

An efficient process for the preparation of 1-(2-methoxyphenoxy)-2,3-epoxypropane, a key intermediate for the synthesis of ranolazine is described.
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64


Ranolazine Intermediate, An Efficient Synthesis of 1-(2-Methoxyphenoxy)-2,3-epoxypropane: Key Intermediate of β-Adrenoblockers

http://pubs.acs.org/doi/suppl/10.1021/op300056k
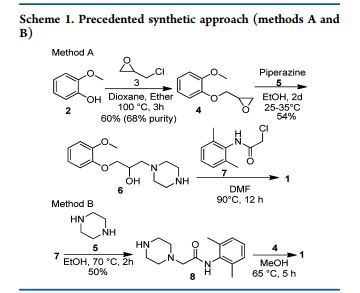
Preparation of 1-(2-Methoxyphenoxy)-2,3-epoxypropane 4.
To a stirring solution of 2-methoxy phenol 2 (10 kg, 80.55 mol) and water (40 L) at about 30 °C was added sodium hydroxide (1.61 kg, 40.25 mol) and water (10 L). After stirring for 30−45 min, epichlorohydrin 3 (22.35 kg, 241.62 mol) was added and stirred for 10−12 h at 25−35 °C. Layers were separated, and water (40 L) was added to the organic layer (bottom layer) containing product. Sodium hydroxide solution (3.22 kg, 80.5 mol) and water (10 L) were added at 27 °C and stirred for 5−6 h at 27 °C.
The bottom product layer was separated and washed with sodium hydroxide solution (3.0 kg 75 mol) and water (30 L). Excess epichlorohydrin (3) was recovered by distillation of the product layer at below 90 °C under vacuum (650−700 mmHg) to give 13.65 kg (94%) of title compound with 98.3% purity by HPLC, 0.2% of 2- methoxy phenol 2, 0.1% of epichlorohydrin 3, 0.1% of chlorohydrin 11, 0.3% of dimer 12 and 0.3% of dihydroxy 13.
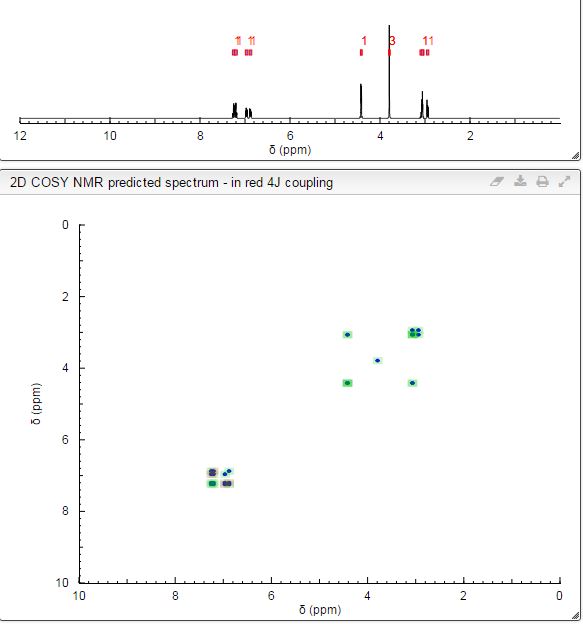
1 H NMR (400 MHz, CDCl3, δ) 6.8−7.0 (m, 4H), 4.3 (dd, J = 5.6 Hz, 5.4 Hz, 1H), 3.8 (dd, J = 5.6 Hz, 5.3 Hz, 1H), 3.7 (s, 3H), 3.2−3.4 (m, 1H), 2.8 (dd, J = 5.6 Hz, 5.4 Hz, 1H), 2.7 (dd, J = 5.6 Hz, 5.3 Hz, 1H);
IR (KBr, cm−1 ) 2935 (C−H, aliphatic), 1594 and 1509 (CC, aromatic), 1258 and 1231 (C−O−C, aralkyl ether), 1125 and 1025 (C−O−C, epoxide);
MS (m/z) 181 (M+ + H).
Compound Details
| Properties | |
| MWt | 180.2 |
| MF | C10H12O3 |
CAS 2210-74-4
| Glycidyl 2-methoxyphenyl ether | |
| Guaiacol glycidyl ether |
1H NMR PREDICT


13C NMR PREDICT



COSY PREDICT

CREDIT……….http://www.molbase.com/en/synthesis_2210-74-4-moldata-95563.html

DR REDDYS LABORATORIES
An Efficient Synthesis of 1-(2-Methoxyphenoxy)-2,3-epoxypropane: Key Intermediate of β-Adrenoblockers
† Innovation Plaza, IPD, R&D, Dr. Reddy’s Laboratories Ltd., Survey Nos. 42, 45,46, and 54, Bachupally, Qutubullapur – 500073, Andhra Pradesh, India
‡ Institute of Science and Technology, Center for Environmental Science, JNT University, Kukatpally, Hyderabad – 500 072, Andhra Pradesh, India
Org. Process Res. Dev., 2012, 16 (10), pp 1660–1664
DOI: 10.1021/op300056k
Publication Date (Web): September 14, 2012
Copyright © 2012 American Chemical Society
*Telephone: +91 4044346000. Fax: +91 4044346285. E-mail: rakeshwarb@drreddys.com.
////////////////1-(2-Methoxyphenoxy)-2,3-epoxypropane, β-Adrenoblockers, ranolazine
COc2ccccc2OCC1CO1
OTHER COMPD


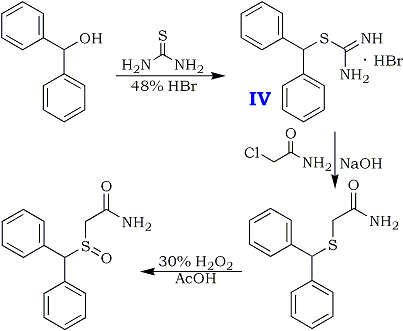
ADRAFINIL
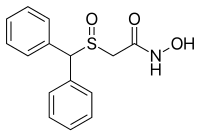
ADRAFINIL
2-((diphenylmethyl)sulfinyl)-acetohydroxamicaci;2-((diphenylmethyl)sulfinyl)-n-hydroxy-acetamid;2-((diphenylmethyl)sulfinyl)-n-hydroxyacetamide;2-(benzhydrylsulfinyl)acetohydroxamicacid;ADRAFINIL;2-[(DIPHENYLMETHYL)SULFINYL]ACETOHYDROXAMIC ACID;CRL 40028;OLMIFON
- CAS 63547-13-7
- MF:C15H15NO3S
- MW:289.35
- EINECS:264-303-1
WATCH THIS POST AS DETAILS LIKE SYNTHESIS ARE UPDATED………….
Adrafinil is touted mainly for its stimulant properties and ability to provide alertness and wakefulness.
- Stay up late/stay awake during normal sleeping hours: Adrafinil may be helpful for night workers who need a kick-start adapting their body’s natural circadian rhythm of wakefulness in the daytime and sleepiness in the evening to their job needs. This can also make it helpful for periodic late-night study sessions. Adrafinil is best taken in the afternoon or evening for nighttime wakefulness.
- Boost energy, alertness, and focus during the day time: Adrafinil can also be used as an energy-boost during waking hours.
- CONTACT SKYPE CATHERINESSPC WICKR
Adrafinil (INN) (brand name Olmifon)[2] is a discontinued wakefulness-promoting agent (or eugeroic) that was formerly used inFrance to promote vigilance (alertness), attention, wakefulness, mood, and other parameters, particularly in the elderly.[3][4] It was also used off-label by individuals who wished to avoid fatigue, such as night workers or others who needed to stay awake and alert for long periods of time. Additionally, “adrafinil is known to a larger nonscientific audience, where it is considered to be a nootropic agent.”[3] Adrafinil is a prodrug; it is primarily metabolized in vivo to modafinil, resulting in very similar pharmacological effects.[3] Unlike modafinil, however, it takes time for the metabolite to accumulate to active levels in the bloodstream. Effects usually are apparent within 45–60 minutes when taken orally on an empty stomach. Adrafinil was marketed in France under the trade name Olmifon[2] until September 2011 when it was voluntarily discontinued.[4]
Pharmacology
Pharmacodynamics
Because α1-adrenergic receptor antagonists were found to block effects of adrafinil and modafinil in animals, “most investigators assume[d] that adrafinil and modafinil both serve as α1-adrenergic receptor agonists.”[3] However, adrafinil and modafinil have not been found to bind to the α1-adrenergic receptor and they lack peripheral sympathomimetic side effects associated with activation of this receptor;[5] hence, the evidence in support of this hypothesis is weak, and other mechanisms are probable.[3] Modafinil was subsequently screened at a variety of targets in 2009 and was found to act as a weak, atypical blocker of the dopamine transporter(and hence as a dopamine reuptake inhibitor), and this action may explain some or all of its pharmacological effects.[6][7][8] Relative to adrafinil, modafinil possesses greater specificity in its action, lacking or having a reduced incidence of many of the common side effects of the former (including stomach pain, skin irritation, anxiety, and elevated liver enzymes with prolonged use).[9][10][11] There is a case report of two patients that adrafinil may increase interest in sex.[3] A case report of adrafinil-induced orofacial dyskinesia exists.[12][13] Reports of this side effect also exist for modafinil.[12]
Pharmacokinetics
In addition to modafinil, adrafinil also produces modafinil acid (CRL-40467) and modafinil sulfone (CRL-41056) as metabolites, which form from metabolic modification of modafinil.
History
Adrafinil was discovered in 1974 by two chemists working for the French pharmaceutical company Laboratoires Lafon who were screening compounds in search of analgesics.[14] Pharmacological studies of adrafinil instead revealed psychostimulant-like effects such as hyperactivity and wakefulness in animals.[14] The substance was first tested in humans, specifically for the treatment of narcolepsy, in 1977–1978.[14] Introduced by Lafon (now Cephalon), it reached the market in France in 1984,[4] and for the treatment of narcolepsy in 1985.[14][15] In 1976, two years after the discovery of adrafinil, modafinil, its active metabolite, was discovered.[14] Modafinil appeared to be more potent than adrafinil in animal studies, and was selected for further clinical development, with both adrafinil and modafinil eventually reaching the market.[14] Modafinil was first approved in France in 1994, and then in the United States in 1998.[15] Lafon was acquired by Cephalon in 2001.[16] As of September 2011, Cephalon has discontinued Olmifon, its adrafinil product, while modafinil continues to be marketed.[4]
Society and culture
Regulation
Athletic doping
Adrafinil and its active metabolite modafinil were added to the list of substances prohibited for athletic competition according to World Anti-Doping Agency in 2004.[17]
New Zealand
In 2005 a Medical Classification Committee in New Zealand recommended to MEDSAFE NZ that adrafinil be classified as a prescription medicine due to risks of it being used as a party drug. At that time adrafinil was not scheduled in New Zealand.[18]
Research
In a clinical trial with clomipramine and placebo as active comparators, adrafinil showed efficacy in the treatment of depression.[3] In contrast to clomipramine however, adrafinil was well-tolerated, and showed greater improvement in psychomotor retardation in comparison.[3] As such, “further investigations of the antidepressive effects of adrafinil are warranted.”[3]
/////////////
SYNTHESIS
Adrafinil (CAS NO.63547-13-7) was discovered in the late 1970s by scientists working with the French pharmaceutical company Group Lafon. First offered in France in 1986 as an experimental treatment for narcolepsy, Lafon later developed modafinil, the primary metabolite of adrafinil. Modafinil possesses greater selective alpha-1 adrenergic activity than adrafinil without the side effects of stomach pain, skin irritations, feelings of tension, and increases in liver enzyme levels.
It is important to monitor the liver of an individual using adrafinil. It can cause liver damage in some instances.
The Adrafinil with CAS registry number of 63547-13-7 is also known as 2-[(Diphenylmethyl)sulfinyl]-N-hydroxyacetamide. The IUPAC name is 2-Benzhydrylsulfinyl-N-hydroxyacetamide. It belongs to product categories of Aromatics Compounds; Aromatics; Intermediates & Fine Chemicals; Pharmaceuticals; Sulfur & Selenium Compounds. This chemical is a light pink solid and its EINECS registry number is 264-303-1. In addition, the formula is C15H15NO3S and the molecular weight is 289.35. This chemical is harmful if swallowed.
Physical properties about Adrafinil are: (1)ACD/LogP: 1.596; (2)ACD/LogD (pH 5.5): 1.60; (3)ACD/LogD (pH 7.4): 1.53; (4)ACD/BCF (pH 5.5): 9.60; (5)ACD/BCF (pH 7.4): 8.34; (6)ACD/KOC (pH 5.5): 175.52; (7)ACD/KOC (pH 7.4): 152.63; (8)#H bond acceptors: 4; (9)#H bond donors: 2; (10)#Freely Rotating Bonds: 6; (11)Index of Refraction: 1.653; (12)Molar Refractivity: 78.858 cm3; (13)Molar Volume: 215.542 cm3; (14)Polarizability: 31.262 10-24cm3; (15)Surface Tension: 67.25 dyne/cm; (16)Density: 1.342 g/cm3
Preparation of Adrafinil: it is prepared by reaction of diphenyl methyl bromide with thiourea. This reaction needs reagent NaOH. After reacting with chloroacetic acid, hydrochloric acid amine and hydrogen peroxide, the product is obtained. The yield is about 73%.

Uses of Adrafinil: it is used as non-amphetamine-type psychostimulant and can wake up and raise awareness. For the elderly arousal disorder and depressive symptoms in symptomatic treatment.
Benzhydrylsulphinyl-acetohydroxamic Acid (Adrafinil)1
Diphenylmethanethiol
15.2 g (0.2 mol) of thiourea and 150 ml of demineralized water are introduced into a 500 ml three-neck flask equipped with a central mechanical stirrer, and with a dropping funnel and a condenser on the (respective) side-necks.The temperature of the reaction mixture is brought to 50°and 49.4g (0.2 mol) of bromodiphenyl- methane are added all at once whilst continuing the heating. After refluxing for about 5 minutes, the solution, which has become limpid, is cooled to 20°C and 200 ml of 2.5 N NaOH are then added dropwise whilst maintaining the said temperature. The temperature is then again kept at the reflex for 30 minutes after which, when the mixture has returned to ordinary temperature (15-25°C), the aqueous solution is acidified with 45 ml of concentrated hydrochloric acid. The supernatant oil is extracted with 250 ml of diethyl ether and the organic phase is washed with 4×80 ml of water and then dried over magnesium sulphate. 39 g of crude diphenylmethane-thiol are thus obtained. Yield 97.5%.
Benzhydryl-thioacetic acid
10 g (0.05 mol) of diphenylmethane-thiol and 2g (0.05 mol) of NaOH dissolved in 60 ml of demineralised water are introduced successively into a 250 ml flask equipped with a magnetic stirrer and a reflux condenser. The reactants are left in contact for 10 minutes whilst stirring, and a solution consisting of 7g (0.075 mol) of chloroacetic acid, 3g (0.075 mol) of NaOH pellets and 60 ml of demineralized water is then added all at once. The aqueous solution is gently warmed to about 50°C for 15 minutes, washed with 50 ml of ether, decanted and acidified with concentrated hydrochloric acid. after filtration, 10.2g of benzhydryl-thioacetic acid are thus obtained. Melting point 129-130°C. Yield 79%.
Ethyl benzhydryl-thioacetate
The following reaction mixture is heated under reflux for 7 hours: 10.2 g (0.0395 mol) of benzhydryl-thioacetic acid, 100 ml of anhydrous ethanol and 2 ml of sulphuric acid. When heating has been completed, the ethanol isevaporated in vacuo; the oily residue is taken up in 100 ml of ethyl ether and the organic solution is then washed with water, with an aqueous sodium carbonate solution and then with water until the wash waters have a neutral pH. After drying over sodium sulphate, the solvent is evaporated. 10.5g of ethyl benzhydryl- thioacetate are thus obtained. Yield 93%.
Benzhydryl-thioacetohydroxamic acid
The following three solutions are prepared:
- Ethyl Benzhydryl-thioacetate 10.8 g (0.0378 mol) in 40 ml methanol
- Hydroxylamine hydrochloride 5.25 g (0.0756 mol) in 40 ml methanol
- Potassium Hydroxide pellets 7.3 g (0.0134 mol) in 40 ml methanol
The solutions are heated, if necessary, until they become limpid, and when the temperatures have again fallen to below 40°C, the solution of potassium hydroxide in methanol is poured into the solution of hydroxylamine hydrochloride in alcohol. Finally, at a temperature of about 5° to 10°C, the solution of ethyl benzhydryl- thioacetate is added in its turn. After leaving the reactants in contact for 10 minutes, the sodium chloride is filtered off the limpid solution obtained is kept for about 15 hours at ordinary temperature. The methanol is then evaporated under reduced pressure, the residual oil is taken up in 100 ml of water and the aqueous solution is acidified with 3 N hydrochloric acid. The hydroxamic acid which has crystallized is filtered off, washed with water and then dried. 9.1 g of product are obtained. Yield = 87.5%. Melting point 118-120°C.
Adrafinil (CRL 40,028)
10.4g (0.038 mol) of benzhydryl-thioacetohydroxamic acid are oxidized at 40°C, over the course of 2 hours, by means of 3.8 ml (0.038 mol) of hydrogen peroxide of 110 volumes strength (33%), in 100 ml of acetic acid.
When the oxidation has ended, the acetic acid is evaporated under reduced pressure and the residual oil is taken up in 60 ml of ethyl acetate. The product which has crystallized is filtered off and then purified by recrystallisation from a 3:2 (by volume) mixture of ethyl acetate and isopropyl alcohol.
8g (73%) of Adrafinil, mp 159-160°C, are thus obtained. H2O Solubility
CLIP

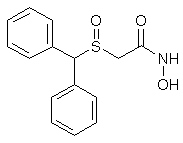
Figure 2: GC/MS extracted ion chromatogram (a) and mass spectrum (b) of derivatized adrafinil in the electron ionization mode (monitoring the m/z 167, 165 and 152 ions; all the four peaks are derivatised adrafinil products).

Figure 4: LC/ESI-MS full scan chromatogram of adrafinil and its metabolites (a) (modafinil acid RT 3.8 min, adrafinil RT 4.0 min, modafinil RT 4.1 min), and LC/ESI-MS full scan mass spectra of modafinil acid (b), adrafinil (c), and (d) modafinil. (b, c and d showing the similar ions at m/z 167, 165, 152 together with the appropriate sodium and potassium adducts).

NMR

1H NMR PREDICT

13C NMR PREDICT


Patent
https://www.google.com/patents/US6180678 below
FIG. 1 shows the structure of adrafinil and its metabolites.
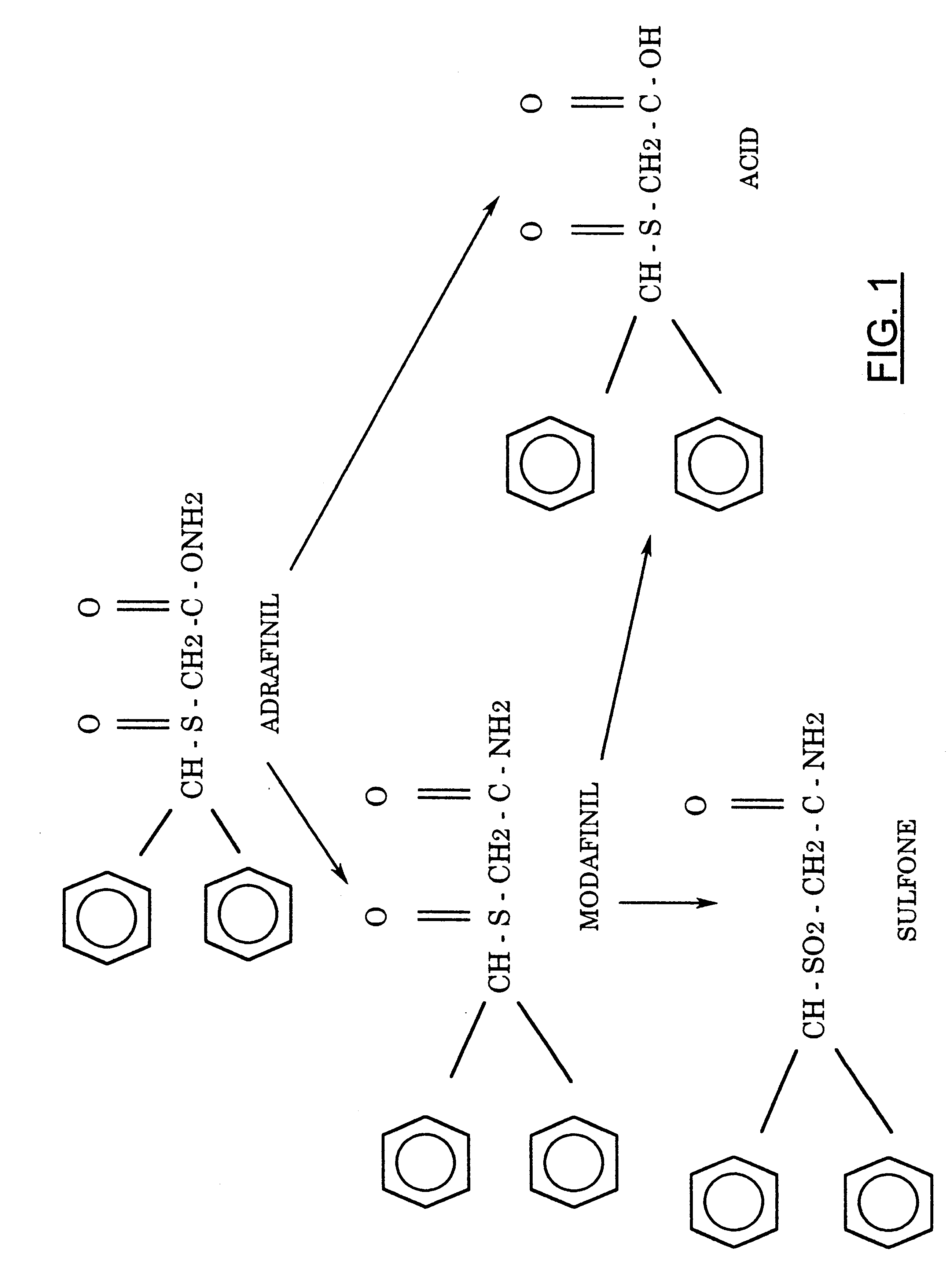
FIG. 2 shows the chemical synthesis of adrafinil.

//////////////
References
- Jump up^ Robertson P, Hellriegel ET (2003). “Clinical pharmacokinetic profile of modafinil”. Clin Pharmacokinet. 42 (2): 123–37. doi:10.2165/00003088-200342020-00002.PMID 12537513.
- ^ Jump up to:a b Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 20–. ISBN 978-3-88763-075-1.
- ^ Jump up to:a b c d e f g h i Milgram, Norton (1999). “Adrafinil: A Novel Vigilance Promoting Agent”.CNS Drug Reviews. 5 (3): 193–212. doi:10.1111/j.1527-3458.1999.tb00100.x. Retrieved2 October 2014.
- ^ Jump up to:a b c d AFSSAPS (2011). “Point d’information sur les dossiers discutés en commission d’AMM Séance du jeudi 1er décembre 2011 – Communiqué”.
- Jump up^ Simon P, Chermat R, Puech AJ (1983). “Pharmacological evidence of the stimulation of central alpha-adrenergic receptors”. Prog. Neuropsychopharmacol. Biol. Psychiatry. 7 (2-3): 183–6. doi:10.1016/0278-5846(83)90105-7. PMID 6310690.
- Jump up^ Zolkowska D, Jain R, Rothman RB, Partilla JS, Roth BL, Setola V, Prisinzano TE, Baumann MH (May 2009). “Evidence for the involvement of dopamine transporters in behavioral stimulant effects of modafinil”. The Journal of Pharmacology and Experimental Therapeutics. 329 (2): 738–46. doi:10.1124/jpet.108.146142.PMC 2672878
 . PMID 19197004.
. PMID 19197004. - Jump up^ Reith ME, Blough BE, Hong WC, Jones KT, Schmitt KC, Baumann MH, Partilla JS, Rothman RB, Katz JL (Feb 2015). “Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter”. Drug and Alcohol Dependence. 147: 1–19. doi:10.1016/j.drugalcdep.2014.12.005. PMC 4297708. PMID 25548026.
- Jump up^ Quisenberry AJ, Baker LE (Dec 2015). “Dopaminergic mediation of the discriminative stimulus functions of modafinil in rats”. Psychopharmacology. 232 (24): 4411–9.doi:10.1007/s00213-015-4065-0. PMID 26374456.
- Jump up^ Ballas, Christos A; Deborah Kim; Claudia F Baldassano; Nicholas Hoeh (July 2002). “Modafinil: past, present and future”. Expert Review of Neurotherapeutics. 2 (4): 449–57.doi:10.1586/14737175.2.4.449. PMID 19810941.
- Jump up^ Alan F. Schatzberg; Charles B. Nemeroff (2009). The American Psychiatric Publishing Textbook of Psychopharmacology. American Psychiatric Pub. pp. 850–. ISBN 978-1-58562-309-9.
- Jump up^ Ballas, Christos A; Kim, Deborah; Baldassano, Claudia F; Hoeh, Nicholas (2002). “Modafinil: past, present and future”. Expert Review of Neurotherapeutics. 2 (4): 449–457.doi:10.1586/14737175.2.4.449. ISSN 1473-7175. PMID 19810941.
- ^ Jump up to:a b Jeffrey K Aronson (31 December 2012). Side Effects of Drugs Annual: A worldwide yearly survey of new data in adverse drug reactions. Newnes. pp. 6–. ISBN 978-0-444-59503-4.
- Jump up^ Thobois S, Xie J, Mollion H, Benatru I, Broussolle E (2004). “Adrafinil-induced orofacial dyskinesia”. Mov. Disord. 19 (8): 965–6. doi:10.1002/mds.20154. PMID 15300665.
- ^ Jump up to:a b c d e f Antonio Guglietta (28 November 2014). Drug Treatment of Sleep Disorders. Springer. pp. 212–. ISBN 978-3-319-11514-6.
- ^ Jump up to:a b Jie Jack Li; Douglas S. Johnson (27 March 2013). Modern Drug Synthesis. John Wiley & Sons. pp. 2–. ISBN 978-1-118-70124-9.
- Jump up^ url=http://www.bloomberg.com/research/stocks/private/snapshot.asp?privcapId=1366624
- Jump up^ World Anti-Doping Agency – 2007 Prohibited List
- Jump up^ MCC Minutes Out of Session Meeting. Medsafe.govt.nz (2013-05-23). Retrieved on 2013-12-18.
External links
- “SID 184744 – PubChem Substance Summary“. PubChem Project. National Center for Biotechnology Information. Retrieved 7 December 2005.
- “Adrafinil – Bank of Automated Data on Drugs“. Bank of Automated Data on Drugs. VIDAL. Archived from the original on 5 October 2008. Retrieved 4 October 2008.
- Milgram, Norton W.; Callahan, Heather; Siwak, Christina (September 1992). “Adrafinil: A Novel Vigilance Promoting Agent” (PDF). CNS Drug Reviews. 5 (3): 193–212.doi:10.1111/j.1527-3458.1999.tb00100.x.

- Thobois, S. P.; Xie, J.; Mollion, H.; Benatru, I.; Broussolle, E. (August 2004). “Adrafinil-induced orofacial dyskinesia”. Movement Disorders. 19 (8): 965–966.doi:10.1002/mds.20154. PMID 15300665.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Olmifon |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral |
| ATC code | N06BX17 (WHO) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 80% |
| Metabolism | 75% (Liver) |
| Metabolites | Modafinil |
| Biological half-life | 1 hour (T1/2 is 12–15 hours for modafinil)[1] |
| Excretion | Kidney |
| Identifiers | |
|
Systematic (IUPAC) name: (±)-2-Benzhydrylsulfinylethanehydroxamic acid
|
|
| Synonyms | CRL-40028 |
| CAS Number | 63547-13-7 |
| PubChem (CID) | 3033226 |
| DrugBank | DB08925 |
| ChemSpider | 2297976 |
| UNII | BI81Z4542G |
| KEGG | D07348 |
| ChEMBL | CHEMBL93077 |
| Chemical and physical data | |
| Formula | C15H15NO3S |
| Molar mass | 289.351 g/mol |
| 3D model (Jmol) | Interactive image |
////////////ADRAFINIL
-
O=S(C(c1ccccc1)c2ccccc2)CC(=O)NO
Update……………..
Adrafinil
-
- Synonyms:CRL-40028
- ATC:N06BX17
- Use:α-adrenoceptor agonist (for symptomatic treatment of vigilance and depressive manifestations), stimulant
- Chemical name:2-[(diphenylmethyl)sulfinyl]-N-hydroxyacetamide
- Formula:C15H15NO3S
- MW:289.36 g/mol
- CAS-RN:63547-13-7
- EINECS:264-303-1
- LD50:>2048 mg/kg (M, i.p.); 1950 mg/kg (M, p.o.)
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 91-01-0 | C13H12O | benzhydrol | Benzenemethanol, α-phenyl- |
| 63547-24-0 | C15H14O3S | (benzhydrylsulfinyl)acetic acid | Acetic acid, [(diphenylmethyl)sulfinyl]- |
| 63547-22-8 | C15H14O2S | (benzhydrylthio)acetic acid | Acetic acid, [(diphenylmethyl)thio]- |
| 79-11-8 | C2H3ClO2 | chloroacetic acid | Acetic acid, chloro- |
| 77-78-1 | C2H6O4S | dimethyl sulfate | Sulfuric acid, dimethyl ester |
| 4237-48-3 | C13H12S | diphenylmethanethiol | Benzenemethanethiol, α-phenyl- |
| 63547-22-8 | C15H14O2S | 2-(diphenylmethylthio)acetic acid | Acetic acid, [(diphenylmethyl)thio]- |
| 63547-44-4 | C15H15NO2S | 2-[(diphenylmethyl)thio]-N-hydroxyacetamide | Acetamide, 2-[(diphenylmethyl)thio]-N-hydroxy- |
| 7803-49-8 | H3NO | hydroxylamine | Hydroxylamine |
| 62-56-6 | CH4N2S | thiourea | Thiourea |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| F | Olmifon | Cephalon |
Formulations
- cps. 300 mg
References
-
- DOS 2 642 511 (Lab. Lafon; appl. 22.9.1976; GB-prior. 2.10.1975).
- US 4 066 686 (Lab. Lafon; 3.1.1978; GB-prior. 2.10.1975).
- US 4 098 824 (Lab. Lafon; 4.7.1978; GB-prior. 2.10.1975).
Title: Adrafinil
CAS Registry Number: 63547-13-7
CAS Name: 2-[(Diphenylmethyl)sulfinyl]-N-hydroxyacetamide
Additional Names: 2-(benzhydrylsulfinyl)acetohydroxamic acid
Manufacturers’ Codes: CRL-40028
Trademarks: Olmifon (Lafon)
Molecular Formula: C15H15NO3S
Molecular Weight: 289.35
Percent Composition: C 62.26%, H 5.23%, N 4.84%, O 16.59%, S 11.08%
Literature References: a-Adrenergic agonist. Prepn, pharmacology: L. Lafon, BE 846880; US 4066686 (1977, 1978 both to Lafon). Mode of action: J. Duteil et al., Eur. J. Pharmacol. 59, 121 (1979); C. Rozé et al., Arch. Int. Pharmacodyn. Ther. 265, 119 (1983). Psychopharmacology in mice: F. A. Rambert et al., J. Pharmacol. 17, 37 (1986).
Properties: Crystals from ethyl acetate-isopropyl alcohol, mp 159-160°. Soly in water <1 g/l. LD50 in mice (mg/kg): <2048 i.p.; 1950 gastric admin (Lafon).
Melting point: mp 159-160°
Toxicity data: LD50 in mice (mg/kg): <2048 i.p.; 1950 gastric admin (Lafon)
Therap-Cat: Treatment of depression.
Keywords: a-Adrenergic Agonist; Antidepressant.


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D

 +91 9323115463
+91 9323115463GLENMARK SCIENTIST , INDIA
web link
web link
New Drug Approvals 13 lakh plus views

 |
Dr. Anthony Melvin Crasto
Principal Scientist, Glenmark Pharma |

ZINPLAVA (BEZLOTOXUMAB), Approved FDA

BEZLOTOXUMAB
Biologic License Application (BLA): 761046
Company: MERCK SHARP DOHME
Drug Name(s):
• ZINPLAVA (BEZLOTOXUMAB)
http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761046s000lbl.pdf
http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2016/761046Orig1s000ltr.pdf
| Drug Name |
Active Ingredient | Approval Date | FDA-approved use on approval date | |
|---|---|---|---|---|
| Zinplava | bezlotoxumab | 10/21/2016 | To reduce the recurrence of Clostridium difficile infection in patients aged 18 years or older Drug Trials Snapshot |
![]()
From Wikipedia, the free encyclopedia
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Human |
| Target | Clostridium difficile |
| Clinical data | |
| ATC code | none |
| Identifiers | |
| CAS Number | 1245634-25-6 |
| ChemSpider | none |
| Chemical and physical data | |
| Formula | C6464H9974N1726O2014S46 |
| Molar mass | 145.6 kg/mol |
Bezlotoxumab (proprietary name Zinplava) is a human monoclonal antibody designed for the prevention of recurrence ofClostridium difficile infection.[1]
Actoxumab and bezlotoxumab are fully human monoclonal antibodies which bind Clostridium difficile (C diff) toxins A and B, respectively.
This drug, along with actoxumab, was developed through Phase II efficacy trials by a partnership between Medarex Inc and MassBiologics of the University of Massachusetts Medical School.[2] The project was then licensed to Merck Sharp & Dohme Corp for further development and commercialization.[3]
A Phase III trial only showed a benefit from bezlotoxumab; the combination of actoxumab and bezlotoxumab worked no better to prevent recurrence of C.difficile associated diarrhea than bezlotoxumab alone.[4]
Progress towards FDA approval
On June 9, 2016, the US FDA’s Antimicrobial Drugs Advisory Committee (formerly known as the Anti-Infective Drugs Advisory Committee)[5] met to discuss bezlotoxumab and voted to recommend approval of Merck’s license application by a vote of 10 to 5, generally expressing a willingness to accept that the trials had proven that bezlotoxumab decreased recurrence of C.diff overall while tempering this acceptance with a robust discussion of whether or not the drug provide more marked benefit in some patient groups and concern over a potential safety signal in the group treated with bezlotoxumab. The data suggested that bezlotoxumab might have the most benefit in sicker, high-risk patients but did show a statistical benefit in all patient subgroups. Although the patient population as a whole contained many very sick individuals and thus there were many adverse events in both the subjects receiving placebo and those receiving bezlotoxumab, the panel focused on a small number of serious events in patients with pre-existing congestive heart failure. In this subset the patients receiving bezlotoxumab appeared to have a higher rate of negative outcomes than the placebo group, although there many have been imbalance in how sick the patients in those groups were.[6][7]
The Prescription Drug User Fee Act (PDUFA) action date for the FDA’s review of bezlotoxumab is July 23, 2016.[8]
Bezlotoxumab gained FDA approval in October 2016: “indicated to reduce the recurrence of Clostridium difficile infection (CDI) in patients 18 years of age or older who are receiving antibiotics for CDI and are at high risk for recurrence.”[9]
Mechanism of TcdB neutralization
By x-ray crystallized structure of N-terminal of Clostridium difficile toxin B (TcdB), the toxin was identified to consist of three domains: a GTD, a cysteine protease and a combined repetitive oligopeptides, CROP domain. The CROP domain consists of four different peptide units, B1, B2, B3 and B4. Bezlotoxumab specifically inhibits the CROP domain of TcdB. It recognizes a specific epitope on toxin TcdB and has high affinity for that region. The GTD domain does not interact with bezlotoxumab, but appears to interact with B1, which is representative of the entire CROP domain. Bezlotoxumab interacts with either B2 andB3 or the overlapping residues region between the two domains. The B4 fragment does not interact with the specific portion of the CROP domain. Characterization of peptide B1 as full CROP domain of TcdB suggests that the antibody specifically react with the B2 region of the CROP domain, leading to the conclusion that TcdB epitope lies within the N-terminus of the CROP domain.[10]

References
- Jump up^ “Statement On A Nonproprietary Name Adopted By The USAN Council – Bezlotoxumab” (PDF). American Medical Association.
- Jump up^ Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD, Leney M, Sloan S, Hay CA, Ambrosino DM (January 2010). “Treatment with monoclonal antibodies against Clostridium difficile toxins”. N. Engl. J. Med. 362 (3): 197–205. doi:10.1056/NEJMoa0907635. PMID 20089970.
- Jump up^ “Merck & Co., Inc., Medarex, Inc. and Massachusetts Biologic Laboratories Sign Exclusive Licensing Agreement for Investigational Monoclonal Antibody Combination for Clostridium Difficile Infection”. Press Release. Merck Sharp & Dohme Corp. April 21, 2009.
- Jump up^ http://www.businesswire.com/news/home/20150920005053/en/Pivotal-Phase-3-Studies-Bezlotoxumab-Merck%E2%80%99s-Investigational
- Jump up^ http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/default.htm
- Jump up^ http://www.medpagetoday.com/Washington-Watch/FDAGeneral/58433?xid=nl_mpt_DHE_2016-06-10&eun=g411987d0r
- Jump up^ http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/ucm505289.htm
- Jump up^ FDA Advisory Panel Gives Nod to Zinplava. June 2016
- Jump up^ FDA Approves Zinplava for Recurrent C. difficile. Oct 25 2016
- Jump up^ Orth P, Hernandez LD, Reichert P, Sheth PR, Beaumont M, Yang XY, Murgolo N, Ermakov G, DiNunzio E, Racine F, Karczewskl J, Secore S, Ingram RN, Mayhood T, Strickland C, Therien AG (June 27, 2014). “Mechanism of Action and Epitopes of Clostridium difficile Toxin B-neutralizing Antibody Bezlotoxumab Revealed by X-ray Crystallography”. Biological Chemistry. 289 (26): 18008–18021. doi:10.1074/jbcM114.560748.
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Human |
| Target | Clostridium difficile |
| Clinical data | |
| ATC code | none |
| Identifiers | |
| CAS Number | 1245634-25-6 |
| ChemSpider | none |
| Chemical and physical data | |
| Formula | C6464H9974N1726O2014S46 |
| Molar mass | 145.6 kg/mol |
///////BEZLOTOXUMAB, FDA 2016, MERCK SHARP DOHME
Generics: FDA´s New Guidance on Prior Approval Supplements
DRUG REGULATORY AFFAIRS INTERNATIONAL

Generics: The US Food and Drug Administration (FDA) recently published a new Guidance regarding Prior Approval Supplements (PAS). Read more about FDA´s Guidance for Industry “ANDA Submissions – Prior Approval Supplements Under GDUFA“.
On October 14, 2016, the US Food and Drug Administration (FDA) published a new Guidance regarding Prior Approval Supplements (PAS).
FDA says that “this guidance is intended to assist applicants preparing to submit to FDA prior approval supplements (PASs) and amendments to PASs for abbreviated new drug applications (ANDAs)”.
Specifically, the guidance describes how the Generic Drug User Fee Amendments of 2012 (GDUFA) performance metric goals apply to:
- A PAS subject to the refuse-to-receive (RTR) standards;
- A PAS that requires an inspection;
- A PAS for which an inspection is not required;
- An amendment to a PAS;
- Other PAS-related matters.
GDUFA is designed to speed the delivery of safe and effective generic drugs to the…
View original post 679 more words
WO 2016181414, IVACAFTOR, NEW PATENT, COUNCIL OF SCIENTIFIC & INDUSTRIAL RESEARCH
![]()


CSIR, Dr. D. Srinivasa Reddy
WO2016181414, PROCESS FOR THE SYNTHESIS OF IVACAFTOR AND RELATED COMPOUNDS
REDDY, Dumbala Srinivasa; (IN).
NATARAJAN, Vasudevan; (IN).
JACHAK, Gorakhnath Rajaram; (IN)
COUNCIL OF SCIENTIFIC & INDUSTRIAL RESEARCH [IN/IN]; Anusandhan Bhawan, Rafi Marg New Delhi 110001 (IN)
The present patent discloses a novel one pot two-step process for the synthesis of ivacaftor and related compounds of [Formula (I)], wherein R1, R2, R3, R4, R5, R6, R7 and Ar1are as described above; its tautomers or pharmaceutically acceptable salts thereof starting from indole acetic acid amides
See Eur J Org Chem, Nov 2015, for an article by the inventors, describing a process for preparing ivacaftor using 4-quinolone-3-carboxylic acid amides. The inventors appear to be based at National Chemical Laboratories of CSIR.
Ivacaftor, also known as N-(2,4-di-tert-butyl-5-hydroxyphenyl)-l,4-dihydro-4-oxoquinoline-3-carboxamide, having the following Formula (A):

Formula (A)
[003] Ivacaftor was approved by FDA and marketed by vertex pharma for the treatment of cystic fibrosis under the brand name KALYDECO® in the form of 150 mg oral tablets. Kalydeco® is indicated for the treatment of cystic fibrosis in patients age 6 years and older who have a G55ID mutation in the CFTR (cystic fibrosis transmembrane conductance regulator)gene.
[004] U.S. 20100267768 discloses a process for preparation of ivacaftor, which involves the coupling of 4-oxo-l,4-dihydro-3- quinoline carboxylic acid with hydroxyl protected phenol intermediate in the presence of propyl phosphonic anhydride (T3P®) followed by deprotection of hydroxyl protection group and optional crystallization with isopropyl acetate. The publication also discloses the use of highly expensive coupling reagent, propyl phosphonic anhydride; which in turn results to an increase in the manufacturing cost. The process disclosed is schematically represented as follows:

[005] Article titled “Discovery of N-(2,4-Di-te -butyl-5-hydroxyphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator” byHadida,S et. al in . Med. Chem., 2014, 57 (23), pp 9776-9795 reportsN-(2,4-di-teri-butyl-5-hydroxyphenyl)-4-oxo- 1 ,4-dihydroquinoline-3-carboxamide (VX-770, 48, ivacaftor), an investigational drug candidate approved by the FDA for the treatment of CF patients 6 years of age and older carrying the G551D mutation.

[006] WO 2014125506 A2 discloses a process for the preparation of ivacaftor in high yield and purity by using novel protected quinolone carboxylic acid compounds as intermediates.
[007] Article titled “Expeditious synthesis of ivacaftor” by Jingshan Shen et. al in Heterocycles, 2014, 89 (4), pp 1035 – 1040 reports an expeditious synthesis for ivacaftor featuring modified Leimgruber-Batcho procedure. The overall yield is 39% over six steps from commercially available 2-nitrobenzoyl chloride.
[008] U.S.2011/064811 discloses a process for preparation of ivacaftor, which involves condensation of 4-oxo-l,4-dihydro-3- quinolone carboxylic acid with 5- amino-2,4-di-(tert-butyl)phenol in the presence of HBTU followed by the formation of ethanol crystalate, which is then treated with diethyl ether to yield ivacaftor as a solid.

[010] U.S. 7,495,103 discloses modulators of ATP-binding cassette transporters such as ivacaftor and a process for the preparation of modulators of ATP-binding cassette transporters such as quinolone compounds. The process includes condensation of 4-oxo-l,4-dihydro-3 -quinolone carboxylic acid with aniline in presence of 2-(lH-7-azabenzotriazol-l-yl)-l,l,3,3-tetramethyluronium hexafluoro phosphate methanaminium (HATU) as shown:
![]()
[011] U.S. 2011/230519 discloses a process for preparation of 4-oxo-l,4-dihydro-3-quinoline carboxylic acid by reaction of aniline with diethylethoxymethylenemalonate at 100-110°C followed by cyclization in phenyl ether at temperature 228-232°C and then hydrolysis, as shown below:

[012] US 7,402,674 B2 discloses 7-Phenylamino-4-quinolone-3-carboxylic acid derivatives, process for their preparation and their use as medicaments.
[013] US 4,981,854 discloses l-aryl-4-quinolone-3 carboxylic acids, processes for their preparation and anti-bacterial agents and feed additives containing these compounds.
Article titled “Ozonolysis Applications in Drug Synthesis” by Van Ornum,S.G. ; Champeau,R.M.; Pariza,R. in Chem. Rev., 2006, 106 (7), pp 2990-3001 reports that ozonolysis for the synthesis of numerous interesting bioactive natural products and pharmaceutical agents.
[014] Article titled “Safe Execution of a Large-Scale Ozonolysis: Preparation of the Bisulfite Adduct of 2-Hydroxyindan-2-carbox-aldehyde and Its Utility in a Reductive Animation” by RaganJ.A. et. al. in Org. Proc. Res. Dev., 2003, 7 (2), pp 155-160 reports various routes to bisulfite adduct, the most efficient of which involved vinyl Grignard addition to 2-indanone followed by ozonolysis and workup with aqueous NaHS03 to effect reduction and bisulfite formation in a single pot. The utility of bisulfite adduct is as an aldehyde surrogate in a reductive amination reaction.

[015] The reported methods for the synthesis of ivacaftor suffered from several drawbacks such as harsh conditions, high temperature reactions and use of large excess of polyphosphoric acid and corrosive phosphoryl chloride etc. Furthermore, synthesis of ivacaftor requires use of high performance liquid chromatography (HPLC) techniques for the separation of ivacaftor and their analogues.
[016] Therefore, development of a simple and efficient synthetic route is in urgent need. Accordingly the present inventors developed environmentally benign, cost effective and short synthetic route for the synthesis of ivacaftor and their analogues.
Example 1:
Procedur A:

To a solution of indole acetic acid (500 mg, 2.85 mmol), aniline (2.85 mmol), HOBt (3.4 mmol) in acetonitrile (10 mL), EDC.HCl (3.4 mmol) followed by DIPEA (11.4 mmol) was added, and mixture was stirred for 16 h at ambient temperature. The
reaction mixture was evaporated to dryness, diluted with EtOAc (25 mL), washed with saturated aqueous NaHC03 solution (5 mL), H20 (5 mL), brine (5 mL), and dried over Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (silica gel 230-400 mesh, ethyl acetate – pet ether) to afford corresponding amide as a colorless solid.
[040] Example 2:
2-(lH-indol-3-yl)-N-phenylacetamide (1) :
Yield: 570 mg; 80%; 1H NMR (200MHz, DMSO-d6) δ = 10.95 (brs, 1 H), 10.14 (s, 1 H), 7.64 (d, J = 7.8 Hz, 3 H), 7.47 – 7.24 (m, 4 H), 7.21 – 6.92 (m, 3 H), 3.76 (s, 2H); MS: 273 (M+Na)+.
[041] Example 3:
5-(2-(lH-indol-3-yl)acetamido)-2,4-di-tert-butylphenyl methyl carbonate (2): Yield: 800 mg; 64%; 1H NMR (200 MHz, DMSO-d6) δ = 11.51 (brs, 1 H), 9.41 (s, 1 H), 8.12 (d, J = 7.6 Hz, 1 H), 7.96 – 7.78 (m, 3 H), 7.71 – 7.42 (m, 3 H), 4.34 (s, 3 H), 4.30 (s, 2 H), 1.79 (s, 9 H), 1.64 (s, 9 H); MS: 459 (M+Na)+.
[042] Example 4:
(S)-2-(lH-indol-3-yl)-N-(l-phenylethyl)acetamide (3):
Yield: 620 mg; 78%; 1H NMR (400MHz ,DMSO-d6)5 = 10.88 (brs, 1 H), 8.48 (d, J = 8.1 Hz, 1 H), 7.59 (d, J = 7.8 Hz, 1 H), 7.39 – 7.26 (m, 5 H), 7.25 – 7.16 (m, 2 H), 7.08 (t, J = 7.3 Hz, 1 H), 7.02 – 6.95 (m, 1 H), 4.96 (t, J = 7.3 Hz, 1 H), 3.59 (s, 2H), 1.38 (d, J = 7.1 Hz, 3 H).
[043] Example 5:
N-(4-Fluorophenyl)-2-(lH-indol-3-yl)acetamide (4):
1H NMR (400 MHz, DMSO-d6) : δ 10.93 (brs, 1H), 10.17 (s, 1H), 7.68 – 7.61 (m, 3H), 7.36 (d, J= 8.1 Hz, 1H), 7.27 (d, J= 2.0 Hz, 1H), 7.15 – 7.13 (m, 3H), 7.11 – 6.99 (m, 1H), 3.73 (s, 2H); 13C NMR (100 MHz, DMSO-d6) : δ 170.1, 159.5, 157.1, 136.6, 136.3, 127.7, 124.4, 121.5, 121.3, 121.2, 119.1, 118.9, 115.8, 115.6, 111.8, 108.9, 34.2; MS: 269 (M+H)+
[044] Example 6:
N-(4-Chlorophenyl)-2-(lH-indol-3-yl)acetamide (5):
1H NMR (200 MHz, DMSO-d6): 510.93 (brs, 1H),10.24 (s, 1H), 7.67 – 7.59 (m, 3H), 7.36 – 7.27 (m, 4H), 7.12 – 6.98 (m, 2H), 3.74 (s, 2H); 13CNMR (100 MHz, DMSO-d6): 5170.4, 138.9, 136.7, 129.1, 127.8, 127.1, 124.5, 121.6, 121.2, 119.2, 119.0, 115.7, 111.9, 108.9, 34.3; MS: 285 (M+H)+.
[045] Example 7:
2-(lH-Indol-3-yl)-N-(p-tolyl)acetamide (6) :
1H NMR (400 MHz, DMSO-d6): 510.91 (brs, 1H), 10.01 (s, 1H), 7.62 (d, J= 7.8 Hz, 1H), 7.50 (d, J= 8.6 Hz, 2H), 7.37 (d, J= 8.1 Hz, 1H), 7.29 – 7.26 (m, 1H), 7.10 – 7.07 (m, 3H), 7.01 – 6.99 (m, 1H), 3.71 (s, 2H), 2.23 (s, 3H); 13C NMR (100 MHz, DMSO-de): 5170.0, 137.4, 136.6, 132.4, 129.5, 127.7, 124.3, 121.4, 119.6, 119.2, 118.8, 111.8, 109.1, 34.2, 20.9; MS: 265 (M+H)+.
[046] Example 8:
N-(4-Ethylphenyl)-2-(lH-indol-3-yl)acetamide (7):
XH NMR (400 MHz, DMSO-d6): 510.91 (brs, 1H), 10.01 (s, 1H), 7.61 (s, 1H), 7.52 (d, J= 8.3 Hz, 2H), 7.36 (d, J= 8.1 Hz, 1H), 7.26 (s, 1H), 7.15 – 7.04 (m, 3H), 6.99 (s, 1H), 2.55 (t, J= 7.5 Hz, 2H), 1.15 (t, J= 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): 5169.9, 138.9, 137.6, 136.6, 128.3, 127.7, 124.3, 121.4, 119.6, 119.2, 118.8, 111.8, 109.1, 40.6, 40.4, 40.2, 40.0, 39.8, 39.6, 39.4, 34.2, 28.0, 16.2; MS: 279 (M+H)+.
[047] Example 9:
2-(lH-Indol-3-yl)-N-(4-propylphenyl)acetamide (8):
1H NMR (400 MHz, DMSO-d6): 58.48 (brs, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.50 – 7.42 (m, 2H), 7.33 – 7.15 (m, 6H), 7.07 (d, J= 8.3 Hz, 2H), 3.92 (s, 2H), 2.52 (t, J= 7.6 Hz, 2H), 1.65 – 1.53 (m, 2H), 0.91 (t, J= 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): 5169.7, 138.9, 136.5, 135.2, 128.8, 126.9, 124.0, 122.8, 120.4, 120.1, 118.7, 111.6, 108.7, 37.4, 34.5, 24.6, 13.7; MS: 315 (M+Na)+.
[048] Example 10:
2-(lH-Indol-3-yl)-N-(4-isopropylphenyl)acetamide (9) :
yield 79% ; 1H NMR (400 MHz, DMSO-d6): δ 10.91 (brs, 1H), 10.01 (s, 1H), 7.62 (d, = 7.8 Hz, 1H), 7.55 – 7.49 (m, = 8.6 Hz, 2H), 7.37 (d, = 8.1 Hz, 1H), 7.26 (d, = 2.0 Hz, 1H), 7.18 – 7.11 (m, = 8.6 Hz, 2H), 7.11 – 7.05 (m, 1H), 7.02 – 6.95 (m, 1H), 2.95 – 2.71 (m, 1H), 1.17 (d, = 6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6): δ 169.9, 143.5, 137.6, 136.6, 127.7, 126.8, 124.3, 121.4, 119.7, 119.2, 118.8, 111.8, 109.2, 24.4; MS: 315 (M+Na)+.
[049] Example 11:
2-(lH-indol-3-yl)-N-(4-(trifluoromethoxy)phenyl)acetamide (10):
Yield 85% ; 1H NMR (400 MHz, CDC13): δ 8.35 (brs., 1 H), 7.44 – 7.38 (m, 2 H), 7.27 – 7.21 (m, 3 H), 7.12 – 7.05 (m, 1H), 7.03 – 6.95 (m, 2H), 6.93 (d, = 8.6 Hz, 2H), 3.75 (s, 2H); 13C NMR (100 MHz, CDC13): δ 170.0, 145.3, 136.5, 136.2, 126.8, 124.1, 123.0, 121.6, 121.2, 120.5, 118.5, 111.7, 108.2, 34.4; MS: 335 (M+Na)+.
[050] Example 12:
N-(2-chloro-5-methoxyphenyl)-2-(lH-indol-3-yl)acetamide (11):
Yield 75% ; XH NMR (200 MHz, DMSO-d6): δ 10.98 (brs, 1H), 9.27 (s, 1H), 7.59 (d, = 7.8 Hz, 1H), 7.53 (d, = 2.9 Hz, 1H), 7.39 – 7.32 (m, 3H), 7.09 – 6.99 (m, 2H), 6.74 (dd, = 3.0, 8.8 Hz, 1H), 3.85 (s, 2H), 3.71 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ 170.4, 160.1, 141.1, 136.7, 130.0, 127.8, 124.4, 121.6, 119.2, 119.0, 111.9, 109.1, 105.4, 55.4, 34.4; MS: 315 (M+Na)+.
[051]Example 13:
N-(2-ethylphenyl)-2-(lH-indol-3-yl)acetamide (12):
Yield 78% ; 1H NMR (400 MHz, CDC13): δ 8.68 (brs, 1H), 7.95 (d, = 8.1 Hz, 1H), 7.67 (d, = 7.8 Hz, 1H), 7.48 – 7.44 (m, 2H), 7.29 – 7.23 (m, 1H), 7.22 – 7.20 (m, 3H), 7.05 (d, = 4.4 Hz, 2H), 2.00 (q, = 7.4 Hz, 2H), 0.67 (t, = 7.6 Hz, 3H); 13C NMR (100 MHz, CDC13): δ 169.9, 136.6, 135.0, 134.3, 128.7, 126.7, 125.1, 124.1, 123.0, 122.5, 120.4, 118.7, 111.6, 108.6, 34.4, 24.2, 13.6.
[052] Example 14:
N-(2-bromophenyl)-2-(lH-indol-3-yl)acetamide(13):
Yield 76%; 1H NMR (200 MHz, DMSO-d6): δ 11.00 (brs, 1H), 9.30 (s, 1H), 7.81 -7.77 (m, 1H), 7.63 – 7.56 (m, 2H), 7.41 – 7.35 (m, 3H), 7.11 – 7.05 (m, 3H), 3.85 (s, 2H);13C NMR (100 MHz, DMSO-d6): δ 169.9, 136.2, 132.5, 128.0, 127.2, 126.4, 125.5, 124.4, 121.2, 118.7, 118.5, 116.4, 111.4, 108.0, 33.2.
[053] Example 15:
N-benzyl-2-(lH-indol-3-yl)acetamide (14):
Yield 85%; 1H NMR (400 MHz, DMSO-d6): δ 10.89 (brs., 1H), 8.40 (t, = 5.7 Hz, 1H), 7.57 (d, = 7.8 Hz, 1H), 7.36 (d, = 8.1 Hz, 1H), 7.32 – 7.18 (m, 6H), 7.08 (t, = 7.5Hz, 1H), 7.03 – 6.90 (m, 1H), 4.28 (d, = 5.9Hz, 2H), 3.60 (s, 2H); 13C NMR (100 MHz, DMSO-de): δ 171.2, 140.1, 136.6, 128.7, 127.7, 127.2, 124.3, 121.4, 119.2, 118.7, 111.8, 109.3, 42.7, 33.2.
[054] Example 16:
2-(lH-indol-3-yl)-N-(4-methoxybenzyl)acetamide(15):
Yield 85% ; 1H NMR (400 MHz, DMSO-d6): δ 10.87 (brs, 1 H), 8.32 (t, = 5.6 Hz, 1 H), 7.55 (d, = 7.8 Hz, 1H), 7.35 (d, = 8.1 Hz, 1H), 7.22 – 7.13 (m, 3H), 7.11 – 7.05 (m, 1 H), 7.00 – 6.94 (m, 1H), 6.84 (d, = 8.6 Hz, 2H), 4.20 (d, = 6.1 Hz, 2H), 3.72 (s, 3H), 3.56 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 171.1, 158.6, 136.6, 132.0, 129.0, 127.7, 124.2, 121.4, 119.2, 118.7, 114.1, 111.8, 109.4, 55.5, 42.1, 33.2.
[055] Example 17:
N,N-dibenzyl-2-(lH-indol-3-yl)acetamide (16):
Yield 70% ; 1H NMR (400 MHz, DMSO-d6): δ 10.91 (brs, 1H), 7.50 (d, = 7.8 Hz, 1H), 7.37 – 7.34 (m, 3H), 7.30 (d, = 6.6 Hz, 1H), 7.25 – 7.19 (m, 3H), 7.17 (t, = 6.6 Hz, 5H), 7.16 (d, = 7.8 Hz, 1H), 7.00 – 6.97 (m, 1H), 4.59 (s, 2H), 4.50 (s, 2H), 3.86 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 171.7, 138.2, 136.6, 129.2, 128.8, 128.1, 127.8, 127.7, 127.5, 127.1, 124.2, 121.5, 119.2, 118.8, 111.8, 108.5, 50.7, 48.4, 31.2.
[056] Example 18:
2-(lH-indol-3-yl)-N-propylacetamide (17):
Yield 75% ; XH NMR (200 MHz, DMSO-d6): δ 10.86 (brs, 1H), 7.88 – 7.80 (m, 1H), 7.56 (d, = 7.6 Hz, 1H), 7.31 (d, = 7.8 Hz, 1H), 7.17 (d, = 2.3 Hz, 1H), 7.06 – 6.92 (m, 2H), 3.48 (s, 2H), 3.00 (q, J = 6.8 Hz, 2H), 1.39 (sxt, / = 7.2 Hz, 2H), 0.88 – 0.75 (t, = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 171.0, 136.6, 127.8, 124.2,
121.4, 119.2, 118.7, 111.8, 109.6, 39.4, 33.3, 22.9, 11.9.
[057] Example 19:
N-hexyl-2-(lH-indol-3-yl)acetamide (18) :
Yield 87% ; 1H NMR (400 MHz, DMSO-d6): δ 10.84 (brs, 1H), 7.83 (brs, 1H), 7.54 (d, = 7.8 Hz, 1H), 7.33 (d, = 8.1 Hz, 1H), 7.21 – 7.13 (m, 1H), 7.06 (t, = 7.6 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 3.47 (s, 2H), 3.03 (q, / = 6.8 Hz, 2H), 1.37 (t, = 6.5 Hz, 2H), 1.30 – 1.15 (m, 6H), 0.84 (t, = 6.7 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 170.9, 136.6, 127.7, 124.2, 121.3, 119.1, 118.7, 111.7, 109.5, 39.06, 33.2, 31.5, 29.6, 26.5, 22.5, 14.4.
[058] Example 20:
Methyl (2-(lH-indol-3-yl)acetyl)-L-alaninate (19):
Yield 79% ; 1H NMR (400 MHz, CDC13): δ 8.53 (brs, 1H), 7.60 (d, = 7.8 Hz, 1H), 7.41 (d, = 8.1 Hz, 1H), 7.25 – 7.23 (m, 1H), 7.19 – 7.14 (m, 2H), 6.27 (d, = 7.3 Hz, 1H), 4.63 (t, = 7.3 Hz, 1H), 3.78 (s, 2H), 3.68 (s, 3H), 1.31 (d, = 7.3 Hz, 3H); 13C NMR (100 MHz, CDC13): δ 173.4, 171.2, 136.4, 127.0, 123.8, 122.5, 119.9, 118.7,
111.5, 108.5, 52.4, 48.0, 33.3, 18.2.
[059] Example 21:
-(6-chloro-lH-indol-3-yl)-N-phenylacetamide(20):

To a solution of 6-Chloro indole 20a (300 mg, 1.98 mmol )in anhydrous THF, Oxalyl chloride (186 μΤ, 276 mg, 2.18 mmol) was added and the mixture stirred at room temperature. After 2 h, N,N-Diisopropylethylamine (758 μΤ, 562 mg, 4.35 mmol) was
introduced to the mixture, followed by the aniline (221.0 mg, 2.37 mmol). The temperature was raised to 45 °C, and heating continued for 18 h. The solvent was evaporated, and then mixture was diluted with EtOAC (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (10 – 20% EtOAc : Petroleum ether) to afford 20b (295 mg, 51% yield) as a yellow coloured solid. IR Omax(film): 3346, 3307,2853, 1724, 1678 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 12.40 (br. s., 1H), 10.68 (s, 1H), 8.79 (d, = 3.2 Hz, 1H), 8.25 (d, = 8.6 Hz, 1H), 7.85 (d, = 7.8 Hz, 2H), 7.62 (d, = 1.7 Hz, 1H), 7.41 – 7.30 (m, 3H), 7.19 – 7.13 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 182.5, 162.5, 140.0, 138.4, 137.4, 129.2, 128.5, 125.4, 124.8, 123.4, 122.9, 120.8, 113.0, 112.3; HRMS (ESI) Calculated for Ci6HnN2OCl[M+H]+: 299.0582, found 299.0580;
A solution of 20b (300 mg, 0.99 mmol) dissolved in MeOH (40 mL) was added to NaBH4 (45 mg, 1.23 mmol). The reaction was stirred for 4h and then added to saturated solution of Na2S04. The reaction mixture was further stirred for lh and then filtered through Celite.The filtrate obtained was concentrated in vacuo, and then mixture was diluted with EtOAc (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was forwarded for next step without further purification.In an N2 atmosphere, TMSC1 (1.272 mL, 9.9 mmol) in CH3CN (40 mL) was added to sodium iodide (1.488 mg, 9.9 mmol) and stirred for 2h. The reaction mixture was cooled to 0 °C and a solution of above crude alcohol (0.99 mmol) in CH3CN (10 mL) was then added drop wise over 30 min, followed by stirring for 3h. The reaction mixture was poured into NaOH (7g in 40 mL of water) and then extracted with ethyl acetate (15×2). The organic layer was washed with aq.Na2S203, dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel (EtOAc:Pet ether) to afford 20 as a off white solid (two steps 38 % ); IR Umax(film): 3273, 3084,2953, 2857, 1629, 1562 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 11.06 (br. s., 1H), 10.13 (br. s., 1H), 7.62 – 7.57 (m, 3H), 7.40 (s, 1H), 7.30 – 7.25 (m, 3H), 7.04 – 6.99 (m, 2H), 3.71 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 170.1,
139.7, 136.9, 129.2, 126.5, 126.3, 125.5, 123.7, 120.6, 119.6, 119.3, 111.5, 109.4, 34.0; HRMS (ESI):Calculated for Ci6Hi4N2OCl[M+H]+: 285.0789, found 285.0786.
[060] Example 22:
2-(5-chloro-lH-indol-3-yl)-N-phenylacetamide(21):

21a 21b 21
To a solution of 5-Chloro indole 21a (300 mg, 1.98 mmol )in anhydrous THF(20 mL), Oxalyl chloride (186 ^L, 276 mg, 2.18 mmol) was added and the mixture stirred at room temperature. After 2 h, N,N-diisopropylethylamine (758 μΕ, 562 mg, 4.35 mmol) was introduced to the mixture, followed by the aniline (221.0 mg, 2.37 mmol). The tempera ture was raised to 45 °C, and heating continued for 18 h. The solvent was evaporated, and then mixture was diluted with EtOAC (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (10 – 20% EtOAc : Petroleum ether) to afford (21b) (305 mg, 53% yield) as a yellow coloured solid. IR rjmax(film): 3346, 3307,2853, 1724, 1678 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 12.40 (br. s., 1H), 10.68 (s, 1H), 8.79 (d, = 3.2 Hz, 1H), 8.25 (d, = 8.6 Hz, 1H), 7.85 (d, = 7.8 Hz, 2H), 7.62 (d, = 1.7 Hz, 1H), 7.42 – 7.30 (m, 3H), 7.20 – 7.14 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 182.4, 162.4, 140.3, 138.4, 135.4, 129.2, 127.9, 124.8, 124.1, 120.8, 114.8, 112.0; HRMS (ESI) Calculated for Ci6HnN2OCl[M+H]+: 299.0582, found 299.0580; A solution of 21b (200 mg, 0.66 mmol) dissolved in MeOH (30 mL) was added to NaBH4 (30 mg, 0.82 mmol). The reaction was stirred for 4h and then added to saturated solution of Na2S04. The reaction mixture was further stirred for lh and then filtered through Celite. The filtrate obtained was concentrated in vacuo, and then mixture was diluted with EtOAc (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was forwarded for next step without further purification. In an N2 atmosphere, TMSC1 (848 mL, 6.6 mmol) in CH3CN (25 mL) was added to sodium iodide (992 mg, 6.6 mmol) and stirred for 2h. The reaction mixture was cooled to 0 °C and a solution of above crude alcohol(0.66 mmol) in CH3CN (5 mL) was then added dropwise over 30 min, followed by stirring for 3h. The reaction mixture was poured into NaOH (5g in 30 mL of water) and then extracted with ethyl acetate(15×2). The organic layer was washed with aq.Na2S203, dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel (EtOAc:Pet ether) to afford 22 as a off white solid (two steps 42 % ); IR Umax(film): 3273, 3084,2955, 2857, 1629, 1562 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 11.13 (br. s., 1H), 10.11 (s, 1H), 7.67 (s, 1H), 7.60 (d, = 7.8 Hz, 2H), 7.39 – 7.27 (m, 4H), 7.13 – 7.02 (m, 2H), 3.16 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 169.9, 139.8, 135.0, 129.2, 128.9, 126.2, 123.6, 121.4, 119.6, 118.6, 113.4, 109.0, 34.0; HRMS (ESI) Calculated for Ci6H14N2OCl[M+H]+: 285.0789, found 285.0786.
[061] Example 23:
2-(l-benzyl-lH-indol-3-yl)-N-phenylacetamide (22):
Yield 79% ; 1H NMR (400 MHz, DMSO-d6): δ 7.67 (d, = 7.8 Hz, 1H), 7.54 (brs, 1H), 7.43 – 7.31 (m, 6H), 7.31 – 7.25 (m, 3H), 7.23 – 7.15 (m, 4H), 7.12 – 7.06 (m, 1H), 5.36 (s, 2H), 3.91 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 169.7, 137.7, 137.2, 137.0, 128.9, 128.9, 127.9, 127.6, 126.9, 124.3, 122.7, 120.2, 119.9, 119.0, 110.2, 107.9, 77.4, 77.1, 76.8, 50.1, 34.5.
[062] Example 24:
Procedure B:

2-(lH-indol-3-yl)-N-phenylacetamidel(100 mg; 0.4 mmol) was dissolved in DCM:MeOH(50 mL; 5: 1), then a stream of 03 was passed through the solution until a blue color developed (10 min). The 03 stream was continued for 4 min. Then surplus O3 was removed by passing a stream of 02 through the solution for 10 min or until the blue colorcompletely vanished. Afterwards pyridine (0.1 mL;1.2mmol) was added to the cold (- 78 °C) mixture. The mixture was allowed to warm to room temperature (1 h) and then Et3N (0.35 mL; 2.4 mmol) were added. After stirring at room temperature overnight the reaction mass was concentrated under reduced pressure to dryness, diluted with EtOAc (30 mL), washed with H20 (5 mL), brine (5 mL), and dried over Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (silica gel 230-400 mesh, MeOH – DCM) to give desired quinolone carboxamide as colorless solid.
[063] Example 25:
4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide (23):
Yield: 65 mg; 62%; XH NMR (200MHz ,DMSO-d6) δ = 12.97 (brs, 1 H), 12.49 (s, 1 H), 8.89 (s, 1 H), 8.33 (d, J = 8.2 Hz, 1 H), 7.91 – 7.69 (m, 4 H), 7.62 – 7.50 (m, 1 H), 7.37 (t, J = 7.8 Hz, 2 H), 7.18 – 7.01 (m, 1 H); MS: 287 (M+Na)+.
[064] Example 26:
2,4-di-tert-butyl-5-(4-oxo-l,4-dihydroquinoline-3-carboxamido)phenyl methyl carbonate (24):
Yield: 35 mg; 34%; 1H NMR (400MHz ,DMSO-d6) δ = 12.96 (brs, 1 H), 12.08 (s, 1 H), 8.94 – 8.82 (m, 1 H), 8.44 – 8.28 (m, 1 H), 7.86 – 7.79 (m, 1 H), 7.78 – 7.73 (m, 1 H), 7.59 (s, 1 H), 7.53 (t, J = 7.5 Hz, 1 H), 7.39 (s, 1 H), 3.86 (s, 3 H), 1.46 (s, 9 H), 1.32 (s, 9 H).
[065] Example 27:
(S)-4-oxo-N-(l-phenylethyl)-l,4-dihydroquinoline-3-carboxamide (25):
Yield: 56 mg; 53%; 1H NMR (500MHz ,DMSO-d6) δ = 12.75 (brs, 1H), 10.54 (d, J = 7.6 Hz, 1H), 8.73 (brs, 1H), 8.28 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 7.9 Hz, 1H), 7.73 -7.68 (m, 1 H), 7.50 (t, J = 7.5 Hz, 1 H), 7.42 – 7.34 (m, 4 H), 7.29 – 7.23 (m, 1 H), 5.18 (t, J = 7.2 Hz, 1 H), 1.50 (d, J = 6.7 Hz, 3 H).
[066] Example 28:
Synthesis of ivacaftor (26):

To a solution of 2,4-di-tert-butyl-5-(4-oxo-l,4-dihydroquinoline-3-carboxamido)phenyl methyl carbonate 5 (30 mg, 0.06mmol) in MeOH (2 mL) was added NaOH (5.3 mg, 0.13mmol) dissolved in H20 (2 mL), and the reaction mixture was stirred at room temperature for 5h. Reaction mass was evaporated to one third of its volume (temperature not exceeding 40°C) and acidified with aq.2N HC1 to pH 2-3. The resulting precipitate was collected by suction filtration give desired compound 7 (19 mg, 76%) as off white solid H NMR (400MHz ,DMSO-d6) δ = 12.88 (d, J = 6.6 Hz, 1 H), 11.81 (s, 1 H), 9.20 (s, 1 H), 8.86 (d, J = 6.6 Hz, 1 H), 8.32 (d, J = 7.8 Hz, 1 H), 7.88 – 7.65 (m, 2 H), 7.51 (t, J = 7.5 Hz, 1 H), 7.16 (s, 1 H), 7.10 (s, 1 H), 1.38 (s,9H), 1.36 (s, 9H).
[067] Example 29:
N-(4-fluorophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (27):
Yield 56% ; 1H NMR (400 MHz, DMSO-d6): δ 12.96 (br. s., 1H), 12.50 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.3 Hz, 1H), 7.86 – 7.72 (m, 4H), 7.54 (t, = 7.3 Hz, 1H), 7.20 (t, = 8.8 Hz, 2H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.2, 159.7, 157.3, 144.6, 139.6, 135.7, 133.5, 126.4, 125.9, 125.8, 121.8, 119.7, 116.1, 115.9, 110.9.
[068] Example 30:
N-(4-chlorophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (28):
Yield 51% ; 1H NMR (400 MHz, DMSO-d6): δ 13.00 (brs., 1H), 12.59 (br. s., 1H), 8.89 (s, 1H), 8.34 (d, = 7.6 Hz, 1H), 7.83 – 7.76 (m, 4H), 7.56 (s, 1H), 7.42 (d, = 7.9 Hz, 2H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.4, 144.7, 139.6, 138.2, 133.5, 129.4, 127.4, 126.4, 125.9, 125.8, 121.6, 119.7, 110.8.
[069] Example 31:
4-oxo-N-(p-tolyl)-l,4-dihydroquinoline-3-carboxamide (29):
Yield 57% ; 1H NMR (400 MHz, DMSO-d6): δ 12.94 (brs., 1H), 12.40 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.8Hz, 1H), 7.82 – 7.80 (m, 1H), 7.76 – 7.7 (m, 1H), 7.63 (d, = 8.3 Hz, 2H), 7.53 (t, = 7.3 Hz, 1H), 7.17 (d, = 8.1 Hz, 2H), 2.29 (s, 3H); 13C NMR (100 MHz, DMSO-de): δ 176.8, 163.1, 144.5, 139.6, 136.8, 133.4, 132.8, 129.9, 126.4, 125.9, 125.7, 120.0, 119.6, 111.1, 20.9; HRMS (ESI):Calculated for Ci7H1502N2[M+H]+: 279.1128, found 279.1127.
[070] Example 32:
N-(4-ethylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (30):
Yield 51% ; 1H NMR (400 MHz, DMSO-d6): δ 12.95 (br. s., 1H), 12.40 (d, = 7.8 Hz, 1H), 8.87 (d, = 6.1 Hz, 1H), 8.33 (d, = 8.1 Hz, 1H), 7.81 – 7.76 (m, 2H), 7.66 – 7.62 (m, = 8.3 Hz, 2H), 7.53 (t, 7 = 7.5 Hz, 1H), 7.22 – 7.17 (m, = 8.3 Hz, 2H), 2.58 (q, = 7.6 Hz, 2H), 1.18 (t, = 7.6 Hz, 3H); 13C NMR (400 MHz, DMSO-d6): δ 181.5, 167.8, 149.3, 144.3, 144.0, 141.7, 138.2, 133.4, 131.1, 130.7, 130.5, 124.8, 124.4, 115.9, 32.8, 20.9.
[071] Example 33:
4-Oxo-N-(4-propylphenyl)-l,4-dihydroquinoline-3-carboxamide (31):
Yield 51%; 1H NMR (500 MHz, DMSO-d6): δ12.93 (brs, 1H), 12.40 (s, 1H), 8.87 (s, 1H), 8.36 – 8.29 (m, 1H), 7.86 – 7.78 (m, 1H), 7.75 (d, J= 7.9 Hz, 1H), 7.68 – 7.61 (m, J= 8.2 Hz, 2H), 7.54 (t, J= 7.6 Hz, 1H), 7.22 – 7.14 (m, J= 8.2 Hz, 2H), 2.55 – 2.51 (m, 2H), 1.64 – 1.53 (m, 2H), 0.90 (t, J= 7.3 Hz, 3H); 13C NMR (500 MHz, DMSO-d6): 176.8, 163.1, 144.5, 139.6, 137.6, 137.0, 133.5, 129.3, 126.4, 125.9, 125.7, 120.0, 119.7, 111.1, 37.2, 24.6, 14.1.
[072] Example 34:
N-(4-isopropylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (32):
Yield 46% ; 1H NMR (500 MHz, DMSO-d6): δ 12.93 (br. s., 1H), 12.40 (br. s., 1H), 8.89 – 8.86 (m, 1H), 8.33(d, = 7.6 Hz, 1H), 7.81 – 7.50 (m, 5H), 7.25 – 7.21 (m, 2H), 2.90-2.83 (m, 1H), 1.22-1. l l(m, 6H); 13C NMR (100 MHz, DMSO-d6): δ 176.8, 163.1, 144.5, 143.9, 139.6, 137.1, 133.4, 127.2, 126.4, 125.9, 125.7, 120.1, 119.6, 111.1, 33.4, 24.4.
[073] Example 35:
4-oxo-N-(4-(trifluoromethoxy)phenyl)-l,4-dihydroquinoline-3-carboxamide(33):
Yield 57% ; 1H NMR (400 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.63 (s, 1H), 8.88 (d, = 4.9 Hz, 1H), 8.32 (d, = 7.8 Hz, 1H), 7.89 – 7.83 (m, = 8.8 Hz, 2H), 7.79 (d, = 7.6 Hz, 1H), 7.77 – 7.73 (m, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.40 – 7.34 (m, = 8.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 176.8, 163.5, 144.7, 144.0, 139.5, 138.5, 133.5, 126.3, 125.9, 125.8, 122.3, 121.4, 119.7, 110.7.
[074] Example 36:
N-(2-chloro-5-methoxyphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(34):
Yield 54% ; XH NMR (400 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.49 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.8 Hz, 1H), 7.83 – 7.75 (m, 1H), 7.56-7.48 (m, 3H), 7.27 – 7.21 (m, 1H), 6.67 (d, = 7.8 Hz, 1H), 3.77 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.4, 160.2, 144.7, 140.4, 139.6, 133.5, 130.3, 126.4, 125.9, 125.8, 119.7, 112.3, 111.0, 109.5, 105.7, 55.5.
[075] Example 37:
N-(2-ethylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(35):
Yield 58% ; 1H NMR (400 MHz, DMSO-d6): δ 12.94 (br. s., 1H), 12.37 (s, 1H), 8.90 (s, 1H), 8.36 (dd, = 8.1, 1.4 Hz, 2H), 8.32 (dd, = 8.1, 1.4 Hz, 2H), 7.82 – 7.74 (m, 1H), 7.53- 7.19 (m, 3H), 7.15 – 7.06(m, 1H), 2.79 (q, = 7.3 Hz, 2H), 1.26 (t, = 7.5 Hz, 3H); 293 (M+H)+.
[076] Example 38:
N-(2-bromophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(36):
Yield 47% ; 1H NMR (200 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.69 (s, 1H), 8.90 (d, = 5.9 Hz, 1H), 8.54 (dd, 7 = 1.4, 8.3 Hz, 1H), 8.34 (d, = 7.6 Hz, 1H), 7.86 – 7.67 (m, 3H), 7.57 – 7.49 (m, 1H), 7.40 (t, = 7.2 Hz, 1H), 7.10 – 7.05 (m, 1H); 13C NMR (100 MHz, DMSO-de): δ 176.7, 163.7, 145.0, 139.5, 137.7, 133.5, 133.1, 128.6, 126.4, 126.0, 125.8, 125.3, 122.9, 119.7, 113.4, 110.8.
[077] Example 39:
N-benzyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(37):
Yield 58% ; 1H NMR (400 MHz, CD3OD-d6): δ 8.82 (s, 1 H), 8.35 (d, = 8.1 Hz, 1 H), 7.79 – 7.77 (m, 1 H), 7.65 (d, = 8.3 Hz, 1 H), 7.52 (t, = 7.6 Hz, 1 H), 7.42 – 7.34 (m, 4 H), 7.31 – 7.26 (m, 1 H), 4.67 (s, 2 H); 13C NMR (400 MHz, DMSO-d6): δ 176.6, 165.0, 144.2, 140.0, 139.5, 133.2, 128.9, 128.7, 127.8, 127.3, 126.6, 125.9, 125.4, 119.5, 111.2, 42.6.
[078] ] Example 40:
N-(4-methoxybenzyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(38):
Yield 56% ; 1H NMR (400 MHz, DMSO-d6): δ 12.73 (br. s., 1H), 10.35 (t, = 5.3 Hz, 1H), 8.78 (d, = 6.1 Hz, 1H), 8.24 (d, = 8.1 Hz, 1H), 7.76 (d, = 7.1 Hz, 1H), 7.73 -7.68 (m, 1H), 7.48 (t, = 7.5 Hz, 1H), 7.28 (d, = 8.3 Hz, 2H), 6.91 (d, = 8.1 Hz, 2H), 4.49 (d, = 5.6 Hz, 2H), 3.74 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 176.6, 164.8, 158.8, 144.1, 139.5, 133.1, 131.9, 129.2, 126.6, 125.8, 125.4, 119.5, 114.3, 111.3, 55.5, 42.0.
[079] Example 41:
N,N-dibenzyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(39):
Yield 43% ; 1H NMR (400 MHz, DMSO-d6): δ 12.21 (br. s., 1H), 8.27 (d, = 4.9 Hz, 1H), 8.21 (d, = 7.6 Hz, 1H), 7.49 – 7.41 (m, 2H), 7.41 – 7.35 (m, 3H), 7.33 – 7.20 (m, 5H), 7.20 – 7.11 (m, 7 = 7.1 Hz, 2H), 4.59 (br. s., 2H), 4.42 (s, 2H).
[080] Example 42:
4-oxo-N-propyl-l,4-dihydroquinoline-3-carboxamide(40):
Yield 47% ;1H NMR (400 MHz, DMSO-d6): δ 12.7 (br.s., 1H)10.05 (t, = 5.5 Hz, 1H), 8.74 (s, 1H), 8.26 (d, = 8.1 Hz, 1H), 7.83 – 7.66 (m, 2H), 7.52 – 7.44 (m, 1H), 3.33 – 3.22 (m, 2H), 1.61 – 1.49 (m, 2H), 0.93 (t, = 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-de): δ 176.6, 164.8, 143.9, 139.5, 133.1, 126.6, 125.9, 125.3, 119.4, 111.4, 39.3, 23.1, 12.0
[081] Example 43:
N-hexyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(41):
Yield 51% ;1H NMR (400 MHz, DMSO-d6): δ 12.68 (m, 1H), 10.02 (t, = 5.5 Hz, 1H), 8.73 (d, = 6.1 Hz, 1H), 8.27 – 8.25 (m, 1H), 7.77 – 7.67 (m, 2H), 7.47 (t, = 7.5 Hz, 1H), 3.33 – 3.29 (m, 2H), 1.56 – 1.45 (m, 2H), 1.34 – 1.25 (m, 6H), 0.88 – 0.82 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 176.6, 164.8, 143.9, 139.5, 133.1, 126.6, 125.9, 125.3, 119.4, 111.4, 38.7, 31.5, 29.8, 26.7, 22.5, 14.4.
[082] Example 44:
Methyl (4-oxo-l,4-dihydroquinoline-3-carbonyl)-L-alaninate(42):
Yield 38% ; 1H NMR (400 MHz, CD3OD): δ 8.74 (s, 1H), 8.47 – 8.29 (m, 1H), 7.86 -7.76 (m, 1H), 7.64 (d, = 8.3 Hz, 1H), 7.58 – 7.44 (m, 1H), 4.69 (d, = 7.3 Hz, 1H), 3.79 (s, 3H), 1.55 (d, = 7.3 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ 177.3, 173.3, 165.5, 143.6, 139.2, 132.9, 126.3, 125.4, 125.2, 118.5, 110.3, 51.5, 47.0, 17.0.
[083] Example 45:
7-chloro-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(43):
Yield 48% ; IR Omax(film): 2920, 2868, 1661, 1601 cm” 1; 1H NMR (400 MHz, DMSO-de): δ 12.91 (br. s., 1H), 12.30 (s, 1H), 8.90 (s, 1H), 8.29 (d, = 8.8 Hz, 1H), 7.80 -7.67 (m, 3H), 7.58 – 7.51 (m, 1H), 7.36 (t, = 7.7 Hz, 2H), 7.09 (t, = 7.3 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 176.3, 162.9, 145.4, 140.3, 139.2, 138.0, 129.5, 128.2, 126.1, 125.1, 123.9, 120.1, 118.8, 111.6.
[084] Example 46:
6-chloro-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(44):
Yield 52% ; 1H NMR (400 MHz, DMSO-d6): δ 13.05 (brs, 1H), 12.27 (s, 1H), 8.88 (s, 1H), 8.21 (d, = 2.2 Hz, 1H), 7.86 – 7.67 (m, 4H), 7.36 (t, = 7.8 Hz, 2H), 7.16 – 7.04 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 175.6, 162.9, 144.9, 139.1, 138.2, 133.5, 130.4, 129.5, 127.5, 124.9, 123.9, 122.0, 120.1, 111.4.
[085] Example 47:
l-benzyl-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(45)
Yield 55% ; 1H NMR (400 MHz, DMSO-d6): δ 12.30 (s, 1H), 9.05 (s, 1H), 8.60 (dd, = 1.7, 8.1 Hz, 1H), 7.82 (d, = 7.8 Hz, 2H), 7.69 – 7.62 (m, 1H), 7.55 – 7.45 (m, 2H), 7.43 – 7.34 (m, 5H), 7.24 – 7.18 (m, 2H), 7.17 – 7.10 (m, 1H), 5.53 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 176.9, 162.9, 148.7, 139.3, 138.7, 134.1, 133.1, 129.4, 128.9, 128.7, 128.0, 127.4, 126.2, 125.5, 123.9, 120.5, 116.9, 112.3, 57.9; HRMS (ESI): Calculated for C23H1802N2Na [M+Na]+: 377.1260, found 377.1259; MS: 355 (M+H)+.
[086] Advantages of invention:
1. Cost-effective process for synthesis.
2. Carried out at environmentally benign conditions.
3. Short synthetic route.
4. Useful for making several related compounds of medicinal

DR SRINIVASA REDDY recieving NASI – Reliance Industries Platinum Jubilee Award (2015) for Application Oriented Innovations in Physical Sciences.
MYSELF WITH HIM

From left to right: Dr. D. Srinivasa Reddy, Shri Y. S. Chowdary, Dr. Harsh Vardhan, Dr. Girish Sahni
- Dr D. Srinivasa Reddy receiving the prestigious “SHANTI SWARUP BHATNAGAR” award at the occasion of the 75th Foundation day of CSIR.

Shanti Swarup Bhatnagar awardees with the honorable Prime Minister of India

NCL PUNE

DSR Group
//////////WO-2016181414, WO 2016181414, IVACAFTOR, new patent, COUNCIL OF SCIENTIFIC & INDUSTRIAL RESEARCH, Anusandhan Bhawan, Rafi Marg New Delhi, INDIA, CSIR, Dr. D. Srinivasa Reddy
IPRAGLIFLOZIN, NEW PATENT, WO2016173551, China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry


WO 2016173551 China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016173551&redirectedID=true
MA, Shuai; (CN).
ZHOU, Weicheng; (CN)
WO2016173551, IPRAGLIFLOZIN PREPARATION METHOD
CHINA STATE INSTITUTE OF PHARMACEUTICAL INDUSTRY [CN/CN]; 4th Floor, Building 1, No.1111 Halley Road,pudong New Area Shanghai 201203 (CN).
SHANGHAI INSTITUTE OF PHARMACEUTICAL INDUSTRY [CN/CN]; No.1320,West Beijing Road,Jing’an District Shanghai 200040 (CN)

MACHINE TRANSLATED FROM CHINESE……
Ignatius column Net (English name: Ipragliflozin) by Astellas Pharma Ltd. (Astellas) new sodium life Pharmaceutical Co., Ltd. (Kotobuki) R & D – glucose cotransporter (Sodium glucose co-transporters, referred to as SGLT-2 ) inhibitor, on January 17, 2014 in the Japanese market for the treatment of patients with type ⅱ diabetes; tradename Suglat, currently provide 25mg and 50mg tablets.
Chemical Name column Ignatius net is (1S) -1,5- dehydration -1-C- [3- (1- benzothien-2-yl-methyl) -4-fluorophenyl] -D-glucose alcohols of the formula the C 21 the H 21 the FO 5 the S, the CAS No. 761423-87-4, as the structure of formula 2, as a column for medicinal Eagle with L- proline net clinical eutectics, such as a structural formula FIG.
Ignatius column in the process of preparation of the net, the chiral synthesis of β glycoside bond synthetic route key points. Currently, Ignatius column net of synthetic methods reported in the literature there are several of these methods for the synthesis of chiral β-glucoside bonds mainly relates to hydroxy-protected D- glucose lactone ester carbonyl nucleophilic addition reaction.
Route One: Patent WO2004080990 synthetic route reported net Ignatius column is as follows:
This route, [1-benzopyran-2-yl (5-bromo-2-fluorophenyl) methoxy] (tert-butyl) dimethylsilane (Compound 10) with n-butyl lithium at -78 deg.] C (or minus 78 deg.] C) the reaction of an organolithium reagent and then with 2,3,4,6-tetra -O- benzyl -D- glucose lactone (compound 9) nucleophilic addition at low temperature -78 ℃ to obtain compound 8, followed by removal of the silicon compound 8 hydroxy group is protected with tetrabutylammonium fluoride (of TBAF) to give compound 7, triethylsilane and then reducing the compound 7 obtained with chiral β glycosidic bond Ignatius column net intermediate 6, the last off at -78 ℃ intermediate ring 6 sugar hydroxyl protecting groups to obtain the desired product – Ignatius column net (compound 2). Compound 10 was prepared by the target product – Ignatius column net synthesis route yield 9.94%, net Ignatius column purity not reported. The disadvantage of this method is that a long synthetic route, after every step of the reaction were purified by column chromatography, and the yield is low. Deprotecting the hydroxy group on two key steps chiral β glycosidic bond synthesis and sugar ring need to be at a low temperature at -78 deg.] C, clearly, it is difficult to meet the needs of industrial production.
Route II: Patent WO2008075736 Ignatius column reported net synthetic route is as follows:
The route of 2- (5-bromo-2-fluorobenzyl) benzothiophene (compound 15) with n-butyl lithium at -43.5 ~ -33.3 ℃ reaction of an organolithium reagent and then with 2,3,4 , 6-tetrafluoro -O- trimethylsilyl -D- glucose lactone (compound 14) nucleophilic addition reactions at -72.6 ~ -65 ℃ to give compound 13, compound 13 and then acetylation, reduction Ignatius column net intermediates prepared with chiral β glycoside bond of 11, finally deacetylated to obtain the desired product of intermediate 11 – Ignatius column net (compound 2). Compound 15 was prepared by the Scheme 2 the desired product in a yield of 72.46%, a purity of compound 2 was 99%. The disadvantage of this method is that the route Ignatius column net synthesis requires at a low temperature of -72.6 ℃ to react and involve nucleophilic addition reaction, a hydroxyl group on the terminal carbon methylation, acetylation of hydroxyl groups on the sugar ring, the end methoxy groups on carbon reduction, the reaction and post-treatment process is very complicated, more difficult to industrial production, and on the terminal carbon-methoxy-reducing agent used in the reduction – t-butyldimethylsilyl more expensive, increasing the whole synthetic route costs.
Route III: Patent WO2015012110 Ignatius column reported net synthetic route is as follows:
On the basis of patent WO2015012110 patent WO2008075736 reported synthetic route for the synthesis net Ignatius column primarily relates to the further improvements: namely: 2- (5-bromo-2-fluorobenzyl) benzothiophene (Compound 15) three butylmagnesium lithium at -12 ~ -26 ℃ organomagnesium reagent prepared by the reaction – compound 16, and then with 2,3,4,6-tetra -O- trimethylsilyl -D- glucose lactone (compound 14) carried out at -12 ~ -16 ℃ nucleophilic addition reaction Ignatius column net key intermediates – compounds 13, this step is nucleophilic addition reaction temperature was raised to -26 ℃, avoid the use of other organic lithium reagent required -78 ℃ low temperature reactions. The disadvantage of this method is that Ignatius column net synthesis still need to involve nucleophilic addition reaction, a hydroxyl group on the terminal carbon methylation, acetylation of hydroxyl groups on the sugar ring, a methoxy group on the terminal carbon reduction reaction and post-treatment very complicated problem is not resolved; in addition, tributyltin lithium magnesium used in the route in the country not commercially available, and can be prepared before the experiment, the manufacturing process is more complex, more difficult to industrial production.
Skilled in the art knows the energy super low temperature chemical reaction operations is considerable. Generally, the reaction temperature at -40 ℃ over the operation of the more conventional reactor in the plant can be relatively easy to do; but lower than the reaction below -40 ℃ the need to use special equipment or a special reactor is required with liquid nitrogen as the cooling source, the higher the cost. For ultra-low temperature improvements often become enlarged or when the process of large-scale, process optimization of key points.
In the background art described in this article about the Ignatius column net three synthetic route, the “connection” between the main synthon mainly related to the organometallic reagents – such as organic lithium or magnesium organic lithium reagents protected hydroxy D- glucose ester carbonyl lactone on nucleophilic substitution reaction with hydroxyl groups to form the corresponding glucose derivative on the terminal carbon; then after hydroxy or derivatives thereof – methoxy reduced to hydrogen, to give the title with β-type hand glycoside bond Ignatius column net key intermediate structure; and finally the removal of hydroxy protecting groups on the pyranose ring to give Ignatius column net. In these types of synthetic route, operation and post-processing reaction steps are more complicated, the cost is high. For example, in Scheme 1 and 2, both the use of ultra-low temperature organolithium reagent – minus 78 ℃; several synthetic route in addition, most of the intermediate purification using column chromatography, such process is not suitable for plant production is amplified. Therefore, an urgent need to find new Ignatius column net synthesis method, and enables industrial production.
(1), from 4-fluoro-3- (2-benzothienyl) phenyl methyl halide (Compound 5) as a starting material, the compound 5 in a suitable solvent, is reacted with an alkyl lithium, followed by reaction with zinc an organic zinc reagent – bis [4-fluoro-3- (2-benzothienyl) methyl phenyl] zinc, and then with 2,3,4,6-tetra -O- pivaloyl bromo -α-D- Generation glucopyranose (compound 4) nucleophilic substitution reaction of intermediate net Ignatius column – compound 3;
(2), compound 3 by an organic base off pivaloyl protecting group to obtain Eagle column net (Compound 2);
Wherein in the 4-fluoro-3- (2-benzothienyl) phenyl methyl halide (Compound 5) Structure X is selected from bromo or iodo;
Synthetic route is as follows:
Example 1, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added n-butyl ether (8mL), stirred and heated to 50 deg.] C 2h, cooling backup. Under nitrogen, was added 2- (5-iodo-2-fluorobenzyl) benzothiophene (2.21g) in toluene (5mL), n-butyl ether (5mL), cooled to -25 deg.] C, was slowly added dropwise 1.6mol / L n-hexyl lithium hexane solution (4.13 ml), to control the internal temperature does not exceed -10 deg.] C, after the addition was complete the reaction was incubated at -20 ℃ 0.5h, a solution of n-butyl ether was added to the backup lithium bromide and zinc bromide, at 10 ℃ reaction was stirred 3h. Was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (3.48 g of) in toluene (10 mL) solution and heated to 80 deg.] C the reaction was stirred 6h, TLC analysis after completion of the reaction, was added 1mol / L dilute hydrochloric acid (7mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, concentrated, and n-heptane (5mL) and methanol (15mL) recrystallized 3.452g 3 of solid compound, yield: 77.65%. Purity: 99.45%. Melting point: 128.9 ~ 130.5 ℃. 1 the H-NMR (CDCl 3 ): [delta] 7.72 (IH, D), 7.64 (IH, D), 7.21-7.30 (4H, m), 7.04 (IH, T), 6.96 (IH, S), 5.40 ( 1H, t), 5.27 (2H , m), 4.36 (1H, d), 4.08-4.21 (4H, m), 3.82 (1H, dd), 1.19 (9H, s), 1.16 (9H, s), 1.11 (9H, s), 0.85 ( 9H, s).
Example 2, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added n-butyl ether (8mL), stirred and heated to 50 deg.] C 2h, cooling backup. Under nitrogen, was added 2- toluene (5mL) (5- iodo-2-fluorobenzyl) benzothiophene (2.21g) in n-butyl ether (5mL), cooled to – 50 ℃, was slowly added dropwise 2.5mol / L n-butyllithium hexane solution (2.64 mL), controlling the internal temperature does not exceed -30 deg.] C, 6h after the addition was complete the reaction was kept at -50 deg.] C, was added a solution of n-butyl ether in said auxiliary zinc bromide and lithium bromide, the reaction was stirred 8h at -20 ℃. Was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (6.954g) in toluene (12mL) solution, heated to 25 deg.] C the reaction was stirred 24h, after completion of the reaction by TLC, was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, concentrated, and n-heptane (5mL) and methanol (15mL) recrystallized 3.237g 3 of solid compound, yield: 72.81%. Purity: 99.36%.
Example 3, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc iodide (1.915g) and lithium iodide (0.803 g) in n-butyl ether was added (10mL), stirred and heated to 50 deg.] C 1.5h, cool reserve. Under nitrogen, was added 2- (5-iodo-2-fluorobenzyl) benzothiophene (2.21g) in toluene (9mL), n-butyl ether (3mL), cooled to -30 deg.] C, was slowly added dropwise 1.6mol / L n-hexyl lithium hexane solution (4.13mL), controlling the internal temperature does not exceed -20 ℃, n-butyl ether solution after the addition was complete the reaction was kept at -30 ℃ at 5h, zinc iodide was added to the backup and lithium iodide the mixture was stirred at 25 ℃ reaction 1h. After addition of 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (4.346g) in toluene (10 mL) solution, the reaction was heated to reflux for 145 ℃ 0.5h, TLC detection completion of the reaction , was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, concentrated, and n-heptane (5mL) and methanol (15mL) recrystallized 3.552 3 g of a solid compound in a yield of 79.9%. Purity: 99.41%.
Example 4, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added n-butyl ether (7mL), stirred and heated to 50 deg.] C 2h, cooling backup. Under nitrogen atmosphere, 2- (5-bromo-2-yl) benzothiophene (1.927g) was added toluene (6mL), n-butyl ether (4mL), cooled to -30 deg.] C, was slowly added dropwise 2.5mol / L n-butyllithium hexane solution (2.88 mL), controlling the internal temperature does not exceed -20 deg.] C, 3h after the addition was complete the reaction was kept at -30 deg.] C, was added a solution of n-butyl ether in said auxiliary zinc bromide and lithium bromide, the reaction was kept at -5 ℃ 4h, was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (4.346g) in toluene (7mL) solution, stirred and heated to 120 ℃ The reaction 4h, after completion of the reaction by TLC, was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, and concentrated under reduced pressure, n-heptane (5mL ) and methanol (15mL) recrystallized 2.783g solid compound 3, yield: 62.6%. Purity: 99.29%.
EXAMPLE 5, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added cyclopentyl ether (8mL), stirred and heated to 50 deg.] C 2h, cooling backup. Under nitrogen, was added 2- (5-iodo-2-fluorobenzyl) benzothiophene (2.21g) in toluene (6mL), cyclopentyl methyl ether (6mL), cooled to -30 deg.] C, was slowly added dropwise 1.6 mol / L hexane solution of n-hexyl lithium (4.5mL), controlling the internal temperature does not exceed -20 ℃, after the addition was complete the reaction was kept at -30 ℃ at 3h, added to the backup lithium bromide and zinc bromide cyclopentylmethyl the ether solution, the reaction incubated at -5 ℃ 4h, was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (4.346g) in toluene (8mL) solution, heated to 120 ℃ reaction was stirred 4h, after completion of the reaction by TLC, was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, and concentrated under reduced pressure, with n-heptane dioxane (5mL) and methanol (15mL) recrystallized 2.088g solid compound 3, yield: 47%. Purity: 99.3%.
6, (1S) -2,3,4,6- four -O- pivaloyl anhydro-1- [3- (1-thiophen-2-yl-methyl) -4 Example Preparation fluorophenyl] glucitol (compound 3) –
Zinc bromide (0.676 g) and lithium bromide (0.261 g) was added methyl t-butyl ether (8mL), was heated to 50 ℃ stirred 3h, cooling backup. Under nitrogen, was added 2- toluene (6mL), methyl t-butyl ether (4mL) (5- iodo-2-fluorobenzyl) benzothiophene (2.21g), cooled to -40 deg.] C, was slowly added dropwise 1.6 mol / L n-hexyl lithium hexane solution (3.94mL), controlling the internal temperature does not exceed -30 ℃, after the addition was complete the reaction was kept at -40 ℃ at 4h, was added to the lithium bromide and zinc bromide spare methyl tert-butyl ether solution, the reaction incubated at 5 ℃ 7H, was added 2,3,4,6-tetra -O- pivaloyl bromo -α-D- glucopyranose (3.48 g of) in toluene (8mL) solution, heated to 90 ℃ reaction was stirred 6h, after completion of the reaction by TLC, was added 1mol / L dilute hydrochloric acid (8mL), water (20 mL), the combined organic phase was washed with water, dried over anhydrous of Na 2 the SO 4 dried, and concentrated under reduced pressure, with n-heptane dioxane (5mL) and methanol (15mL) recrystallized 2.792g solid compound 3, yield: 62.8%. Purity: 99.44%.
Example 7, (1S) -1,5- anhydro-1- [3- (1-methyl-thiophen-2-yl) -4-fluorophenyl] -D-glucitol (Compound 2) preparation
Compound 3 (7.41g) was added methanol (35mL), was added sodium methoxide (2.161g), heated at reflux for 5H reaction, after completion of the reaction by TLC, concentrated and the residue was added methanol (10 mL), water (10 mL), acetic acid ( 3g), was added seed crystal (0.1g), stirred at 5 ℃ crystallization, filtration, the filter cake washed with cold (methanol: (5mL) was washed with 1) solvent to give an off-white solid 3.89g compound 2: water = 1 , yield: 96.2%. Purity: 99.29%. 1 the H-NMR (the CD 3 the OD): [delta] 7.70 (IH, D), 7.63 (IH, D), 7.43 (IH, dd), 7.34-7.38 (IH, m), 7.21-7.26 (2H, m) , 7.08 (1H, t), 7.01 (1H, s), 4.18-4.28 (2H, m), 4.12 (1H, d), 3.88 (1H, dd), 3.70 (1H, dd), 3.30-3.50 (4H , m).
Example 8, (1S) -1,5- anhydro-1- [3- (1-methyl-thiophen-2-yl) -4-fluorophenyl] -D-glucitol (Compound 2) preparatio
Methanol was added (15mL) of the compound 3 (7.41g) was added sodium hydroxide (2g) in water (10 mL) solution was heated to 50 deg.] C the reaction was stirred 10h, TLC detection after completion of the reaction, water (10mL), 2mol / L hydrochloric acid (2mL), stirred at room temperature for crystallization, white solid was suction filtered, the filter cake washed with water (5mL) was washed and dried to give 3.806g of compound 2, yield: 94.1%. Purity: 99.31%.
Preparation 9, Ignatius column eutectic net L- proline (Compound 1) Example
Net Ignatius column (compound 2) (4.04g) was added ethanol (25mL), was added L- proline (1.15 g of), the reaction was heated at reflux for 1h, cooled to room temperature, filtered, the filter cake washed with cold ethanol, and dried to give white solid 4.67g of compound 1. Yield: 90%. Purity: 99.51%. Melting point: 194.0 ~ 202.1 ℃. The MS-ESI (m / Z): 427.16 [the M + of Na] + . 1 the H-NMR (the CD 3 the OD): [delta] 7.75 (IH, D), 7.67 (IH, D), 7.45 (IH, dd), 7.37 (IH, m), 7.24-7.31 (2H, m), 7.10 (1H, t), 7.07 ( 1H, s), 4.23-4.32 (2H, m), 4.13 (1H, d), 3.98 (1H, t), 3.89 (1H, d), 3.71 (1H, dd),3.31-3.50 (5H, m), 3.21-3.27 (1H, m), 2.27-2.34 (1H, m), 2.09-2.17 (1H, m), 1.95-2.02 (2H, m).
Claims
Ignatius one kind of column and net synthesis process, comprising the steps of: (1), from 4-fluoro-3- (2-benzothienyl) methyl-5-phenyl-halide as a raw material, in an appropriate solvent 5 is reacted with an alkyl lithium, followed by reaction with the zinc salt prepared organozinc reagents – bis [4-fluoro-3- (2-benzothienyl) methyl phenyl] zinc, and then with 2,3,4,6-tetra -O- pivaloyl -α-D- glucopyranose 4-bromo nucleophilic substitution reaction of intermediate net Ignatius column 3; (2), compound 3 by an organic base off pivaloyl protecting group prepared net Ignatius column 2; wherein, in the 4-fluoro-3- (2-benzothienyl) methyl-5-phenyl halide of structure X is selected from bromo or iodo; synthesis route is as follows:
////////WO 2016173551, China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry, IPRAGLIFLOZIN, NEW PATENT,
Sacubitril, WO 2016180275, New patent, SUZHOU PENGXU PHARMATECH CO., LTD

Sacubitril, WO 2016180275, New patent, SUZHOU PENGXU PHARMATECH CO., LTD
AHU-377 INTERMEDIATES AND METHOD FOR PREPARING AHU-377 AND AHU-377 INTERMEDIATES PATENT
WO2016180275, new patent, SUZHOU PENGXU PHARMATECH CO., LTD. [CN/CN]; 3rd Floor Building 7, 2358 Chang An Road, Wujiang Suzhou, Jiangsu 215200 (CN)
WANG, Peng; (CN).
LI, Pixu; (CN).
GU, Xiangyong; (CN)

Heart failure is a very high mortality syndrome, for patients with heart failure, so far no drug can significantly improve mortality and morbidity, and thus a new type of therapy is necessary. AHU-377 (CAS No. 149709-62-6) is an enkephalinase inhibitor, which is a prodrug ester groups can be lost through hydrolysis, converted to pharmaceutically active LBQ657, inhibit endorphin enzyme (NEP) the role of the main biological effects of NEP is to natriuretic peptides, bradykinin and other vasoactive peptide degradation failure. AHU-377 and angiotensin valsartan composition according to the molar ratio of 1 LCZ696. LCZ696 is an angiotensin receptor enkephalinase inhibitors, which can lower blood pressure, treat heart failure may become a new drug. Clinical data show, LCZ696 is more effective for the treatment of hypertension than valsartan alone.
Patents US 5,217,996 and US 5,354,892 reported the first synthesis of AHU-377, the synthetic route is as follows:
Reaction with unnatural D-tyrosine derivative as a substrate, more expensive, while the second step in the synthesis is necessary to use Pd-catalyzed Suzuki coupling reaction, whereby preparative route costs than the AHU-377 high.
Patent US 8,115,016 above routes also reported the departure from the pyroglutamate, through multi-step process for preparing a reaction AHU-377, which is more difficult methylation reaction, and the yield is not high. Patent US 8,580,974 also reported a carbonyl group of the a- introducing N, N- dimethyl enamine is converted to methyl, however, there are some problems in the route for constructing methyl chiral centers, are not suitable for scale-up synthesis route as follows:
About the latest AHU377 synthesis intermediates, Patent WO2014032627A1 reported using a Grignard reagent to react with epichlorohydrin, a quicker been important intermediates, synthetic route Compound AHU377 synthesized as follows:
However, the second step of the synthetic route use succinimide nitrogen atoms introduced by Mitsunobu reaction with hydrochloric acid hydrolysis to remove, then converted to Boc protected at the end of the synthesis process AHU377 Boc will have to take off protection, then any connection with succinic anhydride reaction product introduced into the structure of succinic acid portion, so that this method of atom economy and the economy of the steps are low.
Example 1
Synthesis of Compound 2
In inert atmosphere, a solution of three 500mL flask was added compound 1 (10g, 1eq), dissolved after 90mL THF, was added CuI (4.814g, 0.1eq), the system moves to the low temperature in the cooling bath to -20 ℃ when, biphenyl magnesium bromide dropwise addition, the internal temperature was controlled not higher than -10 ℃. Bi closed refrigeration drop, return to room temperature overnight. Completion of the reaction, the reaction solution was poured into saturated the NH 4 of Cl (10vol, 100 mL) was stirred at room temperature for 0.5h. Suction filtered, the filter cake was rinsed with a small amount of EA, and the filtrate was transferred to a separatory funnel carved, and the aqueous phase was extracted with EA (10vol × 2,100mL × 2) and the combined organic phases with saturated NaHC [theta] 3 , the NH 4 of Cl, each Brine 150mL (15vol) washed once, dried over anhydrous over MgSO 4 dried, suction filtered, and concentrated to give a white solid. Product obtained was purified by column 15.2g, yield 78%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.52 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.38-7.25 (m, 8H), 4.62-4.47 ( m, 2H), 4.09 (dd, J = 6.7,3.5Hz, 1H), 3.54 (dd, J = 9.5,3.5Hz, 1H), 3.43 (dd, J = 9.4 , 6.9Hz, 1H), 2.84 ( d, J = 6.6Hz, 2H), 2.38 (s, 1H).
Example 2
Synthesis of Compound 3
In an inert gas, at room temperature was added to the flask 500mL three Ph3P (18.54g, 2eq), 240mL DCM dissolution, butyryl diimide (of 6.44 g), compound 2 (15g), an ice-water bath cooling to 0 ℃ or so, was added dropwise DIAD (14mL) was complete, the reaction go to room temperature.Starting material the reaction was complete, the system was added to water (100 mL) quenched the reaction was stirred for 10min; liquid separation, the aqueous phase was extracted with DCM (100mL × 2), the combined organic phases with saturated Brine 100mL × 2), dried over anhydrous over MgSO 4 dried , filtration, spin dry to give a white solid; product was purified by column 15.4g, yield 82%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.56 (D, J = 7.4Hz, 2H), 7.49 (D, J = 8.0Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.37-7.30 (m, 3H), 7.27 ( d, J = 6.7Hz, 3H), 7.22 (d, J = 8.0Hz, 2H), 4.75 (s, 1H), 4.56 (d, J = 12.0Hz, 1H), 4.45 (d, J = 12.0Hz, 1H ), 4.06 (t, J = 9.6Hz, 1H), 3.70 (dd, J = 10.0,5.2Hz, 1H), 3.23 (dd, J = 13.8,10.3Hz, 1H) , 3.14-3.00 (m, 1H), 2.48 (d, J = 4.0Hz.4H).
Example 3
Synthesis of Compound 4
Protection of inert gas, at room temperature was added to the flask 1L three compound 3 (18.81g), 470mL EtOH was dissolved, was added Pd / C, replaced the H 2 three times, move heated on an oil bath at 60 ℃ reaction. Raw reaction was complete, the system was removed from the oil bath, the reaction solution was suction filtered through Celite and concentrated to give the crude product. It was purified by column pure 11.8g, a yield of 81.2%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.8Hz, 2H), 7.51 (D, J = 7.8Hz, 2H), 7.42 (T, J = 7.5Hz, 2H), 7.33 (T , J = 7.2Hz, 1H), 7.26 (d, J = 7.2Hz, 2H), 4.55 (d, J = 5.2Hz, 1H), 4.06-3.97 (m, 1H), 3.86 (dd, J = 12.0, 3.1Hz, 1H), 3.16 (dd , J = 8.1,2.9Hz, 2H), 2.58 (t, J = 7.0Hz, 4H), 1.26 (s, 2H).
Example 4
Synthesis of Compound 7
Protection of inert gas, at room temperature to a 25mL flask was added three Dess-Martin oxidant (767.7mg), 10mL DCM was dissolved, the system was cooled down to -10 deg.] C, was added 4 (500mg). Starting material the reaction was complete, to the system was added saturated NaHCO3 and Na2S2O3 each 5mL, quench the reaction stirred for 10min; aqueous phase was extracted with DCM (10mL × 3) and the combined organic phases with saturated NaHCO3, Brine 30mL each wash, dried over anhydrous MgSO4, filtration, spin dried to give the crude product used directly in the next reaction cast.
Example 5
Synthesis of Compound 8
Inert gas, at room temperature for three to 500mL flask 7 (497.5mg), 10mL DCM to dissolve an ice water bath to cool, added phosphorus ylide reagent (880.6mg), the system was removed from the ice water bath at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. Liquid separation, the aqueous phase was extracted with DCM (10mL × 2), organic phases were combined, washed with saturated Brine 20mL × 2, dried over anhydrous MgSO4, filtration, spin crude done. Product obtained was purified by column 563mg, 90% yield.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.60-7.53 (m, 2H), 7.51 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.33 (D, J = 7.3Hz, 1H), 7.23 (d , J = 8.1Hz, 2H), 7.13 (dd, J = 9.2,1.5Hz, 1H), 5.26 (td, J = 9.5,6.9Hz, 1H), 4.25-4.05 ( m, 2H), 3.40 (dd , J = 13.7,9.7Hz, 1H), 3.13 (dd, J = 13.8,6.7Hz, 1H), 2.53 (d, J = 2.2Hz, 4H), 1.85 (d, J = 1.4Hz, 3H), 1.30 ( t, J = 7.1Hz, 3H).
Example 6
Synthesis of Compound 9
Protection of inert gas, at room temperature to a 50mL flask was added three 8 (365mg, 1eq), 9mL of ethanol and stirred to dissolve, the system was replaced with hydrogen three times, was added Pd / C (25% w / w) at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. The reaction mixture was suction filtered through Celite and concentrated to give the crude product. Product was purified by column, yield 80.2%, purity 97.2%.
Example 7
Synthesis of Compound 10
Equipped with Compound 9 (100mg) acetic acid A reaction flask (9mL), hydrochloric acid (1mL). The reaction was heated oil bath at 80 deg.] C. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. After saturated NaHCO3 and extracted with EA and concentrated to give crude product. Product obtained was purified by column 90mg, yield 84%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.54 (m, 2H), 7.53-7.48 (m, 2H), 7.41 (dd, J = 10.5,4.9Hz, 2H), 7.31 (dd, J = 8.3 , 6.4Hz, 1H), 7.22 ( d, J = 8.2Hz, 2H), 5.93 (t, J = 9.7Hz, 1H), 4.34-4.00 (m, 3H), 2.91-2.71 (m, 2H), 2.68 -2.57 (m, 2H), 2.55 (ddd, J = 9.4,7.0,4.3Hz, 1H), 2.42 (dt, J = 13.3,6.8Hz, 2H), 1.97-1.74 (m, 1H), 1.64-1.46 (m, 1H), 1.23 ( td, J = 7.1,3.3Hz, 3H), 1.14 (dd, J = 7.1,3.9Hz, 3H)
Example 8
Synthesis of Compound 5
Example 8-1: The reaction flask was added compound 4 (1eq) was added water (2VOL), concentrated hydrochloric acid (2VOL), 110 ℃ reaction was heated in an oil bath overnight, complete conversion of starting material, the HPLC peak area 97%. 10% NaOH solution was added to adjust the pH to about 10, filtration products. Yield 85%.
Example 8-2: The reaction flask was added compound 4 (1eq) was added ethanol (5 vol), water (5 vol), potassium hydroxide (8 eq), was heated in an oil bath overnight at 110 ℃ reaction, complete conversion of the starting material, the HPLC peak area 99%. Water was added (5Vol), filtered to obtain the product. Yield 95%. Product was dissolved in toluene, was added ethanolic hydrochloric acid, the precipitated hydrochloride Compound 5.
NMR data for the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.31 (S, 3H), 7.70-7.61 (m, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.31 (m, 3H), 4.09 (the dq- , J = 42.6,7.1Hz, 1H), 3.62-3.51 (m, 1H), 3.50-3.41 (m, 1H), 3.11-3.00 (m, 1H), 2.95-2.84 (m, 1H), 1.30-1.10 (m, 1H).
EXAMPLE 9
Synthesis of Compound 6
To the reactor was added compound 5, was added absolute ethanol (3vol). Temperature of the outer set 30 ℃ heating, stirring was continued after the temperature reached 25 ℃ 20min. Was added 30% NaOH aqueous solution (1.1eq). External temperature 65 ℃ heating provided, after the internal temperature reached 60 deg.] C was slowly added (of Boc) 2 O (1.1 eq). Stirring 0.5h, reaction monitoring. After completion of the reaction, water was added slowly dropwise (8vol), turn off the heating and natural cooling. The system temperature was lowered to 25 deg.] C and continue stirring for 2h. Filter cake at 50 ℃ blast oven drying to obtain the product.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Synthesis of Intermediate Compound 6 to Compound 10, i.e., the AHU-377, a synthetic route in the background of the present invention, the cited patent application WO2014032627A1 loaded in detail, not in this repeat.
Example 10
Synthesis of Compound 2
Benzyl glycidyl ether preparation (50g) in THF (200mL) was added. Under inert gas protection, the biphenyl magnesium bromide (365mmol) was added to THF (1020mL) was added the reaction flask is placed in a low temperature bath -40 ℃ cooling. Cuprous iodide (O.leq) when the internal temperature dropped to -9 ℃. Continued to decrease the temperature of -23 ℃ dropwise addition of benzyl glycidyl ether in THF was added dropwise to control the internal temperature process of not higher than -15 deg.] C, 47 min when used, the addition was completed the cooling off the reaction was stirred overnight. The cooling system to -20 ℃ quenched with 1N HCl aqueous solution, <10 ℃ Go stirred 30min at room temperature. Liquid separation, the aqueous phase was extracted with THF, the combined THF phases. Respectively saturated ammonium chloride (250mL), saturated brine (250mL) washed. Rotary evaporation to remove THF, and water (200 mL) Continue rotary evaporation 1h, cool to precipitate a solid. Suction crude. Crude n-heptane was added 2Vol beating, suction filtration to obtain the product in a yield of 90 ~ 95%, HPLC peak area 94%. In another column purification was pure, columned yield 88.6%, HPLC 99.1%.
Example 11
Synthesis of Compound 3
Preparation Example 9, said compound taking the embodiment 2 (5g) added to the reaction flask, the reaction flask was added toluene (80mL), phthalimide (2.55 g of) and triphenylphosphine (5.35g of), the nitrogen was replaced protection. An ice-salt bath cooling to -5 deg.] C, was added dropwise DIAD (4.12g), dropwise addition was exothermic, the temperature was raised to 5 ℃. The reaction was continued 1h sampling HPLC test material substantially complete reaction. Join 12g silica spin column done to collect the product (including DIEA derivative).
Example 12
Synthesis of Compound 11
Compound 3 (3g) was added to the reaction flask embodiment taken in Preparation Example 10, was added ethanol (30 mL), with stirring. Was added hydrazine hydrate (2g) was heated in an oil bath reflux 1h, when supplemented with 20mL ethanol was stirred difficulties, the reaction was continued to 2.5h, HPLC showed the starting material the reaction was complete. Add EA / H2O 100mL each liquid separation, the EA phase was washed with water (100mL) and the combined organic phases were washed with water (100mL) and saturated brine (100mL) washed. Anhydrous magnesium sulfate and filtered spin column was done product 1.88g, yield 88%, HPLC 94%.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 7.64 (D, J = 7.2Hz, 2H), 7.57 (D, J = 8.1Hz, 2H), 7.45 (T, J = 7.6Hz, 2H), 7.39-7.32 ( m, 5H), 7.29 (d , J = 8.1Hz, 3H), 4.55-4.43 (m, 2H), 3.38-3.23 (m, 3H), 3.18-3.10 (m, 1H), 2.82-2.74 (m, 1H), 2.61-2.52 (m, 1H ).
Example 13
Synthesis of Compound 11
To the toluene solution of the compound 2 was added phthalimide (1.1 eq), triphenylphosphine (1.3 eq) with stirring. External bath set -10 ℃, to cool the system, the internal temperature dropped to 0 ~ 5 ℃, start dropping DIAD (1.3eq), control the internal temperature -5 ~ 5 ℃. Completion of the dropwise addition, the cooling bath was turned off outside the reaction was stirred at room temperature. The reaction was stirred for 1 to 4 hours. The reaction solution to give compound 3, administered directly in the next reaction. To the above reaction mixture was added hydrazine hydrate (6 eq), heated to 70 ~ 80 ℃, to complete the reaction, filtered hot, the filtrate. Aqueous sodium hydroxide solution (20vol 10%) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. Water was added (20vol) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. The toluene phase was added hydrochloric acid (20vol, 3N), stirred for 0.5h, to form a solid precipitate. Filtration and drying to obtain a product, i.e. compound 11, the hydrochloride salt, yield 60% in two steps.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.46 (S, 3H), 7.63 (dd, J = 16.4,7.7Hz, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.22 (m, 8H ), 4.56 (d, J = 12.1Hz, 1H), 4.48 (d, J = 12.1Hz, 1H), 3.58 (d, J = 7.9Hz, 2H), 3.47 (dd, J = 10.9,6.3Hz, 1H ), 3.11 (dd, J = 13.5,4.9Hz, 1H), 2.92 (dd, J = 13.4,9.1Hz, 1H).
Example 14
Synthesis of Compound 12
Weigh Compound 11 (1.38g) was added to the reaction flask. To the reaction flask plus DCM (14ml) and Et3N (462mg, 0.73ml). Weighed (of Boc) 2O (1.23 g of) was added to DCM (5ml) was dissolved. Room temperature (8 ℃), a solution (of Boc) 2 DCM solution O was added dropwise to the reaction, (2ml) rinsed with DCM. The reaction mixture was stirred at room temperature, detected by HPLC, the reaction ends 4h. Reaction mixture was washed (15ml) 3 times with Brine (15ml) The reaction solution was washed 1 times. Inorganic sulfate, concentrated and purified by column PE:EA = 15:1 give product 560mg, yield 30.8%, HPLC 99.92%.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.49 (D, J = 7.4Hz, 2H), 7.43 (T, J = 7.3Hz, 2H), 7.39-7.28 (m, 5H), 7.24 ( d, J = 9.0Hz, 3H), 5.00-4.80 (br, 1H), 4.51 (q, J = 11.8Hz, 2H), 4.08-3.85 (br, 1H), 3.43 ( d, J = 2.9Hz, 2H) , 3.02-2.77 (m, 2H), 1.42 (s, 9H).
Example 15
Synthesis of Compound 6
Weigh Compound 12 (250mg) and methanol (9ml) was added to the reaction flask. Added Pd / C (138mg, 1 / 4w / w, water content 55%). The H 2replaced 3 times, 50 ℃ stirred and heated. HPLC detection reaction, the reaction end 30h. Filtered off Pd / C, 40 ℃ concentrated under reduced pressure to remove methanol. PE:EA = 3:1 florisil column to give the product 196mg, 100% yield, 99.34% purity.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Method for preparing the AHU-377, characterized by comprising the steps of: (a) Compound (1) S- benzyl glycidyl ether and biphenyl Grignard reagent produced by the reaction of the compound (2) in an organic solvent; ( b) compound (2) with a succinimide or phthalimide Mitsunobu reaction occurs in an organic solvent to form a compound (3); (C) compound (3) in an organic solvent in the role of a catalyst under removal debenzylation protected form compound (4); (D) compound (4) with an oxidizing agent oxidation reaction occurs in an organic solvent to form a compound (7); (E) compound (7) with a phosphorus ylide reagent in an organic solvent to give the compound (8); (F.) compound (8) in an organic solvent in the selective catalytic hydrogenation of the compound (9); and (g) of the compound (9) in an organic solvent in the hydrolysis reaction of the amide compound occurs in the presence of an acid ( 10), i.e., AHU-377;
SUVEN LIFE SCIENCES LTD, WO 2016178064, POLYMORPH OF NINTEDANIB ETHANESULPHONATE, NEW PATENT

NINTEDANIB ETHANESULPHONATE
NEW PATENT
WO2016178064, CLICK FOR PATENT
POLYMORPH OF NINTEDANIB ETHANESULPHONATE, PROCESSES AND INTERMEDIATES THEREOF
SUVEN LIFE SCIENCES LIMITED [IN/IN]; 5th floor, Serene Chamber, Road No.5, Off Avenue No. 7, Banjara Hills, Telangana Hyderabad 500034 (IN)
ARAVA, Veera Reddy; (IN).
GOGIREDDY, Surendra Reddy; (IN).
JASTI, Venkateswarlu; (IN)
DR VEERA ARAVA REDDY

Vice President
Suven Life Sciences Limited, Hyderabad · Research and Development

Surendra Reddy Gogireddy, Sr.Research Associate

JASTI, Venkateswarlu
The present invention provides novel crystalline Form of Nintedanib and process for its preparation. The present invention also provides to a novel process for the preparation of Nintedanib. The present invention further provides to novel intermediates used in the preparation of Nintedanib and process for their preparation.
Nintedanib inhibits multiple receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (nRTKs).The chemical name of Nintedanib is lH-Indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl-(4-methyl-l-iperazinyl)acetyl]amino]phenyl]amino]phenylmethylene] -2-oxo-,methylester, (3Z)-, ethanesulfonate (1 : 1) and is structurally represented by compound of Formula I.
Formula I
Nintedanib is marketed in the United States under the trade name OFEV and is indicated for the treatment of Idiopathic Pulmonary Fibrosis (IPF).
Nintedanib was first described and claimed in U.S. Pat.No. 6,762, 180 and EP 1224 170. These patents disclose a process for the preparation of Nintedanib as depicted in scheme I given below:

U.S.Pat.No. 8,067,617 discloses a process for the preparation of Nintedanib intermediate Enolindole derivative), which is shown in the scheme-II given below:

Scheme-II
U.S.Pat. No. 7,119,093 discloses Nintedanib monoethanesulphonate in crystalline form characterised by X-ray powder diffraction pattern having 2Θ values at 7.70, 8.78, 9.47, 9.82, 11.59, 11.93, 13.15, 13.69, 14.17, 16.32, 16.72, 16.92, 17.43, 17.77, 18.58, 18.81, 19.03, 19.73, 19.87, 20.03, 20.61, 20.83, 21.26, 21.76, 22.05, 22.19, 22.57, 23.10, 23.81, 24.69, 24.78, 24.91, 25.42, 26.24, 26.91, 27.19, 27.61, 27.95, 28.71, 29.25.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, X-ray diffraction pattern, infrared absorption fingerprint and solid state NMR spectrum. One polymorph may give rise to thermal behaviour different from that of another polymorph. Thermal behaviour can be measured in the laboratory by such techniques as capillary melting point, thermo gravimetric analysis (“TGA”) and differential scanning calorimetry (“DSC”), which have been used to distinguish polymorphic forms.
The differences in the physical properties of different polymorphs results from the orientation and intermolecular interactions of adjacent molecules or complexes in the bulk solid. Accordingly, polymorphs are distinct solids sharing the same molecular Formula yet having distinct advantageous physical properties compared to other polymorphs of the same composition or complex. Hence there remains a need for polymorphic forms which have properties suitable for pharmaceutical processing on a commercial scale.
Considering the importance of Nintedanib, there exists a need to develop an alternate and improved process for the preparation of Nintedanib with better yield. Further, the process involved should be simple, convenient and cost-effective for large scale production. The inventors of the present invention during their continuous efforts also developed a novel high melting stable polymorphic form of Nintedanib ethanesulfonate.
EXAMPLES
Example 1: Process for the preparation of Nintedanib Monoethane Sulfonate:
Step-1: Preparation of methyl-3-(hydroxy(phenyl)methylene)-2-oxoindoline-6-carboxylate: To the suspension of methyl 2-oxoindoline-6-carboxylate (50 gm, 0.261 mol) in IPA (350 ml) was added slowly SMO-powder (33.8 gm, 0.626 mol) and stirred for about 15 min. Benzyl chloride (44 g, 0.313 mol) was added after completion of the reaction at a reaction temperature of -5 to -10°C for about 5hrs. The reaction mixture was quenched into ice-water (700 ml) and acidified with Cone. HC1 (2.0-2.5 ml). Filtered the reaction mixture, washed with water (2X100 ml) and dried the precipitate to obtain crude product which can be recrystallized from acetonitrile (28 ml) to obtain methyl-3-(hydroxy(phenyl)methylene)-2- oxoindoline-6-carboxylate pure crystalline solid (32 gm) (61%) (HPLC purity >97%). The filtrate was evaporated in vacuum to give unreacted methyl 2-oxoindoline-6-carboxylate. MR: 216-223°C; IR (KBr, cm“1): 3178, 1711, 1651; 1H-NMR (400 MHz, DMSO): δ 3.80 (s, 3H), 7.17 (s, 1H), 7.28-7.31 (m, 2H), 7.46-7.50 (m, 3H), 7.72 (d, 2H, J = 6.0 Hz), 9.52 (s, 1H), 11.53 (s, 1H); 13C-NMR (100 MHz, DMSO): δ 22.12, 52.41, 101.13, 111.13, 119.23, 123.06, 126.65, 127.06, 128.65, 129.21, 132.26, 134.47, 136.99, 166.58, 172.52 and 175.80; MS: m/z 294 [M]“1
Step-2: Preparation of methyl-3-(acetoxy(phenyl)methylene)-l-acetyl-2-oxoindoline-6-carboxylate (Acetyl derivative):
To the suspension of methyl-3-(hydroxy(phenyl)methylene)-2-oxoindoline-6-carboxylate (45 gm, 0.1512 mol) in acetic anhydride (300 ml) was added pyridine (4.5g) slowly (drop-wise) and stirred the reaction at temperature of 0-5°C for about30 min. After completion of the reaction raised the temperature of the reaction mass to 75-80°C and stirred for about lhr. Cooled the reaction mass and stirred for about 30 min at 25-28°C, filtered, washed with hexane (100ml) and dried the precipitate to obtain methyl-3-(acetoxy(phenyl)methylene)-l-acetyl-2-oxoindoline-6-carboxylate.
MR: 226-229°C; IR (KBr, cm“1): 3413, 1771, 1743, 1717, 1640; 1H-NMR (400 MHz, CDC13): δ 2.38 (s, 3H), 2.62 (s, 3H), 3.92 (s, 3H), 7.44 (m, 3H), 7.62 (d, 2H, J = 7.004 Hz), 7.68 (d, 1H, J = 8.12 Hz), 7.91 (d, 1H, J = 8.0 Hz), 8.90 (s, 1H); 13C-NMR (100 MHz, CDC13): δ 21.08, 21.38, 26.96, 52.25, 52.34, 115.17, 117.18, 121.33, 122.77, 125.82, 126.19, 126.56, 128.15, 128.87, 129.27, 129.34, 130.81, 130.90, 131.47, 131.82, 132.80, 138.55, 160.85, 165.95, 166.38, 166.42, 167.01, 170.67 and 170.76; MS: m/z 380 [M]+1.
Step-3: Preparation of methyl- l-acetyl-3-(((4-(2-chloro-N-methylacetamido)phenyl)amino) (phenyl)methylene)-2-oxoindoline-6-carboxylate) (Chloroacetyl derivative) :
Suspension of methyl-3-(acetoxy(phenyl)methylene)- l-acetyl-2-oxoindoline-6-carboxylate (Acetyl derivative) (49gm, 0.129mol) and N-(4-aminophenyl)-2-chloro-N-methylacetamide(25.66gm, 0.129 mol) in a mixture of methanol (350 ml) and DMF (88 ml) was heated to 60-65°C stirred for about 12hr at the same temperature. After completion of the reaction cooled the reaction mass to room temperature and stirred for about 30min. Filtered the reaction mixture, washed with methanol (2X50ml) and dried the precipitate to obtainmethyl-l-acetyl-3-(((4-(2-chloro-N-ethylacetamido)phenyl)amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate).
MR: 247-250°C; IR (KBr, cm“1): 3432, 1712, 1675, 1591; 1H-NMR (400 MHz, DMSO): δ 2.74 (s, 3H), 3.11 (s, 3H), 3.78(s, 3H), 3.87 (s, 2H), 5.75 (d, 1H, J = 8.08 Hz), 7.01 (d, 2H, J = 7.96 Hz), 7.22 (d, 2H, J = 6.08Hz), 7.36 (d, 1H, J = 8.48 Hz), 7.46 (d, 2H, J = 7.24 Hz), 7.54-7.64 (m, 3H), 8.74 (s, 1H0, 11.92 (s, 1H), 13C-NMR (100MHz, DMSO): δ 27.17, 37.76, 42.48, 52.40, 96.38, 116.17, 117.59, 124.80, 125.33, 125.68, 128.09, 129.00, 130.10, 131.35, 131.97, 134.05, 160.93, 165.63, 166.68, 168.49 and 171.28; MS: m/z 518 [M]+1 and 520 [M]+1.
Step-4: Preparation of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-lyl)acetamide) phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate (Nintedanib free base):
Suspension of methyl- l-acetyl-3-(((4-(2-chloro-N-methylacetamido)phenyl)amino) (phenyl)methylene)-2-oxoindoline-6-carboxylate)(40 gm, 0.077ml) and N-methylpiperidine (23.24 gm, 0.232 mol) in a mixture of DMF (160 ml) was heated to a reaction temperature of 45-50°C for about l-2hrs. The reaction mixture was quenched into ice-water (1.6 Lt) and stirred for about lhr at 15-20°C. Filtered the reaction mixture mass washed with water and dried the precipitate to obtain crystalline crude solid (36 gm). Purified with acetonitrile to obtain Nintedanib free base (34 gm) as a yellow crystals (93.74%) (HPLC purity: >98%). MR: 240-246°C; IR (KBr, cm“1): 3559, 3455, 2940, 2810, 1711, 1657; 1H-NMR (400 MHz, DMSO): δ 2.09 (s, 3H), 2.17 (s, 8H), 2.68 (s, 2H), 3.05 (s, 3H), 3.76 (s, 3H), 5.80 (d, 1H, J = 7.56 Hz), 6.86 (d, 2H, J = 6.72 Hz), 7.11 (d, 1H, J = 6.48 Hz), 7.17 (d, 2H, J = 7.68 Hz), 7.42-7.57 (m, 6H), 10.98 (s, 1H), 12.23 (s, 1H) ; 13C-NMR (100MHz, DMSO): δ 37.17, 46.18, 52.24, 52.79, 55.05, 59.68, 98.10, 109.94, 117.75, 121.96, 124.29, 124.52, 128.06, 128.90, 129.40, 129.92, 130.91, 132.50, 136.72, 140.66, 158.81, 166.84, 169.04 and 170.66; MS: m/z 540 [M]+1.
Step-5: Preparation of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-lyl)acetamide) phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate ethane sulfonate salt:
Suspension of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-l-yl)acetamide)phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate (36 gm, 0.066 mol) in methanol (237 ml) and water (2.88 ml)was heated to 60-65°C and aq. ethane sulfonic acid was added to the reaction mixture. The resulting solution was cooled to 50°C, seeds diluted with isopropanol (237 ml) was added. The reaction mixture was cooled at 0°C for lhr. Filtered the precipitate, washed with mixture of methanol and isopropanol (50 ml), dried to obtain crude Nintedanib monoethane sulfonate (36.6 gm) and crystallized from methanol (5 Vol) to
obtained pure Nintedanib monoethane sulfonate salt as yellow crystals (33 gm) (80%) (HPLC purity >99%).
DSC: 298°C; IR (KBr, cm-1): 3321, 3273, 1710, 1652, 1615, 1515, 1435, 1378, 1289, 1209, 1161, 1087; 1H-NMR (400 MHz, DMSO): δ 1.08 (t, 3H, J = 7.31 Hz), 2.41-2.47 (q, 2H), 2.50-3.16 (broad m, 13H), 3.37 (s, 3H), 3.76 (s, 3H), 5.82 (d, 1H, J = 7.88Hz), 6.87 (d, 2H, J = 7.36 Hz), 7.14-7.20 (m, 3H), 7.49 (s, 1H), 7.49 (d, 2H, J = 6.68 Hz), 7.56-7.63 (m, 3H), 9.45 (s, 1H), 10.99 (s, 1H), 12.25 (s, 1H), 13C-NMR (100 MHz, DMSO): δ 37.15, 42.79, 45.65, 49.40, 52.26, 53.10, 58.04, 98.25, 110.01, 117.78, 121.97, 124.32, 124.59, 128.27, 128.90, 129.36, 130.00, 131.00, 132.52, 136.79, 137.93, 140.00, 158.66, 166.85, 168.47 and 170.65; MS: m/z 540[M]+1.
Example 2: Process for the preparation of Polymorph Form S of Nintedanib monoethanesulf onate :
Crude Nintedanib monoethane sulfonate was dissolved in methanol and heated to 60-64°C for about 15 min. After completion of the reaction cooled to room temperature for about lhr. Filtered the precipitate, washed with mixture of methanol (20ml) and isopropanol (30 ml) and dried to obtain pure crystalline solid (28 gm) (yield: 93.3%) with HPLC purity 99.72% and individual impurities 0.09%, 0.02% and 0.04%.
SUVEN, Chief executive and chairman Venkat Jasti



//////////WO2016178064, POLYMORPH, NINTEDANIB ETHANESULPHONATE, PROCESSES, INTERMEDIATES, suven, new patent
Doxercalciferol, доксэркальциферол , دوكساركالسيفيرول , 度骨化醇

Doxercalciferol
- Molecular FormulaC28H44O2
- Average mass412.648
доксэркальциферол [Russian]
دوكساركالسيفيرول [Arabic]
度骨化醇 [Chinese]
1,3-Cyclohexanediol, 4-methylene-5-[(2E)-2-[(1R,3aS,7aR)-octahydro-7a-methyl-1-[(1R,2E,4R)-1,4,5-trimethyl-2-hexen-1-yl]-4H-inden-4-ylidene]ethylidene]-, (1R,3S,5Z)-
54573-75-0
|
Title: Doxercalciferol CAS Registry Number: 54573-75-0 CAS Name: (1a,3b,5Z,7E,22E)-9,10-Secoergosta-5,7,10(19),22-tetraene-1,3-diol Additional Names: 1a-hydroxyvitamin D2; 1-hydroxyergocalciferol Trademarks: Hectorol (Bone Care) Molecular Formula: C28H44O2 Molecular Weight: 412.65 Percent Composition: C 81.50%, H 10.75%, O 7.75% Literature References: Synthetic vitamin D prohormone. Prepn: H.-Y. P. Lam et al., Science 186, 1038 (1974); eidem, Steroids30, 671 (1977); H. E. Paaren et al., J. Org. Chem. 45, 3253 (1980). Comparative activity and toxicity: G. Sjöden et al., Proc. Soc. Exp. Biol. Med. 178, 432 (1985). Metabolism to bioactive form: J. C. Knutson et al., Endocrinology 136, 4749 (1995). Pharmacology: J. W. Coburn et al., Nephrol. Dial. Transplant. 11, Suppl. 3, 153 (1996). Clinical trial for suppression of secondary hyperparathyroidism in hemodialysis: J. M. Frazao et al., ibid. 13, Suppl. 3, 68 (1998). Properties: Crystals, mp 138-140°. uv max (ethanol): 265 nm (e 18300). LD50 orally in rats: 3.5-6.5 mg/kg (Sjöden). Melting point: mp 138-140° Absorption maximum: uv max (ethanol): 265 nm (e 18300) Toxicity data: LD50 orally in rats: 3.5-6.5 mg/kg (Sjöden) Therap-Cat: Antihyperparathyroid. Keywords: Antihyperparathyroid. |

CLIP

Doxercalciferol (1α-hydroxyvitamin D2) is a commercially approved vitamin D derivative used to treat chronic kidney disease (CKD) patients whose kidneys cannot metabolically introduce a hydroxyl group at C1. A new process for the production of doxercalciferol from ergocalciferol was developed using a continuous photoisomerization of a known vitamin D intermediate as the key step, thus circumventing the limitations of batch photoisomerization processes. Doxercalciferol is produced in an overall yield of about 10% from ergocalciferol.
Doxercalciferol
1H NMR (CDCl3) δ 6.40 (d, 1H, J = 11.2), 6.04 (d, 1H, J = 11.2), 5.35 (s, 1H), 5.15–5.29 (m, 2H), 5.03 (s, 1H), 4.45 (dd, 1H, J = 7.3, 4.0), 4.21–4.31 (m, 1H), 2.81–2.90 (m, 1H), 2.62 (d, 1H, J = 13.3), 2.34 (dd, 1H, J = 13.3, 6.5), 1.83–2.11(m, 6H), 1.42–1.79 (m, 7H), 1.21–1.40 (m, 3H), 1.04 (d, 3H, J = 6.6), 0.94 (d, 3H, J = 6.8), 0.86 (t, 6H, J = 7.3), 0.58 (s, 3H) ppm.

Doxercalciferol (trade name Hectorol) is drug for secondary hyperparathyroidism and metabolic bone disease.[1] It is a synthetic analog of ergocalciferol (vitamin D2). It suppresses parathyroid synthesis and secretion.[2]
PATENT


CLIP

References
 |
|
| Names | |
|---|---|
| IUPAC name
(1S,3R,5Z,7E,22E)-9,10-Secoergosta-5,7,10,22-tetraene-1,3-diol |
|
| Other names
1-Hydroxyergocalciferol; 1-Hydroxyvitamin D2; 1α-Hydroxyergocalciferol; 1α-Hydroxyvitamin D2; Hectorol; TSA 840 |
|
| Identifiers | |
| 54573-75-0 |
|
| 3D model (Jmol) | Interactive image |
| ChEMBL | ChEMBL1200810 |
| ChemSpider | 4444554 |
| DrugBank | DB06410 |
| ECHA InfoCard | 100.170.997 |
| 2790 | |
| PubChem | 5281107 |
| UNII | 3DIZ9LF5Y9 |
| Properties | |
| C28H44O2 | |
| Molar mass | 412.66 g·mol−1 |
| Pharmacology | |
| H05BX03 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
|
/////////////
LUPIN LIMITED, WO 2016181313, NEW PATENT, SOFOSBUVIR
![]()
WO2016181313, A PROCESS FOR THE PREPARATION OF SOFOSBUVIR INTERMEDIATES & ITS POLYMORPH
| LUPIN LIMITED [IN/IN]; Kalpataru Inspire 3rd Floor, Off Western Express Highway Santacruz (East) Mumbai 400 055 (IN) |
SINGH, Girij, Pal; (IN).
SRIVASTAVA, Dhananjai; (IN).
MEHARE, Kishor, Gulabrao; (IN).
MALIK, Vineet; (IN).
DEOKAR, Sharad, Chandrabhan; (IN).
DANGE, Abhijeet, Avinash; (IN)


SUCCESS QUOTIENT: Lupin chairman DB Gupta (sitting) with managing director Kamal K Sharma (centre), directors Vinita Gupta (right) and Nilesh Gupta.
The present invention provides a novel process for preparation N-[(2,3,4,5,6- Pentafluorophenoxy)phenoxyphosphinyl]-L-alanine 1-methylethyl ester (formula 2) and resolving the formula 2 in the presence base to form N-[(S)-(2,3,4,5,6- Pentafluorophenoxy)phenoxyphosphinyl]-L-alanine 1-methylethyl ester (formula 2′).
Sofosbuvir is chemically named as (S)-isopropyl 2-((S)-(((2R,3R,4R,5R)-5-(2,4- dioxo3,4-dihydropyrimidin-l(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran- 2yl)methoxy)-(phenoxy)phosphorylamino)propanoate and is represented by the following chemical structure:

Formula 1
PCT publications WO2011123645 and WO2010135569 describes process for preparation of compound of formula 2′ by reacting isopropyl (chloro(phenoxy)phosphoryl)-L-alaninate and pentaflurophenol in the presence of base.

Formula 2′
Example-1:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium hydride
10.2g of sodium hydride was dissolved in 100 ml anhydrous THF. This solution was slowly added to a solution of pentafluorophenol (50g) in THF (100ml), Reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained solid was dried under vacuum at 45-50°C (yield=55g, confirmed by IR)
Example-2:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium methoxide
2,3,4,5, 6-pentafluorophenol (lOg) was dissolved in methanol (100ml), solution was cooled to 5-10°C. To this was added a solution of sodium methoxide in methanol. The reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained residue was striped with toluene. Obtained solid was dried under vacuum at 45-50°C (yield=l lg)
Example 3:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium hydroxide
2,3,4,5, 6-pentafluorophenol (lOOg) dissolved in methanol (—ml), solution was cooled to 5-10°C. To this was added a solution of sodium hydroxide (— g) in methanol. The reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained residue was striped with dichloromethane. Obtained solid was dried under vacuum at 45-50°C (yield=— g)
Example 4:
Preparation of (2S)-isopropyl-2-((chloro(phenoxy)posphoryl)amino)propanoate:
phenyl phosphodichloridate (30.6g) was dissolved in dichloromethane , to this was added a solution of 1-alanine isopropyl ester free base (19.16g) in dichloromethane at-60°C under nitrogen. Solution of triethylamine (20.7ml) was added to above reaction mass. Reaction mass was stirredat -60°C for 30 min and then temperature was raised to 25 °C. Reaction mass was stirred at 20-25 °C for 60 min & filtered and washed with dichloromethane. Clear filtrate was distilled under reduced pressure obtained residue was stirred with diisopropyl ether & filtered. Clear filtrate was distilled under reduced pressure to get (2S)-isopropyl-2-((chloro(phenoxy)posphoryl)amino)propanoate compound.
Example 5:
Preparation of isopropyl ((perfluorophenoxy)(phenoxy)phosphoryl)-L-alaninate (formula 2):

(Formula 2)
Obtained (2S)-isopropyl-2-((chloro(phenoxy)phosphoryl)amino)propanoate (1.2 mol eq.) was dissolved in dichloromethane and cooled to 0-5°C under nitrogen atmosphere. To this was added solution of sodium 2,3,4,5,6-pentaflurophemolate (1 mol eq.) in tetrahydrofuran . Temperature of reaction mass was raised to 25°C and reaction mass was stirred for 3 hrs. After completion of reaction, reaction mass was distilled under reduced pressure & obtained residue was dissolved I ethyl acetate. Ethyl acetate layer was washed with water, dried over sodium sulfate & distilled off under reduced pressure. Diisopropyl ether was added to obtained residue and stirred for 60 min at 25 °C, obtained mass was filtered & washed with diisopropyl ether. Solid product was dried under vacuum at 40-45 °C .(yield=20g, enantiomer purity=93.45%)
Example 6:
Preparation of (S)-isopropyl 2-(((S)- (perfluorophenoxy)phenoxy)phosphoyl)amino)propanoate (Formula 2′):

Formula 2′
(2S)-isopropyl-2-((chloro(phenoxy)phosphoryl)amino)propanoate (1.2 mol eq.) was dissolved in tetrahydrofuran (3.5 volumes). The reaction mass was cooled to -10°C. Solution of sodium salt of pentafluorophenol (1 mol eq.) in tetrahydrofuran (3.5 volumes) was added dropwise to the reaction mass at -10°C. After completion of the reaction solvent was distilled off. Ethyl acetate and water were added to the reaction mass. Reaction mass was stirred, ethyl acetate layer was separated and washed with sodium bicarbonate solution and brine. Ethyl acetate layer was concentrated under reduced pressure. Reaction mass was stripped with n-hepatane to get crude product. Crude product was dissolved in Methyl tert-butyl ether and n-heptane (1 : 1 ratio). The pH of reaction mass was adjusted to pH 8 by using triethylamine. Reaction mass was stirred overnight. Solid mass was filtered and washed with a mixture of methyl tertiary-butyl ether: n-heptane (1 : 1). The obtained product was dissolved in ethyl-acetate and washed with water and 20% brine solution. Ethyl acetate layer was separated; solvent was distilled off under reduced pressure. Reaction mass was stripped with diisopropyl ether. Di-isopropyl ether was added to the reaction mass. Reaction mass was stirred at 45-50°C. Reaction mass was cooled to 5-10°C and stirred. The titled compound was isolated by filtration and washed with di-isopropyl ether. The titled compound was dried under reduced pressure at 40°C. Yield 66.81%.

Vinita Gupta, CEO, Lupin Pharmaceuticals Inc

Desh Bhandu Gupta- Founder and chairman of Lupin Limited
////////////LUPIN LIMITED, WO 2016181313, NEW PATENT, SOFOSBUVIR
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL


{kind=link}