
Home » Uncategorized (Page 6)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Elsovaptan
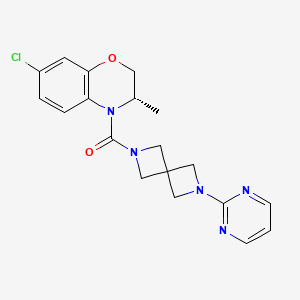
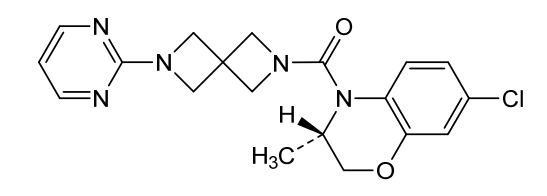
Elsovaptan
CAS 2296801-25-5
MFC19H20ClN5O2 MW385.8 g/mol
[(3S)-7-chloro-3-methyl-2,3-dihydro-1,4-benzoxazin-4-yl]-(6-pyrimidin-2-yl-2,6-diazaspiro[3.3]heptan-2-yl)methanone
[(3S)-7-chloro-3-methyl-2,3-dihydro-4H-1,4-benzoxazin-4-yl][6-(pyrimidin-2-yl)-2,6-diazaspiro[3.3]heptan-2-yl]methanone
vasopressin receptor antagonist, Alzheimer disease, T206306, Y2Z74WGU3J
Elsovaptan is a selective vasopressin receptor antagonist. It is primarily used in scientific research to study the modulation of vasopressin action, particularly in the context of neurodegenerative conditions like Alzheimer’s disease
- Primary Mechanism: Blocks the binding of vasopressin to its receptors, which typically disrupts fluid volume regulation and can lead to vasodilation.
Research Applications
Currently, Elsovaptan is designated as a research-use-only compound and is not approved for human therapeutic or veterinary use.
Its study is often linked to research on:
- Neurodegenerative Diseases: Investigating the role of the vasopressin system in the progression of Alzheimer’s disease.
- Fluid Regulation: Understanding how antagonizing vasopressin affects vascular resistance and blood pressure.
The compound has been listed in recent International Nonproprietary Names (INN) proposals by the World Health Organization (WHO) for pharmaceutical substances, indicating it is an active subject of pharmacological study
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

{kind=link}
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
/////////elsovaptan, ANAX LABS, vasopressin receptor antagonist, Alzheimer disease, T206306, Y2Z74WGU3J
Elisrasib
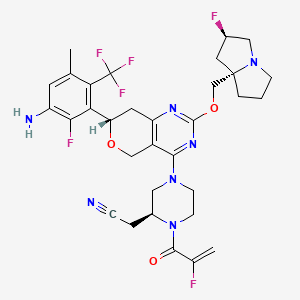
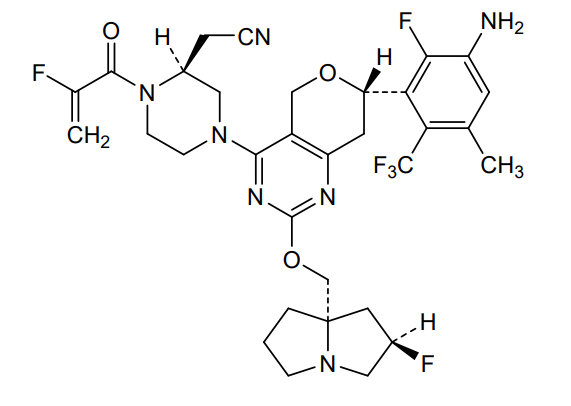
Elisrasib
CAS2914919-85-8
MFC32H35F6N7O3. MW 679.7 g/mol
2-[(2S)-4-[(7S)-7-[3-amino-2-fluoro-5-methyl-6-(trifluoromethyl)phenyl]-2-[[(2R,8S)-2-fluoro-1,2,3,5,6,7-hexahydropyrrolizin-8-yl]methoxy]-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
[(2S)-4-[(7S)-7-[3-amino-2-fluoro-5-methyl-6-(trifluoromethyl)phenyl]-2-{[(2R,7aS)-2-fluorotetrahydro-1Hpyrrolizin-7a(5H)-yl]methoxy}-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, D3S 001, PFW9YLB86H
Elisrasib (D3S-001) is a next-generation, orally available KRAS G12C inhibitor developed by D3 Bio that demonstrates high potency, sustained target engagement, and strong clinical activity in advanced solid tumors, including those resistant to first-generation inhibitors. As of April 2026, clinical trials show it has a 52% objective response rate (ORR) in G12C inhibitor-naive patients and a 30% ORR in refractory populations.
Key Aspects of Elisrasib (D3S-001):
- Mechanism of Action: It is a highly potent, covalent inhibitor that selectively binds the GDP-bound (inactive) form of the KRAS G12C mutant, effectively halting tumor cell proliferation and metastasis.
- Superior Efficacy: Preliminary data suggests elisrasib may be more potent than earlier inhibitors like sotorasib and adagrasib, providing higher target occupancy at lower doses.
- Clinical Performance (AACR 2026 Data):
- Naive Patients: 52% ORR, with a median duration of response (mDOR) of 16.5 months and median progression-free survival (mPFS) of 12.2 months at the 600 mg dose.
- Refractory Patients: 32% ORR, with a mDOR of 15.6 months and mPFS of 8.1 months.
- Targeted Cancers: Clinical trials are focused on KRAS G12C-mutant tumors, specifically non-small cell lung cancer (NSCLC), colorectal cancer (CRC), and other solid tumors.
- Safety Profile: The drug has shown good tolerability and a safe profile in early studies.
Elisrasib is in Phase 1/2 development and was highlighted for its promising results in treating patients with KRAS G12C-mutant tumors
Elisrasib is an orally bioavailable inhibitor of the oncogenic KRAS substitution mutation G12C, with potential antineoplastic activity. Upon oral administration, elisrasib selectively targets the KRAS G12C mutant and inhibits KRAS G12C-mediated signaling. This may halt proliferation and metastasis in susceptible tumor cells. KRAS, a member of the RAS family of oncogenes, serves an important role in cell signaling, division and differentiation. Mutations of KRAS may induce constitutive signal transduction leading to tumor cell proliferation, invasion, and metastasis.
- A Phase 1 Study to Assess Food Effect on the Pharmacokinetics of D3S-001 in Healthy Adult ParticipantsCTID: NCT07093398Phase: Phase 1Status: CompletedDate: 2026-03-25
- A Phase 1/2 Study of D3S-002 as Monotherapy or Combination Therapy in Adult Subjects With Advanced Solid Tumors With MAPK Pathway MutationsCTID: NCT05886920Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2026-03-23
- A Study of D3S-001 Monotherapy or Combination Therapy in Subjects With Advanced Solid Tumors With a KRAS p.G12C MutationCTID: NCT05410145Phase: Phase 1/Phase 2Status: RecruitingDate: 2026-03-12
PAT
SYN
Example 17
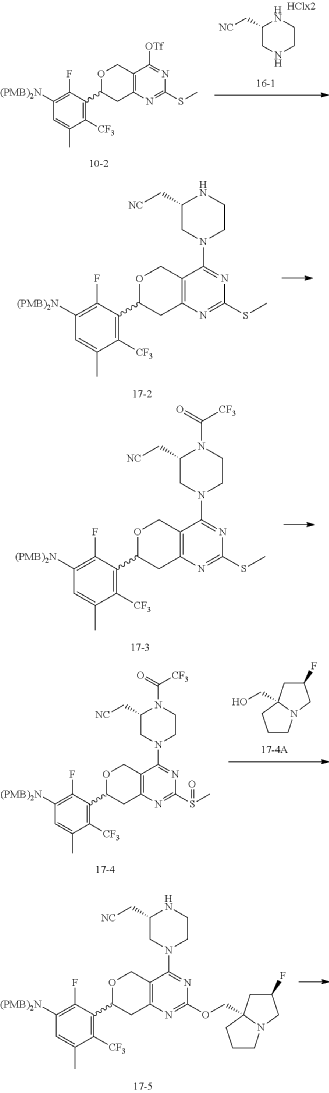
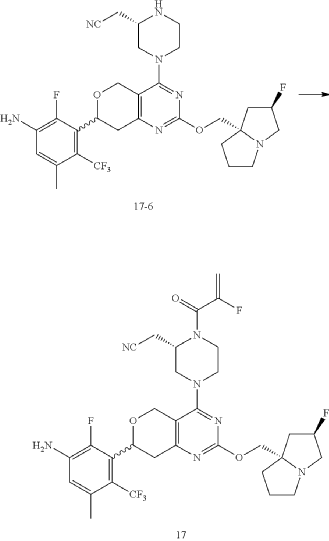
Step 6: Synthesis of Compound 17
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Pyrimidoheterocyclic compounds and application thereofPublication Number: EP-4105211-A1Priority Date: 2020-03-12
- Pyrimidoheterocyclic compounds and application thereofPublication Number: US-2023151004-A1Priority Date: 2020-03-12
//////////elisrasib, anax labs, Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, D3S 001, PFW9YLB86H
Elironrasib
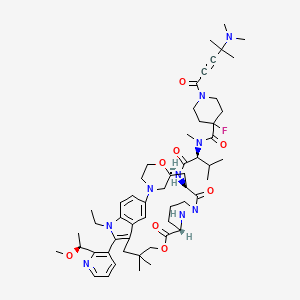
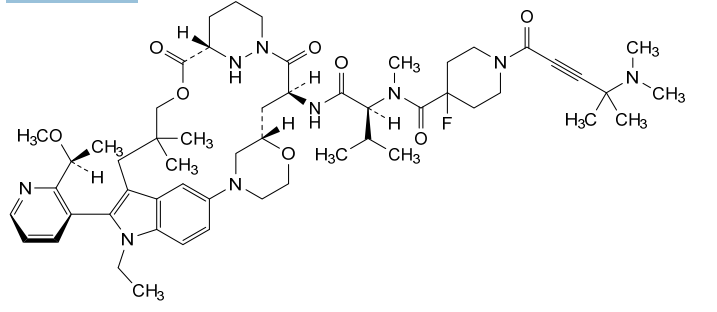
Elironrasib
CAS 2641998-63-0
MFC55H78FN9O8 MW 1012.3 g/mol
1-[4-(dimethylamino)-4-methylpent-2-ynoyl]-N-[(2S)-1-[[(6S,8S,14S)-22-ethyl-21-[2-[(1S)-1-methoxyethyl]-3-pyridinyl]-18,18-dimethyl-9,15-dioxo-5,16-dioxa-2,10,22,28-tetrazapentacyclo[18.5.2.12,6.110,14.023,27]nonacosa-1(26),20,23(27),24-tetraen-8-yl]amino]-3-methyl-1-oxobutan-2-yl]-4-fluoro-N-methylpiperidine-4-carboxamide
- 1-[4-(dimethylamino)-4-methylpent-2-ynoyl]-N-[(2S)-1-[[(6S,8S,14S)-22-ethyl-21-[2-[(1S)-1-methoxyethyl]pyridin-3-yl]-18,18-dimethyl-9,15-dioxo-5,16-dioxa-2,10,22,28-tetrazapentacyclo[18.5.2.12,6.110,14.023,27]nonacosa-1(26),20,23(27),24-tetraen-8-yl]amino]-3-methyl-1-oxobutan-2-yl]-4-fluoro-N-methylpiperidine-4-carboxamide
- 3-Pyridazinecarboxylic acid, N1-[N-[[1-[4-(dimethylamino)-4-methyl-1-oxo-2-pentyn-1-yl]-4-fluoro-4-piperidinyl]carbonyl]-N-methyl-L-valyl-3-[4-[(2R)-1-ethyl-3-(3-hydroxy-2,2-dimethylpropyl)-2-[2-[(1S)-1-methoxyethyl]-3-pyridinyl]-1H-indol-5-yl]-2-morpholinyl]-L-alanyl]hexahydro-, (3–>2)-lactone, (3S)-
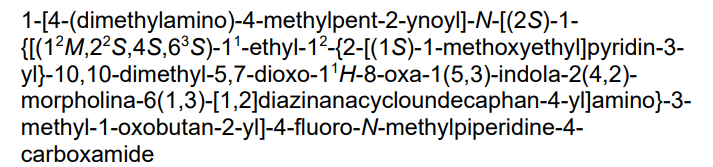
Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, RMC-6291, RMC 6291, 942KVV5CJP
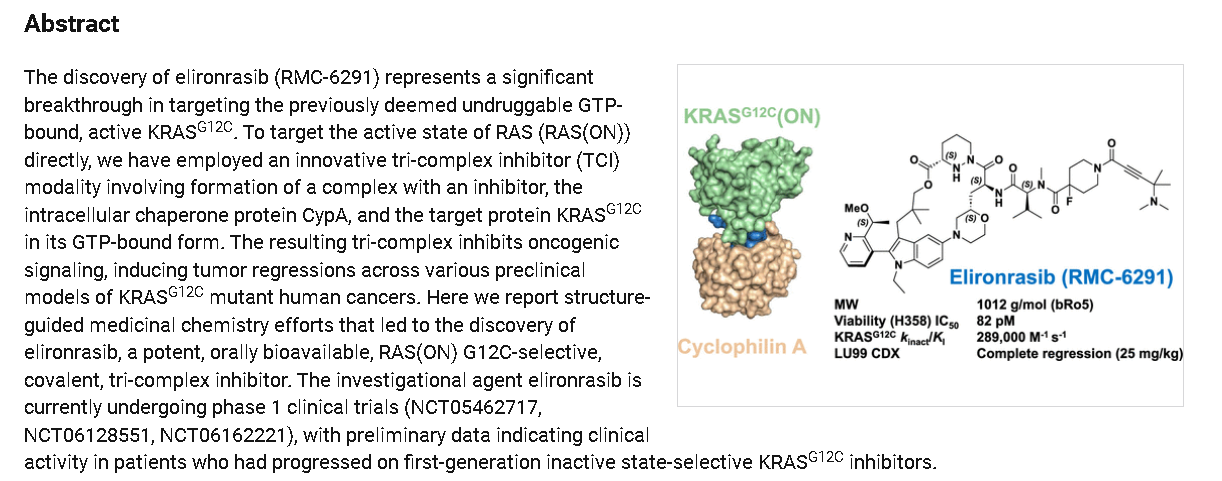
Elironrasib (RMC-6291) is an investigational, orally bioavailable, RAS(ON) G12C-selective inhibitor developed by Revolution Medicines that targets the active GTP-bound form of KRAS G12C. In Phase 1 trials, it showed significant promise in treating advanced KRAS G12C-mutated solid tumors, including non-small cell lung cancer (NSCLC). [1, 2, 3]
Key Clinical Trial Results (as of Oct 2025):
- Response Rate: 42% objective response rate (ORR).
- Disease Control: 79% disease control rate (DCR).
- Durability: Median duration of response was 11.2 months.
- Survival: Median progression-free survival was 6.2 months.
- Overcoming Resistance: Demonstrated efficacy in patients who had previously progressed on first-generation KRAS G12C(OFF) inhibitors.
Mechanism of Action:
Elironrasib acts as a covalent tri-complex inhibitor (TCI). It forms a complex with the intracellular chaperone protein cyclophilin A (CypA) and the active KRAS G12C(GTP) protein, effectively shutting down oncogenic signaling.
Development Status:
- Designation: It has received FDA breakthrough therapy designation for KRAS G12C-mutant NSCLC.
- Trials: Currently in Phase 1 clinical trials (e.g., NCT05462717) to evaluate safety, tolerability, and efficacy, both as a monotherapy and in combination.
- Target Population: Patients with KRAS G12C-addicted solid tumors.
Discovery of Elironrasib (RMC-6291), a Potent and Orally Bioavailable, RAS(ON) G12C-Selective, Covalent Tricomplex Inhibitor for the Treatment of Patients with RAS G12C-Addicted Cancers – PubMed27 Mar 2025 — This information does not constitute medical advice or diagnosis. Elirronrasib (RMC-6291) is a potent, orally bioavailable,
- Revolution Medicines to Present Updated Elironrasib Safety and Efficacy Data in Patients with KRAS G12C Non-Small Cell Lung Cancer Following Treatment with a KRAS(OFF) G12C Inhibitor22 Oct 2025 — This information does not constitute medical advice or diagnosis. Elirronrasib is a RAS(ON) G12C-selective inhibitor being develop…
Revolution Medicines
- Elironrasib May Overcome Resistance to Prior KRAS G12C Inhibition in Non-small Cell Lung Cancer
- OriginatorREVOLUTION Medicines
- ClassAntineoplastics; Morpholines; Piperidines; Pyridazines; Small molecules
- Mechanism of ActionKRAS protein inhibitors
- Phase I/IISolid tumours
- Clinical Phase UnknownNon-small cell lung cancer
- 30 Jan 2026Phase-I/II clinical trials in Solid tumours (Combination therapy, Late-stage disease, Metastatic disease) in USA (PO) (NCT07397338)
- 31 Oct 2025Elironrasib is still in phase I trial in Solid tumours (Late-stage disease, Metastatic disease, Monotherapy) in Australia, Italy, South Korea, Malaysia, Singapore, Spain, Czech Republic, Thailand and USA (PO, Tablet) (NCT05462717)
- 28 Oct 2025No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Metastatic disease, Monotherapy) in Australia, Italy, South Korea, Malaysia, Singapore, Spain, Czech Republic, Thailand (PO, Tablet)
Elironrasib is an orally bioavailable, covalent inhibitor of the active, guanosine triphosphate (GTP)-bound form of the oncogenic KRAS substitution mutation G12C, KRAS G12C(ON), with potential antineoplastic activity. Upon oral administration, elironrasib forms a tri-complex with the intracellular chaperone protein and immunophilin cyclophilin A (CypA) and KRAS G12C(ON). This tri-complex inhibits KRAS G12C(ON)-mediated signaling, which may inhibit tumor cell proliferation. KRAS, a member of the RAS family of oncogenes, serves an important role in cell signaling, division and differentiation. Mutations of KRAS may induce constitutive signal transduction leading to tumor cell growth, proliferation, invasion, and metastasis.
- Study of Elironrasib and Daraxonrasib as Monotherapies and Combination Therapy in Participants With Advanced KRAS G12C Mutant Solid TumorsCTID: NCT06128551Phase: Phase 1/Phase 2Status: RecruitingDate: 2026-04-23
- Dose Escalation and Dose Expansion Study of RMC-6291 Monotherapy in Subjects With Advanced KRASG12C Mutant Solid TumorsCTID: NCT05462717Phase: Phase 1Status: Active, not recruitingDate: 2026-04-08
- Study of RAS(ON) Inhibitors in Combination With Ivonescimab in Patients With Solid TumorsCTID: NCT07397338Phase: Phase 1/Phase 2Status: RecruitingDate: 2026-03-30
- Study of RAS(ON) Inhibitors in Patients With Advanced RAS-mutated NSCLCCTID: NCT06162221Phase: Phase 1/Phase 2Status: RecruitingDate: 2026-03-09
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c02313

SYN
PAT’
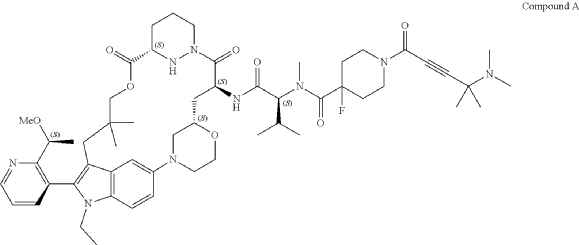
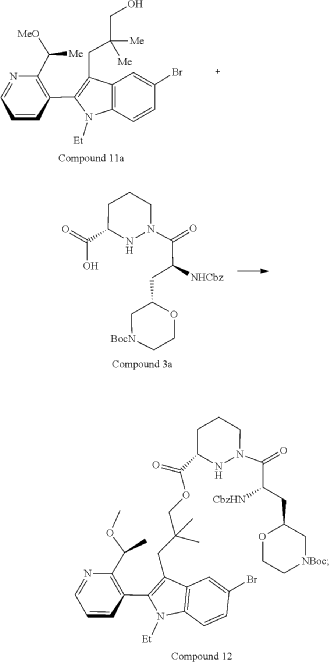
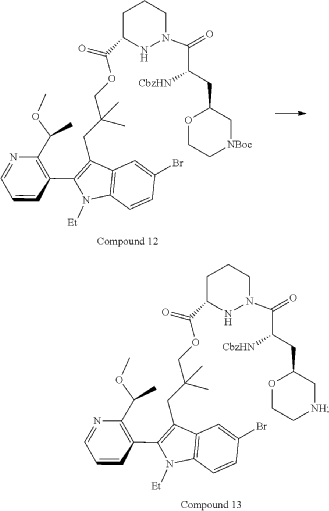
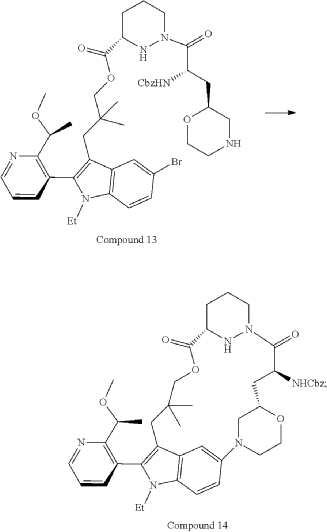
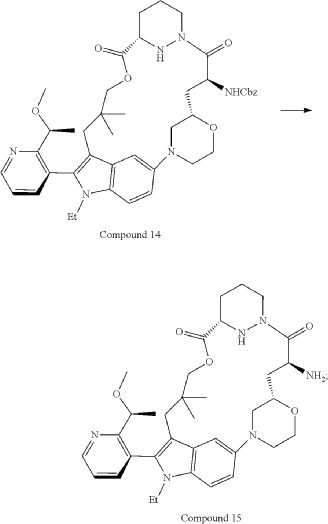
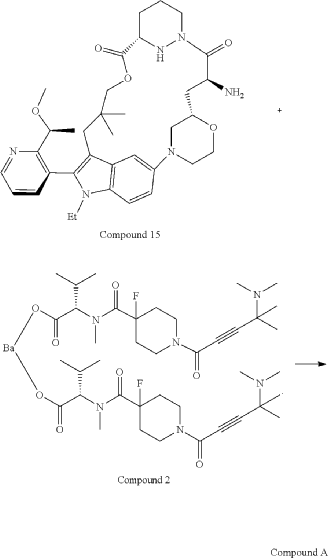
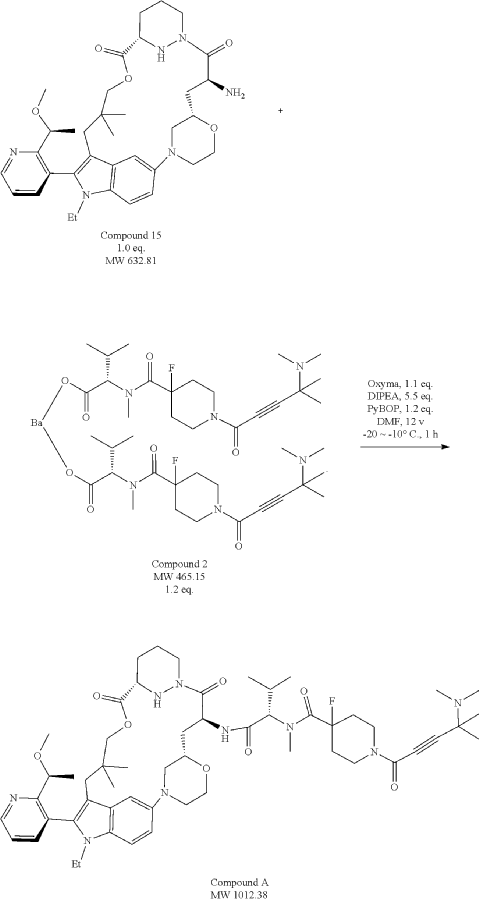
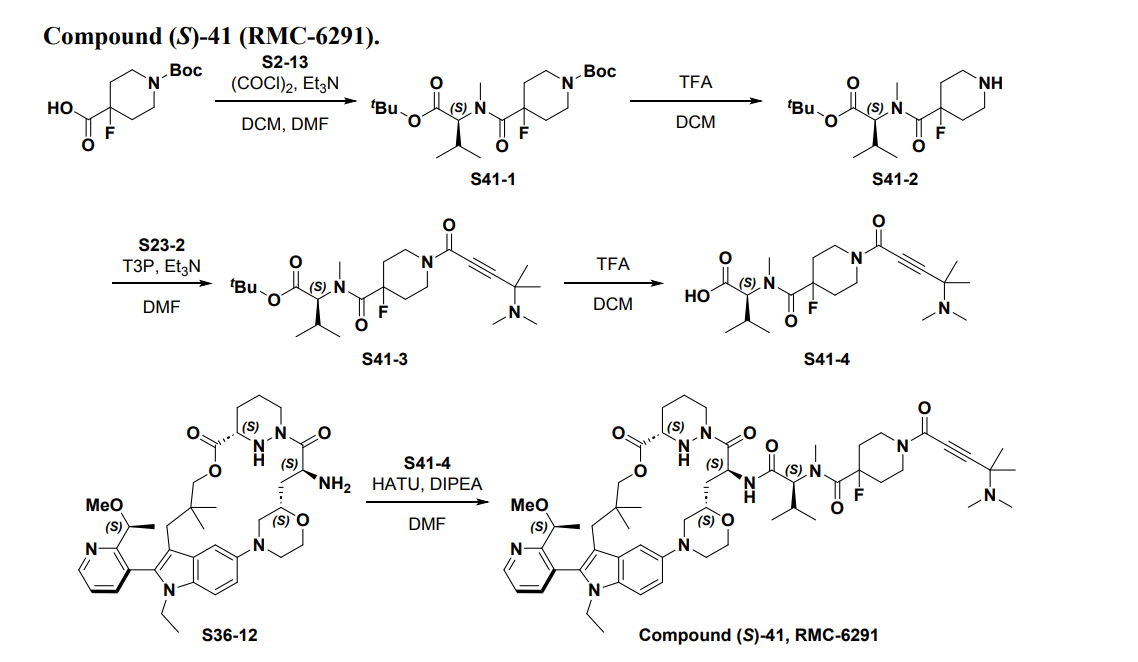
Part 5—Synthesis of Compound A—(12M)-1-(4-(dimethylamino)-4-methylpent-2 ynoyl)-N-((2S)-1-(((22S,63S,4S)-11-ethyl-12-(2-((S)-1-methoxyethyl)pyridin-3-yl)-10,10-dimethyl-5,7-dioxo-61,62,63,64,65,66-hexahydro-11H-8-oxa-2(4,2)-morpholina-1(5,3)-indola-6(1,3) pyridazinacycloundecaphane-4-yl)amino)-3-methyl-1-oxobutan-2 yl)-4-fluoro-N-methylpiperidine-4-carboxamide
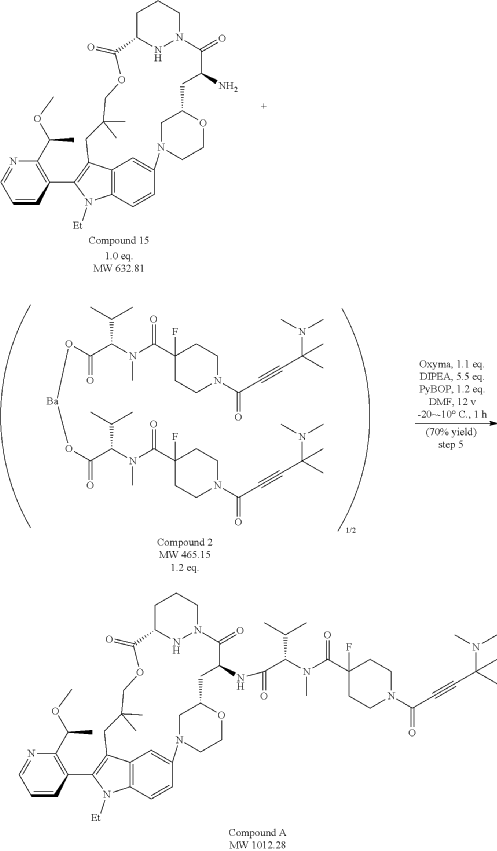
| To a 50 L glass reactor was charged Compound 15 (1.91 kg, 1.0 eq) and DMF (13.9 kg). The mixture was agitated at 20-30° C. until all of the solids were dissolved. Compound 2 (1.70 kg, 1.2 eq) and DMF (3.8 kg) were charged. The mixture was agitated at 20-30° C. until all of the solids were dissolved. DIPEA (2.20 kg, 5.50 eq) was charged at 20-30° C. and the mixture was cooled to −20-−10° C. under agitation. Ethyl cyanoglyoxylate-2-oxime (Oxyma) (0.48 kg, 1.1 eq) was charged to the reactor and the reaction mixture was agitated at −20 to −10° C. for 30 min. PyBOP was charged as a DMF solution (1.89 kg dissolved in 3.62 kg DMF, 1.2 eq) to the reactor at −20 to −10° C. in </=1 h. The reaction mixture was agitated at −20 to −10° C. for 1-3 h. Reaction monitoring by HPLC showed the reaction was complete. |
| The crude product was then further purified by recrystallization with a mixture of EtOAc and n-Heptane to give purified Compound A as a white solid. |
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
References
- Macrocyclic heterocycles and uses thereofPublication Number: US-2023339952-A1Priority Date: 2022-04-20
- Macrocyclic heterocycles and uses thereofPublication Number: WO-2023205701-A1Priority Date: 2022-04-20
- Methods for delaying, preventing, and treating acquired resistance to ras inhibitorsPublication Number: US-2023233569-A1Priority Date: 2020-06-18
- Ras inhibitorsPublication Number: US-2021130303-A1Priority Date: 2019-11-04
- Ras inhibitorsPublication Number: US-2023234929-A1Priority Date: 2019-11-04
- Ras inhibitors
- Publication Number: US-11566007-B2
- Priority Date: 2019-11-04
- Grant Date: 2023-01-31
////////elironrasib, ANAX LABS, Kirsten rat sarcoma viral oncogene homolog inhibitor, antineoplastic, RMC-6291, RMC 6291, 942KVV5CJP
Elecoglipron
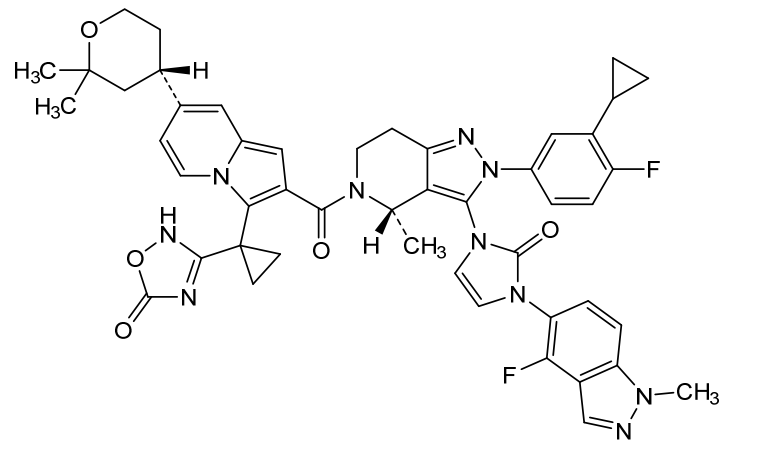
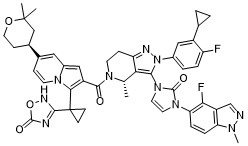
Elecoglipron
CAS 3011682-49-5
MFC48H46F2N10O5 MW880.9 g/mol
3-[1-[2-[(4S)-2-(3-cyclopropyl-4-fluorophenyl)-3-[3-(4-fluoro-1-methylindazol-5-yl)-2-oxoimidazol-1-yl]-4-methyl-6,7-dihydro-4H-pyrazolo[4,3-c]pyridine-5-carbonyl]-7-[(4S)-2,2-dimethyloxan-4-yl]indolizin-3-yl]cyclopropyl]-4H-1,2,4-oxadiazol-5-one
- 1,2,4-Oxadiazol-5(2H)-one, 3-[1-[2-[[(4S)-2-(3-cyclopropyl-4-fluorophenyl)-3-[3-(4-fluoro-1-methyl-1H-indazol-5-yl)-2,3-dihydro-2-oxo-1H-imidazol-1-yl]-2,4,6,7-tetrahydro-4-methyl-5H-pyrazolo[4,3-c]pyridin-5-yl]carbonyl]-7-[(4S)-tetrahydro-2,2-dimethyl-2H-pyran-4-yl]-3-indolizinyl]cyclopropyl]-
- 3-(1-(2-((S)-2-(3-cyclopropyl-4-fluorophenyl)-3-(3-(4-fluoro-1-methyl-1H-indazol-5-yl)-2-oxo-2,3-dihydro-1H-imidazol-1-yl)-4-methyl-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridine-5-carbonyl)-7-((S)-2,2-dimethyltetrahydro-2H-pyran-4-yl)indolizin-3-yl)cyclopropyl)-1,2,4-oxadiazol-5(4H)-one
- 3-[1-[2-[(4S)-2-(3-cyclopropyl-4-fluorophenyl)-3-[3-(4-fluoro-1-methylindazol-5-yl)-2-oxoimidazol-1-yl]-4-methyl-6,7-dihydro-4H-pyrazolo[4,3-c]pyridine-5-carbonyl]-7-[(4S)-2,2-dimethyloxan-4-yl]indolizin-3-yl]cyclopropyl]-4H-1,2,4-oxadiazol-5-one

glucagon-like peptide 1 (GLP-1) receptor agonist, AZD 5004, ECC 5004, G94JJ74N5Y
Elecoglipron (AZD5004 / ECC-5004) is a glucagon-like peptide 1 (GLP-1) receptor agonist. In vitro, AZD5004/ECC5004 binds the human GLP-1 receptor with high affinity (IC₅₀ ~ 2.4 nM), drives G s-coupled cAMP signaling with low-nanomolar potency (EC₅₀ ~ 2–6 nM in HEK293/β-cell cAMP and GSIS assays), shows partial agonism in some cell systems without detectable β-arrestin recruitment or receptor internalization, and potentiates glucose-stimulated insulin secretion (EC₅₀ ~ 5.9 nM). In in vivo non-human primate studies, oral dosing produced robust pharmacodynamic effects — insulin secretion and glucose clearance with estimated EC₅₀ ~ 0.022 nM — and dose-dependent reductions in body-weight gain over long-term dosing, consistent with GLP-1-mediated metabolic effects
PAT
US-11584751-B1
https://patentscope.wipo.int/search/en/detail.jsf?docId=US392022131&_cid=P22-MOWB0S-70559-1
PAT
US-12037339-B2
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Substituted imidazoles as GLP-1 receptor agonistsPublication Number: US-12037339-B2Priority Date: 2020-07-20Grant Date: 2024-07-16
- Substituted imidazoles as GLP-1 receptor agonistsPublication Number: US-11584751-B1Priority Date: 2020-07-20Grant Date: 2023-02-21
//////elecoglipron, anax labs, glucagon-like peptide 1 (GLP-1) receptor agonist, AZD 5004, ECC 5004, G94JJ74N5Y
Egalognastat
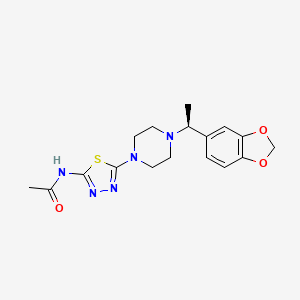
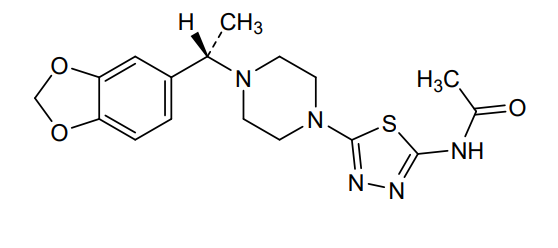
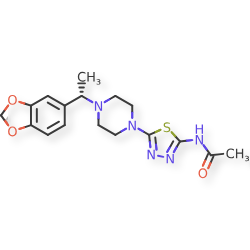
Egalognastat
CAS 1884154-02-2
MF C17H21N5O3S MW375.45
(S)-N-(5-(4-(1-(BENZO(D)(1,3)DIOXOL-5-YL)ETHYL)PIPERAZIN-1-YL)-1,3,4-THIA-DIAZOL-2-YL)ACETAMIDE
ACETAMIDE, N-(5-(4-((1S)-1-(1,3-BENZODIOXOL-5-YL)ETHYL)-1-PIPERAZINYL)-1,3,4-THIADIAZOL-2-YL)-
N-(5-{4-[(1S)-1-(2H-1,3-benzodioxol-5-yl)ethyl]piperazin-1-yl}-1,3,4-thiadiazol-2-yl)acetamide
O-GlcNAcase enzyme inhibitor, ASN90, ASN 90, E9QIS63WUM, ASN 120290
Egalognastat (ASN90) is a potent, selective, and brain-penetrant O-GlcNAcase (OGA) inhibitor (\(IC_{50} = 10.2 \text{ nM}\)). It acts as a disease-modifying agent in pre-clinical studies for neurodegenerative conditions by enhancing protein O-GlcNAcylation, which regulates tau and \(\alpha \)-synuclein pathology. Egalognastat is under investigation for diseases like Alzheimer’s and Parkinson’s.
Key Details on Egalognastat (ASN90):
- Mechanism: As a substrate-competitive OGA inhibitor, it binds to the OGA enzyme and reduces the removal of O-GlcNAc from proteins.
- Disease Targets: It is primarily studied for tauopathies and \(\alpha \)-synucleinopathies.
- Efficacy: Preclinical data shows it raises O-GlcNAcylation of brain proteins and has shown therapeutic potential in models of neurodegeneration.
- Distinction: Unlike earlier sugar-based inhibitors (like Thiamet G), Egalognastat is chemically distinct.
- Status: It is primarily used for research and preclinical development.
- Related Research: Recent studies (2025) have analyzed the potential synaptotoxic effects of OGA inhibitors, including Egalognastat (ASN90) and Ceperognastat, indicating that while they are effective for removing misfolded proteins, they may interfere with synaptic plasticity.
SYN
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2024-08-07
PMID: 39109492
DOI: 10.1021/acs.jmedchem.4c01132
SYN
PAT
PAT


PAT
- Glycosidase inhibitorsPublication Number: US-12187741-B2Priority Date: 2014-08-28Grant Date: 2025-01-07
- Glucosidase inhibitorsPublication Number: ES-2893289-T3Priority Date: 2014-08-28Grant Date: 2022-02-08
- Glycosidase inhibitorsPublication Number: EP-3186243-B1Priority Date: 2014-08-28Grant Date: 2021-07-21
- Glycosidase inhibitorsPublication Number: US-10336775-B2Priority Date: 2014-08-28Grant Date: 2019-07-02
- Glycosidase inhibitorsPublication Number: US-11046712-B2Priority Date: 2014-08-28Grant Date: 2021-06-29
- Glycosidase inhibitorsPublication Number: EP-3868752-A1Priority Date: 2014-08-28
- Glycosidase InhibitorsPublication Number: CN-107108601-APriority Date: 2014-08-28
- Glycosidase inhibitorsPublication Number: US-2019367533-A1Priority Date: 2014-08-28
- Glycosidase inhibitorsPublication Number: WO-2016030443-A1Priority Date: 2014-08-28
- Glycosidase inhibitorsPublication Number: EP-3186243-A1Priority Date: 2014-08-28
- Glycosidase inhibitorPublication Number: JP-2019206554-APriority Date: 2014-08-28
- Glycosidase inhibitorsPublication Number: US-2017298082-A1Priority Date: 2014-08-28
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
////////egalognastat, anax labs, O-GlcNAcase enzyme inhibitor, ASN90, ASN 90, E9QIS63WUM, ASN 120290
Dirozalkib
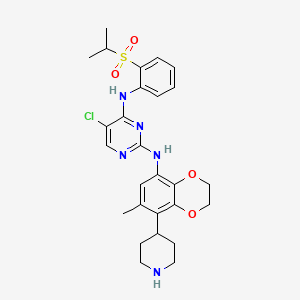
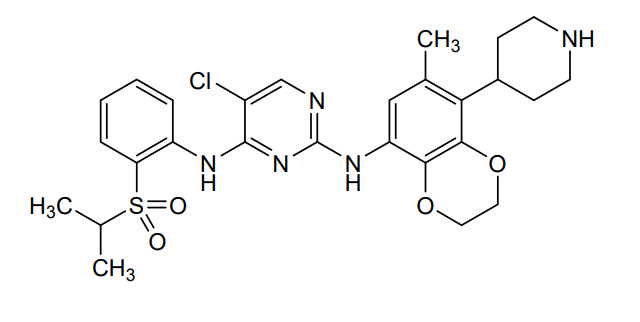
Dirozalkib
CAS 1893419-37-8
MF C27H32ClN5O4S MW558.1 g/mol
5-chloro-2-N-(6-methyl-5-piperidin-4-yl-2,3-dihydro-1,4-benzodioxin-8-yl)-4-N-(2-propan-2-ylsulfonylphenyl)pyrimidine-2,4-diamine

anaplastic lymphoma kinase (ALK) inhibitor, antineoplastic, XZP-3621, XZP 3621, Xuanzhu Biopharmaceutical, 2FH56C28YT
Dirozalkib (XZP-3621) is a novel, potent, and highly selective ALK/ROS1 tyrosine kinase inhibitor developed by Xuanzhu Biopharmaceutical to treat advanced ALK-positive non-small cell lung cancer (NSCLC). It demonstrated high efficacy (47.4% ORR, up to 89.3% in naive patients) in clinical trials and is designed to overcome resistance to earlier inhibitors.
Key Aspects of Dirozalkib
- Indication: Treatment of adult patients with ALK-positive locally advanced or metastatic non-small cell lung cancer (NSCLC).
- Mechanism: Acts as a dual-target ALK/ROS1 tyrosine kinase inhibitor (TKI), effective against ALK fusion-positive cells and various resistance mutations.
- Clinical Efficacy (Phase I/II): In studies, the drug showed significant antitumor activity with an Objective Response Rate (ORR) of 47.4% and an 89.3% ORR in ALK inhibitor-naive patients at 500 mg/day.
- Safety Profile: No dose-limiting toxicities occurred; the maximum tolerated dose was 600 mg/day, with a recommended dose of 500 mg/day. Common adverse events included diarrhea.
- Status: As of early 2026, the NDA (New Drug Application) for Dexitinib (Dirozalkib) was accepted by China’s NMPA, with potential for further market expansion.
- OriginatorXuanzhu Biopharmaceutical
- Class2 ring heterocyclic compounds; Amines; Aniline compounds; Antineoplastics; Chlorinated hydrocarbons; Piperidines; Pyrimidines; Small molecules; Sulfones
- Mechanism of ActionAnaplastic lymphoma kinase inhibitors
- RegisteredNon-small cell lung cancer
- 26 Aug 2025Chemical structure information added.
- 22 Aug 2025Registered for Non-small cell lung cancer (Late-stage disease) in China (PO) – First global approval
- 22 Aug 2025Efficacy and adverse events data from a phase III trial in Non-small cell lung cancer released by Xuanzhu Biopharmaceutical
- A Phase I Study of XZP-3621 in Chinese Patients With ALK or ROS1 Rearrangement Non-small Cell Lung CancerCTID: NCT05055232Phase: Phase 1Status: CompletedDate: 2025-07-24
- Food Effect and Mass Balance Study of XZP-3621 TabletsCTID: NCT05034120Phase: Phase 1Status: CompletedDate: 2025-05-25
- A Study of XZP-3621 in Chinese Patients With ALK Positive NSCLCCTID: NCT05482087Phase: Phase 2Status: Unknown statusDate: 2022-08-01
- A Study to Evaluate and Compare the Efficacy and Safety of XZP-3621 Versus CrizotinibCTID: NCT05204628Phase: Phase 3Status: Unknown statusDate: 2022-01-24
PAT
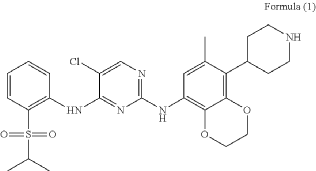
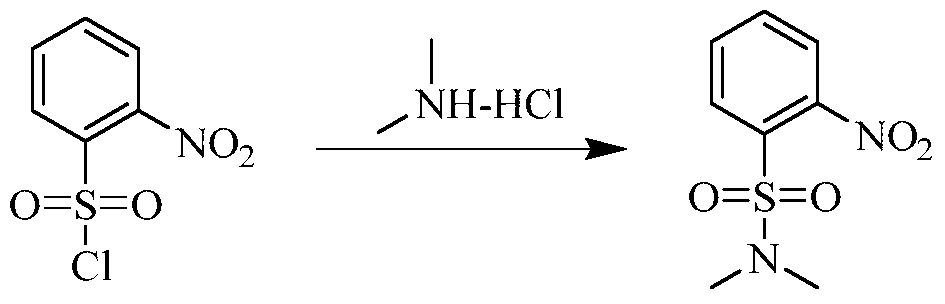
PAT
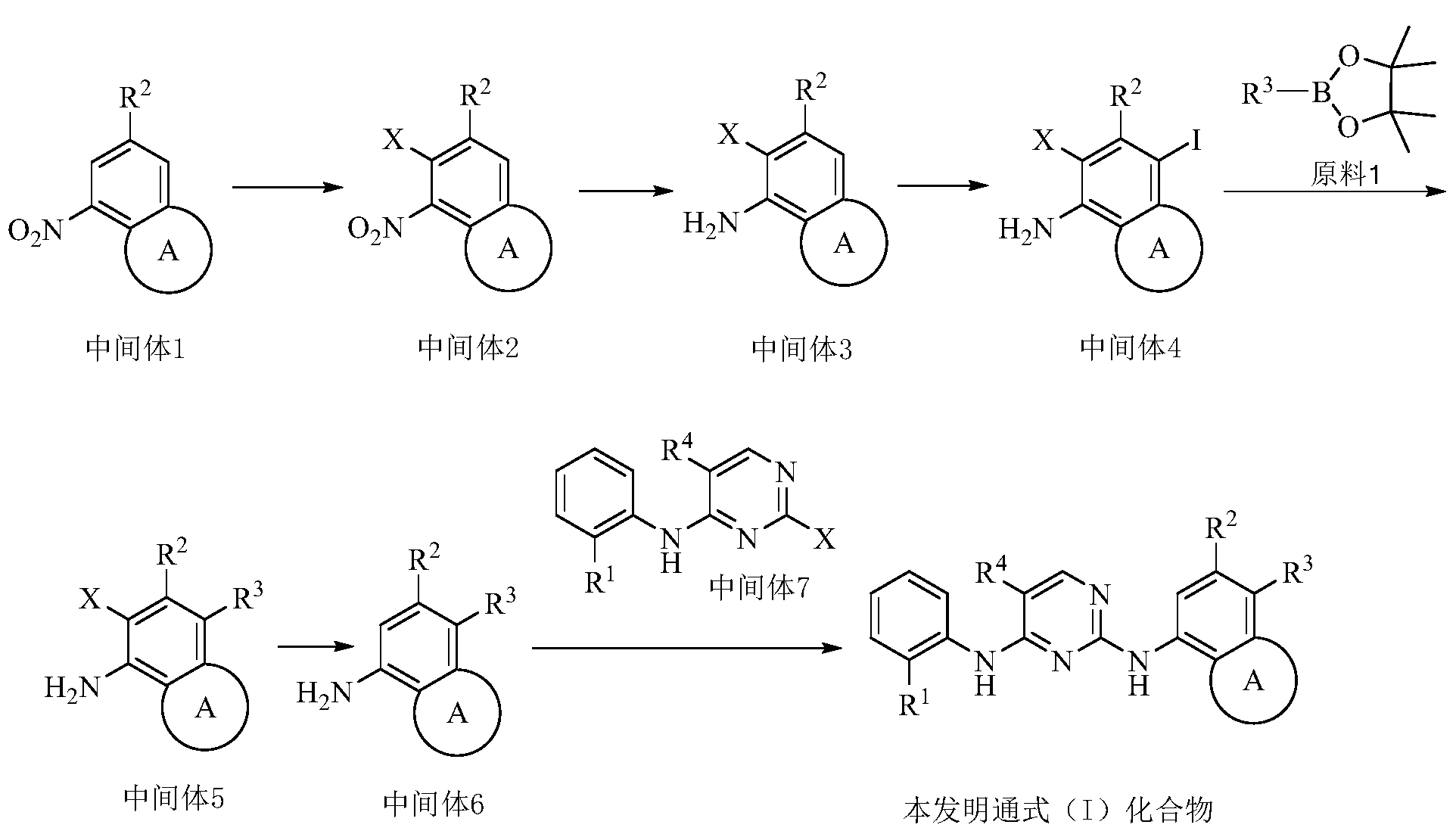
Example 3 Preparation of 2-((5-chloro-2-((7-methyl-8-(piperidin-4-yl)-2,3-dihydrobenzo[b][1,4]dioxin- 5-yl)amino)pyrimidin-4-yl)amino)-N,N-dimethylbenzenesulfonamide (compound 3)
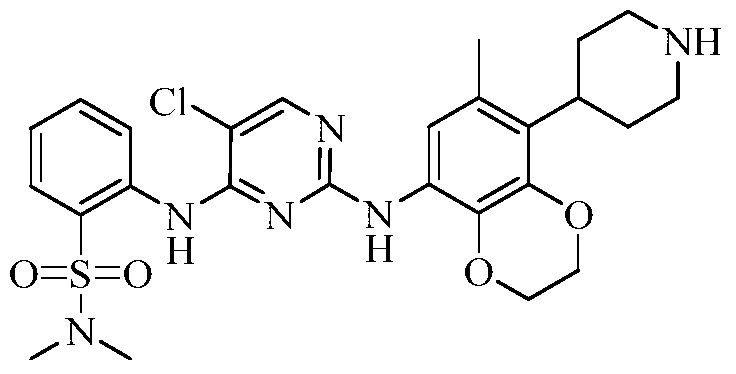
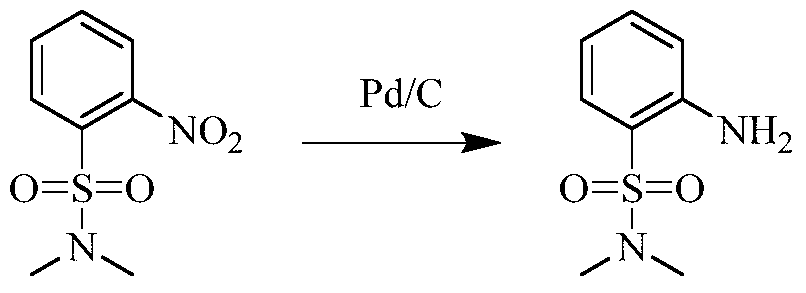
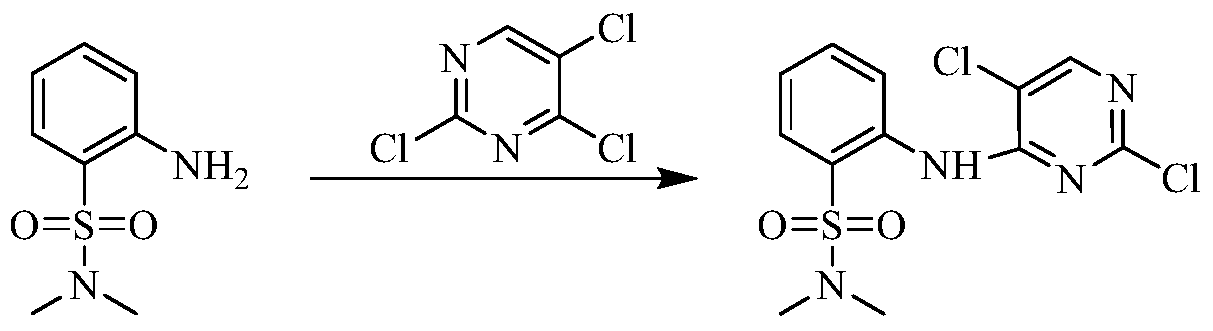
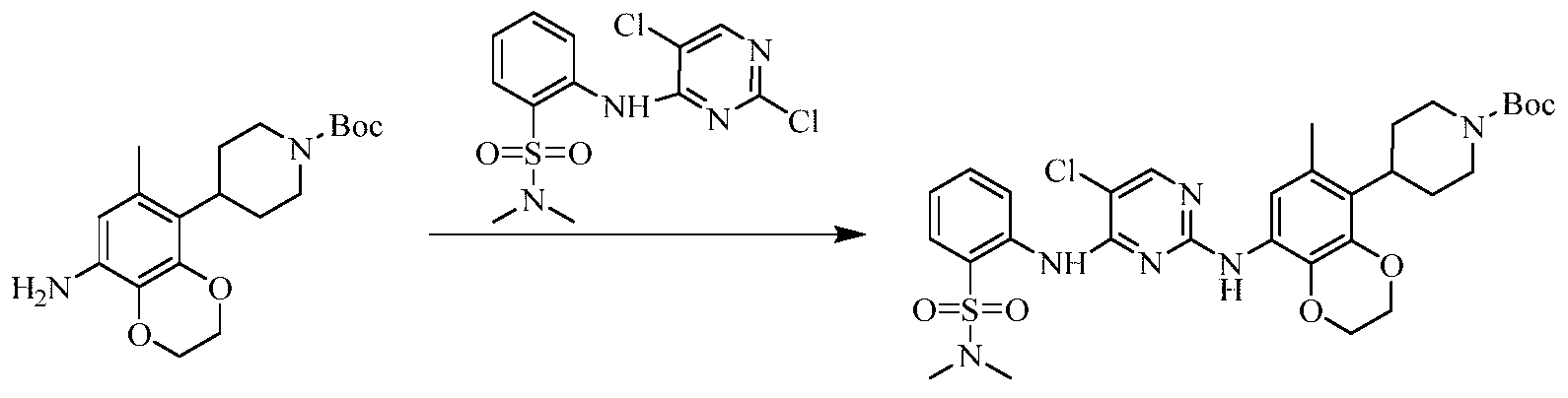
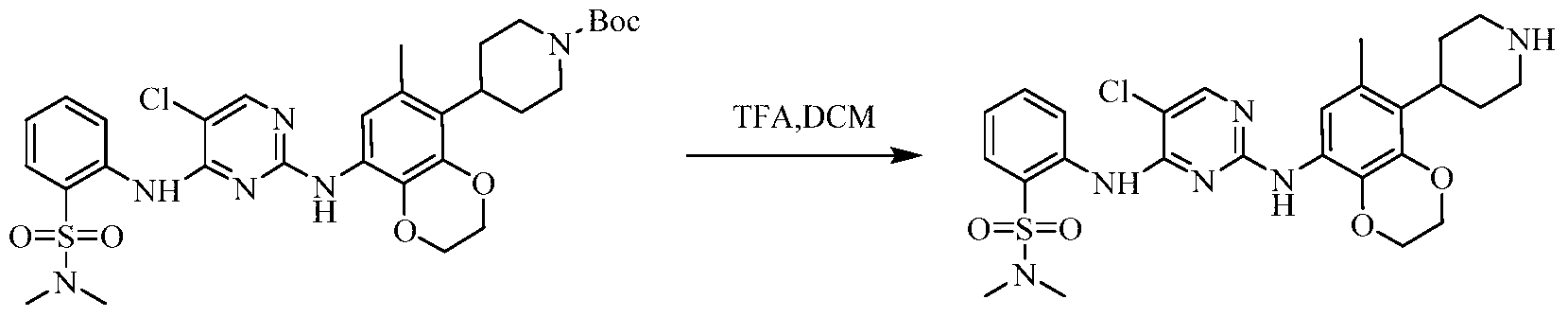
(5) Preparation of 2-((5-chloro-2-((7-methyl-8-(piperidin-4-yl)-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)amino)pyrimidin-4-yl)amino)-N,N-dimethylbenzenesulfonamide
75 mg (0.114 mmol) of tert-butyl 4-(8-((5-chloro-4-((2-(N,N-dimethylaminosulfonyl)phenyl)amino)pyrimidin-2-yl)amino)-6-methyl-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)piperidine-1-carboxylic acid ester was dissolved in dichloromethane (10 mL), and trifluoroacetic acid (1 mL) was added. The mixture was stirred at room temperature for 12 hours. The starting material disappeared as detected by TLC. Water (20 mL) was added, and the mixture was separated. The aqueous phase was extracted twice with dichloromethane (20 mL × 2). The organic phases were combined, dried over anhydrous sodium sulfate, and the solvent was removed by rotary evaporation. The crude product was purified by silica gel column chromatography (methanol:dichloromethane = 1:50) to obtain the final product (30 mg, yield 47.2%).
[0415]Molecular formula:
C26H31ClN6O4S Molecular weight: 559.08 LC-MS (m / z): 280.2 [ M /2+H ] +
[0416]
1H-NMR(400MHz,MeOD)δ:8.44(d,1H,J=1.2),8.11(s,1H),7.86(d,1H,J=1.2),7.56-7.60(m,1H),7.28-7.35(m,2H),4.26(s,4H),3.45-3.48(m,2H),3.06-3.15(m,3H),2.56-2.74(m,8H),2.17(s,3H),1.76-1.80(m,2H).
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: US-10011592-B2Priority Date: 2014-09-29Grant Date: 2018-07-03
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: US-2018086745-A9Priority Date: 2014-09-29
- Polycyclic anaplastic lymphoma kinase inhibitorPublication Number: EP-3202765-A1Priority Date: 2014-09-29
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: KR-20170055555-APriority Date: 2014-09-29
- Crystal form of polycyclic anaplastic lymphoma kinase inhibitorPublication Number: US-2023348443-A1Priority Date: 2020-01-17
- Crystal form of polycyclic anaplastic lymphoma kinase inhibitorPublication Number: US-12441717-B2Priority Date: 2020-01-17Grant Date: 2025-10-14
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: US-2017240534-A1Priority Date: 2014-09-29
- Polycyclic inhibitor of anaplastic lymphoma kinasePublication Number: KR-101909404-B1Priority Date: 2014-09-29Grant Date: 2018-10-17
- Polycyclic anaplastic lymphoma kinase inhibitorPublication Number: WO-2016050171-A1Priority Date: 2014-09-29
- Pharmaceutical composition of anaplastic lymphoma kinase inhibitor and preparation method thereforPublication Number: WO-2025140560-A1Priority Date: 2023-12-28
- CRYSTALLINE FORM OF A POLYCYCLIC ANAPLASIC LYMPHOMA KINASE INHIBITORPublication Number: EP-4092021-A4Priority Date: 2020-01-17
- Polycyclic anaplastic lymphoma kinase inhibitor crystalline formPublication Number: CN-113135905-BPriority Date: 2020-01-17Grant Date: 2023-11-21
- Crystal form of polycyclic anaplastic lymphoma kinase inhibitorPublication Number: WO-2021143819-A1Priority Date: 2020-01-17
- Crystal form of polycyclic anaplastic lymphoma kinase inhibitorPublication Number: EP-4092021-A1Priority Date: 2020-01-17
/////////dirozalkib, anax labs, anaplastic lymphoma kinase (ALK) inhibitor, antineoplastic, XZP-3621, XZP 3621, Xuanzhu Biopharmaceutical, 2FH56C28YT
Deulumateperone
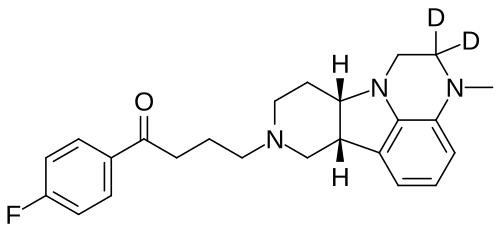
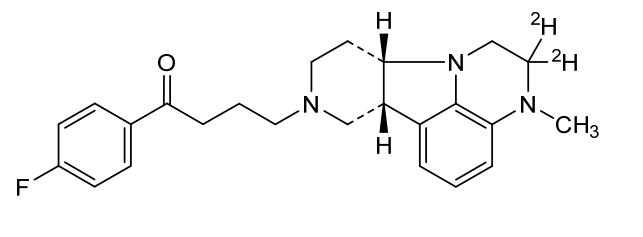
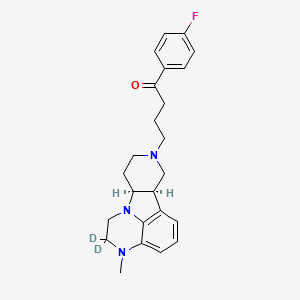
Deulumateperone
CAS 2102683-75-8
MF C24H262H2FN3O MW 395.5 g/mol
4-[(10R,15S)-3,3-dideuterio-4-methyl-1,4,12-triazatetracyclo[7.6.1.05,16.010,15]hexadeca-5,7,9(16)-trien-12-yl]-1-(4-fluorophenyl)butan-1-one
- 1-(4-Fluorophenyl)-4-[(6bR,10aS)-2,3,6b,9,10,10a-hexahydro-2-d-3-methyl-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl-2-d]-1-butanone
- 1-Butanone, 1-(4-fluorophenyl)-4-[(6bR,10aS)-2,3,6b,9,10,10a-hexahydro-2-d-3-methyl-1H-pyrido[3′,4′:4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl-2-d]-

antipsychotic, ITI-1284, ITI 1284, NBA7J58PPP,
Deulumateperone (INNTooltip International Nonproprietary Name; developmental code name ITI-1284) is an experimental antipsychotic of the pyridopyrroloquinoxaline and butyrophenone families as well as a deuterated analogue of lumateperone which is under development as a sublingually administered orally disintegrating tablet (ODT) for the treatment of psychotic disorders, agitation, and generalized anxiety disorder.[2][3][4][1] No recent development has been reported for treatment of depressive disorders, behavioral disorders, and dementia.[2] It is being developed by Intra-Cellular Therapies.[2][3][1] As of January 2025, it has reached phase 2 clinical trials.[2][3]
SYN
PAT
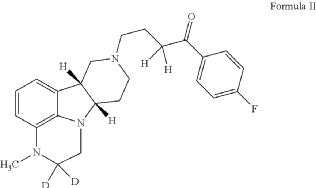
Example 2
2,2-D2-1-(4-fluorophenyl)-4-((6bR,10aS)-3-methyl-2,3,6b,7,10,10a-hexahydro-1H-pyrido[3′,4′: 4,5]pyrrolo[1,2,3-de]quinoxalin-8(9H)-yl)butan-1-one
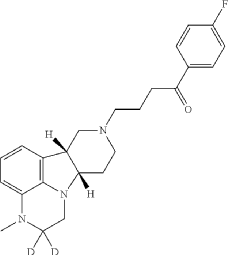
| To a suspension of (6bR, 10aS)-3-Methyl-2-oxo-2,3,6b,9,10,10a-hexahydro-1H,7H-pyrido[3′,4′: 4,5]pyrrolo[1,2,3-de]quinoxaline-8-carboxylic acid ethyl ester (945 mg, 3 mmol) in THF (5 mL) is slowly added BD 3-THF (1.0 M in THF, 10 mL, 10 mmol) at room temperature. After completion of the addition, the reaction mixture is stirred at room temperature overnight and then carefully quenched with D 2O (2.0 mL). The solvent is removed under vacuum and the residue is suspended in HCl (12 N, 9 mL). After stirred at 95° C. for 20 h, the reaction mixture is cooled to room temperature and then adjusted to pH of 12 with 50% NaOH. The mixture is concentrated to dryness to give 2,2-d 2-(6bR, 10aS)-3-Methyl-2,3,6b,7,8,9,10,10a-octahydro-1H-pyrido[3′,4′: 4,5]pyrrolo[1, 2,3-de]quinoxaline as a brown solid, which is used directly for next step without further purification. MS (ESI) m/z 232.2 [M+H] +. |
PAT
- Novel compositions and methodsPublication Number: US-2021315891-A1Priority Date: 2018-08-29
- Transmucosal and subcutaneous compositionsPublication Number: US-10716786-B2Priority Date: 2017-03-24Grant Date: 2020-07-21
- Novel compositions and methodsPublication Number: US-2018271862-A1Priority Date: 2017-03-24
- Transmucosal methods for treating psychiatric and neurological conditionsPublication Number: US-11052083-B2Priority Date: 2017-03-24Grant Date: 2021-07-06
- Novel compositions and methodsPublication Number: US-2020375988-A1Priority Date: 2017-03-24
- Novel compositions and methodsPublication Number: US-2021361648-A1Priority Date: 2017-03-24
- Organic compoundsPublication Number: US-2019231780-A1Priority Date: 2016-03-25
- Organic compoundsPublication Number: US-10688097-B2Priority Date: 2016-03-25Grant Date: 2020-06-23
- Organic compoundsPublication Number: US-11096944-B2Priority Date: 2016-03-25Grant Date: 2021-08-24
- Organic compoundsPublication Number: US-2020352949-A1Priority Date: 2016-03-25
- Organic compounds
- Publication Number: US-2022008423-A1
- Priority Date: 2016-03-25
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- Oguma T, Jino K (2024). “Clinical Pipelines for Alzheimer’s Disease Psychosis and Agitation”. Chemical & Pharmaceutical Bulletin. 72 (7) c23-00416: 610–617. doi:10.1248/cpb.c23-00416. PMID 38945937.
- “Intra-Cellular Therapies”. AdisInsight. 29 January 2025. Retrieved 26 February 2025.
- “Delving into the Latest Updates on ITI-1284 with Synapse”. Synapse. 20 February 2025. Retrieved 26 February 2025.
- “Proposed INN: List 132 International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (4): 1073. 2024.
| Clinical data | |
|---|---|
| Other names | Lumateperone deuterated; Deuterated lumateperone; ITI-1284; ITI1284; ITI-1284-ODT-SL |
| Routes of administration | Sublingual (orally disintegrating tablet)[1] |
| Drug class | Atypical antipsychotic |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2102683-75-8 |
| PubChem CID | 140916642 |
| UNII | NBA7J58PPP |
| KEGG | D13268 |
| Chemical and physical data | |
| Formula | C24H26D2FN3O |
| Molar mass | 395.518 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
////////deulumateperone, anax labs, antipsychotic, ITI-1284, ITI 1284, NBA7J58PPP,
Vepdegestrant
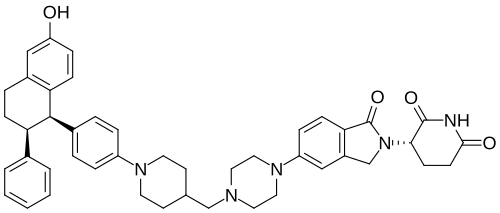
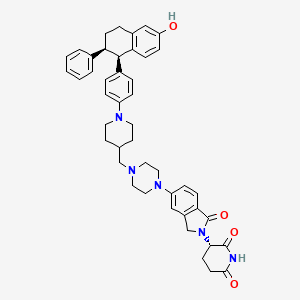
Vepdegestrant
CAS 2229711-08-2
MW 723.9 g/mol, C45H49N5O4
- (S)-3-(5-(4-((1-(4-((1R,2S)-6-Hydroxy-2-phenyl-1,2,3,4-tetrahydronaphthalen-1-yl)phenyl)piperidin-4-yl)methyl)piperazin-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione
- (3S)-3-[5-[4-[[1-[4-[(1R,2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione
- (2(1)R,2(2)S,8(3)S)-2-hydroxy-2(1),2(2),2(3),2-tetrahydro-7(5,2)-isoindola-6(1,4)-piperazina-4(1,4),8(3)-dipiperidina-2(2,1)-naphthalena-1(1),3(1,4)-dibenzenaoctaphane-7(1),8(2),8(7(3)H)-trione
- 2,6-Piperidinedione, 3-(1,3-dihydro-1-oxo-5-(4-((1-(4-((1R,2S)-1,2,3,4-tetrahydro-6-hydroxy-2-phenyl-1-naphthalenyl)phenyl)-4-piperidinyl)methyl)-1-piperazinyl)-2H-isoindol-2-yl)-, (3S)-
(3S)-3-[6-[4-[[1-[4-[(1R,2S)-6-hydroxy-2-phenyl-1,2,3,4-tetrahydronaphthalen-1-yl]phenyl]piperidin-4-yl]methyl]piperazin-1-yl]-3-oxo-1H-isoindol-2-yl]piperidine-2,6-dione
5/1/2026, FDA 2026, APROVALS 2026, Veppanu, ARV 471, WC1U3R1YMI, PF 07850327
To treat estrogen receptor-positive, human epidermal growth factor receptor 2-negative, ESR1-mutated advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy
On May 1, 2026, the FDA approved vepdegestrant (Veppanu), a first-in-class oral PROTAC estrogen receptor (ER) degrader developed by Arvinas and Pfizer, for adults with ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer who have progressed on endocrine therapy. It demonstrated significant progression-free survival (PFS) improvements compared to fulvestrant.
Key Details About Vepdegestrant (Veppanu):
- Mechanism of Action: As an oral PROTAC (Proteolysis-Targeting Chimera), vepdegestrant targets the estrogen receptor for degradation, designed to be more effective than traditional endocrine therapies, particularly in ESR1-mutated tumors.
- Approved Indication: For treating adults with ER+/HER2-, ESR1-mutated advanced/metastatic breast cancer (detected by Guardant360 CDx) after at least one line of endocrine therapy.
- Dosage: The recommended dose is 200 mg taken orally once daily with food.
- Clinical Efficacy (VERITAC-2): In trials, vepdegestrant showed a significantly longer PFS compared to intramuscular fulvestrant.
- Side Effects & Risks: Common side effects include decreased white blood cell counts, increased liver function tests, muscle/bone pain, fatigue, and nausea. Warnings include embryo-fetal toxicity and QTc interval prolongation (heart rhythm issues).
- Companion Diagnostic: Guardant360 CDx was approved alongside the drug to identify patients with ESR1 mutations
Vepdegestrant (developmental code name ARV-471) is an investigational oral proteolysis-targeting chimera (PROTAC) compound that targets the estrogen receptor for protein degradation. It is being developed for the treatment of estrogen receptor-positive, HER2-negative (ER+/HER2-) breast cancer by Arvinas and Pfizer.[1][2][3]
Mechanism of action
Vepdegestrant is designed as a PROTAC that recruits the ubiquitin-proteasome system to target the estrogen receptor for degradation.[4] The compound contains both an E3 ubiquitin ligase-binding moiety and an estrogen receptor-binding domain, intended to bring these proteins into proximity to trigger ubiquitination and subsequent proteasomal degradation of the ER protein.[5] In laboratory studies, vepdegestrant demonstrated ER degradation in ER-positive breast cancer cell lines with reported DC50 values of approximately 1-2 nM.[6]
Vepdegestrant is an orally available hetero-bifunctional molecule and selective estrogen receptor (ER) alpha-targeted protein degrader, using the proteolysis targeting chimera (PROTAC) technology, with potential antineoplastic activity. Vepdegestrant is composed of an ER alpha ligand attached to an E3 ligase recognition moiety. Upon oral administration,vepdegestrant targets and binds to the ER ligand binding domain on ER alpha. E3 ligase is recruited to the ER by the E3 ligase recognition moiety and ER alpha is tagged by ubiquitin. This causes ubiquitination and degradation of ER alpha by the proteasome. This decreases ER alpha protein levels, decreases the expression of ER alpha-target genes and halts ER-mediated signaling. This results in an inhibition of proliferation in ER alpha-overexpressing tumor cells. In addition, the degradation of the ER alpha protein releases the ARV-471 and can bind to additional ER alpha target proteins. ER alpha is overexpressed in a variety of cancers and plays a key role in cancer cell proliferation.
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/slct.202405939
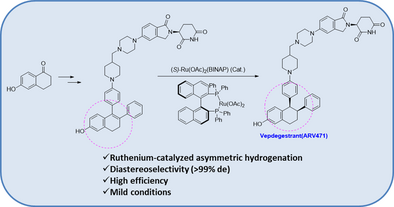
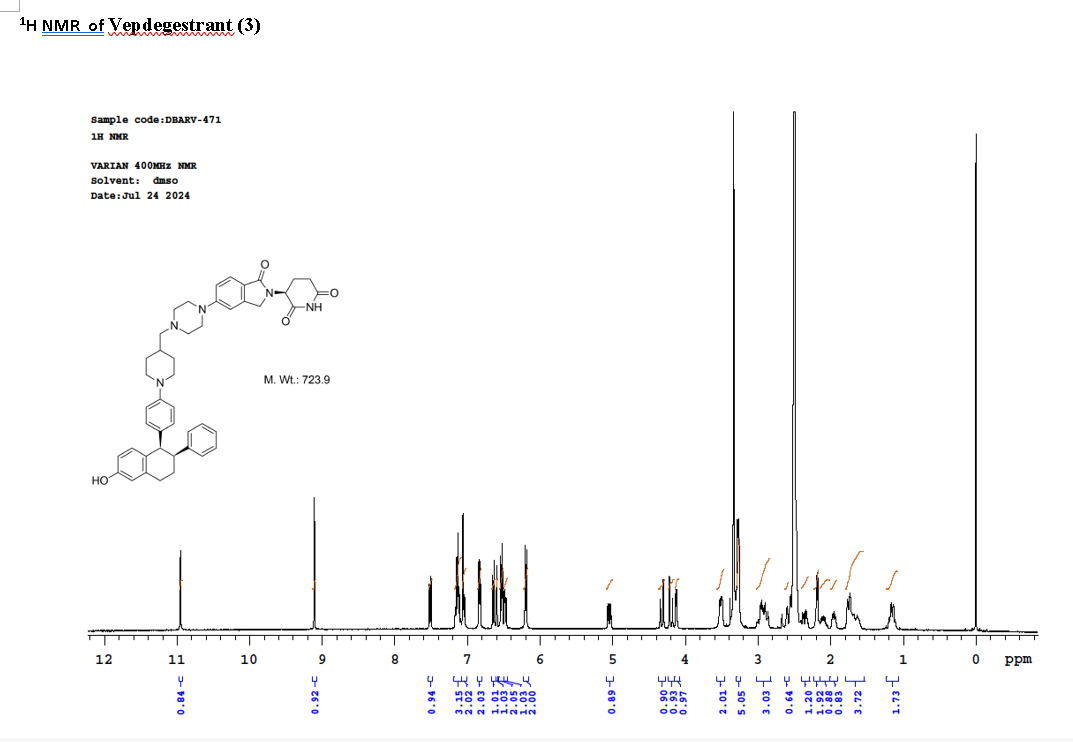
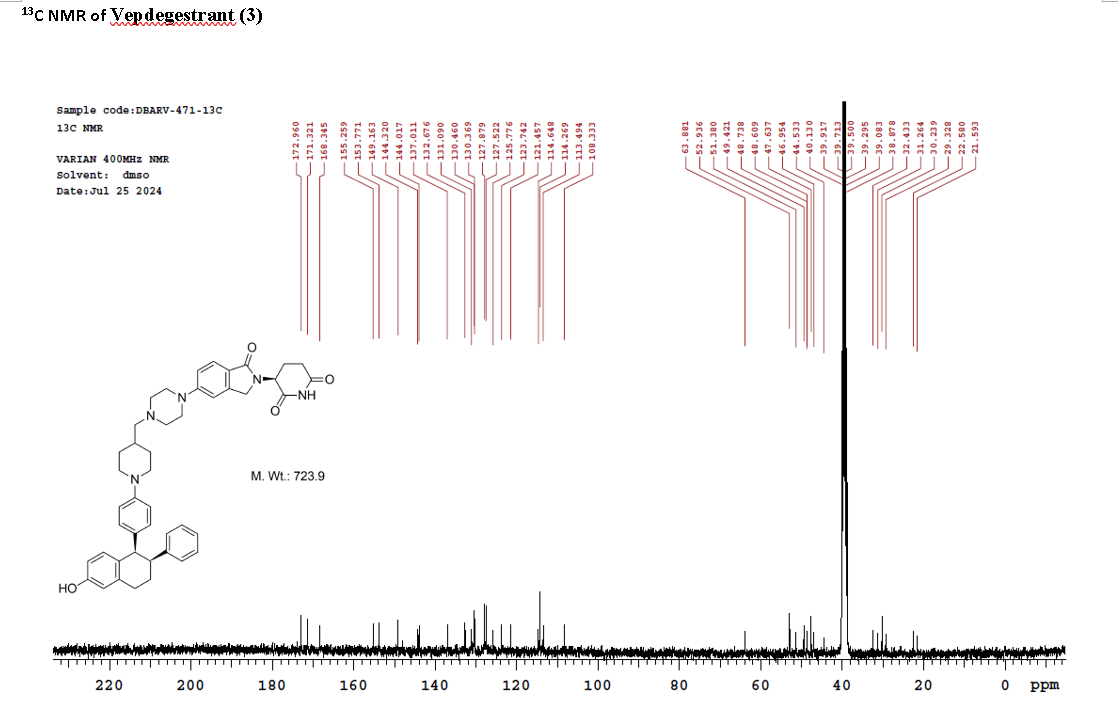
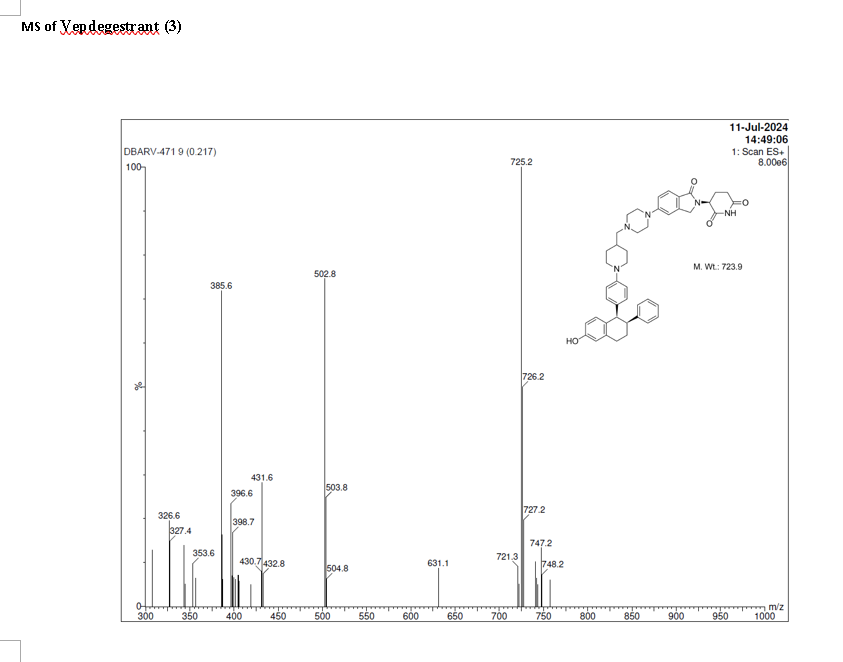
PAT
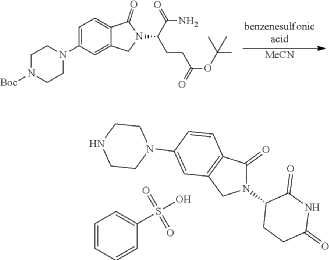
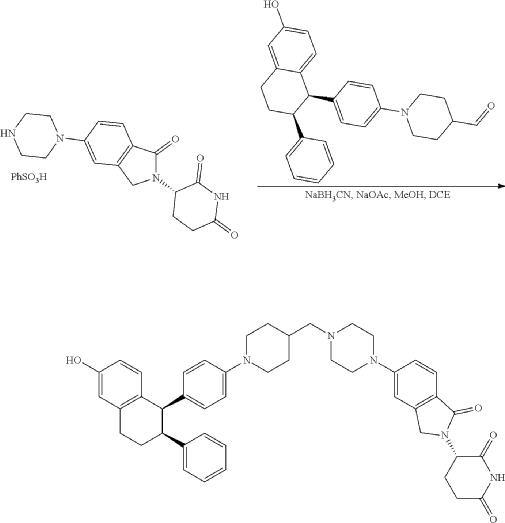
Step 11: Preparation of 3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (Compound (I-b))
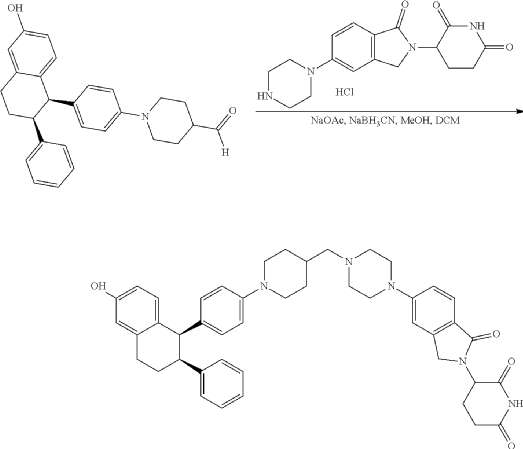
To a solution of 3-(1-oxo-5-piperazin-1-yl-isoindolin-2-yl)piperidine-2,6-dione hydrochloride (319 mg, 0.87 mmol, prepared in Step 17 described for Exemplary Compound 62) in methanol (4 mL) and dichloromethane (4 mL) was added sodium acetate (120 mg, 1.46 mmol, 2 eq). The mixture was stirred at 20° C. for 0.5 h, then to the mixture was added 1-[4-[(1R,2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]piperidine-4-carbaldehyde (300 mg, 0.73 mmol, 1 eq) and sodium cyanoborohydride (137 mg, 2.19 mmol, 3 eq). The mixture was stirred at 20° C. for 12 h. LC-MS showed the starting material was consumed completely and one main peak with desired MW was detected. The reaction mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (Phenomenex luna C 18 column, 250×50 mm, 10 um; mobile phase: [water (0.05% HCl)-acetonitrile]; B %: acetonitrile 10%-40% in 30 min). The desired compound 3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (288.4 mg, 0.37 mmol, 51% yield) was obtained as a white solid of hydrochloride salt. LC-MS (ESI) m/z: 724.4 [M+1] +; 1H NMR (400 MHz, DMSO-d 6) δ 10.97 (s, 1H), 10.83 (s, 0.9H, HCl), 7.60 (d, J=8.5 Hz, 1H), 7.40 (br s, 2H), 7.22-7.11 (m, 5H), 6.83 (d, J=6.0 Hz, 2H), 6.69-6.63 (m, 2H), 6.58-6.47 (m, 3H), 5.07 (dd, J=5.2, 13.2 Hz, 1H), 4.41-4.30 (m, 2H), 4.28-4.21 (m, 1H), 4.00 (d, J=12.7 Hz, 2H), 3.61 (d, J=11.0 Hz, 2H), 3.54-3.36 (m, 6H), 3.16 (br s, 4H), 3.06-2.84 (m, 3H), 2.76-2.53 (m, 1H), 2.43-2.33 (m, 1H), 2.27 (br s, 1H), 2.16-2.04 (m, 3H), 2.02-1.69 (m, 5H).
Synthesis of (3S)-3-[5-[4-[[1-[4-[(1R, 2S)-6-hydroxy-2-phenyl-tetralin-1-yl]phenyl]-4-piperidyl]methyl]piperazin-1-yl]-1-oxo-isoindolin-2-yl]piperidine-2,6-dione (Compound (I-c))
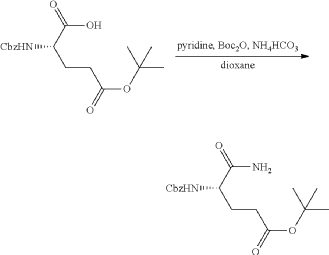
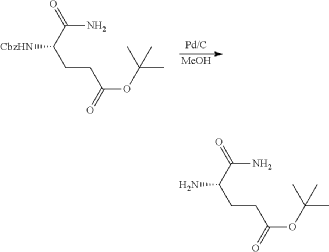
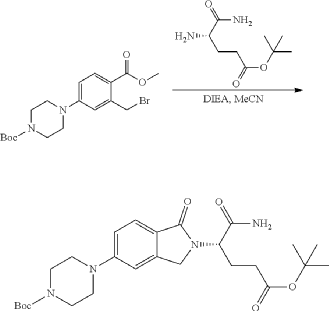
PAT
- Tetralin and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: CN-118834201-APriority Date: 2016-12-01
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: EP-3689868-B1Priority Date: 2016-12-01Grant Date: 2023-09-27
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-10647698-B2Priority Date: 2016-12-01Grant Date: 2020-05-12
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-2025320195-A1Priority Date: 2016-12-01
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-10899742-B1Priority Date: 2016-12-01Grant Date: 2021-01-26
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-11104666-B2Priority Date: 2016-12-01Grant Date: 2021-08-31
- Tetrahydronaphthalene and tetrahydroisoquinoline derivatives as estrogen receptor degradersPublication Number: US-12172981-B2Priority Date: 2016-12-01Grant Date: 2024-12-24
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- Iwata, H.; Naito, Y.; Hattori, M.; Yoshimura, A.; Yonemori, K.; Aizawa, M.; et al. (November 2023). “58P Safety and pharmacokinetics (PK) of vepdegestrant in Japanese patients with estrogen receptor (ER)+/human epidermal growth factor receptor 2 (HER2)- advanced breast cancer: Results from a Japanese phase I study”. Annals of Oncology. 34: S1488–S1489. doi:10.1016/j.annonc.2023.10.193. S2CID 265657144.
- Iwata, H.; Hamilton, E.P.; Ma, C.X.; De Laurentiis, M.; Hurvitz, S.A.; Wander, S.A.; et al. (November 2023). “73TiP Global phase III studies evaluating vepdegestrant in estrogen receptor (ER)+/human epidermal growth factor receptor 2 (HER2)- advanced breast cancer: VERITAC-2 and VERITAC-3”. Annals of Oncology. 34: S1493. doi:10.1016/j.annonc.2023.10.207. S2CID 265654990.
- “Arvinas, Pfizer reworking partnership on ‘Protac’ cancer drug | BioPharma Dive”. http://www.biopharmadive.com. Retrieved 17 September 2025.
- “Estrogen Receptor”. Arvinas. Retrieved 17 September 2025.
- Sakamoto, Kathryn M.; Kim, Kwon B.; Kumagai, Ayumu; Mercurio, Frank; Crews, Craig M.; Deshaies, Raymond J. (18 January 2022). “PROTAC targeted protein degraders: the past is prologue”. Nature Reviews Drug Discovery. 21 (3): 181–200. doi:10.1038/s41573-021-00371-6. PMC 8765495. PMID 35046570.
- “Vepdegestrant (ARV-471) PROTAC ER Degrader”. MedChemExpress. Retrieved 17 September 2025.
- Hamilton, Erika P.; Ma, Cynthia; De Laurentiis, Michelino; Iwata, Hiroji; Hurvitz, Sara A.; Wander, Seth A.; et al. (2024). “VERITAC-2: a Phase III study of vepdegestrant, a PROTAC ER degrader, versus fulvestrant in ER+/HER2- advanced breast cancer”. Future Oncology (London, England). 20 (32): 2447–2455. doi:10.1080/14796694.2024.2377530. ISSN 1744-8301. PMC 11524203. PMID 39072356.
- “A Study to Compare the Efficacy and Safety of Vepdegestrant (ARV-471) Versus Fulvestrant in Participants With Estrogen Receptor-positive, HER2-negative Advanced Breast Cancer (VERITAC-2)”. ClinicalTrials.gov. 30 June 2025. Retrieved 17 September 2025.
- “Arvinas and Pfizer Announce Positive Topline Results from Phase 3 VERITAC-2 Clinical Trial”. Arvinas. Retrieved 17 September 2025.
- “VERITAC-2 Trial Shows Vepdegestrant Significantly Improves Survival in ESR1-Mutant Breast Cancer”. Applied Clinical Trials Online. 24 March 2025. Retrieved 17 September 2025.
- “Arvinas Announces Results from the VERITAC-2 Trial Selected as Late-Breaking Oral Presentation at the 2025 ASCO Annual Meeting”. Arvinas. 23 April 2025. Retrieved 17 September 2025.
- Gough, Sheryl M.; Flanagan, John J.; Teh, Jimmy (15 August 2024). “Oral Estrogen Receptor PROTAC Vepdegestrant (ARV-471) Is Highly Efficacious as Monotherapy and in Combination with CDK4/6 or PI3K/mTOR Pathway Inhibitors in Preclinical ER+ Breast Cancer Models”. Clinical Cancer Research. 30 (16): 3549–3562. doi:10.1158/1078-0432.CCR-23-3465. PMC 11325148. PMID 38819400.
- “FDA Grants Fast Track Status to Vepdegestrant for ER+/HER2– Metastatic Breast Cancer”. Oncology Live. 6 February 2024. Retrieved 17 September 2025.
- “Vepdegestrant Gains FDA Fast Track Designation in ER+/HER2- Breast Cancer”. Targeted Oncology. 6 February 2024. Retrieved 17 September 2025.
- “Arvinas Announces Submission of New Drug Application to U.S. FDA for Vepdegestrant for Patients with ESR1-Mutated ER+/HER2- Advanced or Metastatic Breast Cancer” (Press release). Arvinas. 24 June 2025. Retrieved 17 September 2025.
External links
| Clinical data | |
|---|---|
| Pronunciation | /ˌvɛpdəˈdʒɛstrənt/ VEP-də-JES-trənt |
| Other names | ARV-471 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2229711-68-4 |
| PubChem CID | 134562533 |
| ChemSpider | 114935295 |
| UNII | WC1U3R1YMI |
| ChEMBL | ChEMBL5095210 |
| Chemical and physical data | |
| Formula | C45H49N5O4 |
| Molar mass | 723.918 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
- Targeting the Estrogen Receptor for the Treatment of Breast Cancer: Recent Advances and ChallengesPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-06-28PMID: 37377342DOI: 10.1021/acs.jmedchem.3c00136
- Emerging targeted protein degradation tools for innovative drug discovery: From classical PROTACs to the novel and beyondPublication Name: European Journal of Medicinal ChemistryPublication Date: 2022-03-05PMID: 35092900DOI: 10.1016/j.ejmech.2022.114142
- Structural and Physicochemical Features of Oral PROTACsPublication Name: Journal of Medicinal ChemistryPublication Date: 2024-07-30PMID: 39078401DOI: 10.1021/acs.jmedchem.4c01017
- Discovery of the cereblon-recruiting tubulin PROTACs effective in overcoming Taxol resistance in vitro and in vivoPublication Name: European Journal of Medicinal ChemistryPublication Date: 2024-02-05PMID: 38171146DOI: 10.1016/j.ejmech.2023.116067
- Current advances and development strategies of orally bioavailable PROTACsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2023-12-05PMID: 37708797DOI: 10.1016/j.ejmech.2023.115793
- Discovery of ERD-3111 as a Potent and Orally Efficacious Estrogen Receptor PROTAC Degrader with Strong Antitumor ActivityPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-08-30PMID: 37647546DOI: 10.1021/acs.jmedchem.3c01186
- Expanding Chemical Probe Space: Quality Criteria for Covalent and Degrader ProbesPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-07-05PMCID: PMC10388296PMID: 37403870DOI: 10.1021/acs.jmedchem.3c00550
////////////vepdegestrant, anax lab, approvals 2026, fda 2026, Veppanu, FDA 2026, APROVALS 2026, Veppanu, ARV 471, WC1U3R1YMI, PF 07850327
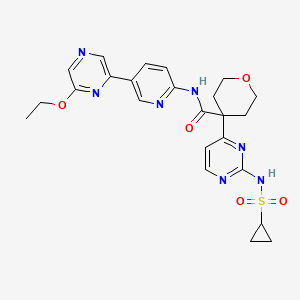
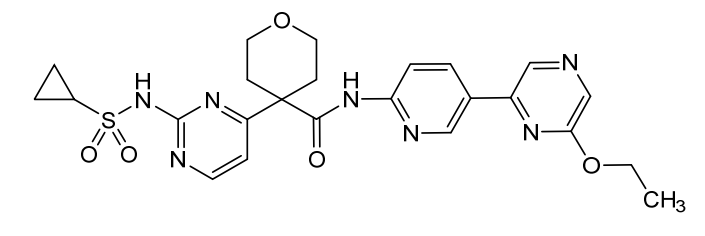
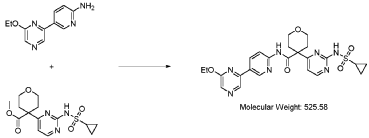
Dencatistat
Dencatistat
CAS 2377000-84-3
MFC24H27N7O5S MW 525.6 g/mol
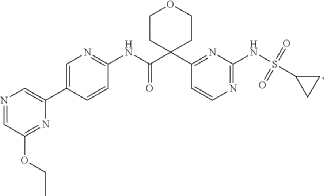
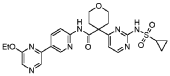
4-[2-(cyclopropylsulfonylamino)pyrimidin-4-yl]-N-[5-(6-ethoxypyrazin-2-yl)-2-pyridinyl]oxane-4-carboxamide
4-[2-(cyclopropanesulfonamido)pyrimidin-4-yl]-N-[5-(6-ethoxypyrazin-2-yl)pyridin-2-yl]oxane-4-carboxamide
CTP synthase 1 inhibitor, antineoplastic, STP 938, CTPS1-IN-2, QG9C9SZZ3T
Dencatistat (formerly known as STP938) is a first-in-class, orally bioavailable cancer drug designed to target specific blood cancers and solid tumours
Dencatistat is an orally bioavailable, small molecule inhibitor of cytidine triphosphate synthase 1 (CTPS1), with potential antineoplastic activity. Upon oral administration, dencatistat targets, binds to and inhibits the activity of CTPS1, thereby decreasing the production of cytidine triphosphate (CTP), an essential building block of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). This may disrupt DNA and RNA synthesis and trigger apoptosis. CTPS1, an enzyme that catalyzes the rate-limiting step in pyrimidine synthesis, plays an important and nonredundant role in B-cell and T-cell proliferation. CTPS1 is required for rapid cell division in certain types of cancers that arise from blood cells.
Mechanism of Action
It works by inhibiting CTPS1 (Cytidine Triphosphate Synthase 1), a key enzyme that cancer cells “addicted” to for DNA synthesis.
- Targeted approach: It aims to kill cancer cells while leaving healthy cells unharmed by exploiting a “synthetic lethal” dependency in certain tumours.
- Precision medicine: It is particularly being tested in patients whose tumours lack CTPS2, a backup enzyme, which makes them highly vulnerable to dencatistat.
🏥 Clinical Status
Developed by Step Pharma, the drug is currently in several clinical trials:
- Lymphoma: Phase 1/2 trials for relapsed or refractory T-cell and B-cell lymphomas.
- Solid Tumours: Phase 1 studies for patients with solid tumours, specifically ovarian and endometrial cancers.
- Essential Thrombocythaemia: A Phase 1b trial for this blood disorder was initiated in 2025.
- Orphan Drug Status: Received FDA Orphan Drug Designation for T-cell lymphoma in May 2025.
- OriginatorStep Pharma
- ClassAnti-inflammatories; Antineoplastics; Antirheumatics; Antithrombotics; Small molecules
- Mechanism of ActionCTPS1 protein inhibitors
- Orphan Drug StatusYes – T-cell lymphoma
- Phase I/IIB-cell lymphoma; T-cell lymphoma
- Phase ISolid tumours; Thrombocytosis
- PreclinicalGraft-versus-host disease; Inflammation
- No development reportedRheumatoid arthritis
- 23 Feb 2026Step Pharma plans phase II trials for Gynaecological cancer
- 10 Feb 2026Preclinical development in Inflammation is till ongoing in France (PO) (Step Pharma pipeline, February 2026)
- 15 Oct 2025Adverse event data from a phase I/II trial in T-cell lymphoma/B-cell lymphoma released by Step Pharma
SYN
US20250177394, Compound CTPS1-IA
PAT
PAT
A. Preparation of Active Ingredient
20 Example A1 – Preparation of crude 4-(2-(cyclopropanesulfonamido)pyrimidin-4-yl)-N-(5- (6-ethoxypyrazin-2-yl)pyridin-2-yl)tetrahydro-2H-pyran-4-carboxamide

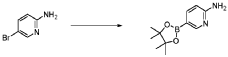
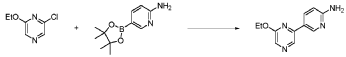
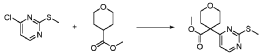
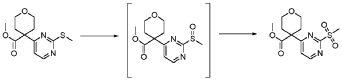
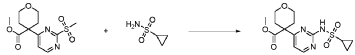
Step 4 – Preparation of crude 4-(2-(cyclopropanesulfonamido)pyrimidin-4-yl)-N-(5-(6- ethoxypyrazin-2-yl)pyridin-2-yl)tetrahydro-2H-pyran-4-carboxamide
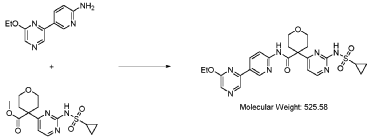
4-(2-(cyclopropanesulfonamido)pyrimidin-4-yl)tetrahydro-2H-pyran-4-carboxylate (1.76 kg, 5.15 mol, 1.00 equiv.) and 5-(6-ethoxypyrazin-2-yl)pyridin-2-amine (1.22 kg, 5.65 mol, 1.10 equiv.) were suspended in a mixture of THF (27.1 L, 15.5 rel. vol.) and DMSO (2.63 L, 1.50 rel. vol.) and stirred until the solids were evenly dispersed. The mixture was concentrated by
STP-P3718PCT
102
distillation at atmospheric pressure and approximately 70 oC to a volume of 15 L. The temperature was adjusted to 20 ± 5 oC, potassium tert-butoxide (6.92 kg 20 wt% solution in THF, 12.3 mol, 2.40 equiv.) was added over 1 h and the reaction mixture stirred at 20 ± 5 oC for 70 minutes until completion. THF (880 mL, 0.500 rel vol.) was charged, followed by acetic acid (780 5 mL, 820 g, 13.6 mol, 2.64 equiv.) over 10 minutes, followed by methanol (4.40 L, 2.50 rel. vol.), followed by water (13.2 L, 7.50 rel. vol.) over 35 minutes. The mixture was stirred at 20 ± 5 oC for 15 minutes and then 16 h at 0 ± 5 oC. The resulting suspension was filtered and washed with water (2 × 8.80 L, 2 × 5.00 rel. vol.), followed by methanol (4.40 L, 2.50 rel. vol.) The filter cake was dried at 35 oC under a flow of nitrogen for 20 h to afford crude 4-(2-10 (cyclopropanesulfonamido)pyrimidin-4-yl)-N-(5-(6-ethoxypyrazin-2-yl)pyridin-2-yl)tetrahydro- 2H-pyran-4-carboxamide (“CTPS1-IA”).
PAT
- Aminopyrimidine derivatives as CTPS1 inhibitorsPublication Number: JP-7428692-B2Priority Date: 2018-03-23Grant Date: 2024-02-06
- Aminopyrimidine derivatives as ctps1 inhibitorsPublication Number: EP-3768674-A1Priority Date: 2018-03-23
- Aminopyrimidine derivative as a CTPS1 inhibitorPublication Number: JP-2021518436-APriority Date: 2018-03-23
- Aminopyrimidine derivatives as ctps1 inhibitorsPublication Number: EP-3768674-B1Priority Date: 2018-03-23Grant Date: 2024-01-03
- Aminopyrimidine derivatives as CTPS1 inhibitorsPublication Number: CN-111868051-APriority Date: 2018-03-23
- Aminopyrimidine derivatives as CTPS1 inhibitorsPublication Number: ES-2974445-T3Priority Date: 2018-03-23Grant Date: 2024-06-27
- CompoundsPublication Number: US-2021024507-A1Priority Date: 2018-03-23
- CompoundsPublication Number: US-2021387965-A1Priority Date: 2018-10-23
- CompoundsPublication Number: US-2023192673-A1Priority Date: 2018-06-04
- Aminopyrimidine derivatives as CTPS1 inhibitorsPublication Number: CN-111868051-BPriority Date: 2018-03-23Grant Date: 2024-04-09
- Aminopyrimidine derivatives as ctps1 inhibitorsPublication Number: WO-2019180244-A1Priority Date: 2018-03-23
- Aminopyrimidine derivatives as ctps1 inhibitorsPublication Number: WO-2019179652-A1Priority Date: 2018-03-23
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
////////dencatistat, anax lab, CTP synthase 1 inhibitor, antineoplastic, STP 938, CTPS1-IN-2, QG9C9SZZ3T
Delocamten
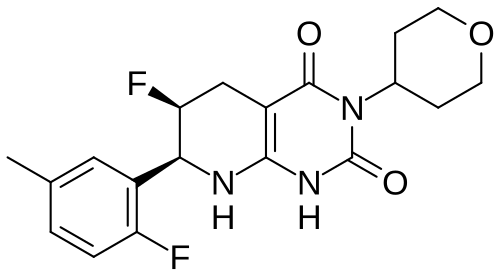
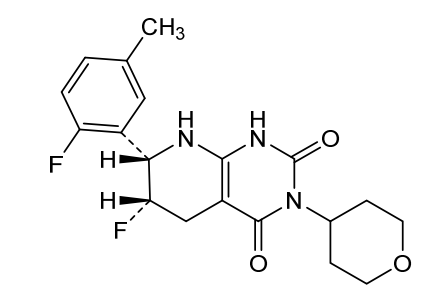
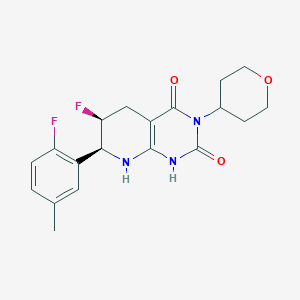
Delocamten
CAS 2417411-02-8
MFC19H21F2N3O3 MW377.4 g/mol
(6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(oxan-4-yl)-5,6,7,8-tetrahydro-1H-pyrido[2,3-d]pyrimidine-2,4-dione
(6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(oxan-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione
cardiac myosin inhibitor, MYK-224; BMS-986435, MYK 224, BMS 986435, IE5886BN8T
Delocamten (development code MYK-224) is a small-molecule cardiac myosin inhibitor developed by Bristol Myers Squibb for hypertrophic cardiomyopathy.[1][2][3]
Delocamten is a small molecule drug. Delocamten is under investigation in clinical trial NCT06122779 (Study to Evaluate Safety, Tolerability and Drug Levels of BMS-986435/MYK-224 in Participants With Heart Failure With Preserved Ejection Fraction (HFpEF)). Delocamten has a monoisotopic molecular weight of 377.16 Da.
SYN
Example 1-3: Preparation of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2, 4 (1H, 3H)-dione (3)
Step 7. Synthesis of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2, 4 (1H, 3H)-dione (3). A mixture of crude 3G (1.0 g, 2.53 mmol) in CH 3CN (15 mL) was put into a microwave reactor with stirring at 120° C. for 30 min. Subsequently, the mixture was concentrated and the residue was purified by preparative HPLC (column: C18 silica gel; mobile phase: CH3CN:H 2O=20:80 (v v) increasing to CH3CN:H 2O=80:20 (v v) within 40 min; detector: UV 254 nm) to give compound 3 (302 mg, 32%), as a white solid, which was identified as Form 1 polymorph (see Example 2). LC-MS (ES, m/z): 378 [M+H] +; 1H NMR (300 MHz, d-DMSO): δ 10.20 (s, 1H), 7.38-7.05 (m, 3H), 6.45 (s, 1H), 5.11-4.81 (m, 3H), 3.89 (dd, J=10.8, 3.9 Hz, 2H), 3.34-3.27 (m, 3H), 2.76-2.48 (m, 4H), 2.28 (s, 3H), 1.39-1.36 (m, 2H); 19F NMR (376 MHz, d 6-DMSO): δ −123.51 (t, J=86.5 Hz), −191.57 (d, J=129.34 Hz).
PAT
Example 1-3: Preparation of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3- (tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2, 4 (1H, 3H)-dione (3).
Scheme 3
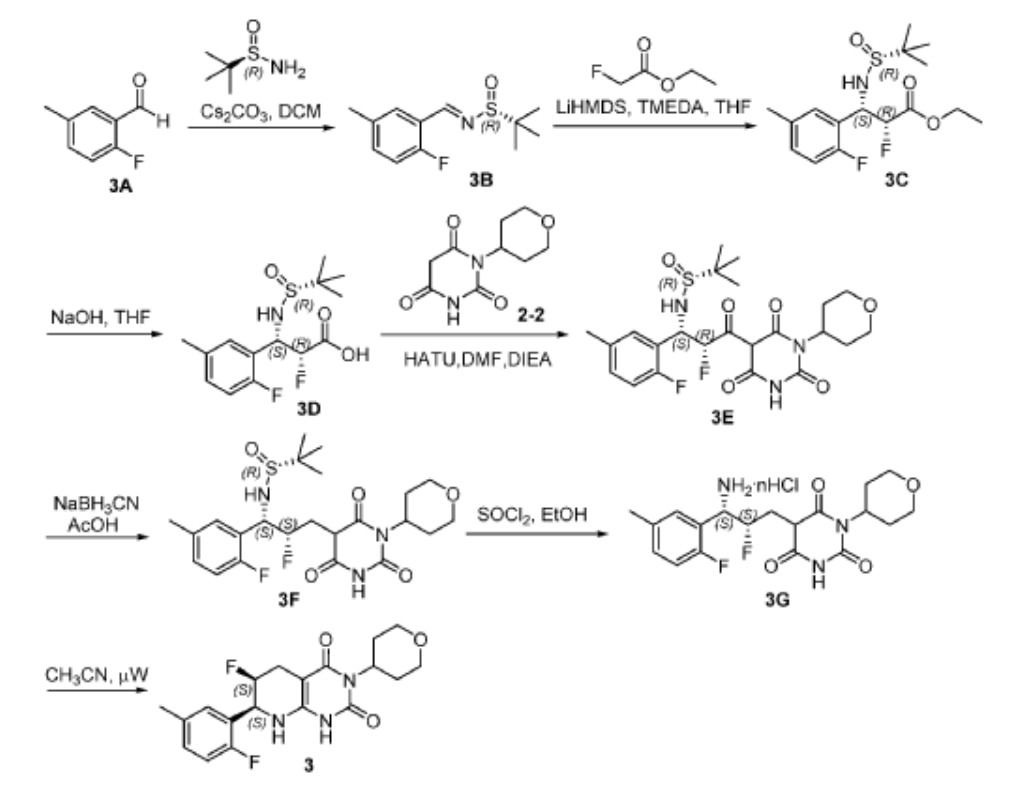
[0165] Step 1. Synthesis of (R,E)-N-(2-fluoro-5-methylbenzylidene)-2-methylpropane-2-sulfinamide (3B). The reaction mixture was filtered and the filtrate was diluted with ether (150 mL). Subsequently, the resulting suspension was filtered. The filtrate was concentrated and the residue was dried in vacuo to give 3B (8.7 g, 97%) as a yellow oil. LC-MS (ES, m/z): 242 [M+H] + ; 1 H NMR (400 MHz, d 6 -DMSO): d 8.87 (s, 1H), 7.76 (m, 1H), 7.29 (m, 1H), 7.03 (m, 1H), 2.37 (d, J = 1.0 Hz, 3H), 1.27 (s, 9H).
[0166] Step 2. Synthesis of ethyl (2R,3S)-3-(((R)-tert-butylsulfinyl)amino)-2-fluoro-3-(2-fluoro-5-methylphenyl)propanoate (3C). To a solution of 3B (4 g, 16.6 mmol), ethyl 2- fluoroacetate (2.6 g, 24.6 mmol), and TMEDA (4.8 mL) in anhydrous THF (40 mL) was added LiHMDS (1 M in THF, 24.6 mL, 24.6 mmol) dropwise at -78 o C over 30 min under an atmosphere of Ar. After stirring at -78 o C for 1 h, the reaction was quenched by adding 1 N aq.
HCl (50 mL), while maintaining the inner temperature of the mixture at < -20 o C. Subsequently, the mixture was concentrated to remove most of the organic solvent and then extracted with EtOAc (100 mL x 3). The combined organic extracts were washed with brine (100 mL) and dried over anhydrous Na2SO4. The solvent was removed and the residue was dried in vacuo to give crude 3C (6.0 g) as a yellow oil, which was used for the next step without further purification. LC-MS (ES, m/z): 348 [M+H] + .
[0167] Step 3. Synthesis of (2R,3S)-3-(((R)-tert-butylsulfinyl)amino)-2-fluoro-3-(2-fluoro-5-methylphenyl)propanoic acid (3D). To a solution of 3C (6.0 g, 17.3 mmol) in THF (40 mL) was added 1N aq. NaOH (34.6 mL, 34.6 mmol) at rt. After stirring at rt for 1 h, the reaction mixture was added ice water (50 mL). The resulting mixture was extracted with EtOAc (100 mL x 2). The aqueous layer was adjusted to pH 5 with sat. aq. citric acid, followed by extraction with EtOAc (100 mL x 3). Subsequently, the combined organic extracts were washed with brine (100 mL) and dried over anhydrous Na 2 SO 4 . The solvent was removed and the residue was purified by preparative HPLC (Column: LC-MS (ES, m/z): 320 [M+H] + ; 1 H NMR (400 MHz, d 6 -DMSO): d 13.57 (br, 1H), 7.55 (dd, J = 7.5, 2.2 Hz, 1H), 7.23– 6.94 (m, 2H), 6.04 (d, J = 10.8 Hz, 1H), 5.37– 4.86 (m, 2H), 2.29 (s, 3H), 1.12 (s, 9H).
[0168] Step 4. Synthesis of (R)-N-((1S,2R)-2-fluoro-1-(2-fluoro-5-methylphenyl)-3-oxo-3- (2,4,6-trioxo-1-(tetrahydro-2H-pyran-4-yl)hexahydropyrimidin-5-yl)propyl)-2-methylpropane-2-sulfinamide (3E). A solution of 3D (700 mg, 2.19 mmol), 2-2 (698 mg, 3.29 mmol), and HATU (1.25 g, 3.29 mmol) in DMF (10 mL) was added DIEA (849 mg, 6.57 mmol) at 0 o C under an atmosphere of Ar. aq. sodium bicarbonate (30 mL) and the resulting solution was extracted with ethyl acetate (50 mL x3). The combined organic extracts were washed with brine (50 mL x 2) and dried over anhydrous Na 2 SO 4 . The solvent was removed and the residue was dried in vacuo to give crude 3E (1.3 g) as a white solid, which was used for the next step without further purification. LC-MS (ES, m/z): 514 [M+H] + ; 1 H NMR (400 MHz, d 6 -DMSO): d 12.16 (br, 1H), 7.66– 7.45 (m, 1H), 7.23– 6.98 (m, 2H), 6.37 (m, 1H), 6.13 (d, J = 10.7 Hz, 1H), 5.22 (m, 1H), 4.79 (m, 1H), 3.94 (m, 2H), 3.35 (t, J = 11.7 Hz, 2H), 2.52– 2.39 (m, 2H), 2.29 (s, 3H), 1.49 (d, J = 12.2 Hz, 2H), 1.04 (s, 9H).
[0169] Step 5. Synthesis of (R)-N-((1S,2S)-2-fluoro-1-(2-fluoro-5-methylphenyl)-3-(2,4,6- trioxo-1-(tetrahydro-2H-pyran-4-yl)hexahydropyrimidin-5-yl)propyl)-2-methylpropane-2-sulfinamide (3F). A solution of crude 3E (1.3 g, 2.53 mmol) in AcOH (10 mL) was added NaBH3CN (398 mg, 6.33 mmol) at 0 o C under an atmosphere of Ar. After stirring at rt for 1 h, the reaction mixture was added ice water (20 mL) and the resulting solution was extracted with EtOAc (50 mL x 3). Next, the combined organic extracts were washed with brine (50 mL) and
dried over anhydrous Na2SO4. The solvent was removed and the residue was dried in vacuo to give crude 3F (1.3 g) as a white solid, which was used for the next step without further purification. LC-MS (ES, m/z): 500 [M+H] + ; 1 H NMR (400 MHz, d 6 -DMSO): d 11.31 (d, J = 28.1 Hz, 1H), 7.41 (d, J = 7.4 Hz, 1H), 7.27– 6.84 (m, 2H), 6.11– 5.78 (m, 2H), 5.08– 4.43 (m, 3H), 3.87 (m, 3H), 2.29 (s, 6H), 1.99 (s, 1H), 1.53– 1.28 (m, 2H), 1.10 (d, J = 2.1 Hz, 10H).
[0170] Step 6. Synthesis of 5-((2S,3S)-3-amino-2-fluoro-3-(2-fluoro-5-methylphenyl)propyl)-1-(tetrahydro-2H-pyran-4-yl)pyrimidine-2, 4, 6 (1H, 3H, 5H)-trione (3G). A solution of crude 3F (1.3 g, 2.60 mmol) in ethanol (10 mL) was added thionyl chloride (334 mg) at 0 o C. After stirring at rt for 1 h, the reaction mixture was concentrated and the residue was dried in vacuo to give crude 3G (1.0 g) as a white solid, which was used for the next step without further purification. LC-MS (ES, m/z): 396 [M+H] + .
[0171] Step 7. Synthesis of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2, 4 (1H, 3H)-dione (3). A mixture of crude 3G (1.0 g, 2.53 mmol) in CH 3 CN (15 mL) was put into a microwave reactor with stirring at 120 o C for 30 min. Subsequently, the mixture was concentrated and the residue was purified by preparative HPLC (column: C18 silica gel; mobile phase: CH3CN:H2O = 20:80 (v/v) increasing to CH3CN:H2O = 80:20 (v/v) within 40 min; detector: UV 254 nm) to give compound 3 (302 mg, 32%), as a white solid, which was identified as Form 1 polymorph (see Example 2). LC-MS (ES, m/z): 378 [M+H] + ; 1 H NMR (300 MHz, d 6 -DMSO): d 10.20 (s, 1H), 7.38– 7.05 (m, 3H), 6.45 (s,1H), 5.11– 4.81 (m, 3H), 3.89 (dd, J = 10.8, 3.9 Hz, 2H), 3.34– 3.27 (m, 3H), 2.76–2.48 (m, 4H), 2.28 (s, 3H), 1.39–1.36 (m, 2H); 19 F NMR (376 MHz, d 6 -DMSO): d -123.51 (t, J = 86.5 Hz), -191.57 (d, J = 129.34 Hz).
PAT
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: EP-4464321-A2Priority Date: 2018-10-29
- Tetrahydropyran-substituted bicyclic pyrimidinedione compounds (THP)Publication Number: ES-2986923-T3Priority Date: 2018-10-29Grant Date: 2024-11-13
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-2024025894-A1Priority Date: 2018-10-29
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-12344607-B2Priority Date: 2018-10-29Grant Date: 2025-07-01
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-2025282779-A1Priority Date: 2018-10-29
- Tetrahydropyran (THP) Substituted Bicyclic Pyrimidinedione CompoundsPublication Number: CN-113056465-APriority Date: 2018-10-29
- Tetrahydropyrane (THP) -substituted bicyclic pyrimidinedione compoundsPublication Number: CN-119977963-APriority Date: 2018-10-29
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: EP-3873904-B1Priority Date: 2018-10-29Grant Date: 2024-07-10
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-11034693-B2Priority Date: 2018-10-29Grant Date: 2021-06-15
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-2022106314-A1Priority Date: 2018-10-29
- Tetrahydropyran (THP)-Substituted Bicyclic-Pyrimidinedione CompoundsPublication Number: JP-2024063091-APriority Date: 2018-10-29
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: TW-202426449-APriority Date: 2018-10-29
- Tetrahydropyrane (THP) -substituted bicyclic pyrimidinedione compoundsPublication Number: CN-113056465-BPriority Date: 2018-10-29Grant Date: 2025-01-28
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: US-2020165247-A1Priority Date: 2018-10-29
- BICYCLIC PYRIMIDINODIONA COMPOUNDS REPLACED WITH TETRAHYDROPYRAN, POLYMORPHIC FORM OF THE SAME AND THE USE OF THE SAME FOR THE TREATMENT OF HCMPublication Number: AR-116880-A1Priority Date: 2018-10-29
- Substituted 5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4-diones for treating cardiac diseasesPublication Number: US-2022340569-A1Priority Date: 2018-10-29
- Tetrahydropyran (thp)-substituted bicyclic-pyrimidinedione compoundsPublication Number: EP-3873904-A1Priority Date: 2018-10-29
- CRYSTALLINE FORMS OF (6S,7S)-6-FLUORO-7-(2-FLUORO-5-METHYLPHENYL)- 3-(TETRAHYDRO-2H-PYRAN-4-YL)-5,6,7,8-TETRAHYDROPYRIDO[2,3- d]PYRIMIDINE-2,4(1H,3H)-DIONEPublication Number: WO-2024026058-A8Priority Date: 2022-07-29
- Crystalline forms of (6s,7s)-6-fluoro-7-(2-fluoro-5-methylphenyl)- 3-(tetrahydro-2h-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3- d]pyrimidine-2,4(1h,3h)-dionePublication Number: EP-4561697-A1Priority Date: 2022-07-29
- Methods of Administering Myosin InhibitorsPublication Number: US-2023338378-A1Priority Date: 2022-04-26
- Methods of treatment with myosin modulatorPublication Number: US-2023158027-A1Priority Date: 2019-11-10
- tetrahydropyran-substituted bicyclic pyrimidinedione compounds (thp)Publication Number: BR-112021008077-A2Priority Date: 2018-10-29
- CRYSTALLINE FORMS OF (6S,7S)-6-FLUORO-7-(2-FLUORO-5-METHYLPHENYL)-3-(TETRAHYDRO-2H-PYRAN-4-YL)-5,6,7,8-TETRAHYDROPYRIDO[2,3-d]PYRIMIDINE-2,4(1H,3H)-DIONEPublication Number: US-2025034129-A1Priority Date: 2023-07-28
- CRYSTALLINE FORMS OF (6S,7S)-6-FLUORO-7-(2-FLUORO-5-METHYLPHENYL)-3-(TETRAHYDRO-2H-PYRANO-4-IL)-5,6,7,8-TETRAHYDROPYRIDE[2 ,3-D]PYRIMIDINE-2,4(1H,3H)-DIONEPublication Number: AR-130058-A1Priority Date: 2022-07-29
- Crystalline forms of (6s,7s)-6-fluoro-7-(2-fluoro-5-methylphenyl)- 3-(tetrahydro-2h-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3- d]pyrimidine-2,4(lh,3h)-dionePublication Number: WO-2024026058-A1Priority Date: 2022-07-29
- Crystalline form of (6S,7S)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2H-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4(1H,3H)-dionePublication Number: CN-119855816-APriority Date: 2022-07-29
- Crystalline forms of (6s,7s)-6-fluoro-7-(2-fluoro-5-methylphenyl)-3-(tetrahydro-2h-pyran-4-yl)-5,6,7,8-tetrahydropyrido[2,3-d]pyrimidine-2,4(1h,3h)-dionePublication Number: TW-202412788-APriority Date: 2022-07-29
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- Lehman, Sarah J.; Crocini, Claudia; Leinwand, Leslie A. (June 2022). “Targeting the sarcomere in inherited cardiomyopathies”. Nature Reviews Cardiology. 19 (6): 353–363. doi:10.1038/s41569-022-00682-0. ISSN 1759-5010. PMC 9119933. PMID 35304599.
- Sebastian, Sneha Annie; Padda, Inderbir; Lehr, Eric J.; Johal, Gurpreet (September 2023). “Aficamten: A Breakthrough Therapy for Symptomatic Obstructive Hypertrophic Cardiomyopathy”. American Journal of Cardiovascular Drugs. 23 (5): 519–532. doi:10.1007/s40256-023-00599-0. PMID 37526885. S2CID 260348901.
- Packard, Elizabeth; de Feria, Alejandro; Peshin, Supriya; Reza, Nosheen; Owens, Anjali Tiku (December 2022). “Contemporary Therapies and Future Directions in the Management of Hypertrophic Cardiomyopathy”. Cardiology and Therapy. 11 (4): 491–507. doi:10.1007/s40119-022-00283-5. PMC 9652179. PMID 36243823.
////////delocamten, ANAX LAB, cardiac myosin inhibitor, MYK-224; BMS-986435, MYK 224, BMS 986435, IE5886BN8T