PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
aLaboratory of Organic Chemistry, Wageningen University, Dreijenplein 8, 6703 HB Wageningen, The Netherlands E-mail: lvy33@163.com
bKey Laboratory of Phytochemical R&D of Hunan Province, Hunan Normal University, Changsha, PR China
Greener ethanol, acetone and ethyl acetate provided better chromatographic resolution in preparative RP-HPLC than the traditional methanol, acetonitrile and tetrahydrofuran.
To make preparative Reversed-Phase High Performance Liquid Chromatography (RP-pHPLC) greener, alternative solvents were considered among others in terms of toxicity, cost, safety, workability, chromatographic selectivity and elution strength. The less toxic solvents ethanol, acetone and ethyl acetate were proposed as possible greener replacements for methanol, acetonitrile and tetrahydrofuran (THF).
For testing their feasibility, five ginkgo terpene trilactones were used as model analytes. The best “traditional” eluent, i.e., methanol–THF–water (2:1:7) was used as the benchmark. A generic two-step chromatographic optimization procedure by UHPLC consisting of (1) a simplex design using the Snyder solvent triangle and (2) HPLC modelling software was used.
In the first step, two ternary mixtures were found (acetone–ethyl acetate–water (20.25:3.75:76) and ethanol–ethyl acetate–water (9.5:7.5:83)), which already gave better results than the benchmark. The second step in which the influence of the gradient time, temperature and ratio of the two best ternary isocratic solvents was studied, led to an optimal 10.5 min gradient and a minimum resolution of 5.76.
In the final step, scale-up from 2.1 to 22 mm i.d. pHPLC columns proceeded successfully. When 0.5 g of the sample was injected, baseline separation was maintained. Chromatographic and absolute purities for products exceeded 99.5% and 95% respectively. This example shows that using less toxic and cheaper solvents for pHPLC can go hand in hand with higher productivity and less waste.
It was isolated from the mold Penicillium citrinum by Akira Endo in the 1970s, and he identified it as a HMG-CoA reductase inhibitor,[1] i.e., a statin. Mevastatin might be considered the first statin drug;[2] clinical trials on mevastatin were performed in the late 1970s in Japan, but it was never marketed.[3] The first statin drug available to the general public was lovastatin.
A British group isolated the same compound from Penicillium brevicompactum, named it compactin, and published their results in 1976.[5] The British group mentions antifungal properties with no mention of HMG-CoA reductase inhibition.
High doses inhibit growth and proliferation of melanoma cells.[6]
Fungal metabolite which is a potent inhibitor of HMG-CoA reductase, the rate controlling enzyme in cholesterol biosynthesis. Isoln from Penicillium citrinum: A. Endo et al.,DE2524355 corresp to US3983140 (1975, 1976 to Sankyo).
Isoln from P. brevicompactum, crystal and molecular structure: A. G. Brown et al.,J. Chem. Soc. Perkin Trans. 11976,1165.
Inhibition of HMG-CoA reductase activity: A. Endo et al.,FEBS Lett.72, 323 (1976); M. S. Brown et al.,J. Biol. Chem.253,1121 (1978).
Therapeutic effects in primary hypercholesterolemia: A. Yamamoto et al.,Atherosclerosis35, 259 (1980).
Total synthesis: N. Y. Wang et al.,J. Am. Chem. Soc.103, 6538 (1981); M. Hirama, M. Uei, ibid.104, 4251 (1982); N. N. Girotra, N. L. Wendler, Tetrahedron Lett.23, 5501 (1982); C.-T. Hsu et al.,J. Am. Chem. Soc.105, 593 (1983); P. A. Grieco et al.,ibid. 1403; D. L. J. Clive et al.,J. Am. Chem. Soc.110, 6914 (1988). Review of syntheses: T. Rosen, C. H. Heathcock, Tetrahedron42,4909-4951 (1986).
Review of mevastatin and related compounds: A. Endo, J. Med. Chem.28, 401-405 (1985).
Properties: Crystals from aq ethanol, mp 152°. [a]D22 +283° (c = 0.48 in acetone). uv max: 230, 237, 246 nm (log e 4.28, 4.30, 4.11).
Melting point: mp 152°
Optical Rotation: [a]D22 +283° (c = 0.48 in acetone)
Endo, Akira; Kuroda M.; Tsujita Y. (December 1976). “ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium”. Journal of Antibiotics (Tokyo)29 (12): 1346–8. doi:10.7164/antibiotics.29.1346. PMID1010803.
Wachtershauser, A.; Akoglu, B; Stein, J (2001). “HMG-CoA reductase inhibitor mevastatin enhances the growth inhibitory effect of butyrate in the colorectal carcinoma cell line Caco-2”. Carcinogenesis22 (7): 1061–7. doi:10.1093/carcin/22.7.1061. PMID11408350.
Brown, Allan G.; Smale, Terry C.; King, Trevor J.; Hasenkamp, Rainer; Thompson, Ronald H. (1976). “Crystal and molecular structure of compactin, a new antifungal metabolite from Penicillium brevicompactum.”. J. Chem. Soc., Perkin Trans. 1 (11): 1165–1170.doi:10.1039/P19760001165. PMID945291.
is related to a new method for producing ML-236B, a precursor of pravastatin sodium, in particular to a method for producing ML-236B lactone form(I), free acid form (II), and sodium salt(III) shown in the following formulae by using a new microorganism isolated from soil. ML-236B is obtained from the culture broth of this microorganism and it is used as a substrate of pravastatin sodium which is a potent cholesterol-lowering agent used in treatment for hypercholesterolemia.
2. Description of the Prior Art
It has been known that heart disease such as myocardial infarction, arteriosclerosis have been caused mainly by hyperlipidemia, especially hypercholesterolemia. It was reported by U.S. Pat. No. 3,983,140 and UK. Patent No. 1,453,425 that a cholesterol-lowering compound called ML-236B produced by a fungus Penicillium sp. had been discovered. ML-236B is produced by soil microorganisms or chemical conversion. It was reported that Penicillium brevicompactin, Penicilmyces sp., Trichoderma longibraiatum, Trichoderma pseudokoningi, Hyphomyces chrisopomus and Penicillium citrium produced ML-236B(David et al., “Biotechnology of filamentous fungi”, p241; JP Publication No. Pyung 4-349034).
Particularly, Sankyo Pharmaceutical Company, Japan, had developed Penicillium citrium SANK 18767 by mutation of a strain Penicillium citrium NRRL-8082 which was reported in 1971. By continuing strain development for 14 years, they had obtained Penicillium citrium Thom SANK 13380. ML-236B productivity had risen from 1.75 mg/l to 42.5 mg/l.
However, the method above described required so much time about 14 years to develop a strain with high ML-236B productivity. It also needed a little long cultivation time, 14 days, and showed relatively low ML-236B productivity.
culturing, in a culture medium, Gliocladium sp. YJ-9515 having the accession number KCTC 0252 BP; and
recovering said at least one compound of ML-236B; wherein the compound of formula I is the lactone form of ML-236B represented by
wherein the compound of formula II is the free acid form of ML-236B represented by
and
wherein the compound of formula III is the sodium salt form of ML-236B represented by
The invention will be described in more detail in the drawings.
FIG. 1 is the IR spectrum of ML-236B obtained from this invention;
and
FIG. 2 is the 13C-NMR spectrum of ML-236B obtained from this invention.
The physical properties such as appearance, melting point. molecular weight, elemental analysis, formular, UV spectrum, IR spectrum, solubility and specific rotation of ML-236B obtained from Example 2, 3 and Comparative Example are described in Table 1.
TABLE 1
COMPARATIVE
Article
EXAMPLE 2, 3
EXAMPLE
Appearance
white crystal
white crystal
Melting point (° C.)
150˜152
150˜152
Molecular weight
calculated 390.2635
experimental 390.2392
experimental 390.2392
Elemental
C 70.74, H 8.77, O 20.49
C 70.74, O 20.49, H 8.77
Analysis (%)
C 70.55 , H 8.69
calculated
C 70.85 , H 8.02
experimental
Formula
C23H34O5
C23H34O5
UV spectrum
230, 237, 246
230, 237, 246
(nm, MeOH)
IR spectrum
3509, 2964, 2938, 2884,
3509, 2964, 2938, 2884,
(cm−1, KBr )
1744, 1698, 1445, 1385,
1744, 1699, 1445, 1385,
1236, 1206, 1182, 1151,
1236, 1206, 1182, 1150,
1077, 1056
1076, 1056
Solubility
methanol, chloroform,
methanol, chloroform,
soluble
ethanol, ethyl acetate
ethanol, ethyl acetate
insoluble
water
water
Specific rotation
+283n
+283n
[α]D
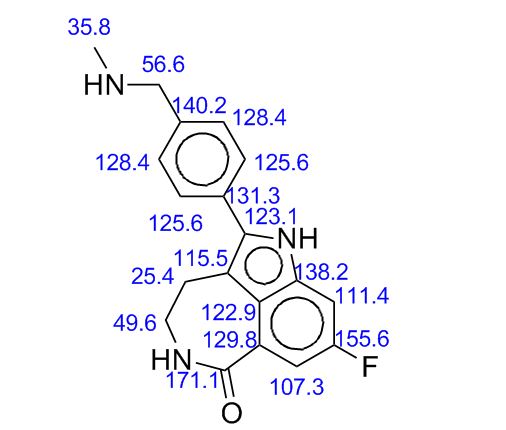
13C NMR data of ML-236B are shown in Table 2 and FIG. 2.
TABLE 2
The
δ c(ppm)
The
δ c(ppm)
number
EX-
COMPAR-
number
EX-
COMPAR-
of
AMPLE
ATIVE
of
AMPLE
ATIVE
carbon
2,3
EXAMPLE
carbon
2,3
EXAMPLE
C-1
171.50
170.67
C-13
124.48
123.33
C-2
39.31
38.44
C-14
134.35
133.38
C-3
63.18
62.12
C-15
128.96
127.96
C-4
36.88
35.84
C-16
133.49
132.37
C-5
77.22
76.26
C-17
31.66
30.70
C-6
33.75
32.82
C-18
14.66
13.64
C-7
24.83
23.78
C-19
—
—
C-8
37.66
36.67
C-20
177.79
176.55
C-9
38.31
37.40
C-21
42.56
41.50
C-10
68.45
67.51
C-22
27.55
26.48
C-11
27.06
26.30
C-23
12.59
11.49
C-12
21.74
20.74
C-24
17.74
16.64
By using a new microorganism which was obtained from this invention, the productivity of pravastatin precursor was elevated highly and the pravastatin precursor could be prepared in a simple way in short time.
Therefore, the present invention could be used effectively in production of pravastatin precursor.
Properties: Odorless, white to off-white, fine or crystalline powder. uv max (methanol): 230, 237, 245 nm. Sol in methanol, water; slightly sol in isopropanol. Practically insol in acetone, acetonitrile, chloroform, ether.
Absorption maximum: uv max (methanol): 230, 237, 245 nm
Pravastatin (marketed as Pravachol or Selektine) is a member of the drug class of statins, used in combination with diet, exercise, and weight-loss for lowering cholesterol and preventing cardiovascular disease.
Medical uses
Pravastatin is primarily used for the treatment of dyslipidemia and the prevention of cardiovascular disease.[1] It is recommended to be used only after other measures such as diet, exercise, and weight reduction have not improved cholesterol levels.[1]
The evidence for the use of pravastatin is generally weaker than for other statins. The antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT), failed to demonstrate a difference in all-cause mortality or nonfatal myocardial infarction/fatal coronary heart disease rates between patients receiving pravastatin 40mg daily (a common starting dose) and those receiving usual care.[2]
Mechanism of action
Pravastatin acts as a lipoprotein-lowering drug through two pathways. In the major pathway, pravastatin inhibits the function of hydroxymethylglutaryl-CoA (HMG-CoA) reductase. As a reversiblecompetitive inhibitor, pravastatin sterically hinders the action of HMG-CoA reductase by occupying the active site of the enzyme. Taking place primarily in the liver, this enzyme is responsible for the conversion of HMG-CoA to mevalonate in the rate-limiting step of the biosynthetic pathway for cholesterol. Pravastatin also inhibits the synthesis of very-low-density lipoproteins, which are the precursor to low-density lipoproteins (LDL). These reductions increase the number of cellular LDL receptors and, thus, LDL uptake increases, removing it from the bloodstream.[6] Overall, the result is a reduction in circulating cholesterol and LDL. A minor reduction in triglycerides and an increase in high-density lipoproteins (HDL) are common.
The present invention relates to a novel process for the preparation of substantially pure l^^^^^δa-hexahydro-beta,delta/6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-/ (beta R, delta R, lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid, sodium salt.
BACKGROUND OF THE INVENTION
US 4,346,227 discloses l,2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid, sodium salt. The compound is also known by the synonyms 3-beta-Hydroxycompactin; Eptastatin and Pravastatin. The compound is used as cholestrerol lowering agent which inhibit the enzyme H G CoA reductase.
The step of conversion of l,2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid to its sodium salt is crilcial. The prior art methods convert the acid form into sodium salt form as final step to afford the sodium salt. The prior art methods for the preparation of sodium salt from the l,2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-t(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)-1-Naphthaleneheptanoic acid are disclosed herein as reference.
WO 98/45410 discloses preparation of 1,2,6,7,8,8a-hexahydro-beta,delta, 6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid sodium by feeding compactin sodium to the microorganism Streptomyces exfoliatus and recovering the hydroxylated compactin sodium (l,2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid sodium salt) by extraction, purification by semi preparative HPLC and crystallization.
The process involves use of HPLC, which is a tedious and expensive technique and cannot be scaled up beyond a limit.
WO 00/46175 discloses a process for preparation of
l/2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-1-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid sodium salt from lactone by hydrolyzing with sodium hydroxide.
Also amine salts can be transformed to sodium salt by treating with sodium hydroxide and/or sodium alkoxide.
When amine salts are employed, it involves an extra step i.e., the preparation of the amine salt.
US 2003/0050502 discloses a process for preparation of sodium salt of a statin by contacting a solution of hydroxy acid of the statin with sodium-2-ethylhexanoate and recovering the corresponding sodium salt.
The process involves use of expensive reagent sodium-2-ethyl hexanoate.
The prior art methods suffer from one or more disadvantages like use of expensive reagents, need of special equipment to carry out the operation or increased number of steps for the preparation of sodium salt of l,2,6,7,8,8a-hexahydro-beta,delta/6-trihydroxy-2-methyl-8-[(2S)-2-me hyl-l-oxobutoxy]-, (beta R,delta
R/lS^δS/δS/δaR)- 1-Naphthaleneheptanoic acid.
The present invention relates to a process, which overcomes all the disadvantages of the prior art and results in substantially pure product in high yields.
Example 1
To a solution of 3,5-Dihydroxy-7-[6-hydroxy-2-methyl-δ-(2-methyl-butyryloxy)-l,2,6,7,δ,δa-hexahydro-naphthalen-l-yl]-heptanoic acid ( 70 g, 0.165 mol) in ethyl acetate (500 ml), solid sodium carbonate (δ.76 g, 0.0δ25 mol) was added and stirred for 2 hours. l,2/6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)-1-Naphthaleneheptanoic acid sodium salt was precipitated. The reaction mixture was filtered and cake was washed with ethyl acetate to get free flowing crystals of l,2,6,7,δ,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)- 1-Naphthaleneheptanoic acid sodium (FORMULA I). Yield: 65 g, δδ% Example 2
To a solution of 3,5~Dihydroxy-7-[6-hydroxy-2-methyl-δ-(2-methyl-butyryloxy)-l,2,6,7,δ,δa-hexahydro-naphthalen-l-yl]-heptanoic acid (10 Kg, 23.6 mol) in isobutyl acetate (60 L), solid sodium carbonate (1.25 Kg, 11.8 moi) was added and stirred for 3 hoursl,2,6,7,8,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)-1-Naphthaleneheptanoic acid sodium salt was precipitated. The reaction mixture was filtered and cake was washed with isobutyl acetate to get free flowing crystals of l,2,6,7,δ,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)- 1-Naphthaleneheptanoic acid sodium (FORMULA I). Yield: 9 Kg, δ5%
Example 3
To a solution of 3,5-Dihydroxy-7-[6-hydroxy-2-methyl-δ-(2-methyl-butyryloxy)-l,2,6,7,δ,δa-hexahydro-naphthalen-l-yl]-heptanoic acid (100. Kg, 236 mol) in butyl acetate (600 L), solid sodium carbonate (12.5 Kg, 118 mol) was added and stirred for 3 hours. l,2,6,7,8,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)-1-Naphthaleneheptanoic acid sodium salt was precipitated. The reaction mixture was filtered and cake was washed with butyl acetate to get free flowing crystals of l,2,6,7,δ,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)- 1-Naphthaleneheptanoic acid sodium (FORMULA I). Yield: 95 Kg, 90%
The invention will be described in more detail in the drawings. Fig. 1 is the IR spectrum of pravastatin sodium Fig. 2 is the13C-NMR spectrum of pravastatin sodium Fig. 3 is the H-NMR spectrum of pravastatin sodium
EXPERIMENTAL EXAMPLE
The physical properties of pravastatin sodium obtained from Example 1 and Comparative Example are described in Table 3.
Table 3
IR spectrum, “C-NMR spectrum, H-NMR spectrum of pravastatin sodium obtained from this invention are represented in Fig. 1, Fig. 2 and Fig. 3, respectively. By using a new microorganism Streptomyces exfoliatus YJ-118 isolated from this invention, ML-236B concentration in culture broth could be raised to 0.5% (w/v) and pravastatin sodium productivity was increased up to 600—1,340 mg/ / much higher than that of other microorganisms (60 mg/ / ) .
EXAMPLE 1
To 125 ml Erlenmeyer flask containing 20 ml seed culture medium(I) that comprises glucose 1%, yeast extract 0.2%, skim milk 0.2%, casein hydrolyte (N-Z amine) 0.5%, pH 7.0. 0.02% (w/v) ML-236B was added and Streptomyces exfoliatus YJ-118 isolated from manufacturing Example was inoculated. The cultivation was done at 27° C., 200 rpm, for 2 days on a rotary shaker. 20 ml of seed culture above was inoculated in 2 l Erlenmeyer flask containing 400 ml production medium(II) that comprises glucose 1.0%, yeast extract 1.0%, polypeptone 0.5%. K2HPO4 0.1%, MgSO4.7H2O 0.05%, NaCl 0.01˜0.1%, pH 7.2 and the flask was cultured at 27° C., 150 rpm. One day after cultivation, 0.05% (w/v) ML-236B (formula II-a) was added every day till the final concentration of ML-236B in culture broth became 0.2% (w/v). The cultivation was continued at 27° C., 150 rpm for 6 days and 0.3% glucose was fed once every two days 2 times in total. After then, the culture broth was adjusted to pH 9.0 and stirred for 3 hr. After centrifugation cell mass was removed and the supernatant was applied to a column of HP-20 500 ml. After washed with water, pravastatin sodium was eluted with 25% acetone solution. Pravastatin sodium fraction was concentrated in vacuo and the residue was applied to semi preparative HPLC(Kromasil C18 resin). Pravastatin sodium was eluted with 35% acetonitrile solution and was obtained as white crystal 1,254 mg (627 mg/l),
References
“Prevachol”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
Pfeffer MA, Keech A, Sacks FM, et al. “Safety and tolerability of pravastatin in long-term clinical trials: prospective Pravastatin Pooling (PPP) Project.” Circulation 2002;105:2341-2346
“Pravastatin”. LactMed. U.S. National Library of Medicine. Retrieved 1 December 2012.
Vaughan, C. J., and A. M. Gotto, Jr. 2004. Update on statins: 2003. Circulation 110: 886–892.
Yoshino G, Kazumi T, Kasama T, et al. (1986). “Effect of CS-514, an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase, on lipoprotein and apolipoprotein in plasma of hypercholesterolemic diabetics”. Diabetes Res. Clin. Pract.2 (3): 179–81. doi:10.1016/S0168-8227(86)80020-1. PMID3091343.
Pravastatin sodium substantially free of pravastatin lactone and epi-pravastatin, and compositions containing same
HMG-CoA reductase inhibitor; bioactive metabolite of mevastatin, q.v. Prepn by microbial hydroxylation: A. Terahara, M. Tanaka, DE3122499; eidem,US4346227 (1981, 1982 both to Sankyo);
N. Serizawa et al.,J. Antibiot.36, 604 (1983).
Structure elucidation: H. Haruyama et al.,Chem. Pharm. Bull.34, 1459 (1986).
HPLC determn in biological fluids: S. Baueret al., J. Chromatogr. B818, 257 (2005).
Effect on serum lipid concentration: N. Nakaya et al.,Atherosclerosis61, 125 (1986);
on hepatic metabolism of cholesterol: E. Reihnér et al.,N. Engl. J. Med.323, 224 (1990).
Clinical comparison with probucol, q.v.: G. Yoshino et al.,Lancet2, 740 (1986).
Clinical reduction of risk of major cardiovascular events in patients with coronary heart disease: LIPID Study Group, N. Engl. J. Med.339, 1349 (1998).
Clinical effect on risk of stroke: H. D. White et al.,ibid.343, 317 (2000).
Derivative Type: Lactone
Molecular Formula: C23H34O6
Molecular Weight: 406.51
Percent Composition: C 67.96%, H 8.43%, O 23.61%
Properties: Colorless plate crystals, mp 138-142°. [a]D22 +194.0° (c = 0.51 in methanol). uv max (methanol): 230, 237, 245 nm.
Melting point: mp 138-142°
Optical Rotation: [a]D22 +194.0° (c = 0.51 in methanol)
Absorption maximum: uv max (methanol): 230, 237, 245 nm
Originally developed at Kyorin, gatifloxacin was first licensed to Gruenenthal in Europe, and that company still maintains rights to the oral and injectable formulations of the product. In October 1996, Kyorin licensed gatifloxacin to BMS, granting the company development and marketing rights in the U.S., Canada, Australia, Mexico, Brazil and certain other markets. In 2006, rights to the compound were returned by BMS. Subsequently, Senju and Kyorin signed a licensing agreement regarding the development of ethical eye drops containing the fluoroquinolone. In April 2000, Sumitomo Dainippon Pharma agreed to comarket the oral formulation in Japan. In August of that year, Allergan in-licensed gatifloxacin from Kyorin, gaining development and commercialization rights to the drug in all territories except Japan, Korea, China and Taiwan. The India-based Lupin Pharmaceuticals signed an agreement in June 2004 with Allergan to promote the ophthalmic solution of gatifloxacin in the pediatric specialty area in the U.S. PediaMed Pharmaceuticals also holds rights to the drug. In 2009, Kyorin licensed the drug candidate to Senju in China.
Gatifloxacin is the common name for (±)-1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid (1), one of the most important broad-spectrum antibacterial agents and a member of the fourth-generation fluoroquinolone family.(1)Fluoroquinolones inhibit the enzyme DNA gyrase (topoisomerase II), which is responsible for the supercoiling of the DNA double helix, preventing the replication and repair of bacterial DNA and RNA.(2) Gatifloxacin (1) reached the market in 1999 under the brand name Tequin for the treatment of respiratory tract infections. The drug is available as tablets and aqueous solutions for intravenous therapy as well as eye drop formulation (Zymar).
To date, there are several processes described for the preparation of gatifloxacin, which can be grouped into two main categories: direct substitution of the 7-position fluorine atom of 1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (2) by 2-methylpiperazine (Scheme 1),(3-5) and through boron chelate-type intermediates to overcome the diminished reactivity induced by the 8-methoxy group, which uses as starting material the ethyl ester derivative 3 (Scheme 2).(6-9)
Masuzawa, K.; Suzue, S.; Hirai, K.; Ishizaki, T. 8-Alkoxyquinolonecarboxylic acid and salts thereof excellent in the selective toxicity and process of preparing the same EP 0 230 295 A3, 1987.
Ruzic, M; Relic, M; Tomsic, Z; Mirtek, M. Process for the preparation of Gatifloxacin and regeneration of degradation products WO 2006/004561 A1, 2006.
Sanchez, J. P.; Gogliotti, R. D.; Domagala, J. M.; Garcheck, S. J.; Huband, M. D.; Sesnie,J. A.; Cohen, M. A.; Shapiro, M. A. J. Med. Chem. 1995, 38, 4478
Takagi, N.; Fubasami, H.; Matsukobo, H.; (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture EP 0 464 823 A1, 1991.
preparation of Gatifloxacin hemihydrate from Ethyl-1- Cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate through boron difluoride chelate. Ethyl-1-cyclopropyl- 6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate is reacted with aqueous hydrofluoroboric acid followed by condensation with 2-methyl piperazine in polar organic solvent resulting in an intermediate l-Cyclopropyl-7- (3-methyl piperazin-1- yl). -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. This intermediate may be further hydrolyzed to yield Gatifloxacin. Gatifloxacin so obtained may needs purification to yield high purity product. However to obtain directly high purity Gatifloxacin it is desirable to isolate the intermediate by cooling to low temperatures . Treating with an alcohol or mixture of alcohols purifies this intermediate. The purified condensed chelate in aqueous ethanol on hydrolysis with triethylamine followed by crystallization in ethanol gives Gatifloxacin hemihydrate with high purity.
Example-I: Preparation of Gatifloxacin • with isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-di luoro-8-methoxy-4-oxo- 1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3- quinόline carboxylate (100g)is suspended in ,40%aq..hydrofluoroboric acid -(1000 ml). Temperature of • the reaction mass is raised and maintained at 95°C to 100°C for 5hrs followed by cooling to 30°C – 35°C. Water (400 ml) is added and maintained at 25°C – 30°C for 2hrs . Product is filtered, washed with water (500 ml) and dried at 40°C – 45°C to constant weight. Dry weight of the product: 101.6 g (Yield: 95.8 %)
100 g of Boron difluoride chelate derivative prepared as above in stage-1 is suspended in acetonitrile (800 ml) , to that 2-methyl piperazine (44.0 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs followed by cooling to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr. The product is filtered and dried at 45°C – 50°C to constant weight. Dry weight of the product: 116.0 g (Yield: 93.9 %) .
The condensed chelate (100 g) prepared as above is suspended in methanol (1500 ml), maintained at 40°C – 45°C for 30 min. The reaction mass is gradually cooled, maintained for 1 hr at -5°C to 0°C. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. Dry weight of the product: 80.0 g (Yield: 80.0 %)
Stage -3: Preparation of Gatifloxacin (Crude)
The pure condensed chelate (100.0 g) prepared as above in stage-2 is suspended in 20% aq. ethanol (1000 ml) , the temperature is raised and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles, distilled under vacuum to remove solvent. Fresh ethanol (200 ml) is added and solvent is removed under vacuum at temperature below 50°C. Ethanol (200 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is mixed at -10°C to -5°C for 1 hr and then filtered. The wet cake is washed with ethanol (25 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 83.3 g (Yield: 91.7 %)
Stage- 4: Purification of crude Gatifloxacin
Crude Gatifloxacin (100.0 g) prepared as above in stage-3 is suspended in methanol (4000 ml), the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. Activated carbon (5 g) is added, maintained for 30 min and the solution is filtered. The filtrate is concentrated to one third of its original volume under vacuum at temperature below 40°C. The reaction mass is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. The dry weight of the pure Gatifloxacin is 76.0 g (Yield: 76.0 %)
Example-II: Preparation of Gatifloxacin without isolation of intermediate (boron difluoride chelate derivative)
Ethyll-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylate (lOOg) is suspended in 40% aq. hydrofluoroboric acid (1000 ml) . Temperature of the reaction mass is raised and maintained at 95°C to 100°C for 5 hrs followed by cooling to 30°C – 35°C. 400 ml DM water is added, maintained at 25°C – 30°C for 2hrs . The product is filtered, washed with DM water (500 ml) and dried at 40°C – 45°C to constant weight. The dry wt is 102.5 g (Yield: 96.6 %)
Stage – 2: Preparation of Gatifloxacin (Crude)
The boron difluoride chelate derivative (100 g) prepared as above in stage-1 is suspended in acetonitrile (800 ml) , 2-methyl piperazine (44 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs. Removed the solvent by vacuum distillation. 20% Aq. ethanol (1000 ml) is added, raised the temperature and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles. The filtrate is distilled under vacuum to remove solvent completely. Fresh ethanol (250 ml) is added and distilled under vacuum at temperature below 50°C. Fresh Ethanol (250 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr and filtered. The wet cake is washed with ethanol (30 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 73.5 g (Yield: 65.4 %)
Stage -3: Purification of crude Gatifloxacin
Crude Gatifloxacin (80.0 g) prepared as above in stage-2 is suspended in methanol (2000 ml) , the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. The reaction mixture is filtered. The filtrate is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the pure Gatifloxacin is 56.0 g (Yield: 70.0 %)
Gatifloxacin is the international common name of l-cyclopropyl-6-fluoro-l, 4-dihydro-8-methoxy- 1- (3-methyl-l-piperazinyl) -4-oxo-3-guinolin-carboxylic acid of formula (I) , with application in medicine and known for its antibiotic activity:
European patent application EP-A-230295 discloses a process for obtaining gatifloxacin that consists on the reaction of compound (II) with 2-
In this process the gatifloxacin is isolated in the form of a hemihydrate after a laborious process of column chromatography and recrystallisation in methanol, which contributes towards making the final yield lower than 20% by weight. Moreover, in said process an undesired by-product is formed, resulting from demethylation at position 8 of the ring. European patent application EP-A-241206 discloses a process for preparing gatifloxacin, whose final steps are as follows:
(III) H ft N Me H DMSO
Gatifloxacin (I)
(IV) This process uses the intermediate compound (III) , which has been prepared and isolated in a separate operation, while the intermediate compound (IV) is also isolated before proceeding to its conversion into gatifloxacin by treatment with ethanol in the presence of triethylamine. The overall yield from these three steps is lower than 40%. These disadvantages — a synthesis involving several steps, low yields, and the need to isolate the intermediate products — hinder the production of gatifloxacin on an industrial scale. There is therefore a need to provide a process for preparing gatifloxacin with a good chemical yield, without the need to isolate the intermediate compounds and that substantially avoids demethylation in position 8 of the ring. The processes termed in English “one pot” are characterised in that the synthesis is carried out in the same reaction vessel, without isolating the intermediate compounds, and by means of successive addition of the reacting compounds. The authors of the present invention have discovered a simplified process for preparing gatifloxacin which does not require isolation of the intermediate compounds .
Example 1: Preparing gatifloxacin from compound (II) 10 g (0.0339 moles, 1 equivalent) of compound
(II) is placed in a flask, 30 ml of acetonitryl (3 volumes) is added and this is heated to a temperature of 76-80° C.
Once reflux has been attained, and being the temperature maintained, 3.28 g (0.0203 moles, 0.6 equivalents) of hexamethyldisilazane (HMDS) is added with a compensated adding funnel. Once addition is completed, the reaction is maintained with stirring for 1 hour at a temperature of 76-80° C. Once this period has elapsed, the reaction mixture is cooled to a temperature ranging between 0 and 15° C, and 5.78 g (0.0407 moles, 1.2 equivalents) of boron trifluoride ethyletherate is added while keeping the temperature below 15° C. Once addition is completed, the temperature is allowed to rise to 15- 25° C and it is kept under these conditions for approximately 2 hours. The pH of the mixture is then adjusted to an approximate value of 9 with triethylamine (approximately 2 ml) . To the resulting suspension is added a solution of 10.19 g (0.1017 moles, 3 equivalents) of 2-methylpiperazine in 28 ml of acetonitryl, while maintaining the temperature between 15 and 25° C. The resulting amber solution is kept with stirring under these conditions for approximately 3 hours . Once the reaction has been completed, the solution is distilled at low pressure until a stirrable paste is obtained. At this point 50 ml of methanol is added, the resulting suspension is raised to a temperature of 63-67° C and is kept under these conditions for approximately 5 hours . Once the reaction has been completed, the mixture is cooled to a temperature of 25-35° C in a water bath, and then at a temperature of 0-5° C in a water/ice bath for a further 1 hour. The resulting precipitate is filtered, washed with cold methanol (2 x 10 ml) and dried at 40° C in a vacuum oven to constant weight. 10.70 g of crude gatifloxacin is obtained, having a water content of 2.95% by weight. The yield of the process is 81.8%.
The crude product is crystallised in methanol by dissolving 20 g of crude gatifloxacin in 1 1 of methanol (50 volumes) at a temperature of 63-67° C. Once all the product has been dissolved, the solution is left to cool to a temperature of 30-40° C, and then to a temperature of 0-5° C in a water/ice bath, maintaining it under these conditions for 1 hour. The resulting suspension is filtered and the solid retained is washed with 20 ml (1 volume) of cold methanol. The solid obtained is dried at 40° C in a vacuum oven to provide 18.65 g of gatifloxacin with a water content of 2.36% by weight.
The overall yield from the compound (II) is 77.7%, with a purity exceeding 99.8% as determined by HPLC chromatography. The content of by-product resulting from demethylation in position 8 of the ring is lower than 0.1% as determined by HPLC chromatography.
An improved process to obtain gatifloxacin (1) through use of boron chelate intermediates has been developed. The methodology involves an initial activation step which accelerates the formation of the first chelate under low-temperature conditions and prevents demethylation of the starting material. To increase the overall yield and to avoid the isolation and manipulation of the resulting intermediates, the process has been designed to be carried out in one pot. As a result, we present here an easy, scaleable and substantially impurity-free process to obtain gatifloxacin (1) in high yield.
A High-Throughput Impurity-Free Process for Gatifloxacin
Department of Research & Development, Química Sintética S.A., c/ Dulcinea s/n, 28805 Alcalá de Henares, and Department of Organic Chemistry, University of Alcalá, 28871 Madrid, Alcalá de Henares, Spain
gatifloxacin (1) as white crystals. Yield 32.3 kg, (93%); purity by HPLC 99.87%; Assay by HPLC 100.8%; mp 167−168 °C(18) (Lit. (J. Med. Chem. 1995, 38, 4478)159−162 °C).
18
DSC analysis showed two endothermic peaks at 166.2 °C (T onset = 164.3 °C) and 190.0 °C (T onset = 188.2 °C) and an exothermic one at 168.1 °C. The shape of this DSC curve is characteristic of a monotropic transition between crystalline forms
Water content by Karl Fischer 3.0%(19) MS m/z 376 (M+ + H);
19
Although there are several hydrates described for gatifloxacin such as, among others, the hemimydrate, sesquihydrate, and pentahydrate(Raghavan, K. S.; Ranadive, S. A.;Gougoutas, J. Z.; Dimarco, J. D.; Parker, W. L.; Dovich, M.; Neuman, A.Gatifloxacin pentahydrate. WO 2002/22126 A1, 2002) , the Gatifloxacin obtained by the present procedure does not seem to form a stoichometric hydrate, but instead it retains moisture.
Thus, the product is usually obtained with a Karl-Fischer value below 1% after drying, but it can absorb moisture until a final content of about 3%. This water content can vary between 2.0% and 3.5%, depending on the relative humidity of the environment. DSC analysis revealed a broad endothermic signal with minimum at 76 °C, while TGA analysis showed that the product loses all the water below 80 °C.
No loss of weight is registered when the product melts, and the weight is constant until the decomposition of the material at about 200 °C. On the basis of these results, it can be said that the water content of the gatifloxacin obtained by the present process is retained moisture instead of water belonging to the lattice. The shape of the derivative of the weight curve at the beginning of the analysis shows that the sample has already lost part of the moisture when the register starts. This is probably due to the sample starting to lose weight when makes contact with the dry atmosphere of the TGA oven that could explain the different values obtained for water content of the analyzed sample by TGA (1.90%) and Karl-Fischer (2.64%) methods.
A Canadian study published in the New England Journal of Medicine in March 2006 claims Tequin can have significant side effectsincluding dysglycemia.[2] An editorial by Dr. Jerry Gurwitz in the same issue called for the Food and Drug Administration (FDA) to consider giving Tequin a black box warning.[3] This editorial followed distribution of a letter dated February 15 by Bristol-Myers Squibb to health care providers indicating action taken with the FDA to strengthen warnings for the medication.[4] Subsequently it was reported on May 1, 2006 that Bristol-Myers Squibb would stop manufacture of Tequin, end sales of the drug after existing stockpiles were exhausted, and return all rights to Kyorin.[5]
Union Health and Family Welfare Ministry of India on 18 March 2011 banned the manufacture, sale and distribution of Gatifloxacin as it caused certain adverse side effects[6]
Gatifloxacin is currently available only in the US and Canada as an ophthalmic solution.
In China it is sold in tablet as well as in eye drop formulations.
Ophthalmic anti-infectives are generally well tolerated. The concentration of the drug observed following oral administration of 400 mg gatifloxacin systemically is approximately 800 times higher than that of the 0.5% Gatifloxacin eye drop. Given as an eye drop, Gatifloxacin Ophthalmic Solution 0.3% & 0.5% cause very low systemic exposures. Therefore, the systemic exposures resulting from the gatifloxacin ophthalmic solution are not likely to pose any risk for systemic toxicities.
The reaction of 1-bromo-2,4,5-trifluoro-3-methoxybenzene (I) with CuCN and N-methyl-2-pyrrolidone at 150 C gives 2,4,5-trifluoro-3-methoxybenzonitrile (II), which by treatment with concentrated H2SO4 yields the benzamide (III) The hydrolysis of (III) with H2SO4 -. water at 110 C affords 2,4,5-trifluoro-2-methoxybenzoic acid (IV), which by reaction with SOCl2 is converted into the acyl chloride (V). The condensation of (V) with diethyl malonate by means of magnesium ethoxide in toluene affords diethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) malonate (VI), which by treatment with p-toluenesulfonic acid in refluxing water gives ethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) acetate (VII). The condensation of (VII) with triethyl orthoformate in refluxing acetic anhydride yields 3-ethoxy -2- (2,4,5-trifluoro-3-methoxybenzoyl) acrylic acid ethyl ester (VIII), which is treated with cyclopropylamine (IX) to afford the corresponding cyclopropylamino derivative (X). The cyclization of (X) by means of NaF in refluxing DMF gives 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (XI), which is hydrolyzed with H2SO4 in acetic acid to yield the corresponding free acid (XII). Finally, this compound is condensed with 2-methylpiperazine (XIII) in hot DMSO.
Title: Gatifloxacin
CAS Registry Number: 112811-59-3
CAS Name: 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid
Trademarks: Tequin (BMS); Zymar (Allergan)
Molecular Formula: C19H22FN3O4
Molecular Weight: 375.39
Percent Composition: C 60.79%, H 5.91%, F 5.06%, N 11.19%, O 17.05%
Literature References: Fluorinated quinolone antibacterial. Prepn: K. Masuzawa et al.,EP230295; eidem,US4980470 (1987, 1990 both to Kyorin); J. P. Sanchez et al.,J. Med. Chem.38, 4478 (1995); of the sesquihydrate: T. Matsumoto et al.,US5880283 (1999 to Kyorin). In vitro antibacterial activity: A. Bauernfeind, J. Antimicrob. Chemother.40, 639 (1997); H. Fukuda et al.,Antimicrob. Agents Chemother.42, 1917 (1998). Clinical pharmacokinetics: M. Nakashima et al.,ibid.39, 2635 (1995). Clinical study in urinary tract infection: H. Nito, 10th Mediterranean Congr. Chemother.1996, 327; in respiratory tract infection: S. Sethi, Expert Opin. Pharmacother.4, 1847 (2003).
Properties: Pale yellow prisms from methanol as hemihydrate, mp 162°.
Melting point: mp 162°
Derivative Type: Sesquihydrate
CAS Registry Number: 180200-66-2
Manufacturers’ Codes: AM-1155
Molecular Formula: C19H22FN3O4.1½H2O
Molecular Weight: 384.40
Percent Composition: C 59.37%, H 6.03%, F 4.94%, N 10.93%, O 18.73%
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic); Quinolones and Analogs
(6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture
Amritsar is one of the largest cities of the Punjab state in India. The city origin lies in the village of Tung, and was named after the lake founded by the fourth Sikh …
After the WHO had released the second draft of the guideline for the design of hold time studies in March already, it now released the final version as part of the Technical Report Series 992. Find out more about the Guideline for Hold Time Studies.
The GMP regulations require that raw materials, packaging materials, intermediate, bulk and finished products need to be stored under suitable conditions. This also includes the definition of maximum hold-times for intermediate and bulk products prior to their further processing. The definition of these times should be justified on the basis of scientific data. This guideline aims at reflecting aspects that…
Erlotinib is an EGFR inhibitor. The drug follows Iressa (gefitinib), which was the first drug of this type. Erlotinib specifically targets the epidermal growth factor receptor (EGFR)tyrosine kinase, which is highly expressed and occasionally mutated in various forms of cancer. It binds in a reversible fashion to the adenosine triphosphate (ATP) binding site of the receptor.[1] For the signal to be transmitted, two EGFR molecules need to come together to form a homodimer. These then use the molecule of ATP to trans-phosphorylate each other on tyrosine residues, which generates phosphotyrosine residues, recruiting the phosphotyrosine-binding proteins to EGFR to assemble protein complexes that transduce signal cascades to the nucleus or activate other cellular biochemical processes. By inhibiting the ATP, formation of phosphotyrosine residues in EGFR is not possible and the signal cascades are not initiated.
Erlotinib hydrochloride (1), chemically named as N-(3-ethynylphenyl)-6,7-bis-(2-meth- oxyethoxy)-4-qumazolimmine monohydro chloride, is an inhibitor of oncogenic and proto- oncogenic protein tyrosine kinases, e.g. epidermal growth factor receptor (EGFR). Erlotinib is therefore useful in the treatment of proliferative disorders and is currently marketed for the treatment of lung cancer and pancreatic cancer.
(Erlotinib Hydrochloride)
(1)
It has been reported that erlotinib hydrochloride can exist in different polymorphic forms. The manufacturing process for many pharmaceuticals is hindered by the fact that the organic compound which is the active ingredient can exist in more than one polymorphic form. It is essential in pharmaceutical development to ensure that the manufacturing process for the preparation of the active ingredient affords a single polymorph with a consistent level of polymorphic purity. If the manufacturing process produces a product with varying degrees of polymorphic purity and/ or or where the process does not control polymorphic inter-conversion, it could lead to serious problems in dissolution and/ or bioavailability in the finished pharmaceutical composition comprising the active ingredient, Erlotinibhydrochloride is disclosed in patent US 5,747,498 and details of the disclosed method for the preparation of erlotinib hydrochloride are described in Scheme 1.
Scheme 1
4-Chloro-6,7-bis-(2-methoxyed oxy)qiunazoline (2) was reacted with 3-emynylaniline (3) or its hydrochloride salt using various solvents and pyridine as a base to yield erlotinib hydrochloride (1) which was treated widi a biphasic mixture consisting of saturated aqueous NaHC03, chloroform and methanol, to formerlotinib base (4). The base (4) obtained in the organic phase was purified by flash chromatography to afford purified erlotinib base. The purified base was further treated with hydrochloric acid in the presence of diethyl ether and chloroform to yield erlotinib hydrochloride.
This isolation of purified erlotinib base required the use of a lengthy workup process including column chromatography and required the chlorinated solvent, chloroform, which is not particularly suitable £01 commercial production of pharmaceuticals. Furthermore, the p irification by column chromatography is neither economical nor feasible at industrial scale. In addition, substantially pure erlotinib could not be obtained.
Two crystalline forms of erlotinib hydrochloride (polymorph A and polymorph B), were characterized by XRPD in patent application, WO 01/34574. Erlotinib hydrochloride can be obtained in form A or in a mixture of polymorph A and B, by refluxing 3-ethynylaniline and 4-chloro-6,7-bis-(2-methoxyemoxy)-qitiiiazoline in a mixture of toluene and acetonitrile. This afforded polymorph A or a mixture of polymorph A and B. It was also disclosed that the formation of polymorph A was favoixred by reducing the amounts of acetonitrile with respect to toluene.
Furthermore, erlotinibhydrochloride polymorph A can be converted into polymorph B by refluxing the polymorph A with alcohol/water. Consequently, in the disclosed methods, there was always contamination of form A with form B and vice-versa. In addition, the products of the reaction are not chemically pure and difficult to purify thereafter. Consequently, these methods are not suitable for preparation of commercial quantities of pure polymorph A.
A process for the preparation of erlotinib hydrochloride, polymorph E by condensation reaction of 3-emynylaiiiline and 4-chloro-6,7-bis-(2-memoxyethoxy)quii azoline in ( , , )- trifiuorotoluene and HC1 was disclosed in U.S. Patent application 2004/0162300. Polymorph E was characterized by XRPD, IR and melting point. However, (α,α,α)- trifluorotoluene is a highly flammable and dangerous solvent for the environment and is not suitable for commercial production. A process for the preparation of erlotinib hydrochloride, polymorph A by reaction of erlotinib base widi aqueous or gaseous HC1 was disclosed in US 2009/0131665. In this method, toluene, a mixture of toluene and methanol, TBME, ethyl acetate, 1-butanol or MIBK were used as a solvent.
However, when DCM, diethyl ether, isopropyl acetate, was used as a solvent, polymorph B was formed. In practice, it has been found that the disclosed methods are inconsistent and afford polymorphic mixtures. In particular, example 1 of US 2009/131665 was repeated and erlotinib hydrochloride was obtained with only 97% purity. In addition, XRPD analysis showed d at the example afforded form B or mixtures of forms A and B. Furthermore, several crystallizations of erlotinib hydrochloride, obtained from repetition of the example, using various solvents and their combinations would not yield a product pure enough to comply with ICH guidelines.
A process for the preparation of a hydrate of erlotinib hydrochloride comprising crystallization of erlotinib hydrochloride using water as solvent, preferably in the absence of organic solvent was disclosed in US 20080167327. This patent also disclosed the process to prepare hemihydrate polymorph form I as well as form II.
A process for the preparation of erlotinib hydrochloride, polymorph M, N and P by reaction of erlotinib base and aqueous or gaseous HC1 dissolved in organic solvents was disclosed in WO 2008/102369.
A process for the preparation of erlotinib hydrochloride by condensation reaction of 4- chloro~6,7-bis-(2-me oxyemoxy)-quinazoline and 3-ethynylaniline in isopropyl alcohol as a solvent and pyridine as a base was disclosed in Molecules Journal (Vol, 11, 286, 2006) but no details on the polymorph were disclosed.
A method for the preparation of erlotinib hydrochloride polymorph A comprising passing hydrochloride gas onto solid erlotinib base containing residual amounts of isopropanol was disclosed in WO 2010/040212. However, in practice it was found that the process did not afford chemically or polymorphically pure product. Repetition of example 1 (page 8) of WO 2010/040212 to prepare erlotinibhydrochloride, by reaction of erlotinib base and gaseous HQ in IPA as a solvent, afforded a mixture of polymorph A and polymorph B (as checked by XRPD).
A process for the preparation of acid salts of erlotinib by reaction of 4-chloro-6,7-bis-(2- memoxyemoxy)-quinazoline and 3-emynykniline or an acid salt of 3-emynylaniline under acidic conditions to form the corresponding erlotinib salt was disclosed in US 2010/0094004.
In order to complete the reaction, several hours (6 hours) of reflux was required and hence it is not a cost effective process. In addition, in practice it was found that the process did not afford chemically or polymorplxLcally pure product. A process £oi the preparation of erlotinib base, polymorph Gl, G2 and G3 was disclosed in WO 2009/002538 and WO 2010/05924.
Scheme 2
A method for the preparation of eiiotinib hydrochloride was disclosed in US 2009/0306377. The method, illustrated in Scheme 2, involves treating 6,7-dimethoxy- 4(3H)-quinazolone (5) with hydrobiOmic acid or pyridine-hydrochloric acid to afford 6,7- dihydroxy-4(3H)-quinazolone (6), which was diacetylated with acetic anhydride to afford diester (7), which was treated with oxalyl chloride/DMF to afford 4-chloro-6,7- ctiacetoxyquinazoline (8). Compound (8) was condensed with 3-e ynylaniline to afford JV- (3-ethynylphenyl)-6,7-dihydfoxy-4-quinazolinamine hydrochloride (9), which was converted into the diol N-(3-emynylphenyl)-6,7-dmyckOxy-4-quinazolinamine (10) by treatment with aqueous ammonia/methanol.
The diol (10) was treated with 2-iodo-ethylmethyl ether to yield compound (4) which on treatment with HC1 afforded erlotinib hydrochloride (1). However, this preparation of erlotinib hydrochloride is a long synthetic route and gives low yields and requires very toxic reagents like pyridine, HBi and controlled reagents like acetic anhydride. Hence, it is not suitable for large scale production. Object of the invention
The priot art processes described above for the preparation of erlotinib and its salts have major disadvantages with respect to the formation and removal of process related chemical and polymorphic impurities; poor commercial viability due to die use of hazardous reactants; expensive, time consuming separation methods such as column chromatography and/ or low yields and purity of final and intermediate products.
As the commercial production of erlotinib hydrochloride is of great importance, for the treatment of cancer, and in view of the above disadvantages associated with the prior art there is a real need for alternative and improved processes for the preparation of erlotinib hydrochloride which do not involve multiple steps and further eliminates the need for cumbersome purification techniques, particularly for the removal of the chemical and polymorphic impurities. The alternative processes must be economical and high yielding and provide erlotinib and its salts with a high degree of chemical and polymorphic purity.
U.S. Patent No. 5,747,498 disclosed 4-(substituted phenylamino) quinazoline derivatives, processes for their preparation, pharmaceutical compositions in which they are present and method of use thereof. These compounds are Tyrosine Kinase Inhibitors and are useful in the treatment of hyperproliferative diseases, such as cancers, in mammals. Among them, erlotinib hydrochloride, chemically N-(3-ethynylphenyl)-6,7-bis(2-methoxy ethoxy)-4-quinazolinamine hydrochloride is a selective inhibitor of the erbB family of oncogenic and protooncogenic protein tyrosine kinases, such as epidermal growth factor receptor (EGFR), and is useful for the treatment of proliferative disorders, such as cancers, particularly non small cell lung cancer, pancreatic cancer, ovarian cancer, breast cancer, glioma, head cancer or neck cancer.
Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, etc. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR).
Solvent medium and mode of crystallization play very important role in obtaining a crystalline form over the other.
Erlotinib hydrochloride can exist in different polymorphic forms, which differ from each other in terms of stability, physical properties, spectral data and methods of preparation.
The U.S. Patent No. 5,747,498 (herein after referred to as the ‘498 patent) makes no reference to the existence of specific polymorphic forms of erlotinibhydrochloride. In this patent, it is disclosed that the compound is isolated according to conventional techniques; more precisely, according to the embodiments exemplified, crude erlotinib hydrochloride residue (obtained by reaction of 4-chloro-6,7-bis-(2-methoxyethoxy)-quinazoline with 3-ethynylaniline or its hydrochloride salt in a solvent such as a d-Cβ-alcohol, dimethylformamide, N-methylpyrrolidin-2-one, chloroform, acetonitrile, tetrahydrofuran, 1,4-dioxane, pyridine or other aprotic solvents, preferably isopropanol) is basified with saturated aqueous NaHCO3 in the presence of methanol and chloroform followed by flash chromatography on silica using 30% acetone in hexane to afford erlotinib free base, which is further treated with hydrochloric acid in the presence of diethyl ether and chloroform to give erlotinib hydrochloride (melting point: 228° – 2300C).
PCT Patent Publication No. WO 99/55683 disclosed erlotinib mesylate anhydrate and hydrate polymorphic forms, their method of preparation and pharmaceutical compositions containing thereof.
PCT Patent Publication No. WO 01/34574 A1 (herein after referred to as the ‘574 patent publication) described two crystalline forms of erlotinib hydrochloride (polymorph A and polymorph B), characterized by powder X-ray diffraction (p-XRD) pattern. The publication further taught that the synthetic procedure described and exemplified in the ‘498 patent produces the erlotinib hydrochloride as a mixture of the polymorphs A and B.
TARCEVA (erlotinib), a kinase inhibitor, is a quinazolinamine with the chemical name N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine. TARCEVA contains erlotinib as the hydrochloride salt that has the following structural formula:
Erlotinib hydrochloride has the molecular formula C22H23N3O4•HCl and a molecular weight of 429.90. The molecule has a pKa of 5.42 at 25oC. Erlotinib hydrochloride is very slightly soluble in water, slightly soluble in methanol and practically insoluble in acetonitrile, acetone, ethyl acetate and hexane.
Aqueous solubility of erlotinib hydrochloride is dependent on pH with increased solubility at a pH of less than 5 due to protonation of the secondary amine. Over the pH range of 1.4 to 9.6, maximal solubility of approximately 0.4 mg/mL occurs at a pH of approximately 2.
Erlotinib is a Human Epidermal Growth Factor Receptor Type 1 /Epidermal Growth Factor Receptor (HER1/EGFR) tyrosine kinase inhibitor.
Erlotinib is described chemically as N-(3-ethynylpheny!)-6,7-bis(2- methoxyethoxy)quinazolin-4-amine, and its hydrochloride salt is represented by the compound of Formula I.
Erlotinib is disclosed in EP0817775 which also a discloses process for its preparation, which involves adding 3-ethynylaniline and 4-chloro-6,7-bis(2-methoxyethoxy)quinazoline in isopropanol containing pyridine and then refluxing the mixture for 4 hours under the atmosphere of dry nitrogen. The solvent is removed and residue is extracted in 10% methanol in CHCI3 and saturated aqueous NaHCO3. N-(3-ethynylphenyl)-6,7-bis(2- methoxyethoxy)quinazolin-4-amine base is separated chromatographically and converted to the hydrochloride salt in a solvent such as CHCI3 using hydrochloric acid.
EP1044969 claims a method for preparing intermediates and compounds covering erlotinib. This patent discloses a process for preparing N-(3-ethynylphenyl)-6,7-bis(2- methoxyethoxy)quinazolin-4-amine which involves stirring 4-[3-[[6,7-bis(2-methoxyethoxy)- 4-quinazolinyl]amino]phenyl]-2-methyl-3-butyn-2-ol with anhydrous sodium hydroxide and 2-methoxyethanol and heating at reflux for 47 hours. The reaction mixture is cooled to 20- 25°C and concentrated HCI is added to it. The resulting mixture is granulated at 20-25°C to crystallize the product.
Indian patent application 902/CHE/2006 discloses a process for preparation of N-(3- ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine hydrochloride. The process involves reacting 3,4-dihydroxy benzaldehyde with substituted ethylmethyl ether in the presence of an inert solvent and base to obtain 3,4-bis(2-methoxyethoxy) benzaldehyde. This 3,4-bis(2-methoxyethoxy) benzaldehyde is converted to 3,4-bis(2-methoxyethoxy) benzaldoxime in the presence of a base and organic solvent and is further dehydrated to 3,4-bis(2-methoxyethoxy) benzonitrile. The benzonitrile so obtained is nitrated to obtain 4,5-bis(2-methoxyethoxy)-2-nitrobenzonitrile which is further reduced to obtain 2-amino- 4,5-bis(2-methoxyethoxy) benzonitrile. N’-(3-ethynylphenyl)-N,N-dimethyl formamidine obtained on formylation of 3-ethynylaniline with N,N-dimethyl formamidine is coupled with 2-amino-4,5-bis(2-methoxyethoxy) benzonitrile to obtain erlotinib free base which on treatment with a polar solvent containing hydrochloric acid gives erlotinib hydrochloride.
Indian patent application 904/CHE/2006 also discloses a process for preparation of N-(3- ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine hydrochloride. The process involves reacting 3,4-dihydroxy benzaldehyde with substituted ethylmethyl ether in the presence of an inert solvent and base to obtain 3,4-bis(2-methoxyethoxy) benzaldehyde. This 3,4-bis(2-methoxyethoxy) benzaldehyde is converted to 3,4-bis(2-methoxyethoxy) benzaldoxime in the presence of a base and organic solvent and is further dehydrated to 3,4-bis(2-methoxyethoxy) benzonitrile. The benzonitrile so obtained is nitrated to obtain 4,5-bis(2-methoxyethoxy)-2~nitrobenzonitrile which is further reduced to get 2-amino-4,5- bis(2-methoxyethoxy) benzonitrile. 2-amino-4,5-bis(2-methoxyethoxy) benzonitrile is formylated with a formylating agent in the presence of formic acid derivative to obtain N’- [2-cyano-4,5-bis(2-methoxyethoxy)phenyl]-N,N-dimethylformamidine which is coupled with an aniline derivative to obtain erlotinib free base which on treatment with a polar solvent containing hydrochloric acid gives erlotinib hydrochloride.
EXAMPLES:
Example – 1a:
Preparation of Erlotinib Hydrochloride : 5.O g of 4-chloro-6,7-bis (2-methoxyethoxy) quinazoline was suspended in 75 ml water and 2.55 g of 3-aminophenyl acetylene was charged at 25 – 300C. Further 1.0 ml 50 % hydrochloric acid was added. The reaction mass was stirred at 25 – 300C for 2 hours. The solid obtained was filtered and washed with water. The product was dried at 40 – 45°C to obtain 6.1 g of erlotinib hydrochloride. In a similar manner, different solvents were used for preparing erlotinib hydrochloride under acidic conditions as given in table 1 below :
Table 1
Example – 2a:
Preparation of Erlotinib Hydrochloride :
5.0 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 75 ml of water and 2.55 g of 3-aminophenyl acetylene was added at 25 – 300C followed by 1.0 ml of 50 % hydrochloric acid. The reaction mass was heated at 35 – 400C for 1 hour. The solid obtained was filtered and washed with water. The product was dried at 40 – 45°C to obtain 5.8 g of erlotinib hydrochloride.
In a similar manner, different solvents were used for preparing erlotinib hydrochloride under acidic conditions as given in table 2 below :
Table 2
Example – 3:
Preparation of Erlotinib Hydrochloride :
5 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 150 ml denatured spirit (SPDS) and 4.6 g of 3-aminophenyl acetylene was charged at 25 – 300C. Further 1.0 ml of methane sulphonic acid was added. The reaction mass was stirred at 25 – 300C for 3 hours. Solid obtained was filtered, washed with SPDS and dried under vacuum. This solid was suspended in water, basified with ammonia and stirred for 10 minutes. The resulting erlotinib base was isolated, washed with water and dried under vacuum. The base was suspended in water and acidified to pH 1.0 – 2.0 using hydrochloric acid. The reaction mixture was stirred for 2 hours, filtered, washed with water and dried at 40 – 450C to obtain 5.8 g of erlotinib hydrochloride.
Example – 4: Preparation of Erlotinib Hydrochloride :
10.0 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 300 ml methanol and 9.2 g of 3-aminophenyl acetylene was charged at 25 – 300C. Further 2.0 ml of benzoic acid was added. The reaction mass was stirred at 25 – 300C for 4 hours. Solid obtained was filtered, washed with methanol and dried under vacuum. This solid was suspended in water and then basified with sodium hydroxide and stirred for 10 minutes. The resulting erlotinib base was isolated, washed with water and dried under vacuum. The base was suspended in water and acidified to pH 1.0 – 2.0 using hydrochloric acid. The reaction mixture was stirred for 2 hours, filtered, washed with water and dried to obtain 11.2 g of erlotinib hydrochloride. Example – 5:
Preparation of Erlotinib Hydrochloride :
15.0 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 450 ml ethanol and 13.8 g of 3-aminophenyl acetylene was added at 25 – 30°C. Further 3.0 g tartaric acid was added. The reaction mass was stirred at 25 – 300C for 6 hours. Solid obtained was filtered, washed with water and dried under vacuum. This solid was suspended in water, basified with potassium hydroxide and stirred for 10 minutes. The resulting erlotinib base was isolated by filtration, washed with ethanol and dried under vacuum. The solid obtained was then suspended in water and acidified to pH 1.0 – 2.0 using hydrochloric acid. The reaction mixture was stirred for 2 hours, filtered, washed with water and dried at 40 – 45°C to obtain 18.3 g of erlotinib hydrochloride.
Example – 6: Preparation of Erlotinib Hydrochloride :
50 g of 4-chloro-6,7-bis(2-methoxyethoxy) quinazoline was suspended in 1500 ml acetonitrile and 46 g of 3-aminophenyl acetylene was added at 25 – 300C, followed by 10 ml acetic acid. The reaction mass was stirred at 25 – 30°C for 30 minutes. Solid obtained was filtered, washed with water and dried under vacuum. This solid was suspended in water, basified with potassium hydroxide and stirred for 10 minutes. The resulting erlotinib base was isolated, washed with acetonitrile and dried under vacuum. The solid obtained was then suspended in water and acidified to pH 1.0 – 2.0 using hydrochloric acid. The reaction mixture was stirred for 2 hours, filtered, washed with water and dried at 40 – 45°C to obtain 63 g of erlotinib hydrochloride.
An efficient, economical and large-scale convergent synthesis of epidermal growth factor receptor- tyrosine kinase inhibitors gefitinib (1, Iressa) and erlotinib (2, Tarceva) approved by U.S. FDA for the treatment of non-small-cell lung cancer is described. The formation of 4-anilinoquinazolines are achieved in a simple one-pot reaction of suitable formamidine intermediates and substituted anilines involving Dimroth rearrangement, thereby avoiding the need to make quinazolin-4(3H)-one intermediates, which require a large experimental inputs. Using this process, we have produced drug candidates 1 with overall yield of 66% from 4-methoxy-5-[3-(4-morpholinyl) propoxy]-2-nitrobenzonitrile (3) and 2 with 63% from 4,5-bis(2-methoxyethoxy)-2-nitrobenzonitrile (6) on a multigram scale.
2 as crude material, which was further recrystallized from ethyl acetate (1 L) and then with methanol (500 mL) to give off-white crystalline compound 2 (350 g, 66% yield). FREE BASE ERLOTINIB
Through a stirred suspension of erlotinib free base 2 (200 g) in methanol (2 L) was passed dry hydrochloric acid gas for ~0.5 h, keeping the temperature of the reaction mass at 15–20 °C. The solid precipitate was filtered and dried at 50 °C to give off-white crystalline material of erlotinib hydrochloride (9) (200 g, 92% yield).
Gefitinib is the first selective inhibitor of epidermal growth factor receptor‘s (EGFR) tyrosine kinase domain. Thus gefitinib is an EGFR inhibitor. The target protein (EGFR) is a family of receptors which includes Her1(erb-B1), Her2(erb-B2), and Her 3(erb-B3). EGFR is overexpressed in the cells of certain types of human carcinomas – for example in lung and breast cancers. This leads to inappropriate activation of the anti-apoptotic Ras signalling cascade, eventually leading to uncontrolled cell proliferation. Research on gefitinib-sensitive non-small cell lung cancers has shown that a mutation in the EGFR tyrosine kinase domain is responsible for activating anti-apoptotic pathways.[1][2] These mutations tend to confer increased sensitivity to tyrosine kinase inhibitors such as gefitinib and erlotinib. Of the types of non-small cell lung cancer histologies, adenocarcinoma is the type that most often harbors these mutations. These mutations are more commonly seen in Asians, women, and non-smokers (who also tend to more often have adenocarcinoma).
The FDA approved Gefitinib in May 2003 for NSCLC a type of lung cancer,[5] Gefitinib is currently marketed in over 64 countries.
In June 2005 the FDA withdrew approval for use in new patients due to lack of evidence that it extended life.[6]
In Europe gefitinib is indicated since 2009 in advanced NSCLC in all lines of treatment for patients harbouring EGFR mutations. This label was granted after gefitinib demonstrated as a first line treatment to significantly improve progression-free survival vs. a platinum doublet regime in patients harbouring such mutations. IPASS has been the first of four phase III trials to have confirmed gefitinib superiority in this patient population.[7] In most of the other countries where gefitinib is currently marketed it is approved for patients with advanced NSCLC who had received at least one previous chemotherapy regime. However, applications to expand its label as a first line treatment in patients harbouring EGFR mutations is currently in process based on the latest scientific evidence.As at August 2012 New Zealand has approved gefitinib as first line treatment for patients with EGFR mutation for naive locally advanced or metastatic, unresectable NSCLC. This publicly funded for an initial 4 month term and renewal if no progression. [8]
In 2014 in the TRANSCOG study Petty et al., demonstarted gefitinib was effective in esophageal cancer patients whose tumours harboured additional copies of the EGFR gene.[9] While gefitinib has yet to be proven to be effective in other cancers, there is potential for its use in the treatment of other cancers where EGFR overexpression is involved.
Erlotinib is another EGFR tyrosine kinase inhibitor that has a similar mechanism of action to gefitinib.
Experimental Uses
In August 2013, the BBC reported that researchers in Edinburgh and Melbourne found, in a small-scale trial of 12 patients, that the effectiveness of Methotrexate for treating ectopic pregnancy was improved when Gefitinib was also administered.[10]
Studies
IPASS (IRESSA Pan-Asia Study) was a randomized, large-scale, double-blinded study which compared Gefitinib vs. carboplatin/ paclitaxel as a first line treatment in advanced NSCLC.[11] IPASS studied 1,217 patients with confirmed adenocarcinoma histology who were former or never smokers. A pre-planned sub-group analyses showed that progression-free survival (PFS) was significantly longer for Gefitinib than chemotherapy in patients with EGFR mutation positive tumours (HR 0.48, 95 per cent CI 0.36 to 0.64, p less than 0.0001), and significantly longer for chemotherapy than Gefitinib in patients with EGFR mutation negative tumours (HR 2.85, 95 per cent CI 2.05 to 3.98, p less than 0.0001). This, in 2009, was the first time a targeted monotherapy has demonstrated significantly longer PFS than doublet chemotherapy.
EGFR Diagnostic tests
Genzyme, QIAGEN, Argenomics S.A. & other companies make tests to detect EGFR mutations, designed to help predict which lung cancer patients may respond best to some therapies, including Gefitinib and Erlotinib.
The tests examine the genetics of tumors removed for biopsy for mutations that make them susceptible to treatment.
The EGFR mutation test may also help AstraZeneca win regulatory approval for use of their drugs as initial therapies. Currently the TK inhibitors are approved for use only after other drugs fail. In the case of gefitinib, the drug works only in about 10% of patients with advanced non-small cell lung cancer, the most common type of lung cancer.
Adverse effects
As gefitinib is a selective chemotherapeutic agent, its tolerability profile is better than previous cytotoxic agents. Adverse drug reactions (ADRs) are acceptable for a potentially fatal disease.
Iressa was approved and marketed from July 2002 in Japan, making it the first country to import the drug.
Gefitinib is an anilinoquinazoline which is useful in the treatment of a certain type of lung cancer (non-small cell lung cancer or NSCLC) that has not responded to chemotherapy. The chemical name for gefitinib is 4-(3′-chloro-4′-fluoroanilino)-7- methoxy-6-(3-morpholinopropoxy) quinazoline. Its structural formula is :
Gefitinib
The earliest known synthesis of gefitinib was first disclosed in the patent application WO 96/33980. The synthetic method employed is depicted in the following reaction scheme 1.
The process involves selective demethylation of 6,7-dimethoxy quinazoline-4-one using methanesulfonic acid and L-methionine to get its 6-hydroxyl derivative, which is protected by acetylation. The acetoxy compound is chlorinated and condensed with chloro-fluoroaniline. Hydrolysis of the acetoxy compound followed by etherification with 3-morpholinopropyl chloride gives crude gefitinib which is purified by column chromatography. The process suffers from many disadvantages as it involves several protection and deprotection steps. The selective demethylation using methionine results in isomeric impurities and has to be purified or else the impurity carries over to subsequent steps in the preparation of gefitinib making it more difficult to isolate a pure product. The process also leads to formation of an N-alkylated impurity at the final stage which must be separated by column chromatography to obtain gefitinib.
Several other approaches are also described in the literature to make gefitinib.
WO 2004/024703 discloses a process for the preparation of gefitinib starting from 3- hydroxy-4-methoxy benzonitrile which involves condensation of 3-hydroxy-4-methoxy benzonitrile with morpholino propyl chloride, nitration, reduction with sodium dithionite to amino compound, hydrolysis of nitrile to amide, cyclisation in the presence of formamide to obtain quinazoline, chlorination with phosphorous oxychloride and finally condensation with chloro-fluoro aniline to obtain gefitinib. The process involves multiple steps and hence is time consuming.
WO 2005/023783 discloses a process for the manufacture of gefitinib starting from 2- amino-4-methoxy-5-(3-morpholinopropoxy)benzonitrile. The process involves a rearrangement reaction of 3-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3- morpholinopropoxy)3,4-dihydroqunazoline-4-imine. The process is not feasible industrially, as the basic raw material is not readily available on a commercial scale and involves the use of excess 3-chloro-4-fluoroaniline which is expensive. A further draw back of the process is in the isomerization of the 4-imine compound which requires anhydrous conditions at high temperature for a longer duration of 96 hours. All the problems associated with this prior art process are overcome by the novel process of the present invention.
WO2005/070909 discloses a process for the preparation of gefitinib starting from isovanillin as depicted in scheme 2
The WO’ 909 process has disadvantages as it forms cis-trans geometrical isomers of the oxime, which have different reactivities. Furthermore, the process uses a large excess of acetic anhydride to convert the oxime to the nitrile at higher temperature.
The patent applications 901 /CHE/2006 and 903/CHE/2006 disclose another route for preparing gefitinib starting from isovanillin. The process involves formation of a formamido compound [N’-[2-cyano-4-{3-(4-morpholinyl)propoxy}phenyl]-N,N-dimethyl formamide], which is unstable and may result in undesired impurities in the final condensation with 3-chloro-4-fluoro aniline, thereby making the process less feasible on an industrial scale. The processes disclosed in the prior art are cumbersome. Therefore, there exists a need for a more economical and efficient method of making gefitinib which is suitable for industrial scale-up.
The process of the present invention avoids use of reagents such as sodium dithionite, acetic anhydride and allows substantial reduction in the number of problems associated with these reagents.
…………………
CLIP
Gefitinib A mixture of compound 1 starting acid 1 is heated in a closed loop to obtain ammonium 2 in 2 classic resonant3 , the 7 – Bit methoxy given electron, so to 6 – position methoxy-electron density is low, so that the acidic conditions demethylase (with methionine and methanesulfonic acid) may optionally occur in the 6 – position, to give compound 4 , 4 of the phenolic hydroxyl group with an acetyl group after protection thionyl chloride to get five , five and six occurred SNAr reaction 7 , 7deacetylated with ammonia and chloride 8 reaction gefitinib.
Detailed Description of the Invention In an embodiment of the present invention, there is provided an improved synthesis of gefitinib from isovanillin , as depicted below in reaction scheme 3.
isovanillin Formula II Formula in
Formula V Formula IV
W
Gefitinib Formula I
An alternate route for the synthesis of gefitinib from isovanillin according to the present invention is depicted below in reaction scheme 4.
Nitration
Step a Step b
Isovanillin Formula VIII Formula IX
Formula XII Formula XI Formula X
Formula XIII Formula XIV
Example 1 :
Preparation of 4-(3′-chloro-4′-fluoroanilino)-7-methoxy-6-(3- morpholinopropoxy)-quinazoline (gefitinib) (formula I)
Methanol (1200 ml) and 6-(3-morpholino propoxy)-7-methoxy-4-chloro quinazoline (200gm) were stirred for 15 minutes at 25-300C, then a solution of 4-fluoro-3- chloroaniline in methanol (213 gm in 400 ml) was charged and refluxed for 6 hours. The reaction mass was cooled to 15-200C, hydrochloric acid (40 ml) was added drop wise, and stirred at 5-100C for 30 minutes. The solid obtained was filtered and washed with chilled methanol (150ml). The solid was dissolved in a mixture of toluene (30 volume) and methanol (5 volume), the reaction mass was concentrated to half the volume and cooled to 5-10°C. The solid obtained was filtered, washed with toluene (200 ml) and dried at 45-50°C to yield the title compound (183 gm, 70% yield).
Example 2: Preparation of 6-(3-morpholino propoxy)-7-methoxy-4- chloroquinazoline (formula VII)
DMF (3 It), 6-(3-chloropropoxy)-7-methoxy-4-chloro quinazoline (200 gm) and morpholine (210 gm), were heated to 70-750C for 6-8 hours. The reaction mass was cooled to room temperature, and methylene chloride (2.5 It) and water (2.5 It) were charged. The layers separated and the aqueous layer extracted with methylene chloride twice (500 ml). The combined methylene chloride layer was washed with water, dried over sodium sulphate (10 gm) and concentrated completely at 35-40°C to yield the title compound (200 gm, 85% yield).
Example 3: Preparation of 6-(3-chloropropoxy)-7-methoxy-4-chloroquinazoline (formula Vl)
6-(3-chloropropoxy)-7-methoxyquinazoline-4-one (400 gm), thionyl chloride (3.2 It) and DMF (100 ml) were refluxed for 7-8 hours. Thionyl chloride was distilled off completely under reduced pressure below 45°C. Methylene chloride (2.5 It) and water (1.5 It) were charged, stirred for 30 minutes at room temperature and the layers separated. The aqueous layer was extracted twice with methylene chloride (500 ml), the combined methylene chloride layer was washed with 1 % sodium bicarbonate solution (1 It), dried over sodium sulphate (20 gm) and concentrated under reduced pressure at 35-40°C. The residue was stirred with isopropyl alcohol (400 ml) at 40-450C for 1 hour, cooled to 0-50C, the solids filtered, washed with chilled isopropyl alcohol (200 ml) and dried under vacuum at 45°C to yield the title compound (406 gm, 95% yield).
Example 4: Preparation of 6-(3-chloropropoxy)-7-methoxyquinazoline-4-one (formula V)
2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid (450gm), formamide (2250 ml) and ammonium formate (200 gm) were heated to 170-1800C for 3-4 hours. The reaction mass was concentrated under reduced pressure at 140-1500C. The residue was stirred in methanol (1000 ml) at 45-50°C and cooled to 5-10°C. The solid obtained was filtered to yield the title compound (420 gm, 90% yield).
Example 5: Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy) benzoic acid (formula IV)
a) Preparation of 3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acid
Methanol (4 It), 3-(3-chloropropoxy)-4-methoxy-6-nitro benzaldehyde (560 gm) and 30% methanolic NaOH solution (5 ml) were heated to 450C. To this reaction mass 35% of H2O2 solution (1200 ml) was added drop wise in 3-4 hours maintaining a pH of 10.5 – 11.5 with 30% methanolic NaOH solution. The reaction mass was quenched into ice water (10 kg) and the pH adjusted to 2.0-3.0 using hydrochloric acid. The solid obtained was filtered, washed with 50% aqueous methanol (500 ml) and dried at 45-500C to yield the title compound (510 gm, 86% yield).
bi) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid – using hydrogen gas
Ethyl acetate (3 It), Pd/C (50 gm) and 3-(3-chloropropoxy)-4-methoxy-6-nitrobenzoic acid (500 gm) were hydrogenated under a hydrogen pressure of 5-6 kg at 35-400C for 3-4 hours. The reaction mass was filtered and the clear filtrate was distilled under reduced pressure at 45-500C. To the residue, hexane (1 It) was charged, stirred at room temperature, the solids filtered and dried at 45-50°C to yield the title compound (403 gm, 90% yield). 5
(bii) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid – using hydrazine hydrate
S-p-chloropropoxyH-methoxy-e-nitrobenzoic acid (100 gm), hydrazine hydrate (50 gms), neutral alumina (20gms), charcoal (10 gms), water (50 ml) and methanol (500
10 ml) were mixed together. The reaction mass was heated to 500C. A solution of ferric chloride (2 gms, 0.012M) in 50 ml methanol was introduced slowly at 55-600C. The reaction mass was filtered over hyflo and the clear filtrate evaporated. The residue obtained was dissolved in 1.0-lit ethyl acetate, washed organic extract with water, evaporated to obtain title compound. (75 gms, 83.6%)
15
(biii) Preparation of 2-amino-4-methoxy-5-(3-chloropropoxy)benzoic acid – using ammonium formate
3-(3-chloropropoxy)-4-methoxy-6-nitro benzoic acid (165 gms), 5% Paladium on carbon (16.5 gms) and DMF (0.66 lit) were mixed together. The reaction mass was heated to
20 400C. Ammonium formate (82.5 gms) was charged in lots maintaining temperature below 500C. The temperature of reaction mass slowly raised to 70°Cand maintained for 2 hours. The reaction mass was cooled to 300C and catalyst was removed by filtration and the clear filtrate evaporated. The residue was dissolved in ethyl acetate (0.825 lit), washed with water and evaporated to yield the title compound. (125 gms,
25 84.5%)
Example 6: Preparation of 3-(3-chloropropoxy)-4-methoxy-6-nitro benzaldehyde (formula III)
5-nitro isovanillin (500 gm), acetonitrile (3.5 Its), K2CO3 (750 gm) and 30 chlorobromopropane (780 gm) were refluxed for 4 hours. The reaction mass was filtered hot, washed with acetonitrile (1 It) and the filtrate was distilled off to remove solvent. The residue was dissolved in methylene chloride (4 It) and washed with water. Water (3 It) was charged to the methylene chloride layer, the pH adjusted to 7.0 to 7.5 with acetic acid, the methylene chloride layer separated, dried over sodium sulphate (50 gm) and distilled out completely under reduced pressure below 400C. The residue was stirred with 2 volumes of n-Hexane at 40-450C, cooled slowly to 0-50C, the solids filtered, washed with n-Hexane (250 ml) and dried at 40-450C to yield the title compound (638 gm, 92% yield).
Example 7: Preparation of 5-nitro isovanillin (formula II)
Isovanillin (500 gm) and acetic acid (1750 ml) were cooled to -5 to O0C. To this solution, nitric acid (750 ml) was charged slowly at -5 to O0C with stirring. The temperature of the reaction mass was slowly raised to 25-300C and maintained for 12 hours. The reaction mass was quenched into ice water (4 kg), the solids filtered and washed with water (2 It). The solids were stirred with a 1% sodium bicarbonate solution (1 It), filtered and dried at 45-500C. The solid was dissolved in 6 volumes of ethyl acetate, ethyl acetate was distilled off up to half the volume and 3 volumes of n-Hexane were charged slowly at 45-50°C. The reaction mass was cooled slowly to 0-5°C, maintained for 1 hour, the solids filtered, washed with 0.5 volumes of 1 :1 mixture of ethyl acetate: n-Hexane and dried at 45-500C to yield the title compound (423 gm, 65 % yield) .
Example 8: Preparation of Methyl-2-hydroxy-3-methoxy benzoate (formula VIII)
a) Preparation of 3-hydroxy-4-methoxy benzoic acid
Methanol (350 ml), isovanillin (50 gm) and 30% methanolic sodium hydroxide solution (1 ml), were heated to 450C. To this solution, 35% hydrogen peroxide solution (107 ml) was charged slowly maintaining pH at 10.5 to 11.5 using methanolic sodium hydroxide solution over a period of 2-3 hours. The reaction mass was quenched into chilled water (1 It) and the pH adjusted to 2-3 using hydrochloric acid. The solids were filtered, washed with 50% aqueous methanol (50 ml) and dried at 45-50°C to yield 3-hydroxy-4- methoxy benzoic acid.
b) Preparation of Methyl-2-hydroxy-3-methoxy benzoate
The solid obtained in step a), was refluxed with 10% methanolic hydrochloric acid solution (250 ml) for 6 hours. The reaction mass was quenched into chilled water (1 It) and repeatedly extracted with methylene chloride (250 ml). The combined methylene chloride layer was washed with water (100 ml * 2) and methylene chloride distilled out completely at 35-40°C. The residue was stirred in hexane (150 ml), at 25-300C. The solid obtained was filtered, washed with hexane (25 ml) and dried at 40-45°C to yield the title compound (50 gm, 83% yield).
Example 9: Preparation of Methyl-5-hydroxy-4-methoxy-2-nitro benzoate (formula
IX)
Methyl-2-hydroxy-3-methoxy benzoate (50 gm) and acetic acid (175 ml) were cooled to
0-5°C. To this solution, 70% nitric acid solution (75 ml) was charged slowly at 0-5°C under stirring and the reaction mass was further stirred for 18 hours. The reaction mass was quenched into chilled water (800 ml) and extracted repeatedly with methylene chloride (400 ml). The combined methylene chloride layer was washed with water, followed by 1% potassium carbonate solution (100 ml), dried over sodium sulphate and methylene chloride distilled off completely at 35-40°C. The residue was dissolved in 10% aqueous methanol (250 ml). The filtrate was gradually cooled to 0-5°C and maintained for 1 hour. The solid obtained was filtered, washed with 10% aqueous methanol (100 ml) and dried at 40-450C to yield the title compound (46 gm, 74% yield).
Example 10: Preparation of Methyl-2-amino-5-hydroxy-4-methoxy benzoate (X) Ethyl acetate (300 ml), methyl-5-hydroxy-4-methoxy-2-nitro benzoate (50 gm) and 10% palladium/carbon (5 gm) were hydrogenated under a hydrogen gas pressure of 5-6 kg for 4 hours. The reaction mass was filtered to remove catalyst. The filtrate was distilled off to remove solvent. The residue obtained was stirred in n-hexane (100 ml) at 0-5°C The solid obtained was filtered and washed with n-hexane (25 ml) to yield the title compound (40 gm, 93% yield).
Example 11 : Preparation of 6-hydroxy-7-methoxy-quinazoline-4-one (formula Xl)
Methyl-2-amino-5-hydroxy-4-methoxy benzoate (50 gm), methanol (400 ml) and formamidine acetate (30 gm) were refluxed for 10 hours. The reaction mass was gradually cooled to 5-10°C and stirred for 1 hour. The solid obtained was filtered and washed with methanol (150 ml) and dried at 50-55°C to yield the title compound (45 gm, 92% yield).
Example 12 : Preparation of Gefitinib
Acetonitrile (500 ml), N-(4-fluoro-3-chloro phenyl)-6-hydroxy-7-methoxy quinazoline-4- amine (50 gm), 3-morpholinopropyl chloride (35 gm) and tetrabutyl ammonium bromide (5 gm) were refluxed for 16 hours. The reaction mass was distilled off to remove acetonitrile completely at 40-45°C. To the residue, water (500 ml) was charged and stirred for 15 minutes at 25-300C. The solid obtained was filtered, washed with methanol (50 ml) and dried at 45-50°C. The crude solid was dissolved in a mixture of toluene (1200 ml) and methanol (200 ml). The reaction mass was distilled off under reduced pressure at 40-450C to 400 ml volume, cooled to 10-150C, stirred for 30 minutes, the solid filtered, washed with toluene (40 ml) and dried to yield gefitinib (28 gm, 40% yield).
The process of preperation of 4-(3-chloro-4-flurophenylamino)-7-methoxy-6-[3-(4-morpholinyl)propoxy]-quinazoline is substantially as herein described with reference to the foregoing example. a) 4-methoxy-3-[3-(4-morpholinyl)-propoxy] benzaldehyde: A mixture of 3-hydroxy -4- methoxy benzaldehyde (20g), 3- morpholinopropyl chloride (28g), potassium carbonate (50g) and DMF (140ml) was stirred and heated to 100°C for 3 hours. The reaction mixture was cooled and filtered. The filtrate was evaporated and the residue obtained was dissolved in ethyl acetate (200ml). The ethyl acetate layer was washed with water, dried over anhydrous sodium sulphate. Evaporation of ethyl acetate yielded 4-methoxy-3-[3-(4-morpholinyl)-propoxy] benzaldehyde (34.8 gms,95%) of the formula II. b) 4-methoxy -3[3-(4-morpholinyl)-propoxy] benzonitrile: The above 34.8 gms of compound was dissolved in 200 ml methanol, and to this added 34.8 g of hydroxylamine hydrochloride and 35 ml of pyridine. The reaction mixture was heated to reflux for 3 hours and then cooled to 10 °C. The material precipitated mass was filtered and the solid mass obtained was washed with chilled methanol (50ml) and dried at room temperature. To this dried material was added 2 volumes of acetic anhydride and heated to 110°C for 4 hours. Then quenched the reaction mass in water and adjusted the pH to 8.0 with sodium bicarbonate and extracted with methylene dichloride. Washed the methylene dichloride layer with water and dried over calcium chloride. On evaporation of the solvent 4-methoxy -3[3-(4-morphoinyl)-propoxy] benzonitrile (31 gms, 90%) of the formula III. NMR spectrum (CDCI3): 5 2.05 (m, 2H), 2.53 (m, 6H), 3.72 (m, 4H), 3.91 (s, 3H). 4.10(mf 2H), 6.89 (d, 1H), 7.25 (d, 1H), 7.28 (dd, 1H). c) 4-Methoxy-5-[3-(4-morpholinyl)-propoxy]- 2-nitro benzonitrile: The above compound (31 gms) of the formula III was dissolved in 30 ml of 70% nitric acid and this was added to 55°C preheated 30 ml of 70% nitric acid slowly over a period of 2 hours under stirring. After completion of the addition, continued stirring at the same temperature for further an hour. Cooled the reaction mixture and quenched in cool water and
adjusted the pH to 8.0. The precipitate obtained was filtered and washed with ice cold water and dried the material at 50°C to get 27 gms yellow solid 4-Methoxy-5-[3-(4-morphoiinyl)-propoxy]- 2-nitro benzonitrile (75%) of the formula IV. NMR spectrum (DMSO-d6): 5 2.18 (m, 2H), 3.26 (m, 4H), 3.53 (m, 4H), 3.99 (s, 3H), 4.04 (m, 2H), 4.29 (m, 2H), 7.73 (s, 1H), 7.91 (s, 1H). d) Synthesis of 2-amino-4-methoxy-5-(3-morpholinopropoxy) benzonitrile: To 4-methoxy-5-[3-(4-morpholinyl) propoxy]-2-nitro benzonitrile (10 g) was added acetic acid (75ml) and water(75ml), stirred the reaction mass for about 10 min, added Iron powder (7g) in portions over a period of 2hrs, Stirred the reaction mixture for about Vi hr at room temperature adjusted PH of the reaction mass to 8 using ammonia solution, extracted the material into ethylacetate, the organic layer was dried over sodium sulfate and concentrated to get product(6g) 1HNMR (CDCI3): 5 2.01 (m, 2H), 2.51 (m. 6H), 3.72 (t, 4H), 3.84 (s, 3H), 3.97 (t, 2H), 4.14 (brs, 2H), 6.23 (s, 1H), 6.85 (s, 1H). (e) Synthesis of N’-(3-chloro-4-fluorophenyl) N,N-dimethyl formamidine. To 3-chloro-4-flouro aniline (10g, 0.0687moles) was added Toluene (40ml), N,N-dimethylformamide dimethyl acetal (18.3ml, 0.1374moles) and acetic acid (0.5ml), heated the reaction mixture to 110°C and stirred for about 2hrs, distilled off toluene to yield dark brown liquid (11g) 1HNMR (CDCI3): 5 3.0 (s, 6H), 6.75 (m, 1H) 6.94 (m, 1H), 6.98 (m, 1H), 7.45 (s, 1H). (f) Synthesis of Gefitinib: To 2-amino-4-methoxy-5-(3-morpholinopropoxy) benzonitrile (5g, 0.0172moles) was added toluene (30ml), N’-(3-chloro-4-fluorophenyl) N, N-dimethyl formamidine (3.44g, 0.0172moles) and acetic acid (0.5ml) refluxed the reaction mixture for about 4hrs cooled the reaction mass to room temperature, toluene layer was separated washed with water and chilled toluene layer to yield crude gefitinib and which was further recrystallized from methano! to get pure off-white crystalline compound(3g) having the mp 194-198°C UV, IR, NMR spectral data together with elemental analysis is in complete agreement with those of standard substance of Gefitinib
An efficient, economical and large-scale convergent synthesis of epidermal growth factor receptor- tyrosine kinase inhibitors gefitinib (1, Iressa) and erlotinib (2, Tarceva) approved by U.S. FDA for the treatment of non-small-cell lung cancer is described. The formation of 4-anilinoquinazolines are achieved in a simple one-pot reaction of suitable formamidine intermediates and substituted anilines involving Dimroth rearrangement, thereby avoiding the need to make quinazolin-4(3H)-one intermediates, which require a large experimental inputs. Using this process, we have produced drug candidates 1 with overall yield of 66% from 4-methoxy-5-[3-(4-morpholinyl) propoxy]-2-nitrobenzonitrile (3) and 2 with 63% from 4,5-bis(2-methoxyethoxy)-2-nitrobenzonitrile (6) on a multigram scale.
Synthesis of Gefitinib[J]. CJPH, 2013, 44(11): 1081-1083..
Synthesis of Gefitinib
1. School of Chemical Engineering, Huaihai Institute of Technology, Lianyungang 222001; 2. Lianyungang Shenghe Biotechnology Limited Company, Lianyungang 222007
Gefitinib was synthesized from 3-hydroxy-4-methoxybenzaldehyde via conversion of aldehyde to nitrile, condensation with N-(3-chloropropy1)morpholine, nitration and reduction to give 2-amino-4-methoxy-5-(3-morpholin-4-ylpropoxy)benzonitrile, which was subjected to amidination with 3-chloro-4-fluoroaniline and cyclization in the presence of formic acid with an overall yield of about 44%.
…………..
Synthesis of Gefitinib
LV Tong-jie,OUYANG Gui-ping,MENG Xiang-bing,LIU Xiao-yu(Key Laboratory of Green Pesticide and Agriculture Bioengineering,Ministry of Education,Research and Development Center for fine Chemicals,Guizhou University,Guizhou Guiyang 550025,China)
The 4-(3-chloro-4-fluorophenylamino)-7-methoxy-6-[3-(4-morpholinyl)pro-poxy]quinazoline(Geifitinib,ZD1839) was synthesized from 3-hydroxy-4-methoxybenzaldehyde,through a seven-step procedure of condensation,conversion of aldehyde to nitrile,n-itration,reduction,cyclization,et al.,and the total yield reached 31.81%.The structure of the compound was characterized by IR,1H-NMR,13C-NMR,and MS.
Sordella R, Bell DW, Haber DA, Settleman J (August 2004). “Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways”. Science305 (5687): 1163–7.doi:10.1126/science.1101637. PMID15284455.
Lynch, Thomas J.; Bell, Daphne W.; Sordella, Raffaella; Gurubhagavatula, Sarada; Okimoto, Ross A.; Brannigan, Brain W.; Harris, Patricia L.; Haserlat, Sara M.; Supko, Jeffrey G.; Haluska, Frank G.; Louis, David N.; Christiani, David C.; Settleman, Jeff; Haber, Daniel A (May 20, 2004). “Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib”. NEJM350 (21): 2129–39. doi:10.1056/nejmoa040938.
Mok TS, Wu YL, Thongprasert S, et al, Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–957. Sebastian M, Schmittel A, Reck, M, First-line treatment of EGFR-mutated nonsmall cell lung cancer: critical review on study methodology, European Respiratory Review. 2014 Mar 1;23(131):92-105.
N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib) represented by Chemical Formula 1 below is a quinazoline derivative useful in treatment of non-small cell lung cancer. The structure of gefitinib is shown in the following Chemical Formula 1.
[Chemical Formula 1]
WO 96/33980 discloses the gefitinib synthesis method as represented in Scheme 1 below.
[Scheme 1]
According to the synthesis method of Scheme 1, 6,7-dimethoxy quinazolin-4-one as a starting material is subjected to selective demethylation, condensation with chlorofluoroaniline and then etherification with 4-(3-morpholinopropyl)chloride, thereby synthesizing gefitinib. Because gefitinib thus synthesized contains an excess of an N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine in the final step, the impurity should be separated via column chromatography, undesirably lowering the yield and making it difficult to achieve commercial production.
To solve such problems, WO 2004/024703 discloses a method of synthesizing gefitinib from a start material of 3-hydroxy-4-methoxy benzonitrile as shown in Scheme 2 below.
[Scheme 2]
In the synthesis method of Scheme 2, a morpholinopropyl group is introduced before forming a quinazoline ring, thus suppressing the production of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine which is the N-alkylated impurity. However, reduction of a nitro compound, formation of a quinazoline ring, and chlorination of the quinazoline ring in the final step to carry out condensation with chlorofluoroaniline are performed in the presence of the morpholinopropyl group, undesirably complicating the reaction process and lengthening the reaction time.
WO 2008/125867 discloses a method of synthesizing gefitinib from a start material of isovanilin as shown in Scheme 3 below.
[Scheme 3]
In the synthesis method of Scheme 3, propoxychloride is introduced before forming the quinazoline ring, thus suppressing the production of the N-alkylated impurity. After a quinazoline ring is formed and a morpholine group is introduced, chlorofluoroaniline is introduced, thus synthesizing gefitinib. However, a chloropropyl group and a morpholine group are separately introduced, instead of the morpholinopropyl group, thus increasing the number of synthesis steps, and also, the nitro reduction and the quinazoline ring reaction are performed in the presence of the chloropropyl group, undesirably causing the production of an impurity.
Additionally, WO 2005/023783 discloses a method of synthesizing gefitinib from imine via a rearrangement reaction, and WO 2005/070909 discloses a method of synthesizing gefitinib by performing nitrilization of oxime and then forming a quinazoline ring.
However, the preparation methods mentioned in the prior techniques produce an excess of impurity or include other routes to suppress the formation of the impurity, undesirably increasing the number of preparation steps and thus resulting in complicated processes and a long synthesis time, and thereby these methods are unsuitable for commercial production.
Therefore, there is required a method of efficiently and simply preparing gefitinib, which may minimize the production of an impurity and be suitable for use in industrial production.
[Citation List]
[Patent Literature]
(Patent Document 1) WO 96/33980
(Patent Document 2) WO 2004/024703
(Patent Document 3) WO 2008/125867
(Patent Document 4) WO 2005/070909
<Example 3-1> Preparation of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib)
About 123.0 g of 4-(3-chloro-4-fluorophenylamino)-7-methoxyquinazolin-6-ol and about 1100.0 mL of N,N-dimethylformamide were placed in a flask. The resulting mixture was suspended with stirring while about 186.0 g of potassium carbonate and about 4.7 g of N,N-dimethylaminopyridine were added. The reaction mixture was cooled to about -10℃, slowly added with about 77.0 g of iodotrimethylsilane while paying attention to heat generation, and stirred at about 15℃ for about 1 hr. Then about 75.5 g of 4-(3-chloropropyl)morpholine was diluted with about 130.0 mL of N,N-dimethylformamide and then slowly added. The reaction mixture was heated to about 80℃ and stirred for about 2 hr. The termination of the reaction was confirmed using HPLC and TLC. The reaction product was cooled to about 20℃, slowly added with 2460.0 mL of purified water, and stirred for 30 min, and the produced solid was filtered. The obtained solid was washed with about 490.0 mL of purified water and then dried in a vacuum at about 50℃ for about 3 hr, yielding about 154.7 g of the title compound N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib) as pale yellow powder.
HPLC purity: 99.21% (N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine: 0.3%)
<Example 3-3> Preparation of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib)
About 8.3 g of the title compound N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib) as pale yellow powder was obtained in the same manner as in Example 3-2, with the exception that dimethylsulfoxide (DMSO) was used as the solvent, instead of N,N-dimethylacetamide (DMAC).
HPLC purity: 98.89% (N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine: 0.7%)
<Example 4-2> Purification of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib)
About 8.6 g of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine obtained in Example 3-2 was suspended with stirring in about 129.0 mL of toluene and about 65.0 mL of anhydrous ethanol, and heated to about 40℃ so as to be thoroughly dissolved. About 1.9 g of neutral activated carbon was added into the resulting solution, stirred for about 1 hr, and filtered to thus remove the activated carbon. The filtrate was concentrated to about 90 mL, and stirred for about 30 min, and the produced solid was filtered. The obtained solid was washed with about 20.0 mL of toluene, and dried at about 40℃ for about 3 hr, thus obtaining about 7.3 g of white gefitinib.
The purified gefitinib was added to about 125.0 mL of anhydrous ethanol to prepare a suspension, which was then refluxed with stirring at about 75℃ so that gefitinib was thoroughly dissolved, and then further stirred for about 1 hr. The solution was gradually cooled to about 20℃ and the produced solid was stirred for about 30 min and then further stirred at about 5℃ for about 1 hr. The obtained solid was filtered, washed with about 7.0 mL of anhydrous ethanol, and dried in a vacuum at about 45℃ for about 5 hr, yielding about 6.5 g of purified white N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib).
HPLC purity: 99.89% (without N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine)
<Example 4-3> Purification of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib)
About 6.2 g of purified white N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazoline-4-amine (gefitinib) was obtained in the same manner as in Example 4-1, with the exception that the gefitinib prepared in Example 3-3 was used.
HPLC purity: 99.87% (N-alkylated impurity, that is, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)-N-(3-morpholinopropyl)quinazoline-4-amine: 0.03%)
Gefitinib is an oral epidermal growth factor receptor tyrosine kinase (EGFR-TK) inhibitor developed by AstraZeneca. It was marketed in Japan in 2002 for the treatment of advanced non-small cell lung cancer. It was approved by the FDA on May 5, 2003 as a third-line treatment for locally advanced or metastatic non-small cell lung. In 2005, Iressa was launched in China under the trade name Iressa for the treatment of locally advanced or metastatic non-small cell carcinoma that had previously received chemotherapy.[0003]Gefitinib has many synthetic routes, mainly three routes developed by the original research company (patent WO9633980) and process optimization and redevelopment based on this. The specific route is as follows:[0004]Route 1:[0005]
[0006]This route uses 6,7-dimethoxyquinazolin-4(3H) ketone as raw material, and selectively demethylates with methanesulfonic acid and L-methionine to obtain 6-hydroxy-7-methoxy-3,4 -Dihydroquinazolin-4-one, which is then acetylated on the phenolic hydroxyl group to obtain the key intermediate 3,4-dihydro-7-methoxy-4-oxoquinazolin-6-ol acetate , And then chlorinated, aminated, hydrolyzed, and etherified to obtain the target product gefitinib.[0007]Route 2:[0008]
[0009]This route uses cheap and easy-to-obtain isovanillin as a raw material, after etherification, nitration, reduction, hydrolysis and cyclization reactions to obtain substituted quinazolinone, and then chlorination with thionyl chloride to obtain 4-chloroquinazoline, Then nucleophilic substitution with 3-chloro-4-fluoroaniline arylamine gives gefitinib.[0010]Route 3:[0011]
[0012]This route also uses cheap and easy-to-obtain isovanillin as a raw material to obtain gefitinib through etherification, nitration, reduction and Dimroth rearrangement reactions and cyclization reactions.[0013]The above methods have their own advantages and disadvantages, but there are no dangerous processes such as nitrification reaction in the first route. Therefore, the first route is a safe and extensive method for preparing gefitinib. Among them, 4-(3-chloro-4-fluoroaniline)-6-acetoxy-7-methoxyquinazoline hydrochloride is the key intermediate of this route. We carefully studied and analyzed the patent, and combined with the experimental situation, we found that the synthesis of this intermediate has problems such as inconvenient amplification and high impurities. After distilling thionyl chloride, the material is easy to agglomerate after being evaporated to dryness. Residual reagents such as thionyl chloride will be wrapped in the agglomerate, which will affect the next step of the reaction. Moreover, the agglomerate is difficult to take out from the reactor, and the yield cannot be determined. Rate, also has an impact on the next feeding. The second step is the amination reaction. The commonly used solvent is isopropanol. The mechanism of the amination reaction is 3-chloro-4-fluoroaniline as a nucleophile to attack 6-acetoxy-4-chloro-7-methoxy The quinazoline salt, thus the substitution reaction occurs, but the solvent isopropanol also has a certain nucleophilicity and participates in the competitive reaction, but the nucleophilicity of isopropanol is weaker than 3-chloro-4-fluoroaniline, therefore, it needs to be inhibited Isopropanol participates in the reaction and reduces side reactions. These problems will affect product purity and scale-up production. Therefore, it is necessary to make reasonable improvements to the method.
Figure 1 is a mass spectrum of 4-(3-chloro-4-fluoroaniline)-6-acetoxy-7-methoxyquinazoline hydrochloride prepared in Example 1;[0034]2 is a hydrogen nuclear magnetic resonance spectrum of 4-(3-chloro-4-fluoroaniline)-6-acetoxy-7-methoxyquinazoline hydrochloride prepared in Example 1.
Example 1[0036](1) Combine 6-acetoxy-7-methoxy-3H quinazolin-4-one (250g, 1.07moL), thionyl chloride (1.75kg) and N,N-dimethylformamide ( 25g, 0.34moL) was placed in a 3L reaction flask, stirred and heated to reflux, and reacted for 6h. After the reaction is complete, cool down. When the internal temperature drops to 60℃, distill under reduced pressure until no droplets flow out. Add toluene ( 1.5kg), stirring and beating at 60°C for 1h, cooling to 20°C, suction filtration, and drying under reduced pressure to obtain 280 g of 6-acetoxy-4-chloro-7-methoxyquinazoline hydrochloride with a yield of 90.7 %.[0037](2) Dissolve 6-acetoxy-4-chloro-7-methoxyquinazoline hydrochloride (280g, 0.97mol) in isopropanol (1.5kg), reduce the temperature to -5°C, and add dropwise 3-chloro-4-fluoroaniline (155g, 1.07mol) in isopropanol, reacted at -10~20℃ for 5h, filtered with suction and dried under reduced pressure to obtain 340g of 4-(3-chloro-4-fluoroaniline)- 6-acetoxy-7-methoxyquinazoline hydrochloride, the yield was 88.3%.
NVP-BEZ235 is a dual inhibitor of phosphatidylinositol 3-kinase (P13K)and the downstream mammalian target of rapamycin (mTOR) by binding to the ATP-binding cleft of these enzymes. It specifically blocks the dysfunctional activation of the P13K pathway and induce G(1) arrest. NPV-BEZ235 has been shown to inhibit VEGF induced cell proliferation and survival in vitro and VEGF induced angiogenesis in vivo. It has also been shown to inhibit the growth of human cancer in animal models.
BEZ-235 is an orally active phosphatidylinositol 3-kinase (PI3K) inhibitor in early clinical trials at Novartis for the treatment of advanced breast cancer, renal cell carcinoma, solid tumors and castration-resistant prostate cancer. Phase I clinical trials were also under way at the company for the treatment of glioma, however, no developments in this indication has been reported. Phase II clinical trials are ongoing at Johann Wolfgang Goethe Universität for the treatment of relapsed or refractory acute leukemia.
PI3Ks perform various functions, promoting cell growth, proliferation, differentiation, motility, survival and intracellular trafficking. Mutations leading to increased activity of PI3Ks, including faulty production or action of PI3K antagonists, have been found in many cancers.
In a suitable lab glass reactor are placed 45.0 g of starting 2[4-(8-bromo-3-methyl-2-oxo-2,3- dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]2-methyl-propionitrile together with 2.25 g of bistriphenylphosphine’palladium dichloride in 445 ml N,N-dimethylformamide. This mixture is heated to 95 0C and then a solution of 22.2 g of 3-quinoline boronic acid in a mixture of 225 ml DMF, 300 ml H2O and 60 g of KHCO3 is added. This mixture is heated for 2 h at 95 0C. Then 1080 ml H2O are added. The product 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl- 2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile precipitates. The mixture is cooled within 1.5 h to 0 – 5 °C. After stirring at that temperature for 2 h the crude product is filtered and washed with 300 ml H2O. This product is dried in vacuo at 60 0C for 18 h, to yield crude product.
40 g of this crude product is dissolved in 200 ml formic acid at 60 0C. 8 g of active charcoal and Smopex 234 are added. The mixture is stirred at 60 0C for 1 h, the charcoal is filtered, the residue washed with 80 ml formic acid and then 175 ml formic acid are distilled off in vacuo. Then 320 ml methanol are added and the mixture is heated at reflux for 3 h. The purified product precipitates from the reaction mixture. The mixture is cooled to 0 – 5 0C within 1 h, then stirred 2 h at that temperature is finally filtered and washed with 80 ml cold methanol. This recrystallisation procedure is repeated again. Finally the twice recrystallised material is dried in vacuo at 60 0C to yield purified 2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin- 3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile.
Example 1a 5-Bromo-2-(2-nitro-vinylamino)-benzoic acid
A suspension of 25 g (16 mmol) of 2-amino-5-bromo-benzoic acid (Fluka, Buchs, Switzerland) in H2O-HCI (37%) (10:1) is stirred for 8 h and then filtered (solution A). 8.17 g (255 mmol) of nitromethane (Fluka, Buchs, Switzerland) are added over 10 min to an ice- bath cooled mixture of 35 g of ice and 15.3 g (382 mmol) of NaOH. After stirring for 1 h at 0 0C and 1 h at rt, the solution is added at 0 0C to 28 g of ice and 42 ml of HCI (37%) (solution B). Solutions A and B are combined and the reaction mixture is stirred for 18 h at rt. The yellow precipitate is filtered off, washed with H2O and dried in vacuo at 400C to give the title compound. ES-MS: 287, 289 (M + H)+, Br pattern; 1H NMR (DMSO-d6): δ 13.7-14.6/br s (1 H), 12.94/d (1 H), 8.07/d (1 H), 8.03/dd (1 H), 7.83/dd (1 H), 7.71/d (1 H), 6.76/d (1 H).
Example 1b 6-Bromo-3-nitro-quinolin-4-ol
29 g (101 mmol) of 5-bromo-2-(2-nitro-vinylamino)-benzoic acid (Example 1a) and 11.9 g (121 mmol) of potassium acetate in 129 ml (152 mmol) of acetic anhydride are stirred for 1.5 h at 120 0C. The precipitate is filtered off and washed with acetic acid until the filtrate is colorless, then is washed with H2O and dried in vacuo to give the title compound. ES-MS: 269, 271 (M + H)+, Br pattern; analytical HPLC: W= 2.70 min (Grad 1).
Example 1c 6-Bromo-4-chloro-3-nitro-quinoline
20 g (74.3 mmol) of 6-bromo-3-nitro-quinolin-4-ol (Example 1b) in 150 ml (1.63 mol) of POCI3 are stirred for 45 min at 120 °C. The mixture is cooled to rt and poured slowly into ice- water. The precipitate is filtered off, washed with ice-cold water, and dissolved in CH2CI2. The organic phase is washed with cold brine, and the aqueous phase is discarded. After drying over MgSO4, the organic solvent is evaporated to dryness to provide the title compound. 1H NMR (CDCI3): J9.20/S (1H), 8.54/d (1H), 8.04/d (1H), 7.96/dd (1H); analytical HPLC: W= 4.32 min (Grad 1).
Example 1d 2-Methyl-2-(4-nitro-phenyl)-propionitrile
O .
To 15 g (92.5 mmol) of (4-nitro-phenyl)-acetonitrile (Fluka, Buchs, Switzerland), 1.64 mg (5.09 mmol) of tetrabutylammonium bromide (Fluka, Buchs, Switzerland) and 43.3 g (305 mmol) of iodomethane in 125 mL of CH2CI2 are added 1O g (250 mmol) of NaOH in 125 ml of water. The reaction mixture is stirred for 20 h at RT. After this time, the organic layer is separated, dried over MgSO4, and evaporated to dryness. The residue is dissolved in diethylether and treated with black charcoal for 30 min, filtered over Celite and evaporated in vacuo to give the title compound as a pale yellow solid. Analytical HPLC: tret= 3.60 minutes (Grad 1).Example 1e (2-(4-Amino-phenyl)-2-methyl-propionitrile
16 g (84.1 mmol) of 2-methyl-2-(4-nitro-phenyl)-propionitrile (Example 1d) and 4.16 g of Raney-Ni are shacked in 160 ml of THF-MeOH (1:1) under 1.1 bar of H2 for 12 h at rt. After completion of the reaction, the catalyst is filtered-off and the filtrate is evaporated to dryness. The residue is purified by flash chromatography on silica gel (hexane-EtOAc 3:1 to 1:2) to provide the title compound as an oil. ES-MS: 161 (M + H)+; analytical HPLC: tret= 2.13 minutes (Grad 1).
Example 1f 2-[4-(6-Bromo-3-nitro-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile
18 g (62.6 mmol) of 6-bromo-4-chloro-3-nitro-quinoline (Example 1c) and 11 g (68.9 mmol) of (2-(4-amino-phenyl)-2-methyl-propionitrile (Example 1e) are dissolved in 350 ml of acetic acid and stirred for 2 h. After this time, water is added and the yellow precipitate is filtered off and washed with H2O. The solid is dissolved in EtOAc-THF (1 :1), washed with sat. aqueous NaHCO3 and dried over MgSO4. The organic phase is evaporated to dryness to give the title compound as a yellow solid. ES-MS: 411 , 413 (M + H)+, Br pattern; analytical HPLC: tret= 3.69 min (Grad 1).
Example 1q 2-[4-(3-Amino-6-bromo-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile
24 g (58.4 mmol) of 2-[4-(6-bromo-3-nitro-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile (Example 1e) is shacked in 300 ml of MeOH-THF (1:1) under 1.1 bar of H2 in the presence of 8.35 g of Raney-Ni for 1 h. After completion of the reaction, the catalyst is filtered off and the filtrate is evaporated to dryness to give the title compound as a yellow foam. ES-MS: 381 , 383 (M + H)+, Br pattern; analytical HPLC: W= 3.21 min (Grad 1).
A solution of 5 g (13.1 mmol) of 2-[4-(3-amino-6-bromo-quinolin-4-ylamino)-phenyl]-2- methyl-propionitrile (Example 1g) and 1.59 g (15.7 mmol) of triethylamine in 120 ml CH2CI2 is added over 40 min to a solution of 2.85 g (14.4 mmol) of trichloromethyl chloroformate (Fluka, Buchs, Switzerland) in 80 ml of CH2CI2 at 00C with an ice-bath. The reaction mixture is stirred for 20 min at this temperature then is quenched with sat. aqueous NaHCO3, stirred for 5 min and extracted with CH2CI2. The organic layer is dried over Na2SO4, filtered and evaporated in vacuo to give crude title compound as a brownish solid. ES-MS: 407, 409 (M + H)+, Br pattern; analytical HPLC: tret= 3.05 min (Grad 1). Example 1i
To a solution of 3.45 g (8.47 mmol) of 2-[4-(8-bromo-2-oxo-2,3-dihydro-imidazo[4,5- c]quinolin-1-yl)-phenyl]-2-methyl-propionitrile (Example 1h), 1.8 g (12.7 mmol) of iodomethane (Fluka, Buchs, Switzerland) and 273 mg (0.847 mmol) of tetrabutylammonium bromide (Fluka, Buchs, Switzerland) in 170 ml of CH2CI2 is added a solution of 508 mg (12.7 mmol) of NaOH (Fluka, Buchs, Switzerland) in 85 ml of H2O. The reaction mixture is stirred for 2 days and 900 mg (6.35 mmol) of iodomethane and 254 mg (6.35 mmol) of NaOH in 5 ml of H2O are added. The reaction mixture is stirred for 1 day at rt . After this time, the reaction is quenched with H2O and extracted with CH2CI2 (2*). The organic layer is washed with brine, dried over Na2SO4, filtered and evaporated in vacuo to give the title compound as a beige solid. ES-MS: 421 , 423 (M + H)+, Br pattern; analytical HPLC: tret= 3.15 min (Grad 1).
Example 2
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]propionitrile p-toluenesulfonate salt
26.5 g of 2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1- yl)-phenyl]propionitrile are placed together with 55 ml formic acid into a glass reactor. This mixture is heated to 60 0C to get a clear solution. This solution is clearfiltered and washed with 36 ml formic acid. Then formic acid is distilled off until the volume of the residual solution is 55 ml. Then a solution of 11.3 g of p-toluenesulfonic acid in 228 ml acetone is added at 50 0C, followed by further addition of 822 ml acetone within 30 minutes. The salt precipitates from the reaction mixture. The mixture is cooled to 0 0C within 2 h, stirred at that temperature for 3 h, is then filtered and washed with 84 ml acetone. The product‘ is dried at 60 0C in vacuo for 18 h to yield 29.8 g (82.4 %) of the 2-Methyl-2-[4-(3-methyl-2-oxo-8- quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile p-toluenesulfonate salt (crystalline form A). The crystalline forms of the present invention are synthesized in accordance with the following examples which are illustrative without limiting the scope of the present invention.
Example 3:
Preparation of form A of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro- imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile
Form A of compound I can be manufactured in the following way: 241 g of free base are dissolved 2.4 I acetic acid at 50 0C. The solution is clearfiltered, washed with 250 ml acetic acid and then at 50 0C 7.2 I of water are added. The free base starts precipitating. The mixture is cooled within 1 h to 25 0C, is then filtered and washed with 10 I H2O. The free base is then dried in vacuo at 50 0C over night to yield 204 g of free base.
6-(4-(aminomethyl)-2-chlorophenoxyl)benzo[c][1,2]oxaborol-1(3H)-ol,
was synthesized at Anacor Pharmaceuticals as described in patent application WO 2010028005
A1
Pro-inflammatory cytokines play a critical role in the development of autoimmune and
inflammatory diseases. Targeting the cytokine environment has proven efficient for averting
inflammation. In this study, we reported that 6-(4-(aminomethyl)-2-
chlorophenoxyl)benzo[c][1,2]oxaborol-1(3H)-ol (AN3485), a benzoxaborole analog, inhibited
TLR2-, TLR3-, TLR4- and TLR5-mediated TNF-α, IL-1β and IL-6 release from human PBMCs
and isolated monocytes with IC50s ranging from 18 to 580 nM, and the inhibition was mediated
at the transcriptional level. Topical administration of AN3485 significantly reduced PMAinduced contact dermatitis and oxazolone-induced delayed-type hypersensitivity in mice,
indicating its capability of penetrating skin and potential topical application in skin
inflammation. Oral administration of AN3485 showed dose-dependent suppression of LPSinduced TNF-α and IL-6 production in mice with an ED90 of 30 mg/kg. Oral AN3485, 35
mg/kg, twice a day, suppressed collagen-induced arthritis in mice over a 20-day period. The
potent anti-inflammatory activity in in vitro and in vivo disease models makes AN3485 an
attractive therapeutic lead for a variety of cutaneous and systemic inflammatory diseases
A new class of boron-containing small molecules has been developed over the past several
years as potential drugs. Different from carbon, boron contains an electrophilic empty p-orbital
which can form transient bonds with nucleophiles in an enzyme active site, which mimics a
tetrahedral transition state of peptide bond cleavage in an enzymatic reaction (Baker et al., 2011).
The benzoxaboroles, in which the boron atom is incorporated into a heteroaromatic ring system,
are able to inhibit a number of important enzymes, including bacterial and fungi Leucyl-tRNA
synthetase (Rock et al., 2007), human phosphodiesterase-4 (PDE4) (Akama et al., 2009) and
HCV NS3/4A protease (Li et al., 2010). Three benzoxaboroles, AN2690 (Tavaborole), AN2728
and AN3365 (GSK’052) are in clinical trials for treatment of onychomycosis, psoriasis/atopic dermatitis and Gram-negative bacterial infection, and have been proven safe in human when
applied topically or systemically
……………………………………………………….
Structure-activity relationships of 6-(aminomethylphenoxy)-benzoxaborole derivatives as anti-inflammatory agent
Bioorg Med Chem Lett 2013, 23(6): 1680
Synthesis of compounds 9a–e. Reagents and conditions: (a) K2CO3, DMSO, 80–90 °C, overnight (33–61%); (b) LAH, THF, 0 °C to rt, 1 h, then 4 M HCl in 1,4-dioxane (43–68%); (c) aq NaOH, MeOH, 50 °C, 2 h (61%), (d) Ac2O, pyridine, rt (79%).
Synthesis of 6-(4-(aminomethyl)-2-chlorophenoxy)benzo[c][1,2]oxaborol-1(3H)-ol (9e): To a solution of 3H-benzo[c][1,2]oxaborole-1,6-diol (8) (300 mg, 2.00 mmol) in DMSO (30 mL) were added K2CO3(828 mg, 6.00 mmol) and 3-chloro-4-fluoro-benzonitrile (7b) (933 mg, 6.00 mmol). The reaction was heated at 90 °C for 7 h. After cooling the reaction mixture to room temperature, EtOAc (50 mL) was added. The organic layer was washed with water (5 × 50 mL). The organic layer was evaporated under vacuum. The residue was purified by reverse phase chromatography to afford 3-chloro-4-(1-hydroxy-1,3-dihydro-benzo[c][1,2]oxaborol-6-yloxy)-benzonitrile (9b) (190 mg, 33%). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.24 (s, 1H), 8.22 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.34 (s, 1H), 7.28 (d, J = 8.2 Hz, 1H), 7.01 (d, J = 8.6 Hz, 1H), 4.99 (s, 2H); ESIMS (m/z): 284 (M−H)−; HPLC: 96.4% (220 nm), 96.0% (maxplot).
To a solution of compound 9b (136 mg, 0.480 mmol) in anhydrous THF (60 mL) was added lithium aluminum hydride (1 M/ether, 1.19 mL, 1.19 mmol) at 0 °C. The reaction was stirred for 2 h. Then the reaction was quenched with 1 M HCl (30 mL). MeOH (50 mL) was added and the solution was filtered. The filtrate was evaporated under vacuum. The residue was purified by reverse phase chromatography (biotage, gradient MeOH/H2O from 10% to 100%). To a suspension of 9e free base in MeOH (5 mL) was added 4 M HCl in 1,4-dioxane (0.2 mL). The mixture became a clear solution then precipitates formed, which were collected by filtration to afford 9e (106 mg, 68%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.19 (s, 1H), 8.18 (br s, 3H), 7.75 (s, 1H), 7.44–7.39 (m, 2H), 7.19–7.10 (m, 3H), 4.98 (s, 2H), 4.03 (q, J = 5.5 Hz, 2H); ESIMS (m/z): 290 (M+H)+; HPLC: 95.9% (220 nm), 96.9% (maxplot).
To a solution of 2-hydroxy-4-methoxy-benzaldehyde (30 g, 197 mmol) in DCM (anhydrous, 120 rnL) was added pyridine (79 mL, 986 mmol) at room temperature. After the mixture was cooled to -10 0C, the Tf2O (50 mL, 296 mmol) was slowly added to the reaction between -10 0C to 0 0C. The addition took about 2.5 hours. After the addition, the stirring was kept for 30 minutes. The EtOAc (200 mL) was added. The organic layer was washed with 1 M HCl (3 X 80 mL), dried over MgSO4, filtered, and evaporated under vacuum. The residue was purified over silica gel, eluting with 5% EtOAc / hexanes to give trifluoro-methanesulfonic acid 2- formyl-5-methoxy-phenyl ester (2) 46 g in 82% yield. 1H NMR (400 MHz,
To a solution of trifluoro-methanesulfonic acid 2-formyl-5-methoxy-phenyl ester (2) (46 g, 160 mmol) in 1,4-dioxane (anhydrous, 360 rnL) were added bis(pinacolato)diboron (82.3 g, 320 mmol), [l,l ‘-bis(diphenylphosphino)ferrocene] palladium(II)chloride (23.7 g, 32 mmol) and KOAc (47.6 g, 480 mmol). The mixture was stirred at room temperature with N2bubbling for 30 minutes. Then the reaction was heated at 100 0C for 3 hours. The solution was filtered, evaporated under vacuum. The residue was purified over silica gel, eluting with 20% EtOAc / hexanes to afford 4-methoxy-2-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzaldehyde (3) 37.8 g in 90% yield. 1H NMR (400 MHz, CHLOROFORM- d) δ ppm 10.34 (s, 1 H), 7.90 (d, J=8.60 Hz, 1 H), 7.26 (s, 1 H), 6.99 (d, J=8.60 Hz, 1 H), 3.86 (s, 3 H), 1.36 (s, 12 H)
Compound 4:
To a clear solution of 4-methoxy-2-(4,4,5,5-tetramethyl-
[l,3,2]dioxaborolan-2-yl)-benzaldehyde (3) (48 g, 180 mmol) in MeOH (anhydrous, 300 mL) was slowly added NaBH4 (6.96 g, 180 mmol). The reaction was stirred at room temperature for 2 hours. Then IM HCl (100 mL) was slowly added. After stirring for overnight, the MeOH was evaporated under vacuum. The solid was filtered, washed with water and air-dried to afford 6-methoxy-3H- benzo[c][l,2]oxaborol-l-ol (4) 23 g in 77% yield. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.11 (s, 1 H), 7.29 (d, J=8.21 Hz, 1 H), 7.23 (d, J=2.34 Hz, 1 H), 7.03 (dd, J=8.40, 2.54 Hz, 1 H), 4.90 (s, 2 H), 3.75 (s, 3 H).
Compound 5:
To a clear solution of 6-methoxy-3H-benzo[c][l,2]oxaborol-l-ol (4) (600 mg, 3.66 mmol) in DCM (anhydrous, 60 mL) was slowly added BBr3 (1M/DCM, 8.05 mL, 8.05 mmol) at -10 0C. The reaction was stirred for 3 hours, with monitoring by NMR. After all 4 had gone, 30 mL of cold water was added. Then 50 mL of EtOAc was added to extract all organic compounds. The organic layer was washed with cold brine, until the pH of aqueous layer changed to pH 7. The organic layer was dried over Na2SO4, filtered, evaporated under vacuum. The residue (-85% HPLC purity) was used directly for the next step reaction without further purification. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.29 (s, 1 H), 9.04 (s, 1 H), 7.17 (d, J=8.21 Hz, 1 H), 7.07 (d, J=2.34 Hz, 1 H), 6.85 (dd, J=8.21, 2.34 Hz, 1 H), 4.85 (s, 2 H). ESMS (m/z): 149 (M- H)“. HPLC: 88.31% (220 nm), 85.02% (maxplot).
Compound 6:
To a solution of 3H-benzo[c][l,2]oxaborole-l,6-diol (5) (300 mg, 2 mmol) in DMSO (30 mL) were added K2CO3 (828 mg, 6 mmol) and 3-chloro-4-fiuoro- benzonitrile (933 mg, 6 mmol). The reaction was heated at 90 0C for 7 hours. After the cooling of reaction solution, EtOAc (50 mL) was added. The organic layer was washed with water (5 X 50 mL). The organic layer was evaporated under vacuum. The residue was purified by reverse phase chromatography to afford 3-chloro-4-(l- hydroxy-l,3-dihydro-benzo[c][l,2]oxaborol-6-yloxy)-benzonitrile (6) 190 mg in 33.3% yield. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.24 (s, 1 H), 8.22 (s, 1 H), 7.77 (d, J=7.81 Hz, 1 H), 7.50 (d, J=8.20 Hz, 1 H), 7.34 (s, 1 H), 7.28 (d, J=8.20 Hz, 1 H), 7.01 (d, J=8.59 Hz, 1 H), 4.99 (s, 2 H). ESMS (m/z): 284 (M-H)“. HPLC: 96.41% (220 nm), 96.0% (maxplot).
(X): IS AN 3485
To a clear solution of 3-chloro-4-(l-hydroxy-l,3-dihydro- benzo[c][l,2]oxaborol-6-yloxy)-benzonitrile (6) (136 mg, 0.48 mmol) in THF
(anhydrous, 60 mL) was added lithium aluminum hydride (lM/ether, 1.19 mL, 1.19 mmol) at 0 0C. The reaction was stirred for 2 hours. Then the reaction was quenched with IM HCl (30 mL). MeOH (50 mL) was added and the solution was filtered. The filtrate was evaporated under vacuum. The residue was purified by reverse phase chromatography (biotage, gradient MeOH / H2O from 10% to 100%) to afford (X) 106 mg (white solid) in 68% yield. 1H NMR (400 MHz, DMSO-J6) δ ppm 9.19 (s, 1 H), 8.18 (br, s, 3 H), 7.75 (s, IH), 7.44-7.39 (m, 2 H), 7.19-7.10 (m, 3 H), 4.98 (s, 2 H), 4.03 (q, J=5.50 Hz, 2 H).
The Cook Islands‘ defence and foreign affairs are the responsibility of New Zealand, which is exercised in consultation with the Cook Islands. In recent times, the …
AG014699, the phosphate salt of AG14447, which has improved aqueous solubility, has been selected for clinical trial.AG014699 is a tricyclic indole poly(ADP-Ribose) polymerase (PARP) inhibitor with potential antineoplastic activity.
M.Wt: 421.3593
Formula: C19H21FN3O5P
CAS No: 459868-92-9
Rucaparib, PF-01367338283173-50-2 cas 6H-Pyrrolo[4,3,2-ef][2]benzazepin-6-one, 8-fluoro-1,3,4,5-tetrahydro-2-[4-[(methylamino)methyl]phenyl]-6H- Azepino[5,4,3-cd]indol-6-one, 8-fluoro-1,3,4,5-tetrahydro-2-[4-[(methylamino)methyl]phenyl] -8-Fluoro-2-[4-[(methylamino)methyl]phenyl]-1,3,4,5- tetrahydro-6H-azepino[5,4,3-cd]indol-6-one;8-Fluoro-2-(4-methylaminomethyl-phenyl)-1,3,4,5-tetrahydro-azepino[5,4,3-cd]indol-6-one8-Fluoro-2-(4-methylaminomethyl-phenyI)-l,3,4,5-tetrahydro-azepino[5,4,3- cd]indol-6-one
WO 2014052550, WO 2014037313, WO 2000042040WO 2004087713WO 2005012305
Rucaparib (AG 014699) is a PARP inhibitor being investigated as a potential anti-cancer agent.
Rucaparib inhibits “the contraction of isolated vascular smooth muscle, including that from the tumours of cancer patients. It also reduces the migration of some cancer and normal cells in culture.”[1]
It is thought that 20% of women with ovarian cancer who are not BRCA positive might also benefit from PARP inhibitors. Clinical trials are beginning (as of April, 2014)
As of November 2012 four clinical trials of rucaparib were recruiting patients.[5]
Inhibition of poly(ADP ribose) polymerase, or PARP, is an exciting new mechanism for the treatment of cancer.(1) The PARP enzyme is responsible for repair of damaged DNA in both normal and tumor cells, and inhibition of this repair mechanism is expected to make the cell more likely to undergo apoptosis. Preclinical work has shown that PARP inhibitors coadministered with a standard chemotherapuetic agent are more effective than the standard treatment aloneRucaparib is a NAD+ ADP-ribosyltransferase inhibitor in phase II clinical development at Cancer Research UK for the treatment of patients with advanced ovarian cancer and in patients with locally advanced or metastatic breast cancer. Clovis Oncology is conducting early clinical evaluation of rucaparib for the treatment of triple negative breast cancer or ER/PR +, HER2 negative with known BRCA1/2 mutations p2 and for the treatment of gBRCA mutation breast cancer.. Pfizer discontinued development of rucaparibin 2011.In 2011, the compound was licensed to Clovis Oncology by Pfizer for the treatment of cancer. In 2012, orphan drug designation was assigned in the U.S. and the E.U. for the treatment of ovarian cancer.
The compound 8-fluoro-2-{4-[(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3- cd]indol-6-one represented by formula
is a small molecule inhibitor of poly(ADP-ribose) polymerase (PARP). 8-Fluoro-2-{4- [(methylamino)methyl]phenyl}-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one and salts thereof, is disclosed in U.S. Patent No. 6,495,541 and PCT Application No. PCT/IB2004/000915, International Publication No. WO 2004/087713, the disclosures of which are incorporated herein by reference in their entireties. U.S. Provisional Patent Applications No. 60/612,459 and 60/679,296, entitled “Polymorphic Forms of the Phosphate Salt of 8-Fluoro-2-{4-[(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H- azepino[5,4,3-cd]indol-6-one,” the disclosures of which are incorporated herein by reference in their entireties, describe novel polymorphic forms of the phosphate salt of 8-fluoro-2-{4- [(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one, and methods for their preparation. U.S. Provisional Patent Applications No. 60/612,458; and 60/683,006, entitled “Therapeutic Combinations Comprising Poly(ADP-Ribose) Polymerases Inhibitor,” the disclosures of which are incorporated herein by reference in its entirety, describe pharmaceutical combinations of 8-fluoro-2-{4- [(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one.
Example IIII:8-Fluoro-2-(4-methylaminomethyl-phenyI)-l,3,4,5-tetrahydro-azepino[5,4,3- cd]indol-6-one
4-(8-fluoro-6-oxo-3,4,5,6-tetrahydro-lH-azepino[5,4,3-cd]indol-2-yl)- benzaldehyde (100 mg, 0.32 mmol; prepared in a manner similar to that described for compound 12 for 2-bromo-8-fluoro-l,3,4,5-tetrahydro-azepino[5,4,3-cd]indol-6-one and 4-formylphenylboronic acid) was reacted with methylamine (1.62 mmol) as described for Compound PPP to yield 8-fluoro-2-(4-methylaminomethyl-phenyl)- l,3,4,5-tetrahydro-azepino[5,4,3-cd]indol-6-one, 32 mg (31%) as a yellow solid: m.p. 1543-155 °C; Η NMR (300 MHz, d6-DMSO) 2.28 (s, 3H), 3.04 (m, 2H), 3.40 (m, 2H), 3.69 (s, 2H), 7.32 (dd, 7= 9.0, 2.4 Hz, IH), 7.44 (m, 3H), 7.57 (d, 7= 8.1 Hz, 2H), 8.25 (br t, IH), 11.67 (br s, IH). HRMS (MALDI MH+) Calcd for C19H18N3OF: 324,1512. Found: 325.1524. Anal. (C19H18N3OF03 H2O) C, H, N.
PAPER
Org. Process Res. Dev., 2012, 16 (12), pp 1897–1904
DOI: 10.1021/op200238p
http://pubs.acs.org/doi/full/10.1021/op200238pNovel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.
To a solution of aqueous sodium hydroxide (40% w/w, 3.6 kg, 2.0 equiv) in water (88 L, 14 L/kg) and methanol (35 L, 5.5 L/kg) was added 12 ……………………………………………………deleted……………………..and dried at 45 °C under vacuum to give 1 as a 1:1 THF solvate (5.57 kg, 14.08 mol, 84% yield);
8-Fluoro-2-(4-methylaminomethyl-phenyl)-1,3,4,5-tetrahydro-azepino[5,4,3-cd]indol-6-one (S)-camphorsulfonate Salt (21)
To a slurry of 1 (5.32 kg, 13.48 mol) in isopropanol (30 L, 5.5 L/kg) and water (39 L, 7.3 L/kg) was added a solution of (S)-camphorsulfonic acid (3.75 kg, 16.18 mol, 1.2 equiv) in water (10.6 L, 2 L/kg). The resultant slurry was then heated to 70 °C and held for 1 h to ensure dissolution. …………………………..deleted…………………..C to give 21 as a white crystalline solid (7.09 kg, 12.76 mol, 95% yield); mp (IPA/water) 303 °C;
The compound 8-fluoro-2-{4-[(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3- cd]indol-6-one represented by formula
is a small molecule inhibitor of poly(ADP-ribose) polymerase (PARP). 8-Fluoro-2-{4- [(methylamino)methyl]phenyl}-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one and salts thereof, is disclosed in U.S. Patent No. 6,495,541 and PCT Application No. PCT/IB2004/000915, International Publication No. WO 2004/087713, the disclosures of which are incorporated herein by reference in their entireties.
U.S. Provisional Patent Applications No. 60/612,459 and 60/679,296, entitled “Polymorphic Forms of the Phosphate Salt of 8-Fluoro-2-{4-[(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H- azepino[5,4,3-cd]indol-6-one,” the disclosures of which are incorporated herein by reference in their entireties, describe novel polymorphic forms of the phosphate salt of 8-fluoro-2-{4- [(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one, and methods for their preparation. U.S. Provisional Patent Applications No. 60/612,458; and 60/683,006, entitled “Therapeutic Combinations Comprising Poly(ADP-Ribose) Polymerases Inhibitor,” the disclosures of which are incorporated herein by reference in its entirety, describe pharmaceutical combinations of 8-fluoro-2-{4- [(methylamino)methyl]phenyl}-1 ,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one.
Example 13. Synthesis of 8-Fluoro-2-(4-methylaminomethyl-phenyl)-1,3.4.5-tetrahvdro-azepinor5.4.3- ccflindol-6-one (15) i
Lactam 14 (14.42 g, 0.038 mol) was dissolved in hydrobromic acid in acetic acid (30%-32%, 140 ml). The reaction solution was stirred for 46 hours at room temperature in a 500ml flask that was connected to an ethanolamine scrubber system. HPLC analysis indicated the completion of the reaction. Ice (30 g) was added to the reaction solution followed by addition of aqueous NaOH (327 ml, 10 M, 3.27 mol) while the temperature was maintained between 25 0C and 35 0C. When addition of NaOH was complete, the pH was 10. The resulting solid was collected by filtration, washed with water (2 x 50 ml). The filter cake was then suspended in water (125 ml) and stirred for 2 hours. The solid was collected by filtration, washed with water (2 x 25 ml) and dried to afford 10.76 g of product (88% yield). 1H NMR (300 MHz, DMSO-d6) δ 2.577(s, 3H), 3.053(m, 2H), 3.406(m, 2H), 4.159(s, 2H), 7.36(dd, 1 H, J= 2.4 Hz and J= 9.3 Hz), 7.44(dd, 1 H, J= 2.4 Hz and J= 11.1 Hz), 7.63(d, 2H, J=8.1 Hz), 7.70(d, 2H, J= 8.1 Hz), 8.265(t, 1H, J= 5.7 Hz), 11.77(s, 1 H). Exact mass calculated for C19H19FN3O: 324.1512. Found: 324.1497.
UPDATES
OriginatorClovis Oncology; Foundation Medicine
ClassDiagnostic agents
Highest Development Phases
RegisteredOvarian cancer
Phase IIIFallopian tube cancer; Peritoneal cancer
Clinical Phase UnknownCancer
Most Recent Events
19 Dec 2016Registered for Ovarian cancer (Diagnosis) in USA
23 Aug 2016Preregistration for Ovarian cancer (Diagnosis) in USA (unspecified route)
05 May 2016Clovis Oncology announces intention to submit PMA application to US FDA
Rucaparib phosphateis in phase Ⅲ clinical trials for the treatment of patients with advanced ovarian cancer, fallopian tube cancer and ovarian cancer. It was granted breakthrough therapy designation by FDA for the treatment of ovarian cancer in 2015.
The compound was originally developed by Pfizer, then licensed to Clovis Oncology by Pfizer in 2011 for the treatment of cancer.
Clovis Oncology receives Breakthrough Therapy designation for rucaparib for treatment of advanced ovarian cancer in patients with BRCA-mutated tumours
7 April 2015 • Author: Victoria White
Clovis Oncology has announced that the U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy designation for the Company’s investigational agent rucaparib as monotherapy treatment of advanced ovarian cancer in patients who have received at least two lines of prior platinum-containing therapy, with BRCA-mutated tumours, inclusive of both germline BRCA (gBRCA) and somatic BRCA (sBRCA) mutations.
2525 28th Street
Suite 100
Boulder, CO 80301 Tel: 303.625.5000
Fax: 303.245.0360
are a biopharmaceutical company focused on acquiring, developing and commercializing cancer treatments in the United States, Europe and other international markets. Our development programs are targeted at specific subsets of cancer, combining personalized medicine with companion diagnostics to direct therapeutics to those patients most likely to benefit from them.
We have three product candidates in clinical development: rociletinib (CO-1686), which is in Phase II development for the treatment of non-small cell lung cancer; rucaparib, which is in Phase II and Phase III clinical trials for the treatment of ovarian cancer; and lucitanib, which is in Phase II clinical trials for the treatment of breast and lung cancers. We have received Breakthrough Therapy designation from the FDA for rociletinib and rucaparib. We maintain global rights for rociletinib and rucaparib, and U.S. and Japanese rights to lucitanib.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....












 :
:






 and
and





























 .
.






 Tandoori chicken at Surjit Food Plaza. amritsar
Tandoori chicken at Surjit Food Plaza. amritsar







 Golden Temple – Harmandir Sahib: Free food for everyone
Golden Temple – Harmandir Sahib: Free food for everyone

%27,_oil_on_canvas_painting_by_Charles_W._Bartlett,_1920,_Honolulu_Academy_of_Arts.jpg)
 tandoori chicken
tandoori chicken





























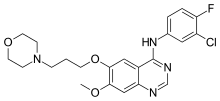














 GEFITINIB
GEFITINIB
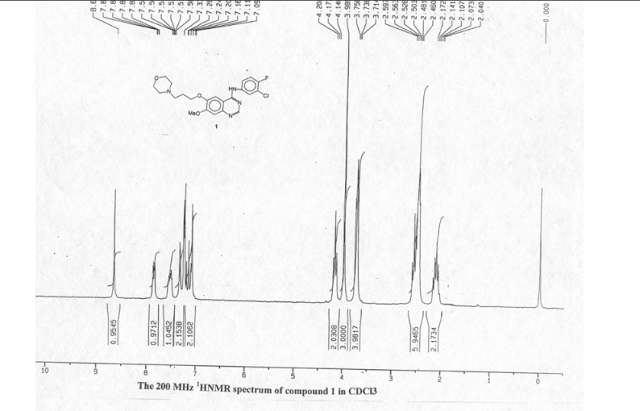
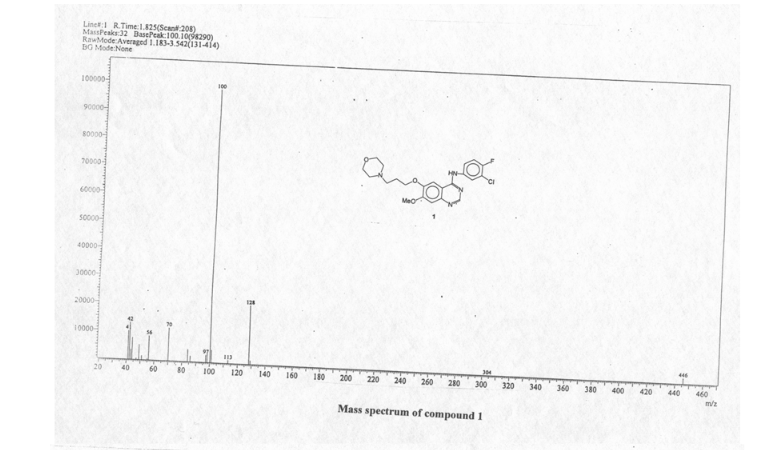



























 AIRPORT
AIRPORT





 FLAG
FLAG









 Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.
Novel PARP inhibitor 1 is a promising new candidate for treatment of breast and ovarian cancer. A modified synthetic route to 1 has been developed and demonstrated on 7 kg scale. In order to scale up the synthesis to multikilogram scale, several synthetic challenges needed to be overcome. The key issues included significant thermal hazards present in a Leimgruber–Batcho indole synthesis, a low-yielding side-chain installation, a nonrobust Suzuki coupling and hydrogen cyanide generation during a reductive amination. In addition to these issues, changing from intravenous to oral delivery required a new salt form and therefore a new crystallization procedure. This contribution describes development work to solve these issues and scaling up of the new process in the pilot plant.