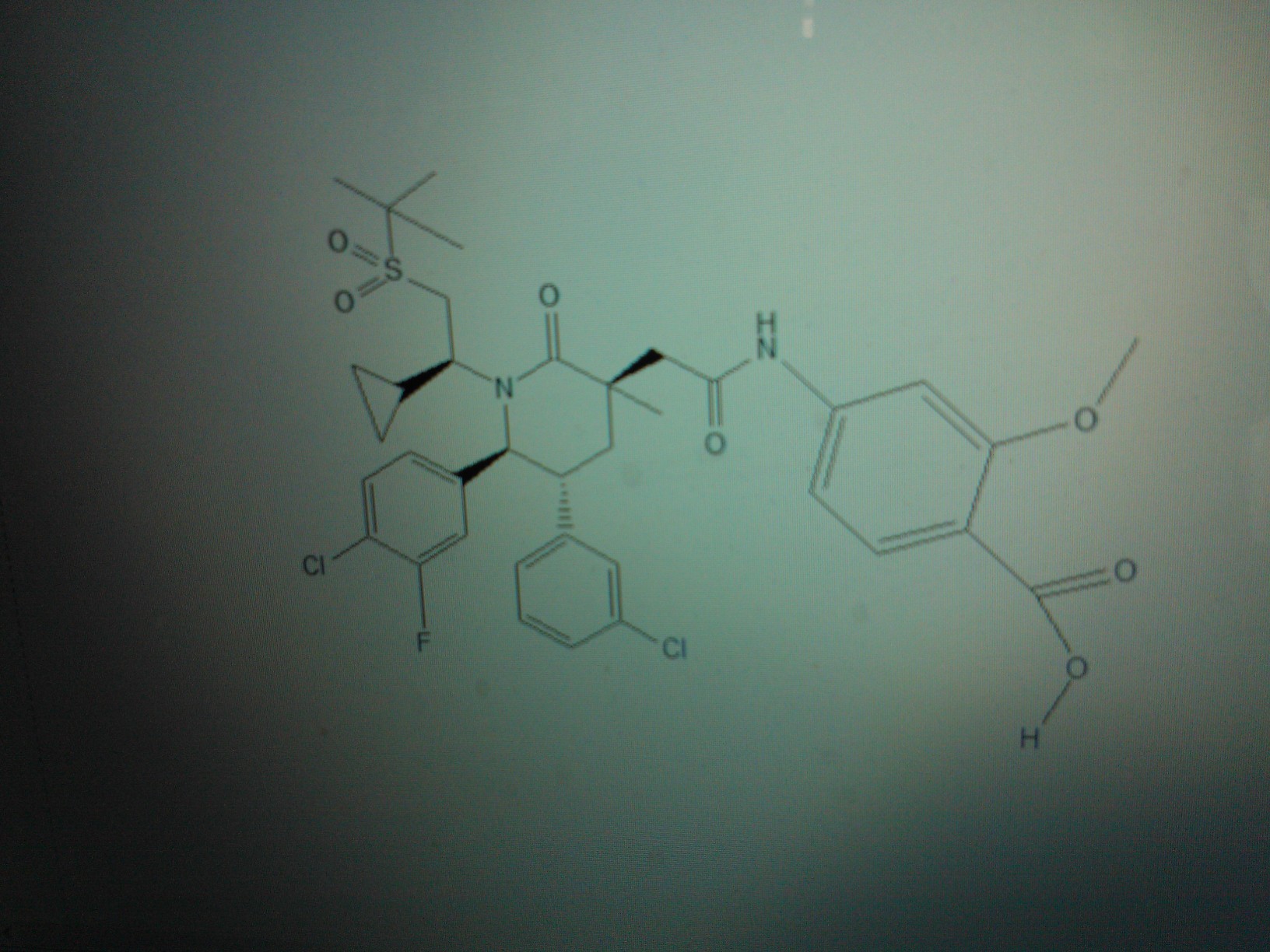
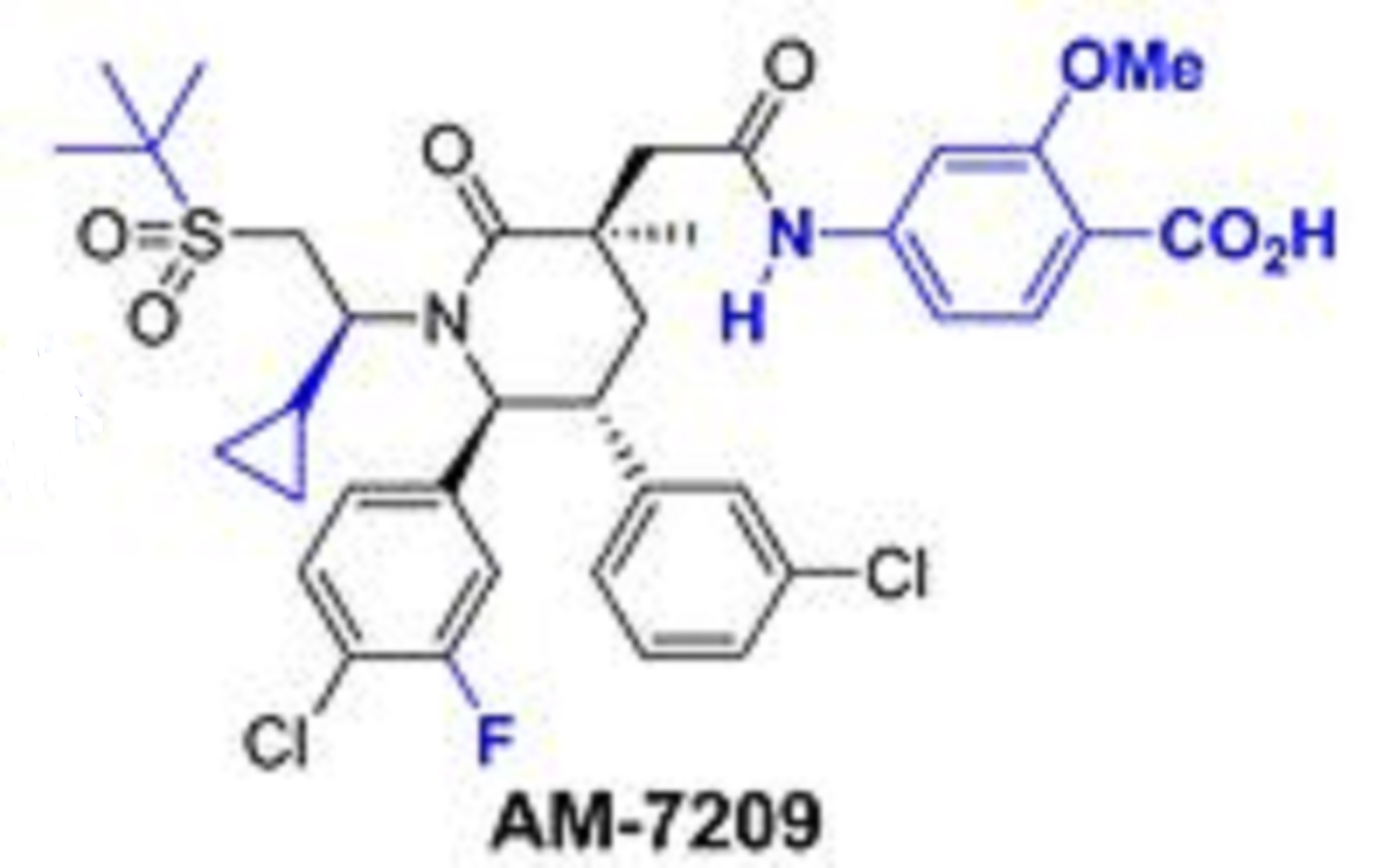
AM 7209

Amgen Inc. INNOVATOR
MF 747.700043 g/mol, C37H41Cl2FN2O7S
cas 1623432-51-8
US8952036
4-({[(3r,5r,6s)-1-[(1s)-2-(Tert-Butylsulfonyl)-1-Cyclopropylethyl]-6-(4-Chloro-3-Fluorophenyl)-5-(3-Chlorophenyl)-3-Methyl-2-Oxopiperidin-3-Yl]acetyl}amino)-2-Methoxybenzoic Acid;
4-[[2-[(3R,5R,6S)-1-[(1S)-2-tert-butylsulfonyl-1-cyclopropylethyl]-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl]acetyl]amino]-2-methoxybenzoic acid
Benzoic acid, 4-[[2-[(3R,5R,6S)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-1-[(1S)-1-cyclopropyl-2-[(1,1-dimethylethyl)sulfonyl]ethyl]-3-methyl-2-oxo-3-piperidinyl]acetyl]amino]-2-methoxy-
4-(2-((3R,5R,6S)-1-((S)-2-(tert-Butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoic Acid
MDM2 inhibitor that is useful as therapeutic agent, particularly for the treatment of cancers
DETAILS COMING…………
p53 is a tumor suppressor and transcription factor that responds to cellular stress by activating the transcription of numerous genes involved in cell cycle arrest, apoptosis, senescence, and DNA repair. Unlike normal cells, which have infrequent cause for p53 activation, tumor cells are under constant cellular stress from various insults including hypoxia and pro-apoptotic oncogene activation. Thus, there is a strong selective advantage for inactivation of the p53 pathway in tumors, and it has been proposed that eliminating p53 function may be a prerequisite for tumor survival. In support of this notion, three groups of investigators have used mouse models to demonstrate that absence of p53 function is a continuous requirement for the maintenance of established tumors. When the investigators restored p53 function to tumors with inactivated p53, the tumors regressed.
p53 is inactivated by mutation and/or loss in 50% of solid tumors and 10% of liquid tumors. Other key members of the p53 pathway are also genetically or epigenetically altered in cancer. MDM2, an oncoprotein, inhibits p53 function, and it is activated by gene amplification at incidence rates that are reported to be as high as 10%. MDM2, in turn, is inhibited by another tumor suppressor, p14ARF. It has been suggested that alterations downstream of p53 may be responsible for at least partially inactivating the p53 pathway in p53WT tumors (p53 wildtype). In support of this concept, some p53WT tumors appear to exhibit reduced apoptotic capacity, although their capacity to undergo cell cycle arrest remains intact. One cancer treatment strategy involves the use of small molecules that bind MDM2 and neutralize its interaction with p53. MDM2 inhibits p53 activity by three mechanisms: 1) acting as an E3 ubiquitin ligase to promote p53 degradation; 2) binding to and blocking the p53 transcriptional activation domain; and 3) exporting p53 from the nucleus to the cytoplasm. All three of these mechanisms would be blocked by neutralizing the MDM2-p53 interaction. In particular, this therapeutic strategy could be applied to tumors that are p53WT, and studies with small molecule MDM2 inhibitors have yielded promising reductions in tumor growth both in vitro and in vivo. Further, in patients with p53-inactivated tumors, stabilization of wildtype p53 in normal tissues by MDM2 inhibition might allow selective protection of normal tissues from mitotic poisons.
The present invention relates to a compound capable of inhibiting the interaction between p53 and MDM2 and activating p53 downstream effector genes. As such, the compound of the present invention would be useful in the treatment of cancers, bacterial infections, viral infections, ulcers and inflammation. In particular, the compound of the present invention is useful to treat solid tumors such as: breast, colon, lung and prostate tumors; and liquid tumors such as lymphomas and leukemias. As used herein, MDM2 means a human MDM2 protein and p53 means a human p53 protein. It is noted that human MDM2 can also be referred to as HDM2 or hMDM2.


PATENT
US8952036
http://www.google.com/patents/US20140243372
Example 4 2-((3R,5R,6S)-1-((S)-2-(tert-Butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetic acid
Step A. Methyl-4-chloro-3-fluorobenzoate
-
A solution of 4-chloro-3-fluoro benzoic acid (450.0 g, 2.586 mol, Fluororochem, Derbyshire, UK) in methanol (4.5 L) was cooled to 0° C. and thionyl chloride (450.0 mL) was added over 30 minutes. The reaction mixture was stirred for 12 hours at ambient temperature. The reaction was monitored by TLC. Upon completion, the solvent was removed under reduced pressure and the residue was quenched with 1.0 M sodium bicarbonate solution (500 mL). The aqueous layer was extracted with dichloromethane (2×5.0 L). The combined organic layer was washed with brine (2.5 L), dried over anhydrous sodium sulfate and concentrated under reduced pressure afforded the title compound as light brown solid. The crude compound was used in the next step without further purification.
-
1H NMR (400 MHz, CDCl3, δ ppm): 7.82-7.74 (m, 2H), 7.46 (dd, J=8.2, 7.5 Hz, 1H), 3.92 (s, 3H).
Step B. 1-(4-chloro-3-fluorophenyl)-2-(3-chlorophenyl)ethanone
-
-
Sodium bis(trimethylsilyl)amide (1 M in tetrahydrofuran, 4 L, 4000 mmol) was added over 1 hour to a solution of 3-chlorophenyl acetic acid (250.0 g, 1465 mmol) in anhydrous tetrahydrofuran (1.75 L) at −78° C. under nitrogen. The resulting reaction mixture was stirred for an additional hour at −78° C. Then, a solution of methyl-4-chloro-3-fluorobenzoate (221.0 g, 1175 mmol, Example 4, Step A) in tetrahydrofuran (500 mL) was added over 1 hour at −78° C., and the resulting reaction mixture was stirred at the same temperature for 2 hours. The reaction was monitored by TLC. On completion, reaction mixture was quenched with 2 N hydrochloric acid (2.5 L) and aqueous phase was extracted with ethyl acetate (2×2.5 L). The combined organic layer was washed with brine (2.5 L), dried over anhydrous sodium sulfate and concentrated under reduced pressure to provide the crude material which was purified by flash column chromatography (silica gel: 100 to 200 mesh, product eluted in 2% ethyl acetate in hexane) to afford the title compound as a white solid.
-
1H NMR (400 MHz, CDCl3, δ ppm): 7.74 (ddd, J=10.1, 8.9, 1.8 Hz, 2H), 7.56-7.48 (m, 1H), 7.26 (t, J=6.4 Hz, 3H), 7.12 (d, J=5.7 Hz, 1H), 4.22 (s, 2H). MS (ESI) 282.9 [M+H]+.
Step C. Methyl 5-(4-chloro-3-fluorophenyl)-4-(3-chlorophenyl)-2-methyl-5-oxopentanoate
-
Methyl methacrylate (125.0 g, 1097 mmol) and potassium tert-butoxide (1 M in tetrahydrofuran, 115 mL, 115 mmol) were sequentially added to a solution of 1-(4-chloro-3-fluorophenyl)-2-(3-chlorophenyl)ethanone (327.0 g, 1160 mmol, Example 4, Step B) in anhydrous tetrahydrofuran (2.61 L), at 0° C. The reaction mixture was stirred for 1 hour at 0° C. and then warmed to ambient temperature and stirred for 12 hours. On completion, the reaction was quenched with water (1.0 L) and extracted with ethyl acetate (2×2.5 L). The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure to get the crude material which was purified by flash column chromatography (silica gel: 60 to 120 mesh, product eluted in 4% ethyl acetate in hexane) affording the title compound (mixture of diastereomers) as light yellow liquid.
1H NMR (400 MHz, CDCl3, δ ppm): 7.74-7.61 (m, 4H), 7.47-7.40 (m, 2H), 7.28-7.18 (m, 6H), 7.16-7.10 (m, 2H), 4.56 (m, 2H), 3.68 (s, 3H), 3.60 (s, 3H), 2.50-2.39 (m, 2H), 2.37-2.25 (m, 2H), 2.10-2.02 (m, 1H), 1.94 (ddd, J=13.6, 9.1, 4.2 Hz, 1H), 1.21 (d, J=7.0 Hz, 3H), 1.15 (d, J=7.0 Hz, 3H). MS (ESI) 383.0 [M+H]+.
Step D. (3S,5R,6R)-6-(4-Chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one and (3R,5R,6R)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one
-
-
In a 2000 mL reaction vessel charged with methyl 5-(4-chloro-3-fluorophenyl)-4-(3-chlorophenyl)-2-methyl-5-oxopentanoate (138.0 g, 360 mmol, Example 4, Step C) (which was cooled on ice for 10 minutes before transferring to a glove bag) anhydrous 2-propanol (500 mL), and potassium tert-butoxide (16.16 g, 144 mmol) were sequentially added while in a sealed glove bag under argon. This mixture was allowed to stir for 30 minutes. RuCl2(S-xylbinap)(S-DAIPEN) (1.759 g, 1.440 mmol, Strem Chemicals, Inc., Newburyport, Mass., weighed in the glove bag) in 30.0 mL toluene was added. The reaction was vigorously stirred at room temperature for 2 hours. The vessel was set on a hydrogenation apparatus, purged with hydrogen 3 times and pressurized to 50 psi (344.7 kPa). The reaction was allowed to stir overnight at room temperature. On completion, the reaction was quenched with water (1.5 L) and extracted with ethyl acetate (2×2.5 L). The organic layer was washed with brine (1.5 L), dried over anhydrous sodium sulfate and concentrated under reduced pressure to get crude material which was purified by flash column chromatography (silica gel; 60-120 mesh; product eluted in 12% ethyl acetate in hexane) to provide a dark colored liquid as a mixture of diastereomers.
-
The product was dissolved in (240.0 g, 581 mmol) in tetrahydrofuran (1.9 L) and methanol (480 mL), and lithium hydroxide monohydrate (2.5 M aqueous solution, 480.0 mL) was added. The reaction mixture was stirred at ambient temperature for 12 hours. On completion, the solvent was removed under reduced pressure and the residue was acidified with 2 N hydrochloric acid to a pH between 5 and 6. The aqueous phase was extracted with ethyl acetate (2×1.0 L). The combined organic layer was washed with brine (750 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide a dark colored liquid, which was used without further purification.
-
A portion of the crude intermediate (25.4 g, predominantly seco acid) was added to a 500 mL round bottom flask, equipped with a Dean-Stark apparatus. Pyridinium p-toluenesulfonate (0.516 g, 2.053 mmol) and toluene (274 mL) were added, and the mixture was refluxed for 1 hour (oil bath temperature about 150° C.). The reaction was cooled to room temperature and concentrated under reduced pressure. The reaction was diluted with saturated aqueous sodium bicarbonate (150 mL), extracted with diethyl ether (2×150 mL), and washed with brine (150 mL). The combined organic layer was dried over magnesium sulfate, filtered and concentrated under reduced pressure. Purification by flash column chromatography (divided into 3 portions, 330 g SiO2/each, gradient elution of 0% to 30% acetone in hexanes, 35 minutes) provided the title compounds as a pale yellow solid and a 1:1.6 mixture of diastereomers at C2. MS (ESI) 353.05 [M+H]+.
-
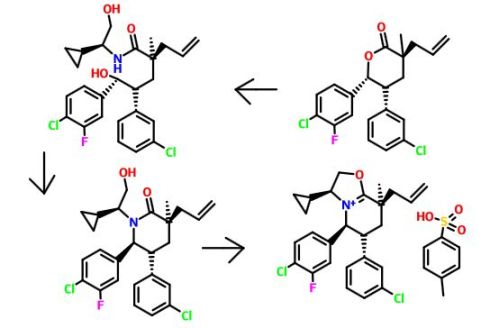
Step E. (3S,5R,6R)-3-Allyl-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one
-
-
(3S,5R,6R)-6-(4-Chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one and (3R,5R,6R)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one (18 g, 51.0 mmol, Example 4, Step D) was added to an oven dried 500 mL round-bottom flask. The solid was dissolved in anhydrous toluene and concentrated to remove adventitious water. 3-Bromoprop-1-ene (11.02 mL, 127 mmol, passed neat through basic alumina prior to addition) in tetrahydrofuran (200 mL) was added and the reaction vessel was evacuated and refilled with argon three times. Lithium bis(trimethylsilyl)amide (1.0 M, 56.1 mL, 56.1 mmol) was added dropwise at −40° C. (dry ice/acetonitrile bath) and stirred under argon. The reaction was allowed to gradually warm to −10° C. and stirred at −10° C. for 3 hours. The reaction was quenched with saturated ammonium chloride (10 mL), concentrated, and the crude product was diluted in water (150 mL) and diethyl ether (200 mL). The layers were separated and the aqueous layer was washed twice more with diethyl ether (200 mL/each). The combined organic layer was washed with brine (100 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure to a residue. The residue was purified by flash chromatography (2×330 g silica gel columns, gradient elution of 0% to 30% acetone in hexanes) to provide the title compound as a white solid. The product can alternatively be crystallized from a minimum of hexanes in dichloromethane. Enantiomeric excess was determined to be 87% by chiral SFC (90% CO2, 10% methanol (20 mM ammonia), 5.0 mL/min, 100 bar (10,000 kPa), 40° C., 5 minute method, Phenomenex Lux-2 (Phenomenex, Torrance, Calif.) (100 mm×4.6 mm, 5 μm column), retention times: 1.62 min. (minor) and 2.17 min. (major)). The purity could be upgraded to >98% through recrystallization in hexanes and dichloromethane.
-
1H NMR (400 MHz, CDCl3, δ ppm): 7.24-7.17 (m, 3H), 6.94 (s, 1H), 6.80 (d, J=7.5 Hz, 1H), 6.48 (dd, J=10.0, 1.9 Hz, 1H), 6.40 (d, J=8.3 Hz, 1H), 5.90-5.76 (m, 1H), 5.69 (d, J=5.2 Hz, 1H), 5.20-5.13 (m, 2H), 3.81 (dd, J=13.9, 6.9 Hz, 1H), 2.62 (dd, J=13.8, 7.6 Hz, 1H), 2.50 (dd, J=13.8, 7.3 Hz, 1H), 1.96 (d, J=8.4 Hz, 2H), 1.40 (s, 3H). MS (ESI) 393.1 [M+H]+.
Step F. (2S)-2-((2R)-3-(4-Chloro-3-fluorophenyl)-2-(3-chlorophenyl)-3-hydroxypropyl)-N—((S)-1-cyclopropyl-2-hydroxyethyl)-2-methylpent-4-enamide
-
-
Sodium methoxide (25% in methanol, 60.7 ml, 265 mmol) was added to a solution of (S)-2-amino-2-cyclopropylethanol hydrochloride (36.5 g, 265 mmol, NetChem Inc., Ontario, Canada) in methanol (177 mL) at 0° C. A precipitate formed during the addition. After the addition was complete, the reaction mixture was removed from the ice bath and warmed to room temperature. The reaction mixture was filtered under a vacuum and the solid was washed with dichloromethane. The filtrate was concentrated under a vacuum to provide a cloudy brown oil. The oil was taken up in dichloromethane (150 mL), filtered under a vacuum and the solid phase washed with dichloromethane to provide the filtrate as a clear orange solution. The solution was concentrated under a vacuum to provide (S)-2-amino-2-cyclopropylethanol as a light brown liquid.
-
(3S,5R,6R)-3-Allyl-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one (32 g, 81 mmol, Example 4, Step E) was combined with (S)-2-amino-2-cyclopropylethanol (26.7 g, 265 mmol) and the suspension was heated at 100° C. overnight. The reaction mixture was cooled to room temperature, diluted with ethyl acetate and washed with 1 N hydrochloric acid (2×), water, and brine. The organic layer was dried over magnesium sulfate and concentrated under vacuum to provide the title compound as a white solid.
-
1H NMR (500 MHz, CDCl3, δ ppm): 0.23-0.30 (m, 2H), 0.45-0.56 (m, 2H), 0.81 (m, 1H), 1.12 (s, 3H), 1.92-2.09 (m, 3H), 2.39 (dd, J=13.6, 7.2 Hz, 1H), 2.86 (br s, 1H), 2.95 (dtd, J=9.5, 6.3, 6.3, 2.9 Hz, 1H), 3.44 (dd, J=11.0, 5.6 Hz, 1H), 3.49 (m, 1H), 3.61 (dd, J=11.0, 2.9 Hz, 1H), 4.78 (d, J=5.6 Hz, 1H), 4.95-5.13 (m, 2H), 5.63 (m, 1H), 5.99 (d, J=6.4 Hz, 1H), 6.94-7.16 (m, 3H), 7.16-7.32 (m, 4H). MS (ESI) 494 [M+H]+.
Step G. (3S,5R,6S)-3-Allyl-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-1-((S)-1-cyclopropyl-2-hydroxyethyl)-3-methylpiperidin-2-one
-
-
A solution of (2S)-2-((2R)-3-(4-chloro-3-fluorophenyl)-2-(3-chlorophenyl)-3-hydroxypropyl)-N—((S)-1-cyclopropyl-2-hydroxyethyl)-2-methylpent-4-enamide (40.2 g, 81 mmol, Example 4, Step F) in dichloromethane (80 mL) was added p-toluenesulfonic anhydride (66.3 g, 203 mmol) in dichloromethane (220 mL) at 0° C., and the reaction mixture was stirred for 10 minutes at same the temperature. 2,6-Lutidine (43.6 mL, 374 mmol, Aldrich, St. Louis, Mo.) was added dropwise via addition funnel at 0° C. The reaction mixture was slowly warmed to room temperature, and then it was stirred at reflux. After 24 hours, sodium bicarbonate (68.3 g, 814 mmol) in water (600 mL) and 1,2-dichloroethane (300 mL) were added in succession. The reaction mixture was heated at reflux for an hour and then cooled to room temperature. The layers were separated and the aqueous layer was extracted with dichloromethane. The combined organic layer was washed with 1 N hydrochloric acid, water, and brine, then concentrated under reduced pressure. The residue was purified by flash chromatography (1.5 kg SiO2 column, gradient elution of 10% to 50% ethyl acetate in hexanes) to provide the title compound as a white solid.
-
1H NMR (500 MHz, CDCl3, δ ppm): 0.06 (m, 1H), 0.26 (m, 1H), 0.57-0.67 (m, 2H), 0.85 (m, 1H), 1.25 (s, 3H), 1.85-2.20 (m, 2H), 2.57-2.65 (m, 2H), 3.09 (ddd, J=11.8, 9.8, 4.8 Hz, 1H), 3.19 (t, J=10.0 Hz, 1H), 3.36 (td, J=10.3, 4.6 Hz, 1H), 3.63 (dd, J=11.0, 4.6 Hz, 1H), 4.86 (d, J=10.0 Hz, 1H), 5.16-5.19 (m, 2H), 5.87 (m, 1H), 6.77 (dd, J=7.7, 1.6 Hz, 1H), 6.80-6.90 (m, 2H), 7.02 (t, J=2.0 Hz, 1H), 7.16 (dd, J=10.0, 7.7 Hz, 1H), 7.21 (dd, J=10.0, 1.6 Hz, 1H), 7.29 (t, J=10.0 Hz, 1H). MS (ESI) 476 [M+H]+.
Step H. (3S,5S,6R,8S)-8-Allyl-5-(4-chloro-3-fluorophenyl)-6-(3-chlorophenyl)-3-cyclopropyl-8-methyl-2,3,5,6,7,8-hexahydrooxazolo[3,2-a]pyridin-4-ium 4-methylbenzenesulfonate
-
-
p-Toluenesulfonic acid monohydrate (30.3 g, 159 mmol, Aldrich, St. Louis, Mo.) was added to a solution of (3S,5R,6S)-3-allyl-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-1-((S)-1-cyclopropyl-2-hydroxyethyl)-3-methylpiperidin-2-one (73.6 g, 154 mmol) in toluene (386 mL). The reaction mixture was heated at reflux using a Dean-Stark apparatus. After 4 hours, the reaction was cooled and concentrated under reduced pressure to provide the title compound as a pale yellow syrup. The crude product was used in next step without further purification.
-
1H NMR (500 MHz, CDCl3, δ ppm): −0.25 to −0.10 (m, 2H), 0.08-0.18 (m, 1H), 0.33-0.50 (m, 2H), 1.57 (s, 3H), 1.92 (dd, J=3.7 and 13.9 Hz, 1H), 2.37 (s, 3H), 2.63 (dd, J=7.3 and 13.7 Hz, 1H), 2.72 (dd, J=7.6 and 13.7 Hz, 1H), 2.93 (t, J=13.7 Hz, 1H), 3.29 (m, 1H), 4.51 (t, J=8.6 Hz, 1H), 4.57-4.63 (m, 1H), 5.33 (d, J=17.1 Hz, 1H), 5.37 (d, J=10.5 Hz, 1H), 5.47 (dd, J=9.1 and 10.0 Hz, 1H), 5.75-5.93 (m, 2H), 6.80 (br s, 1H), 7.08 (s, 1H), 7.16-7.20 (m, 5H), 7.25-7.32 (m, 2H), 7.87 (d, J=8.3 Hz, 2H). MS (ESI) 458 [M+H]+.
-
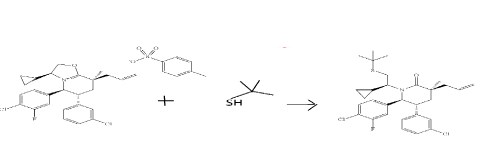
Step I. (3S,5R,6S)-3-Allyl-1-((S)-2-(tert-butylthio)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methylpiperidin-2-one
-
-
2-Methyl-2-propanethiol (15.25 mL, 135 mmol, dried over activated 4 Å molecular sieves) was added to a solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (1.0 M, 135 mL, 135 mmol) at room temperature under argon in a 500 mL round-bottomed flask. The reaction mixture was heated to 60° C. After 30 minutes, a solution of (3S,5S,6R,8S)-8-allyl-5-(4-chloro-3-fluorophenyl)-6-(3-chlorophenyl)-3-cyclopropyl-8-methyl-2,3,5,6,7,8-hexahydrooxazolo[3,2-a]pyridin-4-ium 4-methylbenzenesulfonate (78 g, 123 mmol, Example 4, Step H) in anhydrous tetrahydrofuran (100 mL) was added via cannula. The reaction mixture was heated at 60° C. for 3 hours and then cooled to room temperature. The reaction mixture was quenched with water and extracted thrice with ethyl acetate. The organics were pooled, washed with brine, dried over magnesium sulfate, filtered and concentrated under a vacuum to provide a yellow foam. Purification by flash column chromatography (1.5 kg SiO2 column, gradient elution with 5% to 30% ethyl acetate in hexanes provided the title compound as an off-white foam.
-
1H NMR (400 MHz, CDCl3, δ ppm): −0.89 to −0.80 (m, 1H), −0.15 to −0.09 (m, 1H), 0.27-0.34 (m, 1H), 0.41-0.48 (m, 1H), 1.28 (s, 3H), 1.35 (s, 9H), 1.70-1.77 (m, 1H), 1.86 (dd, J=3.1 and 13.5 Hz, 1H), 2.16 (t, J=13.7, 1H), 2.17-2.23 (m, 1H), 2.60-2.63 (m, 3H), 3.09 (dt, J=3.1 and 10.4 Hz, 1H), 3.62 (t, J=11.1 Hz, 1H), 4.70 (d, J=10.1 Hz, 1H), 5.16 (s, 1H), 5.19-5.21 (m, 1H), 5.82-5.93 (m, 1H), 6.65-6.80 (m, 1H), 6.80-6.83 (m, 1H), 6.84-6.98 (m, 1H), 7.05-7.07 (m, 1H), 7.12-7.18 (m, 2H), 7.19-7.26 (m, 1H). MS (ESI) 548.2 [M+H]+.
Step J. 2-((3R,5R,6S)-1-((S)-2-(tert-Butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetic acid
-
-
Ruthenium(III) chloride hydrate (0.562 mg, 2.493 mmol) was added to a mixture of (3S,5R,6S)-3-allyl-1-((S)-2-(tert-butylthio)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methylpiperidin-2-one (62.17 g, 113 mmol, Example 4, Step I) and sodium periodate (24.67 g) in ethyl acetate (216 mL), acetonitrile (216 mL) and water (324 mL) at 20° C. The temperature quickly rose to 29° C. The reaction mixture was cooled to 20° C. and the remaining equivalents of sodium periodate were added in five 24.67 g portions over 2 hours, being careful to maintain an internal reaction temperature below 25° C. The reaction was incomplete, so additional sodium periodate (13 g) was added. The temperature increased from 22° C. to 25° C. After stirring for an additional 1.5 hours, the reaction mixture was filtered under a vacuum and washed with ethyl acetate. The layers were separated and the aqueous layer was extracted with ethyl acetate. The organics were pooled, washed with brine, dried over magnesium sulfate, filtered and concentrated under a vacuum to provide a dark green foam. Purification by flash column chromatography (1.5 kg SiO2 column, gradient elution of 0% to 20% isopropanol in hexanes) provided an off-white foam. 15% Ethyl acetate in heptanes (970 mL) was added to the foam, and the mixture was heated at 80° C. until the foam dissolved. The solution was then cooled slowly, and at 60° C. the solution was seeded with previously obtained crystalline material. The mixture was cooled to room temperature and then allowed to stand at room temperature for 2 hours before collecting the solid by vacuum filtration to provide a white solid with a very pale pink hue (57.1 g). The mother liquor was concentrated under a vacuum to provide a pink foam (8.7 g). 15% ethyl acetate in heptanes (130 mL) was added to the foam, and it was heated at 80° C. to completely dissolve the material. The solution was cooled, and at 50° C., it was seeded with crystalline material. After cooling to room temperature the solid was collected by vacuum filtration to provide a white crystalline solid with a very pale pink hue.
-
1H NMR (500 MHz, CDCl3, δ ppm): −1.10 to −1.00 (m, 1H), −0.30 to −0.22 (m, 1H), 0.27-0.37 (m, 1H), 0.38-0.43 (m, 1H), 1.45 (s, 9H), 1.50 (s, 3H), 1.87 (dd, J=2.7 and 13.7 Hz, 1H), 1.89-1.95 (m, 1H), 2.46 (t, J=13.7, 1H), 2.69-2.73 (m, 1H), 2.78 (d, J=14.9 Hz, 1H), 2.93 (dd, J=2.0 and 13.7 Hz, 1H), 3.07 (d, J=14.9 Hz, 1H), 3.11 (dt, J=2.7 and 11.0 Hz, 1H), 4.30 (t, J=13.5 Hz, 1H), 4.98 (d, J=10.8 Hz, 1H), 6.75-6.87 (m, 1H), 6.88-6.90 (m, 1H), 6.98 (br s, 1H), 7.02-7.09 (m, 1H), 7.11-7.16 (m, 2H), 7.16-7.25 (m, 1H). MS (ESI) 598.1 [M+H]+.
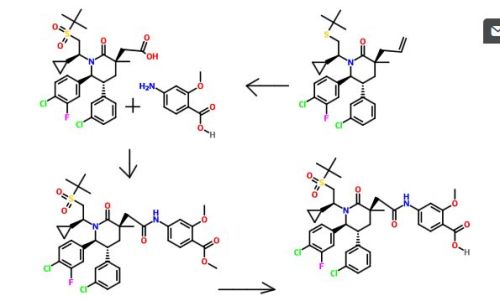
Example 5 4-(2-((3R,5R,6S)-1-((S)-2-(tert-Butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoic acid
Step A. Methyl 4-(2-((3R,5R,6S)-1-((S)-2-(tert-butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoate
-
-
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 76 g, 398 mmol) was added to a mixture of 2-((3R,5R,6S)-1-((S)-2-(tert-butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetic acid (79.4 g, 133 mmol, Example 4, Step J) and methyl 4-amino-2-methoxybenzoate (26.4 g, 146 mmol) in pyridine (332 mL) at 3° C. The mixture was allowed to warm to room temperature and was stirred at room temperature for 16 hours. The reaction mixture was cooled to 0° C. and added to an ice-cold solution of 1 M hydrochloric acid (1 L). Ether (1 L) was added and the layers were agitated and then separated. The organic layer was washed with 1 M hydrochloric acid (6×500 mL), saturated aqueous sodium bicarbonate (500 mL), brine (500 mL), dried over magnesium sulfate, filtered and concentrated under a vacuum to provide an off-white foam.
-
1H NMR (400 MHz, CDCl3, δ ppm): −1.20 to −1.12 (m, 1H), −0.35 to −0.20 (m, 1H), 0.05-0.20 (m, 1H), 0.32-0.45 (m, 1H), 1.45 (s, 9H), 1.48 (s, 3H), 1.86-1.98 (m, 1H), 2.03 (dd, J=2.7 and 13.7 Hz, 1H), 2.43 (t, J=13.7, 1H), 2.64-2.75 (m, 1H), 2.80 (d, J=14.3 Hz, 1H), 2.89-2.96 (m, 2H), 3.24 (dt, J=2.5 and 10.8 Hz, 1H), 3.89 (s, 3H), 3.96 (s, 3H), 4.28-4.36 (m, 1H), 4.98 (d, J=10.8 Hz, 1H), 6.85-6.93 (m, 3H), 6.99 (br s, 1H), 7.06-7.18 (m, 4H), 7.82 (br s, 1H), 7.85 (d, J=8.4 Hz, 1H), 8.81 (br s, 1H). MS (ESI) 761.2 [M+H]+.
Step B. 4-(2-((3R,5R,6S)-1-((S)-2-(tert-Butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoic acid
-
-
A solution of lithium hydroxide monohydrate (18.2 g, 433 mmol) in water (295 mL) was added to a solution of methyl 4-(2-((3R,5R,6S)-1-((S)-2-(tert-butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoate (164.9 g, 217 mmol, Example 5, Step A) in tetrahydrofuran (591 mL) and methanol (197 mL) at room temperature. After stirring for 15 hours at room temperature, a trace amount of the ester remained, so the reaction mixture was heated at 50° C. for 1 hour. When the reaction was complete, the mixture was concentrated under a vacuum to remove the tetrahydrofuran and methanol. The thick mixture was diluted with water (1 L) and 1 M hydrochloric acid (1 L) was added. The resulting white solid was collected by vacuum filtration in a Büchner funnel. The vacuum was removed, and water (1 L) was added to the filter cake. The material was stirred with a spatula to suspend it evenly in the water. The liquid was then removed by vacuum filtration. This washing cycle was repeated three more times to provide a white solid. The solid was dried under vacuum at 45° C. for 3 days to provide the title compound as a white solid.
-
1H NMR (500 MHz, DMSO-d6) δ ppm −1.30 to −1.12 (m, 1H), −0.30 to −0.13 (m, 1H), 0.14-0.25 (m, 1H), 0.25-0.38 (m, 1H), 1.30 (s, 3H), 1.34 (s, 9H), 1.75-1.86 (m, 1H), 2.08-2.18 (m, 2H), 2.50-2.60 (m, 1H), 2.66 (d, J=13.7, 1H), 3.02-3.16 (m, 2H), 3.40-3.50 (m, 1H), 3.77 (s, 3H), 4.05-4.20 (m, 1H), 4.89 (d, J=10.5 Hz, 1H), 6.90-6.93 (m, 3H), 7.19 (d, J=8.8 Hz, 1H), 7.22-7.26 (m, 3H), 7.40-7.50 (m, 1H), 7.54 (br s, 1H), 7.68 (d, J=8.6 Hz, 1H) 10.44 (s, 1H), 12.29 (br s, 1H). MS (ESI) 747.2 [M+H]+.
Patent
WO 2015070224
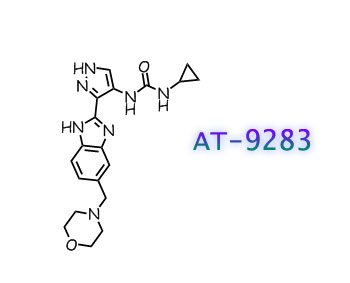
Another particular MDM2 inhibitor is AM-7209 (Compound C herein), which is disclosed in U.S. provisional patent application number 61/770,901, filed February 28, 2013. (See Example No. 5 therein and below). AM-7209 has the following chemical name and structure: 4- (2-((3i?,5i?,65)-l-((5)-2-(tei’i-butylsulfonyl)-l-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)- 5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoic acid

EXAMPLE 4
2-((3R,5R,6S)- l-((S)-2-(tert-Butylsulfonyl)- l-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopip

Step A. Methyl -4-chloro-3-fluorobenzoate

A solution of 4-chloro-3-fluoro benzoic acid (450.0 g, 2.586 mol, Fluorochem, Derbyshire, UK) in methanol (4.5 L) was cooled to 0 °C and thionyl chloride (450.0 mL) was added over 30 minutes. The reaction mixture was stirred for 12 hours at ambient temperature. The reaction was monitored by TLC. Upon completion, the solvent was removed under reduced pressure and the residue was quenched with 1.0 M sodium bicarbonate solution (500 mL). The aqueous layer was extracted with dichloromethane (2 x 5.0 L). The combined organic layer was washed with brine (2.5 L), dried over anhydrous sodium sulfate and concentrated under reduced pressure afforded the title compound as light brown solid. The crude compound was used in the next step without further purification. lH NMR (400 MHz, CDC13, δ ppm): 7.82-7.74 (m, 2H), 7.46 (dd, J= 8.2, 7.5 Hz, 1H), 3.92 (s, 3H).
Step B. l-(4-chloro-3-fluorophenyl)-2-(3-chlorophenyl)ethanone

Sodium bis(trimethylsilyl)amide (1 M in tetrahydrofuran, 4 L, 4000 mmol) was added over 1 hour to a solution of 3-chlorophenyl acetic acid (250.0 g, 1465 mmol) in anhydrous
tetrahydrofuran (1.75 L) at -78 °C under nitrogen. The resulting reaction mixture was stirred for an additional hour at -78 °C. Then, a solution of methyl-4-chloro-3-fluorobenzoate (221.0 g, 1 175 mmol, Example 4, Step A) in tetrahydrofuran (500 mL) was added over 1 hour at -78 °C, and the resulting reaction mixture was stirred at the same temperature for 2 hours. The reaction was monitored by TLC. On completion, reaction mixture was quenched with 2 N hydrochloric acid (2.5 L) and aqueous phase was extracted with ethyl acetate (2 x 2.5 L). The combined organic layer was washed with brine (2.5 L), dried over anhydrous sodium sulfate and concentrated under reduced pressure to provide the crude material which was purified by flash column
chromatography (silica gel: 100 to 200 mesh, product eluted in 2% ethyl acetate in hexane) to afford the title compound as a white solid. ¾ NMR (400 MHz, CDC13, δ ppm): 7.74 (ddd, J= 10.1, 8.9, 1.8 Hz, 2H), 7.56-7.48 (m, 1H), 7.26 (t, J= 6.4 Hz, 3H), 7.12 (d, J= 5.7 Hz, 1H), 4.22 (s, 2H). MS (ESI) 282.9 [M + H]+.
Step C. Methyl 5-(4-chloro-3-fluorop -2-methyl-5-oxopentanoate

Methyl methacrylate (125.0 g, 1097 mmol) and potassium tert-butoxide (1 M in tetrahydrofuran, 1 15 mL, 115 mmol) were sequentially added to a solution of l-(4-chloro-3-fluorophenyl)-2-(3-chlorophenyl)ethanone (327.0 g, 1 160 mmol, Example 4, Step B) in anhydrous tetrahydrofuran (2.61 L), at 0 °C. The reaction mixture was stirred for 1 hour at 0 °C and then warmed to ambient temperature and stirred for 12 hours. On completion, the reaction was quenched with water (1.0 L) and extracted with ethyl acetate (2 x 2.5 L). The combined organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure to get the crude material which was purified by flash column chromatography (silica gel: 60 to 120 mesh, product eluted in 4% ethyl acetate in hexane) affording the title compound (mixture of diastereomers) as light yellow liquid. lH NMR (400 MHz, CDC13, δ ppm): 7.74-7.61 (m, 4H), 7.47-7.40 (m, 2H), 7.28-7.18 (m, 6H), 7.16-7.10 (m, 2H), 4.56 (m, 2H), 3.68 (s, 3H), 3.60 (s, 3H), 2.50-2.39 (m, 2H), 2.37-2.25 (m, 2H), 2.10-2.02 (m, 1H), 1.94 (ddd, J= 13.6, 9.1, 4.2 Hz, 1H), 1.21 (d, J= 7.0 Hz, 3H), 1.15 (d, J= 7.0 Hz, 3H). MS (ESI) 383.0 [M + H]+.
Ste D. (3S,5R,6R)-6-(4-Chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one and (3R,5R,6R)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one

In a 2000 mL reaction vessel charged with methyl 5-(4-chloro-3-fluorophenyl)-4-(3-chlorophenyl)-2-methyl-5-oxopentanoate (138.0 g, 360 mmol, Example 4, Step C) (which was cooled on ice for 10 minutes before transferring to a glove bag) anhydrous 2-propanol (500 mL), and potassium tert-butoxide (16.16 g, 144 mmol) were sequentially added while in a sealed glove bag under argon. This mixture was allowed to stir for 30 minutes. RuCl2(S-xylbinap)(S-DAIPEN) (1.759 g, 1.440 mmol, Strem Chemicals, Inc., Newburyport, MA, weighed in the glove bag) in 30.0 mL toluene was added. The reaction was vigorously stirred at room temperature for 2 hours. The vessel was set on a hydrogenation apparatus, purged with hydrogen 3 times and pressurized to 50 psi (344.7 kPa). The reaction was allowed to stir overnight at room temperature. On completion, the reaction was quenched with water (1.5 L) and extracted with ethyl acetate (2 x 2.5 L). The organic layer was washed with brine (1.5 L), dried over anhydrous sodium sulfate and concentrated under reduced pressure to get crude material which was purified by flash column chromatography (silica gel; 60-120 mesh; product eluted in 12% ethyl acetate in hexane) to provide a dark colored liquid as a mixture of diastereomers.
The product was dissolved in (240.0 g, 581 mmol) in tetrahydrofuran (1.9 L) and methanol (480 mL), and lithium hydroxide monohydrate (2.5 M aqueous solution, 480.0 mL) was added. The reaction mixture was stirred at ambient temperature for 12 hours. On completion, the solvent was removed under reduced pressure and the residue was acidified with 2 N hydrochloric acid to a pH between 5 and 6. The aqueous phase was extracted with ethyl acetate (2 x 1.0 L). The combined organic layer was washed with brine (750 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide a dark colored liquid, which was used without further purification.
A portion of the crude intermediate (25.4 g, predominantly seco acid) was added to a 500 mL round bottom flask, equipped with a Dean-Stark apparatus. Pyridinium / toluenesulfonate (0.516 g, 2.053 mmol) and toluene (274 mL) were added, and the mixture was refluxed for 1 hour (oil bath temperature about 150 °C). The reaction was cooled to room temperature and concentrated under reduced pressure. The reaction was diluted with saturated aqueous sodium bicarbonate (150 mL), extracted with diethyl ether (2 χ 150 mL), and washed with brine (150 mL). The combined organic layer was dried over magnesium sulfate, filtered and concentrated under reduced pressure. Purification by flash column chromatography (divided into 3 portions, 330 g SiCVeach, gradient elution of 0% to 30% acetone in hexanes, 35 minutes) provided the title compounds as a pale yellow solid and a 1 : 1.6 mixture of diastereomers at C2. MS (ESI) 353.05 [M + H]+. Step E. (3S,5R,6R)-3-Allyl-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one

(3S,5R,6R)-6-(4-Chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one and (3R,5R,6R)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one (18 g, 51.0 mmol, Example 4, Step D) was added to an oven dried 500 mL round-bottom flask. The solid was dissolved in anhydrous toluene and concentrated to remove adventitious water. 3-Bromoprop- l-ene (1 1.02 mL, 127 mmol, passed neat through basic alumina prior to addition) in tetrahydrofuran (200 mL) was added and the reaction vessel was evacuated and refilled with argon three times. Lithium bis(trimethylsilyl)amide (1.0 M, 56.1 mL, 56.1 mmol) was added dropwise at -40 °C (dry ice/acetonitrile bath) and stirred under argon. The reaction was allowed to gradually warm to -10 °C and stirred at -10 °C for 3 hours. The reaction was quenched with saturated ammonium chloride (10 mL), concentrated, and the crude product was diluted in water (150 mL) and diethyl ether (200 mL). The layers were separated and the aqueous layer was washed twice more with diethyl ether (200 mL/each). The combined organic layer was washed with brine (100 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure to a residue. The residue was purified by flash chromatography (2 x 330 g silica gel columns, gradient elution of 0% to 30% acetone in hexanes) to provide the title compound as a white solid. The product can alternatively be crystallized from a minimum of hexanes in dichloromethane. Enantiomeric excess was determined to be 87% by chiral SFC (90% C02, 10% methanol (20 mM ammonia), 5.0 mL/min, 100 bar (10,000 kPa), 40 °C, 5 minute method, Phenomenex Lux-2 (Phenomenex, Torrance, CA) (100 mm x 4.6 mm, 5 μιη column), retention
times: 1.62 min. (minor) and 2.17 min. (major)). The purity could be upgraded to > 98% through recrystallization in hexanes and dichloromethane. H NMR (400 MHz, CDC13, δ ppm): 7.24-7.17 (m, 3H), 6.94 (s, 1H), 6.80 (d, J= 7.5 Hz, 1H), 6.48 (dd, J= 10.0, 1.9 Hz, 1H), 6.40 (d, J= 8.3 Hz, 1H), 5.90-5.76 (m, 1H), 5.69 (d, J= 5.2 Hz, 1H), 5.20-5.13 (m, 2H), 3.81 (dd, J= 13.9, 6.9 Hz, lH), 2.62 (dd, J= 13.8, 7.6 Hz, 1H), 2.50 (dd, J= 13.8, 7.3 Hz, 1H), 1.96 (d, J= 8.4 Hz, 2H), 1.40 (s, 3H). MS (ESI) 393.1 [M + H]+.
Step F. (2S)-2-((2R)-3-(4-Chloro-3-fluorophenyl)-2-(3-chlorophenyl)-3-hydroxypropyl)-N-((S)-l-cyclopropyl-2-hydroxyethyl)-2-

Sodium methoxide (25% in methanol, 60.7 ml, 265 mmol) was added to a solution of (S)-2-amino-2-cyclopropylethanol hydrochloride (36.5 g, 265 mmol, NetChem Inc., Ontario, Canada) in methanol (177 mL) at 0 °C. A precipitate formed during the addition. After the addition was complete, the reaction mixture was removed from the ice bath and warmed to room temperature. The reaction mixture was filtered under a vacuum and the solid was washed with
dichloromethane. The filtrate was concentrated under a vacuum to provide a cloudy brown oil. The oil was taken up in dichloromethane (150 mL), filtered under a vacuum and the solid phase washed with dichloromethane to provide the filtrate as a clear orange solution. The solution was concentrated under a vacuum to provide (5)-2-amino-2-cyclopropylethanol as a light brown liquid.
(3S,5R,6R)-3-Allyl-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyltetrahydro-2H-pyran-2-one (32 g, 81 mmol, Example 4, Step E) was combined with (S)-2-amino-2-cyclopropylethanol (26.7 g, 265 mmol) and the suspension was heated at 100 °C overnight. The reaction mixture was cooled to room temperature, diluted with ethyl acetate and washed with 1 N hydrochloric acid (2X), water, and brine. The organic layer was dried over magnesium sulfate and concentrated under vacuum to provide the title compound as a white solid. lH NMR (500
MHz, CDC13, δ ppm): 0.23-0.30 (m, 2H), 0.45-0.56 (m, 2H), 0.81 (m, 1H), 1.12 (s, 3H), 1.92-2.09 (m, 3H), 2.39 (dd, J= 13.6, 7.2 Hz, 1H), 2.86 (br s, 1H), 2.95 (dtd, J= 9.5, 6.3, 6.3, 2.9 Hz, 1H), 3.44 (dd, J= 1 1.0, 5.6 Hz, 1H), 3.49 (m, 1H), 3.61 (dd, J= 1 1.0, 2.9 Hz, 1H), 4.78 (d, J = 5.6 Hz, 1H), 4.95-5.13 (m, 2H), 5.63 (m, 1H), 5.99 (d, J= 6.4 Hz, 1H), 6.94-7.16 (m, 3H), 7.16-7.32 (m, 4H). MS (ESI) 494 [M + H]+.
Ste G. (3S,5R,6S)-3-Allyl-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-l-((S)-l-cyclopropyl-2-hydroxyethyl)-3-

A solution of (2S)-2-((2R)-3-(4-chloro-3-fluorophenyl)-2-(3-chlorophenyl)-3-hydroxypropyl)-N-((S)-l-cyclopropyl-2-hydroxyethyl)-2-methylpent-4-enamide (40.2 g, 81 mmol, Example 4, Step F) in dichloromethane (80 mL) was added / toluenesulfonic anhydride (66.3 g, 203 mmol) in dichloromethane (220 mL) at 0 °C ,and the reaction mixture was stirred for 10 minutes at same the temperature. 2,6-Lutidine (43.6 mL, 374 mmol, Aldrich, St. Louis, MO) was added dropwise via addition funnel at 0 °C. The reaction mixture was slowly warmed to room temperature, and then it was stirred at reflux. After 24 hours, sodium bicarbonate (68.3 g, 814 mmol) in water (600 mL) and 1 ,2-dichloroethane (300 mL) were added in succession. The reaction mixture was heated at reflux for an hour and then cooled to room temperature. The layers were separated and the aqueous layer was extracted with dichloromethane. The combined organic layer was washed with 1 N hydrochloric acid, water, and brine, then concentrated under reduced pressure. The residue was purified by flash chromatography (1.5 kg S1O2 column, gradient elution of 10% to 50% ethyl acetate in hexanes) to provide the title compound as a white solid. lH NMR (500 MHz, CDCI3, δ ppm): 0.06 (m, 1H), 0.26 (m, 1H), 0.57-0.67 (m, 2H), 0.85 (m, 1H), 1.25 (s, 3H), 1.85-2.20 (m, 2H), 2.57-2.65 (m, 2H), 3.09 (ddd, J= 1 1.8, 9.8, 4.8 Hz, 1H), 3.19 (t, J= 10.0 Hz, 1H), 3.36 (td, J= 10.3, 4.6 Hz, 1H), 3.63 (dd, J= 1 1.0, 4.6 Hz, 1H), 4.86 (d, J= 10.0 Hz, 1H), 5.16-5.19 (m, 2H), 5.87 (m, 1H), 6.77 (dd, J= 7.7, 1.6 Hz, 1H), 6.80-6.90 (m, 2H), 7.02 (t, J = 2.0 Hz, 1H), 7, 16 (dd, J= 10.0, 7.7 Hz, 1H), 7.21 (dd, J= 10.0, 1.6 Hz, 1H), 7.29 (t, J= 10.0 Hz, 1H). MS (ESI) 476 [M + H]+.
Step H. (3S,5S,6R,8S)-8-Allyl-5-(4-chloro-3-fluorophenyl)-6-(3-chlorophenyl)-3-cyclopropyl-8-methyl-2,3,5,6,7,8-hex enzenesulfonate

/ Toluenesulfonic acid monohydrate (30.3 g, 159 mmol, Aldrich, St. Louis, MO) was added to a solution of (3S,5R,6S)-3-allyl-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)- l-((S)- l-cyclopropyl-2-hydroxyethyl)-3-methylpiperidin-2-one (73.6 g, 154 mmol) in toluene (386 mL). The reaction mixture was heated at reflux using a Dean-Stark apparatus. After 4 hours, the reaction was cooled and concentrated under reduced pressure to provide the title compound as a pale yellow syrup. The crude product was used in next step without further purification. lH NMR (500 MHz, CDC13, δ ppm): -0.25 to -0.10 (m, 2H), 0.08-0.18 (m, 1H), 0.33-0.50 (m, 2H), 1.57 (s, 3H), 1.92 (dd, J= 3.7 and 13.9 Hz, 1H), 2.37 (s, 3H), 2.63 (dd, J= 7.3 and 13.7 Hz, 1H), 2.72 (dd, J= 7.6 and 13.7 Hz, 1H), 2.93 (t, J= 13.7 Hz, 1H), 3.29 (m, 1H), 4.51 (t, J= 8.6 Hz, 1H), 4.57-4.63 (m, 1H), 5.33 (d, J= 17.1 Hz, 1H), 5.37 (d, J= 10.5 Hz, 1H), 5.47 (dd, J= 9.1 and
10.0 Hz, 1H), 5.75-5.93 (m, 2H), 6.80 (br s, 1H), 7.08 (s, 1H), 7.16-7.20 (m, 5H), 7.25-7.32 (m, 2H), 7.87 (d, J= 8.3 Hz, 2H). MS (ESI) 458 [M + H]+.
Step I. (3S,5R,6S)-3-Allyl- l-((S)-2-(tert-butylthio)-l-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3

2-Methyl-2-propanethiol (15.25 mL, 135 mmol, dried over activated 4 A molecular sieves) was added to a solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (1.0 M, 135 mL, 135 mmol) at room temperature under argon in a 500 mL round-bottomed flask. The reaction mixture was heated to 60 °C. After 30 minutes, a solution of (3S,5S,6R,8S)-8-allyl-5-(4-chloro-3-fluorophenyl)-6-(3-chlorophenyl)-3-cyclopropyl-8-methyl-2,3,5,6,7,8-hexahydrooxazolo[3,2-fl]pyridin-4-ium 4-methylbenzenesulfonate (78 g, 123 mmol, Example 4, Step H) in anhydrous tetrahydrofuran (100 mL) was added via cannula. The reaction mixture was heated at 60 °C for 3 hours and then cooled to room temperature. The reaction mixture was quenched with water and extracted thrice with ethyl acetate. The organics were pooled, washed with brine, dried over magnesium sulfate, filtered and concentrated under a vacuum to provide a yellow foam.
Purification by flash column chromatography (1.5 kg Si02 column, gradient elution with 5% to 30% ethyl acetate in hexanes provided the title compound as an off-white foam. !H NMR (400 MHz, CDCI3, δ ppm): -0.89 to -0.80 (m, 1H), -0.15 to -0.09 (m, 1H), 0.27-0.34 (m, 1H), 0.41-0.48 (m, 1H), 1.28 (s, 3H), 1.35 (s, 9H), 1.70-1.77 (m, 1H), 1.86 (dd, J= 3.1 and 13.5 Hz, 1H), 2.16 (t, J= 13.7, 1H), 2.17-2.23 (m, 1H), 2.60-2.63 (m, 3H), 3.09 (dt, J= 3.1 and 10.4 Hz, 1H), 3.62 (t, J= 1 1.1 Hz, 1H), 4.70 (d, J= 10.1 Hz, 1H), 5.16 (s, 1H), 5.19-5.21 (m, 1H), 5.82-5.93 (m, 1H), 6.65-6.80 (m, 1H), 6.80-6.83 (m, 1H), 6.84-6.98 (m, 1H), 7.05-7.07 (m, 1H), 7.12-7.18 (m, 2H), 7.19-7.26 (m, 1H). MS (ESI) 548.2 [M + H]+.
Step J. 2-((3R,5R,6S)-l-((S)-2-(tert-Butylsulfonyl)- l-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3- -yl)acetic acid

Ruthenium(III) chloride hydrate (0.562 mg, 2.493 mmol) was added to a mixture of (3S,5R,6S)-3-allyl- 1 -((5)-2-(ter?-butylthio)- 1 -cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methylpiperidin-2-one (62.17 g, 1 13 mmol, Example 4, Step I) and sodium periodate (24.67 g) in ethyl acetate (216 mL), acetonitrile (216 mL) and water (324 mL) at 20 °C. The temperature quickly rose to 29 °C. The reaction mixture was cooled to 20 °C and the remaining equivalents of sodium periodate were added in five 24.67 g portions over 2 hours, being careful to maintain an internal reaction temperature below 25 °C. The reaction was incomplete, so additional sodium periodate (13 g) was added. The temperature increased from 22 °C to 25 °C. After stirring for an additional 1.5 hours, the reaction mixture was filtered under a vacuum and washed with ethyl acetate. The layers were separated and the aqueous layer was extracted with ethyl acetate. The organics were pooled, washed with brine, dried over magnesium sulfate, filtered and concentrated under a vacuum to provide a dark green foam. Purification by flash column chromatography (1.5 kg S1O2 column, gradient elution of 0% to 20% isopropanol in hexanes) provided an off-white foam. 15% Ethyl acetate in heptanes (970 mL) was added to the foam, and the mixture was heated at 80 °C until the foam dissolved. The solution was then cooled slowly, and at 60 °C the solution was seeded with previously obtained crystalline material. The mixture was cooled to room temperature and then allowed to stand at room temperature for 2 hours before collecting the solid by vacuum filtration to provide a white solid with a very pale pink hue (57.1 g). The mother liquor was concentrated under a vacuum to provide a pink foam (8.7 g). 15% ethyl acetate in heptanes (130 mL) was added to the foam, and it was heated at 80 °C to completely dissolve the material. The solution was cooled, and at 50 °C, it was seeded with crystalline material. After cooling to room temperature the solid was collected by vacuum filtration to provide a white crystalline solid with a very pale pink hue. lH NMR (500
MHz, CDCI3, δ ppm): -1.10 to -1.00 (m, 1H), -0.30 to -0.22 (m, 1H), 0.27-0.37 (m, 1H), 0.38-0.43 (m, 1H), 1.45 (s, 9H), 1.50 (s, 3H), 1.87 (dd, J= 2.7 and 13.7 Hz, 1H), 1.89-1.95 (m, 1H), 2.46 (t, J= 13.7, 1H), 2.69-2.73 (m, 1H), 2.78 (d, J= 14.9 Hz, 1H), 2.93 (dd, J= 2.0 and 13.7 Hz, 1H), 3.07 (d, J= 14.9 Hz, 1H), 3.1 1 (dt, J= 2.7 and 11.0 Hz, 1H), 4.30 (t, J= 13.5 Hz, 1H), 4.98 (d, J= 10.8 Hz, 1H), 6.75-6.87 (m, 1H), 6.88-6.90 (m, 1H), 6.98 (br s, 1H), 7.02-7.09 (m, 1H), 7.1 1-7.16 (m, 2H), 7.16-7.25 (m, 1H). MS (ESI) 598.1 [M + H]+.
EXAMPLE 5
4- (2-((3R,5R,6S)- l-((S)-2-(tert-Butylsulfonyl)-l-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)- 5- (3-chlorophenyl)-3-methyl-2-o xybenzoic acid

Step A. Methyl 4-(2-((3R,5R,6S)- 1 -((S)-2-(tert-butylsulfonyl)- 1 -cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chloropheny etamido)-2-methoxybenzoate

N-(3-Dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC, 76 g, 398 mmol) was added to a mixture of 2-((3R,5R,6S)-l-((S)-2-(tert-butylsulfonyl)-l-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetic acid (79.4 g, 133 mmol, Example 4, Step J) and methyl 4-amino-2-methoxybenzoate (26.4 g, 146 mmol) in pyridine (332 mL) at 3 °C. The mixture was allowed to warm to room temperature and was stirred at room temperature for 16 hours. The reaction mixture was cooled to 0 °C and added to an ice-cold solution of 1 M hydrochloric acid (1 L). Ether (1 L) was added and the layers were agitated and then separated. The organic layer was washed with 1 M hydrochloric acid (6 x 500 mL), saturated aqueous sodium bicarbonate (500 mL), brine (500 mL), dried over magnesium
sulfate, filtered and concentrated under a vacuum to provide an off-white foam. lH NMR (400 MHz, CDCI3, δ ppm): -1.20 to -1.12 (m, 1H), -0.35 to -0.20 (m, 1H), 0.05-0.20 (m, 1H), 0.32-0.45 (m, 1H), 1.45 (s, 9H), 1.48 (s, 3H), 1.86-1.98 (m, 1H), 2.03 (dd, J= 2.7 and 13.7 Hz, 1H), 2.43 (t, J= 13.7, 1H), 2.64-2.75 (m, 1H), 2.80 (d, J= 14.3 Hz, 1H), 2.89-2.96 (m, 2H), 3.24 (dt, J= 2.5 and 10.8 Hz, 1H), 3.89 (s, 3H), 3.96 (s, 3H), 4.28-4.36 (m, 1H), 4.98 (d, J= 10.8 Hz, 1H), 6.85-6.93 (m, 3H), 6.99 (br s, 1H), 7.06-7.18 (m, 4 H), 7.82 (br s, 1H), 7.85 (d, J= 8.4 Hz, 1H), 8.81 (br s, 1H). MS (ESI) 761.2 [M + H]+.
Step B. 4-(2-((3R,5R,6S 1 -((S)-2-(tert-Butylsulfonyl)- 1 -cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoic acid

A solution of lithium hydroxide monohydrate (18.2 g, 433 mmol) in water (295 mL) was added to a solution of methyl 4-(2-((3R,5R,6S)-l-((S)-2-(tert-butylsulfonyl)-l-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoate (164.9 g, 217 mmol, Example 5, Step A) in tetrahydrofuran (591 mL) and methanol (197 mL) at room temperature. After stirring for 15 hours at room temperature, a trace amount of the ester remained, so the reaction mixture was heated at 50 °C for 1 hour. When the reaction was complete, the mixture was concentrated under a vacuum to remove the
tetrahydrofuran and methanol. The thick mixture was diluted with water (1 L) and 1 M hydrochloric acid (1 L) was added. The resulting white solid was collected by vacuum filtration in a Buchner funnel. The vacuum was removed, and water (1 L) was added to the filter cake. The material was stirred with a spatula to suspend it evenly in the water. The liquid was then removed by vacuum filtration. This washing cycle was repeated three more times to provide a white solid. The solid was dried under vacuum at 45 °C for 3 days to provide the title compound as a white solid. H NMR (500 MHz, DMSO-i¾) δ ppm – 1.30 to -1.12 (m, 1H), -0.30 to -0.13 (m, 1H), 0.14-0.25 (m, 1H), 0.25-0.38 (m, 1H), 1.30 (s, 3H), 1.34 (s, 9H), 1.75-1.86 (m, 1H), 2.08-2.18 (m, 2H), 2.50-2.60 (m, 1H), 2.66 (d, J= 13.7, 1H), 3.02-3.16 (m, 2H), 3.40-3.50 (m, 1H), 3.77 (s, 3H), 4.05-4.20 (m, 1H), 4.89 (d, J= 10.5 Hz, 1H), 6.90-6.93 (m, 3H), 7.19 (d, J= 8.8 Hz, 1H), 7..22-7.26 (m, 3H), 7.40-7.50 (m, 1H), 7.54 (br s, 1H), 7.68 (d, J= 8.6 Hz, 1H) 10.44 (s, 1H), 12.29 (br s, 1H). MS (ESI) 747.2 [M + H]+.
PAPER
Discovery of AM-7209, a Potent and Selective 4-Amidobenzoic Acid Inhibitor of the MDM2–p53 Interaction
Yosup Rew*†, Daqing Sun†, Xuelei Yan†, Hilary P. Beck†, Jude Canon∥, Ada Chen†, Jason Duquette†, John Eksterowicz†, Brian M. Fox†, Jiasheng Fu†, Ana Z. Gonzalez†, Jonathan Houze†, Xin Huang⊥, Min Jiang§, Lixia Jin§, Yihong Li†, Zhihong Li†, Yun Ling§, Mei-Chu Lo†, Alexander M. Long⊥, Lawrence R. McGee†, Joel McIntosh†, Jonathan D. Oliner∥, Tao Osgood∥, Anne Y. Saiki∥, Paul Shaffer⊥, Yu Chung Wang∥, Sarah Wortman‡, Peter Yakowec⊥, Qiuping Ye§, Dongyin Yu∥, Xiaoning Zhao†, Jing Zhou†, Julio C. Medina†, and Steven H. Olson†
†Department of Therapeutic Discovery, ‡Department of Pharmaceutics, and §Department of Pharmacokinetics and Drug Metabolism, Amgen Inc., 1120 Veterans Boulevard, South San Francisco, California 94080, United States
∥ Department of Oncology Research, Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320, United States
⊥ Department of Therapeutic Discovery, Amgen Inc., 360 Binney Street, Cambridge, Massachusetts 02142, United States
J. Med. Chem., 2014, 57 (24), pp 10499–10511
DOI: 10.1021/jm501550p
Publication Date (Web): November 10, 2014
Copyright © 2014 American Chemical Society
Abstract
Structure-based rational design and extensive structure–activity relationship studies led to the discovery of AMG 232 (1), a potent piperidinone inhibitor of the MDM2–p53 association, which is currently being evaluated in human clinical trials for the treatment of cancer. Further modifications of 1, including replacing the carboxylic acid with a 4-amidobenzoic acid, afforded AM-7209 (25), featuring improved potency (KD from ITC competition was 38 pM, SJSA-1 EdU IC50 = 1.6 nM), remarkable pharmacokinetic properties, and in vivo antitumor activity in both the SJSA-1 osteosarcoma xenograft model (ED50 = 2.6 mg/kg QD) and the HCT-116 colorectal carcinoma xenograft model (ED50 = 10 mg/kg QD). In addition, 25 possesses distinct mechanisms of elimination compared to 1
4-(2-((3R,5R,6S)-1-((S)-2-(tert-Butylsulfonyl)-1-cyclopropylethyl)-6-(4-chloro-3-fluorophenyl)-5-(3-chlorophenyl)-3-methyl-2-oxopiperidin-3-yl)acetamido)-2-methoxybenzoic Acid (25)
Compound 25 was prepared as a white solid from 3 according to a procedure similar to that described for the synthesis of 7: 1H NMR (500 MHz, DMSO-d6) δ (ppm) 12.29 (1 H, s, br), 10.44 (1 H, s),. 7.68 (1 H, d, J = 8.6 Hz), 7.54 (1 H, s, br), 7.40–7.50 (1 H, m), 7.22–7.26 (3 H, m), 7.19 (1 H, d, J = 8.8 Hz), 6.90–6.93 (3 H, m), 4.89 (1 H, d, J = 10.5 Hz), 4.05–4.20 (1 H, m), 3.77 (3 H, s), 3.40–3.50 (1 H, m), 3.02–3.16 (2 H, m), 2.66 (1 H, d, J = 13.7 Hz), 2.50–2.60 (1 H, m), 2.08–2.18 (2 H, m), 1.75–1.86 (1 H, m), 1.34 (9 H, s), 1.30 (3 H, s), 0.25–0.38 (1 H, m), 0.14–0.25 (1 H, m), −0.30 to −0.13 (1 H, m), −1.30 to −1.12 (1 H, m);
HRMS (ESI) m/z 747.2063 [M + H]+ (C37H41Cl2FN2O7S requires 747.2068).
AUTHORS

Principal Scientist at ORIC Pharmaceuticals
January 2015 – Present (1 year 1 month)San Francisco Bay Area
Medicinal Chemistry (oncology)
March 2013 – December 2014 (1 year 10 months)San Francisco Bay Area
Medicinal Chemistry (oncology)
1. Led optimization of small molecule inhibitors targeting protein-protein interactions in oncology programs
2. Discovered AM-7209, a back-up clinical candidate of AMG 232 featuring improved potency (KD from ITC competition = 38 pM), by replacing the carboxylic acid with an 4-amidobenzoic acid
March 2009 – February 2013 (4 years)San Francisco Bay Area
Medicinal Chemistry (oncology)
1. Played a critical role in the discovery of AMG 232, a small molecule MDM2 inhibitor in clinical development for the treatment of cancer, by discovering an additional interaction with the Gly58 shelf region
2. Led optimization of piperidinone series lead using a combination of conformational control of both the piperidinone ring and the appended N-alkyl substituent in the MDM2-p53 program
October 2004 – February 2009 (4 years 5 months)San Francisco Bay Area
Medicinal Chemistry (oncology and metabolic disease)
1. Proposed and synthesized the early piperidinone series lead in the MDM2-p53 program (oncology)
2. Designed and synthesized various small molecule enzyme inhibitors (metabolic disease)
October 2002 – September 2004 (2 years)Greater San Diego Area
Total Synthesis of Ramoplanin Aglycons and Their Key Analogues
Advisor: Professor Dale L. Boger

Medicinal Chemist, Executive Director
Experience
2004 – May 2014 (10 years)
| Benzoic acid derivative MDM2 inhibitor for the treatment of cancer [US8952036] |
2014-02-27 |
2015-02-10 |
SEE………..http://apisynthesisint.blogspot.in/2016/01/am-7209.html
/////////
c1(c(ccc(c1)NC(C[C@]2(C[C@@H]([C@H](N(C2=O)[C@H](CS(=O)(=O)C(C)(C)C)C3CC3)c4cc(c(cc4)Cl)F)c5cccc(c5)Cl)C)=O)C(=O)O)OC
CC1(CC(C(N(C1=O)C(CS(=O)(=O)C(C)(C)C)C2CC2)C3=CC(=C(C=C3)Cl)F)C4=CC(=CC=C4)Cl)CC(=O)NC5=CC(=C(C=C5)C(=O)O)OC

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO


































































































 D
D






































![CAS # 72702-95-5, Ponalrestat, Statil, Statyl, 3-[(4-Bromo-2-fluorophenyl)methyl]-3,4-dihydro-4-oxo-1-phthalazineacetic acid](https://i0.wp.com/www.chemblink.com/structures/72702-95-5.gif)