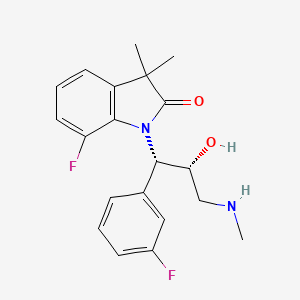
7-fluoro-1-[(1S,2R)-1-(3-fluorophenyl)-2-hydroxy-3-(methylamino)propyl]-3,3-dimethyl-1,3-dihydro-2H-indol-2-one

7-f luoro-1 -[(1 S,2R)-1 -(3-fluorophenyl)-2-hydroxy-3- (methylamino)propyl]-3,3- dimethyl-1 ,3-dihydro-2H-indol-2-one hydrochloride
WAY-315193
| 7-fluoro-1-[(1S,2R)-1-(3-fluorophenyl)-2-hydroxy-3-(methyl amino)propyl]-3,3-dimethyl-1,3-dihydro-2H-indol-2-one; |
| Molecular Formula: |
C20H22F2N2O2 |
| Molecular Weight: |
360.397686 g/mol |
7-Fluoro-1-[(1S,2R)-1-(3-fluorophenyl)-2-hydroxy-3-(methylamino)propyl]-3,3-dimethylindolin-2-one Hydrochloride
| Callain Younghee Kim, Paige Erin Mahaney,Eugene John Trybulski, Puwen Zhang,Eugene Anthony Terefenko, Casey Cameron Mccomas, Michael Anthony Marella, Richard Dale Coghlan, Gavin David Heffernan,Stephen Todd Cohn, An Thien Vu, Joseph Peter Sabatucci, Fei Ye, |
| Applicant |
Wyeth |
Drugs that possess norepinephrine reuptake inhibition, either selectively or in combination with serotonin reuptake inhibition, have been used for multiple indications including major depressive disorder, attention deficit hyperactivity disorder, stress urinary incontinence, vasomotor symptoms, and pain disorders such as diabetic neuropathy and fibromyalgia.1 In the search for new candidates with improvements in both potency and selectivity, one of the lead compounds in the 1-(3-amino- 2-hydroxy-1-phenylpropyl)indolin-2-one series, WAY-315193 (1), was identified.2
Vasomotor symptoms (VMS), referred to as hot flushes and night sweats, are the most common symptoms associated with menopause, occurring in 60% to 80% of all women following ‘ natural or surgically-induced menopause. VMS are likely to be an adaptive response of the central nervous system (CNS) to declining sex steroids. To date, the most effective therapies for VMS are hormone-based treatments, including estrogens and/or some progestins. Hormonal treatments are very effective at alleviating VMS, but they are not appropriate for all women. It is well recognized that VMS are caused by fluctuations of sex steroid levels and can be disruptive and disabling in both males and females. A hot flush can last up to thirty minutes and vary in their frequency from several times a week to multiple occurrences per day. The patient experiences a hot flash as a sudden feeling of heat that spreads quickly from the face to the chest and back and then over the rest of the body. It is usually accompanied by outbreaks of profuse sweating. It may sometimes occur several times an hour, and it often occurs at night. Hot flushes and outbreaks of sweats occurring during the night can cause sleep deprivation. Psychological and emotional symptoms observed, such as nervousness, fatigue, irritability, insomnia, depression, memory loss, headache, anxiety, nervousness or inability to concentrate are considered to be caused by the sleep deprivation following hot flush and night sweats (Kramer et al., In: Murphy et al., 3rd Int’l Symposium on Recent Advances in Urological Cancer Diagnosis and Treatment- Proceedings, Paris, France: SCI: 3-7 (1992)).
Hot flushes may be even more severe in women treated for breast cancer for several reasons: 1) many survivors of breast cancer are given tamoxifen, the most prevalent side effect of which is hot flush, 2) many women treated for breast cancer undergo premature menopause from chemotherapy, 3) women with a history of breast cancer have generally been denied estrogen therapy because of concerns about potential recurrence of breast cancer (Loprinzi, et al., Lancet, 2000, 356(9247): 2059-2063).
Men also experience hot flushes following steroid hormone (androgen) withdrawal. This is true in cases of age-associated androgen decline (Katovich, et al., Proceedings of the Society for Experimental Biology & Medicine, 1990, 193(2): 129-35) as well as in extreme cases of hormone deprivation associated with treatments for prostate cancer (Berendsen, et al., European Journal of Pharmacology, 2001, 419(1): 47-54. As many as one-third of these patients will experience persistent and frequent symptoms severe enough to cause significant discomfort and inconvenience.
The precise mechanism of these symptoms is unknown but generally is thought to represent disturbances to normal homeostatic mechanisms controlling thermoregulation and vasomotor activity (Kronenberg et al., “Thermoregulatory Physiology of Menopausal Hot Flashes: A Review,” Can. J. Physiol. Pharmacol., 1987, 65:1312-1324).
The fact that estrogen treatment (e.g. estrogen replacement therapy) relieves the symptoms establishes the link between these symptoms and an estrogen deficiency. For example, the menopausal stage of life is associated with a wide range of other acute symptoms as described above and these symptoms are generally estrogen responsive.
It has been suggested that estrogens may stimulate the activity of both the norepinephrine (NE) and/or serotonin (5-HT) systems (J. Pharmacology & Experimental Therapeutics, 1986, 236(3) 646-652). It is hypothesized that estrogens modulate NE and 5-HT levels providing homeostasis in the thermoregulatory center of the hypothalamus. The descending pathways from the hypothalamus via brainstem/spinal cord and the adrenals to the skin are involved in maintaining normal skin temperature. The action of NE and 5-HT reuptake inhibitors is known to impinge on both the CNS and peripheral nervous system (PNS). The pathophysiology of VMS is mediated by both central and peripheral mechanisms and, therefore, the interplay between the CNS and PNS may account for the efficacy of dual acting SRI/NRIs in the treatment of thermoregulatory dysfunction. In fact, the physiological aspects and the CNS/PNS involvement in VMS may account for the lower doses proposed to treat VMS (Loprinzi, et al., Lancet, 2000, 356:2059-2063; Stearns et al., JAMA, 2003, 289:2827-2834) compared to doses used to treat the behavioral aspects of depression. The interplay of the CNS/PNS in the pathophysiology of VMS and the presented data within this document were used to support the claims that the norepinephrine system could be targeted to treat VMS.
Although VMS are most commonly treated by hormone therapy (orally, transdermally, or via an implant), some patients cannot tolerate estrogen treatment (Berendsen, Maturitas, 2000, 36(3): 155-164, Fink et al., Nature, 1996, 383(6598): 306). In addition, hormone replacement therapy is usually not recommended for women or men with or at risk for hormonally sensitive cancers (e.g. breast or prostate cancer). Thus, non-hormonal therapies (e.g. fluoxetine, paroxetine [SRIs] and clonidine) are being evaluated clinically. WO9944601 discloses a method for decreasing hot flushes in a human female by administering fluoxetine. Other options have been studied for the treatment of hot flashes, including steroids, alpha- adrenergic agonists, and beta-blockers, with varying degree of success (Waldinger et al., Maturitas, 2000, 36(3): 165-168).
It has been reported that α2-adrenergic receptors play a role in thermoregulatory dysfunctions (Freedman etal., Fertility & Sterility, 2000, 74(1): 20- 3). These receptors are located both pre- and post-synaptically and mediate an inhibitory role in the central and peripheral nervous system. There are four distinct subtypes of the adrenergicα2 receptors, i.e., are 2A, O2B, 0.2c and α2D (Mackinnon et al., TIPS, 1994, 15: 119; French, Pharmacol. Ther., 1995, 68: 175). It has been reported that a non-select 2-adrenoceptor antagonist, yohimbine, induces a flush and an 2-adrenergic receptor agonist, clonidine, alleviates the yohimbine effect (Katovich, et al., Proceedings of the Society for Experimental Biology & Medicine, 1990, 193(2): 129-35, Freedman et al., Fertility & Sterility, 2000, 74(1): 20-3). Clonidine has been used to treat hot flush. However, using such treatment is associated with a number of undesired side effects caused by high doses necessary to abate hot flash described herein and known in the related arts.
Patent
https://www.google.com/patents/WO2005097744A1?cl=en
invention relates to phenylaminopropanol derivatives, compositions containing these derivatives, and methods of their use for the prevention and treatment of conditions ameliorated by monoamine reuptake including, inter alia, vasomotor symptoms (VMS), sexual dysfunction, gastrointestinal and genitourinary disorders, chronic fatigue syndrome, fibromylagia syndrome, nervous system disorders, and combinations thereof, particularly those conditions selected from the group consisting of major depressive disorder, vasomotor symptoms, stress and urge urinary incontinence, fibromyalgia, pain, diabetic neuropathy, and combinations thereof.
EXAMPLE 101 : 7-f luoro-1 -[(1 S,2R)-1 -(3-fluorophenyl)-2-hydroxy-3- (methylamino)propyl]-3,3- dimethyl-1 ,3-dihydro-2H-indol-2-one hydrochloride

[0538] Step 1 : A mixture of 7-fluoro-3, 3-dimethyl-1 ,3-dihydro-2H-indol-2-one (EXAMPLE 99, step 5, 1.0 g; 5.58 mmol) and sodium tert-butoxide (1.0 g, 11.16 mmol) in dry dichloromethane (15 mL) was stirred at room temperature under nitrogen for 20 minutes. Titanium isopropoxide (2.0 mL, 6.70 mmol) was added to a solution of [(2R,3R)-3-(3-fluorophenyl)oxiran-2-yl]methanol (EXAMPLE 47, step 3, 844 mg, 5.02 mmol) in dry dichloromethane (6 mL) and stirred for 20 minutes at room temperature. The epoxide complex was added drop-wise to the mixture of tert- butoxide and allowed to stir for 4 days. The reaction mixture was poured into a 2N aqueous solution of hydrochloric acid and diluted with ethyl acetate. The layers were separated, and the organic layer was washed with water and brine. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give 2.0 g of crude product. The crude product was purified via Isco chromatography (RediSep, silica, gradient of 0% to 100% ethyl acetate in hexane) to yield 600 mg (31 %) of (2S,3S)-7-Fluoro-1 -[1 -(3-fluoro-phenyl)-2,3-dihydroxy-propyl]-3,3-dimethyl- 1 ,3-dihydro-indol-2-one as an oil. MS (ESI) m/z 348 ([M+Hf).
[0539] Step 2: In an analogous manner to EXAMPLE 1 , step 2 (2S, 3S)-toluene- 4-sulfonic acid 3-(7-fluoro-3,3-dimethyl-2-oxo-2,3-dihydro-indol-1 -yl)-3-(3-fluoro- phenyl)-2-hydroxy-propyl ester was prepared from (2S,3S)-7-fluoro-1-[1-(3-fluoro- phenyl)-2,3-dihydroxy-propyl]-3,3-dimethyl-1 ,3-dihydro-indol-2-one. MS (ESI) m/z 502 ([M+Hf).
10 A. Kende, Synth. Comm. 1 : 12 (1982) [0540] Step 3: In an analogous manner to EXAMPLE 5 7-fluoro-1-[(1S,2R)-1-(3- fluorophenyl)-2-hydroxy-3-(methylamino)propyl]-3,3- dimethyl-1 ,3-dihydro-2H-indol- 2-one hydrochloride was prepared from (2S,3S)-toluene-4-sulfonic acid 3-(7-fluoro- 3,3-dimethyl-2-oxo-2,3-dihydro-indol-1-yl)-3-(3-fluoro-phenyl)-2-hydroxy-propyl ester. MS (ESI) m/z 360 ([M+Hf), HRMS: calcd for C20H22F2N2O2 + H+, 361.17221; found (ESI, [M+Hf), 361.1719.
PATENT
https://www.google.com/patents/WO2008024492A2?cl=en
Scheme A
cat.
Scheme B
Acetylbromidθ
EtOH
Scheme C (cat.)
1. TSCI1 TEA1 CH3CN A + B dlbutyltln oxide cat. 1. 33 wt% CH3NH2In EtOH. MeOH
1.35 equiv. 2. NaOH aq., toluene 2. 5N HCI in IPA, toluene
HCI salt
Example 1 :
Preparation of (1 R3/?)-3-(3-fluoropheπv0-2-(hvdroxymethv0oxirane
[0117] A thoroughly dried 5-L jacketed reactor was equipped with a mechanical stirrer, a 500-mL addition funnel, a temperature probe and a nitrogen inlet. The reactor was charged with D-(-)-DIPT (13.0 g 46 mmol), 4-A 5-μm molecular sieves (90 g) and dichloromethane (4.00 L) and then it was purged with nitrogen. The contents of the reactor were cooled to -15°C. Titanium isopropoxide (12.19 g, 43 mmol) was added rapidly to the reaction mixture via the addition funnel and the reaction mixture was further cooled to -200C. A solution of allylic alcohol (127 g, 0.854 mol) in CH2CI2 (380 mL) was added to the reaction mixture via the addition funnel at a rate to keep the temperature in the reactor below -200C. The resulting mixture was allowed to stir at -200C for 10 minutes. A solution of TBHP in CH2CI2 (4.5 M, 380 mL, 1.71 mol) was added to the reaction mixture via the addition funnel at a rate to maintain the temperature below -200C and above -25°C (addition rate 7 ml/min). The reaction mixture was stirred at -200C for 4 hours. Reaction progress was monitored by HPLC: an aliquot was drawn out of the reactor and diluted with MeCN-water. The reaction was deemed complete when the amount of the starting olefin fell below 1 %.
[0118] The reaction mixture was transferred from the reactor into a 6-L flask containing a solution of FeSCU x 7H2O (356 g, 1.28 mol), citric acid monohydrate (93 g, 0.39 mol) and de-ionized water (to the total volume of 1.0 L) chilled in an ice bath to 00C. The rate of transfer was adjusted to maintain the temperature of the mixture below 100C. The flask with the resulting mixture was equipped with a mechanical stirrer and the mixture was stirred for 25 minutes.
[0119] The organic layer was separated and filtered through a pad of Celite. The aqueous phase was extracted with MTBE (2 x 300 mL). Combined organic solutions were cooled to 00C in an ice bath. A 30% solution of NaOH (100 mL) in brine (prepared by dissolving 5 g of NaCI in a solution of NaOH (30.0 g) in 90 mL of water) was cooled in an ice bath to 00C and then added to the combined organic phases. The resulting mixture was stirred rapidly for 2 hours at 00C. Water (400 mL) was added to the mixture and the layers were separated. The aqueous layer was extracted with MTBE (2 x 250 mL). The combined organic layers were dried with Na2SO4 (300 g), the drying agent was filtered off through a paper filter and the filtrate was evaporated on rotary evaporator. The oily residue was mixed with 700 mL of toluene and the solvent was removed on a rotary evaporator. The residue after evaporation: Weight 125.9 g.
HPLC purity (area% 215 nm): 94%, impurities: toluene (3.1 %), starting olefin (1.0%), 3 unknown impurities (< 0.7% each).
1H NMR (CDCI3). Impurities: toluene (1.7 weight%), DIPT (1.1 weight%), t-BuOH (0.4 weight%).
Example 2: Preparation of 7-fluoro-1-r(1 S,2SM-(3-fiuorophenv[)-2,3-dihvdroxypropyπ-3.3- dimethylindolin-2-one
[0120] 7-Fluoro-3,3-dimethyl-1 ,3-dihydro-2H-indol-2-one (60 g, 335 mmol) was mixed under nitrogen with anhydrous dimethylformamide (DMF) (10.8 ml_). To the resulting viscous solution, cooled to 5-7°C, was added via syringe a solution of LiHMDS in THF (1 M in THF, 140 ml, 140 mmol) at a rate to keep the reaction mixture temperature below 7-1O0C (addition of the first 60 ml was very exothermic, later the rate of addition could be increased). The resulting purple-red clear solution was allowed to warm up to 100C.
[0121] In a separate flask, [(2/?,3fl)-3-(3-fluorophenyl)oxiran-2-yl]methanol (59.1 g, 352 mmol, 1.05eq.) was dissolved in 600 ml_ of anhydrous THF, the flask was purged with nitrogen and the solution was cooled to 5-7°C. Titanium isopropoxide (104 ml, 100 g, 584 mmol) was added to the epoxide solution dropwise via syringe maintaining the temperature in the 7-12°C range. The resulting bright-yellow solution was stirred for 40 minutes, allowing it to warm up to room temperature.
[0122] The contents of the second flask, the epoxy-titanium solution, were transferred to the solution of the indolinone salt via cannula maintaining the temperature of the mixture below 15°C. The resulting mixture was stirred at room temperature. The reaction progress was monitored by HPLC: after 20 hours, about 17 area% of indolinone was left, while no epoxide was detectable. Additional amount of the epoxide-titanium isopropoxide complex was prepared from epoxide (9.85 g, 58.4 mmol) and titanium isopropoxide (17.3 ml, 16.6 g, 58.4 mmol) in THF (100 mL) as described above and added slowly to the reaction mixture. The mixture was kept at room temperature for 20 hours longer, at which point HPLC analysis showed 4 area% of the unreacted indolinone and no detectable amount of the epoxide.
[0123] The reaction mixture was transferred into 1.80 L of cold (00C) 2 M aqueous HCI solution (Exotherm. The rate of addition was adjusted to keep the temperature below 15°C). The resulting clear solution was extracted with MTBE (3 x 800 ml), the combined organic phase were washed with brine (800 ml), dried over magnesium sulfate and filtered through a pad of magnesol. The filtrate was evaporated, diluted with toluene (600 ml), and evaporated again to remove maximum amount of solvents. The residue (133 g) contained a sufficiently pure product to be used in the next step without further purification.
HPLC purity (area% at 215 nm): 95%, impurities: indolinone (3.5%).
1H NMR (CDCI3). Impurities: residual solvents (DMF, toluene, MTBE).
Example 3:
Preparation of 7-fluoro-1 -f(SH3-f luorophenylUf S)-oxiran-2-yl)methv0-3.3- dimethylindolin-2-one
[0124] A 2-L round bottom flask, equipped with a mechanical stirrer, a 100-mL addition funnel, a temperature probe and a nitrogen inlet, was charged with a solution of 7-fluoro-1 -[(1 S,2S)-1 -(3-fluorophenyl)-2,3-dihydroxypropyl]-3,3- dimethylindolin-2-one (50.0 g, 144 mmol) in CH2CI2 (500 mL), triethylamine (62 mL, 0.433 mol), solid dibutyltin oxide (716 mg, 2.9 mmol) and DMAP (1.74 g, 14.4 mmol). Tosyl chloride (28.23 g, 148 mmol) was dissolved in CH2CI2 (60 mL) and the solution was added slowly to the reaction mixture (addition rate 5.6 mL/min). Temperature range 200C to 23°C. The reaction flask was cooled in an ice water bath during the addition to keep the temperature below 25°C. After the addition was finished, the bath was removed and the reaction mixture was stirred at room temperature. The reaction progress was monitored by HPLC.
[0125] After about one hour, the amount of the diol fell below 10%. A solution of NaOH, prepared by diluting 72 mL of 10 M aqueous NaOH with 360 mL of deionized water, was added rapidly to the reaction mixture. Solid Bu4N+ CP hydrate (2.05 g, 7.2 mmol) was added and the reaction mixture was stirred rapidly at room temperature. The progress of the epoxide closure was monitored by HPLC. After 2 hour, all fosylate was consumed.
[0126] The layers were separated. The aqueous layer was extracted with 100 mL of CH2CI2. Combined organic solutions were washed with 100 mL portions of 0.5 M aqueous HCI until pH of the washes fell below 5, then with 50 mL of 0.5 M aqueous NaOH, then it was dried with Na2SO-I. The solution was gravity-filtered through a pad of Silica gel (150 g, thickness of the pad 5 cm) prepared in a glass filter funnel. The drying agent and the pad were washed with dichloromethane. The washing continued until no more epoxide was detectable in the eluent (by HPLC). The filtrate was evaporated to dryness on rotary evaporator (room temp. bath).
[0127] The residue after evaporation: weight 42.6 g. HPLC purity 82%, impurities: bis-tosylate (12%), diol (2.5%), indolinone (2.4%). The crude intermediate was used in the next step without further purification.
Example 4:
Preparation of 7-fluoro-1-iY1 S.2fl)-1-(3-fluorophenyl)-2-hvdroxy-3-(methyl amino)propyπ-3,3-dimethylindolin-2-one hydrochloride
[0128] The residue from the previous step (42.6 g) was dissolved in ethanol (160 mL) and the solution was placed into a 1-L round bottom flask equipped with a mechanical stirrer and a temperature probe. Aqueous methylamine (40 weight%, 240 mL, 2.74 mol) was added to the solution and the resulting suspension was stirred at room temperature. The reaction was monitored by HPLC. After 15 hours, the amount of the epoxide fell below 1%. Ethanol was removed on rotary evaporator (bath temperature 27°C). The residue was mixed with MTBE (250 mL) and water (100 mL). The layers were separated. The aqueous layer was extracted with 50 mL of MTBE. Combined organic solutions were washed with 100 mL of water. Small amount of brine was added to speed up the phase separation. The resulting organic solution was extracted with aqueous HCI (200 m L of 2 M solution, then 50 mL of 1 M solution). Combined acidic extracts were washed with 50 mL of MTBE.
[0129] MTBE (200 mL) was added to the aqueous solution. Aqueous NaOH (10 M solution^ 50 mL, 500 mmol) was added to the bi-phasic mixture. The mixture was shaken and the layers were separated. The aqueous layer was extracted with MTBE (100 mL). Combined organic solutions were dried with Na2SO4 (75 g). The drying agent was filtered off and the filtrate was evaporated in vacuum. [0130] The residue (38.0 g) was mixed with 70 ml_ of ethanol and the solvent was removed on rotary evaporator. The residue was re-dissolved in 100 ml_ of ethanol. With magnetic stirring, 2 M HCI in Et2O (57 mL, 114 mmol) was added to the solution. The acidity of the solution was checked by placing a drop of the solution on a wet pH paper to ensure the solution is strongly acidic. The resulting solution was seeded with crystals of 7-fluoro-1-[(1 S,2R)-1-(3-fluorophenyl)-2-hydroxy-3-(methyl amino) propyl]-3,3-dimethyl-1 ,3-dihydro-2H-indol-2-one hydrochloride salt which caused slow crystallization of the salt in about 30 minutes. The slurry was stirred at room temperature for 1 hour.
[0131] The reaction flask was placed into a 0°C bath (equipped with thermostat) and the slurry was stirred magnetically for 21 hours. The cold slurry was filtered through a paper filter. The solid was washed with a 1 :1 mixture of EtOH-Et2O (70 mL) and then was dried on the filter in a stream of air for 2 hours.
[0132] Weight of the crystals 29.7 g (54% from theoretical yield calculated from the diol).
HPLC purity (area% at 215 nm): 98.2%, impurities (relative retention time): 1.05
(0.46%), 0.98 (0.42%), 1.07 (0.15%), 2.05 (0.14%).
Enantiomeric purity 99.4% ee. m.p. 209.5-211.20C.
[a)? = -,10.7°.
1H NMR (D2O, 400 MHz), δ: 7.45-7.30 (m, 3H), 7.16-6.97 (m, 4H), 5.53-5.30 (2H1 broad m), 3.35-3.24 (2H, broad m), 2.82 (s, 3H), 1.41 (s, 3H), 1.27 (broad s, 3H).
Impurities: ethanol (0.3 weight%).
ES+ MS, m/z 361 (MH+).
Anal, calc’d for C20H23CIF2N2O2 (396.9): C, 60.53; H, 5.84; N, 7.06. Found: C, 60.43;
H, 5.69; N, 6.84.
Sn content: 3 ppm.
Example 5:
Preparation of 7-fluoro-3. 3-dimethyl-oxindole via selective C-methylation of 7- fluoro-oxindole [0133] To a stirred slurry of potassium tert-butoxide (185 g, 1.65 mol) in tθtrahydrofuran (1350 mL) was added 7-fluoro-oxindole (50 g, 0.33 mol) and copper (I) bromide-dimethyl sulfide complex (7 g, 0.033 mol). Methyl iodide (150 g, 1.06 mol.) was added to the mixture at 5-100C. The reaction mixture was stirred at 20- 25°C for 1 hour. 10% NH4CI (1000 mL) was added to the reaction mixture. The two layers were separated. The organic layer was concentrated via vacuum distillation at 25-400C to reach a volume of 250 mL. The aqueous layer is extracted with tert- butyl methyl ether (2 x 500 mL). The concentrated organic layer and tert-butyl methyl ether extraction layers were combined and washed with 15% NaCI (250 mL). The organic solution was filtered through silica gel (100 g). Heptane (1250 mL) was added to the filtrate. The mixture was concentrated under atmosphere at 60-950C to reach a volume of 700 mL. The concentrate was cooled to 0-50C from 85-95°C over 2 hours to crystallize. Solid was filtered, washed with heptane (100 mL), and oven- dried to give 41 g (69.4%) of a beige solid 7-fluoro-3, 3-dimethyl-oxindole, 92% w/w purity by HPLC.
Example 6:
Preparation of 3-(3-fluoro-phenyl)-prop-2-en-1-ol
[0134] A 5-L reactor equipped with a mechanical stirrer, thermocouple, and nitrogen inlet was charged with MeOH (1.40 L) and 3-fluorocinnamic acid (0.20 kg, 1.20 mol). To the slurry charged p-TSA (0.023 kg, 0.120 mol) at 200C to 25°C. The suspension was refluxed at 65°C to 68°C for 3-5 hours. The mixture was concentrated via atmospheric distillation to reach a volume of 700 mL. Methanol was then chased off by adding toluene (1.8 L) and was further concentrated to a solution (about 1.5 L). The reaction mixture then washed successively with 5% aqueous NaHCU3 (1.5) and water (1.5 L). The organic mixture was concentrate via atmospheric distillation to a minimum volume of 500 mL. HPLC analysis indicates that the solution strength 53.5% KF 0.17%, 98.8% area HPLC purity of the product. [0135] A 3-L reactor equipped with a mechanical stirrer, thermocouple, and nitrogen inlet was charged with diisobutylaluminum hydride 25% w/w in Toluene (1.56 kg, 1 ,85 L12.75 mol). The solution was cooled to -25°C. To the reactor was then added using FMI pump a solution of 3-(3-Fluoro-phenyl)-acrylic acid methyl ester (0.41 kg, 0.40 L, 1.20 mol) in toluene while maintaining the internal temperature between -15°C to -8°C. The reaction mixture was stirred at -15 to -8°C for 60 minutes. The reaction mixture was then quenched in a 5-L reactor into a solution of concentrated HCI (0.40 L, 0.48 kg; 4.87 mol) in water (0.75 kg) maintaining internal temperature at 400C to 45°C. The biphasic mixture was separated. The lower aqueous layer was washed with Toluene (0.34 kg, 0.40 L). The combined organic phase was successively washed with a 5% aqueous solution of sodium bicarbonate (0.7 L) and 10% brine (0.7 L). The*organic solution was concentrated via atmospheric distillation to reach a volume of 500 mL. HPLC analysis indicates that the solution strength is 53%, 169 g (93% Y), Al: 9 ppm, KF: 0.04%, 99% area HPLC purity of the allylic alcohol.
Example 7:
Preparation of r3-(3-fluoro-phenyl)-oxiranyπ-methanol
[0136] A 3-L reactor equipped with a mechanical stirrer, thermocouple, and nitrogen inlet was charged with toluene (200 mL) and pre-activated molecular sieves powder (4A, 70 g). The resultant slurry was cooled to — 35°C. To the reactor was then added a solution of D-(-)-diisopropyl tartrate (21.6 g, 92.0 mmol) in toluene (25 mL), followed by addition of titanium (IV) isopropoxide (18.7 g, 65.7 mmol). The temperature of the reaction mixture was maintained between -300C to -400C during the addition. To the reactor was then charged with a solution of 3-(3-fluoro-phenyl)- prop-2-en-1-ol (100 g, 657 mmol) in toluene (490 mL) while maintaining the temperature of the reaction mixture between -300C to -400C. The reaction mixture was stirred at -35°C for 30 minutes. To the reactor was then added a solution of 5.5 M tert-butyl hydroperoxide in decane (240 mL, 1310 mmol) while maintaining the temperature of the reaction mixture between -300C to -400C. The reaction mixture was stirred at -35°C for 6 hours, followed by 8 hours at -200C. The reaction mixture was warmed to room temperature and filtered through a thin layer of celite. The filter cake was washed with toluene (2 x 100 ml_). The combined filtrate and washes were cooled to 00C and a solution of 30% sodium hydroxide saturated with sodium chloride (100 mL) was then added. The reaction mixture is stirred at 0°C for 2 hours. To the reaction mixture was then added a solution of sodium metabisulfite (69 g) and citric acid (50 g) in water (600 mL). The biphasic mixture was stirred at room temperature for 1 hour and the phases were separated. The organic phase was successively washed with a 5% sodium bicarbonate (500 mL) and 10% brine (500 mL). The organic solution was then concentrated under vacuum to reach a volume of 500 mL. HPLC analysis indicates that the solution contains 90.3 g (81.7%) of the epoxy alcohol product.
Example 8:
Preparation of r3-(3-fluoro-pheny[)-oxiranyll-methanol
[0137] A 1-L reactor equipped with a mechanical stirrer, thermocouple, and nitrogen inlet is charged with toluene (140 mL) and pre-activated molecular sieves powder (4A, 14 g). The resultant slurry was cooled to -35°C. To the reactor is then added a solution of D-(-)-diisopropyl tartrate (4.31 g, 18.4 mmol) in toluene (20 mL), followed by addition of titanium (IV) isopropoxide (3.74, 13.1 mmol). The temperature of the reaction mixture was maintained between -300C to -400C during the addition. To the reactor is then charged with a solution of 3-(3-fluoro-phenyl)- prop-2-en-1 -ol (20 g, 131 mmol) in toluene (80 mL) while maintaining the temperature of the reaction mixture between — 300C to — 400C. The reaction mixture is stirred at -35°C for 30 minutes. To the reactor is then added a solution of cumene hydroperoxide (88% purity, 45.5 g, 263 mmol) while maintaining the temperature of the reaction mixture between -300C to -400C. The reaction mixture is stirred at – 35°C for 16 hours. A solution of 30% sodium hydroxide saturated with sodium chloride (20 mL) is charged while maintaining temperature of the reaction mixture below -200C. To the reaction mixture is then added a solution of sodium metabisulfite (13.7 g) in water (60 mL) while maintaining the reaction mixture temperature below 25°C. The biphasic mixture is stirred at room temperature for 1 hour. To the reaction mixture is added Celite (70 g) and the mixture is filtered. The filter cake is washed with toluene (2 x 50 mL). The filtrate is successively washed with 5% sodium bicarbonate (100 mL) and 10% brine (100 mL). The organic solution is then concentrated under vacuum to reach a volume of 100 mL.
Example 9:
Preparation of r3-(3-fluoro-phenyl)-oxiranvn-methanol
[0138] A 5-L jacketed reactor equipped with a mechanical stirrer, addition funnel, temperature probe, and nitrogen inlet. All equipment must be rigorously dry. The reactor was charged with D-(-)-DIPT (10.0 mL, 11.0 g, 46 mmol), 4-A, 5-um molecular sieves (49.3 g), dichloromethane (3 L). The flask was purged with nitrogen. The contents of the flask were cooled to 00C. Titanium isopropoxide (9.34 g, 9.73 mL was added rapidly to the flask via an addition funnel. The reaction mixture was cooled to -200C. A solution of allylic alcohol (100 g, 0.657 mol) in CH2CI2 (300 mL) was added to the reaction mixture via an addition funnel while keeping the temperature below -200C.
[0139] The reaction mixture was stirred at -200C for 10 minutes. A solution of TBHP in CH2CI2 (188 mL, 5.7 M) was added to the reaction mixture via an addition funnel while maintaining the temperature between -200C to -25°C. The reaction mixture was stirred at -200C for 4 hours. Reaction progress was monitored by HPLC. A solution prepared from FeSO4 x 6H2O (217 g, 0.79 mol), citric acid monohydrate (72 g, 0.39 mol) and de-ionized water to the total volume of 660 mL, was chilled in an ice bath to 00C.
[0140] The reaction mixture was quenched into the chilled solution of FeSO4 and citric acid in water. The mixture was stirred for 30-60 minutes. The organic layer was checked for the presence of organic peroxides. The layers were separated. The aqueous phase was extracted with MTBE (2 x 200 mL). Combined organic solutions were cooled to 00C in an ice bath. [0141] A 30% solution of NaOH (60 ml_) in brine (prepared by dissolving 5 g of NaCI in a solution of NaOH (30.0 g) in 90 mL of water) was cooled in an ice bath to 00C and then added to the combined organic phases. The resulting mixture was stirred rapidly for 1-2 hours at 00C. Water (300 mL) was added to the mixture. The two layers were separated. The aqueous layer was extracted with MTBE (2 x 250 mL). The combined organic layers were evaporated on. rotary evaporator. HPLC analysis indicates that the solution contains 90.5 g (81.5%) of the epoxy alcohol product with chiral purity 95.6/4.4 and chemical purity 96.5 area %.
Example 10:
Preparation of 7-fluoro-1- f(1S, 2S)-1-(3-fluorophenv0-2,3-dihvdroxypropyl1-3,3- dimethvM . 3- dihvdro-2H-indol-2-one
[0142] To a suspension of 7-fluoro-3, 3-dimethyl-oxindole (35 g, 0.195 mol) in N, N dimethylformamide (36 g, 0.49 mol) and toluene (200 mL) was added (1 M / toluene) lithium bis (trimethylsilyl) amide (585 mL, 0.585 mol). To the resulting mixture was added a solution of (20% / toluene) [3-(3-fluoro-phenyl)-oxiranyl]-methanol (210 g, 0.253 mol) and titanium (IV) isopropoxϊde (72g, 0.253 mol) in toluene (300 mL) at 5- 100C. The reaction mixture was stirred for 3-4 hours at 40-45°C. To the reaction mixture was added 37% HCI (460 g, 2.34 mol) and water (500 mL) at 20-250C to give a bi-phasic mixture. The organic layer was separated. The aqueous layer was extracted with toluene (1000 mL). The combined organic layers were washed with 1N NaOH (200 g), and then with 10% NaCI (200 g). The organic layer was concentrated via atmospheric distillation at 100-1100C to a volume of (1800 mL). The concentrated solution was filtered through silica gel (150 g). The silica gel plug was rinsed with ethyl acetate (850 mL). The filtrate was concentrated via atmospheric distillation at 80-1100C to reach a volume of (250 mL). The concentrate was cooled to 0-50C from 100-1 100C over 4 hours to crystallize. Solid was filtered, washed with heptane (150 mL), and oven-dried to give 50.6 g (74.7%) of a beige solid, 97.4% w/w purity by HPLC.
Example 11 : Preparation of 7-fluoro-1-rf1S.2R)-1-(3-ftuorophenyl)-2-hvdroxy-3-fmethyl amino) propyl1-3.3-dirnethyl-1.3-dihvdro-2H-indol-2-one:
[0143] To the solution of the diol (52 g, 0.144 mol) in MeCN (500 mL) was added Bu2SnO (0.39 g, 1.44 mmol) and TsCI (28.8 g, 0.151 mol). To the resulting solution was added Et3N (29 g, 0.288 mol) dropwise at 0-50C. The reaction was stirred for 1 hour at 0-50C until the tosylation was complete by HPLC. To the reaction containing the mono-tosylate was added a solution of NaOH (58 g, 0.72 mol) in water (400 mL) at 00C. At the end of the epoxide formation, toluene (800 mL) and NaCI (25 g) in water (150 mL) were added to form a bi-phasic reaction mixture. The two layers were separated. The organic layer was washed with 37% w/w HCI (56 g) in water (256 mL) followed by NaCI (50 g) in water (300 mL). The organic layer was diluted with toluene (700 mL) and concentrated to a volume of about 900 mL. The resulting concentrated solution was filtered through a silica gel (200 g) plug. The silica gel plug was eluted with toluene (1.5 L). The combined filtrate was concentrated under vacuum to about 300 mL. Methylamine in EtOH (33 weight %, 245 mL, 2.0 mol) and Ca(OTf)2 (15 g, 43 mmol) were added to the toluene solution. The reaction mixture was stirred at 20-250C for 12 hours then concentrated via vacuum distillation to about 200 mL. MTBE (500 mL) and water (500 mL) were added. The two layers were separated. 37% w/w HCI (160 g,) in water (340 g) was added to the organic layer. Stirred and the two layers were separated. The aqueous organic layer was washed with MTBE (500 mL). To the acidic aqueous layer was charged MTBE (500 mL) then the mixture was cooled to 0-50C and basified with NaOH (50% w/w, 150 g, 100 mL). Reaction mixture was stirred for 20 minutes then the two layers were separated. The organic layer was washed with 15% NaCI (170 mL) then concentrated to about 250 mL via atmospheric distillation. To the MTBE concentrate was added EtOH (2B) (150 mL) followed by HCI (5.7 N in EtOH, 45 mL, 0.26 mol). The mixture was stirred at 20 to 25°C for a minimum of 2 hours and then cooled to 0 to 5°C over 1 hour. The suspension was filtered and washed with MTBE (50 mL) to give 26 g (45%) of an off-white solid.
Example 12:
Preparation of (2EKH3,5-difluorophenvQprop-2-en-1-ol [0144] A 5-L reactor equipped with a mechanical stirrer, thermocouple, and nitrogen inlet was charged with MeOH (1.40 L), 3, 5-difluorocinnamic acid (0.20 kg, 1.09 mol) and p-TSA (0.0207 kg, 0.109 mol) at 200C to 25°C. The suspension was refluxed at 65°C to 68°C for 4-6 hours. The mixture was concentrated via atmospheric distillation to reach a volume of about 700 mL. Methanol was then chased off by adding toluene (1.8 L) and was further concentrated to a solution (about 1.5 L). The reaction was cooled to 500C to 55°C then washed successively with 5% aqueous NaHCO3 (1.5 L) and water (1.5 L). The organic mixture was concentrated via atmospheric distillation to a minimum volume of about 1.5 L. KF 0.17%.
[0145] A 3-L reactor equipped with a mechanical stirrer, thermocouple, and nitrogen inlet was charged with diisobutylaluminum hydride 25% w/w in toluene (1.42 kg, 1.68 L, 2.31 mol). The solution was cooled to -25°C. To the reactor was then added using FMI pump a solution of 3-(3, 5-difluoro-phenyl)-acrylic acid methyl ester (1.4 L, 1.09 mol) in toluene while maintaining the internal temperature between -15°C to -8°C. The reaction mixture was stirred at that temperature for 60 minutes then quenched into a 5-L reactor with a solution of concentrated HCI (0.40 L, 0.48 kg; 4.87 mol) in water (0.70 kg) while maintaining the internal temperature at 400C to 45°C. The biphasic mixture was separated. The lower aqueous layer was washed with toluene (0.34 kg, 0.40 L). The combined organic phase was successively washed with a 5% aqueous solution of sodium bicarbonate (0.70 L) and 10% brine (0.70 L). The organic solution was concentrated via atmospheric distillation to reach a volume of 0.386 Kg, about 500 mL. HPLC analysis indicates that the solution contains 170 g, 91% yield of (2E)-3-(3,5-difluorophenyl)prop-2-en-1-ol. Al: 1 ppm, KF: 0.12%, 99.8% area HPLC purity.
Example 13:
Preparation of r(2ff,3/?)-3-(3.5-difluorophenyl)oxiran-2-vH-methanol [0146] A 3-L reactor equipped with a mechanical stirrer, thermocouple, and nitrogen inlet was charged with toluene (100 mL) and pre-activated molecular sieves powder (4A, 70 g). The resultant slurry was cooled to -35°C. To the reactor was then added a solution of D-(-)-diisopropyl tartrate (19.3 g, 0.082 mol) in toluene (25 mL), followed by addition of titanium (IV) isopropoxide (16.7 g, 0.059 mol). The temperature of the reaction mixture was maintained between -300C to -400C during the addition. To the reactor was then added a solution of 3-(3,5-difluoro-phenyl)- prop-2-en-1-ol (100 g, 0.588 mol) in toluene (250 mL) while maintaining the temperature of the reaction mixture between -300C to -400C. The reaction mixture was stirred at -35°C for 30 min. To the reactor was then added a solution of 5.5 M tert-butyl hydroperoxide in decane (173 g, 1.18 mol) while maintaining the temperature of the reaction mixture between -300C to -400C. The reaction mixture was stirred at -35°C for 6 hours, followed by 8 hours at -25°C. The reaction mixture was warmed to room temperature and filtered through a thin bed of celite (25 g). The filter cake was washed with toluene (2 x 200 mL). The combined filtrate and washes were cooled to 00C and a solution of 30% sodium hydroxide saturated with sodium chloride (100 mL) was then added. The reaction mixture is stirred at 00C for 3 h. To the reaction mixture was then added a solution of sodium metabisulfite (61.5 g) and citric acid (44.5 g) in water (600 mL). The biphasic mixture was stirred at room temperature for 1 hour and the phases were separated. The organic phase was successively washed with a 5% sodium bicarbonate (500 mL) and 10% brine (500 mL). The organic solution was then concentrated under vacuum to reach a volume of about 400 mL. A small portion of the concentrate was taken out for seed generation at 25-300C. To the suspension was then charged 3 volume parts of heptane (300-400 mL). The mixture was cooled to 5-100C then filtered to give 71.3 g, 65% yield of [(2R,3R)-3-(3,5-difluorophenyl)oxiran-2-yl]-methanol as an off-white solid with chiral purity 94 %ee, mp: 48-50°C.
Example 14:
Preparation of 1-r(7S,2S>-1-(3,5-difluorophenyl)-2,3-dihvdroxypropyH-7-fluoro- 3,3-dimethyl-1,3-dihvdro-2H-indol-2-one [0147] To a suspension of dimethyl oxidole (68 g of 74 % strength crude, 280 mmol) in DMF (51 g, 700 mmol.) and toluene (200 rriL), a toluene solution of (Me3Si)2NLi (840 ml_, 1 M, 840 mmol) was added dropwise while keeping the mixture below 100C to give a dark solution. A solution of epoxy alcohol (76 g of 85% strength, 350 mmol) and Ti(OiPr)4 (103 g, 360 mmol) in toluene (400 ml_) was added to the above dark solution at below 100C. The reaction mixture was stirred for 20 hours at 200C before cooling to 00C. A solution of HCI (660 g, 37% in water) in water (750 g) was added at below 200C to give a bi-phasic mixture. The two layers were separated. The organic layer was washed with NaOH (400 ml_, 0.7 N in water, 280 mmol), and brine (230 g). The organic layer was filtered through a silica gel (150 g) plug. The silica gel plug was rinsed with. EtOAc (1100 ml_). The filtrate was concentrated in vacuo at 50°C to a volume of 240 ml_. This concentrate was diluted with CH3CN (300 ml_) to give 1-[(1S,2S)-1-(3,5-difluorophenyl)-2,3-dihydroxypropyl]- 7-fluoro-3,3-dimethyl-1 ,3-dihydro-2H-indol-2-one as a CH3CN solution, 431 g of a 20.8% strength solution, yield: 88%.
Example 15:
Preparation of 1-r(7S..g/?)-1-(3,5-d.fluorophenvπ-2-hvdroxy-3-(methylamϊno) propyπ-7-fluoro-3,3-dimethyl-1,3-dihvdro-2H-indol-2-one
[0148] To the solution of 1-[( 7S,2S)-1 -(3,5-difluorophenyl)-2,3-dihydroxypropyl]-7- fluoro-3,3-dimethyl-1 ,3-dihydro-2H-indol-2-one in acetonitrile (394 g of 20.8% strength solution, 224 mmol) at 200C, tosyl chloride (56g, 269 mmol) and Bu2SnO (1.4 g 5.6 mmol) were added. The reaction mixture was cooled to 5°C, and then Et3N (45 g, 448 mmol) was added dropwise. The reaction mixture was stirred for about 1 hour at 200C until tosylation is complete.
[0149] A solution of NaOH (90 g of 50% w/w solution in water, 1120 mmol) in water (492 g) was added at 5°C. The reaction mixture was stirred for 1 hour. Toluene (1312 ml_) was added to the reaction mixture to give a bi-phasic mixture. The organic layer was separated and washed with HCI (44 g of 37% solution in water, 448 mmol) in water (32OmL) then with brine (400 mL). The organic layer was then concentrated to a volume of (400 mL) under vacuum keeping the temperature below 500C. The concentrate was diluted with toluene (1120 mL). The resulting solution was filtered through a silica gel (320 g) plug. The silica gel plug was eluted with toluene (2400 ml_). The filtrate was concentrated to a volume of 400 ml_ in vacuo keeping the temperature below 500C.
[0150] Methanol (1200 mL) was charged to the mixture then concentrated down to about 400 mL in vacuo while keeping temperature below 500C. To the concentrate was added methanol (1600 mL) and methylamine (252 g of 33 wt % solution in ethanol, 2688 mmol.). The reaction mixture was stirred for 20 hours at 400C until the aminolysis is complete. The mixture was concentrated down to about 400 mL in vacuo. Toluene (960 mL) was added to the concentrate. The mixture was concentrated in vacuo down to about 400 mL.
[0151] HCI (40 g of 5N solution in isopropanol, 224 mmol.) in IPA was added to the mixture. Stirred at 200C for 2 hours. The resulting slurry was filtered then dissolved in acetone (1230 mL) at 400C. Heptane (1640 mL) was added. The resulting solution was concentrated at 700C to a volume of (1230 mL). The resulting slurry was filtered and dried for 18 hours at 55°C to give 46.5 g, 50% overall yield of 1- [(1 S,2R)-1-(3,5-difluorophenyl)-2-hydroxy-3-(methylamino)propyl]-7-fluoro-3,3- dimethyl-1 ,3-dihydro-2H-indol-2-one as a white solid.
Example 16:
Preparation of 7-fluoro-1-fπSH3-fluorophenvθr(2S)-oxiran-2-vπmethyl}-3.3- dimethyl-1.3-dihvdro-2H-indol-2-one
[0152] Diethyl-azodicarboxylate (100 g, 572 mmol) was added dropwise to a solution of 1-[(1S, 2S)-1-(3,5-difluorophenyl)-2,3-dihydroxypropyl]-7-fluoro-3,3- dimethyl-1 ,3-dihydro-2H-indol-2-one (90 g, 260 mmol) and Ph3P (129g, 520 mmol) in toluene (1042 mL) at 250C. The mixture was stirred for 1 hour at 800C. Ph3P (7 g, 26 mmol) was added to the mixture at 800C. The mixture was stirred for 8 hours at 😯0C. Diethyl-azodicarboxylate (9 g, 52 mmol) was added to the mixture at 800C. The mixture was stirred for about 2 hours at 800C until the reaction is complete. Heptane (3120 mL) was added to the reaction mixture at 800C. The mixture was cooled to 100C and then filtered through a silica gel (720 g) plug. The filtrate was discarded. The silica gel plug was rinsed with a solution of ethyl acetate (1100 mL) in heptane (3300 mL). The filtrate was concentrated to dryness at 500C to give 56 g, 80% purity, 52% yield of 7-fluoro-1-{(1 S)-(3-fluorophenyl)[(2S)-oxiran-2-yl]methyl}- 3,3-dimethyl-1 ,3-dihydro-2H-indol-2-one.
Example 17:
Preparation of 7-fluoro-1-rf1S.2R)-1-f3-fluorophenvπ-2-hydroxy-3-(methyl amino) propyll-3,3-dimethyl-1 ,3-dihydro-2H-indol-2-one:
[0153] In a flask with 7-fluoro-1-[(1 S,2S)-1-(3-fluorophenyl)-2,3-dihydroxypropyl]- 3,3-dimethylindolin-2-one (10 g, 0.0288 mol) and para-toluenesulfonic acid (pTSA) (0.0438 g, 0.023 mol) in THF (50 mL), trimethyl orthoacetate (4.15 g, 4.3 mL, 0.0346 moles) was added dropwise. The amber color solution was stirred at room temperature for 1 hour. The reaction mixture was concentrated to oil then THF (50 mL) was added. Cooled to 00C to 5°C then acetyl bromide (8.50 g, 0.0692 mol) was added. The resulting mixture was stirred at room temperature for 3 to 4 hours then concentrated to oil and charged with THF (25 mL) and EtOH 2B (25 mL) followed by K2CO3 -325 (39.8 g, 0.288 mol). The mixture was stirred at room temperature then the mixture was concentrated in vacuo to oil. MTBE (100 mL) and H2O (170 mL) were added to dissolve the oil. The two layers were separated. The aqueous layer was extracted with MTBE (2 x 100 mL). The combined organic layer was concentrated to oil then 33% solution of methylamine in ethanol (15 eq.) was added and stirred at room temperature. At the completion of the reaction, the mixture was concentrated to oil. MTBE 100 mL) and H2O (100 mL) were added. The two layers were separated. The organic layer was extracted with 37% concentrated HCI (30.7g) in H2O (65 g). The lower aqueous layer was extracted with MTBE (100 mL) then cooled to 0-50C. MTBE (100 mL) and a solution of 50% NaOH (30 g) in H2O (30 g) were added to the aqueous layer. The mixture was stirred for 20 minutes at room temperature and the layers were separated. The aqueous layer was back extracted with MTBE (50 mL). The combined organic layer was washed with a 15% NaCI (23 mL) solution. The organic layer was concentrated to give as oil (8.4 g, about 90% by LC/MS, 60% yield). [0154] When ranges are used herein for physical properties, such as molecular weight, or chemical properties, such as chemical formulae, all combinations and subcombinations of ranges specific embodiments therein are intended to be included.
Paper
Organic Process Research & Development 2009, 13, 880–887
Large-Scale Synthesis of a Selective Inhibitor of the Norepinephrine Transporter:
Mechanistic Aspects of Conversion of Indolinone Diol to Indolinone Aminoalcohol
and Process Implications
Asaf Alimardanov,* Alexander Gontcharov, Antonia Nikitenko, Anita W. Chan, Zhixian Ding, Mousumi Ghosh,
Mahmut Levent, Panolil Raveendranath,† Jianxin Ren, Maotang Zhou, Paige E. Mahaney,‡ Casey C. McComas,‡
Joseph Ashcroft, and John R. Potoski
Wyeth Research, 401 North Middletown Road, Pearl RiVer, New York 10965, U.S.A., and Wyeth Research, 500 Arcola Road,
CollegeVille, PennsylVania 19426, U.S.A.
TREATMENT OF GYNECOLOGICAL DISORDERS
WAY-315193 (Wyeth Pharmaceuticals)
Development of a scalable synthesis of WAY-315193 is described.
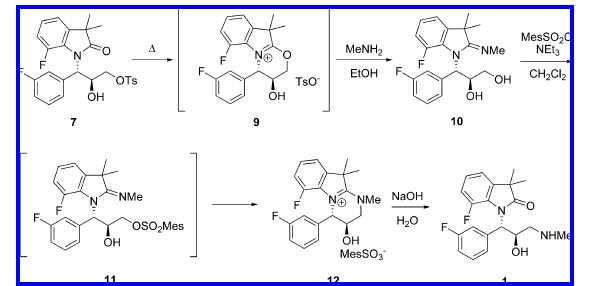
Use of LiHMDS as a base and Ti(O-i-Pr)4 as a Lewis acid was optimal for efficient and reproducible addition of indolinone anion to epoxyalcohol. Conversion of indolinone diol to indolinone aminoalcohol was achieved via monotosylationmethylamination.
The possibility of selective formation of the amidine side product, as well as its utilization for alternative selective preparation of the target aminoalcohol, was demonstrated.

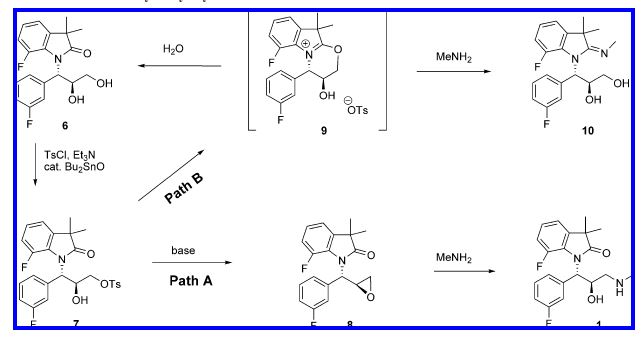
The synthetic route used initially for preparation of 1 is shown in Scheme 1. The key step of the synthesis was the
Sharpless epoxidation of fluorocinnamic alcohol 3 which selectively introduced both relative and absolute configurations at the C-2 and C-3 positions. At the early stages of the project, allylic alcohol 3 was prepared in two steps from commercially available fluorocinnamic acid 2 by treatment with MeI in the presence of Cs2CO3 in acetone, followed by DIBAL reduction at -78 °C. The epoxide 4 was opened with the sodium salt of dimethylfluoroindolinone in DMF to afford the diol. The diol 6 was further elaborated into the final aminoalcohol hydrochloride 1 in 30-34% yield via tosylation with p-toluenesulfonyl chloride (TsCl) in pyridine, isolation of the intermediate monotosylate, treatment with MeNH2, and conversion to HCl salt. Dimethylfluoroindolinone was prepared by reduction and bis-methylation of 7-fluoroisatin by a process developed earlier as described in a prior publication.3



white solid (58% yield). Mp 209-212 °C.
[R]D25°)+10.7°.
1H NMR (D2O, 400 MHz) δ: 7.40-7.25 (m,3H), 7.16-6.97 (m, 4H), 5.47-5.25 (2H, broad m), 3.27-3.20
(2H, broad m), 2.76 (s, 3H), 1.37 (s, 3H), 1.24 (broad s, 3H).
ES+ MS, m/z 361 (MH+). Anal. Calc’d for C20H23ClF2N2O2:C, 60.53; H, 5.84; N, 7.06. Found: C, 60.43; H, 5.69; N, 6.84.
Sn content: <1 ppm. Enantiomeric purity: 99.1% ee. Chiral SFCanalysis conditions: column: Chiralcel OF 250 mm × 4.6 mm;mobile phase: 30% ethanol, 0.4% diethylamine in CO2; detection wavelength: 254 nm; 2 mL/min, 40 °C.
* Corresponding author. E-mail: alimara@wyeth.com.
† Deceased.
‡ Wyeth Research, Collegeville, PA.
(1) (a) For a review on norepinephrine reuptake inhibitors, see: Babu,R. P. K.; Maiti, S. N. Heterocycles 2006, 69, 539. (b) Krell, H. V.;Leuchter, A. F.; Cook, I. A.; Abrams, M. Psychosomatics 2005, 46,379. (c) Hajos, M.; Fleishaker, J. C.; Filipiak-Reisner, J. K.; Brown,M. T.; Wong, E. H. W. CNS Drug ReV. 2004, 10, 23. (d) McCormack,
P. L.; Keating, G. M. Drugs 2004, 64, 2567.
(2) Kim, C. Y.; Mahaney, P. E.; Trybulski, E. J.; Zhang, P.; Terefenko,E. A.; McComas, C. C.; Marella, M. A.; Coghlan, R. D.; Heffernan,G. D.; Cohn, S. T.; Vu, A. T.; Sabatucci, J. P.; Ye, F. Phenylaminopropanol
Derivatives and Methods of Their Use. U.S. Patent 7,517,899,2009.
(3) Wu, Y.; Wilk, B. K.; Ding, Z.; Shi, X.; Wu, C. C.; RaveendranathP.; Durutlic, H. Process for the Synthesis of Progesterone ReceptorModulators. U.S. Patent Publ. Appl. US 2007/027327, 2007.
(4) (a) Gao, Y.; Hanson, R. M.; Klunder, J. M.; Ko, S. Y.; Masamune,H.; Sharpless, K. B. J. Am. Chem. Soc. 1987, 109, 5765. (b) For a recent example of large-scale asymmetric epoxidation, see: Henegar,
K. E.; Cebula, M. Org. Process Res. DeV. 2007, 11, 354.
(5) (a) For indolinone deprotonation for epoxide opening, see: Proudfoot,J. R.; Regan, J. R.; Thomson, D. S.; Kuzmich, D.; Lee, T. W.;Hammach, A.; Ralph, M. S.; Zindell, R.; Bekkali, Y. Preparation ofPropanol and Propylamine Derivatives and Their Use as Glucocorticoid Ligands. WO 2004/063163, 2004. (b) Gillman, K.; Bocchino, D. M.
Preparation of Monosaccharides Prodrugs of Fluorooxindoles Useful in Treatment of Disorders Which are Responsive to the Opening of Potassium Channels. U.S. Patent Publ. Appl. US 2004/0152646, 2004.
(c) For amide deprotonation for epoxide opening, see: Albanese, D.; Landini, D.; Penso, M. Tetrahedron 1997, 53, 4787. (d) Chan, W. N.; Evans, J. M.; Hadley, M. S.; Herdon, H. J.; Jerman, J. C.; Morgan,H. K. A.; Stean, T. O.; Thompson, M.; Upton, N.; Vong, A. K. J. Med.Chem. 1996, 39, 4537.
(6) Bordwell, F. G.; Fried, H. E. J. Org. Chem. 1991, 56, 4218.
(7) (a) Smith, J. G. Synthesis 1984, 629. (b) Parker, R. E.; Isaacs, N. S.Chem. ReV. 1959, 59, 737.
| Patent ID |
Date |
Patent Title |
| US7595338 |
2009-09-29 |
Process for preparing 3, 3-disubstituted oxindoles and thio-oxindoles |
| US2009099164 |
2009-04-16 |
Phenylaminopropanol Derivatives and Methods of Their Use |
| US7517899 |
2009-04-14 |
Phenylaminopropanol derivatives and methods of their use |
| US2009093469 |
2009-04-09 |
Phenylaminopropanol Derivatives and Methods of Their Use |
| US2008146645 |
2008-06-19 |
Process for Preparing Indolinone Phenylaminopropanol Derivatives |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2006118955A2 * |
Apr 27, 2006 |
Nov 9, 2006 |
Wyeth |
Process for preparing 3,3-disubstituted oxindoles and thio-oxindoles |
| WO2006118955A3 * |
Apr 27, 2006 |
Jan 11, 2007 |
Bogdan Kazimierz Wilk |
Process for preparing 3,3-disubstituted oxindoles and thio-oxindoles |
| WO2007041023A1 * |
Sep 27, 2006 |
Apr 12, 2007 |
Wyeth |
1- (1h- indol- 1-yl) -3- (methylamino) -1- phenylpropan-2-ol derivatives and related compounds as modulators of the monoamine reuptake for the treatment of vasomotor symptoms (vms) |
| WO2008024492A2 * |
Aug 22, 2007 |
Feb 28, 2008 |
Wyeth |
Indolinone phenylaminopropanol derivatives and process for the preparation thereof |
| WO2008024492A3 * |
Aug 22, 2007 |
May 15, 2008 |
Wyeth Corp |
Indolinone phenylaminopropanol derivatives and process for the preparation thereof |
| US9403807 |
Dec 2, 2014 |
Aug 2, 2016 |
Merck Sharp & Dohme Corp. |
Mineralocorticoid receptor antagonists |
| WO2005097744A1 * |
Mar 29, 2005 |
Oct 20, 2005 |
Wyeth |
1-(1h-indol-1-yl)-3-(4-methylpiperazin-1-yl)-1-phenyl propan-2-ol derivatives and related compounds as modulators of the norepinephrine (ne) and the serotonine (5-ht) activity and the monoamine reuptake for the treatment of vasomotor symptoms (vms) |
| WO2005097761A1 * |
Mar 29, 2005 |
Oct 20, 2005 |
Wyeth |
Heterocyclic phenylaminopropanol derivatives as modulators of the monoamine reuptake for the treatment of vasomotor symptoms (vms) |
| WO2007041023A1 * |
Sep 27, 2006 |
Apr 12, 2007 |
Wyeth |
1- (1h- indol- 1-yl) -3- (methylamino) -1- phenylpropan-2-ol derivatives and related compounds as modulators of the monoamine reuptake for the treatment of vasomotor symptoms (vms) |
| US20070072897 * |
Sep 27, 2006 |
Mar 29, 2007 |
Wyeth |
Phenylaminopropanol derivatives and methods of their use |
//////////WAY-315193
c1c2c(c(cc1)F)N(C(C2(C)C)=O)[C@@H](c3cc(ccc3)F)[C@@H](CNC)O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO

















 The presentation will load below
The presentation will load below














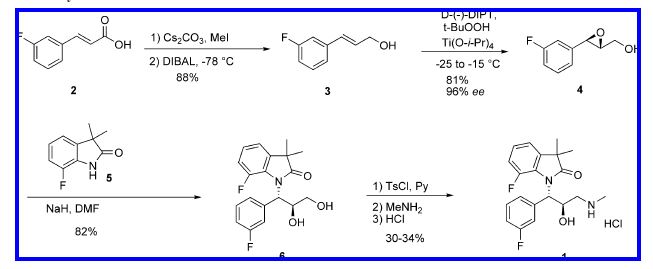





















 On with the show, though. The original synthetic route was provided in the paper and I will provide it to you.
On with the show, though. The original synthetic route was provided in the paper and I will provide it to you.









































 The presentation will load below
The presentation will load below

