
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

DUTOGLIPTIN

Dutogliptin tartrate
Syn name: 1-[N-[3(R)-Pyrrolidinyl]glycyl]pyrrolidin-2(R)-ylboronic acid L-tartrate
Cas number: 890402-81-0
Molecular Formula: C14H26BN3O9
Molecular Weight: 391.18
 DUTOGLIPTIN
DUTOGLIPTIN
[1-[2-(Pyrrolidin-3-ylamino)acetyl]pyrrolidin-2-yl]boronic Acid; [(2R)-1-[2-[[(3R)-Pyrrolidin-3-yl]amino]acetyl]pyrrolidin-2-yl]boronic acid
C10H20BN3O3, 241.0951
852329-66-9
- Dutogliptin
- PHX1149
- UNII-38EAO245ZX
clinical trials
http://clinicaltrials.gov/search/intervention=Dutogliptin
PHX-1149 is a dipeptidyl peptidase IV (CD26; DPP-IV; DP-IV) inhibitor which had been in phase III clinical trials at Phenomix and Forest for the oral, once-daily treatment of type 2 diabetes.
In 2008, the compound was licensed to Forest by Phenomix in North America for development and commercialization; however this license agreement was terminated in 2010. In 2009, the compound was licensed to Chiesi by Phenomix for development and commercialization for the treatment of diabetes type 2 in Europe, Brazil, the Russian Federation and all other members of the Commonwealth of Independent States, Turkey and Northern Africa. Phenomix ceased operations in 2010.
………………………….





http://www.google.com/patents/WO2010107809A2?cl=en
or
http://www.google.com/patents/US20100240611?cl=en
The enzyme dipeptidyl peptidase IV (DPP-IV) is a member of the dipeptidyl peptidase family, which cleaves N-terminal dipeptide residues from proteins, particularly where the dipeptide includes an N-terminal penultimate proline or alanine residue. DPP-IV is believed to be involved in glucose control, as its peptidolytic action inactivates the insulotropic peptides glucagon-like peptide I (GLP-I) and gastric inhibitory protein (GIP).
Inhibition of DPP- IV, such as with synthetic inhibitors in vivo, can serve to increase plasma concentrations of GLP-I and GIP, and thus improve glycemic control in the body. Such synthetic inhibitors would therefore be useful in the treatment of diabetes mellitus and related conditions. Certain such selective DPP-IV inhibitors have been developed, as are disclosed in U.S. Patent 7,317,109, U.S. Patent 7,576,121, U.S. Application Publication Nos. 2007/0060547, 2007/0185061, 2007/0299036, 2008/0182995, 2008/0300413, 2006/0264400, and 2006/0264401, and in International Applications WO2008/027273 and WO2008/144730, the contents of which are incorporated herein by reference. Inhibition of DPP-IV by compounds of the structure of formula (I) is disclosed therein:

Example 1 – Synthesis of (R)-N-( 1 , 1 -Dimethylethoxycarbonyl)(pyrrolidine-2-yl)boronic Acid.

An oven dried 1 L three neck round bottom flask equipped with an overhead stirrer, addition funnel and internal thermocouple was charged with (IS, 2S)-Dimethyl-bis(3,3- dimethylbutyl)cyclohexane-l,2-diamine (approx. 50 g, 161.23 mmol, 1.2 eq), BOC-pyrrolidine (approx. 23.55 ml, 134.35 mmol, 1 eq) and dry toluene (approx. 500 ml) under inert atmosphere. The clear colorless solution was cooled to “ 78° C and a solution of sec-BuLi (approx. 115.16 ml of a 1.4 solution in cyclohexane, 161.23 mmol, 1.2 eq) was added slowly via dropping funnel over approx. 10 minutes (the temperature of the reaction mixture was maintained between approx. – 780C and -650C). The light orange colored solution was stirred for 3.5 hours at approx. -780C, which was then followed by the addition of a solution of trimethylborate (approx. 45.06 ml, 403.05 mmol, 3 eq) in toluene (approx. 75 ml) via dropping funnel over 30 minutes while maintaining the temperature below -650C. The reaction mixture was warmed slowly to room temperature, and stirred for 16 hours at room temperature. The reaction mixture was added into an aqueous sodium hydroxide solution (approx. 670 ml of 2.0 M solution, 1340 mmol, 10 eq) and the resulting cloudy mixture was stirred for 30 minutes before allowing layers to separate. The aqueous phase (product) was transferred to a receiver and backwashed with toluene (approx. 100 ml). The organic phases (chiral amine ligand) were transferred to a receiver for later isolation. The aqueous phase was acidified to pH 5-6 by slow addition of HCl {cone), then extracted with EtOAc (approx. 3 x 500 ml). The organic extracts were combined, dried over Na2SO4 and concentrated until a final volume of approximately 100 ml. Heptane (approx. 300 ml) was added and the concentrated mixture was stirred at room temperature overnight (approx. 15 hours). The resulting white precipitate was filtered and the filter cake was washed with cold heptane. The product was dried at room temperature under vacuum to yield (R)- (pyrrolidine-2-yl)boronic acid (approx. 20.31 g, 94.44 mmol, 70.27 %) as a white solid. [α]25D – 72.5 (c 1, DCM); 94-95 % ee (% ee was determined through chiral HPLC); 1H NMR (400 MHz, D2O) δ 3.40-3.50 (IH), 3.20- 3.30 (IH), 2.90-3.00 (IH), 2.10 (IH), 2.00 (IH), 1.85 (IH), 1.72 (IH), 1.45-1.48 (9H); m/z (ES+) 216.06.
Example 2 – Isolation of the chiral ligand ((1S, 2S)-Dimethyl-bis(3,3-dimethyl butyl) cyclohexane- 1 ,2-diamine)

Water (approx. 300 ml) was added to the first organic extract from the previous workup and cooled to 0° C the mixture was acidified to pH 3 by slow addition of HCl. The resulting cloudy mixture was stirred vigorously before allowing layers to separate. The aqueous phase (product) was transferred to a receiver and backwashed with toluene (approx. 100 ml). The aqueous phase was stirred at O0C and the pH of the solution was adjusted to 12-13 by the addition of sodium hydroxide. The mixture was extracted with toluene (approx. 3 x 500 ml) and the combined organic phases were concentrated under reduced pressure to give the crude chiral diamine (approx. 48.32 g, 155.57 mmol, 96.5%) as light yellow oil. Further purification by vacuum distillation (approx. 120-1300C, house vacuum) yielded the chiral diamine as a colorless oil (approx. 45.57 g, 146.72 mmol) in 91% recovery).Example 3 – Synthesis of (R)-N-(I, l-dimethylethoxycarbonyl)-pinanediol-(Pyrrolidin-2-yl) boronate

A solution of (R)-Pyrrolidine boronic acid (approx. 300 mg, 1.39 mmol) in isopropyl acetate (approx. 10 ml) was treated with (+)-pinanediol (approx. 236.35 mg, 1.39 mmol, 1 eq) and Na2SO4 (approx. 203.25 mg, 1.39 mmol, 1 eq). After 24 hr, the solvent was evaporated to give crude boronic ester (approx. 475.55 mg, 1.36 mmol, 98 %) as a clear oil: 98-99 % de via chiral HPLC; 1U NMR (400 MHz, CDCl3) δ 4.32 (IH), 3.47 (IH), 3.41-3.31 (2H), 3.22-3.05 (IH), 2.38- 2.30 (IH), 2.20-1.75 (8H), 1.45 (9H), 1.41 (3H), 1.28 (3H), .85 (3H); m/z (ES, M+l) 350.28.Example 4 – (R)-N-(Pyrrolidine-2-yl)-pinacol boronate
To a solution of pyrrolidine boronic acid (approx. 456 mg, 2.12 mmol) in isopropyl acetate
(approx. 15 ml) was added pinacol (approx. 251 mg, 2.12 mmol, 1 eq) and Na2SO4 (approx. 310 mg, 2.12 mmol, 1 eq). The mixture was stirred for 24 hr and the solvent was evaporated to yield crude pinacol boronate. The residue was triturated with EtOAc/hexane (approx. 1 : 10) at RT for 1 hr then filtered to give the pinacol boronate (approx. 611 mg, 2.06 mmol, 97 %) as a white solid: . 1H NMR (400 MHz, CDCl3) δ 3.40-2.95 (3H), 1.95-1.50 (4H), 1.40 (9H), 1.20 (12H); m/z (ES+) 298.21. Removal of the Boc-protecting group was achieved by dissolving the white solid pinacol boronate in dry ether (approx. 15 ml), cooling to 0° C in an ice bath followed with addition of 1.5 eq of HCl in dioxane After 8 hours, the solvent was evaporated then triturated in hexane for 1 hr. The white precipitate was filtered and dried to yield the acid salt (approx. 472 mg, 2.02 mmol, 98 %): 1HNMR (CDCl3) δ 3.48 (IH), 3.36 (IH), 3.21 (IH), 2.21 (IH), 2.03 (2H), 1.95 (IH), 1.35 (12H); m/z (ES M+l) 198.21.
Example 5 – Synthesis of (R)-3-(Benzyloxycarbonyl-{2-oxo-2-[(R)-2-((lS,2S,6R,8S)-2,9,9- trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.0^'”]dec-4-yl)-pyrrolidin-l-yl]-ethyl}-amino)- pyrrolidine- 1-carboxylic acid benzyl ester

A mixture of (R)-3-(benzyloxycarbonyl-carboxymethyl-amino)-pyrrolidine- 1-carboxylic acid benzyl ester dicyclohexylamine salt) (approx. 300.Og, 0.505mol), water (approx. 1.5L), 2M aqueous sulfuric acid (approx. 0.75L, 1.5mol) and toluene (approx. 2L) was stirred in a 1OL reactor at room temperature for 15 min. After settling the layers were separated. The aqueous layer was stirred with toluene (approx. 1.0L) for 15 min, and the layers were separated. The combined organic layers were washed with water (approx. 1.5L), and concentrated under vacuum at 450C to 1.5L. To this solution was added N-methylmorpholine (approx. 55.4 mL, 0.505mol) and this mixture was added to a cold solution (approx. 0°-5°C) of ethyl chloroformate (approx. 48.1 mL, 0.505mol) in toluene (approx. 1.0L). The reaction mixture was stirred at 0° – 50C for 15 min and solid (2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02‘6]dec-4-yl)-pyrrolidine hydrochloride) (approx. 144.4g, 0.505mol) was added in one portion followed by addition of N- Methylmorpholine (approx. 110.8 mL, l.Olmol). The mixture was stirred for 30 min at 0°-5°C, and allowed to warm to 20°-25°C. Stirring was continued for an additional 2.5 h. Water (approx. 2.0L) was then added, and the mixture was stirred for an additional 15 min. The layers were separated and the organic layer was subsequently washed with 0.85M aqueous sodium bicarbonate solution (approx. 1.2L), water (approx. 2.0L), and 0.065M citric acid solution (approx. 1.5L). Toluene solution was concentrated under vacuum at 450C, to give 287.3 g (approx. 88.4%) of the title compound. 1H NMR (400 MHz, CDCl3, ppm): mixture of rotomers, 7.35-7.25 (10H,m); 5.22- 4.99 (4H,m); 4.60 (IH, d); 4.22 (IH, dd); 4.11-3.65 (3H, m); 3.60-3.00 (6H, m); 2.32-1.91 (8H, m); 1.89-1.67 (4H, m); 1.42-1.18 (6H, m); 0.84-0.72 (3H, m); m/z (M+H)=644. Example 6 – Synthesis of 2-((R)-Pyrrolidin-3-ylamino)-l-[(R)-2-((lS,2S,6R,8S)-2,9,9-trimethyl- 3,5-dioxa-4-bora-tricyclo[6.1.1.0 ‘ ]dec-4-yl)-pyrrolidin- 1 -yl]-ethanone

a) THF solvateA solution of (R)-3-(Benzyloxycarbonyl-{2-oxo-2-[(R)-2-((l S,2S,6R,8S)-2,9,9-trimethyl-3,5- dioxa-4-bora-tricyclo[6.1.1.02‘”]dec-4-yl)-pyrrolidin- 1 -yl] -ethyl }-amino)-pyrrolidine- 1 – carboxylic acid benzyl ester (approx. 4.76 g, 7.4 mmol) in toluene (approx. 60 mL) was diluted with methanol (approx. 60 mL). 10% Pd/C (wet, 500 mg) was added, and the mixture was hydrogenated at 50 psi for 3 h. The mixture was filtered through celite and washed with methanol (approx. 10 mL). The solution was then concentrated under vacuum to dryness. The residue was dissolved in THF (approx. 10 mL) at 4O0C and crystallized overnight at -1O0C to -15°C. Crystals were filtered, washed with cold THF (approx. 3 mL), and dried under vacuum for 5 h to yield 1.9 g (approx. 68.5%) of the title compound. 1H NMR (400 MHz, D2O, 1 drop TFA), 64.18 – 4.89 (m, IH), 3.93 – 3.85 (m, IH), 3.77 (s, 2H), 3.55 (dd, IH)5 3.45 -3.38 (m, 4H), 3.35 – 3.25 (m, 2H), 3.24 – 3.05 (m, 3H), 2.93 (t, IH), 2.33 – 2.24 (m, IH), 2.15 – 1.42 (m, 16H), 1.09 (s, 3H), 0.94 (s, 3H), 0.78 (d, IH), 0.50 (s, 3H). m/z (ES+) = 376.30.
Thermogravimetric analysis of THF solvate of 2-((R)-Pyrrolidin-3-ylamino)-l-[(R)-2-
((lS,2S,6R,8S)-2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02‘6]dec-4-yl)-pyrrolidin-l-yl]- ethanone was performed as is shown in Figure 5.
X-Ray Diffractogram of THF solvate of 2-((R)-Pyrrolidin-3-ylamino)-l-[(R)-2-((lS,2S,6R,8S)- 2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02‘6]dec-4-yl)-pyrrolidin-l-yl]-ethanone was performed as is shown in Figure 6. b) Non-solvate
A solution of (3-(Benzyloxycarbonyl-{2-oxo-2-[2-(2,9,9-trimethyl-3,5-dioxa-4-bora- tricyclo[6.1.1.02‘6]dec-4-yl)-pyrrolidin-l-yl]-ethyl}-amino]-pyrrolidine-l-carboxylic acid benzyl ester) (approx. 20.Og, 31.Ommol) in toluene (approx. 8OmL) was diluted with methanol (approx. 20 mL). 10% Pd/C (2g, wet) was added, and the mixture was hydrogenated at 50 psi for 3 h. The mixture was filtered through celite and the filter bed was washed with a mixture of toluene (approx. 2OmL) and methanol (approx. 4 mL). The solution was concentrated to 8OmL at 30 -35 0C under vacuum (approx. 90 to 120 mBar). THF (approx. 10OmL) was added and the solution was concentrated to 12OmL at 30 -35 0C under vacuum (approx. 90 to 120 mBar). The mixture was stirred at 35 0C for Ih, resulting in crystallization. The mixture was cooled to 0 0C and held at that temperature for 2h. Crystals were isolated by filtration, washed with a cold mixture of toluene (approx. 20 mL) and THF (approx. 5 mL), and dried under vacuum at 35 0C for 16 h to yield 9.11 g (approx. 24.3 mmol, 78%) of the title compound as a white solid.1H NMR (400 MHz, D20, 1 drop TFA), δ 4.34 (dd, IH, J= 9, 2 Hz), 4.08 (m, IH), 3.99 (s, 2H), 3.74 (dd, IH, J= 13, 8 Hz), 3.52 -3.29 (m, 6H), 3.12 (t, IH, J= 8 Hz), 2.47 (m, IH), 2.27 (m, IH), 2.19 – 2.06 (m, 2H), 2.02 – 1.84 (m, 6H), 1.67 (m, 2H), 1.30 (s, 3H), 1.15 (s, 3H), 1.00 (d, IH, J= 11 Hz), 0.71 (s, 3H). m/z (ES+) = 376.30.
Thermogravimetric analysis of 2-((R)-Pyrrolidin-3-ylamino)-l-[(R)-2-((lS,2S,6R,8S)-2,9,9- trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.0^'”]dec-4-yl)-pyrrolidin-l-yl]-ethanone was performed as is shown in Figure 7.
X-Ray Diffractogram of2-((R)-Pyrrolidin-3-ylamino)-l-[(R)-2-((lS,2S,6R,8S)-2,9,9-trimethyl-
3,5-dioxa-4-bora-tricyclo[6.1.1.0 ‘ ]dec-4-yl)-pyrrolidin-l-yl]-ethanone was performed as is shown in Figure 8.
Example 7 – Synthesis of Dutogliptin Tartrate

A round bottom flask equipped with a magnetic stirrer was charged with 2-(Pyrrolidin-3- ylamino)- 1 -[2-(2,9,9-trimethyl-3,5-dioxa-4-boratricyclo[6.1.1.0]dec-4-yl)-pyrrolidin-l-yl]- ethanone (approx. l:l-Pinanediol borane / THF complex; 2.98 g, 6.67 mmol, leq), (L)-tartaric acid (approx. 1.00 g, 6.67 mmol, 1 eq), and H2O (approx. 15 mL). The mixture was allowed to stir for 1 hour then tert-Butyl methyl ether (approx. 15 ml) and (i?)-N-(l,l- dimethylethoxycarbonyl)(pyrrolidine-2-yl)boronic acid (approx. 1.46 g, 6.80 mmol, 1.02 eq) were added. The bi-phasic mixture was allowed to stir for 20 hours at room temperature before separating the layers. The aqueous phase backwashed with tert-butyl methyl ether (approx. 15 ml) and the organic layers were combined. Lyophilization of the aqueous layer provided dutogliptin tartrate as a white solid (approx. 2.60 g, 6.65 mmol, 99.7%): 1H NMR (400 MHz, D2O, one drop of TFA) δ 4.48 (2H), 3.95-3.88 (IH), 3.81 (2H), 3.59-3.54 (IH), 3.37-3.28 (2H), 3.21-3.16 (2H), 3.11-3.07 (IH), 2.82-2.78 (IH), 2.37-2.28 (IH), 2.04-1.96 (IH), 1.88-1.78 (2H), 1.71-1.60 (IH), 1.50-1.42 (IH); m/z (ES+) 241.10 (-tartrate acid).
| US20060069250 * | Sep 28, 2005 | Mar 30, 2006 | Xiaohu Deng | Synthesis by chiral diamine-mediated asymmetric alkylation |
| US20080182995 * | Oct 31, 2007 | Jul 31, 2008 | Phenomix Corporation | Pyrrolidine compounds and methods for selective inhibition of dipeptidyl peptidase-iv |
| US20080300413 * | Jul 27, 2006 | Dec 4, 2008 | David Alan Campbell | Efficiently preparing boropyrrolidines and derivatives by coupling a (pyrrolidin3-yl-amino-)acetic acid and a 7,9,8-dioxaborotricyclic- (4,3,0,1(2,4))decane; protecting groups avert side reactions; antidiabetic agents |
Regeneron and Sanofi’s dupilumab gets FDA breakthrough therapy status for atopic dermatitis
// // //

Regeneron Pharmaceuticals and Sanofi’s dupilumab has received breakthrough therapy designation from US Food and Drug Administration (FDA) to treat adults with moderate-to-severe atopic dermatitis (AD).
澳格列汀, SP2086, Retagliptin

 澳格列汀, SP2086, Retagliptin 1174122-54-3(Retagliptin), 1174038-86-8 (Retagliptin Hydrochloride), 1256756-88-3(Retagliptin Phosphate) (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7, 8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester Methyl (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo [1,5-a]pyrazine-1-carboxylate, DPP-4 inhibitor Type II diabetes
澳格列汀, SP2086, Retagliptin 1174122-54-3(Retagliptin), 1174038-86-8 (Retagliptin Hydrochloride), 1256756-88-3(Retagliptin Phosphate) (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7, 8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester Methyl (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo [1,5-a]pyrazine-1-carboxylate, DPP-4 inhibitor Type II diabetes
| Jiangsu Hengrui Medicine Co., Ltd |
 Nanjing Changao Pharmaceutical 澳格列汀 is a novel DPP-4 inhibitor (gliptin) for the treatment of type II diabetes. Because Shanghai Sun Sail Pharmaceutical, a wholly owned subsidiary of Nanjing Changao Pharmaceutical, has filed two patents to protect DPP-4 inhibitors (WO2011147207 and CN101786978), it is unknown which one covers this drug. Relevant data’s from WHO showed morbidity rate, disability rate, death rate of diabetes mellitus and overall health level of diabetes mellitus patients have already ranked the third place in non-infectious diseases, diabetes, together with tumors and cardiovascular diseases were the three main diseases which threats human health. Diabetes mellitus is usually classified into type 1 and type 2, there are more than 240 million diabetes patients, and 90% of them are suffering from type 2 diabetes, which also has a 1% growth rate every year, so, type 2 diabetes will be the main new growth point of diabetes drug market. The incidence of diabetes in China is about 5%, the number of patients of which ranks second place in the world just behind India. There are many antidiabetic drugs on the market, insulin injection, metformin, rosiglitazone, pioglitazone are representations of them. However, there is no drug alone can keep the HbA1c level of type 2 diabetes patients within the aimed range in a long term. Even though used in combination, the effect of the drugs will go down year by year after 3-4 years. Adverse reaction is one of the problems of many hypoglycemic drugs, wherein the fatal hypoglycemia is most worried by clinicians; secondly, many oral hypoglycemic drugs, such as sulfonylureas, α-glycosidase inhibitors and thiazolidinediones may all induce weight gain to patients, some of the drugs may also induce cardiovascular diseases. Therefore, developing new type hypoglycemic drugs with brand new mechanism of action, higher safety and effectiveness is an important task that should be completed quickly for the scientists. In the process of constantly finding new methods endocrine hormones were found to play an important role in the pathology and physiology of type 2 diabetes. Dipeptidyl peptidase-IV (DPP-IV) is an important enzyme related to diabetes, inhibiting the action of which to treat type 2 diabetes is a new method with good prospect. DPP-IV inhibitors can indirectly stimulate the secretion of insulin, the action of which is generated by inhibit DPP-IV to stabilize endocrine hormones such as incretin hormones, glucagons-like-peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). GLP-1 is a production expressed by glucagon protogene after eating, and mainly secreted by intestinal mucosa L-cell, and it can stimulate the secretion of insulin by pancreatic β-cells, which plays a significant role in the stability of blood sugar. Experiments prove that GLP-1 has physiological functions as following: acting on pancreatic β-cells in a glucose-dependent manner, facilitating the transcription of insulin genes, increasing the biosynthesis and secretion of insulin, stimulating the proliferation and differentiation of β-cells, inhibiting the apoptosis of β-cells to increasing the number of pancreatic β-cells; inhibiting the secretion of glucagon; inhibiting the appetite and food intake; retarding the emptying of gastric contents, etc., all of these functions are helpful to reduce blood sugar after food intake and to keep blood sugar within constant level. In addition, it won’t cause the danger of severe hypoglycemia. GLP-1 well controlled the blood sugar of type 2 diabetes animal models and patients by multiple mechanisms. However, GLP-1 may lose biological activity through quick degradation by DPP-IV, and the half life of it is shorter than 2 minutes, which utterly limits the clinical use of GLP-1. It was found in researches that DPP-IV inhibitors can totally protect endogenous and even extraneous GLP-1 from inactivation by DPP-IV, improve activated GLP-llevel, and reduce the antagonistic effect of GLP-1 metabolites. Moreover, DPP-IV inhibitors can also delay the incidence of diabetes through stimulating the regeneration of pancreatic β-cells and the improving the glucose tolerance and insulin sensitivity. Dipeptidyl peptidase-IV (DPP-IV) inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with Type 2 diabetes. For reviews on the application of DPP-IV inhibitors for the treatment of Type 2 diabetes, reference is made to the following publications: (1) H.-U.Demuth.et al. “Type 2 diabetes-Therapy with dipeptidyl peptidase IV inhibitors“, Biochim.Biophvs. Acta. 1751:33-44 (2005) and (2) K.Augustyns. et al. “Inhibitors of proline-specific dipeptidyl peptidases: DPP4 inhibitors as a novel approach for the treatment of Type 2 diabetes“, Expert Opin. Ther. Patents, 15:1387-1407 (2005). At present, some DPP-IV inhibitors have been disclosed ( US5462928 , US5543396 , WO9515309 ,WO2003004498 , WO2003082817 , WO2004032836 , WO2004085661 ), including MK-0431 as an DPP-IV inhibitor made by Merck which showed good inhibition activity and selectivity, and which has been on the market by 2006.
Nanjing Changao Pharmaceutical 澳格列汀 is a novel DPP-4 inhibitor (gliptin) for the treatment of type II diabetes. Because Shanghai Sun Sail Pharmaceutical, a wholly owned subsidiary of Nanjing Changao Pharmaceutical, has filed two patents to protect DPP-4 inhibitors (WO2011147207 and CN101786978), it is unknown which one covers this drug. Relevant data’s from WHO showed morbidity rate, disability rate, death rate of diabetes mellitus and overall health level of diabetes mellitus patients have already ranked the third place in non-infectious diseases, diabetes, together with tumors and cardiovascular diseases were the three main diseases which threats human health. Diabetes mellitus is usually classified into type 1 and type 2, there are more than 240 million diabetes patients, and 90% of them are suffering from type 2 diabetes, which also has a 1% growth rate every year, so, type 2 diabetes will be the main new growth point of diabetes drug market. The incidence of diabetes in China is about 5%, the number of patients of which ranks second place in the world just behind India. There are many antidiabetic drugs on the market, insulin injection, metformin, rosiglitazone, pioglitazone are representations of them. However, there is no drug alone can keep the HbA1c level of type 2 diabetes patients within the aimed range in a long term. Even though used in combination, the effect of the drugs will go down year by year after 3-4 years. Adverse reaction is one of the problems of many hypoglycemic drugs, wherein the fatal hypoglycemia is most worried by clinicians; secondly, many oral hypoglycemic drugs, such as sulfonylureas, α-glycosidase inhibitors and thiazolidinediones may all induce weight gain to patients, some of the drugs may also induce cardiovascular diseases. Therefore, developing new type hypoglycemic drugs with brand new mechanism of action, higher safety and effectiveness is an important task that should be completed quickly for the scientists. In the process of constantly finding new methods endocrine hormones were found to play an important role in the pathology and physiology of type 2 diabetes. Dipeptidyl peptidase-IV (DPP-IV) is an important enzyme related to diabetes, inhibiting the action of which to treat type 2 diabetes is a new method with good prospect. DPP-IV inhibitors can indirectly stimulate the secretion of insulin, the action of which is generated by inhibit DPP-IV to stabilize endocrine hormones such as incretin hormones, glucagons-like-peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). GLP-1 is a production expressed by glucagon protogene after eating, and mainly secreted by intestinal mucosa L-cell, and it can stimulate the secretion of insulin by pancreatic β-cells, which plays a significant role in the stability of blood sugar. Experiments prove that GLP-1 has physiological functions as following: acting on pancreatic β-cells in a glucose-dependent manner, facilitating the transcription of insulin genes, increasing the biosynthesis and secretion of insulin, stimulating the proliferation and differentiation of β-cells, inhibiting the apoptosis of β-cells to increasing the number of pancreatic β-cells; inhibiting the secretion of glucagon; inhibiting the appetite and food intake; retarding the emptying of gastric contents, etc., all of these functions are helpful to reduce blood sugar after food intake and to keep blood sugar within constant level. In addition, it won’t cause the danger of severe hypoglycemia. GLP-1 well controlled the blood sugar of type 2 diabetes animal models and patients by multiple mechanisms. However, GLP-1 may lose biological activity through quick degradation by DPP-IV, and the half life of it is shorter than 2 minutes, which utterly limits the clinical use of GLP-1. It was found in researches that DPP-IV inhibitors can totally protect endogenous and even extraneous GLP-1 from inactivation by DPP-IV, improve activated GLP-llevel, and reduce the antagonistic effect of GLP-1 metabolites. Moreover, DPP-IV inhibitors can also delay the incidence of diabetes through stimulating the regeneration of pancreatic β-cells and the improving the glucose tolerance and insulin sensitivity. Dipeptidyl peptidase-IV (DPP-IV) inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with Type 2 diabetes. For reviews on the application of DPP-IV inhibitors for the treatment of Type 2 diabetes, reference is made to the following publications: (1) H.-U.Demuth.et al. “Type 2 diabetes-Therapy with dipeptidyl peptidase IV inhibitors“, Biochim.Biophvs. Acta. 1751:33-44 (2005) and (2) K.Augustyns. et al. “Inhibitors of proline-specific dipeptidyl peptidases: DPP4 inhibitors as a novel approach for the treatment of Type 2 diabetes“, Expert Opin. Ther. Patents, 15:1387-1407 (2005). At present, some DPP-IV inhibitors have been disclosed ( US5462928 , US5543396 , WO9515309 ,WO2003004498 , WO2003082817 , WO2004032836 , WO2004085661 ), including MK-0431 as an DPP-IV inhibitor made by Merck which showed good inhibition activity and selectivity, and which has been on the market by 2006.
-
 sitagliptin
sitagliptin(R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7, 8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester of the following formula is compound A, the code of which is SP2086.


 courtesy yaopha see enlarged image at http://www.yaopha.com/2014/02/10/chemical-structure-and-synthesis-of-hengrui-medicines-diabetes-drug-retagliptin/ …………………………………………………………..
courtesy yaopha see enlarged image at http://www.yaopha.com/2014/02/10/chemical-structure-and-synthesis-of-hengrui-medicines-diabetes-drug-retagliptin/ …………………………………………………………..
- EP2436684A1
- Example 1. Preparation of hydrochloride of compound A (SP2086-HCL)
- (R)-7-[3-t-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester (SM2086-15) (1.35kg, 2.40mol), HCL-ethyl acetate (greater than 2M) (12.3kg) were added into a 100L reaction kettle and stirred to dissolved. The mixture was reacted for more than 2 hours at normal temperature. Detected with TLC to reaction completely before evaporated and pumped to dryness with oil pump to give 1.15∼1.20kg of white to light yellow solid product with [α]
D20
- -28.0∼-33.0° (C=1, methanol), yield 96.0∼100%. The product was hydrochloride of (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7, 8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester (SP2086-HCL). (TLC detection: silica gel GF254plate; developing reagent: chloroform: methanol: ammonia= 40: 1: 0.1; raw material 15: Rf=0.80, product 1: Rf=0.50; ultraviolet visualization).
Example 2. Preparation of phosphate of compound A (SP2086-HPO4)
-
SP2086-HCL(1.20kg, 2.40mol) was added into 100L reaction kettle, and dissolved in dichloromethane (15.2kg), then washed with saturated sodium bicarbonate solution (5.8kg). The aqueous layer was extracted once with dichloromethane ( 6.0 kg). The organic layers were combined and washed once with water (5kg), dried with anhydrous sodium sulphate. The mixture was filtrated and concentrated to dryness under reduced pressure at 40°C to give 1.12 kg of oil. The oil was stirred and dissolved with 30 times amount of isopropanol (26.0kg). A solution of 85% phosphoric acid (305.2g, 2.65mol) in isopropanol (1.22kg) was added immidiately after the oil completely dissolved. The solid was separated out, filtered after stirring for 2 hours and washed with cold isopropanol. The wet product was dried under reduced pressure at 40°C to give 1.16∼1.24kg of white to light yellow solid with a yield of 86.0∼92.0% (the wet product could be directly suspended in isopropanol without drying).
……………………………………… http://www.google.com/patents/EP2230241A1?cl=en Example 1(R)-7-[3-Amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester hydrochloride

Step 1
-
2,2-Dimethyl-5-[2-(2,4,5-trifluoro-phenyl)-acetyl]-[1,3]dioxane-4,6-dione 2,2-Dimethyl-[1,3]dioxane-4,6-dione (5.69 g, 39.5 mmol) was dissolved in 400 mL of dichloromethane under stirring, followed by addition of (2,4,5-trifluoro-phenyl)-acetic acid 1a (7.15 g, 37.6 mmol) and 4-dimethylaminopyridine (7.35 g, 60.2 mmol) in an ice-water bath. Then a suspension of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (8.28 g, 43.2 mmol) in 250 mL of dichloromethane was added dropwise slowly. After stirring at room temperature for 36 hours, the reaction mixture was washed with the solution of 5% potassium bisulfate (250 mL×7) and saturated brine (250 mL×2), dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to obtain the title compound 2,2-dimethyl-5-[2-(2,4,5-trifluoro-phenyl)-acetyl]-[1,3]dioxane-4,6-dione 1b (11.4 g, yield 96%) as a white solid. MS m/z (ESI): 315.5 [M-1]
Step 23-Oxo-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester
-
2,2-Dimethyl-5-[2-(2,4,5-trifluoro-phenyl)-acetyl]-[1,3]dioxane-4,6-dione 1b (15.72 g, 49.6 mmol) was dissolved in 280 mL of ethanol under stirring, then the reaction mixture was heated to 70 °C in an oil bath overnight. After cooling, the mixture was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound 3-oxo-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester 1c (12 g, yield 88%) as a yellow oil. MS m/z (ESI): 259 [M-1]
Step 33-Amino-4-(2,4,5-trifluoro-phenyl)-but-2-enoic acid ethyl ester
-
3-Oxo-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester 1c (24.6 g, 94.5 mmol) was dissolved in 240 mL of methanol, and ammonium acetate (36.4 g, 473 mmol) was added to the solution. The reaction mixture was heated to reflux for 3 hours and monitored by thin layer chromatography until the disappearance of the starting materials. The reaction mixture was concentrated under reduced pressure, then 100 mL of water was added to the residue. The mixture was extracted with ethyl acetate (200 mL×3), and the combined organic phase was washed with 200 mL of saturated brine, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to obtain a light yellow solid. The resulting solid was dissolved in 50 mL of ethyl acetate at 80 °C, then 50 mL of n-hexane and seed-crystal were added to the solution. The mixture was cooled to room temperature, half an hour later, 100 mL of n-hexane was added. The mixture was stored in refrigerator overnight and then filtered under reduced pressure to obtain the title compound 3-amino-4-(2,4,5-trifluoro-phenyl)-but-2-enoic acid ethyl ester 1d(19.5 g, yield 80%) as a white solid. MS m/z (ESI): 260.1 [M+1]Step 43-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester
-
3-Amino-4-(2,4,5-trifluoro-phenyl)-but-2-enoic acid ethyl ester 1d (4.1 g, 15.8 mmol) was added into an autoclave, followed by addition of 70 mL of methanol, di-tert-butyl dicarbonate (3.8 g, 17.4 mmol), chloro(1, 5-cyclooctadiene)rhodium( I ) dimer (32 mg, 0.0632 mmol) and (R)-1-[(S)-2-(diphenyl phosphino)ferrocenyl]-ethyl-tert-butylphosphine (68 mg, 0.126 mmol). The reaction mixture was hydrogenated for 24 hours under 6.67 atmosphere at 30 °C. The mixture was filtered and the filtrate was concentrated under reduced pressure. Then 34 mL of methanol was added to the residue at 50 °C, followed by addition of 12 mL of water until all dissolved. After cooling to room temperature, the mixture was stored in the refrigeratory overnight and then filtered. The solid product was washed with the solvent mixture of methanol/water (v:v = 3:2), dried in vacuo to obtain the title compound 3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester 1e (4 g, yield 70%) as a light yellow solid. MS m/z (ESI): 362.4 [M+1]Step 5(R)-3-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid
-
3-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid ethyl ester 1e (10 g, 27.7 mmol) and sodium hydroxide (3.32 g, 83.1 mmol) were dissolved in the solvent mixture of 100 mL of methanol and 50 mL of water under stirring. The reaction mixture was reacted at 40-45 °C for 1-1.5 hours, then part of the solution was evaporated under reduced pressure. The residue was added with some water, then pH was adjusted to 2-3 with 1 N hydrochloric acid in an ice-water bath. The mixture was extracted with ethyl acetate (200 mLx3), and the combined organic phase was washed with 200 mL of saturated brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure, and then recrystallized from ethyl acetate/n-hexane to obtain the title compound (R)-3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid 1f (9.2 g) as a white solid, which was directly used in the next step. MS m/z (ESI): 332.3 [M-1] Reference: Tetrahedron Asymmetry, 2006, 17(2), 205-209
Step 6C-Pyrazin-2-yl-methylamine
-
Pyrazine-2-carbonitrile 1g (10.5 g, 100 mmol) was dissolved in 150 mL of 1,4-dioxane under stirring, then Raney nickel (1.0 g) was added into a 250 mL autoclave. The reaction mixture was hydrogenated for 8 hours under 40 atmosphere at 60 °C, filtered and concentrated under reduced pressure to obtain the title compound C-pyrazin-2-yl-methylamine 1h (10.7 g, yield 98%) as a brown oil. MS m/z (ESI): 110 [M+1]
Step 72,2,2-Trifluoro-N-pyrazin-2-ylmethyl-acetamide
-
C-Pyrazin-2-yl-methylamine 1h (10.9 g, 100 mmol) was added into a reaction flask, then 20 mL of trifluoroacetic anhydride was added dropwise slowly within an hour at 0 °C in an ice-water bath. The reaction mixture was reacted at room temperature for 2 hours and monitored by thin layer chromatography until the disappearance of the starting materials. Then it was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound 2,2,2-trifluoro-N-pyrazin-2-ylmethyl-acetamide 1i (21.0 g) as a brown oil. MS m/z (ESI): 206.1 [M+1]
Step 83-Trifluoromethyl-imidazo[1,5-a]pyrazine
-
2,2,2-Trifluoro-N-pyrazin-2-ylmethyl-acetamide 1i (21.0 g, 100 mmol) was added into a reaction flask at room temperature, followed by addition of 100 mL of phosphorus oxychloride. After stirring at room temperature for 30 minutes, phosphorous pentoxide (17.8 g, 125 mmol) was added to the solution. The reaction mixture was heated to reflux for 5 hours and monitored by thin layer chromatography until the disappearance of the starting materials. Phosphorus oxychloride was removed, and the reaction system was quenched with deionized water. The mixture was adjusted to pH 5-6 with 20% sodium hydroxide solution in an ice-water bath. The mixture was extracted with ethyl acetate (250 mL×4), and the combined organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound 3-trifluoromethyl-imidazo[1,5-a]pyrazine 1j (12.0 g, yield 65%) as a yellow solid. MS m/z (ESI): 188.0 [M+1] 1H NMR (400 MHz, CDCl3): δ 9.15 (s, 1H), 8.06 (d, 1H), 7.92 (s, 1H), 7.81 (d, 1H)
Step 93-Trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine
-
3-Trifluoromethyl-imidazo[1,5-a]pyrazine 1j (12.0 g, 64.2 mmol) was dissolved in 150 mL of anhydrous ethanol under stirring, then 10% Pd/C (500 mg) was added to the solution. The reaction mixture was stirred at room temperature under a hydrogen atmosphere overnight. The reaction solution was filtered through a pad of coarse silica gel and concentrated under reduced pressure to obtain the title compound 3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine 1k (12.2 g, yield 99%) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 6.84 (s, 1H), 4.10 (m, 4H), 3.26 (m, 2H), 1.81 (s, 1H)
Step 10(R)-[3-Oxo-1-(2,4,5-trifluoro-benzyl)-3-(3-trifluoromethyl-5,6-dihydro-8H-imidazo [1,5-a]pyrazin-7-yl)-propyl]-carbamic acidtert-butyl ester
-
Under a nitrogen atmosphere, 3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid 1k (8.6 g, 45 mmol) and 9.4 mL of triethylamine were dissolved in 300 mL of dichloromethane under stirring. After stirring at room temperature for 5 minutes, 3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine 1f (15.0 g, 45 mmol) and bis(2-oxo-3-oxazolidinyl)phosphonic chloride (17.1 g, 67.3 mmol) were added to the solution successively. The reaction mixture was reacted at room temperature for 2 hours and monitored by thin layer chromatography until the disappearance of the starting materials and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound (R)-[3-oxo-1-(2,4,5-trifluoro-benzyl)-3-(3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazin-7-yl)-propyl]-carbamic acid tert-butyl ester 1l (20.0 g, yield 88%) as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.25 (m, 1H), 7.11 (m, 1H), 7.032 (s, 1H), 4.93 (m, 2H), 4.35 (m, 3H), 4.05 (m, 2H), 2.99 (m, 2H), 2.73 (m, 2H), 1.34 (s, 9H)
Step 11(R)-[3-(1-Bromo-3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazin-7-yl)-3-oxo-1-(2,4,5-trifluoro-benzyl)-propyl]-carbamic acidtert-butyl ester
-
(R)-[3-Oxo-1-(2,4,5-trifluoro-benzyl)-3-(3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazin-7-yl)-propyl]-carbamic acid tert-butyl ester 11 (20.0 g, 39.6 mmol) was dissolved in 300 mL of anhydrous ethanol under stirring, and 1-bromo-2,5-pyrolidinedione (14.1 g, 79.2 mmol) was then added to the solution at room temperature. After stirring for an hour, potassium carbonate (10.9 g, 79.2 mmol) and di-tert-butyl dicarbonate (8.6 g, 39.6 mmol) were added to the mixture, and the mixture was stirred for an hour and monitored by thin layer chromatography until the disappearance of the starting materials. The reaction mixture was filtered through a pad of coarse silica gel to remove potassium carbonate, and then concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound (R)-[3-oxo-1-(2,4,5-trifluoro-benzyl)-3-(1-bromo-3-trifluoromethyl-5,6-dihydro-8H-i midazo [1,5-a]pyrazin-7-yl)-propyl]-carbamic acid tert-butyl ester 1m (20.0 g, yield 86%) as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.063 (m, 1H), 6.88 (m, 1H), 4.72 (s, 1H), 4.56 (s, 1H), 4.13 (m, 3H), 3.88 (m, 2H), 2.94 (m, 2H), 2.62 (m, 2H), 1.36 (s, 9H)
Step 12(R)-7-[3-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester
-
Octacarbonyldicobalt (4.02 g, 11.76 mmol), ethyl chloroacetate (0.71 g, 5.88 mmol), potassium carbonate (1.62 g, 11.76 mmol) and 50 mL of methanol were added into the reaction flask. After stirring for 5 minutes, (R)-[3-oxo-1-(2,4,5-trifluoro-benzyl)-3-(1-bromo-3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazin-7-yl)-propyl]-carbamic acidtert-butyl ester 1m (2.3 g, 3.92 mmol) was added. The reaction mixture was reacted at 60 °C in an oil bath, and the colour of the reaction mixture turned from puce to purple. 2 hours later, Electro-Spray Ionization (ESI) mass spectrometry showed the starting material disappeared. The reaction mixture was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography to obtain the title compound (R)-7-[3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester 1n (1.1 g, yield 50%) as a white solid. MS m/z (ESI): 565.0 [M+1] Reference: Journal of Organometallic Chemistry, 1985, 285(1-3), 293-303
Step 13(R)-7-[3-Amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester hydrochloride
-
[0064](R)-7-[3-tert-Butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester 1n (0.12 g, 2.12 mmol) was added to a solution of 2.2 N hydrochloric acid in 5 mL of ethyl acetate. The reaction mixture was reacted at room temperature for 5 hours and monitored by thin layer chromatography until the disappearance of the starting materials. The reaction mixture was concentrated under reduced pressure to obtain the title compound (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-a]pyrazine-1-carboxylic acid methyl ester hydrochloride 1 (0.12 g, yield 94.3%) as a light yellow solid. MS m/z (ESI): 465.2 [M+1] 1H NMR (400 MHz, CD3OD): δ 7.101-7.08 (m, 1H), 6.906-6.864 (m, 1H), 5.343-4.995 (m, 2H), 4.221-4.093 (m, 5H), 3.954 (s, 3H), 2.978-2.937 (m, 2H), 2.71-2.643 (m, 2H), 2.061 (s, 2H)
| EP2230241A1 * | Nov 27, 2008 | Sep 22, 2010 | Jiangsu Hengrui Medicine Co., Ltd. | Tetrahydro-imidazoý1,5-a¨pyrazine derivatives, preparation methods and medical uses thereof |
| WO2003004498A1 * | Jul 5, 2002 | Jan 16, 2003 | Merck & Co Inc | Beta-amino tetrahydroimidazo (1, 2-a) pyrazines and tetrahydrotrioazolo (4, 3-a) pyrazines as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2005003135A1 * | Jun 18, 2004 | Jan 13, 2005 | Alex Minhua Chen | Phosphoric acid salt of a dipeptidyl peptidase-iv inhibitor |
Zifaxaban, TY-602, Zhifeishaban 知非沙班……Tianjin Institute of Pharmaceutical Research China

Zifaxaban

Zifaxaban
cas 1378266-98-8
rotation (-)
C20 H16 Cl N3 O4 S
C20H16ClN3O4 S, M = 429.87
Tianjin Institute of Pharmaceutical Research
Deep vein thrombosis; Lung embolism
Factor Xa antagonist
TY-602; zhifeishaban; zifaxaban
Chinese J Struc Chem. 2014, 33 (7), 1091-1095.
(S) -5- chloro -N- ((2- oxo _3_ (4_ (2_ oxo _2H_-1-yl) phenyl) oxazolidin-5 -1,3_ yl) methyl) thiophene-2-carboxamide
5-Chloro-N-(5S)-2-oxo-3-[4-(2-oxopyridin-1(2H)-yl)phenyl]oxazolidin-5-ylimethyllthiophene-2-carboxamide]

The title compound(zifaxaban 2, C20H16ClN3O4 S, Mr = 429.87) was synthesized and its crystal structure was determined by single-crystal X-ray diffraction. Zifaxaban crystallizes in monoclinic, space group P21 with a = 5.7900(12), b = 13.086(3), c = 12.889(3) A, β = 100.86(3)°, V = 959.1(3) A3, Z = 2, Dc = 1.489 g/cm3, F(000) = 444, μ = 0.342 mm-1, the final R = 0.0320 and wR = 0.0640 for 2717 observed reflections(I > 2σ(I)).
The absolute configuration of the stereogenic center in the title compound was confirmed to be S by single-crystal X-ray diffraction. Four existing intermolecular hydrogen bonds help to stabilize the lattice and the molecule in the lattice to adopt an L-shape conformation.
Zifaxaban was slightly more active than rivaroxaban 1 in in vitro assay against human FXa and therefore is promising as a drug candidate.
zifaxaban (first disclosed in CN102464658), useful for treating thromboembolic disorders. Zifaxaban, a factor Xa antagonist, is being developed by Tianjin Institute of Pharmaceutical Research, for treating deep vein thrombosis and pulmonary embolism (preclinical, as of November 2014). In May 2014, an IND was filed in China. In June 2014, the institute was seeking to outlicense this product.
In vivo within the cardiovascular, blood coagulation or blood analysis some have formed out of the process of forming a solid mass with the aggregation, called thrombosis, the formation of a solid mass called a thrombus blocks. Thrombosis is an abnormal flow of blood coagulation status due to platelet activation and coagulation factors are activated in accordance therewith.
The blood coagulation was originally a protective mechanism of the organism, there is a mutual antagonism in blood coagulation system and the anti-clotting system. Under physiological conditions, blood clotting factors continue to be activated to produce thrombin, fibrin formation trace, calm on the vascular endothelium, but these traces of fibrin and constantly being activated fibrinolytic system dissolution, while being activated coagulation factors are constantly mononuclear phagocyte system swallowed. The dynamics of the coagulation system and fibrinolysis system, which ensures the blood coagulation potential can also always ensure that the fluid state of the blood.
Sometimes, however, in certain factors can promote the coagulation process, breaking the above dynamic balance triggered the coagulation process, the blood can form a thrombosis or embolism, such as leading to myocardial infarction, stroke, deep vein thrombosis, pulmonary embolism and other thromboembolic disease.
Thromboembolic disease is cardiovascular disease against the most serious diseases, is the first killer of human health. In China, with the improvement and increased aging of the population’s living standards, the incidence of such diseases, mortality, morbidity is increasing every year.
The existing anti-thromboembolic diseases into anti-platelet drugs, anticoagulants and fibrinolytic drugs. Among them, the anti-clotting drugs are the main contents of antithrombotic therapy, mainly thrombin inhibitors and vitamin K antagonists. Heparin and low molecular weight heparin, represented by the presence of oral thrombin inhibitor invalid, non-selective inhibition and high risk of bleeding and other shortcomings. Although warfarin is representative of vitamin K antagonists can be administered orally, but there are narrow therapeutic index, high risk of bleeding and other shortcomings.
Studies have shown that the coagulation process is usually divided into intrinsic coagulation pathway and the extrinsic coagulation pathway. Coagulation process involves a lot of coagulation factors, coagulation factor activated are each the next inactive clotting factor precursor is converted into the activated form. Endogenous, exogenous pathway final summary, the blood coagulation factor X is converted to Xa.
Therefore, theoretically, the direct inhibition of ¾ factor activity should produce effective anti-clotting effect, without the side effects of thrombin inhibitors with. As direct inhibition) (a factor activity on normal hemostasis reaction / adjustment process produces minimal impact. For example, platelets remain low catalytic activity of thrombin on the ability to respond to, and thus does not affect the formation of platelet thrombi, so bleeding integrated minimize the risk of the levy.
research also proved this point. Recently reported a variety of compounds can selectively inhibit efficient Xa, which play a preventive and / or treatment of thromboembolic disease effect (W003000256A1; CN00818966; US2007259913A1; US2007259913A1). Among them, rivaroxaban (Rivaroxaban) was listed in 2008 for hip or knee replacement surgery prophylaxis and treatment of venous thrombosis, with oral, fixed dose and other advantages.
rivaroxaban drawback is the high price of raw materials, low yield preparation, purification of the product is difficult, high production costs. Patent CN00818966 8 reported rivaroxaban synthetic routes as follows:
4

where the first reaction (Preparation of 4- (4-morpholino-3-yl) nitrobenzene) yield of only 17.6%, and rivaroxaban difficult purification.

………………………………
Patent
http://www.google.com/patents/CN103232446A?cl=en
(S) -5- chloro -N- ((2- oxo-3- (4- (2_ oxo -2H- pyridin-1-yl) phenyl) -1, 3_ oxazolidine -5 – yl) methyl) thiophene-2-carboxamide.
[0011] Meanwhile, patent CN201110337461.4 described formula (I) Preparation of the compound:
[0012]

……………………………………..
Patent
CN102464658
http://www.google.com/patents/CN102464658B?cl=en
Example 1
[0046] (S) -5- chloro -N- ((2- oxo-3- (4_ (2_ Batch oxo _2H_ piperidinyl) phenyl) _1,3_ oxazolidin-5-yl) methyl ) thiophene-2-carboxamide (II)
[0048] A, 1- (4- amino-phenyl) -IH- pyridin _2_ -one (Compound VII) is
[0049] The reaction flask was charged with 104g of pyridine -2 (IH) – one (Compound IX), 200g of iodoaniline (compound VIII), 26gCuI, 151g of potassium carbonate, 18g8- hydroxyquinoline, 500mlDMF, nitrogen, heated to reflux, Insulation reaction was stirred 10h. Filtered hot, the filtrate evaporated under reduced pressure to make the solvent, the residue was added ethyl acetate, IL, 0 ° C incubated with stirring lh, filtered and the solid dried, 2L acetonitrile and purified to give 98g dark red solid. Refined liquor was concentrated to 500ml, the ice bath was stirred lh, filtered to give a dark red solid 19g. Total product were 117g, yield 68.9%.
[0050] 1H-NMR (DMSO-Cl6), δ (ppm):… 5 306 (s, 2H), 6 236 (d, 1H), 6 406 (d, 1H), 6 601 (d,. 2H), 6. 977 (d, 2H), 7. 459 (m, 2H).
[0051] B, (R) -2- (2- hydroxy-3- ((2-oxo–2H- pyridin-1-yl) phenyl) amino) propyl) isoindoline-1,3- -dione (Compound V) is
[0052] The reaction flask was added 40gl_ (4- aminophenyl) -IH- pyridin-2-one (Compound VII), 45g (S) _N_ glycidyl phthalimide (Compound VI), 300ml95% ethanol, heating to reflux, the gradual emergence of solid insulation mixing IOh, cooled to room temperature, filtered, and the filter cake washed with ethanol (150ml X 2), and dried to give an off-white solid 38g.
[0053] The mother liquor was taken, evaporated to dryness under reduced pressure, was added 15g (Q-N_ glycidyl phthalimide (Compound VII), 150ml95% ethanol, heated to reflux, stirred incubated 10h, concentrated under reduced pressure, cooled to room temperature , stirred at room temperature for 2h, washed with ethanol and dried to give an off-white solid 33g.
[0054] A total of an off-white solid 71g, yield of 84.8%, without purification, was used directly in the next step.
[0055] 1H-NMR (DMS0_d6), δ (ppm):… 3 053 (m, 1H), 3 194 (m, 1H), 4 644 (m, 2H), 4 020 (m, 1H). , 5. 168 (d, 1H), 5. 851 (t, 1H), 6. 230 (m, 1H), 6. 404 (d, 1H), 6. 665 (d, 2H), 7. 041 ( d, 2H), 7. 435 (m, 1H), 7. 537 (m, 1H), 7. 855 (m, 4H).
[0056] C, ⑶-2- ((2- oxo-3- (4- (2_ oxo _2H_ pyridyl) phenyl) oxazolidin _5_ -1,3_ yl) methyl ) Preparation of isoindoline-1,3-dione (Compound IV) of the
[0057] The reaction flask was charged 50g Compound V, 27gN, N’- carbonyldiimidazole (⑶I), 4_ catalytic amount of dimethylaminopyridine (DMAP), 150mlN, N- dimethylformamide (DMF), stirred for 90 temperature ° C, the reaction was kept for 8 hours to make the solvent was evaporated under reduced pressure, added to IL of water, stirred and dispersed, filtered, washed with water (150mlX “, washed with ethanol (100ml X 1), dried to give a white solid 48g, yield of 90%.
[0058] 1H-NMR (DMSo-CI6), δ (ppm):…. 3 984 (m, 3H), 4 251 (t, 1H), 4 968 (m, 1H), 6 301 (m, 1H), 6. 459 (d, 1H), 7. 423 (d, 2H), 7. 514 (m, 1H), 7. 615 (m, 3H), 7. 892 (m, 4H).
[0059] D, (S) -5- (aminomethyl) -3- (4- (2_ oxo _2H_-1-yl) phenyl) oxazolidin _2_ -1,3_ one hydrochloride (compound III) Synthesis of
[0060] The reaction flask was charged 50g compound IV, 200ml of ethanol, 60ml aqueous methylamine (40%), heated to reflux, stirred incubated 2h, cooled, evaporated under reduced pressure to make the solvent to give a sticky solid.
[0061] added to 300ml of ethanol, 20ml of hydrochloric acid, heated to reflux, stirred incubated lh, cooled to room temperature, incubated with stirring 2h, filtered, washed with ethanol, and dried to obtain;. 34 5g of white solid, yield 88.7%.
1H-NMR (DMS0_d6), δ (ppm):…. 3 240 (m, 2H), 3 980 (m, 1H), 4 255 (m, 1H), 5 028 (m, 1H) , 6. 321 (m, 1H), 6. 475 (d, 1H), 7. 504 (m, 3H), 7. 634 (m, 3H), 8. 561 (s, 1H).
Ε, (S) -5- chloro -N – ((2- oxo-3- (4- (2-oxo–2Η- pyridin-1-yl) phenyl) oxazolidin _1,3_ 5-yl) methyl) thiophene-2-carboxamide Preparation of thiophene (II) of
The reaction flask was charged 15g Compound III, 200ml of tetrahydrofuran, 40ml of water was added with stirring 6. 2g of sodium carbonate was added dropwise 10g5- chloro-thiophene-2-carbonyl chloride (Compound II-1) in tetrahydrofuran IOOml, 30~35 ° C insulation stirred 5h, point board to control the reaction was complete.
to make the solvent was distilled off under reduced pressure, 50ml of water was added, stirring was filtered, the filter cake washed with water and dried to give 18. 5g of white solid.
200ml of acetic acid and purified room temperature overnight, filtered, and the filter cake washed with ethanol and dried to give a white solid 16g, 80% yield.
Melting point: 204 8 ~205 8 ° C;
1H-NMR (DMSo-CI6), δ (ppm):…. 3 623 (t, 2H), 3 893 (m, 1H), 4 230 (t, 1H), 4 871 (m, 1H), 6. 308 (t, 1H), 6. 468 (d, 1H), 7. 193 (d, 1H), 7. 426 (m, 2H), 7. 500 (m, 1H), 7. 637 (m, 4H), 8. 967 (t, 1H);
MS (ESI): m / z = 430 (M + H);
HPLC: rt (%) = 14. 38 (99. 62);
[a] 20d = -37 6 ° (c 0. 3004, DMS0);
WO-2014183667Acetic acid solvate of oxazolidinone derivative, preparation method for the solvate, and application thereof
WO-2014183665Oxazolidinone derivative crystal form I and preparation method and use thereof
WO-2014183666Oxazolidinone derivate crystal form II, preparation method therefor, and application thereo
SEE ABAN SERIES AT…………http://organicsynthesisinternational.blogspot.in/p/aban-series.html
/////////
TORCETRAPIB Revisted



Torcetrapib (CP-529,414, Pfizer) was a drug being developed to treat hypercholesterolemia (elevated cholesterol levels) and prevent cardiovascular disease. Its development was halted in 2006 when phase III studies showed excessive all-cause mortality in the treatment group receiving a combination of atorvastatin (Lipitor) and torcetrapib.
Medical uses
Torcetrapib has not been found to affect either cardiovascular disease or risk of death in those already taking a statin.[1]
Mechanism
Torcetrapib acts (as a CETP inhibitor) by inhibiting cholesterylester transfer protein (CETP), which normally transfers cholesterol from HDL cholesterol to very low density or low density lipoproteins (VLDL or LDL). Inhibition of this process results in higher HDL levels (the “good” cholesterol-containing particle) and reduces LDL levels (the “bad” cholesterol).[vague][citation needed]
Development
The first step in the synthesis was a recently created reaction of amination to p-chlorotrifluoryltoluene, a reaction that was created by Dr. Stephen Buchwald at MIT.[2]
Development of the drug began around 1990; it was first administered in humans in 1999, and manufacturing at production scale began in Ireland in 2005.[3]
Pfizer had previously announced that torcetrapib would be sold in combination with Pfizer’s statin, atorvastatin (Lipitor); however, following media and physician criticism, Pfizer had subsequently planned for torcetrapib to be sold independently of Lipitor.[4]
Clinical trials
A 2004 trial (19 subjects, non-randomised) showed that torcetrapib could increase HDL and lower LDL with and without an added statin.[5]
Nine phase III studies were completed.[6][7][8][9][10][11][12][13][14][15]
Early termination of study
On December 2, 2006 Pfizer cut off torcetrapib’s phase III trial because of “an imbalance of mortality and cardiovascular events” associated with its use.[16] This was a sudden and unexpected event and as late as November 30, 2006 Jeff Kindler, Pfizer’s chief executive, was quoted, “This will be one of the most important compounds of our generation.”[16] In the terminated trial, a 60% increase in deaths was observed among patients taking torcetrapib and atorvastatin versus taking atorvastatin alone.[17] Pfizer recommended that all patients stop taking the drug immediately.[18]
Six studies were terminated early.[6] One of the completed studies found it raised systolic blood pressure and concluded “Torcetrapib showed no clinical benefit in this or other studies, and will not be developed further.”[19]
The drug cost $800m+ to bring into Phase III development.[20]



| A Concise Asymmetric Synthesis of Torcetrapib�, M. Guino, P. H. Phua, J-C. Caille and K. K. Hii, J. Org. Chem., 2007, 72, 6290-6293.doi:10.1021/jo071031gAbstract: Optically active torcetrapib was synthesized in seven steps from achiral precursors without the need for protecting groups, utilizing an enantioselective aza-Michael reaction to achieve asymmetry. |

Example 9 Anhydrous, (-)-(2R,4S)-4-[(3,5-Bιs-trιfluoromethyl-benzyl)-methoxycarbonyl-amιnol-2- ethyl-6-trιfluoromethyl-3,4-dιhydro-2H-quιnolιne-1 -carboxylic acid ethyl ester.
A 2.6g portion of 4(S)-[(3,5-bιs-tπfluoromethyl-benzyl)-methoxycarbonyl-amιno]-2(R)- ethyl-6-tπfluoromethyl-3,4-dιhydro-2H-quιnolιne-1 -carboxylic acid ethyl ester (a mixture of predominantly amorphous material with traces of ethanolate crystalline form; the title compound was also prepared in an analogous manner starting from pure amorphous or pure ethanolate material) was charged to 13 milliliters of hexanes and heated to effect a solution at about 60°C The heat was removed and the reaction was allowed to cool to ambient over a one hour period The reaction was seeded with anhydrous (-)-(2R,4S)-4-[(3,5-bιs-tπfluoromethyl-benzyl)- methoxycarbonyl-amιno]-2-ethyl-6-trιfluoromethyl-3,4-dιhydro-2H-quιnolιne-1 – carboxylic acid ethyl ester and granulated for eighteen hours under ambient conditions. Alternately, the anhydrous crystals may be prepared from hexanes without seeding. The product was collected by filtration and air dried. The isolated product X-ray pattern matched the calculated powder pattern. Density: 1.406 Crystal System: Trigonal
Microscopy: Well formed rods and equant (fractured rods) crystals demonstrating high birefringence when viewed across the C axis. Being in the Trigonal crystal system the crystals do not demonstrate birefringence when viewed down the C axis. The crystals demonstrate a cleavage plane perpendicular to the C axis Fusion Microsocopy In Type A oil dissolution at 50°C.
Dry — clear melt at 86°C.
NMR: No trace of ethanolate
Degree of crystallmity: Highly crystalline Hygroscopicity. Non-hygroscopic at 100% relative humidity over 48 hours.
Appearance: Free flowing white powder
References
- Clark, RW; Sutfin TA, Ruggeri RB, Willauer AT, Sugarman ED, Magnus-Aryitey G, Cosgrove PG, Sand TM, Wester RT, Williams JA, Perlman ME, Bamberger MJ (January 22, 2004). “Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: an initial multidose study of torcetrapib”. Arteriosclerosis, Thrombosis, and Vascular Biology 24 (3): 490–497. doi:10.1161/01.ATV.0000118278.21719.17. PMID 14739125. Retrieved 2006-12-03.
- Clark, RW; Ruggeri RB; Cunningham D; Bamberger MJ (March 2006). “Description of the torcetrapib series of cholesteryl ester transfer protein inhibitors, including mechanism of action”. Journal of Lipid Research 47 (3): 537–552. doi:10.1194/jlr.M500349-JLR200. PMID 16326978. Retrieved 2006-12-03.
- Davidson, MH; McKenny JM; Shear CL; Revkin JH (November 7, 2006). “Efficacy and safety of torcetrapib, a novel cholesteryl ester transfer protein inhibitor, in individuals with below-average high-density lipoprotein cholesterol levels”. Journal of the American College of Cardiology 48 (9): 1774–1781. doi:10.1016/j.jacc.2006.06.067.PMID 17084249. Retrieved 2006-12-03.
- McKenny, JM; Davidson MH; Shear CL; Revkin JH (November 7, 2006). “Efficacy and safety of torcetrapib, a novel cholesteryl ester transfer protein inhibitor, in individuals with below-average high-density lipoprotein cholesterol levels on a background of atorvastatin”. Journal of the American College of Cardiology 48 (9): 1782–1790.doi:10.1016/j.jacc.2006.06.066. PMID 17084250. Retrieved 2006-12-03.
Notes
- Keene, D; Price, C; Shun-Shin, MJ; Francis, DP (Jul 18, 2014). “Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients.”. BMJ (Clinical research ed.) 349: g4379. PMID 25038074.
- Buchwald, Stephen (July 23, 2004). “Research Projects”. Retrieved 2007-10-04.
- “Pfizer Begins Production at Torcetrapib/Atorvastatin Manufacturing Facility” (Press release). Pfizer. June 22, 2005. Retrieved 2006-12-03.
- Berenson, Alex (July 26, 2006). “Heart Pill to Be Sold by Itself”. Business (The New York Times). Retrieved 2006-12-03.
- Brousseau, ME; Schaefer EJ; Wolfe ML; Bloedon LT; Digenio AG; Clark RW; Mancuso JP; Rader DJ (April 8, 2004). “Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol” (abstract). New England Journal of Medicine 350 (15): 1505–1515. doi:10.1056/NEJMoa031766. PMID 15071125. Retrieved 2006-12-03.
- http://clinicaltrials.gov/ct2/results?term=torcetrapib
- http://clinicaltrials.gov/ct2/show/NCT00139061 Phase III Assess HDL-C Increase And Non-HDL Lowering Effect Of Torcetrapib/Atorvastatin Vs. Fenofibrate
- http://clinicaltrials.gov/ct2/show/NCT00134511 Phase III Study To Evaluate The Effect Of Torcetrapib/Atorvastatin In Patients With Genetic High Cholesterol Disorder
- http://clinicaltrials.gov/ct2/show/NCT00134485 Phase III Study To Evaluate The Safety And Efficacy Of Torcetrapib/Atorvastatin In Subjects With Familial Hypercholerolemia
- http://clinicaltrials.gov/ct2/show/NCT00134498 Phase III Study Comparing The Efficacy & Safety Of Torcetrapib/Atorvastatin And Atorvastatin In Subjects With High Triglycerides
- http://clinicaltrials.gov/ct2/show/NCT00267254 Phase III Clinical Trial Comparing Torcetrapib/Atorvastatin To Simvastatin In Subjects With High Cholesterol
- http://clinicaltrials.gov/ct2/show/NCT00138762 Phase III Study of Torcetrapib/Atorvastatin vs Atorvastatin Alone or Placebo in Patients With High Cholesterol
- http://clinicaltrials.gov/ct2/show/NCT00134173 Phase III Coronary IVUS Study to Compare Torcetrapib/Atorvastatin to Atorvastatin Alone in Subjects With Coronary Heart Disease (ILLUSTRATE)
- http://clinicaltrials.gov/ct2/show/NCT00137462 Phase III Lipitor Trial To Study The Effect Of Torcetrpib/Atorvastatin To Atorvastatin Alone.
- http://clinicaltrials.gov/ct2/show/NCT00136981 Phase III Carotid B-Mode Ultrasound Study to Compare Anti-Atherosclerotic Effect of Torcetrpib/Atorvastatin to Atorvastatin Alone. (RADIANCE 1)
- Berenson, Alex (December 3, 2006). “Pfizer Ends Studies on Drug for Heart Disease”. The New York Times. Retrieved 2006-12-03. (registration required)
- Theresa Agovino (Associated Press) (December 3, 2006). “Pfizer ends cholesterol drug development”. Yahoo! News. Retrieved 2006-12-03.[dead link] Each study arm (torcetrapib + atorvastatin vs. atorvastatin alone) had 7500 patients enrolled; 51 deaths were observed in the atorvastatin alone arm, while 82 deaths occurred in the torcetrapib + atorvastatin arm. (Link dead as of 15 January 2007)
- Associated Press (December 2, 2006). “Pfizer cuts off cholesterol drug trials”. Yahoo! News (Yahoo.com). Retrieved 2006-12-03.[dead link] (Link dead as of 15 January 2007)
- Bots et al.; Visseren, Frank L; Evans, Gregory W; Riley, Ward A; Revkin, James H; Tegeler, Charles H; Shear, Charles L; Duggan, William T et al. (July 2007). “Torcetrapib and carotid intima-media thickness in mixed dyslipidaemia (RADIANCE 2 study): a randomised, double-blind trial”. The Lancet 370 (9582): 153–160. doi:10.1016/S0140-6736(07)61088-5.
- Cutler, D. M. (2007-03-29). “The Demise of the Blockbuster?”. The New England Journal of Medicine (Massachusetts Medical Society) 356 (13): 1292–1293. doi:10.1056/NEJMp078020.ISSN 1533-4406. PMID 17392299. Retrieved 2007-04-23.
External links
Troglitazone (Romglizone) an antidiabetic Revisted

Troglitazone, GR-92132X, CI-991, CS-045, Romozin, Prelay, Rezulin, Noscal
CAS 97322-87-7
- CI 991
- CS 045
- Depotox
- GR 92132X
- Noscal
- Rezulin
- Romglizone
- Troglitazone
Withdrawn – 2000
Crystals, m.p. 184-6 °C
Daiichi Sankyo Co., Ltd. INNOVATOR
Troglitazone
 Type-II diabetes mellitus (DM) is characterized by insulin resistance, glucose intolerance, increased hepatic glucose production, and decreased pancreatic insulin secretion. In the past, the drug classes used for type-II DM have targeted the last three of these abnormalities. Sulfonylurea agents bind to ATP-dependent potassium efflux channels to stimulate pancreatic insulin secretion at b-islet cells. The biguanides decrease hepatic glucose production, and thea-glucosidase inhibitors delay carbohydrate digestion to improve glucose tolerance. Until the recent advent of the thiazolidinedione drugs (ciglitazone was first synthesized in 1982), there was no therapy specifically targeting insulin resistance. Drugs of this class all share a common thiazolidine-2-4-dione structure. Marketed drugs of this class include pioglitazone, rosiglitazone, and troglitazone [Figure 1] – the first to reach the market.
Type-II diabetes mellitus (DM) is characterized by insulin resistance, glucose intolerance, increased hepatic glucose production, and decreased pancreatic insulin secretion. In the past, the drug classes used for type-II DM have targeted the last three of these abnormalities. Sulfonylurea agents bind to ATP-dependent potassium efflux channels to stimulate pancreatic insulin secretion at b-islet cells. The biguanides decrease hepatic glucose production, and thea-glucosidase inhibitors delay carbohydrate digestion to improve glucose tolerance. Until the recent advent of the thiazolidinedione drugs (ciglitazone was first synthesized in 1982), there was no therapy specifically targeting insulin resistance. Drugs of this class all share a common thiazolidine-2-4-dione structure. Marketed drugs of this class include pioglitazone, rosiglitazone, and troglitazone [Figure 1] – the first to reach the market.
The “glitazones” act to reduce insulin resistance and also correct hyperglycemia, hyperinsulinemia, and hypertriglyceridemia. Thiazolidinediones bind to the gisoform of the peroxisome proliferator-activated receptor (PPARg), a nuclear transcription factor that regulates the expression of several insulin-responsive genes involved in glucose and lipid metabolism, and the differentiation of fibroblasts into adipose tissue. The net effect is to reduce insulin resistance, mostly through increased glucose uptake by muscle tissue; however, the exact biochemical mechanism is unclear. Effects on lipid metabolism include decreased triglycerides and free fatty acids, and a slight increase or no change in high-density lipoprotein, low-density lipoprotein, and total cholesterol. There also appear to be acute increases in insulin-stimulated glucose uptake that are PPAR-independent. This effect is too rapid to occur via gene transcription, and in the case of troglitazone may result from action of its quinone metabolite. Troglitazone also decreases production of various inflammatory mediators and may antagonize TNFa.
Troglitazone�s most common adverse effect is fluid retention, which may increase preload and induce cardiac hypertrophy. Troglitazone is contraindicated in congestive heart failure, and cases of pulmonary edema have been reported. Troglitazone induces colon polyps in murine models and is therefore contraindicated for patients with familial polyposis coli. Troglitazone and pioglitazone (but not rosiglitazone) induce cytochrome P450 (CYP) 3A4. This enzyme induction can result in decreased drug levels or drug effects with estradiol, terfenadine, cyclosporine, simvistatin, tacrolimus, and other drugs metabolized by CYP 3A4. A small fraction of troglitazone is metabolized by CYP (not 3A4) to an active quinone metabolite, but it is mostly conjugated to sulfate and glucuronide. Troglitazone enhances the anticoagulant effect of warfarin, probably through competitive serum protein binding, and has other drug interactions at the PPAR level. Troglitazone interferes with gemfibrozil’s binding to PPARa, and may decrease NSAID effectiveness by competing for PPARg.
Rezulin� (tradename troglitazone by Parke-Davis) was FDA approved January 29, 1997, and was first marketed in March 1997. Over 600,000 American patients received troglitazone, with an additional 200,000 in Japan. Pre-marketing studies showed 1.9% of patients on troglitazone developed serum alanine aminotransferase levels in excess of three times the upper limit of normal, vs. 0.6% with placebo. Such hepatotoxicity was typically asymptomatic and reversible. A few patients developed overt liver injury, and two liver biopsies among these patients showed hepatocellular injury pattern. It was estimated that only 1 patient in 50,000 to 60,000 would die from liver failure or require liver transplantation. On November 3, 1997, the FDA released a warning regarding 150 adverse events with troglitazone, 35 with acute liver injury, and 3 deaths in Japan from liver failure. The warnings and restrictions about troglitazone were extended in December 1997, July 1998, and June 1999. Troglitazone was voluntarily withdrawn from the US market on March 21, 2000, after it had been demonstrated that Rezulin� was more toxic than either Avandia� (rosiglitazone) or Actos�(pioglitazone).
Troglitazone hepatotoxicity appears to be idiosyncratic. The onset is typically delayed, usually 2-5 months after initiating therapy, although one case was reported after only four doses. Although hypersensitivity has been suggested in several cases, the hallmarks of immune mechanisms, fever, rash, and eosinophilia, are usually absent. Histologic specimens usually show hepatocellular injury, bridging fibrosis and necrosis, intracanalicular cholestasis, and lack of regenerative activity. Samples vary in the amount of WBC infiltration (with or without eosinophils) and steatosis.
Idiosyncratic (or host-dependent) drug reactions are either due to hypersensitivity or to metabolic aberrations. It is not clear whether troglitazone hepatotoxicity is caused by hypersensitivity. Proposed metabolic aberrations include oxidation/reduction reactions with the a-tocopherol moiety on troglitazone (although it is usually considered an antioxidant), reactions from the quinone metabolite (similar to acetaminophen’s quinone metabolite), and genetic variations in cytokines and their receptors, the apoptosis cascade, mitochondrial respiration, and regenerative response. It is unlikely that CYP polymorphisms play a major role, as the incidence of troglitazone hepatotoxicity is too low. Two cases of hepatic toxicity associated with rosiglitazone have also been reported. Although the patients had co-morbidities, exposures to other drugs, and one case may have been due to shock, these cases suggest that hepatotoxicity may be an emerging “class-effect” of thiazolidinediones.
Troglitazone (Rezulin, Resulin, Romozin, Noscal) is an antidiabetic and anti-inflammatory drug, and a member of the drug class of the thiazolidinediones. It was prescribed for patients with diabetes mellitus type 2.[1] It was developed by Daiichi Sankyo Co.(Japan). In the United States, it was introduced and manufactured by Parke-Davis in the late 1990s, but turned out to be associated with an idiosyncratic reaction leading to drug-induced hepatitis. One F.D.A. medical officer evaluating troglitazone, John Gueriguian, did not recommend its approval due to potential high liver toxicity,[2] but a full panel of experts approved it in January 1997.[3] Once the prevalence of adverse liver effects became known, troglitazone was withdrawn from the British market in December 1997, from the United States market in 2000, and from the Japanese market soon afterwards. It didn’t get approval in the rest of Europe.
Approval history
Troglitazone was developed as the first anti-diabetic drug having a mechanism of action involving the enhancement of insulin sensitivity. At the time it was widely believed that such drugs, by addressing the primary metabolic defect associated with Type 2 diabetes, would have numerous benefits including avoiding the risk of hypoglycemia associated with insulin and earlier oral antidiabetic drugs. It was further believed that reducing insulin resistance would potentially reduce the very high rate of cardiovascular disease that is associated with diabetes.[4][5]
Parke-Davis/Warner Lambert submitted the diabetes drug Rezulin for U.S. Food and Drug Administration (F.D.A.) review on July 31, 1996. The medical officer assigned to the review, Dr. John L. Gueriguian, cited Rezulin’s potential to harm the liver and the heart and he questioned its viability in lowering blood sugar for patients with adult-onset diabetes, recommending against the drug’s approval. After complaints from the drugmaker, Gueriguian was removed on November 4, 1996 and his review was purged by the F.D.A.[6][7]Gueriguian and the company had a single meeting, at which Gueriguian used “intemperate” language; The company said it’s objections were based on inappropriate remarks made by Gueriguian.[8] Parke-Davis said at the advisory committee that the risk of liver toxicity was comparable to placebo and that additional data of other studies confirmed this.[9] According to Peter Gøtzsche, when the company provided these additional data one week after approval, they showed a substantial greater risk for liver toxicity.[10]
The F.D.A. approved the drug on January 29, 1997, and it appeared in pharmacies in late March. At the time Dr. Solomon Sobel, a director at the F.D.A., overseeing diabetes drugs, said in a New York Times interview that adverse effects of troglitazone appeared to be rare and relatively mild.[11]
Glaxo Wellcome P.L.C. received approval from the British Medicines Control Agency (MCA) to market troglitazone, as Romozin, in July 1997.[12] After reports of sudden liver failure in patients receiving the drug, the Parke-Davis and the FDA added warnings to the drug label requiring monthly monitoring of liver enzyme levels.[13] Glaxo removed troglitazone from the market in Britain on December 1, 1997.[6] Glaxo had licensed the drug from Sankyo Company of Japan and had sold it in Britain from October 1, 1997.[14][15]
On May 17, 1998, a 55-year old patient named Audrey LaRue Jones died of acute liver failure after taking troglitazone. Importantly, she had been monitored closely by physicians at the National Institutes of Health as a participant in the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) diabetes prevention study.[16][17] This called into question the efficacy of the monitoring strategy. The N.I.H. responded on June 4 by dropping troglitazone from the study.[7][18] Dr. David J. Graham, an F.D.A. epidemiologist charged with evaluating the drug, warned on March 26, 1999 of the dangers of using it and concluded that patient monitoring was not effective in protecting against liver failure. He estimated that the drug could be linked to over 430 liver failures and that patients incurred 1,200 times greater risk of liver failure when taking Rezulin.[7][19] Dr. Janet B. McGill, an endocrinologist who had assisted in the Warner–Lambert’s early clinical testing of Rezulin, wrote in a March 1, 2000 letter to Sen. Edward M. Kennedy (D-Mass.): “I believe that the company . . . deliberately omitted reports of liver toxicity and misrepresented serious adverse events experienced by patients in their clinical studies.”[20]
On March 21, 2000, the F.D.A. withdrew the drug from the market.[21] Dr. Robert I. Misbin, an F.D.A. medical officer, wrote in a July 3, 2000 letter to the House Energy and Commerce Committee of strong evidence that Rezulin could not be used safely, after having been threatened by the FDA with dismissal in March 2000.[6][22] By that time the drug had been linked to 63 liver-failure deaths and had generated sales of more than $2.1 billion for Warner-Lambert.[19] The drug cost $1,400 a year per patient in 1998.[15] Pfizer, which had acquired Warner-Lambert in February 2000, reported the withdrawal of Rezulin cost $136 million.[23]
Lawsuits
In 2009 Pfizer Inc. resolved all but three of 35,000 claims over its withdrawn diabetes drug Rezulin for a total of about $750 million. Pfizer, which acquired rival Wyeth for almost $64 billion, paid about $500 million to settle Rezulin cases consolidated in federal court in New York, according to court filings. The company also paid as much as $250 million to resolve state-court suits. In 2004, it set aside $955 million to end Rezulin cases.[24]
Mode of action
Troglitazone, like the other thiazolidinediones (pioglitazone and rosiglitazone), works by activating peroxisome proliferator-activated receptors (PPARs).
Troglitazone is a ligand to both PPARα and – more strongly – PPARγ. Troglitazone also contains an α-tocopheroyl moiety, potentially giving it vitamin E-like activity in addition to its PPAR activation. It has been shown to reduce inflammation:[25] troglitazone use was associated with a decrease of nuclear factor kappa-B (NF-κB) and a concomitant increase in its inhibitor (IκB). NFκB is an important cellular transcription regulator for the immune response.


rosiglitazone, ciglitazone, darglitazone, englitazone, rosiglitazone, pioglitazone, rosiglitazone, troglitazone
| Systematic (IUPAC) name | |
|---|---|
| (RS)-5-(4-[(6-hydroxy-2,5,7,8-tetramethylchroman-2-yl)methoxy]benzyl)thiazolidine-2,4-dione | |
| Clinical data | |
| Legal status |
?
|
| Pharmacokinetic data | |
| Half-life | 16-34 hours |
| Identifiers | |
| CAS number | 97322-87-7 |
| ATC code | A10BG01 |
| PubChem | CID 5591 |
| IUPHAR ligand | 2693 |
| DrugBank | DB00197 |
| ChemSpider | 5389 |
| UNII | I66ZZ0ZN0E |
| KEGG | D00395 |
| ChEBI | CHEBI:9753 |
| ChEMBL | CHEMBL408 |
| Chemical data | |
| Formula | C24H27NO5S |
| Mol. mass | 441.541 g/mol |
………………….

………………..

A new synthesis of [14C]-labeled CS-045 has been reported: The condensation of 5-acetoxy-2-hydroxy-3,4,6-trimethylacetophenone (I) with phenoxyacetone (II) by means of morpholine and p-toluenesulfonic acid in refluxing benzene gives 6-acetoxy-2,5,7,8-tetramethyl-2-(phenoxymethyl)-3,4-dihydro-2H-benzo[b]pyran-4-one (III), which is reduced with NaBH4 in methanol to the corresponding carbinol (IV). The dehydration of (IV) by means of p-toluenesulfonic acid in refluxing benzene affords 2-acetoxy-2,5,7,8-tetramethyl-2-(phenoxymethyl)-2H-benzo[b]pyran (V), which is hydrogenated with H2 over Pd/C in methanol to give the corresponding 3,4-dihydro derivative (VI). The hydrolysis of (VI) with NaOH in methanol yields the corresponding phenol (VII), which is chloromethylated with paraformaldehyde and dry HCl in dioxane to afford 2-[4-(chloromethyl)phenoxymethyl]-2,5,7,8-tetramethyl-3,4-dihydro-2H-benzo[b]pyran-6-ol (VIII). The protection of (VIII) with chloromethyl methyl ether by means of potassium tert-butoxide in THF gives the corresponding 6-(methoxymethoxy) derivative (IX), which is condensed with [5-14C]-thiazolidine-2,4-dione (X) by means of butyllithium in THF-HMPT to yield 5-[4-[6-(methoxymethoxy)-2,5,7,8-tetramethyl-3,4-dihydro-2H-benzo[b]pyran-2-ylmethoxy]benzyl]-[5-14C]-thiazolidine-2,4-dione (XI). Finally, this compound is deprotected with concentrated HCl in ethylene glycol monomethyl ether at 130 C.
……………….

A new short synthesis of troglitazone has been described: Condensation of the bromoacetal (I) with 4-hydroxybenzaldehyde (II) by means of K2CO3 and NaI in refluxing acetone gives the unsaturated ether (III), which is cyclized with trimethylhydroquinone (IV) by means of bis(trifluoromethylsulfonyl)imide in dichloromethane to yield the 6-hydroxybenzopyran (V). Acylation of (V) with acetic anhydride and DMAP in THF affords the expected acetoxybenzopyran (VI), which is condensed with thiazolidine-2,4-dione (VII) by means of piperidine in toluene to provide the 6-benzylidene-thiazolidine (VIII). The hydrogenation of (VIII) with H2 over Pd/C in methanol gives the corresponding benzyl derivative (IX), which is finally deacetylated with AcOH/HCl/water (3:1:1) in MeOH.
…………..
European Journal of Medicinal Chemistry, 51, 206-215; 2012
http://www.sciencedirect.com/science/article/pii/S0223523412001353

……………………………………….
see Indian Journal of Heterocyclic Chemistry, 15(4), 407-408; 2006
……………………………………………………….
Bioorganic & Medicinal Chemistry Letters, 14(10), 2547-2550; 2004
http://www.sciencedirect.com/science/article/pii/S0960894X04003038


(a) t-Butyldimethylsilyl chloride, imidazole, DMF; (b) LAH, rt, 3 h (75.9%, two steps); (c) 4-fluorobenzaldehyde, KtOBu, DMF, 80 °C, 8 h; (d) 2,4-thiazolidinedione, AcOH, piperidine, toluene, reflux, 4 h (37%, two steps); (e) HCl, MeOH, 15 min; (f) CoCl2, DMG (84%).
………………………
Patent
http://www.google.co.in/patents/US5700820
EXAMPLE-1
A mixture of 70 g of ethyl-3- 4-(6-acetoxy-2,5,7,8-tetramethylchroman-2-ylmethoxy)phenyl!-2-chloropropionate, 13.12 g of thiourea and 80.2 ml of sulpholane was reacted for 80 min., under a nitrogen stream at 115°-120° C. Subsequently, a 656.2 ml Acetic acid, 218.7 ml conc. hydrochloric acid and 109.4 ml water was added to this and the resulting mixture was further heated for 12 hrs at 85°-90° C. The reaction mixture was cooled to room temperature and 196.8 g of sodium bicarbonate was added and once the evolution of carbondioxide had ceased, the solvent was distilled off applying high vacuum. A 10:1 by volume mixture of benzene and ethyl acetate was added to the residue and the crude product was washed with a mixture of equal volumes of a saturated aq. sodium bicarbonate solution & water. The organic layer was dried over anhydrous sodium sulphate and the solvent was distilled off. The resulting crude product was quickly eluted from a silica gel column with 50% ethylacetate-hexane to furnish 40 g of the required 5-{4-(6-hydroxy-2, 5, 7, 8-tetramethylchroman-2-yl-methoxy) benzyl) thiazolidine-2,4-dione (Troglitazone) with a HPLC purity of ˜67-70%. The elution of column was continued further to yield 5- 4-(6-hydroxy-2,5,7,8-tetramethylchroman-2-yl-methoxy)benzyl!2-iminothiazolidine-4-one with HPLC purity of ˜70%.
Lit References:
Oral hypoglycemic agent which improves insulin sensitivity and decreases hepatic glucose production. Prepn: JP Kokai 85 51189; T. Yoshioka et al., US 4572912 (1985, 1986 both to Sankyo); T. Yoshioka et al.,
J. Med. Chem. 32,421 (1989).
Mechanism of action studies: T. P. Ciaraldi et al., Metabolism 39, 1056 (1990); M. Kellerer et al., Diabetes 43, 447 (1994).
Clinical evaluation: T. Kuzuya et al., Diabetes Res. Clin. Pract. 11, 147 (1991).
Clinical metabolic effects: S. L. Suter et al., Diabetes Care 15, 193 (1992).
References
- Fisher, Lawrence (4 November 1997). “Adverse Diabetes Drug News Sends Warner-Lambert Down”. The New York Times. Retrieved 12 December 2012.
- Retired Drugs: Failed Blockbusters, Homicidal Tampering, Fatal Oversights, wired.com
- Cohen, J. S. (2006). “Risks of troglitazone apparent before approval in USA”.Diabetologia 49 (6): 1454–5. doi:10.1007/s00125-006-0245-0. PMID 16601971.
- Henry RR (September 1996). “Effects of troglitazone on insulin sensitivity”. Diabet. Med.13 (9 Suppl 6): S148–50. PMID 8894499.
- Keen H (November 1994). “Insulin resistance and the prevention of diabetes mellitus”. N. Engl. J. Med. 331 (18): 1226–7. doi:10.1056/NEJM199411033311812. PMID 7935664.
- Willman, David (20 December 2000). “NEW FDA: Rezulin Fast-Track Approval and a Slow Withdrawal”. The Los Angeles Times. Retrieved 12 December 2012.
- Willman, David (4 June 2000). “The Rise and Fall of the Killer Drug Rezulin”. The Los Angeles Times. Retrieved 12 December 2012.
- “Report: FDA Removes Medical Officer”.
- Avorn, J (2005). Powerful medicines. New York: Vintage books.
- Gøtzsche, Peter (2013). Deadly medicines and organised crime : how big pharma has corrupted healthcare. London [u.a.]: Radcliffe Publ. p. 185. ISBN 9781846198847.
- Leary, Warren (31 January 1997). “New Class of Diabetes Drug Is Approved”. The New York Times. Retrieved 12 December 2012.
- Sinclair, Neil (31 July 1997). “Glaxo Wellcome gets approval for Romozin”. ICIS News. Retrieved 12 December 2012.
- “www.accessdata.fda.gov”.
- British Broadcasting Corporation (1 December 1997). “Diabetes drug withdrawn from sale”. BBC. Retrieved 12 December 2012.
- Fisher, Lawrence (17 January 1998). “Drug Makers at Threshold of a New Therapy; With a Dose of Biotechnology, Big Change Is Ahead in the Treatment of Diabetes”. The New York Times. Retrieved 12 December 2012.
- Diabetes Prevention Research Group (April 1999). “Design and methods for a clinical trial in the prevention of type 2 diabetes”. Diabetes Care 22 (4): 623–634.doi:10.2337/diacare.22.4.623. Retrieved 12 December 2012.
- Diabetes Prevention Program Research Group (7 February 2002). “Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin”. The New England Journal of Medicine 346 (6): 393–403. doi:10.1056/NEJMoa012512.PMC 1370926. PMID 11832527. Retrieved 12 December 2012.
- Gale, Edwin (January 2006). “Troglitazone: the lesson that nobody learned?”.Diabetologia 49 (1): 1–6. doi:10.1007/s00125-005-0074-6.
- Willman, David (16 August 2000). “FDA’s Approval and Delay in Withdrawing Rezulin Probed”. The Los Angeles Times. Retrieved 12 December 2012.
- Willman, David (10 March 2000). “Fears Grow Over Delay in Removing Rezulin”. The Los Angeles Times. Retrieved 12 December 2012.
- U.S. Food and Drug Administration. “2000 Safety Alerts for Human Medical Products”. U.S. Food and Drug Administration. Retrieved 12 December 2012.
- Willman, David (March 17, 2000). “Physician Who Opposes Rezulin Is Threatened by FDA With Dismissal”. Los Angeles Times.
- Pfizer. “Pfizer Annual Report 2001”. Pfizer. Retrieved 12 December 2012.
- Feeley, Jef (March 31, 2009). “Pfizer Ends Rezulin Cases With $205 Million to Spare”.Bloomberg. Retrieved 6 April 2014.
- Aljada A, Garg R, Ghanim H, et al. (2001). “Nuclear factor-kappaB suppressive and inhibitor-kappaB stimulatory effects of troglitazone in obese patients with type 2 diabetes: evidence of an antiinflammatory action?”. J. Clin. Endocrinol. Metab. 86 (7): 3250–6.doi:10.1210/jc.86.7.3250. PMID 11443197.
External links
- Diabetes Monitor article on troglit
| US4316849 * | 11 Jul 1980 | 23 Feb 1982 | Blasinachim S.P.A. | Process for preparing a crystalline polymorphous type of chenodeoxycholic acid |
| US4572912 * | 28 Aug 1984 | 25 Feb 1986 | Sankyo Company Limited | Treatment of hyperlipemia and hyperglycemia |
| US5248699 * | 13 Aug 1992 | 28 Sep 1993 | Pfizer Inc. | Hydrochloride salt, antidepressant, anorectic |
| US5319097 * | 11 Dec 1991 | 7 Jun 1994 | Imperial Chemical Industries Plc | Pharmaceutical agents |
| AU3255984A * | Title not available | |||
| EP0014590A1 * | 7 Feb 1980 | 20 Aug 1980 | Eli Lilly And Company | Crystalline forms of N-2-(6-methoxy)benzothiazolyl-N’-phenyl urea and process for their preparation |
| EP0022527A1 * | 4 Jul 1980 | 21 Jan 1981 | BLASCHIM S.p.A. | Process for preparing a solvent-free crystalline polymorphous form of chenodeoxycholic acid |
| EP0490648A1 * | 11 Dec 1991 | 17 Jun 1992 | Zeneca Limited | Pharmaceutical agents |
Ragaglitazar ……..Dr. Reddy’s Research Foundation
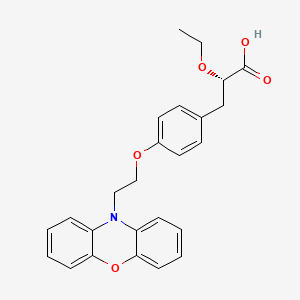
Ragaglitazar
NNC-61-0029, (-) – DRF-2725, NN-622,
(−)DRF 2725
cas 222834-30-2
222834-21-1 (racemate)
Hyperlipidemia; Hypertriglyceridemia; Lipid metabolism disorder; Non-insulin dependent diabetes
PPAR alpha agonist; PPAR gamma agonist
(2S)-2-ETHOXY-3-{4-[2-(10H-PHENOXAZIN-10-YL)ETHOXY]PHENYL}PROPANOIC ACID,
(2S)-2-ethoxy-3-[4-(2-phenoxazin-10-ylethoxy)phenyl]propanoic acid, DRF, 1nyx




……………………………………………………………..

J Med Chem 2001,44(16),2675
http://pubs.acs.org/doi/abs/10.1021/jm010143b

(−)DRF 2725 (6) is a phenoxazine analogue of phenyl propanoic acid. Compound 6 showed interesting dual activation of PPARα and PPARγ. In insulin resistant db/db mice, 6 showed better reduction of plasma glucose and triglyceride levels as compared to rosiglitazone. Compound 6 has also shown good oral bioavailability and impressive pharmacokinetic characteristics. Our study indicates that 6 has great potential as a drug for diabetes and dyslipidemia.

Scheme 1 a
a (a) NaH, DMF, 0−25 °C, 12 h; (b) triethyl 2-ethoxy phosphosphonoacetate, NaH, THF, 0−25 °C, 12 h; (c) Mg/CH3OH, 25 °C, 12 h; (d) 10% aq NaOH, CH3OH, 25 °C, 6 h; (e) (1) pivaloyl chloride, Et3N, DCM, 0 °C, (2) (S)-2-phenyl glycinol/Et3N; (f) 1 M H2SO4, dioxane/water, 90−100 °C, 80 h.
Compound 6 is prepared from phenoxazine using a synthetic route shown in Scheme 1. Phenoxazine 7 upon reaction with p-bromoethoxy benzaldehyde 89 gave benzaldehyde derivative 9. Reacting 9 with triethyl 2-ethoxy phosphonoacetate afforded propenoate 10 as a mixture of geometric isomers. Reduction of 10 using magnesium methanol gave propanoate 11, which on hydrolysis using aqueous sodium hydroxide gave propanoic acid 12 in racemic form. Resolution of 12 using (S)(+)-2-phenyl glycinol followed by hydrolysis using sulfuric acid afforded the propanoic acid 6 in (−) form.
Nate, H.; Matsuki, K.; Tsunashima, A.; Ohtsuka, H.; Sekine, Y. Synthesis of 2-phenylthiazolidine derivatives as cardiotonic agents. II. 2-(phenylpiperazinoalkoxyphenyl)thiazolidine-3-thiocarboxyamides and corresponding carboxamides. Chem. Pharm. Bull. 1987, 35, 2394−2411
(S)-3-[4-[2-(Phenoxazin-10-yl)ethoxy]phenyl]-2-eth-oxypropanoic Acid (6). as a white solid, mp: 89−90 °C.
[α]D 25 = − 12.6 (c = 1.0%, CHCl3).
1H NMR (CDCl3, 200 MHz): δ 1.16 (t, J = 7.0 Hz, 3H), 1.42−1.91 (bs, 1H, D2O exchangeable), 2.94−3.15 (m, 2H), 3.40−3.65 (m, 2H), 3.86−4.06 (m, 3H), 4.15 (t, J = 6.6 Hz, 2H), 6.63−6.83 (m, 10H), 7.13 (d, J = 8.5 Hz, 2H). Mass m/z (relative intensity): 419 (M+, 41), 197 (15), 196 (100), 182 (35), 167 (7), 127 (6), 107 (19).
Purity by HPLC: chemical purity: 99.5%; chiral purity: 94.6% (RT 27.5).

…………………………………
http://www.google.com/patents/US6608194?cl=zh
EXAMPLE 23 (−) 3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxypropanoic acid:

The title compound (0.19 g, 54%) was prepared as a white solid from diastereomer [(2S-N(1S)]-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxy-N-(2-hydroxy-1-phenyl)ethylpropanamide (0.45 g, 0.84 mmol) obtained in example 21b by an analogous procedure to that described in example 22. mp: 89-90° C.
[α]D 25=−12.6 (c=1.0% CHCl3)
1H NMR (CDCl3, 200 MHz): δ 1.16 (t, J=7.02 Hz, 3H), 1.42-1.91 (bs, 1H, D2O exchangeable), 2.94-3.15 (complex, 2H), 3.40-3.65 (complex, 2H), 3.86-4.06 (complex, 3H), 4.15 (t, J=6.65 Hz, 2H), 6.63-6.83 (complex, 10H), 7.13 (d, J=8.54 Hz, 2H).
………………………..
http://www.google.com/patents/EP1049684A1?cl=en
Example 23
(S)-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxypropanoic acid :

The title compound (0.19 g, 54 %) was prepared as a white solid from diastereomer [(2S- N(lS)]-3-[4-[2-(phenoxazin-10-yl)ethoxy]phenyl]-2-ethoxy-N-(2-hydroxy-l- phenyl)propanamide (0.45 g, 0.84 mmol) obtained in example 21b by an analogous procedure to that described in example 22. mp : 89 – 90 °C. [α]D 25 = – 12.6 (c = 1.0 %, CHC13)
*H NMR (CDC13, 200 MHz) : δ 1.16 (t, J = 7.02 Hz, 3H), 1.42 – 1.91 (bs, IH, D20 exchangeable), 2.94 – 3.15 (complex, 2H), 3.40 – 3.65 (complex, 2H), 3.86 – 4.06 (complex, 3H), 4.15 (t, J = 6.65 Hz, 2H), 6.63 – 6.83 (complex, 10H), 7.13 (d, J = 8.54 Hz, 2H).
| Patent | Submitted | Granted |
|---|---|---|
| Benzamides as ppar modulators [US2006160894] | 2006-07-20 | |
| Novel tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US2002077320] | 2002-06-20 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US7119198] | 2006-07-06 | 2006-10-10 |
| Tricyclic compounds and their use in medicine: process for their preparation and pharmaceutical compositions containing them [US6440961] | 2002-08-27 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6548666] | 2003-04-15 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6608194] | 2003-08-19 | |
| CRYSTALLINE R- GUANIDINES, ARGININE OR (L) -ARGININE (2S) -2- ETHOXY -3-{4- [2-(10H -PHENOXAZIN -10-YL)ETHOXY]PHENYL}PROPANOATE [WO0063189] | 2000-10-26 | |
| Pharmaceutically acceptable salts of phenoxazine and phenothiazine compounds [US6897199] | 2002-11-14 | 2005-05-24 |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6939988] | 2005-09-06 |
WO-2014181362
-
wo/2014/181362 a process for the preparation of 3 … – WIPO
patentscope.wipo.int/search/en/WO2014181362Nov 13, 2014 – (WO2014181362) A PROCESS FOR THE PREPARATION OF 3-ARYL-2-HYDROXY PROPANOIC ACID COMPOUNDS …
A process for the preparation of 3-aryl-2-hydroxy propanoic acid compounds
ragaglitazar; saroglitazar
Council of Scientific and Industrial Research (India)
Process for preparing enantiomerically pure 3-aryl-2-hydroxy propanoic acid derivatives (eg ethyl-(S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate), using S-benzyl glycidyl ether as a starting material. Useful as intermediates in the synthesis of peroxisome proliferator activated receptor agonist such as glitazars (eg ragaglitazar or saroglitazar). Appears to be the first filing on these derivatives by the inventors; however see WO2014181359 (for a concurrently published filing) and US8748660 (for a prior filing), claiming synthesis of enantiomerically pure compounds.
- Dolling, U. H.; Davis, P.; Grabowski, E. J. J. Efficient Catalytic Asymmetric Alkylations. 1. Enantioselective Synthesis of (+)-Indacrinone via Chiral Phase-Transfer Catalysis. J. Am. Chem. Soc. 1984, 106, 446–447.
- Andrus, M. B.; Hicken, E. J.; Stephens, J. C. Phase-Transfer Catalyzed Asymmetric Glycolate Alkylation. Org. Lett. 2004, 6, 2289–2292.
- Andrus, M. B.; Hicken, E. J.; Stephens, J. C.; Bedke, D. K. Asymmetric Phase-Transfer Catalyzed Glycolate Alkylation, Investigation of the Scope, and Application to the Synthesis of (-)-Ragaglitazar. J. Org. Chem. 2005, ASAP.
- Henke, B. R. Peroxisome Proliferator-Activated Receptor Dual Agonists for the Treatment of Type 2 Diabetes. J. Med. Chem. 2004, 47, 4118–4127.
- Wilson, T. M.; Brown, P. J.; Sternbach, D. D.; Henke, B. R. The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 2000, 46, 1306–1317.
- Uchida, R.; Shiomi, K.; Inokoshi, J.; Masuma, R.; Kawakubo, T.; Tanaka, H.; Iwai, Y.; Omura, A. Kurasoins A and B, New Protein Farnesyltrasferase Inhibitors Produced by Paecilomyces sp. FO-3684. J. Antibio. 1996, 49, 932–934.
TAK-733……. clinical studies for cancer treatment.

TAK-733
CAS: 1035555-63-5
Synonym: TAK-733; TAK 733; TAK733.
IUPAC/Chemical name:
(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
Chemical Formula: C17H15F2IN4O4
Exact Mass: 504.01060
Molecular Weight: 504.23
Elemental Analysis: C, 40.49; H, 3.00; F, 7.54; I, 25.17; N, 11.11; O, 12.69
Phase I clinical studies for cancer treatment.Takeda Pharmaceutical,
Solid Tumors Therapy
Description of TAK-733: TAK-733 is an orally bioavailable small-molecule inhibitor of MEK1 and MEK2 (MEK1/2) with potential antineoplastic activity. MEK inhibitor TAK-733 selectively binds to and inhibits the activity of MEK1/2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 (MAP2K1/K2) are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.
Current developer: Millennium Pharmaceuticals, Inc./Takeda Pharmaceutical Company Limited.
TAK-733 is being developed at Millennium Pharmaceuticals for the treatment of adult patients with advanced non-hematological malignancies. Phase I clinical trials are ongoing for the treatment of advanced metastatic melanoma. In preclinical studies, the compound has been shown to bind to and potently inhibit MEK.
………………………………….
Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer
- Takeda San Diego;10410 Science Center Drive, San Diego, CA 92121, United States
http://www.sciencedirect.com/science/article/pii/S0960894X11000941

Synthesis of compounds 26 and 27 (Route 4). Reagents and conditions: (a) 1-chloro-2,4-dinitrobenzene, K2CO3, DMF; (b) (R)-O-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)hydroxylamine or 2,2-dimethyl-1,3-dioxan-5-amine, K2CO3 or Cs2CO3, DMF; (c) HCl, THF; (d) Selectfluor, CH3CN,DMF.
TAK-733 exhibited potent enzymatic and cell activity with an IC50 of 3.2 nM against constitutively active MEK enzyme and an EC50 of 1.9 nM against ERK phosphorylation in cells. TAK-733 did not inhibit any other kinases, receptors or ion channels that were tested with inhibitor concentrations up to 10 μM. TAK-733 was found to bind plasma protein moderately (ca. 97% for human and 96% for mouse), and exhibit high permeability and high microsomal stability across species. It did not inhibit P450s up to 30 μM.
The co-crystal structure of TAK-733 in the MEK1 allosteric site has been solved (Fig. 3). As predicted, the pyridone oxygen makes a hydrogen bond with the backbone NH of Ser212. The 2-fluoro-4-iodoaniline moeity sits in the deep lipophilic pocket. The pyrimidinone oxygen makes a hydrogen bond with Lys97, and the propanediol terminal hydroxyl interacts with both Lys97 and the ADP phosphate.

-
Figure 3.
The X-ray co-crystal structure of TAK-733 in the MEK1 allosteric site.

(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
| Molecular Weight: | 504.23 |
| TAK-733 Formula: | C17H15F2IN4O4 |
| CAS Number: | 1035555-63-5 |
TAK-733 is an orally bioavailable small-molecule inhibitor of MEK1 and MEK2 (MEK1/2) with potential antineoplastic activity. MEK inhibitor TAK-733 selectively binds to and inhibits the activity of MEK1/2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 (MAP2K1/K2) are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.
BRAF L597 mutations in melanoma are associated with sensitivity to MEK inhibitors.
Dahlman et al. Cancer Discov. 2012 Jul 13. PMID: 22798288.Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer.
Dong et al. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1315-9. PMID: 21310613.

16: 1652-1659


MEK kinases regulate the pathway that mediates proliferative and anti-apoptotic signaling factors that promote tumor growth and metastasis. TAK-733 is an MEK kinase inhibitor that entered phase I clinical trials for the treatment of cancer. A noteworthy feature of this short synthesis (25% yield overall) is the one-pot, three-step synthesis of the fluoropyridone D, in which the fluorine atom is present at the outset.
The reaction of F with the nosylate G gave a mixture of N- and O-alkylation products (8:1) from which the desired N-alkylation product was isolated by crystallization. The mixture of N-methyl pyrrolidine (NMP) and methanol used in the final deprotection step, helped to ensure formation of the desired polymorph. The nine-step discovery synthesis (3% overall yield) is also presented.
…………………………….
http://www.google.com/patents/WO2008079814A2?cl=en
Example 18: (i?)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione

[0364] To a solution of (i?)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)- 8-methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione (example 6) (1 g, 2.06 mmol, 1 eq) in DMF (19 mL) was added dropwise a mixture of Selectfluor (801 mg, 2.26 mmol, 1.1 eq) in acetonitrile (9 niL) and DMF (5 niL) at ambient temperature while stirring under nitrogen. The reaction mixture was stirred for 10 minutes at ambient temperature and filtered. The filtrate was purified by preparatory LC/MS (30-55% CH3CN in H2O) to give (i?)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (example 18) as an off-white solid. The recovered starting material was subjected to the same reaction conditions to produce a second crop of product. The total yield of product was 347 mg (33% yield). 1H NMR (400 MHz, DMSO-J6) δ ppm 3.36 – 3.40 (m, 1 H) 3.43 – 3.50 (m, 1 H) 3.58 (s, 3 H) 3.61 – 3.72 (m, 1 H) 3.72 – 3.82 (m, 1 H) 4.27 – 4.37 (m, 1 H) 4.78 – 4.87 (m, 1 H) 5.14 (d, J=5.81 Hz, 1 H) 6.93 – 7.03 (m, 1 H) 7.53 (d, J=8.84 Hz, 1 H) 7.65 – 7.74 (m, 1 H) 8.52 (s, 1 H) 10.25 (d, J=LOl Hz, 1 H). [M+H] calc’d for Ci7Hi5F2IN4O4, 505; found, 505.
OTHER ISOMER
Example 19: (5)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione
 CAREFUL PLEASE
CAREFUL PLEASE[0365] To a solution of (5)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)- 8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (example 5) (1.25 g, 2.58 mmol, 1 eq) in DMF (26 mL) was added a mixture of Selectfluor (1.187 g, 3.35 mmol, 1.3 eq) in acetonitrile (26 mL). The reaction mixture was irradiated with microwave at 82 0C for 10 minutes and filtered. The filtrate was purified by preparatory LC/MS (30-55% CH3CN in H2O) to give (5)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione (example 19) as an off-white solid (331 mg, 25% yield). 1H NMR (400 MHz, DMSO-J6) δ ppm 3.36 – 3.42 (m, 1 H) 3.42 – 3.51 (m, 1 H) 3.59 (s, 3 H) 3.63 – 3.72 (m, 1 H) 3.73 – 3.81 (m, 1 H) 4.32 (dd, J=13.14, 3.03 Hz, 1 H) 4.79 (br. s., 1 H) 5.10 (br. s., 1 H) 6.98 (td, J=8.46, 5.31 Hz, 1 H) 7.52 (dd, J=8.46, 1.14 Hz, 1 H) 7.68 (dd, J=10.36, 1.77 Hz, 1 H) 8.51 (s, 1 H) 10.24 (d, J=2.27 Hz, 1 H). [M+H] calc’d for Ci7Hi5F2IN4O4, 505; found, 505.
……………………………………….
http://www.google.com/patents/US8436200?cl=en

The synthetic process of the present invention allows for the preparation of (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione while avoiding the use of a fluorinating reagent in the last step. That is, the present invention provides a valuable process for making (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione characterized by the reaction of a compound of formula (8) with 2-fluoro-4-iodoaniline to give a compound of formula (9). Such a process avoids the use of costly and possible hazardous fluorinating regents in later steps which has significant advantages in large-scale manufacture.
Example 8.1(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
Combine (R)-3-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (24.75 g, 45.58 mmol) and ethanol (250 mL). Add aqueous 9N sulfuric acid (50 mL) over 5 minutes. Heat to 75° C. After 2 hour, cool to ambient temperature and then cool in an ice bath to give a solid. Collect the solid by filtration, rinse with ethanol (3×30 mL), and dry to give the title compound. 1H NMR (400 MHz, DMSO-d6) δ10.24 (s, 1H), 8.52 (s, 1H), 7.69 (dd, 1H, J=10.4, 1.8 Hz), 7.52 (d, 1H, J=8.6 Hz), 6.98 (m, 1H), 5.14 (brs, 1H), 4.83 (brs, 1H), 4.32 (dd, 1H, J=12.9, 2.5 Hz), 3.76 (m, 1H), 3.67 (dd, 1H, J=13.1, 12.9 Hz), 3.58 (s, 3H), 3.46 (ddd, 1H, J=10.9, 5.3, 5.1 Hz), 3.38 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ161.3 (d, J=4.0 Hz), 155.6 (d, J=22.8 Hz), 154.6 (d, J=250 Hz), 152.0, 150.6, 134.3 (d, J=231 Hz), 133.8 (d, J=7.1 Hz), 133.1 (d, J=3.0 Hz), 127.8 (d, J=10.3 Hz), 125.3 (d, J=7.0 Hz), 123.9 (d, J=21.5 Hz), 95.0 (d, J=4.0 Hz), 87.1 (d, J=7.8 Hz), 68.0, 63.8, 50.1, 28.8; 19F NMR (376 MHz, DMSO-d6) δ-124.4, −149.8; MS (M+H)+m/z calcd 505.0. found 505.0.
Google+
|
Information about this agent |
TAK-733 is currently in Phase I clinical trials and is being developed by Millennium Pharmaceuticals, Inc. (a part of Takeda Pharmaceutical Company Limited).
|
References |
1: Acquaviva J, Smith DL, Jimenez JP, Zhang C, Sequeira M, He S, Sang J, Bates RC, Proia DA. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther. 2014 Feb;13(2):353-63. doi: 10.1158/1535-7163.MCT-13-0481. Epub 2014 Jan 7. PubMed PMID: 24398428.
2: Zhang Y, Xue D, Wang X, Lu M, Gao B, Qiao X. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. Mol Med Rep. 2014 Jan;9(1):83-90. doi: 10.3892/mmr.2013.1781. Epub 2013 Nov 7. PubMed PMID: 24213221.
3: Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T, Yoshida S. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013 Dec 1;73(23):7043-55. doi: 10.1158/0008-5472.CAN-13-1825. Epub 2013 Oct 11. PubMed PMID: 24121489.
4: Garraway LA, Baselga J. Whole-genome sequencing and cancer therapy: is too much ever enough? Cancer Discov. 2012 Sep;2(9):766-8. doi: 10.1158/2159-8290.CD-12-0359. PubMed PMID: 22969114.
5: Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, Atefi M, Su Z, Branch S, Lyle PL, Hicks DJ, Bozon V, Glaspy JA, Rosen N, Solit DB, Netterville JL, Vnencak-Jones CL, Sosman JA, Ribas A, Zhao Z, Pao W. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012 Sep;2(9):791-7. Epub 2012 Jul 13. PubMed PMID: 22798288; PubMed Central PMCID: PMC3449158.
6: von Euw E, Atefi M, Attar N, Chu C, Zachariah S, Burgess BL, Mok S, Ng C, Wong DJ, Chmielowski B, Lichter DI, Koya RC, McCannel TA, Izmailova E, Ribas A. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer. 2012 Apr 19;11:22. PubMed PMID: 22515704; PubMed Central PMCID: PMC3444881.
7: Dong Q, Dougan DR, Gong X, Halkowycz P, Jin B, Kanouni T, O’Connell SM, Scorah N, Shi L, Wallace MB, Zhou F. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1315-9. doi: 10.1016/j.bmcl.2011.01.071. Epub 2011 Jan 22. PubMed PMID: 21310613.
| US8030317 | Dec 18, 2007 | Oct 4, 2011 | Takeda Pharmaceutical Company Limited | MAPK/ERK kinase inhibitors |
| US20080255160 | Dec 18, 2007 | Oct 16, 2008 | Qing Dong | Mapk/erk kinase inhibitors |
| WO2008000020A1 | Jun 27, 2007 | Jan 3, 2008 | Gary L Corino | Improved process |
| EP1894932A1 | Jun 10, 2005 | Mar 5, 2008 | Japan Tobacco, Inc. | 5-amino-2,4,7-trioxo-3,4,7,8-tetrahydro-2H-pyrido[2,3-d]pyrimidine derivatives and related compounds for the treatment of cancer |
| US20050222177 * | Jul 29, 2004 | Oct 6, 2005 | Irm Llc | Diseases with abnormal activation of the Abl, BCR-Abl, Bmx, CSK, TrkB, FGFR3, Fes, Lck, B-RAF, C-RAF, MKK6, alpha and beta SAPK2 kinases; antiproliferative; pyrrolo[2,3-d]pyrimidine-7-carboxylic acid [3-phenylcarbamoyl-phenyl]-amides and pyrrolo[3,2-c]pyridine analogs |
EMA Guideline on similar Biological Medicinal Products adopted
![]()

EMA Guideline on similar Biological Medicinal Products adopted
On 23 October, the CHMP adopted the revised Guideline on similar biological medicinal products. Get more details here.

Last year the “Draft Guideline on Similar Biological Medicinal Products” was published by EMA.
After agreement of the revised draft by the Biosimilar Medicinal Products Working Party and Biologics Working Party in July, the CHMP adopted and published the final Guideline on 23 October 2014. They summarized the outline of the document as follows:
“This Guideline outlines the general principles to be applied for similar biological medicinal products (also known as biosimilars) as referred to in Directive 2001/83/EC, as amended, where it is stated that ‘the general principles to be applied [for similar biological medicinal products] are addressed in a guideline taking into account the characteristics of the concerned biological medicinal product published by the Agency’.
This Guideline describes and addresses the application of the biosimilar approach, the choice of the reference product and the principles for establishing biosimilarity.

The scope of the guideline is to fulfil the requirement of section 4, Part II, Annex I to Directive 2001/83/EC, as amended, which states that ‘the general principles to be applied [for similar biological medicinal products] are addressed in a guideline taking into account the characteristics of the concerned biological medicinal product published by the Agency’.”
The date for coming into effect is 30 April 2015 (with the advice: After adoption by CHMP applicants may apply some or all provisions of this guideline in advance of this date.). The document replaces the Guideline on similar biological medicinal products (CHMP/437/04).
For further infromation please see the complete “Guideline on similar biological medicinal products“.
Aplaviroc, AK602, GSK-873140

Aplaviroc
4-(4-{[(3R)-1-butyl-3-[(R)-cyclohexylhydroxymethyl]-2,5-dioxo- 1,4,9-triazaspiro[5.5]undecan-9-yl]methyl}phenoxy)benzoic acid
for the treatment of HIV infection
461023-63-2 of hydrochloride
461443-59-4 (free base)
873140
AK-602
GW-873140
ONO-4128
ono…….innovator
| Ono Pharmaceutical Co., Ltd. |
| Identifiers | |
|---|---|
| CAS number | 461023-63-2 |
| ATC code | None |
| PubChem | CID 3001322 |
| ChemSpider | 2272720 |
| UNII | 98B425P30V |
| KEGG | D06557 |
| ChEMBL | CHEMBL1255794 |
| Chemical data | |
| Formula | C33H43N3O6 |
| Mol. mass | 577.711 g/mol |
Aplaviroc (INN, codenamed AK602 and GSK-873140) is a CCR5 entry inhibitor developed for the treatment of HIV infection.[1][2] It is developed by GlaxoSmithKline.
In October 2005, all studies of aplaviroc were discontinued due to liver toxicity concerns.[3][4] Some authors have claimed that evidence of poor efficacy may have contributed to termination of the drug’s development;[5] the ASCENT study, one of the discontinued trials, showed aplaviroc to be under-effective in many patients even at high concentrations.[6]
Aplaviroc hydrochloride, an orally-effective, long-acting chemokine CCR5 receptor antagonist, had been under development by Ono and GlaxoSmithKline for the treatment of HIV infection. In early 2006, the companies discontinued development of the antagonist based on reports of elevated liver function test values from clinical studies.
Originally developed at Ono, aplaviroc was licensed to GlaxoSmithKline in 2003 for development, manufacturing and marketing. GlaxoSmithKline also obtained rights to evaluate the agent in non-HIV conditions worldwide with the exception of Japan, South Korea and Taiwan.
A low-molecular-weight compound, aplaviroc prevents HIV viral infection by blocking the binding of the virus to the CCR5 receptor
……………….
WO 2002074770
0r
http://www.google.com/patents/EP1378510A1?cl=en
Reference example 3(3)
- (3R)-1-butyl-2,5-dioxo-3-((1R)-1-hydroxy-1-cyclohexyl)-1,4,9-triazaspiro[5.5]undecane • hydrochloride
-
[0136]

TLC:Rf 0.32 (butanol:acetic acid:water = 4:2:1);
NMR (CD3OD): δ 4.16 (d, J = 2.0 Hz, 1H), 3.95 (m, 1H), 3.70 (m, 1H), 3.52 (m, 1H), 3.37 (m, 1H), 3.28 (m, 1H), 3.22-3.13 (m, 2H), 2.46-1.93 (m, 6H), 1.80-1.64 (m, 5H), 1.48-1.15 (m, 6H), 1.02-0.87 (m, 5H);
Optical rotation:[α]D +1.22 (c 1.04, methanol, 26°C).

Example 9(54)
- (3R)-1-butyl-2,5-dioxo-3-((1R)-1-hydroxy-1-cyclohexylmethyl)-9-(4-(4-carboxyphenyloxy)phenylmethyl)-1,4,9-triazaspiro[5.5]undecane • hydrochloride
-
[0359]

TLC:Rf 0.43(chloroform:methanol = 5:1);
NMR (CD3OD):δ 8.05 (d, J = 9.0 Hz, 2H), 7.61 (d, J = 9.0 Hz, 2H), 7.19 (d, J = 9.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H), 4.38 (s, 2H), 4.17 (d, J = 2.1 Hz, 1H), 4.02 (m, 1H), 3.78 (m, 1H), 3.60-3.40 (m, 3H), 3.30-3.10 (m, 2H), 2.56-1.86 (m, 6H), 1.82-1.60 (m, 5H), 1.52-1.16 (m, 6H), 1.06-0.82 (m, 2H), 0.97 (t, J = 7.2 Hz, 3H).

………………….
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
Owing to the special properties of piperazines (increased solubility and H-bond acceptor capability etc.) it is often considered to be a privileged structure and therefore occurs widely, for instance in GlaxoSmithKlines investigational anti-HIV drug aplaviroc (4.37) which, despite being a promising CCR5 receptor antagonist, was discontinued due to hepatotoxicity concerns. In this compound the spirodiketopiperazine unit (4.35) was designed to mimic a type-1 β-turn (4.36) as present in G-protein coupled receptors (Figure 14) [117].
![[1860-5397-9-265-14]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-14.png?scale=2.0&max-width=1024&background=FFFFFF)
The synthesis of aplaviroc and its analogues can be accomplished via the use of an Ugi multicomponent reaction (Ugi-MCR) [118]. The procedure involved the condensation of piperidone 4.38 and butylamine (4.39) followed by reaction of the resulting imine with isocyanide 4.41 and interception of the nitrilium intermediate with the amino acid4.40 (Scheme 47) [119]. This sequence was completed by structural rearrangement and acid-mediated ring closure to produce the spirocyclic diketopiperazine 4.43. Following debenzylation this material was subjected to a reductive amination finally affording aplaviroc analogues (Scheme 47).
![[1860-5397-9-265-i47]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i47.png?scale=2.0&max-width=1024&background=FFFFFF)
- 117 Habashita, H.; Kokubo, M.; Hamano, S.; Hamanaka, N.; Toda, M.; Shibayama, S.; Tada, H.; Sagawa, K.; Fukushima, D.; Maeda, K.; Mitsuya, H. J. Med. Chem. 2006, 49, 4140–4152. doi:10.1021/jm060051s
- Dömling, A.; Huang, Y. Synthesis 2008, 2859–2883. doi:10.1055/s-0030-1257906
ref 118 - Nishizawa, R.; Nishiyama, T.; Hisaichi, K.; Matsunaga, N.; Minamoto, C.; Habashita, H.; Takaoka, Y.; Toda, M.; Shibayama, S.; Tada, H.; Sagawa, K.; Fukushima, D.; Maeda, K.; Mitsuya, H.Bioorg. Med. Chem. Lett. 2007, 17, 727–731. doi:10.1016/j.bmcl.2006.10.084
ref 119
| Patent | Submitted | Granted |
|---|---|---|
| Triazaspiro[5.5]undecane derivative and pharmaceutical composition comprising the same as active ingredient [US7262193] | 2005-09-29 | 2007-08-28 |
| Drugs containing triazaspiro[5.5]undecane derivatives as the active ingredient [US7285552] | 2004-06-03 | 2007-10-23 |
| Triazaspiro[5.5]undecane derivatives and drugs containing the same as the active ingredient [US7053090] | 2004-04-29 | 2006-05-30 |
| WO1998031364A1 * | Jan 20, 1998 | Jul 23, 1998 | Timothy Harrison | 3,3-disubstituted piperidines as modulators of chemokine receptor activity |
| WO2000014086A1 * | Jan 21, 1999 | Mar 16, 2000 | Kyowa Hakko Kogyo Kk | Chemokine receptor antagonists and methods of use therefor |
| WO2002074769A1 * | Mar 18, 2002 | Sep 26, 2002 | Kenji Maeda | Drugs containing triazaspiro[5.5]undecane derivatives as the active ingredient |
References
- Maeda, Kenji; Ogata, Hiromi; Harada, Shigeyoshi et al. (2004). “Determination of binding sites of a unique CCR5 inhibitor AK602 / ONO-4128/ GW873140 on human CCR5” (PDF). Conference on Retroviruses and Opportunistic Infections. Archived from the original on November 3, 2005.
- Nakata, Hirotomo; Maeda, Kenji; Miyakawa, Toshikazu et al. (February 2005). “Potent Anti-R5 Human Immunodeficiency Virus Type 1 Effects of a CCR5 Antagonist, AK602/ONO4128/GW873140, in a Novel Human Peripheral Blood Mononuclear Cell Nonobese Diabetic-SCID, Interleukin-2 Receptor γ-Chain-Knocked-Out AIDS Mouse Model”. Journal of Virology 79 (4): 2087–96.doi:10.1128/jvi.79.4.2087-2096.2005.
- “Aplaviroc (GSK-873,140)”. AIDSmeds.com. October 25, 2005. Retrieved September 5, 2008.[dead link]
- Nichols WG, Steel HM, Bonny T et al. (March 2008). “Hepatotoxicity Observed in Clinical Trials of Aplaviroc (GW873140)”.Antimicrobial Agents and Chemotherapy 52 (3): 858–65. doi:10.1128/aac.00821-07. PMC 2258506. PMID 18070967.
- Moyle, Graeme (December 19, 2006). “The Last Word on Aplaviroc: A CCR5 Antagonist With Poor Efficacy”. The Body.Archived from the original on 6 October 2008. Retrieved September 5, 2008.
- Currier, Judith; Lazzarin, Adriano; Sloan, Louis et al. (2008). “Antiviral activity and safety of aplaviroc with lamivudine/zidovudine in HIV-infected, therapy-naive patients: the ASCENT (CCR102881) study”. Antiviral Therapy (Lond.) 13 (2): 297–306.PMID 18505181.
Further reading
- Horster, S; Goebel, FD (April 2006). “Serious doubts on safety and efficacy of CCR5 antagonists: CCR5 antagonists teeter on a knife-edge”. Infection 34 (2): 110–13. doi:10.1007/s15010-006-6206-1. PMID 16703305.
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
