
Home » Uncategorized (Page 93)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Raw Material Variation into QbD Risk Assessment
DRUG REGULATORY AFFAIRS INTERNATIONAL
Areas of discussion included how expectations for raw material control are evolving within changing regulatory and business paradigms including quality by design (QbD), counterfeiting, complex supply chains, and sourcing changes. discussed risk assessment and mitigation strategies along with supplier risk management plans.

Regulatory Considerations
the lack of a consistent definition of raw materials in regulations pertaining to the pharmaceutical industry. In its Q7 guideline, the International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) defines raw materials as “starting materials, reagents, and solvents intended for use in the production of intermediates or APIs.” However, the term as defined by different speakers could cover a wide range of materials including the following:
• starting or source materials (cell lines, viral or bacterial stocks, media components, chemicals, tissues, serum, water)
• in-process materials (resins, buffers, filters, column housings, tubing, reagents)
• excipients
• packaging components, both…
View original post 3,002 more words
FDA Warning Letter on Data Integrity
DRUG REGULATORY AFFAIRS INTERNATIONAL

The integrity of data is currently in the focus of international authorities In particular the US FDA issued serious violations in Warning Letters to the companies concerned. Read more about the current complaints in a Warning Letter issued to the API manufacturer VUAB Pharma.
For authorities the integrity of data is an essential quality attribute in the manufacture of pharmaceutical products. After some serious deviations international authorities have moved the topic into the centre of their interest. In particular the US FDA issued serious violations in Warning Letters to the companies concerned.

In a current letter to the API manufacturer VUAB Pharma in the Czech Republic the inspector and the authority criticised multiple aspects with regard to “failure to prevent unauthorized access or change to data and to provide controls preventing data omissions”:
- ‘The firm did not retain complete raw data from testing performed to assure the quality…
View original post 408 more words
Development of monographs for Indian pharmacopoeia
DRUG REGULATORY AFFAIRS INTERNATIONAL

Development of monographs for Indian pharmacopoeia
http://www.ipapharma.org/events/speakerpresentation/8-%20Dr%20S%20C%20Mathur.pdf

ALOGLIPTIN
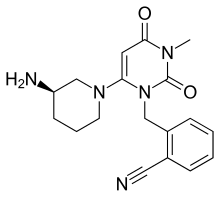
ALOGLIPTIN
Alogliptin is a potent, selective inhibitor of DPP-4 with IC50 of <10 nM, exhibits greater than 10,000-fold selectivity over DPP-8 and DPP-9.
Alogliptin (trade name Nesina in the US[1] and Vipidia in Europe[2]) is an orally administered anti-diabetic drug in the DPP-4 inhibitor class,[3] developed by Syrrx, a company which was acquired by Takeda Pharmaceutical Company in 2005. Like other medications for the treatment of Type 2 diabetes, alogliptin does not decrease the risk of heart attack and stroke. Like other members of the gliptin class, it causes little or no weight gain, exhibits relatively little risk of causing hypoglycemia, and exhibits relatively modest glucose-lowering activity. Alogliptin and other gliptins are commonly used in combination with metformin in patients whose diabetes cannot adequately be controlled with metformin alone.[4]
Clinical study
Alogliptin is a dipeptidyl peptidase-4 inhibitor (DPP-4i) that is designed to slow the inactivation of incretin hormones GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulinotropic peptide). [5]
A randomized clinical trial reporting in 2011 aimed to determine the efficacy and safety of alogliptin versus placebo and vogliboseamong newly diagnosed Type 2 diabetes patients in Japan. The main outcome indicated that alogliptin was statistically superior to both comparitors.[6]
A randomized clinical trial reporting in 2012 aimed to demonstrate that alogliptin was “non-inferior” to a “very low fat/calorie traditional Japanese diet” among newly diagnosed Type 2 diabetes patients in Japan. The outcome indicated that both the drug and dietary treatments comparably impacted indicators of the diabetic condition, such as HbA1c levels and glycemic efficacy. The drug treatment had its impact without changing body mass index (BMI), but the dietary treatment was accompanied by a significant reduction in the BMI.[7]
A randomized clinical trial reporting in 2011 aimed to demonstrate the efficacy of alogliptin as an add-on agent in combination withmetformin and pioglitazone versus simply increasing the dosage of pioglitazone in combination with metformin; in other words, this was a study to look at a three-agent therapy versus a two-agent therapy. The outcome of this study suggested that the addition of alogliptin to metformin and pioglitazone provided superior impact on diabetes biomarkers (e.g. HbA1c) than increasing the dose of pioglitazone in a two agent therapy with metformin.[8]
Reported adverse events
Adverse events appear to be restricted to mild hypoglycemia based on clinical studies.[6][7][8]
Alogliptin is not associated with increased weight, increased risk of cardiovasular events, or heart failure.[9][10]
Market access
In December 2007, Takeda submitted a New Drug Application (NDA) for alogliptin to the United States Food and Drug Adminiistration (USFDA),[11] after positive results from Phase III clinical trials.[1] In September of 2008, the company also filed for approval in Japan,[12] winning approval in April 2010.[11] The company also filed a Marketing Authorization Application (MAA) elsewhere outside the United States, which was withdrawn in June 2009 needing more data.[12] The first USFDA NDA failed to gain approval and was followed by a pair of NDAs (one for alogliptin and a second for a combination of alogliptin and pioglitazone) in July 2011.[11] In 2012, Takeda received a negative response from the USFDA on both of these NDAs, citing a need for additional data.[11]
In 2013 the FDA approved the drug in three formulations: As a stand-alone with the brand-name Nesina. Combined with metforminusing the name Kazano, and when combined with pioglitazone as Oseni.
Diabetes affects millions of people worldwide and is considered one of the main threats to human health in the 21st century. In 2006, the World Health Organization (WHO) estimated that over 180 million people worldwide had diabetes, and the number is projected to double by 2030. Over time, uncontrolled diabetes can damage body systems, including the heart, blood vessels, eyes, kidneys and nerves. According to the WHO, approximately 1.1 million people died from diabetes in 2005, and it is estimated that diabetes-related deaths will increase by more than 50% in the next decade. Globally, the socioeconomic burden of diabetes is substantial.
There are two main types of diabetes, designated type 1 and type 2, with type 2 diabetes accounting for over 90% of all diabetes cases globally. Type 1 diabetes is characterized by insulin deficiency, primarily caused by autoimmune-mediated destruction of pancreatic islet β-cells, and type 2 diabetes is characterized by abnormal insulin secretion and concomitant insulin resistance. To prevent the development of ketoacidosis, people with type 1 diabetes must take exogenous insulin for survival. Although those with type 2 diabetes are not dependent on exogenous insulin as much as subjects with type 1 diabetes, they may require exogenous insulin to control blood glucose levels.
As diabetes has become a global health concern, research interest in the condition has rapidly increased. In addition to studies on prevention, many studies with the aim of developing new interventions for the treatment of diabetes, especially type 2 diabetes, have been conducted. Currently available medications for the treatment and management of type 2 diabetes include metformin, sulfonylureas, thiazolidinediones and insulin. However, these therapies are commonly associated with secondary failure and may cause hypoglycemia. Insulin resistance and progressively worsening hyperglycemia caused by reduced β-cell function are major challenges in managing type 2 diabetes. Evidence suggests that patients with insulin resistance do not develop hyperglycemia until their β-cells are unable to produce enough insulin. New agents that can enhance insulin secretion from islet β-cells in a sustained glucose-dependent manner could therefore hold promise for the treatment of type 2 diabetes.
One promising approach is based on inhibition of the serine protease dipeptidyl- peptidase IV (DPP IV), a postproline dipeptidyl aminopeptidase that belongs to the S9b peptidase family of proteolytic enzymes. It is known that DPP IV plays a key role in maintaining glucose homeostasis by controlling the incretin activity of glucagon-like peptide 1 (GLP-I) and glucose-dependent insulinotropic polypeptide (GIP, also known as gastric inhibitory polypeptide). Inhibition of DPP IV is therefore recognized as a novel therapeutic approach for the treatment of type 2 diabetes.
Recently, a series of DPP IV inhibitors were developed. Among these highly potent compounds, alogliptin benzoate (SYR-322) and its analogs demonstrated encouraging antidiabetic efficacy (EP 1586571 (WO 2005/095381); WO 2008/067465; WO 2007/035379, and US 2004/097510).
Alogliptin benzoate can be prepared as described in EP 1586571 (WO 2005/095381) according to the process set forth in Scheme 1 :

Scheme 1
In accordance with this process, 6-Chlorouracil (1) is alkylated with 2- (bromomethyl)benzonitrile in the presence of NaH and LiBr in a mixture of DMF- DMSO to produce the TV-benzyluracil derivative (2) in 54% yield. Compound (2) is further alkylated with iodomethane and NaH in DMF/THF to give the 1 ,3 disubstituted uracil (3) in 72% yield. Subsequent displacement of chlorouracil (IV) with 3(R)- aminopiperidine dihydrochloride in the presence of either NaHCO3 in hot methanol or K2CO3 in aqueous isopropanol provides alogliptin (4), which is isolated as the corresponding benzoate salt by treatment with benzoic acid in ethanol. The overall yield of this three-stage process is -20-25%. One of the disadvantages of above described process is the difficulty to separate and purify mixtures of solvents with high boiling point (for example, DMF/DMSO) for recycling. Another disadvantage is the usage of hazardous materials such as sodium hydride, which requires anhydrous solvents as a reaction media.
Intermediate 2-((6-chloro-3-methyl-2,4-dioxo-3 ,4-dihydropyrimidin- 1 (2H)-yl)methyl) benzonitrile (3) is alternatively obtained by alkylation of 6-chloro-3 methyluracil with 2-(bromomethyl)benzonitrile by means of diisopropylethylamine in hot NMP (WO 2007/035629). Although this process is more technological than the previously described process (EP 1586571), the overall yield is still moderate (50-55%). The problem of mixed solvents (toluene, NMP, diisopropylethylamine) separation persists for this process as well.

………….
http://www.google.com/patents/EP2410855A1?cl=en
EXAMPLE 1
Preparation of (R)-2-((6-(3 -aminopiperidin-l-yl)-3 -methyl-2,4-dioxo-3 ,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 3):

Scheme 3
Preparation of l-(2-isocyanobenzyl)-3-methylurea
2-cyanobenzylamine hydrochloride (90 g) and Dichloromethane (800 ml) were taken into a round bottomed (RB) flask. Methyl isocyanate (45.6 g) was added at 5°C. Triethylamine (81 g) in Dichloromethane (300 ml) was added at the same temperature and stirred at room temperature for 16h. Water (1 L) was added and stirred for 30 min. The obtained solid was collected by filtration and dried in oven at 50°C for 12h. The yield is 85% and the purity 99.8%.
Preparation of l-(2-isocyanobenzyl)-3-methyIpyrimidine-2,4,6(lH,3H,5H)-trione
a). To a stirred solution of 0.11 mol of sodium ethanolate in 80 ml of ethanol abs. was added 0.1 mol of l-(2-isocyanobenzyl)-3-methylurea and 0.1 mol diethyl malonate. The mixture was refluxed for 3-5 h. The cooled residue was acidified with 0.1 M hydrochloric acid (60 ml). The solid which separated was filtered off and recrystallized from ethanol or any suitable solvent. The yield is 78-85% and purity >95%.
b). In an alternate embodiment, l-(2-isocyanobenzyl)-3-methylurea (30 g), acetic acid (105 ml) and malonic acid (18 g) were mixed and heated to 60°C. Acetic anhydride (60 ml) was added at 60°C and heating was continued for two hours at 80°C. The reaction mixture was poured over ice water (300 ml) and the obtained solid was filtered, washed with water (1×500 ml) and methyl-tert-butylether (100 ml). The yield is 60% with 93.4% purity.
The compound thus prepared can be used for the next step without purification or purified by crystallization or column chromatography.
Preparation of 6-chloro-l-(2-isocyanobenzyl)-3-methylpyriinidine-2,4(lH,3H)- dione
a). l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4,6(lH,3H,5H)-trione (30 g) was mixed with phosphorus oxychloride (300 ml) and cooled to 0°C. Water (9 ml) was added slowly, stirred for 10 min. and heated to reflux at 110°C for 5h. Progress of the reaction was monitored by TLC (50% Ethyl acetate/Hexane). On completion of the reaction, phosphorus oxychloride was distilled off. The crude compound was dissolved in dichloromethane (500 ml) and poured into ice water (500 ml) by small portions. The layers were separated and the aqueous layer was extracted with dichloromethane (200 ml). The combined organic extracts were washed with water and brine, dried over sodium sulphate and concentrated under reduced pressure. The mixture of two isomers (4-chloro and 6-chloro derivatives = 1:1) was isolated and separated by column chromatography using neutral alumina and eluent – 25-50% of ethylacetate and hexane). The off-white solid was obtained, yield – 37%, purity – 99.8%. 1H NMR corresponds to literature data (J. Med. Chem. 2007, 50, 2297-2300).
b). In an alternate embodiment, a solution of l-(2-isocyanobenzyl)-3-methylpyrimidine- 2,4,6(1 H,3H,5H)-trione (18 mmol), phosphorus oxychloride (85 ml), benzyltriethylammonium chloride (16.5 g, 72 mmol) and phosphorus pentachloride (3.8 g, 18 mol) in acetonitrile (80 ml) was refluxed for 4-5 h with stirring. After evaporation under reduced pressure, the resulting oily residue was mixed with methylene chloride (or chloroform) and the mixture was poured into water and ice (50 ml). The layers were separated and the aqueous layer was extracted with dichloromethane (200 ml). The combined organic extracts were washed with water and brine, dried over sodium sulphate and concentrated under reduced pressure. Crude product was crystallized from THF-hexanes to give desired compound in 70.5% yield.
c). In an alternate embodiment, a solution of l-(2-isocyanobenzyl)-3-methylpyrimidine- 2,4,6(1 H,3H,5H)-trione (13.1 mmol) in POCl3 (30 ml) was refluxed for 1-3 h. The solvent was concentrated and then partitioned with CH2Cl2 (100 ml) and water (100 ml). The organic layer was washed with brine, dried over Na2SO4, and concentrated to give 6-chloro compound as a solid (-95%). Compound can be also precipitated from concentrated methylene chloride solution by hexanes and used as a crude for the next step or purified by reslurring in isopropanol, filtered off, washed with isopropanol, and dried under vacuum at 55-60° C.
Preparation of (R)-tert-butyl l-(3-(2-isocyanobenzyI)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate
a). 6-chloro- l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (13 g), Dimethylformamide (130 ml), Potassium carbonate (13 g) and tert-butyl (R)-piperidin- 3-ylcarbamate (10.4 g) were heated to 80°C for 7 hrs. The mixture was then allowed to come to room temperature and poured over ice water (500 ml). The obtained solid was filtered and washed with cold water (500 ml). The solid thus obtained was taken in Methyl-tert-butylether (50 ml) stirred for 10 min. filtered and washed with Hexane (50 ml), to give the N-tert-butyloxycarbonyl protected compound in -75% yield. b). In an alternate embodiment, a flask charged with a stir bar, 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (4.10 mmol), (Λ)-3- terrtnityloxycarbonylaminopiperidine (4.64 mmol), K2CO3 (1.15 g, 8.32 mmol) and DMF (12 mL) was stirred at 75 °C for 6 h. Then, water was added and the mixture was extracted with methylene chloride. The organic layer was washed with brine, dried over Na2SO4, and concentrated to give the N-ter/butyloxycarbonyl protected compound in -93-96% yield.
Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile salts
a). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile hydrochloride
The crude (R)-tert-butyl l-(3-(2-isocyanobenzyl)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate from previous procedure was dissolved in THF and acidified with 6M hydrochloric acid while maintaining the temperature below 15° C. The resultant slurry was cooled to 0-5° C, stirred at this temperature for 3-5 h and then filtered. The filter cake was washed twice with isopropanol and dried in vacuum at 45-5O0C to provide hydrochloride as a white crystalline solid.
b). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile trifluoroacetate
TFA (ImL) was added into the methylene chloride solution of (R)-tert-butyl l-(3-(2- isocyanobenzyl)- 1 -methyl-2,6-dioxo- 1 ,2,3,6-tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate from the above-mentioned procedure. The solution was stirred at room temperature for 1 h and then the mixture was concentrated in vacuo. The residue was dissolved in a small amount of MeOH or isopropanol and the desired salt was precipitated by addition of diisopropyl ether. The solids were filtered off, washed with diisopropyl ether and dried in vacuum at 45-5O0C to provide trifluoroacetate as an off- white powder. c). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile benzoate (Alogliptin)
The crude (R)-tert-butyl l-(3-(2-isocyanobenzyl)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate was dissolved in ethanol. A solution of benzoic acid in ethanol was added and the mixture was slowly heated to 65-70°C. The solution was stirred at this temperature for Ih and was then crystallized by cooling to 0-5° C and stirring for 12 hrs. The solution was filtered, washed with alcohol. The wet cake was then conditioned under nitrogen for 2 hours. The cake was dried for 8 hrs at 40-50° C to provide the benzoic acid salt of alogliptin as a white crystalline solid.
EXAMPLE 2:
Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 4)


Scheme 4 Preparation of 6-amino-l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)- dione
a). l-(2-isocyanobenzyl)-3-methylurea (0.2 mol) and cyanoacetic acid (0.22 mol) were dissolved in acetic anhydride (400 ml), and the mixture was heated at 80°C for 2 hours. Acetic anhydride was distilled off under reduced pressure and water (200 ml) was added. The mixture was cooled to 0-5 0C and 2N NaOH solution (220 ml) was added and stirring was continued for 2 hours. The obtained solids were filtered off, washed with cold methanol and dried under vacuum. The yield of 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione was 72 %.
b). Under nitrogen atmosphere, l-(2-isocyanobenzyl)-3-methylurea (98.4 g) and cyanoacetic acid (80.0 g) was added to N,N-dimethylformamide (836 ml). The mixture was stirred at room temperature and methanesulfonyl chloride (72.8 ml) was added dropwise with stirring at this temperature. The mixture was stirred at room temperature for 4 hrs, cooled with water, and water-isopropanol [2:1 (volume ratio), 1670 ml] was added drop wise. The mixture was stirred under water-cooling for 1 hr, and the precipitated crystals were collected by filtration and dried to give 3-(2-cyano-acetyl)-3- methyl-l-(2-isocyanobenzyl)-urea with 68% yield.
To 3-(2-cyano-acetyl)-3-methyl-l-(2-isocyanobenzyl)-urea (120 g) were added water (962 ml) and 2N aqueous sodium hydroxide solution (24.9 ml), and the mixture was stirred with heating at 80° C for 1 hr. After allowing to cool to room temperature, the crystals were collected by filtration and dried to give 6-amino-l-(2-isocyanobenzyl)-3- methylpyrimidine-2,4(lH,3H)-dione in 76% yield.
c). 6-amino-l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (0.1 mol) was mixed with (R)-piperidin-3-yl-carbamic acid tert.-butyl ester hydrochloride (0.1 mol) of the appropriate amine hydrochloride and (R)-piperidin-3-yl-carbamic acid tert.-butyl ester (0.1 mol). The mixture was heated at 100°C and bubbling continued for 3 hr. Water was added to the cooled mixture and the mixture was extracted with methylene chloride. The organic layer was washed with brine, dried over Na2SO4, and concentrated to give N-tert-butyloxycarbonyl protected compound in ~93-96% yield.
d). Benzoate salt of alogliptin was prepared as described above. While certain embodiments of the invention have been illustrated and described, it will be clear that the invention is not limited to the embodiments described herein. Numerous modifications, changes, variations, substitutions and equivalents will be apparent to those skilled in the art without departing from the spirit and scope of the present invention as described by the claims, which follow.
………………
Patent EP2410855A1
http://www.google.com/patents/EP2410855A1?cl=en

…………..

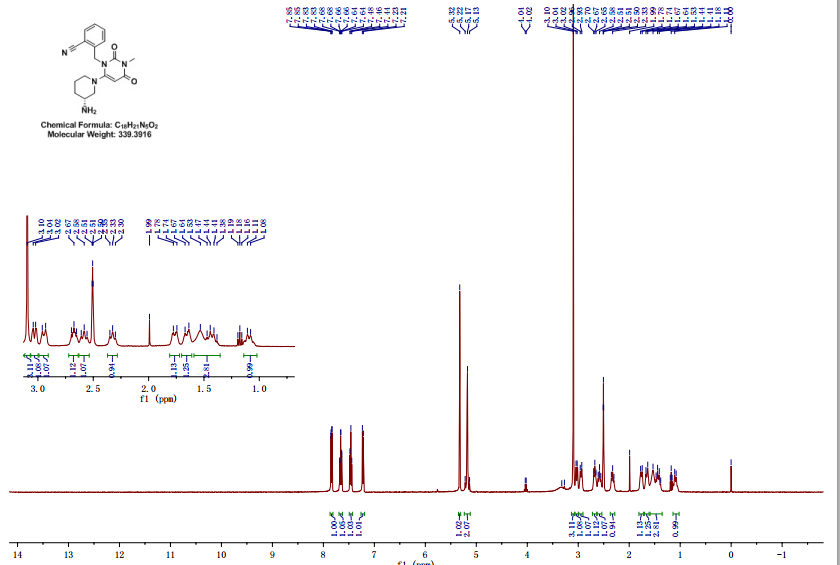
NMR
SOURCE APEXBT

References
- “Takeda Submits New Drug Application for Alogliptin (SYR-322) in the U.S.” (Press release). Takeda Pharmaceutical Company. January 4, 2008. Retrieved January 9, 2008.
- Vipidia: EPAR summary for the public (European Medicines Agency)
- Feng, Jun; Zhang, Zhiyuan; Wallace, Michael B.; Stafford, Jeffrey A.; Kaldor, Stephen W.; Kassell, Daniel B.; Navre, Marc; Shi, Lihong; Skene, Robert J.; Asakawa, Tomoko; Takeuchi, Koji; Xu, Rongda; Webb, David R.; Gwaltney II, Stephen L. (2007). “Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV”. J. Med. Chem.50 (10): 2297–2300.doi:10.1021/jm070104l.PMID 17441705.
- “www.aace.com” (PDF).
- http://www.takeda.com/news/2013/20130618_5841.html
- Seino, Yutaka; Fujita, Tetsuya; Hiroi, Shinzo; Hirayama, Masashi; Kaku, Kohei (September 2011), “Efficacy and safety of alogliptin in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, dose-ranging comparison with placebo, followed by a long-term extension study (abstract only)”, Current Medical Research and Opinion 27 (9): 1781–1792,doi:10.1185/03007995.2011.599371,PMID 21806314, retrieved April 26,2012
- Kutoh, Eiji; Ukai, Yasuhiro (2012),“Alogliptin as an initial therapy in patients with newly diagnosed, drug naïve type 2 diabetes: a randomized, control trial (abstract only)”, Endocrine(January 17, 2012), doi:10.1007/s12020-012-9596-0, PMID 22249941, retrieved April 26, 2012
- Bosi, Emanuele; Ellis, G.C.; Wilson, C.A.; Fleck, P.R. (October 2011), “Alogliptin as a third oral antidiabetic drug in patients with type 2 diabetes and inadequate glycaemic control on metformin and pioglitazone: a 52-week, randomized, double-blind, active-controlled, parallel-group study”, Diabetes, Obesity and Metabolism (October 27, 2011) 13 (12): 1088–1096, doi:10.1111/j.1463-1326.2011.01463.x, retrieved April 26,2012
- White WB, Cannon CP, Heller SR et al. (October 2013). “Alogliptin after acute coronary syndrome in patients with type 2 diabetes”. N. Engl. J. Med. 369(14): 1327–35.doi:10.1056/NEJMoa1305889.PMID 23992602.
- White WB, Zannad F (January 2014). “Saxagliptin, alogliptin, and cardiovascular outcomes”. N. Engl. J. Med. 370 (5): 484.doi:10.1056/NEJMc1313880.PMID 24482824.
- Grogan, Kevin (April 26, 2012),“FDA wants yet more data on Takeda diabetes drug alogliptin”,PharmaTimes (PharmaTimes), PharmaTimes online, retrieved April 26,2012
- “GEN News Highlights: Takeda Pulls MAA for Type 2 Diabetes Therapy”. Genetic Engineering & Biotechnology News. June 4, 2009.
| EP1083172A1 * | May 26, 1998 | Mar 14, 2001 | Rimma Iliinichna Ashkinazi | N-substituted derivatives of 5-oxyiminobarbituric acid |
| US2598936 * | Apr 13, 1950 | Jun 3, 1952 | Searle & Co | Disubstituted cyanoalkanoylureas and thioureas and methods for their production |
| US6066641 * | Dec 12, 1995 | May 23, 2000 | Euro-Celtique S.A. | Aryl thioxanthines |
| US6248746 * | Jan 7, 1999 | Jun 19, 2001 | Euro-Celtique S.A. | 3-(arylalkyl) xanthines |
| US20080194593 * | Jan 11, 2008 | Aug 14, 2008 | Rao Kalla | A2b adenosine receptor antagonists |
| WO1994003456A1 * | Aug 5, 1993 | Feb 17, 1994 | Boehringer Ingelheim Kg | Asymmetrically substituted xanthine with adenosine-antagonistic properties |
| WO2001029010A1 * | Oct 18, 2000 | Apr 26, 2001 | Amjad Ali | Gram-positive selective antibacterial compounds, compositions containing such compounds and methods of treatment |
| WO2007035629A2 * | Sep 15, 2006 | Mar 29, 2007 | Takeda Pharmaceutical | Process for the preparation of pyrimidinedione derivatives |
| WO2007150011A2 * | Jun 22, 2007 | Dec 27, 2007 | Smithkline Beecham Corp | Prolyl hydroxylase inhibitors |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-({6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl}methyl)benzonitrile
|
|
| Clinical data | |
| Trade names | Nesina, Vipidia Kazano, Vipidomet (withmetformin) Oseni, Incresync (withpioglitazone) |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | 20% |
| Metabolism | Limited, hepatic (CYP2D6– and3A4-mediated) |
| Biological half-life | 12–21 hours |
| Excretion | Renal (major) and fecal (minor) |
| Identifiers | |
| CAS Registry Number | 850649-62-6 |
| ATC code | A10BH04 |
| PubChem | CID: 11450633 |
| IUPHAR/BPS | 6319 |
| ChemSpider | 9625485 |
| UNII | JHC049LO86 |
| KEGG | D06553 |
| ChEBI | CHEBI:72323 |
| ChEMBL | CHEMBL376359 |
| Synonyms | SYR-322 |
| Chemical data | |
| Formula | C18H21N5O2 |
| Molecular mass | 339.39 g/mol |
 Alogliptin benzoate
Alogliptin benzoate
| MF: | C18H21N5O2.C7H6O2 |
| MW: | 461.519 |
| Melting Point: | 185-188°C |
| Optical Rotation: | -56.3° (c=1, MeOH) |
Solubility:Soluble in MeOH; Insoluble in ACN
850649-62-6 CAS
Alogliptin
-
- Synonyms:SYR-322
- ATC:A10BH04
- Use:antidiabetic, DPP-4 inhibitor
- Chemical name:2-[[5-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-2H-pyrimidin-1(2H)-yl]methyl]benzonitrile
- Formula:C18H21N5O2
- MW:339.40 g/mol
- CAS-RN:850649-61-5
Derivatives
benzoate
- Formula:C19H19NO2
- MW:293.37 g/mol
- CAS-RN:850649-62-6
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 22115-41-9 | C8H6BrN | 2-(bromomethyl)benzonitril | |
| C12H8ClN3O2 | 2-[[6-chloro-3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl]methyl]benzonitrile | ||
| C13H10ClN3O2 | 2-[[6-chloro-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]benzonitrile | ||
| 4270-27-3 | C4H3ClN2O2 | 6-chloro-2,4(1H,3H)-pyrimidinedione | |
| 74-88-4 | CH3I | methyl iodide | Methane, iodo- |
| 127294-73-9 | C5H12N2 | (3R)-3-piperidinamine |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| J | Nesina | Takeda ,2010 |
Formulations
- tabl. 12.5 and 25 mg
References
-
- Feng, J. et al.: J. Med. Chem. (JMCMAR) 50, 2297-2300 (2007).
- WO 2 005 095 381 (SYRRX; 13.10.2005; appl. 15.12.2004; USA-prior. 15.3.2004).
- WO 2 010 109 468 (MAPI Pharma; 30.9.2010; appl. 25.3.2010; USA-prior. 26.3.2009).
-
solid preparation comprising Alogliptin and Pioglitazone:
- US 20 100 092 551 (Takeda Pharm.; 15.4.2010; appl. 30.1.2008; J-prior. 1.2.2007).
-
solid preparation comprising Alogliptin and Metformin:
- US 20 200 136 127 (Takeda Pharm.; 3.6.2010; appl. 16.7.2008; J-prior. 19.7.2007).
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

TRELAGLIPTIN
1 TRELAGLIPTIN

Trelagliptin succinate (SYR-472)
2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2, 4-dioxopyrimidin-1-yl]methyl]-4-fluorobenzonitrile; butanedioic acid
2-[6-[3(R)-Aminopiperidin-1-yl]-3-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-ylmethyl]-4-fluorobenzonitrile
2- [ [6- [ (3R) -3-amino-l-piperidinyl] -3, 4-dihydro-3- methyl-2, 4-dioxo-l (2H) -pyrimidinyl]methyl] -4-fluorobenzonitrile
succinic acid salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Sponsor/Developer: Takeda Pharmaceuticals and Furiex Pharmaceuticals
Mechanism of action: DPP-4 inhibitor
865759-25-7 cas FREE BASE
1029877-94-8 succinate
- SYR 111472 succinate
- SYR 472
- Syr-472
- Syr111472 succinate
- Trelagliptin succinate
- UNII-4118932Z90
- clinical trials….http://clinicaltrials.gov/search/intervention=SYR+472
Trelagliptin-succinate M. Wt: 475.47
Trelagliptin-succinate Formula: C22H26FN5O6
SYR-472 is an oral dipeptidyl peptidase IV inhibitor originated by Takeda. It is in phase III clinical trials for the treatment of type 2 diabetes.
- Diabetes affects 25.8 million people of all ages, or roughly 8.3 percent of the U.S. population.
- The World Health Organization predicts that there will be 366 million people worldwide affected by diabetes by the year 2030.
- The advent of trelagliptin succinate, a unique once weekly medication for patients with type 2 Diabetes is now the focus of clinical trials and exciting research and development.
- Phase III clinical trials of trelagliptin succinate commenced in September 2011, and are estimated to be complete by the second half of 2013.
TRELAGLIPTIN (SYR-472)
Trelagliptin is a novel DPP-4 inhibitor that is being developed by Takeda. In contrast to alogliplitin, which is once a day, trelagliptin is a once-weekly oral agent which should provide patients with a convenient therapeutic alternative and has the potential to improve compliance. Takeda has commenced Phase III trials of trelagliptin in Japan for the treatment of Type 2 diabetes.

Indication (Phase): Japan—Once-weekly oral treatment for type 2 diabetes (Phase III; study expected to be completed in second half of 2013)


Compound I, A, TRELAGLIPTIN which has the formula:

is a DPP-IV inhibitor that is described in U.S. patent application Ser. No. 11/080,992 filed Mar. 15, 2005 (see Compound 34). Its dosing, administration and biological activities are described in U.S. patent application Ser. No. 11/531,671 filed Sep. 13, 2006. U.S. patent application Ser. No. 11/080,992 and Ser. No. 11/531,671 are incorporated herein by reference in their entirety.
Dipeptidyl peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) (referred herein as “DPP-IV”) is a type II membrane protein and a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (e.g., intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of human disease states, including, but are not limit to, diabetes, particularly type II diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (IFG), metabolic acidosis, ketosis, appetite regulation and obesity; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; AIDS; and cancers.
DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV.
Compound (A) or a salt thereof has been reported as an inhibitor of dipeptidyl peptidase (DPP-IV) , which is an enzyme that decomposes glucagon-like peptide-1 (GLP-1) , a hormone increasing insulin secretion (patent document 1) .
In addition, a method including administering 1 – 250 mg of compound (A) or a salt thereof to a patient once per week (patent documents 2, 3), crystal polymorphs of compound (A) (patent documents 4, 5) , and a preparation of compound (A)
(patent documents 6, 7) have also been reported. Compound (A) and a salt thereof are recommended for oral administration in view of the easiness of self-administration, and a tablet, particularly a tablet in the dosage form for administration once per week, is desired. [0006]
The dosage form of once per week is expected to improve drug compliance of patients, whereas it requires supply of compound (A) or a salt thereof to patients in a high dose as compared to, for example, the dosage form of once per day. Since a solid preparation containing compound (A) or a salt thereof in a high dose increases its size, it may conversely degrade the drug compliance for patients, particularly infants and elderly patients having difficulty in swallowing
……………………..
SYNTHESIS
Compound 34 IS TRELAGLIPTIN

4-Fluoro-2-methylbenzonitrile (31).
A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32).
A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
Alternatively, 32 was made as follows.
4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AIBN (122 g) and heated to 75° C. A suspension of DBH (353 g) in DCE (500 mL) was added at 75° C. portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AIBN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10° C. over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20° C. over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (33).
A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows.
To a solution of 6-chloro-3-methyluracil (750 g) and N,N-diisopropylethylamine (998 mL) in NMP (3 L) was added (at <30° C. over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 60° C. for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5° C. for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2×2.25 L). The material was then dried in a vacuum oven at 40±5° C. for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
TFAsalt OF TRELAGLIPTIN
2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (34).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
FREE BASE OF TRELAGLIPTIN
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 60° C. until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 60° C. and the mixture was allowed to cool to <25° C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2×3.6 L). The filtrate was concentrated at 45° C. under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
HCL salt OF TRELAGLIPTIN
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0° C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35° C. over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 50° C. (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
Succinate salt OF TRELAGLIPTIN

The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purified water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 30° C. over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 60° C. until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57° C. After stirring at 60° C. for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5° C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2×4.2 L). The material was then dried in a vacuum oven at 70±5° C. (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(−)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
Methanesulfonate salt
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75° C. and filtered through #3 Whatman filter paper. The solution was heated back to 75° C. and a 1M solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20° C./hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
…………………………
FORMULATION
COMPD A IS TRELAGLIPTIN
Examples (Comparative Example IA)
Succinate of compound (A) (26.6 mg) was weighed in a glass bottle and used as Comparative Example IA. (Comparative Example 2A)
The succinate of compound (A) and microcrystalline cellulose were uniformly mixed in a mortar at a ratio of 1:10, and the mixture (226.6 mg) was weighed in a glass bottle and used as Comparative Example 2A. (Comparative Example 3A)
The succinate of compound (A) and corn starch were uniformly mixed in a mortar at a ratio of 1:5, and the mixture (126.6 mg) was weighed in a glass bottle and used as Comparative Example 3A. (Example IA) Succinate of compound (A) , mannitol and corn starch according to the formulation of Table IA were uniformly mixed in a fluid bed granulator (LAB-I, POWREX CORPORATION) , and the mixture was granulated by spraying an aqueous solution of dissolved hypromellose 2910, and dried therein. The obtained granules were passed through a sieve -(16M) to give milled granules. To the milled granules were added croscarmellose sodium, microcrystalline cellulose and magnesium stearate, and they were mixed in a bag to give granules for tableting. The granules were punched by a rotary tableting machine (Correct 19K, Kikusui Seisakusho, Ltd.) with a 6.5 mmφ punch to give a plain tablet weighting 121 mg. On the other hand, titanium oxide, yellow ferric oxide and talc were dispersed in a hypromellose 2910 aqueous solution to prepare a film coating liquid. The aforementioned coating liquid was sprayed onto the above-mentioned plain tablet in a film coating machine (Hicoater HCP-75, Freund Corporation), to give 2500 film- coated tablets containing 3.125 mg of compound (A) (free form) per tablet. Table IA

………………………..
POLYMORPHS AND SYNTHESIS
FORM A
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared. For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days. The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE).
[0091] Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR.
[0092] Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1.
Table 1. Characteristic XRPD Peaks (CuKa) of Form A


Characterization Data of Form A of Compound I

8. Amorphous Form
[0137] The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis.
Table A. Approximate Solubilities of Compound I
Compound I having the formula



POLYMORPH SCREEN
Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool;CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation
…………………………
SYNTHESIS
EXAMPLES
1. Preparation of 2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro- 2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile and pharmaceutically acceptable salts


4-Fluoro-2-methylbenzonitrile (3)
[0166] A mixture of 2-bromo-5fluorotoluene ( 2) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was re fluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 3 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H). 2-Bromomethyl-4-fluorobenzonitrile (4)
[0167] A mixture of 4-fluoro-2-methylbenzonitrile (3) (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification.1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro- benzonitrile (6)
[0168] A mixture of crude 3-methyl-6-chlorouracil (5) (0.6 g, 3.8 mmol), 2- Bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO
(10 mL) was stirred at 60 C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=12, 8.4Hz, IH), 7.26 (d, J- 4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for Ci3H9ClFN3O2, 293.68; found 293.68.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, TFA salt (1) (TFA salt of Compound I)

[0169] 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4- fluoro-benzonitrile (5) (300 mg, 1.0 mmol), (i?)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 0C for 2 hrs. The final compound was obtained as a TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, HCl salt

[0170] The TFA salt of Compound I was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0 C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
General procedure for the preparation of salts of Compound I.
[0171] The benzonitrile product may be isolated as the free base if desired, but preferably, the product may be further converted to a corresponding acid addition salt. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of Compound I were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. [0172] The isolation and/or purification steps of the intermediate compounds in the above described process may optionally be avoided if the intermediates from the reaction mixture are obtained as relatively pure compounds and the by-products or impurities of the reaction mixture do not interfere with the subsequent reaction steps. Where feasible, one or more isolation steps may be eliminated to provide shorter processing times, and the elimination of further processing may also afford higher overall reaction yields.
…………………..
TABLET
2. Exemplary formulations comprising succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Provided are examples of tablet formulations that may be used to administer succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (Succinate salt of Compound I) according to the present invention. It is noted that the formulations provided herein may be varied as is known in the art.
The exemplary tablet formulations are as follows:
| 12.5 mg of Compound I (weight of free base form) per tablet | ||||
| Core Tablet Formulation | ||||
| (1) | 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4- | 17.0 | mg | |
| dioxo-3,4-dihydro-2H-pyrimidin-1- | ||||
| ylmethyl]-4-fluoro-benzonitrile (succinate salt) | ||||
| (2) | Lactose Monohydrate, NF, Ph, Eur | 224.6 | mg | |
| (FOREMOST 316 FAST FLO) | ||||
| (3) | Microcrystalline Cellulose, NF, Ph, Eur | 120.1 | mg | |
| (AVICEL PH 102) | ||||
| (4) | Croscarmellose Sodium, NF, Ph, Eur | 32.0 | mg | |
| (AC-DO-SOL) | ||||
| (5) | Colloidal Silicon Dioxide, NF, Ph, Eur | 3.2 | mg | |
| (CAB-O-SIL M-5P) | ||||
| (6) | Magnesium Stearate, NF, Ph, Eur | 3.2 | mg | |
| (MALLINCKRODT, Non-bovine Hyqual) | ||||
| TOTAL | 400.0 | mg | ||
| (per tablet) | ||||
…………..
POLYMORPHS AND SYNTHESIS
EXAMPLES Example 1 Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)

Compound I may be prepared by the follow synthetic route (Scheme 1)

A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)

Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)

Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)

Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)

Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile

The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent.
SUCCINATE SALT OF TRELAGLIPTIN
1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.
……………
patents
1. US 2013172377
2. WO 2011013639
3. WO 2009099172
4.WO 2009099171
5. WO 2008114807
6.WO 2008114800
7. WO 2008033851
8. WO 2007074884
9WO 2007035629
patent document 1: US2005/0261271
patent document 2: US2007/0060530
patent document 3: US2008/0287476
patent document 4: US2008/0227798
patent document 5: US2008/0280931
patent document 6: WO2008/114800
patent document 7: WO2011/013639
| US7906523 * | Oct 30, 2007 | Mar 15, 2011 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605 * | Nov 29, 2007 | Dec 27, 2011 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8188275 * | Oct 30, 2007 | May 29, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8222411 * | Sep 15, 2006 | Jul 17, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US20090275750 * | Sep 15, 2006 | Nov 5, 2009 | Jun Feng | Dipeptidyl peptidase inhibitors |
| WO2013183784A1 | Jun 4, 2013 | Dec 12, 2013 | Takeda Pharmaceutical Company Limited | Solid preparation |
| US20080227798 * | Nov 29, 2007 | Sep 18, 2008 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US20120197018 * | Feb 15, 2012 | Aug 2, 2012 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2007033265A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033266A2 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033350A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetes |
| EP1586571A1 * | Dec 21, 2004 | Oct 19, 2005 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
Pemirolast

Pemirolast (INN) is a mast cell stabilizer used as an anti-allergic drug therapy. It is marketed under the tradenames Alegysal and Alamast.
9-methyl-3-(1H-tetrazol-5-yl)-4H-pyrido-[1, 2-a]-pyrimidin-4-one
It has also been studied for the treatment of asthma.

https://www.google.com/patents/US9006431
Pemirolast is an orally-active anti-allergic drug which is used in the treatment of conditions such as asthma, allergic rhinitis and conjunctivitis. See, for example, U.S. Pat. No. 4,122,274, European Patent Applications EP 316 174 and EP 1 285 921, Yanagihara et al, Japanese Journal of Pharmacology, 51, 93 (1989) and Drugs of Today, 28, 29 (1992). The drug is presently marketed in e.g. Japan as the potassium salt under the trademark ALEGYSAL™.
Commercial pemirolast potassium has the disadvantage that it is known to give rise to sharp plasma concentration peaks in humans (see, for example, Kinbara et al, “Plasma Level and Urinary Excretion of TBX in Humans”, Japanese Pharmacology & Therapeutics, 18(3) (1990), and “Antiallergic agent—ALEGYSAL tablet 5 mg—ALEGYSAL tablet 10 mg—ALEGYSAL dry syrup”, Pharmaceutical Interview Form (IF), Revised in October 2007 (7th version), Standard Commodity Classification No.: 87449). The latter document also reports that the potassium salt of pemirolast is hygroscopic, which is believed to give rise to chemical instability, and possesses a bitter taste.
U.S. Pat. No. 4,122,274 describes a process for the production of salts of pemirolast, including potassium salts and (at Example 14) a sodium salt. As described herein, this technique produces a sodium salt that is physically unstable. Sodium salts of pemirolast are also mentioned (but a synthesis thereof not described) in international patent applications WO 2008/074975 and WO 2008/075028.
COMPARATIVE EXAMPLE 5Recrystallisation of Pemirolast Sodium According to the Method of U.S. Pat. No. 4,122,274
In U.S. Pat. No. 4,122,274, it is stated that the crude title product (pemirolast sodium) was recrystallised from water:ethanol to give pure title product. It is not clear from this level of detail what the ratio of water:ethanol employed was, so several experiments were performed with a view to reproducing the prior art technique.
- (i) Crude sodium salt of pemirolast (480 mg; from Example 4, method (I) above) was recrystallised from water and ethanol (95%) in a 1:1 ratio. The Na salt of pemirolast (480 mg, 1.92 mmol) was dissolved in H2O (8 mL) at 70° C. and EtOH 95% (8 mL) was added. The clear solution was allowed to reach room temperature and the solid material formed was filtered off, washed with a small amount of ethanol and dried in vacuum to give 316 mg of pure sodium salt.
- (ii) Crude sodium salt of pemirolast (500 mg; from Example 4, method (II) above) was dissolved in water (4.9 mL) at 70° C. Thereafter EtOH 95% (ca. 4.0 mL) was added at 70° C. until a solid started to form. Another 0.1 mL of water was added to get everything into solution. The solid material formed upon cooling was collected by filtration and dried under vacuum to give 348 mg of pure sodium salt.
- (iii) Crude sodium salt of pemirolast (300 mg; from Example 4, method (II) above) was recrystallised from water:ethanol (1:1 ratio; 10 mL) at 70° C. The solid material formed upon cooling was collected by filtration and dried under vacuum to give 174 mg of pure sodium salt.
- (iv) Crude sodium salt of pemirolast (300 mg; from Example 4, method (II) above) was recrystallised from water:ethanol (9:1 ratio, 4 mL) at 70° C. The solid material formed upon cooling was collected by filtration and dried under vacuum to give 219 mg of pure sodium salt.
All four samples of pure pemirolast sodium salt had the same physico-chemical properties (Raman spectra and NMR):
1H NMR (D2O) δ: 8.86-8.80 (m, 1H, CH), 8.57 (s, 1H, CH), 7.68-7.59 (m, 1H, CH), 7.22-7.13 (m, 1H, CH), 2.39 (s, 3H, CH3).
The PXRD pattern (measured in respect of Example 5(i) above) is shown in FIG. 3. It was concluded from this that this form of the sodium salt is an amorphous material mixed with a crystalline fraction.
The Raman spectrum was recorded directly after recrystallisation. All samples were then stored under ambient conditions on a shelf in a fume hood. About a month later, a Raman spectrum was recorded, which was significantly different to that recorded earlier. This is shown in FIG. 4, where the lower spectrum accords to the earlier measurement and the upper spectrum accords to the later measurement. In the light of these results, it was concluded that the prior art amorphous form of pemirolast sodium is physically unstable.
The amorphous material was also prepared by drying of the form obtained in accordance with Example 11 below at 40° C. and reduced pressure for 40 hours to yield 12 g of a pale yellow cotton-like amorphous solid.
………………………..
http://www.lookchem.com/Chempedia/Chemical-Technology/Organic-Chemical-Technology/18815.html

1) Firstly, 2-Amino-3-methylpyridine (I) is condensed with ethoxymethylenemalonodinitrile (II) to afford the monocyclic intermediate (III), which is in tautomeric equilibrium with the pyridopyrimidine derivative (IV). Next, the reaction of (IV) with aluminum azide (AlCl3.NaN3) in refluxing THF yields 4-imino-9-methyl-3-(1H-tetrazol-5-yl)-4H-pyrido[1,2-a]pyrimidine (V). Finally, this compound is first hydrolyzed with 1N HCl and then treated with KOH.
2) Compound (IV) can be converted to the final product by a one-pot reaction: (VI) is treated first with NaN3 in refluxing acetic acid, then hydrolyzed with HCl and finally treated with KOH.
………….
EXAMPLE 1
A suspension of 9-methyl-3-(1 H-tetrazol-5-yl)-4H-pyrido-[1,2-a]-pyrimidin-4-one (68.5 g; 0.3 mols) in methanol (420 ml) and water (210 ml) heated at 50° C. is added with a 40% N-methylamine aqueous solution (30 ml, 0.35 mols) to pH=10. The solution is heated at 68-70° C., and acidified with formic acid (21 ml) to pH=3. After completion of the addition the mixture is kept at 68-70° C. for about 15 minutes and then cooled to 20-25° C. The precipitate is filtered, washed with methanol and dried under vacuum at 40° C. to give 9-methyl-3-(1 H-tetrazol-5-yl)-4H-pyrido-[1,2-a]-pyrimidin-4-one with >99.8% HPLC purity (63 g, 92% yield).
EXAMPLE 2
9-Methyl-3-(1 H-tetrazol-5-yl)-4H-pyrido-[1,2-a]-pyrimidin-4-one (63 g, 0.28 mols) is suspended in methanol (1000 ml). The resulting suspension is kept at 45° C. and slowly added with a 45% potassium hydroxide aqueous solution to pH 9-9.5. The suspension is stirred at 45° C. for about 15 minutes and then cooled to 20° C. The precipitate is filtered, washed with methanol and dried under vacuum at 80° C., to obtain Pemirolast Potassium (71.9 g; 0.27 mols, 96% yield) with HPLC purity >99.8%. 1H NMR(D2O, TMS) d (ppm): 2.02 (s, 3H); 6.83 (t, 1H); 7.22 (d, 1H); 8.18 (s, 1H); 8.47 (d, 1H).
References
- Tinkelman DG, Berkowitz RB (February 1991). “A pilot study of pemirolast in patients with seasonal allergic rhinitis”. Ann Allergy 66 (2): 162–5. PMID 1994787.
- Kawashima T, Iwamoto I, Nakagawa N, Tomioka H, Yoshida S (1994). “Inhibitory effect of pemirolast, a novel antiallergic drug, on leukotriene C4 and granule protein release from human eosinophils”. Int. Arch. Allergy Immunol. 103 (4): 405–9. doi:10.1159/000236662. PMID 8130655.
- Abelson MB, Berdy GJ, Mundorf T, Amdahl LD, Graves AL (October 2002). “Pemirolast potassium 0.1% ophthalmic solution is an effective treatment for allergic conjunctivitis: a pooled analysis of two prospective, randomized, double-masked, placebo-controlled, phase III studies”. J Ocul Pharmacol Ther 18 (5): 475–88. doi:10.1089/10807680260362759. PMID 12419098.
- Kemp JP, Bernstein IL, Bierman CW et al. (June 1992). “Pemirolast, a new oral nonbronchodilator drug for chronic asthma”. Ann Allergy 68 (6): 488–91. PMID 1610024.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
9-methyl-3-(1H-tetrazol-5-yl)-4H-pyrido[1,2-a]pyrimidin-4-one
|
|
| Clinical data | |
| Trade names | Alamast |
| AHFS/Drugs.com | monograph |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, ophthalmic |
| Identifiers | |
| CAS Registry Number | 69372-19-6 |
| ATC code | None |
| PubChem | CID: 57697 |
| IUPHAR/BPS | 7329 |
| DrugBank | DB00885 |
| ChemSpider | 51990 |
| UNII | 2C09NV773M |
| KEGG | D07476 |
| ChEMBL | CHEMBL1201198 |
| Chemical data | |
| Formula | C10H8N6O |
| Molecular mass | 228.21 g/mol |
| US4122274 * | May 25, 1977 | Oct 24, 1978 | Bristol-Myers Company | 3-Tetrazolo-5,6,7,8-substituted-pyrido[1,2-a]pyrimidin-4-ones |
| EP0316174A1 | Nov 10, 1988 | May 17, 1989 | Tokyo Tanabe Company Limited | Aqueous preparation of 9-methyl-3-(1H-tetrazol-5-yl)-4H-Pyrido[1,2-a]pyrimidin-4-one potassium salt |
| EP1285921A1 | Jun 25, 2002 | Feb 26, 2003 | Dinamite Dipharma S.p.A. | A process for the preparation of high purity pemirolast |
| JPH0374385A | Title not available | |||
| WO2008074975A1 | Nov 16, 2007 | Jun 26, 2008 | Cardoz Ab | New combination for use in the treatment of inflammatory disorders |
| WO2008075028A1 | Dec 18, 2007 | Jun 26, 2008 | Cardoz Ab | New combination for use in the treatment of inflammatory disorders |
| US4122274 | May 25, 1977 | Oct 24, 1978 | Bristol-Myers Company | 3-Tetrazolo-5,6,7,8-substituted-pyrido[1,2-a]pyrimidin-4-ones | |
| US5254688 * | Jun 19, 1991 | Oct 19, 1993 | Wako Pure Chemical Industries, Ltd. | Process for producing pyrido[1,2-a]pyrimidine derivative | |
| DE243821C | Title not available | ||||
| EP0462834A1 | Jun 20, 1991 | Dec 27, 1991 | Wako Pure Chemical Industries, Ltd | Process for producing pyrido [1,2-a]pyrimidine derivative | |
| WO1993025557A1 | Jun 7, 1993 | Dec 23, 1993 | Smithkline Beecham Plc | Process for the preparation of clavulanic acid |

Pemirolast Potassium (BMY 26517) cas100299-08-9is a histamine H1 antagonist and mast cell stabilizer that acts as an antiallergic agent.
Target: Histamine H1 Receptor
Pemirolast potassium (BMY 26517) is a new oral, nonbronchodilator antiallergy medication that is being evaluated for the therapy of asthma [1]. Pemirolast potassium (BMY 26517) inhibits chemical mediator release from tissue mast cells and is also shown to inhibit the release of peptides including substance P, Pemirolast potassium (BMY 26517) reduces kaolin intake by inhibition of substance P release in rats [2]. Pemirolast potently attenuates paclitaxel hypersensitivity reactions through inhibition of the release of sensory neuropeptides in rats [3]. Pemirolast potassium is used for the treatment of allergic conjunctivitis and prophylaxis for pulmonary hypersensitivity reactions to drugs such as paclitaxel [4].
Molecular formula: C10H7KN6O
Molecular Weight: 266.30
External links
- Mitsubishi Tanabe Pharma Corporation (2007). “ALEGYSAL (English)” (PDF). Retrieved 2008-09-02.
- “DailyMed Announcements”. U.S. National Library of Medicine. 2005. Retrieved 2008-09-02.
Moexipril


RS-10085-197
SPM-925
RS-10085 (free base)

Pharmacology
Moexipril is available as a prodrug moexipril hydrochloride, and is metabolized in the liver to form the pharmacologically active compound moexiprilat. Formation of moexiprilat is caused by hydrolysis of an ethyl ester group.[5] Moexipril is incompletely absorbed after oral administration, and its bioavailability is low.[6] The long pharmacokinetic half-life and persistent ACE inhibition of moexipril allows once-daily administration.[7]
Moexipril is highly lipophilic,[2] and is in the same hydrophobic range as quinapril, benazepril, and ramipril.[7] Lipophilic ACE inhibitors are able to penetrate membranes more readily, thus tissue ACE may be a target in addition to plasma ACE. A significant reduction in tissue ACE (lung, myocardium, aorta, and kidney) activity has been shown after moexipril use.[8]
It has additional PDE4-inhibiting effects.[9]
Side effects
Moexipril is generally well tolerated in elderly patients with hypertension.[10] Hypotension, dizziness, increased cough, diarrhea, flu syndrome, fatigue, and flushing have been found to affect less than 6% of patients who were prescribed moexipril.[3][10]
Mechanism of action
As an ACE inhibitor, moexipril causes a decrease in ACE. This blocks the conversion of angiotensin I to angiotensin II. Blockage of angiotensin II limits hypertension within the vasculature. Additionally, moexipril has been found to possess cardioprotective properties. Rats given moexipril one week prior to induction of myocardial infarction, displayed decreased infarct size.[11] The cardioprotective effects of ACE inhibitors are mediated through a combination of angiotensin II inhibition and bradykininproliferation.[8][12] Increased levels of bradykinin stimulate in the production of prostaglandin E2[13] and nitric oxide,[12] which cause vasodilation and continue to exert antiproliferative effects.[8] Inhibition of angiotensin II by moexipril decreases remodeling effects on the cardiovascular system. Indirectly, angiotensin II stimulates of the production of endothelin 1 and 3 (ET1, ET3)[14] and the transforming growth factor beta-1 (TGF-β1),[15] all of which have tissue proliferative effects that are blocked by the actions of moexipril. The antiproliferative effects of moexipril have also been demonstrated by in vitro studies where moexipril inhibits the estrogen-stimulated growth of neonatal cardiac fibroblasts in rats.[12] Other ACE inhibitors have also been found to produce these actions, as well.
WO 2014202659
http://www.google.com/patents/WO2014202659A1?cl=en
US4344949
http://www.google.co.in/patents/US4344949
References
- Hochadel, Maryanne, ed. (2006). The AARP Guide to Pills. Sterling Publishing Company. p. 640. ISBN 978-1-4027-1740-6. Retrieved2009-10-09.
- Belal, F.F, K.M. Metwaly, and S.M. Amer. “Development of Membrane Electrodes for the Specific Determination of Moexipril Hydrochloride in Dosage Forms and Biological Fluids.” Portugaliae Electrochimica Acta. 27.4 (2009): 463-475.
- Rodgers, Katie, Michael C Vinson, and Marvin W Davis. “Breakthroughs: New drug approvals of 1995 — part 1.” Advanstar Communications, Inc. 140.3 (1996): 84.
- Dart, Richard C. (2004). Medical toxicology. Lippincott Williams & Wilkins. p. 647. ISBN 978-0-7817-2845-4. Retrieved 2009-10-09.
- Kalasz, H, G. Petroianu, K. Tekes, I. Klebovich, K. Ludanyi, et al. “Metabolism of moexipril to moexiprilat: determination of in vitro metabolism using HPLC-ES-MS.” Medicinal Chemistry. 3 (2007): 101-106.
- Jump up^ Chrysant, George S, PK Nguyen. “Moexipril and left ventricular hypertrophy.” Vascular Health Risk Management. 3.1 (2007): 23-30.
- Cawello W, H. Boekens, J. Waitzinger, et al. “Moexipril shows a long duration of action related to an extended pharmacokinetic half-life and prolonged ACE-inhibition.” Int J Clin Pharmacol Ther. 40 (2002): 9-17.
- ^ Jump up to:a b c Chrysant, SG. “Vascular remodeling: the role of angiotensin-converting enzyme inhibitors.” American Heart Journal. 135.2 (1998): 21-30.
- Jump up^ Cameron, RT; Coleman, RG; Day, JP; Yalla, KC; Houslay, MD; Adams, DR; Shoichet, BK; Baillie, GS (May 2013). “Chemical informatics uncovers a new role for moexipril as a novel inhibitor of cAMP phosphodiesterase-4 (PDE4)”. Biochemical Pharmacology 85 (9): 1297–1305. doi:10.1016/j.bcp.2013.02.026. PMC 3625111. PMID 23473803.
- White, WB, and M Stimpel. “Long-term safety and efficacy of moexipril alone and in combination with hydrochlorothiazide in elderly patients with hypertension.” Journal of human hypertension. 9.11 (1995): 879-884.
- Rosendorff, C. “The Renin-Angiotensin System and Vascular Hypertrophy.” Journal of the American College of Cardiology. 28 (1996): 803-812.
- Hartman, J.C. “The role of bradykinin and nitric oxide in the cardioprotective action of ACE inhibitors.” The Annals of Thoracic Surgery. 60.3 (1995): 789-792.
- Jaiswal, N, DI Diz, MC Chappell, MC Khosia, CM Ferrario. “Stimulation of endothelial cell prostaglandin production by angiotensin peptides. Characterization of receptors.” Hypertension. 19.2 (1992): 49-55.
- Phillips, PA. “Interaction between endothelin and angiotensin II.” Clinical and Experimental Pharmacology and Physiology. 26.7. (1999): 517-518.
- Youn, TJ, HS Kim, BH Oh. “Ventricular remodeling and transforming growth factor-beta 1 mRNA expression after nontransmural myocardial infarction in rats: effects of angiotensin converting enzyme inhibition and angiotensin II type 1 receptor blockade.” Basic research in cardiology. 94.4 (1999): 246-253.
////////////
| Systematic (IUPAC) name | |
|---|---|
|
(3S)-2-[(2S)-2-{[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino}propanoyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
|
|
| Clinical data | |
| Trade names | Univasc |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a695018 |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 13-22% |
| Protein binding | 90% |
| Metabolism | Hepatic (active metabolite, moexiprilat) |
| Biological half-life | 1 hour; 2-9 hours (active metabolite) |
| Excretion | 50% (faeces), 13% (urine) |
| Identifiers | |
| CAS Registry Number | 103775-10-6 |
| ATC code | C09AA13 |
| PubChem | CID: 91270 |
| IUPHAR/BPS | 6571 |
| DrugBank | DB00691 |
| ChemSpider | 82418 |
| UNII | WT87C52TJZ |
| KEGG | D08225 |
| ChEMBL | CHEMBL1165 |
| Chemical data | |
| Formula | C27H34N2O7 |
| Molecular mass | 498.568 g/mol |
Aseptic Manufacturing Operation: Chinese Company Zhuhai United Laboratories does not comply with EU GMP
DRUG REGULATORY AFFAIRS INTERNATIONAL


While the focus of attention has been on Indian manufacturers during the last 2 years now also Chinese manufacturers are in the spot light. On 15 June 2015 the National Agency for Medicines and Medical Devices of Romania entered a GMP Non-Compliance Report for Zhuhai United Laboratories into EudraGMDP. Read more about the GMP deviations observed at Zhuhai United.

While the focus of attention has been on Indian manufacturers during the last 2 years now also Chinese manufacturers are again in the spot light. Just recently the EU found serious GMP deviations at an API manufacturer (Huzhou Sunflower Pharmaceuticals) and on 15 June 2015 the National Agency for Medicines and Medical Devices of Romania entered a GMP Non-Compliance Report for Zhuhai United Laboratories Co., LTD located at Sanzao Science &Technology Park, National Hi-Tech Zone, Zhuhai, Guangdong, 519040, China into EudraGMDP.
According to the report issued by the…
View original post 253 more words
Inna Ben-Anat, Global QbD Director of Teva Pharmaceuticals
DRUG REGULATORY AFFAIRS INTERNATIONAL

Meet Inna Ben-Anat, Global QbD Director of Teva Pharmaceuticals. Inna is a key thought leader in Quality by Design for generics.
https://www.linkedin.com/pub/inna-ben-anat/6/47a/670
![]() Ben-Anat, InnaASSOCIATE DIRECTOR, HEAD OF QDD STRATEGY | TEVA PHARMACEUTICALSAssociate Director, Head of QbD Strategy Chemical Engineer with a degree in Quality Assurance and Reliability (Technion-Israel Institute of Technology). QbD Strategy Leader at Teva (USA). Headed the implementation of a global QbD training programme. More than 12 years of pharmaceutical development experience.
Ben-Anat, InnaASSOCIATE DIRECTOR, HEAD OF QDD STRATEGY | TEVA PHARMACEUTICALSAssociate Director, Head of QbD Strategy Chemical Engineer with a degree in Quality Assurance and Reliability (Technion-Israel Institute of Technology). QbD Strategy Leader at Teva (USA). Headed the implementation of a global QbD training programme. More than 12 years of pharmaceutical development experience.

Inna Ben-Anat is a Quality by Design (QbD) Strategy Leader in Teva Pharmaceuticals USA. In this role, Inna has implemented global QbD training program, and is supporting R&D teams in developing Quality by Design strategies, optimizing formulations and processes and assisting develop product specifications. Additionally, Inna supports Process Engineering group with process optimization during scale-up and supports Operations in identification and resolution of any technical issues. Inna has extensive expertise in process development, design…
View original post 1,144 more words
Determining Criticality-Process Parameters and Quality Attributes
DRUG REGULATORY AFFAIRS INTERNATIONAL
Determining Criticality-Process Parameters and Quality Attributes Part I: Criticality as a Continuum
As the pharmaceutical industry tries to embrace the methodologies of quality by design (QbD) provided by the FDA’s process validation (PV) guidance (1) and International Conference on Harmonization (ICH) Q8/Q9/Q10 (2-4), many companies are challenged by the evolving concept of criticality as applied to quality attributes and process parameters. Historically, in biopharmaceutical development, criticality has been a frequently arbitrary categorization between important high-risk attributes or parameters and those that carry little or no risk. This binary designation was usually determined during early development for the purposes of regulatory filings, relying heavily on scientific judgment and limited laboratory studies.
|

With the most recent ICH and FDA guidances…
View original post 8,374 more words