
Home » Uncategorized (Page 6)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
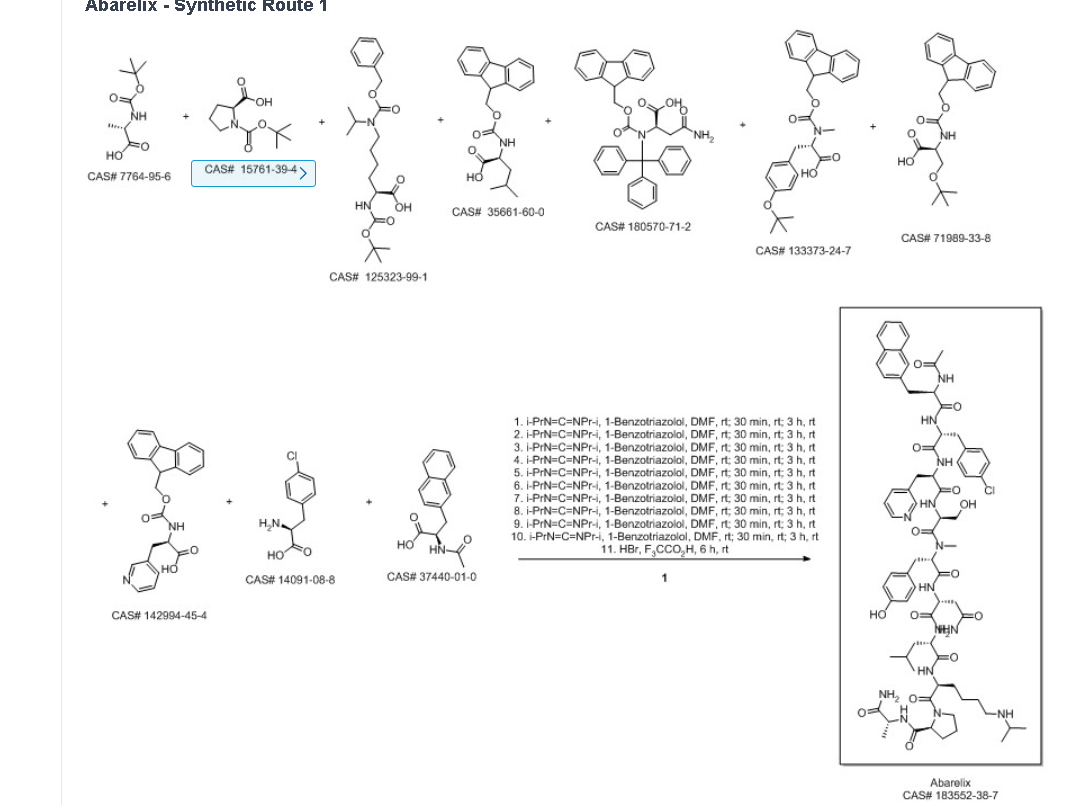
Abarelix
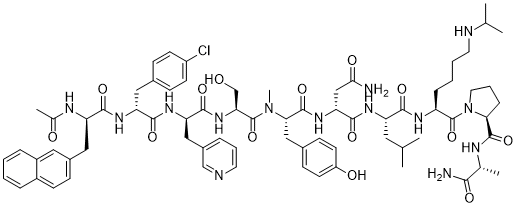
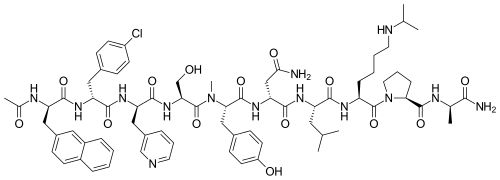
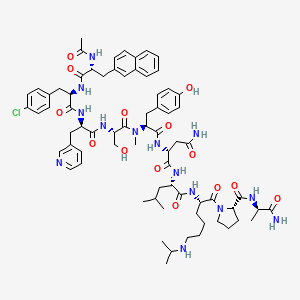
Abarelix
CAS 183552-38-7
785804-17-3 (acetate) 183552-38-7 (free base)
PPI149, PPI-149, PPI 149, R3827, R-3827, R 3827, Abarelix, Abarelix acetate, Plenaxis,
W486SJ5824
Chemical Formula: C72H95ClN14O14
Exact Mass: 1414.6841
Molecular Weight: 1416.06
Ac-D-Nal-[D-(pCl)Phe]-D-Pal-Ser-[Nalpha-Me-Tyr]-D-Asn-Leu-ILys-Pro-DAla-NH2

(2R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-acetamido-3-naphthalen-2-ylpropanoyl]amino]-3-(4-chlorophenyl)propanoyl]amino]-3-pyridin-3-ylpropanoyl]amino]-3-hydroxypropanoyl]-methylamino]-3-(4-hydroxyphenyl)propanoyl]amino]-N-[(2S)-1-[[(2S)-1-[(2S)-2-[[(2R)-1-amino-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxo-6-(propan-2-ylamino)hexan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]butanediamide
(2R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-acetamido-3-naphthalen-2-ylpropanoyl]amino]-3-(4-chlorophenyl)propanoyl]amino]-3-pyridin-3-ylpropanoyl]amino]-3-hydroxypropanoyl]-methylamino]-3-(4-hydroxyphenyl)propanoyl]amino]-N-[(2S)-1-[[(2S)-1-[(2S)-2-[[(2R)-1-amino-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxo-6-(propan-2-ylamino)hexan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]butanediamide
Abarelix is a synthetic decapeptide and antagonist of naturally occurring gonadotropin-releasing hormone (GnRH). Abarelix directly and competitively binds to and blocks the gonadotropin releasing hormone receptor in the anterior pituitary gland, thereby inhibiting the secretion and release of luteinizing hormone (LH) and follicle stimulating hormone (FSH). In males, the inhibition of LH secretion prevents the release of testosterone. As a result, this may relieve symptoms associated with prostate hypertrophy or prostate cancer, since testosterone is required to sustain prostate growth.
Abarelix, sold under the brand name Plenaxis, is an injectable gonadotropin-releasing hormone antagonist (GnRH antagonist) which is marketed in Germany and the Netherlands. It is primarily used in oncology to reduce the amount of testosterone made in patients with advanced symptomatic prostate cancer for which no other treatment options are available.[2][3]
It was originally marketed by Praecis Pharmaceuticals as Plenaxis,[2] and is now marketed by Speciality European Pharma in Germany[4] after receiving a marketing authorization in 2005. The drug was introduced in the United States in 2003, but was discontinued in this country in May 2005 due to poor sales and a higher-than-expected incidence of severe allergic reactions.[5] It remains marketed in Germany and the Netherlands however.[6]
Pat
https://patents.google.com/patent/CN107778354B/en
Example 1: synthesis of peptide resin 1
Dissolving 0.15mol of Fmoc-D-Ala and 0.15mol of HOBt by using a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Taking 0.05mol of MOBHA resin (the substitution value is about 0.6mmol/g), swelling with DMF for 25 minutes, washing and filtering, adding the activated solution, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3 times, wherein the washing time is 3min each time, obtaining Fmoc-D-Ala-MOBHA resin, namely the peptide resin 1, removing Fmoc protection with 20% PIP/DMF solution for 25 minutes before carrying out the next coupling reaction, washing and filtering to obtain the D-Ala-MOBHA resin.
Example 2: synthesis of peptide resin 1
Dissolving 0.15mol of Boc-D-Ala and 0.15mol of HOBt with a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Taking 0.05mol of MOBHA resin (the substitution value is about 0.6mmol/g), swelling with DMF for 25 minutes, washing and filtering, adding an activated Fmoc-D-Ala solution, stirring at room temperature for 3 hours, pumping out the reaction solution, washing 3 times with DMF, washing 3 times with DCM, wherein each washing time is 3min, obtaining Boc-D-Ala-MOBHA resin, namely peptide resin 1, deprotecting with 30% TFA/DCM solution for 30 minutes, neutralizing with DIEA/DCM solution, washing and filtering with DMF and DCM, and obtaining D-Ala-MOBHA resin.
Example 3: synthesis of Abarelix peptide resin
Dissolving 0.15mol of Fmoc-Pro and 0.15mol of HOBt in a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Adding the activated Fmoc-Pro solution into the peptide resin 1 obtained in example 1, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3 minutes each time, removing Fmoc protection with 20% PIP/DMF solution for 25 minutes, washing and filtering to obtain Pro-D-Ala-MOBHA resin.
Boc-Lys (iPr, Z), Fmoc-Leu, Fmoc-D-Asn (Trt), Fmoc-N-Me-Tyr (tBu), Fmoc-Ser (tBu), Fmoc-D-Pal, Fmoc-D-Cpa and Ac-D-Nal are sequentially added in the same method, and the Abarelix peptide resin, Ac-D-Nal-D-Cpa-D-Pal-Ser (tBu) -N-Me-Tyr (tBu) -D-Asn (Trt) -Leu-Lys (iPr, Z) -Pro-D-Ala-MOBHA resin are obtained by washing and filtering.
Example 4: synthesis of Abarelix peptide resin
Dissolving 0.15mol of Boc-Pro and 0.15mol of HOBt by using a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Adding the activated Boc-Pro solution into the peptide resin 1 obtained in example 1, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3min each time, deprotecting with 30% TFA/DCM solution for 30 minutes, neutralizing with DIEA/DCM solution, washing with DMF and DCM, and filtering to obtain Pro-D-Ala-MBHA resin.
Boc-Lys (iPr, Z), Fmoc-Leu, Fmoc-D-Asn (Trt), Fmoc-N-Me-Tyr (tBu), Fmoc-Ser (tBu), Fmoc-D-Pal, Fmoc-D-Cpa and Ac-D-Nal are sequentially added in the same method, and the Abarelix peptide resin, Ac-D-Nal-D-Cpa-D-Pal-Ser (tBu) -N-Me-Tyr (tBu) -D-Asn (Trt) -Leu-Lys (iPr, Z) -Pro-D-Ala-MOBHA resin are obtained by washing and filtering.
Example 5: preparation of crude Abarelix
Taking the abarelix peptide resin prepared in the example 3, adding 8% HBr/TFA solution (acidolysis solution 10mL/g abarelix resin), stirring and reacting for 6 hours, filtering and collecting filtrate, washing the resin with a small amount of TFA for 3 times, combining the filtrates, concentrating under reduced pressure, adding anhydrous ether for precipitation, washing the precipitate with anhydrous ether for 3 times, and draining to obtain white-like powder, namely a crude product of abarelix, wherein the purity of the crude product is 79.3%.
Example 6: preparation of crude Abarelix
Taking the abarelix peptide resin prepared in the example 4, adding 8% HBr/TFA solution (acidolysis solution 10mL/g abarelix resin), stirring and reacting for 6 hours, filtering and collecting filtrate, washing the resin with a small amount of TFA for 3 times, combining the filtrates, concentrating under reduced pressure, adding anhydrous ether for precipitation, washing the precipitate with anhydrous ether for 3 times, and draining to obtain white-like powder, namely a crude product of abarelix, wherein the purity of the crude product is 77.4%.
Example 7: purification and trans-salt conversion of crude Abarelix
Taking the crude Abarelix product obtained in the example 5, dissolving the Abarelix product in 20 percent acetic acid solution, filtering the solution by using a 0.45 mu m microporous membrane, and purifying for later use;
purifying by high performance liquid chromatography, wherein a chromatographic filler is 10 mu m reverse phase C18, a mobile phase system is 0.1% TFA/water solution-0.1% TFA/acetonitrile solution, a chromatographic column with the flow rate of 77mm x 250mm is 90mL/min, eluting by a gradient system, circularly sampling and purifying, sampling a crude product solution in the chromatographic column, starting the mobile phase for elution, collecting a main peak, and evaporating acetonitrile to obtain an abarelix purified intermediate concentrated solution;
taking the Abarelix purified intermediate concentrated solution, and filtering with a 0.45-micrometer filter membrane for later use;
performing salt exchange by high performance liquid chromatography, wherein the mobile phase system is 1% acetic acid/water solution-acetonitrile, the purification is performed by reversed phase C18 with chromatographic packing of 10 μm, the flow rate of a chromatographic column of 77mm × 250mm is 90mL/min, gradient elution and circular sample loading method are adopted, the sample is loaded in the chromatographic column, the mobile phase elution is started, the chromatogram is collected, the change of the absorbance is observed, the main peak of salt exchange is collected and the purity is detected by analyzing the liquid phase, the main peak solutions of salt exchange are combined, the concentration is performed under reduced pressure to obtain the aqueous solution of abarelix acetic acid, and freeze drying is performed to obtain 39.4g abarelix pure product
The total yield was 55.6%, molecular weight: 1417.2, purity: 99.6%, maximum single impurity of 0.13%, no toxic hydantoin degradation products were detected.
Example 8: purification and trans-salt conversion of crude Abarelix
Taking the crude Abarelix product obtained in the example 6, dissolving the Abarelix product by using a purification mobile phase A, and filtering the solution by using a 0.45 mu m microporous filter membrane to purify the Abarelix product for later use;
purifying by high performance liquid chromatography, wherein a chromatographic filler is 10 mu m reverse phase C18, a mobile phase system is 0.1% TFA/water solution-0.1% TFA/acetonitrile solution, a chromatographic column with the flow rate of 77mm x 250mm is 90mL/min, eluting by a gradient system, circularly sampling and purifying, sampling a crude product solution in the chromatographic column, starting the mobile phase for elution, collecting a main peak, and evaporating acetonitrile to obtain an abarelix purified intermediate concentrated solution;
taking the Abarelix purified intermediate concentrated solution, and filtering with a 0.45-micrometer filter membrane for later use;
performing salt exchange by adopting a high performance liquid chromatography, wherein a mobile phase system is 1% acetic acid/water solution-acetonitrile, a chromatographic filler for purification is reversed phase C18 with the diameter of 10 mu m, the flow rate of a chromatographic column with the diameter of 77mm × 250mm is 90mL/min, a gradient elution method and a circular sample loading method are adopted, loading the chromatographic column, starting the mobile phase elution, collecting a spectrum, observing the change of the absorbance, collecting a main salt exchange peak, detecting the purity by using an analysis liquid phase, combining main salt exchange peak solutions, concentrating under reduced pressure to obtain an abarelix acetic acid water solution, and performing freeze drying to obtain 41.7g of an abarelix pure product.
The total yield is 58.9%, molecular weight: 1417.0, purity: 99.5%, maximum single impurity 0.09%, no toxic hydantoin degradation products were detected.
SYN
Ma, Zhonggang; Guo, Dewen; Zeng, Dezhi; Wen, Yongjun. Method for synthesizing abarelix. Assignee Chengdu Shengnuo Biopharm Co., Ltd.. 2018.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
1: Tombal B. New treatment paradigm for prostate cancer: abarelix initiation therapy for immediate testosterone suppression followed by a luteinizing hormone-releasing hormone agonist. BJU Int. 2012 Mar;109(6):E16; author reply E16-7. doi: 10.1111/j.1464-410X.2012.10983.x. PubMed PMID: 22360806.
2: Garnick MB, Mottet N. New treatment paradigm for prostate cancer: abarelix initiation therapy for immediate testosterone suppression followed by a luteinizing hormone-releasing hormone agonist. BJU Int. 2012 Aug;110(4):499-504. doi: 10.1111/j.1464-410X.2011.10708.x. Epub 2011 Nov 16. PubMed PMID: 22093775.
3: Koechling W, Hjortkjaer R, Tankó LB. Degarelix, a novel GnRH antagonist, causes minimal histamine release compared with cetrorelix, abarelix and ganirelix in an ex vivo model of human skin samples. Br J Clin Pharmacol. 2010 Oct;70(4):580-7. doi: 10.1111/j.1365-2125.2010.03730.x. PubMed PMID: 20840449; PubMed Central PMCID: PMC2950992.
4: Retraction statement: Reconstitution of Plenaxis® (Abarelix) 100 mg for injection is more effective with a vortex-like mixer than when performed manually. J Pharm Pract. 2010 Feb;23(1):78. doi: 10.1177/0897190009360369. PubMed PMID: 21507797.
5: Kirby RS, Fitzpatrick JM, Clarke N. Abarelix and other gonadotrophin-releasing hormone antagonists in prostate cancer. BJU Int. 2009 Dec;104(11):1580-4. doi: 10.1111/j.1464-410X.2009.08924.x. Review. PubMed PMID: 20053189.
6: Debruyne F, Bhat G, Garnick MB. Abarelix for injectable suspension: first-in-class gonadotropin-releasing hormone antagonist for prostate cancer. Future Oncol. 2006 Dec;2(6):677-96. Review. PubMed PMID: 17155895.
7: Beer TM, Ryan C, Bhat G, Garnick M; Abarelix Study Group. Dose-escalated abarelix in androgen-independent prostate cancer: a phase I study. Anticancer Drugs. 2006 Oct;17(9):1075-9. PubMed PMID: 17001181.
8: Hogle WP. Abarelix (plenaxis). Clin J Oncol Nurs. 2004 Dec;8(6):663-5. PubMed PMID: 15637961.
9: Mongiat-Artus P, Teillac P. Abarelix: the first gonadotrophin-releasing hormone antagonist for the treatment of prostate cancer. Expert Opin Pharmacother. 2004 Oct;5(10):2171-9. Review. PubMed PMID: 15461552.
10: Wong SL, Lau DT, Baughman SA, Fotheringham N, Menchaca D, Garnick MB. Pharmacokinetics and pharmacodynamics of a novel depot formulation of abarelix, a gonadotropin-releasing hormone (GnRH) antagonist, in healthy men ages 50 to 75. J Clin Pharmacol. 2004 May;44(5):495-502. PubMed PMID: 15102870.
References
- “Abarelix”. PubChem. 2017-07-29.
- “Abarelix”. Drugs.com. Archived from the original on 2018-02-10. Retrieved 2018-01-23.
- Boccon-Gibod L, van der Meulen E, Persson BE (June 2011). “An update on the use of gonadotropin-releasing hormone antagonists in prostate cancer”. Therapeutic Advances in Urology. 3 (3): 127–40. doi:10.1177/1756287211414457. PMC 3159401. PMID 21904569.
- Pharmazeutische Zeitung online: Abarelix (in German)
- Minev B (13 January 2011). Cancer Management in Man: Chemotherapy, Biological Therapy, Hyperthermia and Supporting Measures. Springer Science & Business Media. pp. 182–. ISBN 978-90-481-9704-0.
- “Abarelix”. Drugs.com. Archived from the original on 2019-08-29. Retrieved 2018-08-27.
| Clinical data | |
|---|---|
| Trade names | Plenaxis |
| AHFS/Drugs.com | Monograph |
| Routes of administration | Intramuscular injection |
| Drug class | GnRH analogue; GnRH antagonist; Antigonadotropin |
| ATC code | L02BX01 (WHO) |
| Pharmacokinetic data | |
| Protein binding | 96–99% |
| Identifiers | |
| IUPAC name | |
| CAS Number | 183552-38-7 |
| PubChem CID | 16131215 |
| IUPHAR/BPS | 1188 |
| DrugBank | DB00106 |
| ChemSpider | 10482301 |
| UNII | W486SJ5824 |
| KEGG | D02738 |
| ChEBI | CHEBI:337298 |
| ChEMBL | ChEMBL1252 |
| CompTox Dashboard (EPA) | DTXSID20171443 |
| Chemical and physical data | |
| Formula | C72H95ClN14O14 |
| Molar mass | 1416.09 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////Abarelix, PPI149, PPI-149, PPI 149, R3827, R-3827, R 3827, Abarelix, Abarelix acetate, Plenaxis,
W486SJ5824
O=C(N[C@@H](CC(C)C)C(N[C@@H](CCCCNC(C)C)C(N1[C@H](C(N[C@H](C)C(N)=O)=O)CCC1)=O)=O)[C@H](NC([C@@H](N(C([C@@H](NC([C@H](NC([C@H](NC([C@H](NC(C)=O)CC2=CC=C3C=CC=CC3=C2)=O)CC4=CC=C(Cl)C=C4)=O)CC5=CC=CN=C5)=O)CO)=O)C)CC6=CC=C(O)C=C6)=O)CC(N)=O
Tagtociclib
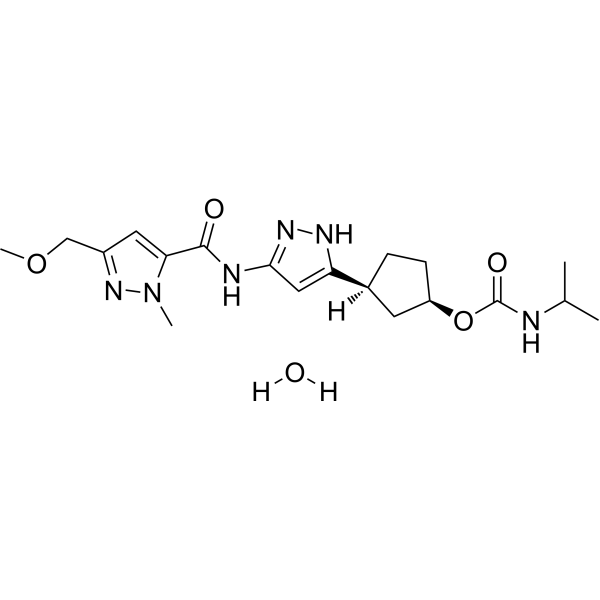
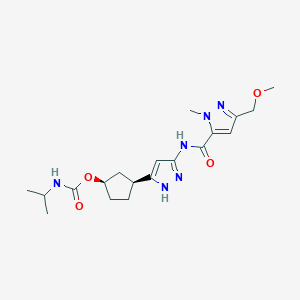
Tagtociclib (PF-07104091), 2460249-19-6, MW 404.5, C19H28N6O4
CAS 2733575-91-0 HYDRATE
| Molecular Weight HYDRATE | 422.48 |
|---|---|
| Formula | C19H30N6O5 |
[(1R,3S)-3-[3-[[5-(methoxymethyl)-2-methylpyrazole-3-carbonyl]amino]-1H-pyrazol-5-yl]cyclopentyl] N-propan-2-ylcarbamate
- (1R,3S)-3-[5-[[[3-(Methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl]amino]-1H-pyrazol-3-yl]cyclopentyl N-(1-methylethyl)carbamate
- (1R,3S)-3-{5-[3-(methoxymethyl)-1-methyl-1H-pyrazole-5carboxamido]-1H-pyrazol-3-yl}cyclopentyl (propan-2yl)carbamate
- (1R,3S)-3-(3-(3-(Methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamido)-1H-pyrazol-5-yl)cyclopentylisopropylcarbamate
- Carbamic acid, N-(1-methylethyl)-, (1R,3S)-3-[5-[[[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl]amino]-1H-pyrazol-3-yl]cyclopentyl ester
PF-07104091 hydrate is a potent and selective CDK2/cyclin E1 and GSK3β inhibitor, with Kis of 1.16 and 537.81 nM, respectively. PF-07104091 hydrate has anti-tumor activity for cyclin E1-amplified cancers. (patent WO2020157652A2).
- OriginatorPfizer
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase 2 inhibitors
Phase IIBreast cancer; Solid tumours
Phase I/IINon-small cell lung cancer; Ovarian cancer; Small cell lung cancer
13 Sep 2024Efficacy, adverse events, pkarmacokinetics and pharmacodynamics data from a phase I/II trial in Solid tumours presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
13 Sep 2024Pharmacodynamics data from a preclinical trial in Breast cancer presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
05 Apr 2024Pharmacodynamics data form preclinical trial in Breast cancer and Ovarian cancer presented at the 115th Annual Meeting of the American Association for Cancer Research (AACR-2024)
Tegtociclib is an orally bioavailable inhibitor of cyclin-dependent kinase 2 (CDK2), with potential antineoplastic activity. Upon administration, tegtociclib selectively targets, binds to and inhibits the activity of CDK2. This may lead to cell cycle arrest, the induction of apoptosis, and the inhibition of tumor cell proliferation. CDKs are serine/threonine kinases that are important regulators of cell cycle progression and cellular proliferation and are frequently overexpressed in tumor cells. CDK2/cyclin E complex plays an important role in retinoblastoma (Rb) protein phosphorylation and the G1-S phase cell cycle transition. CDK2/cyclin A complex plays an important role in DNA synthesis in S phase and the activation of CDK1/cyclin B for the G2-M phase cell cycle transition.
SCHEME
COUPLER
MAIN
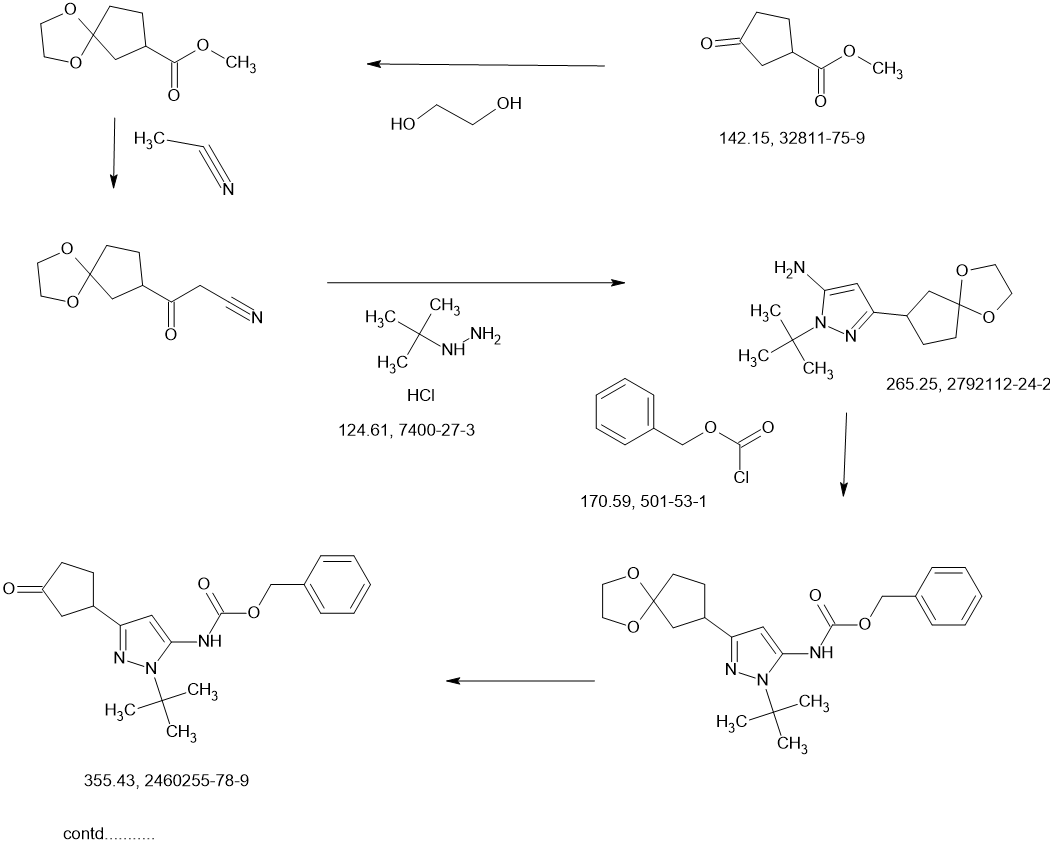
CONTD………….
PATENTS
WO2022018596 78%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022018596&_cid=P22-MDFCVG-44044-1
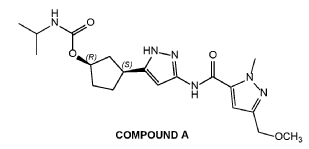
COMPOUND A was prepared as described in Example 13 of U.S. Patent No.
11,014,911.
Preparation of Intermediate 1: benzyl {1-tert-butyl-3-[(1S,3R)-3-hvdroxycvclopentyl]1H-pyrazol-5-yl)carbamate; and Intermediate 2: benzyl {1-tert-butyl-3-[(1R,3S)-3-hydroxycvclopentyl1-1H-pyrazol-5-yl)carbamate.
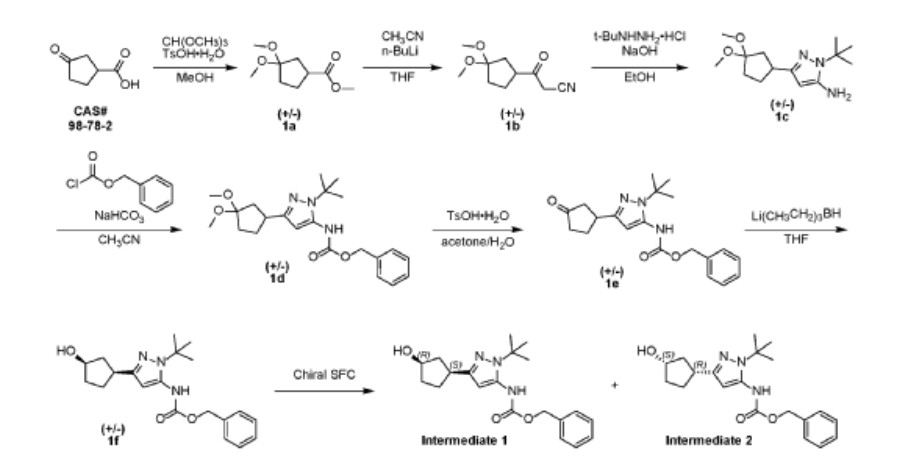
Two parallel reactions, each containing a solution of (±)-3- oxocyclopentanecarboxylic acid (CAS#98-78-2, 900 g, 7.02 mol) in methanol (5 L) at 13 °C were each treated with trimethyl orthoformate (4.47 kg, 42.15 mol, 4.62 L) and 4- toluenesulfonic acid monohydrate (26.72 g, 140.5 mmol). The mixtures were stirred at 13 °C for 25 hours. Each batch was quenched separately with sat. aq NaHCO3 (1 L), then the two batches were combined and concentrated under vacuum to remove most of the methanol. The residue was diluted with ethyl acetate (4 L), and the layers separated. The aqueous layer was further extracted with ethyl acetate (2 x 1 L). The combined organic layers were washed with sat. aq NaCI (3 x 1 L), dried over magnesium sulfate, filtered, and concentrated under vacuum to give (±)-methyl 3,3- dimethoxycyclopentanecarboxylate (1a, 2.5 kg, 13.28 mol, 94%) as a light yellow oil. 1H NMR (400MHz, CHLOROFORM -d) δ = 3.67 (s, 3H), 3.20 (s, 3H), 3.19 (s, 3H), 2.94- 2.82 (m, 1 H), 2.16-2.00 (m, 2H), 1.99-1.76 (m, 4H).
A solution of n-butyllithium (3.44 L of a 2.5 M solution in hexanes, 8.6 mol) was added to a reactor containing THF (3 L) at -65 °C. Anhydrous acetonitrile (453 mL, 353 g, 8.61 mol) was added dropwise, slowly enough to maintain the internal temperature below -55 °C. The mixture was stirred for an additional 1 hour at -65 °C. A solution of (±)-methyl 3,3-dimethoxycyclopentanecarboxylate (1a, 810 g, 4.30 mol) in THF (1 L) was then added dropwise, slowly enough to maintain the internal temperature below -50 °C. After stirring for an additional hour at -65 °C, the reaction was quenched with water (4 L), neutralized with aq HCI (1 M) to pH 7, and extracted with ethyl acetate (3 x 3L). The combined organic layers were washed with sat. aq NaCI (2 x 3L), dried over magnesium sulfate, filtered, and concentrated under vacuum to give crude (±)-3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile (1b, 722 g, 3.66 mol, 85%) as a red oil, which was used without further purification.
Solid sodium hydroxide (131.4 g, 3.29 mol total) was added in portions to a suspension of tert-butylhydrazine hydrochloride (409.4 g, 3.29 mol) in ethanol (3 L) at 16-25 °C. Stirring was continued at 25 °C for 1 hour. A solution of crude (±)-3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile (1b, 540 g, 2.74 mol) in ethanol was added at 25 °C, then the mixture was heated to 75 °C internal and stirred for 30 hours. The reaction was filtered, and the filtrate concentrated under vacuum to give crude product as a red oil. This product was combined with crude from three more identically-prepared batches (each starting with 540 g 1b; 2.16 kg, 10.96 mol total for the 4 batches), and purified by silica gel chromatography (eluting with 0-35% ethyl acetate in petroleum ether), affording (±)-1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-amine (1c, 1.60 kg, 5.98 mol, 54% yield) as a red oil. 1H NMR (CHLOROFORM -d) δ = 5.41 (s, 1 H), 3.50 (br. s., 2H), 3.22 (s, 3H), 3.20 (s, 3H), 3.13 (tt, J=7.9, 9.6 Hz, 1H), 2.25 (dd, J=8.0, 13.3 Hz, 1H), 2.09-2.00 (m, 1H), 1.99-1.91 (m, 1H), 1.83 (dd, J=10.8, 12.8 Hz, 2H), 1.78-1.68 (m, 1H), 1.60 (s, 9H).
Benzyl chloroformate (563.6 mL, 676.3 g, 3.96 mol) was added to a chilled (0-5 °C) solution of (±)-1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-amine (1c, 530 g, 1.98 mol) in acetonitrile (3.5 L). The mixture was stirred at 23 °C for 2 hours, and then solid sodium hydrogen carbonate (532.9 g, 6.34 mol) was added in portions. Stirring was continued at 23 °C for 26 hours. The resulting suspension was filtered and the filtrate concentrated under vacuum to give crude (±)-benzyl [1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 d, 980 g, 1.98 mol max) as a red oil, which was used in the next step without further purification.
A solution of the crude (±)-benzyl [1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 d, 980 g, 1.98 mol max) in acetone (2 L) and water (2 L) at 18 °C was treated with 4-toluenesulfonic acid monohydrate (48.75 g, 256.3 mmol). The mixture was heated to 60 °C internal for 20 hours. After concentration under vacuum to remove most of the acetone, the aqueous residue was extracted with dichloromethane (3 x 3 L). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated under vacuum to a crude red oil. This crude product was combined with crude from two other identically-prepared batches (each derived from 1.98 mol 1c, 5.94 mol total for the 3 batches), and purified by silica gel chromatography (eluting with 0- 50% ethyl acetate in petroleum ether) to give (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 1.6 kg) as a yellow solid. This solid was stirred in 10:1 petroleum ether/ethyl acetate (1.5 L) at 20 °C for 18 hours. The resulting suspension was filtered, the filter cake washed with petroleum ether ( 2 x 500 mL), and the solids dried under vacuum to give (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 1.4 kg, 3.9 mol, 66% combined for the three batches). 1H NMR (DMSO–d6) δ = 9.12 (br. s., 1H), 7.56-7.13 (m, 5H), 6.03 (s, 1 H), 5.12 (s, 2H), 3.41-3.27 (m, 1H), 2.48-2.39 (m, 1H), 2.34-2.10 (m, 4H), 1.98-1.81 (m, 1 H), 1.48 (s, 9H).
A solution of (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 320 g, 0.900 mol) in THF (1.5 L) was degassed under vacuum and purged with dry nitrogen (3 cycles), then cooled to -65 °C internal. A solution of lithium triethylborohydride (1.0 M in THF, 1.80 L, 1.80 mol) was added dropwise at a rate which maintained the internal temperature below -55 °C, then stirring was continued at -65 °C for 1.5 hours. The reaction mixture was quenched with sat. aq NaHCO3 (1.5 L) at -40 to -30 °C. Hydrogen peroxide (30% aqueous, 700 g) was added to the mixture dropwise, while the internal temperature was maintained at -10 to 0 °C. The mixture was stirred at 10 °C for 1 hour, then extracted with ethyl acetate (3 x 2 L). The combined organic layers were washed with sat. aq Na2SO3 (2 x 1 L) and sat. aq NaCI (2 x 1 L). The organics were dried over magnesium sulfate, filtered, and concentrated under vacuum to a crude yellow oil. The crude product from this batch was combined with crude from three other, identically-prepared batches (each starting from 0.900 mol 1 e, for a total of 3.60 mol) for purification. Before chromatography, the combined mixture showed ~3.3:1 cis/trans ratio by NMR. The combined crude product was purified twice by silica gel chromatography, eluting with 0-50% ethyl acetate in dichloromethane), affording (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 960 g) as a light yellow solid, which was further purified by trituration, as described below.
A previous batch of 1f had been obtained from smaller-scale reactions, starting from a total of 120 g 1e (0.34 mol). The columned product from this batch was combined with the columned product from the batch above (which had been derived from 3.60 mol 1 e, for a total of 3.94 mol 1e used for all the combined batches), suspended in 10:1 dichloromethane/methanol (1.5 L), and stirred at 20 °C for 16 hours. The suspension was filtered, and the filter cake washed with petroleum ether (2 x 500 mL). The solids were dried under vacuum to give clean (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 840 g, 2.35 mol, 60% total yield for all the combined batches) as a white solid. 1H NMR (400MHz, DMSO-d6) δ = 9.07 (br. s., 1 H), 7.45-7.27 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.5 Hz, 1 H), 4.21-4.07 (m, 1 H), 2.88 (quin, J=8.6 Hz, 1 H), 2.24-2.13 (m, 1 H), 1.92-1.78 (m, 1 H), 1.78-1.62 (m, 2H), 1.61-1.53 (m, 1 H), 1.47 (s, 9H), 1.52-1.43 (m, 1 H). MS: 358 [M+H]+.
The enantiomers of (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 700 g, 1.96 mol) were separated by chiral SFC.
The product from the first-eluting enantiomer peak (310 g solid) was suspended in methanol/petroleum ether (1 :10, 1 L) and stirred at 25 °C for 1 hour. The suspension was filtered, the filter pad washed with petroleum ether (2 x 500 mL), and the solids dried under vacuum to give benzyl {1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}carbamate (Intermediate 1 , 255 g, 713 mmol, 36%, >99% ee) as a white solid. 1H NMR (400MHz, DMSO -d6) δ = 9.08 (br. s., 1 H), 7.58-7.20 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.4 Hz, 1 H), 4.19-4.09 (m, 1 H), 2.88 (quin, J=8.6 Hz, 1 H), 2.24-2.13 (m, 1 H), 1.91-1.79 (m, 1 H), 1.79-1.61 (m, 2H), 1.61-1.53 (m, 1 H), 1.47 (s, 9H), 1.52-1.44 (m, 1 H). MS: 358 [M+H]+. Optical rotation [α]D +3.76 (c 1.0, MeOH). Chiral purity: >99% ee, retention time 3.371 min. Chiral SFC analysis was performed on a ChiralPak AD-3 150 x 4.6 mm ID, 3 pm column heated to 40 °C, eluted with a mobile phase of CO2 and a gradient of 0-40% methanol+0.05%DEA over 5.5 min, then held at 40% for 3 min; flowing at 2.5 mL/min.
The product from the second-eluting enantiomer peak (300 g solid) was suspended in methanol/petroleum ether (1 :10, 1 L) and stirred at 25 °C for 1 hour. The suspension was filtered, the filter pad washed with petroleum ether (2 x 500 mL), and the solids dried under vacuum to give benzyl {1-tert-butyl-3-[(1R,3S)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}carbamate (Intermediate 2, 255 g, 713 mmol, 36%, 94% ee) as a white solid. 1H NMR (400MHz, DMSO-d6) δ = 9.08 (br. s., 1 H), 7.55-7.19 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.4 Hz, 1 H), 4.23-4.07 (m, 1 H), 2.88 (quin, J=8.7 Hz, 1 H), 2.23-2.14 (m, 1 H), 1.90-1.79 (m, 1 H), 1.77-1.61 (m, 2H), 1.61-1.53 (m, 1 H), 1 .47 (s, 9H), 1.52-1 .44 (m, 1 H). MS: 358 [M+H]+. Optical rotation [α]D -2.43 (c 1 .0, MeOH). Chiral purity: 94% ee, retention time 3.608 min. Chiral SFC analysis was performed on a ChiralPak AD-3 150 x 4.6 mm ID, 3 pm column heated to 40 °C, eluted with a mobile phase of CO2 and a gradient of 0-40% methanol+0.05%DEA over 5.5 min, then held at 40% for 3 min; flowing at 2.5 mL/min.
A sample of the second-eluting enantiomer from a previous batch with [α]D -3.1 (c 1.1, MeOH) and 96% ee was crystalized from dichloroethane/pentane. A crystal structure was obtained by small-molecule X-ray crystallography, which showed (1R,3S) geometry. The absolute stereochemistry of Intermediate 2 was thus assigned (1R,3S) based on its comparable optical rotation and order of elution in the analytical method. Intermediate 1, the enantiomer of Intermediate 2, was thus assigned (1S,3R) stereochemistry.
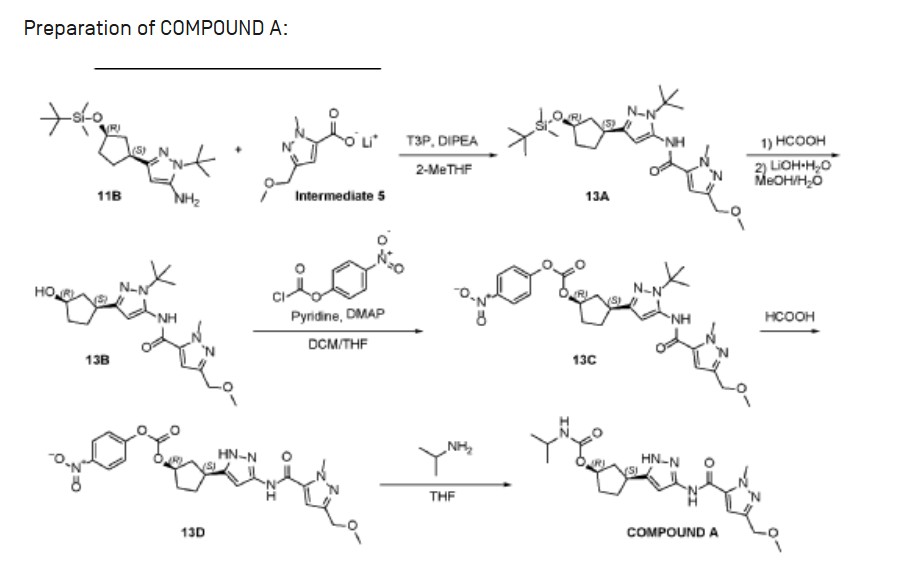
Propylphosphonic anhydride (T3P®, 50 wt% solution in EtOAc, 50.3 g, 79.1 mmol) was added to a room temperature (26 °C) solution of 1-tert-butyl-3-[(1S,3R)-3-{[tert-butyl(dimethyl)silyl]oxy}cyclopentyl]-1H-pyrazol-5-amine (11 B, 8.90g, 26.4 mmol), lithium 3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxylate (Intermediate 5, 5.83 g,
34.3 mmol), and diisopropylethyl amine (10.2 g, 79.1 mmol) in 2-methyltetrahydrofuran (100.0 mL). The resulting mixture was stirred at this temperature for 18 hours. After concentrating the mixture to dryness, the residue was dissolved in dichloromethane (150 mL), and the solution washed sequentially with water (2 x 30 mL), sat. aq NaHCO3 (2 x 30 mL) and sat. aq NaCI (30 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated to give crude N-{1-tert-butyl-3-[(1S,3R)-3-{[tert- butyl(dimethyl)silyl]oxy}cyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H- pyrazole-5-carboxamide (13A, 12.9 g, 100%) as an oil. MS: 490 [M+H]+.
The crude N-{1-tert-butyl-3-[(1S,3R)-3-{[tert-butyl(dimethyl)silyl]oxy}cyclopentyl]- 1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13A, 12.9 g,
26.3 mmol) was dissolved in formic acid (80 mL) and stirred at room temperature (27 °C) for 30 minutes. The mixture was concentrated to dryness, and the residue
dissolved in methanol (80 mL). A solution of lithium hydroxide monohydrate (3.43 g, 81.8 mmol) in water (15 mL) was added, and the mixture stirred at room temperature (27 °C) for 1 hour. The mixture was concentrated to dryness, and the residue was purified by silica gel chromatography (eluting with 0-80% ethyl acetate in petroleum ether) to give N-{1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13B, 8.0 g, 78%) as a yellow gum. MS: 376 [M+H]+.
A solution of N-{1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13B, 8.0 g, 21 mmol) in dichloromethane (80 mL) and THF (80 mL) was treated with DMAP (260 mg, 2.13 mmol), pyridine (5.06 g, 63.9 mmol), and 4-nitrophenyl chloroformate (8.59 g, 42.6 mmol). The resulting yellow suspension was stirred at room temperature for 18 hours. The reaction mixture was concentrated to dryness and purified by silica gel chromatography (eluting with 0-45% ethyl acetate in petroleum ether) to give (1R,3S)-3-[1-tert-butyl-5-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-3-yl]cyclopentyl 4-nitrophenyl carbonate (13C, 10.6 g, 92%) as a light brown gum. MS: 541 [M+H]+.
A solution of (1R,3S)-3-[1-tert-butyl-5-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-3-yl]cyclopentyl 4-nitrophenyl carbonate (13C, 10.6 g, 19.6 mmol) in formic acid (80 mL) was stirred at 70 °C for 18 hours. The solution was concentrated to dryness. The residue was dissolved in dichloromethane (150 mL) and the solution neutralized with sat. aq NaHCO3. The organic layer was washed with water (30 mL) and sat. aq NaCI (30 mL), dried over sodium carbonate, filtered, and concentrated to give crude (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl 4-nitrophenyl carbonate (13D, 8.5 g, 90%, 86% pure by LCMS) as a light yellow glass. MS: 485 [M+H]+.
A room temperature (27 °C) solution of crude (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl 4-nitrophenyl carbonate (13D, 1.7 g, 3.5 mmol) and 2-propylamine (1.04 g, 17.5 mmol) in THF (30 mL) was stirred for 6 hours. The solution was concentrated to dryness, and the residue was combined with the residue from a second batch which had been derived from 1.7 g, 3.5 mmol 13D (total 6.27 mmol 13D consumed for the combined two batches) to give 3.2 g crude product. This product was purified by preparative HPLC on a Phenomenex Gemini C18 250*50mm*10 pm column, eluting with 15-45% water (0.05% ammonium
hydroxide v/v) in acetonitrile. After lyophilization, (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl propan-2 -ylcarbamate (COMPOUND A, 2.06 g, 78%) was obtained as a white crystalline solid monohydrate. MS: 405 [M+H]+. 1H NMR (400MHz, DMSO-d6) d = 12.23 (br s, 1H), 10.73 (br s, 1H), 7.11 (s, 1H), 6.96 (br d, J=7.0 Hz, 1H), 6.41 (br s, 1H), 5.00 (br s, 1H), 4.33 (s, 2H), 4.04 (s, 3H), 3.57 (qd, J=6.6, 13.4 Hz, 1H), 3.26 (s, 3H), 3.17-2.96 (m, 1H), 2.48-2.39 (m, 1H), 2.03 (br d, J=6.8 Hz, 1H), 1.95-1.83 (m, 1H), 1.73 (br d, J=8.5 Hz, 2H), 1.61 (br s, 1 H), 1.03 (br d, J=6.3 Hz, 6H). Optical rotation [α]D +4.8 (c 1.0, MeOH). Chiral purity: >99% ee by chiral analytical SFC. Anal. Calcd for C19H28N6O4-H2O: C, 54.02; H, 7.16; N, 19.89. Found: C, 53.94; H, 7.22; N, 19.81.
PATENT
WO2020157652 EX 13
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020157652&_cid=P22-MDFD2U-50269-1
Example 13: (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}-amino)-1H-pyrazol-5-yl]cyclopentyl propan-2-ylcarbamate
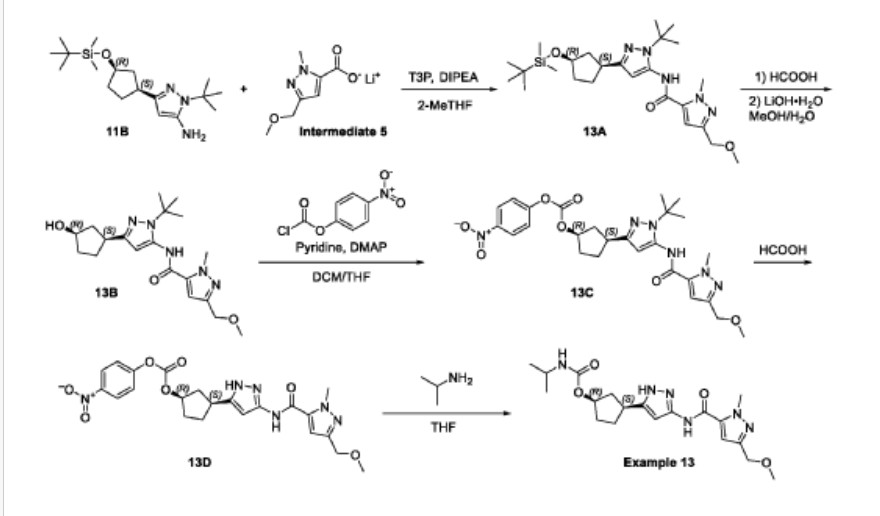
(1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl propan-2-ylcarbamate (Example 13, 2.06 g, 78%) was obtained as a white crystalline solid found to be a monohydrate (Form 1) based on elemental analysis. MS: 405 [M+H]+.1H NMR (400MHz, DMSO-d6) d = 12.23 (br s, 1H), 10.73 (br s, 1H), 7.11 (s, 1H), 6.96 (br d, J=7.0 Hz, 1H), 6.41 (br s, 1H), 5.00 (br s, 1H), 4.33 (s, 2H), 4.04 (s, 3H), 3.57 (qd, J=6.6, 13.4 Hz, 1H), 3.26 (s, 3H), 3.17-2.96 (m, 1H), 2.48-2.39 (m, 1H), 2.03 (br d, J=6.8 Hz, 1H), 1.95-1.83 (m, 1H), 1.73 (br d, J=8.5 Hz, 2H), 1.61 (br s, 1H), 1.03 (br d, J=6.3 Hz, 6H). Optical rotation [a]D +4.8 (c 1.0, MeOH). Chiral purity: >99% ee by chiral analytical SFC. Anal. Calcd for C19H28N6O4-H2O: C, 54.02; H, 7.16; N, 19.89. Found: C, 53.94; H, 7.22; N, 19.81.
The white crystalline solid from above (500 mg) was recrystallized from 9: 1 H2O/CH3CN (2 mL) by heating until dissolved and then allowing the resulting solution to stand at room temperature for 18 h. During the 18 h time period, larger crystals of monohydrate (Form 1) formed. Single crystal X-ray diffraction of a selected crystal from this material provided the structure in FIG.1.
PATENTS
WO2022018667
WO2022174031
WO2022137106
[1]. Douglas Carl BEHENNA, et al. Cdk2 inhibitors. WO2020157652A2.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Tagtociclib, PF-07104091, 2460249-19-6, Tegtociclib, XBD0JF5EHJ, PF 07104091
SPIROBUDIFEN
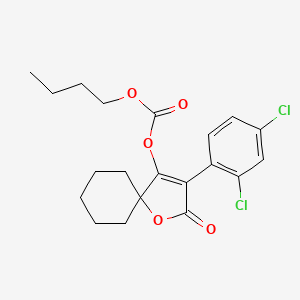
SPIROBUDIFEN
cas 1305319-70-3
413.3 g/mol, C20H22Cl2O5
Butyl 3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl carbonate Butyl 3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl carbonate
- Butyl (3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro(4.5)dec-3-en-4-yl) carbonate
- butyl [3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl] carbonate
Spirobudifen is an oxaspiro compound that is 1-oxaspiro[4.5]dec-3-en-2-one substituted by 2,4-dichlorophenyl and (butoxycarbonyl)oxy groups at positions 3 and 4, respectively. It is an acaricide from Zhejiang Udragon Bioscience. It is a dichlorobenzene, an oxaspiro compound, an organochlorine acaricide and a carbonate ester.
SCHEME
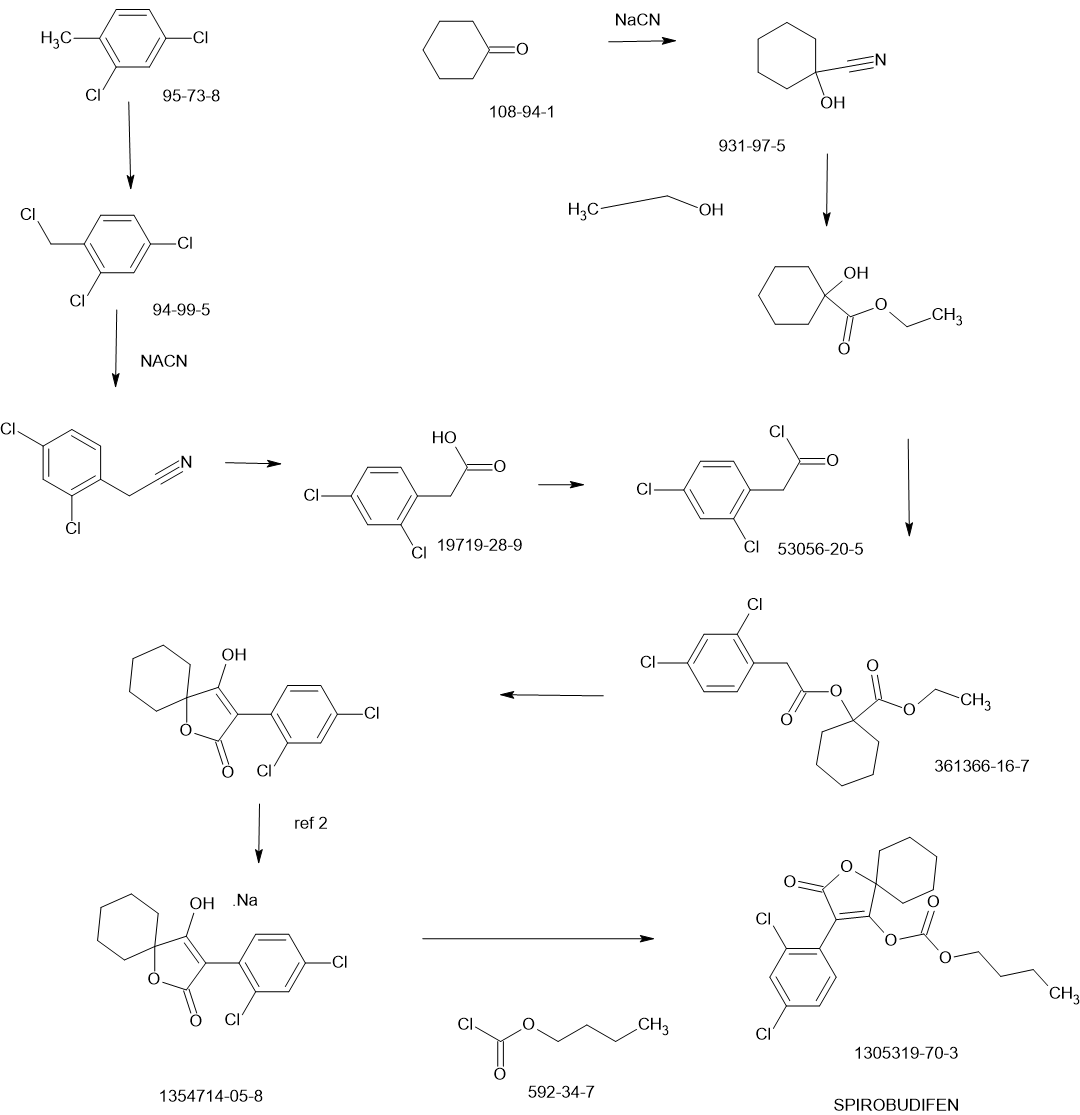
PATENTS
CN112745286
CN102060818
Xiandai Nongyao (2012), 11(1), 15-21
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////SPIROBUDIFEN, 1305319-70-3
SOVESUDIL
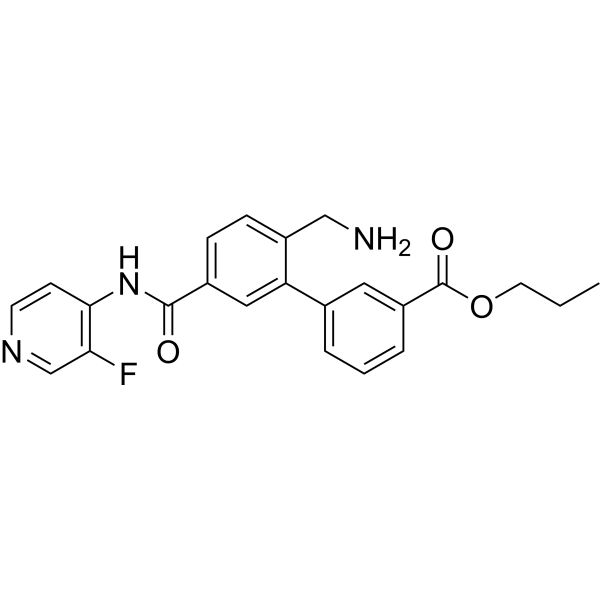
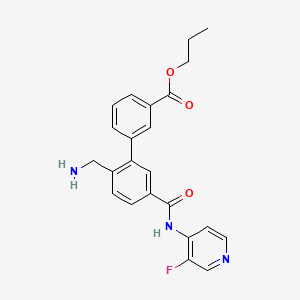
SOVESUDIL
PHP-201; AMA 0076, C23O3R93BM
CAS 1333400-14-8
| Molecular Weight | 407.44 |
|---|---|
| Formula | C23H22FN3O3 |
Sovesudil (PHP-201) is a potent, ATP-competitive, locally acting Rho kinase (ROCK) inhibitor with IC50s of 3.7 and 2.3 nM for ROCK-I and ROCK-II, respectively. Sovesudil lowers intraocular pressure (IOP) without inducing hyperemia.
SCHEME
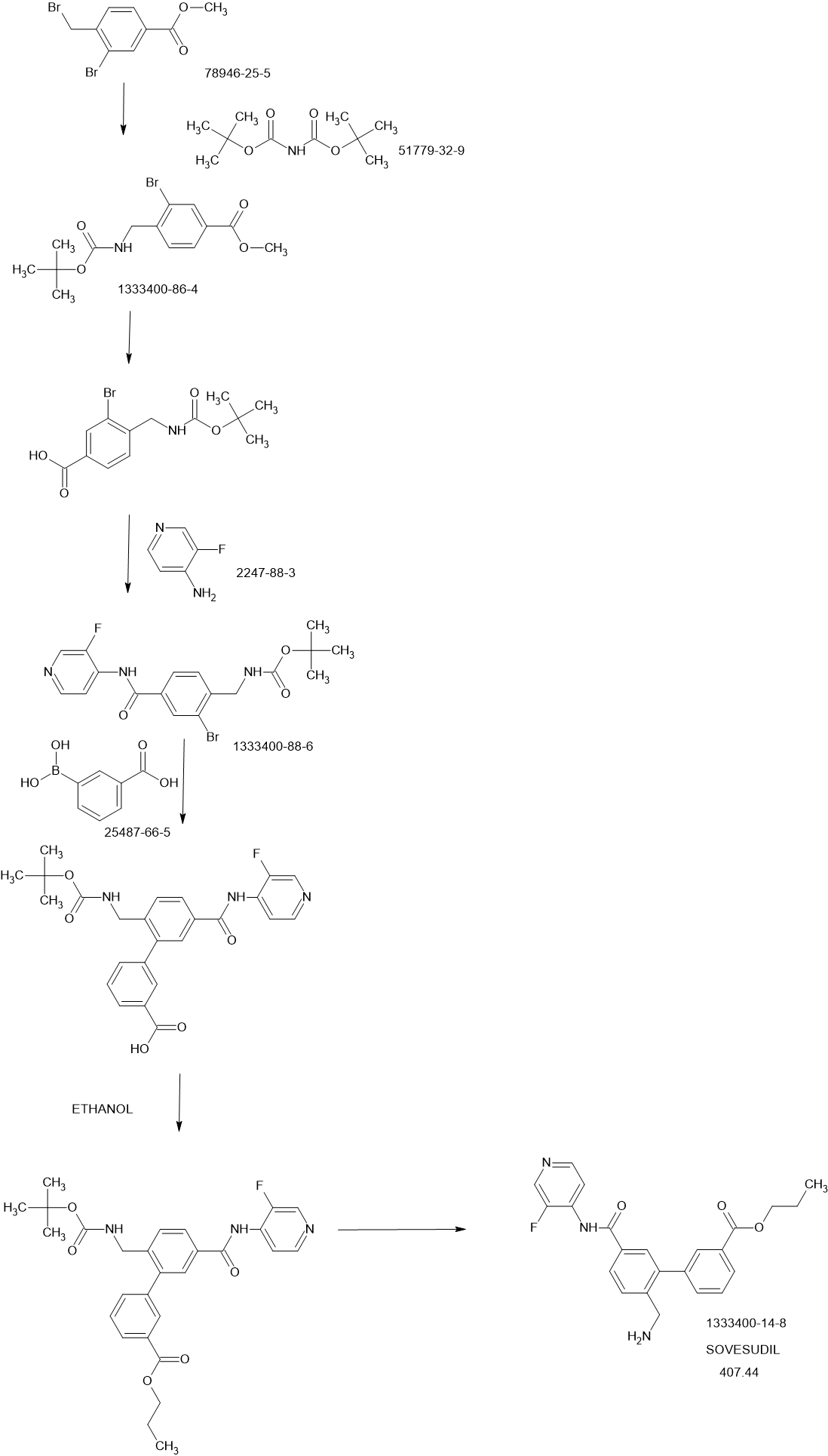
PATENTS
Bioorganic & Medicinal Chemistry Letters (2013), 23(23), 6442-6446
https://www.sciencedirect.com/science/article/abs/pii/S0960894X13011141
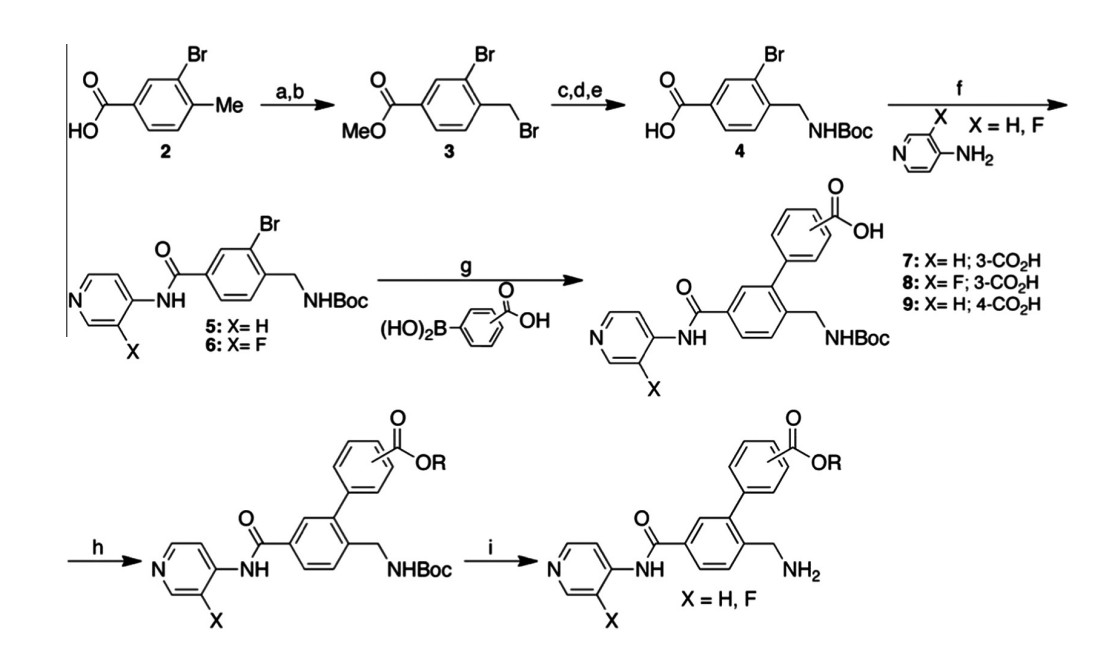
Figure 2. Synthetic scheme for synthesis of compounds 10–35. (a) H2SO4, MeOH, 60 C, 16 h; (b) NBS, AIBN, CCl4, reflux, 16 h; (c) Boc2NH, t-BuOK, DMF, rt, 16 h; (d) DCM/TFA
(50:1), 0 C ? rt, 4 h; (e) NaOH, MeOH, 50 C, 2 h; (f) HATU, DMAP, NEt3, DMA, 30 C, 16 h; (g) Pd(dppf)Cl2, Na2CO3, H2O, DMF, 100 C; 16 h; (h) ROH, TBTU, HOBT, DIEA, DMF,
rt, 16 h or ROH, DCC, DMAP, DCM, rt, 16 h or Me2C@C(Cl)NMe2, THF or DCM, rt, followed by ROH, 16 h; (i) DCM/TFA (7:1), 30 C, 16 h or HCl(g) in DCM, 30 C, 16 h.
PATENT
WO2011107608
- [1]. Van de Velde S, et al. AMA0076, a novel, locally acting Rho kinase inhibitor, potently lowers intraocular pressure in New Zealand white rabbits with minimal hyperemia. Invest Ophthalmol Vis Sci. 2014;55(2):1006-1016. Published 2014 Feb 18. [Content Brief][2]. Ha A, et al. Sovesudil (locally acting rho kinase inhibitor) for the treatment of normal-tension glaucoma: the randomized phase II study [published online ahead of print, 2021 Jul 28]. Acta Ophthalmol. 2021;10.1111/aos.14949. [Content Brief]
////////SOVESUDIL, 1333400-14-8, PHP 201, AMA 0076, C23O3R93BM
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Risevistinel
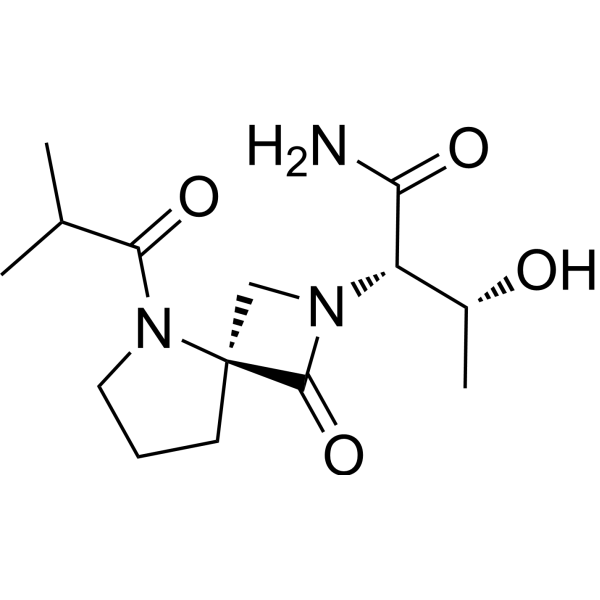
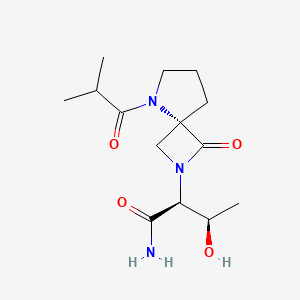
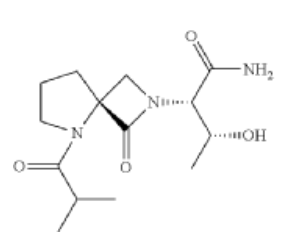
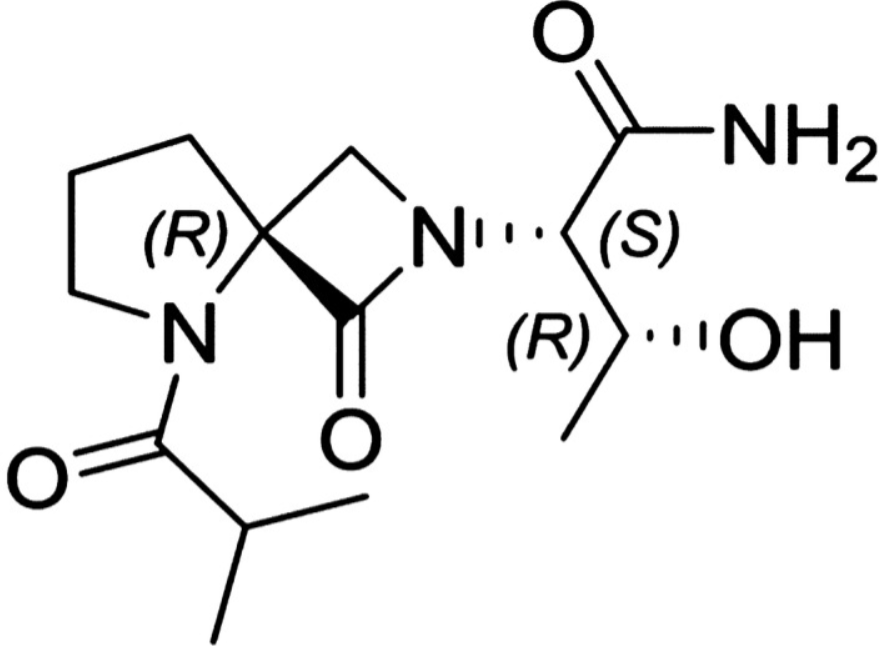
Risevistinel
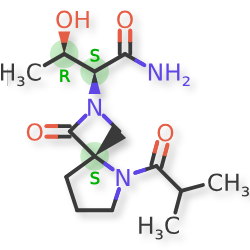
NYX-783 CAS 2591344-26-0, UNII-52TU5MZG22
NYX 2925, 2012536-16-0, X062KF5ZV3
C14H23N3O4, 297.35
UNII-52TU5MZG22,
X062KF5ZV3
(αS,4R)-α-[(1R)-1-Hydroxyethyl]-5-(2-methyl-1-oxopropyl)-1-oxo-2,5-diazaspiro[3.4]octane-2-acetamide
2,5-Diazaspiro[3.4]octane-2-acetamide, α-[(1R)-1-hydroxyethyl]-5-(2-methyl-1-oxopropyl)-1-oxo-, (αS,4S)-
(2S,3R)-3-hydroxy-2-[(4S)-5-(2-methylpropanoyl)-3-oxo-2,5-diazaspiro[3.4]octan-2-yl]butanamide
- NYX-783, NYX 2925
- CS-0113907
- HY-135741
- NYX-2925
Risevistinel (NYX-783) is a positive allosteric modulator of N-methyl-D-aspartate (NMDA) receptor. Nevadistinel can be used to inhibit cognitive impairment associated with neurodegenerative diseases, such as mild cognitive impairment, mild Alzheimer’s disease, Parkinson’s disease, Lewy body disease.
NYX-2925 is an N-methyl-D-aspartate receptor (NMDAR) modulator, and at low concentrations of endogenous agonist (glycine or D-serine) and in the presence of glutamate, NYX-2925 partially activates the NMDAR. NYX-2925 appears to act at a binding site that is distinct from NMDAR agonists or antagonists studied to date, such as D-cycloserine, ketamine, MK-801, or kynurenic acid. The mode of action of NYX-2925 seems to be distinct from that of all existing and emerging drugs that are indicated for the treatment of neuropathic pain. While current medications target individual elements of pain signal transmission or modulation, NYX-2925 can modulate multiple synaptic relays within pain circuits.
NYX-2925 is under investigation in clinical trial NCT04146896 (Efficacy and Safety of NYX-2925 in Subjects With Neuropathic Pain Associated With Diabetic Peripheral Neuropathy (DPN)).
SCHEME
COUPLER
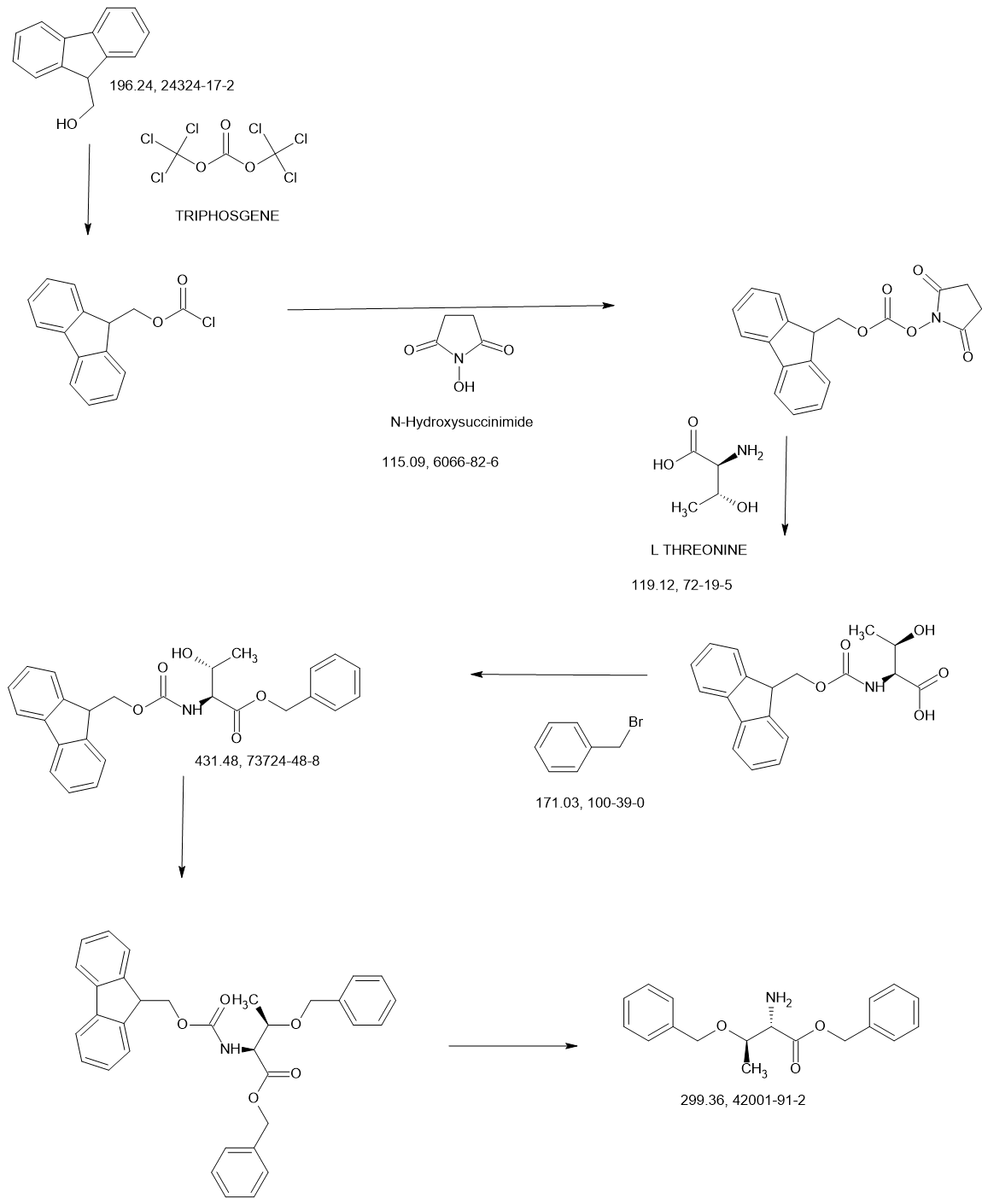
MAIN
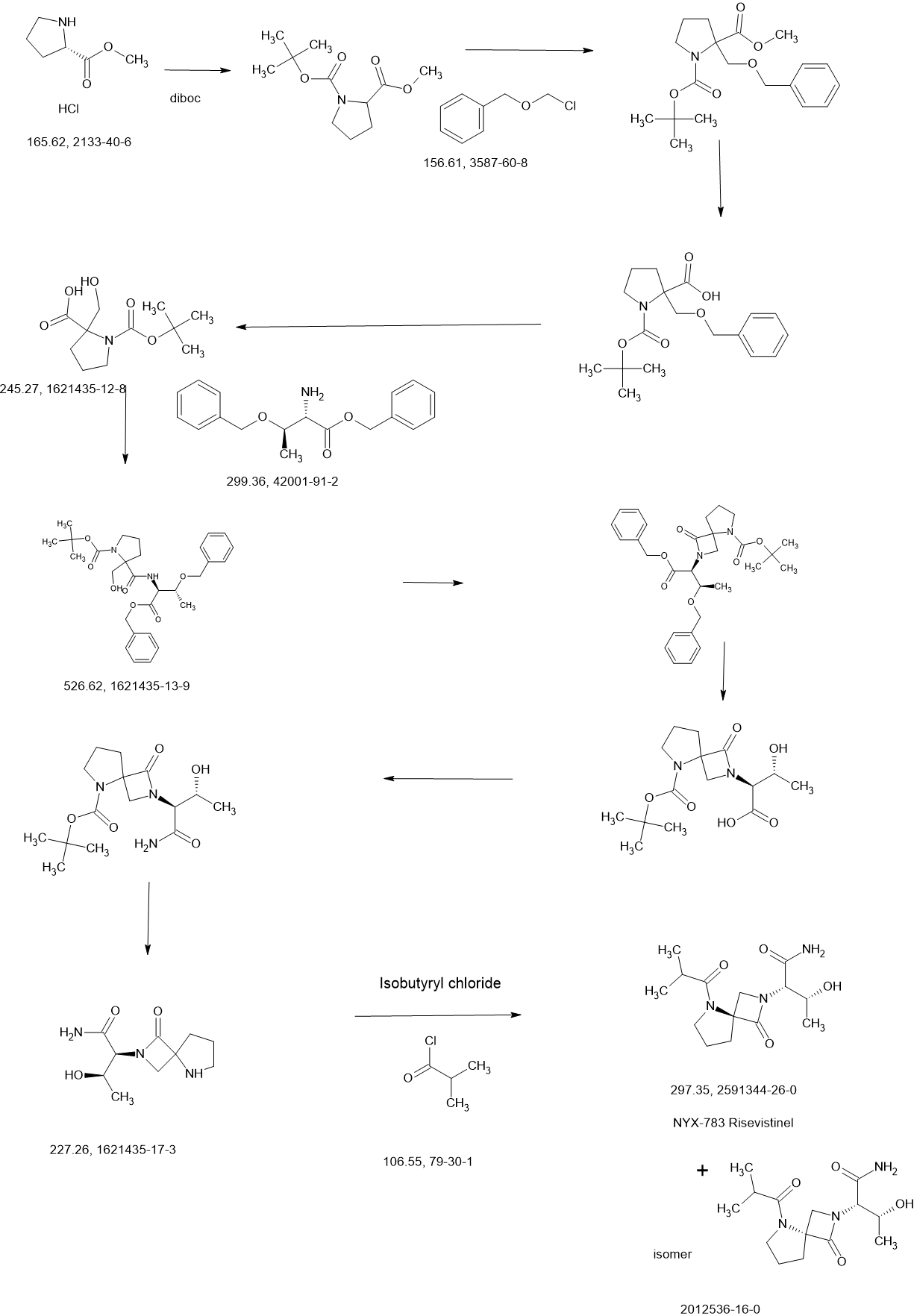
REF
US20210139489 Aptinyx Inc.
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750708&_cid=P11-MD3X0E-31059-1
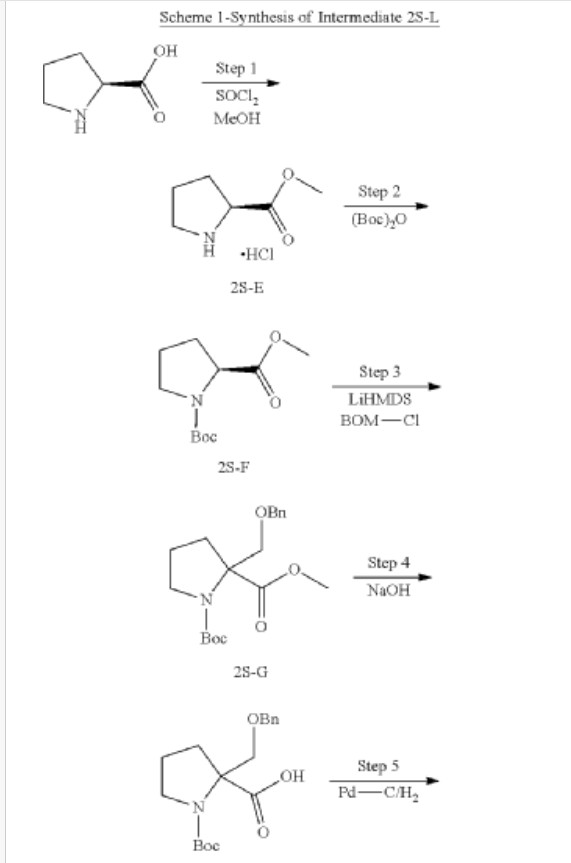
Synthesis of methyl pyrrolidine-2-carboxylate (2S-E)
| 1H-NMR: (500 MHz, DMSO-d 6): δ 4.15-4.13 (m, 1H), 3.65 (s, 3H), 3.35-3.30 (m, 2H), 2.23-2.15 (m, 1H), 1.86-1.78 (m, 3H), 1.41 (s, 9H); |
Synthesis of 1-tert-butyl 2-methyl pyrrolidine-1,2-dicarboxylate (2S-F)
| 1H-NMR: (400 MHz, DMSO-d 6): δ 4.15-4.13 (m, 1H), 3.65 (s, 3H), 3.35-3.30 (m, 2H), 2.23-2.15 (m, 1H), 1.86-1.78 (m, 3H), 1.41 (s, 9H); |
Synthesis of 1-tert-butyl 2-methyl 2-((benzyloxy) methyl) pyrrolidine-1,2-dicarboxylate (2S-G)
| 1H-NMR: (500 MHz, DMSO-d 6): δ 7.36-7.22 (m, 5H), 4.59-4.48 (m, 2H), 4.02-3.88 (m, 1H), 3.63 (s, 3H), 3.49-3.35 (m, 2H), 3.34-3.30 (m, 1H), 2.31-2.23 (m, 1H), 2.04-1.89 (m, 2H), 1.82-1.78 (m, 1H); |
Synthesis of 2-((benzyloxy) methyl)-1-(tert-butoxycarbonyl) pyrrolidine-2-carboxylic acid (2S-H)
Synthesis of 1-(tert-butoxycarbonyl)-2-(hydroxymethyl) pyrrolidine-2-carboxylic acid (2S-I)
| 1H-NMR: (400 MHz, DMSO-d 6): δ 4.66 (br s, 1H), 3.96-3.83 (m, 1H), 3.63-3.59 (m, 1H), 3.49-3.41 (m, 1H), 3.34-3.25 (m, 1H), 2.30-2.17 (m, 1H), 1.95-1.72 (m, 3H), 1.38 (s, 9H). |
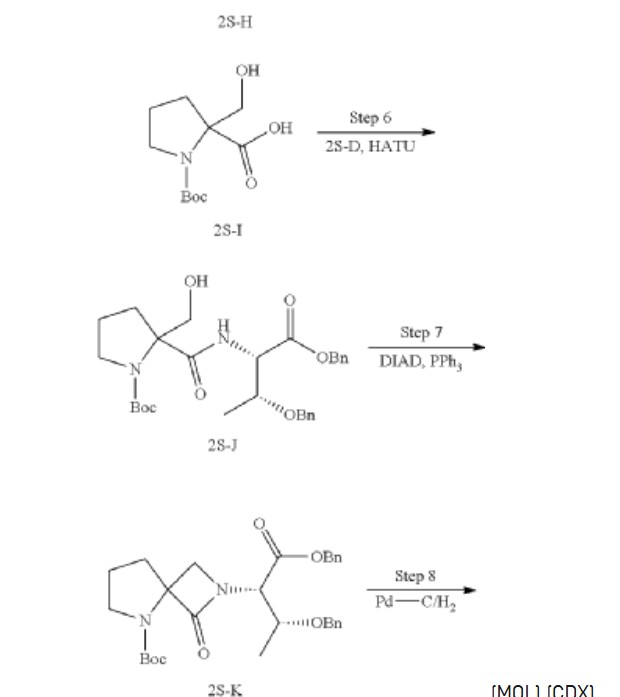
Synthesis of tert-butyl 2-(((2S,3R)-1,3-bis (benzyloxy)-1-oxobutan-2-yl) carbamoyl)-2-(hydroxymethyl) pyrrolidine-1-carboxylate (2S-J)
Synthesis of tert-butyl 2-((2S,3R)-1,3-bis (benzyloxy)-1-oxobutan-2-yl)-1-oxo-2,5-diazaspiro [3.4] octane-5-carboxylate (2S-K)
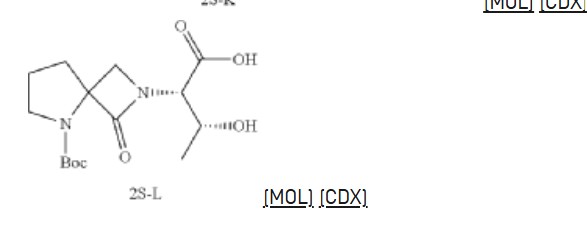
Synthesis of (2S,3R)-2-(5-(tert-butoxycarbonyl)-1-oxo-2,5-diazaspiro [3.4] octan-2-yl)-3-hydroxybutanoic acid (2S-L)
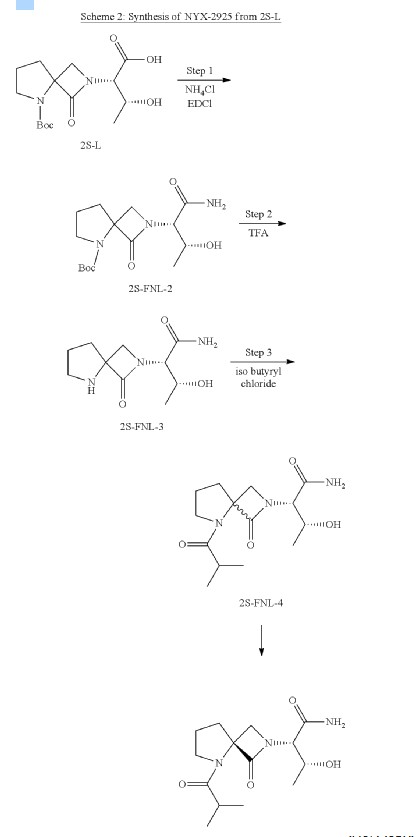
Synthesis of tert-butyl 2-((2S,3R)-1-amino-3-hydroxy-1-oxobutan-2-yl)-1-oxo-2,5-diazaspiro [3.4] octane-5-carboxylate (2S-FNL-2)
Synthesis of (2S,3R)-3-hydroxy-2-(1-oxo-2,5-diazaspiro [3.4] octan-2-yl) butanamide (2S-FNL-3)
| 1H-NMR: (400 MHz, D 2O): δ 4.33-4.29 (m, 2H), 4.09 (d, 1H), 3.95 (d, 1H), 3.57-3.48 (m, 2H), 2.51-2.46 (m, 2H), 2.25-2.19 (m, 2H), 1.31 (d, 3H); |
Synthesis of (2S,3R)-3-hydroxy-2-(5-isobutyryl-1-oxo-2,5-diazaspiro [3.4] octan-2-yl) butanamide (NYX-2925)
PATENT
WO2022086858
WO2021021996
//////Risevistinel, NYX 783, UNII-52TU5MZG22, Aptinyx Inc, NYX 2925, CS 0113907, HY 135741, NYX-2925
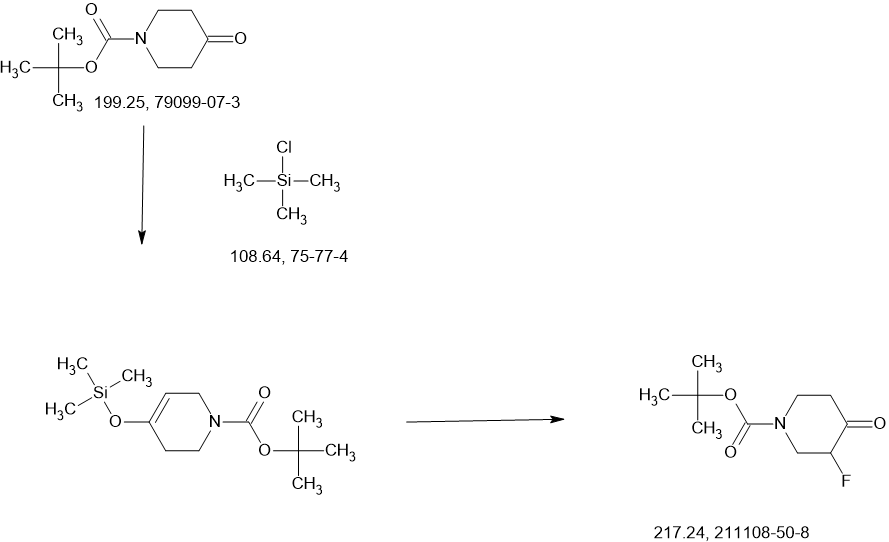
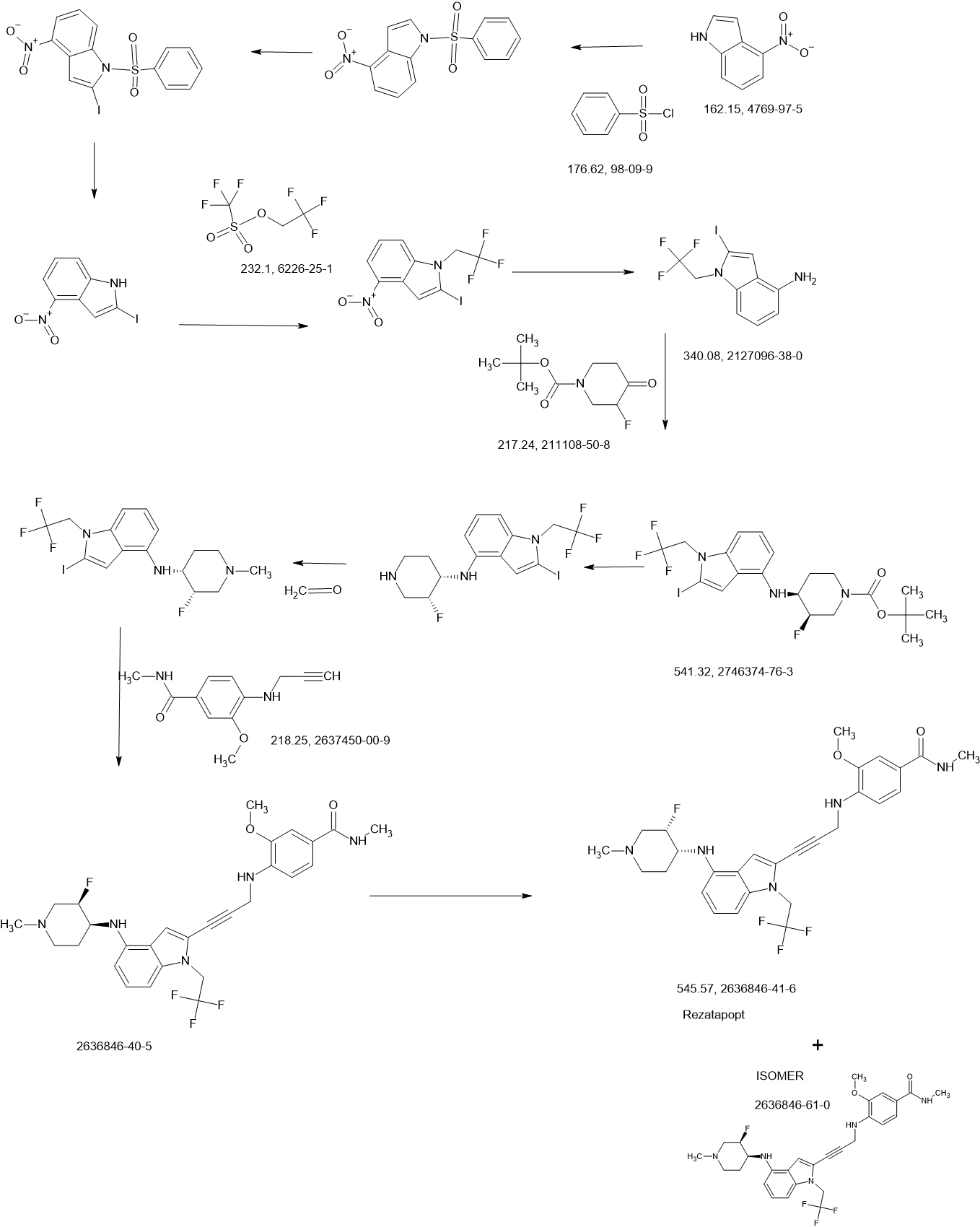
Rezatapopt
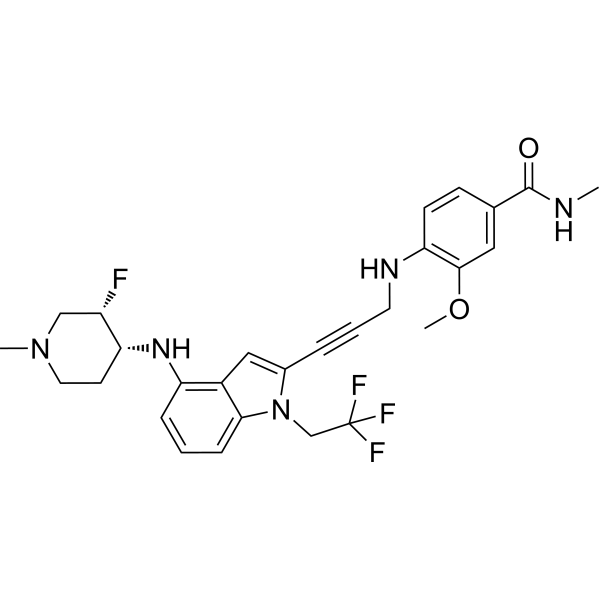
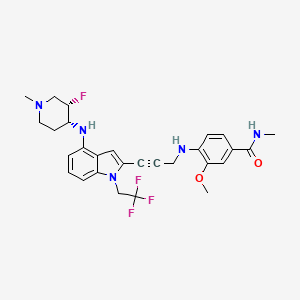
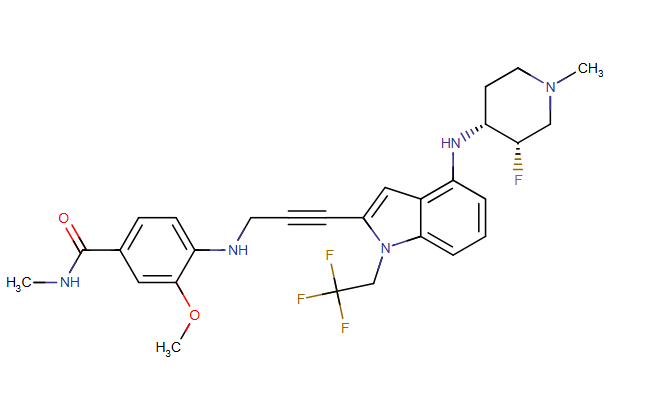
Rezatapopt, PC 14586
CAS 2636846-41-6
| Molecular Weight | 545.57 |
|---|---|
| Synonyms | PC14586 |
| Formula | C28H31F4N5O2 |
| CAS No. | 2636846-41-6 |
4-[3-[4-[[(3S,4R)-3-fluoro-1-methylpiperidin-4-yl]amino]-1-(2,2,2-trifluoroethyl)indol-2-yl]prop-2-ynylamino]-3-methoxy-N-methylbenzamide
- 4-[3-[4-[[(3S,4R)-3-fluoro-1-methylpiperidin-4-yl]amino]-1-(2,2,2-trifluoroethyl)indol-2-yl]prop-2-ynylamino]-3-methoxy-N-methylbenzamide
- Benzamide, 4-[[3-[4-[[(3S,4R)-3-fluoro-1-methyl-4-piperidinyl]amino]-1-(2,2,2-trifluoroethyl)-1H-indol-2-yl]-2-propyn-1-yl]amino]-3-methoxy-N-methyl-
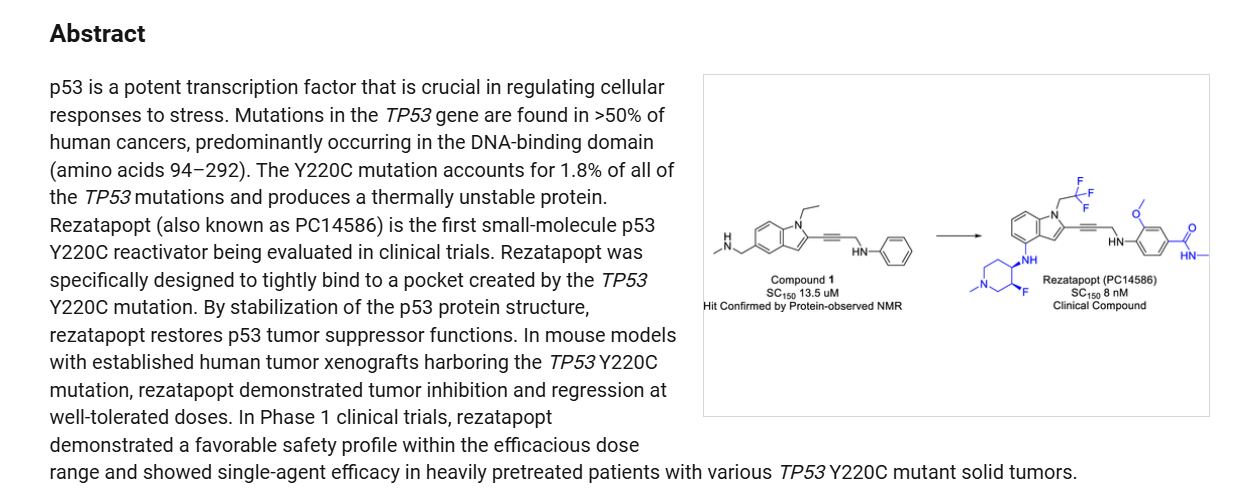
Rezatapopt (PC14586) is an orally active antineoplastic agent. Rezatapopt binds to a pocket created by the TP53 Y220C mutation. Rezatapopt restores p53 tumor suppressor functions by stabilization of the p53 protein structure. Rezatapopt demonstrates tumor inhibition and regression in mouse models with established human tumor xenografts harboring the TP53 Y220C mutation.
SCHEME
COUPLER
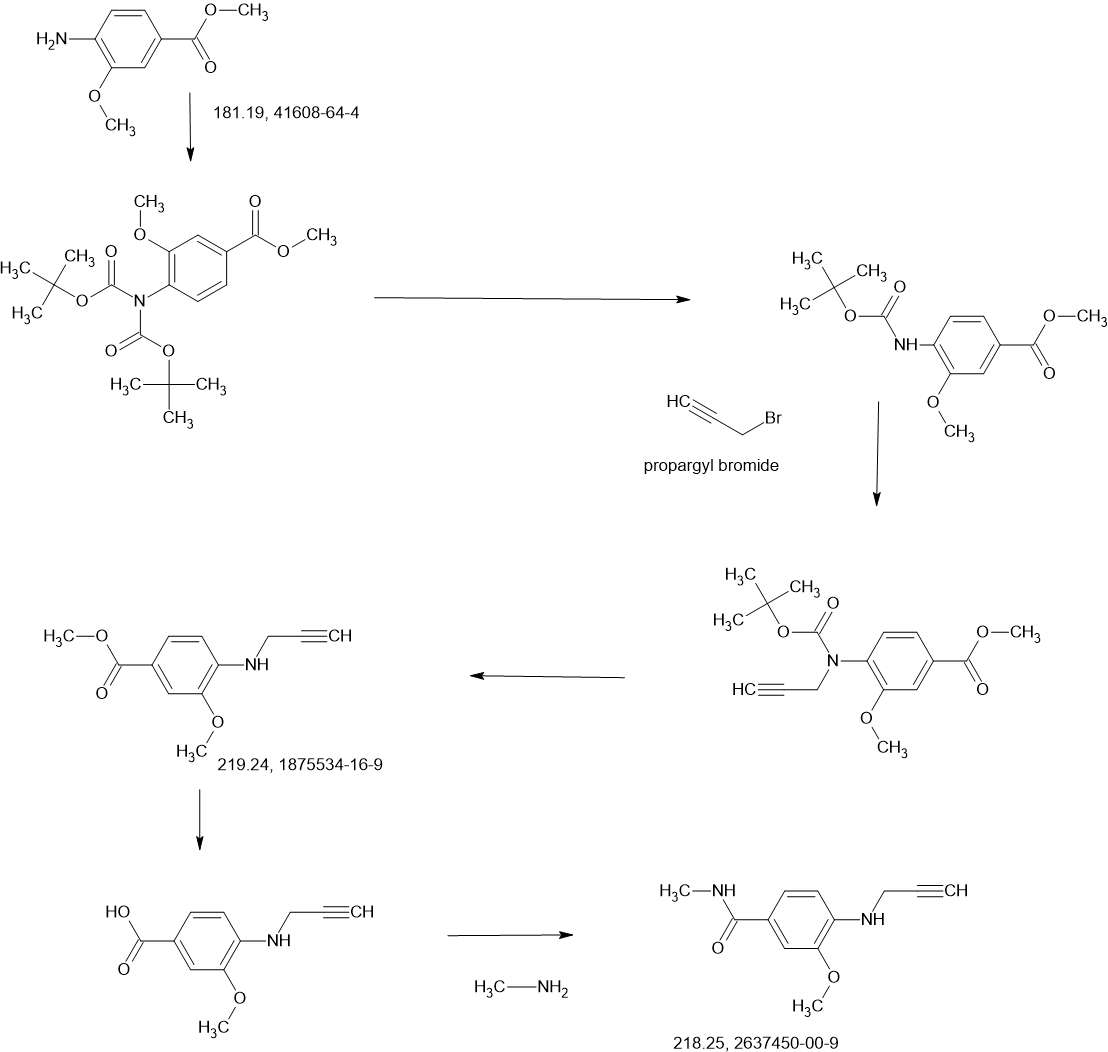
COUPLER
MAIN
REF
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00379
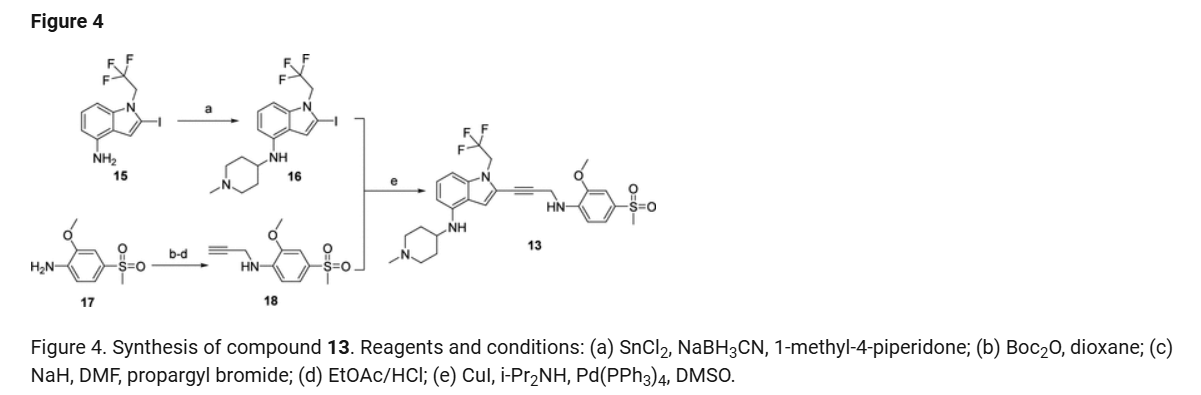
2-Iodo-1-(2,2,2-trifluoroethyl)-1H-indol-4-amine 15 was prepared from 4-nitroindole as described in
WO2017143291. 1
H NMR (400 MHz, dimethylsulfoxide [DMSO]-d6) δ ppm 9.19–10.88 (m, 2 H), 7.63
(d, J=8.34 Hz, 1 H), 7.16–7.25 (m, 1 H), 7.04–7.14 (m, 2 H), 5.14–5.33 (m, 2 H). LCMS (ES+
, m/z):
340.9 [(M+H)+
].
SnCl2.2H2O (398.11 mg, 1.76 mmol, 0.20 eq.) was added to a solution of 2-iodo-1-(2,2,2-trifluoroethyl)-
1H-indol-4-amine 15 (3.00 g, 8.82 mmol, 1.00 eq.) and 1-methylpiperidin-4-one (1.20 g, 10.61 mmol,
1.20 eq.) in MeOH (10.00 mL). The mixture was stirred at 25 °C for 3 hours (h), and then NaBH3CN
(2.77 g, 44.1 mmol, 5.00 eq.) was added, stirring at 25 °C for 69 h. Thin-layer chromatography (TLC)
indicated that the starting material was consumed, and the reaction mixture was filtered. The filtrate was
poured into H2O (200 mL) and extracted with ethyl acetate ([EtOAc] 200 mL2). The combined organic layers were washed with H2O (200 mL), dried over Na2SO4, and concentrated under reduced pressure to give a residue. The crude material was purified by flash column chromatography (Silica gel, petroleum ether (PE) : EtOAc = 0:1) and then by preparative high performance chromatography ([prep-HPLC] column: Phenomenex Luna C18 10040mm5 um; mobile phase: [H2O (0.2% Formic acid-acetonitrile [ACN])]; gradient: 10%–50% acetonitrile over 8.0 minutes) to yield 2-iodo-N-(1-methylpiperidin-4-yl)-1- (2,2,2-trifluoroethyl)-1H-indol-4-amine 16 (2.50 g, 5.72 mmol, 64.93% yield) as a light-brown solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 8.24 (br s, 1 H, formic acid salt), 7.17 (s, 1 H), 6.85–6.95 (m, 1 H), 6.78 (br d, J = 7.99 Hz, 1 H), 6.16 (br d, J = 7.63 Hz, 1 H), 5.44 (br d, J = 2.62 Hz, 1 H), 4.99 (q, J = 8.54 Hz, 2 H), 3.33 (br s, 1 H), 2.85 (br d, J = 9.66 Hz, 2 H), 2.25 (br s, 3 H), 2.07–2.18 (m, 2 H), 1.94 (br d, J = 12.04 Hz, 2 H), 1.46–1.58 (m, 2 H). LCMS (ES+, m/z): 438.0 [(M+H)+]. Boc2O (26.03 g, 119.26 mmol, 6.00 eq.) was added to a solution of 2-methoxy-4-(methylsulfonyl)aniline 17 (4.00 g, 19.88 mmol, 1.00 eq.) in dioxane (40.00 mL) at 25 o C (room temperature). The reaction mixture was stirred at 110 °C for 16 h. TLC and LCMS indicated that the reaction was completed, and it was concentrated in vacuo. The residue was purified by column chromatography (SiO2, PE/EtOAc = 10/1 to 1:1) to yield tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)carbamate (6.00 g, 19.92 mmol, 72.6% purity, 72% yield) as a yellow gum. 1 H NMR (400 MHz, DMSO-d6) δ ppm 8.33 (s, 1 H), 8.03 (d, J = 8.38 Hz, 1 H), 7.47 (dd, J = 8.38, 2.00 Hz, 1 H), 7.44 (d, J = 2.00 Hz, 1 H), 3.91 (s, 3 H), 3.18 (s, 3 H), 1.47 (s, 9 H). LCMS (ES+, m/z): 324.1 [(M+Na)+ ]. NaH (867.27 mg, 60% purity, 21.69 mmol, 3.00 eq.) was added in portions at 0 °C to a mixture of tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)carbamate (3.00 g, 7.23 mmol, 1.00 eq.) in dimethylformamide ([DMF] 30.00 mL) and stirred at 0 °C for 0.5 h. 3-Bromoprop-1-yne (3.23 g, 21.69 mmol, 3.00 eq.) was added to the reaction mixture, stirring at 0 °C for 2.5 h. TLC (Plate 1: PE : EtOAc = 1:1) and LCMS indicated that the starting material was consumed, and the product was detected. The reaction mixture was poured into a saturated solution of NH4Cl (200 mL) at 0 o C and was extracted with EtOAc (200 mL3). The combined organic phase was dried over Na2SO4, filtered, and concentrated
in vacuo. The residue was purified by column chromatography (SiO2, PE : EtOAc = 5:1 to 1:2) to give
tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)(prop-2-yn-1-yl)carbamate (3.00 g, 8.85 mmol,
74% purity, 90% yield) as a light-yellow gum.
1
H NMR (400 MHz, DMSO-d6) δ ppm 7.53–7.56 (m, 1 H), 7.46–7.53 (m, 2 H), 4.10–4.51 (m, 2 H), 3.90
(s, 3 H), 3.27 (s, 3 H), 3.17 (t, J = 2.32 Hz, 1 H), 1.27–1.39 (m, 9 H). LCMS (ES+
, m/z): 283.9 [(M+H-tBu)+].
A solution of 4M HCl/EtOAc (20.00 mL) was added to the solution of tert-butyl (2-methoxy-4-
(methylsulfonyl)phenyl)(prop-2-yn-1-yl)carbamate (3.00 g, 6.54 mmol, 1.00 eq.) in EtOAc (1.00 mL).
The reaction mixture was stirred at 25 °C for 2 h. TLC indicated that the starting material was consumed
completely. The reaction mixture was concentrated in vacuo to yield 2-methoxy-4-(methylsulfonyl)-N-
(prop-2-yn-1-yl)aniline 18 (1.80 g, 7.53 mmol, 85.3% yield, HCl salt) as a yellow solid.
1
H NMR (400 MHz, DMSO-d6) δ ppm 7.38 (dd, J = 8.40, 1.60 Hz, 1 H), 7.22 (d, J = 1.60 Hz, 1 H), 6.75
(d, J = 8.80 Hz, 1 H), 3.99 (d, J = 2.4 Hz, 2 H), 3.87 (s, 3 H) 3.10 (s, 3 H), 3.08 (t, J = 2.31 Hz, 1 H).
LCMS (ES+
, m/z): 240.1 [(M+H)+
].
i-Pr2NH (2.08 g, 20.58 mmol, 2.91 mL, 10 eq.), CuI (392.02 mg, 2.06 mmol, 1 eq), 2-iodo-N-(1-
methylpiperidin-4-yl)-1-(2,2,2-trifluoroethyl)-1H-indol-4-amine 16 (0.9 g, 2.06 mmol, 1 eq.) and
Pd(PPh3)4 (475.71 mg, 411.67 μmol, 0.2 eq.) was added to a solution of 2-methoxy-4-(methylsulfonyl)-N-
(prop-2-yn-1-yl)aniline 18 (622.16 mg, 2.47 mmol, 1.2 eq.) in DMSO (10 mL) at 45 °C under N2. The
reaction mixture was stirred at 45 °C for 1 h. TLC (DCM/MeOH=10:1, Rf = 0.3) indicated that the
starting material was consumed completely. It was poured into ethylenediaminetetraacetic acid ([EDTA]
20 mL) and stirred for 1 h, then extracted with EtOAc (40 mL3). The combined organic phase was washed with brine (40 mL), dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (SiO2, PE : EtOAc = 1:1 to dichloromethane (DCM) / MeOH = 10:1, Rf = 0.3), then by prep-HPLC (column: Phenomenex Luna(2) C18 25050 10u; mobile
phase: [water (0.1% trifluoroacetic acid)-ACN]; B%: 30%–50%, 20 min) to yield compound 13 (0.6 g,
1.09 mmol, 53.08% yield, 99.9% purity) as a light-yellow solid.
1 H NMR (400 MHz, DMSO-d6) δ ppm 1.41–1.54 (m, 2 H), 1.91 (br d, J = 11.00 Hz, 2 H), 1.95–2.08 (m,
2 H) 2.17 (s, 3 H), 2.68–2.80 (m, 2 H), 3.10 (s, 3 H), 3.20–3.29 (m, 1 H), 3.89 (s, 3 H), 4.36 (d,
J = 6.24 Hz, 2 H), 4.92 (q, J = 9.09 Hz, 2 H), 5.49 (d, J = 7.95 Hz, 1 H), 6.15 (d, J = 7.83 Hz, 1 H),
6.50 (t, J = 6.24 Hz, 1 H), 6.68 (d, J = 8.19 Hz, 1 H), 6.89 (d, J = 8.44 Hz, 1 H), 6.99 (t, J = 8.01 Hz,
1 H), 7.09 (s, 1 H), 7.25 (d, J = 1.83 Hz, 1 H), 7.39 (dd, J = 8.31, 1.83 Hz, 1 H). LCMS (ES+, m/z):
549.3 [(M+H)+
]
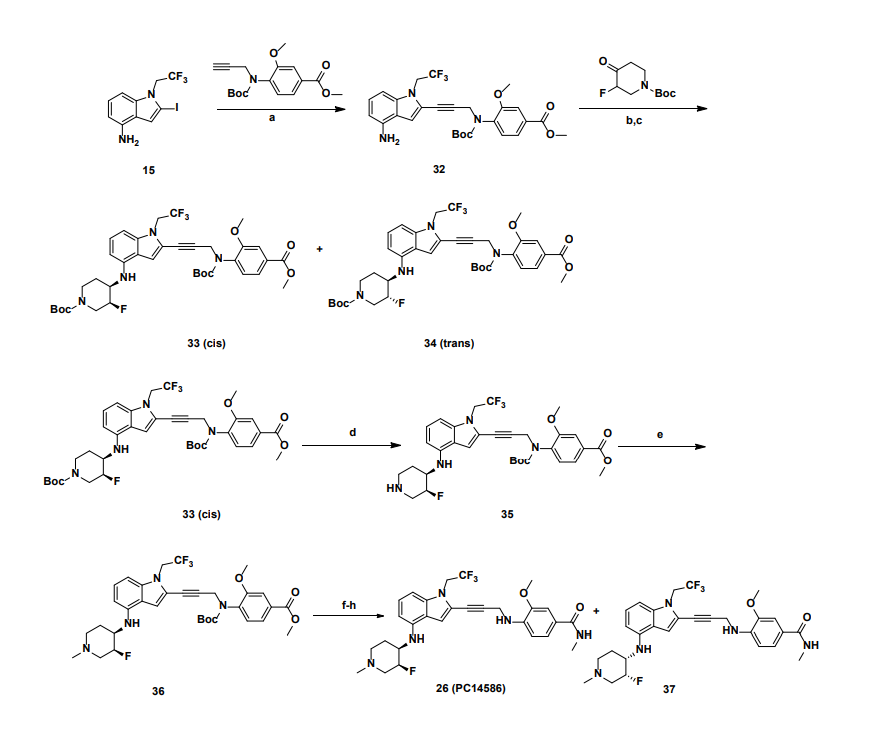
a
Reagents and conditions: (a) Pd(PPh3)4, CuI, diisopropylamine, DMSO, 20 °C, 1 h; (b) TMSCl, DMF, 0 °C, 0.5 h;
(c) BH3.THF, 0 °C, 0.5 h; (d) EtOAc/HCl, 20 °C, 1 h; (e) 10 eq. (CH2O)n, NaBH3CN, MeOH, 20 °C, 16 h; f)
LiOH.H2O, MeOH, 40 °C, 12 h; g) MeNH3Cl, HOBT, EDCI, TEA, DCM, RT, 16 h; h) Chiral SFC separation
PATENTS
WO2023016434 36%
WO2021061643
US20230024905
WO2023016434 Jacobio Pharmaceuticals Co., Ltd.
WO2023225477 PMV Pharmaceuticals, Inc.
US20230024905 PMV Pharmaceuticals, Inc.
WO2021061643 PMV Pharmaceuticals, Inc.
WO2021262483, PMV Pharmaceuticals, Inc.
WO2023196993 PMV Pharmaceuticals, Inc.
WO2021262484 WO2021262541
- [1]. Li Sujing, et al. Heteroarylalkyne compounds for targeting mutant of p53 and their preparation. World Intellectual Property Organization, WO2023016434 A1. 2023-02-16.[2]. Vu BT, et al. Discovery of Rezatapopt (PC14586), a First-in-Class, Small-Molecule Reactivator of p53 Y220C Mutant in Development. ACS Med Chem Lett. 2024 Nov 4;16(1):34-39. [Content Brief][3]. Spiegelberg D, et al. Targeting mutant p53: Evaluation of novel anti-p53R175H monoclonal antibodies as diagnostic tools. Sci Rep. 2025 Jan 6;15(1):1000. [Content Brief][4]. Schram A M, et al. 691TiP PYNNACLE phase II trial of rezatapopt (PC14586) in solid tumors with a TP53 Y220C mutation[J]. Annals of oncology, 2024, 35: S535-S536.
//////////Rezatapopt, PC 14586, 5W59S33KC9
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Resigratinib
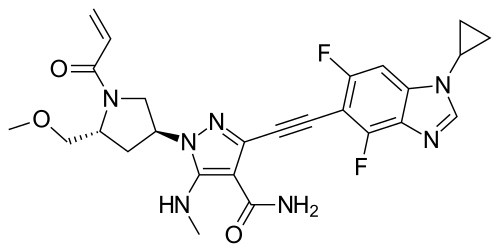
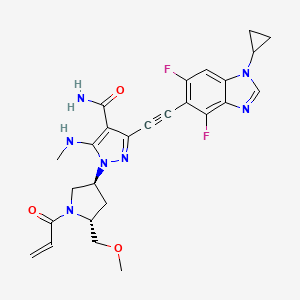
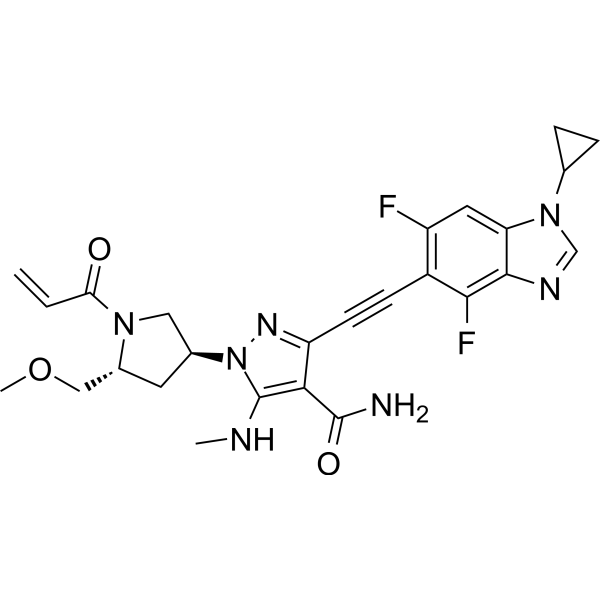
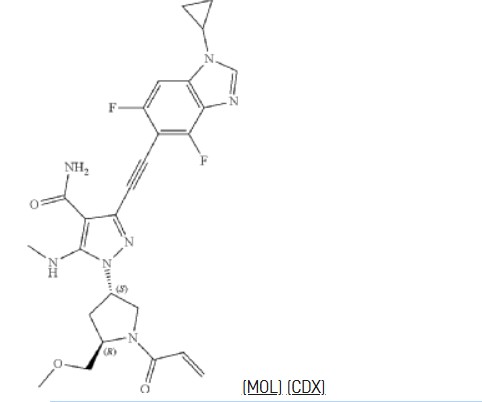
Resigratinib, KIN 3248
CAS 2750709-91-0
C26H27F2N7O3
523.5 g/mol
3-[2-(1-cyclopropyl-4,6-difluorobenzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-prop-2-enoylpyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
- 3-[2-(1-Cyclopropyl-4,6-difluoro-1H-benzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(1-oxo-2-propen-1-yl)-3-pyrrolidinyl]-5-(methylamino)-1H-pyrazole-4-carboxamide
- 3-[2-(1-cyclopropyl-4,6-difluorobenzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-prop-2-enoylpyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
Resigratinib (KIN-3248) is an experimental anticancer medication which acts as a fibroblast growth factor receptor inhibitor (FGFRi) and is in early stage human clinical trials.[1][2][3]
KIN-3248 is a small molecule that targets and inhibits oncogenic fibroblast growth factor receptors (FGFRs). It was designed to mainly target FGFR2 and FGFR3 alterations, which act as oncogenic drivers in 10-20% of cholangiocarcinoma and 20-35% of urothelial cancers, respectively. While effective, disease progression may occur 6 to 8 months after treatment with currently approved FGFR inhibitors is started, and this effect is usually associated with on-target resistance mutations in the kinase domain of FGFR. Therefore, the broad inhibition of FGFR isoforms may be effective against different types of tumors. The safety, tolerability, pharmacokinetics, and preliminary efficacy of KIN-3248 are currently being evaluated in adults with advanced tumors harboring FGFR2 and/or FGFR3 gene alterations. In February 2023, Kinnate Biopharma received Fast Track designation from the FDA for KIN-3248 to treat unresectable, locally advanced or metastatic cholangiocarcinoma (CCA).
SCHEME
COUPLER
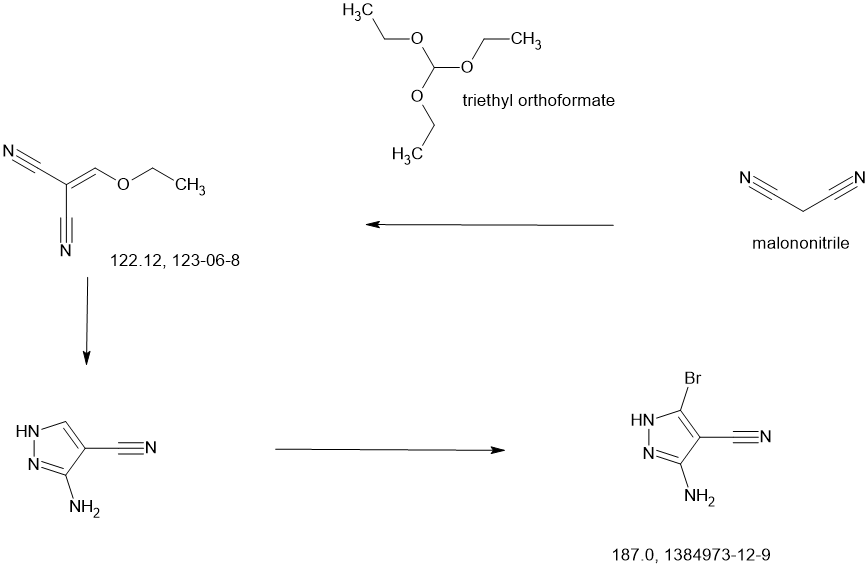
COUPLER
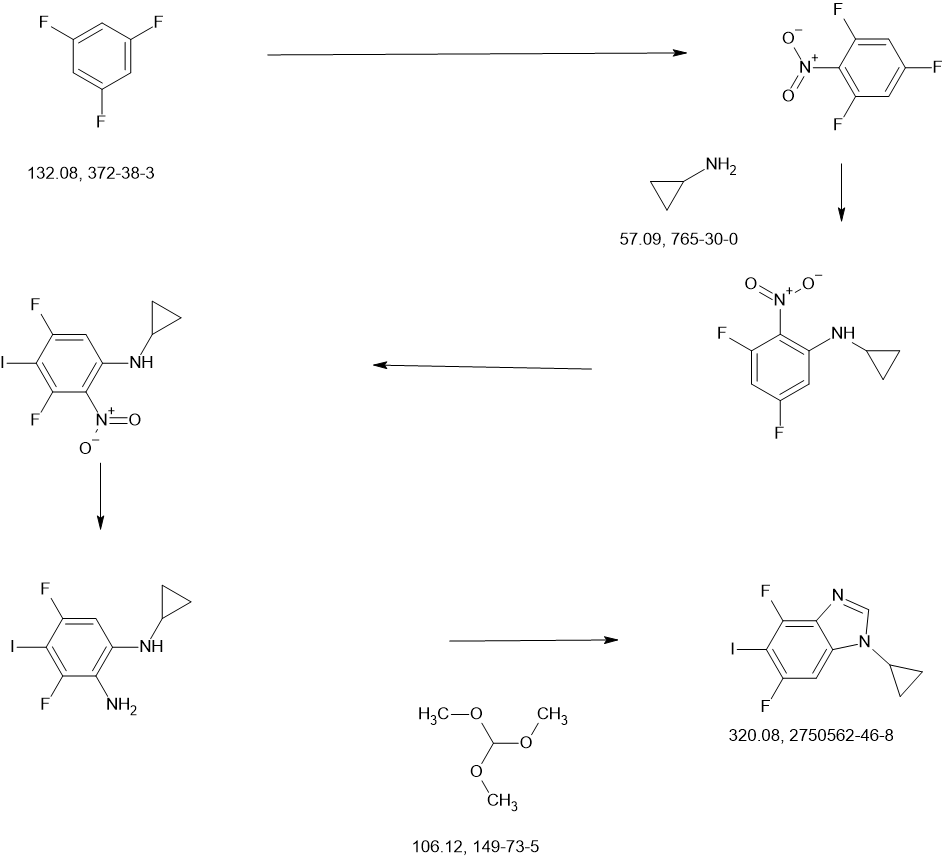
MAIN
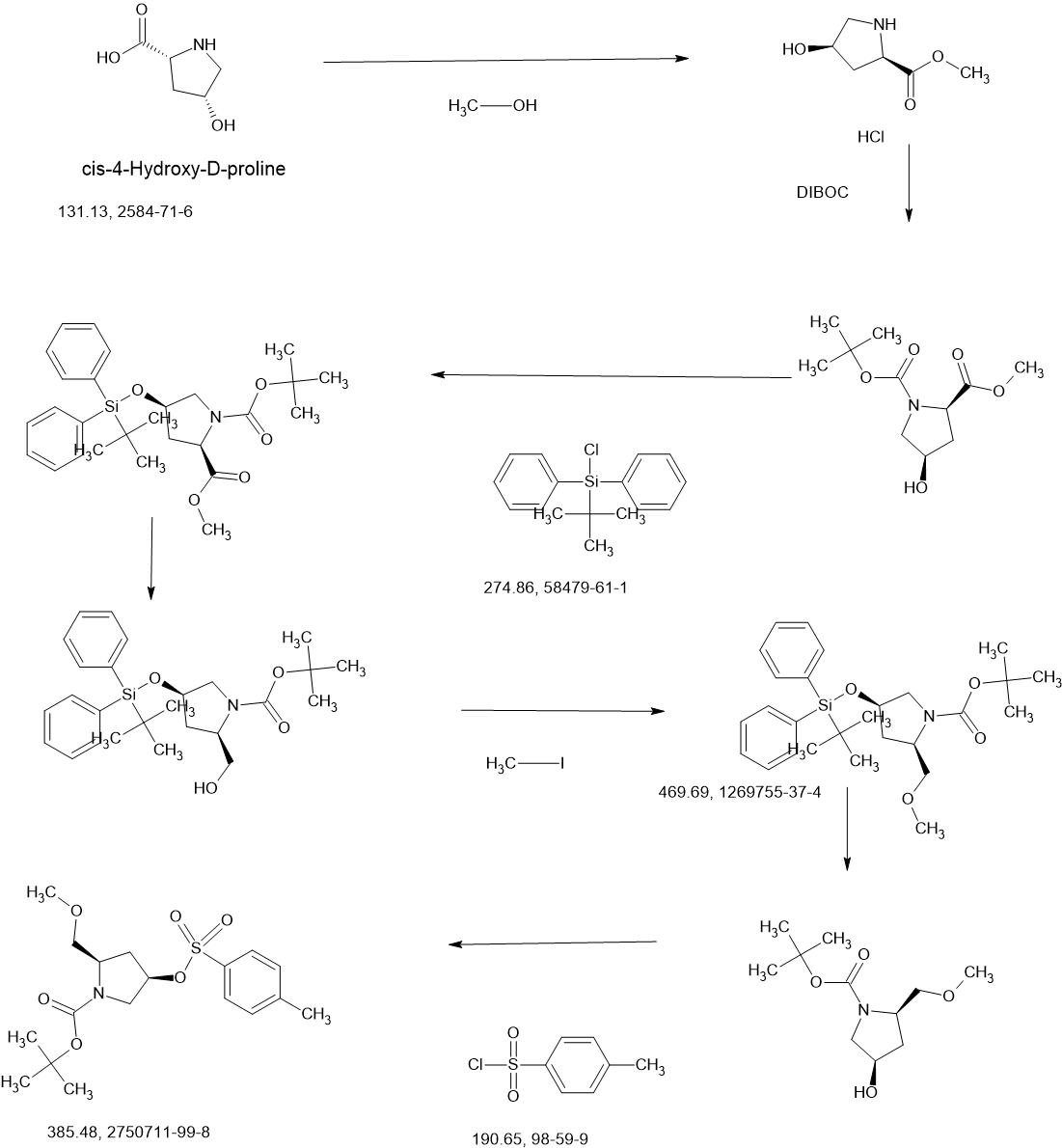
CONTINUED………….
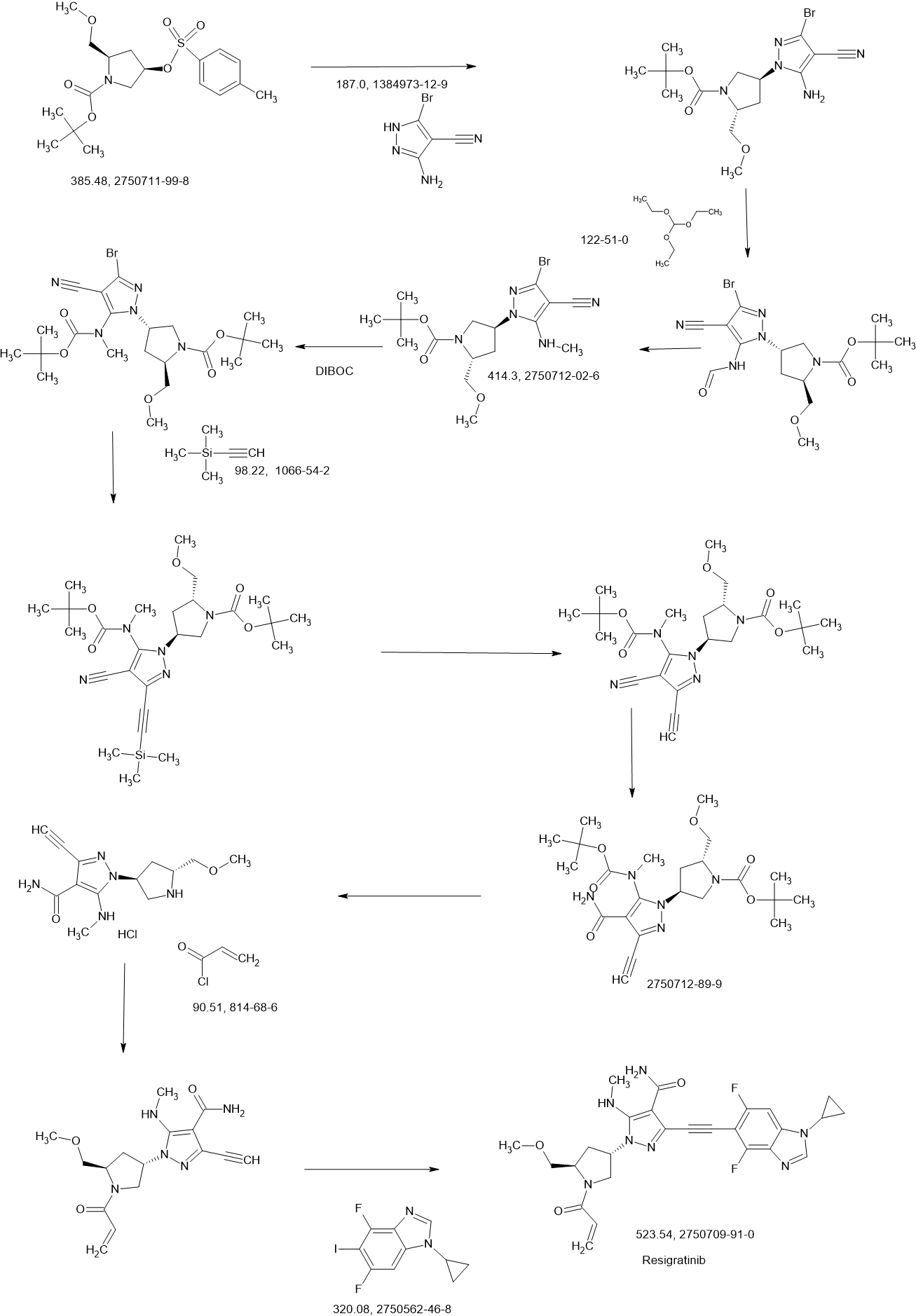
REF
https://patents.google.com/patent/US11345681B1/en
Example 78
3-[2-(1-Cyclopropyl-4,6-difluoro-1,3-benzodiazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
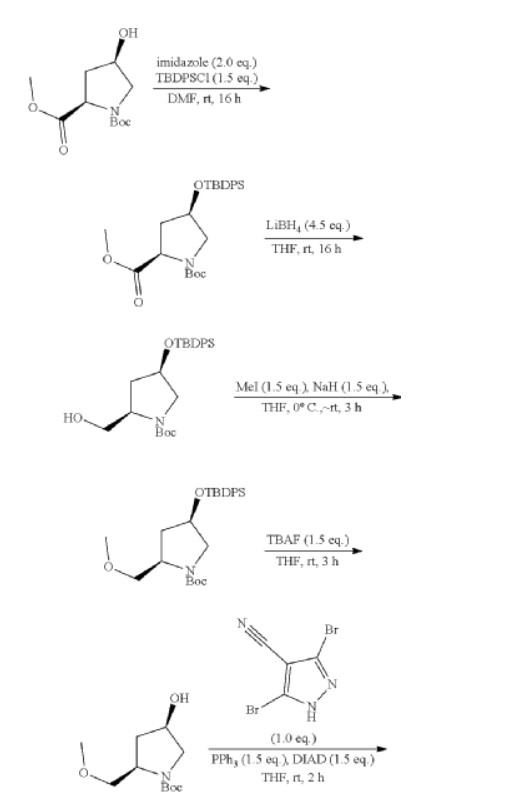
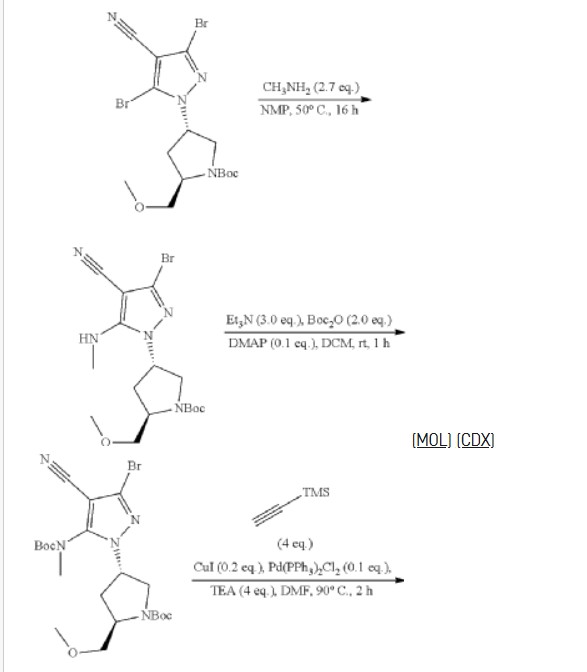


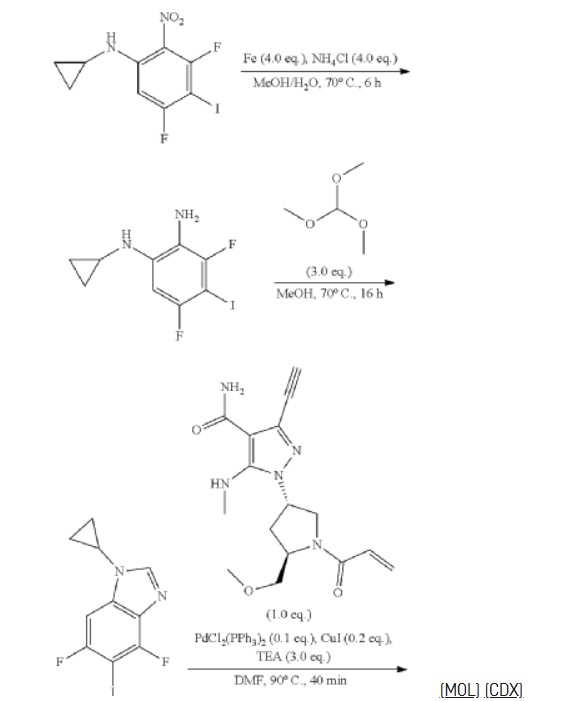
| Step 1: 1-(Tert-butyl) 2-methyl (2R,4R)-4-((tert-butyldiphenylsilyl)oxy)pyrrolidine-1,2-dicarboxylate |
| Step 2: Tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(hydroxymethyl)pyrrolidine-1-carboxylate |
| Step 3: Tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 5: Tert-butyl (2R)-4-(3,5-dibromo-4-cyanopyrazol-1-yl)-2-methoxymethyl)pyrrolidine-1-carboxylate |
| Step 6: Tert-butyl (2S,4R)-4-[3-bromo-4-cyano-5-(methylamino)pyrazol-1-yl]-2-(methoxymethyl)pyrrollidine-1-carboxylate |
| Step 7: (2R,4S)-4-[3-bromo-5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyanopyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 8: Tert-butyl (2R,4S)-4-(5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-[2-(trimethylsilyl)ethynyl]pyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 9: Tert-butyl (2R,4S)-4-(5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 10: Tert-butyl (2R,4S)-4-[5-[(tert-butoxycarbonyl)(methyl)amino]-4-carbamoyl-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 11: 3-Ethynyl-1-[(3S,5R)-5-(methoxymethyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide dihydrochloride |
| Step 12: 3-Ethynyl-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide |
| Step 17: 3-[2-(1-Cyclopropyl-4,6-difluoro-1,3-benzodiazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide |
PATENT
WO2021247969 Kinnate Biopharma Inc EG78
WO2023107980 solid state forms, Kinnate Biopharma Inc
WO2023107979 FGFR kinase inhibitor, Kinnate Biopharma Inc
References
- Franovic A, Mohan A, Uryu S, Wu Q, Jiang P, Miller N, et al. (February 2022). “Activity of KIN-3248, a next-generation pan-FGFR inhibitor, against acquired FGFR-gatekeeper and molecular-brake drug resistance mutations”. Journal of Clinical Oncology. 40 (4_suppl): 461. doi:10.1200/JCO.2022.40.4_suppl.461.
- Harding JJ, Perez CA, Kato S, Sharma M, Garmezy B, Quah CS, et al. (February 2023). “First in human (FIH) phase 1/1b study evaluating KIN-3248, a next-generation, irreversible pan-FGFR inhibitor (FGFRi), in patients (pts) with advanced cholangiocarcinoma (CCA) and other solid tumors harboring FGFR2 and/or FGFR3 gene alterations”. Journal of Clinical Oncology. 41 (4_suppl): TPS637-TPS637. doi:10.1200/JCO.2023.41.4_suppl.TPS637. S2CID 256257314.
- Wang Z, Anderson KS (2022). “Therapeutic Targeting of FGFR Signaling in Head and Neck Cancer”. Cancer Journal (Sudbury, Mass.). 28 (5): 354–362. doi:10.1097/PPO.0000000000000615. PMC 9523489. PMID 36165723.
| Identifiers | |
|---|---|
| CAS Number | 2750709-91-0 |
| PubChem CID | 162381323 |
| UNII | W728TB393W |
| Chemical and physical data | |
| Formula | C26H27F2N7O3 |
| Molar mass | 523.545 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- [1]. Tyhonas JS, et al. Discovery of KIN-3248, An Irreversible, Next Generation FGFR Inhibitor for the Treatment of Advanced Tumors Harboring FGFR2 and/or FGFR3 Gene Alterations. J Med Chem. 2024 Feb 8;67(3):1734-1746. [Content Brief][2]. Balasooriya ER, et al. The Irreversible FGFR Inhibitor KIN-3248 Overcomes FGFR2 Kinase Domain Mutations. Clin Cancer Res. 2024 May 15;30(10):2181-2192. [Content Brief]
/////////Resigratinib, Pan-FGFR Inhibitor KIN-3248, KIN 3248, Pan-fibroblast Growth Factor Receptor Inhibitor KIN-3248, W728TB393W
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Redafamdastat
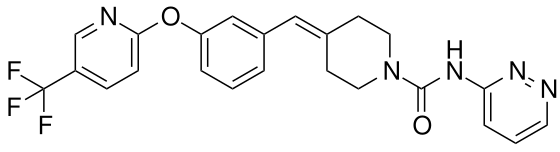
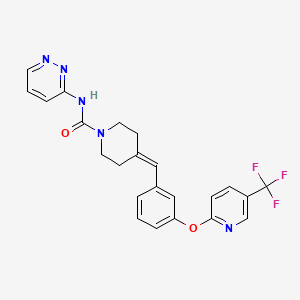
Redafamdastat
cas 1020315-31-4, PF 04457845
JZP-150; JZP150; PF-04457845; PF-4457845; PF04457845; PF4457845, Q7119045
WeightAverage: 455.441
Monoisotopic: 455.156909393
Chemical FormulaC23H20F3N5O2
- 1-Piperidinecarboxamide, N-3-pyridazinyl-4-[[3-[[5-(trifluoromethyl)-2-pyridinyl]oxy]phenyl]methylene]-
- N-pyridazin-3-yl-4-[[3-[5-(trifluoromethyl)pyridin-2-yl]oxyphenyl]methylidene]piperidine-1-carboxamide
Redafamdastat (INNTooltip International Nonproprietary Name; developmental code names JZP-150, PF-04457845) is an inhibitor of the enzyme fatty acid amide hydrolase (FAAH), with an IC50Tooltip half-maximal inhibitory concentration of 7.2 nM, and both analgesic and anti-inflammatory effects in animal studies comparable to those of the cyclooxygenase inhibitor naproxen.[1] It was being developed by Jazz Pharmaceuticals for the treatment of alcoholism, pain, and post-traumatic stress disorder (PTSD) and reached phase 2 clinical trials.[2][3] However, development of the drug was discontinued in December 2023.[2]
SCHEME
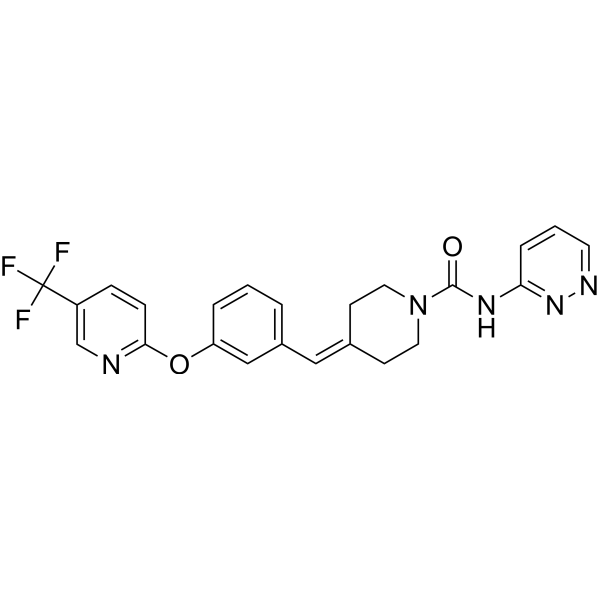
PAPER
ACS Medicinal Chemistry Letters (2011), 2(2), 91-96 86%
PATENT
WO2008047229 86%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008047229&_cid=P11-MCY7EC-33944-1
Example 1a
Synthesis of N-pyridin-3-yl-4-(3-(f5-(trifluoromethyl)pyridin-2-ylloxy)benzylidene)piperidine-1-carboxamide
Phenyl pyridin-3-ylcarbamate
To a stirred solution of 3-aminopyridine (51.7 g, 0.549 moles) in THF (900 mL) at -10 0C was added pyridine (52.1 g, 0.659 moles) in a stream over a 10 min period, followed by the dropwise addition of phenyl chloroformate (90 g, 0.575 moles) over a 20 min period. The reaction tempature increased to 5 0C. A precipitate formed during the addition. The resulting suspension was stirred at temperatures reaching ambient temperature over the next 3 h. The reaction mixture was partitioned between water (2 L) and EtOAc (1.5 L). The aqueous portion was extracted with EtOAc (1 L). The combined organic portions were dried (MgSO4) and concentrated in vacuo to a damp solid residue. This was suspended in
EtOAc:ether (1 :1 , 600 ml_). The resulting suspension was stirred at -10 0C for 2 h and filtered. The solid was rinsed with EtOAσether (1 :1 , 100 ml.) and pressed dry under suction. Further drying in vacuo at 35 0C for 7 h provided 104 g (88%) of product. Analysis, Calcd for Ci2H10N2O2: C, 67.28; H, 4.71 ; N, 13.08. Found: C, 67.15; H, 4.76; N, 12.87.
Step i
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol
3-Hydroxymethyl-phenol (5.00 g, 40.3 mmol, from Lancaster Synthesis), 2-chloro-5-trifluoromethyl-pyridine (7.31 g, 40.3 mmol, from TCI America) and potassium carbonate (6.96 g, 50.3 mmol) were suspended in dimethylformamide (80 mL) and heated to 95 0C. After stirring for 16 h, the solvent was distilled off in vacuo at 65 0C, and a residue was partitioned between water and heptane/ethyl acetate (1 :1 ). The organic layer was separated and the aqueous was extracted again with heptane/ethyl acetate (1 :1 ). The combined organic layer was dried over sodium sulfate, filtered and concentrated to give a residue. The residue was purified by silica gel chromatography (10-60%, EtOAc:heptane) to afford the desired product (5.70 g, 53% yield) as a light yellow oil. 1H NMR (400 MHz, CDCI3) δ ppm 4.73 (s, 2 H) 7.02 (dt, J=8.66, 0.57 Hz, 1 H) 7.04 – 7.11 (m, J=8.06, 2.40, 0.50, 0.50 Hz, 1 H) 7.15 – 7.19 (m, 1 H) 7.25 (ddd, J=8.39, 1.60, 0.80 Hz, 1 H) 7.42 (t, J=7.87 Hz, 1 H) 7.90 (ddd, J=8.67, 2.55, 0.50 Hz, 1 H) 8.43 (td, J=1.68, 0.84 Hz, 1 H).
Step 2
2-(3-Chloromethyl-phenoxy)-5-trifluoromethyl-pyridine
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol from Step 1 (4.68 g, 17.4 mmol), in
dichloromethane (46 mL), was cooled to 0 0C, and treated dropwise with thionyl chloride (1.40 mL, 19.1 mmol). The reaction mixture was allowed to warm to ambient temperature and was stirred for 30 rηjn. Toluene (10 mL) was added and the mixture was concentrated by evaporation to form a residue. The residue was evaporated again from toluene and dried under high vacuum to afford the desired product (4.88 g, 98% yield) as an oil. 1H NMR (400 MHz, CDCI3) δ ppm 4.60 (s, 2 H) 7.03 (d, J=8.70 Hz, 1 H) 7.11 (ddd, J=8.09, 2.35, 0.94 Hz, 1 H) 7.20 (t, J=2.03 Hz, 1 H) 7.26 – 7.31 (m, 1 H) 7.42 (t, J=7.88 Hz, 1 H) 7.91 (dd, J=8.67, 2.53 Hz, 1 H) 8.44 (dd, J=1.51 , 0.90 Hz, 1 H).
Step 3
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester
2-(3-Chloromethyl-phenoxy)-5-trifluoromethyl-pyridine (4.88 g, 17.0 mmol) from Step 2 was treated neat with triethylphosphite (4.36 mL, 25.4 mmol) and heated to 150 0C. After 6 h, the reaction mixture was cooled, treated with an additional 0.5 mL triethylphosphite (2.9 mmol) and reheated to 150 0C. After 6 h, the reaction mixture was removed from the heat and slowly treated with heptane (about 60 mL) while stirring to afford a white solid. The solid was collected by filtration, washed with heptane and dried in a vacuum oven for 16 h at 45 0C to afford a white powder (5.99 g, 91% yield). MS (APCI) M+1= 390.1 ; 1H NMR (400 MHz, CDCI3) δ ppm 1.26 (t, J=7.02 Hz, 6 H) 3.18 (d, J=21.83 Hz, 2 H) 3.99 – 4.10 (m, 4 H) 7.01 (d, J=8.58 Hz, 1 H) 7.03 – 7.08 (m, 1 H) 7.12 (q, J=2.21 Hz, 1 H) 7.19 – 7.24 (m, 1 H) 7.38 (t, J=7.90 Hz, 1 H) 7.90 (dd, J=8.58, 2.53 Hz, 1 H) 8.43 (dd, J=1.66, 0.88 Hz, 1 H).
Step 4
4-[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1 -carboxylic acid tert-butyl ester
[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester (2.3 g, 6.0 mmol) from Step 3 and 1 ,4,7,10, IS-pentaoxacyclopentadecane (15-Crown-5, 0.03 ml_, 0.15 mmol) were combined in THF (10 ml_). The mixture was cooled to 0 °C and sodium hydride (240 mg, 60% dispersion in mineral oil, 6.0 mmol) was added. The reaction was warmed to room temperature, stirred for 30 minutes and then cooled back to 0 0C. A solution of 4-oxo-piperidine-1-carboxylic acid tert-butyl ester (1.2 g, 6.0 mmol) in THF (6 ml_) was added and the reaction was warmed to room temperature. After 16 hours, water was added and the layers were separated. The aqueous layer was extracted with EtOAc (2X200 mL) and the combined organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to a thick oil. Treatment of the oil with hot isopropyl ether (45 mL) provided the title compound as a white solid (1.88 g). 1H NMR (400 MHz, CD3OD) δ ppm 1.46 (s, 9 H) 2.34 (td, J=5.85, 1.18 Hz, 2 H) 2.46 (td, J=5.87, 1.07 Hz, 2 H) 3.37 – 3.44 (m, 2 H) 3.45 – 3.57 (m, 2 H) 6.41 (s, 1 H) 6.92 – 7.04 (m, 2 H) 7.06 – 7.17 (m, 2 H) 7.31 – 7.54 (m, 1 H) 8.08 (ddd, J=8.74, 2.59, 0.56 Hz, 1 H) 8.42 (td, J=1.73, 0.90 Hz, 1 H).
Step 5
2-(3-Piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine hydrochloride
4-[3-(5-Trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1-carboxylic acid tert-butyl ester (1.35 g, 3.11 mmol) from Step 4 was dissolved in CH2CI2 (30 mL) and treated with HCI in diethyl ether (10 mL, 2.0 M, 20 mmol). After 16 hours the reaction was concentrated in vacuo to form a residue and the residue was suspended in acetonitrile (10 mL) to yield a solid. Filtration of the solid provided the title compound as a white solid (1.1 g). 1H NMR (400 MHz, CD3OD) δ ppm 2.62 (td, J=6.11 , 0.91 Hz, 2 H) 2.67 – 2.81 (m, 2 H) 3.14 – 3.21 (m, 2 H) 3.22 – 3.29 (m, 2 H) 6.56 (s, 1 H) 6.99 – 7.09 (m, 2 H) 7.10 – 7.18 (m, 2 H) 7.42 (t, J=7.91 Hz, 1 H) 8.09 (ddd, J=8.74, 2.60, 0.33 Hz, 1 H) 8.41 (td, J=1.63, 0.74 Hz, 1 H).
Step 6
2-(3-Piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine hydrochloride (800 mg, 2.16 mmol, from Step 5), phenyl pyridin-3-ylcarbamate (508 mg, 2.37 mmol) and diisopropylethylamine (0.75 mL, 4.52 mmol) were combined in acetonitrile (10 mL) and stirred at room temperature. After 16 hours, the reaction was concentrated forming a residue and the residue was partitioned between EtOAc and water. The organic layer was separated, washed with 5% NaOH (aq), dried over anhydrous sodium sulfate, filtered and concentrated. Treatment of the residue with hot isopropyl ether and purified from isopropyl ether/methanol provided the title compound as a white solid (574 mg). MS (APCI 10V) AP+ 455.3, 376.2, 335.2, AP- 453.2; 1H NMR (400 MHz, CD3OD) δ ppm 2.46 (td, J=5.86, 0.97 Hz, 2 H) 2.58 (td, J=5.82, 1.16 Hz, 2 H) 3.51 – 3.60 (m, 2 H) 3.61 – 3.70 (m, 2 H) 6.46 (s, 1 H) 6.98 – 7.07 (m, 2 H) 7.09 – 7.19 (m, 2 H) 7.34 (ddd, J=8.41 , 4.81 , 0.65 Hz, 1 H) 7.40 (td, J=7.69, 0.74 Hz, 1 H) 7.91 (ddd, J=8.38, 2.58, 1.44 Hz, 1 H) 8.08 (ddd, J=8.73, 2.61 , 0.55 Hz, 1 H) 8.16 (dd, J=4.84, 1.06 Hz, 1 H) 8.43 (td, J=1.74, 0.91 Hz, 1 H) 8.58 (d, J=1.88 Hz, 1 H).
Example 1b
Large scale synthesis of N-pyridin-3-yl-4-(3-{[5-(trifluoromethyl)pyridin-2-ylloxy)benzylidene)piperidine-1- carboxamide
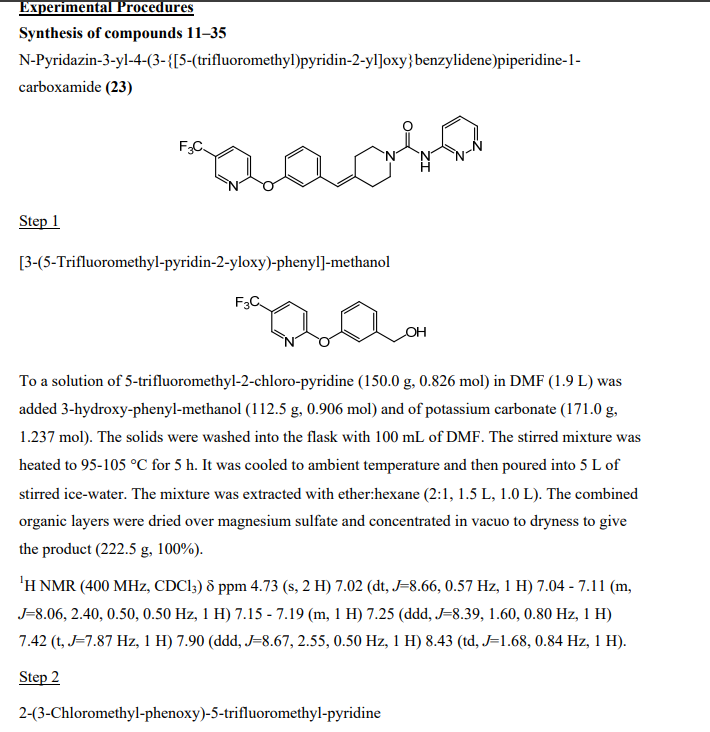
Step 1 : Preparation of r3-(5-Trifluoromethyl-pyridin-2-yloxy)-phenyll-methanol
To a solution of S-trifluoromethyl^-chloro-pyridine (150.0 g, 0.826 mol) in DMF (1.9 L) was added 3-hydroxy-phenyl-methanol (112.5 g, 0.906 mol) and of potassium carbonate (171.0 g, 1.237 mol). The solids were washed into the flask with 100 mL of DMF. The stirred mixture was heated to 95-105 0C for 5 h. It was cooled to ambient temperature and then poured into 5 L of stirred ice-water. The mixture was extracted with etheπhexane (2:1 , 1.5 L, 1.0 L). The combined organic layers were dried over magnesium sulfate and concentrated in vacuo to dryness to give the product (222.5 g, 100%).
Step 2: Preparation of 2-(3-chloromethyl-phenoxy)-5-trifluoromethylpyridine
To a solution of [3-(5-trifluoromethyl-pyridin-2-yloxy)-phenyl]-methanol (281.0 g, 1.044 moles) in dichloromethane (2.0 L) at -5 0C was added dropwise over a 25 min period thionyl chloride (136.6 g, 1.148 mol). A few minutes into the addition, a white substance separated but this went into solution several minutes later. The reaction was stirred at ambient temperature for 1 h and then was concentrated in vacuo to near dryness (357 g). 200 mL of toluene was added to the residue and the solution was again concentrated in vacuo to near dryness. 200 mL of toluene was added and some solid (-8 g) was filtered off. The filtrate was concentrated in vacuo to -390 g of dark yellow liquid.
Step 3: Preparation of [3-(5-trifluoromethyl-pyridin-2-yloxy)-benzyll-phosphonic acid diethyl ester
A solution of 2-(3-chloromethyl-phenoxy)-5-trifluoromethylpyridine (-298 g, -1.036 mol) containing some toluene in triethyl phosphite (267.0 g, 1.551 mol) was heated to 135 °C-140 0C for 7 h. Boiling began at -110 0C and continued throughout the reaction. The solution was left standing at ambient temperature overnight and it solidified. The solid was suspended in etheπhexane (1 :2, 450 mL), and the suspension was stirred at ambient temperature for 3 h and filtered. The solid was rinsed with etherhexane (1 :2, 150 mL) and pressed dry under suction. Further drying in vacuo at 32 0C for 7 h provided 286.3 g (71 % – 2 steps from crude chloride) of product. The filtrate was concentrated in vacuo to remove the low boiling solvents. Triethyl phosphite (36.0 g, 0.217 mol) was added and the solution was heated to 130 0C for 2 h. The reaction was cooled to 100 0C and 300 mL of heptane was added slowly. A solid separated. As the temperature decreased to -30 0C, 150 mL of ether was added. The resulting suspension was left standing at ambient temperature overnight and was filtered. The solid was rinsed with etherheptane (1 :2, 75 mL) and pressed dry under suction. Further drying in vacuo at 32 0C for 7 h afforded and additional 35.7 g (9%) of product. Total yield = 322 g (80%). Anal. Calcd for C17H19 F3NO4P (389.31 ) : C, 52.45; H, 4.92; N, 3.60; F, 14.64; P, 7.96. Found : C, 52.73; H, 5.04; N, 3.58; F, 14.35; P, 7.74; chloride, <0.10%.
Step 4: Preparation of 4-f3-(5-trifluoromethyl-pyridin-2-yloxy)-benzylidenel-piperidine-1-carboxylic acid tert-butyl ester
To a stirred mixture of [3-(5-trifluoromethyl-pyridin-2-yloxy)-benzyl]-phosphonic acid diethyl ester (155.7 g, 0.40 mol) in tetrahydrofuran (800 mL) at -10 0C was added dropwise over a 5 min period 1.0 M tBuOK in tetrahydrofuran (420.0 mL, 0.42 mol). The temperature rose to -3 °C during the addition. The resulting red mixture was stirred between -6 0C and -10 0C for 2.5 h. A solution of tert-butyl 4-oxopiperidine-1-carboxylate (79.7 g, 0.40 mol) in tetrahydrofuran (300 mL) was added dropwise over a 5 min period. The temperature rose to 2 0C. The resulting red mixture was stirred at temperatures reaching 21 0C over the next 16 h. TLC showed product with no phosphonate present. The mixture was poured into 3.5 L of stirred ice-water. The resulting suspension was stirred at ambient temperature for 2.5 h and then was extracted with successive 1.0 L and 0.6 L portions of dichloromethane. The combined extracts were washed with 500 mL of brine, dried over magnesium sulfate and concentrated in vacuo to a thick semi solid residue. 250 mL of methyl t-butyl ether was added. The suspension was stirred at -10 0C for 2 h and filtered. Drying in vacuo at 25 CC for 66 h provided 85 g (49%) of product. The filtrate was concentrated in vacuo to a damp solid residue. This was taken up in 100 mL of methyl t-butyl ether. To the stirred suspension was added 300 mL of heptane and the resulting suspension was stirred at -10 0C for 2 h. The solid was filtered off, rinsed with 50 mL of methyl t-butyl etherheptane (1 :3) and pressed dry under suction. Further drying in vacuo at 34 °C for 6 h provided an additional 34.2 g (19.5%) of product. Total yield = 119.2 g (68.5%).
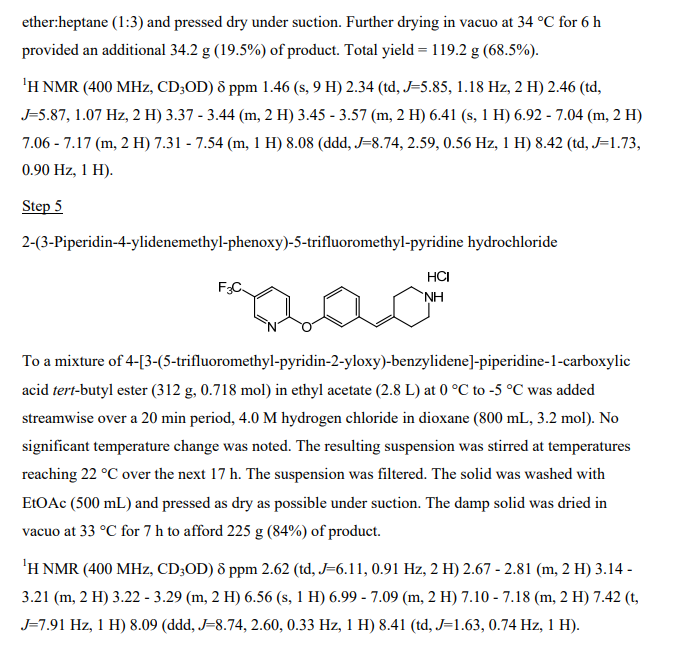
Step 5: Preparation of 2-(3-piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine, hydrochloride
To a mixture of 4-[3-(5-trifluoromethyl-pyridin-2-yloxy)-benzylidene]-piperidine-1-carboxylic acid fert-butyl ester (312 g, 0.718 mol) in ethyl acetate (2.8 L) at 0 0C to -5 0C was added streamwise over a 20 min period, 4.0 M hydrogen chloride in dioxane (800 mL, 3.2 mol). No significant temperature change was noted. The resulting suspension was stirred at temperatures reaching 22 0C over the next 17 h. The suspension was filtered. The solid was washed with EtOAc (500 mL) and pressed as dry as possible under suction. The damp solid was dried in vacuo at 33 0C for 7 h to afford 225 g (84%) of product.
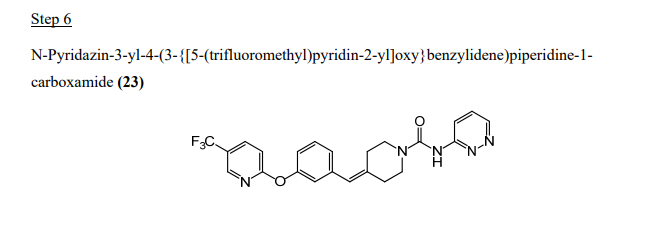
Step 6: Preparation of N-pyridin-3-yl-4-(3-(f5-(trifluoromethyl)pyridin-2-ylloxy}benzylidene)piperidine-1-carboxamide
To a mixture of 2-(3-piperidin-4-ylidenemethyl-phenoxy)-5-trifluoromethyl-pyridine (80.0 g, 0.216 mol) and phenyl pyridin-3-ylcarbamate (48.6 g, 0.227 mol) in acetonitrile (650 mL) was added dropwise diisopropylethyl amine (55.8 g, 0.432 mol). A solution formed after -45 min of stirring. The slightly turbid solution was stirred at ambient temperature for 18 h. TLC showed a prominent product spot with traces of both starting materials and two other fast moving spots. The solution was concentrated in vacuo to a viscous oil. This was partitioned between dichloromethane (600 mL) and water (500 mL). The aqueous layer was extracted with 200 mL of dichloromethane. The combined organic layers were washed with successive portions of 500 mL of 5% sodium hydroxide, and 200 mL of water, then dried over magnesium sulfate and concentrated in vacuo to 139.5 g of a viscous oil. This was dissolved in 350 ml_ of warm (50 0C) methyl t-butyl ether. Soon after a solution formed, solid began separating. The crystallizing mixture was kept at -10 0C for 4 h and filtered. The solid was rinsed with 60 ml_ of methyl t-butyl ether and pressed dry under suction. Further drying in vacuo at 28 0C for 16 h and then at 35 °C for 6 h provided 93.2 g (95%) of product.
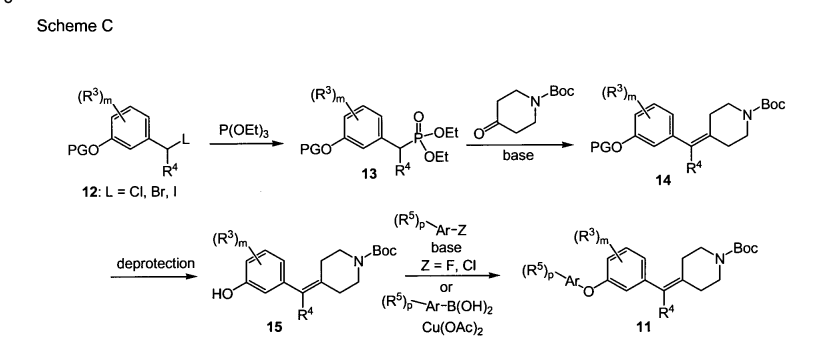
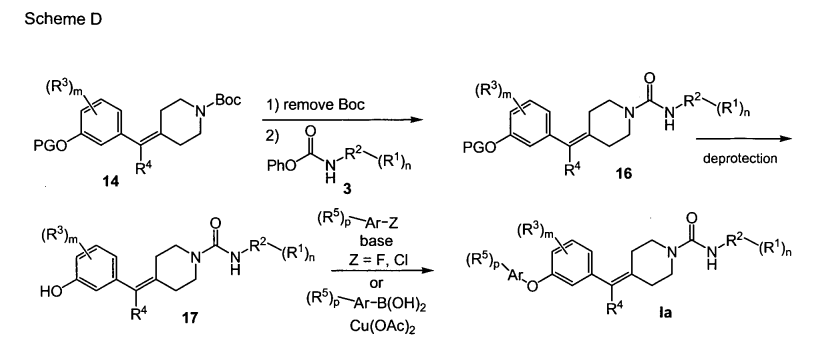
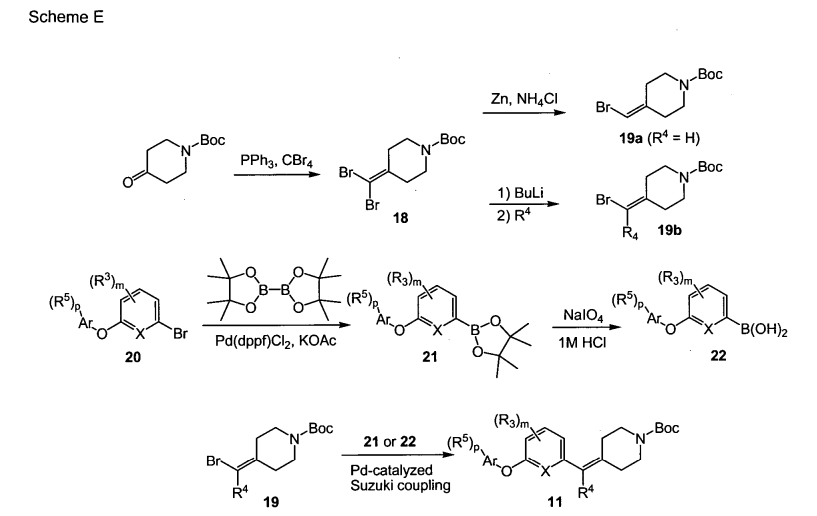
PAPER
ACS Medicinal Chemistry Letters (2011), 2(2), 91-96
https://pubs.acs.org/doi/10.1021/ml100190t
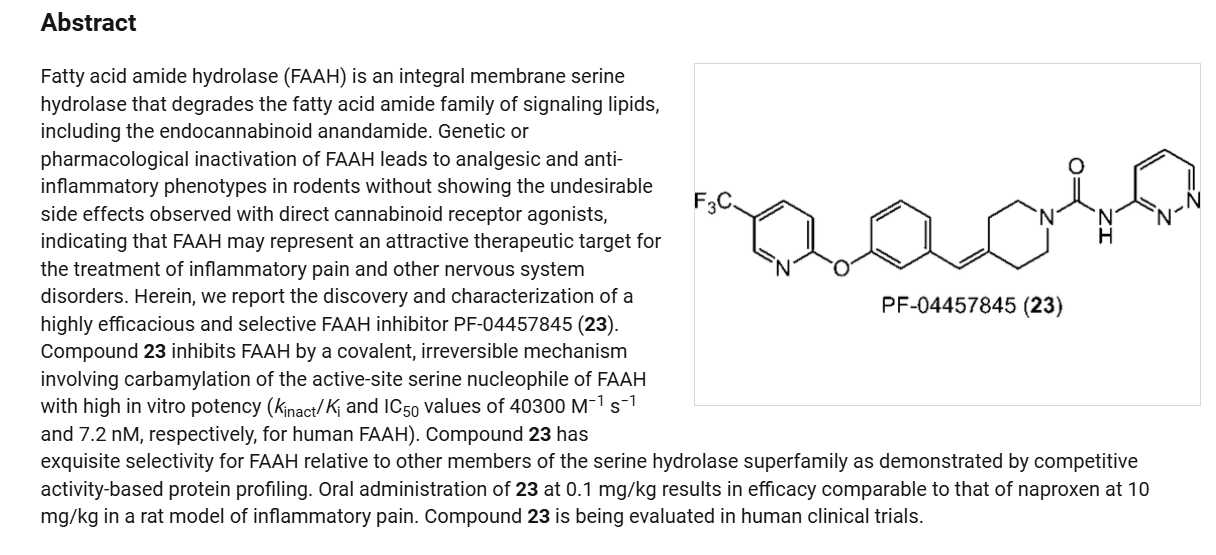





PAPER
Science (Washington, DC, United States) (2017), 356(6342), 1084-1087
Pfizer Products Inc.WO2008047229
References
- ^ Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, et al. (February 2011). “Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor”. ACS Medicinal Chemistry Letters. 2 (2): 91–96. doi:10.1021/ml100190t. PMC 3109749. PMID 21666860.
- ^ Jump up to:a b “JZP 150”. AdisInsight. 26 December 2023. Retrieved 16 August 2024.
- ^ “A Study of JZP150 in Adults With Posttraumatic Stress Disorder – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov.
- [1]. Johnson DS, et al. Discovery of PF-04457845: A Highly Potent, Orally Bioavailable, and Selective Urea FAAH Inhibitor. ACS Med Chem Lett. 2011 Feb 10;2(2):91-96. [Content Brief][2]. Ahn K, et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011 Jul;338(1):114-24. [Content Brief][3]. Buntyn RW, et al. Inhibition of Endocannabinoid-Metabolizing Enzymes in Peripheral Tissues Following Developmental Chlorpyrifos Exposure in Rats. Int J Toxicol. 2017 Jan 1:1091581817725272. [Content Brief]
////////Redafamdastat, PF 04457845, JZP-150, JZP150, PF-04457845, PF-4457845, PF04457845, PF4457845, Q7119045
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
ARAZASETRON BESYLATE
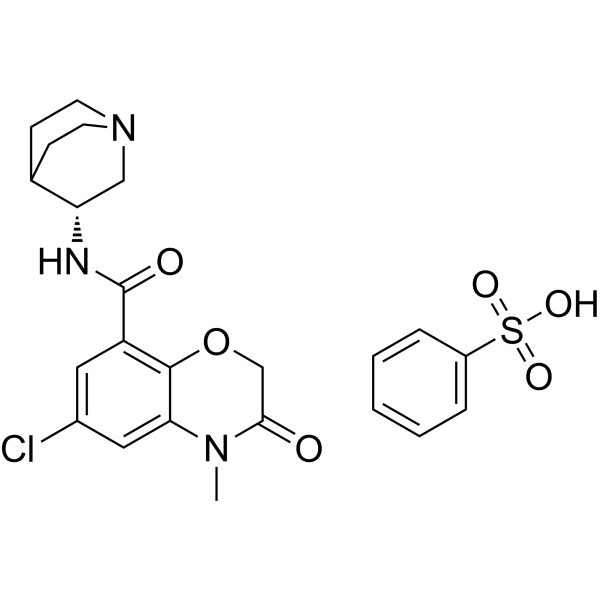
ARAZASETRON BESYLATE
R-Azasetron besylate, SENS-401
Cas 2025360-91-0
C17H20ClN3O3.C6H6O3S, 507.99, UXP39EQ477
2H-1,4-Benzoxazine-8-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-6-chloro-3,4-dihydro-4-methyl-3-oxo-, benzenesulfonate (1:1)
N-[(3R)-1-azabicyclo[2.2.2]octan-3-yl]-6-chloro-4-methyl-3-oxo-3,4-dihydro-2H-1,4-benzoxazine-8-carboxamide; benzenesulfonic acid
X HCL SALT, CAS , 2139305-21-6, Name2H-1,4-Benzoxazine-8-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-6-chloro-3,…
BASE: 2025360-90-9
.HCL SALT CAS, 2566443-39-6
Name 2H-1,4-Benzoxazine-8-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-6-chloro-3,…
BASE: 2025360-90-9
Base CHIRAL 2025360-90-9
- N-(3R)-1-Azabicyclo[2.2.2]oct-3-yl-6-chloro-3,4-dihydro-4-methyl-3-oxo-2H-1,4-benzoxazine-8-carboxamide
(R)-Azasetron besylate (SENS-401) is an orally active calcineurin inhibitor. (R)-Azasetron besylate reduces Cisplatin (HY-17394)-induced hearing loss and cochlear damage.
Arazasetron Besylate is the besylate salt form of the R-enantiomer of azasetron, a benzamide derivative and selective serotonin (5-hydroxytryptamine; 5-HT) receptor and calcineurin antagonist, with potential antinauseant and antiemetic, and otoprotective activities. Upon administration, arazasetron selectively binds to and inhibits 5-HT subtype 3 receptors (5-HT3R) located peripherally on vagus nerve terminals and centrally in the chemoreceptor trigger zone (CTZ) of the area postrema, which may result in suppression of nausea and vomiting. R-azasetron also targets and inhibits the activation of calcineurin, thereby preventing inner ear lesions, nerve degeneration, induction of apoptosis and sensory hair loss. This may prevent hearing loss. Calcineurin activation plays a key role in structural degeneration, swelling, synaptic uncoupling and the induction of apoptosis in the inner ear leading to hair cell loss and hearing loss.
SCHEME
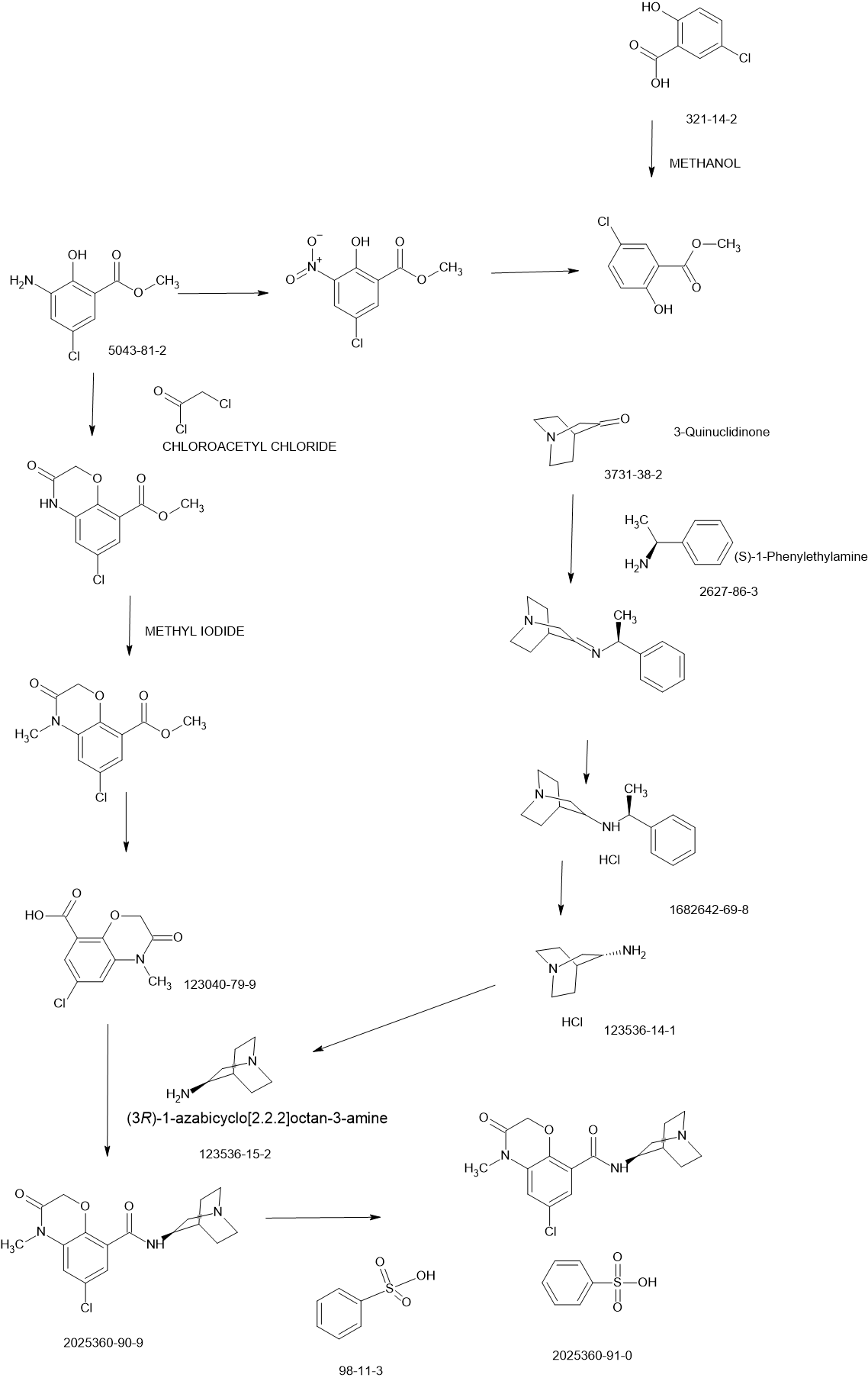
PATENTS
WO2017178645
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017178645&_cid=P22-MCX71S-95827-1
PATENTS
US20190083503
https://patentscope.wipo.int/search/en/detail.jsf?docId=US239434911&_cid=P22-MCX73M-97768-1
PATENTS
WO2010/133663
CN101786963
CN104557906
WO201717864
WO2021014014
WO2023175078
WO2023122719
Base WO2017178645
WO2021014014
WO2023175078
//////////ARAZASETRON BESYLATE, R-Azasetron besylate, UXP39EQ477, SENS-401, SENS 401
PRITELIVIR MESYLATE
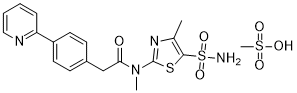
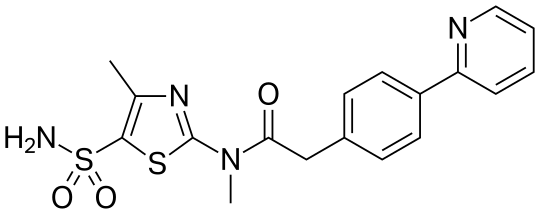
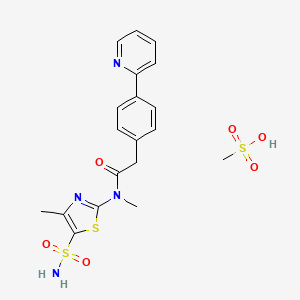
PRITELIVIR MESYLATE
CAS 1428333-96-3
1428321-10-1 HYDRATE
FREE FORM
AIC316 mesylate hydrate; BAY 57-1293 mesylate hydrate
BAY57-1293; BAY 57-1293; BAY-57-1293; BAY571293; BAY 571293; BAY-571293; AIC-316; AIC 316
| Molecular Weight | 516.61 |
|---|---|
| Synonyms | AIC316 mesylate hydrate; BAY 57-1293 mesylate hydrate |
| Formula | C19H24N4O7S3 |
| CAS No. | 1428321-10-1 |
Pritelivir mesylate is an antiviral drug currently under development, specifically targeting herpes simplex virus types 1 and 2 (HSV-1 and HSV-2). It functions by inhibiting the viral helicase-primase enzyme, a crucial component for HSV replication. It is being investigated as a potential treatment for various herpes infections, including those resistant to traditional antivirals like acyclovir.
Key aspects of Pritelivir mesylate:
- Mechanism of Action:Pritelivir is a helicase-primase inhibitor, meaning it blocks the activity of an enzyme essential for the replication of herpes viruses.
- Target Viruses:It is effective against both HSV-1 and HSV-2, the viruses responsible for cold sores and genital herpes, respectively.
- Potential for Resistance:Pritelivir has shown promise in preclinical studies against acyclovir-resistant strains of HSV, making it a potential alternative for patients with drug-resistant infections.
- Clinical Trials:Pritelivir is currently in phase II clinical trials, with ongoing research into its effectiveness and safety.
- Route of Administration:It is being investigated for oral, topical, and vaginal administration.
- Research and Development:Pritelivir is being developed by AiCuris Anti-infective Cures, building upon research from Bayer.
Pritelivir (development codes AIC316 or BAY 57-1293) is a direct-acting antiviral drug in development for the treatment of herpes simplex virus infections (HSV). This is particularly important in immune compromised patients. It is currently in Phase III clinical development by the German biopharmaceutical company AiCuris Anti-infective Cures AG. US FDA granted fast track designation for pritelivir in 2017 and breakthrough therapy designation 2020.
SCHEME
Pritelivir mesylate, an antiviral drug used to treat herpes simplex virus (HSV) infections, is synthesized through a series of chemical reactions, including palladium-catalyzed coupling, ester saponification, and amide coupling reactions. The mesylate salt is then formed by reacting the free base with methanesulfonic acid.
Detailed Synthesis Steps:
- 1. Diaryl Acetic Acid Synthesis:Diaryl acetic acid reagents are synthesized using palladium-catalyzed coupling reactions. These reactions involve the use of organometallic intermediates derived from halo-aryl esters.
- 2. Ester Saponification:The ester group in the synthesized compounds is then converted to a carboxylic acid group through saponification.
- 3. Amide Coupling:The resulting carboxylic acids are coupled with thiazolyl sulfonamides using amide coupling conditions to form the pritelivir molecule.
- 4. Salt Formation:The pritelivir free base is then reacted with methanesulfonic acid to form the mesylate salt, which is the active pharmaceutical ingredient (API).
Key Aspects of Pritelivir Mesylate Synthesis:
- Targeted Mechanism:Pritelivir mesylate inhibits the herpes simplex virus by targeting the viral helicase-primase complex, essential for DNA replication, unlike traditional antivirals that target DNA polymerase.
- Salt and Polymorph Screening:An extensive salt and polymorph screening is performed to optimize the pharmaceutical development of pritelivir, resulting in various salt forms including the mesylate, maleate, and sulfate.
- Solubility and Stability:Pritelivir mesylate is a BCS Class II drug substance with pH-dependent solubility. It exhibits high solubility below pH 3 and poor solubility at neutral pH.
- Formulation Considerations:Due to its limited water solubility, pritelivir mesylate is often formulated with solvents like DMSO, PEG300, Tween-80, and saline or with cyclodextrins like SBE-β-CD.
- Clinical Trials:Pritelivir mesylate is currently under extensive study to evaluate its efficacy and safety profile, with promising results in early clinical trials.
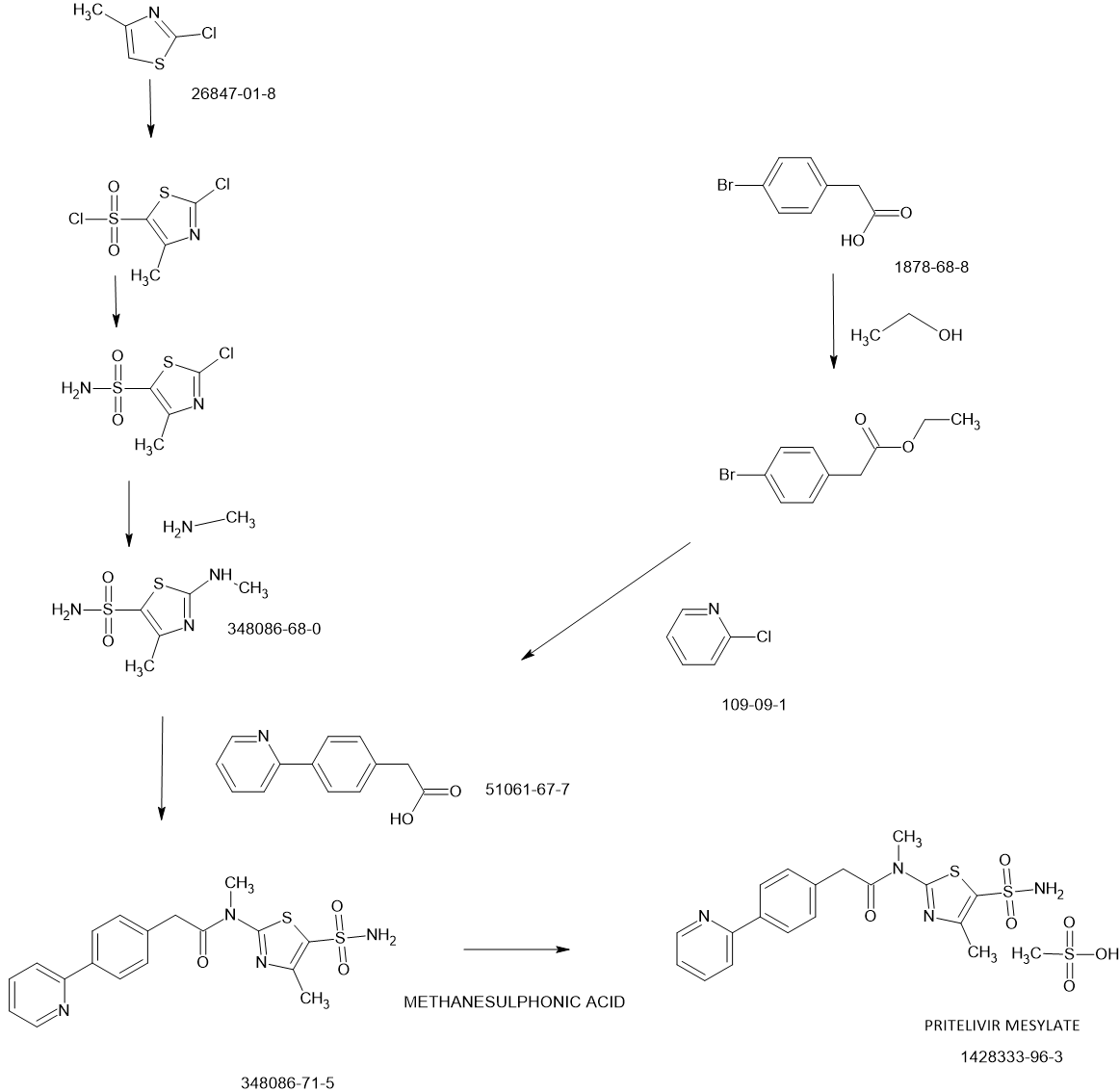
PATENT
WO2018096170
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018096170&_cid=P20-MCVCP5-34284-1
PATENT
WO2018096177
PATENT
https://patents.google.com/patent/WO2018096177A1/en
Likewise, EP 2 598 502 Al describes the crystalline mono mesylate monohydrate salt of N- [5-(aminosulfonyl)-4-methyl- 1 ,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl]-acetar^ in a definite particle size distribution and a specific surface area range, which has demonstrated increased long term stability and release kinetics from pharmaceutical compositions, as well as to pharmaceutical compositions containing said N-[5- (aminosulfonyl)-4-methyl- 1 ,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl]acetaniide mono mesylate monohydrate having the afore-mentioned particle size distribution and specific surface area range.
WO 2013/045479 Al describes an improved and shortened synthesis process of N-[5- (ammosulfonyl)-4-methyl-l,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl]acetaniide and the mesylate salt thereof by using boronic acid derivatives or borolane reagents while avoiding toxic organic tin compounds. Moreover, also the crystalline mesylate monohydrate salt of N- [5 -(aminosulfonyl)-4-methyl- 1 ,3 -thiazol-2-yl] -N-methyl-2- [4-(2-pyridinyl)-phenyl] – acetamide is described therein with increased long-term stability and release kinetics from pharmaceutical compositions thereof.
Said pritelivir is an innovative, highly active and specific inhibitor of herpes simplex virus (HSV) infections. As a compound derived from the chemical class of thiazolylamides, pritelivir is active against both types of herpes simplex virus causing labial and genital herpes, respectively, and retains activity against viruses which have become resistant to marketed drugs. Pritelivir has a mode of action that is distinct from other antiviral agents currently in use for treatment of HSV infections (i.e., the nucleoside analogues acyclovir and its prodrug valacyclovir as well as famciclovir, the prodrug of penciclovix). Whereas nucleoside analogs terminate ongoing DNA chain elongation through inhibition of viral DNA polymerase, pritelivir prevents de novo synthesis of virus DNA through inhibition of the helicase-primase complex. In addition, it does not require activation within an HSV infected cell by viral thymidine kinase and therefore, is also protective to uninfected cells.
With the context of the invention, similar expressions which all would denote the compound pritelivir are “BAY 57-1293”, “AIC090096” and “AIC316”.
Likewise, the terms ”pritelivir”, “BAY 57-1293”, “AIC090096” and “AIC316″ or the compound *’N-[5-(ammosulfonyl)-4-methyl-l,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridm phenyl] -acetamide” would reflect throughout the text a compound having the structural formula:

Synthetic route – Manufacture of N-[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-Yl]-N-methyl- 2-[4-(2-pyridinyl)-phenyl]-acetamide free base hemihydrate
The starting materials (4-pyridine-2-yl-phenyl)-acetic acid (PP-acetic acid; C-023930) and aminothiazole sulfonic acid amide (C-023936) are coupled using standard reaction conditions
(N-Ethyl-N’-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC x HCl), tetrahydrofuran (THF)/N-methylpyrrolidone (NMP) to deliver N-[5-(aminosulfonyl)-4- methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide free base hemihydrate (C-023931). To obtain the hemihydrate, N-[5-(aminosulfonyl)-4-methyl-1,3- thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide hemihydrate free base is recrystallized from THF/water. A flowchart showing the synthesis of N-[5-(aminosulfonyl)- 4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide is provided in below in the reaction scheme below 1.
Description of the manufacturing process of N-r5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide free base hemihydrate
PP-acetic acid and aminothiazole sulfonic acid amide are mixed in THF/NMP, the mixture is cooled and then EDC x HCl is added in portions. The reaction mixture is stirred for several hours, and then added slowly to purified water. The suspension is stirred and filtered; the product cake is washed with purified water and dried at room temperature in a nitrogen stream and then under vacuum. Purified water is added slowly at elevated temperature, the suspension is stirred for several hours. The suspension is cooled to 5°C and stirred further for several hours. The product is isolated by filtration and washed with purified water. The product is dried at 65°C under vacuum until the criterion for water content is reached. A major advantage of the synthesis of free base of N-[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide hemihydrate is the absence impurities related to the presence of mesylate ester that might be present in the N-[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)-phenyl]-acetamide mesylate.
PAPER
By: Carta, Fabrizio ; et al. Journal of Medicinal Chemistry (2017), 60(7), 3154-3164
SULPHURIC ACID FOR SULPHATE A SIMILAR REACTION BUT NOT SAME
PAPER
https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00668
Chemistry of Pritelivir
Synthesis of pritelivir and its analogues is based on the reported methods in the literature (18−20) and presented in Figure 4. A simple retrosynthetic disconnection of the target compound suggests a coupling of the thiazolyl sulfonamide and diaryl acetic acid (Figure 4a). During the course of development an optimized route applying the principles of green chemistry was developed and will be used for the commercial phase.
Figure 4. Synthesis of pritelivir (16) and analogues: (a) disconnection approach to target molecules; (b) synthesis of thiazolyl sulfonamide reagents; (c) synthesis of diaryl acetic acids; (d) synthesis of some representative examples of pritelivir and analogues. (18−20)
The synthesis of the thiazolyl sulfonamide reagents begins with a reaction of chloroacetone (17) and potassium thiocyanide to give an intermediate ketone which was cyclized to the thiazole 18 by treatment with gaseous hydrochloride (Figure 4b). Chlorosulfonylation with chlorosulfonic acid and thionyl chloride resulted in the sulfonyl chloride 19 that was converted to the corresponding sulfonamides 20 and 21 after treatment with ammonia or methylamine, respectively. The 2-chloro substituent in 20 and 21 was converted to the methyl amine in an SNAr reaction to deliver the building blocks 22–23. Diaryl acetic acid reagents were synthesized using palladium catalyzed coupling reactions with organometallic intermediates formed from the corresponding halo-aryl esters (Figure 4c). Ester saponification then delivered the corresponding carboxylic acids (e.g., 26 and 28, Figure 4c). Finally, the target molecules, for instance, 5, 11, 16, and 9, were obtained using amide coupling reaction conditions with corresponding diaryl acetic acids and the thiazolyl sulfonamides (Figure 4d). (18−20)
Medical use
Pritelivir is currently being developed for the treatment of immunocompromised patients with mucocutaneous HSV lesions that are resistant to acyclovir.
HSV in immunocompromised patients
Although HSV infection is very common in the general population, it rarely causes serious disease and is effectively contained by the immune system. In those with a weakened immune system such as transplant recipients, people receiving chemo- or radiotherapy, or HIV patients, an active HSV infection can cause disease in 35-68% of patients and may become severe or even life-threatening.[1]
Standard of care treatments
HSV treatment revolves around the use of nucleoside analogues (NA) which act via the viral DNA polymerase, causing DNA chain termination and prevention of viral replication. First-line treatment is generally acyclovir or its prodrug valacyclovir. Resistance to acyclovir is more common in HSV patients with weakened or suppressed immune systems, affecting between 4 and 25% of cases.[2][3][4][5][6]
Resistance to standard treatments
If HSV drug resistance is mediated by mutation(s) of the viral UL23 gene, which encodes the viral thymidine kinase (TK), then the pyrophosphate analogue foscarnet may be effective as a rescue treatment, since it does not require activation by TK. The use of foscarnet is commonly accompanied by restrictive toxicity, particularly nephrotoxicity.[7] If the virus also acquires resistance to foscarnet, then there is currently no FDA approved treatment.
Clinical research
Completed phase II clinical trials in otherwise healthy patients with genital herpes
- A Double-blind Randomized Placebo Controlled Dose-finding Trial to Investigate Different Doses of a New Antiviral Drug in Subjects With Genital HSV Type 2 Infection.[8][9]
- A Double-blind, Double Dummy, Randomized Crossover Trial to Compare the Effect of “AIC316 (Pritelivir)” 100 mg Once Daily Versus Valacyclovir 500 mg Once Daily on Genital HSV Shedding in HSV-2 Seropositive Adults.[10][11]
Ongoing phase II / phase III clinical trials with pritelivir
A phase II / III multinational, comparator-controlled, clinical trial in immunocompromised patients with acyclovir-resistant mucocutaneous lesions is listed on ClinicalTrials.gov[12] and EudraCT.[13]
Pharmacology
Mechanism of action
Pritelivir is a member of the helicase-primase inhibitors (HPI), a novel class of direct-acting antiviral drugs acting specifically against HSV-1 and HSV-2.[14][15] As the name suggests, the drugs act through inhibition of the viral helicase primase complex, encoded by the UL5 (helicase), UL8 (scaffold protein) and UL52 (primase) genes, which is essential for HSV replication.[16] The helicase primase complex is encoded separately from the viral DNA polymerase (encoded by the UL30 gene). Because HPIs i) do not target the viral DNA polymerase and ii) do not require activation by the viral thymidine kinase enzyme (encoded by the UL23 gene), mutations causing resistance to NAs are not protective against HPIs. Similarly, resistance to HPIs does not confer resistance to NAs.
References
- ^ Wilck, M.B.; Zuckerman, R.A.; A. S. T. Infectious Diseases Community of Practice (2013). “Herpes simplex virus in solid organ transplantation”. Am J Transplant. 13 (Suppl 4): 121–7. doi:10.1111/ajt.12105. PMID 23465005. S2CID 44969727.
- ^ Zuckerman, R.; Wald, A.; A. S. T. Infectious Diseases Community of Practice (2009). “Herpes simplex virus infections in solid organ transplant recipients”. Am J Transplant. 9 (Suppl 4): S104-7. doi:10.1111/j.1600-6143.2009.02900.x. PMID 20070669. S2CID 205846431.
- ^ Frobert, E.; Burrel, S.; Ducastelle-Lepretre, S.; Billaud, G.; Ader, F.; Casalegno, J.S. (2014). “Resistance of herpes simplex viruses to acyclovir: an update from a ten-year survey in France”. Antiviral Res. 111: 36–41. doi:10.1016/j.antiviral.2014.08.013. PMID 25218782.
- ^ Patel, D.; Marchaim, D.; Marcus, G.; Gayathri, R.; Lephart, P.R.; Lazarovitch, T.; Zaidenstein, R.; Chandrasekar, P. (2014). “Predictors and outcomes of acyclovir-resistant herpes simplex virus infection among hematopoietic cell transplant recipients: case-case-control investigation”. Clin Transplant. 28 (1): 1–5. doi:10.1111/ctr.12227. PMID 24033498. S2CID 37729458.
- ^ Danve-Szatanek, C.; Aymard, M.; Thouvenot, D.; Morfin, F.; Agius, G.; Bertin, I. (2004). “Surveillance network for herpes simplex virus resistance to antiviral drugs: 3-year follow-up”. J Clin Microbiol. 42 (1): 242–9. doi:10.1128/JCM.42.1.242-249.2004. PMC 321677. PMID 14715760.
- ^ Chakrabarti, R.; Pillay, D.; Ratcliffe, D.; Cane, P.A.; Collingham, K.E.; Milligan, D.W. (2000). “Resistance to antiviral drugs in herpes simplex virus infections among allogeneic stem cell transplant recipients: risk factors and prognostic significance”. J Infect Dis. 181 (6): 2055–8. doi:10.1086/315524. PMID 10837192.
- ^ SmPC
- ^ NCT01047540
- ^ Wald, A.; Timmler, B.; Magaret, A.; Warren, T.; Trying, S. (2014). “Helicase-primase inhibitor pritelivir for HSV-2 infection”. N Engl J Med. 370 (3): 201–10. doi:10.1056/NEJMoa1301150. PMID 24428466.
- ^ NCT01658826
- ^ Wald, A.; Timmler, B.; Warren, T.; Trying, S.; Johnston, C. (2016). “Effect of Pritelivir Compared With Valacyclovir on Genital HSV-2 Shedding in Patients With Frequent Recurrences: A Randomized Clinical Trial”. JAMA. 316 (23): 2495–2503. doi:10.1001/jama.2016.18189. hdl:1805/14200. PMID 27997653.
- ^ NCT03073967
- ^ 2020-004940-27
- ^ Biswas, S.; Jennens, L.; Field, H.J. (2007). “Single amino acid substitutions in the HSV-1 helicase protein that confer resistance to the helicase-primase inhibitor BAY 57-1293 are associated with increased or decreased virus growth characteristics in tissue culture”. Arch Virol. 152 (8): 1489–500. doi:10.1007/s00705-007-0964-7. PMID 17404685. S2CID 23688945.
- ^ Field, H.J.; Biswas, S. (2011). “Antiviral drug resistance and helicase-primase inhibitors of herpes simplex virus”. Drug Resist Updat. 14 (1): 45–51. doi:10.1016/j.drup.2010.11.002. PMID 21183396.
- ^ Crute, J.J.; Tsurumi, T.; Zhu, L.A.; Weller, S.K.; Olivo, P.D.; Challberg, M.D. (1989). “Herpes simplex virus 1 helicase-primase: a complex of three herpes-encoded gene products”. Proc. Natl. Acad. Sci. U.S.A. 86 (7): 2186–2189. Bibcode:1989PNAS…86.2186C. doi:10.1073/pnas.86.7.2186. PMC 286876. PMID 2538835.
- [1]. Ligat G, et al. Identification of Amino Acids Essential for Viral Replication in the HCMV Helicase-PrimaseComplex. Front Microbiol. 2018 Oct 23;9:2483. [Content Brief][2]. Wald A, et al. Helicase-primase inhibitor Pritelivir for HSV-2 infection. N Engl J Med. 2014 Jan 16;370(3):201-10. [Content Brief][3]. Quenelle DC, et al. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis. Antiviral Res. 2018 Jan;149:1-6. [Content Brief]
| Names | |
|---|---|
| Systematic IUPAC nameN-Methyl-N-(4-methyl-5-sulfamoyl-1,3-thiazol-2-yl)-2-[4-(pyridin-2-yl)phenyl]acetamide | |
| Identifiers | |
| CAS Number | 348086-71-5 |
| 3D model (JSmol) | Interactive image |
| ChemSpider | 430613 |
| KEGG | D12811 |
| PubChem CID | 491941 |
| UNII | 07HQ1TJ4JE |
| CompTox Dashboard (EPA) | DTXSID70188344 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C18H18N4O3S2 |
| Molar mass | 402.49 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).Infobox references | |
///////////PRITELIVIR MESYLATE, AIC316 mesylate hydrate, BAY 57-1293 mesylate hydrate, AIC 316 mesylate hydrate, BAY 57-1293 mesylate hydrate, BAY57-1293, BAY 57-1293, BAY-57-1293, BAY571293, BAY 571293, BAY-571293, AIC-316, AIC 316
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

{kind=link}
{kind=link}
{kind=link}