
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

WO 2016024243, New patent, Dr Reddy’s Laboratories Ltd, Fidaxomicin

![]()
WO 2016024243, New patent, Dr Reddy’s Laboratories Ltd, Fidaxomicin
WO2016024243, FIDAXOMICIN POLYMORPHS AND PROCESSES FOR THEIR PREPARATION
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills, Telangana State, India Hyderabad 500034 (IN)
CHENNURU, Ramanaiah; (IN).
PEDDY, Vishweshwar; (IN).
RAMAKRISHNAN, Srividya; (IN)
Aspects of the present application relate to crystalline forms of Fidaxomicin IV, V & VI and processes for their preparation. Further aspects relate to pharmaceutical compositions comprising these polymorphic forms of fidaxomicin

Fidaxomicin (also known as OPT-80 and PAR-101 ) is a novel antibiotic agent and the first representative of a new class of antibacterials called macrocycles. Fidaxomicin is a member of the tiacumicin family, which are complexes of 18-membered macrocyclic antibiotics naturally produced by a strain of Dactylosporangium aurantiacum isolated from a soil sample collected in Connecticut, USA. The major component of the tiacumicin complex is tiacumicin B. Optically pure R-tiacumicin B is the most active component of Fidaxomicin. The chiral center at C(19) of tiacumicinB affects biological activity, and R-tiacumicin B has an R-hydroxyl group attached at this position. The isomer displayed significantly higher activity than other tiacumicin B-related compounds and longer post-antibiotic activity.
As per WIPO publication number 2006085838, Fidaxomicin is an isomeric mixture of the configurationally distinct stereoisomers of tiacumicin B, composed of 70 to 100% of R-tiacumicin B and small quantities of related compounds, such as S-tiacumicin B and lipiarmycin A4. Fidaxomicin was produced by fermentation of the D aurantiacum subspecies hamdenensis (strain 718C-41 ). It has a narrow spectrum antibacterial profile mainly directed against Clostridium difficile and exerts a moderate activity against some other gram-positive species. Fidaxomicin is bactericidal and acts via inhibition of RNA synthesis by bacterial RNA polymerase at a distinct site from that of rifamycins. The drug product is poorly absorbed and exerts its activity in the gastrointestinal (Gl) tract, which is an advantage when used in the applied indication, treatment of C. difficile infection (CDI) (also known as C. difficile-associated disease or diarrhoea [CDAD]). Fidaxomicin is available as DIFICID oral tablet in US market. Its CAS chemical name is Oxacyclooctadeca-3,5,9, 13, 15-pentaen-2-one, 3-[[[6-deoxy-4-0-(3,5dichloro-2-ethyl-4,6-dihydroxybenzoyl)-2-0-methyl-P-D-manno pyranosyl]oxy]methyl]-12[[6-deoxy-5-C-methyl-4-0-(2-methyl-1 -oxopropyl)- -D-lyxo-hexo pyranosyl]oxy]-1 1 -ethyl-8-hydroxy-18-[(1 R)-1 -hydroxyethyl] -9,13,15-trimethyl-, (3E.5E, 8S.9E.1 1 S.12R.13E, 15E.18S)-. Structural formula (I) describes the absolute stereochemistry of fidaxomicin as determined by x-ray.

(I)
WIPO publication number 2004014295 discloses a process for preparation of Tiacumicins that comprises fermentation of Dactylosporangium aurantiacum NRRL18085 in suitable culture medium. It also provides process for isolation of tiacumicin from fermentation broth using techniques selected from the group consisting of: sieving and removing undesired material by eluting with at least one solvent or a solvent mixture; extraction with at least one solvent or a solvent mixture; Crystallization; chromatographic separation; High-Performance Liquid Chromatography (HPLC); MPLC; trituration; and extraction with saturated brine with at least one solvent or a solvent mixture. The product was isolated from /so-propyl alcohol (IPA) having a melting point of 166-169 °C.
U.S. Patent No. 7378508 B2 discloses polymorphic forms A and B of fidaxomicin, solid dosage forms of the two forms and composition thereof. As per the ‘508 patent form A is obtained from methanol water mixture and Form B is obtained from ethyl acetate.
J. Antibiotics, vol. 40(5), 575-588 (1987) discloses purification of Tiacumicins using suitable solvents wherein tiacumicin B exhibited a melting point of 143-145 °C.
PCT application WO2013170142A1 describes three crystalline forms of Fidaxomicn namely, Form-Z, Form-Z1 and Form-C. IN2650/CHE/2013 describes 6 crystalline polymorphic forms of Fidaxomicin namely, Forms I, Form la, Form II, Form Ha, Form III and Form Ilia).
The occurrence of different crystal forms, i.e., polymorphism, is a property of some compounds. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physico-chemical properties.
Polymorphs are different solid materials having the same molecular structure but different molecular arrangement in the crystal lattice, yet having distinct physico-chemical properties when compared to other polymorphs of the same molecular structure. The discovery of new polymorphs and solvates of a pharmaceutical active compound provides an opportunity to improve the performance of a drug product in terms of its bioavailability or release profile in vivo, or it may have improved stability or advantageous handling properties. Polymorphism is an unpredictable property of any given compound. This subject has been reviewed in recent articles, including A. Goho, “Tricky Business,” Science News, August 21 , 2004. In general, one cannot predict whether there will be more than one form for a compound, how many forms will eventually be discovered, or how to prepare any previously unidentified form.
There remains a need for additional polymorphic forms of fidaxomicin and for processes to prepare polymorphic forms in an environmentally-friendly, cost-effective, and industrially applicable manner.

G.V. Prasad, chairman, Dr Reddy’s Laboratories
EXAMPLES
Example 1 : Preparation of fidaxomicin Form IV:
Fidaxomicin (0.5 g) and a mixture of 1 ,4-Dioxane (10 mL), THF (10 ml) and water (20mL) were charged in Easy max reactor (Mettler Toledo). The reactor was set to temperature cycle with following parameters:
Starting temperature: 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 6 hours.
After completion of temperature cycling process, the slurry was filtered under suction, followed by drying in air tray dryer (ATD) at 40°C to a constant weight to produce crystalline fidaxomicin form-IV.
Example 2: Preparation of fidaxomicin Form V:
Fidaxomicin (1 g) and a mixture of propylene glycol (10 mL) and water (20mL) were charged in Easy max reactor (Mettler Toledo). The reactor was set to temperature cycle with following parameters:
Starting temperature is 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 6 hours.
After completion of temperature cycling process, the slurry was filtered under suction, followed by drying in air tray dryer (ATD) at 40°C to a constant weight to produce crystalline fidaxomicin form-V.
Example 3: Preparation of fidaxomicin Form VI:
Fidaxomicin (0.5 mg) and MIBK (10 mL) were charged in Easy max reactor (Mettler Toledo) and the mixture was heated to 80°C. n-heptane (20 mL) was added to the solution at the same temperature. The mixture was stirred for 1 hour. The reaction mass was then cooled to 25°C. Solid formed was filtered at 25°C and dried at 40°C in air tray dryer (ATD) to a constant weight to produce crystalline fidaxomicin form VI.
Example 4: Preparation of fidaxomicin Form V:
Fidaxomicin (500 mg) and a mixture of R-propylene glycol (5 mL) and water (15 mL) were charged in Easy max reactor (Mettler Toledo). The reactor was set to temperature cycle with following parameters:
Starting temperature is 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 2 hours.
After completion of temperature cycling process, the slurry was filtered and dried at 25°C to produce crystalline fidaxomicin form-V.
Example 5: Preparation of fidaxomicin Form V:
Fidaxomicin (1 g) and a mixture of S-propylene glycol (3 ml_) and water (30 mL) were charged in Easy max reactor (Mettler Toledo). The reactor was set to temperature cycle with following parameters:
Starting temperature is 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 2 hours.
After completion of temperature cycling process, the slurry was filtered and dried at 25°C to produce crystalline fidaxomicin form-V.
Example 6: Preparation of fidaxomicin Form V:
Fidaxomicin (40 g) and a mixture of propylene glycol (400 mL) and water (1600 mL) were charged in Chem glass reactor. The reactor was set to temperature cycle with following parameters:
Starting temperature is 25 °C;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 60 °C over a period of 2 hours;
Cooled to 0 °C over a period of 2 hours;
Temperature raised to 25 °C over a period of 2 hours;
Temperature maintained at 25 °C for 6 hours.
After completion of temperature cycling process, the slurry was filtered under suction, followed by drying in air tray dryer (ATD) at 40°C to a constant weight to produce crystalline fidaxomicin form-V.


The 10-member board at pharmaceutical major Dr Reddy’s thrives on diversity. Liberally sprinkled with gray hairs, who are never quite impressed with powerpoint presentations, “they want information to be pre-loaded so that the following discussions (at the board level) are fruitful,” says Satish Reddy, Chairman, Dr Reddy’s. That said, the company has now equipped its board members with a customized application (that runs on their tablets) to manage board agenda and related processes.
see at

Dr. Reddy’s Laboratories Managing Director and Chief Operating Officer Satish Reddy addressing
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
3-(((6-Deoxy-4-O-(3,5-dichloro-2-ethyl-4,6-dihydroxybenzoyl)-2-O-methyl-β-D-mannopyranosyl)oxy)-methyl)-12(R)-[(6-deoxy-5-C-methyl-4-O-(2-methyl-1-oxopropyl)-β-D-lyxo-hexopyranosyl)oxy]-11(S)-ethyl-8(S)-hydroxy-18(S)-(1(R)-hydroxyethyl)-9,13,15-trimethyloxacyclooctadeca-3,5,9,13,15-pentaene-2-one
|
|
| Clinical data | |
| Trade names | Dificid, Dificlir |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | Minimal systemic absorption[1] |
| Biological half-life | 11.7 ± 4.80 hours[1] |
| Excretion | Urine (<1%), faeces (92%)[1] |
| Identifiers | |
| CAS Number | 873857-62-6 |
| ATC code | A07AA12 |
| PubChem | CID 11528171 |
| ChemSpider | 8209640 |
| UNII | Z5N076G8YQ |
| KEGG | D09394 |
| ChEBI | CHEBI:68590 |
| ChEMBL | CHEMBL1255800 |
| Synonyms | Clostomicin B1, lipiarmicin, lipiarmycin, lipiarmycin A3, OPT 80, PAR 01, PAR 101, tiacumicin B |
| Chemical data | |
| Formula | C52H74Cl2O18 |
| Molar mass | 1058.04 g/mol |
///////////WO-2016024243,WO 2016024243, New patent, Dr Reddy’s Laboratories Ltd, Fidaxomicin
CC[C@H]1/C=C(/[C@H](C/C=C/C=C(/C(=O)O[C@@H](C/C=C(/C=C(/[C@@H]1O[C@H]2[C@H]([C@H]([C@@H](C(O2)(C)C)OC(=O)C(C)C)O)O)\C)\C)[C@@H](C)O)\CO[C@H]3[C@H]([C@H]([C@@H]([C@H](O3)C)OC(=O)C4=C(C(=C(C(=C4O)Cl)O)Cl)CC)O)OC)O)\C
WO 2016024284, New Patent, MIRABEGRON, Wanbury Ltd
WO 2016024284, New Patent, MIRABEGRON, Wanbury Ltd

WANBURY LTD. [IN/IN]; BSEL tech park, B wing, 10th floor, sector 30A opp. Vashi Railway Station, Vashi Navi Mumbai 400703 Maharashtra (IN)
DR. NITIN SHARADCHANDRA PRADHAN; (IN).
DR. NILESH SUDHIR PATIL; (IN).
DR. RAJESH RAMCHANDRA WALAVALKAR; (IN).
MR. NILESH SUBHASH KULKARNI; (IN).
MR. SANTOSH NAMDEV RAWOOL; (IN).
MR. PURUSHOTTAM EKANATH AWATE; (IN)

LEFT , DR K CHANDRAN, DIRECTOR WANBURY
MR ASOK SHINKAR
The present invention relates to a novel process for preparation of Mirabegron of Formula (I) using intermediates of Formula (II), (IIIa), (Illb) and (IV).
The present invention relates to a process for preparation of Mirabegron of Formula
(I).

Formula (I)
The present invention further relates to the preparation of Mirabegron of Formula (I) by using compounds of Formula (II), (Ilia), (Illb) and (IV)

Formula (II)

Formula (IlIa) Formula (Illb)

Formula (IV)
Furthermore, the present invention relates to process for preparation of compound of Formula (II), (Ilia), (Illb) and (IV).
Background of the invention:
Mirabegron is chemically known as 2-amino-N-[4-[2-[[(2R)-2-hydroxy-2-phenylethyl]amino]ethyl]phenyl]-4-thiazoleactamide and is marketed under trade name Myrbetiq.
Mirabegron is a drug used for treatment of overactive bladder. It was first disclosed in US 6,346,532, wherein (R)-Styrene oxide is reacted with 4-nitrophenyl ethyl amine hydrochloride to obtain (R)-l- phenyl-2-[[2-(4-nitrophenyl)ethyl]amino]ethanol, the later is then protected with BOC anhydride and subjected to reduction in the presence of Pd/C to yield N-[2-(4-Aminophenyl)ethyl]-N-[(2R)-2-hydroxy-2-phenylethyljcarbamic acid tert-butyl ester. Thus formed compound was then coupled with (2-amino-l,3-thiazol-4yl) acetic acid to obtain BOC protected Mirabegron which is de-protected to give Mirabegron hydrochloride.
The synthetic route proposed in US 6,346,532 is presented in Scheme-I.
Scheme-I

The major draw-backs of the presented synthetic scheme are as follows:
1. Less atomic efficiency
2. Low yield and extensive impurities formations
3. Use of expensive and sensitive protecting agents
4. Column chromatographic techniques for purifications of intermediates.
One more synthetic route for the preparation of Mirabegron have been proposed US 6,346,532, however it is not exemplified.
US 7,342,117 disclose a process for preparation of Mirabegron. The process involves the step of condensation of 4-nitrophenyl ethylamine and (R)- mandelic acid in presence of tri ethylamine, hydroxybentriazole and l-(3-dimethylaminopropyl)-3-ethyl carbodiimide in N,N-dimethylformamide to obtain compound of Formula (A). The second step involves conversion of compound of Formula (A) to compound of Formula (B) in presence of l,3-dimethyl-2-imidazolidone and borontetrahydro fluoride in tetrahydrofuran. In third step, compound of Formula (B) is subjected to reduction using 10% palladium-carbon in methanol to afford (R)-2-[[2′-(4-aminophenyl)-ethyl amino] -1-phenylethanol (Formula IV), which was further condensed with 2-aminothiazol-4-yl acetic acid in presence of l-(3-dimethylaminopropyl)-3 -ethyl carbodiimide and hydrochloric acid in water to obtain Mirabegron of Formula (I). The schematic representation is as Scheme-II

Another patent application CN103193730, discloses a novel process for preparation of Mirabegron wherein the amino group of 2-aminothiazole-5-acetic acid is protected with a protecting group and is condensed with 4-amino phenyl ethanol to obtain an intermediate (A); which on further oxidation yields intermediate (B). The intermediate B is subjected to reductive amination with (R)-2-amino-l -phenyl ethanol and deprotection, simultaneously to yield Mirabegron. The schematic representation is as Scheme-Ill.

Formula (I)
Scheme-Ill
Other references wherein process for preparation of Mirabegron are disclosed CN103387500 and CN103232352.
Most of the prior art reported for preparation of Mirabegron uses expensive and sensitive protecting agents thereby making process less feasible on industrial scale. Furthermore, the yield and purity of Mirabegron obtained by the processes known in art is not satisfactory. It is well known fact that pharmaceutical products like Mirabegron should have high purity due to the therapeutic advantages and also due to the stringent requirements of regulatory agencies. The purity requirements can be fulfilled either by avoiding the formation of by-products during the process or by purifying the end product of the process. The inventors of present invention have skillfully developed the process to provide Mirabegron with unachieved level of purity. Furthermore, the process of present invention is simple, industrially viable, and economic and avoids unfavorable reaction conditions.
![]()
According to present invention, the process for preparation of compound of Formula (IV), is depicted in Scheme IV

The present invention further relates to a process for preparation of Mirabegron of Formula (I)
The schematic reaction scheme of Mirabegron according to present invention is depicted in Scheme-V.
 Wherein R is -OH or -CI
Wherein R is -OH or -CI
The detail of the invention provided in the following examples is given by the way of illustration only and should not be construed to limit the scope of the present invention.
EXAMPLES
Example 1: Preparation of [2-(formylamino)-l,3-thiazol-4-yl]acetyl chloride; Formula (V); wherein R is -CI
20g of [2-(formylamino)-l,3-thiazol-4-yl]acetic acid was added to 250 ml of methylene dichloride and the mixture was cooled to -10°C followed by lot wise addition of 25g of phosphorous pentachloride. The mixture stirred while maintaining temperature of -10°C for 2-3 hours. After confirming completion of reaction, the product was filtered out, washed with methylene dichloride and dried to obtain 24g (Yield: 92%) of compound of Formula (V); wherein R is -CI
Example 2: Preparation of 4-nitrophenyl-[2-(formylamino)-l,3-thiazol-4-yl]acetate; Formula (IlIa)
2g of p-nitrophenol was added to 40ml of methylene chloride and 4.963g of potassium carbonate, the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of compound of Formula (V) of example 1. After confirming completion of reaction, 5.87g (Yield: 99%) of compound of Formula (Ilia) was isolated. The obtained compound has been identified by;
HNMR(D20 Exchange)
8.614 (S,lH),7.359(d,2H),8.119(d,2H),6.561(S,lH),3.765(S,2H).
Example 3: Preparation of (2-amino-l,3-thiazol-4-yl)acetyl chloride; Formula (VI); wherein R is -CI
5g of (2-amino-l,3-thiazol-4-yl)acetic acid was added to 50 ml of methylene dichloride with few drops of dimethylformamide and 6g of oxalyl chloride at temperature ranging from 0-5°C. the mixture was maintained at 0-5°C for 4-5 hours and after completion of reaction, solid mass was filtered out, washed with methylene dichloride and dried to afford 5g (Yield: 89%) of compound of Formula (VI); wherein R is -CI
Example 4: Preparation of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate; Formula (Illb)
2g of p-nitrophenol was added to 40ml of methylene chloride and 4.96g of potassium carbonate, and the mixture was cooled to 10-15 °C followed by lot wise addition of 3.95g of compound of Formula (VI) prepared in example 3. After confirming completion of reaction, 6.18g (Yield: 99%) of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate of Formula (Illb) was isolated.
The obtained compound has been identified by
HNMR ( D2O Exchange)
7.359(d,2H),8.1 19(d,2H),6.425(S,lH).3.775(S,2H).
Example 5: In-situ preparation of (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol or its hydrochloride salt, of Formula (IV)
Step I – Preparation of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl]-2-phenylethanamide of Formula (IX)
(R)-2-hydroxy-2-phenylacetic acid (75g), triethylamine (50g), hydroxybenzotriazole (HOBt) (33.3g) and l-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (EDC.HC1) (50g) were added to a mixture of 2-(4-nitrophenyl)ethylamine hydrochloride (100g) in Ν,Ν-dimethylformamide (375ml) at 25-30°C. The mixture was stirred for 30 minutes followed by addition of another lot of HOBt (33.3g) and EDC.HC1 (50g) in reaction mixture. The reaction mixture was maintained at 25-30°C for 15 hours under stirring. After completion of reaction, water (1850ml) was added to the reaction mixture and stirred. Subsequently, ethyl acetate (1500ml) was added to the reaction mixture at 25-30°C and stirred. The organic phase was separated from aqueous phase, and was washed sequentially with 1M HC1 solution, 20%aqueous potassium carbonate solution and water. The organic solvent was distilled out under reduced pressure to obtain residue comprising of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl] -2 -phenyl ethanamide of Formula (IX)
Step II – Preparation of (2R)-2-hydroxy-N-[2-(4-aminophenyl)ethyl]-2-phenylethanamide of Formula (X)
The residue from step I, methanol (740ml) and Raney Nickel (14.8g) were charged into an autoclave vessel, 10 kg/cm2 hydrogen gas pressure was applied to the reaction mixture at 25-30°C and the mixture was maintained under stiring 6 hours. Reaction mixture filtered through hyflo bed. Distilled off the solvent completely from the filtrate under reduced pressure to obtain residue comprising (2R)-2-hydroxy-N-[2-(4-aminophenyl)ethyl]-2-phenylethanamide of Formula (X)
Step III – Preparation of (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol dihydrochloride salt, of Formula (IV)
The residue of step II was added in tetrahydrofuran (665ml) and the mixture was cooled to -5 to 0°C. To this cooled mixture was then successively added sodium borohydride (56.26g) and BF3-diethyl ether (466g), and the mixture was stirred for 15 minutes. The temperature of reaction mixture was gradually increased to 50-55°C and was maintained under stirring for 5 hours. After completion of reaction, the reaction mixture was cooled to 0-5°C and 50% sodium hydroxide solution was added till pH is basic. The temperature of reaction mixture is then raised to 25-30°C followed by addition of ethyl acetate (500ml). The organic layer was separated and subjected to distillation to afford a residue. To the residue was added isopropyl alcohol (665ml) and mixture was refluxed for 30 minutes. The mixture was then allowed to cool to 40-45°C, isopropyl alcohol hydrochloride (200ml) was added till pH acidic and mixture was stirred for 2 hours to afford precipitate. The precipitate was filtered out and washed with isopropyl alcohol. The wet cake thus obtained was added to 20% aqueous sodium hydroxide solution (till pH basic) followed by addition of dichloromethane (500ml). The organic layer was separated from aqueous layer and was subjected to distillation under reduced pressure to obtain residue. The residue was taken in toluene (500ml), heated to 55-60°C for 30 minutes and cooled to 10-15°C. The precipitate obtained was filtered, washed with toluene and to the wet cake afforded was added isopropyl alcohol (665ml). The mixture was refluxed for 30 minutes and then cooled to 50-55°C. At 50-55°C slowly isopropyl alcohol hydrochloride (200ml) till pH acidic was added and mixture was stirred for 2 hours to obtain precipitate. The precipitate was filtered out, washed with isopropyl alcohol and dried to get (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol dihydrochloride salt, of Formula (IV)
Yield-70%
HPLC Purity: 98%
Example 6: Alternate method for preparation of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl]-2-phenylethanamide of Formula (IX)
Step I – A mixture of (R)-2-hydroxy-2-phenylacetic acid (lOg), dichloromethane (50ml) and triethylamine (24ml) was cooled to 0-5°C and slowly para-toluene sulfonyl chloride (12.53g) was added to it. The temperature of reaction mixture was raised to 25-30°C and maintained for 12 hours. After completion of reaction, water (100ml) was added to the reaction mixture and the mixture was stirred for 15 minutes. The organic phase was separated and distills out completely under reduced pressure to obtain [(R)-2-hydroxy -2-phenyl acetic tosyl ester].
Yield-56%
Step II – 2-(4-nitrophenyl)ethylamine hydrochloride (6g) was added to dichloromethane (50ml) and stirred for 30 minutes at 25-30°C. The mixture was
then cooled to 0-5 °C and triethylamine (13ml) was added. To say cooled mixture was then slowly added a mixture of (R)-2-hydroxy -2-phenyl acetic tosyl ester (lOg) and dichloromethane (50ml). The temperature of reaction mixture was then raised to reflux temperature and maintained for 5 hours. After completion of reaction, water (50ml) was added to the reaction mixture and the mixture was stirred for 15 minutes. The organic phase was separated and distill out completely under reduced pressure to obtain (R)-2-hydroxy-N-[2-(4-nitrophenyl) ethyl]-2-phenylacetamide
Yield-70%, Purity-96%
Example 7: Preparation of compound of Formula (II) from compound of Formula (V); wherein R is -OH
1.58g of [2-(formylamino)-l,3-thiazol-4-yl]acetic acid of Formula (V) was added solution of (1R )-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) in water (2g of Formula (IV) in 50ml water) followed by addition of 0.66g concentrated hydrochloric acid and 3.27g of l-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride. The mixture was stirred at 25-30°C for 0.5 hours. After completion of reaction, pH was adjusted to 8-9 using aqueous saturated solution of sodium carbonate. The solid precipitated out was filtered, washed with water and dried to obtain 2.1g of compound of Formula (II). (Yield: 72%) The obtained compound has been identified by HNMR
2.502(m,4H),2.599(m,2H),3.685(S,2H),4.9(S, NH protons),7.01(m, 10H, aromatic), 8.54(S,1H), 10.0(S, -OH proton),
HNMR(D20 Exchange) 2.502(m,4H),2.60(m,2H),4.57(m,lH),7.0(m, 10H, aromatic), 8.43(S,1H)
Example 8: Preparation of compound of Formula (II) from compound of Formula (V); wherein R is -CI
lOg of ( 1R)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5), was added to 150ml of acetonitrile with 16.17g of potassium carbonate and the mixture was cooled to 10-15°C. 18.8g of Formula (V) of example 1 was added to above mixture at 10-15°C in lot wise. After completion of reaction, the reaction mixture was concentrated under vacuum and 90ml of water was added for isolation. The product was then filtered out, washed with water and dried to obtain 72g (Yield: 70%) of compound of Formula (II).
Example 9: Preparation of compound of Formula (II) from compound of Formula (IlIa)
5.87g of compound of Formula (IlIa) was added to 40 ml of methylene dichloride with 2.36 g of potassium carbonate and 3.67g of ( 1))-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol (Formula-IV ; prepared by methods known in prior art/ as given in example 5) . The mixture was stirred at 25-30°C for 1 hour. After completion of reaction, the reaction mixture was concentrated followed by addition of 60 ml of water to isolate lg of compound of Formula (II).
Example 10: Insitu preparation of compound of Formula (II) without isolation of compound of Formula (IlIa)
2g of p-nitrophenol was added to 40 ml of methylene chloride with 4.963g of potassium carbonate, and the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of [2-(formylamino)-l,3-thiazol-4-yl]acetyl chloride of Formula (V) of example 1. After confirming complete formation of compound of Formula (Ilia), 2.36g of potassium carbonate and 3.67g of (1R)-2-{[2-(4-aminophenyl)ethyl]amino}-1 -phenyl ethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5) was added insitu, and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, the reaction mixture was concentrated followed by addition of 60 ml of water to isolate lg of compound of Formula (II).
Example 11: Preparation of Mirabegron from compound of Formula (II)
To 2g of compound of Formula (II) was added 30ml of 10% sodium hydroxide and the mixture was stirred at 55-60°C for 3 hours. After completion of reaction, the mixture was cooled to 25-30°C and the solid obtained was filtered, washed with water and dried to yield 1.3g of Mirabegron. (Yield: 70%)
Example 12: Preparation of Mirabegron from compound of Formula (Illb)
6.18g of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate was added to 40ml of methylene dichloride with 2.36g of potassium carbonate and 3.65g of (1R)-2-{ [2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5), and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, solid was filtered out, washed with methylene dichlrode and dried to yield lg of Mirabegron of Formula (I).
Example 13: Insitu preparation of Mirabegron without isolation of compound of Formula (Illb)
To 40ml of methylene chloride was added 2g of p-nitrophenol and 4.96g of potassium carbonate, and the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of compound of Formula (VI) prepared in example 3. After confirming complete formation of compound of Formula (Illb), 2.36g of potassium carbonate and 3.65g of (1R)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5) was added insitu, and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, After completion of reaction, solid was filtered out, washed with methylene dichlrode and dried to yield lg of Mirabegron of Formula (I).
Example 14: Preparation of Mirabegron from compound of Formula (VI); wherein R is -CI
To 20ml of acetone was added 2g of (l/?)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) and 2.15g of potassium carbonate, and the mixture was cooled to 10-15°C followed by addition of (2-amino-l,3-thiazol-4-yl)acetyl chloride of Formula (VI). After completion of reaction, acetone was concentrated under vacuum and 90ml of water was added for for isolation. The product was then filtered out, washed with water and dried to obtain 2g (Yield: 70%) of Mirabegron.



/////WO-2016024284, WO 2016024284, New Patent, MIRABEGRON, Wanbury Ltd
WO 2016024289, NILOTINIB, New Patent by SUN PHARMA
NILOTINIB

WO 2016024289, NILOTINIB, New Patent by SUN
SUN PHARMACEUTICAL INDUSTRIES LTD [IN/IN]; 17/B, Mahal Industrial Estate, Off Mahakali Caves Road, Andheri (east), Mumbai 400093 (IN)
THENNATI, Rajamannar; (IN).
KILARU, Srinivasu; (IN).
VALANCE SURENDRAKUMAR, Macwan; (IN).
SHRIPRAKASH DHAR, Dwivedi; (IN)
The present invention provides novel salts of nilotinib and polymorphs thereof. The acid addition salts of nilotinib with benzenesulfonic acid, butanedisulfonic acid, 1-5- naphthalenedisulfonic acid, naphthalene-1-sulfonic acid and 1-hydroxynaphthoic acid; hydrates and anhydrates thereof.
Nilotinib, 4-methyl-N-[3-(4-methyl-lH-imidazol-l-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl] amino] -benzamide, having the following formula

is marketed under the name Tasigna® in US and Europe. Tasigna contains nilotinib monohydrate monohydrochloride salt and is available as capsules for the treatment of adult patients with newly diagnosed Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase. Tasigna is also indicated for the treatment of chronic phase and accelerated phase Philadelphia chromosome positive chronic myelogenous leukemia (Ph+ CML) in adult patients resistant or intolerant to prior therapy that included imatinib.
Nilotinib is considered a low solubility/low permeability (class IV) compound in the Biopharmaceutics Classification System (BCS). Therefore, dissolution of nilotinib can potentially be rate limiting step for in-vivo absorption. It is soluble in acidic media; being practically insoluble in buffer solutions of pH 4.5 and higher.
WIPO publication 2014059518A1 discloses crystalline forms of nilotinib hydrochloride and methods of the preparation of various crystalline solvates of nilotinib hydrochloride including benzyl alcohol, acetic acid and propylene glycol.
WIPO publication 2011033307A1 discloses nilotinib dihydrochloride and its hydrates and method for their preparation.
WIPO publication 2011163222A1 discloses the preparation of nilotinib salts and crystalline forms thereof. The salts of nilotinib disclosed are hydrochloride, fumarate, 2-chloromandelate, succinate, adipate, L-tartrate, glutarate, p-toluenesulfonate, camphorsulfonate, glutamate, palmitate, quinate, citrate, maleate, acetate, L-malate, L-aspartate, formate, hydrobromide, oxalate and malonate.
WIPO publication number 2011086541A1 discloses a nilotinib monohydrochloride monohydrate salt and methods for preparing.
WIPO publication number 2010054056A2 describes several crystalline forms of nilotinib hydrochloride.
WIPO publication number 2007/015871A1 discloses the preparation of nilotinib salts and crystalline forms thereof. The salts are mixtures of nilotinib and one acid wherein the acids are selected from the group consisting of hydrochloric acid, phosphoric acid, sulfuric acid, sulfonic acid, methane sulfonic acid, ethane sulfonic acid, benzene sulfonic acid, p-toluene sul- fonic acid, citric acid, fumaric acid, gentisic acid, malonic acid, maleic acid, and tartaric acid.
WIPO publication number 2007015870A2 discloses several nilotinib salts including amorphous and crystalline forms of nilotinib free base, nilotinib HC1 and nilotinib sulfate along with their hydrate and solvates.

EXAMPLES:
Example 1: Preparation of nilotinib benzenesulfonate crystalline Form I
Nilotinib base (1 g) was suspended in water (20 ml). A solution of benzenesulfonic acid (0.4 g) in water (3ml) was added and the content was heated at 60 °C for 2-3 h. The mixture was cooled to 25-30 °C, filtered, washed with water (3 x 5 ml) and dried under vacuum for 2 h at 50-55 °C.
1H NMR (500 MHz, DMSO-d6) δ 2.40 (s,3H), 2.42 (s,3H), 7.35-7.37 (m,3H), 7.51-7.66 (m,5H),7.83 (d,lH), 7.96 (s,lH),8.08 (s,lH),8.30 (s,lH) 8.39 (s,lH),8.54 (d,lH), 8.61 (d,lH), 8.64 (s,lH), 8.75 (d,lH), 9.25 (s,lH), 9.34 (d,lH), 9.61 (s,lH), 10.84 (s,lH).
The salt provides an XRPD pattern substantially same as set forth in FIG. 1.
Example 2: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form II
Nilotinib base (100 g) was dissolved in 20 % water in THF solution (2000 ml) at 60-65 °C and insoluble matter was filtered. The filtrate was concentrated under vacuum below 60 °C. Filtered water (1000 ml) was added to the reaction mixture and it was heated at 50-55 °C, followed by addition of 1,4-butanedisulfonic acid -60% aqueous solution (28.6 ml) at same temperature. The content was stirred at 50-55 °C for 2-3h. Reaction mixture as cooled to 25-30 °C and product was filtered, washed with water (200 ml x 2) and dried in air oven at 50-55 °C (yield: 115 g).

Sun Pharma managing director Dilip Shanghvi.
Purity (by HPLC):99.76%
1H NMR (400 MHz,DMSO-d6) δ 1.63-1.66(m,2H), 2.40(d,3H),2.42(s,3H),2.43-2.47(m,2H), 7.51-7.62(m,3H),7.85(dd,lH),7.96(s,lH),8.08(s,lH),8.34(s,lH),8.38(d,lH),8.52-8.55(m,lH), 8.60-8.62 (m,2H), 8.75(d,lH), 9.25(S,1H),9.34(S,1H),9.59(S,1H),10.86(S,1H)
Water content: 7.95 %.
The salt has a XRPD pattern substantially same as set forth in FIG. 2.
Example 3: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form II
Nilotinib base (300 g) was suspended in methanol (3000 ml) and aqueous hydrochloric acid was added to get pH less than 2. Reaction contents were heated at reflux and was filtered and washed with methanol (100 ml). 5% (w/w) NaOH (1200 ml) solution was added at 40-45 °C within 15 min, reaction mixture was stirred for 2h. Product was filtered, washed with water
(300 ml x 3) and dried for lh. Wet material was suspended in water (3000 ml), heated at 50- 55 °C followed by addition of 1,4-butanedisulfonic acid -60% aqueous solution. The reaction mixture was stirred at 50-55°C for 2hrs. Product was filtered at room temperature, washed with water (500 ml x 2) and dried in air oven at 50-55 °C (yield: 293 g).
Purity (by HPLC): 99.88 %
1H NMR (400 MHz,DMSO-d6+TFA-dl) δ 1.75-1.78(m,2H), 2.36(d,3H),2.38(s,3H),2.69- 2.72(m,2H),7.45(d,lH),7.68(d,lH),7.83(s,lH),7.88(dd,lH),7.97(s,lH),8.16-8.19(m,lH), 8.35
(s,2H), 8.63(d,lH),8.68(d,lH),9.04(d,lH),9.21(d,lH),9.53(br s,lH),9.69(d,lH)10.80 (s,lH)
Water content: 6.44 %
Example 4: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form III
Nilotinib butanedisulfonate (210g) was dissolved in acetic acid water mixture (50:50) (2520 ml) at 75-80 °C and was filtered to remove insoluble matter and washed with acetic acid water mixture (50:50) (210 ml). Water (3150ml) was added to the filtrate and stirred first at room temperature and then at 0-5 °C. Product was filtered and washed with water. Material was dried in air oven at 70-75 °C. Dried material was leached with methanol (3438 ml) at reflux temperature, filtered and dried in air oven 70-75°C (yield: 152.6 g)
Purity (by HPLC): 99.89 %
1H NMR (400 MHz,DMSO-d6+TFA-dl) δ 1.73-1.77(m,2H), 2.40(s,6H),2.67-2.70(m,2H), 7.50 (d,lH), 7.70(d,lH), 7.88-7.92(m,2H), 8.07(s,lH),8.23 (dd,lH), 8.34(s,2H), 8.67 (d,lH), 8.72 (d,lH), 9.09(d,lH), 9.23 (s,lH), 9.54(d,lH), 9.74(d,lH), 10.86(s,lH).
Water content: 0.61 %
The salt provides an XRPD pattern substantially same as set forth in FIG. 3.
Example 5: Preparation of crystalline form of nilotinib butanedisulfonate (2: 1)
Crystalline Nilotinib butanedisulfonate (1 g) of Example 2 was suspended in methanol (20 ml) and was stirred at reflux for 60 min. The mixture was cooled to room temperature. Solid was filtered, washed with methanol (2 ml x 3) and dried in air oven at 70-75°C (yield: 0.8 g)
Example 6: Preparation of nilotinib butanedisulfonate (1: 1) crystalline Form IV
Nilotinib base (20 g) was suspended in methanol (800 ml) and 1,4-butanedisulfonic acid -60
% aqueous solution (6 ml) was added at 50-55 °C, and was filtered to remove insoluble matter. Filtrate was stirred at room temperature for 2-3 h. Product formed was filtered, washed with methanol (20 ml x 2) and dried the product in air oven at 70-75 °C (yield: 18.4 g).
Purity (by HPLC):99.86 %
1H NMR (400 MHz,DMSO-d6) δ 1.64-1.68(m,4H), 2.47-2.5 l(m,4H), 2.41(s,3H), 2.42(d,3H), 7.52(d,lH), 7.83-7.89(m,2H), 7.99(s,lH), 8.15(s,lH), 8.36 (d,lH), 8.39(s,lH), 8.65-8.66(m,2H), 8.79(d,lH), 8.89(br s,lH), 9.36(s,lH), 9.41(br s,lH), 9.74(d,lH), 10.91(s,lH).
The salt has XRPD pattern substantially same as set forth in FIG. 4.
Example 7: Preparation of nilotinib 1,5-napthalenedisulfonic acid salt (2: 1) crystalline Form V
Nilotinib base (1 g) was suspended in water (20 ml). A solution of 1,5-napthalenedisulfonic acid (0.4 g; 0.6 eq.) in water (5ml) was added and the content was heated at 50-55 °C for lh. The mixture was cooled to 25-30 °C, filtered and washed with water (10 ml). The product was dried in air oven at 50-55°C (yield: 1.2 g).
1H NMR (400 MHz,DMSO-d6) δ 2.39 (s,3H), 2.42 (s,3H), 7.45-7.61 (m,4H),7.84 (d,lH), 7.97(s,2H),8.08 (m,lH),8.31 (s,lH) 8.38 (s,lH),8.55 (d,lH), 8.63 (s,2H), 8.75 (s,lH), 8.92 (d,lH), 9.26 (s, 1H), 9.34 (s,lH),9.62 (s,lH), 10.85 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 5.
Example 8: Preparation of nilotinib 1,5-napthalenedisulfonic acid salt (1: 1) crystalline Form VI
Nilotinib base (1 g) was suspended in water (20 ml). A solution of 1,5-napthalenedisulfonic acid (0.8 g; 1.2eq) in water (5 ml) was added and the content was heated at 50-55 °C for 1 h. The mixture was cooled to 25-30 °C, filtered, washed with water (10 ml) and dried in air oven at 50-55 °C (yield: 1.4g).
1H NMR(400 MHz,DMSO-d6) δ 2.40 (s,3H),2.41 (s,3H), 7.43-7.52 (m,3H),7.61 (d,lH), 7.85-7.99(m,5H),8.11 (s,lH),8.34 (s,2H), 8.64-8.67 (m,2H), 8.89-8.92 (m,4H),9.40(d,2H), 9.72 (s,lH), 10.87 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 6.
Example 9: Preparation of nilotinib napthalene-1- sulfonic acid salt crystalline Form VII Nilotinib base (1 g) was suspended in water (10 ml) and heated to 50-55 °C. A solution of napthelene-1 -sulfonic acid and methanol (10 ml) was added to it and heated at 70-75 °C for 30 min. The mixture was cooled to 25-30 °C and stirred for 10 min. The product was filtered, washed with water (2 x 2 ml) and dried under vacuum for 1-2 h at 50-55 °C.
1H NMR (400 MHz,DMSO-d6) δ 2.41 (s,3H),2.42 (s,3H), 7.46-7.58 (m,5H), 7.70-8.00 (m,7H)8.11(s,lH)8.31(s,lH),8.37(s,lH),8.63-8.66 (m,3H), 8.81-8.89 (m,2H), 9.31 (s,lH), 9.37 (d,lH), 9.71 (d,lH), 10.86 (s,lH)
The salt has a XRPD pattern substantially same as set forth in FIG. 7.
Example 10: Preparation of nilotinib l-hydroxy-2-napthoic acid salt crystalline Form VIII Nilotinib base (1 g) was suspended in water (20 ml) and heated to 50-55 °C. l-Hydroxy-2-napthoic acid was added to it and the content was heated at 50-55 °C for 1 h. Methanol (5 ml) was added to the mixture and stirred for 30 min. The content was filtered, washed with water (2 x 2 ml) and dried under vacuum for 1 h at 50-55 °C.
1H NMR (400 MHz, DMSO-d6) δ 2.25 (s,3H), 2.41 (s,3H), 7.40-7.92 (m,l lH), 8.23-8.73 (m,8H), 9.24 (s,lH), 9.34(s,lH), 10.70 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 8.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)- 5-(trifluoromethyl)phenyl]-3- [(4-pyridin-3-ylpyrimidin-2-yl) amino]benzamide
|
|
| Clinical data | |
| Trade names | Tasigna |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a608002 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 30%[1] |
| Protein binding | 98%[1] |
| Metabolism | Hepatic (mostly CYP3A4-mediated)[1] |
| Biological half-life | 15-17 hours[1] |
| Excretion | Faeces (93%)[1] |
| Identifiers | |
| CAS Number | 641571-10-0(base) |
| ATC code | L01XE08 |
| PubChem | CID 644241 |
| IUPHAR/BPS | 5697 |
| DrugBank | DB04868 |
| ChemSpider | 559260 |
| UNII | F41401512X |
| KEGG | D08953 |
| ChEBI | CHEBI:52172 |
| ChEMBL | CHEMBL255863 |
| PDB ligand ID | NIL (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C28H22F3N7O |
| Molar mass | 529.5245 g/mol |
//////////////WO 2016024289, WO-2016024289, NILOTINIB, New Patent, SUN
Cc1ccc(cc1Nc2nccc(n2)c3cccnc3)C(=O)Nc4cc(cc(c4)n5cc(nc5)C)C(F)(F)F
Fosfluconazole

Fosfluconazole
Fosfluconazole; 194798-83-9; UNII-3JIJ299EWH; 3JIJ299EWH; NCGC00182029-01;
2-(2,4-difluorophenyl)-1,3-di(1h-1,2,4-triazol-1-yl)propan-2-yl dihydrogen phosphate;
2,4-difluoro-α,α-bis(1H-1,2,4-triazol-1-ylmethyl) benzyl alcohol, dihydrogen phosphate
| Molecular Formula: | C13H13F2N6O4P |
|---|---|
| Molecular Weight: | 386.250688 g/mol |
Research Code:UK-292663, UK 292663, F-FLCZ, F FLCZ
Trade Name:Prodif® PFIZER
MOA:Azole antifungal
Indication:Cryptococcus neoformans; Candidiasis
Status:Approved, Japan PMDA OCT 16 2003
Company:Pfizer (Originator)
Candidiasis,Cryptococcus neoformans, Injection, Solution, Eq. 100 mg/200 mg/400 mg fluconazole per vial
Fosfluconazole (INN) is a water-soluble phosphate prodrug of fluconazole – a triazole antifungal drug used in the treatment and prevention of superficial and systemic fungal infections. The phosphate ester bond is hydrolysed by the action of a phosphatase – an enzyme that removes a phosphate group from its substrate by hydrolysing phosphoric acid monoesters into a phosphate ion and a molecule with a free hydroxyl group (see dephosphorylation).
Fosfluconazole was approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on Oct 16, 2003. It was developed and marketed as Prodif® by Pfizer in Japan.
Fosfluconazole is a water-soluble phosphate prodrug of fluconazole – a triazole antifungal drug. It is indicated for the treatment of candida and cryptococcus infections.
Prodif® is available as solution for intravenous use, containing 100, 200 or 400 mg of free Fosfluconazole per vial. The recommended dose is 50 to 100 mg administered intravenously once daily for candidiasis. Another dose is 50 to 200 mg fluconazole once daily for cryptococcosis.



Reference:1. WO9728169A1 / US6977302B2.
2. Org. Process Res. Dev.2002, 6, 109-112.
http://pubs.acs.org/doi/pdf/10.1021/op010064%2B
2-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazole-1-yl)- 2-propyl dihydrogen phosphate (2). A slurry of dibenzyl 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazole-1-yl)-2-propyl phosphate (30.1 kg, 53.13 mol), 5% palladium-on-carbon catalyst (50% wet, type 5R39, 1.5 kg), and sodium hydroxide (4.36 kg, 108.9 mol) in low-endotoxin water (75.7 L) was hydrogenated at ambient temperature and 414 kPa (60 psi) for 12 h. The slurry was filtered, and the catalyst was washed with low-endotoxin water (9.8 L). After separating the toluene by-product, the aqueous phase was slurried with carbon (3.1 kg) for 30 min. After the carbon was removed by filtration, the aqueous phase was acidified to pH 1.45 by that addition of sulfuric acid (6.69 kg) in low-endotoxin water (25 L) over 2 h. The resulting slurry was granulated at ambient temperature for 1 h and then filtered. The product was sequentially washed with filtered low-endotoxin water (103 L) and filtered acetone (103 L). The product was dried under vacuum at 50 °C for 12 h to give the title compound (18.1 kg, 88%) a white powder: mp 223-224 °C.
1H NMR (DMSO) δ 5.07 (2H, d), 5.24 (2H, d), 6.77-6.83 (1H, m), 7.00-7.18 (2H, m), 7.75 (2H, s), 8.53 (2H, s).
Found: C, 40.28; H, 3.39; N, 21.63;
[MH]+ 387.0786. C13H13F2N6O4P requires: C, 40.43; H, 3.39; N, 21.78; [MH]+ 387.0782.
US6977302
https://www.google.com/patents/US6977302
EXAMPLE 1 1-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propyl dihydrogen phosphate
(a) Dibenzyl 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propyl phosphate
Method A

A solution of 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)propan-2-ol (also known as fluconazole, 10.0 g, 32.6 mmol), 1H-tetrazole (6.85 g, 97.8 mmol), dibenzyl diisopropyl phosphoramidite (22.55 g, 65.2 mmol) in methylene chloride (100 ml) was stirred at room temperature under a nitrogen atmosphere for 2 hours. The mixture was then cooled to 0° C., and a solution of 3-chloroperoxybenzoic acid (13.5 g, 50-55% w/w, 39.1 mmol) in methylene chloride (50 ml) was added maintaining the temperature at 0° C. The resulting mixture was allowed to warm to room temperature for 1 hour before washing with aqueous sodium metabisulphite and sodium bicarbonate. After drying (MgSO4) the solvent was removed and replaced with methyl isobutyl ketone (37 ml) and tert-butyl methyl ether (74 ml). After granulating at −10° C. for 1 hour the product was filtered and washed with ice cold methyl isobutyl ketone and tert-butyl methyl ether (1:3, 15 ml) and dried at 50° C. under vacuum for 18 hours to give the subtitle compound (16.05 g, 87%), m.p. 93° C.
Found: C, 57.12; H, 4.46; N, 14.85. C27H25F2N6O4P requires C, 57.24; H, 4.46; N, 14.84%. m/z 567 (MH+) 1H NMR (300 MHz, CDCl3) δ=4.90 (d, 2H), 4.95 (d, 2H), 5.05 (d, 2H), 5.19 (d, 2H), 6.58-6.73 (m, 2H), 6.88-6.95 (m, 1H), 7.20-7.30 (m, 4H) 7.32-7.38 (m; 6H), 7.80 (s, 2H), 8.36 (s, 2H).
Method B

To stirred ethyl acetate (1530 ml) was added 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)propan-2-ol (also known as fluconazole, 306 g, 1.00 mol) and pyridine (237.3 g, 3.00 mol) before cooling to 0° C. Phosphorus trichloride (137.4 g, 1.00 mol) was added dropwise to the reaction mixture maintaining the temperature between 0-5° C. before allowing the reaction mixture to warm to 15° C. over 30 minutes. Benzyl alcohol (216 g, 2.00 mol) was then added over 30 minutes at 15-20° C. After a further 30 minutes hydrogen peroxide (27.5% w/w in water, 373 g) was added maintaining the temperature at 15-20° C. After 30 minutes the aqueous phase was removed and the organic phase washed with aqueous sodium metabisulphite, dilute hydrochloric acid and water. The solvent was removed at reduced pressure and replaced with methyl isobutyl ketone (850 ml) and tert-butyl methyl ether (1132 ml). After granulating at 20° C. for 1 hour and at 0° C. for 1 hour, the product was filtered and washed with ice cold tert-butyl methyl ether (2×220 ml) and dried at 50° C. under vacuum for 18 hours to give the subtitle compound (358 g, 63%). The melting point and spectroscopic data was identical to that stated in method A.
(b) 2-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propyl dihydrogen phosphate

A slurry of the compound of step (a) (9.80 g, 17.3 mmol), 5% palladium on carbon catalyst (50% wet, 1.0 g) and sodium hydroxide (1.38 g, 34.6 mmol) in water (26 ml) was hydrogenated at room temperature and 414 kPa (60 p.s.i.) for 20 hours. The solution was filtered through a pad of celite (trade mark) and washed with water (5 ml). The toluene was separated and the aqueous phase cooled to 0° C. whereupon sulphuric acid (1.70 g, 17.3 mmol) was added. The resulting slurry was granulated at 0° C. for 1 hour and then filtered, washed with water (2×5 ml) and dried under vacuum at 50° C. to give the title compound (5.80 g, 87%). m.p. 223-224° C.
Found: C, 40.28; H, 3.39; N, 21.63. C13H13F2N6O4P requires C, 40.43; H, 3.39; N, 21.76%. 1H NMR (300 MHz, DMSO) δ=5.07 (d, 2H) 5.24 (d, 2H), 6.77-6.83 (m, 1H), 7.00-7.18 (m, 2H), 7.75 (s, 2H), 8.53 (s, 2H).
EXAMPLE 2 2-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)-2-propyl disodium phosphate

A solution of the compound of Example 1(a) (10.0 g, 17.7 mmol) and sodium acetate (2.90 g, 35.3 mmol) in ethanol (160 ml) and water (20 ml) was hydrogenated over Pearlman’s catalyst (1.00 g) at room temperature and at 345 kPa (50 p.s.i.) for 16 hours. The solution was filtered through a pad of celite (trade mark) and the solvents removed at reduced pressure to leave a thick syrup. This was dissolved in ethanol (100 ml) with the aid of sonication and warmed to reflux. The resulting solution was allowed to cool slowly and granulate for 1 hour at room temperature. The product was filtered, washed with ethanol (10 ml) and dried under vacuum at 50° C. to give the title compound (4.48 g, 59%). m.p. 160-162° C.
1H NMR (300 MHz, D2O) δ=5.01 (d, 2H), 5.40 (d, 2H), 6.60 (m, 1H), 6.79 (m, 1H), 7.11 (m, 1H), 7.63 (s, 2H), 8.68 (s, 2H).
Route 2


Reference:1. CN103864844A.
http://www.google.com/patents/CN103864844A?cl=en
TRANSLATED BY MACHINE…….TEXT MAY VARY
forskolin fluconazole (fosf Iuconazole, Formula I) is fluconazole (Formula IV) of monophosphate prodrugs, fluconazole in the tertiary alcohol into a phosphate ester, not only did not introduce a chiral center, also increased water solubility, because a long time to overcome the low water solubility of fluconazole resulting larger infusion volume defects. After intravenous administration in the role of phosphatases in vivo hydrolysis into fluconazole, pharmacological effect. Blessing from the Central Institute of the United States Secretary of fluconazole Fai end developed, launched in Japan in 2004 I May 15, for the treatment of candidiasis and cryptococcal infections caused deep as true bacteremia, respiratory fungal disease, fungal peritoneum
Inflammation, gastrointestinal fungal disease, fungal urinary tract infections, fungal meningitis.

Synthesis gas itraconazole on forskolin in W09728169, Organic Process Research & Development (200 2), 6 (2), 109-112, CN1789270, Art of Drug Synthesis (2007), 71-82, etc. have been reported in the literature . Which Organic Process Research & Development (2002) described in detail in the first blessing Secretary fluconazole and improved synthetic route for the route problems to adapt to industrial mass production of synthetic routes.
Document Organic Process Research & Development (2002), 6,109-112 discloses the following two synthetic routes.
Route One:

Route two:

The final step is a route to the removal of benzyl group in a methanol solvent by palladium on carbon catalyzed hydrogenation reaction yield was 65%. Since forskolin fluconazole final product insoluble in methanol, and therefore there is a route following shortcomings: a catalyst poisoning, the final product is easy to form methanol solvate, removing the catalyst in the loss of product, the final product are difficult to separate, low yield not suitable for industrial production.
Two routes still using palladium on carbon hydrogenation debenzylation, except that the solvent was changed to sodium hydroxide solution, the product of soluble and stable in aqueous sodium hydroxide solution, after filtering off the catalyst, forskolin fluoro itraconazole by acidification of sodium sulfate can be easily obtained blessing Secretary of fluconazole, the reaction yield of 85-90%.
In the prior art, the removal of benzyl preparation blessing Secretary of fluconazole, the use of a pressure hydrogenation, relatively harsh reaction conditions; and blessing Secretary of fluconazole in water and slightly soluble in methanol, for blessing Secretary fluconazole further refined and purified more difficult. The present invention aims to provide a new and suitable for industrial production methods blessing Secretary fluconazole.
Example 1
2- (2,4-gas-phenyl) -1,3-bis (1H-1, 2,4- two P sat 1-yl) -2-propyl-di-benzyl-pity Cool ( Preparation blessing Secretary fluconazole dibenzyl ester)
Step The method according to CN1210540A in Example 1 A or Method B of (a), was prepared to give the title compound, having 1H-NMR shown in Figure 1 (SOi) MHz, DMS0-D6) spectrum.
Example 2
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas
Itraconazole ammonium salt) Preparation

Formula III blessing Secretary fluconazole two benzyl ester (566g, lmol), 120g of dry Pd / C (containing 5% palladium) and ammonium formate (315g, 5mol) in methanol (6L), and stirred under reflux for 5h , TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (566ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 415g, yield 98.8%.
] lH-Mffi (500MHz, DMS0-D6) δ: 4.87-4.90, 5.58-5.61,6.56-6.60, 6.94-7.03,7.52-7.61,8.96, having 1H-NMR shown in Figure 2 (500MHz, DMS0 -D6) spectrum.
Example 3
2- (2,4-gas-phenyl) -1,3-bis (1H-1, 2,4- two 1-yl) -2-propyl-pity acid dioxide Cool (forskolin
Fluconazole) Preparation of

[0052] Formula II forskolin fluconazole salt (420g, Imol), in water (IL) while stirring, filtered, 2mol / L sulfuric acid aqueous solution (500ml), 5 ° C under stirring for lh, filtered, cold water ( 200ml) wash, 50 ° C under dry blessed Division fluconazole 379g, yield 98%.
1H-Mffi (SOOMHz) DMSO-De) δ:. 5.09-5.12,5.25-5.28,6.80-6.84,7.05-7.16,7.77,8.55,10.32 [0054] Example 4
2_ (2,4_ two gas-phenyl) -1, double 3_ (1Η-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (566g, lmol), 84g of dry Pd / C (5% containing button) and ammonium formate (189g, 3mol) in anhydrous methanol (5L) in the mixture was stirred at reflux for 5h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 410g, yield 97.5%.
Example 5
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (566g, lmol), 30g of dry Pd / C (containing 10% palladium) and ammonium formate (315g, 5mol) in anhydrous methanol (5L) in the mixture was stirred at reflux for 5h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 405g, yield 96.4%.
Example 6
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (566g, lmol), 30g of dry Pd / C (containing 10% palladium) and ammonium formate (315g, 5mol) in ethanol (12L) and stirred was refluxed for 5h, TLC monitoring completion of the reaction, was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 395g, 94% yield.
Example 7
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
forskolin fluconazole dibenzyl ester (566g, lmol), 170g of dry Pd / C (containing 5% of palladium) and ammonium formate (315g, 5mol) in ethanol (16L) was stirred under reflux for 5h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 398g, yield 94.7%.
Example 8
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (566g, lmol), 120g of dry Pd / C (containing 5% palladium) and ammonium formate (315g, 5mol) in isopropanol (12L) in the mixture was stirred at reflux for 5h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 402g, a yield of 95.7%.
Example 9
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
[0071] under nitrogen blessing Secretary fluconazole dibenzyl ester (566g, lmol), 60g of dry Pd / C (containing 5% palladium) and ammonium formate (504g, 8mol) in methanol (8L) in, 50 ° C under stirring reaction 40h, TLC monitoring completion of the reaction, was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added ^ OOml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 398g, yield 94.8%.
Example 10
2_ (2,4_ two gas-phenyl) -1, double 3_ (1H-1, 2,4_ two 1-yl) propyl pity _2_ di press (forskolin gas itraconazole salt) Preparation
Under nitrogen, forskolin fluconazole dibenzyl ester (5668,111101), 8 (^ dry? (1 / (:( containing palladium 5%) and ammonium formate (315g, 5mol) for n-propyl alcohol (12L) in, 60 ° C the reaction was stirred 20h, TLC monitoring completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, ethanol was added (300ml), stirred for beating, and filtered to give a solid forskolin fluconazole salt 398g 95% yield.
Example 11
2- (2,4-gas-phenyl) -1,3-bis (1H-1, 2,4- sit two P-1-yl) -2-propyl-pity acid dioxide Cool (forskolin fluconazole) Preparation of [0077] under nitrogen blessing Secretary fluconazole dibenzyl ester 566 g (Imol) adding 56g of dry Pd / C (containing 5% palladium), methanol 6L, 315 g of ammonium formate, stirring boil under reflux for 5h, TLC after completion of the reaction was filtered, 50 ° C the solvent was distilled off under reduced pressure, addition of IL of water and dissolved with stirring, filtered, 2mol / L sulfuric acid 500mL, 5 ° C with stirring to precipitate lh, filtered, 200mL cold water, 50 ° C drying 365 g, 95% yield.
Example 12 forskolin fluconazole salt and HPLC detection methods blessing Secretary fluconazole:
High performance liquid chromatography (Chinese Pharmacopoeia 2010 edition two Appendix VD): octadecylsilane bonded silica as a filler, Column: Thermo BDS C18 (4.6 X 150mm, 3.5 μ m); methanol as mobile phase A, phosphate buffer (take potassium dihydrogen phosphate 0.68g, set 1000ml water, triethylamine 6ml, adjusted to pH 5.0 with phosphoric acid) as the mobile phase B, a flow rate of 1.0ml / min; column temperature 35 ° C; detection wavelength was 210nm, linear gradient.

After the examination, according to the peak area calculation, purity prepared in Example 2-11 was the implementation of the target product of 99.5%.
| Patent | Submitted | Granted |
|---|---|---|
| Nanoparticulate Anidulafungin Compositions and Methods for Making the Same [US2009238867] | 2009-09-24 | |
| IMIDAZOPYRIDINE SUBSTITUTED TROPANE DERIVATIVES WITH CCR5 RECEPTOR ANTAGONIST ACTIVITY FOR THE TREATMENT OF HIV AND INFLAMMATION [US7790740] | 2008-02-21 | 2010-09-07 |
| Pharmaceutical formulations of cyclodextrins and antifungal azole compounds [US2007082870] | 2007-04-12 | |
| TRIAZOLE DERIVATIVES USEFUL IN THERAPY [EP0880533] | 1998-12-02 | 2002-06-12 |
| Triazole derivatives useful in therapy [US6790957] | 2003-07-31 | 2004-09-14 |
| Process for controlling the hydrate mix of a compound [US7323572] | 2004-01-15 | 2008-01-29 |
| TOPICAL TERBINAFINE FORMULATIONS AND METHODS OF ADMINISTERING SAME FOR THE TREATMENT OF FUNGAL INFECTIONS [US7820720] | 2010-04-29 | 2010-10-26 |
| PHARMACEUTICAL COMPOSITION COMPRISING PHENYLAMIDINE DERIVATIVE AND METHOD OF USING THE PHARMACEUTICAL COMPOSITION IN COMBINATION WITH ANTIFUNGAL AGENT [US8173157] | 2010-04-22 | 2012-05-08 |
| COMPOSITIONS COMPRISING POLYUNSATURATED FATTY ACID MONOGLYCERIDES OR DERIVATIVES THEREOF AND USES THEREOF [US8222295] | 2009-11-26 | 2012-07-17 |
| MASKED CARBOXYLATE NEOPENTYL SULFONYL ESTER CYCLIZATION RELEASE PRODRUGS OF ACAMPROSATE, COMPOSITIONS THEREOF, AND METHODS OF USE [US2009069419] | 2009-03-12 |
| Patent | Submitted | Granted |
|---|---|---|
| Triazole derivatives useful in therapy [US2005130940] | 2005-06-16 | |
| Chemical compounds [US7309790] | 2005-06-16 | 2007-12-18 |
| Combination of voriconazole and an antifungal CYP2C19 inhibitor [US2005182074] | 2005-08-18 | |
| Inhibitors of fungal invasion [US2004106663] | 2004-06-03 | |
| Triazole derivatives useful in therapy [US6977302] | 2004-11-25 | 2005-12-20 |
| Pharmaceuticals [US7691877] | 2007-08-23 | 2010-04-06 |
| SIMPLE PANTOIC ACID ESTER NEOPENTYL SULFONYL ESTER CYCLIZATION RELEASE PRODRUGS OF ACAMPROSATE, COMPOSITIONS THEREOF, AND METHODS OF USE [US7994218] | 2009-03-26 | 2011-08-09 |
| COMPLEX PANTOIC ACID ESTER NEOPENTYL SULFONYL ESTER CYCLIZATION RELEASE PRODRUGS OF ACAMPROSATE, COMPOSITIONS THEREOF, AND METHODS OF USE [US8168617] | 2009-03-19 | 2012-05-01 |
| Purine derivatives [US7642350] | 2006-11-23 | 2010-01-05 |
| IMIDAZOPYRIDINONES [US2009221631] | 2009-09-03 |
IMPURITIES
1









 |
|
| Systematic (IUPAC) name | |
|---|---|
|
{[2-(2,4-Difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl)propan-2-yl]oxy}phosphonic acid
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Legal status |
|
| Routes of administration |
IV |
| Identifiers | |
| CAS Number | 194798-83-9 |
| ATC code | None |
| PubChem | CID 214356 |
| ChemSpider | 185843 |
| UNII | 3JIJ299EWH |
| ChEMBL | CHEMBL1908301 |
| Chemical data | |
| Formula | C13H13F2N6O4P |
| Molar mass | 386.25 g/mol |
| CN1210540A * | Jan 27, 1997 | Mar 10, 1999 | 辉瑞研究开发公司 | Triazole derivatives useful in therapy |
| CN1789270A * | Dec 16, 2005 | Jun 21, 2006 | 西安新安医药科技有限公司 | Mycotic ingection-resisting fosfluconazole hydrate and preparation method thereof |
| CN101890028A * | Feb 22, 2007 | Nov 24, 2010 | 卫材R&D管理有限公司 | Stabilized pharmaceutical composition |
| CN102439018A * | Mar 3, 2010 | May 2, 2012 | 塞普斯制药有限公司 | Fosfluconazole derivatives, synthesis, and use in long acting formulations |
| US20040007689 * | Jun 23, 2003 | Jan 15, 2004 | Pfizer Inc. | Process for controlling the hydrate mix of a compound |
| Reference | ||||
|---|---|---|---|---|
| 1 | * | ARTHUR BENTLEY等: “The Discovery and Process Development of a Commercial Route to the Water Soluble Prodrug, Fosfluconazole“, 《ORGANIC PROCESS RESEARCH & DEVELOPMENT》, vol. 6, no. 2, 18 December 2001 (2001-12-18), XP002491526, DOI: doi:10.1021/op010064+ | ||
| 2 | * | 国大亮 等: “福司氟康唑“, 《齐鲁药事》, vol. 24, no. 1, 30 January 2005 (2005-01-30), pages 60 | ||
| 3 | * | 村上尚道: “fosfluconazole“, 《NEW DRUGS OF THE WORLD:2003》, vol. 33, no. 10, 15 September 2004 (2004-09-15), pages 56 | ||
//////UK-292663, UK 292663, F-FLCZ, F FLCZ, Fosfluconazole, 194798-83-9, UNII-3JIJ299EWH, 3JIJ299EWH, NCGC00182029-01
Fc1ccc(c(F)c1)C(OP(=O)(O)O)(Cn2ncnc2)Cn3ncnc3
Bromuconazole

Bromuconazole
116255-48-2; HSDB 7419
| Molecular Formula: | C13H12BrCl2N3O |
|---|---|
| Molecular Weight: | 377.06388 g/mol |
1-[[4-bromo-2-(2,4-dichlorophenyl)oxolan-2-yl]methyl]-1,2,4-triazole


| Patent | Submitted | Granted |
|---|---|---|
| Phthalamide derivatives [US7132455] | 2006-02-16 | 2006-11-07 |
| Crystal modification II of 2-[2-(1-chloro-cyclopropyl)-3-(2-chlorophenyl)-2-hydroxy-propyl]-2,4-dihydro-3H-1,2,4-triazole-3-thione [US7176226] | 2006-05-18 | 2007-02-13 |
| Anthranilamide insecticides [US7211270] | 2006-03-09 | 2007-05-01 |
| 2-Phenyl-2-substituted-1,3-diketones [US7227043] | 2006-03-16 | 2007-06-05 |
| Biphenyl derivatives and their use as fungicides [US7241721] | 2006-05-11 | 2007-07-10 |
| Cyano anthranilamide insecticides [US7247647] | 2006-05-25 | 2007-07-24 |
| 3-Phenyl substituted 3-substituted-4ketolactams and ketolactones [US7329634] | 2006-05-04 | 2008-02-12 |
| Substituted isoxazoles as fungicides [US7338967] | 2006-04-06 | 2008-03-04 |
| Insecticidal anthranilamides [US7338978] | 2006-04-13 | 2008-03-04 |
| Pyrazolyl carboxanilides for controlling unwanted microorganisms [US7358214] | 2006-04-27 | 2008-04-15 |
//////////////////
C1=CC(=C(C=C1Cl)Cl)C2(CC(CO2)Br)C[N]3C=NC=N3
| Bromuconazole | |
|---|---|
 |
|
| Identification | |
| No CAS | |
| SMILES | |
| InChI | |
| Apparence | cristaux incolores ou poudre sans odeur1. |
| Propriétés chimiques | |
| Formule brute | C13H12BrCl2N3O [Isomères] |
| Masse molaire2 | 377,064 ± 0,017 g/mol C 41,41 %, H 3,21 %, Br 21,19 %, Cl 18,8 %, N 11,14 %, O 4,24 %, |
| Propriétés physiques | |
| T° fusion | 84 °C1 |
| Solubilité | dans l’eau : 0,5 g·l-11 |
| Pression de vapeur saturante | à 25 °C : négligeable1 |
Saperconazole

Saperconazole

The most common systemic fungal infections in humans are blastomycosis, candidosis, aspergillosis, histoplasmosis, coccidioidomycosis, paracoccidioidomycosis, and cryptococcosis.
Fungal diseases are often confined to typical anatomic sites, and many involve a primary focus in the lung, with more characteristic manifestations of specific fungal infections appearing once the infection spreads from a primary site. For example, blastomycosis primarily involves the lungs, and occasionally spreads to the skin. Similarly, the primary form of coccidioidomycosis occurs as an acute, benign, self-limiting respiratory disease, which can then progress to a chronic, often-fatal infection of the skin, lymph glands, liver, and spleen. Other infectious diseases such as paracoccidioidomycosis and candidiasis present in different manners, and depending on the etiology, may exhibit several forms involving internal organs, the lymph nodes, skin, and mucous membranes. Diagnosis of specific fungal diseases can be made by isolation of the causative fungus from various specimens, such as sputum, urine, blood, or the bone marrow, or with certain fungus types, through evidence of tissue invasion.
Many patients suffering from severe systemic fungal infections are hardly, or not at all, able to receive medication via oral administration, as such patients are often in a coma or suffering from severe gastroparesis. As a result, the use of insoluble or sparingly soluble antifungals such as itraconazole free base, which are difficult to administer intravenously to treat such patients, is significantly impeded.
Local or superficial fungal infections are caused by dermatophytes or fungi that involve the outer layers of the skin, nails, or hair. Such infections may present as a mild inflammation, and can cause alternating remissions and eruptions of a gradually extending, scaling, raised lesion. Yeast infections, such as candidiasis and oral candidiasis (thrush), are usually localized to the skin and mucous membranes, with the symptoms varying depending on the site of infection. In many instances, such infections appear as erythematous, often itchy, exudative patches in the groin, axillas, umbilicus, between toes, and on finger-webs. Oral thrush involves an inflamed tongue or buccal mucosa, typically accompanied by white patches of exudate. Chronic mucocutaneous candidiasis is manifested in the form of red, pustular, crusted, thickened lesions on the forehead or nose.Itraconazole or (±)-£is-4-[4-[4-[4-[[2-(2,4-dichlorophenyl)-2-(lH-l-2,4-triazol-l- ylmethyl)- 1 ,3-dioxolan-4-yl]methoxy]phenyl]- 1 -ρiperazinyl]phenyl]-2,4-dihydro-2-( 1 – methyl-propyl)-3H-l,2,4-triazol-3-one, is a broadspectrum antifungal compound developed for oral, parenteral and topical use and is disclosed in US-4,267,179.
The development of effϊcaceous pharmaceutical compositions of itraconazole and saperconazole is hampered considerably by the fact that said compounds are only very sparingly soluble in water. The solubility of both compounds can be increased by complexation with cyclodextrins or derivatives thereof as described in WO 85/02767 and US-4,764,604.
Unexpectedly, it has now been found that each of the individual stereoisomers of itraconazole and saperconazole have greater water solubility than the diastereomeric mixtures of said compounds, in particular when complexed with cyclodextrin or its derivatives. As a result, pharmaceutical compositions having good bioavailability, yet comprising less cyclodextrin as a complexing agent, can be prepared. The present invention is concemced with the stereoisomeric forms of itraconazole (X = CI) and saperconazole (X = F), which may be represented by the formula

cis-©,and the pharmaceutically acceptable acid addition salt forms thereof. The three asterisks indicate the three chiral centers, and ‘cis’ means that the (lH-l,2,4-triazol-l-ylmethyl) moiety and the substituted phenoxy moiety are located at the same side of the plane defined by the 1,3-dioxolane ring.
The four possible stereoisomeric cis forms can be described using various rules of nomenclature. The following tables show the correlation among the C. A. stereochemical descriptor, the absolute configuration at each of the chiral centers and the specific optical
20 rotation [α]jj in 1% methanol (itraconazole; table I) (saperconazole; table H).
Table I

itraconazole

Table π

saperconazole

Itraconazole is a broad-spectrum antifungal agent developed for oral, parenteral and topical use, and is disclosed in U.S. Patent No. 4,267,179. Itraconazole is a synthetic triazole derivative that disrupts the synthesis of ergosterol, the primary sterol of fungal cell membranes. This disruption appears to result in increased permeability and leakage of intracellular content, and at high concentration, cellular internal organelles involute, peroxisomes increase, and necrosis occurs.
As set forth in the USP Dictionary of Drug Names and USAN, itraconazole is defined as 4-[4-[4-[4- [[2-(2,4-dichlorophenyl)-2-(lH-l,2,4-triazol-l-ylmethyl)-l,3-dioxolan-4-yl] methoxy]phenyl]-l-piperazinyl]phenyl]- 2,4-dihydro-2-(l-methylpropyl)-3H-l,2,4-triazol-3-one, or alternatively, as (±)-l-5ec-butyl-4-[/7-[4-[/7-[[(2R*,4S*)-2-(2,4-dichlorophenyl)-2-(lH-l,2,4-triazol-l-ylmethyl)-l,3-dioxolan-4-yl]methoxy]phenyl]-l-piperazinyl]phenyl]-Δ2-l,2,4-triazolin-5-one. There are three asymmetric carbons in itraconazole: one in the sec-butyl side chain on the triazolone and two in the dioxolane ring. As a result, eight possible stereoisomers of itraconazole exist: (R,R,R), (S,S,S), (R,R,S), (S,S,R), (R,S,S), (R,S,R), (S,R,S), and (S,R,R).
(±)Cz‘s-Itraconazole comprises a mixture of only those isomers that describe a “cis” relationship in the dioxolane ring, i.e., the (1Η-1, 2, 4-triazol-l-ylmethyl) moiety and the substituted phenoxy moiety are located on the same side of a plane defined by the 1, 3-dioxolane ring. By convention, the first represented chiral center is at the C-2 position of the dioxolane ring, the second is at the C-4 position of the dioxolane ring, and the third is in the sec-butyl group. Hence, (±)c.s-itraconazole is a mixture of (R,S,S), (R,S,R), (S,R,S) and (S,R,R) isomers.
The four possible stereoisomeric cis forms of itraconazole, and
diastereomeric pairs thereof, are described in more detail in U.S. Patent Nos. 5,474,997 and 5,998,413. In general, the individual stereoisomeric forms of c s-itraconazole have antifungal properties, and contribute to the overall activity of (±)cw-itraconazole.
(±)Ciy-Itraconazole free base is only very sparingly soluble in water, and thus it is extremely difficult to prepare effective pharmaceutical compositions containing the same. A number of means have been used to increase the solubility of itraconazole free base, including complexing or co-formulation with cyclodextrins or derivatives thereof, as described in U.S. Patent No. 4,764,604, U.S. Patent No.5,998,413, and U.S. Patent No. 5,707,975, and coating beads with a film comprising a hydrophilic polymer and itraconazole, as described in U.S. Patent No. 5,633,015.
[0014] Another approach to increase solubility of itraconazole focuses on preparation of the stereoisomers of c s-itraconazole, and in particular (2R, 4S) itraconazole, which may comprise a mixture of two diastereomers ((R,S,S) and
(R,S,R)), as described in U.S. Patent Nos. 5,414,997 and 5,998,413.
Commercially available itraconazole (SPORANOX® brand (±)cis-itraconazole, Janssen Pharmaceutica Products, L.P., Titusville, NJ, U.S.A.) is a free base and a racemic mixture of the cis isomer in the dioxolane ring and is represented by structural formula (I):

(i)
SPORANOX has been approved for use as an antifungal agent for treating immunocompromised and non-immunocompromised patients having: blastomycosis (pulmonary and extrapulmonary); histoplasmosis, including chronic cavitary pulmonary disease and disseminated non-meningeal histoplasmosis; and aspergillosis. In addition, in non-immunocompromised patients, it has been approved for treatment of onychomycosis. See generally, Physician ‘s Desk Reference, 56th ed. (2002). The compound has also been investigated for use in coccidioidomycosis, cryptococcosis, dermatophyte, and candidiasis infections.
Adverse effects associated with the administration of (±)cts-itraconazole free base include nausea, vomiting, anorexia, headache, dizziness, hepatotoxicity, and inhibition of drug metabolism in the liver, leading to numerous, clinically significant, adverse drug interactions. See, Physician ‘s Desk Reference, 56th ed. (2002); Honig et al., J. Clin. Pharmacol. 33:1201-1206 (1993) (terfenadine interaction); Gascon and Dayer, Eur. J. Clin. Pharmacol., 41_:573-578 (1991) (midazolam interaction); and Neuvonen et al, Clin. Pharmacol. Therap., 60:54-61 (1996) (lovastatin interaction). Reactions associated with hypersensitivity, such as urticaria and serum liver enzymes elevation, are also associated with the administration of the drug. A more serious, though less common, adverse effect is hepatotoxicity. See, e.g., Lavrijsen et al., Lancet, 340:251-252 (1992).
In addition, as discussed herein, c/s-itraconazole free base is only very sparingly soluble in water. Thus, due to its relative non-polarity and insolubility, itraconazole free base suffers from two other drawbacks: it cannot be readily formulated in parenteral solution, and it does not effectively penetrate the blood-brain barrier. The latter problem is exacerbated by drug interactions, such as one observed between itraconazole free base and valproate, as described in Villa et al. , Rev. Inst. Med. Trop., Sao Paulo, pp. 231-234 (Jul-Aug 2000), which is incorporated by reference herein in its entirety. In another case of CNS fungal infection, extremely high doses of itraconazole free base were used to treat residual aspergillus infection, as reported by Imai et al., Intern. Med, 38(10):829-832 (1999), which is incorporated by reference herein in its entirety. As a result, numerous therapeutic indications that require rapid achievement of effective blood levels or access to the CNS are difficult to treat or beyond treatment with itraconazole free base.
Furthermore, the emergence of antifungal resistance (e.g., in Aspergillus fumigatus isolates as described by Dannaoui et al., J. Antimicrob. Chemother., 47:333-340 (2001), which is incorporated by reference herein in its entirety) presents an added challenge to the efficacy of itraconazole free base. For those strains of fungi that show resistance, high and relatively constant levels of itraconazole free base must be produced in the target organs of infected patients.
Over the years, a number of formulation routes have been used in order to enhance the adsorption and bioavailability of itraconazole. For example, the currently marketed SPORANOX® solid dosage capsule form of itraconazole free base utilizes sugar-based beads coated with a hydrophilic polymer and an amorphous film of itraconazole. See Physicians Desk Reference, 56th ed., pp.1800- 1804 (2002); and U.S. Patent No. 5,633,015. This dosage form requires up to two capsules three times daily depending on the condition being treated.
Even with the various formulation routes, the dosage amounts and dose frequency for itraconazole can be burdensome to patients. In addition, administration of existing dosage forms of itraconazole have shown significant variability in bioavailability and adsorption, which likely results from food effects. See, Physician ‘s
Desk Reference, 56th ed., pp. 1800-1804 (2002). Thus, it would be desirable to increase bioavailability and adsorption and decrease the per-dose pill count and decrease dosing frequency (e.g., twice a day to once a day) associated with administration of itraconazole in order to provide an improvement over current therapy, particularly with regard to patient compliance, convenience, ease of ingestion, especially with regard to immunocompromized polypharmacy patients (e.g., AIDS or cancer patients).
Posaconazole and Saperconazole Chemistry and Uses
Other related conazoles have also been discovered and used as antifungals. Two of these conazoles that are closely structurally related to itraconazole are posaconazole and saperconazole. Posaconazole (CAS Registry Number: 171228-49-2; CAS Name: 2,5-Anhydro-l ,3,4-trideoxy-2-C-(2,4-difluorophenyl)-4-[[4-[4-[4-[l -[(1 S,2S)- 1 -ethyl-2-hydroxypropyl]- 1 ,5-dihydro-5-oxo-4H- 1 ,2,4-triazol-4-yl]phenyl]- 1 -piperazinyl]phenoxy]methyl]- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)-D-t/Veo-pentitol; Additional Name: (3R-c s)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(l,2,4-triazol-l-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin- 1 -yl]phenyl]-2-[l (S)-ethyl-2(S)-hydroxypropyl]-3,4-dihydro-2H-l,2,4-triazol-3-one) is represented by structural formula (II):

(II)
Saperconazole (CAS Registry Number: 110588-57-3; CAS Name: 4-[4-[4-[4-[[2-(2,4-Difluorophenyl)-2-(lH-l,2,4-triazol-l-ylmethyl)-l,3-dioxolan-4-yl]methoxy]phenyl]- 1 -piperazinyl]phenyl]-2,4-dihydro-2-(l -methylpropyl)-3H- 1 ,2,4-triazol-3-one; Additional Name: (±)-l-sec-butyl-4-[ -[4-| -[[(2R* 4S*)-2-(2,4- difluorophenyl)-2-( 1 H- 1 ,2,4-triazol- 1 -ylmethyl)- 1 ,3 -dioxolan-4-yl]methoxy]phenyl]- 1 -piperazinyl]phenyl]-Δ2-l,2,4-triazolin-5-one) is represented by structural formula (III):

(III)
Consequently, there is a need for soluble forms of conazoles including cis itraconazole, posaconazole and saperconazole that can be readily formulated for use in various modes of administration, including parenteral and oral administration.
A. Preparation of intermediates: Example 1a) utilizing water separator, by 200 parts of glycerin, 90 parts of 1- (2,4-difluorophenyl) -2- (1H-1,2,4- three mixture of 1-yl) ethanone, 600 parts of methanesulfonic acid, 190 parts of benzene was stirred first at reflux for 3 hours, then stirred at room temperature overnight. The reaction mixture was added dropwise a solution of sodium bicarbonate. The product was extracted with chloroform, the extract was washed with water, dried, filtered and evaporated. With 4-methyl-2-pentanone and the residue triturated product was filtered off and dried, yielding 80 parts (67.2%) (cis + trans) -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolane-4-methanol (intermediate 1).
b) by 69 parts of 3,5-dinitrobenzoyl chloride, 80 parts of (cis + trans) -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolane-4-methanol, 400 parts of pyridine and 520 parts of dichloromethane was stirred at room temperature for 3 hours. The reaction mixture was evaporated, and the residue was dissolved in water. The product was extracted with chloroform. The extract was dried, filtered and evaporated. The residue was subjected to silica gel column chromatography, eluting with chloroform / methanol (99:1v / v). Pure fractions were collected, the eluent was evaporated, to give 90 parts (70.4%) of cis -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1 ylmethyl) -1,3-dioxolane-4-methanol 3,5-dinitrobenzoate (residue) (intermediate 2).
c) by 90 parts of (cis) -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxo- dioxolan-4-methanol 3,5-dinitrobenzoate, 16 parts of 50% sodium hydroxide solution, 800 parts of 1,4-dioxane, 400 parts of water and the mixture was stirred at room temperature overnight. The reaction mixture was poured into water and the product was extracted with dichloromethane, extracts washed with water, dried, filtered and evaporated. With 4-methyl-2-pentanone and the residue triturated product was filtered off and dried, yielding 30 parts (56.0%) of cis -2- (2,4-difluorophenyl) -2- (1H-1, 2,4-triazol-1-ylmethyl) -1,3-dioxolane-4-methanol (residue) (intermediate 3).
d) by 11.4 parts of methanesulfonyl chloride, 25 parts of cis -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1, mixture of 1,3-dioxolane-4-methanol, 300 parts of pyridine, 390 parts of dichloromethane was stirred at room temperature for 3 hours. The reaction mixture was evaporated, and the residue was dissolved in chloroform. The organic phase was dried, filtered and evaporated. The residue was triturated with dipropyl ether. The product was filtered off and dried, yielding 29.4 parts (93.2%) of cis -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) – 1,3-dioxolane-4-methanol methanesulfonate (residue) of intermediate 4).
In a similar manner there were also prepared: cis-2- (2,4-difluorophenyl) -2- (1H- imidazol-1-ylmethyl) -1,3-dioxolane-4-methanol mesylate ethanedioate (1/1) (interm. 5).
Example 2a) over 2 hours, dissolved in 100 parts of pyridine 121.2 parts of 2-naphthalenesulfonyl chloride was added dropwise to a stirred, was dissolved in 1300 parts of dichloromethane, and 122.0 parts of (cis + trans ) -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolane-4-methanol and 1.0 parts of N, N- dimethyl-4-pyridin-amine solution. Upon completion, stirring was continued at room temperature overnight. The reaction mixture was washed twice with water, and evaporated under reduced pressure. The residue was purified by silica gel column chromatography, eluting with chloroform. Pure fractions were collected, the eluent was evaporated. The residue was crystallized from 4-methyl-2-pentanone. The product was filtered off and dried, yielding 102.3 parts (51.0%) of cis – [[2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-yl-methyl ) -1,3-dioxolan-4-yl] methyl] -2-naphthalene sulfonate; mp139.5 ℃ (intermediate 6).
Example 3a) at 70 ℃, under nitrogen atmosphere, by 9.0 parts of 4- [4- (4-nitrophenyl) -1-piperazinyl] phenol, 13.6 parts of cis-2- [2,4- difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolane-4-methanol methanesulfonate ester, 6.0 parts of potassium hydroxide and 90 parts of a mixture of DMF was stirred overnight. After cooling, the reaction mixture was diluted with water. The precipitated product was filtered off and purified by silica gel column chromatography, the chloroform / ethyl acetate / hexane / methanol (500:300:200:0.5v / v / v / v) mixture as eluent. Pure fractions were collected, the eluent was evaporated. The residue was crystallized 4-methyl-2-pentanone. The product was filtered off and dried, yielding 6.69 parts (38.5%) of cis -1- [4 – [[2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol – 1- ylmethyl) -1,3-dioxolan-4-yl] methoxy) phenyl] -4- (4-nitrophenyl) piperazine; mp169.8 ℃ (Intermediate 7) .
b) at atmospheric pressure, 50 ℃, with 2 parts of 5% palladium – on-charcoal catalyst by 38.3 parts of cis -1- [4 – [[2- (2,4-difluorophenyl) -2- (1H -1,2,4-triazol-1-ylmethyl) -1,3-dioxolan-4-yl] methoxy] phenyl] -4- (4-nitrophenyl) piperazine, 2 parts of a solution of thiophene (4% solution in methanol) and 600 parts of 2-methoxy-ethanol mixture. After absorption of the calculated amount of hydrogen finished, hot filtered to remove the catalyst, and the filtrate was saturated with water. After cooling, the precipitated product was filtered off, washed with water and 2-propanol and crystallized from 1,4-dioxane. The product was filtered off and dried, yielding 22.7 parts (62.6%) of cis-4- [4- [4 – [[2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolan-4-yl] methoxy] phenyl] -1-piperazinyl] aniline; mp193.0 ℃ (interm. 8).
Example 4a) by 10 parts of 2,4-dihydro-4- [4- [4- [4-methoxyphenyl) -1-piperazinyl] phenyl] -3H-1,2,4- triazol-3-one (U.S. Patent No. 4,267,179 in the implementation of the method in Example ⅩⅦ obtained), 1.5 parts of sodium hydride (50% dispersion), 300 parts of the mixture consisting of dimethyl sulfoxide, at 60 ℃ under a nitrogen atmosphere begging, stirring, until no bubble up. Was then added 5.24 parts of 2-bromopropane, and at 60 ℃, stirring was continued for 1 hour. Further added 1.5 parts of sodium hydride (50% dispersion) and stirring was continued until no more bubble up. Then 5.24 parts of 2-bromopropane was added, and the whole was stirred for 1 hour at 60 ℃. The reaction mixture was cooled, poured into water and the product was extracted with chloroform. The extract was washed with water, dried, filtered and evaporated. The residue was purified by silica gel column chromatography, eluting with chloroform / methanol (99:1v / v). Pure fractions were collected, the eluent was evaporated, the residue was crystallized in 1-butanol, yielding 5.2 parts (47% (2,4-dihydro-4- [4- [4- (4-methoxyphenyl ) -1-piperazinyl] phenyl] -2- (1-methylethyl) -3H-1,2,4- triazol-3-one; mp209.5 ℃ (intermediate 9).
b) by 4.7 parts of 2,4-dihydro-4- [4- [4- (4-methoxyphenyl) -1-piperazinyl] phenyl] -2- (1-methylethyl) -3H-1,2,4- triazol-3-one, a mixture of 75 parts of 48% aqueous hydrobromic acid was stirred at reflux for 3 hours. The reaction mixture was evaporated, and the residue was dissolved in a mixture of methanol and water. With sodium bicarbonate solution, and the whole was, and the product was extracted with chloroform. The extract was dried, filtered and evaporated. The residue was triturated with 2-propanol, yielding 3.9 parts (86%) of 2,4-dihydro-4- [4- [4- (4-hydroxyphenyl) -1-piperazinyl] phenyl] -2 – (1-methylethyl) -3H-1,2,4- triazol-3-one, mp208.4 ℃ (intermediate 10).
PATENT
| EP0006711A1 * | 13 Jun 1979 | 9 Jan 1980 | Janssen Pharmaceutica N.V. | Heterocyclic derivatives of (4-phenylpiperazin-1-yl-aryloxymethyl-1,3-dioxolan-2-yl)-methyl-1H-imidazoles and 1H-1,2,4-triazoles, processes for preparing them and compositions containing them |
| EP0118138A1 * | 24 Jan 1984 | 12 Sep 1984 | Janssen Pharmaceutica N.V. | ((4-(4-(4-Phenyl-1-piperazinyl)phenoxymethyl)-1,3-dioxolan-2-yl)methyl)-1H-imidazoles and 1H-1,2,4-triazoles |
| DE2804096A1 * | 31 Jan 1978 | 3 Aug 1978 | Janssen Pharmaceutica Nv | 1-(1,3-dioxolan-2-ylmethyl)-1h-imidazole und -1h-1,2,4-triazole und deren salze, verfahren zu ihrer herstellung und ihre verwendung bei der bekaempfung pathogener pilze und bakterien |
| Patent | Submitted | Granted |
|---|---|---|
| ORDERED MESOPOROUS SILICA MATERIAL [US2011081416] | 2010-10-15 | 2011-04-07 |
| BENZOYL PEROXIDE COMPOSITION FOR TREATING SKIN [US2011082216] | 2009-10-02 | 2011-04-07 |
| METHODS RELATED TO TIM 3, A TH1-SPECIFIC CELL SURFACE MOLECULE, FOR ACTIVATING ANTIGEN PRESENTING CELLS [US2015044229] | 2014-08-20 | 2015-02-12 |
| METHODS RELATED TO TIM 3, A TH1-SPECIFIC CELL SURFACE MOLECULE, FOR ACTIVATING ANTIGEN PRESENTING CELLS [US2015044230] | 2014-08-20 | 2015-02-12 |
| COSMETIC METHOD FOR INCREASING COLLAGEN EXPRESSION IN SKIN COMPRISING TOPICALLY APPLYING AN EXTRACT OF QUASSIA AMARA [US2015056310] | 2014-08-20 | 2015-02-26 |
| Flexible bone composite [US8771721] | 2013-03-15 | 2014-07-08 |
| Topical formulation [US8513304] | 2012-11-19 | 2013-08-20 |
| Prolonged release bioadhesive therapeutic systems [US8518442] | 2010-07-02 | 2013-08-27 |
| Preparation method for solid dispersions [US8216495] | 2009-03-25 | 2012-07-10 |
| Flexible bone composite [US8221782] | 2011-08-12 | 2012-07-17 |
| Patent | Submitted | Granted |
|---|---|---|
| Crystalline forms of conazoles and methods of making and using the same [US7446107] | 2005-03-31 | 2008-11-04 |
| CIS-itraconazole crystalline forms and related processes, pharmaceutical compositions and methods [US7078526] | 2004-01-29 | 2006-07-18 |
| Novel Saperconazole Crystalline Forms and Related Processes, Pharmaceutical Compositions and Methods [US2007293674] | 2007-12-20 | |
| NOVEL CRYSTALLINE FORMS OF CONAZOLES AND METHODS OF MAKING AND USING THE SAME [US2009088443] | 2009-04-02 | |
| CONTROLLED RELEASE VEHICLES HAVING DESIRED VOID VOLUME ARCHITECTURES [US2014328884] | 2012-12-17 | 2014-11-06 |
| MOLECULES WITH POTENT DHFR BINDING AFFINITY AND ANTIBACTERIAL ACTIVITY [US2014329840] | 2014-05-05 | 2014-11-06 |
| FUNCTIONALLY-MODIFIED OLIGONUCLEOTIDES AND SUBUNITS THEREOF [US2014330006] | 2012-11-15 | 2014-11-06 |
| ASPARTYL-TRNA SYNTHETASE-FC CONJUGATES [US2014335087] | 2012-12-27 | 2014-11-13 |
| GASTRORETENTIVE CONTROLLED RELEASE VEHICLES THAT INCLUDE ETHYLENE COPOLYMERS, ETHYL CELLULOSES, AND/OR THERMOPLASTIC POLYURETHANES [US2014348936] | 2012-12-17 | 2014-11-27 |
| HISTIDYL-TRNA SYNTHETASE-FC CONJUGATES [US2014349369] | 2014-03-14 | 2014-11-27 |
| ASPARTYL-TRNA SYNTHETASES [US2014302075] | 2012-12-06 | 2014-10-09 |
| Rhinosinusitis Prevention and Therapy with Proinflammatory Cytokine Inhibitors [US2014311482] | 2014-01-24 | 2014-10-23 |
| POLYSACCHARIDE ESTER MICROSPHERES AND METHODS AND ARTICLES RELATING THERETO [US2014315720] | 2014-04-04 | 2014-10-23 |
| MODIFIED GREEN TEA POLYPHENOL FORMULATIONS [US2014256616] | 2014-05-19 | 2014-09-11 |
| PLANT-BASED COMPOSITIONS AND USES THEREOF [US2014260466] | 2013-03-15 | 2014-09-18 |
| PLANT-BASED COMPOSITIONS AND USES THEREOF [US2014271928] | 2014-03-14 | 2014-09-18 |
| LIGHT AND ULTRASONIC TRANSDUCER DEVICE [US2014276247] | 2014-03-14 | 2014-09-18 |
| LIGHT AND/OR ULTRASONIC TRANSDUCER DEVICE WITH SENSOR FEEDBACK FOR DOSE CONTROL [US2014276248] | 2014-03-14 | 2014-09-18 |
| PHOTOPROTECTIVE COMPOSITION CONTAINING AN UNMODIFIED GELLING STARCH AND POLYAMIDE PARTICLES [US2014287005] | 2014-03-18 | 2014-09-25 |
| STABILIZED CHEMICAL DEHYDRATION OF BIOLOGICAL MATERIAL [US2014227686] | 2014-04-16 | 2014-08-14 |
| METHODS RELATED TO TIM 3, A TH1-SPECIFIC CELL SURFACE MOLECULE, FOR ACTIVATING ANTIGEN PRESENTING CELLS [US2014242094] | 2014-02-20 | 2014-08-28 |
| NOVEL ENCOCHLEATION METHODS, COCHLEATES AND METHODS OF USE [US2014242153] | 2014-01-30 | 2014-08-28 |
| METHODS OF REDUCING THE PROLIFERATION AND VIABILITY OF MICROBIAL AGENTS [US2010197621] | 2010-08-05 | |
| METHODS OF ADMINISTERING TOPICAL ANTIFUNGAL FORMULATIONS FOR THE TREATMENT OF FUNGAL INFECTIONS [US2010086504] | 2010-04-08 | |
| COMPOSITIONS AND METHODS FOR INCREASING ERYTHROPOIETIN (EPO) PRODUCTION [US2014024699] | 2011-12-09 | 2014-01-23 |
| PROLONGED RELEASE BIOADHESIVE THERAPEUTIC SYSTEMS [US2013310335] | 2013-07-26 | 2013-11-21 |
| Pharmaceutical Composition [US2013315988] | 2011-05-23 | 2013-11-28 |
| Topical Foam Composition [US2013315998] | 2013-08-05 | 2013-11-28 |
| ANTIFUNGAL NAIL COAT AND METHOD OF USE [US2013323189] | 2013-08-09 | 2013-12-05 |
| TOPICAL FORMULATIONS, SYSTEMS AND METHODS [US2013337031] | 2013-03-08 | 2013-12-19 |
Saroglitazar, Lipaglyn by Zydus Cadila

(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid
alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid magnesium salt
(2S)-2-ethoxy-3-[4-[2-[2-methyl-5-(4-methylsulfanylphenyl)pyrrol-1-yl]ethoxy]phenyl]propanoic acid
Benzenepropanoic acid, α-ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]-, (αS)-
ZYH1 compound
Cas no 495399-09-2
Saroglitazar, Lipaglyn
| Molecular Weight | 439.56706 g/mol |
|---|---|
| Molecular Formula | C25H29NO4S |
Cadila Healthcare Ltd innovator
Zydus-Cadila has developed and launched saroglitazar (ZYH-1; Lipaglyn; structure shown), a lipid metabolism modulator, a potent PPAR-alpha agonist with relatively weak PPAR-gamma activity, an insulin sensitizer (glucose-lowering agent), for the once-daily oral treatment of metabolic disorders, including diabetic dyslipidemia and hypertriglyceridemia . The company is also developing saroglitazar for the potential treatment of lipodystrophy, nonalcoholic steatohepatitis (NASH) and type II diabetes.
In June 2013, the Drug Controller General of India (DCGI) approved the drug for launch in India ; in September 2013, the drug was launched. In May 2014, a phase III trial for lipodystrophy was initiated . In January 2015, a phase III study for NASH was initiated .
In February 2015, phase III development was ongoing in type II diabetes . In November 2015, a phase II trial was planned in the US . In June 2016, the US FDA approved the company’s plan to initiate a phase II trial of saroglitazar in patients with NASH .
In June 2012, the company was seeking to outlicense the drug for regional/global partnerships.
By June 2012, an NDA filing had been made for dyslipidemia . In June 2013, the DCGI approved the drug for launch in India . By September 2013, the drug was launched for dyslipidemia and hypertriglyceridemia .
Saroglitazar was approved by the Drug Controller General of India (DCGI) on June 5, 2013. It was developed and marketed as Lipaglyn® by Zydus cadila.
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of the peroxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action on PPARγ improves insulin resistance and consequently lowers blood sugar. It is indicated for for the treatment of diabetic dyslipidemia and hypertriglyceridemia with type 2 diabetes mellitus not controlled by statin therapy.
Lipaglyn® is available as tablet for oral use, containing 4 mg of free Saroglitazar. The recommended dose is 4 mg orally once daily.
Zydus-Cadila has developed and launched saroglitazar for treating diabetic dyslipidemia and hypertriglyceridemia.
In September 2013, saroglitazar was launched in India for treating dyslipidemia and hypertriglyceridemia.
As of March 2015, Zydus-Cadila is developing saroglitazar for treating nonalcoholic steatohepatitis and type II diabetes (both in phase III clinical trials).


Saroglitazar (INN, trade name Lipaglyn) is a drug for the treatment of type 2 diabetes mellitus and dyslipidemia. It is approved for use in India by the Drug Controller General of India.[1] Saroglitazar is indicated for the treatment of diabetic dyslipidemia andhypertriglyceridemia with type 2 diabetes mellitus not controlled by statin therapy. In clinical studies, saroglitazar has demonstrated reduction of triglycerides (TG), LDL cholesterol, VLDL cholesterol, non-HDL cholesterol and an increase in HDL cholesterol a characteristic hallmark of atherogenic diabetic dyslipidemia (ADD). It has also shown favorable Anti-diabetic medication property by reducing the fasting plasma glucose and HBA1c in diabetes patients. The recommended dose of saroglitazar is one tablet of 4 mg once a day.
In February 2013, Saroglitazar became the first glitazar that has been approved by any FDA for clinical use. Saroglitazar is marketed under the trade name Lipaglyn and developed by Zydus Cadila. Saroglitazar (2 and 4 mg q.d.) is currently approved in India by Drug Controller General of India (DCGI ) for the management of diabetic dyslipidemia and hypertriglyceridemia in T2DM not controlled by statin therapy. Lipaglyn provides the option of a once-daily oral therapy for the patients suffering from diabetic dyslipidemia.
Saroglitazar has another first attached to it. It is the first indigenously developed NCE by any Indian company; in this case Zydus Cadila.
Lipaglyn is indicated 4 mg (or 2 mg where such a need arise) oral dose once daily.
Saroglitazar Synthesis
http://ayurajan.blogspot.in/2016/01/saroglitazar.html
WO2003009841A1:
Identification:
| 1H NMR (Estimated) for Saroglitazar |
Experimental: 1H NMR: 1.14 (3H, t, J = 6.9Hz); 2.37 (3H, s); 2.48 (3H, s); 2.92-3.06 (2H, m); 3.32-3.42 (1H, m); 3.57-3.64 (1H, m); 3.9 (2H, t, J=6.36 Hz); 4.0 (1H, dd); 4.28(2H, t, J = 6.2 Hz); 5.9 (1H, d, J = 3.3 Hz); 6.08 (1H, d, J = 3.38 Hz); 6.6 (2H, d, J = 8.5Hz); 7.1(2H, d, J = 8.5Hz); 7.26 (2H, d, J = 8.4Hz); 7.3 (2H, d, J = 8.34Hz)



Details see below
Mechanism of action
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of theperoxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action onPPARγ improves insulin resistance and consequently lowers blood sugar.[2]
Efficacy
Being a dual PPAR agonist, Saroglitazar (Lipaglyn) helps in controlling blood glucose and Lipid parameters especially high triglycerides and high non HDL-Cholesterol.[3] Lipaglyn effectively reduces triglycerides and non HDL-C and controlles high blood sugar, a typical situation in Insulin Resistance condition.[4][5]
Safety
Saroglitazar has not demonstrated any of the adverse effects like weight gain and edema that are usually identified with similar molecules like the glitazone class of drugs.[6] Because it is an insulin sensitizer, Saroglitazar (Lipaglyn) has less potential for hypoglycemia. No major serious adverse events have been reported; however, long-term cardiovascular safety has not been established.[7]
| Saroglitazar, is a drug for the treatment of diabetic dyslipidemia and hypertriglyceridemia with Type 2 diabetes mellitus not controlled by statin therapy. Its trade name is Lipaglyn. It is also a 1,2-Diarylpyrroles derivative, which can be used in the preparation of Nonsteroidal anti-inflammatory drugs (NSAIDs). |
| References: Khanna, I. K., et al.: J. Med. Chem., 40, 1619 (1997) |
_MoA.png)
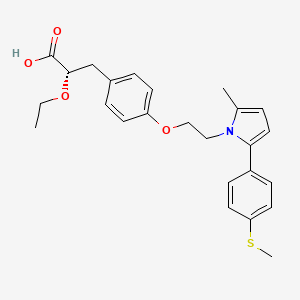
PAPER
A new enantioselective synthesis of (S)-2-ethoxy-3-(4-hydroxyphenyl)propanoic acid esters (EEHP and IEHP), useful pharmaceutical intermediates of PPAR agonists
Tetrahedron Lett 2014, 55(21): 3223
http://www.sciencedirect.com/science/article/pii/S0040403914006200

PATENT
WO 2003009841
http://www.google.co.in/patents/WO2003009841A1?cl=en
PATENT
US 20030236254
http://www.google.com/patents/US20030236254
PATENT
US 20140099333
http://www.google.com/patents/US20140099333
PATENT
http://patentscope.wipo.int/search/en/WO2014174524
 (I)
(I)
The compound as claimed in claim 1 wherein R is -SMe and M+ is Mg+2.
The compound of claim 1 is Saroglitazar.
wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. r .
PATENT
3-Aryl-2-hydroxy propanoic acid derivatives serve as a key intermediate for the synthesis of many pharmaceutically important compounds especially, peroxime proliferator activated receptor (PPAR) agonist.
Optically active 3-aryl-2-alkoxy propanoic acid and its esters, particularly, ethyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (EEHP) and isopropyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (IEHP) are versatile chiral pharmacophores present in many pharmaceutically important compounds, especially in peroxisome proliferator activated receptor (PPAR) agonists that have beneficial effects in treating Type 2 diabetes.
Several PPAR agonists, in particular PPAR α/γ dual agonists, commonly termed as glitazars (Ragaglitazar, Tesaglitazar, Navaglitazar etc.), as shown in the figure below were developed by many pharmaceutical companies that have a potential application in the treatment of Type 2 diabetes and dyslipidemia.
However, many of these drugs were discontinued due to their undesirable side effects, but some of them still have great potential [For example, Saraglitazar (LipaglynTM) developed by Zydus Cadila got approval in India for the treatment of diabetic dyslipidemia or hypertriglyceridemia]. Several PPAR α/γ agonists possessing chiral (S)-l moieties are shown below.

Tesaglitazar Naveglitazar
In addition, these derivatives find an application in photosensitive materials, sweetening agents, treatment of certain eating disorders etc. Therefore, these compounds have attracted a great deal of attention of synthetic chemists and different methods of preparation of the compound of formula (S)-l have been extensively studied.
Generally, the reported protocols for the synthesis involve chiral pool approaches starting from L-tyrosine and its derivatives (Refer WO 02/24625, US 6559335B2, WO 2003/027084), asymmetric synthesis (Org. Lett. 2005, 7, 1947, US 2007/0149804) and resolution processes using chiral amines or enzymes (WO 2000/026200, WO 2001/11073, Org. Process Res. Dev. 2003, 7, 82, Org. Process Res. Dev. 2004, 8, 838, Tetrahedron Asymmetry 2009, 20, 2594).
Some of these methods have disadvantages such as expensive chiral starting materials and catalysts, low enantioselectivity and overall yields, problems associated with the O-alkylation step which often leads to the loss of optical purity, and many others.
The processes described in WO20026200 (Rao et. al.) uses benzyl bromide for benzylation, which is highly lachrymatory. Again, in the processes described, the debenzylation of the final intermediate was done by using Pd/C under pressure, which escalates the process economics.
WO2003024915 describes a process for the preparation 3-aryl-2-hydroxy propanoic acid derivatives from 3-(4-hydroxyphenyl)-2-oxopropanoic acid.
WO 2003008362 describes 3-Aryl-2-hydroxy propanoic acid derivatives of formula I and the preparation thereof.

wherein Rland R2 may be same or different and represent hydrogen or (CI- C6) alkyl.
The process is depicted in Scheme 1 below.
Scheme 1

In another process variant as in Scheme 2, WO’362 discloses a process for the preparation of novel 3-aryl-2 -hydroxy propanol and their derivatives of the formula (I)

wherein OR and OR together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms, which comprises: i) reducing the compound of formula (III) where R represents hydrogen or alkyl group, R3 represents benzyl to a compound of formula (IV) where R3 represents benzyl, ii) cyclizing the compound of formula (IV) to a compound of formula (V) where ORl and OR2 together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms and R3 represents benzyl and iii) debenzylating the compound of formula (V) in the presence of metal catalysts to yield pure compound of formula (I).
Scheme 2

Both the processes described in WO’362 result in poor overall yield and further fail to describe the preparation of compound of formula V using different alkylating agents. This document exemplifies the compound of formula V with similar ether groups as it fails to teach selective alkylation of formula IV.
WO2005019152 discloses an improved process for the preparation of compound of the general formula (la) and (lb).

Wherein, Rl represent H or (C1-C6) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like. R2 represents (Ci-Ce) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t- butyl and the like. R3 represents H, protecting groups such as benzyl, substituted benzyl, (C1-C3) alkyl and like.
The compound of general formula (la) is prepared according to the following schemes 3 and 4.
Scheme 3

Both the processes start with selective O-alkylation or O-aralkylation of L-Tyrosine of formula (2a) using a base, a chelating agent, an alkyl or aralkyl halide in the presence of solvents to obtain the compound of formula (3a), which is diazotized to obtain formula (4a) which upon dialkylation using an excess of alkylating agent and excess base, in presence of suitable solvent to obtain optically pure compound of formula (la). Alternatively, compound of formula (4a) may be selectively esterified to obtain compound of formula (5a), which is subsequently O-alkylated to obtain compound of formula (la) (Scheme 2).
However, the above processes have many disadvantages such as multistep synthesis including protection & deprotection and low overall yield. Further, low temperature diazotization on industrial scale is not viable. Moreover, the starting material is very expensive and hence escalates the process.
In the light of the foregoing, development of a new, alternate enantio-selective synthetic route to these important chiral intermediates, which are simple and can preserve the optical purity at the C-2 carbon of 3-Aryl-2-hydroxy propanoic acid derivatives, is highly desirable. There is a need for an efficient process for synthesis of 3-Aryl-2-hydroxy propanoic acid derivatives of formula (S)-l in high enantiopurity and good overall yield from commercially available starting material.
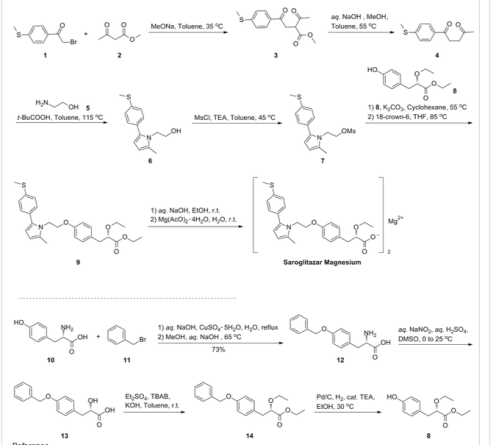
OR

Synthesis of saroglitazar
1. 2-Bromo-1-[4-(methylthio)phenyl]ethanone is condensed with methyl acetoacetate in the presence of NaOMe and Na2SO4 in toluene, to give alpha-keto methyl ester ,
2. This alpha-keto methyl ester ,is hydrolyzed and decarboxylated by means of NaOH in MeOH/toluene at 50 °C giving diketone .
3. Diketone is subjected to Paal-Knorr reaction with ethanolamine in the presence of pivallic acid in toluene at 110 °C to yield pyrrole primary alcohol derivative .
4. Sulfonylation of this pyrrole primary alcohol with MsCl in the presence of Et3N,
5. O-alkylation of mesylate with ethyl 2(S)-ethoxy-3-(4-hydroxyphenyl)propionate in the presence of K2CO3, optionally in the presence of 18-crown-6 in toluene/THF at 80 °C provides ether.
6. Finally, hydrolysis of ethyl ester using NaOH in H2O affords the target saroglitazar.
PATENT
saroglitazar magnesium alongwith its intermediates may be prepared by the reaction scheme- 1, scheme-2 and scheme-3 as shown below, which is also the scope of the present invention.

Scheme-1

EXAMPLES
Example-l:
Preparation of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, sodium methoxide (165 g) and toluene (1000.0 ml) were added under nitrogen environment and cooled to 8°C to 12°C. Methyl acetoacetate (331.55 g) was added dropwise and stirred for 1 hour. 2-bromo-l-(4-methyl sulfonyl phenyl) ethanone (500.0 g) compound (El) in toluene (1500.0 ml) and sodium sulfate
(75.0 g) mixture was stirred for 10 min and filtered at 25° to 35°C. The filtrate as obtained was added dropwise into the previous reaction mixture and stirred at 30°C to 35°C for 30 min. The organic layer was collected and washed with 10% sodium bicarbonate solution. The separated organic layer was collected and washed with water. 2-[2-(4-Methyl sulfanyl-phenyl)-2-oxo-ethyl]-3-oxo-butynic acid methyl ester as obtained in toluene layer is diluted with methanol (2500 ml) and sodium hydroxide solution (89.75 g) in water (2500 ml) was added and heated to 50° to 55°C for 1 hour. The layers were separated and the toluene layer was collected and heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (57.3 g) and ethanol amine (143.9 g) were added and heated to 105° to 1 15°C for removing water azeotropically. The toluene layer was separated and triethyl amine (271.85 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (282.5 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum to obtain the residue. The residue was dissolved in toluene (1500 mL) and used for further process.
ExampIe-2:
Preparation of methanesulfonic acid 2-f2-methyl-5-(4-methylsulfanyl-pheny0-pyrrol- 1-viyethyl ester (Al)

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 4-(methylthio)benzaldehyde (10 g), methyl vinyl ketone (3.63 g), triethylamine (9.95 g) and 3-methyl-5-(2-hydroxyethyl)-4-methyI thiazolium iodide (stetter
catalyst) (2.8 g) were heated to 70°C to 80°C and maintained overnight. The reaction mixture was cooled to room temperature and ethanol (100 mL) was added. The reaction mixture was stirred for 30 min and filtered. The product was washed with ethanol and dried to obtain 1 ,4-diketo compound (CI).
1 ,4-diketo compound (CI) obtained above and toluene (50 mL) were heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (5.7 g) and ethanol amine (14.4 g) were added and heated to 105° to 1 15°C and cooled to 25°C. Triethyl amine (27.2 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (28.3 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum, methanol (2500 ml) was added and heated to 55° to 65 °C and charcoalized for 30 min. The reaction mixture was filtered and washed with methanol. The reaction mixture was cooled to 25° to 35°C and stirred for 30 min. Reaction mass was further cooled to -5° to 5°C and filtered. The wet-cake was washed with methanol and dried to obtain compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.2).
Example-3:
Purification of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 70 g methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l -yl]-ethyl ester (Al) and 420 mL ethyl acetate were added at 25°C. The reaction mixture was stirred for 30 min to obtain clear solution. 3.5 g charcoal was added and stirred for 30 min. The reaction mixture was filtered and washed with ethyl acetate. The filtrate was concentrated and 315 mL methanol was added. The reaction mixture was stirred for 2 hours at 25°C and cooled to 0°C. The product precipitated was filtered and washed with methanol to obtain crystalline
compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.3).
Example-4:
Preparation of saroglitazar magnesium (T)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) (100.0 g) and toluene (1300.0 ml) were charged and reaction mixture was heated to 45° to 55°C. Potassium carbonate (58.0 g) was added and stirred for 30 min. Toluene solution of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol- 1 -yl]-ethyl ester (Al) (150.24 g) obtained in example- 1, 18-Crown-6 (5.0 g) and THF (200.0 ml) were added and heated to 75°C to 85°C for 36 hour, The reaction mixture was cooled to 25° to 35°C and water (1000.0 ml) was added and stirred for 15 min. The separated aqueous layer was treated with toluene (200.0 ml) and stirred for 15 min. The organic, layers were combined and washed with caustic solution (600.0 ml). The separated organic layer was washed with water (600.0 ml) and characoalized with HP-120 (5.0 g) charcoal and stirred for 30 min and filtered. The filtrate was added sodium hydroxide 20.14 g solution in water (200.0 ml) and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with n-butyl acetate. The separated aqueous layer was added magnesium acetate tetrahydrate solution (90.0 g) in water (100.0 ml) and stirred for 1 hour. The aqueous layer was extracted with methylene dichloride (2000 ml). The separated organic layer was washed with sodium chloride solution and charcoalized. The charcoalized solution was filtered and filtrate was distilled to remove toluene completely. The residue was diluted with toluene (1000 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
The reaction of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al) and 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) may also be performed in similar manner as above in absence of phase transfer catalyst 18-Crown-6.
ExampIe-5:
Preparation of saroglitazar (S)-(-)-phenyl ethylamine salt:

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, residue-A obtained in example- 1 and ethanol (400 mL) were stirred for 15 min. Sodium hydroxide 20.14 g solution in water (200.0 ml) was added and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with isopropyl acetate (400 mL). The separated aqueous layer was diluted with isopropyl acetate (500 mL) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with (S)-(-)-phenyl ethylamine (55.94 g) and stirred for 2 hours at 25°C and 30 min at 45°C. The reaction mixture was cooled to 0°C and stirred for 2 hours, filtered and washed with isopropyl acetate. The wet-cake was dried to obtain saroglitazar phenyl ethylamine salt.
ExampIe-6:
Preparation of saroglitazar magnesium from saroglitazar (SH-)-phenyl ethylamine salt:
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, saroglitazar phenyl ethylamine wet-cake obtained in example-7 and isopropyl acetate (800 mL) were added at 25°C. The reaction mixture was diluted with water (400.0 ml) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with sodium hydroxide solution (20.14 g) in water (200 mL) and stirred for 30 min. The separated aqueous layer was treated with magnesium acetate tetrahydrate (2.29 g) in water (5 mL) solution and stirred for 60 min. The reaction mixture was extracted with methylene dichloride (800 mL). The methylene dichloride was complete removed by distillation under vacuum below 40°C to obtain the residue. The residue was diluted with methylene dichloride (50 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain substantially amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).

PATENT
WO 2015029066
Dwivedi, Shri Prakash Dhar; Singh, Ramesh Chandra; Patel, Vikas; Desai, Amar Rajendra
Cadila Healthcare Ltd
Polymorphic form of pyrrole derivative and intermediate thereof
Pyrrole derivative of present invention is chemically 2-ethoxy-3-(4-(2-(2-methyl-5-(4-(methylthio)phenyl)-lH-pyrrol-l-yl)ethoxy)pKenyl)propanoate, which may be optically active or racemic and its pharmaceutically acceptable salts, hydrates, solvates, polymorphs or intermediates thereof. The INN name for pyrrole derivative is Saroglitazar® which is magnesium salt of pyrrole compound of Formula (I), having below chemical structure.

The present invention relates to Saroglitazar free acid of Formula (IA) or its pharmaceutically acceptable salts, pharmaceutically acceptable solvates, pharmaceutically acceptable esters, stereoisomers, tautomers, analogs and derivs. thereof. The present invention also provides an amorphous form of saroglitazar free acid and processes of prepn. thereof. The present invention also provides pharmaceutical compn. comprising an amorphous form saroglitazar magnesium.
Amorphous forms of saroglitazar free acid and its salt form are claimed. Also claims the process for the synthesis the same compound. Useful for treating obesity, hyperlipidemia and hypercholesteremia. Picks up from WO2015011730, claiming the stable composition comprising saroglitazar magnesium or its derivatives. Zydus-Cadila has developed and launched saroglitazar for treating diabetic dyslipidemia and hypertriglyceridemia.
In September 2013, saroglitazar was launched for treating dyslipidemia and hypertriglyceridemia.
As of March 2015, Zydus-Cadila is developing saroglitazar for treating nonalcoholic steatohepatitis and type II diabetes (both in phase III clainical trials).
Pyrrole derivative of present invention is chemically 2-ethoxy-3-(4-(2-(2-methyl- 5-(4-(methylthio)phenyl)-lH-pyrrol-l-yl)ethoxy)ph’enyl)propanoate, which may be optically active or racemic and its pharmaceutically acceptable salts, hydrates, solvates, polymorphs or intermediates thereof. The INN name for pyrrole derivative is Saroglitazar® which is magnesium salt of pyrrole compound o f saroglitazar,
the process comprising: 5WO 2015/029066 PCT/IN2014/000551 (a) dissolving saroglitazar magnesium of Formula (I) in one or more organic solvents to obtain a solution, (b) adding the solution in one or more o f anti-solvent at temperature from about -80°C to about 150°C to obtain saroglitazar magnesium o f Formula (I); and (c) obtaining the amorphous saroglitazar magnesium by removal of anti-solvent.
Example-1: Preparation of saroglitazar magnesium (Ί) In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 2-ethoxy-3-(4-hydroxy-phenyl)- propionic acid ethyl ester (A) (100.0 g) and cyclohexane (1300.0 ml) were charged and reaction mixture was heated to 45° to 55°C. Potassium carbonate (58.0 g) was added and stirred for 30 min. methanesulfonic acid 2-[2-methyl-5-(4-methyIsulfanyl-phenyl)-pyrroll-yl]-ethyl ester (A l) (150.24 g) and THF (200.0 ml) were added and heated to 75°C to 85°C for 36 hour. The reaction mixture was cooled to 25° to 35°C and water (1000.0 ml) was added and stirred for 15 min. The separated aqueous layer was treated with cyclohexane (200.0 ml) and stirred for 15 min. The organic layers were combined and washed with caustic solution (600.0 ml). The separated organic layer was washed with water (600.0 ml) and characoalized with (5.0 g) charcoal and stirred for 30 min and filtered. The filtrate was distilled to remove cyclohexane and the residue was collected (residue-A). The residue-A as obtained was treated with ethanol (400.0 ml) and stirred for 15 min. Sodium hydroxide 20.14 g solution in water (200.0 ml) was added and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with n-butyl acetate. The separated aqueous layer was added magnesium acetate tetrahydrate solution (90.0 g) in water (100.0 ml) and stirred for I hour. The aqueous layer was extracted with methylene dichloride (200 ml). The separated organic layer was washed with sodium chloride solution and charcoalized. The charcoalized solution was filtered and filtrate was distilled to remove methylene dichloride completely. The residue was diluted with methylene dichloride (1000 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain substantially amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.I).
PATENT
| WO/2015/011730 |
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015011730
The present invention relates to the stable pharmaceutical composition of a suitable hypolipidemic agent. Preferably, the present invention discloses novel formulations of the compound of formula (I), or pharmaceutically acceptable salts of compounds of formula (I). More particularly the present invention relates to the stable pharmaceutical composition of compounds of formula (I) comprising compounds of formula (I) or its pharmaceutically acceptable salts, wherein the pH of the formulation is maintained above 7. formula (I)
The compounds of formula (I) are new synthetic compounds having hypolipidemic activity. The compounds of formula (I) are used primarily for triglyceride lowering, with concomitant beneficial effect on glucose lowering and cholesterol lowering.
The structural formula of compounds of formula (I) is shown below.

wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. Preferably, R is selected from alkylthio or thioalkyl groups; most preferably R represents -SCH3.The Mg+2 salt is preferred. The compounds of formula (I) are generally insoluble in water, but freely soluble in dimethyl sulfoxide, dichloromethane & slightly soluble in methanol and IPA.
1H NMR PREDICT


13c NMR PREDICT


“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
/////////////////
O=C(O)[C@@H](OCC)Cc3ccc(OCCn1c(ccc1C)c2ccc(SC)cc2)cc3
REFERENCES
- “Zydus Group launches new diabetic drug”. The Times of India. Jun 6, 2013.
- “Lipaglyn (Saroglitazar) for Treating Hypertriglycerdemia in Type II Diabetes, India”. Drug Development and Technology.
- “The nuances of atherogenic dyslipidemia in diabetes: focus on triglycerides and current management strategies.”. Indian Heart Journal.
- “Observational Study of Effects of Saroglitazar on Glycaemic and Lipid Parameters on Indian Patients with Type 2 Diabetes”. SCIENTIFIC REPORTS.
- “From ‘Make in India’ to ‘Made in India’: the saroglitazar story.”. Indian Heart Journal.
- “Observational study to evaluate the safety and efficacy of saroglitazar in Indian diabetic dyslipidemia patients.”. Indian Heart Journal.
- Munigoti, SrinivasaP; Harinarayan, CV (2014). “Role of Glitazars in atherogenic dyslipidemia and diabetes: Two birds with one stone?”. Indian Journal of Endocrinology and Metabolism 18 (3): 283. doi:10.4103/2230-8210.131134. PMC 4056123.PMID 24944919.
Indian Pat. Appl. (2015), IN 2013MU02905
WO 2015033357
WO 2015150565
WO 2015001573
IN 2013MU02828
WO 2015029066
IN 2013MU01910
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003009841A1 * | Jul 25, 2002 | Feb 6, 2003 | Cadila Healthcare Ltd | Novel pyrroles having hypolipidemic hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them and their use in medicine |
| WO2012104869A1 | Jan 30, 2012 | Aug 9, 2012 | Cadila Healthcare Limited | Treatment for lipodystrophy |
| INMU19102013A | Title not available | |||
| US6987123 | Aug 10, 2001 | Jan 17, 2006 | Cadila Healthcare Limited | Heterocyclic compounds, their preparation, pharmaceutical compositions containing them and their use in medicine |
| US7041837 | Jul 19, 2002 | May 9, 2006 | Cadilla Healthcare Limited | Heterocyclic compounds having hypolipidemic, hypocholesteremic activities process for their preparation and pharmaceutical compositions containing them and their use in medicine |
| US7323491 | Mar 1, 2004 | Jan 29, 2008 | Cadila Healthcare Limited | Heterocyclic compounds, their preparation, pharmaceutical compositions containing them and their use in medicine |
| US8110598 | Feb 7, 2012 | Cadila Healthcare Limited | Heterocyclic compounds, their preparation, pharmaceutical compositions containing them and their use in medicine | |
| US8212057 | Jul 25, 2002 | Jul 3, 2012 | Cadila Healthcare Limited | Pyrroles having hypolipidemic hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them and their use in medicine |
| US20110275669 | Nov 10, 2011 | Cadilla Healthcare Limited | Novel pyrroles having hypolipidemic hypocholesteremic activities, process for their preparation and pharmaceutical compositions containing them and their use in medicine |
 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Cadila banks on diabetes drug
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet …http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp
Mumbai, June 5: Cadila Healthcare will launch a homegrown drug against diabetes by the third quarter of this year.
The Drug Controller General of India has approved its drug — Lipaglyn — to treat “diabetic dyslipidemia”.
Diabetic dyslipidemia is a condition where a person is diabetic and has elevated levels of total cholesterol. Over 80 per cent of diabetic patients are dyslipidemic.
http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp
Zydus Cadila said it is looking for partnership to market its new chemical entity (NCE) Lipaglyn, to be used for treating a type of diabetes in developed and developing markets. “Lipaglyn is the first glitazar to be approved in the world and the first NCE discovered and developed indigenously by an Indian pharma company.
The new drug is expected to be launched in Q3 of this fiscal in the country,” Zydus Cadila Chairman and Manging Director Pankaj Patel told reporters.
The company has spent USD 250 million in developing Lipaglyn and aims to spend another USD 150-200 million to launch the drug in overseas markets in next 3-5 years period, Patel said, adding that the company is looking for marketing partnerships.
“We expect this to be a blockbuster drug, which means over USD 1 billion sales a year, when the drug is sold globally, he said. The market for this drug is estimated at Rs 100 crore in the local market over the next three years and having market potential size of over USD 30 billion in the world market, he said.
Zydus Cadila took about eight years to develop the molecule and conducted clinical trials on more than 1,000 patients in India, Patel said, adding that the company is yet to finalise the price, but believes that it will be reasonably priced in the local market.
The company said that the Indian drug regulator Drug Controller General of India (DCGI) has approved Lipaglyn to be used for treating ‘diabetic dyslipidemia’.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
|
|
| Clinical data | |
| Trade names | Lipaglyn |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 495399-09-2 |
| ATC code | None |
| PubChem | CID 60151560 |
| ChemSpider | 32079086 |
| Chemical data | |
| Formula | C25H29NO4S |
| Molar mass | 439.56 g/mol |
by WORLD DRUG TRACKER
DR ANTHONY
do not miss out on updates
see my update at https://newdrugapprovals.org/2015/03/09/saroglitazar-magnesium-new-patent-wo-2015029066-cadila-healthcare-ltd/ 9 may 2015
Lipaglyn (Saroglitazar) won a lot of support at the 75th Anniversary Conference of the American Diabetes Association. Lipaglyn is currently under Phase III clinical development for treatment of Non Alcoholic SteatoHepatitis (NASH), a serious liver disease and an unmet healthcare need, globally. There is currently no drug approved for treating NASH. Lipaglyn is already approved in India for the treatment of diabetic dyslipidemia


Speaking on the development, Mr. Pankaj R. Patel, Chairman and Managing Director, Zydus Cadila said, “These new robust scientific data on the safety and efficacy of Lipaglyn
(Saroglitazar) being presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) reflect our continued commitment to millions of patients living with Diabetes, Dyslipidemia, Non-alcoholic fatty liver disease (NAFLD) and Non-alcoholic steatohepatitis (NASH).”
Zydus Cadila, a leading global healthcare provider, today announced that new scientific and clinical data on Saroglitazar will be presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, USA from 5thto 9th June, 2015. Several analyses of real-world patient data of Saroglitazar will also be presented. The abstracts are available on theADA website.
Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.

Pankaj Patel, chairman and MD, Cadila Healthcare Ltd

Zydus is an innovation-led global healthcare provider that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 19,000 people worldwide including over 1200 scientists engaged in research and is dedicated to creating healthier communities globally.
With a strong research pipeline of NCEs, biologics and vaccines, the group became India’s first pharmaceutical company to launch its own indigenously researched therapy Lipaglyn which is also the world’s first approved therapy for diabetic dyslipidaemia. Exemptia, the world’s first biosimilar of Adalimumab is also a product of Zydus innovation. Zydus also collaborates with partners to support and make therapies affordable and accessible to communities across the world.
As a leading healthcare provider, it aims to become a global research-based pharmaceutical company by 2020.
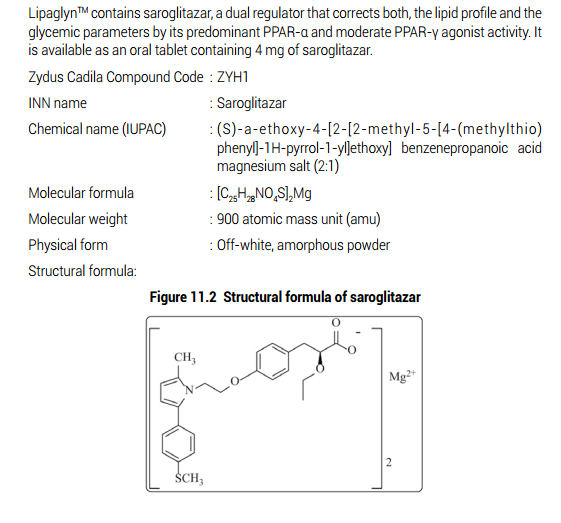




Pankaj R. Patel (left), Chairman & Managing Director, Zybus Cadila,

Ganesh Nayak, Chief Operating Officer and Executive Director, Zydus Cadila


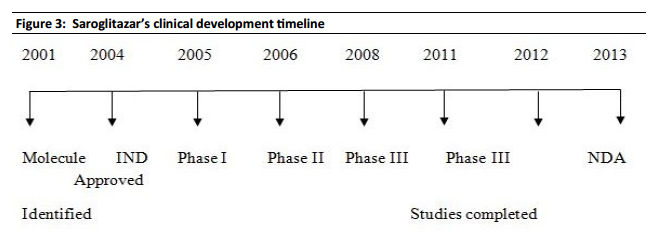
Zydus Cadila has announced a breakthrough in the anti-diabetic drug Lipaglyn. Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.
The Zydus Group announced a breakthrough in its research efforts with Lipaglyn (Saroglilazar), a novel drug targeted at bridging an unmet healthcare need for treating Diabetic Dyslipidemia or Hypertriglyeeridemia in Type II diabetes, not controlled by statins alone. The drug has been approved for launch in India by the Drug Controller General of India (DCGI). With a novel action that offers lipid and glucose lowering effects in one molecule, Lipaglyn is the first Glitazar to be approved anywhere in the world.
“Lipaglyn provides patients suffering from diabetic dyslipidemia the option of a once-daily oral therapy that has a beneficial effect on both lipid parameters as well as glycemic control,” said Pankaj R. Fatel, Chairman and Managing Director, Zydus Cadila. “It has always been our dream to take a molecule right from the concept stage up to its launch. Today, we have realized this dream. It is an important breakthrough and I would like to dedicate this to all the Indian research scientists in the Held of drug discovery,” Patel added,
Diabetic Dyslipidemia is a condition where a person is diabetic and has elevated levels of the total cholesterol, the “bad” low-density lipoprotein (LDL) cholesterol and the triglycerides and a decrease in the “good” high-density lipoprotein (HDL) cholesterol concentration in the blood. Optimal LDL cholesterol levels ibr adults with diabetes are less than 100 mg/dh, optimal HDL cholesterol levels are equal to or greater than 40 mg/dL, and desirable triglycerides levels are less than 150 mg/dLT LipaglynrM, a non-thiazoKdinedione, is the first therapy to be approved for this condition,
World over, it is estimated that 30% of all deaths occur due lo cardiovascular diseases (CVD). In India, one out of every five persons is at serious risk of developing CVD, Research has shown that diabetes is one of the major risk factors of CVD. India has a population of nearly 65 million diabetics and 77 million prc-diabctics, 85 – 97% of the diabetes patients suffer from dyslipidemia or lipid abnormalities. Hence, addressing the problem of diabetes and dyslipidemia is crucial in tackling the health risk posed by CVD.
Discovered by the Zydus Research Centre, the dedicated NCE research arm of the Zydus group, LipaglynrM is a best-in-class innovation, designed to have a unique cellular mechanism of action following an extensive structure-activity relationship study initiated in the year 2000, Lipaglyn1M has a predominant affinity to PPAR alpha isoform and moderate affinity to PPAR gamma isoform of PPAR nuclear receptor subfamily. The molecule has shown beneficial effects on lipids and glyeemic control without side effects. This molecule underwent extensive pre-clinical characterisation and the I.ND was submitted in the year 2004,
As a part of the clinical development programme, extensive Phase-I, Phase-II and Phase-Ill clinical trials were conducted to evaluate the phamacokinetics, pharmacodynamics, efficacy and safety of Lipaglyn. The new drug application for Lipaglyn1 was based on a comprehensive clinical development programme spanning eight years.
Results from the first Phase III programme with Pioglitazone as a comparator drug in diabetes patients showed that the 4 mg dose of Lipaglyn led to a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and also showed a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbAlc) thereby confirming its beneficial effects of both lipid and glyeemic control in diabetic patients,
In the second Phase III study, Lipaglyn was studied in diabetic dyslipidemic patients insufficiently controlled with statin therapy. The results from this study confirmed that Lipaglyn had a pronounced beneficial effect on both the lipid and glyeemic parameters in these subjects.
In both the studies, Lipaglyn was well tolerated and had a better safety profile than the comparators. Importantly Lipaglyn1 M has a non-renal route of elimination, and did not show adverse events like edema, weight gain, myopathies or derangement of liver and/or kidney functions, thus making it sale and efficacious. LipaglynIM is recommended for once daily administration as 4 mg tablets.
Zydus will offer a dedicated LipaglynIM support programme to patients and earegivers, The programme shall provide important support and information regarding access, adherence, education and thereby help patients to start and appropriately manage their disease and therapy over time.
About Lipaglyn
Lipaglyn[TM] (Saroglitazar) was launched in September 2013 in India, for treating Hypertriglyceridemia and Diabetic Dyslipidemia in Patients with Type 2 Diabetes not controlled by statins. Since then, more than 80,000 patients are availing this drug with a prescriber base over 3500 diabetologists, cardiologists and physicians. Lipaglyn[TM] helps in a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and has also shown a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbA1c), thereby confirming its beneficial effects on both lipid and glycemic control in diabetic patients. Lipaglyn[TM] is a prescription medicine, and can be taken only under the advice and guidance of a registered medical practitioner.
About Zydus
Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies, including small molecule drugs, biologic therapeutics and vaccines. The group employs over 16,500 people worldwide including over 1200 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global research based pharmaceutical company by 2020.
References
Zydus to present new scientific data on Lipaglyn in the US
New Delhi, Jun 8 (UNI) Healthcare services provider, Zydus Cadila today said the new scientific and clinical data on Lipaglyn (Saroglitazar) will be presented at the 75th annual scientific sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, US from 5th to 9th June,2015.
Read more at http://www.uniindia.com/news/business-economy/zydus-to-present-new-scientific-data-on-lipaglyn-in-the-us/84440.html
READ …..https://newdrugapprovals.org/2013/06/07/cadila-banks-on-diabetes-druglipaglynsaroglitazar/
http://lipaglyn.com/downloads/Lipaglyn_Product_Monograph.pdf
http://www.ijpcs.net/sites/default/files/IJPCS_3_1_02_0.pdf
http://onlinelibrary.wiley.com/doi/10.1002/prp2.136/pdf
////////////
CCO[C@@H](Cc1ccc(cc1)OCCn2c(ccc2c3ccc(cc3)SC)C)C(=O)O
CCOC(CC1=CC=C(C=C1)OCCN2C(=CC=C2C3=CC=C(C=C3)SC)C)C(=O)O
Zydus Cadila’s, Lipaglyn (Saroglitazar) won a lot of support at the 75th Anniversary Conference of the American Diabetes Association
Lipaglyn (Saroglitazar) won a lot of support at the 75th Anniversary Conference of the American Diabetes Association. Lipaglyn is currently under Phase III clinical development for treatment of Non Alcoholic SteatoHepatitis (NASH), a serious liver disease and an unmet healthcare need, globally. There is currently no drug approved for treating NASH. Lipaglyn is already approved in India for the treatment of diabetic dyslipidemia
Speaking on the development, Mr. Pankaj R. Patel, Chairman and Managing Director, Zydus Cadila said, “These new robust scientific data on the safety and efficacy of Lipaglyn
(Saroglitazar) being presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) reflect our continued commitment to millions of patients living with Diabetes, Dyslipidemia, Non-alcoholic fatty liver disease (NAFLD) and Non-alcoholic steatohepatitis (NASH).”
Zydus Cadila, a leading global healthcare provider, today announced that new scientific and clinical data on Saroglitazar will be presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, USA from 5thto 9th June, 2015. Several analyses of real-world patient data of Saroglitazar will also be presented. The abstracts are available on theADA website.
Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.
Pankaj Patel, chairman and MD, Cadila Healthcare Ltd
Zydus is an innovation-led global healthcare provider that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 19,000 people worldwide including over 1200 scientists engaged in research and is dedicated to creating healthier communities globally.
With a strong research pipeline of NCEs, biologics and vaccines, the group became India’s first pharmaceutical company to launch its own indigenously researched therapy Lipaglyn which is also the world’s first approved therapy for diabetic dyslipidaemia. Exemptia, the world’s first biosimilar of Adalimumab is also a product of Zydus innovation. Zydus also collaborates with partners to support and make therapies affordable and accessible to communities across the world.
As a leading healthcare provider, it aims to become a global research-based pharmaceutical company by 2020.
Pankaj R. Patel (left), Chairman & Managing Director, Zybus Cadila,
Ganesh Nayak, Chief Operating Officer and Executive Director, Zydus Cadila
Zydus Cadila has announced a breakthrough in the anti-diabetic drug Lipaglyn. Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.
The Zydus Group announced a breakthrough in its research efforts with Lipaglyn (Saroglilazar), a novel drug targeted at bridging an unmet healthcare need for treating Diabetic Dyslipidemia or Hypertriglyeeridemia in Type II diabetes, not controlled by statins alone. The drug has been approved for launch in India by the Drug Controller General of India (DCGI). With a novel action that offers lipid and glucose lowering effects in one molecule, Lipaglyn is the first Glitazar to be approved anywhere in the world.
“Lipaglyn provides patients suffering from diabetic dyslipidemia the option of a once-daily oral therapy that has a beneficial effect on both lipid parameters as well as glycemic control,” said Pankaj R. Fatel, Chairman and Managing Director, Zydus Cadila. “It has always been our dream to take a molecule right from the concept stage up to its launch. Today, we have realized this dream. It is an important breakthrough and I would like to dedicate this to all the Indian research scientists in the Held of drug discovery,” Patel added,
Diabetic Dyslipidemia is a condition where a person is diabetic and has elevated levels of the total cholesterol, the “bad” low-density lipoprotein (LDL) cholesterol and the triglycerides and a decrease in the “good” high-density lipoprotein (HDL) cholesterol concentration in the blood. Optimal LDL cholesterol levels ibr adults with diabetes are less than 100 mg/dh, optimal HDL cholesterol levels are equal to or greater than 40 mg/dL, and desirable triglycerides levels are less than 150 mg/dLT LipaglynrM, a non-thiazoKdinedione, is the first therapy to be approved for this condition,
World over, it is estimated that 30% of all deaths occur due lo cardiovascular diseases (CVD). In India, one out of every five persons is at serious risk of developing CVD, Research has shown that diabetes is one of the major risk factors of CVD. India has a population of nearly 65 million diabetics and 77 million prc-diabctics, 85 – 97% of the diabetes patients suffer from dyslipidemia or lipid abnormalities. Hence, addressing the problem of diabetes and dyslipidemia is crucial in tackling the health risk posed by CVD.
Discovered by the Zydus Research Centre, the dedicated NCE research arm of the Zydus group, LipaglynrM is a best-in-class innovation, designed to have a unique cellular mechanism of action following an extensive structure-activity relationship study initiated in the year 2000, Lipaglyn1M has a predominant affinity to PPAR alpha isoform and moderate affinity to PPAR gamma isoform of PPAR nuclear receptor subfamily. The molecule has shown beneficial effects on lipids and glyeemic control without side effects. This molecule underwent extensive pre-clinical characterisation and the I.ND was submitted in the year 2004,
As a part of the clinical development programme, extensive Phase-I, Phase-II and Phase-Ill clinical trials were conducted to evaluate the phamacokinetics, pharmacodynamics, efficacy and safety of Lipaglyn. The new drug application for Lipaglyn1 was based on a comprehensive clinical development programme spanning eight years.
Results from the first Phase III programme with Pioglitazone as a comparator drug in diabetes patients showed that the 4 mg dose of Lipaglyn led to a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and also showed a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbAlc) thereby confirming its beneficial effects of both lipid and glyeemic control in diabetic patients,
In the second Phase III study, Lipaglyn was studied in diabetic dyslipidemic patients insufficiently controlled with statin therapy. The results from this study confirmed that Lipaglyn had a pronounced beneficial effect on both the lipid and glyeemic parameters in these subjects.
In both the studies, Lipaglyn was well tolerated and had a better safety profile than the comparators. Importantly Lipaglyn1 M has a non-renal route of elimination, and did not show adverse events like edema, weight gain, myopathies or derangement of liver and/or kidney functions, thus making it sale and efficacious. LipaglynIM is recommended for once daily administration as 4 mg tablets.
Zydus will offer a dedicated LipaglynIM support programme to patients and earegivers, The programme shall provide important support and information regarding access, adherence, education and thereby help patients to start and appropriately manage their disease and therapy over time.
About Lipaglyn
Lipaglyn[TM] (Saroglitazar) was launched in September 2013 in India, for treating Hypertriglyceridemia and Diabetic Dyslipidemia in Patients with Type 2 Diabetes not controlled by statins. Since then, more than 80,000 patients are availing this drug with a prescriber base over 3500 diabetologists, cardiologists and physicians. Lipaglyn[TM] helps in a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and has also shown a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbA1c), thereby confirming its beneficial effects on both lipid and glycemic control in diabetic patients. Lipaglyn[TM] is a prescription medicine, and can be taken only under the advice and guidance of a registered medical practitioner.
About Zydus
Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies, including small molecule drugs, biologic therapeutics and vaccines. The group employs over 16,500 people worldwide including over 1200 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global research based pharmaceutical company by 2020.
References
Zydus to present new scientific data on Lipaglyn in the US
New Delhi, Jun 8 (UNI) Healthcare services provider, Zydus Cadila today said the new scientific and clinical data on Lipaglyn (Saroglitazar) will be presented at the 75th annual scientific sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, US from 5th to 9th June,2015.
Read more at http://www.uniindia.com/news/business-economy/zydus-to-present-new-scientific-data-on-lipaglyn-in-the-us/84440.html
READ …..https://newdrugapprovals.org/2013/06/07/cadila-banks-on-diabetes-druglipaglynsaroglitazar/
http://lipaglyn.com/downloads/Lipaglyn_Product_Monograph.pdf
http://www.ijpcs.net/sites/default/files/IJPCS_3_1_02_0.pdf
http://onlinelibrary.wiley.com/doi/10.1002/prp2.136/pdf
//////
Should Equipment Status Identification Labels be retained with the Batch Record?

Should Equipment Status Identification Labels be retained with the Batch Record?
Keeping equipment status identification labels with the batch record provides additional confirmation during the review process. But is it required?
Keeping equipment status identification labels with the batch record or other files is often done to provide additional confirmation during review of the record. It supports verification that certain equipment was cleaned before usage for manufacturing. But is it required?
The U.S. Food and Drug Administration FDA has answered this question in an Q&A Document. Assuming each major piece of equipment has a unique “Cleaning and Use Log” that is adequately retained, these “quick reference” equipment labels can be discarded according the agency. FDA sees “no value in the retention of such labels in addition to the required equipment log or batch record documentation. The labels serve a valuable, temporary purpose of positively identifying the current status of equipment and the material under process. Any status label should be correct, legible, readily visible, and associated with the correct piece of equipment. The information on the temporary status label should correspond with the information recorded in the equipment cleaning and use log, or the previous batch record for non-dedicated equipment.”
However, as said before, it might be useful keeping these labels in a batch record. Many companies are doing so; not because it is a requirement but it is a helpful and reliable practice.
/////////
Selurampanel, BGG 492
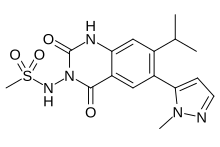
Selurampanel, BGG492,
cas 912574-69-7
Chemical Formula: C16H19N5O4S
Exact Mass: 377.1158
UNII-7WG1MR7DAR;
N-(7-isopropyl-6-(1-methyl-1H-pyrazol-5-yl)-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)methanesulfonamide
N-[7-Isopropyl-6-(1-methyl-1H-pyrazol-5-yl)-2,4-dioxo-1,2,3,4-tetrahydroquinazolin-3-yl]methanesulfonamide
PHASE 2 , FOR EPILEPSY, TITINUS
NOVARTIS INNOVATOR
Selurampanel (INN, code name BGG492) is a drug closely related to the quinoxalinedione series which acts as a competitive antagonist of the AMPA and kainate receptors and, as of 2015, is being investigated in clinical trials by Novartis for the treatment ofepilepsy.[1][2][3] It has also been studied in the acute treatment of migraine, and was found to produce some pain relief, but with a relatively high rate of side effects.[4]
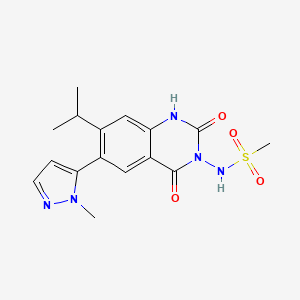
PATENT
Example 44: N-[7-IsopropyI-6-(l-methyl-lH-pyrazol-4-yl)-2,4-dioxo-l,4-dihydro-2H-quinazoIin-3-yl]-methanesulfonamide
2-Amino-4-isopropyl-5-(2-methyl-2H-pyrazol-3-yl)-benzoic acid methyl ester

The 2-amino-5-iodo-4-isopropyl-benzoic acid methyl ester required for the coupling reaction described below was prepared according to the procedures described in WO 2004/033435 Al.
The l-methyl-5-tributylstannanyl-lH-pyrazole required for the coupling reaction was prepared according to the procedure described above.
2-Amino-5-iodo-4-isopropyl-benzoic acid methyl ester (300 mg, 0.94 mmol) and l-methyl-5-tributylstannanyl-lH-pyrazole (523 mg, 1.5 equiv) were weighed in air and added in a flame-dried flask. [Bistriphenylphosphine]dichloropalladium (67.3 mg, 0.1 equiv) was added and the flask was closed by a septum. Dioxane (1 mL) was added and the mixture was stirred for 18 h (TLC control) at 100 0C. The mixture was dissolved with EtOAc, filtered and evaporated to dryness. The crude product was purified by flash chromatography (hexanes to EtOAc / hexanes (4:6)) to yield 2-amino-4-isopropyl-5-(2-methyl-2H- pyrazol-3-yl)-benzoic acid methyl ester (169 mg, 66%) as a yellow solid. (ESI-MS: m/z 21 A [M+H]+, rt 5.20 min).
2-(4-Chloro-phenoxycarbonylamino)-4-isopropyl-5-(2-methyl-2H-pyrazol-3-yl)-benzoic acid methyl ester

4-Chlorophenyl-chloroformate (88 μL, 1.1 equiv) was added to a solution of 2-amino-4-isopropyl-5-(2~ methyl-2H-pyrazol-3-yl)-benzoic acid methyl ester (156 mg, 0.57 mmol) in dioxane (1.5 mL). The mixture was stirred for 2 h (TLC control) at 80 0C. The mixture was evaporated to dryness. The obtained yellow solid was used in the next step without further purification, (rt 6.77 min)
N-[7-Isopropyl-6-(2-methyl-2H-pyrazol-3 -yl)-2,4-dioxo- 1 ,4-dihydro-2H-quinazolin-3 -yl] -methanesulfonamide

CH3SO2NHNH2 (79.5 mg, 1.1 equiv) and J-Pr2NEt (225 μL, 2 equiv) were added to a solution of 2-(4-chloro-phenoxycarbonylamino)-4-isopropyl-5-(2-methyl-2H-pyrazol-3-yl)-benzoic acid methyl ester (281 mg, 0.65 mmol) in dioxane (8 mL). The mixture was stirred for 16 h (TLC control) at 80 0C. The mixture was evaporated to dryness. The crude product was purified by flash chromatography (MeOH / DCM (1:9)) to provide N-[7-isopropyl-6-(2-methyl-2H-pyrazol-3 ~yl)-2,4-dioxo- 1 ,4-dihydro-2H-quinazolin-3 -yl]-methanesulfonamide as a white solid (120 mg, 48%) (ESI-MS: m/z 378 [M+H]+, rt 4.20 min).
| Patent | Submitted | Granted |
|---|---|---|
| Substituted 1H-quinazoline-2,4-diones useful as AMPA receptor ligands [US7655666] | 2008-06-26 | 2010-02-02 |
| N-(2,4-dioxo-6-(tetrahydrofuran-2-yl)-7-(trifluoromethyl)-1,4-dihydro-2H-quinazolin-3-yl)methanesulfonamide [US8012988] | 2010-06-10 | 2011-09-06 |
| 2,4-DIOXO-1,4-DIHYDRO-2H-QUINAZOLIN-3-YL-SULFONAMIDE DERIVATIVES [US2013053381] | 2011-05-18 | 2013-02-28 |
| Use of 1H-quinazoline-2,4-diones [US2013090346] | 2012-09-05 | 2013-04-11 |
| Use of 1H-quinazoline-2,4-diones [US2013096145] | 2011-06-24 | 2013-04-18 |
| Use of 1H-quinazoline-2,4-diones [US2014163050] | 2014-02-12 | 2014-06-12 |
| FOMULATION COMPRISING 1 H-QUINAZOLINE-2, 4-DIONE AMPA RECEPTOR ANTAGONISTS, IN THE FORM OF IMMEDIATE RELEASE TABLETS AND PREPARATION THEREOF [US2012263791] | 2010-12-21 | 2012-10-18 |
| Use of 1H-Quinazoline-2,4-Diones [US2014018376] | 2010-10-20 | 2014-01-16 |
| 1-H-QUINAZOLINE-2, 4-DIONES FOR USE IN THE TREATMENT OF NEURONAL CEROID LIPOFUSCINOSIS [US2012122903] | 2010-07-23 | 2012-05-17 |
References
- Faught, Edward (2014). “BGG492 (selurampanel), an AMPA/kainate receptor antagonist drug for epilepsy”. Expert Opinion on Investigational Drugs 23 (1): 107–113.doi:10.1517/13543784.2014.848854. ISSN 1354-3784.
- Belcastro, Vincenzo; Verrotti, Alberto (2015). “Novel Molecular Targets for Drug-Treatment of Epilepsy”: 183–199.doi:10.1007/978-3-319-12283-0_10.
- Hanada, Takahisa (2014). “The AMPA receptor as a therapeutic target in epilepsy: preclinical and clinical evidence”. Journal of Receptor, Ligand and Channel Research: 39.doi:10.2147/JRLCR.S51475. ISSN 1178-699X.
- Gomez-Mancilla B, Brand R, Jürgens TP, et al. (February 2014). “Randomized, multicenter trial to assess the efficacy, safety and tolerability of a single dose of a novel AMPA receptor antagonist BGG492 for the treatment of acute migraine attacks”. Cephalalgia 34 (2): 103–13.doi:10.1177/0333102413499648. PMID 23963355.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[7-Isopropyl-6-(2-methylpyrazol-3-yl)-2,4-dioxo-1H-quinazolin-3-yl]methanesulfonamide
|
|
| Identifiers | |
| CAS Number | 912574-69-7 |
| ATC code | None |
| PubChem | CID 45381907 |
| ChemSpider | 32698379 |
| Chemical data | |
| Formula | C16H19N5O4S |
| Molar mass | 377.418 g/mol |
see……..http://apisynthesisint.blogspot.in/2016/02/selurampanel-bgg-492.html
////Selurampanel, BGG492, 912574-69-7
CC(C)c1cc2c(cc1c3ccnn3C)c(=O)n(c(=O)[nH]2)NS(=O)(=O)C
CS(=O)(NN1C(NC2=C(C=C(C3=CC=NN3C)C(C(C)C)=C2)C1=O)=O)=O
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

