
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Enzomenib
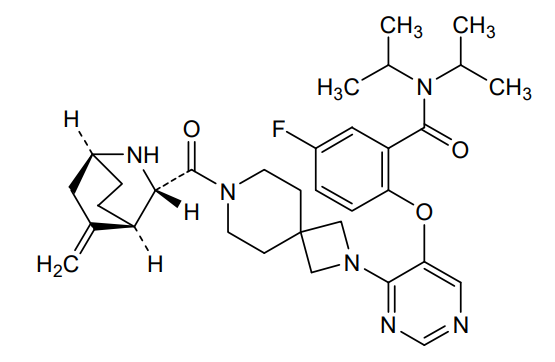
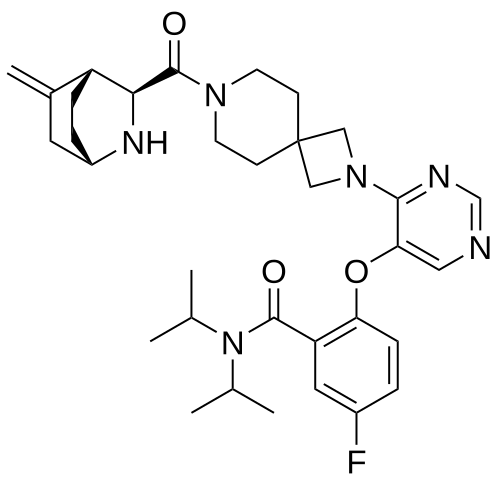
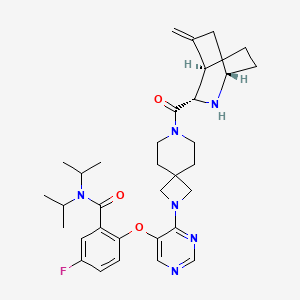
Enzomenib
CAS 2412555-70-3
MF C33H43FN6O3 MW 590.7 g/mol
5-fluoro-2-[4-[7-[(1S,3S,4R)-5-methylidene-2-azabicyclo[2.2.2]octane-3-carbonyl]-2,7-diazaspiro[3.5]nonan-2-yl]pyrimidin-5-yl]oxy-N,N-di(propan-2-yl)benzamide
5-fluoro-2-[(4-{7-[(1S,3S,4R)-5-methylidene-2-azabicyclo[2.2.2]octane-3-carbonyl]-2,7-
diazaspiro[3.5]nonan-2-yl}pyrimidin-5-yl)oxy]-N,Ndi(propan-2-yl)benzamide
menin-MLL (mixed-lineage leukemia) protein, interaction inhibitor, antineoplastic, DSP-5336, Fast Track, Orphan Drug designations
Enzomenib is an investigational new drug that is being evaluated for the treatment of acute leukemia.[1] It is a small molecule inhibitor that targets the interaction between menin and mixed-lineage leukemia (MLL) proteins.[2] Enzomenib particularly in patients with KMT2A (MLL) rearrangements or NPM1 mutations.[3]
The U.S. Food and Drug Administration (FDA) has granted both Fast Track and Orphan Drug designations to Enzomenib.[4]
Enzomenib is an orally bioavailable, small molecule inhibitor of menin, with potential antineoplastic activity. Upon oral administration, enzomenib targets and binds to the nuclear protein menin, thereby preventing the interaction between the two proteins menin and menin-mixed lineage leukemia (MLL; myeloid/lymphoid leukemia; KMT2A) and the formation of the menin-MLL complex. This reduces the expression of downstream target genes and results in an inhibition of the proliferation of MLL-rearranged leukemic cells. The menin-MLL complex plays a key role in the survival, growth, transformation and proliferation of certain kinds of leukemia cells.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US295244745&_cid=P21-MGISYZ-31333-1
Example 3 to 19
| The following compounds of Examples 3 to 19 were prepared according to a similar method to Example 1 by using each corresponding starting compound. |

PAT
Optically active azabicyclo derivatives
Publication Number: JP-7614262-B2
Priority Date: 2018-08-27
Grant Date: 2025-01-15
- Optically active azabicyclo derivativesPublication Number: CN-112585140-BPriority Date: 2018-08-27Grant Date: 2023-07-04
- Optically active azabicyclo ring derivativePublication Number: JP-2023134729-APriority Date: 2018-08-27
- Chiral azabicyclyl compound derivativePublication Number: TW-I815954-BPriority Date: 2018-08-27Grant Date: 2023-09-21
- Optically active azabicyclo ring derivativePublication Number: US-11911381-B2Priority Date: 2018-08-27Grant Date: 2024-02-27
- Optically active azabicyclo ring derivativePublication Number: US-2024148727-A1Priority Date: 2018-08-27
- Optically active azabicyclic derivativePublication Number: AU-2019327006-A1Priority Date: 2018-08-27
- Optically active azabicyclic derivativePublication Number: EP-3845533-A1Priority Date: 2018-08-27
- Optically active azabicyclo ring derivativePublication Number: US-2021338668-A1Priority Date: 2018-08-27
- Optically active azabicyclo ring derivativePublication Number: US-11369605-B2Priority Date: 2018-08-27Grant Date: 2022-06-28
- Optically active azabicyclo ring derivativePublication Number: US-2022288072-A1Priority Date: 2018-08-27
- Optically active azabicyclo ring derivativePublication Number: US-2020157114-A1Priority Date: 2018-08-27
- Optically active azabicyclic derivativePublication Number: WO-2020045334-A1Priority Date: 2018-08-27
- Optically active azabicyclo ring derivativesPublication Number: JP-2020105191-APriority Date: 2018-08-27
- Chiral azabicyclyl compound derivativePublication Number: TW-202024082-APriority Date: 2018-08-27
- Optically active azabicyclo ring derivativePublication Number: US-10815241-B2Priority Date: 2018-08-27Grant Date: 2020-10-27



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Enzomenib – Sumitomo Pharma”. AdisInsight. Springer Nature Switzerland AG.
- Dempke WC, Desole M, Chiusolo P, Sica S, Schmidt-Hieber M (September 2023). “Targeting the undruggable: menin inhibitors ante portas”. Journal of Cancer Research and Clinical Oncology. 149 (11): 9451–9459. doi:10.1007/s00432-023-04752-9. PMC 11798168. PMID 37103568.
- “Sumitomo Pharma Presents New Clinical Data on DSP-5336 at the European Hematology Association 2024 Congress”. Sumitomo Pharma Co., Ltd. 14 June 2024.
- Flaherty C (15 July 2024). “FDA Grants Fast Track Designation to DSP-5336 in KMT2A/NMP1+ AML”. OncLive.
| Clinical data | |
|---|---|
| Other names | DSP-5336 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2412555-70-3 |
| PubChem CID | 146430058 |
| DrugBank | DB18514 |
| ChemSpider | 129534736 |
| UNII | VW83Y2JLZ5 |
| ChEMBL | ChEMBL5314915 |
| Chemical and physical data | |
| Formula | C33H43FN6O3 |
| Molar mass | 590.744 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////enzomenib, Interaction inhibitor, antineoplastic, DSP 5336, Fast Track, Orphan Drug designations
Envudeucitinib
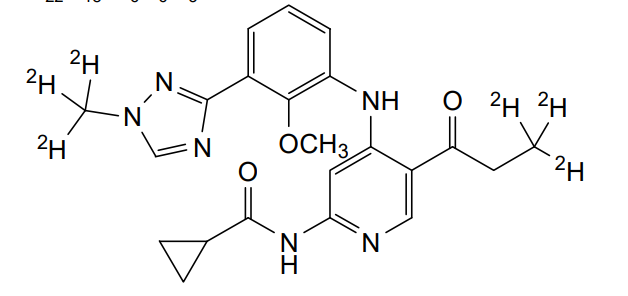
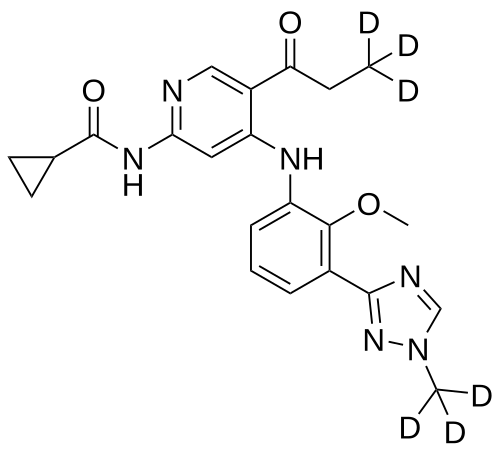
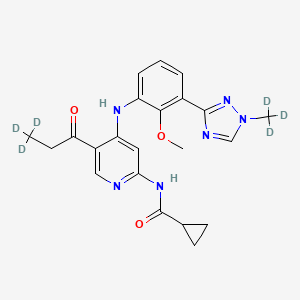
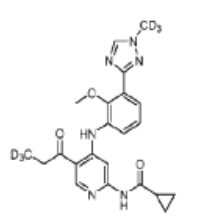
Envudeucitinib
CAS 2417135-66-9
MF C22H18[2]H6N6O3 MW426.5 g/mol
N-[4-{2-methoxy-3-[1-(2H3)methyl-1H-1,2,4-triazol-3-yl]anilino}-5-(3,3,3-2H3)propanoylpyridin-2-yl] cyclopropanecarboxamide
N-(4-(2-methoxy-3-(1-(trideuteriomethyl)-1,2,4-triazol-3-yl)anilino)-5-(3,3,3-trideuteriopropanoyl)pyridin-2-yl)cyclopropanecarboxamide
N-[4-[2-methoxy-3-[1-(trideuteriomethyl)-1,2,4-triazol-3-yl]anilino]-5-(3,3,3-trideuteriopropanoyl)pyridin-2-yl]cyclopropanecarboxamide
Janus kinase inhibitor, anti-inflammatory, Fronthera U.S. Pharmaceuticals, psoriasis, FTP 637
Envudeucitinib is an investigational new drug that is being evaluated for the treatment of psoriasis. It is a selective tyrosine kinase 2 (TYK2) inhibitor developed by Fronthera U.S. Pharmaceuticals LLC and now owned by Alumis, Inc. for the treatment of autoimmune diseases. Envudeucitinib targets the TYK2 signaling pathway, which plays a crucial role in regulating multiple pro-inflammatory cytokines such as IL-12, IL-23, and type I interferons.[1][2]
PAT
- Crystalline forms of a tyk2 inhibitor and uses thereofPublication Number: WO-2024081603-A1Priority Date: 2022-10-10
- Crystalline forms of a tyk2 inhibitor and uses thereofPublication Number: WO-2024059529-A1Priority Date: 2022-09-12
- Tyk2 inhibitors and uses thereofPublication Number: WO-2023227946-A1Priority Date: 2022-05-27
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024081603&_cid=P11-MGGDZU-88200-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023227946&_cid=P11-MGGE36-91523-1
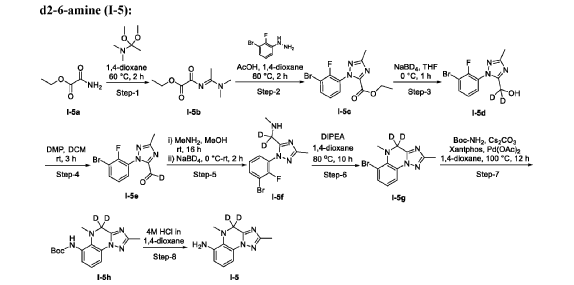
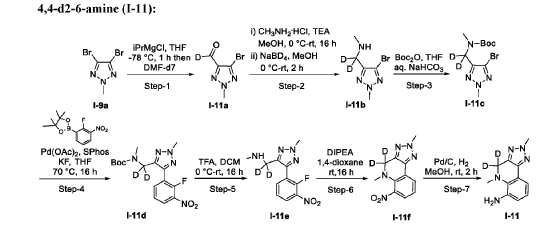
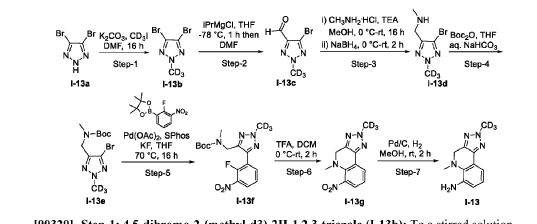
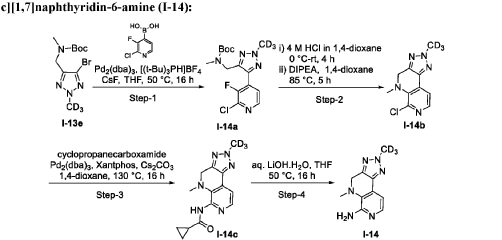
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | FTP-637 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2417135-66-9 |
| PubChem CID | 158715582 |
| IUPHAR/BPS | 13205 |
| UNII | KD2MDJ4GAB |
| KEGG | D13123 |
| Chemical and physical data | |
| Formula | C22H18D6N6O3 |
| Molar mass | 426.506 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Deng L, Wan L, Liao T, Wang L, Wang J, Wu X, et al. (August 2023). “Recent progress on tyrosine kinase 2 JH2 inhibitors”. International Immunopharmacology. 121 110434. doi:10.1016/j.intimp.2023.110434. PMID 37315371.
- Loo WJ, Turchin I, Prajapati VH, Gooderham MJ, Grewal P, Hong CH, et al. (2023). “Clinical Implications of Targeting the JAK-STAT Pathway in Psoriatic Disease: Emphasis on the TYK2 Pathway”. Journal of Cutaneous Medicine and Surgery. 27 (1_suppl): 3S – 24S. doi:10.1177/12034754221141680. PMID 36519621.
////////Envudeucitinib, Janus kinase inhibitor, anti-inflammatory, Fronthera U.S. Pharmaceuticals, psoriasis, FTP 637
Darbinurad
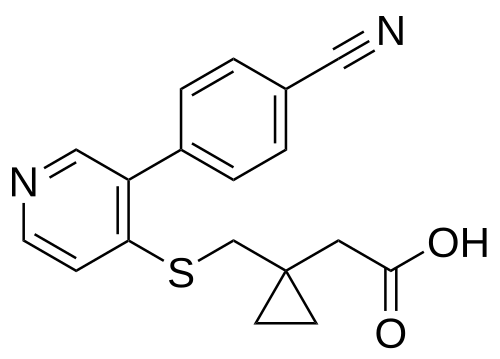
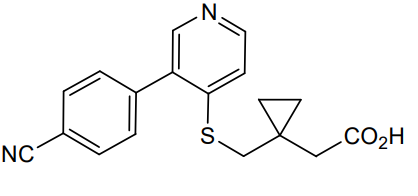
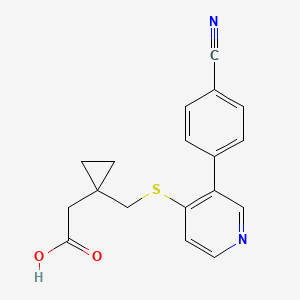
Darbinurad
CAS 1877347-38-0
MF C18H16N2O2S MW 324.4 g/mol
[1-({[3-(4-cyanophenyl)pyridin-4-yl]sulfanyl}methyl)cyclopropyl]acetic
acid
- 1-[[[3-(4-Cyanophenyl)-4-pyridinyl]thio]methyl]cyclopropaneacetic acid
- Cyclopropaneacetic acid, 1-[[[3-(4-cyanophenyl)-4-pyridinyl]thio]methyl]-
2-[1-[[3-(4-cyanophenyl)-4-pyridinyl]sulfanylmethyl]cyclopropyl]acetic acid
urate transporter inhibitor, AYFFM7L5F0
Darbinurad is a investigational new drug that is being evaluated for the treatment of gout. It is a selective urate transporter 1 (URAT1) inhibitor that blocks the reabsorption of uric acid within the renal proximal tubule, thereby reducing serum uric acid concentrations.[1][2]
| Uric acid is the final metabolite of diet and purine in human body. In vivo environment (pH 7.4, 37 degrees), uric acid is present in blood mainly in the form of sodium salt of uric acid, the serum uric acid value of normal people is generally lower than 6 mg/dL. When uric acid in serum exceeds 7 mg/dL (Shi, et al., Nature 2003, 425: 516-523), sodium salt of uric acid will crystallize out and precipitate on joints and other parts of the body, and result in disorders such as gout, urinary stones, kidney stones, etc. Patients with gout are often accompanied with other complications, including hypertension, diabetes, hyperlipidemia, dyslipidemia, atherosclerosis, obesity, metabolic disease, nephropathy, cardiovascular disease, and respiratory disease, etc. (Rock, Et al., Nature Reviews Rheumatology 2013, 9: 13-23). In 2002, Japanese scientists Endou group reported that anion transport channel protein URAT1 is a major protein responsible for reabsorption of uric acid in kidney, they also found that the blood uric acid in people with URAT1 gene mutation (causing the synthesis of such protein being interrupted, inducing nonfunctional proteins) is only one-tenth of that in normal people (Enomoto et. al., Nature 2002 417: 447-452). These findings in human genetics demonstrate that URAT1 anion transport protein in kidney plays very important role in concentration of uric acid in blood, and indicates that URAT1 is a very good and specific target of a drug for reducing blood uric acid. |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US209029213&_cid=P21-MGDFSK-15618-1
Example 12: Synthesis of Compound 20
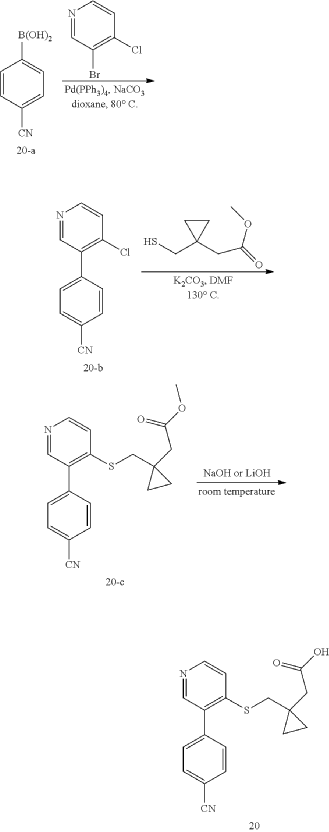
Step 1: Synthesis of 4-(4-chloropyridin-3-yl)benzonitrile (20-b)
Step 2: Synthesis of methyl 2-(1-(((3-(4-cyanophenyl)pyridin-4-yl)thio) methyl)cyclopropyl)acetate (20-c)
Step 3: Synthesis of 2-(1-(((3-(4-cyanophenyl)pyridin-4-yl)thio)methyl) cyclopropyl)acetic acid (20)
PAT
Carboxylic acid compound, method for preparation thereof, and use thereof
Publication Number: KR-102474640-B1, Priority Date: 2014-08-13, Grant Date: 2022-12-05
- Diaryl imidazole compound and pest control agentPublication Number: EP-3181552-B1Priority Date: 2014-08-13Grant Date: 2020-10-21
- Carboxylic acid compound and its preparation method and usePublication Number: CN-106573908-BPriority Date: 2014-08-13Grant Date: 2021-02-05
- Diarylimidazole compound and pest control agentPublication Number: EP-3766871-A1Priority Date: 2014-08-13
- Diarylimidazole compound and harmful organism control agentPublication Number: US-2022000112-A1Priority Date: 2014-08-13
- Carboxylic acid compound, method for preparation thereof, and use thereofPublication Number: EP-3181557-B1Priority Date: 2014-08-13Grant Date: 2023-03-01
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | D-0120 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1877347-38-0 |
| PubChem CID | 118902135 |
| ChemSpider | 128992995 |
| UNII | AYFFM7L5F0 |
| Chemical and physical data | |
| Formula | C18H16N2O2S |
| Molar mass | 324.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Kaufmann D, Chaiyakunapruk N, Schlesinger N (November 2024). “Optimizing gout treatment: A comprehensive review of current and emerging uricosurics”. Joint Bone Spine. 92 (2) 105826. doi:10.1016/j.jbspin.2024.105826. PMID 39622367.
- “Darbinurad”. PatSnap.
/////////Darbinurad
PharmmaEx Mumbai INDIA 3-4 October 2025

Congratulations Pharmmaexians,
We have signed as our Chief Guest Dr Anthony Melvin Crasto Advisor AfricurePharma Row2Tech Glenmark IPCA AdvectProc Niper-G Dept Pharma Min Chem and Fert Govt of India .
Thanks and Regards
Shivam Sharma
PharmmaEx Mumbai
3rd and 4th October 2025
Bombay Exhibition Centre Nesco Goregaon, .Mumbai India
Imlunestrant
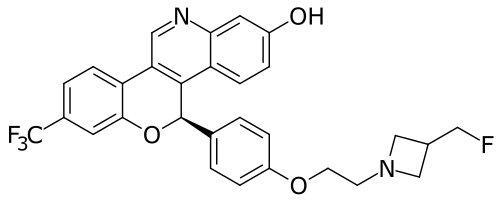
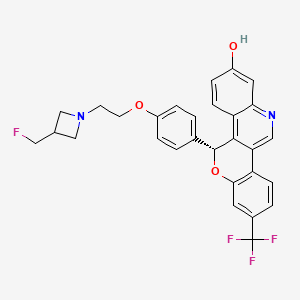

Imlunestrant
CAS 2408840-26-4
as tosylate: 2408840-41-3
(5R)-5-[4-[2-[3-(fluoromethyl)azetidin-1-yl]ethoxy]phenyl]-8-(trifluoromethyl)-5H-chromeno[4,3-c]quinolin-2-ol
- (5r)-5-(4-(2-(3-(fluoromethyl)azetidin-1-yl)ethoxy)phenyl)-8-(trifluoromethyl)-5h-(1)benzopyrano(4,3-c)quinolin-2-ol
- 5h-(1)benzopyrano(4,3-c)quinolin-2-ol, 5-(4-(2-(3-(fluoromethyl)-1-azetidinyl)ethoxy)phenyl)-8-(trifluoromethyl)-, (5r)-
MF C29H24F4N2O3 MW 524.516
FDA 9/25/2025, Inluriyo, LY3484356, LY-3484356, To treat estrogen receptor-positive, human epidermal growth factor receptor 2-negative, estrogen receptor-1-mutated advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy
Imlunestrant, sold under the brand name Inluriyo, is an anti-cancer medication used for the treatment of breast cancer.[1] It is an is an estrogen receptor antagonist.[1] It is used as the salt, imlunestrant tosylate.[2] It is taken by mouth.[1] It was developed by Eli Lilly and Company.[2]
The most common adverse events and laboratory abnormalities include decreased hemoglobin, musculoskeletal pain, decreased calcium, decreased neutrophils, increased AST, fatigue, diarrhea, increased ALT, increased triglycerides, nausea, decreased platelets, constipation, increased cholesterol, and abdominal pain.[2]
Imlunestrant was approved for medical use in the United States in September 2025.[2]
SYN
- Imlunestrant with or without Abemaciclib in Advanced Breast CancerPublication Name: The New England journal of medicinePublication Date: 2025-03-27PMID: 39660834DOI: 10.1056/nejmoa2410858
- Targeting the Estrogen Receptor for the Treatment of Breast Cancer: Recent Advances and ChallengesPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-06-28PMID: 37377342DOI: 10.1021/acs.jmedchem.3c00136
- Novel endocrine therapies: What is next in estrogen receptor positive, HER2 negative breast cancer?Publication Name: Cancer Treatment ReviewsPublication Date: 2023-06PMID: 37146385DOI: 10.1016/j.ctrv.2023.102569
- Oral Selective Estrogen Receptor Degraders (SERDs) as a Novel Breast Cancer Therapy: Present and Future from a Clinical PerspectivePublication Name: International Journal of Molecular SciencesPublication Date: 2021-07-22PMCID: PMC8345926PMID: 34360578DOI: 10.3390/ijms22157812
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US281655517&_cid=P12-MG7DCV-14904-1
Example 1A
5-(4-{2-[3-(Fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol, Isomer 1Separate the two enantiomers of 5-(4-{2-[3-(fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol by chiral SFC with the following conditions: Column: LUX® Cellulose-1, 5×25 cm; eluting with a mobile phase of 30% iPrOH (with 0.5% DMEA) in CO 2; column temperature: 40° C.; flow rate: 300 g/minute; UV detection wavelength: 270 nm to give Example 1A as the first eluting enantiomer (Isomer 1). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 1 by chiral analytical SFC, >99% ee, t (R): 1.30 minutes; column: CHIRALCEL® OD-H, 4.6×150 mm; eluting with a mobile phase of 30% MeOH (0.2% IPA) in CO 2; column temperature: 40° C.; flow rate: 5 mL/minute; UV detection wavelength: 225 nm. Isolate the title compound of Example 1B to give the second eluting enantiomer (Isomer 2). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 2 by chiral analytical SFC, 98% ee, t (R): 2.03 minutes; column: CHIRALCEL® OD-H, 4.6×150 mm; eluting with a mobile phase of 30% MeOH (0.2% IPA) in CO 2; column temperature: 40° C.; flow rate: 5 mL/minute; UV detection wavelength: 225 nm.
Alternate Preparation Example 1B
Crystalline 5-(4-{2-[3-(Fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020014435&_cid=P12-MG7DHN-18354-1
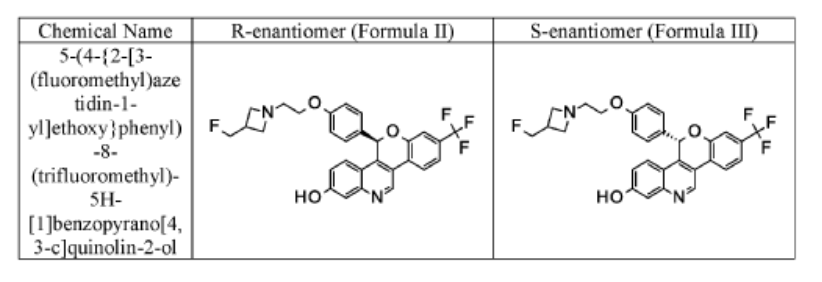
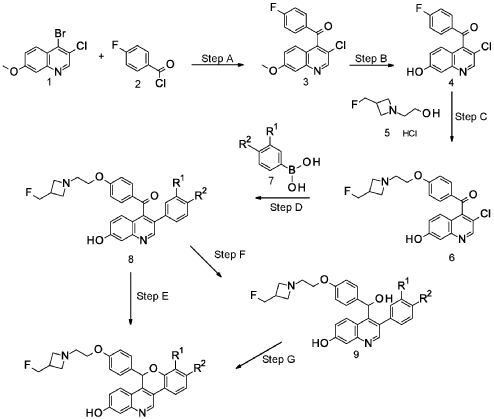
EXAMPLE 1
Racemic 5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [ 1 ]benzopyrano[4,3 -c]quinolin-2-ol
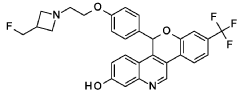
Cool a solution of (4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl){3-[2-fluoro-4-(trifluoromethyl)phenyl]-7-hydroxyquinolin-4-yl}methanone (5.27 g, 9.71 mmol) in 1,4-dioxane (100 mL) to 5 °C. Add lithium triethylborohydride (1 M in THF, 30.0 mL, 30.0 mmol). Remove the cooling bath and stir for 1.5 hours at room temperature. Quench the mixture with water. Add saturated NH4Cl solution and EtOAc. Separate the layers and extract the aqueous layer with EtOAc. Combine the organic extracts, dry over anhydrous MgS04, filter, and concentrate the filtrate. Dissolve the crude residue in THF (100 mL).
Add sodium hydride (60% in mineral oil, 1.94 g, 48.5 mmol). Reflux the solution for 1.5 hours. Add additional sodium hydride (60% in mineral oil, 1.94 g, 48.5 mmol), then reflux for an additional 30 minutes. Cool the solution to room temperature and quench with water. Add EtOAc and saturated NH4Cl solution. Separate the layers and extract the aqueous layer with EtOAc. Combine the organic extract, dry over anhydrous MgS04, filter, and concentrate the filtrate. Purify the residue by silica gel column chromatography eluting with a gradient of 5-7% MeOH in DCM to give the title compound (3.70 g, 72%) as a light yellow foam. ES/MS (m/z): 525.2 (M+H).
Prepare the following compounds in a manner essentially analogous to the method of Example 1, with the following variations in procedure. For the reduction, use 3 to 5 equivalents of lithium triethylborohydride with reaction times from 30 minutes to one hour and drying of the organic layers over magnesium sulfate or sodium sulfate. ETse the crude residue directly or purify by silica gel column chromatography eluting with a gradient of 0-5-7.5-10% MeOH in DCM before cyclization. Complete the cyclization by refluxing in THF for up to 16 hours, or in DMF, from 2 hours at room temperature for Ex 2, to 2 hours at 85 °C for Ex 8. Extract with DCM or EtOAc and dry organic layers over magnesium sulfate or sodium sulfate. Purify by silica gel column chromatography using up to 10% (MeOH or 7 M ammoniated MeOH) in DCM (Ex 2: gradient 0-10% MeOH in DCM; Ex 5: gradient 4-10% 7 M ammoniated MeOH in DCM; Ex 8: gradient 5-7.5% 7 M ammoniated MeOH in DCM) or by high pH reversed phase HPLC as noted.
EXAMPLE 1A
-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 1
and
EXAMPLE 1B
5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
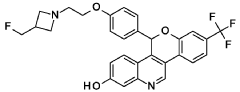
Separate the two enantiomers of 5-(4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[l]benzopyrano[4,3-c]quinolin-2-ol by chiral SFC with the following conditions: Column: LUX® Cellulose-l, 5 x 25 cm; eluting with a mobile phase of 30% iPrOH (with 0.5% DMEA) in C02; column temperature: 40 °C; flow rate: 300 g/minute; UV detection wavelength: 270 nm to give Example 1 A as the first eluting enantiomer (Isomer 1). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 1 by chiral analytical SFC, >99% ee, /(R>: 1.30 minutes; column: CHFRALCEL® OD-H, 4.6 x 150 mm; eluting with a mobile phase of 30% MeOH (0.2% IP A) in C02; column temperature: 40 °C; flow rate: 5 mL/minute; UV detection wavelength: 225 nm. Isolate the title compound of Example 1B to give the second eluting enantiomer (Isomer 2). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 2 by chiral analytical SFC, 98% ee, /(R>: 2.03 minutes; column: CHIRALCEL® OD-H, 4.6 x 150 mm; eluting with a mobile phase of 30% MeOH (0.2% IP A) in C02; column temperature: 40 °C; flow rate: 5 mL/minute; UV detection wavelength: 225 nm.
Alternate Preparation EXAMPLE 1B
Crystalline 5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
Stir 5-(4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[l]benzopyrano[4,3-c]quinolin-2-ol, 4-methylbenzenesulfonic acid, Isomer 2 (23.8 g, 0.034 mol) in water (250 mL) at 1000 rpm. Add NaOH (76 pL) and stir the solution for 2 hours. Add DCM (600 mL). Separate the mixture, dry the DCM extract with magnesium sulfate, filter the material through a syringe filter (0.45 pm), and concentrate to dryness. Allow the material to sit under a N2 stream over a weekend. Add 1 : 1 EtOH/water (80 mL) and stir the mixture with sonication. Collect a tan solid by filtration on a nylon membrane to give the title compound (10.47 g, 0.02 mol, 59%).
PAT
- Selective estrogen receptor degradersPublication Number: US-2023234960-A1Priority Date: 2018-07-12
- Selective estrogen receptor degraderPublication Number: CN-112424205-BPriority Date: 2018-07-12Grant Date: 2023-10-31
- selective estrogen receptor degraderPublication Number: CN-117379428-APriority Date: 2018-07-12
- Selective estrogen receptor degradersPublication Number: US-11993608-B2Priority Date: 2018-07-12Grant Date: 2024-05-28
- Selective estrogen receptor degradersPublication Number: US-12128040-B2Priority Date: 2018-07-12Grant Date: 2024-10-29
PAT
https://patents.google.com/patent/US11926634B2/en
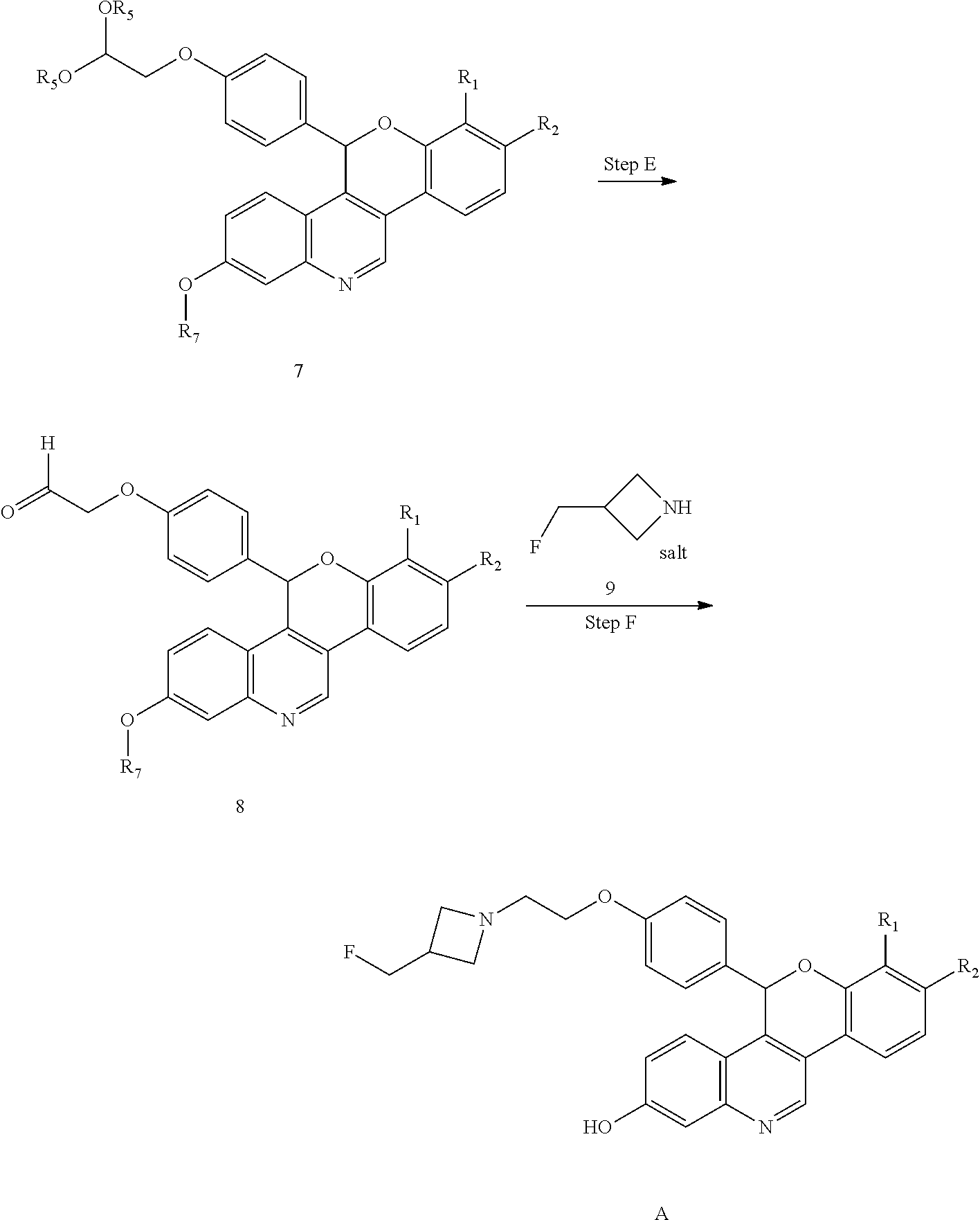
Selective estrogen receptor degraders (SERDs) bind to the estrogen receptor (ER) and downregulate ER-mediated transcriptional activity. The degradation and downregulation caused by SERDs can be useful in the treatment of various proliferative immune mediated disorders, cell proliferation disorders, including cancers such as breast cancer, ovarian cancer, endometrial cancer, prostate cancer, uterine cancer, gastric cancer, and lung cancer as well as mutations due to emerging resistance. Some small molecule examples of SERDs have been disclosed in the literature (see, e.g., WO2005073204, WO2014205136, and WO2016097071). Nonetheless, there is a need for new SERDs to treat ER-positive cancers, such as breast cancer, gastric cancer, and/or lung cancer.
As described in U.S. Pat. No. 10,654,866 (the ‘866 patent) a series of SERDs of the following formula have been discovered, along with pharmaceutically acceptable salts thereof:
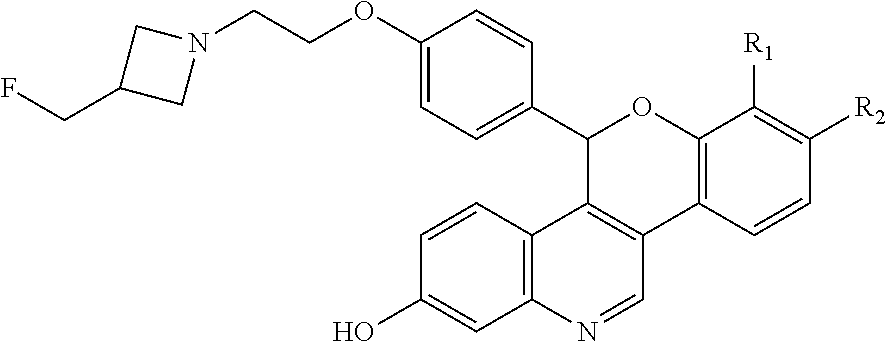
wherein one of R1 and R2 are independently Cl, F, —CF3, or —CH3, and the other is H.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Trade names | Inluriyo |
| Other names | LY3484356, LY-3484356 |
| AHFS/Drugs.com | Inluriyo |
| License data | US DailyMed: Imlunestrant |
| Routes of administration | By mouth |
| Drug class | Estrogen receptor antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2408840-26-4as tosylate: 2408840-41-3 |
| PubChem CID | 146603228 |
| DrugBank | DB19043 |
| ChemSpider | 115010421 |
| UNII | 9CXQ3PF69Uas tosylate: F7UDT90EW5 |
| KEGG | D12216as tosylate: D12217 |
| ChEMBL | ChEMBL5095183 |
| Chemical and physical data | |
| Formula | C29H24F4N2O3 |
| Molar mass | 524.516 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/218881s000lbl.pdf
- “FDA approves imlunestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer”. U.S. Food and Drug Administration (FDA). 25 September 2025. Retrieved 27 September 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “U.S. FDA approves Inluriyo (imlunestrant) for adults with ER+, HER2-, ESR1-mutated advanced or metastatic breast cancer” (Press release). Eli Lilly. 25 September 2025. Retrieved 27 September 2025 – via PR Newswire.
- World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
Further reading
- Jhaveri, Komal L.; Jeselsohn, Rinath; Lim, Elgene; Hamilton, Erika P.; Yonemori, Kan; Beck, J. Thaddeus; et al. (June 2022). “A phase 1a/b trial of imlunestrant (LY3484356), an oral selective estrogen receptor degrader (SERD) in ER-positive (ER+) advanced breast cancer (aBC) and endometrial endometrioid cancer (EEC): Monotherapy results from EMBER”. Journal of Clinical Oncology. 40 (16_suppl): 1021. doi:10.1200/JCO.2022.40.16_suppl.1021. S2CID 249445691.
- Jhaveri, Komal; O’Shaughnessy, Joyce; Andre, Fabrice; Goetz, Matthew P.; Harbeck, Nadia; Martín, Miguel; et al. (March 2023). “Abstract OT1-01-02: EMBER-4: A phase 3 adjuvant trial of imlunestrant vs standard endocrine therapy (ET) in patients with ER+, HER2- early breast cancer (EBC) with an increased risk of recurrence who have previously received 2 to 5 years of adjuvant ET”. Cancer Research. 83 (5_Supplement): OT1–01–02-OT1-01–02. doi:10.1158/1538-7445.SABCS22-OT1-01-02.
- Neven, P.; Stahl, N.; Losada, M.J. Vidal; Jimenez, M. Martin; Kaufman, P.A.; Harbeck, N.; et al. (October 2023). “273P A preoperative window-of-opportunity (WOO) study of imlunestrant in ER+, HER2- early breast cancer (EBC): Final analysis from EMBER-2”. Annals of Oncology. 34: S292 – S293. doi:10.1016/j.annonc.2023.09.470. S2CID 264385454.
External links
- Clinical trial number NCT04975308 for “A Study of Imlunestrant, Investigator’s Choice of Endocrine Therapy, and Imlunestrant Plus Abemaciclib in Participants With ER+, HER2- Advanced Breast Cancer (EMBER-3)” at ClinicalTrials.gov
/////////Imlunestrant, FDA 2025, APPROVALS 2025, Inluriyo, CANCER, LY3484356, LY 3484356, 9CXQ3PF69U
Dapolsertib
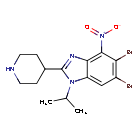
Dapolsertib
CAS 1616359-00-2
MF C15H18Br2N4O MW 446.14 g/mol
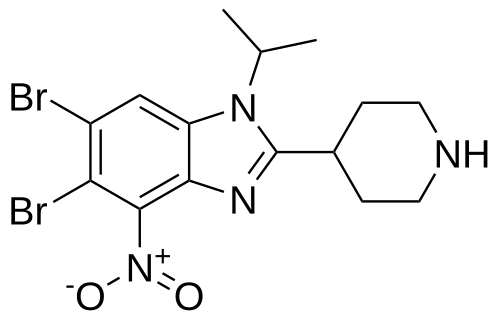
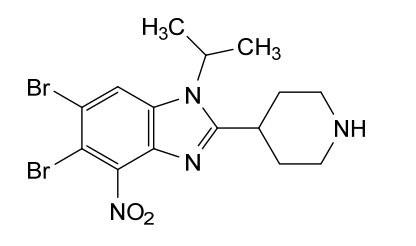
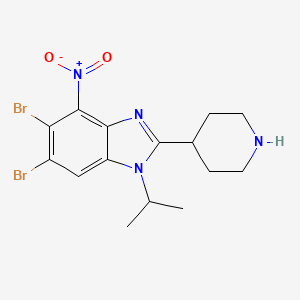
5,6-dibromo-4-nitro-2-piperidin-4-yl-1-propan-2-ylbenzimidazole
5,6-dibromo-4-nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzimidazole
serine/ threonine kinase inhibitor, antineoplastic
Ryvu Therapeutics SA, MEN1703, SEL24-B489
- SEL24-B489
- SEL-24 free base
- 9M7X64VTLI
- SEL-24
Dapolsertib is an investigational new drug that is being evaluated for the treatment of cancer. It is dual inhibitor of PIM family of serine/threonine protein kinases and mutant forms of FMS-related tyrosine kinase 3 (FLT3) that is being developed by Ryvu Therapeutics SA.[1]
Dapolsertib is an orally available inhibitor of PIM family serine/threonine protein kinases and mutant forms of FMS-related tyrosine kinase 3 (FLT3; STK1) with potential antineoplastic activity. Upon oral administration, dapolsertib binds to and inhibits the kinase activities of PIM-1, -2 and -3, and mutant forms of FLT3, which may result in the interruption of the G1/S phase cell cycle transition, an inhibition of cell proliferation, and an induction of apoptosis in tumor cells that overexpress PIMs or express mutant forms of FLT3. FLT3, a tyrosine kinase receptor that is overexpressed or mutated in various cancers, plays a role in signaling pathways that regulate hematopoietic progenitor cell proliferation, and in leukemic cell proliferation and survival. PIM kinases, downstream effectors of many cytokine and growth factor signaling pathways, including the FLT3 signaling pathway, play key roles in cell cycle progression and apoptosis inhibition and may be overexpressed in various malignancies.
- MEN1703 (SEL24) in Participants With Acute Myeloid LeukemiaCTID: NCT03008187Phase: Phase 1/Phase 2Status: CompletedDate: 2025-04-29
- MEN1703 (SEL24) to Treat Relapsed or Refractory Aggressive B-cell Non-Hodgkin Lymphoma (JASPIS-01)CTID: NCT06534437Phase: Phase 2Status: RecruitingDate: 2025-04-11
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014096388&_cid=P12-MG5YKY-59978-1
3.9. Compounds of Example 26:
3.9. Compounds of Example 26:
5,6-dibromo-4-nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzodiazole (Example 26A):
4,5-dibromo-1-N-(propan-2-yl)benzene-1,2-diamine (2,8g, 9,lmmol) and
isonipeconic acid (1,17g, 9,lmmol) were taken up in phosphoric acid (17,82g, 0,18mol). The resulting mixture was stirred at 180°C for 3,5 hours. The mixture was allowed to cool to RT and diluted with water to 200ml. The solution was basified to pH 14.0 using solid NaOH. The resulting precipitate was then filtered off and washed repeatedly with MeOH. The filtrate was concentrated in-vacuo. The product was purified on Al2O3 (basic) using DCM/MeOH/NH3 sat. in MEOH (25: 15: 1). The obtained product (8,7mmol, 3,9g) was dissolved in cone. H2SO4 (30ml). Next KNO3 (8,7mmol, 0,89g) was added in one portion at 0° C. The resulting mixture was stirred at 0°C for 3h and at RT overnight. Then the mixture was poured onto ice. The product was filtered and washed with water.The product was purified on on Al2O3 (basic) using DCM/MeOH/NH3 sat. in MEOH (25: 15: 1) to afford 5,6-dibromo-4- nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzodiazole (1,9g). 1H NMR (600 MHz, DMSO) δ 8.74 (bs, 1H), 8.48 (s, 1H), 8.35 (bs, 1H), 4.94 (hept, J = 6.8 Hz, 1H), 3.52 – 3.46 (m, 1H), 3.42 – 3.37 (m, 2H), 3.08 (bs, 2H), 2.07 – 1.96 (m, 4H), 1.60 (d, J = 6.9 Hz, 6H). m/z 446,8; rt 2,7min.
5,6-dibromo-4-nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzodiazole (Example 26A):
4,5-dibromo-1-N-(propan-2-yl)benzene-1,2-diamine (2,8g, 9,lmmol) and
isonipeconic acid (1,17g, 9,lmmol) were taken up in phosphoric acid (17,82g, 0,18mol). The resulting mixture was stirred at 180°C for 3,5 hours. The mixture was allowed to cool to RT and diluted with water to 200ml. The solution was basified to pH 14.0 using solid NaOH. The resulting precipitate was then filtered off and washed repeatedly with MeOH. The filtrate was concentrated in-vacuo. The product was purified on Al2O3 (basic) using DCM/MeOH/NH3 sat. in MEOH (25: 15: 1). The obtained product (8,7mmol, 3,9g) was dissolved in cone. H2SO4 (30ml). Next KNO3 (8,7mmol, 0,89g) was added in one portion at 0° C. The resulting mixture was stirred at 0°C for 3h and at RT overnight. Then the mixture was poured onto ice. The product was filtered and washed with water.The product was purified on on Al2O3 (basic) using DCM/MeOH/NH3 sat. in MEOH (25: 15: 1) to afford 5,6-dibromo-4- nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzodiazole (1,9g). 1H NMR (600 MHz, DMSO) δ 8.74 (bs, 1H), 8.48 (s, 1H), 8.35 (bs, 1H), 4.94 (hept, J = 6.8 Hz, 1H), 3.52 – 3.46 (m, 1H), 3.42 – 3.37 (m, 2H), 3.08 (bs, 2H), 2.07 – 1.96 (m, 4H), 1.60 (d, J = 6.9 Hz, 6H). m/z 446,8; rt 2,7min.
PAT
Novel benzimidazole derivatives as kinase inhibitors
Publication Number: WO-2014096388-A2
Priority Date: 2012-12-21
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: KR-20150095908-APriority Date: 2012-12-21
- Benzimidazole derivatives as kinase inhibitorsPublication Number: US-10174013-B2Priority Date: 2012-12-21Grant Date: 2019-01-08
- Novel Benzimidazole Derivatives as Kinase InhibitorsPublication Number: US-2015336967-A1Priority Date: 2012-12-21
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: US-2017152249-A1Priority Date: 2012-12-21
- Benzimidazole derivatives as Kinase InhibitorsPublication Number: US-9388192-B2Priority Date: 2012-12-21Grant Date: 2016-07-12
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: EP-2935244-B1Priority Date: 2012-12-21Grant Date: 2018-06-27
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: ES-2688395-T3Priority Date: 2012-12-21Grant Date: 2018-11-02
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: JP-2016503779-APriority Date: 2012-12-21
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: JP-6169185-B2Priority Date: 2012-12-21Grant Date: 2017-07-26
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: KR-101779272-B1Priority Date: 2012-12-21Grant Date: 2017-09-18
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | MEN1703, SEL24-B489 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1616359-00-2 |
| PubChem CID | 76286825 |
| IUPHAR/BPS | 13204 |
| ChemSpider | 81367232 |
| UNII | 9M7X64VTLI |
| ChEMBL | ChEMBL4467168 |
| Chemical and physical data | |
| Formula | C15H18Br2N4O2 |
| Molar mass | 446.143 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Wu M, Li C, Zhu X (December 2018). “FLT3 inhibitors in acute myeloid leukemia”. Journal of Hematology & Oncology. 11 (1) 133. doi:10.1186/s13045-018-0675-4. PMC 6280371. PMID 30514344.
//////////Dapolsertib, antineoplastic, MEN1703, SEL24-B489, MEN 1703, SEL24 B489, Ryvu Therapeutics SA
Crelosidenib
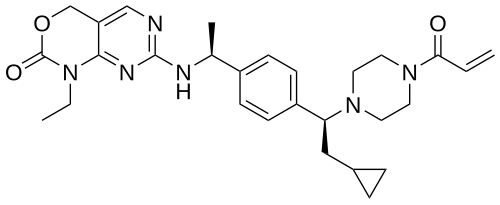
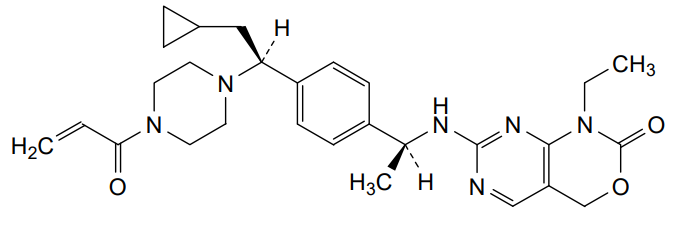
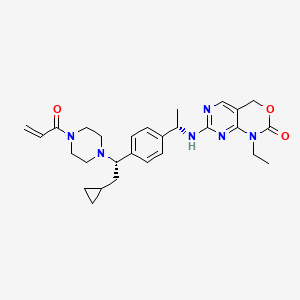
Crelosidenib
CAS 2230263-60-0
7-{[(1S)-1-(4-{(1S)-1-[4-(prop-2-enoyl)piperazin-1-yl]-2-cyclopropylethyl}phenyl)ethyl]amino}-1-ethyl-1,4-dihydro-2Hpyrimido[4,5-d][1,3]oxazin-2-one
isocitrate dehydrogenase 1 (IDH1) inhibitor, antineoplastic
MF C28H36N6O3 MW 504.6 g/mol
- LY3410738
- 7-[[(1S)-1-[4-[(1S)-2-cyclopropyl-1-(4-prop-2-enoylpiperazin-1-yl)ethyl]phenyl]ethyl]amino]-1-ethyl-4H-pyrimido[4,5-d][1,3]oxazin-2-one
- 7-(((1S)-1-(4-((1S)-2-cyclopropyl-1-(4-prop-2-enoylpiperazin-1-yl)ethyl)phenyl)ethyl)amino)-1-ethyl-4H-pyrimido(4,5-d)(1,3)oxazin-2-one
Crelosidenib is an investigational new drug that is being evaluated for the treatment of cancer. It acts as a selective inhibitor of isocitrate dehydrogenase 1 (IDH1), an enzyme that plays a crucial role in cellular metabolism and is frequently mutated in various cancers, including cholangiocarcinoma.[1][2]
Crelosidenib is an orally available inhibitor of mutant form of the isocitrate dehydrogenase type 1 (IDH1; IDH-1; IDH1 [NADP+] soluble), including the substitution mutation at arginine (R) in position 132, IDH1(R132), with potential antineoplastic activity. Upon oral administration, crelosidenib specifically and covalently binds to and modifies a single cysteine (Cys269) in the allosteric binding pocket of mutant forms of IDH1, thereby inactivating IDH1. This inhibits the formation of the oncometabolite 2-hydroxyglutarate (2HG) from alpha-ketoglutarate (a-KG). This depletes 2-HG levels, prevents 2HG-mediated signaling and leads to both an induction of cellular differentiation and an inhibition of cellular proliferation in tumor cells expressing mutant forms of IDH1. In addition, crelosidenib has the ability to cross the blood-brain barrier (BBB). IDH1 mutations, including IDH1(R132) mutations, are highly expressed in certain malignancies, including gliomas; they initiate and drive cancer growth by both blocking cell differentiation and catalyzing the formation of 2HG.
Syn
example 2 [US11001596B2]
https://patentscope.wipo.int/search/en/detail.jsf?docId=US289829390&_cid=P12-MG4UBU-88518-1
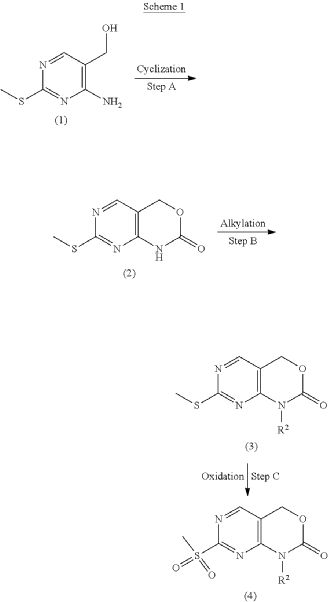
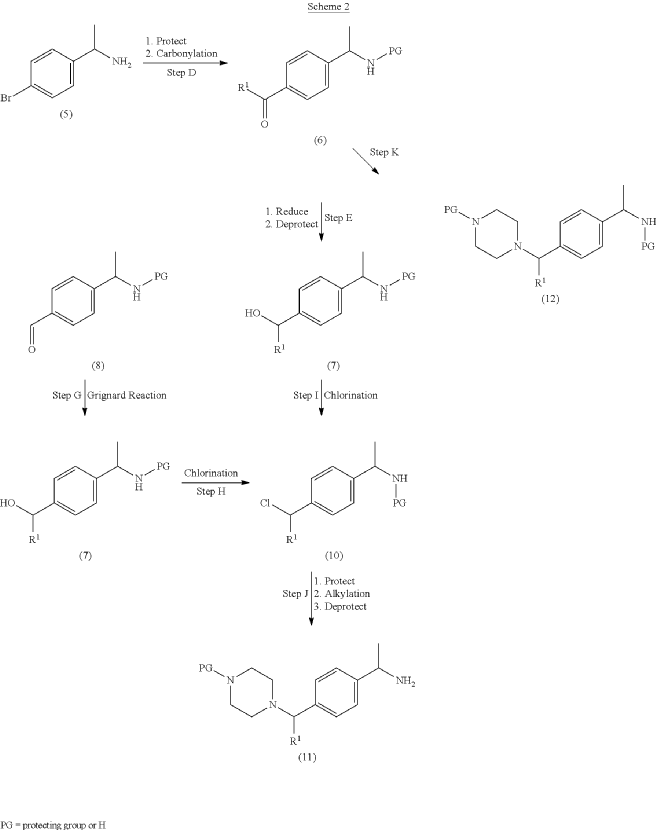
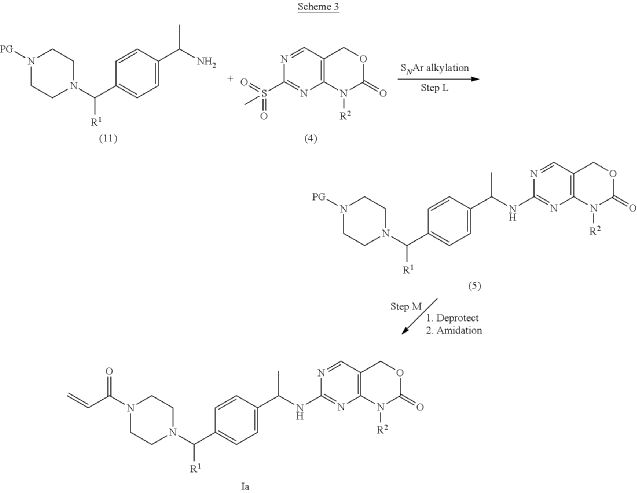
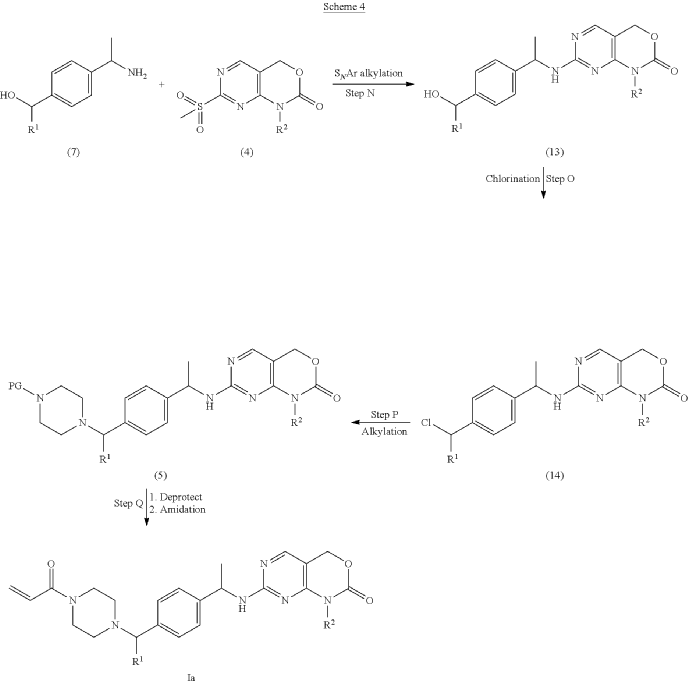

PAT
- 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant idh1 and idh2 inhibitorsPublication Number: US-2021206780-A1Priority Date: 2016-12-16
- 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant idh1 and idh2 inhibitorsPublication Number: US-2021230185-A1Priority Date: 2016-12-16
- 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant idh1 and idh2 inhibitorsPublication Number: CA-3045303-CPriority Date: 2016-12-16Grant Date: 2022-05-17
- 7-phenylethylamino-4H-pyrimido [4,5-D ] [1,3] oxazin-2-one compounds as inhibitors of mutant IDH1 and IDH2Publication Number: CN-110072867-BPriority Date: 2016-12-16Grant Date: 2022-07-08
- Mutant IDH1 and IDH2 inhibitorsPublication Number: CN-115109075-APriority Date: 2016-12-16
- Pyrido[4,3-d][1,3]oxazin-2-one compounds as mutant idh1 and idh2 inhibitorsPublication Number: EP-3763717-B1Priority Date: 2016-12-16Grant Date: 2023-03-08
- 7-phenylethylamino-4H-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitorsPublication Number: US-11629156-B2Priority Date: 2016-12-16Grant Date: 2023-04-18
- 7-phenylethylamino-4H-pyrimido[4,5-d][1,3]oxazin-2-one compounds as mutant IDH1 and IDH2 inhibitorsPublication Number: US-11649247-B2Priority Date: 2016-12-16Grant Date: 2023-05-16
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | LY3410738 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2230263-60-0 |
| PubChem CID | 135125140 |
| IUPHAR/BPS | 12340 |
| ChemSpider | 115009279 |
| UNII | A4DU555RMD |
| KEGG | D12708 |
| Chemical and physical data | |
| Formula | C28H36N6O3 |
| Molar mass | 504.635 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Zarei M, Hue JJ, Hajihassani O, Graor HJ, Katayama ES, Loftus AW, et al. (February 2022). “Clinical development of IDH1 inhibitors for cancer therapy”. Cancer Treatment Reviews. 103 102334. doi:10.1016/j.ctrv.2021.102334. PMID 34974243.
- Demir T, Moloney C, Mahalingam D (July 2024). “Emerging targeted therapies and strategies to overcome resistance in biliary tract cancers”. Critical Reviews in Oncology/Hematology. 199 104388. doi:10.1016/j.critrevonc.2024.104388. PMID 38754771.
.///////////Crelosidenib, Antineoplastic, cholangiocarcinoma, LY3410738, LY 3410738
Copper (64Cu) adarulatide tetraxetan
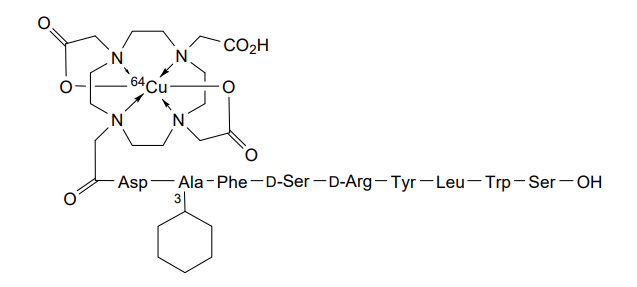
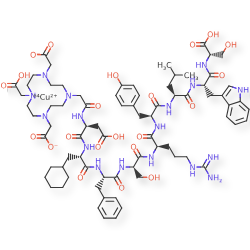
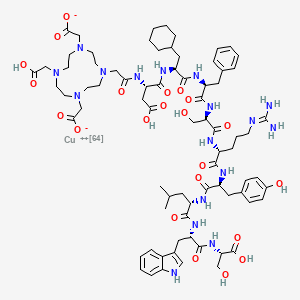
Copper (64Cu) adarulatide tetraxetan
| Adarulatide tetraxetan copper Cu-64 |
CAS 2841388-40-5
MF C76H10764CuN17O22, MF1,674.7
Cuprate(3-)-64Cu, [N-[2-[4,10-bis[(carboxy-κO)methyl]-7-(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl-κN1,κN4,κN7,κN10 ]acetyl]-L-α-aspartyl-3-cyclohexyl-L-alanyl-L-phenylalanyl-D-seryl-D-arginyl-L-tyrosyl-L-leucyl-L-tryptophanyl-L-serinato(5-)]-, hydrogen (1:3)
2-[4-[2-[[(2S)-3-carboxy-1-[[(2S)-1-[[(2S)-1-[[(2R)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(1S)-1-carboxy-2-hydroxyethyl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-cyclohexyl-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-2-oxoethyl]-7-(carboxylatomethyl)-10-(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;copper-64(2+)
diagnostic imaging agent, QM8HMM6RJP
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Copper (64Cu) adarulatide tetraxetan. diagnostic imaging agent, QM8HMM6RJP
Claziprotamide
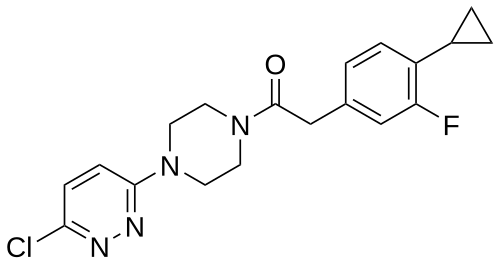
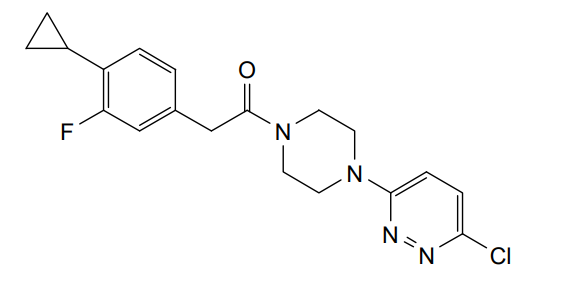
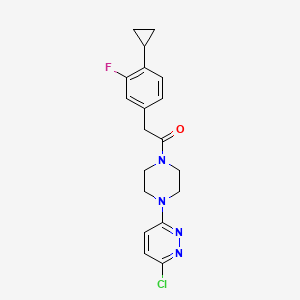
Claziprotamide
CAS 2361124-03-8, BBP 671
MF C19H20ClFN4O MW374.8 g/mol
1-[4-(6-chloropyridazin-3-yl)piperazin-1-yl]-2-(4-cyclopropyl-3-fluorophenyl)ethan-1-one
1-[4-(6-chloropyridazin-3-yl)piperazin-1-yl]-2-(4-cyclopropyl-3-fluorophenyl)ethan-1-one
pantothenate kinases 1 and 3 (PanK1 and PanK3) positive allosteric modulator
Claziprotamide is an investigational new drug that is being evaluated for the treatment of rare metabolic disorders such as pantothenate kinase-associated neurodegeneration (PKAN) and neurodegeneration with brain iron accumulation (NBIA). It acts as a positive allosteric modulator (PAM) of pantothenate kinases 1 and 3 (PANK1 and PANK2) which are critical for coenzyme A biosynthesis and cellular metabolism.[1][2]
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US319558593&_cid=P22-MG32VL-67777-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019133635&_cid=P22-MG32PO-63930-1
SCHEME 4B.
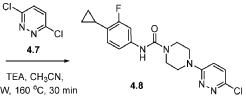
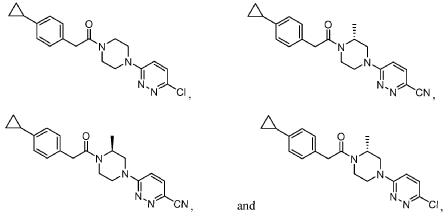
[00184] In one aspect, compounds of type 4.8, and similar compounds, can be prepared according to reaction Scheme 4B above. Thus, compounds of type 4.6 can be prepared by a urea bond formation reaction between an appropriate amine, e.g., 4.2 as shown above, and an appropriate isocyanate, e.g., 4.5 as shown above. Appropriate amines and appropriate isocyanates are commercially available or prepared by methods known to one skilled in the art. The nucleophilic substitution is carried out in the presence of an appropriate solvent, e.g., diethyl ether, for an appropriate period of time, e.g., 3 hours. The nucleophilic substitution is followed by a deprotection reaction. The deprotection reaction is carried out in the presence of an appropriate deprotecting agent, e.g., trifluoroacetic acid, in an appropriate solvent, e.g., dichloromethane, for an appropriate period of time, e.g., 1 hour. Compounds of type 4.8 can be prepared by an arylation reaction of appropriate amine, e.g., 4.6 as shown above, and an appropriate aryl halide, e.g., 4.7 as shown above. Appropriate aryl halides are commercially available or prepared by methods known to one skilled in the art. The arylation reaction is carried out in the presence of an appropriate base, e.g., triethylamine, in an appropriate solvent, e.g., acetonitrile, at an appropriate temperature, e.g, 160 °C, for an appropriate period of time, e.g., 30 minutes using microwave irradiations. As can be appreciated by one skilled in the art, the above reaction provides an example of a generalized approach wherein compounds similar in structure to the specific reactants above (compounds similar to compounds of type 3.6, 4.1, 4.2, and 4.3), can be substituted in the reaction to provide 4-aryl-N-phenylpiperazine-l -carboxamide derivatives similar to Formula 4.4.
PAT
- Methods of treating disorders associated with castorPublication Number: US-2023321092-A1Priority Date: 2017-12-27
- Small molecule modulators of pantothenate kinasesPublication Number: US-11891378-B2Priority Date: 2017-12-27Grant Date: 2024-02-06
- Small molecule modulators of pantothenate kinasesPublication Number: US-2024287039-A1Priority Date: 2017-12-27
- Small molecule modulator of pantothenate kinasePublication Number: KR-102728619-B1Priority Date: 2017-12-27Grant Date: 2024-11-08
- Small molecule modulators of pantothenate kinasesPublication Number: US-2021061788-A1Priority Date: 2017-12-27
- Methods of treating disorders associated with castorPublication Number: US-11547709-B2Priority Date: 2017-12-27Grant Date: 2023-01-10
- Small molecule modulators of pantothenate kinasesPublication Number: AU-2018395222-B2Priority Date: 2017-12-27Grant Date: 2023-06-08
- Small molecule modulators of pantothenate kinasePublication Number: CN-111818922-BPriority Date: 2017-12-27Grant Date: 2023-06-13
- Small molecule modulators of pantothenate kinasePublication Number: JP-7352565-B2Priority Date: 2017-12-27Grant Date: 2023-09-28
- Small molecule modulators of pantothenate kinasesPublication Number: EP-3731844-A1Priority Date: 2017-12-27
- Small molecule modulators of pantothenate kinasePublication Number: KR-20200130242-APriority Date: 2017-12-27
- MODULATORS OF SMALL MOLECULES OF PANTOTENATE KINASESPublication Number: BR-112020012875-A2Priority Date: 2017-12-27
- Small molecule modulator of pantothenate kinasePublication Number: JP-2021508739-APriority Date: 2017-12-27
- Methods of treating disorders associated with castorPublication Number: US-2021023081-A1Priority Date: 2017-12-27
- Treatment of organic acidemias or pantothenate kinase associated neurodegeneration with modulators of pantothenate kinasesPublication Number: WO-2023230560-A1Priority Date: 2022-05-26
- Methods of treating disorders associated with castorPublication Number: WO-2022133034-A1Priority Date: 2020-12-16
- Small molecule modulators of pantothenate kinasesPublication Number: WO-2019133635-A1Priority Date: 2017-12-27
- Small molecule modulators of pantothenate kinasesPublication Number: AU-2018395222-A1Priority Date: 2017-12-27
- Small molecule modulators of pantothenate kinasePublication Number: CN-111818922-APriority Date: 2017-12-27
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | BBP-671 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2361124-03-8 |
| PubChem CID | 142616838 |
| ChemSpider | 129431674 |
| UNII | 74N47PKZ3K |
| PDB ligand | Y92 (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C19H20ClFN4O |
| Molar mass | 374.84 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Tangallapally R, Subramanian C, Yun MK, Edwards A, Sharma LK, Yang L, et al. (August 2024). “Development of Brain Penetrant Pyridazine Pantothenate Kinase Activators”. Journal of Medicinal Chemistry. 67 (16): 14432–14442. doi:10.1021/acs.jmedchem.4c01211. PMC 11345825. PMID 39136313.
- “Claziprotamide”. PatSnap.
/////////Claziprotamide, BBP 671, ORPHAN DRUG
Camibirstat
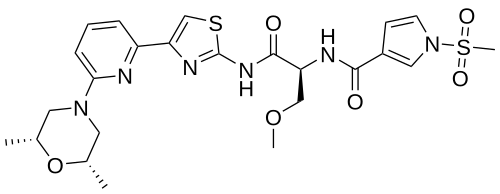
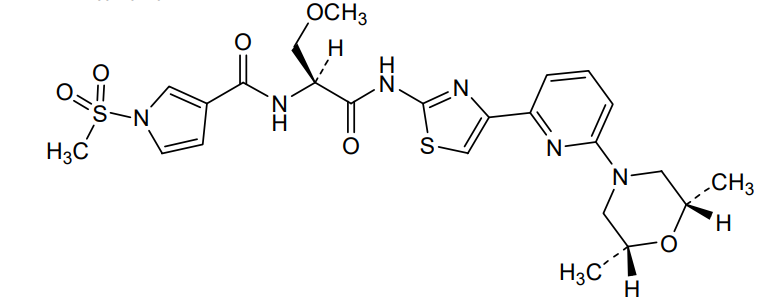
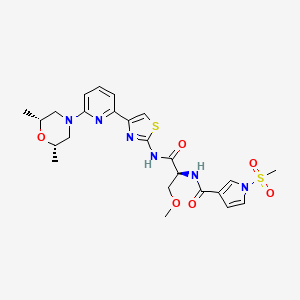
Camibirstat
CAS 2671128-05-3
N-{(2S)-1-[(4-{6-[(2R,6S)-2,6-dimethylmorpholin-4-yl]pyridin-2-yl}-1,3-thiazol-2-yl)amino]-3-methoxy-1-oxopropan-2-yl}-1-(methanesulfonyl)-1H-pyrrole-3-carboxamide
ATPase inhibitor, antineoplastic
MW C24H30N6O6S2 MF 562.7 g/mol
- 1H-Pyrrole-3-carboxamide, N-((1S)-2-((4-(6-((2R,6S)-2,6-dimethyl-4-morpholinyl)-2-pyridinyl)-2-thiazolyl)amino)-1-(methoxymethyl)-2-oxoethyl)-1-(methylsulfonyl)-
- N-[(2S)-1-[[4-[6-[(2S,6R)-2,6-dimethylmorpholin-4-yl]pyridin-2-yl]-1,3-thiazol-2-yl]amino]-3-methoxy-1-oxopropan-2-yl]-1-methylsulfonylpyrrole-3-carboxamide
- FHD 286
Camibirstat is an investigational new drug that is being evaluated for the treatment of cancer. It is a small molecule that acts as a selective inhibitor of SMARCA2 and SMARCA4, which are key components of the SWI/SNF chromatin remodeling complex.[1]
It is being developed by Foghorn Therapeutics.[2]
Camibirstat is an orally bioavailable, allosteric, small molecule inhibitor of transcription activator BRG1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 4; SMARCA4) and BRM (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2; SMARCA2), with potential antineoplastic activity. Upon oral administration, camibirstat targets, binds to, and inhibits the activity of BRG1 and/or BRM, the primary ATPase components and mutually exclusive subunits of the BRG1/BRM-associated factor (BAF) complexes. This may lead to the inhibition of the SWI/SNF chromatin remodeling complex, disrupt chromatin remodeling and gene expression, and result in the downregulation of oncogenic pathways and the inhibition of tumor cell proliferation. BAF is an important regulator of transcriptional programs and gene expression. Mutations in BAF or its transcription factor partners are found in certain diseases including cancers.
PAT
- Compounds and uses thereofPublication Number: US-2024101550-A1Priority Date: 2020-01-29
- Compounds and their usesPublication Number: JP-7561195-B2Priority Date: 2020-01-29Grant Date: 2024-10-03
- Compounds and uses thereofPublication Number: TW-I859406-BPriority Date: 2020-01-29Grant Date: 2024-10-21
- Compounds and uses thereofPublication Number: IL-295100-APriority Date: 2020-01-29
- Compounds and uses thereofPublication Number: KR-20220133258-APriority Date: 2020-01-29
- Compounds and uses thereofPublication Number: US-11485732-B2Priority Date: 2020-01-29Grant Date: 2022-11-01
- Compounds and uses thereofPublication Number: JP-2023512039-APriority Date: 2020-01-29
- Compounds and uses thereofPublication Number: US-2023129003-A1Priority Date: 2020-01-29
- Compounds and uses thereofPublication Number: WO-2021155262-A1Priority Date: 2020-01-29
- Compounds and uses thereofPublication Number: TW-202136252-APriority Date: 2020-01-29
- Compounds and uses thereofPublication Number: AU-2021213811-A1Priority Date: 2020-01-29
- Compound and use thereofPublication Number: CN-115023226-APriority Date: 2020-01-29
- Compounds and uses thereofPublication Number: EP-4096664-A1Priority Date: 2020-01-29
- Methods of treating cancersPublication Number: WO-2021236080-A1Priority Date: 2020-05-20
- Methods of treating cancersPublication Number: EP-4153176-A1Priority Date: 2020-05-20
- ways to treat cancerPublication Number: CN-115867314-APriority Date: 2020-05-20
- how to treat cancerPublication Number: JP-2023535124-APriority Date: 2020-05-20
- Compounds and uses thereofPublication Number: US-2021230154-A1Priority Date: 2020-01-29
- Treatment options with inhibitors of BRG1 and BRM enzyme activityPublication Number: CN-117337179-APriority Date: 2021-03-19
- Therapeutic regimens of an inhibitor of the enzymatic activity of brg1 and brmPublication Number: EP-4308124-A1Priority Date: 2021-03-19
- Treatment regimens for inhibitors of BRG1 and BRM enzymatic activityPublication Number: JP-2024511383-APriority Date: 2021-03-19
- Therapeutic regimens of an inhibitor of the enzymatic activity of brg1 and brmPublication Number: US-2024189318-A1Priority Date: 2021-03-19
- Methods of treating cancersPublication Number: US-2022079940-A1Priority Date: 2020-05-20
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US331910582&_cid=P11-MG1TKU-39131-1
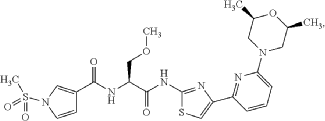
Example 1. Preparation of N—((S)-1-((4-(6-(cis-2,6-dimethylmorpholino)pyridin-2-yl)thiazol-2-yl)amino)-3-methoxy-1-oxopropan-2-yl)-1-(methylsulfonyl)-1H-pyrrole-3-carboxamide
| N—((S)-1-((4-(6-(cis-2,6-dimethylmorpholino)pyridin-2-yl)thiazol-2-yl)amino)-3-methoxy-1-oxopropan-2-yl)-1-(methylsulfonyl)-1H-pyrrole-3-carboxamide was synthesized as shown in Scheme 1 below. |
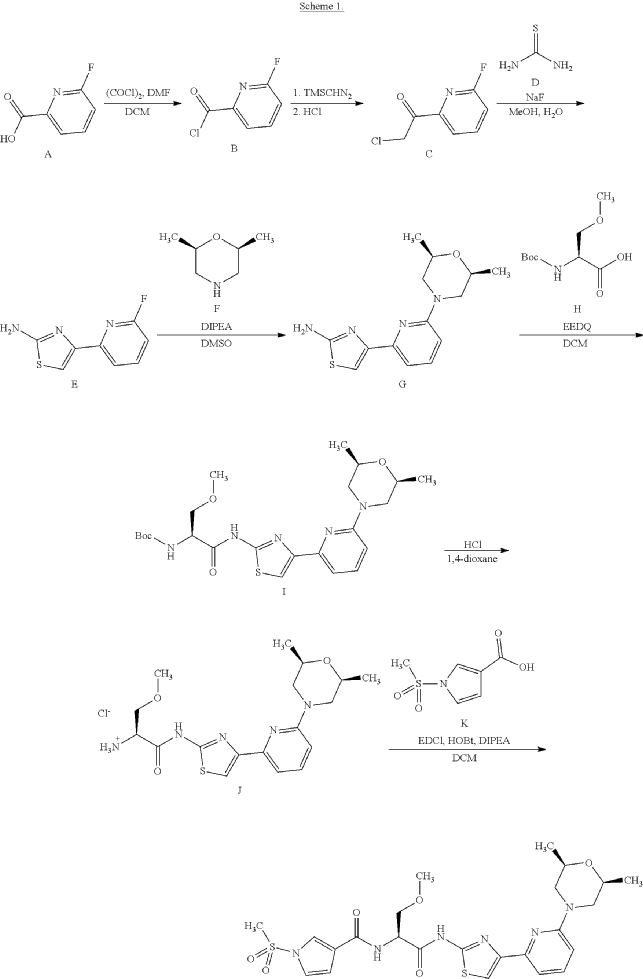
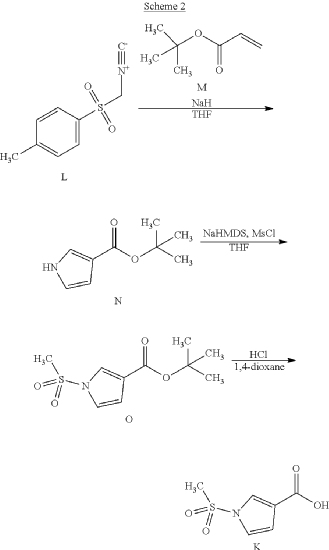
Step 7: Preparation of N—((S)-1-((4-(6-(cis-2,6-dimethylmorpholino)pyridin-2-yl)thiazol-2-yl)amino)-3-methoxy-1-oxopropan-2-O-1-(methylsulfonyl)-1H-pyrrole-3-carboxamide
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | FHD286 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2671128-05-3 |
| PubChem CID | 156818030 |
| ChemSpider | 115010237 |
| UNII | QHA5XLA4SA |
| ChEMBL | ChEMBL5095181 |
| Chemical and physical data | |
| Formula | C24H30N6O6S2 |
| Molar mass | 562.66 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Yu L, Wu D (July 2024). “SMARCA2 and SMARCA4 Participate in DNA Damage Repair”. Frontiers in Bioscience (Landmark Edition). 29 (7): 262. doi:10.31083/j.fbl2907262. PMID 39082357.
- “Camibirstat”. PatSnap.
/////////////Camibirstat, ATPase inhibitor, antineoplastic, Foghorn Therapeutics, FHD 286
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}