID4021345 |
| Chemical and physical data | |
| Formula | C10H13N5O3S |
| Molar mass | 283.31 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Ateganosine, nucleoside analogue, antineoplastic, 6-THIO-2′-DEOXYGUANOSINE, 2′-Deoxythioguanosine, TGdR, Thioguanine deoxyriboside, KR0RFB46DF, fast track designation, NSC-71261, NSC 71261
Bimokalner
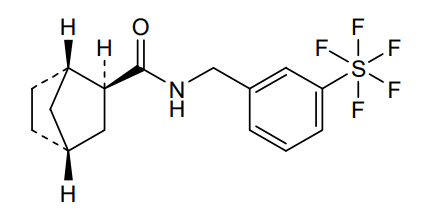
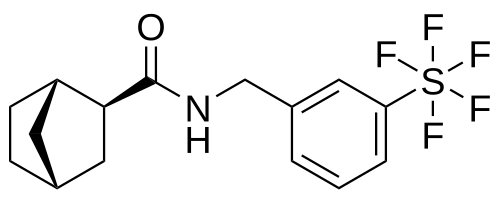
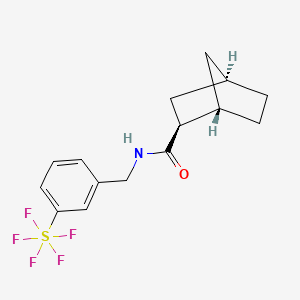
Bimokalner
CAS 2243284-19-5
MF C15H18F5NOS MW 355.4 g/mol
- KEY5KKX6QY
- orb2663976
- (1S,2S,4R)-N-[[3-(pentafluoro-λ6-sulfanyl)phenyl]methyl]bicyclo[2.2.1]heptane-2-carboxamide
(1S,2S,4R)-N-{[3-(pentafluoro-λ6sulfanyl)phenyl]methyl} bicyclo[2.2.1]heptane-2-carboxamide
voltage-gated potassium channel (Kv7.4) agonist
Bimokalner is an investigational new drug under evaluation for preventing and treating hearing loss caused by cisplatin treatment. It is a voltage-gated potassium channel agonist targeting Kv7.4 and is being developed by Acousia Therapeutics GmbH.[1][2]
PAT
Compounds useful as potassium channel openers, Publication Number: US-11884642-B2, Priority Date: 2017-02-28, Grant Date: 2024-01-30
- Novel Compounds Useful As Potassium Channel OpenersPublication Number: KR-20210134826-APriority Date: 2017-02-28
- Novel compounds useful as potassium channel openersPublication Number: KR-102382795-B1Priority Date: 2017-02-28Grant Date: 2022-04-05
- Novel Compounds Useful As Potassium Channel OpenersPublication Number: KR-102443685-B1Priority Date: 2017-02-28Grant Date: 2022-09-15
- Compounds useful as potassium channel openersPublication Number: CN-114105942-BPriority Date: 2017-02-28Grant Date: 2024-07-12
- Novel compounds useful as potassium channel openers.Publication Number: JP-7474289-B2Priority Date: 2017-02-28Grant Date: 2024-04-24
- Compounds useful as potassium channel openersPublication Number: US-11034665-B2Priority Date: 2017-02-28Grant Date: 2021-06-15
- Novel compounds useful as potassium channel openersPublication Number: US-2021261518-A1Priority Date: 2017-02-28
- Novel compounds useful as potassium channel openersPublication Number: AU-2018227005-B2Priority Date: 2017-02-28Grant Date: 2021-11-11
- Compounds useful as potassium channel openersPublication Number: CN-110312710-BPriority Date: 2017-02-28Grant Date: 2022-02-15
- New compounds useful as potassium channel openersPublication Number: CN-114105942-APriority Date: 2017-02-28
- Pentacyclothienyl and indanyl urea derivatives as potassium channel openersPublication Number: EP-3567034-A1Priority Date: 2017-02-28
- New Compounds Useful as Potassium Channel OpenersPublication Number: KR-20190105058-APriority Date: 2017-02-28
- Novel compounds useful as potassium channel openersPublication Number: US-2020157072-A1Priority Date: 2017-02-28
- Novel compounds useful as potassium channel openersPublication Number: WO-2018158256-A2Priority Date: 2017-02-28
- Pentacyclothienyl and indanyl urea derivatives as potassium channel openersPublication Number: EP-3567034-B1Priority Date: 2017-02-28Grant Date: 2020-10-28
PAT
(1R,2R,4S)-rel-N-(3-(pentafluorosulfanyl)benzyl)bicyclo[2.2.1]heptane-2-carboxamide
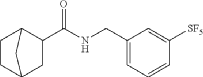
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Bimokalner”. PatSnap.
- Tavanai E, Rahimi V, Khalili ME, Falahzadeh S, Motasaddi Zarandy M, Mohammadkhani G (2024). “Age-related hearing loss: An updated and comprehensive review of the interventions”. Iranian Journal of Basic Medical Sciences. 27 (3): 256–269. doi:10.22038/IJBMS.2023.72863.15849. PMC 10849199. PMID 38333758.
| Clinical data | |
|---|---|
| Other names | ACOU085 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2243284-19-5 |
| PubChem CID | 135309173 |
| UNII | KEY5KKX6QY |
| Chemical and physical data | |
| Formula | C15H18F5NOS |
| Molar mass | 355.37 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Bimokalner, Acousia Therapeutics, KEY5KKX6QY, orb 2663976
Asengeprast
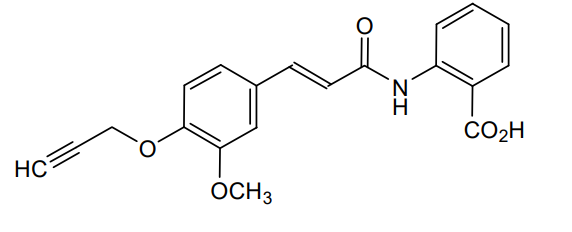
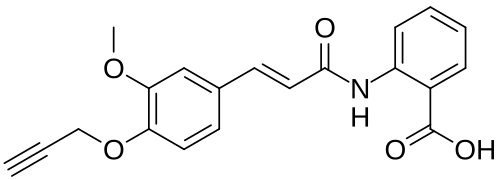
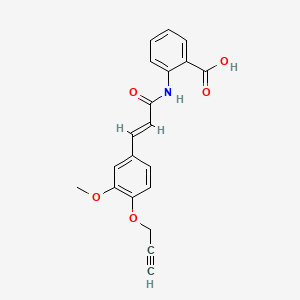
Asengeprast
CAS 1001288-58-9
FT011, FT 011, orphan drug status, systemic sclerosis, SHP-627, SHP 627,
Fast Track
2-[[(E)-3-(3-methoxy-4-prop-2-ynoxyphenyl)prop-2-enoyl]amino]benzoic acid
2-[(2E)-3-{3-methoxy-4-[(prop-2-yn-1-yl)oxy]phenyl}prop-2-enamido]benzoic acid G protein-coupled receptor 68 (GPR68) antagonist,
anti-inflammatory
MF C20H17NO5 MW 351.4 g/mol. C6V7ZU2NPR
Asengeprast (development code FT011) is an experimental scleroderma drug candidate.[1] It is a small molecule inhibitor of the G-protein coupled receptor GPR68 with antifibrotic activity.[2] It is being developed by Certa Therapeutics.
The European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) has granted orphan drug status to FT011, for systemic sclerosis (SSc).[3]
Asengeprast has been reported to attenuate fibrosis and chronic heart failure in experimental diabetic cardiomyopathy.[4] Asengeprast can also inhibit kidney fibrosis and prevent kidney failure.[5] It was developed by structure-activity optimization of the antifibrotic activity of cinnamoyl anthranilates, by assessment of their ability to prevent TGF-beta-stimulated production of collagen.[6]
Effects of FT011 in Systemic Sclerosis, CTID: NCT04647890
Phase: Phase 2, Status: Completed, Date: 2023-12-20
SYN
Evaluation and optimization of antifibrotic activity of cinnamoyl anthranilates
Publication Name: Bioorganic & Medicinal Chemistry Letters
Publication Date: 2009-12-15
PMID: 19879136
DOI: 10.1016/j.bmcl.2009.09.120
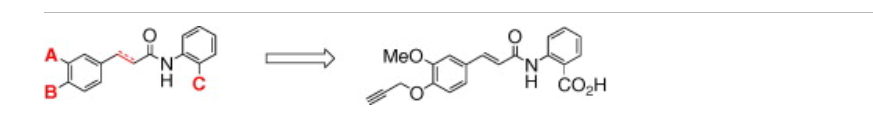
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018144620&_cid=P21-MFTHV7-45829-1
PAT
Publication Number: WO-2008003141-A1
Priority Date: 2006-07-05
- Tranilast analogues (substituted cinnamoyl anthranilate compounds) for treatment of conditions associated with firbrosisPublication Number: NZ-574028-APriority Date: 2006-07-05
- Therapeutic CompoundsPublication Number: US-2010130497-A1Priority Date: 2006-07-05
- Therapeutic compoundsPublication Number: US-2014357628-A1Priority Date: 2006-07-05
- Therapeutic compoundsPublication Number: US-8765812-B2Priority Date: 2006-07-05Grant Date: 2014-07-01
- Therapeutic compoundsPublication Number: US-9561201-B2Priority Date: 2006-07-05Grant Date: 2017-02-07
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Asengeprast Ligand page”. IUPHAR/BPS Guide to PHARMACOLOGY.
- “Certa Therapeutics website”.
- Inácio P (23 July 2024). “Certa’s FT011 granted orphan drug status in Europe for SSc”. Scleroderma News.
- Zhang Y, Edgley AJ, Cox AJ, Powell AK, Wang B, Kompa AR, et al. (May 2012). “FT011, a new anti-fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathy”. European Journal of Heart Failure. 14 (5): 549–562. doi:10.1093/eurjhf/hfs011. PMID 22417655.
- Gilbert RE, Zhang Y, Williams SJ, Zammit SC, Stapleton DI, Cox AJ, et al. (2012). “A purpose-synthesised anti-fibrotic agent attenuates experimental kidney diseases in the rat”. PLOS ONE. 7 (10): e47160. Bibcode:2012PLoSO…747160G. doi:10.1371/journal.pone.0047160. PMC 3468513. PMID 23071743.
- Zammit SC, Cox AJ, Gow RM, Zhang Y, Gilbert RE, Krum H, et al. (December 2009). “Evaluation and optimization of antifibrotic activity of cinnamoyl anthranilates”. Bioorganic & Medicinal Chemistry Letters. 19 (24): 7003–7006. doi:10.1016/j.bmcl.2009.09.120. PMID 19879136.
| Chemical structure of asengeprast (FT011) | |
| Clinical data | |
|---|---|
| Other names | FT011 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1001288-58-9 |
| PubChem CID | 23648966 |
| ChemSpider | 24664633 |
| UNII | C6V7ZU2NPR |
| ChEMBL | ChEMBL1075834 |
| Chemical and physical data | |
| Formula | C20H17NO5 |
| Molar mass | 351.358 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- FT011, a Novel Cardiorenal Protective Drug, Reduces Inflammation, Gliosis and Vascular Injury in Rats with Diabetic RetinopathyPublication Name: PLOS ONEPublication Date: 2015-07-29PMCID: PMC4519240PMID: 26222724DOI: 10.1371/journal.pone.0134392
- A new anti-fibrotic drug attenuates cardiac remodeling and systolic dysfunction following experimental myocardial infarctionPublication Name: International Journal of CardiologyPublication Date: 2013-09-30PMID: 23219315DOI: 10.1016/j.ijcard.2012.11.067
- Attenuation of Armanni–Ebstein lesions in a rat model of diabetes by a new anti-fibrotic, anti-inflammatory agent, FT011Publication Name: DiabetologiaPublication Date: 2012-12-16PMID: 23242170DOI: 10.1007/s00125-012-2805-9
- A Purpose-Synthesised Anti-Fibrotic Agent Attenuates Experimental Kidney Diseases in the RatPublication Name: PLoS ONEPublication Date: 2012-10-10PMCID: PMC3468513PMID: 23071743DOI: 10.1371/journal.pone.0047160
- FT011, a new anti‐fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathyPublication Name: European Journal of Heart FailurePublication Date: 2012-05PMID: 22417655DOI: 10.1093/eurjhf/hfs011
///////////Asengeprast, FT011, FT 011, orphan drug status, systemic sclerosis, SHP-627, SHP 627, C6V7ZU2NPR, Fast Track
Asandeutertinib
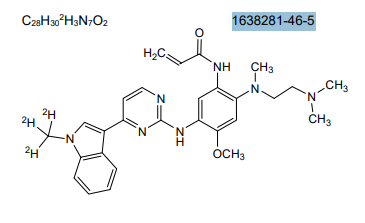
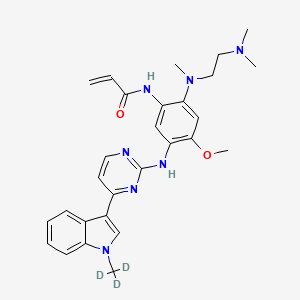
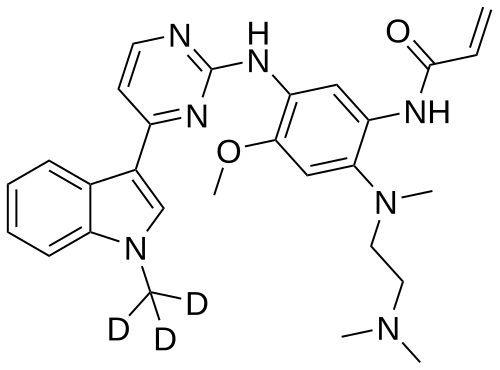
Asandeutertinib, Osimertinib-d3
CAS 1638281-46-5
- 9EKD2E8BM5
- N-(2-(2-(dimethylamino)ethyl-methylamino)-4-methoxy-5-((4-(1-(trideuteriomethyl)indol-3-yl)pyrimidin-2-yl)amino)phenyl)prop-2-enamide
- N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[1-(trideuteriomethyl)indol-3-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide
N-[2-{2-(dimethylamino)ethylamino}-4-methoxy-5-({4-[1-(2H3)methyl-1H-indol-3-yl]pyrimidin-2-
yl}amino)phenyl]prop-2-enamide
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, antineoplastic
MF C28H30. 2H3. N7O2, C28H30D3N7O2 MW 502.6 g/mol
Asandeutertinib is an investigational new drug that is being evaluated for the treatment of cancer. It is an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) with antineoplastic properties.[1][2] Developed by TYK Medicines, this small molecule drug is currently being investigated for the treatment of non-small cell lung cancer (NSCLC), particularly in patients with EGFR mutations.[1][3]
PAT
- 2-(2,4,5-substituted aniline) pyrimidine derivative, pharmaceutical composition and use thereofPublication Number: US-10414756-B2Priority Date: 2014-08-15Grant Date: 2019-09-17
- 2-(2,4,5-substituted aniline) pyrimidine derivative, pharmaceutical composition and use thereofPublication Number: US-2018016258-A1Priority Date: 2014-08-15
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US210080627&_cid=P21-MFT3HT-86141-1
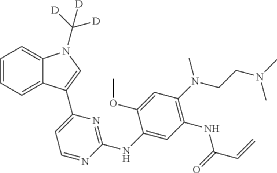
Embodiment 3A
N-(2-{2-dimethylaminoethyl-methylamino}-4-methoxy-5-{[4-(1-(D3-methyl)indol-3-yl)pyrimidin-2-yl]amino}phenyl)-2-acrylamide
| Under ice bath condition, to N 1-(2-dimethylaminoethyl)-5-methoxy-N 1-methyl-N 4-[4-(1-[D 3-methylindol]-3-yl)pyrimidin-2-yl]phenyl-1,2,4-triamine (intermediate 3, 20 g) in THF (200 mL) and water (20 mL), was added 6.9 g NaOH. Acryloyl chloride 4.05 g was added while stirring, the reaction mixture was stirred for 30 min at room temperature, then stirred for 1 h at room temperature. After the result of TLC showed that the reaction was complete, 200 mL water and 20 mL aqueous ammonia were added into the reaction mixture, the solid was precipitated and filtered out. The solid was collected and washed with water, dried for 8 h at 50° C. to deliver the title compound (yield 87%). |
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Asandeutertinib”. PatSnap.
- “Asandeutertinib”. IUPHAR/BPS Guide to PHARMACOLOGY.
- Han B, Zhang W, Wu L, Chen B, Zhao Y, Liu J, et al. (October 2024). “P1. 12A. 07 A Phase 1 Study of TY-9591 in Advanced Non-Small Cell Lung Cancer (NSCLC) Patients with EGFR Positive Mutation”. Journal of Thoracic Oncology. 19 (10): S195. doi:10.1016/j.jtho.2024.09.353.
| Clinical data | |
|---|---|
| Other names | Runnor-9591, TY 9591 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1638281-46-5 |
| PubChem CID | 87056175 |
| IUPHAR/BPS | 13201 |
| ChemSpider | 129431787 |
| UNII | 9EKD2E8BM5 |
| Chemical and physical data | |
| Formula | C28H30D3N7O2 |
| Molar mass | 502.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////////Asandeutertinib, antineoplastic, 9EKD2E8BM5, Osimertinib-d3
Admilparant
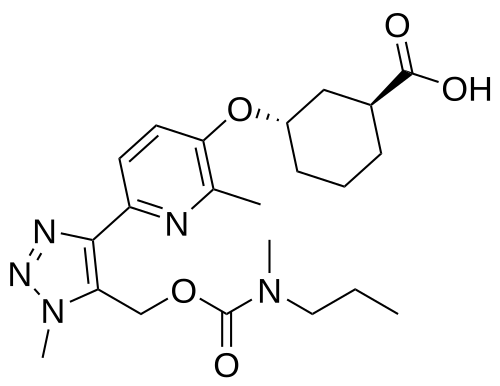
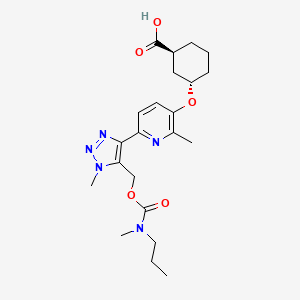
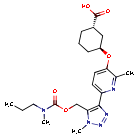
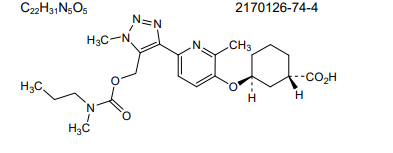
Admilparant, (BMS-986278)
CAS 2170126-74-4
MF C22H31N5O5 MW 445.5 g/mol
(1S,3S)-3-({2-methyl-6-[1-methyl-5-({[methyl(propyl)carbamoyl]oxy}methyl)-1H-1,2,3-triazol-4-l]pyridin-3-yl}oxy)cyclohexane-1-carboxylic acid
lysophosphatidic acid receptor 1 (LPA1) antagonist
- 4UN9AOU6G8
- BMS986278
- (1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic acid
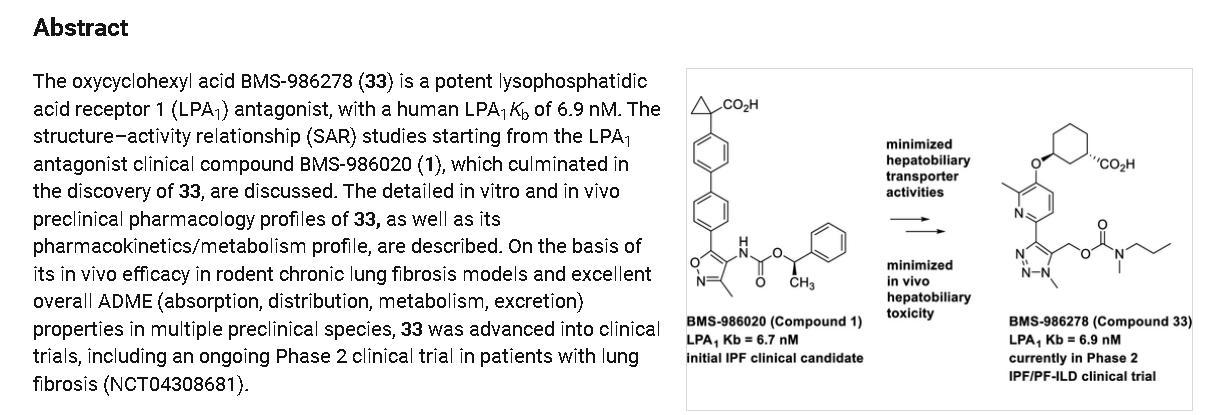
Admilparant is an investigational new drug being developed by Bristol-Myers Squibb for the treatment of idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF). It is a first-in-class lysophosphatidic acid receptor 1 (LPA1) antagonist.[1][2]
As of 2024, admilparant is in Phase III clinical trials for both IPF and PPF.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry, Publication Date: 2021-10-28, PMID: 34709814
DOI: 10.1021/acs.jmedchem.1c01256
(1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)-oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic Acid (33). Compound 33 was prepared using the same
synthetic sequence as 25, except that intermediate 42 was reacted with
N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine. 1H NMR (500 MHz, DMSO-d6, 100 °C) δ 11.99−11.46 (m,1H), 7.82 (d, J = 8.3 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 5.65 (s, 2H),
4.89−4.62 (m, 1H), 4.10 (s, 3H), 3.12 (br t, J = 7.2 Hz, 2H), 2.79 (s,3H), 2.69 (tt, J = 9.4, 4.4 Hz, 1H), 2.44 (s, 3H), 2.03 (dt, J = 13.8, 4.5Hz, 1H), 1.92−1.86 (m, 1H), 1.86−1.79 (m, 2H), 1.74−1.68 (m, 1H),
1.68−1.58 (m, 2H), 1.58−1.51 (m, 1H), 1.43 (dq, J = 14.4, 7.1 Hz,2H), 0.76 (br t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6, 100°C) δ 175.4, 154.7, 150.1, 147.7, 143.9, 141.4, 129.6, 120.0, 118.6, 71.8,
54.5, 49.5, 37.4, 34.4, 33.4, 31.6, 28.7, 27.2, 19.8, 19.4, 18.6, 10.1. m/z446 [M + H]+
. HPLC/UV purity: 99.9% using the following reverse phase chromatographic conditions: Agilent HPLC; Phenomenex Kinetex-C-18; 100 (L) × 4.6 mm2 (i.d.) column; 2.6 μm particle size; wavelength, 220−380 nm; flow rate, 1.0 mL/min; temperature, 35°C; injection volume, 4 μL of 0.25 mg/mL in 1:1 MeCN:H2O; mobilephase A, H2O−0.05% TFA; mobile phase B, MeCN−0.05% TFA; gradient elution, starting at 10−80% B over 10 min and ending at 95% Bafter an additional 4 min; retention time = 8.28 min. Stereoisomeric purity was >99.5% using the following chiral chromatographic conditions: UPC2 Analytical SFC, ChromegaChiral CC4; 250 (L) ×4.6 mm2 (i.d.); 5 μm column; flow rate, 3 mL/min; temperature, 40 °C;injection volume, 10 μL of 0.25 mg/mL in MeCN:MeOH (1:1);mobile phase, 30% MeOH and 70% CO2 at 120 bar retention time =6.05 min. Accurate mass, [M + H]+ at m/z = 446.2398 (−2.03 ppmfrom theoretical for C22H32N5O5). [α]20D = +28.24° (MeOH, c = 0.51).
Elem. Anal. (theoretical): C, 59.31; H, 7.01; N, 15.72. Found: C, 59.35;H, 6.78; N, 15.69. UV (MeOH) at 254 nm (ε = 17,856), 290 nm (ε =7,519), and 296 nm (ε = 8,288). Concentration: adjusted for purity,
0.05154840 g/L or 0.0001157047 mol/L. Melting point = 152−154°C. Accurate mass, [M + H]+ at m/z 466.2398 (−2.03 ppm fromtheoretical for C22H32N5O5).
synthetic sequence as 25, except that intermediate 42 was reacted with N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine
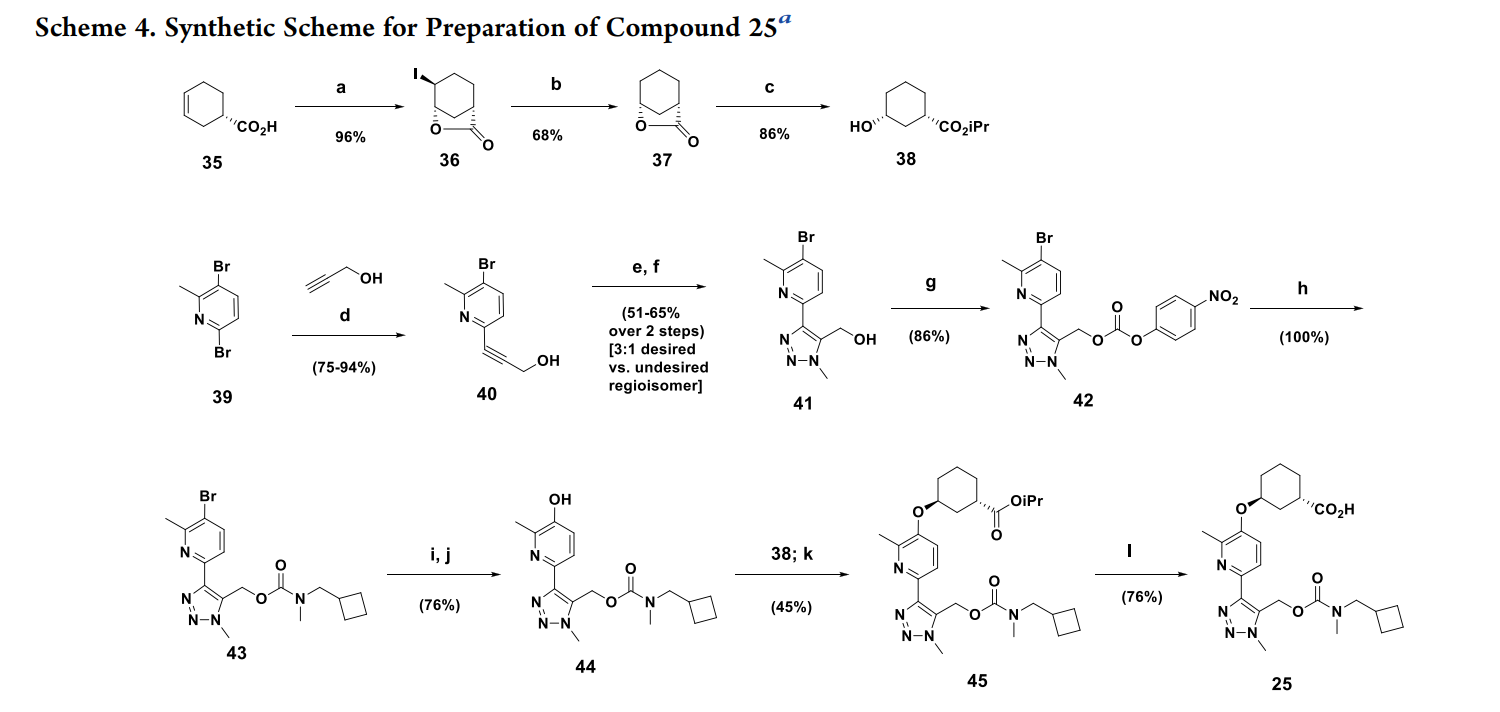
a
Reagents and conditions: (a) I2 (1.1 equiv)/KI (2.5 equiv)/NaHCO3 (3 equiv)/water (96%); (b) H2 (50 psi)/ Pd/C (cat)/Et3N (2 equiv)/EtOAc (68%); (c) CH3COCl (2.5 equiv)/iPrOH (87−95%); d) (Ph3P)2PdCl2 (5%)/ Et3N/CuI (5%)/RT (75−94%); (e) Ru(II)-(Ph3P)2(Me5Cyp)Cl (5%)/TMSCH2N3/dioxane 50 °C/15 h; (f) Bu4NF/0 °C to RT (51−65% over 2 steps; 3:1 desired:undesired regioisomer); (g) 4-nitrophenyl chloroformate/pyridine/CH2Cl2 (86%); (h) N-cyclobutyl N-methylamine/iPr2NEt/CH2Cl2 (100%); (i) B2(pin)2/KOAc/PdCl2(dppf)/THF/80 °C; (j) NaH2BO4/H2O/RT (76% over 2 steps); (k) 38; 1,1′-(azodicarbonyl)dipiperidine/Bu3P/toluene/50 °C (45%); (l)LiOH/H2O/MeOH (76%).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US208146892&_cid=P20-MFS2PF-83792-1
PATENT
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2022249443-A1Priority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: US-RE49352-EPriority Date: 2016-06-21Grant Date: 2023-01-03
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: AU-2021209334-B2Priority Date: 2016-06-21Grant Date: 2023-06-01
- Carbamoyloxymethyltriazole cyclohexylates as LPA antagonistsPublication Number: JP-7312295-B2Priority Date: 2016-06-21Grant Date: 2023-07-20
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2023390249-A1Priority Date: 2016-06-21
- Carbamoyloxymethyltriazolylcyclohexanes as LPA antagonistsPublication Number: CN-109963843-BPriority Date: 2016-06-21Grant Date: 2022-03-11
- Carbamoyloxymethyltriazole cyclohexyl acid as LPA antagonistPublication Number: CN-114601830-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acid as an LPA antagonistPublication Number: KR-102377340-B1Priority Date: 2016-06-21Grant Date: 2022-03-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-20220038537-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-102463621-B1Priority Date: 2016-06-21Grant Date: 2022-11-03
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Admilparant (BMS-986278): Idiopathic Pulmonary Fibrosis Likelihood of Approval”. Pharmaceutical Technology. 25 December 2023. Retrieved 2024-11-23.
- Corte TJ, Behr J, Cottin V, Glassberg MK, Kreuter M, Martinez FJ, et al. (October 2024). “Efficacy and Safety of Admilparant, an LPA1 Antagonist in Pulmonary Fibrosis: A Phase 2 Randomized Clinical Trial”. American Journal of Respiratory and Critical Care Medicine. 211 (2): 230–238. doi:10.1164/rccm.202405-0977OC. PMID 39393084.
- Splete H (16 September 2024). “Admilparant Affects Biomarkers in Pulmonary Fibrosis”. Medscape. Retrieved 2024-11-23.
| Clinical data | |
|---|---|
| Other names | BMS-986278 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2170126-74-4 |
| PubChem CID | 132232205 |
| DrugBank | DB18011 |
| ChemSpider | 115009679 |
| UNII | 4UN9AOU6G8 |
| KEGG | D12657 |
| ChEMBL | ChEMBL5087506 |
| Chemical and physical data | |
| Formula | C22H31N5O5 |
| Molar mass | 445.520 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
/////////Admilparant, BMS 986278, PHASE 3, Bristol-Myers Squibb, idiopathic pulmonary fibrosis, 4UN9AOU6G8
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}