………………….
…………………………………..
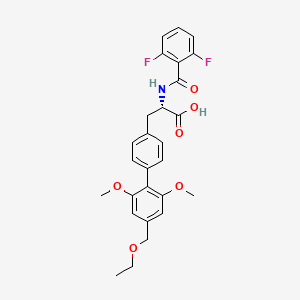
Example 12: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
The product obtained in Example 10 (200 mg) was hydrolyzed in a similar manner as described in Example 3 to give the title compound (160 mg) . The product obtained in Example 11 (220 mg) was also hydrolyzed in a similar manner as described in Example 3 to give the title compound (167 mg) . mp. 156-158°C; IR (Nujol) 1735, 1655 cm“1; MS (ESI) m/z 498 (M-H) .
…………………….
PATENT
https://www.google.com/patents/WO2003072536A1?cl=en
OUT LINE
phenylalanine derivative of the formula (I) :
wherein X1 is a halogen atom, X2 is a halogen atom, Q is a group of the formula -CH2– or -(CH2)2– and Y is a lower alkyl group, or a pharmaceutically acceptable salt thereof, which has excellent inhibitory activity against α4 integrin-mediated cell adhesion.
Thus, the present invention relates to a process for preparing a compound of the formula (I) :
wherein the symbols are the same as defined above, or a pharmaceutically acceptable salt thereof, comprising : (1) coupling a compound of the formula (VI) :
wherein Z is a leaving group, R1NH is a protected amino group and C02R is a protected carboxyl group with a compound of the formula (V) :
wherein the symbols are the same as defined above, removing the protecting group from the protected amino group, and if necessary, converting the resulting compound into a salt, to yield a compound of the formula (IV) :
wherein the symbols are the same as defined above, or a salt thereof,
(2) condensing the compound (IV) or a salt thereof with a compound of the formula (III) :
wherein the symbols are the same as defined above, a salt or a reactive derivative thereof to yield a compound of the formula (II) :
Ethyl (ocS) – – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4- hydroxybenzene propionate and ethyl (otS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzene propionate are described in J. Med. Chem. , 33: 1620 (1990) and JP-A-7- 157472, respectively. 4-Bromo-3, 5-dimethoxybenzyl alcohol is described in, for example, J. Med. Chem. , 20: 299 (1977), and can also be prepared according to the following process.
Firstly, 4-bromo-3, 5-dihydroxybenzoic acid is methylated to give methyl 4-bromo-3, 5-dimethoxybenzoate, which is then reduced to yield 4-bromo-3, 5-dimethoxy benzyl alcohol. The methylation can be carried out by reacting with dimethyl sulfate in the presence of a base in a suitable solvent (e.g., ethyl acetate). The reduction can be carried out by reacting with an reducing agent (e.g., lithium alminium hydride, sodium borohydride and calcium borohydride) in a suitable solvent (e.g., tetrahydrofuran) .
EXAMPLES
The following Examples are provided to further illustrate the process of preparation according to the present invention. In the following examples, some compounds may be referred to by different compound name depending on the nomenclature, as illustrated below.
Ethyl (αS) -α-amino-4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate
Another name: ethyl (2S) -2-amino-3- [4- (4-ethoxymethyl- 2, 6-dimethoxyphenyl) phenyl]propanoate
Ethyl (αS) – [ [1, 1-dimethylethoxy] carbonyl] amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate
Another name 1: ethyl (2S) -2- [ (t-butoxycarbonyl) – amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) – phenyl]propanoate
Another name 2: Ethyl N- (t-butoxycarbonyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
Ethyl (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionate Another name 1: Ethyl (2S) -2- [ (2, 6- difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6- di ethoxyphenyl) phenyl] propanoate
Another name 2: Ethyl N- [2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
(ocS) – – [ (2, 6-Difluorobenzoyl) amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionic acid
Another name 1: (2S) -2- [ (2, 6-difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) phenyl]propanoic acid
Another name 2: N- [ 2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
EXAMPLE 1 (1) Under nitrogen atmosphere, pyridine (130.3 g) and trifluoromethanesulfonic anhydride (170.4 g) were added dropwise to a solution of ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-hydroxybenzenepropionate
(170.0 g) in dichloromethane (1.7 L) at 10 ° C or below. After stirring for 1 hour at the same temperature, water
(850 ml) was added dropwise to the mixture and the mixture was stirred for 2 hours at the same temperature. The organic layer was washed with 10 % aqueous citric acid solution and aqueous saturated sodium hydrogen carbonate solution, and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy)benzenepropionate (242.5 g) as oil . MS (m/z) : 441 (M+) (2) Under nitrogen atmosphere, to a mixture of ethyl (αS)- – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzenepropionate (66.2g), 4- ethoxymethyl-2, 6-dimethoxyphenylboric acid (54.0 g) , triphenylphosphine (9.83 g) and N-methylpyrrolidone (330 ml) were added palladium acetate (1.68 g) and diisopropylamine (24.9 g ), and the mixture was heated at 90 °C. After stirring for 1 hour at the same temperature, the mixture was cooled and toluene and water were added. The organic layers were washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [[ (1, 1-dimethylethoxy) carbonyl] amino] – 4′ -ethoxymethyl-2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionate (90.1 g) as oil.
The product was dissolved in ethanol (330 ml) , and after addition of p-toluenesulfonic acid monohydrate (28.5 g) , the mixture was stirred for 2 hours at 75 °C. After cooling to room temperature, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was dissolved in ethyl acetate with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS)-α- amino-4′ -ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4- propionate p-toluenesulfonate (63.4 g) .
MS (m/z) : 387 (M+-p-toluenesulfonic acid), M.p. 127-129°C
(3) To a mixture of ethyl (αS) -α-amino-4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate p- toluenesulfonate (29.0 g) , sodium hydrogen carbonate (15. 2 g) , water (290 ml) and ethyl acetate (290 ml) was added dropwise 2, 6-difluorobenzoyl chloride (9. 6 g) at 15 °C or below and the mixture was stirred for 30 minutes at the same temperature. The ethyl acetate layer was washed with saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo. The residue was recrystallized from isopropanol-water to yield ethyl (αS) -oi- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (26.4 g) . MS (m/z) : 527 (M+) , M.p. 87-89°C (4) To a solution of sodium hydroxide (2.9 g) in water- tetrahydrofuran (317 ml-159 ml) was added ethyl (oιS)-α- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate (31.7 g) at 15°C and the mixture was stirred for 4 hours at the same temperature. After neutralizing with IN HC1, the organic solvent was removed in vacuo. The aqueous layer was cooled, the crystalline precipitates were collected by filtration and recrystallized from ethanol-water to yield (αS) -a- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionic acid (28.8 g) . MS (m/z): 499 (M+) , M.p. 154-155°C
EXAMPLE 2 (1) Under nitrogen atmosphere, a mixture of ethyl (oιS)-o:- [[ (1, 1-dimethylethoxy) carbonyl] amino] -4-bromobenzene propanoate (11.17 g) , 4-ethoxymethyl-2, 6- dimethoxyphenylboronic acid (10.80 g ), palladium acetate (0.34 g), triphenylphosphine (1.57 g) , anhydrous potassium carbonate (12.44 g) , iV-methylpyrrolidone (56 ml) and water (11 ml) was stirred for 50 minutes at 80 °C. After completion of the reaction, the mixture was cooled to room temperature and extracted with ethyl acetate and water. The organic layer was washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution, dried over magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure to yield ethyl (αS)-α- [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate (20.4 g) as oil. The product was dissolved in ethanol (100 ml) , and after addition of p-toluenesulfonic acid monohydrate (5.7 g) , the mixture was stirred for 1.5 hours at 75 °C. After cooling, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was suspended in toluene with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS) – -amino-4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate p- toluenesulfonate (13.80 g) . (2) The compound obtained in the above step (1) was treated in the same manner as described in Example 1 (2) to (4) to yield (αS) -a- [ [2 , 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionic acid. The physicochemical data were the same as that obtained in Example 1.
EXAMPLE 3
To a solution of ethyl (αS) -α- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (500 g ) in water (12.6 ml) and dioxane (50 ml) was added hydrochloric acid (12.4 g) and the mixture was stirred for 60 hours at 60 “C. The organic solvent was removed in vacuo and the aqueous layer was cooled. The crystalline precipitates were collected by filtration and recrystallized from ethanol- water to yield (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionic acid (426 mg) . The physicochemical data were the same as that obtained in Example 1.
REFERENCE EXAMPLE 1
(1) To a mixture of 4-bromo-3, 5-dimethoxybenzylalcohol (44.5 g) , triethylammonium benzyl chloride (2.05 g) and 20% aqueous sodium hydroxide solution (288 g) was added diethyl sulfate (41.7 g) under ice-cooling, and the mixture was stirred overnight at 25-30 °C. After stirring for 1 hour at 70 °C, the mixture was cooled and extracted with toluene. The toluene layer was washed with water and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield 4-bromo-3, 5- dimethoxybenzyl ethyl ether (49.5 g) as colorless oil. MS (m/z): 276 (M++2) , 274 (M+)
(2) Under nitrogen atmosphere, to a solution of 4-bromo- 3, 5-dimethoxybenzyl ethyl ether (440.0 g) in tetrahydrofuran (4.0 L) was added dropwise n-butyl lithium (1.6 M n-hexane solution, 1.1 L) at -60°C. After stirring for 15 minutes at the same temperature, trimethyl borate (249.3 g) was added. The temperature of the mixture was gradually elevated, followed by stirring for 1 hour under ice-cooling. To the mixture was added dropwise 10% aqueous sulfuric acid solution (835 g ) . The mixture was extracted with ethyl acetate and the organic layer was washed with water and saturated aqueous NaCl solution. After drying over magnesium sulfate, the solvent was removed in vacuo. The residue was dissolved in isopropyl ether with heating and cooled. The crystalline precipitates were collected by filtration and dried to yield 4-ethyoxymethyl-2, 6- dimetoxyphenylboronic acid (312.9 g) . M.p. 59-61°C
REFERENCE EXAMPLE 2
(1) To a suspension of 4-bromo-3, 5-dihydroxybenzoic acid (95.0 kg) in ethyl acetate (950 L) were added anhydrous potassium carbonate (270.8 kg) and dimethyl sulfate (174.7 kg) . The mixture was heated at 50-80 ‘C for about 4 hours and partitioned by adding water. The organic layer was washed with water and saturated aqueous NaCl solution and concentrated under reduced pressure. The residue was suspended into methanol, stirred under heating and cooled. The crystalline precipitates were collected by filtration and dried to yield methyl 4-bromo-3, 5-dimethoxybenzoate (98.8 kg) as pale yellow crystals. MS (m/z): 277 (M++2) , 275 (M+) , M.p. 120-122°C
(2) To a solution of calcium chloride (46.5 kg) in ethanol (336 L) were added tetrahydrofuran (672 L) and methyl 4- bromo-3, 5-dimethoxybenzoate (96.0 kg) to obtain a suspension. To the suspension was added sodium borohydride
(31.7 kg) by portions at room temperature, and the mixture was stirred for about 9 hours at temperature of room temperature to 45 °C. The reaction mixture was added dropwise to aqueous HC1 solution and stirred for about 16 hours at room temperature. Organic solvent was removed in vacuo, and water (1440 L) was added to the residue and stirred for 1 hour at 50 °C. After cooling, the crystalline precipitates were collected by filtration and dried to yield 4-bromo-3, 5-dimethoxybenzyl alcohol (83.3 kg) as colorless crystals. MS (m/z): 249 (M++2), 247 (M+) , M.p. 100-102°C.
INDUSTRIAL APPLICABILITY The process for preparation of the present invention makes it possible to afford a compound of the formula (I) or a pharmaceutically acceptable salt thereof with high- purity, in a high yield and inexpensively, and, therefore, the process of the present invention is industrially very useful.













 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
amcrasto@gmail.com




 .
.


 MANUDEVI
MANUDEVI








 .
. .
.













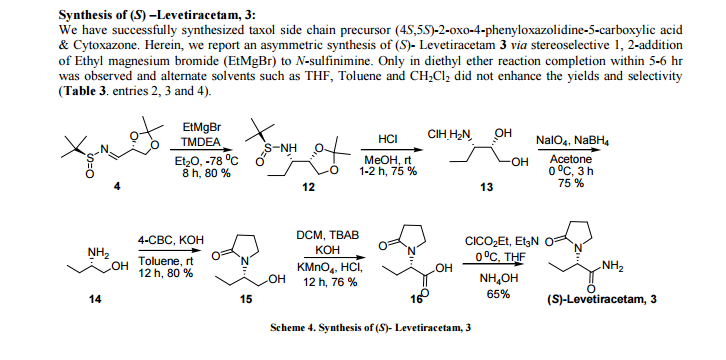








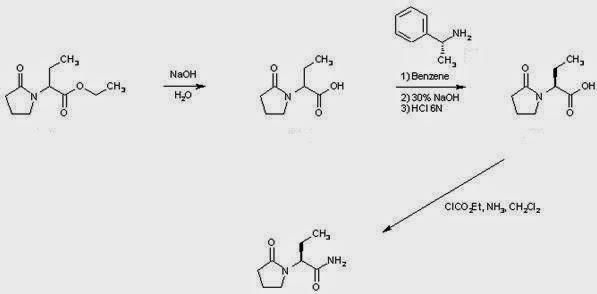






 US 7902380, Levetiracetam
US 7902380, Levetiracetam




 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..


 dalat city
dalat city hanoi
hanoi







 .
.











 .
.









 ROSE HIPS
ROSE HIPS




















 Japan
Japan