
Home » Uncategorized (Page 8)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
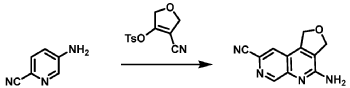
Bretisilocin
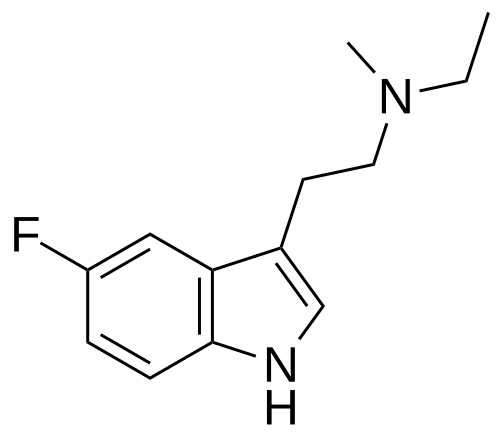
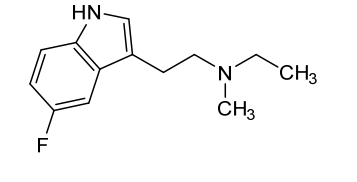
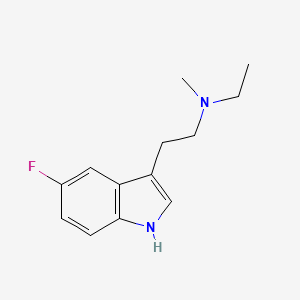
Bretisilocin
CAS2698331-35-8
MF C13H17FN2 MW220.29 g/mol
N-ethyl-2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine
serotonin (5-HT2A) receptor agonist, GM-2505, GM 2505, 5-Fluoro-N-methyl-N-ethyltryptamine, 5F-MET, 5-F-MET, 5-Fluoro-MET, DS425RQ8SX
Bretisilocin, also known by its developmental code name GM-2505 and as 5-fluoro-N-methyl-N-ethyltryptamine (5F-MET or 5-fluoro-MET), is a serotonergic psychedelic of the tryptamine family which is under development for the treatment of major depressive disorder.[1][7][2][3] It is an analogue of dimethyltryptamine (DMT) and is the 5-fluorinated derivative of methylethyltryptamine (MET).[8] Bretisilocin’s route of administration is intravenous infusion.[1][2][3][4]
The drug acts as a potent and well-balanced serotonin 5-HT2A and 5-HT2C receptor agonist, serotonin 5-HT2B receptor partial agonist or antagonist, and serotonin releasing agent.[2][9][8][10] It produces psychedelic-like effects in animals and similarly produces robust hallucinogenic effects in humans.[9][3] The duration of bretisilocin is 60 to 90 minutes and is intermediate between the durations of DMT and psilocybin.[6][11][4][12][8][2] It has been regarded by its developer as an “improved version of DMT”.[12]
Bretisilocin was first described in the literature by 2022.[9][10] It is under development by Gilgamesh Pharmaceuticals.[1] As of June 2025, the drug is in phase 2 clinical trials for the treatment of major depressive disorder.[1] Bretisilocin was acquired from Gilgamesh Pharmaceuticals by AbbVie in a deal worth up to $1.2 billion in August 2025.[13][14] It was encountered as a novel recreational designer drug in 2026.[5]
Chemistry
Bretisilocin, also known as 5-fluoro-N-methyl-N-ethyltryptamine, is a substituted tryptamine derivative.[8] It is a derivative of dimethyltryptamine (DMT) and methylethyltryptamine (MET) as well as of 5-fluorotryptamine (5-FT).[6][8]
Synthesis
The chemical synthesis of bretisilocin has been described.[10]
Analogues
Some analogues of bretisilocin include 5-fluoro-DMT, 5-fluoro-DET, 5-fluoro-EPT, 5-chloro-DMT, 5-bromo-DMT, 5-fluoro-AMT, 5-fluoro-AET, 5-MeO-MET, and 7-F-5-MeO-MET, among others.
History
Bretisilocin was first described in the scientific literature by at least 2022.[9][10] It was patented by Jason Wallach and colleagues at the University of the Sciences in Philadelphia that year.[10] The drug was encountered as a novel recreational designer drug in March 2026.[5]
Society and culture
Names
Bretisilocin is the generic name of the drug and its INNTooltip International Nonproprietary Name.[16] It is also known by its developmental code name GM-2505.[1][9][3]
Legal status
Canada
Bretisilocin is not a controlled substance in Canada as of 2025.[17]
United States
Bretislocin is not an explicitly controlled substance in the United States.[18] However, it could be considered a controlled substance under the Federal Analogue Act if intended for human consumption.
Research
Bretisilocin is under development as a potential pharmaceutical drug by Gilgamesh Pharmaceuticals.[1] As of June 2025, it is in phase 2 clinical trials for the treatment of major depressive disorder.[1] A phase 2a trial of bretisilocin for major depressive disorder has been completed and the efficacy and safety data for the trial have been released.[1][19][20][21] The drug has since been acquired from Gilgamesh Pharmaceuticals by AbbVie in a deal worth up to $1.2 billion.[13][14] In 2026 bretisilocin entered European Medicines Agency’s priority medicines (PRIME) scheme for major depressive disorder.[22][23]
SYN
Example 12: N-ethyl-2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine (12)
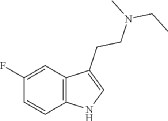
Synthesis of N-[2-(5-fluoro-1H-indol-3-yl)ethyl]formamide
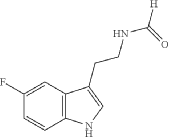
To a solution of 5-fluorotryptamine hydrochloride (3 g, 14.0 mmol) in H 2O (200 mL) with stirring was added KOH until a precipitate was obtained. The aqueous mixture was extracted with EtOAc (3×70 mL), the organic phases were pooled, washed with brine, dried over anhydrous Na 2SO 4, and concentrated in vacuo. Residual EtOAc was removed by azeotropic distillation with ethyl formate (3×20 mL). The resulting 5-fluorotryptamine free base was transferred to a 30 mL oven-dried microwave vessel containing 3 Å molecular sieves (3.3 g). Ethyl formate (20 mL, 248 mmol) was added to the microwave vessel and the mixture was reacted for 2.5 h at 80° C. with 150 W in a microwave reactor. Upon completion, ethyl formate was removed under reduced pressure to provide N-[2-(5-fluoro-1H-indol-3-yl)ethyl]formamide (1.7 g, 8.24 mmol, 58.9% yield). The product was used in the subsequent reaction without further purification.
To a solution of 5-fluorotryptamine hydrochloride (3 g, 14.0 mmol) in H 2O (200 mL) with stirring was added KOH until a precipitate was obtained. The aqueous mixture was extracted with EtOAc (3×70 mL), the organic phases were pooled, washed with brine, dried over anhydrous Na 2SO 4, and concentrated in vacuo. Residual EtOAc was removed by azeotropic distillation with ethyl formate (3×20 mL). The resulting 5-fluorotryptamine free base was transferred to a 30 mL oven-dried microwave vessel containing 3 Å molecular sieves (3.3 g). Ethyl formate (20 mL, 248 mmol) was added to the microwave vessel and the mixture was reacted for 2.5 h at 80° C. with 150 W in a microwave reactor. Upon completion, ethyl formate was removed under reduced pressure to provide N-[2-(5-fluoro-1H-indol-3-yl)ethyl]formamide (1.7 g, 8.24 mmol, 58.9% yield). The product was used in the subsequent reaction without further purification.
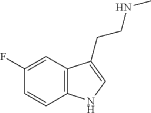
Synthesis of 2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine
Synthesis of N-ethyl-2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine (12)
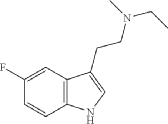
N-ethyl-2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine (12) was synthesized in a similar manner as described above for N-(2-(5-fluoro-1H-indol-3-yl)ethyl)-N-propylpropan-1-amine (5), starting from 2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine (0.7 g, 3.64 mmol), and acetaldehyde (0.96 g, 21.8 mmol), to provide the title compound as a colorless oil after purification by column chromatography using silica gel as a stationary phase and 20% EtOH/EtOAc (1% Et 3N v/v) as the mobile phase (0.62 g, 2.81 mmol, 77.2% yield), and subsequently the corresponding HCl salt as a white crystalline solid. HR-ASAP-MS: m/z 221.1442 (theoretical [M+H] +, C 13H 18FN 2 +), m/z 221.1449 (observed, Δ=−3.2 ppm). 1H-NMR (400 MHz, d 6-DMSO) δ 11.15 (s, 1H), 10.74 (s, 1H), 7.44 (dd, J=10.1, 2.5 Hz, 1H), 7.36 (dd, J=8.8, 4.6 Hz, 1H), 7.33 (d, J=2.3 Hz, 1H), 6.93 (dt, J=9.2, 2.5 Hz, 1H), 3.31-3.13 (m, 4H), 3.13-3.05 (m, 2H), 2.78 (d, J=3.1 Hz, 3H), 1.26 (t, J=7.3 Hz, 3H). 13C-NMR (101 MHz, d 6-DMSO) δ 156.74 (d, J=231.1 Hz, 1C), 132.88 (s, 1C), 126.96 (d, J=10.0 Hz, 1C), 125.42 (s, 1C), 112.48 (d, J=9.9 Hz, 1C), 109.51 (s, 1C), 109.32 (d, J=26.0 Hz, 1C), 103.13 (d, J=23.1 Hz, 1C), 54.37 (s, 1C), 49.82 (s, 1C), 38.13 (s, 1C), 19.68 (s, 1C), 8.79 (s, 1C). 19F-NMR (377 MHz, d 6-DMSO) δ−124.79 (s, 1F).
PAT
PAT
Methods of treating mood disorders
Publication Number: US-2022041551-A1
Priority Date: 2020-02-18
- Methods and compositions relating to psychedelics and serotonin receptor modulatorsPublication Number: EP-4313030-A1Priority Date: 2021-04-01
- Methods and compositions related to hallucinogens and serotonin receptor modulatorsPublication Number: KR-20240037873-APriority Date: 2021-04-01
- Halogenated psilocybin derivatives and methods of usingPublication Number: US-2023293558-A1Priority Date: 2020-09-01
- Methods of treating mood disordersPublication Number: US-2022241243-A1Priority Date: 2020-02-18
- Methods of treating mood disordersPublication Number: US-11440879-B2Priority Date: 2020-02-18Grant Date: 2022-09-13
- Fluorinated tryptamine compounds, analogues thereof, and methods using samePublication Number: EP-4347559-A1Priority Date: 2021-06-02
- Methods and compositions relating to psychedelics and serotonin receptor modulatorsPublication Number: WO-2022212854-A1Priority Date: 2021-04-01
- Methods and compositions relating to psychedelics and serotonin receptor modulatorsPublication Number: US-2024197681-A1Priority Date: 2021-04-01
- Methods and compositions relating to psychedelics and serotonin receptor modulatorsPublication Number: TW-202304423-APriority Date: 2021-04-01
- Methods and compositions relating to psychedelics and serotonin receptor modulatorsPublication Number: AU-2022246909-A1Priority Date: 2021-04-01



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
References
- “GM 2505”. AdisInsight. 5 June 2025. Retrieved 29 July 2025.
- Marek GJ, Makai-Bölöni S, Umbricht D, Christian EP, Winters J, Dvorak D, et al. (2025). “A novel psychedelic 5-HT 2A receptor agonist GM-2505: The pharmacokinetic, safety, and pharmacodynamic profile from a randomized trial healthy volunteer”. Journal of Psychopharmacology 02698811251378512. doi:10.1177/02698811251378512. hdl:1887/4298848. PMID 41099491.
- Hughes Z, Christian E, Dvorak D, Umbricht D, Winters J, Raines S, et al. (December 2023). “ACNP 62nd Annual Meeting: Poster Abstracts P1 – P250: P238. Subjective and Pharmacodynamic Effects of the Novel 5-HT2A Receptor Agonist GM-2505 in Healthy Volunteers Show High Translatability From Rodent Data and Hold Promise for Future Development in Patients With Depression”. Neuropsychopharmacology. 48 (Suppl 1). Springer Science and Business Media LLC: 63–210 (202–203). doi:10.1038/s41386-023-01755-5. PMC 10729595. PMID 38040809.
- Umbricht D, Christian E, Winters J, Raines S, Hughes ZA, Leong W, et al. (2024). “Pharmacokinetic, pharmacodynamic and subjective and effects of the novel 5-HT2A receptor agonist GM-2505 in healthy volunteers”. Neuroscience Applied. 3 104845. doi:10.1016/j.nsa.2024.104845.
- “Бретисилоцин (5F-MET)”. АИПСИН (in Russian). Retrieved 18 March 2026.
- Peplow M (22 June 2024). “Should Next-Generation Psychedelics Skip the Trip?”. Scientific American. Retrieved 20 February 2025.
Gilgamesh is also working on GM-2505, a 5-HT2A agonist that is structurally related to psilocybin and DMT. GM-2505 completed a phase 1 trial late last year and should enter phase 2 for major depressive disorder this year. Its psychedelic effect lasts 60 to 90 minutes — long enough for patients to “explore the altered state of consciousness that might be needed for long-term durable efficacy,” Krugel says, yet within a timeframe that is manageable for healthcare systems. “Personally, I believe that the hallucinogenic effects are an important component, as multiple hallucinogenic compounds have demonstrated durable, transformational changes from a single dose in human studies,” he adds.
- Witkin JM, Golani LK, Smith JL (April 2023). “Clinical pharmacological innovation in the treatment of depression”. Expert Review of Clinical Pharmacology. 16 (4): 349–362. doi:10.1080/17512433.2023.2198703. PMID 37000975.
GM-2505 is a dual-acting compound with both agonist activity at 5-HT 2A receptors and a releaser of 5-HT. […]
- “Methods of treating mood disorders”. Google Patents. 2022. Retrieved 14 November 2024.
- Hughes Z, Klein A, Austin E, Dvorak D, Gatti S, Kiss L, et al. (December 2022). “ACNP 61st Annual Meeting: Poster Abstracts P1 – P270: P254. Gm-2505 is a Novel 5-Ht2a Receptor Agonist and 5-Ht Releaser That Induces Rapid, Robust, and Durable Antidepressant Effects at Doses Associated With Decreased Power in Low Frequency EEG Bands in Rats”. Neuropsychopharmacology. 47 (Suppl 1): 63–219 (209–209). doi:10.1038/s41386-022-01484-1. PMC 9714397. PMID 36456693.
- WO 2022/256554, Wallach J, Dybek M, “Fluorinated Tryptamine Compounds, Analogues Thereof, and Methods Using Same”, published 8 December 2022, assigned to University of the Sciences in Philadelphia[…] Synthesis of N-ethyl-2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine (12) [structure] N-ethyl-2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine (12) was synthesized […] […] Table 1. Selected compounds of the present invention. […] [Compound 12:] […] Table 3. Functional Activity of Compounds at 5-HT2A (Ca2+), 5-HT2B (Ca2+), 5-HT2c (Ca2+), and 5-HT1A (cAMP inhibition) […]
- Hughes Z, Klein A, Dvorak D, Austin E, Kiss L, Marek G, et al. (2023). “22. GM-2505 has Rapid Onset Antidepressant Activity and Causes Dose-Dependent Changes in qEEG With Increasing 5-HT2A Receptor Occupancy”. Biological Psychiatry. 93 (9): S102–S103. doi:10.1016/j.biopsych.2023.02.262.
- Gunther M (31 January 2023). “Gilgamesh Tweaks Known Psychedelics To Improve Therapies”. Lucid News – Psychedelics, Consciousness Technology, and the Future of Wellness. Retrieved 20 February 2025.
- Taylor NP (25 August 2025). “AbbVie tunes in to Gilgamesh’s story, inking $1.2B deal for psychedelic program”. Fierce Biotech. Retrieved 15 October 2025.
- Psychedelic Alpha (25 August 2025). “AbbVie to Acquire Gilgamesh’s Bretisilocin for Up to $1.2B”. Psychedelic Alpha. Retrieved 15 October 2025.
- Halberstadt AL, Geyer MA (2018). “Effect of Hallucinogens on Unconditioned Behavior”. Behavioral Neurobiology of Psychedelic Drugs. Curr Top Behav Neurosci. Vol. 36. pp. 159–199. doi:10.1007/7854_2016_466. ISBN 978-3-662-55878-2. PMC 5787039. PMID 28224459.
- https://iris.who.int/bitstream/handle/10665/380497/9789240107038-eng.pdf “bretisilocinum bretisilocin N-ethyl-2-(5-fluoro-1H-indol-3-yl)-N-methylethan-1-amine serotonin (5-HT2A) receptor agonist”
- “Controlled Drugs and Substances Act”. Department of Justice Canada. Retrieved 19 January 2026.
- Orange Book: List of Controlled Substances and Regulated Chemicals (January 2026) (PDF), United States: U.S. Department of Justice: Drug Enforcement Administration (DEA): Diversion Control Division, January 2026
- Psychedelic Alpha (27 May 2025). “Gilgamesh’s Next-Gen Psychedelic GM-2505 Prints Impressive Results in Phase 2a Major Depressive Disorder Study”. Psychedelic Alpha. Retrieved 29 July 2025.
- Taylor NP (27 May 2025). “Gilgamesh links psychedelic to 94% remission rate in midphase depression trial”. Fierce Biotech. Retrieved 29 July 2025.
- Dunne R (31 May 2025). “Gilgamesh’s psychedelic drug demonstrates exceptional efficacy for treating depression”. Mugglehead Investment Magazine. Retrieved 29 July 2025.
- Psychedelic Access and Research European Alliance (2026-03-19). “Bretisilocin Becomes First Psychedelic in EMA PRIME Scheme for Depression”. Drug Policy Tracker. Retrieved 2026-03-29.
- European Medicines Agency (EMA) (2026-03-18). “New PRIME tools to accelerate development of medicines in the EU”. http://www.ema.europa.eu. Retrieved 2026-03-19.
External links
- 5-Fluoro-MET (Bretisilocin; GM-2505) – Isomer Design
- 5-f-met bretisilocin – Bluelight
- Bretisilocin (5-Fluoro-MET) – r/ResearchChemicals – Reddit Search
| Clinical data | |
|---|---|
| Other names | GM-2505; GM2505; 5-Fluoro-N-methyl-N-ethyltryptamine; 5F-MET; 5-F-MET; 5-Fluoro-MET |
| Routes of administration | Intravenous,[1][2][3][4] intranasal[5] |
| Drug class | Serotonergic psychedelic; Hallucinogen; Serotonin 5-HT2A and 5-HT2C receptor agonist; Serotonin 5-HT2B receptor partial agonist or antagonist; Serotonin releasing agent |
| Legal status | |
| Legal status | Investigational |
| Pharmacokinetic data | |
| Onset of action | IVTooltip Intravenous injection: 10–20 minutes (peak)[2] |
| Elimination half-life | 45 (40–50) minutes[2][3] |
| Duration of action | IVTooltip Intravenous injection: 60–90 minutes[2][6] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2698331-35-8 |
| PubChem CID | 156836209 |
| ChemSpider | 129221851 |
| ChEMBL | ChEMBL5028766 |
| Chemical and physical data | |
| Formula | C13H17FN2 |
| Molar mass | 220.291 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////bretisilocin, serotonin (5-HT2A) receptor agonist, GM-2505, GM 2505, 5-Fluoro-N-methyl-N-ethyltryptamine, 5F-MET, 5-F-MET, 5-Fluoro-MET, DS425RQ8SX
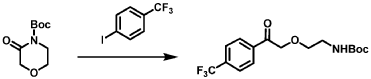
Blixeprodil
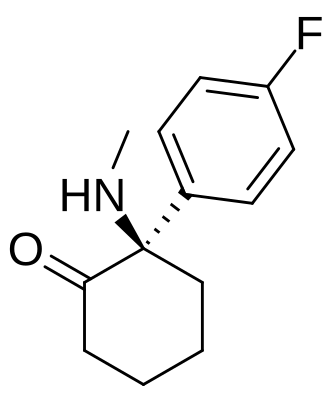
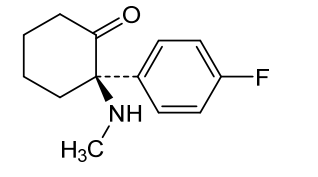
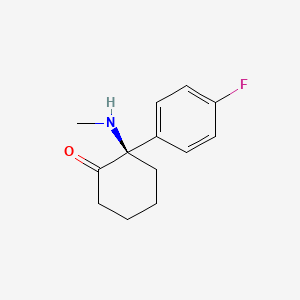
Blixeprodil
CAS 2881017-49-6
MF C13H16FNO MW 221.27 g/mol
Cyclohexanone, 2-(4-fluorophenyl)-2-(methylamino)-, (2R)-
(2R)-2-(4-fluorophenyl)-2-(methylamino)cyclohexan-1-one
N-methyl-D-aspartate (NMDA) receptor antagonist, GM-1020, GM1020, (R)-4-Fluorodeschloroketamine, (R)-4-FDCK, (R)-4FDCK, S2MGG2PC5K
Blixeprodil,[5] also known by its developmental code name GM-1020 or as (R)-4-fluorodeschloroketamine ((R)-4-FDCK), is an NMDA receptor antagonist related to ketamine which is under development for the treatment of major depressive disorder, bipolar depression, and other depressive disorders.[1][6][2][3][7][8] It is taken by mouth.[1][2][3]
The drug is orally active, in contrast to the poor oral bioavailability of ketamine.[3] Its oral bioavailability is >60%.[4][9] The time to peak levels of blixeprodil is 1.5 hours and its elimination half-life is 4.3 hours.[4] In a clinical study comparing it with the serotonergic psychedelic bretisilocin (GM-2505), both blixeprodil and bretisilocin produced hallucinogenic effects.[10]
Blixeprodil shows antidepressant-like effects in rodents.[3][11][4][9] It appears to have a greater separation between antidepressant-like and ataxia-inducing doses than ketamine in rodents and hence might have better tolerability.[3][7][9] Whereas ketamine shows only 3-fold separation between antidepressant-like and ataxic doses, there was 13-fold separation for blixeprodil, and it did not produce hyperlocomotion at doses >20-fold higher than the minimum antidepressant-like dose.[9] In relation to the preceding, blixeprodil is claimed to be non-dissociative at therapeutic doses.[2][4] However, dissociative and other related effects have been observed at low incidences and at higher doses.[4]
The drug is a close analogue of ketamine, with a 4-fluoro group instead of a 2-chloro group on the phenyl ring and in (2R)-enantiopure form.[12] Hence, blixeprodil is related to arketamine ((R)-ketamine); it is said to “bet” on the notion that arketamine is importantly involved in the antidepressant effects of ketamine, in spite of arketamine having less propensity for inducing dissociation.[13]
Blixeprodil is being developed by Gilgamesh Pharmaceuticals.[1][6][2] As of July 2024, it is in phase 2 clinical trials for major depressive disorder and bipolar depression and is in phase 1 trials for other depressive disorders.[1][6][2]
SYN
Example 15: Preparation of Compounds 117rac and 18rac
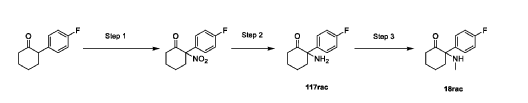
Step 1: Preparation of 2-(4-fluorophenyl)-2-nitrocyclohexan-1-one
[0364] A mixture of 2-(4-fluorophenyl)cyclohexan-1-one (5 g, 26.01 mmol, 1 eq), ceric ammonium nitrate (CAN, 28.52 g, 52.02 mmol, 2 eq), and Cu(OAc)2 (945 mg, 5.20 mmol, 0.2 eq) in DCE (50 mL) was stirred at 85 °C for 12 hrs. The mixture was cooled, filtered and concentrated. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate=100/1 to 0/1) to afford 2-(4-fluorophenyl)-2-nitrocyclohexan-1-one (2.5 g, 10.54 mmol, 40.52% yield) as a yellow oil.1H NMR (400MHz, CHLOROFORM-d) δ = 7.47 – 7.29 (m, 2H), 7.22 – 7.04 (m, 2H), 3.12 (ddd, J = 3.6, 10.0, 14.0 Hz, 1H), 2.86 – 2.76 (m, 1H), 2.75 – 2.62 (m, 1H), 2.61 – 2.47 (m, 1H), 2.08 – 1.86 (m, 3H), 1.80 (dt, J = 3.6, 9.2 Hz, 1H).
Step 2: Preparation of 2-amino-2-(4-fluorophenyl)cyclohexan-1-one (117rac)
[0365] A mixture of 2-(4-fluorophenyl)-2-nitrocyclohexan-1-one (3 g, 12.65 mmol, 1 eq) and Zn (19.85 g, 303.51 mmol, 24 eq) in AcOH (25 mL) was stirred at 20 °C for 12 hrs. The mixture was cooled, filtered, and concentrated. The residue was dissolved in DCM, washed with sat.
NaHCO3, H2O, and brine, dried over Na2SO4, filtered, and concentrated. The residue was
purified by silica gel (PE:EA = 50:1 – 8:1) to afford 2-amino-2-(4-fluorophenyl)cyclohexan-1- one (1.5 g, 7.24 mmol, 57.23% yield) (117rac) as a brown oil. LCMS (RT = 1.336 min, MS calc.: 207.11, [M+H]+ = 208.1) 1H NMR (400MHz, CHLOROFORM-d) δ = 7.26 – 7.19 (m, 2H), 7.11 – 7.01 (m, 2H), 2.87 – 2.73 (m, 1H), 2.50 – 2.42 (m, 1H), 2.41 – 2.29 (m, 1H), 2.04 – 1.96 (m, 1H), 1.93 (s, 2H), 1.83 – 1.63 (m, 4H); 3C NMR (101 MHz, CHLOROFORM-d) δ = 213.28, 163.27, 160.82, 137.67, 137.63, 127.99, 127.91, 116.16, 115.95, 65.93, 39.71, 28.08, 22.61
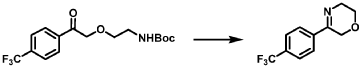
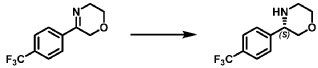
Step 3: Preparation of 2-(4-fluorophenyl)-2-(methylamino)cyclohexan-1-one (18rac)
[0366] A mixture of 2-amino-2-(4-fluorophenyl)cyclohexan-1-one (1.3 g, 6.27 mmol, 1 eq) and methyl trifluoromethanesulfonate (1.03 g, 6.27 mmol, 1 eq) in hexafluoroisopropanol (HFIP, 130 mL) was stirred at 0 – 25 °C for 12 hrs under N2 atmosphere. The mixture was filtered and concentrated. The residue was adjusted to pH = 7 with sat. Na2CO3 (20 ml). The aqueous phase was extracted with EA (50 mL x 2). The combined organic phase was washed with brine (50 mL x 2), dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by prep-HPLC (column: Welch Xtimate C18250*70 mm, 10 μm; mobile phase: A: water(0.05% NH3H2O), B: ACN; B%: 18% – 48%, 32 min) to afford 2-(4-fluorophenyl)-2- (methylamino)cyclohexan-1-one (590 mg, 4.02 mmol, 42.45% yield) (18rac) as a white solid. LCMS (RT = 1.415 min, MS calc.: 221.12, [M+H]+ = 222.1); 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.26 – 7.17 (m, 2H), 7.07 (br t, J = 8.4 Hz, 2H), 2.92 – 2.74 (m, 1H), 2.50 – 2.26 (m, 3H), 2.12 – 1.93 (m, 4H), 1.90 – 1.63 (m, 4H); 13C NMR (101 MHz, CHLOROFORM-d) δ = 211.15, 163.20, 160.75, 134.68, 134.65, 128.99, 128.91, 115.79, 115.58, 69.37, 39.70, 35.85, 28.87, 27.70, 22.21.
SYN
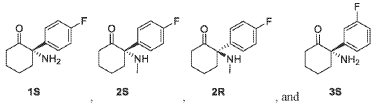
Example 1: Preparation of Compounds 1 and 2 and Their Enantiomers.
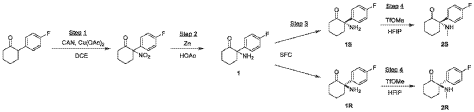
Step 1: Preparation of 2-(4-fluorophenyl)-2-nitrocyclohexan-1-one
[0110] A mixture of 2-(4-fluorophenyl)cyclohexan-1-one (14 g, 72.83 mmol, 1 eq), CAN (79.85 g, 145.66 mmol, 72.59 mL, 2 eq), and Cu(OAc)2 (2.65 g, 14.57 mmol, 0.2 eq) in DCE (140 mL) was stirred at 85 °C for 12 h. On completion, the mixture was filtered and concentrated. The residue was purified by column chromatography (SiO2, Petroleum ether/Ethyl acetate=100/1 to 0/1) to afford 2-(4-fluorophenyl)-2-nitrocyclohexan-1-one (6.1 g, 25.71 mmol, 35.31% yield) as a yellow solid.1H NMR (400 MHz, CHLOROFORM-d) δ = 7.41 – 7.31 (m, 2H), 7.16 (t, J=8.4 Hz, 2H), 3.11 (ddd, J=3.6, 10.4, 14.0 Hz, 1H), 2.87 – 2.76 (m, 1H), 2.73 – 2.64 (m, 1H), 2.60 -2.48 (m, 1H), 2.02 – 1.88 (m, 3H), 1.84 – 1.72 (m, 1H).
Step 2: Preparation of 2-amino-2-(4-fluorophenyl)cyclohexan-1-one (1)
[0111] To a mixture of 2-(4-fluorophenyl)-2-nitrocyclohexan-1-one (5.6 g, 23.61 mmol, 1 eq) in AcOH (10 mL) was added Zn (15.44 g, 236.06 mmol, 10 eq) in several portions and the resulting mixture was stirred at 30 °C for 12 h. On completion, the mixture was filtered and concentrated. The residue was dissolved in DCM (20 mL), washed with sat. aq. NaHCO3 (10 mL), H2O (5 mL), and brine (10 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified by prep-HPLC (column: Agela DuraShell C18 (250 mm*80 mm, 10 μm); mobile phase: A: water (NH4HCO3), B: ACN; B%: 35%, 20 min) to afford 2-amino-2-(4-fluorophenyl)cyclohexan-1-one (2.9 g, 13.99 mmol, 59.28% yield, 1) as a brown oil.1H NMR (400 MHz, CHLOROFORM-d) δ = 7.52 – 7.40 (m, 2H), 7.32 (br s, 1H), 7.34 – 7.20 (m, 2H), 2.93 – 2.92 (m, 1H), 3.08 – 2.92 (m, 1H), 2.74 – 2.63 (m, 1H), 2.63 – 2.50 (m, 1H), 2.28 – 2.16 (m, 1H), 2.10 (br s, 2H), 2.04 – 1.85 (m, 4H).
Note: The free base of this compound is unstable and dimerizes over time. It should be stored frozen or quickly converted to the HCl salt to prevent this.
Step 3: Preparation of (S)-2-amino-2-(4-fluorophenyl)cyclohexan-1-one (1S) and (R)-2-amino-2-(4-fluorophenyl)cyclohexan-1-one (1R)
[0112] The racemate 1 (2.9 g) was separated by SFC (column: DAICEL CHIRALPAK AD (250 mm*30 mm, 10 μm); mobile phase: A: CO2, B: 0.1% NH3H2O in ETOH; B%: 27%, multi-injection process with 6-min spacing between injections) to afford ENT-1 free base (RT = 2.266 min, 1.1 g, 1.62 mmol, 1S_FB) as a yellow oil and ENT-2 free base (RT = 2.945 min, 1.1 g, 1.28 mmol, 1R_FB) as a yellow oil.
[0113] A portion of each free base was further purified by prep-HPLC (column: Welch Xtimate C18 (100 mm*25 mm, 3 μm); mobile phase: A: water (0.04% HCl), B: ACN; B%: 1% – 20%, 8 min) to afford ENT-1 HCl (RT = 2.266 min, 272 mg, HCl salt, 1S) as a white solid and ENT-2 HCl (RT = 2.945 min, 283 mg, HCl salt, 1R) as a white solid.
[0114] ENT-1 HCl, RT = 2.266 min (assigned here as the S isomer, 1S); LCMS (RT = 1.449 min, MS calc.: 207.1, [M+H]+ = 208.1); 1H NMR (400MHz, DMSO-d6) δ = 8.83 (br s, 3H), 7.50 – 7.42 (m, 2H), 7.41 – 7.32 (m, 2H), 3.03 (br dd, J=2.4, 14.0 Hz, 1H), 2.45 – 2.27 (m, 2H), 2.21 -2.05 (m, 1H), 1.97 (td, J=2.8, 9.6 Hz, 1H), 1.81 (br d, J=11.6 Hz, 1H), 1.71 – 1.47 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ = 206.52, 164.22, 161.76, 130.78, 130.69, 130.08, 130.05, 116.90, 116.68, 66.26, 34.75, 27.52, 21.53; ENT-2 HCl, RT = 2.945 min (assigned here as the R isomer, 1R); LCMS (RT = 1.449 min, MS calc.: 207.1, [M+H]+ = 208.0); 1H NMR (400MHz, DMSO-d6) δ = 8.84 (br s, 3H), 7.49 – 7.42 (m, 2H), 7.40 – 7.33 (m, 2H), 3.03 (br dd, J=1.6, 14.0 Hz, 1H), 2.45 – 2.27 (m, 2H), 2.23 – 2.06 (m, 1H), 1.97 (dt, J=2.8, 6.1 Hz, 1H), 1.81 (br d, J=11.6 Hz, 1H), 1.70 – 1.46 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ = 206.50, 164.22, 161.76, 130.78, 130.70, 130.08, 130.05, 116.89, 116.68, 66.26, 34.75, 27.51, 21.52.
[0115] The retention times above, which identify the enantiomers, were determined using the free bases using the following chiral analytical method: column: Chiralpak AD-3 (150 mm×4.6 mm I.D., 3 μm); mobile phase: A: CO2 B: EtOH (0.1% IPAm, v/v); gradient (Time (min)/A%/B%): 0.0/90/10, 0.5/90/10, 3.5/50/50, 4.5/50/50, 5.0/90/10; flow rate: 2.5 mL/min; column temp.: 35 °C; ABPR: 2,000 psi.
Step 4: Preparation of (S)-2-(4-fluorophenyl)-2-(methylamino)cyclohexan-1-one (2S) and (R)-2-(4-fluorophenyl)-2-(methylamino)cyclohexan-1-one (2R)
[0116] Compound 1S_FB (540 mg, 2.61 mmol, 1 eq) and methyl trifluoromethanesulfonate (427.59 mg, 2.61 mmol, 285.06 μL, 1 eq) were combined in hexafluoroisopropanol (40 mL) at 0
°C under N2 atmosphere and then the mixture was allowed to warm to 25 °C and stirred for 12 h. On completion, the residue was adjusted to pH 7 with sat. aq. Na2CO3 (10 mL) and the combined organic phase was washed with brine (100 mL * 2), dried over Na2SO4, filtered, and concentrated in vacuum. The residue was purified by prep-HPLC (column: Waters Xbridge C18 (150 mm*50 mm, 10μm); mobile phase: A: water (10 mM NH4HCO3), B: ACN; B%: 30% – 50%, 10 min) to afford 2S (260 mg, 1.18 mmol, 45.10% yield) as a white solid. Compound 2R was prepared by the same procedure starting from 1R_FB (590 mg, 2.85 mmol) in hexafluoroisopropanol (60 mL) (other quantities scaled based on molar equivalents) and obtained as an off-white solid (260 mg, 1.18 mmol, 41.27% yield).
[0117] 2S (assigned here as the S isomer) (free base); LCMS (RT = 1.427 min, MS calc.: 221.1, [M+H]+ = 222.1); 1H NMR (400MHz, CHLOROFORM-d) δ = 7.21 (dd, J = 5.4, 8.8 Hz, 2H), 7.10 – 7.02 (m, 2H), 2.85 – 2.74 (m, 1H), 2.49 – 2.37 (m, 1H), 2.36 – 2.25 (m, 1H), 2.22 (br s, 1H), 2.03 (s, 3H), 1.96 (dt, J = 3.2, 5.8 Hz, 1H), 1.88 – 1.64 (m, 4H); 13C NMR (101 MHz, CHLOROFORM-d) δ = 211.25, 163.22, 160.76, 134.80, 134.77, 128.98, 128.90, 115.80, 115.59, 69.38, 39.73, 35.92, 28.92, 27.72, 22.24; 2R (assigned here as the R isomer) (free base); LCMS (RT = 1.415 min, MS calc.: 221.1, [M+H]+ = 222.1); 1H NMR (400MHz, CHLOROFORM-d) δ = 7.25 – 7.17 (m, 2H), 7.11 – 7.02 (m, 2H), 2.85 – 2.75 (m, 1H), 2.48 – 2.38 (m, 1H), 2.35 – 2.19 (m, 2H), 2.04 (s, 3H), 1.97 (br dd, J = 2.8, 6.1 Hz, 1H), 1.89 – 1.66 (m, 4H); 13C NMR (101 MHz, CHLOROFORM-d) δ = 211.24, 163.22, 160.77, 134.78, 134.74, 128.99, 128.91, 115.81, 115.60, 69.38, 39.73, 35.91, 28.91, 27.72, 22.24.
PAT
- Methods of treating neuropsychiatric disordersPublication Number: US-2024216339-A1Priority Date: 2021-06-08
- Arylcyclohexylamine derivatives and their use in the treatment of psychiatric disordersPublication Number: US-11344510-B2Priority Date: 2019-12-26Grant Date: 2022-05-31
- Arylcyclohexylamine derivatives and their use in the treatment of psychiatric disordersPublication Number: US-2022409555-A1Priority Date: 2019-12-26
- Arylcyclohexylamine derivatives and their use in the treatment of psychiatric disordersPublication Number: WO-2021134086-A1Priority Date: 2019-12-26
- A treatment for patients with mood disorders using n-ethyl-2-(5-fluoro-1h-indol-3-yl)- n-methylethan-1-amine or a pharmaceutically acceptable salt thereofPublication Number: WO-2025111597-A1Priority Date: 2023-11-22
- Methods of treating psychiatric disorders or pain using (r)-2-(4-fluorophenyl)-2-(methylamino)cyclohexan-1-one or pharmaceutically acceptable salts thereofPublication Number: WO-2025106879-A1Priority Date: 2023-11-15
- (ampa-pam)-nmda receptor antagonist combination therapy for treatment of mental conditions and disordersPublication Number: WO-2023154450-A2Priority Date: 2022-02-11
- Arylcyclohexylamine derivatives and their use in the treatment of psychiatric disordersPublication Number: US-2024300886-A1Priority Date: 2021-06-25
- Arylcyclohexylamine derivatives and their use in the treatment of psychiatric disordersPublication Number: EP-4358946-A1Priority Date: 2021-06-25
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- “GM 1020”. AdisInsight. 12 July 2024. Retrieved 20 February 2025.
- Peplow M (June 2024). “Next-generation psychedelics: should new agents skip the trip?”. Nature Biotechnology. 42 (6): 827–830. doi:10.1038/s41587-024-02285-1. PMID 38831049.
Other companies are confident that they can further reduce or even erase those effects without losing therapeutic efficacy. Gilgamesh, for example, is taking that approach with ketamine, DMT and psilocybin. In the case of ketamine, says Kruegel, the dissociative side effects require that the subjects remain under supervision. So Gilgamesh designed a ketamine analog called GM-1020 that has no dissociative effects (distortions in sight, sound and feelings of detachment) and that also has better oral bioavailability than ketamine itself. After completing a phase 1 trial last year, the company began dosing patients with GM-1020 in a phase 2 trial for major depressive disorder in March. “The hope is that the psychoactive effects will be limited enough that this can eventually be taken at home,” says Kruegel.
- Klein AK, Austin EW, Cunningham MJ, Dvorak D, Gatti S, Hulls SK, et al. (May 2024). “GM-1020: a novel, orally bioavailable NMDA receptor antagonist with rapid and robust antidepressant-like effects at well-tolerated doses in rodents”. Neuropsychopharmacology. 49 (6): 905–914. doi:10.1038/s41386-023-01783-1. PMC 11039472. PMID 38177696.
- Marek G, Umbricht D, Christian E, Winters J, Raines S, Kiss L, et al. (December 2023). “ACNP 62nd Annual Meeting: Poster Abstracts P251 – P500: P352. GM-1020: An Oral NMDA Receptor Antagonist for Depression Demonstrates Target Engagement at Doses That Do Not Cause Dissociation, Ataxia or Sedation in a Phase 1 Single Ascending Dose Study”. Neuropsychopharmacology. 48 (Suppl 1): 211–354 (269–269). doi:10.1038/s41386-023-01756-4. PMC 10729596. PMID 38040810.
- https://iris.who.int/bitstream/handle/10665/380497/9789240107038-eng.pdf “blixeprodilum blixeprodil (2R)-2-(4-fluorophenyl)-2-(methylamino)cyclohexan-1-one N-methyl-D-aspartate (NMDA) receptor antagonist”
- “Delving into the Latest Updates on GM-1020 with Synapse”. Synapse. 15 February 2025. Retrieved 20 February 2025.
- Klein A, Dvorak D, Austin E, Marek G, Sporn J, Hughes Z, et al. (2023). “531. GM-1020 is a Novel, Orally Bioavailable NMDA Antagonist With Improved Separation Between Antidepressant and Ataxic Doses Compared to Ketamine”. Biological Psychiatry. 93 (9): S308–S309. doi:10.1016/j.biopsych.2023.02.771.
- Hughes Z (December 2024). “ACNP 63rd Annual Meeting: Panels, Mini-Panels and Study Groups: 19.4 Translational Profile of GM-1020, a Novel Orally Bioavailable NMDA Receptor Antagonist That Achieves Robust Target Engagement Without Dissociation or Sedation”. Neuropsychopharmacology. 49 (Suppl 1): 1–64 (25–25). doi:10.1038/s41386-024-02010-1. PMC 11627185. PMID 39643632.
- Kiss L, Klein A, Austin E, Dvorak D, Gatti S, Papp M, et al. (December 2022). “ACNP 61st Annual Meeting: Poster Abstracts P1 – P270: P215. GM-1020: A Novel, Orally Bioavailable NMDA Receptor Antagonist With Rapid and Robust Antidepressant Effects and Reduced Ataxia in Rodents”. Neuropsychopharmacology. 47 (Suppl 1): 63–219 (185–186). doi:10.1038/s41386-022-01484-1. PMC 9714397. PMID 36456693.
- Dvorak D, Christian E, Hughes Z, Klein A, Austin E, Kiss L, et al. (2024). “ACNP 63rd Annual Meeting: Poster Abstracts P1-P304: P87. GM-1020 (NMDA Antagonist) Vs GM-2505 (5-HT2A Agonist) – Distinct Mechanisms, Same Outcome?”. Neuropsychopharmacology. 49 (S1): 65–235. doi:10.1038/s41386-024-02011-0. ISSN 0893-133X. PMC 11627186. Retrieved 19 January 2026.
- Trunnell ER, Baines J, Farghali S, Jackson T, Jayne K, Smith R, et al. (August 2024). “The need for guidance in antidepressant drug development: Revisiting the role of the forced swim test and tail suspension test”. Regulatory Toxicology and Pharmacology. 151 105666. doi:10.1016/j.yrtph.2024.105666. PMID 38942190.
- Sá VL, de Jesus Santos G, da Fonseca Fraga I, da Silva JM, Santos, MG, et al. (2015). Avaliação farmacológica de um análogo a um antagonista do receptor N-Metil-D-Aspartato [Pharmacological evaluation of an analogue of an N-Methyl-D-Aspartate receptor antagonist] (PDF). I Congresso de Ciências Farmacêuticas do Interior Baiano.
[Translated:] […] ketamine has low oral availability and a narrow therapeutic index, generating adverse effects such as dissociation, cognitive impairment, sedation, and ataxia, which limits the acceptance of the drug in the treatment of depression. The preclinical characterization through in vitro and in vivo studies of GM-1020 ((R)-2-(4-fluorophenyl)-2-(methylamino)cyclohexan-1-one) may indicate a new therapy that presents bioavailability when administered orally and absence of undesirable motor effects.
- Gunther M (31 January 2023). “Gilgamesh Tweaks Known Psychedelics To Improve Therapies”. Lucid News – Psychedelics, Consciousness Technology, and the Future of Wellness. Retrieved 20 February 2025.
| Clinical data | |
|---|---|
| Other names | GM-1020; GM1020; (R)-4-Fluorodeschloroketamine; (R)-4-FDCK; (R)-4FDCK |
| Routes of administration | Oral[1][2][3] |
| Drug class | NMDA receptor antagonist[1][2][3] |
| Pharmacokinetic data | |
| Bioavailability | >60%[4] |
| Elimination half-life | 4.3 hours[4] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2881017-49-6 |
| PubChem CID | 156552274 |
| Chemical and physical data | |
| Formula | C13H16FNO |
| Molar mass | 221.275 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////blixeprodil, N-methyl-D-aspartate (NMDA) receptor antagonist, GM-1020, GM1020, (R)-4-Fluorodeschloroketamine, (R)-4-FDCK, (R)-4FDCK, S2MGG2PC5K
Birelentinib
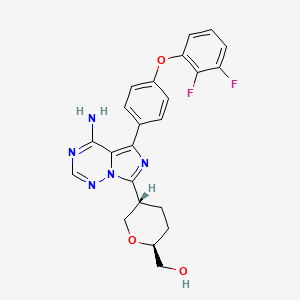
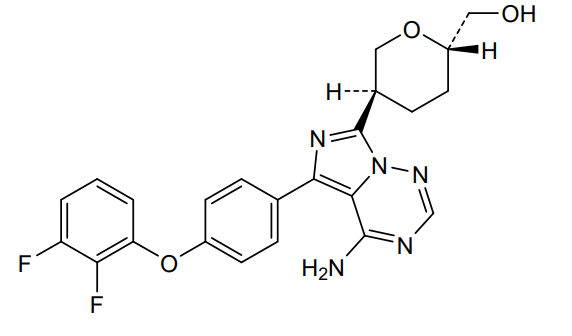
Birelentinib
CAS 2662512-15-2
MF C23H21F2N5O3 MW453.4 g/mol
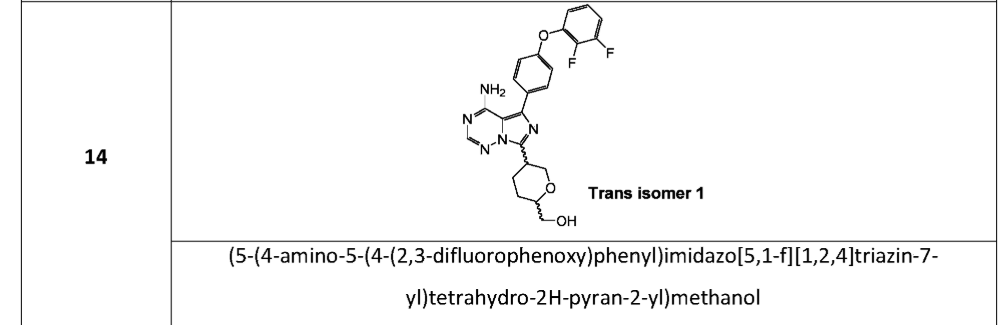
[(2S,5S)-5-[4-amino-5-[4-(2,3-difluorophenoxy)phenyl]imidazo[5,1-f][1,2,4]triazin-7-yl]oxan-2-yl]methanol
[(2S,5S)-5-{4-amino-5-[4-(2,3-difluorophenoxy)phenyl]imidazo[5,1-f][1,2,4]triazin-7-yl}oxan-2-yl]methanol
tyrosine kinase inhibitor, antineoplastic, DZD8586, DZD 8586, Fast Track designation, BTK-IN-30, Z2F599L9GD
Birelentinib (also known as DZD8586) is a first-in-class, non-covalent dual inhibitor of LYN (lymphocyte-specific protein tyrosine kinase) and BTK (Bruton’s tyrosine kinase).
It is currently being developed by Dizal Pharmaceutical as an oral therapy for various B-cell malignancies.
Clinical Status and FDA Designations
As of late 2025, birelentinib has received significant attention for its potential in treating resistant blood cancers:
- Fast Track Designation: In August 2025, the U.S. FDA granted Fast Track designation to birelentinib for adult patients with relapsed or refractory (R/R) chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL).
- Target Population: It is specifically intended for those who have failed at least two prior therapies, including a covalent BTK inhibitor and a BCL-2 inhibitor.
- Key Trials: It is being evaluated in multiple studies, including the Phase 3 Tai-Shan6 trial comparing it against standard treatments like bendamustine and rituximab.
Unique Therapeutic Properties
Birelentinib is designed to overcome common drug resistance mechanisms found in existing treatments:
- Overcoming Resistance: It targets both BTK-dependent pathways (including the common C481X mutation) and BTK-independent B-cell receptor (BCR) signaling pathways.
- Blood-Brain Barrier (BBB) Penetration: A notable feature is its ability to fully penetrate the blood-brain barrier, which may offer therapeutic benefits for patients with central nervous system (CNS) involvement.
- Efficacy: Early Phase 1/2 data presented at the ASH Annual Meeting and EHA Congress in 2025 showed an Objective Response Rate (ORR) of 84.2% in heavily pretreated patients
Birelentinib is an orally bioavailable non-covalent dual inhibitor of tyrosine-protein kinases Lyn (LYN) and BTK (Bruton’s tyrosine kinase; Bruton agammaglobulinemia tyrosine kinase), with potential antineoplastic activity. Upon oral administration, birelentinib targets and inhibits both LYN and BTK, thereby blocking both BTK-dependent and BTK-independent B-cell antigen receptor (BCR) signaling pathways. This prevents the proliferation of malignant B-cells in which the BCR signaling pathway is overactivated. Birelentinib is able to cross the blood-brain barrier (BBB) and thus potentially useful in the treatment of central nervous system (CNS) metastases
SYN’
SYN
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
Publication Number: WO-2021136219-A1
Priority Date: 2020-01-02
/////////birelentinib, tyrosine kinase inhibitor, antineoplastic, DZD8586, DZD 8586, Fast Track designation, BTK-IN-30, Z2F599L9GD
Betovumeline
Betovumeline
CAS 1314018-44-4
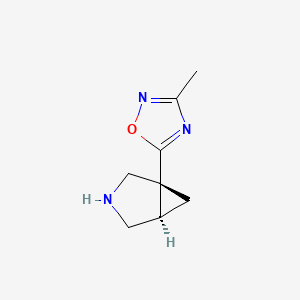
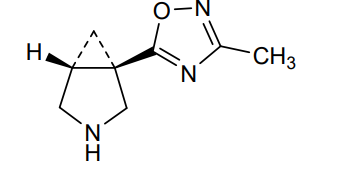
MF C8H11N3O MW 165.19 g/mol
(1R,5R)-1-(3-Methyl-1,2,4-oxadiazol-5-yl)-3-azabicyclo[3.1.0]hexane
(1R,5R)-1-(3-methyl-1,2,4-oxadiazol-5-yl)-3-azabicyclo[3.1.0]hexane
muscarinic receptor agonist, ML 007, MPL 0527, 5UXW4B47R9
Betovumeline (also known as ML-007 or MPL-0527) is a muscarinic receptor agonist currently being developed for the treatment of neurological and neuropsychiatric disorders.
It is specifically designed to target muscarinic receptors in the brain, which play a critical role in cognitive and motor functions.
Key Characteristics
- Mechanism of Action: It acts as an agonist for muscarinic acetylcholine receptors (mAChR).
- Research Focus: It is primarily being investigated for its potential in treating neurological disorders, such as schizophrenia or Alzheimer’s disease-related cognitive impairment.
- Chemical Detail: Its chemical structure is (1R,5R)-1-(3-methyl-1,2,4-oxadiazol-5-yl)-3-azabicyclohexane.
- Development Stage: It is an investigational drug, meaning it is currently for research use only and has not yet been approved for general medical or human use.
SYN
SYN
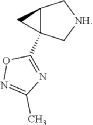
“Compound 1” refers to 1-(3-methyl-[1,2,4]oxadiazol-5-yl)-(1R,5R)-3-aza-bicyclo[3.1.0]hexane having the following structural formula:
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Cognition Enhancing Compounds and Compositions, Methods of Making, and Methods of TreatingPublication Number: US-2016199356-A1Priority Date: 2010-09-08
- Cognition enhancing compounds and compositions, methods of making, and methods of treatingPublication Number: US-9403806-B1Priority Date: 2010-09-08Grant Date: 2016-08-02
- Cognition enhancing compounds and compositions, methods of making, and methods of treatingPublication Number: US-9738633-B2Priority Date: 2010-09-08Grant Date: 2017-08-22
- Cognition enhancing compounds and compositions, methods of making, and methods of treatingPublication Number: US-9174972-B2Priority Date: 2010-09-08Grant Date: 2015-11-03
- Methods of treating neurological disordersPublication Number: US-2025032460-A1Priority Date: 2021-11-24
- Methods of treating neurological disordersPublication Number: CN-118510500-APriority Date: 2021-11-24
- Cognition Enhancing Compounds and Compositions, Methods of Making, and Methods of TreatingPublication Number: US-2013274299-A1Priority Date: 2010-09-08
- Cognition Enhancing Compounds and Compositions, Methods of Making, and Methods of TreatingPublication Number: US-2018099956-A1Priority Date: 2010-09-08
- Cognition Enhancing Compounds and Compositions, Methods of Making, and Methods of TreatingPublication Number: US-2016340348-A1Priority Date: 2010-09-08
////////betovumeline, muscarinic receptor agonist, ML 007, MPL 0527, 5UXW4B47R9
Becondogrel
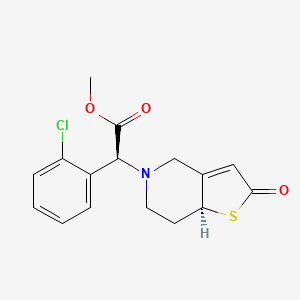
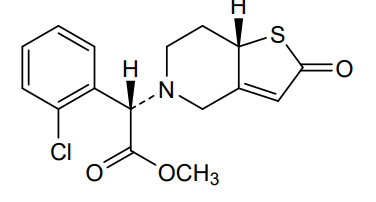
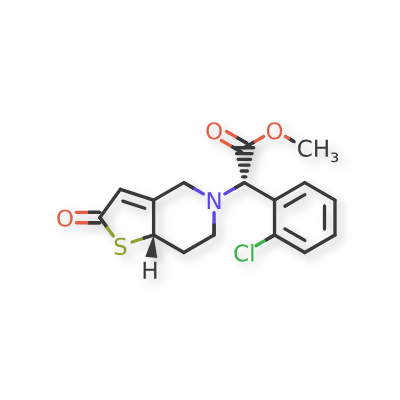
Becondogrel
CAS 1416696-44-0
MF C16H16ClNO3S, MW 337.821
2-OXO-CLOPIDOGREL, (.ALPHA.S,7AS)-
(4S)-2-Oxoclopidogrel
Methyl (S)-(2-chlorophenyl)[(7aS)-2-oxo-2,6,7,7atetrahydrothieno[ 3,2-c]pyridin-5(4H)-yl]acetate
methyl (S)-(2-chlorophenyl)[(7aS)-2-oxo-2,6,7,7atetrahydrothieno[3,2-c]pyridin-5(4H)-yl]acetate
platelet aggregation inhibitor, D7X6820P1N, Copidogrel oxide, (4S)-2-Oxoclopidogrel
Becondogrel is an antiplatelet medication and an irreversible P2Y12 receptor antagonist. It is chemically known as 2-oxoclopidogrel, which is a direct metabolic intermediate of the widely used drug clopidogrel (Plavix).
Key Characteristics
- Mechanism of Action: It prevents blood cells (platelets) from sticking together, which helps inhibit the formation of blood clots (thrombosis).
- Relationship to Clopidogrel: Standard clopidogrel is a prodrug that requires two metabolic steps in the liver to become active. Becondogrel is designed to bypass the first of these steps, potentially reducing the individual variability in effectiveness seen with clopidogrel due to genetic differences in liver enzymes (CYP450).
- Clinical Status: As of early 2025, becondogrel was included in the World Health Organization’s (WHO) proposed International Nonproprietary Name (INN) list, indicating its development for medical use
SYN
- [EP3290423B1]
- https://patentscope.wipo.int/search/en/detail.jsf?docId=EP213389219&_cid=P22-MNPFAY-18756-1
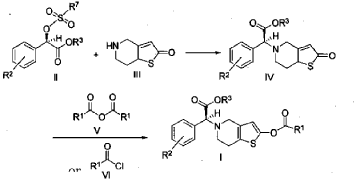
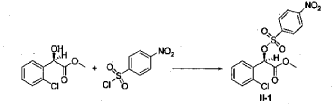
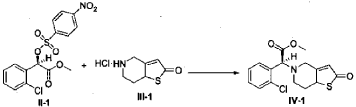
Example 3
(2S)-Methyl 2-(2-oxo-7,7a-dihydrothieno[3,2-c]pyridin-5(2H,4H,6H)-yl)-2-(2-chlorophenyl)-acetate (IV-1)
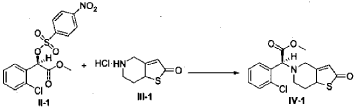
[0042] 58.1 g (0.15 mol) of (R)-methyl 2-(2-chlorophenyl)-2-(4-nitrophenylsulfonyloxy)-acetate (II-1), 32.3 g (0.17 mol) of 5,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2(4H)-one hydrochloride (III-1), and 37.8g (0.38 mol) of potassium bicarbonate were added to 500 ml of acetonitrile. The reaction was stirred under a nitrogen atmosphere at room temperature for 26 hrs. The reaction solution was allowed to stand and the insoluble material was filtered off, to obtain a dark red mother liquor. The solvent was evaporated under reduced pressure, and 35.4 g of an oil product was obtained after purification by flash column chromatography (petroleum ether:ethyl acetate = 4:1). Yield 70%. Recrystalization from ethanol afforded 18.1 g of a pure product (IV-1) as a white solid. mp: 146-148°C, ee = 97.5%, [α] D 19 = +114.0° (c 0.5, MeOH); 1H-NMR (300 MHz, CDCl 3) δ 1.79-1.93 (m, 1 H), 2.30-2.40 (m, 1 H), 2.56-2.70 (m, 1 H), 3.00-3.27 (m, 2 H), 3.72 (s, 3 H), 3.79-3.93 (m, 1 H), 4.12-4.19 (m, 1 H), 4.89 (d, 1 H, J= 5.6 Hz), 6.00 (d, 1 H, J = 5.2 Hz), 7.26-7.50 (m, 4 H); 13C-NMR (75 MHz, CDCl 3) δ 33.9, 34.0, 49.0, 49.7, 51.1, 51.6, 52.2, 52.4, 67.3, 76.6, 77.0, 77.4, 126.6, 126.8, 127.2, 129.8, 130.1, 132.7, 134.8, 167.2, 167.4, 170.8, 198.6; ESI-MS m/ z 338.1 [M+H] +; HRMS Calcd for C 16H 17NO 3SCl [M+H] + m/ z 338.0618, found 338.0626.
SYN
- [US2022028976]
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023204830&_cid=P22-MNPFG3-21704-1
PAT
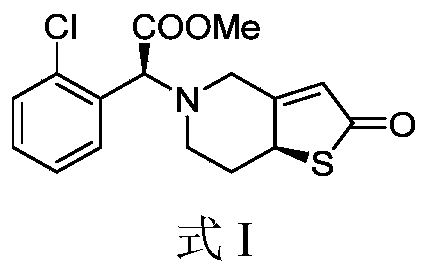
. The chemical name of the compound with the Equation I structure is: (S)-2-(2-chlorophenyl)-2-((S)-2-oxo-2,6,7,7a-tetrahydrothiophene[3,2-c]and pyridine-5(4H))yl)methyl acetate.
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Synthesis of Biologically Active Piperidine Metabolites of Clopidogrel: Determination of Structure and Analyte Development.Publication Name: The Journal of organic chemistryPublication Date: 2015-07-07PMID: 26151079DOI: 10.1021/acs.joc.5b00632
- Antiplatelet Agents Aspirin and Clopidogrel Are Hydrolyzed by Distinct Carboxylesterases, and Clopidogrel Is Transesterificated in the Presence of Ethyl AlcoholPublication Name: The Journal of Pharmacology and Experimental TherapeuticsPublication Date: 2006-12PMID: 16943252DOI: 10.1124/jpet.106.110577
- Contribution of Hepatic Cytochrome P450 3A4 Metabolic Activity to the Phenomenon of Clopidogrel ResistancePublication Name: CirculationPublication Date: 2004-01-20PMID: 14707025DOI: 10.1161/01.cir.0000112378.09325.f9
PAT
- Solid preparation containing oxidized clopidogrel and preparation method thereforPublication Number: EP-4628079-A1Priority Date: 2022-12-02
- Method for treating blood plasma sample containing clopidogrel oxide, and measurement methodPublication Number: EP-4394372-A1Priority Date: 2021-08-23
- Crystalline form b of tetrahydrothienopyridine compound, preparation method, composition and application thereofPublication Number: US-2022289761-A1Priority Date: 2019-05-17
- B crystal form of tetrahydrothienopyridine compound, preparation method therefor, composition and applicationPublication Number: WO-2020233226-A1Priority Date: 2019-05-17
- B crystal form of tetrahydrothienopyridine compound, preparation method therefor, composition and applicationPublication Number: EP-3985009-A1Priority Date: 2019-05-17
- Optically active 2-hydroxy tetrahydrothienopyridine derivatives, preparation method and use in manufacture of medicament thereof
- Publication Number: EP-3290423-B1
- Priority Date: 2010-02-02
- Grant Date: 2021-07-21
//////////becondogrel, platelet aggregation inhibitor, D7X6820P1N, Copidogrel oxide, (4S)-2-Oxoclopidogrel
Balomenib
Balomenib
CAS 2939850-17-4
MF C33H34F3N7O2 MW617.7 g/mol
4-methyl-1-[[(2S)-5-oxomorpholin-2-yl]methyl]-5-[[2-[6-(2,2,2-trifluoroethyl)quinazolin-4-yl]-2,7-diazaspiro[3.5]nonan-7-yl]methyl]indole-2-carbonitrile
- 1H-Indole-2-carbonitrile, 4-methyl-1-[[(2S)-5-oxo-2-morpholinyl]methyl]-5-[[2-[6-(2,2,2-trifluoroethyl)-4-quinazolinyl]-2,7-diazaspiro[3.5]non-7-yl]methyl]-
- 4-methyl-1-[(2S)-5- oxomorpholin-2- yl]methyl]-5- [[2-[6-(2,2,2- trifluoroethyl) quinazolin-4-yl]-2,7- diazaspiro[3.5]nonan- 7-yl]methyl]indole-2- carbonitrile
4-methyl-1-{[(2S)-5-oxomorpholin-2-yl]methyl}-5-({2-[6-(2,2,2-trifluoroethyl)quinazolin-4-yl]-2,7-diazaspiro[3.5]nonan-7-
yl}methyl)-1H-indole-2-carbonitrile
menin inhibitor, antineoplastic, ZE63-0302, 3BEG4BWN8E
Balomenib (also known as ZE63-0302) is an oral, small-molecule menin inhibitor currently in clinical development for metabolic and oncological conditions. It works by disrupting the protein-protein interaction between menin and KMT2A (formerly MLL), a mechanism that plays a critical role in both pancreatic beta-cell function and certain types of leukemia.
Key Therapeutic Areas
- Type 2 Diabetes (T2D): Balomenib is being investigated as a potentially disease-modifying treatment to improve pancreatic beta-cell function and survival. As of late 2025, it has advanced into Phase 1b clinical trials specifically for adults with T2D to evaluate its effects on fasting glucose, insulin dynamics, and HbA1c.
- Oncology (AML): It is also a candidate for treating acute myeloid leukemia (AML) with KMT2A rearrangements or NPM1 mutations. Preclinical data suggests it may be more effective against resistance mutations than earlier menin inhibitors.
Development and Safety
- Corporate Development: The drug was originally developed by Eilean Therapeutics. It is now the lead program for Clywedog Therapeutics, which is merging with Barinthus Biotherapeutics to focus on metabolic diseases.
- Safety Profile: Early trial results indicate a favorable safety profile. Notably, it was designed to minimize QTc prolongation (heart rhythm issues), a side effect common in other menin inhibitors.
- Сlinical Study Aiming to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single and Multiple Ascending Doses of ZE63-0302 in Healthy VolunteersCTID: NCT06780124Phase: Phase 1Status: CompletedDate: 2026-01-22
- Study to Assess Safety, Tolerability, PK, and PD of Multiple Doses of ZE63-0302 Administrated Orally in T2DM Patients.CTID: NCT07234864Phase: Phase 1Status: RecruitingDate: 2026-01-22
SYN
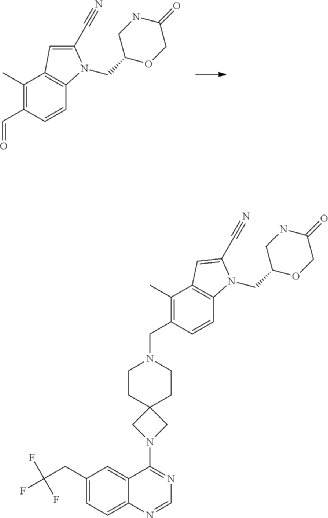
Example 46. 4-Methyl-1-{[(2S)-5-oxomorpholin-2-yl]methyl}-5-({2-[6-(2,2,2-trifluoroethyl)quinazolin-4-yl]-2,7-diazaspiro[3.5]non-7-yl}methyl)-1H-indole-2-carbonitrile (Compound 102)
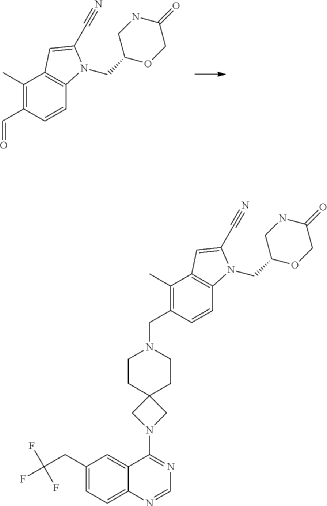
Compound was prepared using procedure described in the Example 45 and 5-formyl-4-methyl-1-{[(2S)-5-oxomorpholin-2-yl]methyl}-1H-indole-2-carbonitrile P177 instead of 5-formyl-4-methyl-1-{[(2R)-5-oxomorpholin-2-yl]methyl}-1H-indole-2-carbonitrile P176. Compound 102 was obtained with yield 49%. 1H NMR (400 MHz, DMSO-d 6), δ: 8.46 (s, 1H), 7.99 (m, 2H), 7.73 (m, 2H), 7.52 (m, 1H), 7.46 (d, J=5.6 Hz, 1H), 7.31 (d, J=4.8 Hz, 1H), 4.54 (m, 1H), 4.20 (m, 2H), 4.05 (m, 1H), 3.90 (m, 4H), 3.52 (m, 2H), 3.35 (m, 1H), 3.17 (m, 1H), 2.39 (m, 2H), 1.79 (m, 4H). LCMS (ESI) [MH] +: 618.
PAT
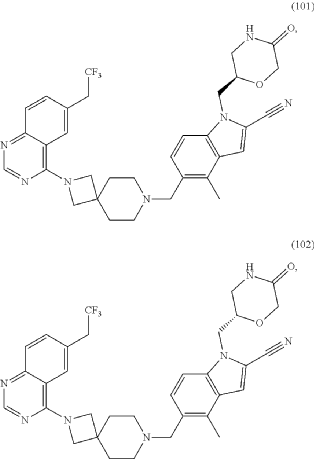
Example 46. 4-Methyl-1-{[(2S)-5-oxomorpholin-2-yl]methyl}-5-({2-[6-(2,2,2-trifluoroethyl)quinazolin-4-yl]-2,7-diazaspiro[3.5]non-7-yl}methyl)-1H-indole-2-carbonitrile (Compound 102)
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Inhibitors of menin-mll interactionPublication Number: WO-2023107696-A2Priority Date: 2021-12-09
- Inhibitors of menin-mll interactionPublication Number: US-2025163061-A1Priority Date: 2021-12-09
- Inhibitors of menin-mll interactionPublication Number: EP-4444300-A2Priority Date: 2021-12-09
///////////balomenib, menin inhibitor, antineoplastic, ZE63-0302, 3BEG4BWN8E
Atebimetinib
Atebimetinib
CAS 2669009-92-9
MF C23H27FN4O6S MW506.5 g/mol
[4-[(dimethylamino)methyl]-3-[[2-fluoro-3-(methylsulfamoylamino)phenyl]methyl]-2-oxochromen-7-yl] N,N-dimethylcarbamate
4-[(dimethylamino)methyl]-3-({2-fluoro-3-[(methylsulfamoyl)amino]phenyl}methyl)-2-oxo-2H-1-benzopyran-7-yl
dimethylcarbamate
MEK tyrosine kinase inhibitor, antineoplastic, IMM-104, IMM 104, Fast Track designation, TEL9243A3N
Atebimetinib (IMM-104) is an investigational oral, deep cyclic inhibitor (DCI) that targets the MAP kinase (MAPK) pathway in solid tumors, particularly RAS-mutant pancreatic cancer. Designed for rapid, pulsatile inhibition to minimize resistance and side effects, it is currently in Phase 2a trials, having shown promising, durable tumor shrinkage and high 1-year survival rates.
Key Aspects of Atebimetinib:
- Mechanism of Action: As a DCI, it works differently from standard inhibitors by targeting MAPK with a short half-life, allowing for rapid “pulsing” that suppresses tumor growth while permitting healthy cells to recover, thus improving tolerability.
- Targeted Cancers: Primarily aimed at RAS-mutant advanced or metastatic solid tumors, including pancreatic ductal adenocarcinoma (PDAC).
- Clinical Trial Results: In a Phase 2a study (NCT05585320), the combination of atebimetinib with modified chemotherapy showed a 64% overall survival (OS) rate at 12 months for first-line pancreatic cancer patients.
- Fast Track Designation: In 2024, the FDA granted fast track designation for atebimetinib to treat patients with pancreatic adenocarcinoma who have progressed after one line of therapy.
- Advantage over Traditional Inhibitors: It is designed to avoid typical MAP kinase inhibitor adverse events like pyrexia (fever) while overcoming the rapid resistance often seen in other therapies.
Atebimetinib is being developed by Immuneering Corporation.
Development Status
- FDA Designations: In 2024, the FDA granted atebimetinib Fast Track designation for patients with pancreatic adenocarcinoma (PDAC) who have progressed after one line of treatment.
- Future Plans: A global registrational Phase 3 trial, named MAPKeeper 301, is planned to begin dosing patients in mid-2026.
Clinical Trial Results (Phase 2a)
Recent data from the Phase 2a trial (as of early 2026) showed significant survival benefits when combined with modified chemotherapy (gemcitabine and nab-paclitaxel) for first-line pancreatic cancer:
- Overall Survival (OS): Reported at 94% at 6 months, 86% at 9 months, and 64% at 12 months. This is roughly double the 1-year survival rate typically seen with standard chemotherapy alone (~35%).
- Progression-Free Survival (PFS): Median PFS reached 8.5 months.
- Disease Control Rate: Approximately 81% of patients achieved disease control.
SYN
WO2023076991 COMBINATION THERAPY FOR TREATING ABNORMAL CELL GROWTH
SYN
WO2025010293 MEK IMMUNE ONCOLOGY INHIBITOR PHARMACEUTICAL COMPOSITIONS
EXAMPLE 1A
Synthesis of Compound A
[0198] Compound A was prepared in 1 step:
[0199] 4-(bromomethyl)-3-(2-fluoro-3-((N-methylsulfamoyl)amino)benzyl)-2-oxo-2H-chromen-7-yl dimethylcarbamate (22.22 g, 34.79 mmol) was suspended in methanol. Dimethylamine 2M was added and the formed reaction mixture was stirred until full conversion was observed. After full conversion the reaction was concentrated under reduced pressure. IM HC1 was added to the residue and the water layer was extracted with CH2CI2. The water layer was made basic with solid Na CCE. The basic water layer was extracted with CH2CI2. The organic layer from the basic extraction was washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure to obtain the title compound (13.23 g, 25.7 mmol, yield: 74%) as a light yellow amorphous solid.
[0200] Yield: Compound A was isolated as a light yellow solid (74% over 1 step). Analysis: LCMS (Method T): tR = 1.53 min; m/z calculated for [M+H]+ = 507.2, found = 507.2; 1H NMR (400 MHz, DMSO) d 9.38 (s, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.28 (td, J = 8.0, 1.6 Hz, 1H), 7.25 – 7.18 (m, 2H), 7.15 (dd, J = 8.8, 2.4 Hz, 1H), 7.00 (t, J = 7.9 Hz, 1H), 6.90 – 6.77 (m, 1H), 4.04 (s, 2H), 3.64 (s, 2H), 3.06 (s, 3H), 2.93 (s, 3H), 2.52 (d, J = 4.9 Hz, 3H), 2.19 (s, 6H).
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Mek immune oncology inhibitor pharmaceutical compositionsPublication Number: WO-2025010293-A2Priority Date: 2023-07-03
- Inhibiting mitogen-activated protein (map)/erk kinase (mek)1 and mek2 and related methods of treatmentPublication Number: WO-2024220440-A1Priority Date: 2023-04-17
- Methods of treating cancer with a ras mutationPublication Number: WO-2024186693-A1Priority Date: 2023-03-03
- Combination therapy for treating abnormal cell growthPublication Number: WO-2023235356-A1Priority Date: 2022-06-03
- Combination therapy for treating abnormal cell growthPublication Number: WO-2023147297-A2Priority Date: 2022-01-25
- Methods of treating abnormal cell growthPublication Number: WO-2023081676-A1Priority Date: 2021-11-02
- Combination therapy for treating abnormal cell growthPublication Number: WO-2023076991-A1Priority Date: 2021-10-28
- Mek inhibitors and therapeutic uses thereofPublication Number: US-2023119327-A1Priority Date: 2020-01-10
//////atebimetinib, FLAX LAB, antineoplastic, IMM-104, IMM 104, Fast Track designation, TEL9243A3N
Anvumetostat
Anvumetostat
CAS 2790567-82-5
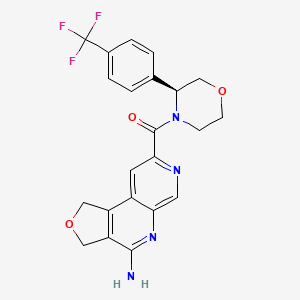
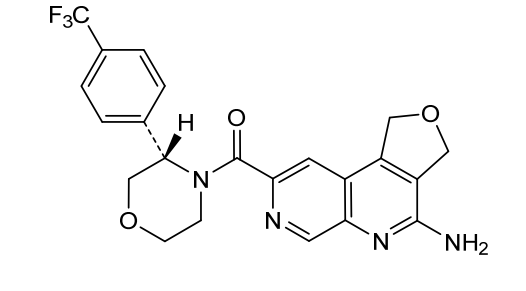
MF C22H19F3N4O3 MW444.4 g/mol
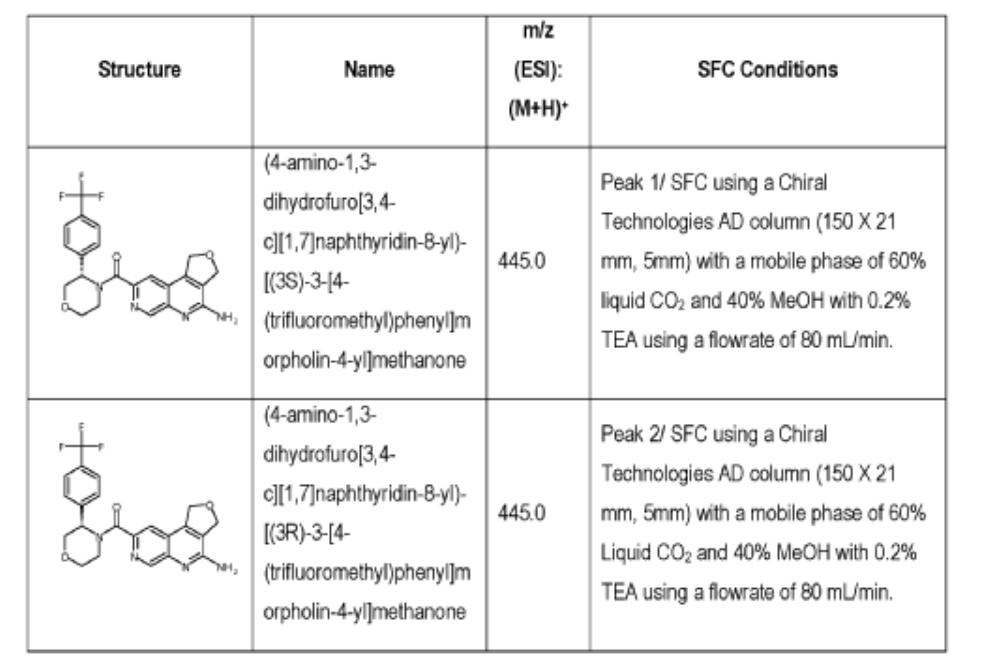
(4-amino-1,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone
(4-amino-1,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl){(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl}methanone
antineoplastic, AMG 193, QAT649EJ5E, PRMT5-IN-27,
Anvumetostat (also known as AMG 193) is an orally available, small-molecule inhibitor of protein arginine methyltransferase 5 (PRMT5), primarily being developed for the treatment of advanced solid tumours with MTAP-null (methylthioadenosine phosphorylase-deficient) mutations.
Mechanism of Action
- Targeting PRMT5: It is a potent and selective MTA-cooperative inhibitor of PRMT5.
- Synthetic Lethality: In cells where the MTAP gene is deleted (a common occurrence in various cancers), a metabolite called MTA (methylthioadenosine) accumulates. Anvumetostat selectively binds to the PRMT5-MTA complex, inhibiting its methyltransferase activity.
- Cellular Impact: By blocking PRMT5, the drug reduces the methylation of arginine residues in histones (H2A, H3, and H4), which can lead to decreased growth or death of cancer cells.
Clinical Development
Anvumetostat was initially developed by Amgen, Inc. and is currently in clinical trials. Institute (.gov) +1
- Current Status: As of early 2026, it is in Phase 2 of global research and development.
- Study Focus: Trials are evaluating its efficacy both as a monotherapy and in combination with other treatments for adult patients with metastatic or locally advanced MTAP-null cancers.
Key Identifiers
- Alternate Names: AMG 193, AMG-193.
- Chemical Class: Orally bioavailable small molecule.
- Genetic Biomarker: Specifically targets cancers with MTAP-null status
Anvumetostat is an orally available small molecule inhibitor of protein arginine methyltransferase 5 (PRMT5), with potential antiproliferative and antineoplastic activities. Upon oral administration, anvumetostat selectively binds to PRMT5 and inhibits its function. By inhibiting its methyltransferase activity, levels of both monomethylated and dimethylated arginine residues in histones H2A, H3 and H4 are decreased. This modulates the expression of genes involved in several cellular processes, including cellular proliferation. This may increase the expression of antiproliferative genes and/or decrease the expression of genes that promote cell proliferation, which may lead to decreased growth of rapidly proliferating cells, including cancer cells. PRMT5, a type II methyltransferase that catalyzes the formation of both omega-N monomethylarginine (MMA) and symmetric dimethylarginine (sDMA) on histones and a variety of other protein substrates involved in signal transduction and cellular transcription, is overexpressed in several neoplasms. Elevated levels are associated with decreased patient survival. Methylthioadenosine phosphorylase (MTAP) is deleted in certain cancer cells leading to an accumulation of methylthioadenosine (MTA). As MTA inhibits PRMT5, MTAP-null cancer cells are specifically sensitive to PRMT5 inhibitors.
SYN
[0163] Examples 481 and 482: (4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4- (trifluoromethyl)phenyl)morpholino)methanone
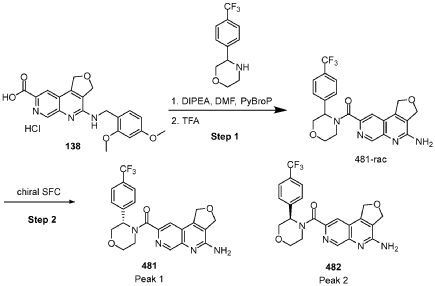
[0164] Step 1: To a solution of 3-(4-(trifluoromethyl)phenyl)morpholine (0.100 g, 0.432 mmol, Enamine), 4-((2,4-dimethoxybenzyl)amino)-l,3-dihydrofuro[3,4-c][l,7]naphthyridine-8-carboxylic acid hydrochloride (138) (0.271 g, 0.649 mmol) and l,l’-dimethyltriethylamine (0.559 g, 0.755 mL, 4.32 mmol, Sigma- Aldrich Corporation) in DMF (4 mL) was added bromotripyrrolidinophosphonium hexafluorophosphate (0.202 g, 0.432 mmol, Sigma-Aldrich Corporation) and the resulting mixture was heated at 50 °C for 30 min. The reaction was brought to rt, diluted with water, sat.NaHCCh and extracted with EtOAc (3x). The combined organics were dried over Na2SO4, filtered and concentrated. The residue was then chromatographed on silica gel using 0-50% 3:1 EtOAc/EtOH in heptane to afford (4-((2,4-dimethoxybenzyl)amino)- 1 ,3 -dihy drofuro [3 ,4-c] [ 1 ,7]naphthyridin-8-y 1) (3 – (4 -(trifluoromethyl)phenyl)morpholino)methanone (0.160 g, 0.269 mmol, 62.2% yield) as a light yellow solid, m/z (ESI): 595 (M+H)+.
[0165] To a solution of (4-((2,4-dimethoxybenzyl)amino)-l,3-dihydrofuro[3,4-c] [l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone (0.160 g, 0.269 mmol, 62.2 % yield) in DCM (2 mL) was added TFA (14.80 g, 10 mL, 130 mmol, Aldrich) and the resulting mixture was heated at 50 °C for 1 h. The reaction was concentrated, washed with 10% Na2CO3 and extracted with DCM. The combined organics were concentrated and chromatographed on silica gel using 0-50% 3:1 EtOAc/EtOH in heptane to afford (4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone as the TFA salt (0.078 g, 0.140 mmol, 32.3% yield) as an off-white solid, m/z (ESI): 445 (M+H)+.
[0166] Step 2: (S)-(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4- (trifluoromethy l)phenyl)morpholino)methanone and (R)-(4-amino- 1 ,3 -dihy drofuro [3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone
(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone 2,2,2-trifluoroacetate were separated via preparative SFC using a Chiral Technologies AD column (150 x 21 mm, 5mm) with a mobile phase of 60% Liquid CO2 and 40% MeOH with 0.2% TEA using a flowrate of 80 mL/min to generate peak 1, (S)-(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone with an ee of >99%, and peak 2, (R)-(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone with an ee of 99.28%. Peak assignment determined by
SFC with AD column with 60% Liquid CO2 and 40% MeOH with 0.2% TEA and absolute
stereochemistry was arbitrarily assigned.
Peak 1: (S)-(4-amino-l,3-dihydrofuro[3,4-c][l,7]naphthyridin-8-yl)(3-(4-(trifluoromethyl)phenyl)morpholino)methanone (481) as a white solid . m/z (ESI): 445 (M+H)+. NMR 1H (400 MHz, DMSO-d6) 5 ppm 8.67 – 9.03 (m, 1 H), 7.85 (s, 1 H), 7.77 (br s, 4 H), 7.07 (br s, 2 H), 5.75 (s, 1 H), 5.37 (br s, 2 H), 5.04 (br s, 2 H), 4.46 – 4.61 (m, 1 H), 3.89 (br dd, J=12.2, 3.3 Hz, 4 H), 3.58 (br d, ./=5,8 Hz, 1 H). 19F NMR (377 MHz, DMSO-d6 ) 5 ppm -60.90 (br s, 3 F).
Peak 2: (R)-(4-amino- 1 ,3 -dihy drofuro [3 ,4-c] [ 1 ,7]naphthyridin-8-yl)(3 -(4-(trifluoromethyl)phenyl)morpholino)methanone (482) as a white solid, m/z (ESI): 445 (M+H)+. NMR 1H (400 MHz, DMSO-d6) 5 ppm 8.88 (br s, 1 H), 7.85 (s, 1 H), 7.77 (br d, J=1.7 Hz, 4 H), 7.07 (br s, 2 H),
5.69 – 5.78 (m, 1 H), 5.37 (br s, 2 H), 5.04 (br s, 2 H), 4.45 – 4.61 (m, 1 H), 3.89 (br dd, J=12.4, 3.3 Hz, 4 H), 3.51 – 3.64 (m, 1 H). 19F NMR (DMSO-d6, 377 MHz) 5 -60.90 (s, 3 F).
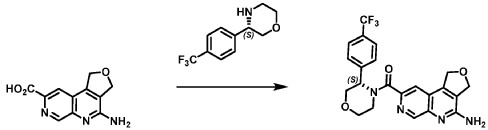
SYN
Example 4. Synthesis of Compound I – (4-amino-1 ,3-di hydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone
Reaction Scale 1
[0137] 4-Amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridine-8-carboxylic acid (1.0 kg, 4.3 mol, 1.0 equiv), (3S)-3-[4-(trifluoromethyl)phenyl]morpholine (1.2 kg, 5.2 mmol, 1.2- equiv), and DMF, (6.6 kg, 7.0 V) were charged to a clean, dry reactor. To the mixture was added triethylamine (1.1 Kg, 13.8 mol, 2.6 equiv). The mixture was cooled to 10 ± 5 °C and O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate (TBTU) (1.67 kg, 5.2 mol, 1.2 equiv) was added slowly. Next, an additional amount of DMF (0.94 Kg, 1 V) was added. The reaction mixture was warmed to 25 ± 5 °C and stirred over 18 hours. Water (1 .0 kg, 1 V) was charged followed by MeCN (1 .6 kg, 2 V) and the reaction mass was warmed to 45 °C. Next, water (7.0 Kg, 7 V) was added over 30 min. A seed lot of 4-amino-1 ,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (10 g, 22 mmol, 0.01 equiv), was charged and the mixture was stirred at 45 °C for over 2 hours before being cooled to 20 °C over 10 hours. Water (12.0 kg, 12 V) was added over 2 hours at 20 °C and further stirred for over 4 hours before being filtered. The reactor was rinsed with a mixture of 10% DMF in water (9.83 kg, 10 V) and the resulting rinse mixture was used to wash the cake. The reactor was rinsed with a mixture of water (10.0k kg, 10 V) and the resulting rinse mixture was used to wash the cake. This rinsing and washing protocol was repeated once more with water (10.0k kg, 10V). The cake was dried under vacuum with a stream of nitrogen to afford (4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-
(trifluoromethyl)phenyl]morpholin-4-yl]methanone. LCMS: 445.20 1H NMR (400 MHz, DMS0-d6 at 130 °C): 8.87 (s, 1 H), 7.80 (s, 1 H), 7.73 (d, 0=8.7 Hz, 2H), 7.71 (d, 0=8.7 Hz, 2H), 6.58 (br s, 2H), 5.72 (br s, 1 H), 5.38 (m, 2H), 5.09 (t, 0=3.5 Hz, 2H), 4.44 (br d, 0=12.3 Hz, 1 H), 4.08 (br d, 0=13.4 Hz, 1 H), 3.96 (dd, 0=12.3, 3.7 Hz, 1 H), 3.86 (br dd, 0=11 .4, 3.0 Hz, 1 H), 3.66 (td, 0=11 .4, 3.0 Hz, 1 H), 3.28 (m, 1 H).
Reaction Scale 2
[0138] 4-Amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridine-8-carboxylic acid (85.0 g, 352.2 mmol, 1.0 equiv), (3S)-3-[4-(trifluoromethyl)phenyl]morpholine (99.6 g, 422.6 mmol, 1.2- equiv), and DMF, (674 mL, 8.7 mol, 7.9 V) were charged to a clean, dry 5 L reactor. To the mixture was added 1 -methylimidazole (75.2 g, 916.2 mmol, 2.6 equiv). The mixture was cooled to 0 °C and N,N,N’,N’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) (118.6 g, 422.6 mmol, 1.2 equiv) was added slowly. Next, an additional amount of DMF (170 mL, 2 V) was added at 0 °C. The reaction mixture was warmed to 25 °C and stirred overnight. Next, the reaction mass was warmed to 45 °C and 2-methyltetrahydrofuran, (169.2 mL, 2 V) was added followed by slow addition of water (850 mL, 10 V) over 30 min by addition funnel. A seed lot of 4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (1.6 g, 3.5 mmol, 0.1 equiv), was charged as a slurry in a 1 :1 v/v of DMF and water (31 .3 mL) and the mixture was stirred at 45 °C for approximately 12 hrs. Water (510 mL, 6 V) was added over 1 h 10 min by addition funnel and the mixture was further stirred at 45°C for 30 min before being filtered. The reactor was rinsed with water (340 mL, 4 V) and the resulting rinse mixture was used to wash the cake. This rinsing and washing protocol was repeated twice more. The cake was dried under vacuum with a stream of nitrogen to afford (4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone. LCMS: 445.20 1H NMR (400 MHz, DMSO-d6 at 130 °C): 8.87 (s, 1 H), 7.80 (s, 1 H), 7.73 (d, J=8.7 Hz, 2H), 7.71 (d, J=8.7 Hz, 2H), 6.58 (br s, 2H), 5.72 (br s, 1 H), 5.38 (m, 2H), 5.09 (t, J=3.5 Hz, 2H), 4.44 (br d, J=12.3 Hz, 1 H), 4.08 (br d, J=13.4 Hz, 1 H), 3.96 (dd, J=12.3, 3.7 Hz, 1 H), 3.86 (br dd, J=11.4, 3.0 Hz, 1 H), 3.66 (td, J=11.4, 3.0 Hz, 1 H), 3.28 (m, 1 H).
Reaction Scale 3:
[0139] 4-Amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridine-8-carboxylic acid (Compound A’) (20.0 g, 86.5 mmol, 1.0 equiv) was added to dimethylsulfoxide (400 mL) at 20 °C. To the mixture was added 1 ,T-carbonyldiimidazole (15.4 g, 95.2 mmol, 1.1 equiv) and the mixture was heated to 60 °C for 1 hour. A solution of (S)-3-(4-(trifluoromethyl)phenyl)morpholin-4-ium chloride (25.5 g, 95.2 mmol, 1.1 equiv) and dimethylsulfoxide (40 mL) was added, and the mixture was heated to 80 °C for 11 hours. The reaction mixture was cooled to 35 °C, then water (265 mL) was added, then the batch was cooled to 20 °C. The reaction was filtered, washed with 40% water:DMSO (80 mL), then washed with water (100 mL). The cake was dried under vacuum with a stream of nitrogen to afford (4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (Compound I). LCMS: 445.20 1H NMR (400 MHz, DMSO-d6 at 130 °C): 8.87 (s, 1 H), 7.80 (s, 1 H), 7.73 (d, J=8.7 Hz, 2H), 7.71 (d, J=8.7 Hz, 2H), 6.58 (br s, 2H), 5.72 (br s, 1 H), 5.38 (m, 2H), 5.09 (t, >3.5 Hz, 2H), 4.44 (br d, >12.3 Hz, 1H), 4.08 (br d, >13.4 Hz, 1 H), 3.96 (dd, >12.3, 3.7 Hz, 1 H), 3.86 (br dd, >11 .4, 3.0 Hz, 1 H), 3.66 (td, >11 .4, 3.0 Hz, 1 H), 3.28 (m, 1 H).
Recrystallization of Compound I
[0140] A clean, dry 5 L reactor was charged with (4-amino-1 ,3-dihydrofuro[3,4-c][1 ,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (279.7 g, 0.6 mol, 1.0 equiv) followed by acetone (6.2 L,
22 V). The mixture was stirred at 40 °C for 15 minutes before cooling to 25 °C. The reactor was discharged into a flask and the reactor was rinsed with acetone and the process stream was polish-filtered back into the reactor.
The reactor jacket was set to 65 °C and the reaction volume was reduced to approximately 6 V by distillation at atmospheric pressure, crystallization was observed. The reaction temperature was set to cool to 20 °C over two hours. Heptane (2.8 L, 10 V) was added over two hours. The slurry was filtered and the cake was washed twice with a 4:1 Heptane/acetone mix (750 mL, 3 V each) and dried under vacuum with a nitrogen purge to afford (4-amino-1,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl] methanone.

ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Process for synthesizing naphthyridine derivatives and intermediates thereofPublication Number: EP-4396170-A1Priority Date: 2021-08-30
- PRMTS inhibitorsPublication Number: CN-116888120-APriority Date: 2020-12-16
- Prmts inhibitorsPublication Number: WO-2022132914-A1Priority Date: 2020-12-16
- Prmts inhibitorsPublication Number: EP-4263545-A1Priority Date: 2020-12-16
- Mta-cooperative prmt5 inhibitors for use in the treatment of cancerPublication Number: EP-4572760-A1Priority Date: 2022-08-15
- Cancer treatments using mta-cooperative prmt5 inhibitorsPublication Number: WO-2023196545-A1Priority Date: 2022-04-08
- Process for the synthesis of naphthyridine derivatives and intermediates thereofPublication Number: CN-117897379-APriority Date: 2021-08-30
- Process for synthesizing naphthyridine derivatives and intermediates thereofPublication Number: WO-2023034786-A1Priority Date: 2021-08-30
- Process for Synthesizing Naphthyridine Derivatives and Intermediates ThereofPublication Number: US-2024360147-A1Priority Date: 2021-08-30
- Prmt5 inhibitor for use in cancer therapyPublication Number: WO-2024170488-A1Priority Date: 2023-02-13
- Cancer treatments using a prmt5 inhibitor and a mat2a inhibitorPublication Number: WO-2024118897-A1Priority Date: 2022-11-30
- Cancer treatments using a prmt5 inhibitor and a mat2a inhibitorPublication Number: EP-4626435-A1Priority Date: 2022-11-30
- MTA synergizes with PRMT5 inhibitors for cancer treatmentPublication Number: CN-119730853-APriority Date: 2022-08-15
- Mta-cooperative prmt5 inhibitors for use in the treatment of cancerPublication Number: WO-2024038004-A1Priority Date: 2022-08-15
////////anvumetostat, ANAX LAB, antineoplastic, AMG 193, QAT649EJ5E, PRMT5-IN-27,
Andamertinib
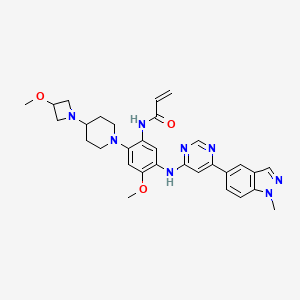
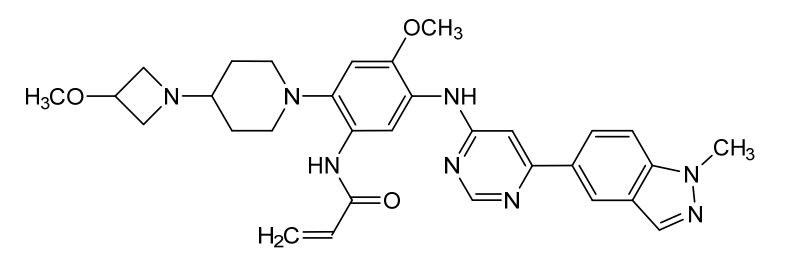
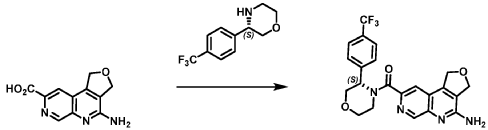
Andamertinib
CAS 2254145-43-0
MF C31H36N8O3 MW568.7 g/mol
N-[4-methoxy-2-[4-(3-methoxyazetidin-1-yl)piperidin-1-yl]-5-[[6-(1-methylindazol-5-yl)pyrimidin-4-yl]amino]phenyl]prop-2-enamide
N-(4-methoxy-2-[4-(3-methoxyazetidin-1-yl)piperidin-1-yl]-5-{[6-(1-methyl-1H-indazol-5-yl)pyrimidin-4-yl]amino}phenyl)prop-2-enamide
epidermal growth factor receptor tyrosine kinase inhibitor, antineoplastic, PLB 1004, 5X3KAG7ZBW
Andamertinib (also known as PLB1004) is an investigational, orally bioavailable, and irreversible small-molecule inhibitor of the epidermal growth factor receptor (EGFR). It is primarily being developed to treat non-small cell lung cancer (NSCLC) with specific genetic mutations.
Key Clinical & Therapeutic Features
- Target Mutations: It specifically targets EGFR exon 20 insertion (ex20ins) mutations, which are often resistant to standard first- and second-generation EGFR inhibitors.
- Broad Selectivity: Beyond ex20ins, it also shows activity against classical mutations like Del19, L858R, and the resistance mutation T790M.
- Brain Penetration: Andamertinib is designed to cross the blood-brain barrier, making it potentially effective for patients with brain metastases.
- Clinical Performance: In phase 2 studies (e.g., the KANNON study), it demonstrated a confirmed objective response rate (ORR) of 42.7% and a median progression-free survival of 6.2 months in pretreated patients.
Regulatory Status (as of Early 2026)
- China: A New Drug Application (NDA) was accepted by the National Medical Products Administration (NMPA) in May 2025 and granted priority review for treating NSCLC with EGFR ex20ins mutations.
- Global: It remains in various stages of clinical trials globally, including studies for first-line treatment and combination therapies with other agents like vebreltinib.
Andamertinib is an orally bioavailable, mono-anilino-pyrimidine, mutant-selective epidermal growth factor receptor (EGFR) inhibitor, with potential antineoplastic activity. Upon oral administration, andamertinib targets, binds to and irreversibly inhibits the activity of various EGFR mutations, including exon 20 insertion (Ex20ins) activating mutations, the gatekeeper mutation T790M, ExDel19, and L858R. This prevents EGFR-mediated signaling, induces cell death and inhibits tumor growth in tumor cells expressing these EGFR mutations. EGFR, a receptor tyrosine kinase mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization.
SYN

SYN
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Aminopyrimidine compound, preparation method therefor and use thereofPublication Number: US-11352352-B2Priority Date: 2017-06-13Grant Date: 2022-06-07
- Aminopyrimidine compound, preparation method therefor and use thereofPublication Number: EP-3640248-A1Priority Date: 2017-06-13
- Aminopyrimidine compound, preparation method therefor and use thereofPublication Number: US-2020087296-A1Priority Date: 2017-06-13
- Aminopyrimidine derivatives, preparation method therefor and use thereofPublication Number: EP-3640248-B1Priority Date: 2017-06-13Grant Date: 2023-08-23
//////////andamertinib, FLAX LAB, antineoplastic, PLB 1004, 5X3KAG7ZBW
Amsulostat
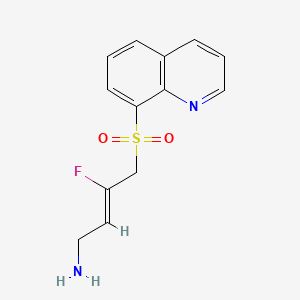
Amsulostat
CAS 2409963-83-1
MF C13H13FN2O2S MW280.32
2-Buten-1-amine, 3-fluoro-4-(8-quinolinylsulfonyl)-, (2Z)-
(2Z)-3-fluoro-4-(quinolin-8-ylsulfonyl)but-2-en-1-amine
(Z)-3-fluoro-4-quinolin-8-ylsulfonylbut-2-en-1-amine
(2Z)-3-fluoro-4-(quinolin-8-ylsulfonyl)but-2-en-1-amine
protein-lysine-oxidase (LOX) inhibitor, antifibrotic, PXS 5505, LOX inhibitor PXS-5505, LOX-IN-3, pan-LOX inhibitor PXS-5505, DO94E28WYW, SNT 5505
Amsulostat is an orally available, small-molecule, irreversible inhibitor of all lysyl oxidases (LOX) family members, with potential antifibrotic activity. Upon oral administration, amsulostat targets, binds to and inhibits the activity of all enzymes in the LOX family. This prevents the post-translational oxidative deamination of lysine residues on target proteins, including collagen and elastin, and reduces the formation of deaminated lysine (allysine), the formation of inter- and intramolecular cross-linkages and may prevent remodeling of the extracellular matrix (ECM), thereby reducing fibrotic tissue formation in certain chronic fibrotic diseases. LOX is often upregulated in fibrotic tissue and plays a key role in fibrosis.
Amsulostat (formerly PXS-5505) is an orally available, investigational, pan-lysyl oxidase (pan-LOX) inhibitor designed by Syntara to treat fibrotic diseases and solid tumors. It works by preventing collagen cross-linking and remodeling of the extracellular matrix, effectively reducing fibrosis. The drug is currently in Phase 2 clinical trials for myelofibrosis, showing promise in reducing symptom burden and spleen volume, and is also being studied for myelodysplastic syndrome (MDS) and pancreatic cancer.
Key Aspects of Amsulostat:
- Mechanism of Action: Irreversibly inhibits all LOX family members (LOX, LOXL1-4), reducing fibrotic tissue.
- Clinical Status (Myelofibrosis): Phase 2a data showed 73% of patients (who were suboptimal responders to ruxolitinib) achieved
reduction in total symptom score, with significant spleen volume reduction.
- Clinical Status (Other Cancers): Phase 2 trials (AZALOX) are evaluating its use in myelodysplastic syndrome (MDS) and chronic myelomonocytic leukemia (CMML). It is also being tested in combination with chemotherapy for pancreatic cancer to improve drug delivery to tumors.
- Regulatory Status: Has received Orphan Drug Designation for primary myelofibrosis from the FDA (USA) and EMA (Europe).
- Safety Profile: Clinical trials have reported it is well-tolerated with no treatment-related serious adverse events in early findings.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Amsulostat’s ability to target the stiff, fibrotic environment surrounding tumors makes it a promising “add-on” therapy to increase the effectiveness of existing cancer treatments, including chemotherapy and immunotherapy.
An orally available, small-molecule, irreversible inhibitor of all lysyl oxidases (LOX) family members, with potential antifibrotic activity. Upon oral administration, amsulostat targets, binds to and inhibits the activity of all enzymes in the LOX family. This prevents the post-translational oxidative deamination of lysine residues on target proteins, including collagen and elastin, and reduces the formation of deaminated lysine (allysine), the formation of inter- and intramolecular cross-linkages and may prevent remodeling of the extracellular matrix (ECM), thereby reducing fibrotic tissue formation in certain chronic fibrotic diseases. LOX is often upregulated in fibrotic tissue and plays a key role in fibrosis.
SYN
syn

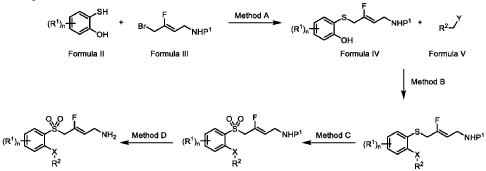
(Z)-3-fluoro-4-(quinolin-8-ylsulfonyl)but-2-en-1-amine dihydrochloride (Compound 33)
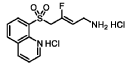
[0282] White solid; m.p.150-153 °C; 1H NMR (300 MHz, CD3OD) d ppm: 9.18 (d, J = 4.7 Hz, 1H), 8.70 (dd, J = 8.4, 2.6 Hz, 1H), 8.57 (d, J = 7.4 Hz, 1H), 8.45 (d, J = 8.5 Hz, 1H), 7.99– 7.68 (m, 2H), 5.22 (dt, J = 32.9, 7.4 Hz, 1H), 5.00 (d, J = 19.4 Hz, 2H), 3.60 (d, J = 7.7 Hz, 2H); LCMS: for C13H13FN2O2S calculated 280.1, found 281.1 [M+1]+.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: EP-3829520-B1Priority Date: 2018-08-03Grant Date: 2024-11-27
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: US-2025082593-A1Priority Date: 2018-08-03
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: US-2021353571-A1Priority Date: 2018-08-03
- Lox enzyme inhibiting methods and compositionsPublication Number: EP-4138824-A1Priority Date: 2020-04-21
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: WO-2020024017-A1Priority Date: 2018-08-03
- Inhibitors of haloallylamine sulfone derivatives of lysyl oxidase and uses thereofPublication Number: KR-20210045984-APriority Date: 2018-08-03
- Halide allylamine derivate inhibitor for amine oxidase and its usePublication Number: TW-202019877-APriority Date: 2018-08-03
- Haloallylamine sulfone derivative inhibitors of lysyl oxidases and uses thereofPublication Number: US-12178791-B2Priority Date: 2018-08-03Grant Date: 2024-12-31
- Bithiazol deratives as inhibitors of lysyl oxidasesPublication Number: EP-4333843-A1Priority Date: 2021-05-13
- Inhibitors of lysyl oxidasesPublication Number: US-11712437-B2Priority Date: 2021-05-13Grant Date: 2023-08-01
- Method for selecting cancer patients for whom combination therapy of retinoid with cancer therapeutic agent is effective, and combination drug of retinoid with cancer therapeutic agentPublication Number: CN-115997122-APriority Date: 2020-06-26
- Method for selecting cancer patient for which combination therapy of retinoid and cancer treatment agent will be effective, and combined drug of retinoid and cancer treatment agentPublication Number: EP-4173639-A1Priority Date: 2020-06-26
- Method for selecting cancer patients for whom combination therapy with retinoid and cancer therapeutic agent is effective, and combination medicament with retinoid and cancer therapeutic agentPublication Number: US-2023301950-A1Priority Date: 2020-06-26
////////////amsulostat, ANAX LAB, protein-lysine-oxidase (LOX) inhibitor, antifibrotic, PXS 5505, LOX inhibitor PXS-5505, LOX-IN-3, pan-LOX inhibitor PXS-5505, DO94E28WYW, SNT 5505