
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

WO 2016015596, Omarigliptin, Sunshine Lake Pharma Co Ltd, New patent


(WO2016015596) PROCESS FOR PREPARING 2, 3-DISUBSTITUTED-5-OXOPYRAN COMPOUND
SUNSHINE LAKE PHARMA CO., LTD. [CN/CN]; Northern Industrial Area, Songshan Lake Dongguan, Guangdong 523000 (CN)
SUN, Guodong; (CN).
LIU, Yongjun; (CN).
WEI, Mingjie; (CN).
LAI, Cailang; (CN).
LI, Dasheng; (CN).
ZHANG, Shouhua; (CN).
WANG, Zhongqing; (CN)
To a mixture of methanol (42 mL) and tert-butyl ( (1R, 2S) -1- (2, 5-difluorophenyl) -1-hydroxypent-4-yn -2-yl) carbamate (7.0 g) cooled to-5℃ was added a solution of KOH (3.2 g) in methanol (28 mL) dropwise. After dropwise addition, the resulting mixture was stirred for 30 minutes, then iodine (5.7 g) was added to the mixture. The reaction mixture was stirred at 0 ℃ for 10 minutes, followed by 25 ℃ for 6 hours, and then quenched with water (140 mL) . Then the mixture was stirred at 25 ℃ for 2 hours. The precipitate was collected by filtration and washed sequentially with methanol/water (40 mL, v: v=1: 1) . The resulting solid was dried at 45 ℃ in vacuo to give the title compound as awhite solid (8.8 g, purity: 95.0%)

//////////WO 2016015596, Omarigliptin, Sunshine Lake Pharma Co Ltd, NEW PATENT
WOCKHARDT, WO 2016016766, ISAVUCONAZONIUM SULPHATE, NEW PATENT

![]()
(WO2016016766) A PROCESS FOR THE PREPARATION OF ISAVUCONAZONIUM OR ITS SALT THEREOF
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
KHUNT, Rupesh Chhaganbhai; (IN).
RAFEEQ, Mohammad; (IN).
MERWADE, Arvind Yekanathsa; (IN).
DEO, Keshav; (IN)
The present invention relates to a process for the preparation of stable Isavuconazonium or its salt thereof. In particular of the present invention relates to process for the preparing of isavuconazonium sulfate, Isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide has purity more than 90%. The process is directed to preparation of solid amorphous form of isavuconazonium sulfate, isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide. The present invention process of Isavuconazonium or its salt thereof is industrially feasible, simple and cost effective to manufacture of isavuconazonium sulfate with the higher purity and better yield.
Habil Khorakiwala, chairman of Indian generic drugmaker Wockhardt
Isavuconazonium sulfate is chemically known l-[[N-methyl-N-3-[(methylamino) acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl)thiazol-2-yl]butyl]-lH-[l,2,4]-triazo-4-ium Sulfate and is structurally represented by formula (I):

Formula I
Isavuconazonium sulfate (BAL8557) is indicated for the treatment of antifungal infection. Isavuconazonium sulfate is a prodrug of Isavuconazole (BAL4815), which is chemically known 4-{2-[(lR,2R)-(2,5-Difluorophenyl)-2-hydroxy-l-methyl-3-(lH-l ,2,4-triazol-l-yl)propyl]-l ,3-thiazol-4-yl}benzonitrile compound of Formula II

Formula II
US Ppatent No. 6,812,238 (referred to herein as ‘238); 7,189,858 (referred to herein as ‘858); 7,459,561 (referred to herein as ‘561) describe Isavuconazonium and its process for the preparation thereof.
The US Pat. ‘238 patent describes the process of preparation of Isavuconazonium chloride hydrochloride.
The US Pat. ‘238 described the process for the Isavuconazonium chloride hydrochloride, involves the condensation of Isavuconazole and [N-methyl-N-3((tert-butoxycarbonyl methylamino) acetoxymethyl) pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester. The prior art reported process require almost 15-16 hours, whereas the present invention process requires only 8-10 hours. Inter alia prior art reported process requires too many step to prepare isavuconazonium sulfate, whereas the present invention process requires fewer steps.
Moreover, the US Pat. ‘238 describes the process for the preparation Isavuconazonium hydrochloride, which may be used as the key intermediate for the synthesis of isavuconazonium sulfate, compound of formula I. There are several drawbacks in the said process, which includes the use of anionic resin to prepare Isavuconazonium chloride hydrochloride, consequently it requires multiple time lyophilization, which makes the said prior art process industrially, not feasible.
The inventors of the present invention surprisingly found that Isavuconazonium or a pharmaceutically acceptable salt thereof in yield and purity could be prepared by using substantially pure intermediates in suitable solvent.
Thus, an object of the present invention is to provide simple, cost effective and industrially feasible processes for manufacture of isavuconazonium sulfate. Inventors of the present invention surprisingly found that isavuconazonium sulfate prepared from isavuconazonium iodide hydrochloride, provides enhanced yield as well as purity.
The process of the present invention is depicted in the following scheme:

Formula I
Formula-IA
The present invention is further illustrated by the following example, which does not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present application.
Examples
Example-1: Synthesis of l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino) acetoxymethyl]pyridin-2-yl]carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3 – [4-(4-cyanophenyl)thiazol-2-yl]butyl] – 1 H-[ 1 ,2,4] -triazo-4-ium iodide
Isavuconazole (20 g) and [N-methyl-N-3((tert-butoxycarbonylmethylamino)acetoxy methyl)pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester (24.7 g) were dissolved in acetonitrile (200ml). The reaction mixture was stirred to add potassium iodide (9.9 g). The reaction mixture was stirred at 47-50°C for 10-13 hour. The reaction mixture was cooled to room temperature. The reaction mass was filtered through celite bed and washed acetonitrile. Residue was concentrated under reduced pressure to give the crude solid product (47.7 g). The crude product was purified by column chromatography to get its pure iodide form (36.5 g).
Yield: 84.5 %
HPLC Purity: 87%
Mass: m/z 817.4 (M- 1)+
Example-2: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride
l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide (36.5 g) was dissolved in ethyl acetate (600 ml). The reaction mixture was cooled to -5 to 0 °C. The ethyl acetate hydrochloride (150 ml) solution was added to reaction mixture. The reaction mixture was stirred for 4-5 hours at room temperature. The reaction mixture was filtered and obtained solid residue washed with ethyl acetate. The solid dried under vacuum at room temperature for 20-24 hrs to give 32.0 gm solid.
Yield: 93 %
HPLC Purity: 86%
Mass: m/z 717.3 (M-HC1- 1)
Example-3: Preparation of Strong anion exchange resin (Sulfate).
Indion GS-300 was treated with aqueous sulfate anion solution and then washed with DM water. It is directly used for sulfate salt.
Example-4: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium Sulfate
Dissolved 10.0 g l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride in 200 ml deminerahzed water and 30 ml methanol. The solution was cooled to about 0 to 5°C. The strong anion exchange resin (sulfate) was added to the cooled solution. The reaction mixture was stirred to about 60-80 minutes. The reaction was filtered and washed with 50ml of demineralized water and methylene chloride. The aqueous layer was lyophilized to obtain
(8.0 g) white solid.
Yield: 93 %
HPLC Purity: > 90%
Mass: m/z 717.4 (M- HS04) +

////////WOCKHARDT, WO 2016016766, ISAVUCONAZONIUM SULPHATE, NEW PATENT
LUPIN, SOFOSBUVIR, NEW PATENT, WO 2016016865
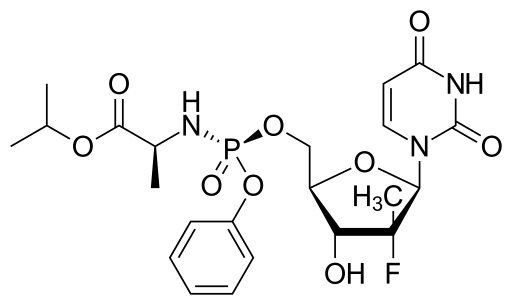
![]()
(WO2016016865) A PROCESS FOR THE PREPARATION OF NUCLEOSIDE PHOSPHORAMIDATE
LUPIN LIMITED [IN/IN]; 159 CST Road, Kalina, Santacruz (East), State of Maharashtra, Mumbai 400 098 (IN)
ROY, Bhairab, Nath; (IN).
SINGH, Girij, Pal; (IN).
SHRIVASTAVA, Dhananjai; (IN).
MEHARE, Kishor, Gulabrao; (IN).
MALIK, Vineet; (IN).
DEOKAR, Sharad, Chandrabhan; (IN).
DANGE, Abhijeet, Avinash; (IN)
The present invention pertains to process for preparing nucleoside phosphoramidates and their intermediates. Phosphoramidates are inhibitors of RNA-dependent RNA viral replication and are useful as inhibitors of HCV NS5B polymerase, as inhibitors of HCV replication and for treatment of hepatitis C infection in mammals. One of the recently approved phosphoramidate by USFDA is Sofosbuvir [1190307-88-0]. Sofosbuvir is a component of the first all-oral, interferon-free regimen approved for treating chronic hepatitis C. The present invention provides novel intermediate, its process for preparation and use for the preparation of Sofosbuvir. The present invention also gives one pot process for preparation of Sofosbuvir.
Hepatitis C virus (HCV) infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals. There are limited treatment options for individuals infected with hepatitis C virus. The current approved therapeutic option is the use of immunotherapy with recombinant interferon- [alpha] alone or in combination with the nucleoside analog ribavirin.
US 7964580 (‘580) is directed towards novel nucleoside phosphoramidate prodrug for the treatment of hepatitis C virus infection.
US’580 patent claims Sofosbuvir and rocess for preparation of Sofosbuvir of Formula 1.

Formula 1
Process for preparation of Sofosbuvir as per US ‘580 patent involve reaction of compound of Formula 4″ with a nucleoside 5’

Compound 4″ nucleoside 5′
Wherein X’ is a leaving group, such as CI, Br, I, tosylate, mesylate, trifluoroacetate, trifluroslfonate, pentafluorophenoxide, p-nitro-phenoxide.
Objects of the invention
The object of the present invention is to provide a novel intermediate of Formula 2

Formula 2
wherein X’ is a leaving group selected from 1-hydroxybenzotriazole, 5-(Difluoromethoxy)-lH-benzimidazole-2-thiol, 2-Mercapto-5-methoxybenzimidazole, cyanuric acid, 2-oxazolidinone, 2-Hydroxy Pyridine. The above leaving group can be optionally substituted with n-alkyl, branched alkyl, substituted alkyl; cycloalkyl; halogen; nitro; or aryl, which includes, but not limited to, phenyl or naphthyl, where phenyl or naphthyl are further optionally substituted with at least one of Ci-C6 alkyl, C2-C6 alkenyl, C2-C6 alkynyl, Ci-C6 alkoxy, F, CI, Br, I, nitro, cyano, Ci-C6 haloalkyl, -N(Rr)2, Ci-C6 acylamino, -NHS02Ci-C6 alkyl, -S02N(Rr)2, COR1″, and -S02Ci-C6 alkyl; (Rr is independently hydrogen or alkyl, which includes, but is not limited to, Ci-C2o alkyl, Ci-Cio alkyl, or Ci-C6 alkyl, R1” is -OR1 or -N(Rr)2).
Another object of the present invention is to provide a process to prepare the intermediate of Formula 2.
Another object of the present invention is use of the intermediate of Formula 2 in the preparation of Sofosbuvir of Formula 1.

Formula 1

Example 1:
Process for the preparation of S-oxazolidinone derivative of Formula 2

Step-1 Preparation of phosphorochloridate solution:
Dichloromethane (DCM 400ml) was charged in round bottom flask flushed with nitrogen. Phenyl phosphodichloridate (18.30ml) was added in one portion in the flask. The flask was cooled to -60°-70°C with a dry ice-acetone bath. Solution of L-alanine isopropyl ester hydrochloride (20.6gm)) in DCM (50ml) was added to the reaction flask. To this was added a solution of triethylamine (11.20ml) in MDC (100 ml) was added over a course of 60 minutes, while maintaining internal temperature below -70 °C throughout the addition. After completion of reaction, temperature of reaction mass was raised to room temperature.
100ml THF was charged in another round bottom flask flushed with nitrogen followed by the addition of S-4-phenyloxazolidnone (lOgm). Triethyl-amine (11.2ml) & LiCl (2.85gm) were added to the above flask. The reaction mass was stirred for 15-30 min at room temperature and was cooled to 0-5 °C. Phosphorochloridate solution from step-1 was added drop- wise to the reaction flask in 15-45 min maintaining reaction temperature at 0-5 °C. The reaction mass was stirred for 30-60min at 0°-5°C. The reaction progress was monitored on thin layer chromatography. After completion of the reaction, the reaction temperature was raised to room temperature. Agitation was resumed for an additional 30min. The reaction mass was filtered and concentrated under reduced pressure. To this was added diisopropyl ether (400ml) and aqueous saturated ammonium chloride solution and reaction mass was stirred for 10-15 minutes. Organic layer was separated and was washed with water (100ml) & dried over sodium sulfate and concentrated under vacuum. Cyclohexane (50ml) was charged to the obtained oily mass and reaction mass was stirred till solid precipitated out. Solid was filtered and washed with cyclohexane and dried under vacuum (8.80gm MP 56.5°-56.6°C). The obtained product was characterized by mass, NMR & IR. 1H NMR (DMSO-d6) δ 1.142 -1.18
(m, 9H), 3.85-3.92 (m, 1H), 4.72-4.89(m, 2H), 5.31-5.32(d, 1H), 6.25-6.3 (m, 1H), 6.95-7.31 (m, 10H); MS, m/e 433 (M+l) +
Example 2: Process for the preparation of 2-hydroxy pyridine derivatives of formula 2:

Anhydrous dichloromethane (DCM) 700ml was charged in round bottom flask flushed with nitrogen. The flask was cooled to -60° to -70°C in a dry ice acetone bath. Phenyl phosphodichloridate (76.04 gm) was added in one portion in the flask at -65°C. Solution of L-alanine isopropyl ester hydrochloride (60.56 gm) in DCM (50 ml) was added to the reaction mass. Solution of triethylamine (72.44gm) in DCM (50ml) was added to the reaction mass over a course of 60 minutes, while maintaining internal temperature below -70°C throughout the addition. The resulting white slurry was agitated for additional 60 minutes. Then the temperature of reaction mass was raised to room temperature. Reaction mass was stirred for 60 min & TLC was checked. Reaction mass was filtered and rinsed with anhydrous dichloromethane (2 XI 00 mL). The filtrate was concentrate under vacuum to 20 V and reaction mass was filtered, washed with DCM (15ml). The filtrate was transferred to RBF. The reaction mass was cooled to 0°-10°C. A solution of 2-hydroxy-3-nitro-5- (trifluoromethyl) pyridine (15.gm) in DCM (100ml) & triethyl amine (21.89gm) was added to the reaction mass. Temperature of reaction mass was raised to 20-30°C. Reaction mass was stirred overnight. Reaction was monitored using TLC. After completion, the reaction mass was filtered and washed with DCM (30ml). Filtrate was washed with water (150 ml x 2). Organic layer was concentrated under vacuum and degased. Diisopropyl ether (200ml) was charged to reaction mass and reaction mass was stirred for 15 minutes , filtered and washed with methyl ter-butyl ether (MTBE 30ml). Filtrate was concentrated under vacuum and dried. (8.68gm, MP-125.5°-131.5°C). Obtained compound was characterized by Mass, NMR & IR. 1H NMR (DMSO-d6) δ 1.07 -1.27 (m, 9H), 4.04-4. l l(m, 1H), 4.73-4.79(m, 1H), 6.76-7.43 (m, 5H), 9.00-9.02 (d, 2H); MS, m/e 478 (M+l) +; FTIR, 1203, 1409, 1580, 1732, 3217.
Other 2-hydroxy pyridine derivatives of Formula 2 were prepared by following the process disclosed in example 2-
2-Hydroxy-5-fluoropyridine derivative of Formula 2;-1H NMR (DMSO-d6) δ 1.09 -1.23 (m, 9H), 3.02-3.06 (m, lH), 3.85-4.01 (m,lH), 4.79-4.87(m, 1H), 6.4-6.52 (m,lH), 7.10-7.89 (m,6H); MS, m/e 383 (M+l) +,
2-Hydroxy-5-nitropyridine derivative of Formula 2:- 1H NMR (DMSO-d6) δ 1.06 -1.22 (m, 9H),4.0-4.02 (m,lH), 4.7-4.8(m,lH), 6.5-6.6 (m,lH),7.12-7.42 (m,6H),8.66-8.68 (d, lH),9.07-9.13(d,lH); MS, m/e 410 (M+l) +
2-Hydroxy-3, 5-dinitropyridine derivative of Formula 2:- 1H NMR (DMSO-d6) δ 1.11 -1.24 (m, 9H), 3.04-3.09(m,lH), 4.8-4.86(m,lH), 7.09-7.39 (m,5H),8.97-9.06 (d,2H)
Example 3: Process for the preparation of Sofosbuvir by coupling of isopropyl(((3-nitro-5-(trifluromethyl)pyridin-2-yl)oxy)phenoxy)phosphoryl-L-alaninate with 1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-3-methyltetrahydrofuran-2-yl)pyrimidine-2,4(lH,3H)-dione :

To a solution of l-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-3-methyltetrahydrofuran-2-yl)pyrimidine-2,4(lH,3H)-dione (0.2gm) in THF (4 ml), tert- butylmagnesium chloride (0.80ml, 1.7 M solution in THF) was added dropwise at room temperature and reaction mass was stirred for 30 minutes. A solution of pyridine derivative from example 2 (0.36gm) in THF (4ml) was added dropwise to the reaction mass at room temperature. Completion of reaction was monitored using TLC. After completion of reaction, reaction mass was quenched by using saturated ammonium chloride solution (10ml). Reaction mass was extracted with ethyl acetate (50ml). Organic layer was separated, dried over magnesium sulfate and concentrated under vacuum. The resulting residue was purified by column chromatography on silica gel & obtained solid product was characterized. MS, m/e 530.2 (M+l) +.
/////////LUPIN, SOFOSBUVIR, NEW PATENT, WO 2016016865
Canagliflozin , New patent, WO 2016016774, SUN PHARMACEUTICAL INDUSTRIES LIMITED


WO2016016774, CRYSTALLINE FORMS OF CANAGLIFLOZIN
SUN PHARMACEUTICAL INDUSTRIES LIMITED [IN/IN]; Sun House, Plot No. 201 B/1 Western Express Highway Goregaon (E) Mumbai, Maharashtra 400 063 (IN)
SANTRA, Ramkinkar; (IN).
NAGDA, Devendra, Prakash; (IN).
THAIMATTAM, Ram; (IN).
ARYAN, Satish, Kumar; (IN).
SINGH, Tarun, Kumar; (IN).
PRASAD, Mohan; (IN).
GANGULY, Somenath; (IN).
WADHWA, Deepika; (IN)
The present invention relates to crystalline forms of canagliflozin, processes for their preparation, and their use for the treatment of type 2 diabetes mellitus. A crystalline Form R1of canagliflozin emihydrate. The crystalline Form R1 of canagliflozin hemihydrate of claim 1, characterized by an X-ray powder diffraction peaks having d-spacing values at about 3.1, 3.7, 4.6, and 8.9 A

The present invention relates to crystalline forms of canagliflozin, processes for their preparation, and their use for the treatment of type 2 diabetes mellitus.
Canagliflozin hemihydrate, chemically designated as (l<S)-l,5-anhydro-l-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol hemihydrate, is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. Its chemical structure is represented by Formula I.

Formula I
U.S. Patent Nos. 7,943,582 and 8,513,202 disclose crystalline forms of canagliflozin hemihydrate.
PCT Publication No. WO 2009/035969 discloses a crystalline form of
canagliflozin, designated as I-S.
PCT Publication No. WO 2013/064909 discloses crystalline complexes of canagliflozin with L-proline, D-proline, and L-phenylalanine, and the processes for their preparation.
PCT Publication No. WO 2014/180872 discloses crystalline non-stoichiometric hydrates of canagliflozin (HxA and HxB), and the process for their preparation.
PCT Publication No. WO 2015/071761 discloses crystalline Forms B, C, and D of canagliflozin.
Chinese Publication Nos. CN 103980262, CN 103936726, CN 103936725, CN 103980261, CN 103641822, CN 104230907, CN 104447722, CN 104447721, and CN 104130246 disclose different crystalline polymorphs of canagliflozin.
In the pharmaceutical industry, there is a constant need to identify critical physicochemical parameters of a drug substance such as novel salts, polymorphic forms, and co-crystals, that affect the drug’s performance, solubility, and stability, and which may play a key role in determining the drug’s market acceptance and success.
The discovery of new forms of a drug substance may improve desirable processing properties of the drug, such as ease of handling, storage stability, and ease of purification. Accordingly, the present invention provides novel crystalline forms of canagliflozin having enhanced stability over known crystalline forms of canagliflozin.

EXAMPLES
Example 1 : Preparation of a crystalline Form Rl of canagliflozin hemihydrate
Amorphous canagliflozin (5 g) was suspended in an aqueous solution of sodium formate (80 mL of a solution prepared by dissolving 137.7 g of sodium formate in 180 mL of de-ionized water). The suspension was stirred at room temperature for 20 hours to obtain a reaction mixture. De-ionized water (100 mL) was added to the reaction mixture, and then the reaction mixture was stirred for 1.5 hours. De-ionized water (50 mL) was added to the reaction mixture, and then the reaction mixture was stirred for 30 minutes. The reaction mixture was filtered, then washed with de-ionized water (300 mL), and then dried under vacuum for 12 hours to obtain a solid. The solid was further dried under vacuum at 60°C for 6 hours.
Yield: 4.71 g
Example 2: Preparation of a crystalline Form R2 of canagliflozin monohydrate
Amorphous canagliflozin (5 g) was suspended in an aqueous solution of sodium formate (80 mL of a solution prepared by dissolving 137.7 g of sodium formate in 180 mL of de-ionized water). The suspension was stirred at room temperature for 20 hours to obtain a reaction mixture. De-ionized water (100 mL) was added to the reaction mixture, and then the reaction mixture was stirred for 1.5 hours. De-ionized water (50 mL) was added to the reaction mixture, and then the reaction mixture was stirred for 30 minutes. The reaction mixture was filtered, then washed with de-ionized water (300 mL), and then dried under vacuum for 12 hours at room temperature.
Yield: 4.71 g
Example 3 : Preparation of a crystalline Form R2 of canagliflozin monohydrate
Canagliflozin hemihydrate (0.15 g; Form Rl obtained as per Example 1) was suspended in de-ionized water (3 mL). The suspension was stirred at room temperature for 24 hours. The reaction mixture was filtered, then dried at room temperature under vacuum for 5 hours.
Yield: 0.143 g
Example 4: Preparation of a crystalline Form R3 of canagliflozin hydrate
Amorphous canagliflozin (100 g) was suspended in an aqueous solution of sodium formate (1224 g of sodium formate in 1600 mL of de-ionized water). The suspension was stirred at room temperature for 20 hours to obtain a reaction mixture. De-ionized water
(2000 mL) was added to the reaction mixture, and then the reaction mixture was stirred for one hour. De-ionized water (1000 mL) was added to the reaction mixture, and then the reaction mixture was stirred for another one hour. The reaction mixture was filtered, then washed with de-ionized water (6000 mL), and then dried under vacuum for 30 minutes to obtain a solid. The solid was then dried under vacuum at 30°C to 35°C until a water content of 8% to 16% was attained.
Yield: 100 g

./////////////Canagliflozin , New patent, WO 2016016774, SUN PHARMACEUTICAL INDUSTRIES LIMITED
Patiromer

Patiromer
1260643-52-4 FREE FORM
CAS 1208912-84-8
(C10 H10 . C8 H14 . C3 H3 F O2 . 1/2 Ca)x
2-Propenoic acid, 2-fluoro-, calcium salt (2:1), polymer with diethenylbenzene and 1,7-octadiene
RLY5016
RELYPSA INNOVATOR
Patiromer is a powder for suspension in water for oral administration, approved in the U.S. as Veltassa in October, 2015. Patiromer is supplied as patiromer sorbitex calcium which consists of the active moiety, patiromer, a non-absorbed potassium-binding polymer, and a calcium-sorbitol counterion. Each gram of patiromer is equivalent to a nominal amount of 2 grams of patiromer sorbitex calcium. The chemical name for patiromer sorbitex calcium is cross-linked polymer of calcium 2-fluoroprop-2-enoate with diethenylbenzene and octa-1,7-diene, combination with D-glucitol. Patiromer sorbitex calcium is an amorphous, free-flowing powder that is composed of individual spherical beads.
Veltassa is a powder for suspension in water for oral administration. The active ingredient is patiromer sorbitex calcium which consists of the active moiety, patiromer, a non-absorbed potassium-binding polymer, and a calcium-sorbitol counterion.

Each gram of patiromer is equivalent to a nominal amount of 2 grams of patiromer sorbitex calcium. The chemical name for patiromer sorbitex calcium is cross-linked polymer of calcium 2-fluoroprop-2-enoate with diethenylbenzene and octa-1,7-diene, combination with D-glucitol.
Mechanism of Action
Veltassa is a non-absorbed, cation exchange polymer that contains a calcium-sorbitol counterion. Veltassa increases fecal potassium excretion through binding of potassium in the lumen of the gastrointestinal tract. Binding of potassium reduces the concentration of free potassium in the gastrointestinal lumen, resulting in a reduction of serum potassium levels.

Treatment of Hyperkalemia
Hyperkalemia is usually asymptomatic but occasionally can lead to life-threatening cardiac arrhythmias and increased all-cause and in-hospital mortality, particularly in patients with CKD and associated cardiovascular diseases (Jain et al., 2012; McMahon et al., 2012; Khanagavi et al., 2014). However, there is limited evidence from randomized clinical trials regarding the most effective therapy for acute management of hyperkalemia (Khanagavi et al., 2014) and a Cochrane analysis of emergency interventions for hyperkalemia found that none of the studies reported mortality or cardiac arrhythmias, but reports focused on PK (Mahoney et al., 2005). Thus, recommendations are based on opinions and vary with institutional practice guidelines (Elliot et al., 2010; Khanagavi et al., 2014). Management of hyperkalemia includes reducing potassium intake, discontinuing potassium supplements, treatment of precipitating risk factors, and careful review of prescribed drugs affecting potassium homeostasis. Treatment of life-threatening hyperkalemia includes nebulized or inhaled beta-agonists (albuterol, salbutamol) or intravenous (IV) insulin-and-glucose, which stimulate intracellular potassium uptake, their combination being more effective than either alone. When arrhythmias are present, IV calcium might stabilize the cardiac resting membrane potential. Sodium bicarbonate may be indicated in patients with severe metabolic acidosis. Potassium can be effectively eliminated by hemodialysis or increasing its renal (loop diuretics) and gastrointestinal (GI) excretion with sodium polystyrene sulfonate, an ion-exchange resin that exchanges sodium for potassium in the colon. However, this resin produces serious GI adverse events (ischemic colitis, bleeding, perforation, or necrosis). Therefore, there is an unmet need of safer and more effective drugs producing a rapid and sustained PK reduction in patients with hyperkalemia.
In this article we review two new polymer-based, non-systemic oral agents, patiromer calcium (RLY5016) and zirconium silicate (ZS-9), under clinical development designed to induce potassium loss via the GI tract, particularly the colon, and reduce PK in patients with hyperkalemia.
1. Patiromer calcium
This metal-free cross-linked fluoroacrylate polymer (structure not available) exchanges cations through the gastrointestinal (GI) tract. It preferentially binds soluble potassium in the colon, increases its fecal excretion and reduces PK under hyperkalemic conditions.
The development program of patiromer includes several clinical trials. An open-label, single-arm study evaluated a titration regimen for patiromer in 60 HF patients with CKD treated with ACEIs, ARBs, or beta blockers (clinicaltrials.gov identifier: NCT01130597). Another open-label, randomized, dose ranging trial determined the optimal starting dose and safety of patiromer in 300 hypertensive patients with diabetic nephropathy treated with ACEIs and/or ARBs, with or without spironolactone (NCT01371747). The primary outcomes were the change in PK from baseline to the end of the study. Unfortunately, the results of these trials were not published.
In a double-blind, placebo-controlled trial (PEARL-HF, NCT00868439), 105 patients with a baseline PK of 4.7 mmol/L and HF (NYHA class II-III) treated with spironolactone in addition to standard therapy were randomized to patiromer (15 g) or placebo BID for 4 weeks (Pitt et al., 2011). Spironolactone, initiated at 25 mg/day, was increased to 50 mg/day on day 15 if PK was ≤5.1 mmol/L. Patients were eligible for the trial if they had either CKD (eGFR <60 ml/min) or a history of hyperkalemia leading to discontinuation of RAASIs or beta-blockers. Compared with placebo, patiromer decreased the PK (-0.22 mmol/L, while PK increased in the placebo group +0.23 mmol/L, P<0.001), and the incidence of hyperkalemia (7% vs. 25%, P=0.015) and increased the number of patients up-titrated to spironolactone 50 mg/day (91% vs. 74%, P=0.019). A similar reduction in PK and hyperkalemia was observed in patients with an eGFR <60 ml/min. Patiromer produced more GI adverse events (flatulence, diarrhea, constipation, vomiting: 21% vs 6%), hypokalemia (<4.0 mmol/L: 47% vs 10%, P<0.001) and hypomagnesaemia (<1.8 mg/dL: 24% vs. 2.1%), but similar adverse events leading to study discontinuation compared to placebo. Unfortunately, recruited patients had normokalemia and basal eGFR in the treatment group was 84 ml/min. Thus, this study did not answer whether patiromer is effective in reducing PK in patients with CKD and/or HF who develop hyperkalemia on RAASIs.
A two-part phase 3 study evaluated the efficacy and safety of patiromer in the treatment of hyperkalemia (NCT01810939). In a single-blind phase (part A) 243 patients with hyperkalemia and CKD (102 with HF) on RAASIs were treated with patiromer BID for 4 weeks: 4.2 g in patients with mild hyperkalemia (5.1-<5.5 mmol/L, n=92) and 8.4 g in patients with moderate-to-severe hyperkalemia (5.5-<6.5 mmol/L, n=151). Part B was a placebo-controlled, randomized, withdrawal phase designed to confirm the maintained efficacy of patiromer and the recurrent hyperkalemia following that drug’s withdrawal. Patients (n=107) who completed phase A with a normal PK were randomized to continue on patiromer (27 with HF) or placebo (22 with HF) besides RAASIs for 8 weeks. The primary endpoint was the difference in mean PK between the patiromer and placebo groups from baseline to the end of the study or when the patient first had a PK <3.8 or ≥5.5 mmol/L. In part A patiromer produced a rapid reduction in PK that persisted throughout the study in patients with and without HF (-1.06 and -0.98 mmol/L, respectively; both P<0.001 vs. placebo); three-fourths of patients in both groups had normal PK (3.8-<5.1 mmol/L) at 4 weeks. In part B patiromer reduced PK (-0.64 mmol/L) in patients with or without HF (P<0.001). As compared with placebo, fewer patients, with or without HF, presented recurrent hyperkalemia in the patiromer group or required RAASI discontinuation regardless of HF status (Pitt, 2014). Patiromer was well-tolerated, with a safety profile similar to placebo even in HF patients. The most common adverse events were nausea, diarrhea, and hypokalemia.
INDICATIONS AND USAGE
Veltassa is a potassium binder indicated for the treatment of hyperkalemia.
Veltassa should not be used as an emergency treatment for lifethreatening hyperkalemia because of its delayed onset of action.
Patiromer (USAN, trade name Veltassa) is a drug used for the treatment of hyperkalemia (elevated blood potassium levels), a condition that may lead to palpitations and arrhythmia (irregular heartbeat). It works by binding potassium in the gut.[1][2]

Medical uses
Patiromer is used for the treatment of hyperkalemia, but not as an emergency treatment for life-threatening hyperkalemia, because it acts relatively slowly.[2] Such a condition needs other kinds of treatment, for example calcium infusions, insulin plus glucose infusions, salbutamol inhalation, and hemodialysis.[3]
Typical reasons for hyperkalemia are renal insufficiency and application of drugs that inhibit the renin–angiotensin–aldosterone system (RAAS) – e.g. ACE inhibitors, angiotensin II receptor antagonists, or potassium-sparing diuretics – or that interfere with renal function in general, such as nonsteroidal anti-inflammatory drugs (NSAIDs).[4][5]
Adverse effects
Patiromer was generally well tolerated in studies. Side effects that occurred in more than 2% of patients included in clinical trials were mainly gastro-intestinal problems such as constipation, diarrhea, nausea, and flatulence, and also hypomagnesemia (low levels of magnesium in the blood) in 5% of patients, because patiromer binds magnesium in the gut as well.[2][6]
Interactions
No interaction studies have been done in humans. Patiromer binds to many substances besides potassium, including numerous orally administered drugs (about half of those tested in vitro). This could reduce their availability and thus effectiveness,[2] wherefore patiromer has received a boxed warning by the US Food and Drug Administration (FDA), telling patients to wait for at least six hours between taking patiromer and any other oral drugs.[7]
Pharmacology
Mechanism of action
Patiromer works by binding free potassium ions in the gastrointestinal tract and releasing calcium ions for exchange, thus lowering the amount of potassium available for absorption into the bloodstream and increasing the amount that is excreted via the feces. The net effect is a reduction of potassium levels in the blood serum.[2][4]
Lowering of potassium levels is detectable 7 hours after administration. Levels continue to decrease for at least 48 hours if treatment is continued, and remain stable for 24 hours after administration of the last dose. After this, potassium levels start to rise again over a period of at least four days.[2]
Pharmacokinetics
Patiromer is not absorbed from the gut, is not metabolized, and is excreted in unchanged form with the feces.[2]
Physical and chemical properties
The substance is a cross-linked polymer of 2-fluoroacrylic acid (91% in terms of amount of substance) with divinylbenzenes (8%) and 1,7-octadiene (1%). It is used in form of its calcium salt (ratio 2:1) and with sorbitol (one molecule per two calcium ions or four fluoroacrylic acid units), a combination called patiromer sorbitex calcium.[8]
-
2-fluoroacrylic acid
-

o-divinylbenzene
-

p-divinylbenzene
-

1,7-octadiene


Patiromer sorbitex calcium is an off-white to light brown, amorphous, free-flowing powder. It is insoluble in water, 0.1 M hydrochloric acid, heptane, and methanol.[2][8]
Hyperkalemia Is a Clinical Challenge
Hyperkalemia may result from increased potassium intake, impaired distribution between the intracellular and extracellular spaces, and/or conditions that reduce potassium excretion, including CKD, hypertension, diabetes mellitus, or chronic heart failure (HF) (Jain et al., 2012). Additionally, drugs and nutritional/herbal supplements (Table 1) can produce hyperkalemia in up to 88% of hospitalized patients by impairing normal potassium regulation (Hollander-Rodríguez and Calvert, 2006; Khanagavi et al., 2014).
 Although the prevalence of hyperkalemia in the general population is unknown, it is present in 1-10% of hospitalized patients depending on how hyperkalemia is defined (McMahon et al., 2012; Gennari, 2002). Hyperkalemia is a common problem in patients with conditions that reduce potassium excretion, especially when treated with beta-adrenergic blockers that inhibit Na+,K+-ATPase activity or RAAS inhibitors (RAASIs) [angiotensin-converting-enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), mineralocorticoid receptor antagonists or renin inhibitors] that decrease aldosterone excretion (Jain et al., 2012; Weir and Rolfe, 2010). The incidence of hyperkalemia with RAASIs in monotherapy is low (≤2%) in patients without predisposing factors, but increases with dual RAASIs (5%) and in patients with risk factors such as CKD, HF, and/or diabetes (5-10%) (Weir and Rolfe, 2010). Thus, hyperkalemia is a key limitation to fully titrate RAASIs in these patients who are most likely to benefit from treatment. Thus, we need new drugs to control hyperkalemia in these patients while maintaining the use of RAASIs.
Although the prevalence of hyperkalemia in the general population is unknown, it is present in 1-10% of hospitalized patients depending on how hyperkalemia is defined (McMahon et al., 2012; Gennari, 2002). Hyperkalemia is a common problem in patients with conditions that reduce potassium excretion, especially when treated with beta-adrenergic blockers that inhibit Na+,K+-ATPase activity or RAAS inhibitors (RAASIs) [angiotensin-converting-enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), mineralocorticoid receptor antagonists or renin inhibitors] that decrease aldosterone excretion (Jain et al., 2012; Weir and Rolfe, 2010). The incidence of hyperkalemia with RAASIs in monotherapy is low (≤2%) in patients without predisposing factors, but increases with dual RAASIs (5%) and in patients with risk factors such as CKD, HF, and/or diabetes (5-10%) (Weir and Rolfe, 2010). Thus, hyperkalemia is a key limitation to fully titrate RAASIs in these patients who are most likely to benefit from treatment. Thus, we need new drugs to control hyperkalemia in these patients while maintaining the use of RAASIs.
History
Studies
In a Phase III multicenter clinical trial including 237 patients with hyperkalemia under RAAS inhibitor treatment, 76% of participants reached normal serum potassium levels within four weeks. After subsequent randomization of 107 responders into a group receiving continued patiromer treatment and a placebo group, re-occurrence of hyperkalemia was 15% versus 60%, respectively.[9]
Approval
The US FDA approved patiromer in October 2015.[7] The drug is not approved in Europe as of January 2016.
PATENT
PATENT
CLIP
https://www.oatext.com/polymer-and-heterocyclic-compounds-their-utility-and-application-as-drug.php
The Structure of some commercially available polymer sequestrant drugs, were as follows:
Were sorbitol, which is frequently dosed with SPS as a laxative the risk of swelling of above drugs Leeds to some improvements to the above drudge polymers to increase of its capacity and reducing its swelling property sevelamer is changed into cross liked N,N, N,N-tetrakis (3-aminopropyl) butane-1,4-diamin (Schemes 6, 7) as illustrated below support the safety profile in clinical studies of up to 52 weeks it is approved for treatment of hyperphosphatemia by FDA in 1998.
Scheme 6. Showing the network formation of patiromer amine residue
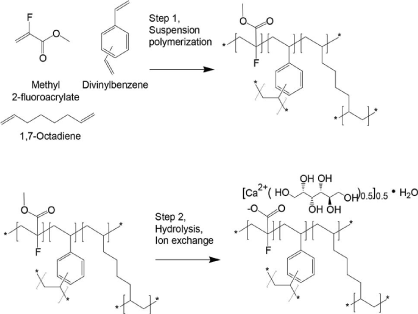
Scheme 7. Synthetic steps of Patiromer
Patiromer is a non-absorbed, potassium-sequestering polymer which is a crosslinked form of poly (fluoroacrylic acid).The fluorine substituent lowers the pKa of the acid group in patiromer compared to acrylic acid such that a higher proportion of acid groups are available for ion binding.
Suspension polymerization during patiromer manufacture allows for the generation of monodisperse uniform polymer particles, with spherical shape, controlled size distribution, and low swelling. The bead particles have a median diameter of around 100 µm. Patiromer was approved by the FDA for the treatment of hyperkalemia in 2015 based on clinical studies showing effective potassium lowering and acceptable safety profile in clinical studies of up to 52 weeks duration.
PATENT
https://patents.google.com/patent/WO2017109658A1/en

wherein m is the number of 2-fluoro-2-propanoate groups and



wherein m, n, p and * are as defined above, characterized in that the polymerization reaction to obtain the compound of formula (III) is carried out in the presence of a water-soluble radical initiator and of an inert dispersing agent.
References
- 1 Henneman, A; Guirguis, E; Grace, Y; Patel, D; Shah, B (2016). “Emerging therapies for the management of chronic hyperkalemia in the ambulatory care setting”. American Journal of Health-System Pharmacy 73 (2): 33–44. doi:10.2146/ajhp150457. PMID 26721532.
- 2FDA Professional Drug Information for Veltassa.
- 3Vanden Hoek TL, Morrison LJ, Shuster M, Donnino M, Sinz E, Lavonas EJ, Jeejeebhoy FM, Gabrielli A; Morrison; Shuster; Donnino; Sinz; Lavonas; Jeejeebhoy; Gabrielli (2010-11-02). “Part 12: cardiac arrest in special situations: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care”. Circulation 122 (18 Suppl 3): S829–61. doi:10.1161/CIRCULATIONAHA.110.971069. PMID 20956228.
- 4Esteras, R.; Perez-Gomez, M. V.; Rodriguez-Osorio, L.; Ortiz, A.; Fernandez-Fernandez, B. (2015). “Combination use of medicines from two classes of renin-angiotensin system blocking agents: Risk of hyperkalemia, hypotension, and impaired renal function”. Therapeutic Advances in Drug Safety 6 (4): 166. doi:10.1177/2042098615589905. PMID 26301070.
- 5Rastegar, A; Soleimani, M (2001). “Hypokalaemia and hyperkalaemia”. Postgraduate Medical Journal 77 (914): 759–64. doi:10.1136/pmj.77.914.759. PMC 1742191. PMID 11723313.
- 6Tamargo, J; Caballero, R; Delpón, E (2014). “New drugs for the treatment of hyperkalemia in patients treated with renin-angiotensin-aldosterone system inhibitors — hype or hope?”. Discovery medicine 18 (100): 249–54. PMID 25425465.
- 7″FDA approves new drug to treat hyperkalemia”. FDA. 21 October 2015.
- 8RxList: Veltassa.
- 9Weir, Matthew R.; Bakris, George L.; Bushinsky, David A.; Mayo, Martha R.; Garza, Dahlia; Stasiv, Yuri; Wittes, Janet; Christ-Schmidt, Heidi; Berman, Lance; Pitt, Bertram (2015). “Patiromer in Patients with Kidney Disease and Hyperkalemia Receiving RAAS Inhibitors”. New England Journal of Medicine 372 (3): 211. doi:10.1056/NEJMoa1410853. PMID 25415805.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-Fluoropropenoic acid, cross-linked polymer with diethenylbenzene and 1,7-octadiene
|
|
| Clinical data | |
| Trade names | Veltassa |
| AHFS/Drugs.com | entry |
| Legal status |
|
| Routes of administration |
Oral suspension |
| Pharmacokinetic data | |
| Bioavailability | Not absorbed |
| Metabolism | None |
| Onset of action | 7 hrs |
| Duration of action | 24 hrs |
| Excretion | Feces |
| Identifiers | |
| CAS Number | 1260643-52-4 1208912-84-8 (calcium salt) |
| ATC code | None |
| PubChem | SID 135626866 |
| DrugBank | DB09263 |
| UNII | 1FQ2RY5YHH |
| KEGG | D10148 |
| ChEMBL | CHEMBL2107875 |
| Synonyms | RLY5016 |
| Chemical data | |
| Formula | [(C3H3FO2)182·(C10H10)8·(C8H14)10]n
[Ca91(C3H2FO2)182·(C10H10)8·(C8H14)10]n (calcium salt) |
////
Mr. Glenn Saldanha Chairman & and Managing Director, Glenmark Pharmaceuticals Limited, conferred ‘India Pharma Leader Award’ by the Government of India

Mr. Glenn Saldanha Chairman & and Managing Director, Glenmark Pharmaceuticals Limited, conferred ‘India Pharma Leader Award’ by the Government of India
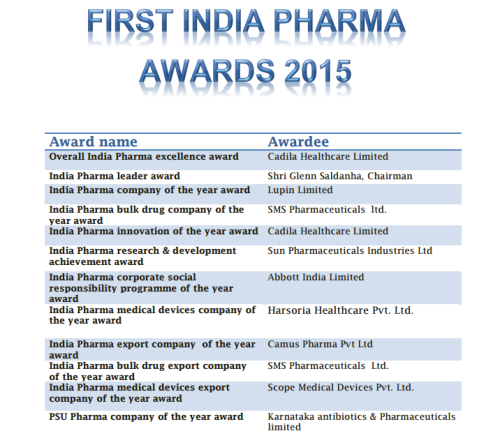
Indian Ministry for Chemicals and Fertilizers on Thursday conferred 1st India Pharma awards to 12 Indian drug companies under various categories to motivate Indian Pharma and medical devices industries.
As per reports, Union Minister for Chemicals and Fertilizers Ananth Kumar conferred 1st India Pharma awards in Bengaluru on Thursday evening.
Speaking on the occasion, Ananth Kumar said that the Pharma Industry in the country is growing at a higher rate than GDP and needs to be complimented for this.
“Indian government would like domestic Pharma industry to be global leaders,” he said, adding that the government and the Pharma entrepreneurs will work together as team Pharma India, with the aim of serving millions of ailing people. He also assured full support to the industry.
The awards constituted by Department of Pharmaceuticals were given to outstanding Pharma Industries to motivate Indian Pharma and medical devices industries. The winner of the awards are:
CATEGORY OF AWARD NAME OF THE COMPANY
OVERALL INDIA PHARMA EXCELLENCE AWARD CADILA HEALTHCARE LIMITED
INDIA PHARMA LEADER AWARD GLENN SALDANA, CHAIRMAN & MANAGING DIRECTOR, GLENMARK PHARMACEUTICALS LIMITED
INDIA PHARMA COMPANY OF THE YEAR AWARD LUPIN LIMITED
INDIA PHARMA BULK DRUG COMPANY OF THE YEAR AWARD SMS PHARMACEUTICALS LTD
INDIA PHARMA INNOVATION OF THE YEAR AWARD CADILA HEATHCARE LIMITED
INDIA PHARMA RESEARCH AND DEVELOPMENT ACHIEVEMENT AWARD SUN PHARMACEUTICALS INDUSTRIES LTD
INDIA PHARMA CORPORATE SOCIAL RESPONSIBILITY PROGRAMME OF THE YEAR AWARD ABBOTT INDIA LIMTED
INDIA PHARMA MEDICAL DEVICES COMPANY OF THE YEAR AWARD HARSORIA HEALTHCARE PVT LTD
INDIA PHARMA EXPORT COMPANY OF THE YEAR AWARD CAMUS PHARMA PVT LTD
INDIA PHARMA BULK DRUG EXPORT COMPANY OF THE YEAR SMS PHARMACEUTICALS LTD
INDIA PHARMA MEDICAL DEVICES EXPORT COMPANY OF THE YEAR AWARD SCOPE MEDICAL DEVICES PVT LTD
SPECIAL AWARD: PHARMA PSU COMPANY OF THE YEAR AWARD KARNATAKA ANTIBIOTICS AND PHARMACEUTICALS LIMITED, A PSU UNDER DEPARTMENT OF PHARMACEUTICALS
CLIP
India Pharma Awards given by Minister of Chemicals and …
pib.nic.in/newsite/PrintRelease.aspx?relid=134291
Jan 8, 2016 – OVERALL INDIA PHARMA EXCELLENCE AWARD. CADILA HEALTHCARE LIMITED. INDIA PHARMA LEADER AWARD. SHRI GLENN …
Press Information Bureau
Government of India
Ministry of Chemicals and Fertilizers
08-January-2016 12:49 IST
India Pharma Awards given by Minister of Chemicals and Fertilizers
The Union Minister for Chemicals and Fertilizers, Shri Ananth Kumar gave away the 1st India Pharma awards in Bengaluru on Thursday evening. The awards constituted by Department of Pharmaceuticals were given to outstanding Pharma Industries to motivate Indian Pharma and medical devices industries. The winner of the awards are:
| CATEGORY OF AWARD | NAME OF THE COMPANY |
| OVERALL INDIA PHARMA EXCELLENCE AWARD | CADILA HEALTHCARE LIMITED |
| INDIA PHARMA LEADER AWARD | SHRI GLENN SALDANA, CHAIRMAN & MANAGING DIRECTOR, GLENMARK PHARMACEUTICALS LIMITED |
| INDIA PHARMA COMPANY OF THE YEAR AWARD | LUPIN LIMITED |
| INDIA PHARMA BULK DRUG COMPANY OF THE YEAR AWARD | SMS PHARMACEUTICALS LTD |
| INDIA PHARMA INNOVATION OF THE YEAR AWARD | CADILA HEATHCARE LIMITED |
| INDIA PHARMA RESEARCH AND DEVELOPMENT ACHIEVEMENT AWARD | SUN PHARMACEUTICALS INDUSTRIES LTD |
| INDIA PHARMA CORPORATE SOCIAL RESPONSIBILITY PROGRAMME OF THE YEAR AWARD | ABBOTT INDIA LIMTED |
| INDIA PHARMA MEDICAL DEVICES COMPANY OF THE YEAR AWARD | HARSORIA HEALTHCARE PVT LTD |
| INDIA PHARMA EXPORT COMPANY OF THE YEAR AWARD | CAMUS PHARMA PVT LTD |
| INDIA PHARMA BULK DRUG EXPORT COMPANY OF THE YEAR | SMS PHARMACEUTICALS LTD |
| INDIA PHARMA MEDICAL DEVICES EXPORT COMPANY OF THE YEAR AWARD | SCOPE MEDICAL DEVICES PVT LTD |
| SPECIAL AWARD: PHARMA PSU COMPANY OF THE YEAR AWARD | KARNATAKA ANTIBIOTICS AND PHARMACEUTICALS LIMITED, A PSU UNDER DEPARTMENT OF PHARMACEUTICALS |
Speaking on the occasion the Shri Ananth Kumar said that the Pharma Industry in the country is growing at a higher rate than GDP and needs to be complimented for this. He said that the government would like domestic Pharma industry to be global leaders. He said that the government and the Pharma entrepreneurs will work together as team Pharma India, with the aim of serving millions of ailing people. Shri Ananth Kumar assured full support to the industry.

References
Click to access First%20India%20Pharma%20Awards%202015.pdf
http://pib.nic.in/newsite/PrintRelease.aspx?relid=134291
http://www.glenmarkpharma.com/common/pdf/Glenn_Saldanha-Profile.pdf
Click to access First%20India%20Pharma%20Awards%202015%20%20Final.pdf
Click to access First%20India%20Pharma%20Awards%202015.pdf
////
WO 2016012938, New patent, LINACLOTIDE, DR. REDDY’S LABORATORIES LIMITED,
WO2016012938, IMPROVED PROCESS FOR PREPARATION OF AMORPHOUS LINACLOTIDE
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No 3, Banjara Hills, Telangana, INDIA Hyderabad 500034 (IN)
KALITA, Dipak; (IN).
NIVRUTTI, Ramrao Jogdand; (IN).
BALAKUMARAN, Kesavan; (IN).
DESHMUKH, Shivshankar; (IN).
VUTUKURU, Naga Chandra Sekhar; (IN).
KASINA, Vara Prasad; (IN).
NALAMOTHU, Sivannarayana; (IN).
VILVA, Mohan Sundaram; (IN).
KHAN, Rashid Abdul Rehman; (IN).
TIRUMALAREDDY, Ramreddy; (IN).
MUSTOORI, Sairam; (IN)
![]()
The present application relates to an improved process for the formation of disulfide bonds in linaclotide. The present application also relates to an improved process for the purification of linaclotide.
The present application relates to an improved process for the preparation of amorphous linaclotide. Specifically, the present application relates to an improved process for the formation of disulfide bonds in linaclotide. The present application further relates to a purification process for the preparation of amorphous linaclotide.
INTRODUCTION
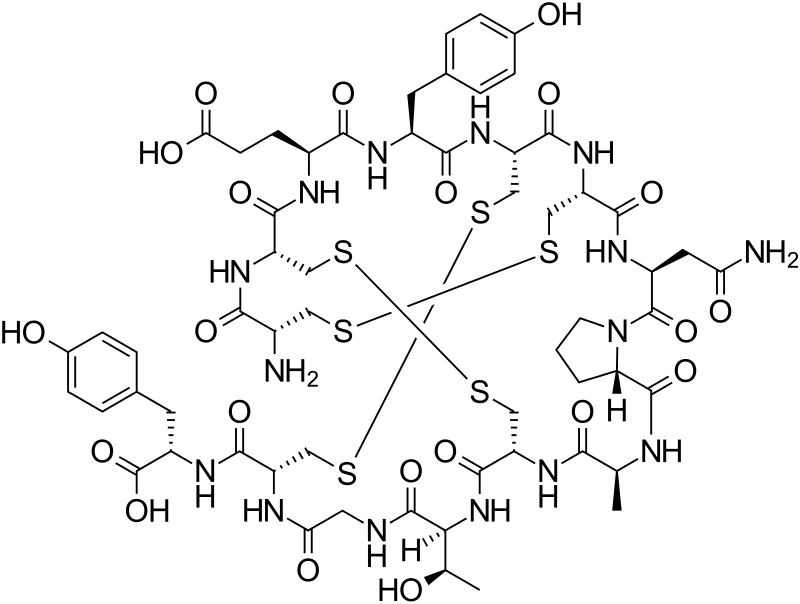
Linaclotide is a 14-residue peptide which is an agonist of the guanylate cyclase type-C receptor. Linaclotide may be used for the treatment of chronic constipation and irritable bowel syndrome. Structurally, linaclotide has three disulfide bonds and they are present between Cys1-Cys6, Cys2-Cys-10 and Cys5-Cys13. The structure of linaclotide is shown below:
1 2 3 4 5 6 7 8- 9 10 11 12 13 14

Benitez et al. Peptide Science, 2010, Vol. 96, No. 1 , 69-80 discloses a process for the preparation of linaclotide. The process involves the use of 2-chlorotrityl (CTC) resin and 9-fluorenylmethoxycarbonyl (Fmoc) chemistry. The Cys residues are protected by Trt (trityl) group. The amino acids are coupled to one another using 3 equivalents of 1 -[bis(dimethylamino)methylene]-6-chloro-1 H-benzotriazolium hexafluorophosphate 3-oxide (HCTU) as coupling agent and 6 equivalents of diisoprpylethylamine (DIEA) as base in dimethylformamide (DMF). The Fmoc group is removed using piperidine-DMF (1 :4). The Cys residues are incorporated using 3 equivalents of Ν,Ν’-diisopropylcarbodiimide (DIPCDI) as coupling agent and 3 equivalents of 1 -hydroxybenzotriazole (HOBt) as an activating agent. After the elongation of the peptide chain, the peptide was cleaved from the solid support (CTC resin) by first treating with 1 % trifluoroacetic acid (TFA) and then with a mixture of TFA, triisoprpylsilane (TIS) and water in the ratio of 95:2.5:2.5. The disulfide bonds are prepared by subjecting the linear peptide to air oxidation in sodium dihydrogen phosphate (100 mM) and guanidine hydrochloride buffer (2 mM).
US2010/261877A1 discloses a process for purification of linaclotide. The process involves first purification of crude peptide by reverse-phase chromatographic purification followed by concentrating the purified pools and dissolving the purified linaclotide in aqueous-isopropanol or aqueous-ethanol and spray-drying the solution to afford pure Linaclotide.
The synthesis of a peptide containing disulfide bridges is difficult for two main reasons; one is potential risk of racemization during the formation of linear chain and the other is mis-folding of the disulfide bridges. Hence, there is a need in the art to a cost-effective process for the preparation of pure linaclotide.
EXAMPLES
Example 1 : Preparation of Crude Linaclotide using polyvinyl polymer bound complex of sulfur trioxide-pyridine
The linear chain of peptide of formula (I) (0.1 g) and polyvinyl polymer bound complex of sulfur trioxide-pyridine (0.062 g) was charged in water (100 mL). The pH of the reaction mass was adjusted to 8.5 to 9 by addition of ammonium hydroxide. The reaction mass was stirred at 25 °C for 15 hours and trifluoroacetic acid (2 mL) was added to the reaction mass to adjust the pH up to 2-2.5. The reaction mass was stirred for 3 hours at the same temperature to afford crude linaclotide.
HPLC Purity: 59.92%
Example 2: Preparation of Crude Linaclotide using DMSO in water
The pH of water (100 ml_) was adjusted to 9.1 by the addition of aqueous ammonia. DMSO (1 ml_) and linear chain of peptide of formula (I) (100 mg) were charged. The reaction mass was stirred for 17 hours at 25 °C and acidified with trifluoroacetic acid to pH 1 .9 and stirred for 8 hours at the same temperature to afford crude linaclotide.
HPLC Purity: 57%
Example 3: Preparation of Crude Linaclotide using DMSO in water
The pH of water (1500 ml_) was adjusted to 9 by the addition of aqueous ammonia. DMSO (15 ml_) and linear chain of peptide of formula (I) (15 g) were charged. The reaction mass was stirred for 17 hours at 25 °C and acidified with acetic acid to pH 1 .9 and stirred for 8 hours at the same temperature to obtain crude linaclotide.
HPLC Purity: 46.02%
Example 4: Preparation of Crude Linaclotide in water
To a mixture of water (1900 mL) and ammonium sulfate (26.4 g), ammonium hydroxide was added drop wise to adjust the pH up to 8.5. Linear chain of peptide of formula (I) (26.4 g) was added and the reaction mass was stirred for 8 hours at 25 °C. Trifluoroacetic acid (20 mL) was added drop wise and the reaction mixture was stirred for 15 hours at 25 °C to afford crude linaclotide.
HPLC Purity: 63.38%
Example 5: Preparation of Crude Linaclotide using a complex of pyridine-sulfur trioxide
Linear chain of peptide of formula (I) (0.2 g) was added to water (250 mL) and the pH of the reaction mass was adjusted to 8.91 by the drop wise addition of aqueous ammonia. A complex of pyridine-sulfur trioxide (0.124 g) was added to the reaction mass and stirred for 16 hours at 25 °C. Another lot of complex of pyridine-sulfur trioxide (0.124 g) was added to the reaction mass and stirred for 5 hours at 25 °C to afford crude linaclotide.
Example 6: Preparation of Crude Linaclotide using guanidine hydrochloride
To a solution of sodium bicarbonate (0.89 g) in water (100 mL), cysteine (0.363 g), cysteine (0.072 g) and guanidine hydrochloride (9.50 g) were charged. Acetonitrile (15 mL) and linear chain of peptide of formula (I) (0.1 g) was added to the reaction mass.
The reaction mass was stirred for 3 hours at 25 °C and trifluoroacetic acid (2 mL) was added. The reaction mass was stirred for 18 hours at the same temperature. Another lot of trifluoroacetic acid (2 mL) was added to the reaction mass and stirred for 18 hours at the same temperature to afford crude linaclotide.
Example 7: Preparation of Crude Linaclotide using Clear-OX™
Pre-conditioned Clear-Ox™ (0.5 g) was added to a solution of ammonium sulfate (1 .32 g) in water (100 mL) of pH 8.5, adjusted by addition of ammonium hydroxide. The linear chain of peptide of formula (I) (0.1 g) was added to the reaction mass and stirred for 3 hours at 25 °C. Another lot of Pre-conditioned Clear-Ox™ (0.5 g) was added to the reaction mass and stirred for 1 .30 hours. Trifluoroacetic acid (2 mL) was added to the reaction mass and stirred for 16 hours at the same temperature to afford crude linaclotide.
HPLC Purity: 67.5%
Example 8: Preparation of Crude Linaclotide using reduced Glutathione
To a mixture of ammonium sulphate (5.28 g) in water (400 mL) and isopropyl alcohol (400 mL), reduced glutathione (0.248 g) was added and the pH was adjusted to 8.5 by using aqueous ammonia. The linear chain of peptide of formula (I) (0.81 g) was added to the reaction mixture and stirred at ambient temperature for 17 hours. Isopropyl alcohol was evaporated under vacuum to afford crude linaclotide.
HPLC Purity: 69.56%%
Example 9: Preparation of Crude Linaclotide using DMSO and air bubbling
To a mixture of water (95 mL) and ammonium sulfate (1 .32 g), ammonium hydroxide was added drop wise to adjust the pH up to 8.5. Linear chain of peptide of formula (I) (0.1 g) and DMSO (5 mL) was added and the reaction mass was stirred for 20 hours at 25 °C with continuous air bubbling. Trifluoroacetic acid (2 mL) was added to the reaction mass and stirred for 19 hours with continuous air bubbling at the same temperature to afford the title product.
HPLC Purity: 59.1 1 %
Example 10: Preparation of Crude Linaclotide using solid supported TEMPO
To a mixture of water (100 mL) and silica bound TEMPO (0.01 g), linear chain of peptide of formula (I) (0.1 g) and sodium hypochlorite solution (1 mL) were added and the reaction mass was stirred 18 hours at 25 °C. Another lot of sodium hypochlorite solution (0.5 mL) was added to the reaction mass and stirred for further 7 hours at the same temperature to afford title product.
HPLC Purity: 42.70%………………see more in patent
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
L-Cysteinyl-L-cysteinyl-L-glutamyl-L-tyrosyl-L-cysteinyl-L-cysteinyl-L-asparaginyl-L-prolyl-L-alanyl-L-cysteinyl-L-threonylglycyl-L-cysteinyl-L-tyrosine cyclo(1-6),(2-10),(5-13)-tris(disulfide)
|
|
| Clinical data | |
| Trade names | Linzess |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 851199-59-2 |
| ATC code | A06AX04 |
| PubChem | CID 16158208 |
| IUPHAR/BPS | 5017 |
| ChemSpider | 17314504 |
| UNII | N0TXR0XR5X |
| KEGG | D09355 |
| Chemical data | |
| Formula | C59H79N15O21S6 |
| Molar mass | 1526.74 g/mol |
///////WO 2016012938, DR. REDDY’S LABORATORIES LIMITED , Telangana, INDIA , Hyderabad, LINACLOTIDE, new patent
smiles O=C(O)[C@@H](NC(=O)[C@H]4NC(=O)CNC(=O)[C@@H](NC(=O)[C@H]2NC(=O)[C@@H](NC(=O)[C@H]5N(C(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](N)CSSC1)CSSC2)CCC(=O)O)Cc3ccc(O)cc3)CSSC4)CC(=O)N)CCC5)C)[C@H](O)C)Cc6ccc(O)cc6
WO 2016012539, Tadalafil , New patent, KRKA, D.D., NOVO MESTO

WO 2016012539, A PROCESS FOR THE PREPARATION OF CGMP-PHOSPHODIESTERASE INHIBITOR AND ORAL PHARMACEUTICAL FORMULATION COMPRISING TADALAFIL CO-PRECIPITATES
KRKA, D.D., NOVO MESTO [SI/SI]; Smarjeska cesta 6 8000 Novo mesto (SI)
BARIC, Matej; (SI).
BENKIC, Primoz; (SI).
BOMBEK, Sergeja; (SI).
KRASOVEC, Dusan; (SI).
SKRABANJA, Vida; (SI).
VRECER, Franc; (SI).
BUKOVEC, Polona; (SI).
HUDOVORNIK, Grega; (SI).
KROSELJ, Vesna; (SI)
![]()
The present Invention relates to an improved process for preparation of tadalafil and crystallization and/or purification thereof, wherein the processes are conducted at increased pressure. The invention relates also to a process for preparation of tadalafil co-precipitates and to a solid pharmaceutical composition comprising tadalafil co-precipitates and at least one water soluble diluent and/or water insoluble non-swellable diluent, wherein the composition is substantially free of water insoluble swellable diluents
The present invention relates to a process for the preparation of CGMP-phosphodiesterase inhibitor, particularly tadalafil, a method for production co-precipitate thereof and to solid oral pharmaceutical formulations comprising tadalafil co-precipitate.
Tadalafil, chemically known as (6R-trans)-6-(1,3-benzodioxol-5-il)-2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino.1′, 2′:1,6]pyrido[3,4-b]indole-1,4-dione, is a potent and selective inhibitor of the cyclic guanosine monophosphate (cGMP) – specific phosphodiesterase enzyme PDE5. It is shown below as structural formula I:

Tadalafil is marketed under the tradename CIALIS* and is used for the treatment of erectile dysfunction. The product is available as a film-coated tablet for oral administration containing 2.5, 5, 10 and 20 mg of active ingredient and the following inactive ingredients: lactose monohydrate, hydroxypropylcellulose, sodium lauryl sulfate, croscarmellose sodium, microcrystaliine cellulose, magnesium stearate, hypromellose, triacetin, titanium dioxide (E171), iron oxide (E172) and talc.
Tadalafil is practically insoluble in water and very slightly soluble in organic solvent such as ethanol, methanol and acetone.
Problems associated with low solubility of tadalafil in ethanol and most of other organic solvents resulted in the need of large quantities of solvents required to perform synthesis and crystallization of tadalafil at industrial scale, which have unwanted technological, environmental and economical impact.
US Patent No. 5 859 006 describes the synthesis of the tadalafil and its intermediate (A) which involves reacting D-tryptophan methyl ester with a piperonal in the presence of dichloromethane and trifluoroacetic acid which provides a mixture of desired cis and undesired trans isomer of intermediate A with poor selectivity. The isomers are further separated by column chromatography. The cis isomer is further reacted with chloroacetyl chloride in chloroform, providing another intermediate of tadalafil (B) which reacted with methylamine to give tadalafil of formula (1) in methanol slurry requiring an additional purification step by flash chromatography.
An improved process in the synthesis of tadalafil via modified Pictet-Spengler reaction is described in WO 04/011463 in which D-tryptophan methyl ester hydrochloride and piperonal are condensed in anhydrous isopropyl alcohol to provide hydrochloride of intermediate A. After isolation of desirable cis isomer, the product is further reacted with chloroacetyl chloride and then with methylamine in THF to give tadalafil.
Therefore there still exists a need for an improved process for a synthesis and purification of tadalafil, which would overcome the disadvantages of the prior art processes.
Low solubility of tadalafil in aqueous solutions is further disadvantageous because in vivo absorption is typically dissolution rate-limited which may result in poor bioavailability of the drug. Different approaches in the processes of preparation of pharmaceutical compositions have been applied to overcome the poor solubility.
For example, EP 1 200 092 Bl describes a pharmaceutical composition of free drug particulate form of tadalafil wherein at least 90% of the particles have a particle size of less than about 40 μm as well as composition comprising tadalafil, wherein the compound is present as solid particles not embedded in polymeric co-precipitate. Apparently, preferably at least 90% of the particles have a particle size of less than 10 μm. The technological drawback of such small particles is possible chargeability and secondary agglomeration due to increased surface energy which can cause problems during the micronization and further processing.
WO 2008/134557 describes another approach to overcome the low-solubility problem by pharmaceutical composition comprising starch and tadalafil characterized by particle size having d(90) greater than 40 μm wherein the weight ratio of starch to tadalafil is 4.5 to 1 or greater. Apparently, the preferred ratio is at least 15 to 1.
Yet another approach to overcome the low-solubility problem is to use a “co-precipitate” of tadalafil and a carrier or excipient. For example, EP 828 479 Bl describes a solvent based process wherein tadalafil and a carrier are co-precipitated with a medium in which the tadalafil and carrier are substantially insoluble. EP 828 479 describes a solvent based process wherein tadalafil and hydroxypropyl methylcellulose phthalate are co-precipitated in weakly acidic medium from a combination of non-aqueous water miscible solvent and water. However, pharmaceutical composition prepared according to EP 828479 exhibit deviations in release rate of tadalafil which was due to poor reproducibility of a process for preparation of co-precipitate. It was found that precipitation in acidic media causes unwanted degradation of hydroxypropyl methylcellulose phthalate and that precipitation at higher temperatures does not produce desired product.
WO 2008/005039 also describes a solid composite including tadalafil being in intimate contact with a carrier. The carriers include hydrophilic polymers such as povidone, cellulose derivatives, polyethylene glycol and polymethacrylates. The compositions are prepared by combining tadalafil with hydrophilic polymer and removal of the solvent by evaporation.
WO 2010/115886 describes an adsorbate comprising poorly soluble active ingredient with a particulate and/or porous carrier wherein the adsorbate is prepared by using non-polar solvent. Apparently, the solvents used are selected from the group of chlorinated hydrocarbon (dichloromethane or trichloromethane), diisopropylether and hexane, which is also the main drawback of this solution.
Co-precipitates of phosphodiesterase-5-inhibitor and copolymer of different acrylic acid derivatives are described in WO 2011/012217. The procedures described involve the use of tetrahydrofurane.
Poor solubility can also be solved with co-crystals. WO 2010/099323 discloses crystalline molecular complexes of tadalafil with co-former selected from the group of a short to medium chain organic acids, alcohols and amines.
WO 2012/107541 and WO 2012/107092 disclose co-granulate of tadalafil with cyclodextrines.
WO 2014/003677 discloses a pharmaceutical composition comprising solid dispersion particles containing tadalafil and a dispersing component, which composition further comprises a solubilizer.
Based on the above, there is still a need for an improved dosage form containing tadalafil and improved technological process for the preparation thereof.
The process for preparing tadalafil according to a preferred embodiment of the present invention is disclosed in Scheme 1.
Scheme 1

Example 1: Synthesis of tadalafil intermediate B via intermediate A
D-tryptophan methyl ester hydrochloride (9g) and piperonai (6g) was suspended in acetonitrile (60mL). The reaction mixture was stirred and heated at about 105*C for three to five hours in an autoclave. The reaction suspension was cooled to ambient temperature and aqueous solution (60m L) of sodium carbonate (4.1g) was added. The mixture was then cooled in an ice bath and the solution of chloroacetyl chloride (5.1mL) in acetonitrile was slowly added to the reaction mixture. A solid was obtained, filtered and washed twice with aqueous solution of acetonitrile. The crude product was dried, and intermediate B (13.4g) with a purity of 97% (HPLC area%) was obtained.
Example 1A:
D-tryptophan methyl ester hydrochloride (8.2kg) and piperonai (5.1kg) was suspended in acetonitrile (55L). The reaction mixture was stirred and heated at about to 105″C for three hours in the reactor vessel. The reaction suspension was cooled to ambient temperature and aqueous solution (55L) of sodium carbonate (4.8kg) was added. The mixture was then cooled in an ice bath and the solution of chloroacetyl chloride (5.2L) was slowly added to the reaction mixture at 5-10°C. A solid was obtained, centrifuged and washed twice with aqueous solution of acetonitrile (2x 121). The crude product was dried at temperature up to 50″C, and intermediate B (12.3kg) with a purity of 98% (HPLC area%) was obtained.
Comparative example 1:
D-tryptophan methyl ester hydrochloride (9.0g) and piperonai (5.84g) was suspended in acetonitrile (60mL). The reaction mixture was stirred and heated at about to 80-85’C for 15-20 hours in the reactor vessel. The reaction suspension was cooled to 0-10°C. The Intermediate A was then isolated on centrifuge and was dried at temperature up to 60°C.
The isolated dried Intermediate A (12,8g) was charged into reactor and suspended with ethyl acetate. The aqueous solution (60mL) of sodium carbonate (5.3g) was added to precooied suspension of Intermediate A. The chloroacetyl chloride (3.4mL) was slowly added to the above reaction mixture. The solid was obtained, centrifuge and washed twice with water (2x 10mL). The crude product was dried at temperature up to 70°C, and intermediate B (11.8g) with a purity of 99% (HPLC area%) was obtained.
Example 2: Synthesis oftadalafil
Intermediate B (4g) obtained in Example 1 and 40% aqueous methylamine solution (1.6mL) were dissolved in 70% aqueous solution of 2-propanol (120mL) while heating in a closed reaction vessel above the reflux temperature (110-120°C) for two to five hours. The solution was hot filtered and cooled on an ice bath. The precipitated product was filtered and dried. The purity of the product was 99.9% (HPLC area%) and the particle distribution of the product was D(90) of about 144 microns.
Example 2A: Synthesis of tadalaf il
Intermediate B (12.3kg) obtained in Example 1A and 40% aqueous methylamine solution (4.76L) were dissolved in 70% aqueous solution of 2-propanol (402L) while heating in a closed reaction vessel above the reflux temperature (110-120°C) for three hours. The solution was hot filtered and cooled on an ice bath. The precipitated product was filtered and dried. The final product (9.8kg) with a purity of more than 99.99% (HPLC area%) and the particle distribution of the product was D(90) of about 155 microns was obtained.
Comparative example 2:
Intermediate B (10g) obtained in the above comparative example 1 and 31% ethanolic methylamine solution (12.3mL) were suspended in absolute ethanol (150mL). The suspension
was heated up to 55°C for 3 – 6 hours. The suspension was cooled on an ice bath. The product was filtered and dried. The crude product (8.22g) with a purity of more than 99.9% (HPLC area%) was obtained and crystallized from hot DMSO solution. The product Is crystallized with addition of water.
Example 3: Recrystallization of tadalaf il
Tadalafil (700g) (99% purity) was suspended in 70% aqueous solution of 2-propanol (24.6L) and suspension was heated to about 110°C in an autoclave at pressure of 0.31MPa until the material was dissolved. The obtained solution was then hot filtrated and cooled to about 10°C. The isolated tadalafil (660g) has a purity of 99.95% (HPLC area%) and the particle distribution D(90) of about 144 microns.
Example 3A: Recrystallization of tadalafil
Tadalafil (5g) (99% purity) was suspended in 70% aqueous solution of acetone (lOOmL) and suspension was heated to about 90°C in an autoclave at pressure of 0.28MPa until the material was dissolved. The obtained solution was then hot filtrated and cooled to about 10°C. The isolated tadalafil (4.44g) has a purity of 99.99% (HPLC area%).
Example 3B: Recrystallization of tadalafil
Tadalafil (4g) (99% purity) was suspended in 70% aqueous solution of acetonitrile (lOOmL) and suspension was heated to about 85°C in an autoclave at pressure of 0.2MPa until the material was dissolved. The obtained solution was then hot filtrated and cooled to about 10°C. The isolated tadalafil (3g) has a purity of 99.99% (HPLC area%).
Example 3C: Recrystallization of tadalafil
Tadalafil (5g) (99% purity) was suspended in 70% aqueous solution of tetrahydrofuran (60mL) and suspension was heated to about 120″C in an autoclave at pressure of 0.3MPa until the material was dissolved. The obtained solution was then hot filtrated and cooled to about 10°C. The isolated tadalafil has a purity of 99.99% (HPLC area%).
Comparative example 3:
Tadalafil (lg) (99% purity) was suspended in 2-propanol (200mL) and suspension was heated up to reflux temperature until the material was dissolved. The obtained solution was then hot filtrated and cooled to about lO’C. The crystallized tadalafil was centrifuged and dried in an oven at temperature up to 70°C.
Comparative Example 4: Preparation of tadalafil co-precipitate with HPMCP HP-50, Precipitation at higher temperature
Tadalafil (100 g) and hydroxypropyl methylcellulose phthalate (100 g) were dissolved in a mixture of acetone (2430m L) and water (270mL) at reflux temperature. Solution was hot filtered and added to 0.25 M HCI in water (4150mL) at 65°C. Precipitate was collected by vacuum filtration, washed with water and dried in vacuum tray dryer up to 70°C. Dry material was milled by a pin mill. HPLC assay of tadalafil was 48.5 %; average particle size of co-precipitate was 53 μm, specific surface area 2.5 m2/g-
Example 5: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (1 kg) and hydroxypropyl methylcellulose phthalate (1 kg) were dissolved in mixture of acetone (20L) and water (3 L) at 54°C and under pressure O.lMPa. Solution was hot filtered and added to water (42 L) at 2°C. Suspension was heated up to reflux and acetone was distilled off. Tadalafil co-precipitate was collected by pressure filtration and dried in vacuum dryer. Dry material was milled by a pin mill. HPLC assay of tadalafil was 53.5%.
Example 6: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (1 kg) and hydroxypropyl methylcellulose phthalate (1 kg) were dissolved in mixture of acetone (20 L) and water (3 L) at 54°C and under pressure O.lMPa. Solution was hot filtered and added to water (42 L) at 2°C. Suspension was heated up to reflux and acetone was distilled off. Tadalafil co-precipitate was collected by centrifuge and dried in a fluid bed dryer. Dry material was milled by a pin mill. HPLC assay of tadalafil was 52.5 %.
3
Example 7: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (0.786 kg) and hydroxypropyl methylcellulose phthaiate (1.140 kg) were dissolved in a mixture of acetone (24L) and water (2.3 L) at 54°C and under pressure 0.1MPa. Solution was filtered hot and added to water (42 L) at 2°C. Suspension was collected by centrifuge and dried in a vacuum tray dryer up to 70°C. Dry material was milled by a pin mill. HPLC assay of tadalafil was 43.5 %, average particle size of co-precipitate was 49 μm, specific surface area 31.0 m2/g-
Example 8: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (2 g) and hydroxypropyl methylcellulose phthaiate HP 50 (2 g) were dissolved in a mixture of acetone (48.5mL) and water (5.5mL) at reflux temperature. To obtained solution crospovidone (lg) was added. Obtained suspension was co-precipitated in water (83mL) at 2°C. Obtained material was collected with a vacuum filter and dried in vacuum dryer up to 90°C. HPLC assay of tadalafil 39.9%. Yield was 90%.
Example 9: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (2 g) and hydroxypropyl methylcellulose phthaiate HP 50 (2 g) were dissolved in a mixture of acetone (54mL) and methanol (19mL) at reflux temperature. To obtained solution crospovidone (lg) was added. Obtained suspension was co-precipitated in heptane (83mL) at 0°C. Obtained material was collected with a vacuum filter and dried in vacuum dryer up to 50°C. HPLC assay of tadalafil was 36.1 %. Yield was 90%.
Example 10: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadalafil (2 g) and hydroxypropyl methylcellulose phthaiate HP 50 (2 g) were dissolved in a mixture of aceton (54mL) and methanol (19mL) at reflux temperature. Obtained solution was co-precipitated in heptane (83mL) at 0°C. Obtained material was collected with a vacuum filter and dried in vacuum dryer up to 50°C. HPLC assay of tadalafil was 36.1 %. Yield was 90%.
Example 11: Preparation of tadalafil co-precipitate with HPMCP HP-50
Tadaiafil (1.3 kg) and hydroxypropyl methylcellulose phthalate {1.53 kg) were dissolved in mixture of acetone (32 L) and water (4 L) at 54°C and 1000 mbar. Solution was hot filtered and added to water (54 L) at 2°C. Tadalafil co-precipitate was collected by decanter centrifuge and dried in a vacuum drier. Dry material (2.4kg) was milled in a pin mill. HPLC assay of tadalafil was 48.8 %; average particle size of co-precipitate was 54 μm and specific surface area 26.1 m2/g<
Example 12: Preparation of tadalafil co-precipitate with hydroxypropyl cellulose
Tadalafil (3g) and Klucel ELF (3g) was dissolved in a mixture of acetone (73mL) and water (8mL) at 50°C. Solution was hot filtered and added to 125mL water at 90°C. After that acetone was distilled off at 65°C and suspension was stirred for additional hour. Precipitated material was filtered using preheated filter funnel and dried at 80°C. Yield 3.8 g, HPLC assay was 50.0%.
Example 13: Preparation of tadalafil co-precipitate with hydroxypropyl cellulose
Tadaiafil (3g) and Klucel ELF (3g) was dissolved in a mixture of acetone (73mL) and water (8m L) at 50°C. Solution was hot filtered and added to 125m L water at 90°C with dissolved lactose (14g) at 90°C. After that acetone was distilled off at 65°C and suspension was stirred for additional hour. Precipitated material was filtered using preheated filter funnel and dried at 80°C. Yield 5 g, HPLC assay was 48.8%.
Examples of tablets prepared according to the present Invention
Example Fl: Tablets containing tadalafil co-precipitate with HPMCP HP-50 prepared in accordance with Example 11 with water soluble mannitol and without swellable water insoluble diluents

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with mannitol, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Example F2: Tablets containing tadalafil co-precipitate with HPC prepared in accordance with Example 13 with water soluble mannitol and without swellable water insoluble diluents

Tadalafil co-precipitate with HPC was homogeneously mixed with mannitol, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Example F3: Tablets containing tadalafil co-precipitate with HPMCP with water soluble spray-dried lactose and without swellable water insoluble diluents

Tadaiafil co-precipitate with HPMCP was homogeneously mixed with spray-dried lactose, starch 1500 and sodium lauryi sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets.
Example F4: Tablets containing tadalafil co-precipitate with HPMCP with water insoluble non-swellable anhydrous dibasic calcium phosphate and without swellable water insoluble diluents

Tadalafil co-precipitate with HPMCP was homogeneously mixed with calcium phosphate, croscarmellose sodium and sodium lauryi sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets.
Comparative examples of tablets containing microcrvstalline cellulose
Comparative example F5: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water soluble mannitol and water insoluble swellable microcrvstalline cellulose as diluent 
Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with mannitol, microcrystalline cellulose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F6: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water soluble lactose anhydrous and water insoluble swellable microcrystalline cellulose as diluent

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with lactose anhydrous, microcrystalline cellulose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F7: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water soluble lactose monohydrate and spray dried lactose and water insoluble swellable microcrystalline cellulose as diluent

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with lactose monohydrate, spray dried lactose, microcrystalline cellulose, croscarmeilose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F8: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water insoluble non-swellable calcium phosphate and water insoluble swellable microcrystalline cellulose as diluent

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with calcium phosphate, microcrystalline cellulose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F9: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with only water insoluble swellable microcrystalline cellulose as diluent

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with microcrystalline cellulose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example is shown in Figure 1.
Comparative example F10: Tablets containing tadalafil co-precipitate with HPMCP HP-50 with water insoluble swellable microcrystalline cellulose and cellactose as diluents

Tadalafil co-precipitate with HPMCP HP-50 was homogeneously mixed with microcrystalline cellulose, cellactose, croscarmellose sodium and sodium lauryl sulphate. The magnesium stearate was added and mixed. The resultant blend was compressed into tablets. Dissolution profile of the example F10 is shown in Figure 2, together with dissolution profiles of the same sample, taken after two months at 22°C and 60% RH.
In comparison, dissolution profile of composition according to invention is unaffected by storage at 40°C/75% for one month (Figure 2).
The aforementioned tablet formulations were film-coated with a film-coating dispersion containing:

Figures 1 and 2 show dissolution profiles of tablet formulations comprising tadalafil co-precipitates prepared according to listed examples. Dissolution conditions comprise: basket apparatus (USP I), 100 RPM, 0.1M HCI + 0.2% SDS, 900 mL
![]()
Krka, tovarna zdravil, d.d., Novo mesto


/////////WO 2016012539, KRKA, D.D., NOVO MESTO, tadalafil, new patent
When can a Chemical Substance be qualified as a “New Active Substance”? The New Reflection Paper of the EMA gives Information

When can a Chemical Substance be qualified as a “New Active Substance”? The New Reflection Paper of the EMA gives Information
A chemical structure with a therapeutic moiety for which no authorisation dossier has been submitted so far and which is – from a chemical structure point of view – not related to any other authorised substances is per se a “NAS” (New Active Substance). But what about a physiologically active molecule present for example in different salts or esters? In which cases do the different derivatives of an effective substance have the NAS status?
The EMA provides clarification to these questions in a new Reflection Paper which was published on 19 January this year. The document entitled “Reflection paper on the chemical structure and properties criteria to be considered for the evaluation of new active substance (NAS) status of chemical substances” describes the criteria according to which isomers, mixtures of isomers, complexes, derivatives, esters, ethers, salts and other solid forms of physiologically active molecules can be classified as “NAS “. If an applicant claims the NAS status of a substance to the regulatory authority in the centralised (CP) or decentralised procedure (MRP/DCP), the authority will first check whether the claim is justified. Afterwards – in case of a positive decision – the usual review of the application dossier will be performed.
The applicant can refer to the criteria described in this Reflection Paper to substantiate his/ her claim of a NAS status. In general, the evidence has to be brought for the derivative in question that it differs significantly in properties with regard to efficacy and /or safety from the already approved active substance.
The scope of this Reflection Papers covers neither biological and biotechnological active substances nor active substances to be included in radiopharmaceuticals.
//////
New Website ECA Validation Group: Version 02 of ECA´s Good Practice Guide on Validation online available

The ECA Validation Group was founded in autumn 2011 by representatives of the pharmaceutical industry after ECA´s 4th European GMP Conference. The mission of the group is to assemble knowledge on Validation, for example by continuously developing ECA´s Process Validation Good Practice Guide. Now the Validation Group launched a new website.
Since the ECA Foundation was established back in 1999 its mission has been to provide support to the Pharmaceutical Industry and Regulators to promote the move towards a harmonised set of GMP and regulatory guidelines by providing information and interpretation of new or updated guidances. For that purpose the ECA has initiated and established various working and interest groups concentrating on different topics.
The ECA Validation Group was founded in autumn 2011 by representatives of the pharmaceutical industry after ECA´s 4th European GMP Conference. This group’s mission is to assemble knowledge on Validation, for example by continuously developing ECA´s Process Validation Good Practice Guide.
Now the group launched its new website to provide members and those interested with information and practical tools. Here’s what you can find on the new website:
- Current News
- A news archive
- Training Courses and Validation Conferences
- ECA´s Process Validation Good Practice Guide
- Discussion Forum
- Presentations
- Useful links
- Q&A section
- Membership information
Members of the group have now the opportunity to download the version 2 of ECA´s Good Practice Guide on Validation free of charge. On 174 pages the revised Good Practice Guide comprises the main elements of the new validation approach (“what to do”). On the other hand, it also serves as a supporting guide for the implementation (“how to do”).
To find out more we invite you to visit the ECA´s Validation Group new website.
//////
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

