
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Tambiciclib
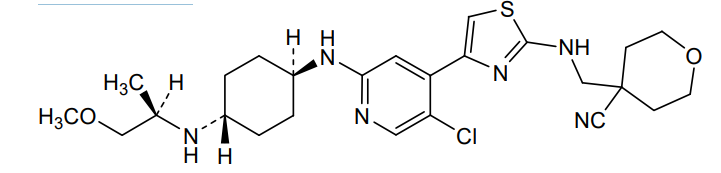
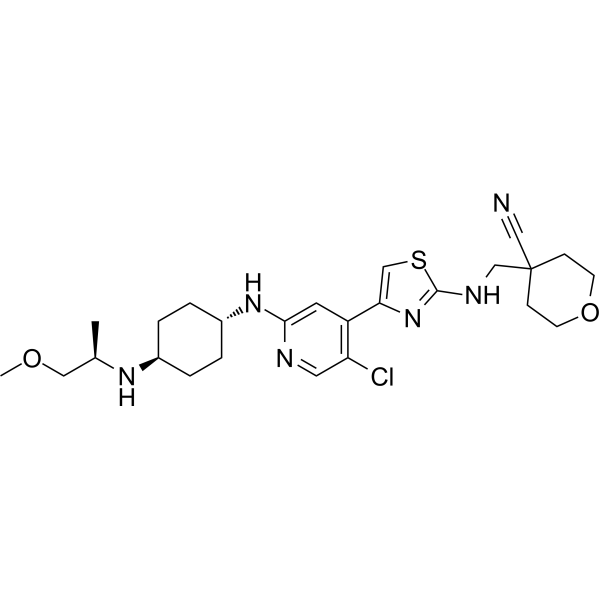
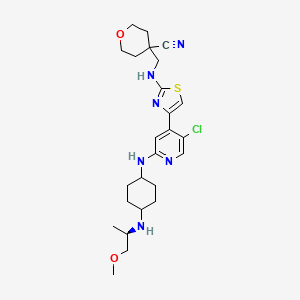
Tambiciclib
CAS 2247481-08-7
MF C25H35ClN6O2S, 519.10
4-[[[4-[5-chloro-2-[[4-[[(2R)-1-methoxypropan-2-yl]amino]cyclohexyl]amino]-4-pyridinyl]-1,3-thiazol-2-yl]amino]methyl]oxane-4-carbonitrile
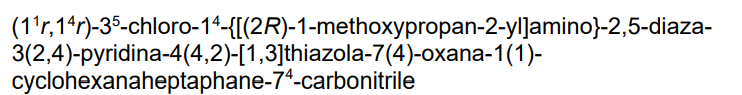
cyclin-dependent kinase inhibitor, antineoplastic, GFH 009, JSH 009, XDZ7VK8CXC, Orphan Drug , Acute myeloid leukaemia, Peripheral T-cell lymphoma
Tambiciclib (GFH009, JSH-009) is an orally active, highly potent and selective CDK9 inhibitor (IC50 = 1 nM), demonstrating >200-fold selectivity over other CDKs, >100-fold selectivity over DYRK1A/B, and excellent selectivity over 468 kinases/mutants. Tambiciclib demonstrates potent in vitro and in vivo antileukemic efficacy in acute myeloid leukemia (AML) mouse models by inhibiting RNA Pol II phosphorylation, downregulating MCL1 and MYC, and inducing apoptosis. Tambiciclib can be used for AML research.
Tambiciclib is a selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon administration, tambiciclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins. This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII). It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
- OriginatorGenFleet Therapeutics
- DeveloperGenFleet Therapeutics; Sellas Life Sciences Group
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase 9 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia; Peripheral T-cell lymphoma
- Phase IIAcute myeloid leukaemia
- Phase I/IIDiffuse large B cell lymphoma; Haematological malignancies; Peripheral T-cell lymphoma
- Phase ISolid tumours
- PreclinicalColorectal cancer; T-cell prolymphocytic leukaemia
- 13 Oct 2025Preclinical trials in T-cell prolymphocytic leukaemia (Combination therapy) in USA (Parenteral)
- 13 Oct 2025Preclinical trials in T-cell prolymphocytic leukaemia (Monotherapy) in USA (Parenteral)
- 13 Oct 2025Pharmacodynamics data from preclinical studies in T-cell prolymphocytic leukaemia released by SELLAS Life Sciences
CLINICAL
- A Study of GFH009 in Combination With Zanubrutinib in Subjects With Relapsed or Refractory DLBCLCTID: NCT06375733Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-08-12
- A Study of GFH009 Monotherapy in Patients with Relapsed or Refractory Peripheral T-cell Lymphoma (PTCL)CTID: NCT05934513Phase: Phase 1/Phase 2Status: RecruitingDate: 2024-12-13
Publication Name: European Journal of Medicinal Chemistry
Publication Date: 2018-10-05
PMID: 30253346
DOI: 10.1016/j.ejmech.2018.09.025
SYN
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018192273&_cid=P12-MJ18VV-17351-1
Example 1: Synthesis of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino) cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4- carboxynitrile
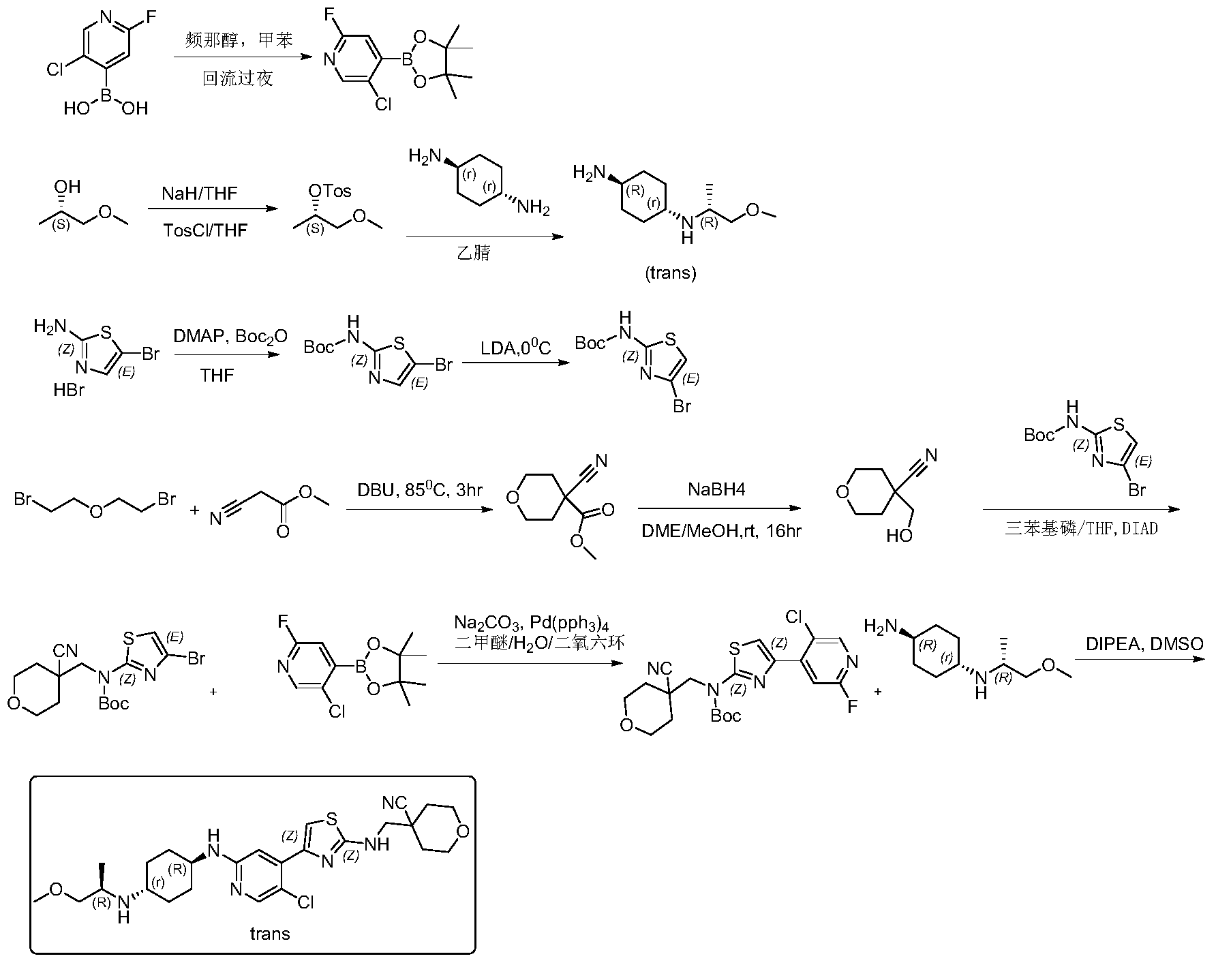
Step 1: Synthesis of 5-chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhecyclopentan-2-yl)pyridine
[0102]5-Chloro-2-fluoropyridine-4-boronic acid (0.7 g, 4.46 mmol) and pinacol (0.63 g, 5.35 mmol) were added to 50 mL of toluene, and the mixture was refluxed at 120 °C overnight. TLC showed a small amount of starting material remaining. The reaction mixture was cooled to room temperature and concentrated, then dried by an oil pump to give 0.92 g of a white solid compound, 5-chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhexacyclopentan-2-yl)pyridine, yield 80%, MS (ESI): m/z 258.1 (M+H) + .
[0103]Step 2: Synthesis of (S)-1-methoxypropyl-2-yl-4-toluenesulfonyl ester
[0104]60% sodium hydride (NaH) (6.52 g, 283 mmol) was added to anhydrous tetrahydrofuran (THF) (200 mL). The mixture was cooled to 0 °C in an ice bath under nitrogen protection, and (S)-(+)-1-methoxy-2-propanol (21 g, 233 mmol) was added dropwise. After the addition was complete, the mixture was brought to room temperature and stirred for 1.5 hours. The reaction mixture was then cooled back to 0 °C, and a tetrahydrofuran (THF) solution of p-toluenesulfonyl chloride (45.3 g, 283 mmol) (200 mL) was added dropwise. After the addition was complete, the mixture was stirred overnight at room temperature. TLC showed that the starting material had reacted completely. The reaction mixture was diluted with ethyl acetate (500 mL), and the reaction was quenched by adding water (500 mL) dropwise while cooling in an ice bath. The mixture was separated, and the aqueous phase was extracted once more with ethyl acetate (200 mL). The combined organic phases were washed with water (200 mL) and then with saturated brine (200 mL). The crude product was dried with anhydrous sodium sulfate, filtered, and concentrated to obtain 43 g of a pale yellow oily substance. Column separation (petroleum ether/ethyl acetate = 5/1) yielded 37 g of (S)-1-methoxypropyl-2-yl-4-toluenesulfonyl ester, a pale yellow oily substance, with a yield of 65.1%. MS (ESI): m/z 245.1 (M+H) + .
[0105]Step 3: Synthesis of (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine
[0106](S)-1-methoxypropyl-2-yl 4-toluenesulfonyl ester (5 g, 20.5 mmol) and trans-1,4-cyclohexanediamine (5.84 g, 51.2 mmol) were added to 50 mL of acetonitrile and heated to 90 °C overnight. The reaction was monitored by TLC until complete. After cooling, the reaction solution was filtered, the filtrate was concentrated, and the residue was dissolved in dichloromethane and separated by silica gel stirring column (dichloromethane/methanol = 10/1) to give 2.5 g of the pale yellow liquid compound (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine, yield 65%, MS (ESI): m/z 187.3 (M+H) + .
[0107]Step 4: Synthesis of tert-butyl 5-bromothiazol-2-ylcarbamate
[0108]105 g (403 mmol) of 5-bromothiazol-2-amine hydrobromide was suspended in 500 mL of tetrahydrofuran. Dimethylaminopyridine (2.41 g, 20 mmol) was added, resulting in a white turbidity. A tetrahydrofuran solution of di-tert-butyl dicarbonate (105.6 g, 484.6 mmol) was slowly added dropwise, and the reaction was allowed to proceed at room temperature for two days. The reaction solution was concentrated and dissolved in 300 mL of dichloromethane. The solution was mixed with silica gel and separated by column chromatography (petroleum ether/ethyl acetate = 10/1-6/1 gradient elution) to give 45 g of off-white solid, yield 40%. MS (ESI): m/z 278.98 (M+H) + .
[0109]Step 5: Synthesis of tert-butyl 4-bromothiazol-2-ylcarbamate
[0110]A 200 mL solution of diisopropylamine (64 mL, 446 mmol) in tetrahydrofuran was added to a dry three-necked flask. Under nitrogen protection, the mixture was cooled to 0 °C, and n-butyllithium (2.5 M, 173 mL, 431.7 mmol) was added dropwise. The reaction was allowed to proceed for 1 hour after the addition was complete. Then, a 400 mL solution of 5-bromothiazol-2-ylcarbamate in tetrahydrofuran was added dropwise at 0 °C. The reaction was allowed to proceed for 2 hours after the addition was complete. TLC showed that the reaction was complete. At 0℃, ice water (5 mL) was slowly added dropwise to quench the reaction. After stirring for 30 minutes, saturated ammonium chloride (500 mL) aqueous solution was added. The mixture was separated, and the aqueous layer was extracted with dichloromethane (2 × 300 mL). The organic layers were combined, washed with saturated brine, dried with anhydrous sodium sulfate, filtered, concentrated, and recrystallized from petroleum ether:ethyl acetate = 30:1. 31 g of tert-butyl 4-bromothiazol-2-ylcarbamate was obtained as a white solid, yield 77.5%. MS (ESI): m/z 278.98 (M+H) + .
[0111]Step Six: Synthesis of Methyl 4-cyano-tetrahydro-2H-pyran-4-carbonate
[0112]Methyl cyanoacetate (39.1 g, 395.3 mmol) and 2,2-dibromoethyl ether (100 g, 434.8 mmol) were added to 600 mL of dimethylformamide, followed by DBU (90 g, 593 mmol). The mixture was heated to 85 °C and reacted for 3 hours. TLC showed that the starting material reacted completely. The solid was filtered off, washed with ethyl acetate (2 × 300 mL), and the mother liquor was concentrated to obtain a brown oily substance. The oil was distilled under reduced pressure at an internal temperature of 65-70 °C, and the fraction collected was a colorless liquid. Crystallization was observed to give 42 g of a white solid, 4-cyano-tetrahydro-2H-pyran-4-carbonate. Yield: 62.8%, MS (ESI): m/z 178.2 (M+H) + .
[0113]Step 7: Synthesis of 4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile
[0114]4-Cyano-tetrahydro-2H-pyran-4-carbonate methyl ester (42 g, 248.4 mmol) was dissolved in 400 mL of ethylene glycol dimethyl ether and 40 mL of methanol. The mixture was cooled to 0 °C in an ice bath, and sodium borohydride (11.1 g, 149 mmol) was added in portions. After the addition was complete, the mixture was allowed to rise to room temperature and stirred for 16 hours. The reaction was completed by TLC. The reaction solution was concentrated, and methanol was added to quench excess sodium borohydride. The solution was then concentrated again. Column chromatography (petroleum ether/ethyl acetate = 5/1) yielded 28 g of 4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile, a pale yellow oil, yield: 79.5%, MS (ESI): m/z 142.1 (M+H) + .
[0115]Step 8: Synthesis of tert-butyl (4-bromothiazolyl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate
[0116]4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile, 4-bromothiazol-2-ylcarbamate tert-butyl ester, and triphenylphosphine were added to anhydrous tetrahydrofuran (THF) and cooled to 0°C. Diisopropyl azodicarbonate (DIAD) was added dropwise. The mixture was stirred at room temperature for 10 minutes, then heated to 40°C overnight. The reaction solution was concentrated, and the residue was dissolved in dichloromethane. The solution was mixed with silica gel and separated by column chromatography (petroleum ether/ethyl acetate = 50/1, 30/1, 20/1) to obtain (4-bromothiazol-2-yl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate tert-butyl ester, a white solid of 365 mg, yield 50%. MS (ESI): m/z 402.1 (M+H) + .
[0117]Step Nine: Synthesis of tert-butyl (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate
[0118]5-Chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhexacyclopentan-2-yl)pyridine and sodium carbonate were added to a mixture of dimethyl ether/H₂O
/ dioxane. The system was purged with nitrogen twice. Then, tert-butyl (4-bromothiazolyl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate and tetraphenylphosphine palladium Pd(pph 3 )
4 were added . The system was purged with nitrogen three times. The temperature was then raised to 70°C and the reaction was carried out for 6 hours. TLC showed that only half of the starting material remained. Heating was then stopped and the reaction was terminated. The reaction solution was cooled to room temperature, ethyl acetate and methanol were added, and the mixture was filtered. The filter cake was washed with ethyl acetate, the filtrate was concentrated, and the residue was dissolved in dichloromethane. The residue was washed with saturated brine, separated, and the organic phase was dried over anhydrous sodium sulfate. The mixture was filtered, and silica gel was added for mixing. The sample was separated by column chromatography (petroleum ether/ethyl acetate = 30/1) to give 3.2 g of (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate, a white foamy solid, with a yield of 55%. MS (ESI): m/z 453.1 (M+H) + .
[0119]Step 10: Synthesis of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino)cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4-carboxynitrile
[0120]The tert-butyl carbamate (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate (3.2 g, 7.1 mmol) and (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine (3.9 g, 21.2 mmol) and diisopropylethylamine (DIPEA) were added to 30 mL of dimethyl sulfoxide. Under nitrogen protection, the mixture was heated to 100-110 °C and reacted for two days. The reaction was monitored by TLC and LCMS. The starting material (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate tert-butyl ester had completely disappeared, with some BOC-free intermediate remaining. The reaction was stopped, and the reaction solution was cooled and diluted with ethyl acetate (60 mL). Water (150 mL) was added under ice bath. The mixture was separated, and the aqueous layer was extracted again with ethyl acetate (2 × 50 mL). The organic layers were combined, washed with saturated brine (100 mL), dried with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product of yellowish-brown oil. Column separation (acetonitrile/water/trifluoroacetic acid = 80/20/0.001) yielded 700 mg of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino)cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4-carboxynitrile, a pale yellow solid. Yield: 19.1%. ¹H NMR (400 MHz, CDCl₃
) )δ8.06(s,1H),7.38(s,1H),6.97(s,1H),5.92(brs,1H),4.45(d,J=8.0Hz,1H),4.02(dd,J 1=2.8Hz, J2=12Hz,2H),3.71-3.74(m,4H),3.54-3.56(m,1H),3.35(s,3H),3.21-3.25(m,2 H),3.00-3.05(m,1H),2.50-2.60(m,1H),2.15(d,J=9.6Hz,2H),2.04-2.07(m,1H),1.95(d ,J=12.8Hz,3H),1.74-1.82(m,3H),1.10-1.30(m,4H),1.00(d,J=.4Hz,3H),MS(ESI):m/z 519.3(M+H) + .
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US376039987&_cid=P12-MJ18R0-12787-1
PAT
- A novel cyclin-dependent kinase CDK9 inhibitorPublication Number: CN-108727363-BPriority Date: 2017-04-19Grant Date: 2020-06-19
- Inhibitor of cyclin-dependent kinase CDK9Publication Number: US-10952999-B2Priority Date: 2017-04-19Grant Date: 2021-03-23
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: EP-3613737-B1Priority Date: 2017-04-19Grant Date: 2021-12-29
- Pharmaceutical combination and use thereof in treatment of cancerPublication Number: WO-2024239512-A1Priority Date: 2023-05-22
- Polymorph of cdk9 inhibitor and preparation method for polymorph and use thereofPublication Number: WO-2020244612-A1Priority Date: 2019-06-06
- Polymorphic substance of CDK9 inhibitor and preparation method and application thereofPublication Number: CN-113966332-APriority Date: 2019-06-06
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: EP-3613737-A1Priority Date: 2017-04-19
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: US-2020078343-A1Priority Date: 2017-04-19



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
//Tambiciclib, cyclin-dependent kinase inhibitor, antineoplastic, GFH 009, JSH 009, XDZ7VK8CXC, Orphan Drug , Acute myeloid leukaemia, Peripheral T-cell lymphoma
Talorasib
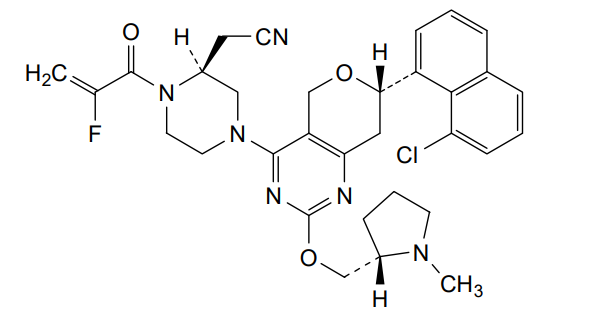
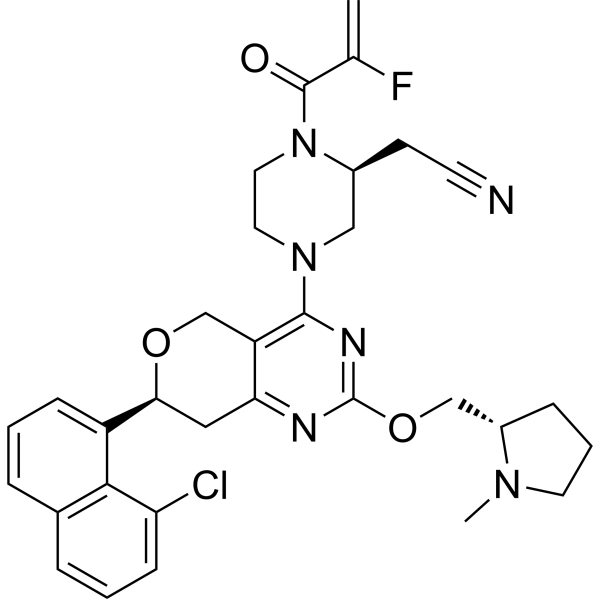

Talorasib
CAS 2648584-48-7
MFC32H34ClFN6O3 MW605.10
- (2S)-4-[(7S)-7-(8-Chloro-1-naphthalenyl)-7,8-dihydro-2-[[(2S)-1-methyl-2-pyrrolidinyl]methoxy]-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoro-1-oxo-2-propen-1-yl)-2-piperazineacetonitrile
- [(2S)-4-[(7S)-7-(8-chloronaphthalen-1-yl)-2-{[(2S)-1-methylpyrrolidin-2-yl]methoxy}-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
- 2-Piperazineacetonitrile, 4-[(7S)-7-(8-chloro-1-naphthalenyl)-7,8-dihydro-2-[[(2S)-1-methyl-2-pyrrolidinyl]methoxy]-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoro-1-oxo-2-propen-1-yl)-, (2S)-
[(2S)-4-[(7S)-7-(8-chloronaphthalen-1-yl)-2-{[(2S)-1-methylpyrrolidin-2-yl]methoxy}-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
Kirsten rat sarcoma viral oncogene homologue (KRAS)inhibitor, antineoplastic, 727W6T7DPK
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN380619664&_cid=P20-MJ0TAW-52678-1
| Preparation Example 1: Synthesis of the compound shown in formula (I) |
| (1) Synthesis of Compound 1 |
| Synthetic route of compound 1: |
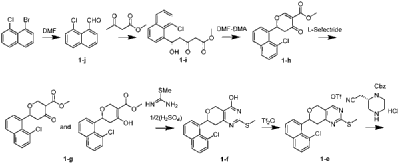
| Synthesis of compound 1-j |
| 1-Bromo-8-chloronaphthalene (500 mg, 2.07 mmol) was dissolved in THF (20 mL), cooled to -78 °C, and n-BuLi (2.5 M, 1.66 mL, 4.14 mmol) was added dropwise under nitrogen protection. After the addition was complete, the mixture was stirred at -78 °C for 10 min, and then DMF (800 μL, 10.35 mmol) was added dropwise at -78 °C. After the addition was complete, the reaction mixture was stirred at -78 °C for 30 min, then heated to room temperature and stirred for 2 h. The reaction was quenched with 50 mL of saturated ammonium chloride solution and extracted with ethyl acetate (50 mL * 2). The organic phase was washed with saturated brine (50 mL * 2), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by rapid column chromatography (EA/PE = 1/10) to give compound 1-j (330 mg, 84% yield) as a white solid. LC-MS (ESI): m/z=191.0[M+H] + ; 1 H NMR (400MHz, CDCL 3 ):δ11.31(s,1H),8.03(dd,1H,J 1 =1.2Hz,J 2 =8.4Hz), 7.92(dd,1H,J 1 =1.2Hz,J 2 =7.2Hz),7.86(1H,J=8.4Hz),7.70(dd,1H,J 1 =1.2Hz,J 2 =7.6Hz), 7.59(t,1H,J=7.6Hz), 7.47(t,1H,J=8Hz). |
| Synthesis of compound 1-i |
| At room temperature, NaH (60%, 242 mg, 6.05 mmol) was added to 6 mL of THF. Then, methyl acetoacetate (543 μL, 5.04 mmol) was added under nitrogen atmosphere at room temperature. The mixture was stirred for 30 minutes under nitrogen atmosphere at room temperature, and then n-BuLi (2.5 M, 2.4 mL, 6.05 mmol) was added dropwise at -15 °C to -10 °C. After the addition was complete, the mixture was maintained at this temperature for 30 minutes, and then a 10 mL solution of compound 1-j (320 mg, 1.68 mmol) in THF was added dropwise. After the addition was complete, the mixture was stirred at low temperature (-10 °C to 0 °C) for 2 hours, then quenched with saturated ammonium chloride solution (50 mL), and then extracted with ethyl acetate (50 mL x 2). The organic phase was washed with saturated brine (50 mL * 2), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by rapid column chromatography (EA/DCM = 1/10) to give compound 1-i (510 mg, 99% yield) as a white solid. LC-MS (ESI): m/z = 329.1 [M + Na] ⁺ ; 1H NMR (400 MHz, CDCl₂) 3 ): δ8.06(d,1H,J=6.4Hz),7.79(d,2H,J=8Hz),7.58(dd,1H,J 1 =7.6Hz,J 2 =1.6Hz),7.53(t,1H,J=7.6Hz),7.34(t,1H,J=7.6Hz),6.91(dd,1H,J 1 =9.2Hz, J 2 =2.4Hz),3.74(s,3H),3.54(s,2H),3.36(dd,1H,J 1 =18Hz,J 2 =1.6Hz),3.24(d,1H,J=3.6Hz),2.85-2.75(m,1H). |
| Synthesis of compound 1-h |
| Compound 1-i (510 mg, 1.66 mmol) was dissolved in DCM (18 mL) at room temperature, followed by the addition of DMF-DMA (245 μL, 1.83 mmol) under nitrogen atmosphere at room temperature. After stirring the reaction mixture for 45 minutes at room temperature, BF was added. 3 Et 2 O (232 μL, 1.83 mmol). After addition, the mixture was stirred at room temperature for 1 hour, then diluted with 100 mL of ethyl acetate. The organic phase was then sequentially quenched with saturated NaHCO3. 3 The sample was washed with a solution (100 mL) and saturated saline solution (100 mL * 2), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude compound 1-h (520 mg). The crude product required no purification and was used directly in the next reaction. LC-MS (ESI): m/z = 317.1 [M+1] + . |
| Synthesis of compound 1-g |
| Compound 1-h (520 mg, 1.64 mmol) was dissolved in THF (20 mL) at room temperature, and then tri-sec-butylborohydride (1 M, 1.64 mL, 1.64 mmol) was added dropwise under nitrogen atmosphere at -78 °C. After addition, the mixture was stirred at -78 °C for 1 hour, the reaction was quenched with saturated ammonium chloride solution (50 mL), extracted with ethyl acetate (50 mL * 2), the organic matter was washed with saturated brine (50 mL * 2), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by rapid column chromatography (PE/EA = 4/1) to give compound 1-g (338 mg, 65% yield) as a yellow oil. LC-MS (ESI): m/z = 319.0 [M+1] + . |
| Synthesis of compound 1-f |
| Compound 1-g (338 mg, 1.06 mmol) was dissolved in methanol (20 mL) at room temperature. Then, under nitrogen atmosphere at 0 °C, sodium methoxide (286 mg, 5.3 mmol) and compound 2-methyl-2-mercaptourea sulfate (265 mg, 0.954 mmol) were added sequentially. After the addition was complete, the mixture was brought to room temperature and stirred for 20 hours. The pH of the reaction solution was adjusted to 5 with 1 N dilute hydrochloric acid, and a solid precipitated. The solid was filtered, the filter cake was washed with water (5 mL * 2), and the solid was collected and dried under vacuum to give crude product 1-f (313 mg) as a white solid. LC-MS (ESI): m/z = 359.1 [M+1] + . |
| Synthesis of compound 1-e |
| Compound 1-f (313 mg, 0.87 mmol) was dissolved in DCM (10 mL) at room temperature. Then, under nitrogen atmosphere in an ice-water bath, DIPEA (431 μL, 2.61 mmol) and trifluoromethanesulfonic anhydride (219 μL, 1.31 mmol) were added sequentially. After addition, the reaction mixture was stirred in an ice-water bath for 2 hours, quenched with saturated sodium bicarbonate solution (50 mL), extracted with DCM (50 mL x 2), and the organic phase was treated with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was purified by rapid column chromatography (EA/PE = 1/10) to give compound 1-e (83 mg, 16% yield in 2 steps) as a white solid. LC-MS (ESI): m/z = 491.0 [M+1] + . |
| Synthesis of compound 1-d |
| Compound 1-e (83 mg, 0.169 mmol) was dissolved in DMF (10 mL) at room temperature, followed by the sequential addition of DIPEA (84 μL, 0.507 mmol) and (S)-2-cyanomethylpiperazine-1-carboxylate hydrochloride (59.9 mg, 0.203 mmol). After addition, the mixture was stirred for 1 hour at 100 °C under nitrogen protection, cooled to room temperature, quenched with saturated brine (50 mL), and extracted with ethyl acetate (50 mL x 2). The organic phase was washed with saturated brine (50 mL x 3), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was purified by rapid column chromatography (EA/PE = 1/1) to give compound 1-d (101 mg, 99% yield) as a white solid. LC-MS (ESI): m/z = 600.2 [M+1] + . |
| Synthesis of compound 1-c |
| Compound 1-d (101 mg, 0.168 mmol) was dissolved in ethyl acetate (10 mL) at room temperature, followed by the addition of MCPBA (85%, 88.4 mg, 0.437 mmol) at room temperature. After addition, the mixture was stirred at room temperature for 2 hours, quenched with saturated sodium bicarbonate solution (20 mL), extracted with ethyl acetate (25 mL x 2), and the organic phase was treated with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was purified by rapid column chromatography (EA/PE = 1/4) to give compound 1-c (88 mg, 82% yield) as a white solid. LC-MS (ESI): m/z = 632.1 [M+1] + . |
| Synthesis of compound 1-b |
| Compound 1-c (88 mg, 0.139 mmol) was dissolved in toluene (10 mL) at room temperature. The reaction mixture was then cooled to 0 °C, and N-methylprolyl (29 μL, 0.243 mmol) and t-BuONa (27 mg, 0.278 mmol) were added sequentially. After the addition was complete, the reaction mixture was stirred for 0.5 hours under nitrogen in an ice-water bath, quenched with water (20 mL), and extracted with ethyl acetate (30 mL * 2). The organic phase was treated with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product. The crude product was purified by rapid column chromatography (MeOH/DCM = 1/10) to give compound 1-b (78 mg, 84% yield) as a white solid. LC-MS (ESI): m/z = 667.3 [M+1] + . |
| Synthesis of compound 1-a |
| Compound 1-b (72 mg, 0.108 mmol) was dissolved in methanol (50 mL) at room temperature. The reaction solution was then cooled to -78 °C, purged twice with nitrogen, and then Pd/C (150 mg) and ZnBr were added. 2 (24.3 mg, 0.108 mmol), the reaction mixture was purged with hydrogen three times, brought to room temperature, and stirred under hydrogen atmosphere for 5 hours. The reaction mixture was filtered and concentrated to obtain a crude product, which was then purified by a rapid separation column (MeOH/DCM = 1:4) to give compound 1-a (20 mg, 35% yield) as a white solid. LC-MS (ESI): m/z = 533.0 [M+1] + . |
| Synthesis of Compound 1 |
| At room temperature, compound 2-fluoroacrylic acid (5.1 mg, 0.0563 mmol) was dissolved in DMF (2 mL). Then, at 0 °C, HATU (25.6 mg, 0.0675 mmol) and DIPEA (18.6 μL, 0.113 mmol) were added sequentially. After the addition was complete, the reaction mixture was stirred at 0 °C under nitrogen for 20 minutes. Then, a DMF solution of compound 1-a (20 mg, 0.0375 mmol) (3 mL) was added to the above reaction mixture. The mixture was brought to room temperature and stirred for another 5 hours. The reaction mixture was quenched with saturated brine (20 mL), extracted with ethyl acetate (25 mL * 2), washed with saturated brine (50 mL * 3), treated with anhydrous sodium sulfate, filtered, and concentrated to obtain the crude product. The crude product was purified by PREP-TLC (MeOH/DCM = 1/10) to obtain compound 1 (6 mg, 26% yield) as a white solid. LC-MS (ESI): m/z=605.2[M+1] + ; 1 H NMR (400MHz, CDCl 3 ): δ7.99-7.93(m,1H),7.83(t,2H,J=8.8Hz),7.62-7.49(m,2H),7.36(t,1H,J=7.6Hz),6.5 5-6.44(m,1H),5.51-5.31(m,1H),5.25(d,1H,J=16.8Hz),5.02-4.93(m,1H),4.82(dd,1H,J 1 =2.4Hz, J 2 =13.6Hz),4.48-4.38(m,1H),4.32-4.19(m,1H),4.17-4.04(m,1H),4. 00(d,1H,J=14Hz),3.87-3.70(m,1H),3.66-3.36(m,2H),3.31-3.16(m ,2H),3.14-2.98(m,1H),2.96-2.69(m,4H),2.59(d,3H,J=18Hz),2.52 -2.34(m,1H),2.15-2.06(m,1H),1.87-1.74(m,2H),0.93-0.76(m,2H). |
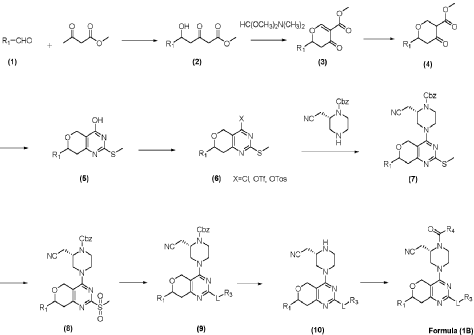
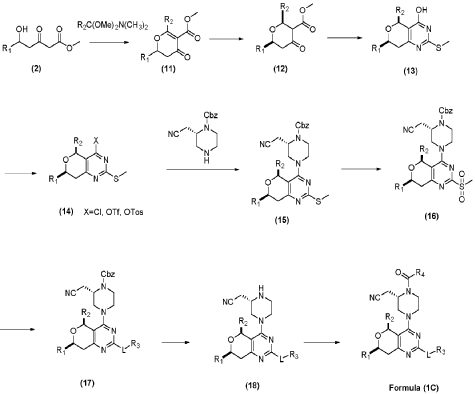
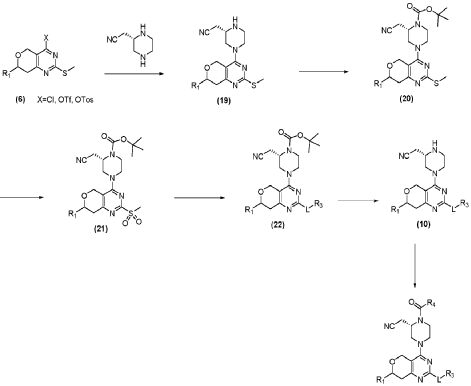
| (2) Resolution of compound 1 |
| Synthesis of compounds 1-1 and 1-2 |
| |
| The challenge lay in obtaining the compound shown in formula (I) through chiral resolution of compound 1. Despite trying various conditions, the two isomers of compound 1 could not be separated on a thin-layer chromatography plate, making separation impossible by thin-layer chromatography. Even in HPLC, the separation of the two isomers of compound 1 was poor, making separation impossible by preparative HPLC. Finally, chiral resolution had to be resorted to. After trying several conditions (as shown in Table 1 below), chiral resolution condition 9 was finally found, which enabled the separation of the compound shown in formula (I) and its diastereomers. |
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022081655&_cid=P20-MJ0T2K-48115-1
PAT
- Substituted dihydropyranopyrimidine compounds as kras inhibitorsPublication Number: US-2022112204-A1Priority Date: 2020-10-14
- Substituted dihydropyranopyrimidine compounds as kras inhibitorsPublication Number: WO-2022081655-A1Priority Date: 2020-10-14
- Oxygen-containing heterocyclic compound, preparation method and application thereofPublication Number: WO-2021109737-A1Priority Date: 2019-12-02
- Oxygen-containing heterocyclic compound, preparation method and application thereofPublication Number: EP-4015520-A1Priority Date: 2019-12-02
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////talorasib, antineoplastic, 727W6T7DPK
Talogreptide mesaroxetan


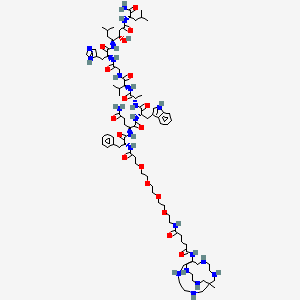
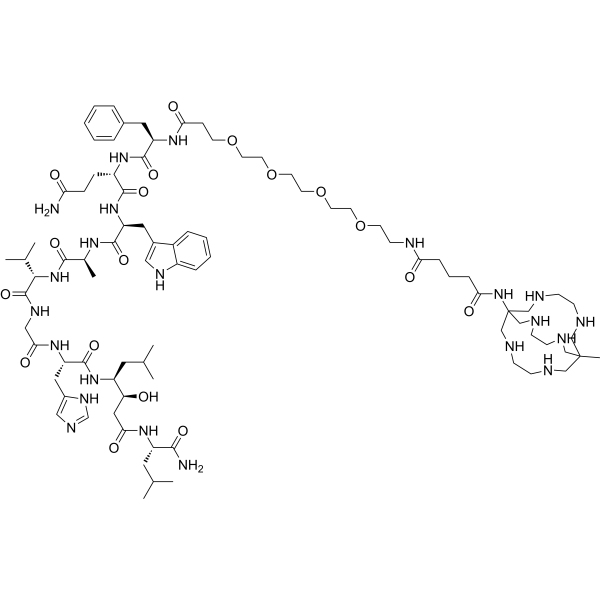
Talogreptide mesaroxetan
CAS 1801418-23-4
MF C86H140N22O18 MW1770.17
{MeCOSar}-PEG4-{d-Phe}-Gln-Trp-Ala-Val-Gly-His-{Sta}-Leu-NH2
(2S)-N-[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[[(2S)-1-[[(3S,4S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-3-hydroxy-6-methyl-1-oxoheptan-4-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]-2-[[(2R)-2-[3-[2-[2-[2-[2-[[5-[(8-methyl-3,6,10,13,16,19-hexazabicyclo[6.6.6]icosan-1-yl)amino]-5-oxopentanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]-3-phenylpropanoyl]amino]pentanediamide
N-{21-[(8-methyl-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosan-1-yl)amino] -17,21-dioxo-4,7,10,13-tetraoxa-16-azahenicosan-1-oyl}-D-phenylalanyl-L-glutaminyl-L-tryptophyl-L-alanyl-Lvalylglycyl-L-histidyl-(3S,4S)-4-amino-3-hydroxy-6-methylheptanoyl-L-leucinamide
diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Talogreptide mesaroxetan (CAS 1801418-23-4) is a synthetic peptide, a complex molecule used as a diagnostic imaging agent with potential antitumor effects, targeting G-protein coupled receptors (GRPr) often overexpressed in cancers, allowing for specific tumor visualization in PET scans, particularly for metastatic disease detection, known for its high specificity and contrast for imaging tumors like those expressing GRPr.
Key Characteristics:
- Type: A peptide-based diagnostic agent, often labeled with radioisotopes like Copper-64 ($^{64}$Cu) for Positron Emission Tomography (PET) imaging, notes Patsnap Synapse.
- Structure: It’s a modified peptide sequence incorporating elements like PEG4 and specific amino acids, MedchemExpress.com.
- Function: Binds strongly to GRPr, helping to highlight tumors and metastatic sites.
- Application: Used in research to create high-contrast PET scans for better tumor detection and monitoring, showing promise in visualizing lymph node metastasis.
In Simple Terms:
Imagine it as a “smart tracer” that seeks out specific cancer cells. When attached to a radioactive tag, it lights up tumors on a PET scan, helping doctors see cancer more clearly, notes Patsnap Synapse.
Syn
WO2024086891
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024086891&_cid=P10-MIZEJM-53111-1
67Cu radioisotope
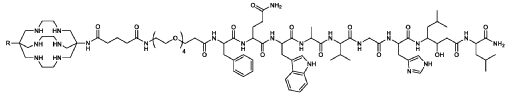
where R is CH3C(0)-;
(67CU-SAR-BBN)
Paper
Molecular Pharmaceutics (2015), 12(8), 2781-2790
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Talogreptide mesaroxetan, diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Suvadronabinol
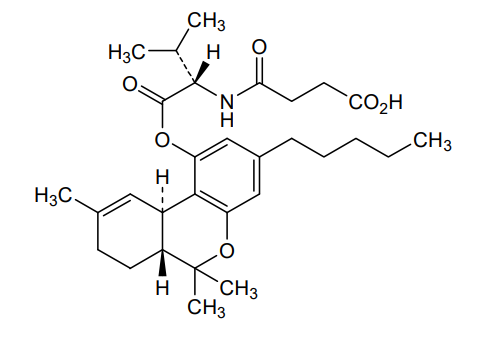
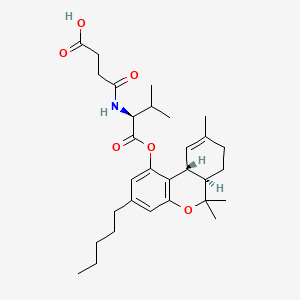
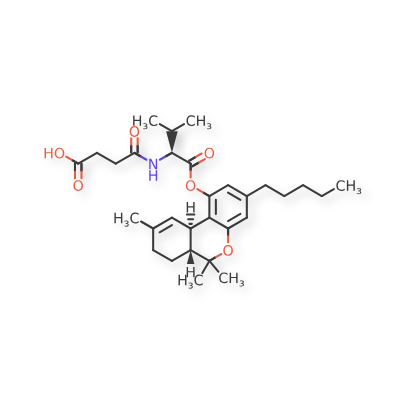
Suvadronabinol
CAS 1225194-84-2
MF C30H43NO6 MW513.7 g/mol
4-{[(2S)-3-methyl-1-oxo-1-{[(6aR,10aR)-6,6,9-trimethyl-3-pentyl6a,7,8,10a-tetrahydro-6H-dibenzo[b,d]pyran-1-yl]oxy}butan-2-yl]amino}-4-oxobutanoic acid
4-{[(2S)-3-methyl-1-oxo-1-{[(6aR,10aR)-6,6,9-trimethyl-3-pentyl-6a,7,8,10a-tetrahydro-6H-dibenzo[b,d]pyran-1-yl]oxy}butan-2-yl]amino}-4-oxobutanoic acid
cannabinoid receptor agonist, DB 21741, XV9S3R9XJC
Suvadronabinol (DB21741) is a potent, synthetic small-molecule cannabinoid receptor type 1 (CB1) agonist, initially developed for therapeutic potential in areas like appetite stimulation, pain, or weight management, acting similarly to cannabis compounds but with specific design, currently in preclinical research stages, noted for its high selectivity and potency.
Key Characteristics:
- Type: Small Molecule Drug.
- Mechanism: A highly selective agonist for the cannabinoid receptor type 1 (CB1).
- Development: Originally developed by Elsohly Laboratories, it’s in preclinical R&D, with status as an experimental compound.
- Molecular Weight: Approximately 513.31 Da.
- CAS Number: 1225194-84-2.
Potential Applications (Research Areas):
- Appetite Stimulation & Weight Loss: Similar to dronabinol, it targets pathways involved in metabolism and appetite.
- Pain Management: As a cannabinoid, it interacts with the endocannabinoid system, which plays a role in pain perception.
Status:
- It’s an investigational compound, meaning it’s still under study and not yet approved for medical use.
In essence, Suvadronabinol is a targeted synthetic cannabinoid designed to interact with the body’s CB1 receptors, showing promise in preclinical research for conditions where cannabinoid effects are desired, but it’s not a widely available or established medicine.
SYN
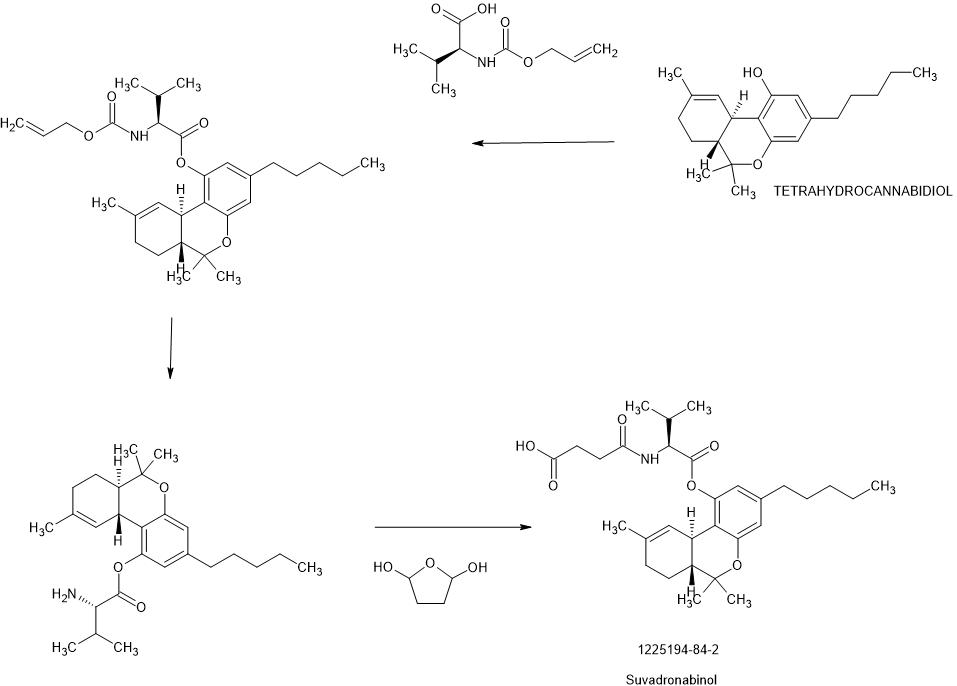
SYN
US20150045282
SYN
WO2010051541
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010051541&_cid=P22-MIXZ0J-96045-1
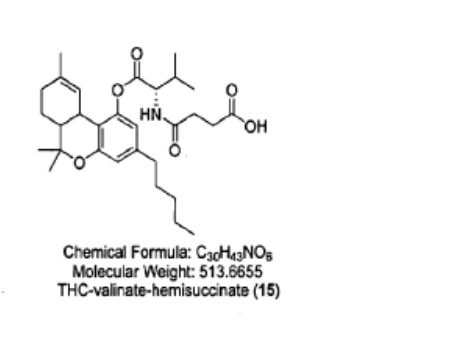
Example 10: Preparation of THC-valinate-hemisuccinate (15):
Compound 15 was also prepared using scheme II, where the starting material was compound 6 (THC-valinate). Product 15 was purified using column chromatography (>85% yield) and confirmed by mass spectroscopy in the positive ionization mode (M+NlV = 531) (Fig 17). The structure of product 15 was also confirmed by spectral analysis 1H-NMR and 13C-NMR (see Fig 18 for 13C-NMR assignments).
Spectral analysis of Δ9-THC prodrugs prepared above: Identity and purity of the synthesized prodrugs was established by spectral means including 1H-NMR, 13C-NMR and 2D-NMR such as COSY, HMQC, HMBC, as well as other spectroscopic means (IR1 UV and MS). The synthetic protocols outlined above yielded prodrugs with ≥95% purity.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////Suvadronabinol, cannabinoid receptor agonist, DB 21741, XV9S3R9XJC
Surzetoclax
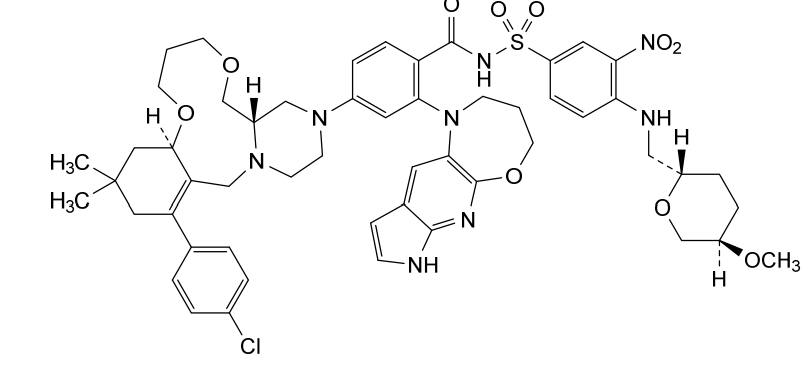

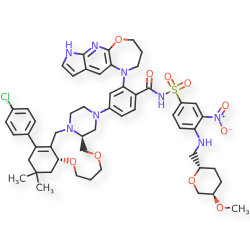
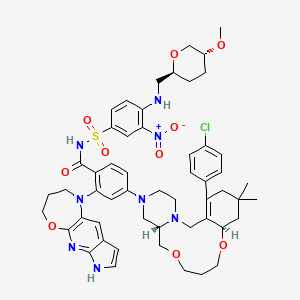
Surzetoclax
CAS 2858632-01-4
MF C53H63ClN8O10S, 1039.64
NAMES
4-[(4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl1,2,4a,5,8,9,10a,11,13,15-decahydro-7H,12Hpyrazino[2,1-g][1,5,8]benzodioxaazacycloundecin3(4H)-yl]-2-(3,4-dihydro-2Hpyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1(7H)-yl)-N-[4-({[(2S,5R)-5-methoxyoxan-2-yl]methyl}amino)-3-nitrobenzene-1-sulfonyl]benzamide
4-(14-(4-chlorophenyl)-12,12-dimethyl-1,2,4a,5,8,9,10a,11,13,15-decahydro-7H,12H-benzo[f]pyrazino[2,1-c][1,8]dioxa[4]azacycloundecin-3(4H)-yl)-2-(3,4-dihydro-2H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1(7H)-yl)-N-((4-((((2S,5R)-5-methoxytetrahydro-2H-pyran-2-yl)methyl)amino)-3-nitrophenyl)sulfonyl)benzamide
D-erythro-Hexitol, 1,5-anhydro-6-[[4-[[[4-[(4aS,10aR)-14-(4-chlorophenyl)-1,2,4a,5,8,9,10a,11,13,15-decahydro-12,12-dimethyl-7H,12H-pyrazino[2,1-g][1,5,8]benzodioxaazacycloundecin-3(4H)-yl]-2-(3,4-dihydro-2H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1(7H)-yl)benzoyl]amino]sulfonyl]-2-nitrophenyl]amino]-3,4,6-trideoxy-2-O-methyl-
1,5-Anhydro-6-[[4-[[[4-[(4aS,10aR)-14-(4-chlorophenyl)-1,2,4a,5,8,9,10a,11,13,15-decahydro-12,12-dimethyl-7H,12H-pyrazino[2,1-g][1,5,8]benzodioxaazacycloundecin-3(4H)-yl]-2-(3,4-dihydro-2H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1(7H)-yl)benzoyl]amino]sulfonyl]-2-nitrophenyl]amino]-3,4,6-trideoxy-2-O-methyl-D-erythro-hexitolB-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, ABBV 453, C3TU3CHH6L, Bcl-2-IN-16
Surzetoclax, also known as ABBV 453; is a highly potent and selective BCL-2 inhibitor with a Ki of approximately 0.07 nM. It induces apoptosis in BCL-2–dependent hematologic cancer cells, showing EC50 values typically below 10 nM in sensitive models. In vivo, Surzetoclax causes rapid tumor regression in xenograft models of non-Hodgkin lymphoma (NHL) and chronic lymphocytic leukemia (CLL). It is orally bioavailable and demonstrates dose-dependent target engagement with favorable pharmacokinetics. Compared to Venetoclax, Surzetoclax was designed to reduce risks of tumor lysis syndrome and other dose-limiting toxicities.
Surzetoclax is a small molecule drug. The usage of the INN stem ‘-toclax’ in the name indicates that Surzetoclax is a B-cell lymphoma 2 (Bcl-2) inhibitor. Surzetoclax has a monoisotopic molecular weight of 1038.41 Da.
- A Study to Assess Adverse Events and Change in Disease Activity of Oral ABBV-453 Alone or in Combination With Subcutaneous and/or Oral Antimyeloma Agents in Adult Participants With Multiple Myeloma (MM)CTID: NCT06953960Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-12-01
- A Study to Assess the Adverse Events and Change in Disease Activity in Adult Participants With Relapsed or Refractory Multiple Myeloma Receiving Oral ABBV-453 TabletsCTID: NCT05308654Phase: Phase 1Status: Active, not recruitingDate: 2025-08-14
- A Study Assessing Adverse Event and How Oral ABBV-453 Moves Through the Body in Adult Participants With Relapsed or Refractory (R/R) Chronic Lymphocytic Leukemia (CLL)/Small Lymphocytic Lymphoma (SLL)CTID: NCT06291220Phase: Phase 1Status: Active, not recruitingDate: 2025-06-06
SYN
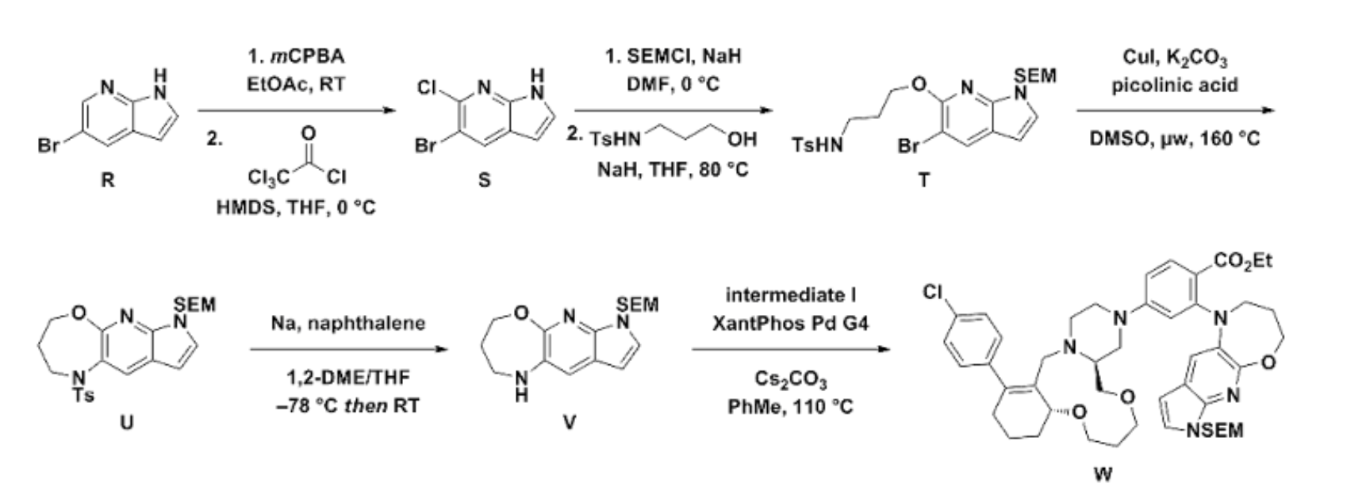
The synthesis of surzetoclax (ABBV-453), a complex, next-generation BCL-2 inhibitor, can be accomplished through a patented 27-step convergent route or a more streamlined, AI-assisted method that involves the modular assembly of three key fragments.
Patented Synthesis (Literature Route)
The published synthesis (described in patent WO 2023/141536 A1) is a 27-step convergent route with a 12-step longest linear sequence. The molecule is assembled from three main components: a 6,11,6-fused tricyclic core, a trans-1,2-disubstituted tetrahydropyran (THP) unit, and a 5,6,7-fused heteroaromatic system.
Key Steps and Intermediates:
- Core Tricycle Assembly: The 6,11,6-fused macrocycle is formed by a sequence initiated from dimedone, involving a macrocyclization step using an 11-membered ring bis-triflate intermediate.
- THP Fragment Construction: The THP moiety’s synthesis includes an enzymatic resolution step using porcine pancreatic lipase to establish the required stereochemistry.
- Heteroaromatic System: The 7-azaindole-oxazepane tricycle is formed via a microwave-assisted, copper-mediated cyclization reaction.
- Final Coupling: The fragments are joined through Buchwald–Hartwig and N-sulfonylamide coupling reactions to yield the final surzetoclax molecule.
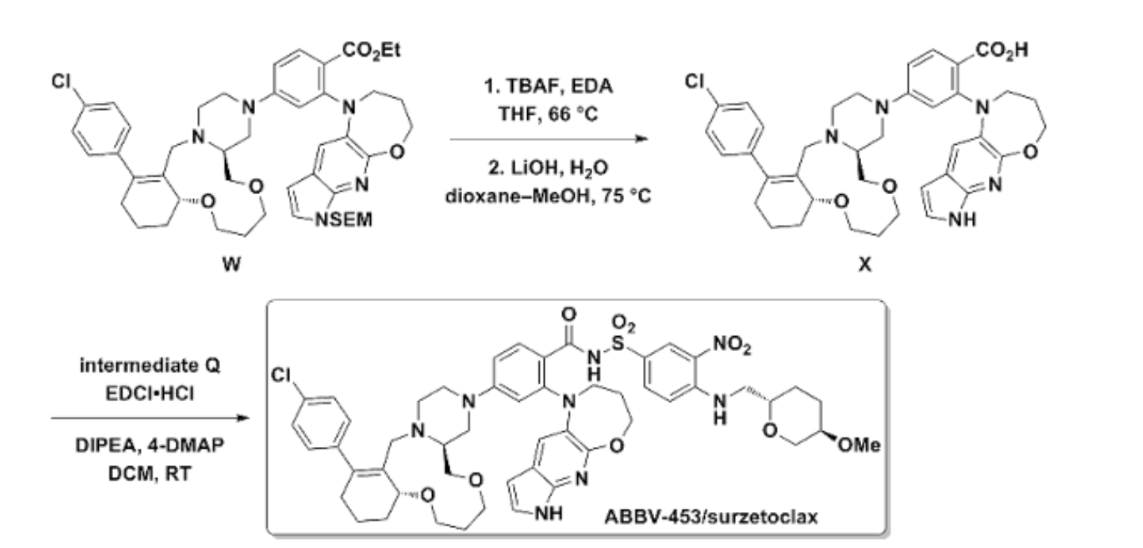
AI-Assisted Synthesis (ChemAIRS Route)
A more efficient, human-directed AI retrosynthesis approach developed by Chemical.AI offers a more convergent and experimentally practical alternative. This route also uses three fragments but employs different, more efficient coupling strategies.
Key Features of the Revised Route:
- Fragment 25a (Core Tricycle): Assembled in 10 steps using more accessible starting materials and a Mizoroki-Heck coupling to incorporate an aryl group.
- Fragment 12a (THP Motif): The synthesis is streamlined to four steps from simple building blocks, utilizing a Mitsunobu reaction to introduce the azide precursor instead of the patent’s longer tosylation/displacement sequence.
- Fragment 12b (Azaindole-Oxazepane Tricycle): An efficient nickel-photoredox C–N coupling is used to form an intermediate that then undergoes a base-promoted cyclization.
- Final Assembly: Fragments 12a and 12b are joined using a palladium-catalyzed amidocarbonylation, followed by deprotection and a final SNAr coupling with fragment 25a to form surzetoclax.
The AI-assisted route achieves greater modularity and adaptability for potential scale-up compared to the patented process.
PAT
1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepine bcl-2 inhibitors
Publication Number: US-11964990-B2
Priority Date: 2022-01-21
Grant Date: 2024-04-23
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US403063458&_cid=P10-MIWJNM-88215-1
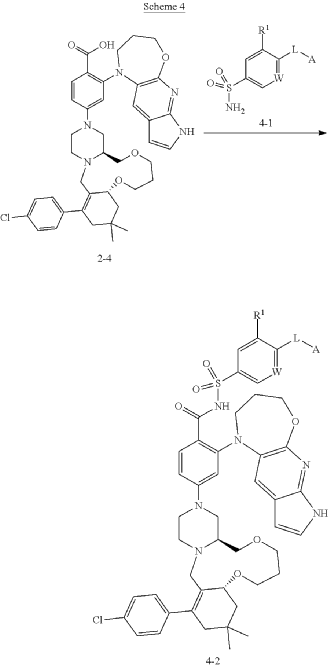
Example 25
4-((4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl-1,2,4a,5,8,9,10a,11,13,15-decahydro-7H,12H-benzo[f]pyrazino[2,1-c][1,8]dioxa[4]azacycloundecin-3(4H)-yl)-2-(3,4-dihydro-2H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1(7H)-yl)-N-((4-((((2S,5R)-5-methoxytetrahydro-2H-pyran-2-yl)methyl)amino)-3-nitrophenyl)sulfonyl)benzamide
Example 25A
(S)-(3,4-dihydro-2H-pyran-2-yl)methanol
Example 25B
(S)-2-((benzyloxy)methyl)-3,4-dihydro-2H-pyran
Example 25C
(3R,6S)-6-((benzyloxy)methyl)tetrahydro-2H-pyran-3-ol
Example 25D
(2S,5R)-2-((benzyloxy)methyl)-5-methoxytetrahydro-2H-pyran
Example 25E
((2S,5R)-5-methoxytetrahydro-2H-pyran-2-yl)methyl 4-methylbenzenesulfonate
Example 25F
(2S,5R)-2-(azidomethyl)-5-methoxytetrahydro-2H-pyran
Example 25G
4-((((2S,5R)-5-methoxytetrahydro-2H-pyran-2-yl)methyl)amino)-3-nitrobenzenesulfonamide
Example 25H
7,7-dimethyl-4,6,7,8-tetrahydro-2H,5H-1,3-benzodioxin-5-one
Example 251
4′-chloro-2-(hydroxymethyl)-5,5-dimethyl-5,6-dihydro-[1,1′-biphenyl]-3(4H)-one
Example 25J
4′-chloro-2-(chloromethyl)-5,5-dimethyl-5,6-dihydro-[1,1′-biphenyl]-3(4H)-one
Example 25K
(R)-4′-chloro-2-(chloromethyl)-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-3-ol
Example 25L
tert-butyl (S)-4-(((R)-4′-chloro-3-hydroxy-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)-3-(hydroxymethyl)piperazine-1-carboxylate
Example 25M
tert-butyl (4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl-1,2,4a,5,8,9,10a,11,13,15-decahydro-7H,12H-benzo[f]pyrazino[2,1-c][1,8]dioxa[4]azacycloundecine-3(4H)-carboxylate
Example 25N
(4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl-1,2,3,4,4a,5,7,8,9,10a,11,12,13,15-tetradecahydrobenzo[f]pyrazino[2,1-c][1,8,4]dioxaazacycloundecine
Example 250
ethyl 2-bromo-4-((4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl-1,2,4a,5,8,9,10a,11,12,13-decahydrobenzo[f]pyrazino[2,1-c][1,8,4]dioxaazacycloundecin-3(4H,7H,15H)-yl)benzoate
Example 25P
5-bromo-1H-pyrrolo[2,3-b]pyridine 7-oxide
Example 25Q
5-bromo-6-chloro-1H-pyrrolo[2,3-b]pyridine
Example 25R
5-bromo-6-chloro-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridine
Example 25S
N-(3-((5-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrolo[2,3-b]pyridin-6-yl)oxy)propyl)-4-methylbenzenesulfonamide
Example 25T
1-tosyl-7-((2-(trimethylsilyl)ethoxy)methyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepane
Example 25U
7-((2-(trimethylsilyl)ethoxy)methyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepane
Example 25V
ethyl 4-((4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl-1,2,4a,5,8,9,10a,11,12,13-decahydrobenzo[f]pyrazino[2,1-c][1,8,4]dioxaazacycloundecin-3(4H,7H,15H)-yl)-2-(7-((2-(trimethylsilyl)ethoxy)methyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1-yl)benzoate
Example 25W
ethyl 4-((4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl-1,2,4a,5,8,9,10a,11,12,13-decahydrobenzo[f]pyrazino[2,1-c][1,8,4]dioxaazacycloundecin-3(4H, 7H,15H)-yl)-2-(2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1-yl)benzoate
Example 25X
4-((4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl-1,2,4a,5,8,9,10a,11,12,13-decahydrobenzo[f]pyrazino[2,1-c][1,8,4]dioxaazacycloundecin-3(4H,7H,15H)-yl)-2-(2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1-yl)benzoic acid
Example 25Y
4-((4aS,10aR)-14-(4-chlorophenyl)-12,12-dimethyl-1,2,4a,5,8,9,10a,11,13,15-decahydro-7H,12H-benzo[f]pyrazino[2,1-c][1,8]dioxa[4]azacycloundecin-3(4H)-yl)-2-(3,4-dihydro-2H-pyrrolo[3′,2′:5,6]pyrido[2,3-b][1,4]oxazepin-1(7H)-yl)-N-((4-((((2S,5R)-5-methoxytetrahydro-2H-pyran-2-yl)methyl)amino)-3-nitrophenyl)sulfonyl)benzamide
SYN
https://www.chemical.ai/blog/dl7xc1h1477b1hp21ajpef87z13c73
Thanks and CREDIT, https://www.chemical.ai/chemairs



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////Surzetoclax, antineoplastic, ABBV 453, C3TU3CHH6L, Bcl-2-IN-16
Surlorian

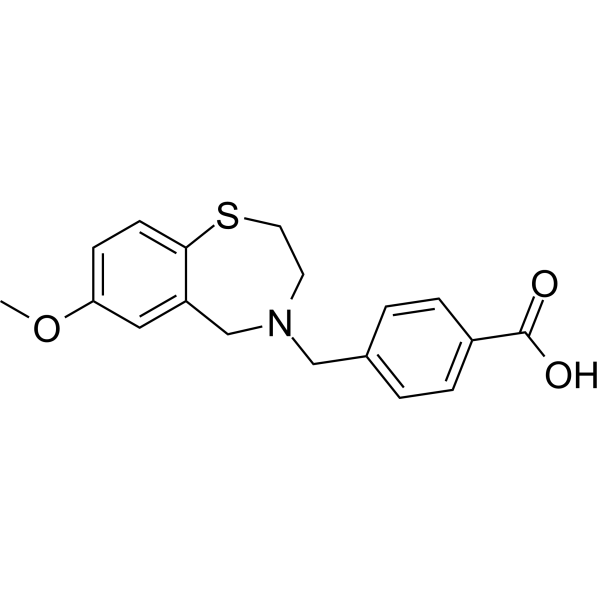
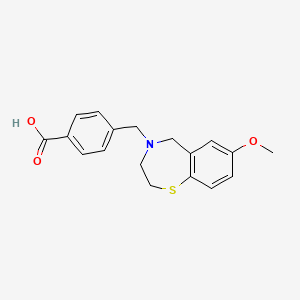
Surlorian
CAS 1467605-57-7
MFC18H19NO3S, 329.41
4-[(7-methoxy-2,3-dihydro-1,4-benzothiazepin-4(5H)-yl)methyl]benzoic acid
ryanodine receptor (RyR) stabilizer, ARM 210, RYCAL DMD, s48168, S 48168, 1033GN605L
Surlorian (ARM210) is a novel drug candidate, specifically a Ryanodine Receptor (RyR) stabilizer developed by RyCarma Therapeutics, designed to treat heart failure by repairing leaky RyRs in heart and skeletal muscles, aiming to improve both cardiac function and muscle weakness by restoring normal calcium regulation. It’s an allosteric modulator, a “Rycal,” that fixes these crucial calcium channels without blocking them, addressing a core issue in heart failure and related muscle diseases.
Key Aspects:
- Mechanism: It stabilizes damaged RyR channels, which become leaky due to stress (like in heart failure), preventing unregulated calcium leakage from the sarcoplasmic reticulum (SR).
- Target: Acts systemically on RyRs in both cardiac (heart) and skeletal (muscle) cells.
- Benefits: Aims to improve heart contraction/relaxation and alleviate skeletal muscle weakness, a common heart failure symptom.
- Therapeutic Area: Heart failure, potentially addressing underlying calcium dysregulation.
- Status: In clinical development, with Phase II trials underway as of mid-2025, according to AdisInsight.
- Chemical Info: Molecular Formula: C18H19NO3S; CAS No: 1467605-57-7.
In essence, Surlorian offers a new approach to heart failure by fixing the fundamental calcium handling problem in muscles, rather than just managing symptoms.
- OriginatorARMGO Pharma
- DeveloperNational Institute of Neurological Disorders and Stroke; RyCarma Therapeutics; Servier
- ClassAntiarrhythmics; Heart failure therapies; Small molecules
- Mechanism of ActionRyanodine receptor calcium release channel modulators
- Orphan Drug StatusYes – Polymorphic catecholergic ventricular tachycardia; Congenital structural myopathies; Duchenne muscular dystrophy
- Phase IIPolymorphic catecholergic ventricular tachycardia
- Phase ICardiac-arrhythmias; Congenital structural myopathies; Heart failure
- No development reportedDuchenne muscular dystrophy; Limb girdle muscular dystrophies; Sarcopenia; X-linked bulbo-spinal atrophy
- 04 Sep 2025Chemical structure information added.
- 02 Apr 2025Surlorian is still in phase I trial for Congenital structural myopathies in USA (PO) (NCT04141670)
- 02 Apr 2025Phase-I clinical trials in Heart failure (unspecified route), prior to April 2025 (RyCarma Therapeutics pipeline, April 2025)
- Treatment of an Inherited Ventricular ArrhythmiaCTID: NCT05122975Phase: Phase 2Status: TerminatedDate: 2024-09-19
- S 48168 (ARM 210) for the Treatment of RYR1-related Myopathies (RYR1-RM)CTID: NCT04141670Phase: Phase 1Status: CompletedDate: 2024-08-22
SYN
EXAMPLE 1: PREPARATION OF 4-[(7-METHOXY-2,3-DIHYDRO-1,4-BENZOTHIAZEPIN-4(5H)YL)METHYL]BENZOIC ACID
| 4-[(7-methoxy-2,3-dihydro-1,4-benzothiazepin-4(5H)yl)methyl]benzoic acid was prepared as described below. |
Stage 1: 7-methoxy-2,3,4,5-tetrahydrobenzo[f][1,4]thiazepine (“Amine”)

2-(4-Methoxyphenylthio)ethanamine (1)
Benzyl 2-(4-methoxyphenylthio)ethylcarbamate (2)
Benzyl 7-methoxy-2,3-dihydrobenzo[f][1,4]thiazepine-4(5H)-carboxylate (3)
7-Methoxy-2,3,4,5-tetrahydrobenzo[f][1,4]thiazepine hydrobromide (Amine)
Stage 2: −[(7-methoxy-2,3-dihydro-1,4-benzothiazepin-4(5H)yl)methyl]benzoic acid

In Scheme 2, L is a leaving group, which is, by way of example, a halogen or a sulfonate (OSO 2R′ wherein R′is alkyl or aryl, e.g., OMs (mesylate) or OTs (tosylate)). Amine (4) (1 mmol) was dissolved dichloromethane. To the solution was added alkylation reagent (5) (1 mmol), followed by N,N-diisopropylethylamine (2 mmol). The mixture was stirred at room temperature overnight. The solution was loaded onto a silica gel column directly and eluted with hexane/EtOAc (2:1, v/v) to afforded the desired product.
SYN
PAT
- Agents for treating disorders involving modulation of ryanodine receptorsPublication Number: US-2014088171-A1Priority Date: 2012-04-18
- Agents for treating disorders involving modulation of ryanodine receptorsPublication Number: US-2014378437-A1Priority Date: 2012-04-18
- Agents for treating disorders involving modulation of ryanodine receptorsPublication Number: US-8853198-B2Priority Date: 2012-04-18Grant Date: 2014-10-07
- Agents for treating disorders involving modulation of ryanodine receptorsPublication Number: WO-2013156505-A1Priority Date: 2012-04-18
- Drugs for treating diseases involved in the modulation of ryanodine receptorsPublication Number: JP-2015514736-APriority Date: 2012-04-18
- Drugs for treating diseases involved in the modulation of ryanodine receptorsPublication Number: JP-5965542-B2Priority Date: 2012-04-18Grant Date: 2016-08-10
- Agents for treating disorders involving modulation of ryanodine receptorsPublication Number: KR-101731459-B1Priority Date: 2012-04-18Grant Date: 2017-04-28
- Agents for treating disorders involving modulation of ryanodine receptorsPublication Number: KR-20150003347-APriority Date: 2012-04-18
- Agents for treating disorders involving modulation of ryanodine receptorsPublication Number: US-2013281512-A1Priority Date: 2012-04-18

- ARM-210 hemifumarate
- ZHR6WM1ADJ
- UNII-ZHR6WM1ADJ
- 1467606-11-6
- Surlorian fumarate (USAN)
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Surlorian, ryanodine receptor (RyR) stabilizer, ARM 210, RYCAL DMD, s48168, S 48168, 1033GN605L
Sumecigrel, Vicagrel
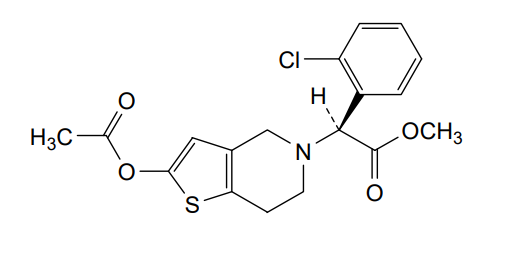
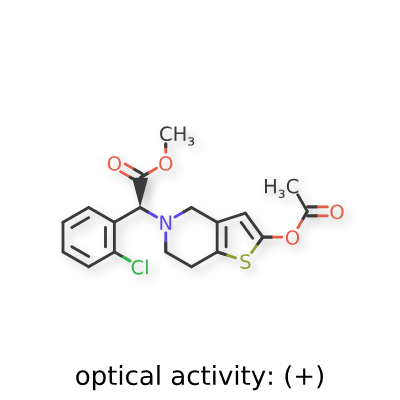
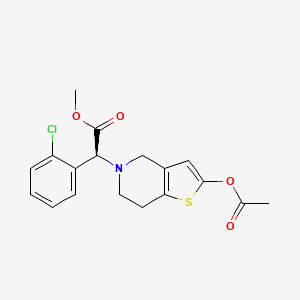
Sumecigrel, VICAGREL
CAS 1314081-53-2
MF C18H18ClNO4S MW379.858
- (S)-2-(2-ACETOXY-6,7-DIHYDROTHIENO(3,2-C)PYRIDINE-5(4H)-YL)-2-(2-CHLOROPHENYL)ACETIC ACID METHYL ESTER
- METHYL (.ALPHA.S)-2-(ACETYLOXY)-.ALPHA.-(2-CHLOROPHENYL)-6,7-DIHYDROTHIENO(3,2-C)PYRIDINE-5(4H)-ACETATE
- METHYL (2S)-2-(2-ACETYLOXY-6,7-DIHYDRO-4H-THIENO(3,2-C)PYRIDIN-5-YL)-2-(2-CHLOROPHENYL)ACETATE
- METHYL (S)-(2-(ACETYLOXY)-6,7-DIHYDROTHIENO(3,2-C)PYRIDIN-5(4H)-YL)(2-CHLOROPHENYL)ACETATE
- THIENO(3,2-C)PYRIDINE-5(4H)-ACETIC ACID, 2-(ACETYLOXY)-.ALPHA.-(2-CHLOROPHENYL)-6,7-DIHYDRO-, METHYL ESTER, (.ALPHA.S)-
- VICAGREL
methyl (S)-2-(acetyloxy)-6,7-dihydrothieno[3,2- c]pyridin-5(4H)-ylacetate
platelet aggregation inhibitor, 8A63K3TN0U, VICAGREL
- Pharmacokinetic/Pharmacodynamic Study of Vicagrel Capsules and Clopidogrel Tablets in Healthy CYP2C19 Normal MetabolizersCTID: NCT07067775Phase: Phase 1Status: CompletedDate: 2025-09-09
- Efficacy and Safety Study of Vicagrel in Patients With Acute Coronary Syndrome (ACS) Undergoing Percutaneous Coronary Intervention (PCI)CTID: NCT06577519Phase: Phase 3Status: RecruitingDate: 2024-10-01
- PK/PD Study of Vicagrel and Clopidogrel in Healthy Subjects With Different CYP2C19 MetabolizersCTID: NCT05162053Phase: Phase 1Status: CompletedDate: 2023-11-03
- A Clinical Trial to Evaluate the Effect of Food on PK and PD of Vicagrel Capsules in Healthy Adult SubjectsCTID: NCT04919551Phase: Phase 1Status: CompletedDate: 2021-11-01
- The Efficacy, Safety and Pharmacokinetic of Antiplatelet Therapy for VicagrelCTID: NCT03599284Phase: Phase 2Status: CompletedDate: 2019-09-23
- Pharmacokinetics and Pharmacodynamics of Vicagrel in Healthy Adult Subjects of Different CYP2C19
- CTID: NCT03942458
- Phase: Phase 1
- Status: Completed
- Date: 2019-09-19
Sumecigrel (also known as
vicagrel) is an investigational small molecule drug classified as a P2Y12 inhibitor and antiplatelet agent. It is currently under clinical development for the treatment of various cardiovascular and peripheral conditions.
Key Information
- Therapeutic Class: Antiplatelet agent; P2Y12 inhibitor. These types of drugs work by preventing platelets in the blood from sticking together and forming clots, which is a key process in conditions like heart attack and stroke.
- Developer: Jiangsu Vcare PharmaTech.
- Clinical Status: It is currently in the pre-registration phase for acute coronary syndrome (ACS). It has also been under investigation for ischemic stroke and peripheral arterial disease.
- Synonyms: The drug is also widely referred to by its USAN (United States Adopted Name) and INN (International Nonproprietary Name) designation, vicagrel.
Chemical Details
- Formula:
C18H18ClNO4SC sub 18 H sub 18 ClNO sub 4 SC18H18ClNO4S.
- CAS Number: 1314081-53-2.
- UNII: 8A63K3TN0U.
For more detailed information regarding its regulatory status, you can check the official precisionFDA or PubChem databases.
SYN
SYN
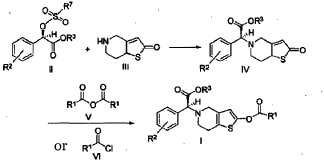
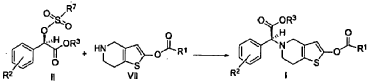
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN85433897&_cid=P22-MITPA1-71386-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP75374721&_cid=P22-MITPA1-71386-1
Example 3
(2S)-Methyl 2-(2-oxo-7,7a-dihydrothieno[3,2-c]pyridin-5(2H,4H,6H)-yl)-2-(2-chlorophenyl)-acetate (IV-1)
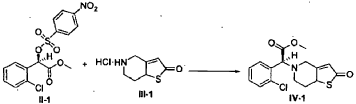
[0036] 58.1 g (0.15 mol) of (R)-methyl 2-(2-chlorophenyl)-2-(4-nitrophenylsulfonyloxy)-acetate ( II-1), 32.3 g (0.17 mol) of 5,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2(4H)-one hydrochloride ( III-1), and 37.8g (0.38 mol) of potassium bicarbonate were added to 500 ml of acetonitrile. The reaction was stirred under a nitrogen atmosphere at room temperature for 26 hrs. The reaction solution was allowed to stand and the insoluble material was filtered off, to obtain a dark red mother liquor. The solvent was evaporated under reduced pressure, and 35.4 g of an oil product was obtained after purification by flash column chromatography (petroleum ether:ethyl acetate = 4:1). Yield 70%. Recrystalization from ethanol afforded 18.1 g of a pure product (IV-1) as a white solid. mp: 146-148°C, ee = 97.5%, [α] D 19 = +114.0° (c 0.5, MeOH); 1H-NMR (300 MHz, CDCl 3) δ 1.79-1.93 (m, 1 H), 2.30-2.40 (m, 1 H), 2.56-2.70 (m, 1 H), 3.00-3.27 (m, 2 H), 3.72 (s, 3 H), 3.79-3.93 (m, 1 H), 4.12-4.19 (m, 1 H), 4.89 (d, 1 H, J= 5.6 Hz), 6.00 (d, 1 H, J = 5.2 Hz), 7.26-7.50 (m, 4 H); 13C-NMR (75 MHz, CDCl 3) δ 33.9, 34.0, 49.0, 49.7, 51.1, 51.6, 52.2, 52.4, 67.3, 76.6, 77.0, 77.4, 126.6, 126.8, 127.2, 129.8, 130.1, 132.7, 134.8, 167.2, 167.4, 170.8, 198.6; ESI-MS m/ z 338.1 [M+H] +; HRMS Calcd for C 16H 17NO 3SCl [M+H] + m/ z 338.0618, found 338.0626.
Reference Example 4
(S)-Methyl 2-(2-benzoyloxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)-acetate (I-1)
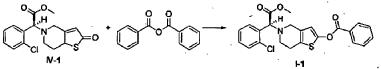
[0038] (2S)-Methyl 2-(2-chlorophenyl)-2-(2-oxo-5,6,7,7a-tetrahydrothieno[3,2-c]pyridinyl)acetate ( IV– 1) (113 mg) was dissolved in acetonitrile (10 ml), 0.10 ml of triethylamine was added, and 151 mg of benzoic anhydride was added dropwise at 0°C, and then the mixture was warmed to room temperature and reacted for 2 hrs. The reaction solution was poured into water (30 ml), the aqueous phase was extracted with ethyl acetate (50 ml x 3), and the organic phase was washed with saturated aqueous sodium bicarbonate solution and saturated saline, dried over anhydrous sodium sulfate, and evaporated, to obtain a crude product, which was subjected to flash column chromatography (petroleum ether:ethyl acetate = 40 : 3), to obtain (S)-methyl 2-(2-benzoyloxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)-acetate (I-1) (77 mg). Yield 52%, mp: 84-86°C, ee = 93.5% (chiral HPLC analysis conditions: Chiralpak IC 4.6 mm x 250 mm; column temperature: 25° C; mobile phase: 90% n-hexane/10% isopropanol/0.1% diethylamine; flow rate: 0.5 ml/min; and detection wavelength: UV 254 nm), [α] D20 = +34.00° (c 0.50, MeOH); 1H-NMR (300 MHz, CDCl 3) δ 2.82-2.93 (m, 4 H), 3.57-3.68 (m, 2 H), 3.73 (s, 3 H), 4.95 (s, 1 H), 6.42 (s, 1 H), 7.26-8.17 (m, 9 H); 13C-NMR (75 MHz CDCl 3) δ 25.0, 48.2, 50.4, 52.2, 67.8, 112.1, 125.9, 127.2, 128.5, 128.6, 129.5, 129.8, 130.0, 130.2, 133.9, 134,7, 149.9, 163.5; ESI-MS m/ z 442.1 [M+H] +; HRMS Calcd for C 23H 21NO 4SCl [M+H] +m/ z 442.0891, found 442.0880.
Example 5
(S)-Methyl 2-(2-acetoxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)-acetate (I-2)
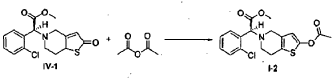
[0040] Following the method described in Example 4, (2S)-methyl 2-(2-chlorophenyl)-2-(2-oxo-5, 6, 7, 7a-tetrahydrothieno[3,2-c]pyridinyl)acetate (IV-1) (6.5 g) was reacted with acetic anhydride (3.6 ml), to prepare (S)-methyl 2-(2-acetoxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)-acetate ( I-2) (6.8 g). Yield 93%. Recrystallization from ethanol afforded a white solid, mp: 73-75°C, ee = 98.9% (chiral HPLC analysis conditions: Chiralpak IC 4.6 mm x 250 mm; column temperature: 25°C; mobile phase: 92% n-hexane/8% tetrahydrofuran/0.1% diethylamine; flow rate: 0.5 ml/min; and detection wavelength: UV 254 nm), [α] D23 = +45.00°(c = 1.0, CH 3OH); 1H-NMR (300 MHz, CDCl 3) δ 2.26 (s, 3 H), 2.65-2.90 (m, 4 H), 3.47-3.69 (m, 2 H), 3.72 (s, 3 H), 4.92 (s, 1 H), 6.26 (s, 1 H), 7.24-7.70 (m, 4 H); 13C-NMR (75 MHz, CDCl 3) δ 20.2, 24.5, 47.6, 49.8, 51.6, 67.3, 111.5, 125.3, 126.6, 128.8, 128.9, 129.3, 129.4, 133.3, 134.2, 149.1, 167.2, 170.7; ESI-MS m/ z 380.0 [M+H] +; HRMS Calcd for C 18H 19NO 4SCl [M+H] +m/ z 380.0723, found 380.0737.
Reference Example 6
(R)-Methyl 2-(2-acetoxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)-acetate (I-2′)
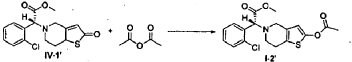
[0042] Following the method described in Example 4, (2R)-methyl 2-(2-chlorophenyl)-2-(2-oxo-7,7a-dihydrothieno[3.2-c]pyridin-5(2H,4H,6H)-yl)-acetate ( IV-1′) (prepared following Examples 1-3) was reacted with acetic anhydride, to prepare (R)-methyl 2-(2-acetoxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)-acetate ( I-2′), ee = 98.2% (chiral HPLC analysis conditions were the same as those in Example 5), [α] D 23 =-44.00° (c= 1.0, CH 3OH).
Reference Example 7
(S)-Methyl 2-(2-propanoyloxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)-acetate (I-3)
[0043] Following the method described in Example 4, (2S)-methyl 2-(2-chlorophenyl)-2-(2-oxo-7, 7a-dihydrothieno[3.2-c]pyridin-5(2H, 4H,6H)-yl)-acetate ( IV-1) (338 mg) was reacted with propionic anhydride (0.27 ml), to prepare (S)-methyl 2-(2-propanoyloxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2-chlorophenyl)-acetate ( I-3) (267 mg).Yield 66%, ee = 96.5% (chiral HPLC analysis conditions were the same as those in Example 4), [α] D20 = + 36.00°( c 0.50, MeOH); 1H-NMR (300 MHz, CDCl 3) δ 1.23 (t, 3 H, J = 7.4 Hz), 2.55 (q, 2 H, J= 7.7 Hz), 2.76-2.78 (m, 2 H), 2.87-2.88 (m, 2 H), 3.53 (d, 1 H, J = 14.2 Hz), 3.65 (d, 1 H, J = 13.6 Hz), 3.72 (s, 3 H), 4.91 (s, 1 H), 6.26 (s, 1 H), 7.26-7.69 (m, 4 H); 13C-NMR (75 MHz, CDCl 3) δ 8.8, 21.1, 25.0, 27.4, 48.2, 50.3, 52.2, 67.8, 106.2, 111.7, 125.6, 127.2, 129.1, 129.5, 129.8, 130.0, 123.7, 149.8, 171.2; ESI-MS m/ z 394.1 [M+H] +; HRMS Calcd for C 19H 21NO 4SCl [M+H] +m/ z 394.0883, found 394.0880.
PAT
- Optically active 2-hydroxytetrahydrothienopyridine derivatives, preparation method and use in manufacture of medicament thereofPublication Number: KR-102215429-B1Priority Date: 2010-02-02Grant Date: 2021-02-16
- Optically active 2-hydroxy tetrahydrothienopyridine derivatives, preparation method and use in manufacture of medicament thereofPublication Number: EP-3290423-B1Priority Date: 2010-02-02Grant Date: 2021-07-21
- Optically active 2-hydroxy tetrahydrothienopyridine derivatives, preparation method and use in manufacture of medicament thereofPublication Number: US-2015011583-A1Priority Date: 2010-02-02
- Optically active 2-hydroxy tetrahydrothienopyridine derivatives, preparation method and use in manufacture of medicament thereofPublication Number: US-2017121341-A1Priority Date: 2010-02-02
- Optically active 2-hydroxy tetrahydrothienopyridine derivatives, preparation method and use in manufacture of medicament thereofPublication Number: US-2019055260-A1Priority Date: 2010-02-02
- Optically active 2-hydroxy tetrahydrothienopyridine derivatives, preparation method and use in manufacture of medicament thereofPublication Number: US-8772489-B2Priority Date: 2010-02-02Grant Date: 2014-07-08
- Optically active 2-hydroxy tetrahydrothienopyridine derivatives, preparation method and use in manufacture of medicament thereofPublication Number: WO-2011095049-A1Priority Date: 2010-02-02
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Sumecigrel, platelet aggregation inhibitor, 8A63K3TN0U, VICAGREL
Soquelitinib
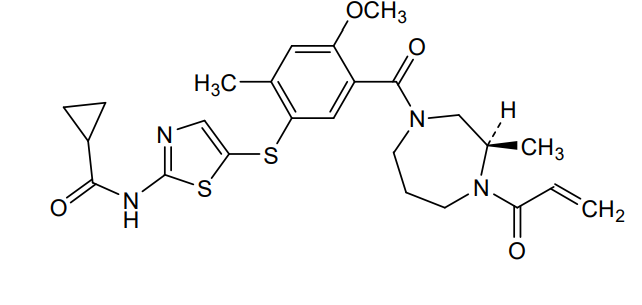
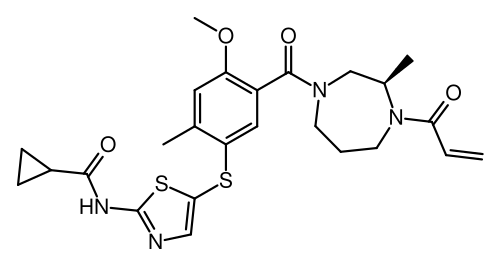
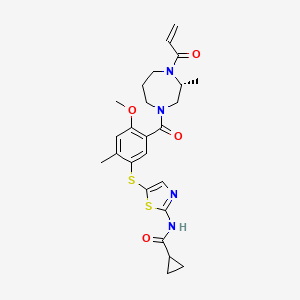
Soquelitinib
CAS 2226636-04-8
MF C25H30N4O4S2, 514.7 g/mol
N-[5-({4-methoxy-2-methyl-5-[(3R)-3-methyl-4-(prop-2-enoyl)-1,4-diazepane-1-carbonyl]phenyl}sulfanyl)-1,3-thiazol-2-yl]cyclopropane-1-carboxamide
tyrosine kinase inhibitor, antineoplastic, CPI818, CPI-000818, CPI596, CP I818, CPI 000818, CP I596, 6I5H17AN3I,
Soquelitinib (CPI-818) is an experimental drug which acts as a selective inhibitor of the enzyme interleukin-2-inducible T-cell kinase (ITK). It is in clinical trials for the treatment of T-cell lymphoma.[1][2]
Soquelitinib is an orally available, small-molecule, irreversible inhibitor of interleukin-2 inducible T-cell kinase (ITK) with potential immunomodulatory and antineoplastic activities. Upon oral administration, soquelitinib selectively and covalently binds to the cysteine residue at position 442 (CYS-442) of ITK, thereby disrupting ITK-mediated signal transduction, while sparing tyrosine-protein kinase TXK (resting lymphocyte kinase, RLK) activity. This may abrogate T-cell receptor (TCR) signaling through ITK and inhibit TCR-induced proliferation of malignant T-cells. Additionally, inhibiting ITK activation may prevent the upregulation of GATA-3, a transcription factor that drives T-helper 2 (Th2) cell differentiation and is overexpressed in certain T-cell lymphomas. Thus, selective inhibition of ITK may inhibit Th2 responses without affecting T-helper 1 (Th1)-dependent immunity. ITK, a member of the Tec family of non-receptor protein tyrosine kinases plays a significant role in the T-cell development, differentiation and production of pro-inflammatory cytokines.
- Safety, Tolerability, and Preliminary Efficacy of Soquelitinib in Participants With Moderate to Severe ADCTID: NCT06345404Phase: Phase 1Status: RecruitingDate: 2025-07-22
- Study of the ITK Inhibitor Soquelitinib to Reduce Lymphoproliferation and Improve Cytopenias in Autoimmune Lymphoproliferative Syndrome (ALPS)-FAS PatientsCTID: NCT06730126Phase: Phase 2Status: RecruitingDate: 2025-05-31
- Soquelitinib vs Standard of Care in Participants With Relapsed/Refractory Peripheral T-cell Lymphoma Not Otherwise Specified, Follicular Helper T-cell Lymphomas, or Systemic Anaplastic Large-cell LymphomaCTID: NCT06561048Phase: Phase 3Status: RecruitingDate: 2025-04-17
- A Dose Escalation Study Evaluating CPI-818 in Relapsed/Refractory T-Cell LymphomaCTID: NCT03952078Phase: Phase 1Status: Active, not recruitingDate: 2025-04-16
Syn
- US11008314,
- https://patentscope.wipo.int/search/en/detail.jsf?docId=US278926237&_cid=P10-MISM56-82578
- SIMILAR
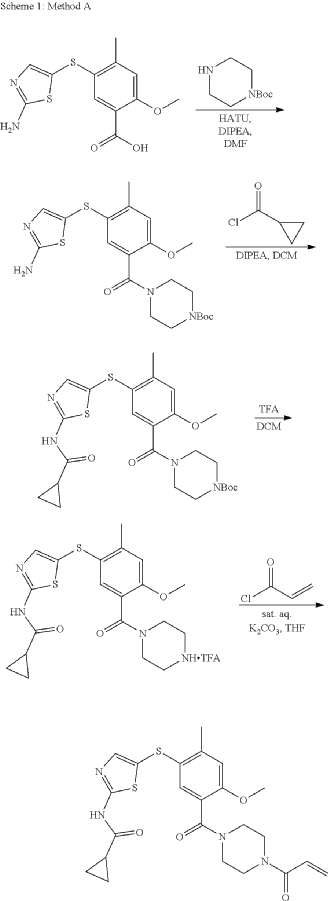

Syn
- WO2018089261 COMPD 44
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018089261&_cid=P10-MISM0C-78029-1
SYN
Embodiment B23. A method for an Th2/ITK-mediated disease in a patient in need thereof, the method comprising administering to the patient about 250 mg to about 1,000 mg per day of a compound of Formula (A) or a pharmaceutically acceptable salt thereof, wherein the compound of Formula (A) is:
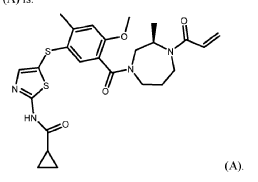
REF
https://www.nature.com/articles/s44386-024-00002-1
Pat
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: US-2022363676-A1Priority Date: 2016-11-03
- Compounds and methods for modulating Interleukin-2-inducible T-cell kinasePublication Number: US-11897874-B2Priority Date: 2016-11-03Grant Date: 2024-02-13
- Itk inhibitors for increasing th1 cell activityPublication Number: WO-2023196278-A1Priority Date: 2022-04-05
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: US-2019375743-A1Priority Date: 2016-11-03
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: WO-2018089261-A2Priority Date: 2016-11-03
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: US-11008314-B2Priority Date: 2016-11-03Grant Date: 2021-05-18
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: EP-3534899-B1Priority Date: 2016-11-03Grant Date: 2022-06-01
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Khodadoust MS, Feldman TA, Yoon DH, Yannakou CK, Radeski D, Kim YH, et al. (November 2020). “Cpi-818, an oral interleukin-2-inducible T-cell kinase inhibitor, is well-tolerated and active in patients with T-cell lymphoma”. Blood. 136: 19–20. doi:10.1182/blood-2020-137782.
- Hsu LY, Rosenbaum JT, Verner E, Jones WB, Hill CM, Janc JW, et al. (December 2024). “Synthesis and characterization of soquelitinib a selective ITK inhibitor that modulates tumor immunity”. npj Drug Discovery. 1 (1) 2: 1–4. doi:10.1038/s44386-024-00002-1.
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2226636-04-8 |
| PubChem CID | 134517711 |
| DrugBank | DB18749 |
| ChemSpider | 129629996 |
| UNII | 6I5H17AN3I |
| KEGG | D12762 |
| Chemical and physical data | |
| Formula | C25H30N4O4S2 |
| Molar mass | 514.66 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////////Soquelitinib, tyrosine kinase inhibitor, antineoplastic, CPI818, CPI-000818, CPI596, CP I818, CPI 000818, CP I596, 6I5H17AN3I,
Sitokiren
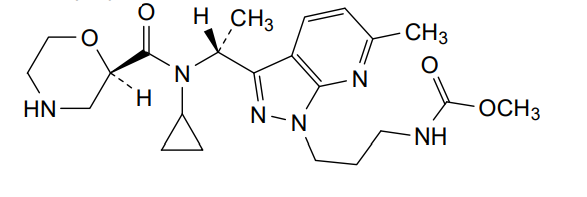
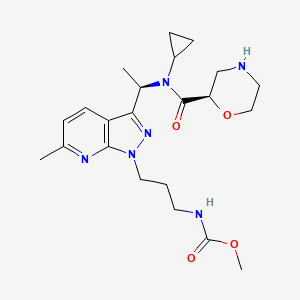
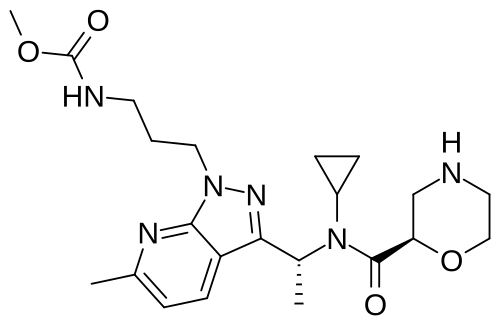
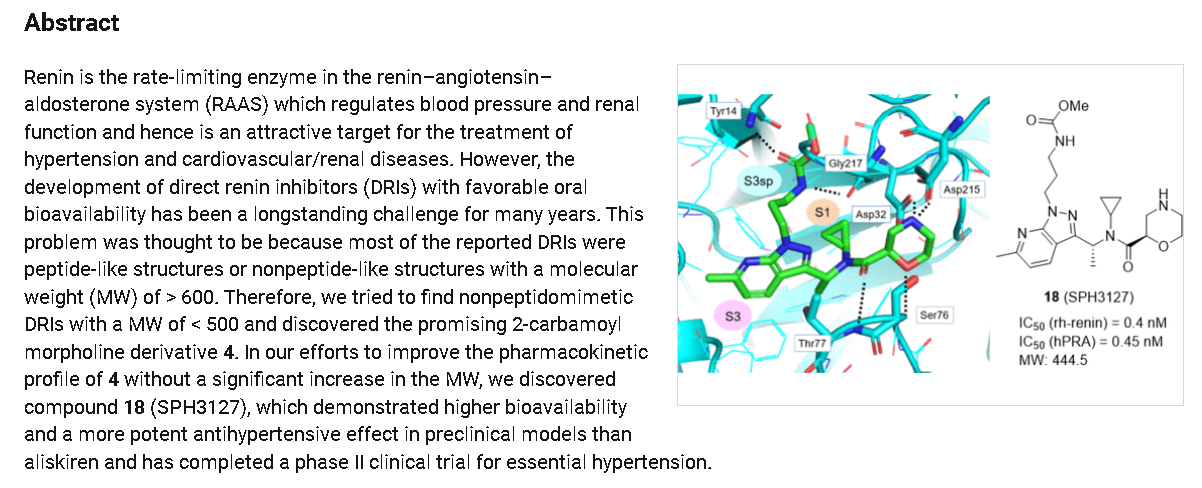
Sitokiren
CAS 1399849-02-5,
MF C22H32N6O4, 444.5 g/mol
methyl N-[3-[3-[(1R)-1-[cyclopropyl-[(2R)-morpholine-2-carbonyl]amino]ethyl]-6-methylpyrazolo[5,4-b]pyridin-1-yl]propyl]carbamate
methyl [3-(3-{(1R)-1-[(2R)-N-cyclopropylmorpholine-2-carboxamido]ethyl}-6-methyl-1H-pyrazolo[3,4-
b]pyridin-1-yl)propyl]carbamate
renin inhibitor, SPH 3127, C2M78A9V6Z
Sitokiren, also known as SPH3127, isa highly potent, orally active direct renin inhibitor developed by Mitsubishi Tanabe Pharma Corp. that was initially investigated for hypertension and cardiovascular diseases. Recent research has shown it also has a strong anti-inflammatory effect, particularly in the gut, making it a potential candidate for treating conditions like inflammatory bowel disease (IBD).
What it is
- Direct renin inhibitor: Sitokiren directly inhibits the enzyme renin, which is the rate-limiting step in the renin-angiotensin-aldosterone system (RAAS).
- Chemical properties: It is a small molecule with the chemical formula
C22H32N6O4
- Developed by: Mitsubishi Tanabe Pharma Corp..
- Alternative name: SPH3127 is another name for sitokiren.
How it works
- Blocks the RAAS: By inhibiting renin, it prevents the RAAS from over-activating.
- Potential benefits: This inhibition may help in managing blood pressure and has also shown promise in suppressing inflammation in the gut, which is a key factor in IBD.
Current research and potential applications
- Hypertension: Sitokiren was initially developed for its potential to treat hypertension, and preclinical models have shown it to be more potent than the approved drug aliskiren.
- Inflammatory bowel disease (IBD): Studies using sitokiren in mouse models have demonstrated its ability to reduce inflammation and protect against damage in colitis, suggesting it could be a novel therapeutic for IBD.
SPH-3127 is under investigation in clinical trial NCT05359068 (Study to Evaluate the Efficacy and Safety of SPH3127 in Patients With Mild-moderate Essential Hypertension).
SPH3127 is a small-molecule renin inhibitor developed by Shanghai Pharmaceuticals for hypertension and kidney disease. It is believed to be more potent than aliskiren.[1][2][3]
SYN
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.2c00834
Methyl N-[3-(3-{(1S)-1-[cyclopropyl-((2R)-morpholine-2-carbonyl)amino]ethyl}-6-methyl
pyrazolo[3,4-b]pyridin-1-yl)propyl]carbamate (18-diastereomer). This isomer was separated
from a mixture of the corresponding diastereomers using NH-silica gel column chromatography
as the more polar isomer. 1H NMR (400 MHz, DMSO-d6) : 0.20 (m, 1H), 0.51−0.74 (m, 3H),
1.70 (d, J = 7.0 Hz, 3H), 1.94 (m, 2H), 2.57 (s, 3H), 2.60−2.75 (m, 3H), 2.79 (m, 1H), 2.87 (dd,J = 2.4, 12.5 Hz, 1H), 3.00 (m, 2H), 3.47 (m, 1H), 3.51 (s, 3H), 3.79 (d, J = 10.9 Hz, 1H),
4.30−4.46 (m, 2H), 4.66 (dd, J = 2.1, 9.4 Hz, 1H), 5.84 (q, J = 7.0 Hz, 1H), 7.02 (d, J = 8.2 Hz,
1H), 7.16 (m, 1H), 7.83 (d, J = 8.2 Hz, 1H). MS (APCI) m/z: 445.1 [M + H]+. Purity and
diastereomeric excess measured by chiral HPLC: 98.37%, 81.23% de (column: Chiralpak IC (4.6
mm × 250 mm, elution: hexane/EtOH/diethylamine, 50:50:0.1 (v/v), flow rate: 0.5 mL/min,
column temperature: 25 °C, retention time: 29.40 min).
SYN
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022047730&_cid=P12-MIR63I-81994-1
PAT
Nitrogen-containing saturated heterocyclic compound
Publication Number: US-9278944-B2
Priority Date: 2011-03-16
Grant Date: 2016-03-08
https://patentscope.wipo.int/search/en/detail.jsf?docId=US95781978&_cid=P12-MIR64F-82901-1
PAT
- Salt of morpholine derivative and crystalline form thereof, as well as preparation method, pharmaceutical composition and use of the samePublication Number: EP-3398946-B1Priority Date: 2015-12-29Grant Date: 2022-05-04
- Nitrogen-containing saturated heterocyclic compoundPublication Number: EP-2687518-B1Priority Date: 2011-03-16Grant Date: 2017-11-01
- Nitrogen-containing saturated heterocyclic compoundPublication Number: US-10155731-B2Priority Date: 2011-03-16Grant Date: 2018-12-18
- Nitrogen-containing saturated heterocyclic compoundPublication Number: US-2014011807-A1Priority Date: 2011-03-16
- Nitrogen-containing saturated heterocyclic compoundPublication Number: US-2016145220-A1Priority Date: 2011-03-16
- Methods to treat inflammatory bowel diseasePublication Number: US-2023398123-A1Priority Date: 2020-09-04
- Salt of morpholine derivative and crystalline form thereof, as well as preparation method, pharmaceutical composition and use of the samePublication Number: EP-3398946-A1Priority Date: 2015-12-29
- Salts of morpholine derivative, crystal forms thereof, processes for producing the same, pharmaceutical compositions including the same, and use thereofPublication Number: US-10519150-B2Priority Date: 2015-12-29Grant Date: 2019-12-31
- Salts of morpholine derivative, crystal forms thereof, processes for producing the same, pharmaceutical compositions including the same, and use thereofPublication Number: US-2019016718-A1Priority Date: 2015-12-29
- Salt of morpholine derivative and its crystal form, manufacturing method thereof, pharmaceutical composition and use thereofPublication Number: TW-I705065-BPriority Date: 2015-12-29Grant Date: 2020-09-21
- Methods to treat inflammatory bowel diseasePublication Number: WO-2022047730-A1Priority Date: 2020-09-04
- Application of nitrogen-containing saturated heterocyclic compoundPublication Number: WO-2022048614-A1Priority Date: 2020-09-04
- Methods to treat inflammatory bowel diseasePublication Number: WO-2022048618-A1Priority Date: 2020-09-04
- Application of nitrogen-containing saturated heterocyclic compoundPublication Number: EP-4209218-A1Priority Date: 2020-09-04
- Application of nitrogen-containing saturated heterocyclic compoundPublication Number: US-2023330093-A1Priority Date: 2020-09-04
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Iijima, Daisuke; Sugama, Hiroshi; Takahashi, Yoichi; Hirai, Miki; Togashi, Yuko; Xie, Jianshu; Shen, Jingkang; Ke, Ying; Akatsuka, Hidenori; Kawaguchi, Takayuki; Takedomi, Kei; Kashima, Akiko; Nishio, Masashi; Inui, Yosuke; Yoneda, Hikaru; Xia, Guangxin; Iijima, Toru (25 August 2022). “Discovery of SPH3127: A Novel, Highly Potent, and Orally Active Direct Renin Inhibitor”. Journal of Medicinal Chemistry. 65 (16): 10882–10897. doi:10.1021/acs.jmedchem.2c00834. PMID 35939295. S2CID 251400126.
- Zhang, Leduo; Mao, Yu; Gao, Zhiwei; Chen, Xiaoyan; Li, Xin; Liu, Yanjun; Xia, Guangxin (February 2020). “The Nonclinical Pharmacokinetics and Prediction of Human Pharmacokinetics of SPH3127, a Novel Direct Renin Inhibitor”. European Journal of Drug Metabolism and Pharmacokinetics. 45 (1): 15–26. doi:10.1007/s13318-019-00573-9. PMID 31494843. S2CID 201848935.
- Jing, Shan; Xu, Ranchi; Yang, Kexu; Liu, Wenfang; Zhang, Leduo; Ke, Ying; Xia, Guangxin; Lin, Yang (April 2021). “Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of SPH3127: A Phase I, Randomized, Double-Blind, Placebo-Controlled Trial”. Clinical Therapeutics. 43 (4): 735.e1–735.e14. doi:10.1016/j.clinthera.2021.01.025. PMID 33653620. S2CID 232104329.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1399849-02-5 |
| PubChem CID | 117877477 |
| ChemSpider | 76799450 |
| UNII | C2M78A9V6Z |
| ChEMBL | ChEMBL4110551 |
| Chemical and physical data | |
| Formula | C22H32N6O4 |
| Molar mass | 444.536 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////Sitokiren, renin inhibitor, SPH 3127, C2M78A9V6Z
Setidegrasib
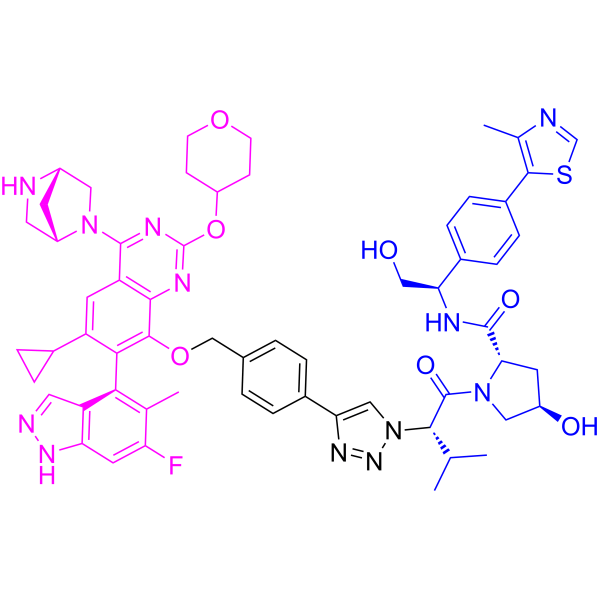
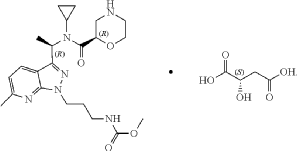
Setidegrasib
CAS 2821793-99-9
MF C60H65FN12O7S MW1117.30
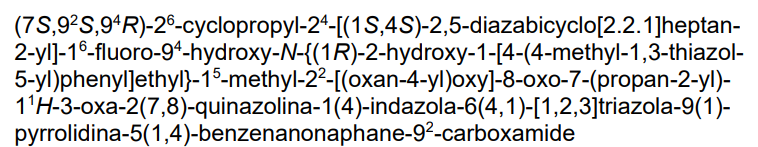
(2S,4R)-1-[(2S)-2-[4-[4-[[6-cyclopropyl-4-[(1S,4S)-2,5-diazabicyclo[2.2.1]heptan-2-yl]-7-(6-fluoro-5-methyl-1H-indazol-4-yl)-2-(oxan-4-yloxy)quinazolin-8-yl]oxymethyl]phenyl]triazol-1-yl]-3-methylbutanoyl]-4-hydroxy-N-[(1R)-2-hydroxy-1-[4-(4-methyl-1,3-thiazol-5-yl)phenyl]ethyl]pyrrolidine-2-carboxamide
Kirsten rat sarcoma viral oncogene homologue (KRAS) degradation
inducer, antineoplastic, ASP-3082, ASP 3082, 3NQ4ME292X, KRAS G12D inhibitor 17
Setidegrasib (KRAS G12D inhibitor 17, ASP3082) is a PROTAC KRAS degrader (DC50: 37 nM). Setidegrasib induces the degradation of G12D-mutation KRAS protein. Setidegrasib suppresses p-ERK, p-AKT, p-S6 levels in AsPC-1 cells. Setidegrasib exhibits anti-tumor activity in various cancer xenograft models in mice. Setidegrasib can be used for the study of KRAS(G12D)-mutated solid tumors. (Blue: VHL ligase ligand (HY-168699); Black: linker (HY-168698); Pink: G12D ligand (HY-168700)).
Setidegrasib is a small molecule drug. The usage of the INN stem ‘-rasib’ in the name indicates that Setidegrasib is a Ras protein inhibitor. Setidegrasib has a monoisotopic molecular weight of 1116.48 Da.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022173032&_cid=P21-MIPU3D-50779-1
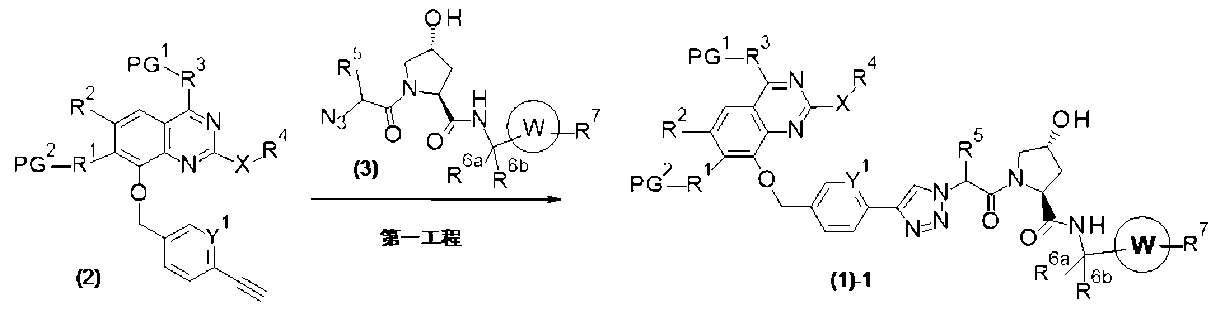
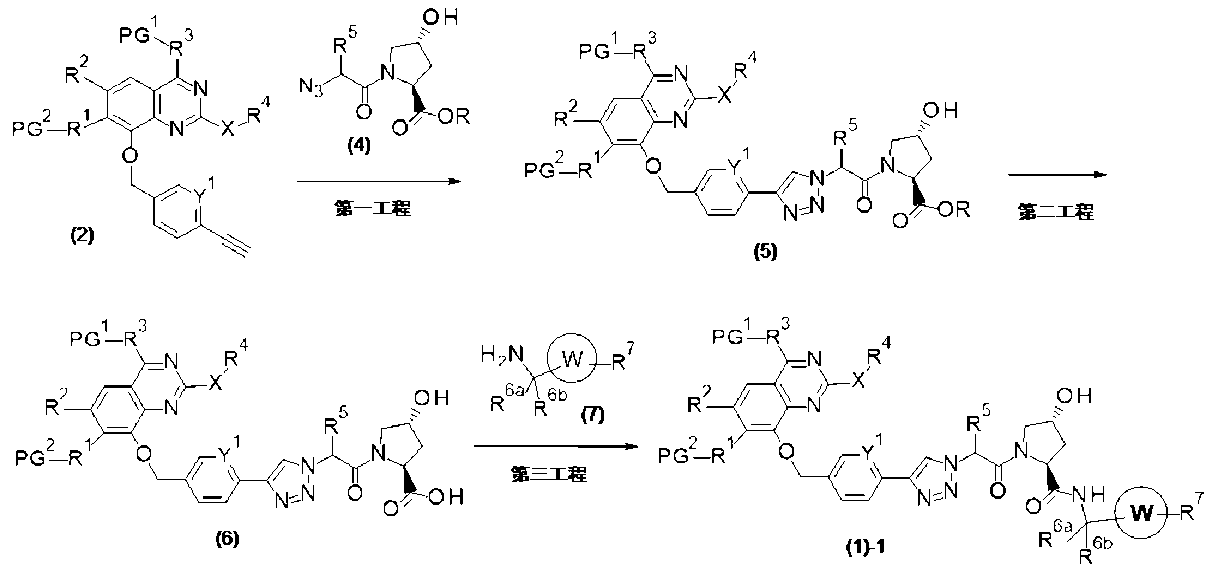
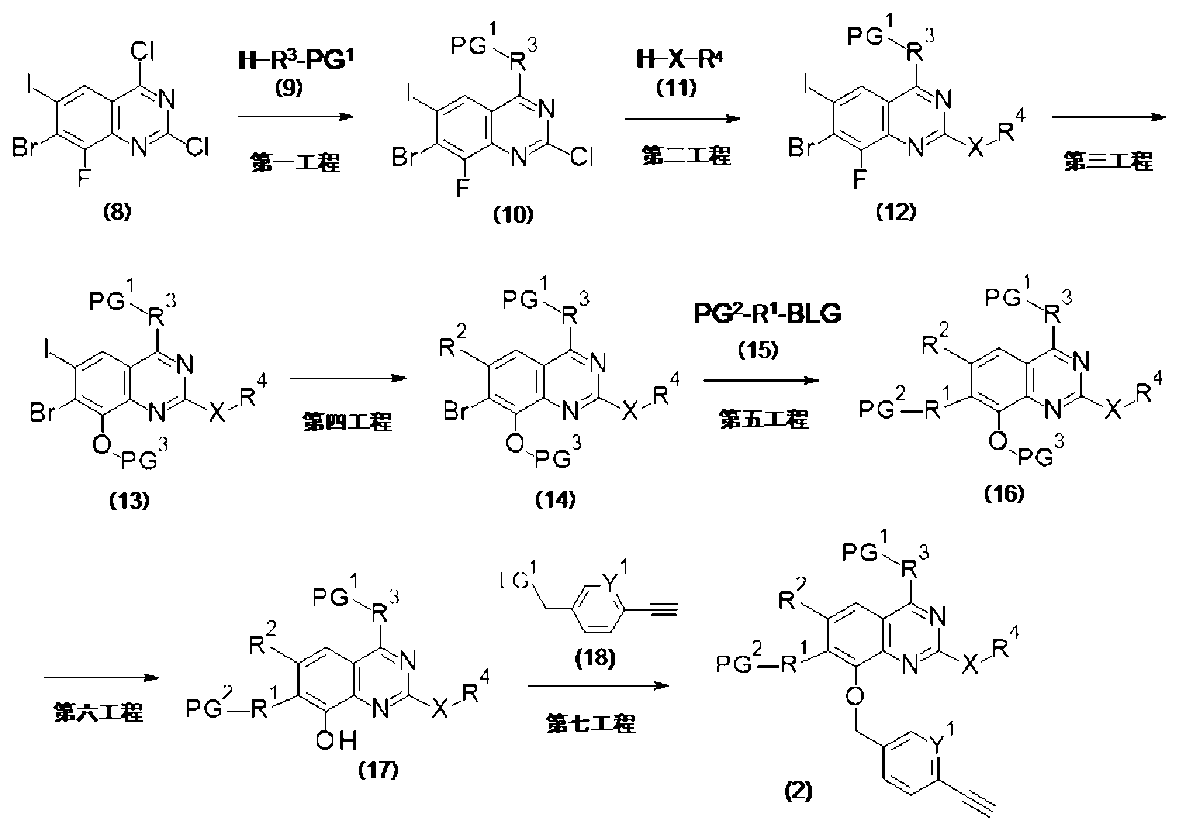
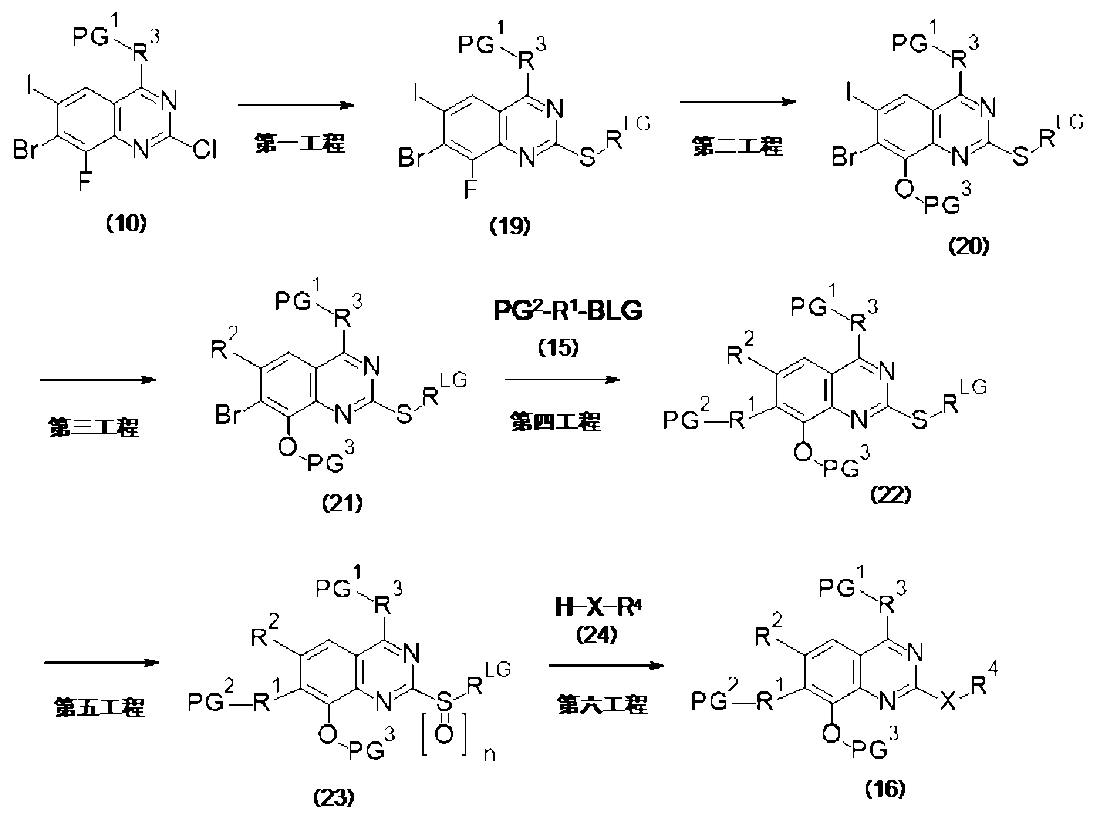
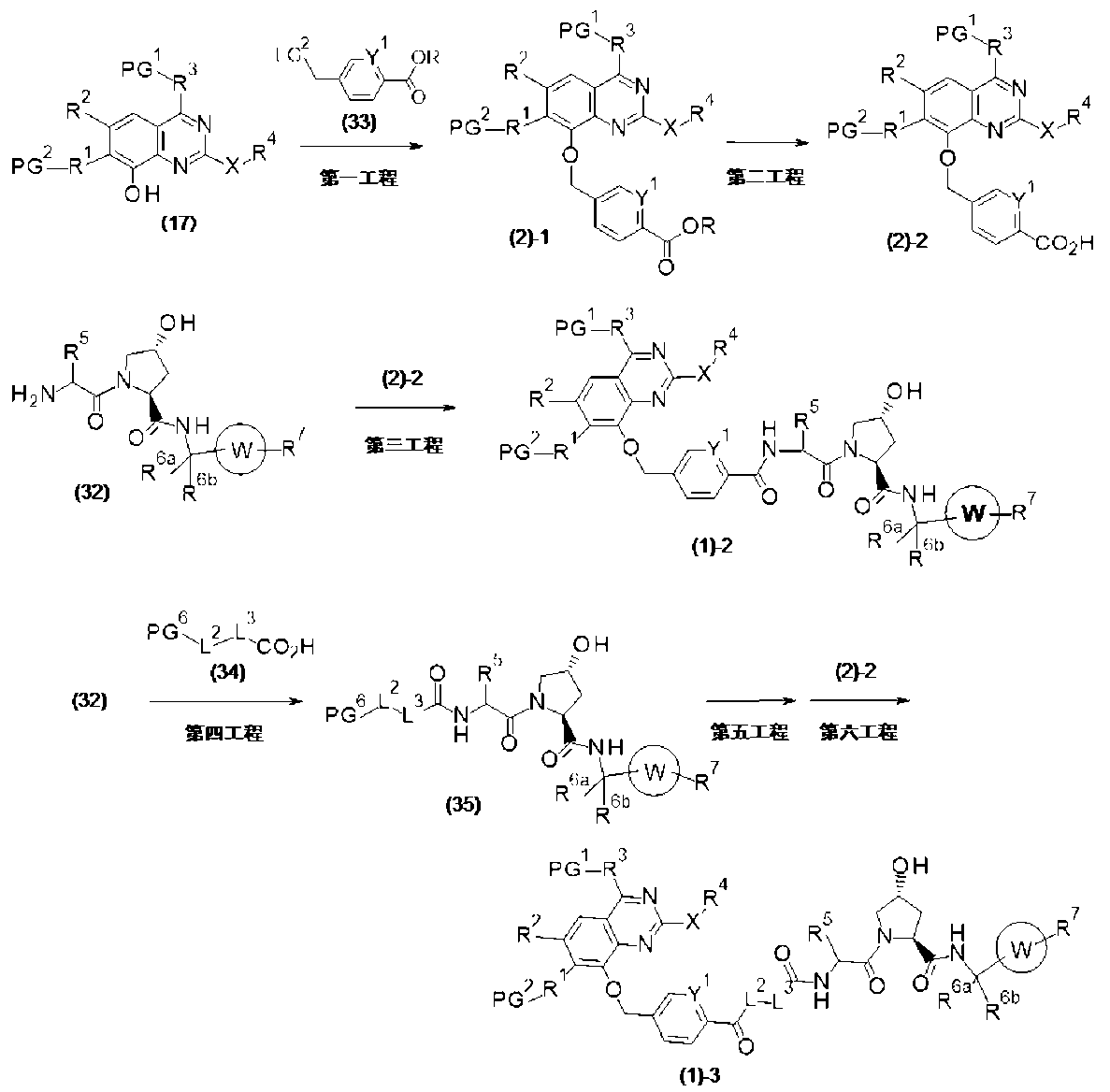
PAT
- Combination of anticancer agents comprising a bifunctional compound with g12d mutant kras inhibitory activityPublication Number: WO-2024033537-A1Priority Date: 2022-08-12
- Combination of anticancer agents comprising a bifunctional compound with g12d mutant kras inhibitory activityPublication Number: WO-2024033538-A1Priority Date: 2022-08-12
- Quinazoline compound for inducing degradation of g12d-mutation kras proteinPublication Number: WO-2022173032-A1Priority Date: 2021-02-15
- Quinazoline compound for inducing degradation of g12d-mutation kras proteinPublication Number: EP-4293024-A1Priority Date: 2021-02-15
- Quinazoline compound for inducing degradation of g12d mutant kras proteinPublication Number: US-2024182483-A1Priority Date: 2021-02-15
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. Yoshinari, et al. Preparation of quinazoline-linked (4R)-4-hydroxy-L-prolinamide compounds for inducing degradation of G12D-mutation KRAS protein: World Intellectual Property Organization, WO2022173032[P]. 2022-08-18.[2]. Yoshinari T, et al. Discovery of KRAS(G12D) selective degrader ASP3082. Commun Chem. 2025 Aug 23;8(1):254. [Content Brief]
////////Setidegrasib, antineoplastic, ASP-3082, ASP 3082, 3NQ4ME292X, KRAS G12D inhibitor 17
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}