
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

New FDA Requirements for the Development of Herbal Medicinal Products
DRUG REGULATORY AFFAIRS INTERNATIONAL
The previous FDA guideline for herbal medicinal products from 2004 is supposed to be replaced by a new version. In August 2015, the FDA has presented the draft of the revised guideline. Find out more about the FDA Guideline Botanical Drug Development.
In August 2015, the FDA has published a draft of the guideline “Botanical Drug Development”. This guideline addresses issues arising from the particular nature of herbal medicinal products. After its finalization it is supposed to replace the previous guideline from June 2004.
The general approach in the development of herbal medicinal products remained unchanged since 2004. But due to the better understanding of herbal medicinal products and the experience gained during the review of the approval documents for herbals (NDAs/New Drug Applications and INDs/Investigational New Drug Applications), specific recommendations could be adjusted. Still, new sections will be supplemented to better address the late development phase.
The…
View original post 34 more words
Genotoxic impurities: the new ICH M7 addendum to calculation of compound-specific acceptable intakes
DRUG REGULATORY AFFAIRS INTERNATIONAL

Genotoxic impurities: the new ICH M7 addendum to calculation of compound-specific acceptable intakes
The draft for a guideline ICH M7(R1) published recently supplements the ICH-M7 guideline published last year. Read more about the calculation of compound-specific acceptable intakes of genotoxic impurities.
The final document of the ICH-Guideline M7 was published in June 2014. It describes the procedure for evaluating the genotoxic potential of impurities in medicinal products (see also our news Final ICH M7 Guideline on Genotoxic Impurities published dated 23 July 2014).
An important approach to the risk characterisation of impurities is the TTC concept (TTC = threshold of toxicological concern). According to this approach the exposure to a mutagenic impurity having the concentration of 1.5 µg per adult person per day is considered to be associated with a negligible risk. It can be used as default evaluation approach to most pharmaceuticals for long-term treatment (> 10 years)…
View original post 200 more words
TR 700, TR 701FA, Tedizolid phosphate

“TR-700”
5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)-pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one
US Patent Publication No. 20070155798, which is hereby incorporated by reference in its entirety, recently disclosed a series of potently anti-bacterial oxazolidinones including

wherein R═H, PO(OH)2, and PO(ONa)2.

(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate, CAS 856867-55-5

DISODIUM SALT

CAS 856867-39-5
- C17 H16 F N6 O6 P . 2 Na
- 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-[(phosphonooxy)methyl]-, sodium salt (1:2), (5R)-
-
- DA 7218, Tedizolid phosphate disodium salt
In addition, improved methods of making the free acid are disclosed in U.S. patent application Ser. No. 12/577,089, which is assigned to Trius Therapeutics, Inc., and which is incorporated herein by reference
crystalline (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate 1 (R═PO(OH)2), was more stable and non-hygroscopic than the salt forms that were tested. In addition, unlike typical crystallizations, where the crystallization conditions, such as the solvent and temperature conditions, determine the particular crystalline form, the same crystalline form of 1 (R═PO(OH)2) was produced using many solvent and crystallization conditions. Therefore, this crystalline form was very stable, was made reproducibly, and ideal for commercial production because it reduced the chances that other polymorphs would form contaminating impurities during production. However, in all preliminary testing, the free acid crystallized as fine particles, making filtering and processing difficult.
To overcome difficulties in filtering and processing crystalline (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate 1 (R═PO(OH)2), processes described herein result in significantly reduced filtering time, avoid more toxic solvents, and significantly increased ease of preparing dosage forms such as tablets. It has been found that implementing various processes can control the particle size distribution of the resulting material, which is useful for making the crystalline form, and for commercial production and pharmaceutical use. Surprisingly, the process for increasing the particle size reduces the amount of the dimer impurity, in comparison to the process for making the free acid disclosed in U.S. patent application Ser. No. 12/577,089. Thus, various methods of making and using the crystalline form are also provided.
In addition, by using methods of making the free acid disclosed in U.S. patent application Ser. No. 12/577,089, which is assigned to the same assignee as in the present application, and by using the crystallization methods described herein, a crystalline free acid having at least 96% purity by weight may be formed that comprises a compound having the following formula:

(hereinafter “the chloro impurity”), i.e., (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one in an amount less than 1%.
Similarly, by using methods of making the free acid disclosed in U.S. patent application Ser. No. 12/577,089, which is assigned to the same assignee as in the present application, and by using the crystallization methods described herein, a crystalline free acid having at least 96% purity by weight may be formed that comprises a compound having the following formula:
(hereinafter “TR-700”), i.e., 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)-pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one, in an amount less than 1%.
The crystalline free acid may have one or more of the attributes described herein.
In some aspects, a purified crystalline (R)-3-(4-(2-(2-methyltetrazol-5-yl)-pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate, i.e., the free acid, has a purity of at least about 96% by weight. In some embodiments, the crystalline free acid has a median volume diameter of at least about 1.0 μm.
BRIEF DESCRIPTION OF THE DRAWINGS……http://www.google.com/patents/US8426389
FIG. 1 the FT-Raman spectrum of crystalline 1 (R═PO(OH)2).

FIG. 2 shows the X-ray powder pattern of crystalline 1 (R═PO(OH)2).
http://www.google.com/patents/US8426389
FIG. 3 shows the differential scanning calorimetry (DSC) thermogram of crystalline 1 (R═PO(OH)2).
http://www.google.com/patents/US8426389
FIG. 4 shows the 1H NMR spectrum of 1 (R═PO(OH)2).

FIG. 5 depicts the TG-FTIR diagram of crystalline 1 (R═PO(OH)2).
http://www.google.com/patents/US8426389
FIG. 6 is a diagram showing the dynamic vapor sorption (DVS) behavior of crystalline 1 (R═PO(OH)2).
FIG. 7 is a manufacturing process schematic for 1 (R═PO(OH)2) (TR-701 FA) in a tablet dosage form.

FIG. 8 is a manufacturing process schematic for 1 (R═PO(OH)2) (TR-701 FA) Compounding Solution for Lyophilization.

FIG. 9 is a manufacturing process schematic for 1 (R═PO(OH)2) (TR-701 FA) for Injection, 200 mg/vial: sterile filtering, filling, and lyophilization.
FIG. 10 is a representative particle size distribution of crystalline free acid without regard to controlling particle size distribution as also described herein.
FIG. 11 is a representative particle size distribution of crystalline free acid made using laboratory processes to control particle size described herein.
FIG. 12 is a representative particle size distribution of crystalline free acid made using scaled up manufacturing processes to control particle size described herein.
These impurities include

i.e., 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)-pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one (“TR-700”) and/or

i.e., (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one (“chloro impurity”).
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4128654 | Feb 10, 1978 | Dec 5, 1978 | E. I. Du Pont De Nemours And Company | 5-Halomethyl-3-phenyl-2-oxazolidinones |
| US4250318 | Aug 9, 1978 | Feb 10, 1981 | Delalande S.A. | Novel 5-hydroxymethyl oxazolidinones, the method of preparing them and their application in therapeutics |
| US4340606 | Oct 23, 1980 | Jul 20, 1982 | E. I. Du Pont De Nemours And Company | 3-(p-Alkylsulfonylphenyl)oxazolidinone derivatives as antibacterial agents |
| US4461773 | Jan 5, 1984 | Jul 24, 1984 | E. I. Dupont De Nemours And Company | P-Oxooxazolidinylbenzene compounds as antibacterial agents |
| US4476136 | Feb 24, 1982 | Oct 9, 1984 | Delalande S.A. | Aminomethyl-5 oxazolidinic derivatives and therapeutic use thereof |
| US4948801 | Jul 29, 1988 | Aug 14, 1990 | E. I. Du Pont De Nemours And Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| US5523403 | May 22, 1995 | Jun 4, 1996 | The Upjohn Company | Tropone-substituted phenyloxazolidinone antibacterial agents |
| US5565571 | Apr 28, 1994 | Oct 15, 1996 | The Upjohn Company | Substituted aryl- and heteroaryl-phenyloxazolidinones |
| US5652238 | Sep 27, 1994 | Jul 29, 1997 | Pharmacia & Upjohn Company | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| US5688792 | Aug 16, 1994 | Nov 18, 1997 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US6365751 | Apr 17, 2001 | Apr 2, 2002 | Zeneca Ltd. | Antibiotic oxazolidinone derivatives |
| US6627646 * | Jul 17, 2001 | Sep 30, 2003 | Sepracor Inc. | Norastemizole polymorphs |
| US6689779 | May 18, 2001 | Feb 10, 2004 | Dong A Pharm. Co., Ltd. | Oxazolidinone derivatives and a process for the preparation thereof |
| US7129259 | Dec 1, 2004 | Oct 31, 2006 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141583 | Apr 23, 2001 | Nov 28, 2006 | Astrazeneca Ab | Oxazolidinone derivatives with antibiotic activity |
| US7144911 | Dec 24, 2003 | Dec 5, 2006 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| US7202257 | Jul 6, 2004 | Apr 10, 2007 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US7396847 | Sep 9, 2002 | Jul 8, 2008 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7462633 | Jun 29, 2004 | Dec 9, 2008 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US7473699 | Feb 25, 2003 | Jan 6, 2009 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7498350 | Nov 24, 2003 | Mar 3, 2009 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US7816379 | Dec 17, 2004 | Oct 19, 2010 | Dong-A Pharm. Co., Ltd. | Oxazolidinone derivatives |
| US20020115669 | Aug 29, 2001 | Aug 22, 2002 | Wiedeman Paul E. | Oxazolidinone chemotherapeutic agents |
| US20030166620 | May 18, 2001 | Sep 4, 2003 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| US20040180906 | Dec 24, 2003 | Sep 16, 2004 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20050038092 | Jun 29, 2004 | Feb 17, 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20050107435 | Sep 9, 2002 | May 19, 2005 | Gravestock Michael B. | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US20050288286 | Jul 6, 2004 | Dec 29, 2005 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20060116386 | Nov 24, 2003 | Jun 1, 2006 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US20060116400 | Nov 24, 2003 | Jun 1, 2006 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline derivatives as antibacterial agents |
| US20060270637 | Feb 24, 2004 | Nov 30, 2006 | Astrazeneca Ab | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents |
| US20070155798 | Dec 17, 2004 | Jul 5, 2007 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20070185132 | Jun 29, 2004 | Aug 9, 2007 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereo |
| US20070191336 | Dec 23, 2004 | Aug 16, 2007 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070203187 | Jan 22, 2007 | Aug 30, 2007 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20070208062 | May 24, 2005 | Sep 6, 2007 | Astrazeneca Ab | 3-(4-(2-dihydroisoxazol-3-ylpyridin-5-yl)phenyl)-5-triazol-1-ylmethyloxazolidin-2-one derivatives as mao inhibitors for the treatment of bacterial infections |
| US20080021012 | May 24, 2005 | Jan 24, 2008 | Astrazeneca Ab | 3-[4-{6-Substituted Alkanoyl Pyridin-3-Yl}-3-Phenyl]-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones As Antibacterial Agents |
| US20080021071 | May 24, 2005 | Jan 24, 2008 | Astrazeneca Ab | 3-{4-(Pyridin-3-Yl) Phenyl}-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20080064689 | May 24, 2004 | Mar 13, 2008 | Astrazeneca Ab | 3-[4-(6-Pyridin-3-Yl)-3-Phenyl] -5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20090018123 | Jun 19, 2006 | Jan 15, 2009 | Milind D Sindkhedkar | Oxazolidinones Bearing Antimicrobial Activity Composition and Methods of Preparation |
| US20090192197 | Jul 30, 2009 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives | |
| US20100093669 | Oct 9, 2009 | Apr 15, 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| US20100227839 | Sep 9, 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin- 5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate | |
| AU2004299413A1 | Title not available | |||
| AU2009200606A1 | Title not available | |||
| CA2549062A1 | Dec 17, 2004 | Jun 30, 2005 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| CN101982468A | Dec 17, 2004 | Mar 2, 2011 | 东亚制药株式会社 | Novel oxazolidinone derivatives and pharmaceutical compositions comprising the derivatives |
| EP0312000A1 | Oct 12, 1988 | Apr 19, 1989 | The Du Pont Merck Pharmaceutical Company | Aminomethyl oxooxazolidinyl aroylbenzene derivatives useful as antibacterial agents |
| EP0352781A2 | Jul 27, 1989 | Jan 31, 1990 | The Du Pont Merck Pharmaceutical Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| EP1699784A1 | Dec 17, 2004 | Sep 13, 2006 | Dong-A Pharmaceutical Co., Ltd. | Novel oxazolidinone derivatives |
| EP2305657A2 | Dec 17, 2004 | Apr 6, 2011 | Dong-A Pharmaceutical Co., Ltd. | Oxazolidinone derivatives |
| EP2435051A1 | May 27, 2010 | Apr 4, 2012 | Trius Therapeutics | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
| IN236862A1 | Title not available | |||
| JPS5799576A | Title not available | |||
| KR20110071107A | Title not available | |||
| NZ547928A | Title not available | |||
| NZ575842A | Title not available | |||
| WO1993009103A1 | Oct 5, 1992 | May 13, 1993 | Upjohn Co | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| WO1993023384A1 | Apr 21, 1993 | Nov 25, 1993 | Upjohn Co | Oxazolidinones containing a substituted diazine moiety and their use as antimicrobials |
| WO1995007271A1 | Aug 16, 1994 | Mar 16, 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO1995014684A1 | Sep 27, 1994 | Jun 1, 1995 | Michel R Barbachyn | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| WO2001094342A1 | May 18, 2001 | Dec 13, 2001 | Cho Jong Hwan | Novel oxazolidinone derivatives and a process for the preparation thereof |
| WO2002081470A1 | Apr 3, 2002 | Oct 17, 2002 | Astrazeneca Ab | Oxazolidinones containing a sulfonimid group as antibiotics |
| WO2003022824A1 | Sep 9, 2002 | Mar 20, 2003 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| WO2003035648A1 | Oct 23, 2002 | May 1, 2003 | Astrazeneca Ab | Aryl substituted oxazolidinones with antibacterial activity |
| WO2003047358A1 | Dec 2, 2002 | Jun 12, 2003 | Vaughan Leslie Crow | Cheese flavour ingredient and method of its production |
| WO2003072575A1 | Feb 25, 2003 | Sep 4, 2003 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring) methyl-oxazolidinone derivatives and their use as antibacterial agents |
| WO2003072576A2 | Feb 25, 2003 | Sep 4, 2003 | Astrazeneca Ab | Oxazolidinone derivatives, processes for their preparation, and pharmaceutical compositions containing them |
| WO2004048350A2 | Nov 24, 2003 | Jun 10, 2004 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| WO2004083205A1 | Mar 16, 2004 | Sep 30, 2004 | Astrazeneca Ab | Antibacterial 1, 3- oxazolidin -2- one derivatives |
| WO2005005398A2 | Jun 29, 2004 | Jan 20, 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| WO2005051933A1 | Nov 23, 2004 | Jun 9, 2005 | Vijay Kumar Kaul | An improved process for the synthesis of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester, a key intermediate for oxazolidinone antimicrobials and compounds prepared thereby |
| WO2005058886A1 | Dec 17, 2004 | Jun 30, 2005 | Dong A Pharm Co Ltd | Novel oxazolidinone derivatives |
| WO2005116017A1 | May 24, 2005 | Dec 8, 2005 | Astrazeneca Ab | Process for the preparation of aryl substituted oxazolidinones as intermediates for antibacterial agents |
| WO2006038100A1 | Oct 6, 2005 | Apr 13, 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007023507A2 | Jun 19, 2006 | Mar 1, 2007 | Milind D Sindkhedkar | Oxazolidinones bearing antimicrobial activity composition and methods of preparation |
| WO2007138381A2 | Oct 13, 2006 | Dec 6, 2007 | Delorme Daniel | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| WO2010042887A2 | Oct 9, 2009 | Apr 15, 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| WO2010091131A1 | Feb 3, 2010 | Aug 12, 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| WO2010138649A1 | May 27, 2010 | Dec 2, 2010 | Trius Therapeutics, Inc. | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
Wockhardt, WO 2007023507, N-[[3-[3,5-difluoro-4-[4-(tetrazol-2-yl)piperidin-1-yl]phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide


Cas 928156-95-0,
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-(2H-tetrazol-2-yl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
| C18H21F2N7O3 | |
| Molecular Weight: | 421.401246 g/mol |
|---|
N-[[3-[3,5-difluoro-4-[4-(tetrazol-2-yl)piperidin-1-yl]phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide

Example- 14 and 15
(S)-N- { 3- [4-(4-(2H-tetrazol-2-yl)-piperidin- 1 -yl)-3 , 5-difluorophenyl] -2-oxo-oxazolidin-
5-ylmethyl }-acetamide and
(S)-N- { 3- [4-(4-(l H-tetrazol- 1 -yl)-piperidin- 1 -yl)-3 , 5-difluorophenyl] -2-oxo-oxazolidin-
5-ylmethyl }-acetamide

and

A mixture of (S)-N-{3-[4-methanesulphonyloxy piperidin-l-yl)-3,5-difluorophenyl]-2- oxo-oxazolidin-5-ylmethyl}-acetamide (1.12 mM), tetrazole (1.68 mM), and K2CO3 (1.68 mM) in DMF (6 ml) was heated for 22 hrs at 850C. The resulting mixture was poured into ice-water mixture, stirred for 30 min. And the separated solid was purified by column chromatography to obtain two isomeric products in 18% and 12% yields respectively. Isomer A: M.P. 234-2370C; MS(M+1)- 422 ; M.F. C18H21F2N7O3 Isomer B: M.P. 214-2170C; MS(M+1)- 422 ; M.F. C18H2JF2N7O3
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
Our New Drug Discovery team has developed a number of lead molecules, mainly in the area of anti-infectives; these are currently at various stages of development.
Of these molecules, the most advanced of the New Chemical Entities (NCE) is WCK 771, which has commenced Phase II human clinical trials.
WCK 771 is a broad-spectrum antibiotic, which has proven effective in treating diverse staphylococcal infections like MRSA and VISA.
Other lead molecules at various stages of pre-clinical trials are: WCK 2349, WCK 4873 and WCK 4086.
http://www.wockhardt.com/how-we-touch-lives/new-drug-discover.aspx


///////
Wockhardt, WO 2015136473, sodium (2S, 5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate

WO-2015136473
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015136473&redirectedID=true
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
Our New Drug Discovery team has developed a number of lead molecules, mainly in the area of anti-infectives; these are currently at various stages of development.
Of these molecules, the most advanced of the New Chemical Entities (NCE) is WCK 771, which has commenced Phase II human clinical trials.
WCK 771 is a broad-spectrum antibiotic, which has proven effective in treating diverse staphylococcal infections like MRSA and VISA.
Other lead molecules at various stages of pre-clinical trials are: WCK 2349, WCK 4873 and WCK 4086.
http://www.wockhardt.com/how-we-touch-lives/new-drug-discover.aspx
WO-2015136473
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015136473&redirectedID=true
Process for the synthesis of sodium (2S, 5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate (disclosed in WO2014135929) is claimed. Used as an intermediate in the synthesis of several antibacterial compounds. For a concurrent filing see WO2015136387, claiming the combination of an antibacterial agent with sulbactam.
In September 2015, Wockhardt’s pipeline lists several antibacterial programs, including WCK-771 and WCK-2349 (both in phase II), WCK-5107 (phase I), and also investigating iv and oral second generation oxazolidinones, WCK-4873, and iv and oral formulation of WCK-4086 (in preclinical stage) for treating the bacterial infection.
For a prior filing see WO2015125031, claiming the combination of an antibacterial agent (eg cefepime or cefpirome) and nitrogen containing bicyclic compound, useful for treating bacterial infection.
A compound of Formula (I), chemically known as sodium (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate, can be used as an intermediate in the synthesis of several antibacterial compounds and is disclosed in PCT International Patent Application No. PCT/IB2013/059264. The present invention discloses a process for preparation of a compound of Formula (I).

Scheme 1
Example 1
Synthesis of sodium (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicvclor3.2.11octane-2- carboxylate
Step 1; Preparation of -Γl-Γ(feΓt-butyldimethylsilyl -oxymethyll-5-Γdimethyl(oxido -λ-4-sulfanylidenel-4-oxo-pentyll-carbamic acid tert-butyl ester (III):
To a suspension of trimethylsulfoxonium iodide (180.36 gm, 0.819 mol) in tetrahydrofuran (900 ml), sodium hydride (32.89 g, 0.819 mol, 60% in mineral oil) was charged in one portion at 30°C temperature. The reaction mixture was stirred for 15 minutes and then dropwise addition of dimethylsulphoxide (1.125 ml) was done over a period of 3 hours at room temperature to provide a white suspension. The white suspension was added to a pre-cooled a solution of 2-(feri-butyldimethylsilyl-oxymethyl)-5-oxo-pyrrolidine-l-carboxylic acid tert-buty\ ester (II) (225 g, 0.683 mol, prepared as per J. Org Chem.; 2011, 76, 5574 and WO2009067600) in tetrahydrofuran (675 ml) and triethylamine (123.48 ml, 0.887 mol) mixture at -13°C by maintaining the reaction mixture temperature below -10°C. The resulting suspension was stirred for additional 1 hour at -10°C. The reaction mixture was carefully quenched by addition of saturated aqueous ammonium chloride (1.0 L) at -10°C to 10°C. The reaction was extracted by adding ethyl acetate (1.5 L). The layers were separated and aqueous layer was re-extracted with ethyl acetate (500 ml x 3). The combined organic layer was washed successively with saturated aqueous sodium bicarbonate (1.0 L), water (2.0 L) followed by saturated aqueous sodium chloride solution (1.0 L). Organic layer was dried over sodium sulfate and evaporated under vacuum to provide 265 g of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-buty\ ester (III) as an yellow oily mass.
Analysis:
Mass: 422.3 (M+l); for Molecular weight: 421.68 and Molecular Formula: ![]()
1H NMR (CDC13): δ 4.77 (br d, 1H), 4.38 (br s, 1H), 3.58 (br s, 3H), 3.39 (s, 3H), 3.38 (s, 3H), 2.17-2.27 (m, 2H), 1.73-1.82 (m, 2H), 1.43 (s, 9H), 0.88 (s, 9H), 0.01 (s, 3H), 0.04 (s, 3H).
Step 2: Preparation of 5-r4-benzyloxyimino-l-(fert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyll-carbamic acid tert- butyl ester (IV):
To a suspension of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-butyl ester (III) (440.0 g, 1.045 mol) in tetrahydrofuran (6.6 L), O-benzhydroxylamine hydrochloride (200.0 g, 1.254 mol) was charged. The reaction mixture was heated to 50°C for 2.5 hours. The reaction mixture was filtered through pad of celite and filtrate was concentrated to provide a residue. The residue was dissolved in ethyl acetate (5.0 L) and washed successively with saturated aqueous sodium bicarbonate (1.5 L), water (1.5 L) and saturated aqueous sodium chloride (1.5 L). Organic layer was dried over sodium sulfate. Solvent was evaporated under vacuum to yield 463.0 g of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) as an oily mass.
Analysis:
Mass: 486.1 (M+l); for Molecular weight: 485.4 and Molecular Formula: ![]()
1H NMR (CDCI3): δ 7.26-1 6 (m, 5H), 5.10 (s, 2H), 4.66 (br d, 1H), 3.58-4.27 (m, 2H), 3.56-3.58 (m, 3H), 2.40-2.57 (m, 2H), 1.68-1.89 (m, 2H), 1.44 (s, 9H), 0.89 (s, 9H), 0.02 (s, 3H), 0.04 (s, 3H).
Step 3: Preparation of 5-5-benzyloxyimino-2-(fert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V):
To a solution of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) (463.0 g 0.954 mol) in tetrahydrofuran (6.9 L), was charged potassium feri-butoxide (139.2 g, 1.241 mol) in portions over a period of 30 minutes by maintaining temperature -10°C. The resulting suspension was stirred for additional 1.5 hours at -10°C to -5°C. The reaction mixture was quenched by addition of saturated aqueous ammonium chloride (2.0 L) at -5°C to 10°C. The organic layer was separated and aqueous layer was extracted with ethyl acetate (1.0 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride solution (2.0 L). Organic layer was dried over sodium sulfate, and then evaporated under vacuum to yield 394.0 g of 5-5-benzyloxyimino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-butyl ester (V) as an yellow oily mass.
Analysis:
Mass: 449.4 (M+l) for Molecular weight: 448.68 and Molecular Formula: C24H4oN204Si;
1H NMR (CDC13): δ 7.25-1 3 (m, 5H), 5.04-5.14 (m, 2H), 4.35 (br s, 1H), 3.95 (br s, 1H), 3.63-3.74 (br d, 2H), 3.60-3.63 (m, 1H), 2.70-2.77 (m, 1H), 2.33-2.41 (m, 1H), 1.79-1.95 (m, 2H), 1.44 (s, 9H), 0.88 (s, 9H), 0.03 (s, 3H), 0.04 (s, 3H).
Step 4: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI):
To a solution of 5-5-benzyloxyimino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V) (394.0 g, 0.879 mol) in dichloromethane (5.0 L) and glacial acetic acid (788 ml), was charged sodium cyanoborohydride (70.88 g, 1.14 mol) one portion. The resulting reaction mixture was stirred at temperature of about 25 °C to 30°C for 2 hours. The mixture was quenched with adding aqueous solution of sodium bicarbonate (1.3 kg) in water (5.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (2.0 L). The combined organic layer washed successively with water (2.0 L), saturated aqueous
sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to provide a residue. The residue was purified by silica gel column chromatography to yield 208 g of (25,5i?5)-5-benzyloxyamino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-buty\ ester (VI) as pale yellow liquid.
Analysis:
Mass: 451.4 (M+l); for Molecular weight: 450.70 and Molecular Formula: C24H42N204Si;
1H NMR (CDC13): δ 7..26-7.36 (m, 5H), 4.90-5.50 (br s, 1H), 4.70 (dd, 2H), 4.09-4.25 (m, 2H), 3.56-3.72 (m, 2H), 2.55-3.14 (m, 2H), 1.21-1.94 (m, 4H), 1.45 (s, 9H), 0.89 (s, 9H), 0.05 (s, 6H).
Step 5: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine (VII):
To a solution of 5-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI) (208 g, 0.462 mol) in dichloromethane (3.0 L), boron trifluoride diethyletherate complex (114.15 ml, 0.924 mol) was charged in one portion. The resulting reaction mixture was stirred at temperature of about 25°C to 35°C temperature for 2 hours. The reaction mixture was quenched with saturated aqueous sodium bicarbonate (2.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride (1.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to yield 159 g of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) as a yellowish syrup.
Analysis:
Mass: 351.3 (M+l); for Molecular weight: 350.58 and Molecular Formula: C19H34N202Si.
Step-6: Preparation of (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-r3.2.11octane (VIII):
Part 1; Preparation of (2S,5RS)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
To a solution of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) (159.0 g, 0.454 mol) in a mixture of acetonitrile (2.38 L) and diisopropylethylamine (316.5 ml, 1.81 mol) was added triphosgene (59.27 gm, 0.199 mol) dissolved in acetonitrile (760 ml) at -15°C over 30 minutes under stirring. The resulting reaction mixture was stirred for additional 1 hour at -10°C. The reaction mixture was quenched by addition of saturated aqueous sodium bicarbonate (2.0 L) at -5°C to 10°C. Acetonitrile was evaporated from the reaction mixture under vacuum and to the left over aqueous phase, dichloromethane (2.5 L) was added. The organic layer was separated and aqueous layer extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed successively with water (2.0 L), saturated aqueous sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum and the residue was passed through a silica gel bed to yield 83.0 g of diastereomeric mixture (25, 5i?5)-6-benzyloxy-2-(feri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio as a yellow liquid.
Part-2: Separation of diastereomers to prepare (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
A mixture of diastereomers (2S,5Z?S)-6-benzyloxy-2-(teri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio (47.0 gm, 0.125 mol), was dissolved in n-hexane (141 ml) and stirred at temperature of about 10°C to 15°C for 1 hour. Precipitated solid was filtered and washed with n-hexane (47 ml) to provide 12.0 g of diastereomerically pure (25,5i?)-6-benzyloxy-2-(tert-butyl-dimethylsilyl-oxymethyl)-7-oxo- 1,6-diaza-bicyclo-[3.2.1] octane (VIII) as a white crystalline material.
Analysis:
Mass: 377.3 (M+l); for Molecular weight: 376.58 and Molecular Formula: ![]()
1H NMR (CDCI3): δ Ί -Ί.ΑΑ (m, 5H), 4.95 (dd, 2H), 3.76-3.85 (ddd, 2H), 3.37-3.40 (m, 1H), 3.28-3.31 (m, 2H), 2.89 (brd, 1H), 1.90-2.02 (m, 2H), 1.62- 1.74 (m, 2H), 1.56 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
Diastereomeric purity as determined by HPLC: 99.85%
Step-7: Preparation of (25,5R)-6-benzyloxy-2-hvdroxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane (IX):
To a solution of (25,5i?)-6-benzyloxy-2-(ieri-butyl-dimethylsilyl-oxymethyl)-7-oxo- l,6-diaza-bicyclo-[3.2.1]octane (VIII) ( 12.0 g, 31.9 rnmol) in tetrahydrofuran (180 ml) was charged tetra 7? -butyl ammonium fluoride (38.0 ml, 38 mmol, 1 M in tetrahydrofuran) at room temperature. The reaction mixture was stirred for 2 hours. It was quenched with saturated aqueous ammonium chloride ( 100 ml). The organic layer was separated and aqueous layer extracted with dichloromethane (150 ml x 3). The combined organic layer was washed with saturated aqueous sodium chloride (150 ml), dried over sodium sulfate and evaporated under vacuum to yield 24.0 g of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l ,6-diaza-bicyclo-[3.2.1]octane (IX) as a yellow liquid. The compound of Formula (IX) was purified by silica gel (60-120 mesh) column chromatography using a mixture of ethyl acetate and hexane as an eluent.
Analysis:
Mass: 263.1 (M+l); for Molecular weight: 262.31 and Molecular Formula: C14H18N203
1H NMR (CDCb): δ 7.34-7.42 (m, 5H), 4.95 (dd, 2H), 3.67-3.73 (m, 1H), 3.53-3.60 (m, 2H), 3.32-3.34 (m, 1H), 2.88-3.01 (m, 2H), 2.09 (brs, 1H), 1.57-2.03 (m, 2H), 1.53- 1.57 (m, 1H), 1.37- 1.40 (m, 1H).
Step 8: Preparation of sodium salt of (25, 5R)-6-benzyloxy-7-oxo-l,6-diaza-bicvclor3.2.11-octane-2-carboxylic acid (I):
Step I:
Compound of Formula (IX) obtained in step 8 above was used without any further purification. To the clear solution of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane (IX) (24.0 g, 31.8 mmol) (quantities added based upon theoretical basis i.e 8.3 g ) in dichloromethane (160 ml), was added Dess-Martin reagent (24.1 g, 57.24 mmol) in portions over 15 minutes. The resulting suspension was stirred for 2 hours at 25°C. The reaction was quenched by adding a solution, prepared from saturated aqueous sodium hydrogen carbonate solution (160 ml) and 72.0 g of sodium thiosulfate. Diethyl ether (160 ml) was added to the reaction mixture and it was stirred for 5-10 minutes and filtered through celite. Biphasic layer from filtrate was separated. Organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (160 ml) followed by saturated aqueous sodium chloride solution (160 ml). Organic layer was dried over sodium sulfate and evaporated to dryness at 30°C to obtain 20.0 g of intermediate aldehyde, which was used immediately for the next reaction.
Step II:
To the crude intermediate aldehyde (20.0 g, 31.6 mmol) (quantities added based upon theoretical yield i.e. 8.2 g) obtained as above, was charged i-butyl alcohol (160 ml) and cyclohexene (10.8 ml, 110.6 mmol). The reaction mixture was cooled to temperature of about 10°C to 15°C. To this mixture was added clear solution prepared from sodium hypophosphate (14.8 g, 94.8 mmol) and sodium chlorite (5.7 g, 63.2 mmol) in water (82.0 ml) over a period of 30 minutes by maintaining temperature between 10°C to 15°C. The reaction mixture was further stirred for 1 hour and was quenched with saturated aqueous ammonium chloride solution. The reaction mixture was subjected to evaporation under vacuum at 40°C to remove i-butyl alcohol. Resulting mixture was extracted with dichloromethane (3 x 150 ml). Layers were separated. Combined organic layer was washed with aqueous brine solution, dried over sodium sulfate and evaporated to dryness under vacuum to obtain 16.0 g of crude residue. To this residue was added acetone (83 ml) to provide a clear solution and to it was added dropwise a solution of sodium 2-ethyl hexanoate (4.5 g) in acetone (24 ml). The reaction mixture was stirred for 15 hours at 25°C to 30°C to provide a suspension. To the suspension was added diethyl ether (215 ml) and stirred for 30 minutes. Resulting solid was filtered over suction, and wet cake was washed with cold acetone (83 ml) followed by diethyl ether (83 ml). The solid was dried under vacuum at 40°C to provide 3.6 g of off-white colored, non-hygroscopic sodium salt of (25, 5i?)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]-octane-2-carboxylic acid (I).
Analysis:
Mass: 275.2 as M-1 (for free acid) for Molecular Weight: 298 and Molecular Formula: ![]()
NMR (DMSO-d6): δ 7.43-7.32 (m, 5H), 4.88 (q, 2H), 3.48 (s, IH), 3.21 (d, IH), 2.73 (d, IH), 2.04-2.09 (m, IH), 1.77-1.74 (m, IH), 1.65-1.72 (m, IH), 1.55-1.59 (m, IH);
Purity as determined by HPLC: 97.47%;
[a]D25: -42.34° (c 0.5, water).
///////
GSK2334470

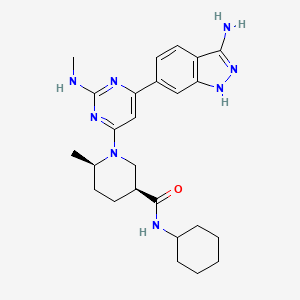
GSK2334470
GSK2334470; 1227911-45-6; GSK-2334470; GSK 2334470;
(3S,6R)-1-[6-(3-Amino-1H-indazol-6-yl)-2-(methylamino)-4-pyrimidinyl]-N-cyclohexyl-6-methyl-3-piperidinecarboxamide
(3S.6/?V1-r6-(3-Amino-1 H-indazol-6-ylV2-(methylaminoV4-pyrimidinyll-Λ/-cvclohexyl-6- methyl-3-piperidinecarboxamide
| Molecular Weight | 462.59 |
| Formula | C25H34N8O |
| CAS Number | 1227911-45-6 |
![]()
Phosphoinositide Dependent Kinase (PDK) 1 Inhibitors
[α]20D = – 32.6 o (c 1.17, MeOH)
[α] D = -27.6 (Concentration = 1.16, Solvent = Methanol)
SOL………DMSO to 100 mM
ethanol to 100 mM
nmr……http://www.chemietek.com/Files/Line2/Chemietek,%20GSK2334470%20(1),%20NMR-DMSO.pdf
http://file.selleckchem.com/downloads/nmr/S708702-GSK2334470-HNMR-Selleck.pdf


GSK2334470 is a potent and selective PDK1 (3-Phosphoinositide dependent protein kinase-1) inhibitor. GSK2334470 blocks the phosphorylation of known PDK1 substrates, but surprisingly find that the potency and kinetics of inhibition vary for different PDK1 targets. GSK2334470 subsequent activation of PDK1 substrates S6K1, SGK and RSK in HEK293, U87 and mouse embryonic fibroblast cell lines.
GSK2334470 inhibited activation of an Akt1 mutant lacking the PH domain (pleckstrin homology domain) more potently than full-length Akt1, suggesting that GSK2334470 is more effective at inhibiting PDK1 substrates that are activated in the cytosol rather than at the plasma membrane. GSK2334470 also suppressed T-loop phosphorylation and activation of RSK2 (p90 ribosomal S6 kinase 2), another PDK1 target activated by the ERK (extracellular-signal-regulated kinase) pathway.
GSK2334470 is a highly specific and potent inhibitor of PDK1 (3-Phosphoinositide dependent protein kinase-1) with IC50 of 10 nM. It does not suppress activity on other 96 kinases, including Aurora, ROCK, p38 MAPK and PI3K. GSK2334470 has been used in cells to ablate T-loop phosphorylation and activate SGK, S6K1 and RSK as well as suppress the activation of Akt.
PATENT
WO 2010059658
http://www.google.com/patents/WO2010059658A1?cl=en
Example 78
(3S.6/?V1-r6-(3-Amino-1 H-indazol-6-ylV2-(methylaminoV4-pyrimidinyll-Λ/-cvclohexyl-6- methyl-3-piperidinecarboxamide
To (3S,6R)-1-[6-(4-cyano-3-fluorophenyl)-2-(methylamino)-4-pyrimidinyl]-Λ/-cyclohexyl-6- methyl-3-piperidinecarboxamide (260 mg, 0.58 mmol) in EtOH (10 ml.) as a suspension at room temperature in a microwave vial was added hydrazine monohydrate (807 uL, 16.7 mmol, 30 equiv) in one portion. The mixture was capped and heated at 100 0C for 48 hours. A duplicate run was performed. The crude reactions from both runs were combined, and concentrated in vacuo. The residue was taken up in 10 ml. of water. The resulting suspension was sonicated briefly, and filtered. The solids collected were dried under vacuum at room temperature over P2O5 for 18 hours, and then at 65 0C under vacuum for another 18 hours to afford the title compound (410 mg) as a cream-colored solid. LC-MS (ES) m/z = 463 [M+H]+. 1H NMR (400 MHz, CD3OD): δ 1.16 – 1.32 (m, 3H),1.29 (d, J = 6.8 Hz, 3H), 1.34 – 1.45 (m, 2H), 1.65 – 1.68 (m, 1 H), 1.76 – 1.81 (m, 5H), 1.85 – 1.92 (m, 2H), 1.97 – 2.05 (m, 1 H), 2.35 – 2.42 (m, 1 H), 2.97 (s, 3H), 3.1 1 – 3.15 (m, 1 H),3.64 – 3.70 (m, 1 H), 4.45 – 4.65 (bs, 1 H), 4.72 – 4.92 (bs, 1 H), 6.45 (s, 1 H), 7.52 (dd, J =8.5, 1.14 Hz, 1 H), 7.75 (d, J = 8.3 Hz, 1 H), 7.85 (s, 1 H).
ntermediate 112
Cis- methyl-6-methyl-3-piperidinecarboxylate

A solution of cis-3-methyl 1-(phenylmethyl)-6-methyl-1 ,3-piperidinedicarboxylate (69 g, 237 mol) in EtOH (50 mL) and EtOAc (300 mL) was added to a slurry of 10% Pd/C (3.7 g) in EtOAc (30 mL) and EtOH (10 mL) EtOH under nitrogen in a Parr Shaker bottle. The mixture was hydrogenated under 65 psi at room temperature for 4 hours. The mixture was filtered through celite, and washed with EtOAc. The filtrate was concentrated in vacuo to give 37 g of the title compound as a liquid. LC-MS (ES) m/z = 158 [M+H]+.
Intermediate 113
Methyl (3S,6f?)-6-methyl-3-piperidinecarboxylate L-(+)-tartaric acid salt 
L-(+)-Tartaric acid salt A suspension of L-(+)-tartaric acid (39 g, 260 mmol, 1.05 equiv) in IPA (200 ml.) and water (13 mL) water was heated in a water bath at 600C until all dissolved. To this hot stirred solution was added neat racemic methyl (3S,6R)-6-methyl-3-piperidinecarboxylate (39 g, 248 mmol), followed by addition of 25 mL of IPA rinse. The resulting mixture was heated to 60 0C, resulting in a clear solution, and then cooled to room temperature, while the hot water bath was removed. This hot solution was seeded with a sample of methyl (3S,6R)-6-methyl-3-piperidinecarboxylate L-(+)-tartaric acid salt that had a chiral purity of 98% ee, and aged at ambient temperature (with the water bath removed) for 20 minutes. The mixture turned into an oily texture with seeds still present. To the mixture was added 5 mL of water, and heated in the warm water bath at 43 0C. The mixture became clear with the seeds still present. The heating was stopped, and the mixture was stirred in the warm water bath. After 20 minutes, the mixture gradually turned into a paste. After another 10 min, the water bath was removed, and the mixture was stirred at ambient temperature for another 1 hour. The resulting paste was filtered. The cake was washed with 50 mL of IPA, giving 62 g of wet solids. This cake was taken up in 150 mL of IPA and 8 mL of water, and stirred as a slurry while being heated in a water bath to 60 0C (internal temp 55 0C) for 5 minutes. The heating was turned off while the mixture was still stirred in the warm water bath. After 30 min, the mixture was filtered. The cake was washed with 100 mL of IPA. Drying under house vacuum at room temperature for 48 hours gave 46.7 g of solids. An analytical sample was derivatised to the corresponding N-Cbz derivative (as in the preparation of intermediate 1 11 ), which was determined by chiral HPLC (methods used to analyze the resolution of intermediate 11 1 above) to have 85% ee. This material was taken up in IPA (420 mL) and water (38 mL) as a suspension. The mixture was heated in a water bath to 65 0C, at which time the mixture became a clear solution. The heating bath was removed. The mixture was seeded and aged at ambient temp for 20 hours. The solids formed were filtered, and washed with 100 mL of IPA. The solids collected were dried under house vacuum at room temperature for 24 h, and then under vacuum at room temperature for another 24 hours to give 28.5 g of the title compound. An analytical sample was converted to the N-Cbz derivative. The ee was determined to be 97.7%. LC-MS (ES) m/z = 158 [M+H]+.
Intermediate 114 4,6-Dichloro-Λ/-methyl-2-pyrimidinamine

Methylamine (2M solution, 113 ml_, 217 mmol, 2.05 equiv) was charged to a 1 L 3-neck flask fitted with a magnetic stirrer and a thermometer. The mixture was chilled in an ice bath. To this stirred solution was added via addition funnel a solution of 4,6-dichloro-2-(methylsulfonyl)pyrimidine (25 g, 1 10 mmol) in EtOAc (250 ml.) portionwise over a 25 minutes period. The temp was between 5-10 0C. After completion of addition, the ice bath was removed, and the mixture was stirred for 1 hour at ambient temperature. LCMS showed conversion complete. The suspension was filtered, and washed with EtOAc. The filtrate was concentrated in vacuo. The residue was partitioned between water (100 ml.) and EtOAc (450 ml_). The organic was washed with brine, dried over MgSO4, filtered and concentrated in vacuo to give white solids, which were triturated in 150 ml. of CH2CI2. These solids were collected by filtration and washing with cold CH2CI2 (50 ml_). Drying under house vacuum at room temperature for 20 hours, and then high vacuum at room temperature for 3 hours gave 9.31 g of the title compound as a solid. LC-MS (ES) m/z = 179 [M+H]+.
Intermediate 121 (3S,6/?)-1-r6-Chloro-2-(methylamino)-4-pyrimidinyll-Λ/-cvclohexyl-6-methyl-3-piperidinecarboxamide

To a suspension of (3S,6/?)-1-[6-chloro-2-(methylamino)-4-pyrimidinyl]-6-methyl-3-piperidinecarboxylic acid (3.05 g, 10.71 mmol) in CH2CI2 (50 ml.) at room temperature was added Hunig’s base (2.70 ml_, 15.43 mmol, 1.3 equiv) and cyclohexylamine (1.60 ml_, 14.2 mmol, 1.2 equiv), and the resulting mixture was chilled in an ice bath. To this stirred solution was added HATU (4.96 g, 13.1 mmol, 1.1 equiv) in one portion, and the resulting suspension was stirred in the ice bath for 30 minutes. LCMS showed conversion complete. The mixture was diluted with CH2CI2 (50 ml.) and filtered through celite. The filtrate was washed water (2 X 25 ml.) and then brine. The organic was dried over Na2SO4, filtered, and concentrated in vacuo. Silica gel column chromatography using gradient elution of 1 % EtOAc in CHCI3 to 50% EtOAc in CHCI3 afforded the title compound (4.26 g) as a foam. LC-MS (ES) m/z = 366 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2011), 54(6), 1871-1895.
http://pubs.acs.org/doi/full/10.1021/jm101527u

Phosphoinositide-dependent protein kinase-1(PDK1) is a master regulator of the AGC family of kinases and an integral component of the PI3K/AKT/mTOR pathway. As this pathway is among the most commonly deregulated across all cancers, a selective inhibitor of PDK1 might have utility as an anticancer agent. Herein we describe our lead optimization of compound 1toward highly potent and selective PDK1 inhibitors via a structure-based design strategy. The most potent and selective inhibitors demonstrated submicromolar activity as measured by inhibition of phosphorylation of PDK1 substrates as well as antiproliferative activity against a subset of AML cell lines. In addition, reduction of phosphorylation of PDK1 substrates was demonstrated in vivo in mice bearing OCl-AML2 xenografts. These observations demonstrate the utility of these molecules as tools to further delineate the biology of PDK1 and the potential pharmacological uses of a PDK1 inhibitor.
REFERENCES
Najafov, et al., Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem.J. (2011), 433 (2) 357.
For a PDK1 inhibitor, the substrate matters.
Knight ZA. Biochem J. 2011 Jan 15;433(2):e1-2. PMID: 21175429.
Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1.
Najafov A, et al. Biochem J. 2011 Jan 15;433(2):357-69. PMID: 21087210.

Jeffrey Axten
Director, Medicinal Chemistry, Virtual Proof of Concept DPU at GlaxoSmithKline
/////
Improved one-pot synthesis of N, N-diisopropyl-3-(2-Hydroxy-5-methylphenyl)-3-phenyl propanamide; a key intermediate for the preparation of racemic Tolterodine

Tolterodine is chemically known as (R)-N,N-disiopropyl-3-(2-hydroxy-5-methyl phenyl)-3-phenyl propyl amine. Tolterodine acts as a muscarinic receptor antagonist. It is useful in the treatment of urinary incontinence [1]. Tolterodine tartrate acts by relaxing the smooth muscle tissues in the walls of the bladder by blocking cholinergic receptors[2]. Tolterodine tartrate [3] is marketed by Pharmacia & Upjohn in the brand name of Destrol®.
The present invention relates to a novel process for the preparation of N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide (4); a key intermediate for the preparation of Tolterodine (1). Some different approaches have been published [4–8] for the preparation of N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide (4). These methods involve multistep synthesis using hazardous, expensive reagents and some of the methods [6] involve activators like Grignard reagents, LDA, n-butyl lithium, Lewis acids. Hence there is a need to develop an alternative, plant friendly procedure for the preparation of N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide (4) from 3,4-dihydro-6-methyl-4-phenylcoumarin (2) (Fig1).


Ring opening reactions of dihydrocoumarins are well known in literature[9–11]. But in the present invention, we have described a new methodology (Scheme 1 & Scheme2) for the preparation ofN,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide (4) by using inexpensive and commercially vailable starting materials like 3, 4-dihydro-6-methyl 4-phenylcoumarin (2), which was synthesized from p-cresol and trans-cinnamic acid [12].

Scheme 1
N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide 4.

Scheme 2
N-Isopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide 5.
3,4-Dihyhydro-6-methyl 4-phenylcoumarin (2) reacts with diisopropylamine (6) in presence of acetic acid gives N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide (4) at room temperature. This process of compound 4 is very useful for commercialization of Tolterodine 1 in plant.
General procedure for the synthesis of compounds 4-4c & 5-5c
To a solution of 3,4-dihyhydro-6-methyl 4-phenylcoumarin 2 (10 g, 42 mmol) in diisopropylether (200 mL), N,N-diisopropylamine (33.95 g, 336 mmol) and acetic acid (10 g, 168 mmol) were added at room temperature. The suspension was stirred for 16 h at room temperature. The reaction mass was concentrated, the resulting residue was crystallized with D.M.Water (50 mL) and diisopropyl ether (50 mL) mixture to gave N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide 4 (10.6 g, 75% yield).
N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide 4
IR (KBr) cm-1: 3024 (Aromatic C-H, str.), 2949, 2904, 2869 (Aliphatic C-H, str.), 1630 (C═O, str.), 1609, 1555, 1510 (C═C, str.), 1469, 1459 (CH2 bending), 1270 (C-N, str.), 1072 (C-O, str.), 788, 769 (Aromatic CH Out-of-plane bend). 1H NMR (300 MHz, DMSO-d6) δ 1.04 (d, 12H), 2.089 (s, 3H), 2.79 (m, 2H), 3.037 (m, 2H), 4.702 (t, 1H), 6.6 (d, 1H), 6.75 (d, 2H), 7.127-7.246 (m, 5H). 13C NMR (125 MHz, DMSO-d6) δ 19.39, 20.36, 45.69, 115.33, 125.70, 127.20, 128.15, 130.60, 144.43, 152.23, 173.37. MS m/z: 340 [(M + H)+].
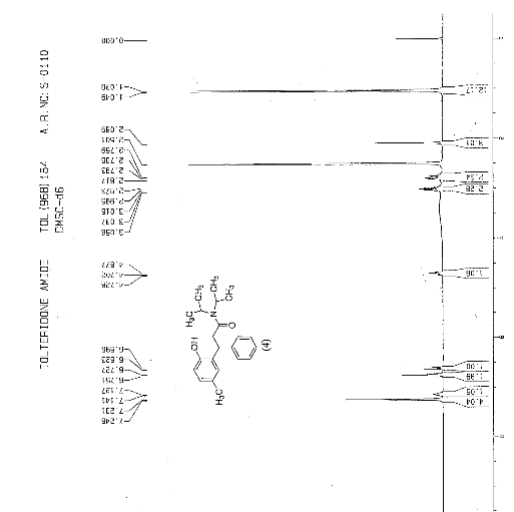

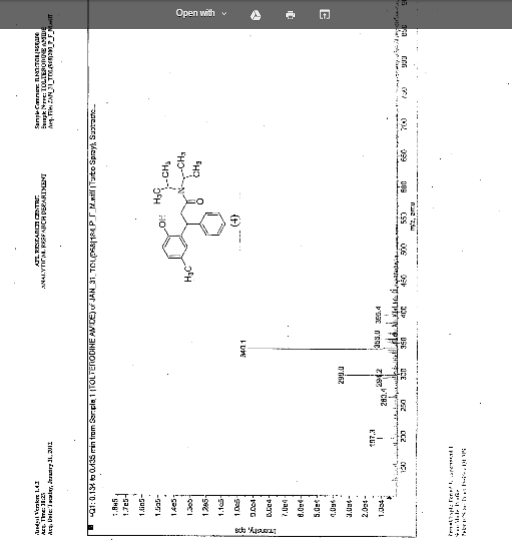
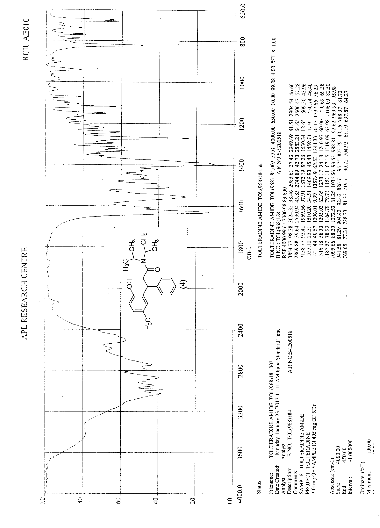
Improved one-pot synthesis of N, N-diisopropyl-3-(2-Hydroxy-5-methylphenyl)-3-phenyl propanamide; a key intermediate for the preparation of racemic Tolterodine
2Engineering Chemistry Department, AU college of Engineering, Andhra University, Visakhapatnam 530003, Andhra Pradesh, India
The electronic version of this article is the complete one and can be found online at:http://www.sustainablechemicalprocesses.com/content/2/1/2
http://www.sustainablechemicalprocesses.com/content/2/1/2/additional

Srinivas garaga
scientist at Aurobindo Pharma
Chemical Research and Development Department, Aurobindo Pharma Ltd

/////
Chlorzoxazone
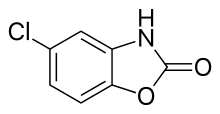
Chlorzoxazone
| Chlorzoxazone; Paraflex; Chlorzoxazon; Myoflexin; Solaxin; 95-25-0; |
5-chloro-3H-1,3-benzoxazol-2-one
A centrally acting central muscle relaxant with sedative properties. It is claimed to inhibit muscle spasm by exerting an effect primarily at the level of the spinal cord and subcortical areas of the brain. (From Martindale, The Extra Pharmacopoea, 30th ed, p1202)
| Property | Value | Source |
|---|---|---|
| melting point | 191-191.5 | Marsh, D.F.; US. Patent 2,895,877; July 21, 1959; assigned to McNeil Laboratories, Inc. |
Marsh, D.F.; US. Patent 2,895,877; July 21, 1959; assigned to McNeil Laboratories, Inc.
Chlorzoxazone (Paraflex) is a centrally acting muscle relaxant used to treat muscle spasm and the resulting pain or discomfort. It acts on the spinal cord by depressing reflexes. It is sold as Muscol or Parafon Forte, a combination of chlorzoxazone and acetaminophen (Paracetamol). Possible side effects include dizziness, lightheadedness, malaise, nausea, vomiting, and liver dysfunction. Used with acetaminophen it has added risk of hepatoxicity, which is why the combination is not recommended. It can also be administered for acute pain in general and for tension headache (muscle contraction headache).
Synthesis

Chlorzoxazone, 5-chloro-2-benzoxazolione, is synthesized by a hetercyclization reaction of 2-amino-4-chlorophenol with phosgene.
2
The Chlorzoxazone with CAS registry number of 95-25-0 is also known as 2-Benzoxazolol, 5-chloro-. The IUPAC name is 5-Chloro-3H-1,3-benzoxazol-2-one. It belongs to product categories of Oxazole&Isoxazole; Intermediates & Fine Chemicals; Pharmaceuticals. Its EINECS registry number is 202-403-9. In addition, the formula is C7H4ClNO2 and the molecular weight is 169.57. This chemical should be stored in sealed containers in cool, dry place and away from oxidizing agents.
Physical properties about Chlorzoxazone are: (1)ACD/LogP: 2.19; (2)# of Rule of 5 Violations: 0; (3)ACD/LogD (pH 5.5): 2.19; (4)ACD/LogD (pH 7.4): 2.15; (5)ACD/BCF (pH 5.5): 27.16; (6)AACD/KOC (pH 7.4): 340.79; (7)#H bond acceptors: 3; (8)#H bond donors: 1; (9)#Freely Rotating Bonds: 0; (10)Index of Refraction: 1.603; (11)Molar Refractivity: 39.18 cm3; (12)Molar Volume: 114 cm3; (13)Surface Tension: 50 dyne/cm; (14)Density: 1.486 g/cm3; (15)Flash Point: 157.5 °C; (16)Enthalpy of Vaporization: 60.3 kJ/mol; (17)Boiling Point: 336.9 °C at 760 mmHg; (18)Vapour Pressure: 5.58E-05 mmHg at 25 °C.
Preparation of Chlorzoxazone: it is prepared by reaction of 5-chloro-2-hydroxy-benzamide. The reaction needs reagents iodobenzene diacetate, KOH and solvent methanol at the temperature of 0 °C. The yield is about 68%.

References
- D.F. Marsh, U.S. Patent 2,895,877 (1959).
- Dong DL, Luan Y, Feng TM, Fan CL, Yue P, Sun ZJ, Gu RM, Yang BF. (2006). “Chlorzoxazone inhibit contraction of rat thoracic aorta”. Eur J Pharmacol 545 (2–3): 161–6. doi:10.1016/j.ejphar.2006.06.063. PMID 16859676.
- Park J, Kim K, Park P, Ha J (2006). “Effect of high-dose aspirin on CYP2E1 activity in healthy subjects measured using chlorzoxazone as a probe”. J Clin Pharmacol 46 (1): 109–14. doi:10.1177/0091270005282635. PMID 16397290.
- Wan J, Ernstgård L, Song B, Shoaf S (2006). “Chlorzoxazone metabolism is increased in fasted Sprague-Dawley rats”. J Pharm Pharmacol 58 (1): 51–61. doi:10.1211/jpp.58.1.0007. PMC 1388188. PMID 16393464.
- PARAFON DSC (chlorzoxazone) tablet, Daily Med, U.S. National Library of Medicine
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-chloro-3H-benzooxazol-2-one
|
|
| Clinical data | |
| Trade names | Parafonforte |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a682577 |
| Routes of administration |
oral |
| Pharmacokinetic data | |
| Bioavailability | well absorbed |
| Protein binding | 13–18% |
| Metabolism | hepatic |
| Biological half-life | 1.1 hr |
| Excretion | urine (<1%) |
| Identifiers | |
| CAS Registry Number | 95-25-0 |
| ATC code | M03BB03 |
| PubChem | CID: 2733 |
| IUPHAR/BPS | 2322 |
| DrugBank | DB00356 |
| ChemSpider | 2632 |
| UNII | H0DE420U8G |
| KEGG | D00771 |
| ChEBI | CHEBI:3655 |
| ChEMBL | CHEMBL1371 |
| Chemical data | |
| Formula | C7H4ClNO2 |
| Molecular mass | 169.565 g/mol |
/////
As of September 2015, updated Requirements apply to the Application of a CEP!
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
![]()
As of September 2015, updated Requirements apply to the Application of a CEP!
The EDQM recently revised its certification policy. Read more here about what you now need to consider when applying for a Certificate of Suitability (CEP).
The EDQM recently published a revised version of its certification policy document titled “Content of the dossier for chemical purity and microbiological quality“. The revision takes into account the new regulatory developments in Europe that are reflected in many revised and, to some extent, new guidelines of the EMA, ICH as well as in some revised general chapters and monographs of the European Pharmacopoeia (see the summary of these guidance documents under “References” at the end of the policy document).
The aim of the policy document is to provide CEP applicants with a guideline for preparing the authorisation dossier and for compiling all the documents required for this…
View original post 850 more words
Gnidia glauca

Search of complementary and alternative medicine has gained a thrust in the recent decade due to the pronounced side effects and health hazards of the chemically synthesized drugs. Hereby, a comprehensive knowledge about the traditionally used medicinal plants is indispensable for exploration of its novel bioactive components. One of such comparatively less explored medicinal plant is Gnidia glauca. Although, it has folkloric, traditional phytomedicinal and agrochemical applications in various parts of the world, still there are no available scientific validations or evidences to support the fact. In African medicine it is used for treatment of abdominal pain, cancers, wounds, snake bites, sore throat and burns. It is also well known for its piscicidal, insecticidal, molluscicidal and even homicidal activity for its use as arrow poisons. Similarly, its antineoplastic activity is reported to be remarkably superior [1]. However, till date there is no comprehensive information on the plant.

In view of the background, herein we present the first commentary on complete research carried out till date on G. glauca and its promises as complementary and alternative medicine (Figure 1).

Metabolic enzymes, like α-amylase and α-glucosidase are considered as key targets for discovery of antidiabetic drugs. Ethanolic, methanolic and ethyl acetate extracts of G. glauca flowers showed an excellent inhibitory potential (~70 % and above) against α-amylase while only methanol extract of leaf showed high inhibition against α-glucosidase [11].
- Rao SB, Jayanthi M, Yogeetha R, Ramakrishnaiah H, Nataraj J (2013) Free radical scavenging activity and reducing power of Gnidia glauca (Fresen.) Gilg. J App Pharm Sci 3: 203-207.
- Naik ST, Maheswarappa V (2007) Prospects of using plant extracts in management of pineapple heart rot. Karnataka J AgricSci 20: 180-182.
- Junaid S, Dileep N, Rakesh KN, Pavithra GM, Vinayaka KS, et al. (2013) Anticariogenic activity of Gnidia glauca (Fresen.) Gilg, Pothosscandens L. and ElaegnuskologaSchlecht. J App Pharm Sci 3: 020-023.
- Prashith KTR, Vivek MN, Junaid S, Rakesh KN, Dileep N, et al. (2014) Inhibitory activity of Polyalthialongifolia, Anaphalislawii andGnidia glauca against Colletotrichumcapsici and urinary tract pathogens. SciTechnol Arts Res J 3: 26-30.
- Kareru PG, Kenji GM, Gachanja AN, Keriko JM, Mungai G (2006) Traditional medicines among the Embu and Mbeere peoples of Kenya. Afr J Tradit Complement Altern Med 4: 75-86.
- Amarajeewa BWRC, Mudalige AP, Kumar V (2007) Chemistry and mosquito larvicidal activity of Gnidia glauca. Proceedings of the Peradeniya University Research Sessions, Sri Lanka, 12: 101-102.
- Kupchan SM, Shizuri Y, Sumner WC Jr, Haynes HR, Leighton AP, et al. (1976) Isolation and structural elucidation of new potent antileukemicditerpenoid esters from Gnidia species. J Org Chem 41: 3850-3853.
- Ghosh S,Derle A,Ahire M, More P,Jagtap S, et al. (2013) Phytochemical analysis and free radical scavenging activity of medicinal plants Gnidia glauca andDioscoreabulbifera. PLoS One 8: e82529.
- Ghosh S,Ahire M, Patil S, Jabgunde A, BhatDusane M, et al. (2012) Antidiabetic Activity of Gnidia glauca and Dioscoreabulbifera: Potent Amylase and Glucosidase Inhibitors. Evid Based Complement Alternat Med 2012: 929051.
- Ghosh S, Patil S, Ahire M, Kitture R, Gurav DD, et al. (2012) Gnidia glauca flower extract mediated synthesis of gold nanoparticles and evaluation of its chemocatalytic potential. J Nanobiotechnology 10: 17.
- Khadke SS, Pachauri DR, Mahajan SD.(2011) An acute oral toxicity study of Gnidia glauca (Fresen.) Gilg. in albino rats as per OECD guideline 425. Int J PharmTech Res 3: 787-791.




Professor B.A. Chopade
Fogarty Fellow Illinois University, Chicago, USAVice- Chancellor
Aurangabad-431004
Maharashtra State, India
Ph.No. : (office) (0240)-2403111 Fax No. (0240)-2403113/335

Balu A Chopade
Professor B.A. Chopade has been working as a Vice-Chancellor of Dr. Babasaheb Ambedkar Marathwada University, Aurangabad, Maharashtra from 04/06/2014. He has been working as Professor of Microbiology and Coordinator of University of Potential Excellence Programme (UPE Phase I & II) of UGC in Biotechnology at University of Pune. He was Director of Institute of Bioinformatics and Biotechnology (IBB), University of Pune from 2006 to 2012. He has established and developed IBB as a unique national institute and as a centre of excellence in research, innovation and teaching in biotechnology in India. He has successfully established an innovative benchmarking of publications in peer reviewed international journals of repute by undergraduate students at IBB. He was Head of the Department of Microbiology, University of Pune from 1994, 1996-2000 and 2003-2006. He has 35 years of experience in research, innovation, teaching and administration at the University of Pune.
Professor Chopade has several national and international academic honors and professional distinctions to his credits. He was the Government of India Scholar at the University of Nottingham, England and obtained a Ph.D. degree in microbiology and molecular genetics (1983-1986). He was also the recipient of the most prestigious Fogarty International NIH Research Fellowship Award from Govt. of USA for Post-Doctoral Research at the University of Illinois at Chicago (1994-1996) in genetic engineering. He is also recipient of International Award in Microbiology from International Union of Microbiological Societies (IUMS) in 1986. He has had very distinguished academic career and has carved his career entirely on the basis of merit and academic excellence.He was also coordinator of ALIS link programme between British Council London and Department of Microbiology, University of Pune (1994-1997).
He has published more than 100 research papers in peer reviewed international and national journals with high impact factor. The total impact factor of his research is more than 260, with h-index 26 and i10-index 52. His work is cited more than 2002 times (www.scholar.google.com). He has obtained 2 USA and 8 Indian patents. His research work has been cited by Nobel Laureate Professor Arthur Kornberg from University of Stanford, California, USA. His pioneering work on e-DNA and Acinetobacter vesicles is also cited by “Nature” journal from England. His work also has been cited in 3 textbooks of microbiology published from USA and Europe. He has presented more than 150 papers in International and National Conferences and has given large number of plenary lectures. He has successfully supervised 27 Ph.D., 4 M.Phil. and 10 Post Doctoral scholars for their research. Currently 2 Post-Doctoral Fellows and 4 Ph.D. students are working with him. His 3 students had obtained Young Scientist Awards in 1993 at Stockholm, from International Congress of Chemotherapy (ICC), Europe. His research area includes microbial and molecular genetics, biotechnology and nanomedicine. He is on editorial board of Wealth of India Publication series, from CSIR New Delhi, as well as number of research journals. He has obtained research grants and funding of more than rupees 10 crores from national and international funding agencies. He has successfully completed 32 major research projects from various National and International funding agencies. He has developed a new herbal medicine “Infex” which is manufactured by Shrushti Herbal Pharma Ltd., Bangalore. He is a pioneer in the area of Industry-Academia interactions and entrepreneurship in biotechnology and microbiology at IBB, University of Pune.
He was a visiting scientist at the Pasteur Institute, Paris, France and King’s College, University of London in 1990. He has received number of awards and most notable are: Pradnya Bhushan Dr. Babasaheb Ambedkar Award(2014) Aurangabad. Bronze Medal, International Genetically Engineered Machines (iGEM), Massachusetts Institute of Technology (MIT), USA (2009), Pradnyavant Award (2011) by Undalkar Foundation, Karad. Maharashtra, Best teacher award by Pune Municipal Corporation (1993); Best research paper awards in microbial and molecular genetics (1988 & 2002) by Association of Microbiologist of India; He was recipient of Wadia Oration award (2008) by Institution of Engineers, India. Best research paper award in Bioinformatics (2009) by SBC, India. Summer Fellowship of Indian Academy of Sciences, Bangalore (2001). His biography is published by American Biographical Institute, USA (2000) and International Biographical Centre Cambridge (1991). He is member of American Society for Microbiology, USA and Society for General Microbiology, England since 1984. He is also a life member of number of national organizations like Association of Microbiologist of India (AMI) and Biotechnology Society of India (BSI), Society of Biological Chemists of India (SBC) Indian Science Congress (ISC). He is recipient of Marcus’s Who’s Who in Science and Engineering U.S.A. (2001), Marcus’s Who’s Who of the World, U.S.A. (2000), Marcus’s Who’s Who in Medicine, U.S.A. (2002), Marcus’s Who’s Who in Education, U.S.A. (2002).
He has been working on various authorities of University of Pune, as well as many State and Central Universities in India. Such as, Chairman, Board of Studies in Microbiology from 1997-2000 & 2005-2007. Member, BOS in Biotechnology (2005-2006, 2012-2017), Member of Academic Council (1997-2000 & 2000-2005) and Board of College and University Development (BCUD) of University of Pune from 1997-2000 & 2000-2005. Member, Faculty of Science, University of Pune (1997-2000, 2003-2005) and Member, Board of Teaching and Research (BUTR), (1997-2002). Member, Board of studies in Biochemistry and Molecular Biology, Central University Pondichery (2001-2003). Member, Board of Studies in Biochemistry and Molecular Biology, Shivaji University Kolhapur (2009-2014). Member, Board of Studies in Life Sciences, North Maharashtra University, Jalgaon (1994-1999). Member, Faculty of Science, Bharti Vidyapeeth Pune (2013-2018). Member, Faculty of Science, North Maharashtra University, Jalgaon (1990-1999).
He was chairman of large number of committees of UGC, New Delhi such as 11th Plan Research Committee, Research Projects and Deemed University Status since 2008. Chairman, International Travel Grants, (2008-2013). He was Chairman of State Eligibility Test (SET) in Microbiology for Govt. of Maharashtra and Goa from (1997-2000). He has active an involvement in the national and international scientific organizations. He has been involved in University administration in the various capacities for more than 33 years, as a chairman and member of large number of development, finance, examination and administration committees of University of Pune.
He is member of research and recognition committees of numerous state and central universities in India. He also worked as a coordinator of DBT Potential Excellence Programme at the Department of Microbiology, University of Pune (1994-1998). He is nominee of Department of Biotechnology, Government of India for Reliance Industries limited Mumbai, Biorefinery of Somaiya Group of Industries in Karnataka and Agharkar Research Institute (ARI) Pune.
His vision for Dr.Babasaheb Ambedkar Marathwada University (BAMU), Aurangabad is to transform it as one of the best research and innovation Universities in India and subsequently develop as a world class University.
///////Gnidia glauca, Phytochemistry, Ethnomedicine, Dr. Babasaheb Ambedkar Marathwada University, Aurangabad, Maharashtra, india, Balu A Chopade
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

