
Home » x ray contrast agent
Category Archives: x ray contrast agent
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Teprosulvose
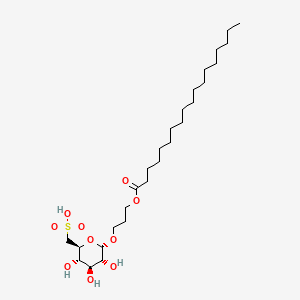
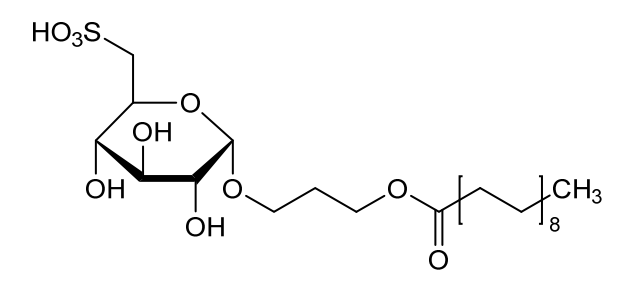
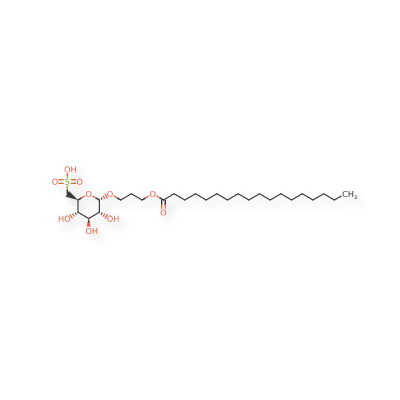
Teprosulvose
CAS 1983131-47-0
MF C27H52O10S MW568.761
| Sulfoquynovosylacylpropanediol [(2S,3S,4S,5R,6S)-3,4,5-trihydroxy-6-(3-octadecanoyloxypropoxy)oxan-2-yl]methanesulfonic acid |
3-(octadecanoyloxy)propyl 6-deoxy-6-sulfo-α-D-glucopyranoside
radiosensitizer (veterinary use), WV7377RGM8, SQAP
Teprosulvose (CAS 1983131-47-0) is a novel synthetic glycolipid, specifically a sulfoquinovosylacylpropanediol (SQAP). It is primarily developed for use in veterinary medicine as a radiosensitizer, intended to enhance the effectiveness of radiation therapy in treating malignant tumors.
1. Chemical Identity and Structure
- USAN/INN Name: Teprosulvose
- Systemic Name: 3-(octadecanoyloxy)propyl 6-deoxy-6-sulfo-$\alpha$-D-glucopyranoside
- Molecular Weight: 568.76 g/mol
- Structure: It consists of a glucose derivative (6-deoxy-6-sulfo-$\alpha$-D-glucopyranoside) linked via a propyl bridge to a long-chain fatty acid (stearic acid/octadecanoic acid).
Regulatory Data
Teprosulvose is currently in the investigational stage, primarily focused on veterinary oncology.
- USAN/INN Status: The name “Teprosulvose” was officially adopted by the USAN Council in 2024 (File LM-156).
- Classification: Radiosensitizer.
- Target Application: Adjuvant therapy for malignant tumors in animals (e.g., canine or feline cancers).
- Current Status: It has not yet received full FDA or EMA approval for human use. In the U.S., it is typically handled under Investigational New Animal Drug (INAD) protocols for clinical trials in veterinary patients.
Note: Because it is a specialized veterinary investigative agent, detailed safety data (LD50, pharmacokinetics) is generally found in specific FDA Freedom of Information (FOI) summaries or peer-reviewed veterinary oncology journals rather than standard human drug databases.
INN List 131 (WHO): Teprosulvose was officially included in the World Health Organization’s International Nonproprietary Names (INN) list in 2024. This confirms its unique status as a distinct drug substance.
USAN Council: The United States Adopted Names Council assigned the name in 2024, classifying it as a radiosensitizer.
FDA Status: It is currently under investigation (INAD) for canine oral melanoma and other solid tumors in veterinary medicine. Human clinical trial data is not yet widely available as the primary focus remains on the “Veterinary First” pathway.
Mechanism of Action: It is a potent inhibitor of DNA polymerase $\alpha$ and $\beta$. By inhibiting the repair of radiation-induced DNA damage, it effectively “locks in” the damage to tumor cells while sparing normal tissue due to differential uptake.
PAT
US Patent 10,206,942 (and related continuations): Covers the use of SQAP compounds in combination with radiation.
WO 2017/023812: International filing regarding the composition and therapeutic application of these glycolipids.
PAT
PAT
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-7973145-B2Priority Date: 2007-07-20Grant Date: 2011-07-05
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-2009209475-A1Priority Date: 2007-07-20
- Novel sulfonated sugar derivative, and use thereof for medicinal agentPublication Number: EP-2130834-A1Priority Date: 2007-07-20
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-2010298246-A1Priority Date: 2007-07-20
- Novel sulfonated sugar derivatives and their use as pharmaceuticalsPublication Number: JP-4435861-B2Priority Date: 2007-07-20Grant Date: 2010-03-24
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

//////////teprosulvose, radiosensitizer (veterinary use), WV7377RGM8, SQAP
Filricianine




Filricianine
CAS 2140857-94-7
MF C45H52N2O12S3, 909.1
3H-Indolium, 3-(3-carboxypropyl)-2-[2-[3-[2-[1,3-dihydro-3,3-dimethyl-1-(3-sulfopropyl)-2H-indol-2-ylidene]ethylidene]-2-(4-sulfophenoxy)-1-cyclohexen-1-yl]ethenyl]-3-methyl-1-(3-sulfopropyl)-, inner salt
3-[(2Z)-3-(3-carboxypropyl)-2-[(2E)-2-[3-[(E)-2-[3,3-dimethyl-1-(3-sulfopropyl)indol-1-ium-2-yl]ethenyl]-2-(4-sulfophenoxy)cyclohex-2-en-1-ylidene]ethylidene]-3-methylindol-1-yl]propane-1-sulfonate
3-[(3RS)-3-(3-carboxypropyl)-2-{(1Ξ)-2-[(3Ξ)-3-{(2Ξ)-2-[3,3-dimethyl-1-(3-sulfopropyl)- 1,3-dihydro-2H-indol-2-ylidene]ethylidene}-2-(4-sulfophenoxy)cyclohex-1-en-1-yl]ethen-1-yl}-3-methyl-3H-indol-1-ium-1-yl]propane-1-sulfonate
diagnostic imaging agent, CI4MD9KLX8

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////filricianine, diagnostic imaging agent, CI4MD9KLX8
Alizulatide vixocianine




Alizulatide vixocianine
CAS 2924859-51-6
MF C115H145N17O25S, 2,197.55
L-Serine, N-[6-[2-[7-[1,3-dihydro-1,1-dimethyl-3-(4-sulfobutyl)-2H-benz[e]indol-2-ylidene]-1,3,5-heptatrien-1-yl]-1,1-dimethyl-1H-benz[e]indolio]-1-oxohexyl]-L-α-glutamyl-L-α-glutamyl-L-α-aspartyl-3-cyclohexyl-L-alanyl-L-phenylalanyl-D-seryl-D-arginyl-L-tyrosyl-L-leucyl-L-tryptophyl-, inner salt
4-[2-[7-[3-[6-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2R)-1-[[(2R)-5-carbamimidamido-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(1S)-1-carboxy-2-hydroxyethyl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-cyclohexyl-1-oxopropan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-6-oxohexyl]-1,1-dimethylbenzo[e]indol-3-ium-2-yl]hepta-2,4,6-trienylidene]-1,1-dimethylbenzo[e]indol-3-yl]butane-1-sulfonate

diagnostic imaging agent, 8M3Q8XZ6MJ
Alizulatide vixocianine is a polypeptide that can be discovered through polypeptide screening. Polypeptide screening is a research tool mainly based on immunoassay methods to identify active polypeptides. It can be applied to protein interaction, functional analysis, antigenic epitope screening, especially in the fields of active molecule research and development.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////alizulatide vixocianine, diagnostic imaging agent, 8M3Q8XZ6MJ
Zopocianine



Zopocianine
CAS 2206660-94-6, NA SALT 2206660-95-7
MF C74H93N7O27S4, 1,640.83
L-Tyrosine, N-[[[(1S)-1,3-dicarboxypropyl]amino]carbonyl]-L-g-glutamyl-3-[2-(2-aminoethoxy)ethoxy]propanoyl-L-phenylalanyl-O-[6-[2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt
N-{[(1S)-1,3-dicarboxypropyl]carbamoyl}-L-γ-glutamyl3-[2-(2-aminoethoxy)ethoxy]propanoyl-L-phenylalanylO-[(6Ξ)-2-{(1Ξ)-2-[3,3-dimethyl-1-(4-sulfobutyl)-5-
sulfonato-3H-indol-1-ium-2-yl]ethen-1-yl}-6-{(2Ξ)-2-
[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2Hindol-2-ylidene]ethylidene}cyclohex-1-en-1-yl]-Ltyrosine
diagnostic imaging agent, UD9V5S9M7A, OTL 0078, OTL 78
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////zopocianine, diagnostic imaging agent, UD9V5S9M7A, OTL 0078, OTL 78
Talogreptide mesaroxetan


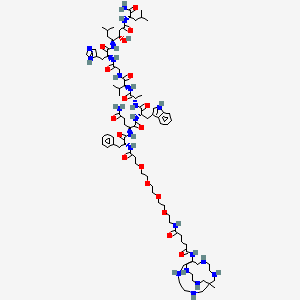
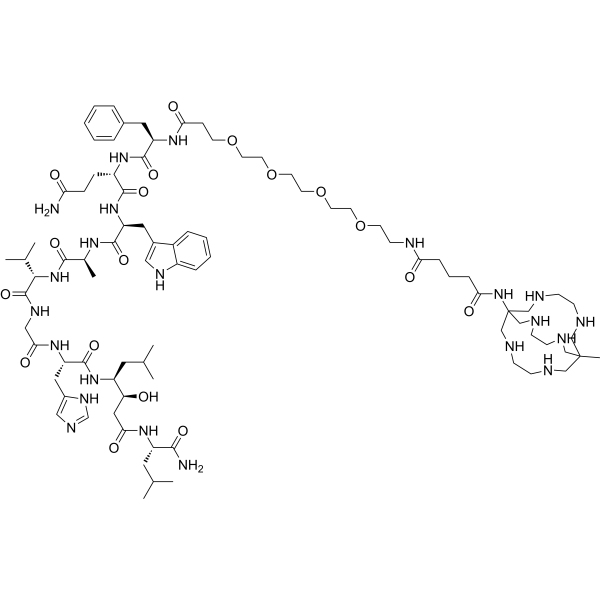
Talogreptide mesaroxetan
CAS 1801418-23-4
MF C86H140N22O18 MW1770.17
{MeCOSar}-PEG4-{d-Phe}-Gln-Trp-Ala-Val-Gly-His-{Sta}-Leu-NH2
(2S)-N-[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[[(2S)-1-[[(3S,4S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-3-hydroxy-6-methyl-1-oxoheptan-4-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]-2-[[(2R)-2-[3-[2-[2-[2-[2-[[5-[(8-methyl-3,6,10,13,16,19-hexazabicyclo[6.6.6]icosan-1-yl)amino]-5-oxopentanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]-3-phenylpropanoyl]amino]pentanediamide
N-{21-[(8-methyl-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosan-1-yl)amino] -17,21-dioxo-4,7,10,13-tetraoxa-16-azahenicosan-1-oyl}-D-phenylalanyl-L-glutaminyl-L-tryptophyl-L-alanyl-Lvalylglycyl-L-histidyl-(3S,4S)-4-amino-3-hydroxy-6-methylheptanoyl-L-leucinamide
diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Talogreptide mesaroxetan (CAS 1801418-23-4) is a synthetic peptide, a complex molecule used as a diagnostic imaging agent with potential antitumor effects, targeting G-protein coupled receptors (GRPr) often overexpressed in cancers, allowing for specific tumor visualization in PET scans, particularly for metastatic disease detection, known for its high specificity and contrast for imaging tumors like those expressing GRPr.
Key Characteristics:
- Type: A peptide-based diagnostic agent, often labeled with radioisotopes like Copper-64 ($^{64}$Cu) for Positron Emission Tomography (PET) imaging, notes Patsnap Synapse.
- Structure: It’s a modified peptide sequence incorporating elements like PEG4 and specific amino acids, MedchemExpress.com.
- Function: Binds strongly to GRPr, helping to highlight tumors and metastatic sites.
- Application: Used in research to create high-contrast PET scans for better tumor detection and monitoring, showing promise in visualizing lymph node metastasis.
In Simple Terms:
Imagine it as a “smart tracer” that seeks out specific cancer cells. When attached to a radioactive tag, it lights up tumors on a PET scan, helping doctors see cancer more clearly, notes Patsnap Synapse.
Syn
WO2024086891
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024086891&_cid=P10-MIZEJM-53111-1
67Cu radioisotope
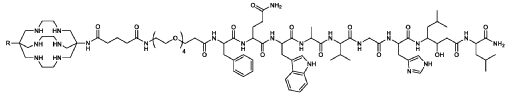
where R is CH3C(0)-;
(67CU-SAR-BBN)
Paper
Molecular Pharmaceutics (2015), 12(8), 2781-2790
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Talogreptide mesaroxetan, diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Mangaciclanol
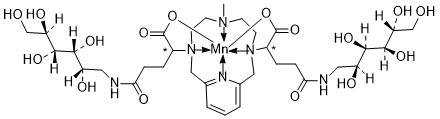
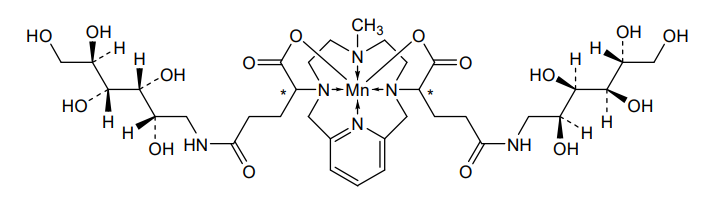
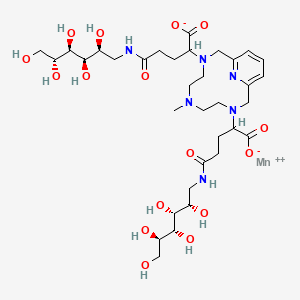
Mangaciclanol
Cas 2169771-05-3
MF C34H56MnN6O16 MW859.8 g/mol
- ANU6AE7NAP
- [[1,1′-[(6-Methyl-3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(15),11,13-triene-3,9-diyl-kappaN3,kappaN6,kappaN9,kappaN15)bis[[4-(carboxy-kappaO)-1-oxo-4,1-butanediyl]imino]]bis[1-deoxy-D-glucitolato]](2-)]manganese
2-[9-[1-carboxylato-4-oxo-4-[[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]amino]butyl]-6-methyl-3,6,9,15-tetrazabicyclo[9.3.1]pentadeca-1(15),11,13-trien-3-yl]-5-oxo-5-[[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]amino]pentanoate;manganese(2+)

diagnostic imaging agent, ANU6AE7NAP
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////mangaciclanol, diagnostic imaging agent, ANU6AE7NAP
Digadoglucitol




Digadoglucitol
DA-52534
CAS 2098944-37-5
μ-[2,2′,2”,2”’,2””,2””’-({[(2S,3R,4R,5R)-2,3,4,5,6- pentahydroxyhexyl]azanediyl}bis{[2-(hydroxy-κO)propane3,1-diyl]-1,4,7,10-tetraazacyclododecane-10,1,4,7-tetraylκ4 N1 ,N4 ,N7 ,N10})hexa(acetato-κO)]digadolinium diagnostic agent
C40H69Gd2N9O19
MW 1,294.536
F2Q2ZU6CAU
- Digadoglucitol free acid
- USL2PB6YFS
- 1-[Bis[2-hydroxy-3-[4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododec-1-yl]propyl]amino]-1-deoxy-D-glucitol
- 2098944-28-4 FREE ACID
- D-Glucitol, 1-[bis[2-hydroxy-3-[4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododec-1-yl]propyl]amino]-1-deoxy-

SCHEME

PATENT
Bracco Imaging SpA
WO2017098044
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017098044&_cid=P12-MAUH6W-49653-1



PATENT
WO2023006722
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023006722&_cid=P12-MAUHGF-56290-1
PATENT
WO2022023240
.///////////Digadoglucitol, F2Q2ZU6CAU, X RAY CONTRAST AGENT, DA-52534, DA 52534
Iomeprol



Iomeprol
- 78649-41-9
- Iomeprolum
- Iomeron
- Iomeprolo
WeightAverage: 777.089
Monoisotopic: 776.8541
Chemical FormulaC17H22I3N3O8
FDA APPROVED, 11/27/2024, Iomervu, For use as a radiographic contrast agent
1-N,3-N-bis(2,3-dihydroxypropyl)-5-[(2-hydroxyacetyl)-methylamino]-2,4,6-triiodobenzene-1,3-dicarboxamide
- N1,N3-Bis(2,3-dihydroxypropyl)-5-(2-hydroxy-N-methylacetamido)-2,4,6-triiodoisophthalamide
- 1-N,3-N-bis(2,3-dihydroxypropyl)-5-[(2-hydroxyacetyl)-methylamino]-2,4,6-triiodobenzene-1,3-dicarboxamide
- DTXCID2028987
- E7337
- N,N’-bis(2,3-dihydroxypropyl)-5-[glycoloyl(methyl)amino]-2,4,6-triiodoisophthalamide
Iomeprol, sold under the brand name Imeron among others, is a medication used as a radiocontrast agent in X-ray imaging.[1][2][3]
Iomeprol was approved for medical use in the United States in November 2024.[1][4][5]
Experimental Properties
| Property | Value | Source |
|---|---|---|
| melting point (°C) | 285-291 | https://www.chemos.de/import/data/msds/GB_en/78649-41-9-A0017152-GB-en.pdf |
| boiling point (°C) | 198 | https://datasheets.scbt.com/sds/eghs/en/sc-211652.pdf |
the first synthesis method is to take 5-amino-2, 4, 6-triiodoisophthalic acid as a starting material, methylate amino to chlorinate the starting material to prepare diacyl chloride, then use acetoxy acetyl chloride to carry out acylation reaction, then carry out amidation reaction with 3-amino-1, 2-propylene glycol, and finally use sodium hydroxide to hydrolyze the product to obtain iomeprol, as shown in the synthesis scheme 1
U.S. Patent No. 4,364,921 discloses three preparation processes of iopromide. One of them is shown in the following reaction scheme 1.
[Reaction scheme 1]

U.S. patent No. 4,364,921 are those basically under the same concept as shown in the following reaction schemes 3 and 4.
[Reaction scheme 3]

[Reaction scheme 4]

References:

M F C Co., Ltd.;Park Yung-ho;Hwang Seong-gwan;Park Jang-ha;Seo Rak-seok;Kim Gyeong-deok KR2020/77762, 2020, A Location in patent:Paragraph 0133-0136
Yield:78649-41-9 95%
Reaction Conditions:
with sodium hydroxide in N,N-dimethyl acetamide at 10 – 60; for 16 h;
Steps:
6 Synthesis of Iomeprole
Prepared N,N-bis(2,3-dihydroxypropyl)-5-(2-hydroxyacetamido)-2,4,6-triiodoisophthalamide 20 g (0.026 mol) in a reactor and 60 g of N,N-dimethylacetamide was added, followed by heating and stirring at 60 °C to dissolve.1.3 g (0.033 mol) of caustic soda was added to the reactor and stirred to dissolve. The reactor was cooled to 10 °C or less, and 4.5 g (0.032 mol) of iodomethane was added to the reactor, followed by stirring at 25 °C for 14 to 16 hours. After confirming the completion of the reaction, acetic acid was added to neutralize the reaction solution.The solvent was removed through concentration under reduced pressure at 60 to 65 °C and 60 g of ethanol was added to crystallize. The wet body was obtained by filtration and dried under reduced pressure in an oven at 60 °C for 12 hours to obtain iomeprol (yield 95%, purity 99.8%).
Iomeprol (CAS NO.: ), with its systematic name of , N,N’-bis(2,3-dihydroxypropyl)-5-((hydroxyacetyl)methylamino)-2,4,6-triiodo-, could be produced through many synthetic methods.
Following is one of the synthesis routes:
Firstly, 5-Amino-2,4,6-triiodo-1,3-benzenedicarboxylic acid (I) is treated with chloride to produce the dichloride (II). Secondly, compound (II) is treated with acetoxyacetyl chloride to afford compound (III), which is methylated with iodomethane. Lastly, condensation with 3-amino-1,2-propanediol of the N-methyl derivative (IV) thus obtained, followed by deacetylation with alkali metal hydroxide, yields iomeprol.

PATENT
https://patents.google.com/patent/WO2009134030A1/en
The process for preparing iopromide of formula (1) according to the present invention is shown in the following reaction scheme 5.
[Reaction scheme 5]

Step 1
5-amino-2,4,6-triiodoisophthalic acid dichloride of formula (2) is used as a starting material. The compound of formula (2) is reacted with methoxyacetyl chloride in dimethylacetamide solvent to produce 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid dichloride of formula (3) which is then used without an additional purification procedure for the next step. The compound of formula (3) is reacted with 2,3-dihydroxypropylamine in dimethylacetamide solvent in the presence of triethylamine to form 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride of formula (4), as shown in the following reaction scheme 6.
[Reaction scheme 6]

In the above reaction scheme, if 2,3-dihydroxypropylamine is used preferably in 0.6 to 1 equivalents, more preferably in 0.7 equivalents, the compound of formula (4) can be obtained in reasonable yield with minimizing the generation of the compound of formula (5) which is a bismer by-product.
In addition, since the unreacted compound of formula (3) existing in the filtrate obtained along with the compound of formula (4) can be recycled to the next batch without an additional recovery procedure, the loss of the yield of iopromide which occurs during the removal procedure of the bismer by-product generated in a large amount in conventional process can be prevented ultimately.
Step 2
The compound of formula (4) is reacted with acetic anhydride in acetic acid solvent in the presence of sulfuric acid as a catalyst to convert into the compound of formula (19). Sulfuric acid of preferably 0.01 to 0.2 moles, more preferably 0.05 to 0.1 moles per 1 molar reaction is added at a temperature of preferably from 0 to 30°C, more preferably from 5 to 25°C. Acetic anhydride of preferably 0.19 to 1 L, more preferably 0.35 to 0.7 L per 1 molar reaction is used.
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid-N,N’-bis-(2,3-diacetoxypropyl) diamide of the compound of following formula (21), which is generated by a simultaneous conversion of the bismer by-product of formula (5) already produced in the step 1 with the conversion of the compound of formula (4) into the compound of formula (19), is easily removed by the simple crystallization procedure of the compound of formula (19). That is, the by-product of the compound of formula (21) can be removed even without an additional removal procedure. It consequently means that the bismer by-product of the compound of formula (5) which is hard to remove according to conventional process can be effectively removed in the present invention even without any additional purification procedures.
[Formula 5]

[Formula 21]

Step 3
The compound of formula (19) is reacted with 2,3-dihydroxy-N-methypropylamine in dimethylacetamide solvent in the presence of triethylamine as a base to convert into the compound of formula (20). By the hydrolysis of the compound of formula (20) in aqueous NaOH solution without further purification therefor, iopromide of formula (1) can be obtained.
The present invention will be explained more specifically by the following examples. However, the examples are not intended to limit the scope of the present invention thereto.
Example 1: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride (Formula 4)
5-amino-2,4,6-triiodoisophthalic acid dichloride (13.7 kg, 23 mol) was dissolved in dimethylacetamide (17.2 kg) and the mixture was cooled to 15 ℃ . Methoxyacetyl chloride (3.74 kg, 34.5 mol) was added dropwise thereto for 2 hours, and then the mixture was stirred for 15 hours. After confirming the disappearance of the starting material by HPLC analysis for reaction, methylene chloride (45.7 kg) and water (11.5 kg) were subsequently added to the reaction mixture upon being stirred, and then the stirring was stopped and the layers became separated. The obtained organic layer was washed with aqueous sodium bicarbonate solution and concentrated by distillation under reduced pressure. To a solution of the obtained concentrate dissolved in dimethylacetamide (43.1 kg), triethylamine (1.95 kg, 19.32 mol) was added and then a solution of 2,3-dihydroxypropylamine (1.47 kg, 16.13 mol) dissolved in dimethylacetamide (10.78 kg) was added dropwise thereto for 5 hours with maintaining 0 to 5 ℃ . After additional 3 hours with stirring, the reaction mixture was concentrated by distillation under reduced pressure and to the concentrate, methylene dichloride (213.33 kg) was added dropwise for 5 hours to form solid. The solid was filtered and the title compound was obtained as a white solid (10.98 kg, yield 66.1 %).
1 H NMR(dmso-d6, 500MHz) 10.2,10.06(2s, 1H); 8.79, 8.71, 8.63 (3t, 1H); 4.5~4.0(br, 2H); 4.04, 4.00(2s, 2H); 3.71~3.66(m, 1H); 3.48, 3.47(2s, 3H); 3.40~3.36(m, 2H); 3.36~3.27(m, 1H); 3.2~3.09 (m, 1H)
Example 2: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (Formula 19)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-dihydroxypropyl)amide chloride (9.97 kg, 13.8 mol) was dispersed in acetic acid, and then anhydrous acetic acid (7.45 kg) was added thereto and the mixture was cooled to 5 ℃ . Sulfuric acid (135 g) was slowly added thereto and the mixture was stirred for 1 hour. To the obtained clear solution, sodium acetate trihydrate (376 g) was added and dissolved at 0 to 5 ℃ . And then water (96.6 kg) was added for 3 hours with maintaining 0 to 10 ℃ to produce solid. The produced solid was filtered and the title compound was obtained as a white solid (10.04 kg, yield 90.2 %).
1 H NMR(dmso-d6, 500MHz) 10.12, 9.99(2s, 1H); 8.91, 8.80(2t, 1H); 5.10~5.06(m, 1H); 4.32~4.27(m, 1H); 4.20~4.16(m, 1H); 4.01, 4.00(2s, 2H); 3.52~3.37(m, 2H); 3.47, 3.46(2s, 3H); 2.02(s, 6H)
Example 3: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-dihydroxypropyl)]diamide (iopromide)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (7.23 kg, 8.97 mol) was dissolved in dimethylacetamide (12.6 kg), and triethylamine (1.95 kg, 19.32 mol) was added thereto and a solution of 2,3-dihydroxy-N-methylpropylamine (943 g, 8.97 mol) dissolved in dimethylacetamide (4.2 kg) was added dropwise thereto at room temperature. After additional 2 hours with stirring, the solution was concentrated by distillation under reduced pressure. To an aqueous solution of the obtained 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-diacetoxypropyl)]diamide dissolved in water, a solution of sodium hydroxide (897 g, 22.43 mol) dissolved in water was added and the reaction mixture was stirred for 10 hours with maintaining 20 to 25 ℃ . The reaction solution was passed through cation exchange resin column and anion exchange resin column to produce a colorless and transparent aqueous solution. The obtained aqueous solution was distilled under reduced pressure to remove water completely, crystallized in ethanol and filtered to obtain a white crystalline iopromide (6.032 kg, yield 85 %).
1 H NMR(dmso-d6, 500MHz) 10.07, 10.03, 9.97, 9.90(4s,1H); 8.66, 8.57, 8.52(3t, 1H); 4.76~4.74(m, 1H); 4.72, 4.67(2t, 1H); 4.59~4.58(m, 1H); 4.54~4.44(m, 1H); 4.00(s, 2H); 3.89~3.88(m, 1H); 3.69~3.68(m, 2H); 3.47(s, 3H); 3.44~3.38(m, 4H); 3.23~3.17(m, 3H), 2.85~2.83(4s, 3H)
Example 4: Synthesis of 5-methoxyacetylamino-2,4,6-triiodoisophthalic acid [(2,3-dihydroxy-N-methylpropyl)-(2,3-diacetoxypropyl)]diamide (Formula 20)
5-methoxyacetylamino-2,4,6-triiodoisophthalic acid (2,3-diacetoxypropyl)amide chloride (80.65 g, 0.1 mol) was dissolved in dimethylacetamide (140.5 g), and then triethylamine (11.13 g, 0.11 mol) was added thereto and a solution of 2,3-dihydroxy-N-methylpropylamine (10.5 g, 0.1 mol) dissolved in dimethylacetamide (46.9 g) was added dropwise thereto at room temperature. After additional 2 hours with stirring, the produced solid was filtered and then the filtrate was concentrated by distillation under reduced pressure. To the obtained concentrate, diethyl ether was added dropwise to form solid. The formed solid was filtered and the title compound was obtained as a white solid (85.8 g, yield 98 %).
1 H NMR(dmso-d6, 500MHz) 10.10, 10.06, 10.00, 9.92(4s,1H); 8.93, 8.83, 8.78(3m, 1H); 5.09(br, 1H); 4.78~4.74(m, 1H); 4.62~4.58(m, 1H); 4.34~4.26(m, 1H); 4.22~4.16(m, 1H); 4.00(s, 2H); 3.89(br, 1H); 3.72~3.65,(m, 1H); 3.49~3.40(br, 2H);3.47(s, 3H); 3.48~3.38(m, 2H); 3.22~3.12,(m, 1H); 3.07~3.04(m, 1H); 2.87~2.82(m, 2H); 2.03(s, 3H); 2.02(s, 3H)
PATENT
https://patents.google.com/patent/RU2563645C2/ru
Scheme 3

Example 1Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 60
° C.In a 2 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was heated to 60 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 2 h. The reaction mixture was maintained at 60 °C for an additional 4 h, during which time the pH spontaneously remained in the range of 5-5.5. The red solution was cooled to 25°C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached a stable negative value in the range from 0 to -20 mV.During quenching of the reaction mixture, the pH was maintained at 5 by adding minimal amounts of 30% (w/w) aqueous NaOH solution.HPLC analysis (the results of which are shown in Fig. 1) showed the degree of conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (by area % on the HPLC chromatogram), and the resulting solution was used in the next stage of the synthesis without any further processing.Example 2Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 40
° CIn a 2 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, solid I
2 (250.6 g, 0.988 mol) was added in one portion to an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution, 0.816 mol; pH 9.6) heated to 40 °C. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g, 0.494 mol) was slowly added over 3 h. The reaction mixture was then heated for 2 h at 40°C, 1 h at 50°C and 1 h at 60°C, during which time the pH spontaneously remained in the range of 5-5.5. The resulting red solution was cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution during quenching, which was carried out by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of -20 to -50 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 3Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a stock solution heated to 30
° C and quenching the reaction mixture with bisulfite at pH5 in the final stepIn a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g of solution; 0.816 mol; pH 9.6) was diluted with H
2 O (1054 g), heated to 30 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h. The temperature of the reaction mixture was raised to 60°C and maintained at this temperature for a further 4 h, with the pH spontaneously remaining in the range of 5-5.5. The resulting red solution was cooled to 25°C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5 by adding 30% (w/w) aqueous NaOH solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of 0 to -20 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 4Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups using a starting solution heated to 30
° C and quenching the reaction mixture with bisulfite at pH7 in the final stepIn a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of the sodium salt of 3,5-disubstituted phenol 1 corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was diluted with H
2 O (1054 g), heated to 30 °C and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. When the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h. The temperature of the reaction mixture was raised to 60°C and maintained at this temperature for a further 4 h, during which time the pH spontaneously remained in the range of 5-5.5. The resulting red solution was cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution during quenching, which was carried out by adding 18% (w/w) aqueous sodium bisulfite solution until the color was lost and the oxidation-reduction potential, measured with a suitable electrode, reached stable negative values in the range of -20 to -50 mV.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 5Preparation of a compound of formula 2, in which both substituents R and R’ are -NH-CH
2 -CH(OH)CH
2 OH groups, using a starting solution at room temperature (approximately 20
° C)In a 4 L four-necked jacketed reactor equipped with a mechanical stirrer, reflux condenser and combination pH/temperature electrode, an aqueous solution of 3,5-disubstituted phenol 1 sodium salt corresponding to a phenol concentration of 22.8% (w/w) (1175 g solution; 0.816 mol; pH 9.6) was first diluted with H
2 O (1054 g) maintaining the temperature at 20 °C, and then solid I
2 (250.6 g; 0.988 mol) was added in one portion. The resulting solution was then heated to 40 °C and when the pH spontaneously dropped to 5, a 50% (w/w) aqueous solution of HIO
3 (173.6 g; 0.494 mol) was slowly added over 4 h . The temperature of the reaction mixture was raised to 60°C over 2 h and maintained at this temperature for a further 3 h, during which time the pH spontaneously remained in the range of 5-5.5. The red solution was then cooled to 25°C, the pH was adjusted to 7 and maintained at this level by adding 30% (w/w) aqueous NaOH solution, and quenched with sodium bisulfite (18% (w/w) aqueous solution) until color loss and stable negative values (in the range of -20 to -50 mV) of the oxidation-reduction potential were reached, measured with suitable redox electrodes.HPLC analysis showed the conversion of the starting compound to 3,5-disubstituted-2,4,6-triiodophenol 2b to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further treatment.Example 6Preparation of a compound of formula 2 in which the substituent R is a -NH-CH
2 -CH(OH)CH
2 OH group and R’ is -NH-CH(CH
2 OH)
2 using a starting solution heated to 60
° C.In a 1 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser, and combination pH/temperature electrode, N-(2,3-dihydroxypropyl)-N’-[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-1,3-benzenedicarboxamide (100.3 g, 0.305 mol) was dissolved in H
2 O (430 g) and converted to the corresponding sodium salt by adding 30% (w/w) NaOH (40.6 g, 0.305 mol) (pH 9.5). The solution was heated to 60 °C and solid I
2 (93.1 g, 0.367 mol) was added in one portion; When the pH spontaneously dropped to 5, 50% (w/w) aqueous HIO3 (64.5 g, 0.183 mol) was added slowly over 2 h
. The reaction temperature was maintained at 60 °C for a further 4 h, during which time the pH of the reaction mixture spontaneously remained in the range 5-5.5. The resulting red solution was cooled to 25 °C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5, by adding 30% (w/w) aqueous NaOH until colourlessness and a stable negative oxidation-reduction potential, measured with a suitable oxidation-reduction electrode, in the range 0 to -20 mV.HPLC analysis (Fig. 2) showed the degree of conversion of the starting compound to N-(2,3-dihydroxypropyl)-N’-[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-2,4,6-triiodo-1,3-benzenedicarboxamide >98% (by area % on the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any additional processing.Example 7Preparation of a compound of formula 2 in which both substituents R and R’ are -NH-CH(CH
2 OH)
2 groups using a starting solution heated to 60
° C.In a 0.5 L jacketed four-neck reactor equipped with a mechanical stirrer, reflux condenser, and combination pH/temperature electrode, N,N’-bis[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-1,3-benzenedicarboxamide (50 g, 0.152 mol) was dissolved in H
2 O (215 g) and converted to the corresponding sodium salt by adding 30% (w/w) NaOH (20.3 g, 0.152 mol) (pH 9.5). The solution was heated to 60 °C and solid I
2 (46.4 g, 0.183 mol) was added in one portion; When the pH spontaneously dropped to 5, 50% (w/w) aqueous HIO3 (32.2 g, 0.091 mol) was added slowly over 2 h
. The reaction temperature was maintained at 60 °C for an additional 4 h, during which time the pH of the reaction mixture spontaneously remained in the range 5-5.5. The resulting red solution was cooled to 25 °C and quenched by adding 18% (w/w) aqueous sodium bisulfite solution, maintaining the pH at 5, with 30% (w/w) aqueous NaOH until colorlessness and a stable negative oxidation-reduction potential (in the range of -20 to -50 mV), as measured by a suitable oxidation-reduction electrode.HPLC analysis showed the conversion of the starting compound to N,N’-bis[2-hydroxy-1-(hydroxymethyl)ethyl]-5-hydroxy-2,4,6-triiodo-1,3-benzenedicarboxamide to be >98% (based on area % of the HPLC chromatogram), and the resulting solution was used in the next step of the synthesis without any further processing.Comparative example 1This test was carried out to evaluate the efficiency of the iodination reaction disclosed by Patil et al., in ARKIVOC, 2006, 104 and Tetrahedron Lett., 2005, 46, 7179.In a 50 mL three-necked round-bottomed flask equipped with a thermometer and a reflux condenser, solid 3,5-disubstituted phenol 1 (16.4 g, 50 mmol) was suspended in ethanol (30 mL). Then, to the resulting suspension, heated to 38-40 °C, solid I
2 (15.2 g, 60 mmol) was added in one portion and a solution of HIO
3 (5.3 g, 30 mmol) in H
2 O (3 mL) was added in the indicated order over 5 min. The resulting dark brown mixture was stirred at 38-40 °C for about 1 h and then the change in appearance of the reaction mixture was recorded, which turned into a clear dark brown solution. The reaction mixture was maintained at the above temperature for a total of 3.5 h, then cooled to room temperature, which caused crystallization of the pale yellow solid product. After 15 h at room temperature, the solid was isolated by filtration and dried to give the desired 3,5-disubstituted-2,4,6-triiodophenol (12.1 g, 17 mmol). Yield 34.3%.During the iodination reaction, the reaction mixture was analyzed by HPLC. In particular, the first analysis was performed 1.5 hours after the start of iodination (the reaction time was chosen based on the literature sources cited), and its results are shown in Fig. 3, and the second analysis was performed after another 2 hours (the total reaction time was 3.5 hours), and its results are shown in Fig. 4. The results show that even after 3.5 hours, the conversion is not complete and a significant amount (13% based on the area on the HPLC chromatogram) of the original substrate is still present. On the other hand, a longer reaction time leads to the formation of a significant amount of impurities, which are decay products, which are easily detected after 3.5 hours of reaction (Fig. 4). This is certainly a factor that adversely affects the reaction yields. However, the low reaction yields can also be attributed to the solubility of 3,5-disubstituted-2,4,6-triiodophenol 2b in an alcohol medium, as confirmed by the analysis of the mother liquor shown in Fig. 5, which interferes with the quantitative isolation of the iodination product.In this regard, the increase in both the reaction yield and the product purity due to the use of an aqueous medium and reaction conditions, expressed in the above results, is evident from a comparison of Figs. 3-5 with Figs. 1 and 2, which show chromatograms (HPLC) of crude reaction solutions (Examples 1 and 6, respectively) obtained using the method of the present invention.
PATENT
https://patents.google.com/patent/KR20200032280A/en
[Scheme 3]

Example One.
Iomeproletic Produce
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) 5g (1 equivalent) and 3.6 g (5 equivalents) of calcium chloride was added to 25 g of methanol together, and dissolved at reflux at room temperature or 70 ° C. for 60 minutes.
After cooling the temperature of the solution to 10 ℃ to 15 ℃ 0.3g (0.62 equivalents) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred for 3 hours at the same temperature until the reaction was completed.
After completion of the reaction, 1 mL of HCl (35%) was added to acidify, 25 mg of 2-butanol was added, stirred at a temperature of 70 to 80 ° C. for 2 hours, cooled, filtered and washed with 2-butanol to obtain an iomeprole crude product.
The above prepared omeprolol was added to a mixture of 25 mL of methanol and 10 mL of water, heated to 50 ° C. to dissolve, put 20 mL of 2-butanol, refluxed at 90 ° C. for 3 hours, cooled to room temperature, and the resulting crystal was filtered.
After washing with 2-butanol and drying under reduced pressure at 90 ° C. for 12 hours, 4.17 g of Iomeprole (HPLC: 99.3%) was obtained.
Example 2.
Preparation of Iomeprole
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) 5g (1 equivalent) and 3.6 g (5 equivalents) of calcium chloride was added to 25 g of methanol together and refluxed at 70 ° C for 30 minutes to dissolve.
After cooling the solution to 0-5 ° C., 0.3 g (0.62 equivalents) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred for 7 hours at the same temperature until the reaction was completed.
After completion of the reaction, 1 mL of HCl (35%) was added to acidify, 30 mL of 2-butanol was added, refluxed at a temperature of 70-80 ° C. for 2 hours, stirred, filtered and washed with 2-butanol to obtain an iomeprole crude product.
After adding the above prepared omeprolol to a mixture of 25 mL of methanol and 10 mL of water, the temperature was raised to 50 ° C. to dissolve, 20 mL of 2-butanol was added, refluxed at 90 ° C. for 3 hours, cooled to room temperature, and the resulting crystal was filtered. .
After washing with 2-butanol and drying under reduced pressure at 90 ° C. for 12 hours, 4.21 g of Iomeprole (HPLC: 99.1%) was obtained.
Comparative example 1 (Inorganic chloride Non-addition )
After adding 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) to 25 g of methanol It was refluxed for 30 minutes.
After the temperature of the turbid solution was cooled to 10 ° C to 15 ° C, 0.3 g (0.62 equivalent) of calcium hydroxide was added and stirred at the same temperature for 1 hour.
2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred at the same temperature for 5 hours.
As a result of reactivity review by HPLC, synthesis of iomeprole progressed 5%, 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6- Triiodoisophthalamide (1b) was found to be 90% or more remaining, so that the reactivity was very low.
Comparative example 2 (Inorganic base Non-addition )
5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6-triiodoisophthalamide (1b) and 3.6 g of calcium chloride are methanol After adding to 25 g, the mixture was refluxed for 30 minutes to dissolve.
After the temperature of the solution was cooled from 10 ° C to 15 ° C, 2.48 g (3 equivalents) of dimethyl sulfate was added to the reaction solution and stirred at the same temperature for 5 hours.
As a result of reactivity review by HPLC, synthesis of iomeprole proceeds 0.5%, 5- (2-hydroxyacetamido) -N, N’-bis (2, 3-dihydroxypropyl) -2, 4, 6- Triiodoisophthalamide (1b) was found to be very low reactivity with more than 99% remaining.
PATENT
https://patents.google.com/patent/CN102363600B/en
.Synthetic route is seen formula 1:

Embodiment
Embodiment 1
5-methylamino-2,4,6-triiodo m-phthaloyl chloride synthetic:
In the there-necked flask that agitator and reflux condensing tube are housed, 76g(0.133mol under the room temperature) 5-methylamino-2,4,6-triiodo m-phthalic acid is dissolved in 250mL(2.53mol) in the ethyl acetate, after waiting to stir, add 29mL(0.4mol) sulfur oxychloride, be warming up to 50 ℃ of acyl chloride reaction temperature then, stir and finished reaction in 6 hours.After treating that ethyl acetate and sulfur oxychloride boil off under the decompression, residue boils off solvent and washs through frozen water after adding the 100mL ethyl acetate, dry product 68.9g, yield is that 84.9%(is with 5-methylamino-2,4,6-triiodo m-phthalic acid meter), m.p.167 ~ 169 ℃.
Embodiment 2
5-methylamino-2,4,6-triiodo m-phthaloyl chloride synthetic:
The acyl chloride reaction temperature is 80 ℃, and in 3 hours reaction times, all the other are operated with embodiment 1.
Embodiment 3
5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride synthetic:
In reaction flask, add 36.6g(0.06mol under the room temperature) 5-methylamino-2,4,6-triiodo m-phthaloyl chloride and 80mL N,N-dimethylacetamide, heating in water bath to 50 ℃, after the stirring and dissolving, be cooled to 10 ℃, begin to drip 10.2g(0.09mol) chloroacetyl chloride, dropwising the back heats up 50 ℃, stirred 3 hours, and be cooled to 10 ℃, be added dropwise to the 150mL frozen water.Filter, through the frozen water washing, dry product 38.7g, yield be 94%(with 5-methylamino-2,4,6-triiodo m-phthaloyl chloride meter), m.p.194 ~ 196 ℃.
Embodiment 4
5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride synthetic:
The chlorine acylation temperature is 90 ℃, and in 1 hour reaction times, all the other are operated with embodiment 3.
Embodiment 5
5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
In reaction flask, add 41.2g(0.06mol under the room temperature) 5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride and 80mL N, the N-N,N-DIMETHYLACETAMIDE after the stirring and dissolving, is cooled to 10 ℃, add 16g(0.16mol) triethylamine and 14.6g(0.16mol) 3-amino-1, the 2-propylene glycol is heated to 20 ℃ then, stirs and finishes reaction in 15 hours, be chilled to after-filtration below 10 ℃, after evaporated under reduced pressure, residue is dissolved in the 160mL methyl alcohol with filtrate, adds 200mL water and stirs evenly, leave standstill, with the solid filtering of separating out, the washing, dry product 43.4g, yield is that 91%(is with 5-[N-methyl-2-chloracetyl amido]-2,4,6-triiodo m-phthaloyl chloride meter), m.p.204 ~ 207 ℃.
Embodiment 6
5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
The amidate action temperature is 50 ℃, and in 8 hours reaction times, all the other are operated with embodiment 5.
Embodiment 7
5-[N-methyl-glycolamide base]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide synthetic:
In reaction flask, add 47.7g(0.06mol) 5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2,3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide, 250mL water, stir and to add 26g(0.32mol down) sodium acetate, back flow reaction is after 24 hours, the pressure reducing and steaming solvent.The residue dissolve with methanol filters, and filtrate boils off methyl alcohol, and vacuum-drying gets solid, is dissolved in the 150mL water, adds gac 1.8g, and reflux 30min filters.Filtrate is successively respectively by 732 Zeo-karbs and 717 anionite-exchange resin, and pressure reducing and steaming solvent again is after the resistates vacuum-drying, add 180mL ethanol and carry out recrystallization, get white solid 40.1g, HPLC detects purity greater than 99.0%, and yield is that 86%(is with 5-[N-methyl-2-chloracetyl amido]-N, N ‘-two (2, the 3-dihydroxypropyl)-2,4,6-three iodo-1,3-benzenedicarboxamide meter), m.p.〉280 ℃. 1H-NMR?(DMSO-D 6)δ(ppm):2.91(s,4H),3.21~3.36(m,4H),?3.41~3.65(m,4H),?3.85(s,2H),?3.98(m,2H),?4.1~?4.5(m,5H),?8.2~?8.4(d,?2H); 13C-NMR(D 2O)δ(ppm)33.2,44.7,60.7,64.6,71.2,90.1,99.8,99.9,145.5,150.5,150.6,171.3,171.4,173.9。
PATENT
https://patents.google.com/patent/CN114213273A/en
synthetic route is as follows:

Example 1
The synthesis process of iomeprol with 5-amido-N, N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodo-1, 3-benzenedicarboxamide as initial material includes successive chloroacetylation, methylation, hydrolysis and hydroxylation to synthesize iomeprol product, and includes the following steps:

the specific process comprises 4 reaction steps:
s1, chloroacetylation (preparation of 5- (2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
12g of 5-amino-N.N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodoisophthalamide was dissolved in 24g N, N-dimethylacetamide and stirred at room temperature. 12g of chloroacetyl chloride were added while controlling the temperature below 60 ℃. After the addition, the temperature is kept between 50 and 60 ℃, the stirring is carried out for 4 hours, and after the reaction is finished, the vacuum concentration is carried out below 65 ℃ until the reaction is dried. 36ml of ethyl acetate and 36ml of a 5% aqueous solution of sodium hydrogencarbonate were added to the concentrate to conduct extraction. The aqueous layer after separation was extracted twice with 10ml of ethyl acetate. The organic solutions thus extracted were mixed, 15ml of 5% saline was added thereto, and the mixture was washed, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 18.5g of an oily substance.
S2, methylation reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
dissolving 18.5g of the oily substance in the previous step by using 37ml of acetone, cooling to 0 ℃, adding 3.8g of potassium carbonate, keeping the temperature and stirring, dropwise adding 5.8g of dimethyl sulfate while stirring, keeping the temperature and reacting for 8 hours, after the reaction is finished, carrying out vacuum concentration to dryness at 60 ℃, dissolving the concentrated solution in 50ml of ethyl acetate and 50ml of water, and adding 10ml of ethyl acetate into the separated water layer for secondary extraction. The organic solutions thus extracted were mixed, 15ml of 5% saline was added thereto, and the mixture was washed, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 19.3g of an oily substance.
S3, ester hydrolysis reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide):
the above oily substance was dissolved in 20ml of methanol, 6.0g of water was added, 30.0g of 30% aqueous sodium hydroxide solution was added, and the reaction was carried out at 15 ℃ for 4 hours, and after the completion of the reaction, methanol was distilled off under reduced pressure to obtain an aqueous solution containing the objective compound.
S4 hydroxylation reaction (preparation of 5- (N-methyl-2-hydroxyacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide, i.e. iomeprol):
adding 6.0g of sodium acetate into the aqueous solution, heating to reflux for 24 hours, distilling under reduced pressure to remove the solvent, adding methanol into the residue to dissolve, filtering, evaporating the methanol from the filtrate, dissolving the residue in pure water, adding activated carbon, heating to reflux for 30 minutes, filtering, sequentially passing the filtrate through cation resin and anion resin, evaporating to remove the solvent, adding 70ml of ethanol to recrystallize, filtering, drying under reduced pressure at 50 ℃ to obtain 8.5g of white solid, wherein the HPLC purity is 99.2%, and the molar yield is 64%.
Example 2
S1, chloroacetylation (preparation of 5- (2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
500g of 5-amino-N.N’ -bis (2, 3-dihydroxypropyl) -2,4, 6-triiodoisophthalamide was dissolved in 1000g N, N-dimethylacetamide and stirred at room temperature. 500g of chloroacetyl chloride was added while controlling the temperature below 60 ℃. After the addition, the temperature is kept between 50 and 60 ℃, the stirring is carried out for 4 hours, and after the reaction is finished, the vacuum concentration is carried out below 65 ℃ until the reaction is dried. 1500ml of ethyl acetate and 1500ml of 5% aqueous sodium bicarbonate solution were added to the concentrate to extract. The aqueous layer after separation was extracted twice with 480ml of ethyl acetate. The organic solutions thus extracted were mixed, and 650ml of 5% brine was added thereto, followed by washing, and after recovering the organic solution layer, magnesium sulfate was added to remove water, followed by filtration and distillation under reduced pressure to obtain 772.6g of an oil.
S2, methylation reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-bis (2-chloroacetoxy) propyl) -isophthalamide):
dissolving 772.6g of the oily matter in the previous step by 1545ml of acetone, cooling to 0 ℃, adding 158.5g of potassium carbonate, keeping the temperature and stirring, dropwise adding 241.5g of dimethyl sulfate while stirring, keeping the temperature and reacting for 8 hours, after the reaction is finished, vacuum concentrating at 60 ℃ to dryness, dissolving the concentrated solution in 1500ml of ethyl acetate and 1500ml of water, and adding 480ml of ethyl acetate into the separated water layer for secondary extraction. The organic solutions thus extracted were mixed, and then 650ml of 5% saline was added thereto, followed by washing, and after recovering the organic solution layer, magnesium sulfate was added thereto to remove water, followed by filtration and distillation under reduced pressure to obtain 805.7g of an oily substance.
S3, ester hydrolysis reaction (preparation of 5- (N-methyl-2-chloroacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide):
the above oil was dissolved in 800ml of methanol, 250g of water was added, 1250g of a 30% aqueous solution of sodium hydroxide was added, and the reaction was carried out at 15 ℃ for 4 hours, and after the completion of the reaction, methanol was distilled off under reduced pressure to obtain an aqueous solution containing the objective compound.
S4 hydroxylation reaction (preparation of 5- (N-methyl-2-hydroxyacetamido) -2,4, 6-triiodo-N, N’ -bis (2, 3-dihydroxypropyl) -isophthalamide, i.e. iomeprol):
adding 250g of sodium acetate into the aqueous solution, heating to reflux for 24 hours, distilling under reduced pressure to remove the solvent, adding methanol into the residue to dissolve, filtering, evaporating the filtrate to remove the methanol, dissolving the residue in pure water, adding activated carbon, heating to reflux for 60 minutes, filtering, sequentially passing the filtrate through cation resin and anion resin, evaporating to remove the solvent, adding 3000ml of ethanol to recrystallize, filtering, drying under reduced pressure at 50 ℃ to obtain 365.0g of white solid, wherein the HPLC purity is 99.1%, and the molar yield is 66%.
Compared with two synthesis methods in patent EP0026281A1, the synthesis method of iomeprol developed by the invention does not use thionyl chloride, reduces the difficulty of waste gas treatment, does not use acetoxy acetyl chloride, and ensures the process stability. Meanwhile, the used starting material (compound II) is a common intermediate of other contrast agents iohexol and ioversol, so that corresponding supporting construction is reduced.
Therefore, the synthetic route designed by the invention has the advantages of mild reaction conditions, stable quality, high yield, low cost, environmental protection and suitability for industrial production.
The embodiments are described in a progressive manner, each embodiment focuses on differences from other embodiments, and the same or similar parts among the embodiments are referred to each other. The device disclosed by the embodiment corresponds to the method disclosed by the embodiment, so that the description is simple, and the relevant points can be referred to the method part for description.
Side effects
It is classified as a water-soluble, nephrotrophic, low osmolar X-ray contrast medium.[2] Low osmolar non-ionic agents are better tolerated and less likely to cause side effects than the high osmolar ionic agents.[2]
Society and culture
Iomeprol is not metabolized in the human body but excreted in unchanged form.[medical citation needed] It is decomposed slowly and can therefore accumulate in the environment.[6]
Legal status
Iomeprol was approved for medical use in the United States in November 2024.[1][4]
Brand names
Iomeprol is sold under the brand name Iomervu.[1]
References
- ^ Jump up to:a b c d e https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/216017s000,216017s000lbl.pdf
- ^ Jump up to:a b c Rossiter D (2014). South African medicines formulary (11th ed.). Rondebosch, South Africa: Health and Medical Pub. Group .of the South African Medical Association. ISBN 978-1-875098-30-9. OCLC 869772940.
- ^ Haberfeld H, ed. (2020). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Iomeron 300 mg J/ml-Infusionsflasche.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Pfundstein P, Martin C, Schulz W, Seitz W, Ruth KM, Wille A, et al. (January 2015). “IC-ICP/MS-Analytik”. GIT Labor-Fachzeitschrift (in German): 29–31.
- Dooley M, Jarvis B: Iomeprol: a review of its use as a contrast medium. Drugs. 2000 May;59(5):1169-86. doi: 10.2165/00003495-200059050-00013. [Article]
- Katayama H, Spinazzi A, Fouillet X, Kirchin MA, Taroni P, Davies A: Iomeprol: current and future profile of a radiocontrast agent. Invest Radiol. 2001 Feb;36(2):87-96. doi: 10.1097/00004424-200102000-00004. [Article]
- Rosati G: Clinical pharmacology of iomeprol. Eur J Radiol. 1994 May;18 Suppl 1:S51-60. doi: 10.1016/0720-048x(94)90094-9. [Article]
- EMC Summary of Product Characteristics: Iomeron (iomeprol) solution for injection [Link]
- FDA Approved Drug Products: IOMERVU (iomeprol) injection, for intra-arterial or intravenous use [Link]
- Medsafe NZ: IOMERON® (iomeprol) safety data sheet [Link]
| showvteContrast media (V08) |
|---|
| Clinical data | |
|---|---|
| Trade names | Iomervu, others |
| License data | US DailyMed: Iomeprol |
| Routes of administration | Intravenous, intra-arterial |
| ATC code | V08AB10 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Metabolism | none |
| Elimination half-life | 109±20 min |
| Excretion | Kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 78649-41-9 |
| PubChem CID | 3731 |
| DrugBank | DB11705 |
| ChemSpider | 3600 |
| UNII | 17E17JBP8L |
| KEGG | D01719 |
| ChEBI | CHEBI:31710 |
| CompTox Dashboard (EPA) | DTXSID1049061 |
| Chemical and physical data | |
| Formula | C17H22I3N3O8 |
| Molar mass | 777.089 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////Iomeprol, Iomervu, FDA 2024, APPROVALS 2024, 78649-41-9, Iomeprolum, Iomeron, Iomeprolo, UNII-17E17JBP8L, E-7337, E 7337, E-7337, E7337, XRAY CONTRAST AGENT
Lutetium (177Lu) chloride


Lutetium (177Lu) chloride
塩化ルテチウム (177Lu)
| Formula | Lu. 3Cl |
|---|---|
| CAS | 16434-14-3 |
| Mol weight | 281.326 |
2022/9/15 EMA 2022, Illuzyce
EndolucinBeta
(177Lu)lutetium(3+) trichloride
| Diagnostic aid, Radioactive agent |
Lutetium 177 is an isotope of a rare-earth lanthanide metal lutetium. Radioactive decay of Lu 177 produces electrons with low energies making the isotope suitable for treatment of metastatic disease. A complex of Lu177 and somatostatin analog DOTA-TATE was approved by the FDA for the treatment of somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumors, including foregut, midgut, and hindgut neuroendocrine tumors in adults. It is marketed under a tradename Lutathera. Lutetium in the complex with other carriers – phosphonates and monoclonal antibodies – was investigated in clinical trials as radiotherapy to prostate, ovarian, renal and other types of cancer.Lutetium (177Lu) chloride is a radioactive compound used for the radiolabeling of pharmaceutical molecules, aimed either as an anti-cancer therapy or for scintigraphy (medical imaging).[5][6] It is an isotopomer of lutetium(III) chloride containing the radioactive isotope 177Lu, which undergoes beta decay with a half-life of 6.65 days.
Medical uses
Lutetium (177Lu) chloride is a radiopharmaceutical precursor and is not intended for direct use in patients.[5] It is used for the radiolabeling of carrier molecules specifically developed for reaching certain target tissues or organs in the body. The molecules labeled in this way are used as cancer therapeutics or for scintigraphy, a form of medical imaging.[5] 177Lu has been used with both small molecule therapeutic agents (such as 177Lu-DOTATATE) and antibodies for targeted cancer therapy[8][9]
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Lumark, EndolucinBeta, Illuzyce |
| AHFS/Drugs.com | Lumark UK Drug Information EndolucinBeta UK Drug Information |
| License data | EU EMA: by INN |
| Pregnancy category | AU: X (High risk)[1][2] |
| ATC code | None |
| Legal status | |
| Legal status | AU: Unscheduled [3][4]EU: Rx-only [5][6][7]In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 16434-14-3 |
| PubChem CID | 71587001 |
| DrugBank | DBSALT002634 |
| ChemSpider | 32700269 |
| UNII | 1U477369SN |
| KEGG | D10828 |
| CompTox Dashboard (EPA) | DTXSID20167745 |
| Chemical and physical data | |
| Formula | Cl3Lu |
| Molar mass | 281.32 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILES[Cl-].[Cl-].[Cl-].[177Lu+3] |
Contraindications
Medicines radiolabeled with lutetium (177Lu) chloride must not be used in women unless pregnancy has been ruled out.[5]
Adverse effects
The most common side effects are anaemia (low red blood cell counts), thrombocytopenia (low blood platelet counts), leucopenia (low white blood cell counts), lymphopenia (low levels of lymphocytes, a particular type of white blood cell), nausea (feeling sick), vomiting and mild and temporary hair loss.[5]
Society and culture
Legal status
Lutetium (177Lu) chloride (Lumark) was approved for use in the European Union in June 2015.[5] Lutetium (177Lu) chloride (EndolucinBeta) was approved for use in the European Union in July 2016.[6]
On 21 July 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Illuzyce, a radiopharmaceutical precursor.[10] Illuzyce is not intended for direct use in patients and must be used only for the radiolabelling of carrier medicines that have been specifically developed and authorized for radiolabelling with lutetium (177Lu) chloride.[10] The applicant for this medicinal product is Billev Pharma ApS.[10] Illuzyce was approved for medical use in the European Union in September 2022.[7]
References
- ^ “Lutetium (177Lu) Chloride”. Therapeutic Goods Administration (TGA). 21 January 2022. Archived from the original on 5 February 2022. Retrieved 5 February 2022.
- ^ “Updates to the Prescribing Medicines in Pregnancy database”. Therapeutic Goods Administration (TGA). 12 May 2022. Archived from the original on 3 April 2022. Retrieved 13 May 2022.
- ^ “TGA eBS – Product and Consumer Medicine Information Licence”. Archived from the original on 5 February 2022. Retrieved 5 February 2022.
- ^ http://www.ebs.tga.gov.au/servlet/xmlmillr6?dbid=ebs/PublicHTML/pdfStore.nsf&docid=1C7A40803A3A3F94CA2587D4003CE48A&agid=(PrintDetailsPublic)&actionid=1 Archived 30 July 2022 at the Wayback Machine[bare URL PDF]
- ^ Jump up to:a b c d e f g “Lumark EPAR”. European Medicines Agency (EMA). Archived from the original on 25 October 2020. Retrieved 7 May 2020. Text was copied from this source under the copyright of the European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c “EndolucinBeta EPAR”. European Medicines Agency (EMA). Archived from the original on 28 October 2020. Retrieved 7 May 2020. Text was copied from this source under the copyright of the European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Illuzyce EPAR”. European Medicines Agency (EMA). 18 July 2022. Archived from the original on 22 September 2022. Retrieved 21 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Lundsten S, Spiegelberg D, Stenerlöw B, Nestor M (December 2019). “The HSP90 inhibitor onalespib potentiates 177Lu‑DOTATATE therapy in neuroendocrine tumor cells”. International Journal of Oncology. 55 (6): 1287–1295. doi:10.3892/ijo.2019.4888. PMC 6831206. PMID 31638190.
- ^ Michel RB, Andrews PM, Rosario AV, Goldenberg DM, Mattes MJ (April 2005). “177Lu-antibody conjugates for single-cell kill of B-lymphoma cells in vitro and for therapy of micrometastases in vivo”. Nuclear Medicine and Biology. 32 (3): 269–78. doi:10.1016/j.nucmedbio.2005.01.003. PMID 15820762.
- ^ Jump up to:a b c “Illuzyce: Pending EC decision”. European Medicines Agency. 21 July 2022. Archived from the original on 30 July 2022. Retrieved 30 July 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Lutetium chloride Lu-177”. Drug Information Portal. U.S. National Library of Medicine.
.///////////Lutetium (177Lu) chloride, EMA 2022, EU 2022, APPROVALS 2022, Illuzyce, EndolucinBeta, 塩化ルテチウム (177Lu),

NEW DRUG APPROVALS
ONE TIME
$10.00
Gadopiclenol

![Chemical structure of gadopiclenol [gadolinium chelate of 2,2′,2″-(3,6,9-triaza-1(2,6)-pyridinacyclodecaphane-3,6,9-triyl)tris(5-((2,3-dihydroxypropyl)amino)-5-oxopentanoic acid)]. The PCTA parent structure is shown in red. Two water molecules are included to show the coordination in solution.](https://www.researchgate.net/profile/Jean-Marc-Idee/publication/334838366/figure/fig1/AS:797490152476678@1567147877999/Chemical-structure-of-gadopiclenol-gadolinium-chelate-of.jpg)
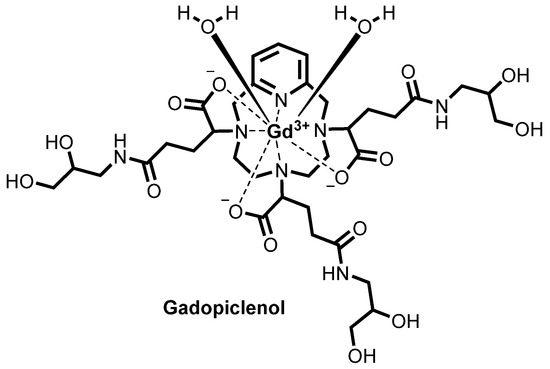
Gadopiclenol
ガドピクレノール;
| Formula | C35H54N7O15. Gd |
|---|---|
| CAS | 933983-75-6 |
| Mol weight | 970.0912 |
FDA APPROVED 2022/9/21, Elucirem
Diagnostic agent (MR imaging), WHO 10744, P 03277, UNII: S276568KOY
EluciremTM; G03277; P03277; VUEWAY
(alpha3,alpha6,alpha9-Tris(3-((2,3-dihydroxypropyl)amino)-3-oxopropyl)-3,6,9,15-tetraazabicyclo(9.3.1)pentadeca-1(15),11,13-triene-3,6,9-triacetato(3-)-kappaN3,kappaN6,kappaN9,kappaN15,kappaO3,kappaO6,kappaO9)gadolinium
- OriginatorGuerbet
- ClassDiagnostic agents; Gadolinium-containing contrast agents; Macrocyclic compounds; Propylamines; Pyridines
- Mechanism of ActionMagnetic resonance imaging enhancers
- RegisteredCNS disorders
- Phase IIIUnspecified
- Phase IILiver cancer
- 21 Sep 2022Registered for CNS disorders (Diagnosis) in USA (IV)
- 13 Jun 2022Guerbet plans to launch Gadopiclenol in Europe
- 13 Jun 2022The European Medicines Agency (EMA) accepts brand name EluciremTM for Gadopiclenol
PATENT
https://patents.google.com/patent/WO2020030618A1/en
MRI contrast agents used in daily diagnostic practice typically include gadolinium complex compounds characterized by high stability constants that guarantee against the in vivo release of the free metal ion (that is known to be extremely toxic for living organisms).
Another key parameter in the definition of the tolerability of a gadolinium-based contrast agent is the kinetic inertness (or kinetic stability) of Gd(III)-complex, that is estimated through the half-life (ti/2) of the dissociation (i.e. decomplexation) of the complex.
A high inertness becomes crucial in particular for those complex compounds having lower thermodynamic stability and/or longer retention time before excretion, in order to avoid or minimize possible decomplexation or transmetallation reactions.
EP1931673 (Guerbet) discloses PCTA derivatives of formula

and a synthetic route for their preparation.
EP 2988756 (same Applicant) discloses a pharmaceutical composition comprising the above derivatives together with a calcium complex of 1,4,7, 10-tetraazacyclododecane- 1,4,7, 10-tetraacetic acid. According to the EP 2988756, the calcium complex compensates the weak thermodynamic stability observed for PCTA-based gadolinium complexes, by forming, through transmetallation, a strong complex with free lanthanide ion, thereby increasing the tolerability of the contrast agent.
Both EP1931673 and EP 2988756 further refer to enantiomers or diastereoisomers of the claimed compounds, or mixture thereof, preferentially chosen from the RRS, RSR, and RSS diastereoisomers. Both the above patents disclose, among the specific derivatives, (a3, a6, a9)-tris(3- ((2,3-dihydroxypropyl)amino)-3-oxopropyl)-3,6,9,15-tetraazabicyclo(9.3.1)pentadeca- l(15),l l,13-triene-3,6,9-triacetato(3-)-(KN3,KN6,KN9,KN15,K03,K06,K09)gadolinium, more recently identified as gadolinium chelate of 2,2′,2″-(3,6,9-triaza-l(2,6)- pyridinacyclodecaphane-3,6,9-triyl)tris(5-((2,3-dihydroxypropyl)amino)-5-oxopentanoic acid), (CAS registry number: 933983-75-6), having the following formula

otherwise identified as P03277 or Gadopiclenol.
For Gadopiclenol, EP1931673 reports a relaxivity of 11 mM _1s _1Gd 1 (in water, at 0.5 T, 37°C) while EP 2988756 reports a thermodynamic equilibrium constant of 10 14 9 (log Kterm
= 14.9).
Furthermore, for this same compound a relaxivity value of 12.8 mM _1s 1 in human serum (37°C, 1.41 T), stability (log Kterm) of 18.7, and dissociation half-life of about 20 days (at pH 1.2; 37°C) have been reported by the proprietor (Investigative Radiology 2019, Vol 54, (8), 475-484).
The precursor for the preparation of the PCTA derivatives disclosed by EP1931673 (including Gadopiclenol) is the Gd complex of the 3,6,9,15-tetraazabicyclo- [9.3.1]pentadeca-l(15),l l,13-triene-tri(a-glutaric acid) having the following formula

Gd(PCTA-tris-glutaric acid)
herein identified as “Gd(PCTA-tris-glutaric acid)”. In particular, Gadopiclenol is obtained by amidation of the above compound with isoserinol.
As observed by the Applicant, Gd(PCTA-tris-qlutaric acid) has three stereocenters on the glutaric moieties (identified with an asterisk (*) in the above structure) that lead to a 23 = 8 possible stereoisomers. More particularly, the above structure can generate four pairs of enantiomers, schematized in the following Table 1
Table 1

Isomer RRR is the mirror image of isomer SSS and that is the reason why they are called enantiomers (or enantiomer pairs). As known, enantiomers display the same physicochemical properties and are distinguishable only using chiral methodologies, such as chiral chromatography or polarized light.
On the other hand, isomer RRR is neither equal to nor is it the mirror image of any of the other above six isomers; these other isomers are thus identified as diastereoisomers of the RRR (or SSS) isomer. Diastereoisomers may display different physicochemical properties, (e.g., melting point, water solubility, relaxivity, etc.).
Concerning Gadopiclenol, its chemical structure contains a total of six stereocenters, three on the glutaric moieties of the precursor as above discussed and one in each of the three isoserinol moieties attached thereto, identified in the following structure with an asterisk (*) and with an empty circle (°), respectively:

This leads to a total theoretical number of 26 = 64 stereoisomers for this compound. However, neither EP1931673 nor EP 2988756 describe the exact composition of the isomeric mixture obtained by following the reported synthetic route, nor does any of them provide any teaching for the separation and characterization of any of these isomers, or disclose any stereospecific synthesis of Gadopiclenol. Summary of the invention
The applicant has now found that specific isomers of the above precursor Gd(PCTA- tris-glutaric acid) and of its derivatives (in particular Gadopiclenol) possess improved physico-chemical properties, among other in terms of relaxivity and kinetic inertness.
An embodiment of the invention relates to a compound selected from the group consisting of:
the enantiomer [(aR,a’R,a”R)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15- tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9-triacetato(3-)- Kl\l3,Kl\l6,Kl\l9,Kl\ll5,K03,K06,K09]-gadolinium (RRR enantiomer) having the formula (la):

the enantiomer [(aS,a’S,a”S)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15-tetraazabicyclo- [9.3.1]pentadeca-l(15),ll,13-triene-3,6,9-triacetato(3-)KN3,KN6,KN9,KN15,K03,K06,K09]- gadolinium (SSS enantiomer) having the formula (lb):

the mixtures of such RRR and SSS enantiomers, and a pharmaceutically acceptable salt thereof.
Another embodiment of the invention relates to an isomeric mixture of Gd(PCTA-tris- glutaric acid) comprising at least 50% of the RRR isomer [(aR,a’R,a”R)-a,a’,a”-tris(2- carboxyethyl)-3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9- triacetato(3-)-KN3,KN6,KN9,KN15,K03,K06,K09]-gadolinium, of formula (la), or of the SSS isomer [(aS,a’S,a”S)-a,a’,a”-tris(2-carboxyethyl)-3,6,9,15- tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene-3,6,9-triacetato(3-)- Kl\l3,Kl\l6,Kl\l9,Kl\ll5,K03,K06,K09]-gadolinium of formula (lb), or of a mixture thereof, or a pharmaceutically acceptable salt thereof. Another aspect of the invention relates to the amides obtained by conjugation of one of the above compounds or isomeric mixture with an amino group, e.g. preferably, serinol or isoserinol.
An embodiment of the invention relates to an amide derivative of formula (II A)
F( N RI R2)3 (II A)
in which :
F is:
a RRR enantiomer residue of formula Ilia

a SSS enantiomer residue of formula Illb

or a mixture of such RRR and SSS enantiomer residues;
and each of the three -NRIR2 group is bound to an open bond of a respective carboxyl moiety of F, identified with a full circle (·) in the above structures;
Ri is H or a Ci-Ce alkyl, optionally substituted by 1-4 hydroxyl groups;
R2 is a Ci-Ce alkyl optionally substituted by 1-4 hydroxyl groups, and preferably a C1-C3 alkyl substituted by one or two hydroxyl groups.
Another embodiment of the invention relates to an isomeric mixture of an amide derivative of Gd(PCTA-tris-glutaric acid) having the formula (II B)
F'( N RI R2)3 (II B)
in which :
F’ is an isomeric mixture of Gd(PCTA-tris-glutaric acid) residue of formula (III)

said isomeric mixture of the Gd(PCTA-tris-glutaric acid) residue comprising at least 50 % of an enantiomer residue of the above formula (Ilia), of the enantiomer residue of the above formula (Illb), or of a mixture thereof; and each of the -NR1R2 groups is bound to an open bond of a respective carboxyl moiety of F’, identified with a full circle (·) in the above structure, and is as above defined for the compounds of formula (II A).
EXPERIMENTAL PART
HPLC characterization of the obtained compounds.
General procedures
Procedure 1: HPLC Characterization of Gd(PCTA-tris-glutaric acid) (isomeric mixture and individual/enriched isomers).
The HPLC characterization of the Gd(PCTA-tris-glutaric acid) obtained as isomeric mixture from Example 1 was performed with Agilent 1260 Infinity II system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC system HPLC equipped with quaternary pump, degasser, autosampler,
PDA detector ( Agilent 1260 Infinity II system)
Stationary phase: Phenomenex Gemini® 5pm C18 lloA
Mobile phase: H2O/HCOOH 0.1% : Methanol
Elution : Gradient Time (min) H2O/HCOOH 0.1% Methanol
0 95 5
5 95 5
30 50 50
35 50 50
40 95 5
Flow 0.6 mL/min
Temperature 25 °C
Detection PDA scan wavelenght 190-800nm
Injection volume 50 pL
Sample Cone. 0.2 mM Gd(PCTA-tris-glutaric acid) complex
Stop time 40 min
Retention time GdL = 18-21 min.
Obtained HPLC chromatogram is shown in Figure 1
The HPLC chromatogram of the enriched enantiomers pair C is shown in Figure 2.
Procedure 2: HPLC Characterization of Gadopiclenol (isomeric mixture) and compounds obtained by coupling of enantiomers pair C with R, S, or racemic isoserinol.
The HPLC characterization of Gadopiclenol either as isomeric mixture obtained from Example 2, or as the compound obtained by conjugation of enantiomers pair C of the Gd(PCTA-tris-glutaric acid) with R, S, or racemic isoserinol was performed with Thermo Finnigan LCQ DECA XPPIus system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC system HPLC equipped with quaternary pump, degasser, autosampler,
PDA and MS detector (LCQ Deca XP-Plus – Thermo Finnigan )
Stationary phase: Phenomenex Gemini 5u C18 110A
Mobile phase: H2O/TFA 0.1% : Acetonitrile/0.1%TFA
Elution : Gradient Time (min) H2O/TFA 0.1% Acetonitrile/0.1%TFA
0 100 0
5 100 0
22 90 10
26 90 10
Flow 0.5 mL/min
Temperature 25 °C
Detection PDA scan wavelenght 190-800nm
MS positive mode – Mass range 100-2000
Injection volume 50 pL
Sample cone. 0.2 mM Gd complex
Stop time 26 min
Retention time GdL = 20-22min.
Obtained HPLC chromatograms are shown in Figure 6.
Procedure 3: Chiral HPLC method for the separation of enantiomers of the compound C
A specific chiral HPLC method was set up in order to separate the RRR and SSS enantiomers of the enantiomers pair C (compound VI), prepared as described in Example 3. The separation and characterization of the enantiomers were performed with Agilent 1200 system or Waters Alliance 2695 system. The experimental setup of the HPLC measurements are summarized below.
Analytical conditions
HPLC System HPLC equipped with quaternary pump, degasser, autosampler,
PDA detector
Stationary phase SUPELCO Astec CHIROBIOTIC 5 pm 4.6x250mm
Mobile phase H2O/HCOOH 0.025% : Acetonitrile
Elution : isocratic 2% Acetonitrile for 30 minutes
Flow 1 mL/min
Column Temperature 40°C
Detection 210-270 nm. Obtained HPLC chromatogram is shown in Figure 5a) compared to the chromatograms of the pure RRR enantiomer (compound XII of Example 5, Tr. 7.5 min.) and the pure SSS enantiomer (Compound XVII of Example 6, Tr. 8.0 min), shown in figure 5b) and 5c), respectively.
Example 1: Synthesis of Gd(PCTA-tris-glutaric acid) (isomeric mixture)
Gd(PCTA-tris-glutaric acid) as an indiscriminate mixture of stereoisomers has been prepared by using the procedure reported in above mentioned prior-art, according to the following synthetic Scheme 1 :
Scheme 1

a) Preparation of Compound II
Racemic glutamic acid (33.0 g, 0.224 mol) and sodium bromide (79.7 g, 0.782 mol) were suspended in 2M HBr (225 ml_). The suspension was cooled to -5°C and NaN02 (28.0 g, 0.403 mol) was slowly added in small portions over 2.5 hours, maintaining the inner temperature lower than 0 °C. The yellow mixture was stirred for additional 20 minutes at a temperature of -5°C; then concentrated sulfuric acid (29 ml.) was dropped in the mixture. The obtained dark brown mixture was warmed to RT and then extracted with diethyl ether (4×150 ml_). The combined organic phases were washed with brine, dried over Na2S04 and concentrated to a brown oil (21.2 g), used in the following step without further purification. The oil was dissolved in ethanol (240 ml_), the resulting solution was cooled in ice and thionyl chloride (14.5 ml_, 0.199 mol) was slowly added. The slightly yellow solution was stirred at RT for 2 days. Then the solvent was removed in vacuum and the crude oil was dissolved in dichloromethane (200 ml.) and washed with 5% aq. NaHCC>3 (4×50 ml_), water (1×50 ml.) and brine (1×50 ml_). The organic phase was concentrated and purified on silica eluting with petroleum ether-ethyl acetate 3: 1, obtaining 19.5 g of pure product. (Yield 33%).
b) Preparation of Compound IV
A solution of Compound II (17.2 g, 0.0645 mol) in acetonitrile (40 ml.) was added to a suspension of 3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-l(15),l l,13-triene (pyclen) Compound (III) (3.80 g, 0.018 mol) and K2CO3 (11.2 g, 0.0808 mol) in acetonitrile (150 ml_). The yellow suspension was heated at 65 °C for 24 h, then the salts were filtered out and the organic solution was concentrated. The orange oil was dissolved in dichloromethane and the product was extracted with 1M HCI (4 x 50 ml_). The aqueous phases were combined, cooled in ice and brought to pH 7-8 with 30% aq. NaOH. The product was then extracted with dichloromethane (4 x 50 ml.) and concentrated to give a brown oil (10.1 g, yield 73%). The compound was used in the following step without further purification.
c) Preparation of compound V
Compound IV (9.99 g, 0.013 mol) was dissolved in Ethanol (40 ml.) and 5M NaOH (40 ml_). The brown solution was heated at 80 °C for 23 h. Ethanol was concentrated; the solution was cooled in ice and brought to pH 2 with cone HCI. The ligand was purified on resin Amberlite XAD 1600, eluting with water-acetonitrile mixture, obtaining after freeze- drying 5.7 g as white solid (yield 73%). The product was characterized in HPLC by several peaks.
d) Preparation of compound VI
Compound V (5.25 g, 0.0088 mol) was dissolved in deionized water (100 ml.) and the solution was brought to pH 7 with 2M NaOH (20 ml_). A GdCh solution (0.0087 mol) was slowly added at RT, adjusting the pH at 7 with 2M NaOH and checking the complexation with xylenol orange. Once the complexation was completed, the solution was concentrated and purified on resin Amberlite XAD 1600 eluting with water-acetonitrile gradient, in order to remove salts and impurities. After freeze-drying the pure compound was obtained as white solid (6.79 g, yield 94%). The product was characterized in HPLC; the obtained HPLC chromatogram, characterized by several peaks, is shown in Figure 1 A compound totally equivalent to compound VI, consisting of an isomeric mixture with a HPLC chromatogram substantially superimposable to that of Figure 1 is obtained even by using (S)-methyl a-bromoglutarate obtained starting from L-glutamic acid.
Example 2: Synthesis of Gadopiclenol (isomeric mixture)
Gadopiclenol as an indiscriminate mixture of stereoisomers has been prepared as disclosed in EP11931673 B1 by coupling the isomeric mixture of Gd(PCTA-tris-glutaric acid) obtained from Example 1 with racemic isoserinol according to the following synthetic Scheme 2:
Scheme 2

Preparation of compound VII
Compound VI (0.90 g, 0.0011 mol) obtained from Example 1 was added to a solution of racemic isoserinol (0.40 g, 0.0044 mol) in water adjusted to pH 6 with cone. HCI. Then N- ethyl-N’-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI-HCI) (1.0 g, 0.0055 mol) and hydroxybenzotriazole (HOBT) (0.12 g, 0.00088 mol) were added and the resulting solution was stirred at pH 6 and RT for 24 h. The product was then purified on preparative HPLC on silica C18, eluting with water/acetonitrile gradient. Fractions containing the pure compound were concentrated and freeze-dried, obtaining a white solid (0.83 g, yield 78%). The product was characterized in HPLC; the obtained HPLC chromatogram is shown in Figure 4a.
Example 3: Isolation of the enantiomers pair related to the peak C.
Compound VI obtained as described in Example 1 (step d) (1.0 g, 0.0013 mol) was dissolved in water (4 ml.) and the solution was acidified to pH 2-3 with cone. HCI. The obtained solution was loaded into a pre-packed column of silica C18 (Biotage® SNAP ULTRA C18 120 g, HP-sphere C18 25 pm) and purified with an automated flash chromatography system eluting with deionized water (4 CV) and then a very slow gradient of acetonitrile. Fractions enriched of the enantiomers pair related to the peak C were combined, concentrated and freeze-dried obtaining a white solid (200 mg).
The HPLC chromatogram of the obtained enriched enantiomers pair C is shown in Figure 2.
Corresponding MS spectrum (Gd(H4L)+:752.14 m/z) is provided in Figure 3
Example 4: Coupling of the enantiomers pair C with isoserinol.
a) Coupling of the enantiomers pair C with R-isoserinol.
Enriched enantiomers pair C collected e.g. as in Example 3 (34 mg, titer 90%, 0.040 mmol) was dissolved in deionized water (5 ml_), and R-isoserinol (16 mg, 0.17 mmol) was added adjusting the pH at 6 with HCI 1M. Then, EDCI-HCI (39 mg, 0.20 mmol) and HOBT (3 mg, 0.02 mmol) were added and the solution was stirred at RT at pH 6 for 48 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (21 mg, yield 54%).
The HPLC chromatogram of the obtained product is shown in Figure 6b.
b) Coupling of the enantiomers pair C with S-isoserinol
Enriched enantiomers pair C collected e.g. as in Example 3 (55 mg, titer 90%, 0.066 mmol) was dissolved in deionized water (5 mL), and S-isoserinol (34 mg, 0.29 mmol) was added adjusting the pH at 6 with 1M HCI. Then, EDCI-HCI (64 mg, 0.33 mmol) and HOBT (4.5 mg, 0.033 mmol) were added and the solution was stirred at RT at pH 6 for 48 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (52 mg, yield 81%).
HPLC chromatogram of the obtained product is shown in Figure 6c.
c) Coupling of the enantiomers pair C with racemic isoserinol.
The enriched enantiomers pair C collected e.g. as in Example 3 (54 mg, titer 90%, 0.065 mmol) was dissolved in deionized water (5 mL), and racemic isoserinol (27 mg, 0.29 mmol) was added adjusting the pH at 6 with 1M HCI. Then, EDCI-HCI (62 mg, 0.32 mmol) and HOBT (4.3 mg, 0.032 mmol) were added and the solution was stirred at RT at pH 6 for 24 h. The solution was concentrated and loaded to pre-packed silica C18 column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with water/acetonitrile gradient using an automated flash chromatography system. Fractions containing the pure product, or showing a major peak at the HPLC with area greater than 90%, were combined, concentrated and freeze-dried giving a white solid (60 mg, yield 95%).
HPLC chromatogram of the obtained product is shown in Figure 6d. Example 5: Stereoselective synthesis of the RRR Gd(PCTA-tris-glutaric acid) (compound XII).
RRR enriched Gd(PCTA-tris-glutaric acid) acid has been prepared by following the synthetic Scheme 3 below
Scheme 3

comprising :
a) Preparation of Compound VIII
The preparation was carried out as reported in Tetrahedron 2009, 65, 4671-4680.
In particular: 37% aq. HCI (50 pL) was added to a solution of (S)-(+)-5- oxotetrahydrofuran-2-carboxylic acid (2.48 g, 0.019 mol) (commercially available) in anhydrous methanol (20 ml_). The solution was refluxed under N2 atmosphere for 24 h. After cooling in ice, NaHCC>3 was added, the suspension was filtered, concentrated and purified on silica gel with hexanes/ethyl acetate 1 : 1. Fractions containing the pure product were combined and concentrated, giving a colorless oil (2.97 g, yield 89%).
b) Preparation of Compounds IX and X
Compound VIII (445 mg, 2.52 mmol) obtained at step a) was dissolved in anhydrous dichloromethane (6 ml.) and triethylamine (0.87 ml_, 6.31 mmol) was added. The solution was cooled at -40°C and then (triflic) trifluoromethansulfonic anhydride (0.49 ml_,2.91 mmol) was slowly added. The dark solution was stirred at -40°C for 1 h, then a solution of Compound III (104 mg, 0.506 mmol) in anhydrous dichloromethane (3 ml.) and triethylamine (1 ml_, 7.56 mmol) were added and the solution was slowly brought to RT and stirred at RT overnight. The organic solution was then washed with 2M HCI (4x 10 ml_), the aqueous phase was extracted again with dichloromethane (3 x 10 ml_). The organic phases were combined and concentrated in vacuum, obtaining 400 mg of a brown oil that was used in the following step with no further purification.
c) Preparation of Compound XI
Compound X (400 mg, 0.59 mmol) was dissolved in methanol (2.5 ml.) and 5M NaOH (2.5 ml_). The brown solution was heated at 80°C for 22 h to ensure complete hydrolysis. Methanol was concentrated, the solution was brought to pH 1 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with deionized water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (64 mg, yield 18 %). The HPLC showed a major peak.
d) Compound XII
Compound XI (32 mg, 0.054 mmol) was dissolved in deionized water (4 mL) and the pH was adjusted to 7 with 1M NaOH. GdCl3-6H20 (20 mg, 0.054 mmol) was added and the pH was adjusted to 7 with 0.1 M NaOH. The clear solution was stirred at RT overnight and the end of the complexation was checked by xylenol orange and HPLC. The HPLC of the crude showed the desired RRR isomer as major peak: about 80% in area %. The mixture was brought to pH 2 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-sphere C18 25 pm), eluting with deionized water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (36 mg, yield 90%).
By reaction of the collected compound with isoserinol e.g. by using the procedure of the Example 2, the corresponding RRR amide derivative can then be obtained.
Example 6: stereoselective synthesis of the SSS Gd(PCTA-tris-glutaric acid) (compound XVII).
SSS enriched Gd(PCTA-tris-glutaric acid) acid has been similarly prepared by following the synthetic Scheme 4 below Scheme 4

comprising :
a) Preparation of Compound XIII
37% aq. HCI (100 pl_) was added to a solution of (R)-(-)-5-oxotetrahydrofuran-2- carboxylic acid (5.0 g, 0.038 mol) (commercially available) in anhydrous methanol (45 ml_). The solution was refluxed under N2 atmosphere for 24 h. After cooling in ice, NaHC03 was added, the suspension was filtered, concentrated and purified on silica gel with hexanes/ethyl acetate 1 : 1. Fractions containing the pure product were combined and concentrated, giving a colorless oil (6.7 g, yield 99%).
b) Preparation of Compounds XIV and XV
Compound XIII (470 mg, 2.67 mmol) was dissolved in anhydrous dichloromethane (6 ml.) and trimethylamine (0.93 ml_, 6.67 mmol) was added. The solution was cooled down at -40°C and then trifluoromethanesulfonic anhydride (0.50 ml_, 3.07 mmol) was slowly dropped. The dark solution was stirred at -40°C for 1 h, then Compound III (140 mg, 0.679 mmol) and trimethylamine (0.93 ml_, 6.67 mmol) were added and the solution was slowly brought to RT overnight. The organic solution was then washed with water (3 x 5 ml.) and 2M HCI (4 x 5 ml_). The aqueous phase was extracted again with dichloromethane (3 x 10 ml_). the organic phases were combined and concentrated in vacuum, obtaining 350 mg of a brown oil that was used in the following step with no further purification. c) Preparation of Compound XVI
Compound XV (350 mg, 0.514 mmol) was dissolved in methanol (4.5 ml.) and 5M NaOH (4.5 ml_). The obtained brown solution was heated at 80°C for 16 h to ensure complete hydrolysis. Methanol was concentrated, the solution was brought to pH 2 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-SPHERE C18 25 pm), eluting with a water/acetonitrile gradient. Fractions containing the pure product were combined, concentrated and freeze-dried (52 mg, yield 17%). The HPLC showed a major peak.
d) Preparation of Compound XVII
Compound XVI (34 mg, 0.057 mmol) was dissolved in deionized water (5 mL) and the pH was adjusted to 7 with 1 M HCI. GdCl3-6H20 (20 mg, 0.0538 mmol) was added and the pH was adjusted to 7 with 0.1 M NaOH. The solution was stirred at RT overnight and the end of complexation was checked by xylenol orange and HPLC. The HPLC of the crude showed the desired SSS isomer as major peak: about 85% in area %. The solution was brought to pH 2.5 with concentrated HCI and purified through an automated flash chromatography system with a silica C18 pre-packed column (Biotage® SNAP ULTRA C18 12 g, HP-SPHERE C18 25 pm), eluting with a water/acetonitrile gradient. Fractions containing the pure product SSS were combined, concentrated and freeze-dried (39 mg, yield 87%).
Example 7: Kinetic studies of the dissociation reactions of Gd(PCTA-tris- glutaric acid) (isomeric mixture) in 1.0 M HCI solution (25°C)
The kinetic inertness of a Gd(III)-complex is characterized either by the rate of dissociation measured in 0.1-1.0 M HCI or by the rate of the transmetallation reaction, occurring in solutions with Zn(II) and Cu(II) or Eu(III) ions. However, the dissociation of lanthanide(III)-complexes formed with macrocyclic ligands is very slow and generally proceeds through a proton-assisted pathway without the involvement of endogenous metal ions like Zn2+ and Cu2+.
We characterized the kinetic inertness of the complex Gd(PCTA-tris-glutaric acid) by the rates of the dissociation reactions taking place in 1.0 M HCI solution. The complex (isomeric mixture from Example 1) (0.3 mg) was dissolved in 2.0 mL of 1.0 M HCI solution and the evolution of the solution kept at 25 °C was followed over time by HPLC. The HPLC measurements were performed with an Agilent 1260 Infinity II system by use of the analytical Procedure 1.
The presence of a large excess of H+ ([HCI] = 1.0 M), guarantees the pseudo-first order kinetic conditions.
GdL + yH÷ ^ Gd3+ + HyL y=7 and 8 (Eg. 1) where L is the protonated PCTA-tri-glutaric acid, free ligand, and y is the number of protons attached to the ligand.
The HPLC chromatogram of Gd(PCTA-tris-glutaric acid) is characterized by the presence of four signals (A, B, C and D) having the same m/z ratio (Gd(H4L)+ :752.14 m/z) in the MS spectrum. Each of these peaks is reasonably ascribable to one of the 4 pairs of enantiomers generated by the three stereocenters on the three glutaric arms of the molecule, formerly identified in Table 1. The HPLC chromatogram of this complex in the presence of 1.0 M HCI changes over time: in particular, the areas of peaks A, B, C, and D decrease, although not in the same way for the different peaks, while new signals corresponding to non-complexed diastereoisomers are formed and grow over time. Differences in the decrease of the integral areas of the peaks can be interpreted by a different dissociation rate of the enantiomer pairs associated to the different peaks.
In the presence of [H + ] excess the dissociation reaction of enantiomer pairs of Gd(PCTA-tris-glutaric acid) can be treated as a pseudo-first-order process, and the rate of the reactions can be expressed with the following Eq. 2, where kA, kB, kc and kD are the pseudo-first-order rate constants that are calculated by fitting the area-time data pair, and [A]t, [B]t, [C]t and [D]t are the total concentration of A, B, C and D compounds at time t.

The decrease of the area values of signals of A, B, C, and D has been assessed and plotted over time. Area values of A, B, C and D signals as a function of time are shown in Figure 7.
Area value at time t can be expressed by the following equation:
A. = A + (A0 – A )e kxt
(Eg. 3)
where At, A0 and Ae are the area values at time t, at the beginning and at the end of the reactions, respectively, kx pseudo-first-order rate constants (/fX=/fA, kB, kc and kD) characterizing the dissociation rate of the different enantiomer pairs of Gd(PCTA-tris-glutaric acid) complex were calculated by fitting the area – time data pairs of Figure 7 to the above equation 3. kx rate constants and half-lives (ti/2= In2/ x) are thus obtained, as well as the average the half-life value for the isomeric mixture of Gd(PCTA-tris-glutaric acid), calculated by considering the percentage composition of the mixture. Obtained values are summarized in the following Table 2, and compared with corresponding values referred in the literature for some reference contrast agents. (Gd-DOTA or DOTAREM™). Table 2. Rate constants ( kx ) and half-lives (ti/2= In2/ x) characterizing the acid catalyzed dissociation of the different stereoisomers of Gd(PCTA-tris-glutaric acid), Dotarem® and Eu(PCTA) in 1.0 M HCI (pH 0) ( 25°C)
A B C D
Ms 1) (4.5±0.1) x105 (1.1±0.1)x104 (1.6±0.1)x10-6 (1.2±0.1)x10-5 fi/2 (hour) 4.28 ± 0.03 1.76 ± 0.02 120 ± 3 15.8 ± 0.5
fi/2 (hour)

average
Dotarem a
k, (S‘1) 8.0×10-6
fi/2 (hour) 23 hour
Eu(PCTA) b
*1 (s·1) 5.08X10·4
fi/2 (hour) 0.38 hour
a) Inorg. Chem. 1992, 31 ,1095-1099.
b) Tircso, G. et al. Inorg Chem 2006, 45 (23), 9269-80.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
A gadolinium-based paramagnetic contrast agent, with potential imaging enhancing activity upon magnetic resonance imaging (MRI). Upon administration of gadopiclenol and placement in a magnetic field, this agent produces a large magnetic moment and creates a large local magnetic field, which can enhance the relaxation rate of nearby protons. This change in proton relaxation dynamics, increases the MRI signal intensity of tissues in which this agent has accumulated; therefore, contrast and visualization of those tissues is enhanced compared to unenhanced MRI.
FDA Approves New MRI Contrast Agent Gadopiclenol
September 22, 2022
https://www.diagnosticimaging.com/view/fda-approves-new-mri-contrast-agent-gadopiclenol
Requiring only half of the gadolinium dose of current non-specific gadolinium-based contrast agents (GBCAs), gadopiclenol can be utilized with magnetic resonance imaging (MRI) to help detect lesions with abnormal vascularity in the central nervous system and other areas of the body.
Gadopiclenol, a new magnetic resonance imaging (MRI) contrast agent that offers high relaxivity and reduced dosing of gadolinium, has been approved by the Food and Drug Administration (FDA).1
Approved for use with MRI in adults and pediatric patients two years of age or older, gadopiclenol is a macrocyclic gadolinium-based contrast agent that aids in the diagnosis of lesions with abnormal vascularity in the brain, spine, abdomen, and other areas of the body.
Recently published research demonstrated that gadopiclenol provides contrast enhancement and diagnostic efficacy at half of the gadolinium dosing of other gadolinium-based contrast agents (GBCAs) such as gadobutrol and gadobenate dimeglumine.2
Co-developed by Bracco Diagnostics and Guerbet, gadopiclenol will be manufactured and marketed as Vueway™ (Bracco Diagnostics) and Elucirem™ (Guerbet).1,3
Alberto Spinazzi, M.D., the chief medical and regulatory officer at Bracco Diagnostics, said gadopiclenol is “a first of its kind MRI agent that delivers the highest relaxivity and highest kinetic stability of all GBCAs on the market today.”
Reference
1. Bracco Diagnostics. Bracco announces FDA approval of gadopiclenol injection, a new macrocyclic high-relaxivity gadolinium-based contrast agent which will be commercialized as VUEWAY™ (gadopiclenol) injection and VUEWAY™ (gadopiclenol) phamarcy bulk package by Bracco. Cision PR Newswire. Available at: https://www.prnewswire.com/news-releases/bracco-announces-fda-approval-of-gadopiclenol-injection-a-new-macrocyclic-high-relaxivity-gadolinium-based-contrast-agent-which-will-be-commercialized-as-vueway-gadopiclenol-injection-and-vueway-gadopiclenol-pharmacy-bulk-p-301630124.html . Published September 21, 2022. Accessed September 21, 2022.
2. Bendszus M, Roberts D, Kolumban B, et al. Dose finding study of gadopiclenol, a new macrocyclic contrast agent, in MRI of central nervous system. Invest Radiol. 2020;55(3):129-137.
3. Guerbet. Guerbet announces U.S. Food and Drug Administration (FDA) approval of Elucirem™ (gadopiclenol) injection for use in contrast-enhanced MRI. Cision PR Newswire. Available at: https://www.prnewswire.com/news-releases/guerbet-announces-us-food-and-drug-administration-fda-approval-of-elucirem-gadopiclenol-injection-for-use-in-contrast-enhanced-mri-301630085.html . Published September 21, 2022. Accessed September 21, 2022.
////Gadopiclenol, FDA 2022, APPROVALS 2022, ガドピクレノール, WHO 10744, P 03277, EluciremTM, G03277; P03277, VUEWAY, Guerbet

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}