
Home » Uncategorized (Page 84)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Flow Chemistry India 2016, 21-22 January 2016, Mumbai, India

Flow Chemistry India 2016Date: Thursday, 21 January 2016 – Friday, 22 January 2016
|
SELECTBIO INDIA
http://selectbiosciences.com/conferences/index.aspx?conf=FCINDIA16&se=india
Register…………..http://selectbiosciences.com/conferences/registration.aspx?conf=FCINDIA16&se=india

venue
Hotel Ramada Powai and Convention Centre, Mumbai, India


Professor & Research Chair, Nelson Mandela Metropolitan University |

Professor, Synthetic Organic Chemistry , The University of Tokyo |

Deputy Director and Chair, National Chemical Laboratory |

Professor, Eindhoven University of Technology |

Scientific Director, University of Lyon |

Chairman, Flow Chemistry Society
Professor, University of Warsaw |






Maninderjit Singh Ahluwalia
Overview
SELECTBIO INDIA is delighted to welcome you all at the 4th International Conference Flow Chemistry India 2016 to be held in Mumbai on January 21-22, 2016 under the auspices of the Flow Chemistry Society. The society aims to unite and represent those who are actively working on this rapidly developing field. This meeting is dedicated to the integration of flow chemistry into everyday practice throughout the world by delivering the latest knowledge and making it available for the entire chemistry community.
Society members save 25% on the registration fee and non-members will receive their first year’s membership included in the fee.
Running alongside the conference will be an exhibition covering the latest technological advances in the area of flow chemistry.
Who Should Attend
• Scientists, Chemists, Chemical Engineers and Researchers working in Pharmaceutical and Fine Chemicals Research and Development including Drug Discovery, Medicinal Chemistry and Chemical Process Development
• Scientists, Chemists and Chemical Engineers working in Pharmaceutical and Fine Chemical Bulk Manufacturing Units
• Corporate Management, Scientists, Managers responsible for development of Pharmaceutical and Fine Chemicals R & D and Manufacturing activities
• Scientists, Chemists & Engineers belonging to the fields of Inorganic, Organic, Medicinal, Natural Products, Analytical, High-throughput and Process Chemistry in the Academic research as well as in Applied research and development in the area of Agrochemical, Petrochemical and Fragrance industry
• Scientists working in or interested in applications of Flow Chemistry in Material science, Green chemistry, Nanotechnology, Biotechnology, Theoretical Chemistry, Information technology and Flow synthesis instruments including Engineering & Automation
Conference Package – Includes Registration, 2 Nights Accommodation, Dinner & Airport Transfers (Valid up to January 5, 2016 only)
Call for Posters
You can also present your research on a poster while attending the meeting. Submit an abstract for consideration now!
Poster Submission Deadline: 30 November 2015
Agenda Topics
- Advances in Micro & Continuous Flow Reactors, Systems & Processes
- Applications in Pharmaceutical Industry & API Synthesis
- Engineering Aspects of Flow Chemistry
- Flow Reactor – Choosing the Right One
- Photochemistry & Multistep Synthesis in Flow
- Quality Issue and QbD in Flow Chemistry
- Scale up – From Micro to Commercial Scale
- Yield Improvement, Cost Cutting and Waste Reduction in Flow Chemistry
Sponsorship and Exhibition Opportunities
http://selectbiosciences.com/conferences/index.aspx?conf=FCINDIA16&se=india

Workshop Tutor
Charlotte Wiles
CEO CHEMTRIX
A Workshop on “Flow Chemistry Demonstrations (Lab & Plant Scale) for Chemical and Pharmaceutical Industry-” will be held one day prior to the training course i.e. on 20th January, 2016 from 10:00 am – 05:00 pm in Mumbai. This workshop is supported by Process Intensification will be jointly conducted by :
Dr. Dinesh Kudav (Mumbai University); Dr. Charlotte Wiles (Chemtrix BV-Neth); Mr. Wouter Stam (Flowid, NV-Neth); Mr. Manjinder Singh (CIPLA & VP-FCS-India Chapter); Dr. Viktor Gyollai, (AM Technology-UK); Dr. Prashant Kini (UPL Ltd.); Mr. Kumar Oza (Pi & TCPL); Mr. Madhav Sapre (Pi & Sharon Bio); et al .
This workshop is specially designed to demonstrate application/capabilities of Flow Chemistry running “live” reactions in Continuous Flow Reactors. The reactions likely to be demonstrated using Flow Chemistry includes :• Nitration • Organometallic reaction• Oxidation • Bi-phasic reaction• Nano-Particle preparation in Flow• Biocatalytic Reaction with enhanced enzyme life.
This workshop is free for the registered delegates of Flow Chemistry India 2016 Conference and Continuous Flow Reactors Training Course.
You can visit Mumbai city
Taj hotel, mumbai
Gateway of india
 Food in mumbai
Food in mumbai
 mumbai skyline
mumbai skyline

The Bandra-Worli Sea Link is a cable-stayed bridge that connects central Mumbai with its western suburbs

get in if you can
The Mumbai Suburban Railway system carries more than 6.99 million commuters on a daily basis. It has the highest passenger densities of any urban railway …

Chhatrapati shivaji in mumbai india
British-victoria terminus


VADA PAV


SELECTBIO CONFERENCES PICS










/////////
WCK Series by Wockhardt for treating the bacterial infection

Which WCK is it, WCK-4873 , WCK-4086, WCK ? for treating the bacterial infection.
(Not sure) will arrive at correct one….keep watching………..

CAS 1627163-98-7
- C14 H16 N2 O4 . Na,

- 1,6-Diazabicyclo[3.2.1]octane-2-carboxylic acid, 7-oxo-6-(phenylmethoxy)-, sodium salt (1:1), (1R,2S,5R)-
- SODIUM (2S, 5R)-6-(BENZYLOXY)-7-OXO-1,6-DIAZABICYCLO[3.2.1]OCTANE-2-CARBOXYLATE
sodium (2S,5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate…..WO2014135929

Patent
http://www.google.com/patents/WO2015136473A1?cl=en

EXAMPLES
The following examples illustrate the embodiments of the invention that are presently best known. However, it is to be understood that the following are only exemplary or illustrative of the application of the principles of the present invention. Numerous modifications and alternative compositions, methods, and systems may be devised by those skilled in the art without departing from the spirit and scope of the present invention. The appended claims are intended to cover such modifications and arrangements. Thus, while the present invention has been described above with particularity, the following examples provide further detail in connection with what are presently deemed to be the most practical and preferred embodiments of the invention.
Example 1
Synthesis of sodium (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicvclor3.2.11octane-2- carboxylate
Step 1; Preparation of -Γl-Γ(feΓt-butyldimethylsilyl -oxymethyll-5-Γdimethyl(oxido -λ-4-sulfanylidenel-4-oxo-pentyll-carbamic acid tert-butyl ester (III):
To a suspension of trimethylsulfoxonium iodide (180.36 gm, 0.819 mol) in tetrahydrofuran (900 ml), sodium hydride (32.89 g, 0.819 mol, 60% in mineral oil) was charged in one portion at 30°C temperature. The reaction mixture was stirred for 15 minutes and then dropwise addition of dimethylsulphoxide (1.125 ml) was done over a period of 3 hours at room temperature to provide a white suspension. The white suspension was added to a pre-cooled a solution of 2-(feri-butyldimethylsilyl-oxymethyl)-5-oxo-pyrrolidine-l-carboxylic acid tert-buty\ ester (II) (225 g, 0.683 mol, prepared as per J. Org Chem.; 2011, 76, 5574 and WO2009067600) in tetrahydrofuran (675 ml) and triethylamine (123.48 ml, 0.887 mol) mixture at -13°C by maintaining the reaction mixture temperature below -10°C. The resulting suspension was stirred for additional 1 hour at -10°C. The reaction mixture was carefully quenched by addition of saturated aqueous ammonium chloride (1.0 L) at -10°C to 10°C. The reaction was extracted by adding ethyl acetate (1.5 L). The layers were separated and aqueous layer was re-extracted with ethyl acetate (500 ml x 3). The combined organic layer was washed successively with saturated aqueous sodium bicarbonate (1.0 L), water (2.0 L) followed by saturated aqueous sodium chloride solution (1.0 L). Organic layer was dried over sodium sulfate and evaporated under vacuum to provide 265 g of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-buty\ ester (III) as an yellow oily mass.
Analysis:
Mass: 422.3 (M+l); for Molecular weight: 421.68 and Molecular Formula: ![]()
1H NMR (CDC13): δ 4.77 (br d, 1H), 4.38 (br s, 1H), 3.58 (br s, 3H), 3.39 (s, 3H), 3.38 (s, 3H), 2.17-2.27 (m, 2H), 1.73-1.82 (m, 2H), 1.43 (s, 9H), 0.88 (s, 9H), 0.01 (s, 3H), 0.04 (s, 3H).
Step 2: Preparation of 5-r4-benzyloxyimino-l-(fert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyll-carbamic acid tert- butyl ester (IV):
To a suspension of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-butyl ester (III) (440.0 g, 1.045 mol) in tetrahydrofuran (6.6 L), O-benzhydroxylamine hydrochloride (200.0 g, 1.254 mol) was charged. The reaction mixture was heated to 50°C for 2.5 hours. The reaction mixture was filtered through pad of celite and filtrate was concentrated to provide a residue. The residue was dissolved in ethyl acetate (5.0 L) and washed successively with saturated aqueous sodium bicarbonate (1.5 L), water (1.5 L) and saturated aqueous sodium chloride (1.5 L). Organic layer was dried over sodium sulfate. Solvent was evaporated under vacuum to yield 463.0 g of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) as an oily mass.
Analysis:
Mass: 486.1 (M+l); for Molecular weight: 485.4 and Molecular Formula: ![]()
1H NMR (CDCI3): δ 7.26-1 6 (m, 5H), 5.10 (s, 2H), 4.66 (br d, 1H), 3.58-4.27 (m, 2H), 3.56-3.58 (m, 3H), 2.40-2.57 (m, 2H), 1.68-1.89 (m, 2H), 1.44 (s, 9H), 0.89 (s, 9H), 0.02 (s, 3H), 0.04 (s, 3H).
Step 3: Preparation of 5-5-benzyloxyimino-2-(fert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V):
To a solution of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) (463.0 g 0.954 mol) in tetrahydrofuran (6.9 L), was charged potassium feri-butoxide (139.2 g, 1.241 mol) in portions over a period of 30 minutes by maintaining temperature -10°C. The resulting suspension was stirred for additional 1.5 hours at -10°C to -5°C. The reaction mixture was quenched by addition of saturated aqueous ammonium chloride (2.0 L) at -5°C to 10°C. The organic layer was separated and aqueous layer was extracted with ethyl acetate (1.0 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride solution (2.0 L). Organic layer was dried over sodium sulfate, and then evaporated under vacuum to yield 394.0 g of 5-5-benzyloxyimino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-butyl ester (V) as an yellow oily mass.
Analysis:
Mass: 449.4 (M+l) for Molecular weight: 448.68 and Molecular Formula: C24H4oN204Si;
1H NMR (CDC13): δ 7.25-1 3 (m, 5H), 5.04-5.14 (m, 2H), 4.35 (br s, 1H), 3.95 (br s, 1H), 3.63-3.74 (br d, 2H), 3.60-3.63 (m, 1H), 2.70-2.77 (m, 1H), 2.33-2.41 (m, 1H), 1.79-1.95 (m, 2H), 1.44 (s, 9H), 0.88 (s, 9H), 0.03 (s, 3H), 0.04 (s, 3H).
Step 4: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI):
To a solution of 5-5-benzyloxyimino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V) (394.0 g, 0.879 mol) in dichloromethane (5.0 L) and glacial acetic acid (788 ml), was charged sodium cyanoborohydride (70.88 g, 1.14 mol) one portion. The resulting reaction mixture was stirred at temperature of about 25 °C to 30°C for 2 hours. The mixture was quenched with adding aqueous solution of sodium bicarbonate (1.3 kg) in water (5.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (2.0 L). The combined organic layer washed successively with water (2.0 L), saturated aqueous
sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to provide a residue. The residue was purified by silica gel column chromatography to yield 208 g of (25,5i?5)-5-benzyloxyamino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-buty\ ester (VI) as pale yellow liquid.
Analysis:
Mass: 451.4 (M+l); for Molecular weight: 450.70 and Molecular Formula: C24H42N204Si;
1H NMR (CDC13): δ 7..26-7.36 (m, 5H), 4.90-5.50 (br s, 1H), 4.70 (dd, 2H), 4.09-4.25 (m, 2H), 3.56-3.72 (m, 2H), 2.55-3.14 (m, 2H), 1.21-1.94 (m, 4H), 1.45 (s, 9H), 0.89 (s, 9H), 0.05 (s, 6H).
Step 5: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine (VII):
To a solution of 5-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI) (208 g, 0.462 mol) in dichloromethane (3.0 L), boron trifluoride diethyletherate complex (114.15 ml, 0.924 mol) was charged in one portion. The resulting reaction mixture was stirred at temperature of about 25°C to 35°C temperature for 2 hours. The reaction mixture was quenched with saturated aqueous sodium bicarbonate (2.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride (1.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to yield 159 g of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) as a yellowish syrup.
Analysis:
Mass: 351.3 (M+l); for Molecular weight: 350.58 and Molecular Formula: C19H34N202Si.
Step-6: Preparation of (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-r3.2.11octane (VIII):
Part 1; Preparation of (2S,5RS)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
To a solution of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) (159.0 g, 0.454 mol) in a mixture of acetonitrile (2.38 L) and diisopropylethylamine (316.5 ml, 1.81 mol) was added triphosgene (59.27 gm, 0.199 mol) dissolved in acetonitrile (760 ml) at -15°C over 30 minutes under stirring. The resulting reaction mixture was stirred for additional 1 hour at -10°C. The reaction mixture was quenched by addition of saturated aqueous sodium bicarbonate (2.0 L) at -5°C to 10°C. Acetonitrile was evaporated from the reaction mixture under vacuum and to the left over aqueous phase, dichloromethane (2.5 L) was added. The organic layer was separated and aqueous layer extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed successively with water (2.0 L), saturated aqueous sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum and the residue was passed through a silica gel bed to yield 83.0 g of diastereomeric mixture (25, 5i?5)-6-benzyloxy-2-(feri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio as a yellow liquid.
Part-2: Separation of diastereomers to prepare (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
A mixture of diastereomers (2S,5Z?S)-6-benzyloxy-2-(teri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio (47.0 gm, 0.125 mol), was dissolved in n-hexane (141 ml) and stirred at temperature of about 10°C to 15°C for 1 hour. Precipitated solid was filtered and washed with n-hexane (47 ml) to provide 12.0 g of diastereomerically pure (25,5i?)-6-benzyloxy-2-(tert-butyl-dimethylsilyl-oxymethyl)-7-oxo- 1,6-diaza-bicyclo-[3.2.1] octane (VIII) as a white crystalline material.
Analysis:
Mass: 377.3 (M+l); for Molecular weight: 376.58 and Molecular Formula: ![]()
1H NMR (CDCI3): δ Ί -Ί.ΑΑ (m, 5H), 4.95 (dd, 2H), 3.76-3.85 (ddd, 2H), 3.37-3.40 (m, 1H), 3.28-3.31 (m, 2H), 2.89 (brd, 1H), 1.90-2.02 (m, 2H), 1.62- 1.74 (m, 2H), 1.56 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
Diastereomeric purity as determined by HPLC: 99.85%
Step-7: Preparation of (25,5R)-6-benzyloxy-2-hvdroxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane (IX):
To a solution of (25,5i?)-6-benzyloxy-2-(ieri-butyl-dimethylsilyl-oxymethyl)-7-oxo- l,6-diaza-bicyclo-[3.2.1]octane (VIII) ( 12.0 g, 31.9 rnmol) in tetrahydrofuran (180 ml) was charged tetra 7? -butyl ammonium fluoride (38.0 ml, 38 mmol, 1 M in tetrahydrofuran) at room temperature. The reaction mixture was stirred for 2 hours. It was quenched with saturated aqueous ammonium chloride ( 100 ml). The organic layer was separated and aqueous layer extracted with dichloromethane (150 ml x 3). The combined organic layer was washed with saturated aqueous sodium chloride (150 ml), dried over sodium sulfate and evaporated under vacuum to yield 24.0 g of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l ,6-diaza-bicyclo-[3.2.1]octane (IX) as a yellow liquid. The compound of Formula (IX) was purified by silica gel (60-120 mesh) column chromatography using a mixture of ethyl acetate and hexane as an eluent.
Analysis:
Mass: 263.1 (M+l); for Molecular weight: 262.31 and Molecular Formula: C14H18N203
1H NMR (CDCb): δ 7.34-7.42 (m, 5H), 4.95 (dd, 2H), 3.67-3.73 (m, 1H), 3.53-3.60 (m, 2H), 3.32-3.34 (m, 1H), 2.88-3.01 (m, 2H), 2.09 (brs, 1H), 1.57-2.03 (m, 2H), 1.53- 1.57 (m, 1H), 1.37- 1.40 (m, 1H).
Step 8: Preparation of sodium salt of (25, 5R)-6-benzyloxy-7-oxo-l,6-diaza-bicvclor3.2.11-octane-2-carboxylic acid (I):
Step I:
Compound of Formula (IX) obtained in step 8 above was used without any further purification. To the clear solution of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane (IX) (24.0 g, 31.8 mmol) (quantities added based upon theoretical basis i.e 8.3 g ) in dichloromethane (160 ml), was added Dess-Martin reagent (24.1 g, 57.24 mmol) in portions over 15 minutes. The resulting suspension was stirred for 2 hours at 25°C. The reaction was quenched by adding a solution, prepared from saturated aqueous sodium hydrogen carbonate solution (160 ml) and 72.0 g of sodium thiosulfate. Diethyl ether (160 ml) was added to the reaction mixture and it was stirred for 5-10 minutes and filtered through celite. Biphasic layer from filtrate was separated. Organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (160 ml) followed by saturated aqueous sodium chloride solution (160 ml). Organic layer was dried over sodium sulfate and evaporated to dryness at 30°C to obtain 20.0 g of intermediate aldehyde, which was used immediately for the next reaction.
Step II:
To the crude intermediate aldehyde (20.0 g, 31.6 mmol) (quantities added based upon theoretical yield i.e. 8.2 g) obtained as above, was charged i-butyl alcohol (160 ml) and cyclohexene (10.8 ml, 110.6 mmol). The reaction mixture was cooled to temperature of about 10°C to 15°C. To this mixture was added clear solution prepared from sodium hypophosphate (14.8 g, 94.8 mmol) and sodium chlorite (5.7 g, 63.2 mmol) in water (82.0 ml) over a period of 30 minutes by maintaining temperature between 10°C to 15°C. The reaction mixture was further stirred for 1 hour and was quenched with saturated aqueous ammonium chloride solution. The reaction mixture was subjected to evaporation under vacuum at 40°C to remove i-butyl alcohol. Resulting mixture was extracted with dichloromethane (3 x 150 ml). Layers were separated. Combined organic layer was washed with aqueous brine solution, dried over sodium sulfate and evaporated to dryness under vacuum to obtain 16.0 g of crude residue. To this residue was added acetone (83 ml) to provide a clear solution and to it was added dropwise a solution of sodium 2-ethyl hexanoate (4.5 g) in acetone (24 ml). The reaction mixture was stirred for 15 hours at 25°C to 30°C to provide a suspension. To the suspension was added diethyl ether (215 ml) and stirred for 30 minutes. Resulting solid was filtered over suction, and wet cake was washed with cold acetone (83 ml) followed by diethyl ether (83 ml). The solid was dried under vacuum at 40°C to provide 3.6 g of off-white colored, non-hygroscopic sodium salt of (25, 5i?)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]-octane-2-carboxylic acid (I).
Analysis:
Mass: 275.2 as M-1 (for free acid) for Molecular Weight: 298 and Molecular Formula: ![]()
NMR (DMSO-d6): δ 7.43-7.32 (m, 5H), 4.88 (q, 2H), 3.48 (s, IH), 3.21 (d, IH), 2.73 (d, IH), 2.04-2.09 (m, IH), 1.77-1.74 (m, IH), 1.65-1.72 (m, IH), 1.55-1.59 (m, IH);
Purity as determined by HPLC: 97.47%;
[a]D25: -42.34° (c 0.5, water).


Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.

/////////
New molecules from Wochkardt to treat bacterial infections

WCK ?
( Not sure) Keep watching this post………..
TRANS-SULFURIC ACID MONO-{2-[5-(3-AZETIDINYLAMINO)-METHYL-[1,3,4]- OXADIAZOL-2-YL]-7-OXO-1,6-DIAZABICYCLO[3.2.1] OCT-6-YL} ESTER TRIFLUOROACETATE
trans-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[1,3,4]- oxadiazol-2-yl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl}ester trifluoroacetate
(25,5R)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[ l,3,4]-oxadiazol-2-yl)-7-oxo- l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester
2-(1 ,3,4-OXADIAZOL-2-YL)-7-OXO-1 ,6-DIAZABICYCLO[3.2.1 ]OCTANE DER
(25,5R)-Sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[i,3,41-oxadiazol-2-yl)-7-oxo-l,6-diaza- bicvclo[3.2.11 oct-6-yll ester
PCT International Patent Application No. PCT/US2013/034562.
Indian Patent Application No. 1635/MUM/2014
Molecular Weight: 488.3 and Molecular Formula:![]()

![]()
Scheme 1. Typically, compound of Formula (I) is prepared from sodium salt of 6-benzyloxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylic acid (III).
The sodium salt of 6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylic acid
(III) is reacted with 3-(ier^butoxycarbonyl-hydrazinocarbonylmethyl-amino)-azetidine-1-carbamic acid tert-buty\ ester (II) in presence of coupling agent at a temperature ranging from -15°C to 60°C for about 1 hour to about 24 hours to provide an intermediate compound of Formula (IV). Typical, non-limiting examples of coupling agent include EDC hydrochloride, dicyclohexylcarbodiimide, diisopropylcarbodiimide (DIC), (benzotriazol-l-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), O-(benzotriazol- 1 -yl)-N,N,N’ ,Ν’ -tetramethyluroniumhexafluorophosphate (HBTU), O-(benzotriazol-l-yl)- Ν,Ν,Ν’,Ν’-tetramethyluroniumtetrafluoroborate (TBTU), 0-(7-azabenzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU), O-(6-ahlorobenzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HCTU), 0-(3,4-dihydro-4-oxo-l,2,3-benzotriazine-3-yl)-N,N,N’,N’-tetramethyl uronium tetrafluoroborate(TDBTU), 3-(diethylphosphoryloxy)- 1 ,2,3-benzotriazin-4(3H)-one (DEPBT), carbonyldiimidazole (CDI), pivalyl chloride, HOBt and the like. In some embodiments, compound of Formula (II) is reacted with a compound of Formula (III) in presence of EDC hydrochloride and HOBt at a temperature of about 25°C to about 35°C for about 15 hours to provide an intermediate compound of Formula (IV). In some embodiments, a compound of Formula (II) is reacted with a compound of Formula (III) in presence of suitable solvent such as dimethylformamide, water or a mixture thereof.
The compound of Formula (IV) is cyclized to provide a compound of Formula (V). The cyclization of a compound of Formula (IV) is effected by treating with a reagent such as p-toluene sulfonyl chloride, p-nitrobenzene sulfonyl chloride, methane sulfonyl chloride or triphenylphosphine in a suitable solvent such as toluene, chloroform, dichloromethane, or N,N-dimethyl formamide at a temperature ranging from about -10° C to about 70°C for about 15 minutes to about 4 hours to provide 1,3,4-oxadiazole intermediate compound of Formula (V). In some embodiments, a compound of Formula
(IV) is cyclized in presence of triphenylphosphine, iodine and triethylamine, at a temperature of about -10°C to about 0°C for about 30 minutes to provide a compound of Formula (V). In some embodiments, compound of Formula (IV) is cyclized to a compound of Formula (V) in presence of dichloromethane as solvent.

Sulfonation

Scheme 1
Example 1
Synthesis of traras-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]- oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl]ester trifluoroacetate (I)
Step 1; Preparation of traras-{3-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane-2-carbonyl)-hydrazinocarbonyl]-2-oxo-ethyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (IV):
A solution of 3-(ier^butoxycarbonyl-hydrazinocarbonylmethyl-amino)-azetidine-1-carbamic acid tert-butyl ester (II) (2.8 g, 0.008 mol) in dimethylformamide (7 ml) was added to a stirred solution of sodium salt of 6-benzyloxy-7-bicyclo [3.2.1] octane-2-carboxylic acid (III) (2.43 g 0.008 mol) in water (41 ml). To this EDC.HCl (2.32 g, 0.012 mol) and HOBt (1.09 g, 0.008 mol) was added and stirred for 15 hours. Dichloro methane (50 ml) was added and layers were separated. Organic layer was dried over sodium sulfate and concentrated. The residue (6.1 gm) was purified by silica gel column chromatography using mixture of acetone and hexane as eluent to afford 3.4 g of ir ns-3-({2-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazino]-2-oxo-ethyl}-teri-butoxy carbonyl-amino)-azetidine-l -carboxylic acid tert-butyl ester (IV) in 70% yield.
Analysis:
Mass: 603.3 (M+l); for Molecular Weight: 602.6; Molecular Formula: ![]()
1H NMR (400 MHz, CDC13): δ 8.45. (bs, IH), 8.20 (bs, IH) 7.38-7.45 (m, 5H), 5.04 (d, IH), 4.91 (d, IH), 4.13 (m, 2H), 3.97-4.04 (m, 5H), 3.30 (s, IH), 3.07 (s, 2H), 2.91 (d, IH), 2.31 (m, IH), 2.20 (d, IH), 1.93-2.00 (m, 2H), 1.45 (s, 18H).
Step 2: Preparation of tr «s-{2-[5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-2-oxo-ethyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (V):
Triethyl amine (3.6 ml, 0.026 mol) was added to a cooled (0 °C) solution of iodine (1.62 gm, 0.0063 mol) and triphenylphosphine (1.67 g, 0.0063 mol) in dichloromethane (64 ml). After stirring for 15 minutes a solution of 3-({2-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazino]-2-oxo-ethyl}-fert-butoxycarbonyl- amino)-azetidine-l-carboxylic acid tert-butyl ester (IV) (3.2 g, 0.0053 mol) in dichloromethane (16 ml) was added. Reaction mixture was stirred at -10°C to 0°C for another 30 minutes. Dichloromethane was concentrated and ethyl acetate (35 ml) was added; stirred and filtered to remove triphenylphosphine oxide. Filtrate was concentrated and purified by silica gel column chromatography using a mixture of methanol and chloroform as eluent to obtain 4.5 g of 3-{ [5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4] oxadiazol-2-yl-methyl]-tert-butoxycarbonyl-amino}-azetidine- 1 -carboxylic acid tert-buty\ ester (V).
Analysis:
Mass: 585.4 (M+l); for Molecular Weight: 584.6 and Molecular Formula: ![]()
1H NMR (400 MHz, CDC13): δ 7.64-7.68 (m, 6H), 7.52-7.56 (m, 3H) 7.42-7.48 (m, 7H), 7.36-7.38 (m, 2H), 5.07 (d, IH), 4.92 (d, 2H), 4.72 (s, IH), 4.68 (s, 2H), 4.15 (s, 2H), 4.01 (s, 2H), 3.36 (s, IH), 2.91 (d, IH), 2.79 (d, IH), 2.27-2.30 (m, 2H), 2.11-2.14 (m, IH), 1.97-1.99 (m, IH), 1.42 (s, 18H).
Step 3: Preparation of tr «s-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (VI):
Palladium on carbon (10%) was added to a stirred solution of 3-{ [5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl-methyl]-feri-butoxy carbonyl-amino}-azetidine-l -carboxylic acid tert-butyl ester (V) (4.5 g) in methanol (45 ml). Resulting suspension was stirred under hydrogen gas pressure of about 50 psi for 15 hours at 25°C. The reaction mixture was filtered through celite bed and washed using additional methanol (5 ml). The filtrate was concentrated to obtain 3.5 g of ir ns-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-teri-butoxy carbonyl-amino)-azetidine-l -carboxylic acid tert-butyl ester (VI) in 92% yield.
Analysis:
Mass: 495.4 (M+l); for Molecualr Weight: 494.5 and Molecular Formula: ![]()
1H NMR (400 MHz, DMSO): δ 9.86 (s, 1H), 7.51-7.62 (m, 12H), 4.70 (s, 2H), 4.58 (d, 1H), 3.99 (d, 2H), 3.65 (s, 2H), 2.92 (d, 1H), 2.67 (d, 1H), 2.31 (s, 1H), 2.00-2.11 (m, 2H), 1.84 (m, 1H), 1.31 (s, 18H).
Step-4: Preparation of traras-tetrabutyl ammonium salt-methyl-{2-[5-(7-oxo-6-sulphooxy-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-tert-butoxycarbonyl-amino )-azetidine-l-carboxylic acid fert-butyl ester (VII):
Sulfur trioxide-pyridine complex (3.17 g, 0.019 mol) and triethyl amine (4.5 ml, 0.033 mol) was added to a stirred solution of ir ns- {2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-butyl ester (VI) (2.62 g, 0.0066 mol) in dichloromethane (20 ml). The reaction mixture was stirred for 2 hours. Aqueous solution of 0.5 N potassium dihydrogen phosphate (50 ml) followed by ethyl acetate (40 ml) was added, stirred for 10 minutes and aqueous layer was separated. Aqueous layer was again extracted with the mixture of dichloromethane (10 ml) and ethyl acetate (20 ml). Combined organic layers were concentrated. The residue was dissolved in water (50 ml), washed with diethyl ether (2 x 25 ml) to remove triphenylphosphine oxide (a side product carried from the step-2) and extracted with dichloromethane (2 x25 ml). Dichloromethane was dried over sodium sulfate and concentrated to give 2.7 g of residue (87%). This residue was again dissolved in dichloromethane (50 ml) followed by addition of triethylamine (5.70 ml, 0.042 mol). Tetrabutylammonium hydrogen sulphate (1.27 g, 0.0037 mol) was added and stirred for 2 hours. Water (30 ml) was added to the reaction mixture and layers were separated. Dichloromethane layer was dried on sodium sulfate and solvent was concentrated under vacuum. The residue (2.7 g) was purified by silica gel column chromatography using methanol and chloroform as eluent to get 2.1 g of irans-tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy- 1 ,6-diaza-
bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-buty\ ester (VII) in 48% yield.
Analysis:
Mass: 575.4 (M+l) as free sulfonic acid; for Molecular Weight: 816.6 and Molecular Formula: C22H34N6O10S. Ci6H36N;
1H NMR (400 MHz, CDC13): δ 4.63-4.69 (m, 5H), 4.40 (s, 2H), 4.16 (s, 2H), 4.02 (s, 2H), 3.28-3.32 (m, 12H), 3.23 (s, 1H), 2.84 (d, 1H), 2.24-2.32 (m, 2H), 2.02-2.04 (m, 1H), 1.63-1.71 (m, 12H), 1.46-1.56 (m, 12H), 1.44 (s, 18H), 0.99-1.02 (m, 18H).
Step 5: Preparation of traras-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl]ester trifluoroacetate (I)
irans-Tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy- 1 ,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-butyl ester (VII) (2.1 g, 0.003 mol) was cooled to 0°C and to this was added trifluoro acetic acid cooled at 0°C in 15 minutes and the reaction mixture was stirred for 3 hours. The obtained reaction mixture was concentrated under high vacuum. Diethyl ether (20 ml) was added and solid precipitated was stirred and diethyl ether was decanted. This treatment was repeated twice. Solid separated was dried and dichloromethane (20 ml) was added and stirred; solid was allowed to settle and dichloromethane was decanted. Again this treatment was repeated twice and the solid was dried to get 1 g of irans-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo [3.2.1]oct-6-yl]ester trifluoroacetate (I) in 76% yield.
Analysis:
Mass: 375.2 (M+l) as free sulfonic acid; for Molecular Weight: 488.3 and Molecular Formula:![]()
CF3COOH;
1H NMR (400 MHz, DMSO): δ 4.64 (d, IH), 4.06 (s, 3H), 3.92 (s, 2H), 3.81-3.86 (m, IH), 3.73 (s, 2H), 2.94-2.97 (d, IH), 2.70 (d, IH), 2.16 -2.19 (m, IH), 1.88-2.14 (m, 2H), 1.86-1.88 (m, IH);
19F NMR (DMSO-d6): δ -74.41 (CF3COOH);
1 C NMR (DMSO-de as a TFA salt): δ 165.4, 165.1, 164.9, 159.2-158.2 (TFA-C), 57.7, 52.6 (2C), 52.3, 49.3, 46.1, 40.4, 20.1, 19.7.
PATENT
WO2015110963


Example-1
(25,5R)-Sulfuric acid mono-r2-(5-azetidin-3-ylmethyl-ri,3,41-oxadiazol-2-yl)-7-oxo-l,6-diaza- bicvclor3.2.11 oct-6-yll ester:

Step-1: Preparation of (25,5R)-2-{N’-[2-(5)-iV-tert-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazino carbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of sodium (2S, 5R)-7-oxo-6-benzyloxy-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (8.45 g, 28.3 mmol) (prepared according to the process disclosed in PCT/IB2013/059264) in water (100 ml) was added 3-(N-feri-butoxycarbonyl-azetidin-3-yl)-acetic acid hydrazide (5.9 g, 25.7 mmol), EDC hydrochloride (7.47 g, 38.6 mmol) and N-hydroxybenzotriazole (3.47 g, 25.7 mmol) at 25°C to 35°C under stirring. The reaction mixture was stirred for 18 hours. Precipitated solid was filtered under suction and washed with water (100 ml). It was dried to provide 10.01 g of (25,5R)-2-{N’-[2-(S)-N-fert-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazinocarbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane in 80% yield.
Analysis:
Mass: 486.4 (M-l), for Molecular Formula of C24H33N5O6;
Purity as determined by HPLC: 89.90%.
Step-2: Preparation of (25,5R)-2-(5-(/V-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of (25,5i?)-2-{N’-[2-(5)-N-ieri-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazinocarbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane (4 gm, 8.21 mmol) in chloroform (70 ml) was added p-toluenesulfonylchloride (2.34 gm, 12.3 mmol) followed by dnsopropylethylamine (4.4 ml, 24.6 mmol). The reaction mixture was heated under stirring at 75°C for 18 hours. The reaction mixture was concentrated under vacuum and the resulting mass was purified by using silica gel column chromatography, to provide (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane in 3.3 g quantity in 86% yield as a solid.
Analysis:
Mass: 470.4 (M+l), for Molecular Formula of C^HsiNsOs;
1H NMR: (CDCb): δ 7.36-7.44 (m, 5H), 5.08 (d, 1H), 4.93 (d, 1H), 4.68-4.71 (m, 1H), 4.10-4.15 (m, 2H), 3.68-3-72 (m, 2H), 3.37 (s, 1H), 3.13-3.15 (m, 2H), 2.90-3.11 (m, 2H), 2.77 (d, 1H), 2.25-2.31 (m, 2H), 2.10-2.19 (m, 1H), 1.87- 1.97 (m, 1H), 1.43 (s, 9H).
Step-3: Preparation of (25,5R)-2-(5-(iV-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To the solution of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane ( 3.3 g, 7.0 rnmol) in methanol (35 ml) was subjected to catalytic hydrogenolysis using 10% palladium on charcoal (350 mg) under atmospheric hydrogen gas pressure at 25°C to 35°C for 2 hours. The reaction mixture was filtered through celite bed and was washed with methanol (30 ml). The filtrate was concentrated under vacuum below 35°C to provide 2.7 g of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-hydroxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane, which was used immediately for the next reaction.
Analysis:
Mass: 378.4 (M-l), for Molecular Formula of CnH^NsOs.
Step-4: Preparation of tetrabutylammonium salt of (2S,5R)-2-(5-(V-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)- 6- hydroxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane (2.7 gm, 7.12 mmol) in dichloromethane (50 ml) was added triethylamine (5 ml, 35 mmol) followed by sulfur trioxide pyridine complex (2.26 g 14.2 mmol) under stirring at 25°C to 35°C. The reaction mixture was stirred for 2 hours. To the reaction mixture was added aqueous 0.5 N potassium dihydrogen phosphate solution (100 ml). It was stirred for about 30 minutes and tetrabutyl ammonium hydrogen sulfate (2.17 gm 6.4 mmol) was added. It was stirred for 2 hours. Layers were separated and organic layer was concentrated under vacuum to provide a crude mass, which was purified by silica gel column chromatography to furnish 2.1 g of tetrabutylammonium salt of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ 1 ,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo- 1 ,6-diaza-bicyclo[3.2.1] octane as solid in 43% yield.
Analysis:
Mass: 458.3 (M- l), as a free sulfonic acid, for Molecular Formula of C17H25N5O8S. N(C4H9)4; Purity as determined by HPLC: 94.87%.
Step-5: Preparation of (25,5R)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[l,3,4]-oxadiazol-2-yl)- 7- oxo-l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester:
To the solution of tetrabutylammonium salt of (25,5i?)-2-(5-(N-feri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane (1.0 g, 2.2 mmol) in dichloromethane (5 ml) was charged trifluoroacetic acid (5 ml) with syringe at – 10°C under stirring. The reaction mixture was stirred for 1 hour. The mixture was evaporated under vacuum by maintaining temperature below 35 °C, to provide a residue, which was suspended in diethyl ether (25 ml) twice. The suspension was filtered and the solid was suspended further in dichloromethane (50 ml) and stirred for 30 minutes. The suspension was filtered and dried to afford the 310 mg of (25,5i?)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[ l,3,4]-oxadiazol-2-yl)-7-oxo- l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester as a solid in 60% yield.
Analysis:
Mass: 358.2 (M-l), for Molecular Formula of C^HnNsOeS;
1H NMR (DMSO-d6): δ 8.50 (br s, IH), 8.62 br s, IH), 4.60 (d, IH), 4.05 (s, 3H), 3.82-3.84 (m, IH), 3.21-3.27 (m, 4H), 2.93-2.96 (m, IH), 2.75 (d, IH), 2.12-2.17 (m, IH), 1.96-2.05 (m, 2H), 1.82-1.88 (m, IH).
Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.
///////
Zidebactam, WCK 5107 in PHASE 1 FROM WOCKHARDT

Zidebactam, WCK 5107
![]()
Useful for treating bacterial infections
CAS 1436861-97-0, UNII: YPM97423DB, Wockhardt Biopharm
Molecular Formula, C13-H21-N5-O7-S
Molecular Weight, 391.4029
Disclosed in PCT International Patent Application No. PCT/IB2012/054290D
- 01 Aug 2015 Phase-I clinical trials in Bacterial infections (In volunteers, Combination therapy) in USA (IV) (NCT02532140)
trans- sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(2S, 5R)-sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(1R,2S,5R)-l,6-Diazabicyclo [3.2.1] octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-[2-[(3R)-3-piperidinylcarbonyl]hydrazide]
trans- sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(2S, 5R)-sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(lR,2S,5R)-l,6-Diazabicyclo [3.2.1] octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-[2-[(3R)-3 -piperidinylcarbonyl] hydrazide]
1,6-Diazabicyclo(3.2.1)octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-(2-((3R)-3-piperidinylcarbonyl)hydrazide), (1R,2S,5R)-

Zidebactam potassium
cas is 1706777-49-2
Zidebactam sodium ………..below

Cas 1706777-46-9
Sodium;[(2S,5R)-7-oxo-2-[[[(3R)-piperidine-3-carbonyl]amino]carbamoyl]-1,6-diazabicyclo[3.2.1]octan-6-yl] sulfate
UNII-NHY7N0Y9DG; NHY7N0Y9DG; Zidebactam sodium; Zidebactam sodium, (-)-; 1,6-Diazabicyclo(3.2.1)octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-(2-((3R)-3-piperidinylcarbonyl)hydrazide), sodium salt (1:1), (1R,2S,5R)-; 1706777-46-9;
| Molecular Formula: | C13H20N5NaO7S |
|---|---|
| Molecular Weight: | 413.381969 g/mol |
In September 2015, the drug was reported to be in phase I clinical trial.One of the family members US09132133, claims a combination of sulbactam and WCK-5107.
Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate.
Treatment of infections caused by resistant bacteria remains a key challenge for the clinician community. One example of such challenging pathogen is Acinetobacter baumannii (A. baumannii), which continues to be an increasingly important and demanding species in healthcare settings. The multidrug resistant nature of this pathogen and its unpredictable susceptibility patterns make empirical and therapeutic decisions more difficult. A. baumannii is associated with infections such as pneumonia, bacteremia, wound infections, urinary tract infections and meningitis.
Therefore, there is a need for development of newer ways to treat infections that are becoming resistant to known therapies and methods. Surprisingly, it has been found that a compositions comprising cefepime and certain nitrogen containing bicyclic compounds (disclosed in PCT/IB2012/054290) exhibit unexpectedly synergistic antibacterial activity, even against highly resistant bacterial strains.

PATENT
http://www.google.com/patents/WO2013030733A1?cl=en

Scheme-1

function with Boc group)
o ormua –
Scheme-2
Example-2 trans-sulfuric acid mono-r2-(N,-r(R)-piperidin-3-carbonyll-hvdrazinocarbonyl)-7-oxo-l,6- diaza-bicyclo Γ3.2.11 oct-6-νΠ ester
Step-1: Preparation of trans-3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-1 of Example- 1 above, and by using trans-6- benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylic acid (25 gm, 0.084 mol), N,N- dimethyl formamide (625 ml), EDC hydrochloride (24 gm, 0.126 mol), HOBt (16.96 gm, 0.126 mol), (R)-N-tert-butoxycarbonyl-piperidin-3-carboxylic acid hydrazide (21.40 gm , 0.088 mol) to provide the title compound in 17.0 gm quantity, 41% yield as a white solid.
Analysis: MS (ES+) CzsHasNsOe = 502.1 (M+l);
I^NMR (CDCI3) = 8.40 (br s, IH), 7.34-7.44 (m, 5H), 5.05 (d, IH), 4.90 (d, IH), 4.00 (br d, IH), 3.82 (br s, IH), 3.30 (br s, IH), 3.16-3.21 (m, IH), 3.06 (br d, IH), 2.42 (br s, IH), 2.29-2.34 (m, IH), 1.18-2.02 (m, 4H), 1.60-1.75 (m, 4H), 1.45-1.55 (m, 2H),1.44 (s, 9H).
Step-2: Preparation of trans-3-[N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-2 of Example- 1 above, and by using trans-3- [N ‘ -(6-benzyloxy-7-oxo- 1 ,6-diaza-bicyclo [3.2.1 ]octane-2-carbonyl)-hydrazinocarbonyl] -(R)- piperidin-l-carboxylic acid tert-butyl ester (16.5 gm , 0.033 mol), methanol (170 ml) and 10% palladium on carbon (3.5 gm) to provide the title compound in 13.5 gm quantity as a pale pink solid and it was used for the next reaction immediately.
Analysis: MS (ES+) CiglfeNsOe = 411.1 (M+l);
Step-3: Preparation of tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza- bicyclo [3.2.1] octane-2-carbonyl)-hydrazinocarbonyl] -(R)-piperidin- 1 -carboxylic acid tert- butyl ester:
By using the procedure described in Step-3 of Example- 1 above, and by using trans-3- [N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)- piperidin-1 -carboxylic acid tert-butyl ester (13.5 gm , 0.033 mol), pyridine (70 ml) and pyridine sulfur trioxide complex (26.11 gm, 0.164 mol), 0.5 N aqueous potassium dihydrogen phosphate solution (400 ml) and tetrabutylammonium sulphate (9.74 gm, 0.033 mol) to provide the title compound in 25 gm quantity as a yellowish solid, in quantitative yield.
Analysis: MS (ES-)
as a salt = 490.0 (M-l) as a free sulfonic acid;
Step-4: trans-sulfuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7- oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl]ester:
By using the procedure described in Step-4 of Example- 1 above, and by using tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester (24 gm , 0.032 mmol), dichloromethane (60 ml) and trifluoroacetic acid (60 ml) to provide the title compound in 10 gm quantity as a white solid, in 79% yield.
Analysis: MS (ES-)= C13H21N5O7S = 390.2 (M-l) as a free sulfonic acid;
HXNMR (DMSO-d6) = 9.97 (d, 2H), 8.32 (br s, 2H), 4.00 (br s, IH), 3.81 (d, IH), 3.10-3.22 (m, 3H), 2.97-3.02 (m, 2H), 2.86-2.91 (m, IH), 2.65-2.66 (m, IH), 1.97-2.03 (m, IH), 1.57-1.88 (m, 7H).
-32.6°, (c 0.5, water).
PATENT
http://www.google.com/patents/WO2015059643A1?cl=en

Both, cefepime and a compound of Formula (I) may be present in the composition in their free forms or in the form of their pharmaceutically acceptable derivatives (such as salts, pro-drugs, metabolites, esters, ethers, hydrates, polymorphs, solvates, complexes, or adducts).
Individual amounts of a compound of Formula (I) or a stereoisomer or a pharmaceutically acceptable derivative thereof, and cefepime or pharmaceutically acceptable derivative thereof in the composition may vary depending on clinical requirements. In some embodiments, a compound of Formula (I) or a stereoisomer or a pharmaceutically acceptable derivative thereof in the composition is present in an amount from about 0.01 gram to about 10 gram. In some other embodiments, cefepime or a pharmaceutically acceptable derivative thereof in the composition is present in an amount from about 0.01 gram to about 10 gram.
PATENT
http://www.google.com/patents/WO2015063653A1?cl=en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015110885
Formula (I)

(a) hydrogenolysis of a compound of Formula (II) to obtain a compound of Formula (III);

convertin a compound of Formula (III) to a compound of Formula (IV);



Example 1
Synthesis of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (25, 5R)-6-hydroxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III):
(25, 5i?)-6-benzyloxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazino-carbonyl] -l,6-diazabicyclo[3.2.1]octane (II) (130 g, 0.259 mol) was dissolved in methanol (1040 ml) to obtain a clear solution. To this solution, was added 10% palladium on carbon (13 g, 0.26 mol). The suspension was stirred under 230-250 psi hydrogen atmosphere at temperature of about 30 °C for about 2 hour. The catalyst was filtered over celite bed and catalyst containing bed was washed with additional methanol (400 ml). The methanolic solution was re-filtered through fresh celite bed and washed with methanol (100 ml). The filtrate was concentrated under vacuum at temperature of about 30°C to obtain the off white solid as product. The so obtained solid was stirred with cyclohexane (750 ml). The solid was then filtered and washed with cyclohexane (320 ml) and dried under suction to obtain 107 g of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo [3.2.1]octane (III).
Analysis:
Mass: 412.4 (M+l); for Molecular Formula of C18H29N5O6 and Molecular Weight of 411.5; and
Purity as determined by HPLC: 98.02%.
Step-2: Preparation of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1] octane (IV):
A solution of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) (106 g, 0.26 mol) in dichloromethane was charged with triethyl amine (110 ml, 0.78 mol) under stirring. To this clear solution was added pyridine sulfur trioxide complex (82.5 g, 0.53 mol) under nitrogen atmosphere and stirred at temperature of about 30°C for about 2 hour. The reaction mixture was diluted with 0.5 N aqueous potassium dihydrogen phosphate solution (2100 ml) followed by ethyl acetate (2100 ml). The turbid solution was stirred for 15 minute and then the layers were separated. The aqueous layer was washed with dichloromethane (530 ml) and then with ethyl acetate (1060 ml). Tetrabutyl ammonium sulfate (79 g, 0.23 mol) was added to the separated aqueous layer and stirred for 12 hour. The extraction of the product was done using dichloromethane as solvent (1150 ml x 2). The organic layer was dried over sodium sulfate and then evaporated under vacuum at temperature below 40°C to furnish 108 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo
[3.2.1] octane (IV).
Analysis:
Mass: 490.3 (M-l) as free sulfonic acid; for Molecular Formula of Ci8H28N509S.N(C4H9)4 and Molecular weight of 733.0; and
Purity as determined by HPLC: 86.50 %.
Step-3: Preparation of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) (88 g, 0.12 mol) was dissolved in dichloromethane (225 ml). The reaction mass was cooled to about -10°C and to this trifluoroacetic acid (225 ml) was added slowly. The reaction mixture was stirred for 1 hour at temperature of about -10°C. The solvent was removed under high vacuum at about 30°C. The residue (280 g) was stirred with diethyl ether (1320 ml) for 1 hour. The precipitated solid was filtered and the cake was washed with fresh diethyl ether (440 ml). This process was repeated with fresh diethyl ether (1320 ml + 440 ml). The obtained white solid was dried at temperature of about 30°C and suspended in acetone (1320 ml). The pH of the suspension was adjusted to 6.5-7.0 using 10% solution of sodium 2-ethyl hexanoate in acetone. The resulting suspension was filtered under suction and the wet cake was washed with acetone (440 ml) to provide the crude solid. The solid was further dried under vacuum at 40°C to yield 40 g of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I).
Analysis:
Mass: 392.2 (M+l); for Molecular formula of C13H21N5O7S and Molecular Weight of 391.4;
Purity as determined by HPLC: 92.87%; and
Melting point as determined by DSC: 274°C.
Example 2
Synthesis of Pure (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (25, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III):
The procedure for the synthesis of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) is same as given in Step- 1 of Example 1.
Step-2: Preparation of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1] octane (IV):
A solution of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) (106 g, 0.26 mol) in dichloromethane was charged with triethylamine (110 ml, 0.78 mol) under stirring to provide a clear solution. To this clear solution was added pyridine sulfur trioxide complex (82.5 g, 0.53 mol) under nitrogen atmosphere and stirred at temperature of about 30 °C for 2 hours. The reaction mixture was diluted with 0.5 N aqueous potassium dihydrogen phosphate solution (2100 ml) followed by ethyl acetate (2100 ml). The turbid solution was stirred for 15 minutes and then the layers were separated. The aqueous layer was washed with dichloromethane (530 ml) and then with ethyl acetate (1060 ml) respectively. Tetrabutyl ammonium sulfate (79 g, 0.23 mol) was added to the separated aqueous layer and stirred for 12 hours. The extraction of the product was done using dichloromethane as solvent (1150 ml x 2). Aliquot of the organic layer was dried over sodium sulfate for purity check. Considering the purity of the product as obtained above, silica gel (530 g) was added to the dichloromethane layer and stirred for 1 hour. This was filtered and again silica was taken in dichloromethane (3200 ml) and stirred for 45 minutes and filtered. Combined dichloromethane layer was filtered through the celite bed again and washed with additional 200 ml dichloromethane. The solvent was removed to obtain 88 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-!, 6-diaza-bicyclo[3.2.1]octane (IV) as white foam.
Analysis:
Mass: 490.3 (M-l) as a free sulfonic acid; for Molecular Formula of Ci8H28N509S.N(C4H9)4 and Molecular Weight of 733.0; and
Purity as determined by HPLC: 98.34%.
Step-3: Preparation of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
The above obtained tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) having purity of more than 98% (88 g, 0.12 mol) was dissolved in dichloromethane (225 ml). The reaction mass was cooled to temperature of about -10°C and to this trifluoroacetic acid (225 ml) was added slowly. The reaction mixture was stirred for 1 hour at about -10°C. The solvent was removed under high vacuum at temperature of about 30°C. The residue (280 g) was stirred with diethyl ether (1320 ml) for 1 hour. The precipitated solid was filtered and the cake was washed with fresh diethyl ether (440 ml). This process was repeated with fresh diethyl ether (1320 ml + 440 ml). The obtained white solid was dried at about 30°C and suspended in acetone (1320 ml). The pH of the suspension was adjusted to 6.5-7.0 using 10% solution of sodium 2-ethyl hexanoate in acetone. The resulting suspension was filtered under suction and the wet cake was washed with acetone (440 ml) to provide the crude solid. The solid was further dried under vacuum at 40°C to yield 40 g of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I).
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4; and
Purity as determined by HPLC: 98.7%.
Recovery of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1] octane (IV):
The silica recovered from the Step-2 was stirred with dichloromethane containing 2%
methanol (2000 ml) for one hour. Silica was filtered, washed with additional same composition of solvents (500 ml). Combined dichloromethane was filtered through the celite bed and washed with same composition of solvents (200 ml), evaporated to afford 1 1 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l , 6-diaza-bicyclo[3.2.1] octane (IV) as off white solid.
Repeating Step-3 with the above obtained tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2- [((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1 , 6-diaza-bicyclo [3.2.1] octane (IV) produced additional 7 g of compound of Formula (I).
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of CnH^NsOvS and Molecular Weight of 391.4;
Purity as determined by HPLC: 98.7%; and
Assay as determined by HPLC: 104% against reference standard of compound of Formula (I).
Example 3
Preparation of amorphous form of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1, 6-diaza-bicyclo[3.2. l]octane (I) :
Tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) (60 g, 0.081 mol), obtained in Step-2 of Example-2 was dissolved in dichloromethane (150 ml, 2.5 volume) to obtain a clear solution. Reaction mass was cooled to about -10°C and to it trifluoroacetic acid (150 ml) was slowly added. The reaction mixture was stirred for 1 hour at about – 10°C. The solvent was removed under high vacuum at about 30°C. Diethyl ether (600 ml x 3) was added to the residue ( 184 g) and stirred for 15 minute every time. The solvent was decanted off and the residue was washed with acetonitrile (600 ml x 3). This process was also repeated with dichloromethane (600 ml x 3). The off white solid was
isolated and dried under high vacuum at about 35 °C for 3 hour to obtain 33 g of amorphous form of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I). The XRD is shown in Figure 1.
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4;
HPLC purity: 92.26%; and
Melting point as determined by DSC: 210°C (loss of moisture below 100°C).
Example 4
Preparation of crystalline form of (25, 5R)-7-oxo-6-sulpho-oxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
The (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I) obtained as white solid (40 g) in Step-3 of Example 2 was dissolved in demineralised water (40 ml) to obtain a clear solution. To this isopropyl alcohol (280 ml) was added under stirring at room temperature. The obtained turbid solution became sticky initially then slowly started to convert into white solid, stirring continued for about 17 hours at temperature of about 30°C. The precipitated solid was filtered and washed with water: isopropyl alcohol mixture (20 ml: 140 ml). White solid was dried under high vacuum at temperature of about 45 °C for 5 hours to get 34 g of crystalline form of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1] octane (I).
Analysis:
Mass: 392.2 (M+l) for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4;
Purity as determined by HPLC: 98.7%;
Assay as determined by HPLC: 104% against reference standard of compound of Formula (I); and
Melting point as determined by DSC: 278°C (9% loss of moisture at 143-152°C).
X-ray powder diffraction pattern comprising a peak selected from the group consisting of 10.31 (± 0.2), 10.59 (± 0.2), 12.56 (± 0.2), 13.84 (± 0.2), 15.65 (± 0.2), 18.19 (± 0.2), 18.51(± 0.2), 20.38 (± 0.2), 20.65 (± 0.2), 24.30 (± 0.2), 24.85 (± 0.2) and 25.47 (± 0.2) degrees 2 theta.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014135931
Scheme 1.


Formula (I)
preparation of a compound of Formula (I), comprising:

Formula (I)
(a) reacting a compound of Formula (II) with a compound of Formula (III) to obtain a compound of Formula (IV);

Formula (II) Formula (III)

Formula (IV)
(b) hydrogenolysis of a compound of Formula (IV) to obtain a compound of Formula

X. Formula (V)
(c) sulfonating a compound of Formula (V) to obtain a compound of Formula (VI); and

Formula (VI)
(d) converting a compound of Formula (VI) into a compound of Formula (I).
Example -1
Preparation of (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (II):
Step-1: Preparation of (R)-Ethyl-N-Boc-piperidine-3-carboxylate (VIII)
To a solution of (R)-N-Boc-piperidine-3-carboxylic acid (1 kg. 4.36 mol) in N,N-dimethylacetamide (3 L) was charged potassium carbonate (0.664 kg, 4.80 mol) under mechanical stirring and the resulting suspension was stirred for 30 minutes at room temperature. To the reaction mass, ethyl iodide (0.75 kg, 4.80 mol) was charged via addition funnel and the reaction mass was stirred for 15 minutes at room temperature followed by at 50°C for 1 hour. The reaction was monitored using TLC (ethyl acetate: hexane 1:1). After the reaction was complete, the reaction mass was allowed to cool to room temperature and diluted with ethyl acetate (5 L). The suspension was filtered under suction and the wet cake was washed with ethyl acetate (5 L). The filtrate was stirred with 5% w/v sodium thio sulfate (15 L) and layers were separated. The aqueous layer was re-extracted with additional ethyl acetate (5 L). The combined organic layer was washed with water (5 L) and dried over sodium sulfate. The organic layer was evaporated under vacuum to provide semi-solid which solidifies upon standing as (R)-ethyl-N-Boc-piperidine-3-carboxylate in 1.1 kg quantity in 99.5% yield.
Analysis:
NMR: (CDC13): 4.63 (q, 2H), 3.90 (d, 1H), 2.87-2.95 (m, 2H), 2.73 (td, 1H), 2.32-2.39 (m, 1H), 1.66-2.01 (m, 2H), 1.52-1.68 (m, 2H), 1.39 (s, 9H), 1.19 (t, 3H).
Mass: (M+l): 258.1 for C13H23N04;
Step-2: Preparation of (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (II):
(R)-N-Boc-ethyl-piperidine-3-carboxylate (1.1 kg, 4.28 mol) was liquefied by warming and transferred to a round bottom flask (10 L), to this was charged hydrazine hydrate (0.470 kg, 9.41 mol) and stirring was started. The reaction mixture was stirred at about 120°C to 125°C for 5 hours. As the TLC showed (Chloroform: methanol 9:1) completion of reaction, the reaction mixture was cooled to room temperature and diluted with water (5.5 L) followed by dichloromethane (11 L) and was stirred for 20 minutes. The layers were separated and aqueous layer was extracted with additional dichloro methane (5.5 L). Combined organic layer was washed with water (2.75 L). The organic layer was dried over sodium sulfate and evaporated under vacuum to provide a thick gel which upon stirring and seeding in the presence of cyclohexane (5.5 L) provided white solid. The suspension was filtered and wet cake was washed with fresh cyclohexane (0.5 L). The cake was dried at 35°C under vacuum to provide (R)-N-Boc-piperidine-3-carboxylic acid hydrazide as a white solid in 0.90 kg quantity in 87% yield.
Analysis
NMR: (CDC13): 7.42 (br s, 1H), 3.92 (d, 1H), 3.88 (s, 2H), 3.54-3.65 (br s, 1H), 3.17 (br t, 1H), 2.98 (br s, 1H), 2.22-2.32 (br s, 1H), 1.82-1.90 (br m, 2H), 1.76 (s, 1H), 1.60-1.70 (m, 1H), 1.45 (s, 9H).
Mass (M+l): 244.1 for C11H21N303.
Specific rotation: [ ]25D = -53.5° (c 0.5, Methanol).
HPLC purity: 99%
Example 2
Preparation of (2S, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)- hydrazinocarbonyl] -l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (2S, 5R)- 6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1 ,6-diaza-bicyclo [3.2.1 ] octane(IV) :
Sodium (2S, 5R)-7-oxo-6-benzyloxy-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylate (III, 200 gm, 0.67 mol; prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) was dissolved in water (2.8 L) to obtain a clear solution under stirring at room temperature. To the clear solution was added successively, (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (171 gm, 0.70 mol), EDC hydrochloride (193 gm, 1.01 mol), and HOBt (90.6 gm, 0.67 mol) followed by water (0.56 L) under stirring at 35°C. The reaction mixture was stirred at 35°C for 20 hours. As maximum precipitation was reached, TLC (acetone: hexane 35:65) showed completion of reaction. The suspension was filtered under
suction and the wet cake was washed with additional water (2 L). The wet cake was suspended in warm water (10 L) and stirred for 5 hours. It was filtered under suction and dried under vacuum at 45°C to furnish (2S, 5R)-6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (IV) as a white powder in 270 gm quantity in 87% yield.
Analysis
NMR: (CDC13): 8.40 (br s, 1H), 7.34-7.44 (m, 5H), 5.05 (d, 1H), 4.90 (d, 1H), 4.00 (br d, 1H), 3.82 (br s, 1H), 3.30 (br s, 1H), 3.16-3.21 (m, 1H), 3.06 (br d, 1H), 2.42 (br s, 1H), 2.29-2.34 (m, 1H), 1.18-2.02 (m, 4H), 1.60-1.75 (m, 4H), 1.45-1.55 (m, 2H),1.44 (s, 9H).
Mass: (M+l) = 502.1 for C25H35N506
HPLC purity: 98.4%
Step-2: Preparation of (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2. l]octane (V):
(2S,5R)-6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino-carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (153 gm, 0.305 mol) was dissolved in methanol (1.23 L) to obtain a clear solution. To this solution, was added 10% Pd-C (15.3 gm, 50% wet) catalyst. The suspension was stirred for 3 hours under 100 psi hydrogen atmosphere at 35°C. As reaction showed completion on TLC (TLC system methanol: chloroform 10:90), the catalyst was filtered through celite under suction. The catalyst was washed with additional methanol (600 ml). The filtrate was evaporated under vacuum below 40°C to provide a crude residue. The residue was stirred with cyclohexane (1.23 L) for 1 hour. The solid was filtered at suction and the wet cake was washed with additional cyclohexane (0.25 L) to furnish (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (V) in 125 gm quantity as a solid in quantitative yield. The product being unstable was used immediately for the next reaction.
Analysis:
NMR: (CDC13): 9.0 (br s, 2H), 4.01 (br d, 2H), 3.80 (br s, 1H), 3.74 (br s, 1H), 3.48 (s, 1H), 3.13-3.26 (m, 3H), 2.96 (br s, 1H), 2.47 (br s, 1H), 2.28-2.32 ( br dd, 1H), 2.08 (br s, 1H), 1.90-2.0 (m, 3H),1.65-1.80 (m, 3H) 1.44 (s, 9H).
Mass: (M-l): 410.3 for C18H29N506
HPLC purity: 96.34%
Step-3: Preparation of Tetrabutyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]- 1 ,6-diaza-bicyclo[3.2.1 ] octane (VI) :
A solution of (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (113 gm, 0.274 mol), in dichloromethane (1.13 L) was charged with triethylamine (77 ml, 0.548 mol) under stirring to provide a clear solution. To the clear solution, was added pyridine sulfur trioxide complex (57 gm, 0.356 mol) under stirring at 35°C. The reaction mixture was stirred for 3 hours. The reaction mixture was worked up by adding 0.5 M aqueous potassium dihydrogen phosphate (1.13 L) followed by ethyl acetate (2.26 L) and the biphasic mixture was stirred for 15 minutes at 35°C. Layers were separated. Aqueous layer was re-extracted with dichloromethane ethyl acetate mixture (1:2 v/v, 2.26 L twice). Layers were separated. To the aqueous layer, was added solid tetrabutyl ammonium hydrogen sulfate (84 gm, 0.247 mol) and stirring was continued for 3 hours at room temperature. Dichloromethane (1.13 L) was added to the reaction mixture. Layers were separated. The aqueous layer was re-extracted with additional dichloromethane (0.565 L). Layers were separated. To the combined organic layer was added silica gel (226 gm) and the suspension was stirred for 1 hour. Suspension was filtered and silica gel was washed with dichloromethane (1 L). The combined filtrate was evaporated under vacuum to provide solid mass. To the solid mass was added cyclohexane (0.9 L) and stirred till complete solidification occurred (about 1 to 2 hours). The suspension was filtered under suction and the wet cake was dried under vacuum below 40°C to furnish tetrabutyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (VI) as a white solid in 122 gm quantity in 60% yield.
Analysis
NMR: (CDC13): 8.50 (br s, 2H), 4.32 (br s, 1H), 3.97 (d, 2H), 3.15-3.37 (m, 12H), 2.43 (br s, 1H), 2.33 (d, 1H), 2.10-2.2 (br m, 1H), 1.84-1.95 (m, 3H), 1.60-1.73 (m, 13H), 1.39-1.48 (m, 19H), 0.98 (t, 12H).
Mass: (M-l): 490.4 as a free sulfonic acid for C18H28N509S.N(C4H9)4;
HPLC purity: 96.3%
Step-4: Synthesis of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2. l]octane (I):
Tetra-butyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (113 gm, 0.154 mol) was dissolved in dichloromethane (280 ml) and to the clear solution was slowly added trifluoroacetic acid (280 ml) between 0 to 5°C. The reaction mixture was stirred between 0 to 5°C for 1 hour. The solvent and excess trifluoroacetic acid was evaporated under vacuum below 40°C to approximately 1/3 of it’s original volume to provide pale yellow oily residue. The oily residue was stirred with diethyl ether (2.25 L) for 1 hour to provide a suspension. The precipitate was filtered under suction and transferred to a round bottom flask, to it was added diethyl ether (1.1 L) under stirring. The suspension was stirred for 30 minutes and filtered under suction to provide a solid. The solid was charged in a round bottom flask and to it was added acetone (1.130 L). The pH of suspension was adjusted to 4.5 to 5.5 by adding 10% solution of sodium-2-ethyl hexanoate in acetone carefully. The resulting suspension was filtered under suction and the wet cake was washed with acetone (550 ml) to provide a crude solid. The obtained solid was dried under vacuum below 40°C to furnish 65 gm of a crude mass. The crude mass was dissolved in water (65 ml) under stirring and to the clear solution was added isopropyl alcohol (455 ml). The suspension was stirred for 24 hours and filtered under suction. The wet cake was washed with isopropyl alcohol (225 ml) and dried under vacuum below 40°C to provide a crystalline (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I) free from impurities in 48 gm quantity in 80% yield.
Analysis:
NMR: (DMSO-d6) = 9.97 (d, 2H), 8.32 (br s, 2H), 4.00 (br s, IH), 3.81 (d, IH), 3.10-3.22 (m, 3H), 2.97-3.02 (m, 2H), 2.86-2.91 (m, IH), 2.65-2.66 (m, IH), 1.97-2.03 (m, IH), 1.57-1.88 (m, 7H).
Mass: (M-l): 390.3 for C13H21N507S
HPLC purity: 95.78%
Specific rotation: [(X]25D: – 32.6° (c 0.5, water)
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 10.28 (+ 0.2), 10.57 (± 0.2), 12.53 (± 0.2), 13.82 (± 0.2), 15.62 (± 0.2), 18.16 (± 0.2), 18.49 (± 0.2), 20.35 (+ 0.2), 20.64 (± 0.2), 21.33 (+ 0.2), 22.99 (+ 0.2), 23.18 (+ 0.2), 24.27 (± 0.2), 24.81 (+ 0.2), 25.45 (± 0.2), 29.85 (+ 0.2), 30.45 (± 0.2), 32.39 (+ 0.2), 36.84 (± 0.2).
REFERENCES
Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of WCK-5107 Alone and in Combination With Cefepime (NCT02532140) https://clinicaltrials.gov/show/NCT02532140
ClinicalTrials.gov Web Site 2015, September 01, To evaluate the safety,tolerability and pharmacokinetics of single intravenous doses of WCK 5107 alone and in combination with cefepime in healthy adult human subjects.
| WO2013030733A1 * | Aug 24, 2012 | Mar 7, 2013 | Wockhardt Limited | 1,6- diazabicyclo [3,2,1] octan-7-one derivatives and their use in the treatment of bacterial infections |
| WO2014135931A1 * | Oct 12, 2013 | Sep 12, 2014 | Wockhardt Limited | A process for preparation of (2s, 5r)-7-oxo-6-sulphooxy-2-[((3r)-piperidine-3-carbonyl)-hydrazino carbonyl]-1,6-diaza-bicyclo [3.2.1]- octane |
| IB2012054290W | Title not available |
Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.
///////see………http://apisynthesisint.blogspot.in/2015/11/wck-5107-in-phase-1-from-wockhardt.html
SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
C1C[C@H](CNC1)C(=O)NNC(=O)[C@@H]2CC[C@@H]3C[N@]2C(=O)N3OS(=O)(=O)O
or
O=C(NNC(=O)[C@@H]2CC[C@@H]1CN2C(=O)N1OS(=O)(=O)O)[C@@H]3CCCNC3
C1CC(CNC1)C(=O)NNC(=O)C2CCC3CN2C(=O)N3OS(=O)(=O)[O-].[Na+]
Atagabalin

Atagabalin
Trans-dimethyl gababutin; UNII-JT7957Q2FB; 223445-75-8;
2-[(3S,4S)-1-(aminomethyl)-3,4-dimethylcyclopentyl]acetic acid
2-[(3S,4S)-1-(aminomethyl)-3,4-dimethyl-cyclopentyl]acetic acid
3,4-trans-2-(1-(aminomethyl)-3,4-dimethylcyclopentyl)acetic acid
Cyclopentaneaceticacid, 1-(aminomethyl)-3,4-dimethyl-, (3S,4S)-
Pfizer Inc. INNOVATOR
Atagabalin (PD-0200,390) is a drug developed by Pfizer and related to gabapentin, which similarly binds to the α2δ calcium channels (1 and 2).[1] It was under development as a treatment for insomnia,[2][3][4] but was discontinued following unsatisfactory trial results.
Gabapentin (Neurontin®) (1) was launched as an add-on therapy for epilepsy in 1994. Utility against neuropathic pain and anxiety have been reported preclinically and efficacy against neuropathic pain has been demonstrated clinically in humans. Pregabalin (Lyrica®) (2), has superior potency and pharmacokinetics to gabapentin and has been approved for the management of neuropathic pain associated with diabetic peripheral neuropathy, post-herpetic neuralgia, adjunctive treatment of partial seizures, and fibromyalgia in the US.

Gabapentin and pregabalin are thought to mediate their pharmacological actions through binding to the α2δ subunit of a voltage gated calcium channeland it has been shown that gabapentin and pregabalin bind to this α2δ subunit with IC50 values of 140 nM and 80 nM, respectively. We have recently disclosed our initial SAR investigations around five-membered ring gabapentin analogues, which we have termed gababutins.In that Letter, we investigated a range of 3-substituted gababutin analogues and identified the 3-(R)-methyl gababutins (3) and (4). Both (3) and (4) bind to the gabapentin binding site with high affinity but have different in vivo profiles, with (3) being effective on oral dosing in models of anxiety and (4) being effective on oral dosing in models of neuropathic pain.
SYNTHESIS

PATENT
WO 1999021824
http://www.google.co.in/patents/WO1999021824A1?cl=en
synthesis of 3-oxo-2,8-diazaspiro[4,5]decane-
8-carboxylic acid tert-butyl ester (P. W. Smith et al., J. Med. Chem., 1995;38:3772). The compounds may also be synthesized by the methods outlined by G. Satzinger et al., (Ger Offen 2,460,891; US 4,024,175, and Ger Offen 2,611,690; US 4,152,326) (General Schemes 3 and 4). The compounds may also be synthesized by the route outlined by G. Griffiths et al., Helv. Chim. Ada, 1991 ;74:309 (General Scheme 5). General Scheme 1

(i) Ethyl cyanoacetate, piperidine (Cope et al., J. Am. Chem. S c.,1941 ;63:3452); (ii) NaCN, EtOH/H2O; (iii) EtOH, HCl; (iv) H2O/H+; (v) H2, Rh/C, MeOH; (vi) HCl.
General Scheme 2


(i) Ph3P=CHCO2Me; (ii) MeNO2, 1,1,3,3-tetramethylguanidine; (iii) Raney nickel, EtOH/H2O; (iv) HCl.
General Scheme 3

(i) Ethylcyanoacetate, ammonia then H3θ+; (ii) H2SO4; (iii) AC2O; (iv) MeOH; (v) Curtius Reaction; (vi) HCl, H2O then anion exchange.
General Scheme 4

(i) Ethylcyanoacetate, ammonia then H3O “; (ii) H2SO4; (iii) AC2O; (iv) H2NOH; (v) PhSO2Cl; (vi) Et3N, MeOH; (vii) HCl, H O then anion exchange.
General Scheme 5


(i) Ethyl cyanoacetate, piperidine (Cope et al., J. Am. Chem. Soc, 1941 ;63:3452); (ii) NaCN, EtOH/H2O; (iii) BnOH, HCl; (iv) H2O/H+; (v) H2, Rh/C, MeOH.
EXAMPLE 1
Reagents: (i) Triethylphosphonoacetate, NaH; (ii) MeNO2,Bu4N+F”; (iϋ) H2, Ni; (iv) HCl Synthesis of (trans)-(3,4-Dimethyl-cyclopentylidene)-acetic acid ethyl ester (2)
NaH (60% dispersion in oil, 737 mg, 18.42 mmol) was suspended in dry tetrahydrofuran (50 mL) and cooled to 0°C. Triethylphosphonoacetate (3.83 mL, 19.30 mmol) was added and the mixture stirred at 0°C for 15 minutes. The ketone (1) (1.965 g, 17.54 mmol) in THF (10 mL) was then added and the mixture allowed to warm to room temperature. After 2 hours, the mixture was partitioned between diethyl ether (200 mL) and water (150 mL). The organic phase was separated, washed with brine, dried (MgSO4) and the solvent removed in vacuo.
The residue was purified by flash chromatography (silica, ethyl acetate:heptane 1 :9) to give 3.01 g (94%) of (2) as a colorless oil.
*H NMR 400 MHz (CDCI3): δ 1.01 (3H, d, J = 6 Hz), 1.03 (3H, d, J = 6 Hz), 1.26
(3H, t, J = 7 Hz), 1.49 (2H, m), 2.07 (1H, m), 2.24 (1H, m), 2.61 (1H, m), 4.13 (2H, q, J = 7 Hz), 5.72 (1H, s).
MS (CI+) m/e: 183 ([MH+], 18%).
Synthesis of (trans)-(3,4-Dimethyl-l-nitromethyl-cyclopentyl)-acetic acid ethyl ester (3)
The unsaturated ester (2) (2.95 g, 16.2 mmol) was dissolved in tetrahydrofuran (10 mL) and stirred at 70°C with nitromethane (1.9 mL, 35.2 mmol) and tetrabutylammonium fluoride (1.0 M in tetrahydrofuran, 22 mL, 22.0 mmol). After 6 hours, the mixture was cooled to room temperature, diluted with ethyl acetate (50 mL), and washed with 2N HCl (30 mL) followed by brine (50 mL). The organic phase was collected, dried (MgSO4) and the solvent removed in vacuo. The residue was purified by flash chromatography (silica, ethyl acetate :heptane 1 :9) to give 1.152 g (29%) of a clear oil. !H NMR 400 MHz (CDCI3): δ 0.98 (6H, d, J = 6 Hz), 1.10-1.39 (5H, m), 1.47
(2H, m), 1.87 (1H, m), 2.03 (1H, m), 2.57 (2H, ABq, J = 16, 38 Hz), 4.14 (2H, q, J = 7 Hz), 4.61 (2H, ABq, J = 12, 60 Hz).
MS (ES+) m/e: 244 ([MH+], 8%).
IR (film) v ein-1 : 1186, 1376, 1549, 1732, 2956. Synthesis of (±)-(trans)-7,8-Dimethyl-spiro[4.4]nonan-2-one (4)
The nitroester (3) (1.14 g, 4.7 mmol) was dissolved in methanol (50 mL) and shaken over Raney nickel catalyst under an atmosphere of hydrogen (40 psi) at 30°C. After 5 hours, the catalyst was removed by filtration through celite. The solvent was removed in vacuo to give 746 mg (95%) of a pale yellow oil which solidified on standing.
! H NMR 400 MHz (CDC13): δ 0.98 (6H, d, J = 6 Hz), 1.32 (2H, m), 1.46 (2H, m), 1.97 (2H, m), 2.27 (2H, ABq, J = 16, 27 Hz), 3.23 (2H, s), 5.62 (1H, br s). MS (ES+) m/e: 168 ([MH+], 100%). IR Cfilπ v cm-1 : 1451, 1681, 1715, 2948, 3196.
Synthesis of (±)-(trans)-(l-Aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid hydrochloride (5)
The lactam (4) (734 mg, 4.40 mmol) was heated to reflux in a mixture of 1 ,4-dioxan (5 mL) and 6N HCl (15 mL). After 4 hours, the mixture was cooled to room temperature, diluted with water (20 mL), and washed with dichloromethane
(3 x 30 mL). The aqueous phase was collected and the solvent removed in vacuo. The residue was triturated with ethyl acetate to give 675 mg (69%) of a white solid after collection and drying.
ΪH NMR 400 MHz (d6-DMSO): δ 0.91 (6H, d, J = 6 Hz), 1.18 (2H, m), 1.42 (2H, m), 1.72 (1H, m), 1.87 (1H, m), 2.42 (2H, ABq, J = 16, 24Hz), 2.90 (2H, ABq,
J = 12, 34 Hz), 8.00 (3H, br s), 12.34 (1H, br s).
MS (ES+) m/e: 186 ([MH-HC1J+, 100%).
PATENT
WO 2002000209
PATENT
http://www.google.co.in/patents/WO1999021824A1?cl=en
PATENT
WO 2007010387
http://www.google.com/patents/WO2007010387A2?cl=en

21 22

Scheme IH
PAPER
Synthesis and in vivo evaluation of 3,4-disubstituted gababutins
Bioorganic&Medicinal Chemistry Letters (2010), 20, (1), 248-251.
The synthesis of 3,4-trans-dimethyl cyclopentanone (14), is detailed in Scheme 1.

-
Scheme 1.
Reagents and conditions: (i) (−)-menthol, pyridine, CH2Cl2; (ii) butadiene, TiCl4, toluene, −10 °C (100% yield, 65% de) or butadiene, Et2AlCl, toluene, −60 °C (64% yield, 95% de); (iii) LiAlH4, THF; recrystallisation from acetone; (iv) pyridine, MsCl, 0 °C, 18h (82%); (v) LiAlH4, diethyl ether, 40 °C, 2h (98%); (vi) KMnO4, nBu4NBr, H2O–CH2Cl2, rt, 18h; then SO2, 0 °C (82%); (vii) methanol, cH2SO4, rt, 18h (90%) (viii) KOtBu, THF, 75 °C, 3h (100%); (ix) DMSO, H2O, 140 °C, 4 h (86%).

-
Scheme 3.
Reagents and conditions: (i) triethylphosphonoacetate, NaH, THF, 0 °C to rt (95%); (ii) MeNO2, TBAF, THF, reflux (65%); (iii) H2, Ni, MeOH; (iv) 6 N HCl, 1,4-dioxane, reflux (69% from nitroester).
References
- 2 Corrigan B, Feltner DE, Ouellet D, Werth JL, Moton AE, Gibson G (August 2009). “Effect of renal impairment on the pharmacokinetics of PD 0200390, a novel ligand for the voltage-gated calcium channel alpha-2-delta subunit”. British Journal of Clinical Pharmacology 68 (2): 174–80. doi:10.1111/j.1365-2125.2009.03444.x. PMC 2767279. PMID 19694735.
- 3 Quintero JE, Pomerleau F, Huettl P, Johnson KW, Offord J, Gerhardt GA (May 2011). “Methodology for rapid measures of glutamate release in rat brain slices using ceramic-based microelectrode arrays: Basic characterization and drug pharmacology”. Brain Research 1401: 1–9. doi:10.1016/j.brainres.2011.05.025. PMID 21664606.
- 4 Kjellsson MC, Ouellet D, Corrigan B, Karlsson MO (June 2011). “Modeling Sleep Data for a New Drug in Development using Markov Mixed-Effects Models”. Pharmaceutical Research 28 (10): 2610–27. doi:10.1007/s11095-011-0490-x. PMID 21681607.
| Patent | Submitted | Granted |
|---|---|---|
| Pyrazolo[4,3-d]pyrimidines as Phosphodiesterase Inhibitors [US7572799] | 2005-11-03 | 2009-08-11 |
| Substituted morpholine compounds for the treatment of central nervous system disorders [US7659394] | 2005-11-03 | 2010-02-09 |
| Therapeutic pyrazolo[3,4-B]pyridines and indazoles [US7423054] | 2006-06-01 | 2008-09-09 |
| Amide derivatives as ion-channel ligands and pharmaceutical compositions and methods of using the same [US7312233] | 2006-09-14 | 2007-12-25 |
| Compounds useful in therapy [US7482375] | 2006-10-26 | 2009-01-27 |
| Therapeutic pyrazolo[3,4-b]pyridines and indazoles [US7485636] | 2006-09-28 | 2009-02-03 |
| Substituted N-sulfonylaminophenylethyl-2-phenoxyacetamide compounds as VR1 receptor antagonists [US7566739] | 2006-09-14 | 2009-07-28 |
| Amide derivatives as ion-channel ligands and pharmaceutical compositions and methods of using the same [US7576099] | 2006-08-31 | 2009-08-18 |
| Substituted sulfonylaminoarylmethyl cyclopropanecarboxamide as VR1 receptor antagonists [US7622589] | 2006-09-21 | 2009-11-24 |
| Alpha 2 Delta Ligands for Fibromyalgia and Other Disorders [US2009203782] | 2009-08-13 |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
[(3S,4S)-1-(aminomethyl)-3,4-dimethylcyclopentyl]acetic acid
|
|
| Identifiers | |
| CAS Registry Number | 223445-75-8 |
| ATC code | None |
| PubChem | CID: 9794485 |
| ChemSpider | 7970252 |
| UNII | JT7957Q2FB |
| ChEMBL | CHEMBL593430 |
| Chemical data | |
| Formula | C10H19NO2 |
| Molecular mass | 185.263 g/mol |
//////C[C@H]1CC(C[C@@H]1C)(CC(=O)O)CN
READ IMAGABALIN, PD 217074
USP revises Chapter on Pharmaceutical Water
DRUG REGULATORY AFFAIRS INTERNATIONAL
.jpg)
Changes to the fundamental monograph on pharmaceutical water <1231> Water for Pharmaceutical Purposes from the US-American Pharmacopeia have been published for comments in the Pharmacopeial Forum 41(5). The revision presented in the current draft mainly has a structural nature. The content of the monograph has been reorganised in 9 new chapters which aim at improving readibility and searchability of the content searched:
1. INTRODUCTION
2. SOURCE WATER CONSIDERATIONS
3. WATERS USED FOR PHARMACEUTICAL MANUFACTURING AND TESTING PURPOSES
4. VALIDATION AND QUALIFICATION OF WATER PURIFICATION, STORAGE, AND DISTRIBUTION SYSTEMS
5. DESIGN AND OPERATION OF PURIFIED WATER AND WATER FOR INJECTION SYSTEMS
6. SAMPLING
7. CHEMICAL EVALUATIONS
8. MICROBIAL EVALUATIONS
9. ALERT AND ACTION LEVELS AND SPECIFICATIONS
The draft document is available for free on the website of the USP Pharmacopeial Forum. You only need to register for free. The deadline for comments is 20 November 2015.
http://www.gmp-compliance.org/enews_5070_USP-revises-Chapter–1231–on-Pharmaceutical-Water_n.html
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorobiphenyl-2-carboxamide
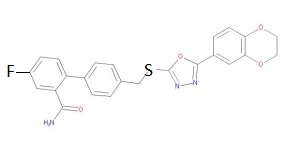
Cas 1820758-44-8
C24 H18 F N3 O4 S
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorobiphenyl-2-carboxamide


Glycogen synthase kinase-3 (GSK-3) is a constitutively active, ubiquitous serine/threonine kinase that takes part in a number of physiological processes ranging from glycogen metabolism to apoptosis. GSK-3 is a key mediator of various signaling pathways, such as the Wnt and the insulin/AKT signaling pathways.
Therefore, dysregulation of GSK-3 has been linked to various human diseases, such as cancer, diabetes, and neurodegenerative diseases.Two related isoforms of GSK-3 exist in mammals, GSK-3α and -β, which share a sequence identity within their catalytic domains of 98%.
Beyond the catalytic domains they show significant differences. Although these isoforms are structurally related, they are not functionally equivalent, and one cannot compensate for loss of the other.
The debate on the respective contributions of the isoforms GSK-3α and GSK-3β on the pathogenesis of different diseases is ongoing.
Various studies indicate that the therapies of certain diseases benefit from specific targeting of GSK-3α and GSK-3β. GSK-3α was recently identified as a differentiation target in acute myeloid leukemia (AML). AML is a hematopoietic malignancy defined by uncontrolled proliferation and disrupted myeloid differentiation. AML is the second most common form of leukemia in adults.
The current treatment of AML with conventional chemotherapy is very aggressive yet ineffective for the majority of patients with the disease.Thus, alternative targeted treatment approaches for AML are highly desirable. GSK-3α recently emerged as a potential target in this disease.
PAPER

The challenge for glycogen synthase kinase-3 (GSK-3) inhibitor design lies in achieving high selectivity for one isoform over the other. The therapy of certain diseases, such as acute myeloid leukemia (AML), may require α-isoform specific targeting. The scorpion shaped GSK-3 inhibitors developed by our group achieved the highest GSK-3α selectivity reported so far but suffered from insufficient aqueous solubility. This work presents the solubility-driven optimization of our isoform-selective inhibitors using a scorpion shaped lead. Among 15 novel compounds, compound 27 showed high activity against GSK-3α/β with the highest GSK-3α selectivity reported to date. Compound 27 was profiled for bioavailability and toxicity in a zebrafish embryo phenotype assay. Selective GSK-3α targeting in AML cell lines was achieved with compound 27, resulting in a strong differentiation phenotype and colony formation impairment, confirming the potential of GSK-3α inhibition in AML therapy
Evaluation of Improved Glycogen Synthase Kinase-3α Inhibitors in Models of Acute Myeloid Leukemia
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01200
http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.5b01200/suppl_file/jm5b01200_si_001.pdf
compound 27 as a colorless solid. HPLC: 96%, tR = 6.93 min.
1H NMR (DMSO-d6, 500 MHz, 300 K): δ (ppm) = 4.32 (td, J = 5.2 Hz, J = 3.7 Hz, 4H), 4.60 (s, 2H), 7.05 (d, J = 8.4 Hz, 1H), 7.25 (dd, J = 9.1 Hz, J = 2.7 Hz, 1H), 7.31 (td, J = 8.6 Hz, J = 2.8 Hz, 1H), 7.38 (m, 3H), 7.41 (d, J = 2.0 Hz, 1H), 7.45 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H), 7.49 (d, J = 8.2 Hz, 2H), 7.73 (s, 1H).
13C NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.1, 64.4, 114.3 (d, JC–F = 21 Hz), 115.0, 115.9 (d, JC–F = 21 Hz), 115.9, 118.1, 120.0, 128.6 (2C), 128.8 (2C), 132.0 (d, JC–F = 8 Hz), 134.8, 135.5, 138.9, 139.0 (d, JC–F = 7 Hz), 143.8, 146.7, 160.9 (d, JC–F = 247 Hz), 162.7, 164.9, 169.5.
EI-MS: m/z = 463 (100, [M+]), 464 (26, [M+ + H]), 465 (7, [M+ + 2H].
ABOUT Boris Schmidt
Boris Schmidt
-
Clemens Schöpf-Institute of Chemistry and BiochemistryDarmstadt, Hessen, Germany*Phone: +49 6151 163075. Fax: +49 6151 163278.E-mail: Schmidt_boris@t-online.de.
………………………………………….
ABOUT Theresa Neumann
-
Clemens Schöpf-Institute of Chemistry and BiochemistryDarmstadt, Hessen, Germany
////////FC(C=C1C(N)=O)=CC=C1C(C=C2)=CC=C2CSC3=NN=C(O3)C4=CC5=C(OCCO5)C=C4
New 5-Substituted-N-(piperidin-4-ylmethyl)-1H-indazole-3-carboxamides: Potent Glycogen Synthase Kinase-3 (GSK-3) Inhibitors in Model of Mood Disorders
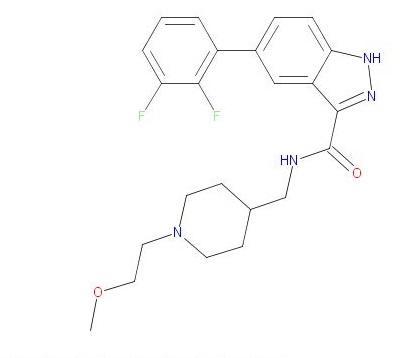
CAS 1452582-16-9, 428.47, C23 H26 F2 N4 O2
1H-Indazole-3-carboxamide, 5-(2,3-difluorophenyl)-N-[[1-(2-methoxyethyl)-4-piperidinyl]methyl]-
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
![]()
1 H-indazole-3-carboxamide compounds acting as glycogen synthase kinase 3 beta (GSK-33) inhibitors and to their use in the treatment of GSK-33-related disorders such as (i) insulin-resistance disorders; (ii) neurodegenerative diseases; (iii) mood disorders; (iv) schizophrenic disorders; (v) cancerous disorders; (vi) inflammation, (vii) substance abuse disorders; (viii) epilepsies; and (ix) neuropathic pain.
Protein kinases constitute a large family of structurally related enzymes, which transfer phosphate groups from high-energy donor molecules (such as adenosine triphosphate, ATP) to specific substrates, usually proteins. After phosphorylation, the substrate undergoes to a functional change, by which kinases can modulate various biological functions.
In general, protein kinases can be divided in several groups, according to the substrate that is phosphorylated. For example, serine/threonine kinase phosphorylates the hydroxyl group on the side chain of serine or threonine aminoacid.
Glycogen synthase kinases 3 (GSK-3) are constitutively active multifunctional enzymes, quite recently discovered, belonging to the serine/threonine kinases group.
Human GSK-3 are encoded by two different and independent genes, which leads to GSK-3a and GSK-33 proteins, with molecular weights of about 51 and 47 kDa, respectively. The two isoforms share nearly identical sequences in their kinase domains, while outside of the kinase domain, their sequences differ substantially (Benedetti et al., Neuroscience Letters, 2004, 368, 123-126). GSK-3a is a multifunctional protein serine kinase and GSK-33 is a serine-threonine kinase.
It has been found that GSK-33 is widely expressed in all tissues, with widespread expression in the adult brain, suggesting a fundamental role in neuronal signaling pathways (Grimes and Jope, Progress in Neurobiology, 2001, 65, 391-426). Interest in glycogen synthase kinases 3 arises from its role in various physiological pathways, such as, for example, metabolism, cell cycle, gene expression, embryonic development oncogenesis and neuroprotection (Geetha et al., British Journal Pharmacology, 2009, 156, 885-898).
GSK-33 was originally identified for its role in the regulation of glycogen synthase for the conversion of glucose to glycogen (Embi et al., Eur J Biochem, 1980, 107, 519-527). GSK-33 showed a high degree of specificity for glycogen synthase.
Type 2 diabetes was the first disease condition implicated with GSK- 3β, due to its negative regulation of several aspects of insulin signaling pathway. In this pathway 3-phosphoinositide-dependent protein kinase 1 (PDK-1 ) activates PKB, which in turn inactivates GSK-33. This inactivation of GSK-33 leads to the dephosphorylation and activation of glycogen synthase, which helps glycogen synthesis (Cohen et al., FEBS Lett, 1997, 410, 3-10). Moreover, selective inhibitors of GSK-33 are expected to enhances insulin signaling in prediabetic insulin- resistant rat skeletal muscle, thus making GSK-33 an attractive target for the treatment of skeletal muscle insulin resistance in the pre-diabetic state (Dokken et al., Am J. Physiol. Endocrinol. Metab., 2005, 288, E1 188-E1 194).
GSK-33 was also found to be a potential drug target in others pathological conditions due to insulin-resistance disorders, such as syndrome X, obesity and polycystic ovary syndrome (Ring DB et al., Diabetes, 2003, 52: 588-595).
It has been found that GSK-33 is involved in the abnormal phosphorylation of pathological tau in Alzheimer’s disease (Hanger et al., Neurosci. Lett, 1992, 147, 58-62; Mazanetz and Fischer, Nat Rev Drug Discov., 2007, 6, 464-479; Hong and Lee, J. Biol. Chem., 1997, 272, 19547- 19553). Moreover, it was proved that early activation of GSK-33, induced by apolipoprotein ApoE4 and β-amyloid, could lead to apoptosis and tau hyperphosphorylation (Cedazo-Minguez et al., Journal of Neurochemistry, 2003, 87, 1 152- 1 164). Among other aspect of Alzheimer’s disease, it was also reported the relevance of activation of GSK-33 at molecular level (Hernandez and Avila, FEBS Letters, 2008, 582, 3848-3854).
Moreover, it was demonstrated that GSK-33 is involved in the genesis and maintenance of neurodegenerative changes associated with Parkinson’s disease (Duka T. et al., The FASEB Journal, 2009; 23, 2820- 2830).
Accordingly to these experimental observations, inhibitors of GSK-33 may find applications in the treatment of the neuropathological consequences and the cognitive and attention deficits associated with tauopathies; Alzheimer’s disease; Parkinson’s disease; Huntington’s disease (the involvement of GSK-33 in such deficits and diseases is disclosed in Meijer L. et al., TRENDS Pharm Sci, 2004; 25, 471 -480); dementia, such as, but not limited to, vascular dementia, post-traumatic dementia, dementia caused by meningitis and the like; acute stroke; traumatic injuries; cerebrovascular accidents; brain and spinal cord trauma; peripheral neuropathies; retinopathies and glaucoma (the involvement of GSK-33 in such conditions is disclosed in WO 2010/109005).
The treatment of spinal neurodegenerative disorders, like amyotrophic lateral sclerosis, multiple sclerosis, spinal muscular atrophy and neurodegeneration due to spinal cord injury has been also suggested in several studies related to GSK-33 inhibition, such as, for example in Caldero J. et al., “Lithium prevents excitotoxic cell death of motoneurons in organotypic slice cultures of spinal cord”, Neuroscience. 2010 Feb 17;165(4):1353-69, Leger B. et al., “Atrogin-1 , MuRF1 , and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients”, Muscle Nerve, 2009 Jul; 40(1 ):69-78, and Galimberti D. et al., “GSK33 genetic variability in patients with Multiple Sclerosis”, Neurosci Lett. 201 1 Jun 1 5;497(1 ):46- 8. Furthermore, GSK-33 has been linked to the mood disorders, such as bipolar disorders, depression, and schizophrenia.
Inhibition of GSK-33 may be an important therapeutic target of mood stabilizers, and regulation of GSK-33 may be involved in the therapeutic effects of other drugs used in psychiatry. Dysregulated GSK-33 in mood disorder, bipolar disorder, depression and schizophrenia could have multiple effects that could impair neural plasticity, such as modulation of neuronal architecture, neurogenesis, gene expression and the ability of neurons to respond to stressful, potentially lethal conditions (Jope and Ron, Curr. Drug Targets, 2006, 7, 1421- 1434).
The role of GSK-33 in mood disorder was highlighted by the study of lithium and valproate (Chen et al., J. Neurochem., 1999, 72, 1327- 1330; Klein and Melton, Proc. Natl. Acad. Sci. USA, 1996, 93, 8455-8459), both of which are GSK-33 inhibitors and are used to treat mood disorders. There are also existing reports from the genetic perspective supporting the role of GSK-33 in the disease physiology of bipolar disorder (Gould, Expert. Opin. Ther. Targets, 2006, 10, 377-392).
It was reported a decrease in AKT1 protein levels and its phosphorylation of GSK-33 at Serine-9 in the peripheral lymphocytes and brains of individuals with schizophrenia. Accordingly, this finding supports the proposal that alterations in AKT1 -GSK-33 signaling contribute to schizophrenia pathogenesis (Emamian et al., Nat Genet, 2004, 36, 131- 137).
Additionally, the role of GSK-33 in cancer is a well-accepted phenomenon.
The potential of small molecules that inhibit GSK-33 has been evidenced for some specific cancer treatments (Jia Luo, Cancer Letters, 2009, 273, 194-200). GSK-33 expression and activation are associated with prostate cancer progression (Rinnab et al., Neoplasia, 2008, 10, 624-633) and the inhibition of GSK3b was also proposed as specific target for pancreatic cancer (Garcea et al., Current Cancer Drug Targets, 2007, 7, 209-215) and ovarian cancer (Qi Cao et al., Cell Research, 2006, 16 671 -677). Acute inhibition of GSK-33 in colon-rectal cancer cells activates p53-dependent apoptosis and antagonizes tumor growth (Ghosh et al., Clin Cancer Res 2005, 1 1 , 4580-4588).
The identification of a functional role for GSK-33 in MLL-associated leukaemia suggests that GSK-33 inhibition may be a promising therapy that is selective for transformed cells that are dependent on HOX overexpression (Birch et al., Cancer Cell, 2010, 1 7, 529-531 ).
GSK-33 is involved in numerous inflammatory signalling pathways, for example, among others GSK-33 inhibition has been shown to induce secretion of the anti-inflammatory cytokine IL-1 0. According to this finding, GSK-33 inhibitors could be useful to regulate suppression of inflammation (G. Klamer et al., Current Medicinal Chemistry, 2010, 17(26), 2873-2281, Wang et al., Cytokine, 2010, 53, 130-140).
GSK-33 inhibition has been also shown to attenuate cocaine-induced behaviors in mice. The administration of cocaine in mice pretreated with a GSK-33 inhibitor demonstrated that pharmacological inhibition of GSK3 reduced both the acute behavioral responses to cocaine and the long- term neuroadaptations produced by repeated cocaine (Cocaine-induced hyperactivity and sensitization are dependent on GSK3, Miller JS et al. Neuropharmacology. 2009 Jun; 56(8):1 1 16-23, Epub 2009 Mar 27).
The role of GSK-33 in the development of several forms of epilepsies has been demonstrated in several studies, which suggest that inhibition of GSK-33 could be a pathway for the treatment of epilepsy (Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy, Lohi H et al., Hum Mol Genet. 2005 Sep 15;14(18):2727-36 and Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease, Purl R et al., J Biol Chem. 2009 Aug 21 ;284(34) 22657-63). The relationship between GSK-33 inhibition and treatment of neuropathic pain has been demonstrated in Mazzardo-Martins L. et al., “Glycogen synthase kinase 3-specific inhibitor AR-A014418 decreases neuropathic pain in mice: evidence for the mechanisms of action”, Neuroscience. 2012 Dec 13;226, and Xiaoping Gu et al., “The Role of Akt/GSK33 Signaling Pathway in Neuropathic Pain in Mice”, Poster A525, Anesthesiology 2012 October 13-17, 2012 Washington.
A review on GSK-33, its function, its therapeutic potential and its possible inhibitors is given in “GSK-33: role in therapeutic landscape and development of modulators” (S. Phukan et al., British Journal of Pharmacology (2010), 160, 1- 19).
WO 2004/014864 discloses 1 H-indazole-3-carboxamide compounds as selective cyclin-dependant kinases (CDK) inhibitors. Such compounds are assumed to be useful in the treatment of cancer, through a mechanism mediated by CDK2, and neurodegenerative diseases, in particular Alzheimer’s disease, through a mechanism mediated by CDK5, and as anti-viral and anti-fungine, through a mechanism mediated by CDK7, CDK8 and CDK9.
Cyclin-dependant kinases (CDKs) are serine/threonine kinases, first discovered for their role in regulating the cell cycle. CDKs are also involved in regulating transcription, mRNA processing, and the differentiation of nerve cells. Such kinases activate only after their interaction and binding with regulatory subunits, namely cyclins.
Moreover, 1 H-indazole-3-carboxamide compounds were also described as analgesics in the treatment of chronic and neuropathic pain (see, for example, WO 2004/074275 and WO 2004/101 548) and as 5-HT4 receptor antagonists, useful in the treatment of gastrointestinal disorders, central nervous system disorders and cardiovascular disorders (see, for example, WO 1994/101 74).
Patent
WO 2013124158
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
SEE ENTRY 8

DMSO-de; δ 13.09 (s, 1 H), 8.23-8.42 (m, 2H), 7.72 (dd, J=0.82, 8.69 Hz, 1 H), 7.55 (td, J=1.76, 8.74 Hz, 1 H), 7.24-7.49 (m, 3H), 3.40 (t, J=6.04 Hz, 2H), 3.22 (s, 3H), 3.18 (d, J=6.40 Hz, 2H), 2.84 (d, J=11.53 Hz, 2H), 2.42 (t, J=5.95 Hz, 2H), 1.82- 2.02 (m, 2H), 1.41 -1.71 (m, 3H), 1.06-1.31 (m, 2H)
PAPER

Hit Optimization of 5-Substituted-N-(piperidin-4-ylmethyl)-1H-indazole-3-carboxamides: Potent Glycogen Synthase Kinase-3 (GSK-3) Inhibitors with in Vivo Activity in Model of Mood Disorders
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01208

http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01208
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
Angelini S.p.A., Angelini Research Center,
![]()
/////
COCCN1CCC(CNC(=O)c2n[nH]c3ccc(cc23)c4cccc(F)c4F)CC1
AZD 1080
 .
.
AZD 1080
2-Hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]-1H-indole-5-carbonitrile
2-hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]1H-indole-5-carbonitrile
AZD1080 is a selective, orally active, brain permeable GSK3 inhibitor, inhibits human GSK3α and GSK3β with Ki of 6.9 nM and 31 nM, respectively, shows >14-fold selectivity against CDK2, CDK5, CDK1 and Erk2.
Cas 612487-72-6, AZD1080,
AZD-1080, a glycogen synthase kinase 3 (GSK-3) inhibitor, had been in early clinical trials for the treatment of Alzheimer’s type dementia by AstraZeneca
| Astrazeneca Ab |
PATENTS
WO 2003082853
http://www.google.com/patents/WO2003082853A1?cl=en
PAPER
Organic Process Research & Development (2008), 12(3), 540-543.
http://pubs.acs.org/doi/abs/10.1021/op800020r

A mild and robust method for the large-scale palladium-catalysed cyanation of aryl bromides has been developed. The reaction is sensitive to cyanide poisoning of the catalyst, and it was found that the order of adding the reagents had a strong impact on the performance of the reaction. Addition of the cyanide source to a preheated mixture of the other reagents was critical for achieving a robust and scaleable process. This improved protocol allowed the reaction to be run to full conversion within 3 h at 50 °C on a 6.7 kg scale. Furthermore, it led to the identification of several new efficient catalysts for the reaction.
2-hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]1H-indole-5-carbonitrile (2) (5.2 kg, 15.6 mol), 90% yield with a purity of >90% by HPLC. 1H NMR (d6-DMSO, 400 MHz) δ 14.79 (broad s, 1H), 10.86 (broad s, 1H), 8.08 (s, 1H), 7.95 (s, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.27 (dd,J = 8.0, 0.9 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 3.57 (t, J = 4.4 Hz, 4H), 3.36 (s, 2H), 2.36 (broad s, 4H); 13C NMR (d6-DMSO, 100 MHz) δ 168.8, 148.6, 141.8, 137.0, 136.1, 125.4, 123.9, 122.3, 121.1, 118.8, 118.3, 108.7, 101.3, 84.6, 66.1, 58.4, 52.8. MS (ES) m/z [M + 1] 335.
PAPER
Topics in Organometallic Chemistry (2012), 42(Organometallics as Catalysts in the Fine Chemical Industry), 125-134.
http://link.springer.com/chapter/10.1007%2F3418_2011_25
PATENT
https://www.google.co.in/patents/WO2007089193A1?cl=en

In the above scheme, preferably Rl is bromo and X is chloro.
Synthesis of 2-Hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl] lH-indole-5-carbonitrile citrate
Example 14
2-Hydroxy-3-r5-(moφholin-4-ylmethyl)pyridin-2-yl1 lH-indole-5-carbonitrile citrate salt 2-Hydroxy-3-[5-(moφholin-4-ylmethyl)pyridin-2-yl] lH-indole-5-carbonitrile (5.14 kg, 15.4 mol) was suspended in ethanol (54 L) at room temperature. The suspension was heated to an inner temperature of 700C and a solution of citric acid (3.424 kg, 17.82 mol, 1.300 eq)) in water (103 L) was added keeping the inner temperature above 650C. The mixture was heated to reflux. After this the resulting solution was mixed with activated charcoal (0.412 kg) and reflux continued for 3.5 h after which the reaction mixture was clear filtered at 830C followed by cooling to room temperature over 20 h. After filtration the precipitate was washed twice with a cold mixture of ethanol/water (6.9 L/13.7 L). Drying under vacuum at 5O0C gave 6.648 kg, 82.2% yield of 2-hydroxy-3-[5-(morpholin- 4-ylmethyl)pyridin-2-yl]lH-indole-5-carbonitrile citrate having a purity of at least 98%. The palladium content was less than 1 ppm and the zinc content was lower than 10 ppm. 1H NMR (Jd-DMSO3 400 MHz) δ 14.7 (br s, 1 H), 11.55 (s, 1 H), 10.98 (s, IH), 8.31 (s, 1 H), 8.08 (br d, J= 1.84Hz, IH), 8.02 (s, IH), 7.90 (br d, J = 8.92Hz, 1 H), 7.31 (d, J = 8.0 Hz, 1 H), 7.02 (d, J= 8.0Hz), 4.28 (s, 2 H), 3.97 (m, 2 H), 3.94 (m, 2H), 3.35 (m, 9H), 3.32 (m, 2H) ppm; 13C NMR (d6-DMSO, 400MHz) δ 168.9, 148.5, 142.7, 139.8, 137.5,126.4, 124.9, 124.8, 120.9, 119.4, 118.4, 113.3, 109.0, 101.6, 85.7, 63.1, 55.5, 50.3, 40.1, 39.9, 39.7, 39.2, 39.0, 38.8ppm; MS (ES) m/z [M++l] 335.
LIK 066, Licogliflozin diprolinate
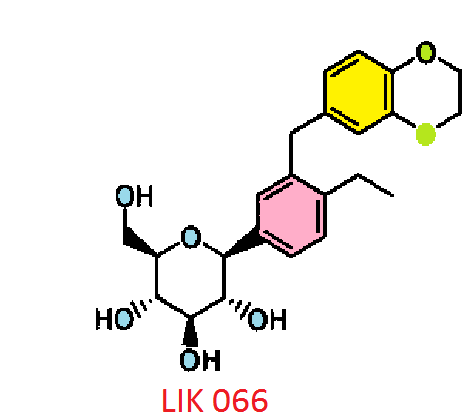
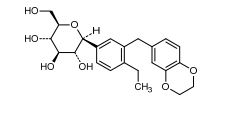
Licogliflozin
LIK 066
Licogliflozin diprolinate

LIK-066, a new flozin on the horizon
C23 H28 O7 . 2 C6 H11 N O, 642.7795, 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C…WO2011048112
CAS 1291095-45-8, (1S)-1,5-anhydro-1-C-[3-[(2,3-dihydro-1,4-benzodioxin-6-yl)methyl]-4-ethylphenyl]-D-glucitol (1:1) WITH L-Proline, compd., 1:1 Proline Co-crvstal , 1:1 Proline Co-crvstal …..…WO2011048112
CAS BASE 1291094-73-9, 416.46, C23 H28 O7
(1S)-1,5-Anhydro-1-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-D-glucitol bis[1-[(2S)-pyrrolidin-2-yl]ethanone]
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Sodium glucose transporter-2 inhibitor
SGLT 1/2 inhibitor
Novartis Ag innovator
Clinical trial……..https://clinicaltrials.gov/ct2/show/NCT01915849
https://clinicaltrials.gov/ct2/show/NCT02470403
- 10 Jun 2015 Novartis initiates enrolment in a phase II trial for Type 2 diabetes mellitus in USA (NCT02470403)
- 02 Apr 2014 Novartis terminates a phase II trial in Type-2 diabetes mellitus in USA, Poland, Argentina, Hungary, Puerto Rico and South Africa (NCT01824264)
- 01 Jan 2014 Novartis completes a phase II trial in Type 2 diabetes mellitus in USA (NCT01915849)
Licogliflozin, a SGLT-1/2 inhibitor, is in phase II clinical development at Novartis for the treatment of metabolic disorders, for the treatment of heart failure in patients with type 2 diabetes, for the treatment of obesity and for the treatment of polycystic ovary syndrome (PCOS) in overweight and obese women. Phase II trials for the treatment of type 2 diabetes had been discontinued.
EMA/415156/2014 European Medicines Agency decision P/0183/2014 of 24 July 2014 on the agreement of a paediatric investigation plan and on the granting of a deferral and on the granting of a waiver for (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3- dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran3,4,5-triol (2:1) (LIK066) (EMEA-001527-PIP01-13) in accordance with Regulation (EC) No 1901/2006 of the European Parliament and of the Council
1. Opinion of the Paediatric Committee on the agreement of a Paediatric Investigation Plan and a deferral and a waiver. 2014, EMEA-001527-PIP01-13 (here) [ Novartis revealed the IUPAC name here].
Where name is given
http://www.who.int/medicines/publications/druginformation/issues/DrugInformation2017_Vol31-4/en/


http://www.who.int/medicines/publications/druginformation/issues/PL_118.pdf?ua=1
SEE ALSO


LIK-066 is in phase II clinical studies at Novartis for the treatment of type 2 diabetes.
In June 2014, the EMA’s PDCO adopted a positive opinion on a pediatric investigation plan (PIP) for LIK-066 for type 2 diabetes
Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition. Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
Type 1 diabetes, also known as Insulin Dependent Diabetes Mellitus (IDDM), is characterized by loss of the insulin-producing β-cells of the islets of Langerhans of the pancreas leading to a deficiency of insulin. Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) – is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin). Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from β-cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
Chronic elevation of blood glucose level also leads to damage of blood vessels. In diabetes, the resultant problems are grouped under “microvascular disease” (due to damage of small blood vessels) and “macro vascular disease” (due to damage of the arteries). Examples of microvascular disease include diabetic retinopathy, neuropathy and nephropathy, while examples of macrovascular disease include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
Diabetic retinopathy, characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US. Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy. Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide. A high glycemic diet (i.e., a diet that consists of meals that give high postprandial blood sugar) is known to be one of the causative factors contributing to the development of obesity.
Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level. Therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
Glucose is unable to diffuse across the cell membrane and requires transport proteins. The transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase. Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
SGLT belongs to the sodium/glucose co-transporter family SLCA5. Two different SGLT isoforms, SGLT1 and SGLT2, have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different. SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
As a consequence, glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments. Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule. In healthy human, more than 99% of plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1 % of the total filtered glucose being excreted in urine. It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule. A drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient’s needs for both blood glucose control, while preserving insulin secretion. In addition, SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
Indeed small molecule SGLT2 inhibitors have been discovered and the anti-diabetic therapeutic potential of such molecules has been reported in literature [T-1095 (Diabetes, 1999, 48, 1794-1800, Dapagliflozin (Diabetes, 2008, 57, 1723-1729)].
SYNTHESIS
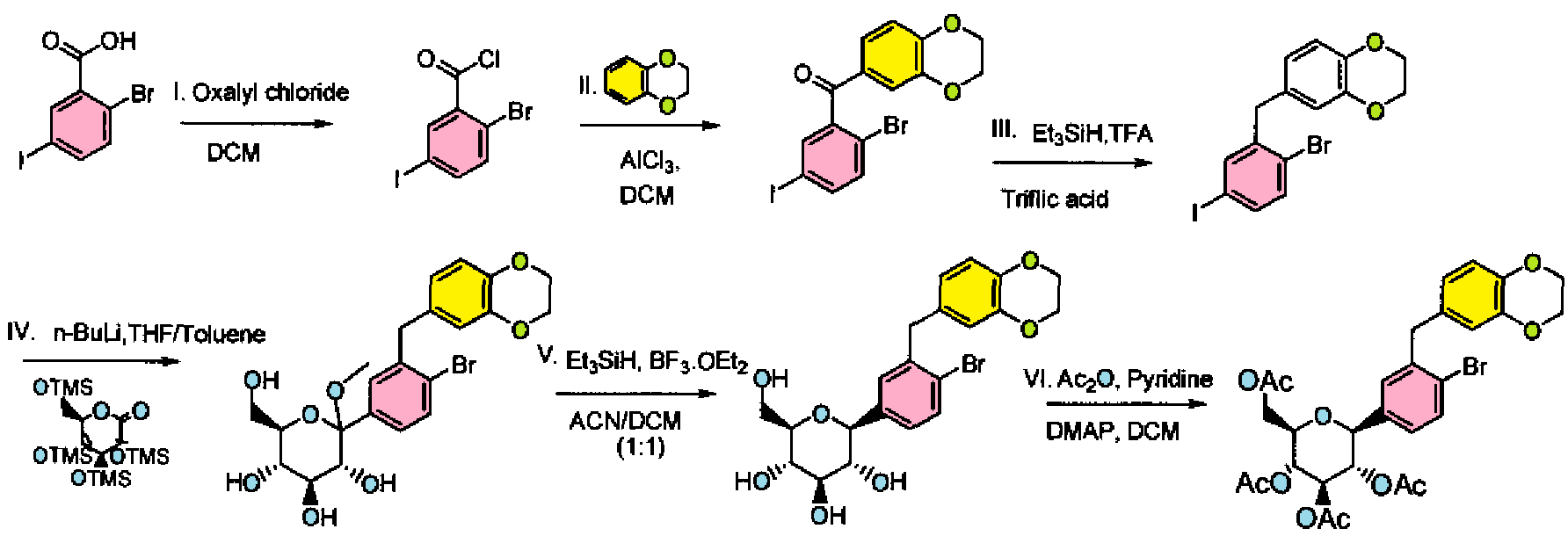
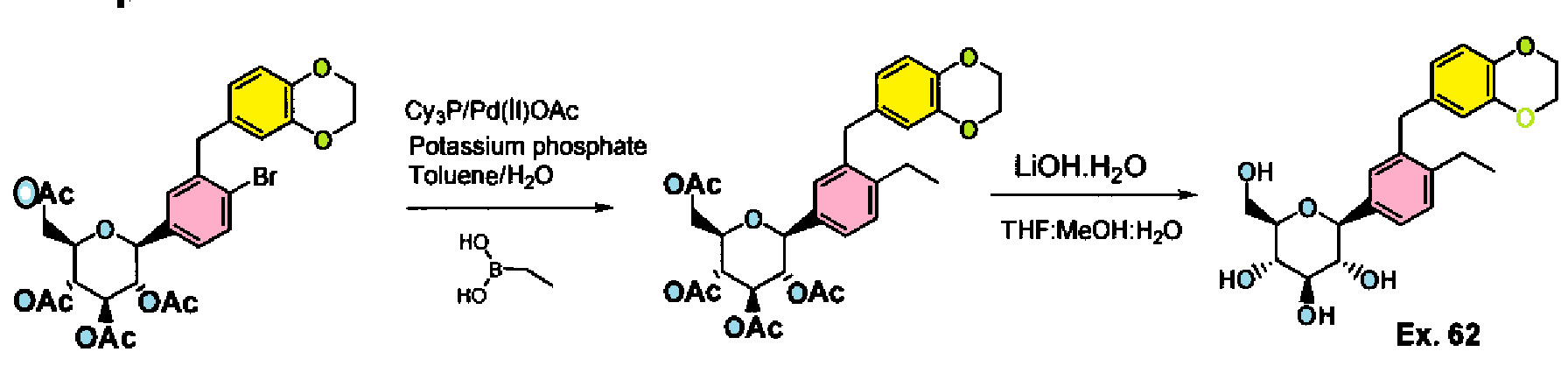
PATENT
WO 2011048112
https://www.google.com/patents/WO2011048112A1?cl=en
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
Example 61-62:

Ex. 61
Example 61 : Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 mL), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Example 62: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 mL) and washed with brine (75 mL), brine containing 5 mL of 5% aqueous KHS04 (75 mL), and brine (20 mL) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.59)
H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J – 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m z 434.2 (M+18).
PICK UP IDEAS FROM HERE
Examples 57-58:

Ex. 57 Ex. 58
Step I: To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
Step II: To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2). The dichloromethane layers were combined and washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL X 2), and brine (200 mL), then dried over sodium sulfate and concentrated. The solid product was triturated with hexanes, and the triturated product was dried under vacuum to furnish (2-bromo-5-iodo-phenyl)-(2,3- dihydro-benzo[1 ,4]dioxin-6-yl)-methanone (30 g).
1H N R (400 MHz, DMSO-D6): δ 4.29-4.37 (m, 4H), 7.02 (d, J = 8.4 Hz, 1 H), 7.16 (d, J = 2.4 Hz, 1 H), 7.18-7.19 (m, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.77-7.81 (m, 1 H), 7.82 (d, J = 2.0 Hz, 1 H).
Step III: To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution (200 mL X 2), water (200 mL), and brine (200 mL), then dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish 6-(2-bromo-5-iodo-benzyl)-2,3- dihydro-benzo[1 ,4]dioxine (26.5 g). H NMR (400 MHz, DMSO-D6): δ 3.90 (s, 4H), 4.2 (s, 2H), 6.65 (dd, J = 8.4 Hz, J = 2.0 Hz, H), 6.68 (d, J = 2.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, H), 7.39 (d, J = 8.4 Hz, 1 H), 7.50 (dd, J = 8.4 Hz, J = 2.4 Hz 1 H), 7.67 (d, J = 2.8 Hz, 1 H).
Step IV: To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C. The reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C. After stirring for 1 h, 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq. NaHC03 solution (~75 mL) and extracted with ethyl acetate (250 mL X 3), dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish (3R,4S,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (28.4 g)
Example 57: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-1 ,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
Step V: To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C. After stirring for 4 h at 10°C, the reaction was quenched with saturated aqueous sodium bicarbonate (~ 100 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 X 150 mL). The organic layers were combined and dried over sodium sulfate, concentrated to furnish (3R,4R,5S,6R)-2- [4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol (28.4 g). Crude product was used for next reaction without purification. Example 58: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2!3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate Step V: To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature. After stirring for 2 h, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (500ml) and washed with 1 N HCI (200 mL X 2) followed by brine (200ml), then dried over sodium sulfate and
concentrated. The resulting crude compound was dissolved in ethanol (320 mL) at 65 °C and allowed to cool to room temperature while stirring. Light yellow solid formed was filtered and washed with cold ethanol (150 mL) followed by hexane (200 mL) to get acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin- 6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester powder (22.5 g, purity 98%).
COCRYSTAL
Example 75: 1:1 Proline Co-crvstal with f2S.3R.4R.5S.6R¾-2-r3-f2.3-Dihvdro- benzori.41dioxin-6-ylmethyl)-4-ethyl-phenvn-6-hvdroxymethyl-tetrahydro-pyran- 3.4.5-triol
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62) was completely amorphous initially but formed a crystalline complex with proline. This was confirmed by powder X-ray diffraction (PXRD) analysis. The stiochiometry of Example 62 and L- proline in the co-crystal prepared by method 1 was found to be 1 :1 by NMR
spectroscopy & HPLC. Characterization data for co-crystals of Example 62 and proline prepared by method 1 is shown in Table 3. Relative intensities of the most prominent powder x-ray diffraction peaks for co-crystals of Example 62 and proline are shown in Table 3A.
Table 3

Table 3A

3.70 15.78 18.36 25.18
9.68 10.68 18.88 36.33
11.07 21.21 20.42 69.29
14.26 14.81 21.18 27.94
14.80 22.97 22.50 12.25
15.40 4 98 23.78 33.08
16.12 8.45 24.56 6.92
16.59 18.78 25.79 21.69
17.31 100.0 27.46 8.90
17.60 20.35 31.97 7.65
17.98 47.20 32.46 5.98
1:1 Proline Co-crvstal
Example 77: 1:1 Proline Co-crvstal with (2S.3R.4R.5S.6Ri-2-f3-(2.3-Dihvdro- benzoh .41dioxin-6-ylmethvh-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 2:
1 :1 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1500mg,3.6mmol), L- proline (415mg, 3.6mmol) and ethanol (23 ml_) were added to a 50 mL 3-neck round bottom flask equipped with nitrogen purging, magnetic stirring bar,
thermometer pocket & calcium chloride guard tube and the mixture was stirred at 25-30°C for 30 min., then heat to reflux. A clear solution was observed which was refluxed for 30 min., then slowly cool to 25-30°C causing percipitation. Di- isopropyl ether (DIPE, 23 mL) was added while maintaining the mixture at 25-30°C and stirring continuously for additional one to two hours at the same temperature. The precipitate was collected by filtration using vacuum (Nitrogen atmosphere), and the filter cake was washed with ethanol-DIPE mixture (1 :1 v/v, 10ml) followed by DIPE (23 mL). The product was vacuum dried at 65-70°C for 5-6 hrs.
1:1 Proline Co-crvstal (ΔΗ 53 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig. 1. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 2.
2:1 Proline Co-crvstal
Example 78: 2:1 Proline Co-crvstal with f2S.3R.4R.5S.6R>-2-r3-f2.3-Pihvdro-benzof1.41dioxin-6-ylmethvH-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 3: 1 :2 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1 kg) was added to 15 L of ethanol with agitation while maintaining the mixture at 20-25 °C. The mixture was stirred for 10 min at 20-25 °C, then L-proline (537 gm) was added while maintaining the mixture at 20-25 °C. The mixture was stirred at this temperature for 30 min., then heated to reflux and refluxed for 30 min. The mixture was slowly cooled to 25-30°C then stired for 1 hr. DIPE (15 L) was added while maintaining the temperature at 25-30 °C and the mixture was stirred at this temperature for 1 hr. The precipitated product was collected by filtration and the product was washed with DIPE (5 L). The product was air dried at 65-70 °C to yield 1.22 kg
(79%) of a 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C (ΔΗ 85 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig.
3. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 4. Relative
intensities of the most prominent powder x-ray diffraction peaks for the 1 :2 co- crystals of Example 62 and proline are shown in Table 5.
Table 5

PATENT
WO 2012140597
http://www.google.co.in/patents/WO2012140597A1?cl=en
. TABLE 2:

Intermediate 2: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-

Intermediate 2
Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- 4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for 1 h at same temperature, reaction was quenched with the addition of 1 N HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic layers were washed with brine (10 ml), dried over sodium sulfate, concentrated and purified by preparative HPLC to give 220 mg of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J = 6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H), 3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H), 4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57 (dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m, 3H). MS (ES) m/z 553.3 (M+1 ).
Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2 mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it was stirred at room temperature for 20 min. Reaction mixture was cooled to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5 ml). The reaction mixture was stirred at same temperature for 5 min and then at room temperature for 2 h. Reaction mixture was cooled to 0°C and quenched by the addition of 10% sodium bisulfite solution (5 ml). This was extracted with ether (3 X 10 ml). Combined organic layer was washed with brine (5 ml), dried over sodium sulfate and concentrated to give 700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6- [3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro- pyran-2-ylmethyl ester.
To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.
![]()
PATENT
SEE INDIAN PATENT
IN 2009DE02173
Glycoside derivatives and uses thereof
REFERENCES
Pediatric investigation plan (PIP) decision: (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2:1) ( LIK066) (EMEA-001527-PIP01-13)
European Medicines Agency (EMA) Web Site 2014, July 24
Safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) assessment of LIK066 in healthy subjects and in patients with type 2 diabetes mellitus (T2DM) (NCT01407003)
ClinicalTrials.gov Web Site 2011, August 07
IN 2009DE02173
| WO2001016147A1 | 24 Aug 2000 | 8 Mar 2001 | Kissei Pharmaceutical | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 | 2 Oct 2000 | 19 Apr 2001 | Bruce Ellsworth | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 | 15 Mar 2001 | 20 Sep 2001 | Hideki Fujikura | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 | 29 Mar 2001 | 11 Oct 2001 | Squibb Bristol Myers Co | O-aryl glucoside sglt2 inhibitors and method |
| WO2003020737A1 | 5 Sep 2002 | 13 Mar 2003 | Squibb Bristol Myers Co | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003043985A1 | 20 Nov 2002 | 30 May 2003 | Andrew Thomas Bach | Heterocyclic compounds and methods of use |
| WO2004018491A1 | 21 Aug 2003 | 4 Mar 2004 | Nobuhiko Fushimi | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004078163A2 | 26 Feb 2004 | 16 Sep 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004080990A1 | 12 Mar 2004 | 23 Sep 2004 | Kazuhiro Ikegai | C-glycoside derivatives and salts thereof |
| WO2004099230A1 | 30 Apr 2004 | 18 Nov 2004 | Eikyu Yoshiteru | Monosaccharide compounds |
| WO2004103995A1 | 19 May 2004 | 2 Dec 2004 | Gary Michael Ksander | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2005011592A2 | 29 Jul 2004 | 10 Feb 2005 | Janssen Pharmaceutica Nv | Substituted indazole-o-glucosides |
| WO2005021566A2 | 20 Aug 2004 | 10 Mar 2005 | Barsoumian Edward Leon | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085237A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085265A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006011502A1 | 27 Jul 2005 | 2 Feb 2006 | Chugai Pharmaceutical Co Ltd | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006054629A1 | 17 Nov 2005 | 26 May 2006 | Kissei Pharmaceutical | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2008016132A1 | 3 Aug 2007 | 7 Feb 2008 | Daiichi Sankyo Co Ltd | Benzyl phenyl glucopyranoside derivative |
| WO2011048112A1 * | 19 Oct 2010 | 28 Apr 2011 | Novartis Ag | Glycoside derivatives and uses thereof |
| US20030114390 * | 4 Oct 2002 | 19 Jun 2003 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| US20040018998 | 21 Sep 2001 | 29 Jan 2004 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US20060009400 | 28 Jun 2005 | 12 Jan 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948 | 15 Jul 2005 | 26 Jan 2006 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349 | 27 Jul 2005 | 2 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060035841 | 9 Aug 2005 | 16 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031 | 30 Sep 2005 | 6 Apr 2006 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060293252 | 14 Aug 2006 | 28 Dec 2006 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014 | 26 Jul 2007 | 31 Jan 2008 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015032272A1 * | 19 Aug 2014 | 12 Mar 2015 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9034921 | 1 Jun 2012 | 19 May 2015 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
INVENTORS OF LIK 066
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
| BEBERNITZ, Gregory, Raymond; (US). BOCK, Mark, G.; (US). REDDY, Dumbala Srinivas; (IN). HAJARE, Atul Kashinath; (IN). VYAVAHARE, Vinod; (IN). BHOSALE, Sandeep Bhausaheb; (IN). KURHADE, Suresh Eknath; (IN). SALUNKHE, Videsh; (IN). SHAIKH, Nadim, S.; (IN). BHUNIYA, Debnath; (IN). PALLE, P., Venkata; (IN). FENG, Lili; (US). LIANG, Jessica; (US) |
 Mark G Bock
Mark G Bock
BEBERNITZ, Gregory, Raymond….PIC NOT AVAILABLE

Dr. Srinivasa Reddy
 NADEEM SHAIKH
NADEEM SHAIKH
 Venkata Palle
Venkata Palle
ONLY FEW…………………….
//////Licogliflozin diprolinate
see……..http://medcheminternational.blogspot.in/2015/11/lik-066-novartis-for-treatment-of-type.html
