
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO









 PM, MODI
PM, MODI


 Photos: India’s first Mars orbiter
Photos: India’s first Mars orbiter India launches mission to Mars
India launches mission to Mars











Abstract
As a part of our continued efforts to discover new COX inhibitors, a series of 3-methyl-1-phenylchromeno[4,3-c]pyrazol-4(1H)-ones were synthesized and evaluated for in vitro COX inhibitory potential. Within this series, seven compounds (3a–d, 3h, 3k and 3q) were identified as potential and selective COX-2 inhibitors (COX-2 IC50’s in 1.79–4.35 μM range; COX-2 selectivity index (SI) = 6.8–16.7 range). Compound 3b emerged as most potent (COX-2 IC50 = 1.79 μM; COX-1 IC50 >30 μM) and selective COX-2 inhibitor (SI >16.7). Further, compound 3b displayed superior anti-inflammatory activity (59.86% inhibition of edema at 5 h) in comparison to celecoxib (51.44% inhibition of edema at 5 h) in carrageenan-induced rat paw edema assay. Structure–activity relationship studies suggested that N-phenyl ring substituted with p-CF3 substituent (3b, 3k and 3q) leads to more selective inhibition of COX-2. To corroborate obtained experimental biological data, molecular docking study was carried out which revealed that compound 3b showed stronger binding interaction with COX-2 as compared to COX-1.
Authors
- a Department of Natural Products, National Institute of Pharmaceutical Education and Research, Sector-67, S.A.S. Nagar (Mohali) 160062, Punjab, India
- b Department of Pharmacoinformatics, National Institute of Pharmaceutical Education and Research, Sector-67, S.A.S. Nagar 160062, Punjab, India
Sanjay Corresponding author. Tel.: +91 172 2214683; fax: +91 172 2214692.
CLICK……….
Cyclooxygenase (COX) or prostaglandin endoperoxide synthase (PGHS), catalyzes the conversion of arachidonic acid to inflammatory mediators such as prostaglandins (PGs), prostacyclins and thromboxanes. COX exists in mainly two isoforms: COX-1 and COX-2. Nonsteroidal anti-inflammatory drugs (NSAIDs), widely used for relief of fever, pain and inflammation, act by inhibiting COX catalyzed biosynthesis of inflammatory mediators.
However, the therapeutic use of classical NSAIDs is associated with well-known side effects at the gastrointestinal level (mucosal damage, bleeding) and, less frequently, at the renal level.
Two decades after the discovery of COX isoforms, it was recognized that selective inhibition of COX-2 might be endowed with improved anti-inflammatory properties and reduced gastrointestinal toxicity profiles than classical NSAIDs.
Overall, these selective COX-2 inhibitors (coxibs) have fulfilled the hope of possessing reduced risk in gastrointestinal events, but unfortunately cardiovascular concerns regarding the use of these agents have emerged that led to the withdrawal of rofecoxib (Vioxx) and valdecoxib (Bextra) from the market in 2004 and 2005, respectively.
Ongoing safety concerns pertaining to the use of non-selective NSAIDs have spurred development of coxibs with improved safety profile.
……………………………………………………………………………………………..

cas no 1616882-93-9
- mf……….C18 H11 F3 N2 O2
- [1]Benzopyrano[4,3-c]pyrazol-4(1H)-one, 3-methyl-1-[4-(trifluoromethyl)phenyl]-
3-Methyl-1-(4-(trifluoromethyl)phenylchromeno[4,3-c]pyrazol-4(1H)-one

Scheme 1.
Reagent and conditions: (a) Piperidine, rt, 20 min; (b) ArNHNH2, EtOH, reflux, 5 h; (c) K2CO3, acetone, reflux, 24 h.
COMPD IS
| 3b | R1=H | R2= H | 4-CF3-C6H4 | 90 |
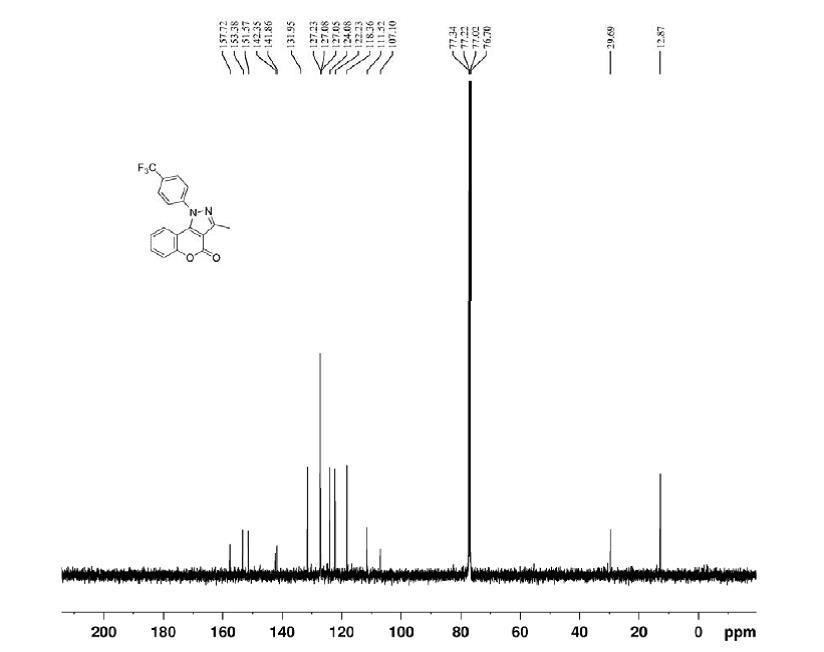
- 3-Methyl-1-(4-(trifluoromethyl)phenylchromeno[4,3-c]pyrazol-4(1H)-one (3b):
- White solid; yield 90%; mp: 224–225 °C;
- 1H NMR (CDCl3, 400 MHz): δ ppm 7.89 (d, 2H, J = 8.32 Hz, Ar-H), 7.73 (d, 2H, J = 8.24 Hz, Ar-H), 7.45–7.52 (m, 2H, H-6, H-7), 7.16 (dd, 1H, J = 1.4, 8.2 Hz, H-9), 7.10 (td, 1H, J = 1.56, 7.38 Hz, H-8), 2.69 (s, 3H, CH3);
- 13C NMR (CDCl3, 100 MHz): δ ppm 157.7, 153.3, 151.5, 142.3, 141.8, 131.9, 127.2, 127.1, 127.0, 124.0, 122.2, 118.3, 111.5, 107.1, 12.8;
- HRMS (ESI) m/z: Calcd for C18H11F3N2O2Na [M + Na]+ 367.0670; found 367.0676.
Synthetic Communications (2014), 44(13), 1914-1923
- DOI:
- 10.1080/00397911.2013.879184
Jagdeep Grovera, Somendu Kumar Roya & Sanjay Madhukar Jachaka*
pages 1914-1923
http://www.tandfonline.com/doi/abs/10.1080/00397911.2013.879184#.VCI5f0DgXXM
Abstract
Unprecedented cyclization was observed during N-sulfonylation of 3-[1-(phenylhydrazono)-ethyl]-chromen-2-one in pyridine, affording 3-methyl-1-phenylchromeno[4,3-c]pyrazol-4(1H)-ones. To avoid use of noxious pyridine, reaction was tried in different basic conditions and the best results were obtained with potassium carbonate in acetone. A wide range of substrates bearing either electron-donating or electron-withdrawing substituents on phenylhydrazine ring were compatible with the developed methodology. Rapid access of starting material, 3-acetylcoumarin, excellent yields of products, and use of environmentally benign base and solvent for the cyclization make this strategy an efficient and convenient method for synthesis of 3-methyl-1-phenylchromeno[4,3-c]pyrazol-4(1H)-ones.



References
1. Jones, G.; Willett, P.; Glen, R. C.; Leach, A. R.; Taylor, R. J. Mol. Biol. 1997, 267, 727.
2. Bernstein, F. C.; Koetzle, T. F.; Williams, G. J. B.; Meyer, E. F.; Brice, M. D.; Rodgers, J. R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. J. Mol. Biol. 1977, 112, 535.






{kind=link}