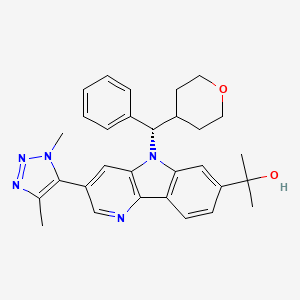

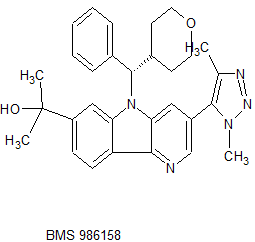
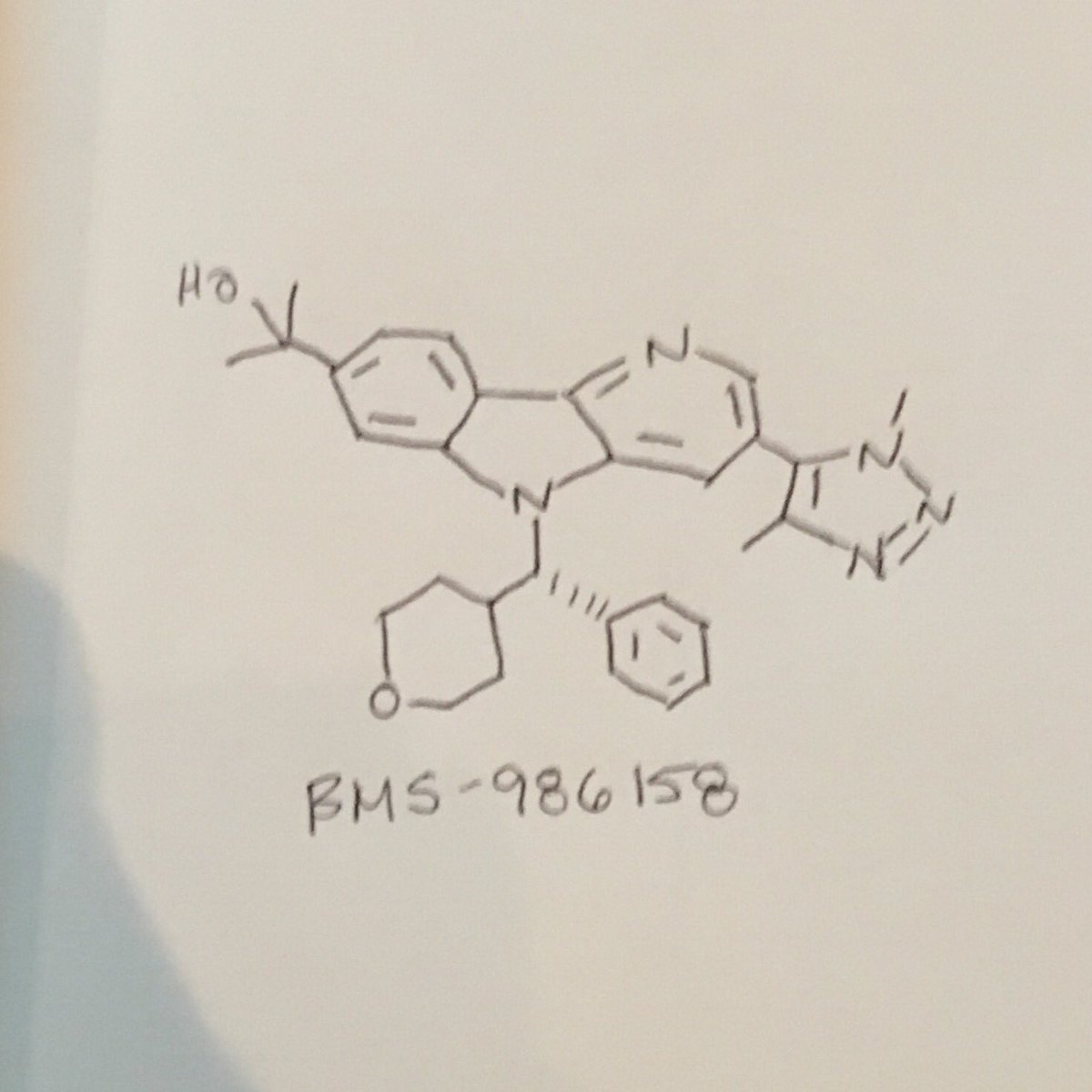
BMS 986158
MF C30H33N5O2, MW495.627 g/mol
CAS 1800340-40-2
5H-Pyrido[3,2-b]indole-7-methanol, 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-α,α-dimethyl-5-[(S)-phenyl(tetrahydro-2H-pyran-4-yl)methyl]-
MOA:Bromodomain and extraterminal domain protein inhibitor
Indication:Solid tumoursStatus:
Phase II :Bristol-Myers Squibb (Originator)
Phase I/IISolid tumours
- Originator Bristol-Myers Squibb
- Class Antineoplastics; Small molecules
- Mechanism of Action Bromodomain and extraterminal domain protein inhibitors
- 01 Jun 2015 Phase-I/II clinical trials for Solid tumours (Late-stage disease, Metastatic disease) in Canada (NCT02419417)
- 02 Apr 2015 Bristol-Myers Squibb plans a phase I/IIa trial for Solid tumours (Late-stage disease) in USA, Australia and Canada (NCT02419417)
The genomes of eukaryotic organisms are highly organized within the nucleus of the cell. The long strands of duplex DNA are wrapped around an octomer of histone proteins to form a nucleosome. This basic unit is then further compressed by the aggregation and folding of nucleosomes to form a highly condensed chromatin structure. A range of different states of condensation are possible, and the tightness of this structure varies during the cell cycle, being most compact during the process of cell division. There has been appreciation recently that chromatin templates form a fundamentally important set of gene control mechanisms referred to as epigenetic regulation. By conferring a wide range of specific chemical modifications to histones and DNA (such as acetylation, methylation, phosphorylation, ubiquitinylation and SUMOylation) epigenetic regulators modulate the structure, function and accessibility of our genome, thereby exerting a huge impact in gene expression.
Histone acetylation is most usually associated with the activation of gene transcription, as the modification loosens the interaction of the DNA and the histone octomer by changing the electrostatics. In addition to this physical change, specific proteins bind to acetylated lysine residues within histones to read the epigenetic code. Bromodomains are small (-110 amino acid) distinct domains within proteins that bind to acetylated lysine residues commonly but not exclusively in the context of histones. There is a family of around 50 proteins known to contain bromodomains, and they have a range of functions within the cell. The BET family of bromodomain containing proteins
comprises 4 proteins (BRD2, BRD3, BRD4 and BRD-T) which contain tandem bromodomains capable of binding to two acetylated lysine residues in close proximity, increasing the specificity of the interaction.
BRD2 and BRD3 are reported to associate with histones along actively
transcribed genes and may be involved in facilitating transcriptional elongation (Leroy et al, Mol. Cell. 2008 30(1):51-60), while BRD4 appears to be involved in the recruitment of the pTEF-I3 complex to inducible genes, resulting in phosphorylation of RNA polymerase and increased transcriptional output (Hargreaves et al, Cell, 2009 138(1): 1294145). All family members have been reported to have some function in controlling or executing aspects of the cell cycle, and have been shown to remain in complex with chromosomes during cell division – suggesting a role in the maintenance of epigenetic memory. In addition some viruses make use of these proteins to tether their genomes to the host cell chromatin, as part of the process of viral replication (You et al., Cell, 2004 117(3):349-60).
Recent articles relating to this target include Prinjha et al., Trends in
Pharmacological Sciences, March 2012, Vol. 33, No. 3, pp. 146-153; Conway, ACS Med. Chem. Lett., 2012, 3, 691-694 and Hewings et al, J. Med. Chem., 2012, 55, 9393-9413.
Small molecule BET inhibitors that are reported to be in development include GSK-525762A, OTX-015, TEN-010 as well as others from the University of Oxford and Constellation Pharmaceuticals Inc.
Hundreds of epigenetic effectors have been identified, many of which are chromatin-binding proteins or chromatin-modifying enzymes. These proteins have been associated with a variety of disorders such as neurodegenerative disorders, metabolic diseases, inflammation and cancer. Thus, these compounds which inhibit the binding of a bromodomain with its cognate acetylated proteins, promise new approaches in the treatment of a range of autoimmune and inflammatory diseases or conditions and in the treatment of various types of cancer.
| |
| Inventors |
Derek J. Norris, George V. Delucca, Ashvinikumar V. Gavai, Claude A. Quesnelle, Patrice Gill, Daniel O’MALLEY, Wayne Vaccaro, Francis Y. Lee, Mikkel V. DEBENEDETTO, Andrew P. Degnan, Haiquan Fang, Matthew D. Hill, Hong Huang, William D. Schmitz, JR John E. STARRETT, Wen-Ching Han, John S. Tokarski, Sunil Kumar MANDAL |
| Applicant |
Bristol-Myers Squibb Company |

PATENT
WO 2015100282
Examples 54 & 55
2-[3-(Dimethyl-lH-l,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2- b] indol-7-yl] pr opan-2-ol

Enantiomer A, Example 54 Enantiomer B, Example 55
Step 1 : 2-C hloro-5-(l ,4-dimethyl- 1H- 1 ,2,3-triazol-5-yl)pyridin-3-amine
To a 100 mL round bottom flask containing 5-bromo-2-chloropyridin-3-amine (2.90 g, 14.0 mmol), l,4-dimethyl-5-(tributylstannyl)-lH-l,2,3-triazole (2.70 g, 6.99 mmol) [Seefeld, M.A. et al. PCT Int. AppL, 2008, WO2008098104] and Pd(PPh3)4 (0.61 g, 0.52 mmol) in DMF (20 mL) was added cuprous iodide (0.20 g, 1.05 mmol) and Et3N (1.9 mL, 14.0 mmol). The reaction mixture was purged with N2 for 3 min and then heated at 100 °C for 1 h. After cooling to room temperature, the mixture was diluted withl0% LiCl solution and extracted with EtOAc (2x). The combined organics were washed with sat. NaCl, dried over MgS04, filtered and concentrated. CH2C12 was added, and the resulting precipitate was collected by filtration. The mother liquor was concentrated and purified using ISCO silica gel chromatography (40 g column, gradient from 0% to 100% EtOAc/CH2Cl2). The resulting solid was combined with the precipitate and triturated with cold EtOAc to give the title compound (740 mg, 47%) as a light tan solid. LCMS (M+H) = 224.1; HPLC RT = 1.03 min (Column: Chromolith ODS S5 4.6 x 50 mm; Mobile Phase A: 10:90 MeOH: water with 0.1% TFA; Mobile Phase B: 90: 10 MeOH:water with 0.1% TFA; Temperature: 40 °C; Gradient: 0-100% B over 4 min; Flow: 4 mL/min).
Step 2: Methyl 3-((2-chloro-5-(l,4-dimethyl-lH-l,2,3-triazol-5-yl)pyridin-3-yl)amino)benzoate
Following a procedure analogous to that described in Step 2 of Example 1, 2-chloro-5-(l ,4-dimethyl-lH-l,2,3-triazol-5-yl)pyridin-3-amine (740 mg, 3.31 mmol) was converted to the title compound (644 mg, 54%). 1H NMR (400 MHz, CDC13) δ 7.94 (t, J=1.9 Hz, 1H), 7.88 (d, J=2.1 Hz, 1H), 7.83 (dt, J=7.8, 1.3 Hz, 1H), 7.49 (t, J=7.9 Hz, 1H), 7.40 (d, J=2.1 Hz, 1H), 7.36 (ddd, J=8.0, 2.3, 0.9 Hz, 1H), 6.38 (s, 1H), 3.99 (s, 3H), 3.93 (s, 3H), 2.34 (s, 3H); LCMS (M+H) = 358.2; HPLC RT = 2.34 min (Column:
Chromolith ODS S5 4.6 x 50 mm; Mobile Phase A: 10:90 MeOH:water with 0.1% TFA; Mobile Phase B: 90: 10 MeOH:water with 0.1% TFA; Temperature: 40 °C; Gradient: 0-100% B over 4 min; Flow: 4 mL/min).
Step 3: Methyl 3-(l,4-dimethyl-lH-l,2,3-triazol-5-yl)-5H-pyrido[3,2-6]indole-7-carboxylate
Following a procedure analogous to that described in Step 3 of Example 1 , methyl 3-((2-chloro-5-(l,4-dimethyl-lH-l,2,3-triazol-5-yl)pyridin-3-yl)amino)benzoate (2.82 g, 7.88 mmol) was converted to the title compound (1.58 g, 62%). 1H NMR (500 MHz, DMSO-de) δ 11.93 (s, 1H), 8.62 (d, J=1.8 Hz, 1H), 8.36 (dd, J=8.2, 0.6 Hz, 1H), 8.29 -8.22 (m, 1H), 8.16 (d, J=1.8 Hz, 1H), 7.91 (dd, J=8.2, 1.4 Hz, 1H), 4.02 (s, 3H), 3.94 (s, 3H), 2.31 (s, 3H); LCMS (M+H) = 322.3; HPLC RT = 1.98 min (Column: Chromolith ODS S5 4.6 x 50 mm; Mobile Phase A: 10:90 MeOH:water with 0.1% TFA; Mobile Phase B: 90: 10 MeOH:water with 0.1% TFA; Temperature: 40 °C; Gradient: 0-100% B over 4 min; Flow: 4 mL/min).
Alternate synthesis of Methyl 3-(l,4-dimethyl-lH-l,2,3-triazol-5-yl)-5H-pyrido[3,2-b] indole-7-carboxylate
A mixture of methyl 3-bromo-5H-pyrido[3,2-b]indole-7-carboxylate (Step 2 of Example 40, 3.000 g, 9.83 mmol), l,4-dimethyl-5-(tributylstannyl)-lH-l,2,3-triazole (4.18 g, 10.82 mmol), copper (I) iodide (0.281 g, 1.475 mmol), Pd(Ph3P)4 (0.738 g, 0.639 mmol) and triethylamine (2.74 mL, 19.66 mmol) in DMF (25 mL) was purged under a nitrogen stream and then heated in a heating block at 95 °C for 2 hours. After cooling to room temperature the reaction mixture was diluted with water and extracted into ethyl acetate. Washed with water, NH4OH, brine and concentrated. The residue was triturated with 100 mL CHC13, filtered off the solid and rinsed with CHC13 to give. 1.6 g of product. The filtrate was loaded unto the ISCO column (330 g column, A: DCM; B:
10%MeOH/DCM, 0 to 100% gradient) and chromatographed to give an additional 0.7 g. of methyl 3 -( 1 ,4-dimethyl- 1 H- 1 ,2,3 -triazol-5 -yl)-5H-pyrido [3 ,2-b]indole-7-carboxylate (2.30 g total, 7.16 mmol, 72.8 % yield).
Step 4: Methyl 3-(l,4-dimethyl-lH-l,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Following a procedure analogous to that described in Step 4 of Example 1 , methyl 3-(l,4-dimethyl-lH-l,2,3-triazol-5-yl)-5H-pyrido[3,2-¾]indole-7-carboxylate (80 mg, 0.25 mmol) was converted to the title compound (65 mg, 53%) after purification by prep HPLC (Column: Phen Luna C 18, 30 x 100 mm, 5 μιη particles; Mobile Phase A: 5:95 acetonitrile: water with 0.1% TFA; Mobile Phase B : 95 : 5 acetonitrile: water with 0.1% TFA; Gradient: 10-100% B over 14 min, then a 2-min hold at 100% B; Flow: 40 mL/min). 1H NMR (400 MHz, CDC13) δ 8.51 (d, J=1.8 Hz, 1H), 8.50 (s, 1H), 8.47 (d, J=8.1 Hz, 1H), 8.10 (dd, J=8.1, 1.1 Hz, 1H), 7.63 (d, J=1.8 Hz, 1H), 7.46 (d, J=7.3 Hz, 2H), 7.40 – 7.30 (m, 3H), 5.62 (d, J=10.6 Hz, 1H), 4.11 – 4.03 (m, 4H), 3.92 – 3.83 (m, 4H), 3.56 (td, J=l 1.9, 1.8 Hz, 1H), 3.35 (td, J=l 1.9, 1.9 Hz, 1H), 3.18 – 3.05 (m, 1H), 2.30 (s, 3H), 2.04 (d, J=13.0 Hz, 1H), 1.71 – 1.58 (m, 1H), 1.50 – 1.37 (m, 1H), 1.09 (d, J=12.8 Hz, 1H); LCMS (M+H) = 496.3; HPLC RT = 2.93 min (Column: Chromolith ODS S5 4.6 x 50 mm; Mobile Phase A: 10:90 MeOH:water with 0.1% TFA; Mobile Phase B: 90: 10 MeOH:water with 0.1% TFA; Temperature: 40 °C; Gradient: 0-100% B over 4 min; Flow: 4 mL/min).
Step 5 : 2- [3-(Dimethyl- lH-1 ,2,3-triazol-5-yl)-5- [oxan-4-yl(phenyl)methyl] -5H-pyrido [3,2-6] indol-7-yl] pr opan-2-ol,
Following a procedure analogous to that described in Step 5 of Example 1 , methyl 3-(l ,4-dimethyl- IH- 1 ,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate (65 mg, 0.13 mmol) was converted to racemic 2-[3-(dimethyl-lH-l,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-¾]indol-7-yl]propan-2-ol, which was separated by chiral prep SFC (Column: Chiralpak IB 25 x 2 cm, 5 μιη; Mobile Phase: 70/30 C02/MeOH; Flow: 50 mL/min);to give Enantiomer A (24 mg, 36%) and Enantiomer B (26 mg, 38%). Enantiomer A: 1H NMR (500 MHz, CDC13) 5 8.44 (d, J=1.8 Hz, IH), 8.36 (d, J=8.2 Hz, IH), 7.98 (s, IH), 7.56 (d, J=1.7 Hz, IH), 7.47 – 7.41 (m, 3H), 7.37 – 7.32 (m, 2H), 7.31 – 7.28 (m, IH), 5.59 (d, J=10.5 Hz, IH), 4.06 (dd, J=11.8, 2.8 Hz, IH), 3.90 – 3.84 (m, 4H), 3.55 (td, J=11.9, 2.0 Hz, IH), 3.35 (td, J=11.9, 2.0 Hz, IH), 3.15 – 3.04 (m, IH), 2.30 (s, 3H), 2.04 (d, J=13.6 Hz, IH), 1.92 (s, IH), 1.75 (s, 6H), 1.69 – 1.58 (m, IH), 1.47 – 1.38 (m, IH), 1.12 (d, J=13.4 Hz, IH); LCMS (M+H) = 496.4; HPLC RT = 2.46 min (Column: Chromolith ODS S5 4.6 x 50 mm; Mobile Phase A: 10:90 MeOH:water with 0.1% TFA; Mobile Phase B: 90: 10 MeOH:water with 0.1% TFA; Temperature: 40 °C; Gradient: 0- 100% B over 4 min; Flow: 4 mL/min). SFC RT = 5.50 min (Column: Chiralpak IB 250 x 4.6 mm, 5 μιη; Mobile Phase: 70/30 C02/MeOH; Flow: 2 mL/min); SFC RT = 1.06 min (Column:
Chiralcel OD-H 250 x 4.6 mm, 5 μιη; Mobile Phase: 50/50 C02/(1 : 1 MeOH/CH3CN); Flow: 2 mL/min); [a]D2° = -117.23 (c = 0.08, CHC13). Enantiomer B: 1H NMR (500 MHz, CDC13) δ 8.44 (d, J=l .8 Hz, IH), 8.36 (d, J=8.2 Hz, IH), 7.98 (s, IH), 7.56 (d, J=1.7 Hz, IH), 7.47 – 7.41 (m, 3H), 7.37 – 7.32 (m, 2H), 7.31 – 7.28 (m, IH), 5.59 (d, J=10.5 Hz, IH), 4.06 (dd, J=11.8, 2.8 Hz, IH), 3.90 – 3.84 (m, 4H), 3.55 (td, J=11.9, 2.0 Hz, IH), 3.35 (td, J=l 1.9, 2.0 Hz, IH), 3.15 – 3.04 (m, IH), 2.30 (s, 3H), 2.04 (d, J=13.6 Hz, IH), 1.92 (s, IH), 1.75 (s, 6H), 1.69 – 1.58 (m, IH), 1.47 – 1.38 (m, IH), 1.12 (d, J=13.4 Hz, IH); LCMS (M+H) = 496.4; HPLC RT = 2.46 min (Column: Chromolith ODS S5 4.6 x 50 mm; Mobile Phase A: 10:90 MeOH:water with 0.1% TFA; Mobile Phase B: 90: 10 MeOH:water with 0.1% TFA; Temperature: 40 °C; Gradient: 0-100% B over 4 min; Flow: 4 mL/min). SFC RT = 8.30 min (Column: Chiralpak IB 250 x 4.6 mm, 5 μιη; Mobile Phase: 70/30 C02/MeOH; Flow: 2 mL/min); SFC RT = 2.83 min (Column: Chiralcel OD-H 250 x 4.6 mm, 5 μιη; Mobile Phase: 50/50 C02/(1 : 1 MeOH/CH3CN); Flow: 2 mL/min); [a]D2° = +88.78 (c = 0.10, CHC13).
Alternate Synthesis of Examples 54
2-[3-(Dimethyl-lH-l,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2- b] indol-7-yl] propan-2-ol.
Enantiomer A, Example 54
Step 1: (S)-methyl 3-(l,4-dimethyl-lH-l,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
The enantiomers of phenyl(tetrahydro-2H-pyran-4-yl)methanol ( 2.0 g, 10.4 mmol) [Orjales, A. et al. J. Med. Chem. 2003, 46, 5512-5532], were separated on preperative SFC. (Column: Chiralpak AD 5 x 25 cm, 5 μιη; Mobile Phase: 74/26
C02/MeOH; Flow: 270 mL/min; Temperature 30°C). The separated peaks were concentrated and dried under vacuum to give white solids. Enantiomer A: (S)-phenyl(tetrahydro-2H-pyran-4-yl)methanol: (0.91 g, 45.5%) SFC RT = 2.32 min
(Column: Chiralpac AD 250 x 4.6 mm, 5 μιη; Mobile Phase: 70/30 C02/MeOH; Flow: 3 mL/min); Temperature 40°C. Enantiomer B: (R)-phenyl(tetrahydro-2H-pyran-4-yl)methanol. (0.92 g, 46%) SFC RT = 3.09 min (Column: Chiralpac AD 250 x 4.6 mm, 5 μιη; Mobile Phase: 70/30 C02/MeOH; Flow: 3 mL/min); Temperature 40°C.
Following a procedure analogous to that described in Step 4 of Example 1 except using toluene (120mL) as the solvent, methyl 3-(l ,4-dimethyl-lH-l,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate (4 g, 12.45 mmol) and (R)-phenyl(tetrahydro-2H-pyran-4-yl)methanol (Enantiomer B above, 5.86 g, 30.5 mmol) was converted to the title compound (5.0 g, 81%). HPLC RT = 2.91 min (Column: Chromolith ODS S5 4.6 x 50 mm; Mobile Phase A: 10:90 MeOFLwater with 0.1% TFA; Mobile Phase B: 90: 10 MeOFLwater with 0.1% TFA; Temperature: 40 °C; Gradient: 0- 100% B over 4 min; Flow: 4 mL/min).
Step 2. (S)-2-[3-(Dimethyl-lH-l,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido [3,2-b] indol-7-yl] propan-2-ol
A 500 mL round bottom flask containing (S)-methyl 3-(l,4-dimethyl-lH-l,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate (5.0 g, 10.09 mmol) in THF (150 mL) was cooled in an ice/MeOH bath. MeMgBr, (3M in Et20, 17.0 mL, 51.0 mmol) was added slowly over 4 min. The resulting solution was stirred for 2 h and then quenched carefully with sat. NH4C1. The reaction mixture was diluted with 10% LiCl solution extracted with EtOAc. The organic layer was dried over MgS04, filtered and concentrated. The crude material was purified using ISCO silica gel chromatography (120 g column, gradient from 0%> to 6%>
MeOH/CH2Cl2). The product was collected and concentrated then dissolved in hot MeOH(35mL). To the mixture was added 15mL water and the mixture was cooled to room temperature. The resulting white precipitate was collected by filtration with 2: 1 MeOH/water rinse then dried under vacuum to give the title compound (3.2 g, 62%>). 1H
NMR (500 MHz, CDC13) δ 8.40 (d, J=1.8 Hz, 1H), 8.33 (d, J=8.2 Hz, 1H), 7.93 (s, 1H), 7.53 (d, J=l .8 Hz, 1H), 7.46 (d, J=7.3 Hz, 2H), 7.42 (dd, J=8.2, 1.4 Hz, 1H), 7.37 – 7.31 (m, 2H), 7.30 – 7.28 (m, 1H), 5.56 (d, J=10.5 Hz, 1H), 4.06 (d, J=8.9 Hz, 1H), 3.89 – 3.83 (m, 1H), 3.55 (td, J=11.9, 2.1 Hz, 1H), 3.35 (td, J=11.9, 2.1 Hz, 1H), 3.10 (q, J=10.8 Hz, 1H), 2.39 (s, 3H), 2.23 (s, 3H), 2.03 (d, J=14.2 Hz, 1H), 1.89 (s, 1H), 1.74 (s, 6H), 1.68 -1.59 (m, 1H), 1.46 – 1.36 (m, 1H), 1.12 (d, J=12.2 Hz, 1H); LCMS (M+H) = 496.3; HPLC RT = 2.44 min (Column: Chromolith ODS S5 4.6 x 50 mm; Mobile Phase A: 10:90 MeOH: water with 0.1% TFA; Mobile Phase B: 90: 10 MeOH: water with 0.1%
TFA; Temperature: 40 °C; Gradient: 0-100% B over 4 min; Flow: 4 mL/min); SFC RT = 2.01 min (Column: Chiralcel OD-H 250 x 4.6 mm, 5 μιη; Mobile Phase: 60/40 C02/(1 : 1 MeOH/CH3CN); Flow: 2 mL/min). SFC RT = 1.06 min (Column: Chiralcel OD-H 250 x 4.6 mm, 5 μιη; Mobile Phase: 50/50 C02/(1 : 1 MeOH/CH3CN); Flow: 2 mL/min).
| Patent ID |
Patent Title |
Submitted Date |
Granted Date |
| US9458156 |
Tricyclic compounds as anticancer agents |
2014-12-23 |
2016-10-04 |
3rd speaker at #MEDI 1st time disclosures is Ashvin Gavai of @bmsnews talking about an oral BET inhibitor to treat cancer #ACSSanFran

//////////
CC(C)(O)c2cc3n(c1cc(cnc1c3cc2)c4c(C)nnn4C)[C@@H](C5CCOCC5)c6ccccc6

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO





















![Graphical abstract: Identification of 4-N-[2-(4-phenoxyphenyl)ethyl]quinazoline-4,6-diamine as a novel, highly potent and specific inhibitor of mitochondrial complex I](http://pubs.rsc.org/services/images/RSCpubs.ePlatform.Service.FreeContent.ImageService.svc/ImageService/image/GA?id=C6MD00655H "Graphical abstract")