
Home » Uncategorized (Page 8)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gamcemetinib



Gamcemetinib
CAS 1887069-10-4
CC-99677 , OS2IR8TV1O
| Molecular Weight | 469.94 |
|---|---|
| Formula | C22H20ClN5O3S |
- (10R)-3-[[2-Chloro-5-(ethoxymethyl)-4-pyrimidinyl]oxy]-9,10,11,12-tetrahydro-10-methyl-8H-[1,4]diazepino[5′,6′:4,5]thieno[3,2-f]quinolin-8-one (ACI)
- (10R)-3-{[2-chloro-5-(ethoxymethyl)pyrimidin-4-yl]oxy}-10-methyl-9,10,11,12-tetrahydro-8H-[1,4]diazepino[5′,6′:4,5]thieno[3,2-f]quinolin-8-one
- BMS 986371
- BMS-986371
- CC 99677
- CC-99677
(R)-3-((2-Chloro-5-(ethoxymethyl)pyrimidin-4-yl)oxy)-10-methyl-9,10,11,12-tetrahydro-8H-[1,4]diazepino[5′,6′:4,5]thieno[3,2-f]quinolin-8-one
- OriginatorCelgene Corporation
- ClassAnti-inflammatories
- Mechanism of ActionMAP-kinase-activated kinase 2 inhibitors
- Orphan Drug StatusNo
- 14 Nov 2024Efficacy and adverse events data from a phase II trial in Ankylosing Spondylitis presented at the ACR Convergence 2024 (ACR-2024)
- 27 Mar 2024Pharmacokinetics and adverse events data from a phase I trial (In volunteers) presented at the 125th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics 2024 (ASCPT-2024)
- 26 Oct 2023Discontinued – Phase-I for Inflammation (In volunteers) in USA, United Kingdom (PO) prior to October 2023 (Bristol-Myers Squibb pipeline, October 2023)
Gamcemetinib (CC-99677) is a potent, covalent, and irreversible inhibitor of the mitogen-activated protein (MAP) kinase-activated protein kinase-2 (MK2) pathway in both biochemical (IC50=156.3 nM) and cell based assays (EC50=89 nM). Gamcemetinib is extracted from patent WO2020236636, compound 1.
SCHEME
SIDECHAIN

SIDECHAIN

MAIN

REF
WO2018170203
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018170203&_cid=P11-MBK7CL-38003-1



PATENT
WO2018170199 CELGENE
WO2018170203
US20160075720
WO2020236636, compound 1
////////////Gamcemetinib, BMS 986371, BMS-986371, CC 99677, CC-99677, OS2IR8TV1O
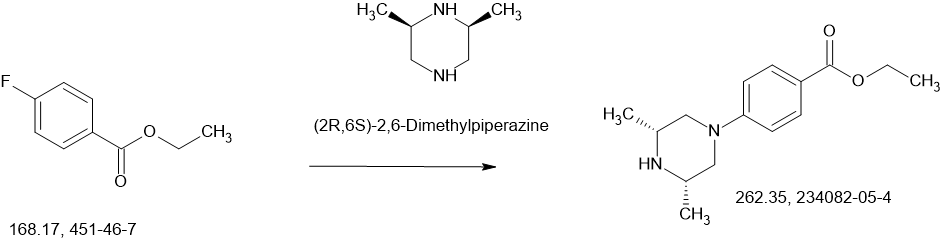
Fexagratinib
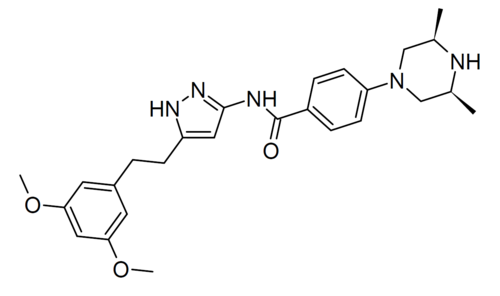
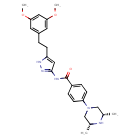
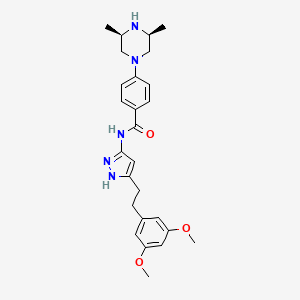
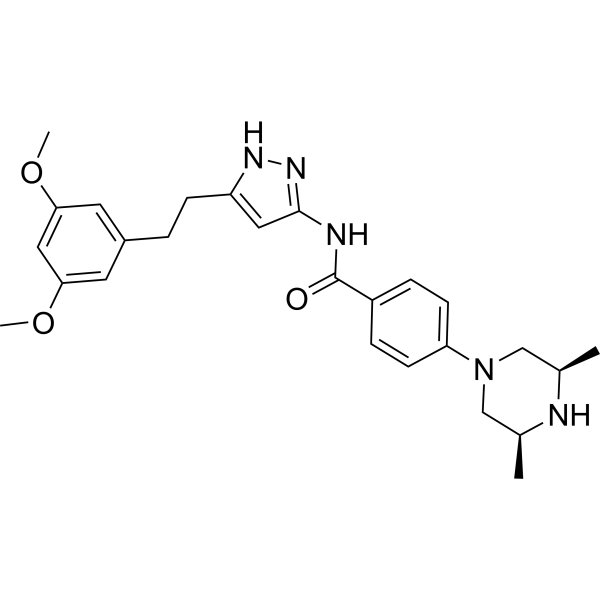
Fexagratinib
AZD 4547; ADSK091 cas 1035270-39-3
WeightAverage: 463.582
Monoisotopic: 463.258339943
Chemical FormulaC26H33N5O3
N-(5-(2-(3,5-DIMETHOXYPHENYL)ETHYL)-1H-PYRAZOL-3-YL)-4-((3R,5S)-3,5-DIMETHYLPIPERAZIN-1-YL)BENZAMIDE
N-{5-[2-(3,5-dimethoxyphenyl)ethyl]-1H-pyrazol-3-yl}-4-[(3R,5S)-3,5-dimethylpiperazin-1-yl]benzamide
- OriginatorAstraZeneca
- DeveloperAbbisko Therapeutics; AstraZeneca; Dust Diseases Authority; Institute of Respiratory Health; National Cancer Institute (USA); University of Glasgow; University of Leeds; University of Wisconsin-Madison
- ClassAntineoplastics; Benzamides; Phenyl ethers; Piperazines; Pyrazoles; Small molecules
- Mechanism of ActionType 1 fibroblast growth factor receptor antagonists; Type 3 fibroblast growth factor receptor antagonists; Type-2 fibroblast growth factor receptor antagonists
- Phase IIGastric cancer; Lymphoma; Multiple myeloma; Solid tumours; Urogenital cancer
- PreclinicalSkin cancer
- No development reportedLiver cancer
- DiscontinuedBladder cancer; Breast cancer; Glioblastoma; Head and neck cancer; Lung cancer; Mesothelioma; Non-small cell lung cancer; Oesophageal cancer
- 13 Sep 2024Pharmacodynamics data from the preclinical studies in Solid tumours presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
- 28 Feb 2024No recent reports of development identified for preclinical development in Liver-cancer in China (PO)
- 23 Jan 2024Preclinical trials in Solid tumours (Monotherapy) in China (PO) (Abbisko Therapeutics pipeline, January 2024)
Fexagratinib (AZD4547) is an experimental drug which acts as an inhibitor of the fibroblast growth factor receptors, having high affinity for FGFR1, FGFR2 and FGFR3 and weaker activity at FGFR4. It has reached clinical trials in humans against several forms of cancer, but has had only limited use as a medicine due to an unfavorable side effect profile, though it may have some applications in combination with other drugs. However it is still widely used in cancer research.[1][2][3][4][5]
SCHEME
SIDECHAIN
MAIN
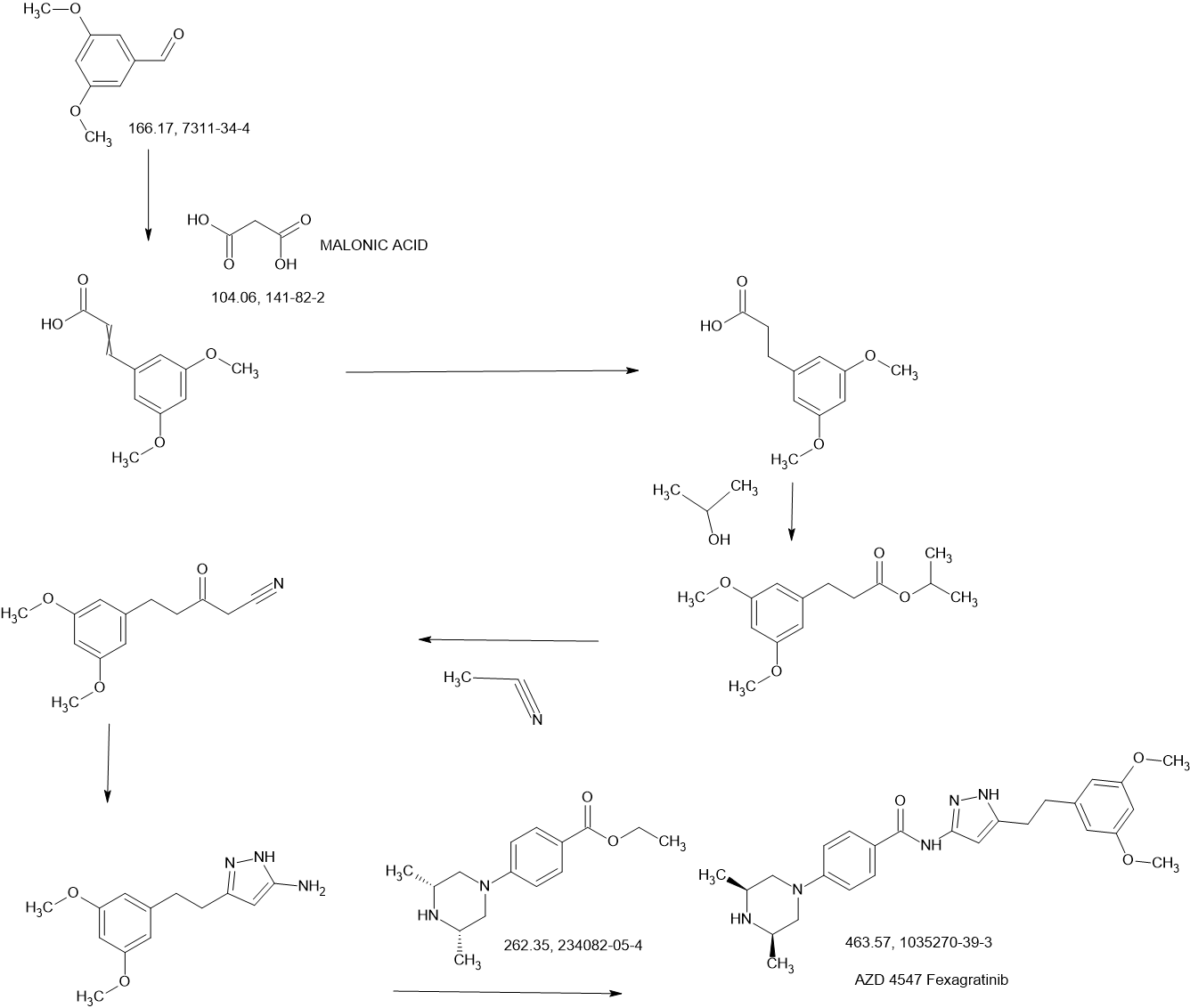
SYN
| At present, the preparation of AZD4547 mainly includes the following methods: |
| (1) Patent application WO2008075068A1 discloses a preparation method comprising the following steps: |
| |
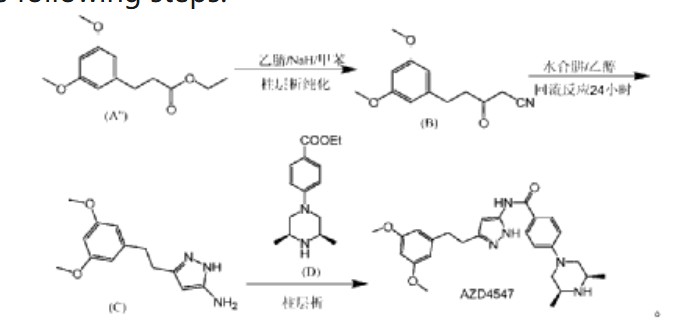
| In the preparation method, AZD4547 is prepared by three-step reactions using ethyl 3-(3,5-dimethoxyphenyl)propionate as a raw material, wherein the first step reaction needs to be purified by column chromatography, and the yield is only 42%; the second step reaction requires reflux reaction for 24 hours, hydrazine hydrate is prone to explosion in high-temperature reactions, and hydrazine hydrate is a highly toxic and genotoxic reagent, and direct high-temperature reaction is not friendly to humans and the environment; the third step reaction also requires column chromatography purification, and the total yield of the three-step reaction for preparing AZD4547 is only 21.08%; therefore, the multi-step reactions of the preparation method require column chromatography operations, have poor safety, low yield, are not suitable for industrialization, and cannot solve the problem of drug accessibility. |
| (2) Patent application CN111072638A discloses another preparation method, comprising the following steps: |
| |
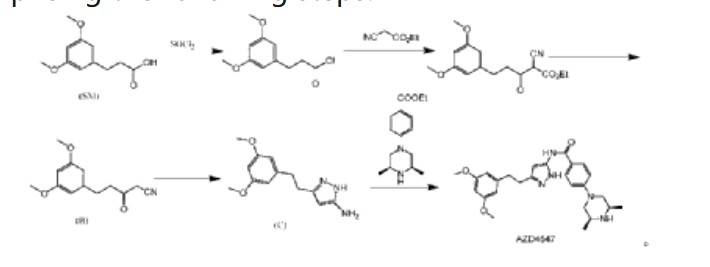
| In this preparation method, 3-(3,5-dimethoxyphenyl)propionic acid is used as the starting material, and AZD4547 is prepared through a five-step reaction with a total yield of 42.5%. In this preparation method, highly toxic reagent ethyl cyanoacetate and expensive reagents palladium carbon, stannous chloride, and Raney nickel are required, and it is not suitable for industrial production. |
| (3) In addition, patent application WO2016137506A1 discloses a method for preparing AZD4547 key intermediate 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine, as follows: |
| |
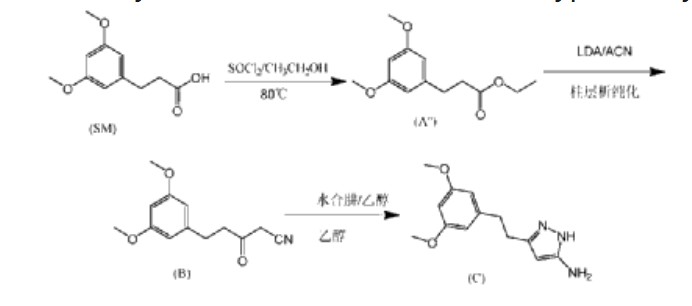
| In this preparation method, the first step of the reaction uses ethanol reflux reaction, the second step of the reaction uses a large amount of solvent, and needs to be reacted at an ultra-low temperature of -78°C. After the reaction is completed, column chromatography purification is required, which is not suitable for industrial application. |
CN115819239
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN394502634&_cid=P11-MBA7JR-97597-1
| Example 1 |
| Add isopropanol (300 mL) and 3-(3,5-dimethoxyphenyl)propionic acid (60.0 g, 0.285 mol) to a 1L three-necked reaction bottle, raise the temperature to 40±5°C, and stir for 5 to 10 minutes to dissolve. Add SOCl dropwise at 40±5°C. 2 (37.3g, 0.314mol), the dropping time is ≥0.5 hours (the dropping process is obviously exothermic), after the dropping is completed, the temperature is raised to 60±5℃, the reaction is stirred for 1 hour, and the reaction of the raw materials is complete when HPLC is detected. The reaction solution is cooled to 35±5℃, the temperature is controlled below 50℃ and the solution is concentrated under reduced pressure until there is no obvious fraction, methyl tert-butyl ether (300mL) is added to dissolve, 5% potassium carbonate aqueous solution is added under ice bath to adjust the pH value of the reaction solution to 8-9, the temperature is controlled at 25±5℃ and stirred for 0.5 hours, the solution is allowed to stand and the organic phase is separated, washed with saturated brine, and the solution is concentrated under reduced pressure at 45℃ to dryness to obtain 72.1g of light yellow oily 3-(3,5-dimethoxyphenyl) propionic acid isopropyl ester, purity: 94%, yield: 94.3%. |
| 1HNMR(DMSO-d 6 ,400MHz)δ6.384-6.378(d,2H),6.318-6.306(t,1H),4.925-4.831(m,1H),3.706(s,6H),2.787-2.749(t,2H),2.571-2.533(t,2H),1.164-1.148(d,6H)。 |
| Example 2 |
| Under nitrogen protection, add isopropyl 3-(3,5-dimethoxyphenyl)propionate (20.0 g, 0.079 mol), anhydrous acetonitrile (80 ml), and anhydrous tetrahydrofuran (100 ml) to a 500 ml three-necked reaction bottle, cool the reaction solution to an internal temperature of about -20 ° C, slowly add lithium diisopropylamide (83 ml, 0.166 mol, 2M THF solution), and add the solution dropwise for about 25 minutes. Continue stirring for 5-10 minutes, and detect the reaction of the raw materials by HPLC. After the reaction mixture was completely dried, acetic acid solution (15 ml) was added to quench the reaction, the mixture was concentrated under reduced pressure, water (100 ml) was added, the pH was adjusted to neutral with 25% aqueous sodium carbonate solution, ethyl acetate (200 ml) was added for extraction (HPLC chart see Figure 2), the organic layer was concentrated under reduced pressure until there was no fraction, ethanol (200 ml) was added, stirred and slurried, filtered, and the filter cake was dried in vacuum at 45°C to obtain 14.8 g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile with a purity of 98% and a yield of 76%. |
| 1HNMR(DMSO-d 6 ,400MHz)δ6.370-6.364(s,2H),6.320-6.309(s,1H),4.038(s,2H),3.709(s,6H),2.851-2.815(t,2H),2.739-2.702(t,2H)。 |
| After preliminary separation, the HPLC, LCMS and 1 HNMR spectra of the impurity-containing mother liquor are shown in Figures 3-5. After analysis, the main impurity is generated by the self-polymerization of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile, and the structure of the impurity compound [compound of formula (B’)] is as follows: |
| |
| Example 3 |
| Under nitrogen protection, 3-(3,5-dimethoxyphenyl)propionic acid isopropyl ester (11.29 g, 0.045 mol), anhydrous acetonitrile (40 ml) and anhydrous tetrahydrofuran (50 ml) were added to a 500 ml three-necked reaction bottle, the reaction solution was cooled to -20°C, diisopropylamide lithium tetrahydrofuran solution (47 ml, 0.094 mol) was slowly added dropwise, and the addition was completed in about 25 minutes. The reaction was continued with stirring for 5-10 minutes. HPLC detected that the raw material reaction was complete, anhydrous ethanol (20 ml) was added to quench the reaction, and 2-methyltetrahydrofuran (50 g) was added for extraction. The pH of the aqueous layer was adjusted to neutral with hydrochloric acid, filtered, and the filter cake was dried in vacuo at 45°C to obtain 9.28 g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile, purity: 99.5%, yield: 88.0%. |
| Example 4 |
| Under nitrogen protection, a tetrahydrofuran solution containing isopropyl 3-(3,5-dimethoxyphenyl)propionate (120.0 g, 0.4756 mol), anhydrous acetonitrile (380 g, 9.25 mol), and anhydrous tetrahydrofuran (270 g) were added to a 3L three-necked reaction flask. The mixture was stirred until the internal temperature dropped to about -20°C. At this temperature, a tetrahydrofuran solution of lithium diisopropylamide (500 ml, 1 mol) was slowly added dropwise. After the addition was completed, the mixture was stirred at about -20°C for 1 minute. -2 hours, HPLC detected that the raw material was completely converted, ethanol (474g) was added to the reaction to quench the reaction, and the reaction was concentrated under reduced pressure. Purified water was added, the internal temperature was controlled at 0-15°C, HCl was slowly added, the pH was adjusted to 7.0, and a large amount of solid was precipitated. The reaction was stirred for 30 minutes and filtered, and the mixture was rinsed with purified water and ethanol in turn. The mixture was dried in vacuo at 45°C to obtain 98.7g of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile with a purity of 99.6% and a yield of 89.0%. |
| In addition, the inventors investigated the effects of the reaction raw materials, anhydrous acetonitrile, alkaline reagent, and reaction temperature on the reaction. The purity and reaction phenomena in the HPLC test were as follows: |
| |
| |
| From the above experimental investigation factors and experimental phenomena, it can be seen that the types of ester groups of different reaction raw materials, the amount of acetonitrile and the alkaline reagent have the following effects on the reaction: |
| (1) Effect of the type of ester group in the reaction raw materials on the reaction |
| When the reaction raw material is a compound of formula (A’) having a methyl ester group, a sticky mass will be formed during the reaction, affecting stirring, and the purity of the reaction is not high. Specifically, at the beginning of the reaction, a sticky mass appears in the reaction liquid, affecting stirring. As the reaction proceeds, the reaction liquid gradually becomes sticky, and even sticks to the wall, making it impossible to stir. |
| When the reaction raw material is a compound of formula (A”) having an ethyl ester group, the reaction purity is increased to 87%, but sticky lumps are still formed during the reaction, affecting stirring. The specific situation is similar to that when the reaction raw material is a compound of formula (A’) having a methyl ester group. |
| The appearance of viscous clumps during process scale-up can easily lead to incomplete reactions, and may even cause dangerous situations such as entanglement of stirring blades and burning of motors. Therefore, the above two preparation processes are not suitable for industrial scale-up production. |
| When the ester structure of the reaction raw material is changed to isopropyl ester, the reaction liquid is homogeneously clear without sticky micelles, and the reaction control purity is increased to more than 97%, which is suitable for industrial scale-up production. The inventors analyzed that the above experimental phenomenon may be due to the higher stability of the isopropyl ester structure, which reduces the formation of side reactions. |
| (2) Effect of acetonitrile dosage on the reaction |
| In Experiment 6 and Experiment 3 of the present invention, when the molar ratio of acetonitrile to the reaction raw material increased from 10eq to 20eq, the reaction control purity increased from 90.4% to 97.2%. |
| Comparing Experiment 2 and Experiment 3 in Experiment 1, when the molar ratio of acetonitrile to the reaction raw material increased from 1.2eq to 25eq, the reaction control purity increased from 60.8% to 92.8%. |
| (3) Effect of the selection and dosage of alkaline reagents on the reaction |
| From the above experimental results, it can be seen that the reaction control purity of NaHMDS, LDA and n-BuLi is relatively high. |
| The optimal molar ratio of alkaline reagent to reaction raw materials is 2.1eq. A molar ratio lower than 2eq may result in incomplete reaction. |
| Example 5 |
| Step 1: Synthesis of 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile (compound of formula (B)) |
| Under nitrogen protection, add isopropyl 3-(3,5-dimethoxyphenyl)propionate (20.0 g, 0.079 mol), anhydrous acetonitrile (80 ml), and anhydrous tetrahydrofuran (100 ml) into a 500 ml three-necked reaction bottle, cool the reaction solution to -20°C, slowly add lithium diisopropylamide (83 ml, 0.166 mol, 2M THF solution) dropwise, add for about 25 min, stir and react for 5-10 minutes, HPLC detection shows that the raw material reaction is complete, add anhydrous ethanol (40 ml), concentrate under reduced pressure to a viscous state, add anhydrous ethanol (60 ml) to prepare an ethanol solution, and directly put into the next step reaction. |
| Step 2: Preparation of 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (compound of formula (C)) |
| Add acetic acid (26.0 g, 0.436 mol), ethanol (100 ml), and 80% hydrazine hydrate (15.0 g, 0.238 mol) to a 500 ml three-necked reaction bottle, heat to an internal temperature of about 68°C, slowly add the product ethanol solution (18.5 g, 0.079 mol) obtained in step 1 to the mixed solution of acetic acid and hydrazine hydrate at this temperature, add for about 40 minutes, stir and react at an internal temperature of about 68°C for 1 hour, and HPLC detection shows that 5-(3,5-dimethoxyphenyl)-3-oxopentanonitrile is completely converted; the reaction solution is concentrated under reduced pressure, water (100 ml), ethyl acetate (200 ml), and about 25% Na 2 CO 3 (40ml) adjust the pH value of the water layer to 7-8; separate the water layer, wash the layers with saturated brine (20ml), concentrate the organic layer under reduced pressure until there is no fraction, add isopropyl acetate (100ml) and reduce the pressure to bring it to a viscous state, add isopropyl acetate (120ml) and heat to dissolve, cool and crystallize, filter at about 10°C, and dry under vacuum at 50°C to obtain 16.3g of 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine with a purity of 99.6% and a total yield of 83% in two steps. |
| 1HNMR(DMSO-d 6 ,400MHz)δ6.370-6.364(s,2H),6.320-6.309(s,1H),4.038(s,2H),3.709(s,6H),2.851-2.815(t,2H),2.739-2.702(t,2H)。 |
| In addition, the inventors investigated the effect of the amount of acetic acid used in this step of the reaction on the reaction, and the purity was controlled by HPLC as follows: |
| |
| In addition, the inventors also investigated that the solid compound of formula (B) obtained after purification of the product in step 1 was reacted with hydrazine hydrate in the presence of acetic acid, and the compound of formula (B) was also completely converted to obtain a high-purity compound of formula (C). |
| Example 6 |
| 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (100.0 g, 0.4044 mol), ethyl 4-((3R,5S)-3,5-dimethylpiperazin-1-yl)benzoate (132.5 g, 0.5050 mol), and 2-methyltetrahydrofuran (1300 ml) were added to the reaction kettle, heated to 50-55°C and stirred for 1 hour, filtered through diatomaceous earth, and the filtrate was added to a clean reaction kettle, heated to atmospheric distillation with water, and the reaction temperature was controlled at 78°C-88°C, and 25% KO-tAm toluene solution (490.0 g) was slowly added dropwise for about 2 hours. After the addition was completed, the reaction temperature was adjusted to 83-88°C and stirred for 3-6 hours. Sampling was performed to detect whether the reaction of the raw materials was complete. The reaction system was cooled to 30-60°C, water (8 ml) was slowly added to quench the reaction, and the mixture was stirred at 30-60°C for 0.5 hour, then cooled to about 25°C, water (400 ml) was added, stirred and allowed to stand for stratification, the organic phase was separated, water (200 ml) was added, the mixture was heated to about 50°C and stirred for 0.5 hour, the water layer was separated, and this was repeated 2-3 times until the pH of the water layer was 7.0-9.5; the organic layer was concentrated under reduced pressure to remove part of the solvent, the residue was heated to 80-90°C and stirred for 1 hour, slowly cooled to 20-30°C, stirred for 2-5 hours, filtered, rinsed twice with ethyl acetate, and dried in vacuo at 45°C to obtain 155.6 g of a white amorphous solid product (AZD4547) with a purity of 98.5% and a yield of 83%. |
| 1HNMR(DMSO-d 6 ,400MHz)δ12.067(s,1H),10.275(s,1H),7.888-7.867(d,2H),6.943-6.922(d,2H),6.437-6.409(m,3H),6.317(s,1H),3.712-3.692(m,8H),2.861-2.803(m,6H),2.230-2.174(m,3H),1.036-1.020(d,6H)。 |
| Example 7 |
| 3-(3,5-dimethoxyphenethyl)-1H-pyrazole-5-amine (10.0 g, 0.040 mol), ethyl 4-((3R,5S)-3,5-dimethylpiperazin-1-yl)benzoate (12.1 g, 0.047 mol), anhydrous tetrahydrofuran (170 ml) were added to the reaction bottle, heated and distilled at atmospheric pressure until about 100 ml remained, cooled to -30°C to -20°C, and NaHMDS (0.125 mol, 63 ml, 2M THF solution) was slowly added dropwise. The temperature of the reaction system was controlled at about -25°C and stirred for 20 minutes. HPLC detected that the raw materials were basically reacted. Water (30 ml) was slowly added under temperature control to quench, and glacial acetic acid (about 10 ml) was added to neutralize. The temperature was raised to about 0°C and stirred, and 20% Na 2 CO 3 (10ml), concentrated under reduced pressure until there is no fraction, added ethyl acetate (120ml) to the residue, heated to about 45°C, stirred and separated, the organic phase was separated, added with saturated brine (30ml), washed once, concentrated under reduced pressure to leave about (30ml), then added ethyl acetate (30ml), concentrated under reduced pressure again, repeated twice, a large amount of solid precipitated, added ethyl acetate to a material volume of about 50ml, stirred at 0-10°C for 1 hour, filtered, and dried in vacuo at 45°C to obtain 16.87g of white amorphous solid product (AZD4547) with a purity of 99.8% and a yield of 91%. |
Heterocycles (2020), 100(2), 276-282 ,
CN111072638
PATENT
CN115819239
Nature Catalysis (2021), 4(5), 385-394
Shandong Huagong (2021), 50(7), 19-21
CN111072638
Heterocycles (2020), 100(2), 276-282
Physical Chemistry Chemical Physics (2020), 22(17), 9656-9663
Journal of Chemical Theory and Computation (2019), 15(2), 1265-1277
Journal of Medicinal Chemistry (2017), 60(14), 6018-6035
Bioorganic & Medicinal Chemistry Letters (2016), 26(20), 5082-5086
WO2016089208
WO2008075068
References
- ^ Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al. (April 2012). “AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family”. Cancer Research. 72 (8): 2045–2056. doi:10.1158/0008-5472.CAN-11-3034. PMID 22369928.
- ^ Katoh M, Nakagama H (March 2014). “FGF receptors: cancer biology and therapeutics”. Medicinal Research Reviews. 34 (2): 280–300. doi:10.1002/med.21288. PMID 23696246.
- ^ Katoh M (July 2016). “FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review)”. International Journal of Molecular Medicine. 38 (1): 3–15. doi:10.3892/ijmm.2016.2620. PMC 4899036. PMID 27245147.
- ^ Zengin ZB, Chehrazi-Raffle A, Salgia NJ, Muddasani R, Ali S, Meza L, et al. (February 2022). “Targeted therapies: Expanding the role of FGFR3 inhibition in urothelial carcinoma”. Urologic Oncology. 40 (2): 25–36. doi:10.1016/j.urolonc.2021.10.003. PMID 34840077.
- ^ Zarei P, Ghasemi F (2024). “The Application of Artificial Intelligence and Drug Repositioning for the Identification of Fibroblast Growth Factor Receptor Inhibitors: A Review”. Advanced Biomedical Research. 13: 9. doi:10.4103/abr.abr_170_23. PMC 10958741. PMID 38525398.
| Identifiers | |
|---|---|
| showIUPAC name | |
| CAS Number | 1035270-39-3 |
| PubChem CID | 51039095 |
| IUPHAR/BPS | 7707 |
| DrugBank | DB12247 |
| ChemSpider | 26333104 |
| UNII | 2167OG1EKJ |
| ChEBI | CHEBI:63453 |
| ChEMBL | ChEMBL3348846 |
| PDB ligand | 66T (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID80145887 |
| ECHA InfoCard | 100.206.232 |
| Chemical and physical data | |
| Formula | C26H33N5O3 |
| Molar mass | 463.582 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////Fexagratinib, AZD 4547, ADSK091
ETRIPAMIL



ETRIPAMIL
CAS 1593673-23-4
AS ACETATE 512.64 CAS 2891832-59-8
HCL SALT 2560549-35-9
WeightAverage: 452.595
Monoisotopic: 452.267507647
Chemical FormulaC27H36N2O4
12/12/2025, FDA 2025, APPROVALS 2025
Benzoic acid, 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]methylamino]ethyl]-, methyl ester
methyl 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]-methylamino]ethyl]benzoate
- Methyl 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]methylamino]ethyl]benzoate
- (-)-MSP 2017
- MSP 2017
- OriginatorMilestone Pharmaceuticals
- DeveloperCorxel Pharmaceuticals; Milestone Pharmaceuticals
- ClassAmines; Antiarrhythmics; Benzoates; Esters; Ischaemic heart disorder therapies; Small molecules
- Mechanism of ActionCalcium channel antagonists
- PreregistrationParoxysmal supraventricular tachycardia
- Phase IIAtrial fibrillation
- Phase IUnspecified
- No development reportedAngina pectoris
- 14 May 2025Milestone Pharmaceuticals has patent protection for etripamil in the USA
- 28 Mar 2025Milestone pharmaceuticals plans to request a Type A meeting with USFDA to discuss the issues raised in the complete response letter
- 28 Mar 2025USFDA has issued a Complete Response Letter (CRL) regarding New Drug Application (NDA) for Etripamil for Paroxysmal supraventricular tachycardia
Etripamil has been used in trials studying the treatment of Paroxysmal Supraventricular Tachycardia (PSVT).
Etripamil (MSP-2017) is a short-acting, L-type calcium-channel antagonist. Etripamil inhibits calcium influx through slow calcium channels, thereby slowing AV node conduction and prolonging the AV node refractory period. Etripamil increases heart rate and decreases systolic blood pressure. Etripamil can be used in the study of paroxysmal supraventricular tachycardia (PSVT).
To treat episodes of paroxysmal supraventricular tachycardia
SCHEME
SIDE CHAIN

MAIN

SYN
US20180110752/ U.S. Patent No. 10,117,848,
EXAMPLES
Example 1: Synthesis methyl 3-(2-((4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl)(methyl)amino)ethyl)benzoate
Part I: Synthesis of 5-Bromo-2-(3,4-dimethoxyphenyl)-2-isopropylpentanenitrile
Part II: Synthesis of methyl 3-(2-(methylamino)ethyl)benzoate
Part III: Reaction of Compound II with Compound III Produced Compound I
| Analysis of the product by mass spectrometry revealed a peak with a mass-to-charge ratio (m/z) of 453, corresponding to the M+H molecular ion of compound I. |
Example 2: Concentrated Solution of Acetate Salt of Compound I
| A concentrated aqueous solution of the acetate salt of compound I is formed according to the following protocol: |
| This protocol readily can be adapted to provide a concentrated solution of the methanesulfonate salt of compound I. |
PRED BY CHIRAL SEPERATION
US20230065401
WO2016165014
EP4119137 chiral sepn done
[0034] In one embodiment the present invention is a kit for treating a cardiac arrhythmia (e.g., PSVT or atrial fibrillation), angina, or a migraine in a subject in need thereof wherein the kit comprises a nasal delivery system comprising two doses of a therapeutically effective amount of compound I having a structure according to the formula:
and instructions for nasally administering to the subject (i) a first dose, and, optionally, (ii) a second dose of an aqueous composition comprising a pharmaceutically acceptable acetate or methanesulfonate salt of compound I, or a racemate or enantiomer thereof, wherein the acetate or methanesulfonate salt of compound I, or the racemate or enantiomer thereof, is dissolved in the aqueous composition at a concentration of 350 mg/mL± 50 mg/mL, and wherein the second dose of the compound is to be administered between 5 minutes and 60 minutes after the first dose.
Cross ref U.S. Patent No. 10,117,848,
[0336]
- 1. A method of treating a cardiac arrhythmia in a subject in need thereof with a therapeutically effective amount of compound I having a structure according to the formula:
the method comprising nasally administering to the subject (i) a first dose, and (ii) a second dose of an aqueous composition comprising a pharmaceutically acceptable acetate or methanesulfonate salt of compound I, or a racemate or enantiomer thereof, wherein the acetate or methanesulfonate salt of compound I, or the racemate or enantiomer thereof, is dissolved in the aqueous composition at a concentration of 350 mg/mL ± 50 mg/mL, and wherein the second dose of the compound is administered between 5 minutes and 25 minutes after the first dose.
PATENT
Journal of the American College of Cardiology (2018), 72(5), 489-497
American Heart Journal (2022), 253, 20-29
Expert Opinion on Investigational Drugs (2020), 29(1), 1-4
EP4119137 WO2016165014
EP-2170050-B1
US-9737503-B2
US-4968717-A
EP-0231003-A2
- [1]. Stambler BS, et al. Etripamil Nasal Spray for Rapid Conversion of Supraventricular Tachycardia to Sinus Rhythm. J Am Coll Cardiol. 2018 Jul 31;72(5):489-497. [Content Brief][2]. Milestone Pharmaceuticals Announces USAN Approval of Generic Name “Etripamil” for its Phase 2 Clinical Development Product for the Treatment of Paroxysmal Supraventricular Tachycardia.[3]. Ascah A, et al. Cardiovascular and Pharmacokinetic Profiles of Intravenous Etripamil in Conscious Telemetered Cynomolgus Monkeys. Int J Toxicol. 2025 Apr 1:10915818251327963. [Content Brief][4]. Pion J, et al. Preclinical Safety Evaluation of Etripamil Nasal Spray in Cynomolgus Macaques (Macaca fascicularis) to Assess for Safety in Patients With Paroxysmal Supraventricular Tachycardia. Int J Toxicol. 2024 Sep-Oct;43(5):503-510. [Content Brief]
//////////ETRIPAMIL, (-)-MSP 2017, MSP 2017, FDA 2025, APPROVALS 2025
ELUBIOL


ELUBIOL
Dichlorophenyl imidazoldioxolan
CAS 67914-69-6
- Elubiol
- 67914-69-6
- OristaR DCI
- Dichlorophenyl imidazoldioxolan
- (+/-)-Dichlorophenyl imidazoldioxolan
AMY 925
C27H30Cl2N4O5, 561.5 g/mol
ethyl 4-[4-[[(2R,4S)-2-(2,4-dichlorophenyl)-2-(imidazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazine-1-carboxylate
Elubiol (Dichlorophenyl imidazoldioxolan) has moderate sebum-inhibiting activity and can be used in the treatment of oily skin or dandruff.
SCHEME

PATENT
DE2804096
https://patentscope.wipo.int/search/en/detail.jsf?docId=DE102084041&_cid=P20-MB323Q-91006-1
PATENT
US4358449
https://patentscope.wipo.int/search/en/detail.jsf?docId=US37288536&_cid=P20-MB3265-92366-1
PATENT
CN102070620
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84648943&_cid=P20-MB329Q-94515-1
| Example 1 |
| According to the method of the present invention, bacteriostatic ester (±) cis-4-[4-[[2-(2,4-dichlorophenyl)-2-(1-H-imidazolemethyl)-1,3-dioxolane-4-yl]methoxy]phenyl]-1-piperazinecarboxylic acid ethyl ester is prepared, comprising the following steps: |
| 1. Condensation reaction |
| In a dry 500ml three-necked flask, add 473g of dimethyl sulfoxide, 130g of active lipid, 50g of N-(4-hydroxyphenyl)piperazine, and 21g of potassium hydroxide. Control the temperature at 30℃ and keep the reaction for 24 hours. After the reaction, add 520g of purified water. After the addition is completed, cool to 5℃, stir and keep warm for 2h, and filter to obtain the antibacterial ester condensate. The condensation yield is about 85%. |
| 2. Esterification reaction |
| In a three-necked flask, 322g of dichloromethane, 50g of antibacterial ester condensate, and 52g of potassium carbonate were added, and then 11.9g of ethyl chloroformate was slowly added. After the addition was completed, the temperature was controlled at 25°C and the reaction was kept warm for 4 hours. After the reaction was completed, 108g of purified water was slowly added. After the addition was completed, stirring was continued for 2h. The organic layer was washed three times with purified water until the pH reached 7. After washing, dichloromethane was evaporated under reduced pressure. After evaporation, 60ml of methyl isobutyl ketone was added and the temperature was kept at 0-5°C for 2-4h. The antibacterial ester was obtained by suction filtration, and the esterification yield was about 80%. |
| Heat the antibacterial ester and dissolve it in 8 times the amount of acetone, add 0.5 times the amount of activated carbon, reflux and keep warm for 0.5 hours, cool down to no reflux, filter and remove the activated carbon, concentrate the filtrate to 5 times the weight of the antibacterial ester, add water and cool down to 0-5°C after concentration, keep warm for 1-3 hours under stirring, and filter to obtain an off-white crystalline powder. After analysis, the antibacterial ester content is greater than 97%. |
PATENT
CN101665490
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN83857361&_cid=P20-MB32BE-95479-1


| Synthesis of 4-(4-hydroxyphenyl)piperazine: |
| Example 1 |
| In a 1000ml reaction bottle, under nitrogen protection, add 500g of water, 178.5g of dichloroethylamine hydrochloride and 109g of p-hydroxyaniline, heat to 100°C, add 160g of 50% sodium hydroxide solution (80g of sodium hydroxide dissolved in 80g of water), reflux for 10 hours. Then cool to 35°C, add 400g of methanol, adjust the pH value to 8 with ammonia water, filter, and dry the filter cake in vacuum at 40°C to obtain 128g of 4-(4-hydroxyphenyl)piperazine (HPLC content greater than 98%), with a yield of 71.9%. |
| Example 2 |
| In a 1000ml reaction bottle, under nitrogen protection, add 500g of water, 178.5g of dichloroethylamine hydrochloride and 218g of p-hydroxyaniline, heat to 70°C, add 112g of 50% potassium hydroxide solution (56g of potassium hydroxide dissolved in 56g of water), and react for 5 hours. Then cool to 35°C, add 400g of methanol, adjust the pH value to 8 with ammonia water, filter, and dry the filter cake in vacuum at 40°C to obtain 112g of 4-(4-hydroxyphenyl)piperazine (HPLC content greater than 98%), with a yield of 62.9%. |
| Example 3 |
| In a 1000ml reaction bottle, under nitrogen protection, add 500g of water, 312g of dichloroethylamine hydrobromide and 150g of p-hydroxyaniline, stir at room temperature (25°C), add 200g of 50% potassium bicarbonate solution (100g of potassium bicarbonate dissolved in 100g of water), react for 1 hour, then cool to 35°C, add 400g of methanol, adjust the pH value to 8 with ammonia water, filter, and dry the filter cake in vacuum at 40°C to obtain 87g of 4-(4-hydroxyphenyl)piperazine (HPLC content greater than 98%), with a yield of 48.8%. |
| Example 4 |
| In a 1000ml reaction bottle, under nitrogen protection, add 500g of water, 452g of dichloroethylamine hydroiodide and 327g of p-hydroxyaniline, heat to 100°C, add 480g of 50% sodium hydroxide solution (240g of sodium hydroxide dissolved in 240g of water), reflux for 10 hours. Then cool to 35°C, add 600g of methanol, adjust the pH value to 8 with ammonia water, filter, and dry the filter cake in vacuum at 40°C to obtain 154g of 4-(4-hydroxyphenyl)piperazine (HPLC content greater than 98%), with a yield of 86.5%. |
| Synthesis of Ethyl [4-(4-Hydroxyphenyl)]-1-piperazinecarboxylate |
| Example 5 |
| In a 2000 ml reaction bottle, add 178 g of 4-(4-hydroxyphenyl)piperazine, 150 g of sodium bicarbonate and 500 g of acetone, cool to -20°C with ice brine, add 110 g of ethyl chloroformate dropwise, and keep the temperature in the bottle not higher than zero degrees. After the addition is complete, heat to room temperature and react for 5 hours; |
| Add 700g of water, stir for 1 hour and filter. Add the filter cake obtained by filtration to a 1000ml reaction bottle, add 300g of 75% ethanol solution by volume, heat to dissolve, cool to zero degrees with ice brine, filter, and dry the filter cake in vacuum at 40°C to obtain 146g of [4-(4-hydroxyphenyl)]-1-piperazinecarboxylic acid ethyl ester (HPLC content greater than 99%), 58.4%. |
| Example 6 |
| In a 2000ml reaction bottle, add 178g of 4-(4-hydroxyphenyl)piperazine, 180g of sodium carbonate and 500g of acetone, cool to -10°C with ice brine, add 165g of ethyl chloroformate dropwise, and keep the temperature in the bottle not higher than zero degrees. After the addition is complete, heat to 50 degrees and react for 1 hour; |
| Add 700g of water, stir for 1 hour and filter. Add the filter cake obtained by filtration to a 1000ml reaction bottle, add 300g of 75% ethanol solution by volume, heat to dissolve, cool to zero degrees with ice brine, filter, and dry the filter cake in vacuum at 40°C to obtain 156g of [4-(4-hydroxyphenyl)]-1-piperazinecarboxylic acid ethyl ester (HPLC content greater than 99%), 62.4%. |
| Example 7 |
| In a 2000ml reaction bottle, add 178g of 4-(4-hydroxyphenyl)piperazine, 400g of potassium bicarbonate and 1000g of acetone, cool to 0°C with ice brine, add 440g of ethyl chloroformate dropwise, and keep the temperature in the bottle not higher than zero degrees. After the addition is completed, react at about 0°C for 10 hours; |
| Add 1000g of water, stir for 1 hour and filter. Add the filter cake obtained by filtration to a 1000ml reaction bottle, add 500g of 75% ethanol solution by volume, heat to dissolve, cool to zero degrees with ice brine, filter, and dry the filter cake in vacuum at 40°C to obtain 216g of [4-(4-hydroxyphenyl)]-1-piperazinecarboxylic acid ethyl ester (HPLC content greater than 99%), 86.4%. |
| Example 8 |
| In a 2000ml reaction bottle, add 178g of 4-(4-hydroxyphenyl)piperazine, 140g of triethylamine, and 500g of acetone; cool to -10°C with ice brine, add 110g of ethyl chloroformate dropwise, and keep the temperature in the bottle not higher than zero degrees. After the addition is complete, react at -10°C for 10 hours; |
| Add 700g of water, stir for 1 hour and filter. Add the filter cake obtained by filtration to a 1000ml reaction bottle, add 300g of 75% ethanol solution by volume, heat to dissolve, cool to zero degrees with ice brine, filter, and dry the filter cake in vacuum at 40°C to obtain 126g of [4-(4-hydroxyphenyl)]-1-piperazinecarboxylic acid ethyl ester (HPLC content greater than 99%), 50.4%. |
| Synthesis of Ketoconazole Derivatives: |
| Example 9 |
| In a 1000ml reaction bottle, add 45g of cis-[2-(2,4-dichlorophenyl)-2(1H-imidazol-1-yl-methyl)-1,3-dioxopentyl]-4-methyl-p-toluenesulfonate, 25g of ethyl [4-(4-hydroxyphenyl)]-1-piperazinecarboxylate, 5.6g of potassium hydroxide and 180g of dimethyl sulfoxide; react at 25°C for 20 hours. After the reaction, add 450g of ice water to the reaction bottle to reduce the temperature in the reaction bottle to 10°C, and filter; wash the filter cake with water until it is neutral and dry; obtain 42g of crude ketoconazole derivative (HPLC content is 94%). |
| In a 1000ml reaction bottle, add 42g of crude ketoconazole derivative and 350g of ethyl acetate, heat to dissolve, add 0.5g of activated carbon, reflux for half an hour, filter, wash the filter cake with hot ethyl acetate, combine the ethyl acetate, and concentrate to 230g; cool naturally to room temperature, then continue to cool to 0°C with ice water, and keep warm for 1 hour, filter, and vacuum dry to obtain 39g of white powder (HPLC content greater than 99%), with a yield of 73.6%. |
| Example 10 |
| In a 1000 ml reaction bottle, add 45 g of cis-[2-(2,4-dichlorophenyl)-2(1H-imidazol-1-yl-methyl)-1,3-dioxolane]-4-methyl-p-toluenesulfonate, 50 g of ethyl [4-(4-hydroxyphenyl)]-1-piperazinecarboxylate, 11.2 g of sodium hydroxide and 200 g of dioxane; react at 50° C. for 10 hours. After the reaction, add 450 g of ice water to the reaction bottle to reduce the temperature in the reaction bottle to 10° C. and filter; wash the filter cake with water until it is neutral and dry; obtain 41 g of crude ketoconazole derivative (HPLC content is 94%). |
| In a 1000ml reaction bottle, add 41g of crude ketoconazole derivative and 340g of ethyl acetate, heat to dissolve, add 0.5g of activated carbon, reflux for half an hour; filter, wash the filter cake with hot ethyl acetate, combine ethyl acetate, and concentrate to 230g; cool naturally to room temperature, then continue to cool to 0°C with ice water, and keep warm for 1 hour, filter, and vacuum dry to obtain 37g of white powder (HPLC content greater than 99%), with a yield of 69.8%. |
| Embodiment 11 |
| In a 1000ml reaction bottle, add 45g of cis-[2-(2,4-dichlorophenyl)-2(1H-imidazol-1-yl-methyl)-1,3-dioxopentyl]-4-methyl-p-toluenesulfonate, 100g of ethyl [4-(4-hydroxyphenyl)]-1-piperazinecarboxylate, 22.4g of sodium methoxide and 300g of tetrahydrofuran; react at 0°C for 50 hours. After the reaction, add 500g of ice water to the reaction bottle to reduce the temperature in the reaction bottle to 10°C, filter; wash the filter cake with water until neutral and dry; obtain 49g of crude ketoconazole derivative (HPLC content is 94%). |
| In a 1000ml reaction bottle, add 49g of crude ketoconazole derivative and 350g of ethyl acetate, heat to dissolve, add 0.5g of activated carbon, reflux for half an hour; filter, wash the filter cake with hot ethyl acetate, combine ethyl acetate, and concentrate to 250g; cool naturally to room temperature, then continue to cool to 0°C with ice water, and keep warm for 1 hour, filter, and vacuum dry to obtain 43.9g of white powder (HPLC content greater than 99%), with a yield of 82.8%. |
| Example 12 |
| In a 1000ml reaction bottle, add 45g of cis-[2-(2,4-dichlorophenyl)-2(1H-imidazol-1-yl-methyl)-1,3-dioxopentyl]-4-methyl-p-toluenesulfonate, 42g of ethyl [4-(4-hydroxyphenyl)]-1-piperazinecarboxylate, 15g of sodium ethoxide and 300g of N,N-dimethylformamide; react at 10°C for 30 hours. After the reaction, add 500g of ice water to the reaction bottle to reduce the temperature in the reaction bottle to 10°C, and filter; wash the filter cake with water until it is neutral and dry; obtain 51.2g of crude ketoconazole derivative (HPLC content is 94%). |
| In a 1000ml reaction bottle, add 51.2g of crude ketoconazole derivative and 400g of ethyl acetate, heat to dissolve, add 0.5g of activated carbon, reflux for half an hour; filter, wash the filter cake with hot ethyl acetate, combine ethyl acetate, and concentrate to 250g; cool naturally to room temperature, then continue to cool to 0°C with ice water, and keep warm for 1 hour, filter, and vacuum dry to obtain 44.8g of white powder (HPLC content greater than 99%), with a yield of 84.5%. |
REF
[1]. Pierard GE, et al. Modulation of sebum excretion from the follicular reservoir by a dichlorophenyl-imidazoldioxolan. Int J Cosmet Sci. 1996 Oct;18(5):219-27. [Content Brief]
////////////ELUBIOL, AMY 925, Dichlorophenyl imidazoldioxolan, OristaR DCI
Elfucose



Elfucose
Cas 87-96-7
Chemical Formula: C6H12O5
Exact Mass: 164.07
Molecular Weight: 164.157
L-fucopyranose (6-deoxy-L-galactopyranose)
(3S,4R,5S,6S)-6-methyloxane-2,3,4,5-tetrol
- 6-Deoxy-L-galactose (ACI)
- Fucose, L- (8CI)
- (-)-Fucose
- 46: PN: US20220380460 SEQID: 47 claimed sequence
- 6-Desoxygalactose
- L-(-)-Fucose
- L-Fucose
- L-Galactomethylose
- L-Galactopyranose, 6-deoxy-
- CERC 803
- Elfucose
- Fucose
- NSC 1219
- congenital glycosylation disorders
- 6-Deoxy-L-galactopyranose
- L-galactomethylose
- 87-96-7
- Fucose, L-
- 6-deoxy-galactose
Fucose is under investigation in clinical trial NCT03354533 (Study of ORL-1F (L-fucose) in Patients With Leukocyte Adhesion Deficiency Type II).
L-fucopyranose is the pyranose form of L-fucose. It has a role as an Escherichia coli metabolite and a mouse metabolite. It is a L-fucose and a fucopyranose.
SCHEME

PATENT
WO2016150629
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016150629&_cid=P10-MB1MYE-34318-1
Examples
The invention will now be illustrated in more detail by the following non-limiting examples.
Example 1: Production of L-fucose by biocatalytic oxidation of L-fucitol with galactose oxidase in the presence of peroxidase and catalase
A solution of L-fucitol (6.0 mL aqueous solution containing 600 mg L-fucitol, CAS 13074-06-1, Santa Cruz Biotechnology) was added to a round-bottom three-neck bottle (50 mL), followed by the addition of 1.2 mL K2HPO4 / KH2PO4 ( 1000 mM , pH=7.0) and 0.095 mL catalase (from bovine liver, SIGMA, 21,300 U/mg, 34 mg/mL), 0.120 mL peroxidase (from horseradish, 173 U/mg solid, SIGMA ) and 2.218 mL galactose oxidase (38.4 mg/mL, 2,708 U/mL). The resulting solution was purged with O 2 at room temperature until all L-fucitol was converted to L-fucose. The reaction was monitored by HLPC. The final product was isolated and analyzed by 1 H and 13C NMR. The results are summarized in Table 1.
[Table 1]
Reaction time [h] Conversion [%]
0
3,5 54,0
24 95,8
29 96,0
PATENT
WO2010022244
WO2007021879
////////////Elfucose, 6-Deoxy-L-galactose, Fucose, L- , (-)-Fucose, 6-Desoxygalactose, L-(-)-Fucose, L-Fucose, L-Galactomethylose, L-Galactopyranose, 6-deoxy-, CERC 803, Elfucose, Fucose, NSC 1219, congenital glycosylation disorders, 6-Deoxy-L-galactopyranose, L-galactomethylose, 87-96-7, Fucose, L-, 6-deoxy-galactose
Deupsilocin



Deupsilocin, Psilocin-d10
Psilocin-D10- Deupsilocin
- Psilocine-d10
| Molecular Formula | C12H16N2O |
| Molecular Weight | 214.3299 |
CAS 1435934-64-7
3-[2-[Di(methyl-d3)amino]ethyl-1,1,2,2–d4]-1H-indol-4-ol
3-[2-[bis(trideuteriomethyl)amino]-1,1,2,2-tetradeuterioethyl]-1H-indol-4-ol
| 1H-Indol-4-ol, 3-[2-[di(methyl-d3)amino]ethyl-1,1,2,2-d4]- |
Many mental health disorders, as well as neurological disorders, are impacted by alterations, dysfunction, degeneration, and/or damage to the brain’s serotonergic system, which may explain, in part, common endophenotypes and comorbidities among neuropsychiatric and neurological diseases. Many therapeutic agents that modulate serotonergic function are commercially available, including serotonin reuptake inhibitors, selective serotonin reuptake inhibitors, antidepressants, monoamine oxidase inhibitors, and, while primarily developed for depressive disorders, many of these therapeutics are used across multiple medical indications including, but not limited to, depression in Alzheimer’s disease and other neurodegenerative disease, chronic pain, existential pain, bipolar disorder, obsessive compulsive disorder, anxiety disorders and smoking cessation. However, in many cases, the marketed drugs show limited benefit compared to placebo, can take six weeks to work and for some patients, and are associated with several side effects including trouble sleeping, drowsiness, fatigue, weakness, changes in blood pressure, memory problems, digestive problems, weight gain and sexual problems.
The field of psychedelic neuroscience has witnessed a recent renaissance following decades of restricted research due to their legal status. Psychedelics are one of the oldest classes of psychopharmacological agents known to man and cannot be fully understood without reference to various fields of research, including anthropology, ethnopharmacology, psychiatry, psychology, sociology, and others. Psychedelics (serotonergic hallucinogens) are powerful psychoactive substances that alter perception and mood and affect numerous cognitive processes. They are generally considered physiologically safe and do not lead to dependence or addiction. Their origin predates written history, and they were employed by early cultures in many sociocultural and ritual contexts. After the virtually contemporaneous discovery of (5R,8R)-(+)-lysergic acid-N,N-diethylamide (LSD) and the identification of serotonin in the brain, early research focused intensively on the possibility that LSD and other psychedelics had a serotonergic basis for their action. Today there is a consensus that psychedelics are agonists or partial agonists at brain serotonin 5-hydroxytryptamine 2 A (5-HT2A) receptors, with particular importance on those expressed on apical dendrites of neocortical pyramidal cells in layer V, but also may bind with lower affinity to other receptors such as the sigma-1 receptor. Several useful rodent models have been developed over the years to help unravel the neurochemical correlates of serotonin 5-HT2A receptor activation in the brain, and a variety of imaging techniques have been employed to identify key brain areas that are directly affected by psychedelics.
Psychedelics have both rapid onset and persisting effects long after their acute effects, which includes changes in mood and brain function. Long lasting effects may result from their unique receptor affinities, which affect neurotransmission via neuromodulatory systems that serve to modulate brain activity, i.e., neuroplasticity, and promote cell survival, are neuroprotective, and modulate brain neuroimmune systems. The mechanisms which lead to these long-term neuromodulatory changes are linked to epigenetic modifications, gene expression changes and modulation of pre- and post-synaptic receptor densities. These, previously under-researched, psychedelic drugs may potentially provide the next-generation of neurotherapeutics, where treatment resistant psychiatric and neurological diseases, e.g., depression, post-traumatic stress disorder, dementia and addiction, may become treatable with attenuated pharmacological risk profiles.
Although there is a general perception that psychedelic drugs are dangerous, from a physiologic safety standpoint, they are one of the safest known classes of CNS drugs. They do not cause addiction, and no overdose deaths have occurred after ingestion of typical doses of classical psychotic agents, such as LSD, psilocybin, or mescaline (Scheme 1). Preliminary data show that psychedelic administration in humans results in a unique profile of effects and potential adverse reactions that need to be appropriately addressed to maximize safety. The primary safety concerns are largely psychologic, rather than physiologic, in nature. Somatic effects vary but are relatively insignificant, even at doses that elicit powerful psychologic effects. Psilocybin, when administered in a controlled setting, has frequently been reported to cause transient, delayed headache, with incidence, duration, and severity increased in a dose-related manner [Johnson et al., Drug Alcohol Depend, 2012, 123 (1-3):132-140]. It has been found that repeated administration of psychedelics leads to a very rapid development of tolerance known as tachyphylaxis, a phenomenon believed to be mediated, in part, by 5-HT2A receptors. In fact, several studies have shown that rapid tolerance to psychedelics correlates with downregulation of 5-HT2A receptors. For example, daily LSD administration selectively decreased 5-HT2 receptor density in the rat brain [Buckholtz et al., Eur. J. Pharmacol., 1990, 109:421-425. 1985; Buckholtz et al., Life Sci. 1985, 42:2439-2445].
SCHEME

PATENT
Mindset Pharma Inc., US11591353
https://patentscope.wipo.int/search/en/detail.jsf?docId=US376433397&_cid=P10-MARMO8-36145-1
PATENT
WO2021155470
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021155470&_cid=P10-MARMST-39096-1
PATENT
Cybin IRL Limited, WO2023247665
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023247665&_cid=P10-MARMVV-41020-1
PATENT
WO2023078604
WO2022195011
| Classic psychedelics and dissociative psychedelics are known to have rapid onset antidepressant and anti-addictive effects, unlike any currently available treatment. Randomized clinical control studies have confirmed antidepressant and anxiolytic effects of classic psychedelics in humans. Ketamine also has well established antidepressant and anti-addictive effects in humans mainly through its action as an NMDA antagonist. Ibogaine has demonstrated potent anti-addictive potential in pre-clinical studies and is in the early stages of clinical trials to determine efficacy in robust human studies [Barsuglia et al., Prog Brain Res, 2018, 242:121-158; Corkery, Prog Brain Res, 2018, 242:217-257]. |
/////////Deupsilocin, Psilocin-d10, KXD3HS8D6X, Psilocin-D10, Deupsilocin, Psilocine-d10

{kind=link}