
Home » Uncategorized (Page 5)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Fosrolapitant



Fosrolapitant
CAS 2573694-38-7
MF C27H29F6N2O8P MW654.5 g/mol
phosphonooxymethyl (5S,8S)-8-[[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy]methyl]-2-oxo-8-phenyl-1,9-diazaspiro[4.5]decane-9-carboxylate
(phosphonooxy)methyl (5S,8S)-8-({(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy}methyl)-2-oxo-8-phenyl-1,7-diazaspiro[4.5]decane-7-carboxylate
neurokinin 1 (NK1) receptor antagonist, HR20013, HR 20013, M5QGY92X8B
Fosrolapitant (HR20013) is a novel, intravenous, highly selective neurokinin-1 (NK-1) receptor antagonist used for the prevention of chemotherapy-induced nausea and vomiting (CINV), particularly for cisplatin-based regimens. As a prodrug, it is rapidly converted to rolapitant, offering a long half-life (~180 h). It is often combined with palonosetron and dexamethasone for high efficacy.
Fosrolapitant is a small molecule drug. The usage of the INN stem ‘-pitant’ in the name indicates that Fosrolapitant is a neurokinin NK1 (substance P) receptor antagonist. Fosrolapitant has a monoisotopic molecular weight of 654.16 Da.
Key Aspects of Fosrolapitant:
- Mechanism: Acts as an NK-1 receptor antagonist to prevent nausea/vomiting.
- Administration: Intravenous (IV) formula, often combined as a fixed-dose with palonosetron (HR20013).
- Metabolism: Completely converted to rolapitant in the body, which has a prolonged half-life of approximately 180 hours.
- Clinical Efficacy: In trials (e.g., PROFIT trial), it demonstrated high effectiveness in preventing CINV in patients receiving highly emetogenic chemotherapy.
- Safety Profile: Common adverse events in trials included constipation (22.7%), increased blood pressure (18.2%), abdominal distension (13.6%), and injection site reactions (9.1%).
Fosrolapitant is designed to improve convenience and patient compliance in managing acute and delayed nausea and vomiting associated with cancer treatments.
HR20013 for Nausea and Vomiting Associated With Moderate Emetic Risk Anticancer Agents
CTID: NCT06554184
Phase: Phase 3
Status: Completed
Date: 2025-11-17
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020259675&_cid=P20-MKT4GS-85082-1

Under N2 protection, compound 3 (1.95 g, 2.543 mmol, 1 eq) dissolved in dichloromethane (40 mL) was added to a 100
mL single-necked flask. Trifluoroacetic acid (1.45 mL, 19.52 mmol, 8 eq) was slowly added under ice water cooling. The mixture was stirred until the reaction was complete, concentrated, and 2.29 g of oil was obtained. After separation and purification, 1.39 g of white foamy solid was obtained, with a yield of 83.5%.
[0129]
1H-NMR(400MHz,CD 3OD):δ(ppm)7.89(s,2H),7.86(s,1H),7.41-7.27(m,5H),5.66(d,J=12Hz,1H),5.50-5.47(m,1H),4.60(d,J=8Hz,1H),4.20-3.88(m,3H),2.51-2.10(m,5H),1.86-1.66(m,3H),1.44-1.31(m,4H).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US380158929&_cid=P20-MKT480-75882-1
Example 1

STEP 1

Compound 1 (2.43 g, 4.86 mmol, 1 eq) was weighed and dissolved in dichloromethane (36 mL) in a 100 mL three-necked flask under N 2 atmosphere. Diisopropylethylamine (5 g, 38.76 mmol, 8 eq) was added and the mixture was cooled to −30° C. Trimethylchlorosilane (1.36 g, 12.52 mmol, 2.6 eq) was added and the mixture was stirred at room temperature for 2 h. The reaction mixture was cooled to −25° C. A solution of chloromethyl chloroformate (0.77 g, 6 mmol, 1.23 eq) in dichloromethane was added dropwise and the mixture was stirred under controlled temperature at −20° C.˜−5° C. until completion of the reaction. The reaction solution was poured into ice water, put to separation, and extracted with dichloromethane. Water and 1 N hydrochloric acid solution were added and put to separation. The organic layer was then successively washed with brine, saturated aqueous solution of sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and concentrated to give 3.0 g yellow jelly with a yield of 104%.


| Compound 3 (1.95 g, 2.543 mmol, 1 eq) was added into a 100 mL single-necked flask and dissolved in dichloromethane (40 mL) under N 2 atmosphere. Trifluoroacetic acid (1.45 mL, 19.52 mmol, 8 eq) was added slowly under ice water cooling. The reaction mixture was stirred until completion of the reaction, and then concentrated to give 2.29 g oil which was then purified purified to give 1.39 g white foamy solid with a yield of 83.5%. |
PAT
- Use of nk1 antagonist prodrug compound in combination with 5-ht3 receptor antagonistPublication Number: US-2024016822-A1Priority Date: 2020-12-25
- Use of nk1 antagonist prodrug compound in combination with 5-ht3 receptor antagonistPublication Number: EP-4268818-A1Priority Date: 2020-12-25
- Neurokinin-1 antagonistPublication Number: EP-3991730-A1Priority Date: 2019-06-28
- Neurokinin-1 antagonistPublication Number: WO-2020259675-A1Priority Date: 2019-06-28
- Neurokinin-1 antagonistsPublication Number: TW-202115064-APriority Date: 2019-06-28



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////fosrolapitant, neurokinin 1 (NK1) receptor antagonist, HR20013, HR 20013, M5QGY92X8B
Evetifator


Evetifator
CAS 2278265-85-1
MF C20H19ClF3N3O4 MW457.8 g/mol
2-(4-chlorophenoxy)-N-[3-[5-[3-(trifluoromethoxy)cyclobutyl]-1,3,4-oxadiazol-2-yl]-1-bicyclo[1.1.1]pentanyl]acetamide
2-(4-chlorophenoxy)-N-(3-{5-[(1s,3s)-3-(trifluoromethoxy)cyclobutyl]-1,3,4-oxadiazol-2-yl}bicyclo [1.1.1]pentan-1-yl)acetamide
eukaryotic translation initiation factor 2B (eIF2B) activator, DNL-343, DNL 343, FYL3Y9D7SK
Evetifator (also known as DNL343) is a potent, selective, and brain-penetrant small molecule activator of eukaryotic initiation factor 2B (eIF2B). As of 2026, it is primarily being investigated for the treatment of neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS).
Key Characteristics and Function
- Mechanism of Action: It acts as an eIF2B activator. eIF2B is a critical regulator of protein synthesis; by activating it, the drug aims to address the Integrated Stress Response (ISR) which, when chronically activated, leads to neurodegeneration.
- Pharmacological Profile:
- Potency: It shows an
IC50cap I cap C sub 50𝐼𝐶50 of 3.2 nM in cellular reporter assays.
- Brain Penetration: It is specifically designed to cross the blood-brain barrier to target the central nervous system (CNS).
- Potency: It shows an
- A Study to Evaluate the Bioavailability and Safety of DNL343 in Healthy VolunteersCTID: NCT04581772Phase: Phase 1Status: CompletedDate: 2021-06-11
- A Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of DNL343 in Healthy VolunteersCTID: NCT04268784Phase: Phase 1Status: CompletedDate: 2022-02-07
- HEALEY ALS Platform Trial – Regimen G DNL343CTID: NCT05842941Phase: Phase 2/Phase 3Status: CompletedDate: 2025-02-04
- A Study to Determine the Safety, Pharmacokinetics, and Pharmacodynamics of DNL343 in Participants With Amyotrophic Lateral SclerosisCTID: NCT05006352Phase: Phase 1Status: CompletedDate: 2024-09-19
- HEALEY ALS Platform Trial – Master ProtocolCTID: NCT04297683Phase: Phase 2/Phase 3Status: Active, not recruitingDate: 2025-12-31
SYN
WO 2019/032743
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019032743&_cid=P10-MKOU1U-66006-1

EXAMPLE 3
2-(4-chlorophenoxy)-N-[3-[5-[cis-3-(trifluoromethoxy)cyclobutyl]-1,3,4-oxadiazol-2-yl]-1- bicyclo[1.1.1]pentanyl]acetamide
[0257] 2-(4-chlorophenoxy)-N-[1-(hydrazinecarbonyl)-3-bicyclo[1.1.1]pentanyl]acetamide (200 mg, 0.65 mmol), 3-cis-(trifluoromethoxy)cyclobutanecarboxylic acid (131 mg, 0.71 mmol; 8:1 to 10:1 ratio of cis- to trans-) and triethylamine (NEt3) (0.45 mL, 3.23 mmol) were dissolved in EtOAc (2.6 mL) and T3P solution (0.58 mL, 1.94 mmol, 50 % in EtOAc) was added. The resulting reaction mixture was heated to 100 °C overnight, cooled to rt and was diluted with sat. aq. NaHCO3 solution (10 mL) and EtOAc (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude reaction mixture was purified employing reverse-phase prep-HPLC to deliver the desired product as a clear oil. 1H-NMR (400 MHz; CDCl3): δ 7.33-7.29 (m, 2H), 7.03 (s, 1H), 6.91-6.87 (m, 2H), 4.76-4.69 (m, 1H), 4.44 (s, 2H), 3.39-3.30 (m, 1H), 2.92-2.84 (m, 2H), 2.74-2.68 (m, 2H), 2.67 (s, 6H). LC-MS m/z: = 458.20 [M+H]+.
[0258] Alternatively, a mixture of 2-(4-chlorophenoxy)acetic acid (50 mg, 0.27 mmol), 2-(4-chlorophenoxy)acetic acid (50 mg, 0.27 mmol), NEt3 (123 mg, 1.21 mmol) and T3P (185 mg, 0.29 mmol, 50% purity) in DCM (1 mL) was stirred at 0 °C for 1 h. To the mixture was added 1-[5-[3-cis-(trifluoromethoxy)cyclobutyl]-1,3,4-oxadiazol-2-yl]bicyclo[1.1.1]pentan-3-amine HCl salt (8:1 to 10:1 favoring the cis- diastereomer) (70 mg, 0.24 mmol) at 0 °C. The mixture was stirred at 25 °C for 12 h. To the reaction was added sat. aq. NaHCO3 (4 mL). The aqueous phase was extracted with DCM (5 mL, 3 mL). The combined organic phase was washed with brine (10 mL), dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure to provide the title compound.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022133236&_cid=P10-MKOTTO-58939-1
2-(4-chlorophenoxy)-N-[3-[5-[cA-3-(trifluoromethoxy)cyclobutyl]-l,3,4-oxadiazol-2-yl]-l-bicyclo[l.l.l]pentanyl]acetamide, designated herein as Compound I, has the following formula:

Example 1. Synthesis of Compound I
2-(4-chlorophenoxy)-/V-[l-(hydrazinecarbonyl)-3-bicyclo[l.l.l]pentanyl]acetamide
[0131] To a suspension of methyl 3-[[2-(4-chlorophenoxy)acetyl]amino]bicyclo[l.l.l]pentane-l-carboxylate (270 mg, 0.87 mmol) in EtOH (0.25-0.1M) was added hydrazine hydrate (131 mg, 2.6 mmol) in EtOH (3.5 mL) and the reaction mixture was heated at 90 °C overnight. The reaction mixture
was cooled to rt often causing the product to crystallize out of solution. This solid was collected by removal of the supernatant. If the product did not crystallize, the solution was concentrated, and the crude product was sufficiently pure to use in subsequent steps.
LC-MS m/z: = 310.1 [M+H]+.
2-(4-chlorophenoxy)-N-[3-[5-[cis-3-(trifluoromethoxy)cyclobutyl]-1,3,4-oxadiazol-2-yl]-1-bicyclo[1.1.1]pentanyl]acetamide
[0132] 2-(4-chlorophenoxy)-N-[1-(hydrazinecarbonyl)-3-bicyclo[1.1.1]pentanyl]acetamide (200 mg, 0.65 mmol), 3-cis-(trifluoromethoxy)cyclobutanecarboxylic acid (131 mg, 0.71 mmol; 8:1 to 10:1 ratio of cis- to trans-) and triethylamine (NEt3) (0.45 mL, 3.23 mmol) were dissolved in EtOAc (2.6 mL) and T3P solution (0.58 mL, 1.94 mmol, 50 % in EtOAc) was added. The resulting reaction mixture was heated to 100 ºC overnight, cooled to rt and was diluted with sat. aq. NaHCO3 solution (10 mL) and EtOAc (10 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (3 x 10 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude reaction mixture was purified employing reverse-phase prep-HPLC to deliver the desired product as a clear oil.1H-NMR (400 MHz; CDCl3): δ 7.33-7.29 (m, 2H), 7.03 (s, 1H), 6.91-6.87 (m, 2H), 4.76-4.69 (m, 1H), 4.44 (s, 2H), 3.39-3.30 (m, 1H), 2.92-2.84 (m, 2H), 2.74-2.68 (m, 2H), 2.67 (s, 6H). LC-MS m/z: = 458.20 [M+H]+.
[0133] Alternatively, a mixture of 2-(4-chlorophenoxy)acetic acid (50 mg, 0.27 mmol), NEt3 (123 mg, 1.21 mmol) and T3P (185 mg, 0.29 mmol, 50% purity) in DCM (1 mL) was stirred at 0 °C for 1 h. To the mixture was added 1-[5-[3-cis-(trifluoromethoxy)cyclobutyl]-1,3,4-oxadiazol-2-yl]bicyclo[1.1.1]pentan-3-amine HCl salt (8:1 to 10:1 favoring the cis- diastereomer) (70 mg, 0.24 mmol) at 0 °C. The mixture was stirred at 25 °C for 12 h. To the reaction was added sat. aq. NaHCO3 (4 mL). The aqueous phase was extracted with DCM (5 mL, 3 mL). The combined organic phase was washed with brine (10 mL), dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure to provide the title compound.
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023250107&_cid=P10-MKOTXL-62392-1
Example 14: Preparation of 2-(4-chlorophenoxy)-N-(3-(5-((ls,3s)-3-(trifluoromethoxy)cyclobutyl)- l,3,4-oxadiazol-2-yl)bicyclo[l.l.l]pentan-l-yl)acetamide (I)

[0377] XI- la, 2-(4-chlorophenoxy)acetic acid (XII- la), and 2-MeTHF were charged to a reactor under N2 condition and cooled to 0 ~ 5 °C. TEA was added while maintaining an internal temperature of not more than about 10 °C under N2 condition and rinsed with 2-methyltetrahydrafuran (2-MeTHF). The contents were agitated at about 0 ~ 5 °C for not less than about 20 minutes. Diphenylphosphinic chloride in 2-MeTHF solution is added slowly while maintaining an internal temperature of not more than about 10 °C under N2 condition and rinsed with 2-MeTHF. The contents were warmed to about 20 ~ 25 °C and then agitated for not less than about 1 hour until the reaction was completed. The contents were cooled to about 0 ~ 5 °C and then aqueous 10% K2CO3 was added while maintaining an internal temperature of not more than about 10 °C. After phase separation, the organic layer was successively washed with aqueous 10% K2CO3 and 5% K2CO3. The organic layer was concentrated to a target volume 3 V. 2-MeTHF was added and then the contents were concentrated to a target volume 3 V to control the water content to not more than about 0.3 w/w%. IPA was added and then the contents were heated to about 60 ~ 70 °C to
dissolve all solids. The contents were filtered at about 60 ~ 70 °C through cartridge filter and rinsed with pre-heated IPA (60 ~ 70 °C). The filtrate was concentrated to a target volume 4 V. IPA was added and concentrated to a target volume 4 V to control the residual 2-MeTHF relative to IPA to not more than 1 % by GC. The contents were adjusted to about 20 ~25 °C. n-Heptane was added and then heated to about 60 ~ 80 °C to dissolve all solids. The contents were adjusted to about 62 ~ 70 °C. Seed crystal was charged and agitated for not less than about 0.5 hour. n-Heptane was added while maintaining an internal temperature of about 60 ~ 65 °C. The contents were cooled to about 0 ~ 5 °C over 12 hour (5 °C per hour). The slurry was agitated for not less than 2 hours. The slurry was filtered and washed with precooled IPA/n-heptane mixture. The wet cake was dried at 25 °C under vacuum. If any individual impurity except 2-(4-chlorophenoxy)-N-(3-(5-(trans-3-(trifluoromethoxy)cyclobutyl)-l,3,4-oxadiazol-2-yl)bicyclo[l.l.l]pentan-l-yl)acetamide was more than 0.12%, recrystallization was performed.
[0378] ’H NMR (600 MHz, MeCN-d3): 7.65 (s, 1H), 7.3
PAT
- Compounds, compositions and methodsPublication Number: JP-2023052166-APriority Date: 2017-08-09
- Compounds, compositions, and methodsPublication Number: CN-111094233-BPriority Date: 2017-08-09Grant Date: 2024-03-15
- Modulators of eukaryotic initiation factor 2B, compositions and methodsPublication Number: US-11236100-B2Priority Date: 2017-08-09Grant Date: 2022-02-01
- Compounds, compositions, and methodsPublication Number: CN-118239938-APriority Date: 2017-08-09
- Solid forms of a compoundPublication Number: US-2024059662-A1Priority Date: 2020-12-18
- Modulators of eukaryotic initiation factor 2B, compositions and methodsPublication Number: US-2022411433-A1Priority Date: 2017-08-09
- COMPOUNDS, COMPOSITIONS AND METHODSPublication Number: WO-2019032743-A1Priority Date: 2017-08-09
- Modulators of eukaryotic initiation factor 2B, compositions and methodsPublication Number: US-11851440-B2Priority Date: 2017-08-09Grant Date: 2023-12-26
- Compounds, compositions and methodsPublication Number: US-2021147435-A1Priority Date: 2017-08-09
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
//////evetifator, ANAX, eukaryotic translation initiation factor 2B (eIF2B) activator, DNL-343, DNL 343, FYL3Y9D7SK
Evategrel



Evategrel
CAS 2760609-74-1
MF C21H26ClNO7S MW 472.0 g/mol
(2Z)-2-[(4R)-1-[(1S)-1-(2-chlorophenyl)-2-methoxy-2-oxoethyl]-4-(propan-2-yloxycarbonyloxymethylsulfanyl)piperidin-3-ylidene]acetic acid
(Z)-[(4R)-1-[(1S)-1-(2-chlorophenyl)-2-methoxy-2-oxoethyl]-4-{[({[(propan-2-yl)oxy]carbonyl}oxy)methyl]sulfanyl}piperidin-3-ylidene]acetic acid
platelet aggregation inhibitor, CG-0255, CG 0255, 9FKJ76ZX22
Evategrel (CG-0255) is a promising new antiplatelet drug, a thioether prodrug, designed to improve upon clopidogrel (Plavix) by offering faster action, consistent potency, and overcoming resistance, with both oral and intravenous (IV) formulations available for emergency use. It works by rapidly converting to the same active metabolite as clopidogrel (H4) through simple hydrolysis, bypassing the CYP enzymes that can cause variability and resistance with clopidogrel. Clinical trials show it’s well-tolerated, potent, and has potential to become a superior P2Y12 inhibitor for preventing blood clots in cardiovascular conditions.
Key Features
- Fast & Potent: Achieves significant platelet inhibition (IPA) within 15-30 minutes.
- Consistent Activation: Relies on liver carboxylesterases, avoiding CYP2C19 variability, leading to less individual response difference.
- Dual Formulation: First P2Y12 inhibitor with both IV (for emergencies/surgery) and oral forms.
- Overcomes Resistance: Specifically designed to address clopidogrel resistance issues.
- Low Drug-Drug Interactions: Expected to have minimal interactions.
How it Works
- Prodrug: Evategrel is inactive when administered.
- Hydrolysis: Liver esterase enzymes quickly break it down (hydrolyze it) in a single step.
- Active Metabolite: This process creates H4, the same active antiplatelet molecule as clopidogrel’s active form.
- Platelet Inhibition: H4 blocks the P2Y12 receptor on platelets, preventing them from clumping (aggregating).
Development & Potential
- Developed by China-based CureGene.
- Shows promise as a “best-in-class” P2Y12 antagonist, potentially benefiting patients with acute coronary syndromes (ACS) or those undergoing PCI (percutaneous coronary intervention).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023144782&_cid=P10-MKNELX-27983-1

Synthesis Example 1


Steps 11 and 12. Synthesize la-1 and la-2

The solution of 1–13 (1.8 g, 3.4 mmol) in TFA (10 mL) was stirred at room temperature for 30 minutes. After stirring, the reaction mixture was added to a saturated NaHCO3 solution (100 mL), followed by collection with EtOAc (100 mL * 3). The combined organic layers were washed with saturated NaHCO3 , dried over Na2SO4 , and filtered. The filtrate was concentrated under reduced pressure, and the residue was purified by reversed-phase column chromatography (C18, CH3CN / H2O = 80/20 ) to give la (550 mg, 34% yield). la was further purified by chiral column chromatography to give la-1 and la-2.
the:
LC-MS [M+l]+ = 472.1

57.59 (s, 1H), 7.38 (d, J = 4 Hz, 1H), 7.32-7.26 (m, 2H), 5.86 (s, 1H), 5.22 (dd, 12.2, 2.6 Hz, 1H), 5.00-4.83 (m, 3H), 4.50 (dd, J = 66.2, 11.9 Hz, 1H), 3.82 (s, 1H), 3.70 (d, J = 4.9 Hz, 3H), 3.52 (dd, J = 37.9, 12.9 Hz, 1H), 2.92-2.64 (m, 2H), 2.45-2.30 (m, 1 H), 1.95-1.84 (m, 1H), 1.30 ( 6.2 Hz, 6H)O
la-1:
NMR (400 MHz, CDC13) 6 7.65 (s, 1H), 7.46 – 7.43 (m, 1H), 7.33 (dd, J = 6.3, 2.7 Hz, 2H), 5.91 (s, 1H), 5.27 (d, J = 12.3 Hz, 1H), 5.04 – 4.87 (m, 3H), 4.49 (d, J = 13.7 Hz, 1H), 3.88 (s, 1H), 3.75 (s, 3H), 3.58 (d, J = 14.0 Hz, 1H), 2.87 (s, 2H), 2.44 (s, 1H), 1.95 (dd, J = 14.2, 3.3 Hz, 1H), 1.35 (d, J = 6.2 Hz, 6H)O
la-2:
NMR (400 MHz, CDCh) 5 7.63 (s, 1H), 7.44 (dt, J = 8.2, 3.1 Hz, 1H), 7.35 – 7.31 (m,
2H), 5.92 (s, 1H), 5.25 (d, J = 12.3 Hz, 1H), 5.07 (s, 1H), 4.94 (td, J = 12.5, 6.5 Hz, 2H), 4.68 (d, J = 13.4 Hz, 1H), 3.87 (s, 1H), 3.76 (s, 3H), 3.50 (d, J = 13.4 Hz, 1H), 2.90 (s, 1H), 2.75 (d, J = 12.3 Hz, 1H), 2.44 (s, 1H), 1.96 (d, 7 = 13.2 Hz, 1H), 1.34 (d, J = 6.3 Hz, 6H) =
PAT
- Pharmaceutical composition of antiplatelet drug, and use thereofPublication Number: WO-2023144782-A1Priority Date: 2022-01-28
- Antiplatelet drugs and uses thereofPublication Number: US-2023295089-A1Priority Date: 2020-07-29
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////evategrel, platelet aggregation inhibitor, CG-0255, CG 0255, 9FKJ76ZX22
Emestedastat



Emestedastat
CAS 1346013-80-6
MF C19H19N5O2S MW381.5 g/mol
[(1R,3r,5S)-3-hydroxy-3-(pyrimidin-2-yl)-8-azabicyclo[3.2.1]octan-8-yl][5-(1H-pyrazol-4-yl)thiophen-3-yl]methanone
11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor, UE-2343, UE 2343, 106ELK29GH
Emestedastat (proposed brand name Xanamem; developmental code name UE-2343) is a steroidogenesis inhibitor which is under development for the treatment of major depressive disorder, Alzheimer’s disease, and fragile X syndrome.[1][2] It specifically acts as a centrally penetrant inhibitor of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) and thereby inhibits the synthesis of the glucocorticoid steroid hormone cortisol.[1][3][4][2] As of August 2024, emestedastat is in phase 2 clinical trials for major depressive disorder and Alzheimer’s disease and is in the preclinical stage of development for fragile X syndrome.[1][2] Clinical effectiveness for Alzheimer’s disease has been mixed.[2] It was originated by the University of Edinburgh and is being developed by Actinogen Medical.[1]
- Phase I MAD, Fed-Fasted, CSF Study of UE2343 in Healthy SubjectsCTID: NCT02616445Phase: Phase 1Status: CompletedDate: 2025-01-22
- Effect of 10 mg Xanamem on Dementia Due to Alzheimer’s DiseaseCTID: NCT06125951Phase: Phase 2Status: Active, not recruitingDate: 2025-12-02
- A Phase I Study of Oral UE2343 in Healthy SubjectsCTID: NCT01770886Phase: Phase 1Status: CompletedDate: 2013-07-17
- Xanamem™ in Healthy Elderly SubjectsCTID: NCT03830762Phase: Phase 1Status: CompletedDate: 2025-01-22
- OriginatorUniversity of Edinburgh
- DeveloperActinogen Medical
- ClassAntidementias; Azabicyclo compounds; Ketones; Pyrazoles; Pyrimidines; Small molecules; Thiophenes
- Mechanism of Action11-beta-hydroxysteroid dehydrogenase type 1 inhibitors
- Phase II/IIIAlzheimer’s disease
- Phase IIMajor depressive disorder
- No development reportedFragile X syndrome
- 15 Sep 2025Meeting similar to that of Type C will be held for Alzheimer’s disease (AD) with European Medicines Agency and subsequently with the UK MHRA and other regulators in 2026
- 27 Aug 2025Pharmacokinetics data from a phase I pharmacokinetics trial in volunteers released by Actinogen
- 28 Jul 2025No recent reports of development identified for preclinical development in Fragile-X-syndrome in Australia (PO)
Syn
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022094668&_cid=P20-MKKJLN-11715-1


SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021062472&_cid=P20-MKKJLN-11715-1
PAT
- medical cognitive therapyPublication Number: CN-114761005-APriority Date: 2019-09-30
- Medicinal cognitive treatmentsPublication Number: CA-3152902-A1Priority Date: 2019-09-30
- Therapeutic editingPublication Number: AU-2020395113-A1Priority Date: 2019-12-02
- Medicinal cognitive treatmentsPublication Number: EP-4037684-A1Priority Date: 2019-09-30
- Medicinal cognitive treatmentsPublication Number: US-2023000843-A1Priority Date: 2019-09-30
- Medicinal cognitive treatmentsPublication Number: WO-2021062472-A1Priority Date: 2019-09-30
- Cognitive pharmacological treatmentPublication Number: JP-2022550221-APriority Date: 2019-09-30
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
References
- “UE 2343”. AdisInsight. 28 August 2024. Retrieved 9 October 2024.
- Seckl J (January 2024). “11β-Hydroxysteroid dehydrogenase and the brain: Not (yet) lost in translation”. Journal of Internal Medicine. 295 (1): 20–37. doi:10.1111/joim.13741. PMID 37941106.
- Bachurin SO, Gavrilova SI, Samsonova A, Barreto GE, Aliev G (March 2018). “Mild cognitive impairment due to Alzheimer disease: Contemporary approaches to diagnostics and pharmacological intervention”. Pharmacological Research. 129: 216–226. doi:10.1016/j.phrs.2017.11.021. PMID 29170097.
- Canet G, Hernandez C, Zussy C, Chevallier N, Desrumaux C, Givalois L (2019). “Is AD a Stress-Related Disorder? Focus on the HPA Axis and Its Promising Therapeutic Targets”. Frontiers in Aging Neuroscience. 11 269. doi:10.3389/fnagi.2019.00269. PMC 6776918. PMID 31611783.
///////////emestedastat, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor, UE-2343, UE 2343, 106ELK29GH
Direclidine



Direclidine
CAS 1803346-98-6
MF C19H30N4O2 MW346.5 g/mol
ethyl 2-[4-(2-methylpyrazol-3-yl)piperidin-1-yl]-6-azaspiro[3.4]octane-6-carboxylate
ethyl (2r,4s)-2-[4-(1-methyl-1H-pyrazol-5-yl)piperidin-1-yl]-6-azaspiro[3.4]octane-6-carboxylate
muscarinic M4 receptor positive allosteric modulator, TXB4V44U24, NBI-1117568, NBI 1117568
Direclidine (INNTooltip International Nonproprietary Name;[2] developmental code names NBI-1117568, HTL-0016878)[1] is an investigational antipsychotic drug for schizophrenia[3] that was out-licensed from Nxera Pharma to Neurocrine Biosciences, a United States-based pharmaceutical company.[4][1][5] It is an oral small molecule.[6][7]
Direclidine (NBI-1117568) is an investigational, oral drug in Phase 3 trials for schizophrenia, developed by Neurocrine Biosciences, working as a selective M4 muscarinic receptor agonist to treat psychosis by modulating dopamine indirectly. It’s an innovative small molecule with potential for neuropsychiatric disorders, showing promise in early trials and aiming to offer a new treatment approach beyond traditional antipsychotics.
Key aspects of Direclidine:
- Drug Class: Small molecule, muscarinic M4 receptor agonist, antipsychotic.
- Mechanism: Acts as a selective agonist for the M4 receptor, which indirectly helps improve dopamine levels, targeting schizophrenia symptoms.
- Developer: Originally from Nxera Pharma, licensed to Neurocrine Biosciences.
- Status: In Phase 3 clinical trials for schizophrenia.
- Significance: Offers a novel mechanism for treating schizophrenia, potentially with fewer side effects than current treatments.
- Other Uses: Also being explored for bipolar mania and other neuropsychiatric conditions.
Development Timeline & Data:
- Positive Phase 2 data was announced in August 2024, showing promise for its use in schizophrenia.
- Topline data from ongoing Phase 3 studies is expected around 2027.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015118342&_cid=P20-MKJ4D7-04370-1


Typical procedure for the preparation of piperidines via sodium triacetoxyborohydride reductive amination, Boc-deprotection and ethylcarbamate formation as exemplified by the preparation of Example 1 -7, ethyl 2-[4-(1 -methyl-1 H-imidazol-2-yl)piperidin-1 -yl]-6-aza s p i ro [3.4] octa n e -6 -ca rb oxy late

Example 1-7
4-(1 -Methylimidazol-2-yl)piperidine hydrochloride (0.244 g, 1 .21 mmol) and 6-Boc-2-oxo-6-azaspiro[3,4]octane (0.273 g, 1 .21 mmol) were dissolved in DCM (10 mL) at rt and titanium isopropoxide (0.4 mL, 2.42 mmol) was added. The reaction mixture was stirred at rt for 1 h. The reaction mixture was cooled to -5 °C, then STAB (0.513 g, 2.42 mmol) and acetic acid (27 pL, 480 pmol) were added and the reaction mixture was stirred overnight under nitrogen while warming to rt. The reaction mixture was quenched with the addition of NaHC03 (sat aq.) (10 mL) and diluted with DCM then filtered through a pad of celite. The layers were separated and the aqueous layer was extracted with DCM. The combined DCM layers were washed with brine, then dried over MgS04. The solvents were removed in vacuo, and the residue was purified by column chromatography (normal phase, [Biotage SNAP cartridge KP-sil 25g , 40-63 μΠΊ, 60 A, 50 mL per min, gradient 1 % to 10% MeOH in DCM]) to give an inseparable mixture of isomers of terf-butyl 2-[4-(1 -methyl-1 H-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate (0.330 g, 72%) as a yellow gum.
LCMS (Method A): m/z 374 (M+H)+ (ES*), at 1.68 min, UV inactive.
Terf-butyl 2-[4-(1-methyl-1 H-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate (0.326 g, 0.87 mmol) was dissolved in 4 M hydrogen chloride in dioxane (1 .2 mL, 5.2 mmol). The reaction mixture was stirred at rt for 18 h. The volatiles were then removed in vacuo and the residue dissolved DCM (17 mL) and triethylamine (0.49 mL, 3.49 mmol). Ethyl chloroformate (125 μί, 1.31 mmol) was added dropwise and the solution stirred at rt for 18 h. The mixture was then poured into NaHC03 (aq) (75 mL) and DCM (75 mL), extracted (2 x 75 mL) , and the combined DCM extracts washed with brine (20 mL) then dried over MgSO*. After concentration, the residue was purified by column chromatography (normal phase, [Biotage SNAP cartridge KP-sil 25 g, 40-63 μΐη, 60 A, 50 mL per min, gradient 1 % to 10% MeOH in DCM]) to provide ethyl 2-[4-(1 -methyl-1 H-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate as a brown oil as a mixture of diastereomers (0.25 g, 83%). Preparative HPLC was used to separate the diastereomers, using a Phenomenex Gemini-N C18 column, 150 x 21 mm, eluting with 38 to 48% MeCN/H20 at 18 mL/min and collecting fractions by monitoring at 218 nm to give ethyl 2-[4-(1 -methyl-1 H-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate, Example 1-7 Isomer 1 , (0.044 g, 15%) as a colourless oil and ethyl 2-[4-(1 -methyl-1 /-/-imidazol-2-yl)piperidine]-6-azaspiro[3.4]octane-6-carboxylate, Example 1 -7 Isomer 2, (0.031 g, 10%) as a colourless oil. The data for Isomer 2 are in Table 3
PAT
- Pharmaceutical compoundsPublication Number: EP-4413985-A2Priority Date: 2014-02-06
- BICYCLIC AZA COMPOUNDS AS MUSCARINIC RECEPTOR AGONISTSPublication Number: FI-3406609-T3Priority Date: 2014-02-06Grant Date: 2024-09-10
- Bicyclic aza compounds as muscarinic receptor agonistsPublication Number: EP-3406609-B1Priority Date: 2014-02-06Grant Date: 2024-06-26
- Bicyclic aza compounds as muscarinic m1 receptor agonists.Publication Number: WO-2015118342-A1Priority Date: 2014-02-06
- Bicyclic aza compounds as muscarinic m1 receptor and/or m4 receptor agonistsPublication Number: US-2018179184-A1Priority Date: 2014-02-06
- Bicyclic aza compounds as muscarinic m1 receptor and/or m4 receptor agonistsPublication Number: US-2017240530-A1Priority Date: 2014-02-06
- Bicyclic aza compounds as muscarinic m1 receptor and/or m4 receptorPublication Number: US-2020325118-A1Priority Date: 2014-02-06
- Bicyclic aza compounds as muscarinic receptor agonistsPublication Number: ES-2986327-T3Priority Date: 2014-02-06Grant Date: 2024-11-11
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Overview
It is a selective muscarinic acetylcholine M4 receptor agonist that indirectly modulates dopamine as the basis for its putative improvement of schizophrenia.[6] In April 2016, the compound was out-licensed from Nxera Pharma to Allergan, an Irish pharmaceutical company, as part of Nxera’s wider muscarinic agonist portfokio. By September 2017, it had advanced to Phase I clinical trial for the indication of “neuropsychiatric symptoms associated with Alzheimer’s disease and other dementias”[8] Following Allergan’s acquisition by AbbVie, the license was returned to Nxera in January 2021.[9] In November 2021, the compound was newly out-licensed to Neurocrine Biosciences, a U.S. pharmaceutical company.[5] It has been under development as a treatment for schizophrenia, and is currently in Phase III clinical trials.[10][11]
History
- 2016
- April: The rights to develop Nxera Pharma’s muscarinic agonist portfolio, including NBI-1117568 were transferred to Allergan.[9]
- 2017
- September: Allergan initiated Phase I clinical trials for NBI-1117568 to treat “neurobehavioral symptoms related to Alzheimer’s disease and other conditions.”[8]
- 2021
- 2022
- October: Phase II clinical trial of NBI-1117568 for the treatment of adults with schizophrenia was initiated.[12]
- 2024
- 2025
- May: Neurocrine Biosciences initiated a Phase III registrational program for NBI-1117568 for the treatment of adults with schizophrenia.[14]
Clinical trials
Phase II clinical trial
The Phase II clinical trial was conducted in 15 sites across the U.S. with 200 adult patients diagnosed with schizophrenia.[15] The primary endpoint was assessed by the change in the total score of the Positive and Negative Syndrome Scale (PANSS) after six weeks of treatment. The 20 mg once-daily group showed a statistically significant improvement of 7.5 points compared to the placebo group (improvement of 18.2 points from baseline, p = 0.011, effect size = 0.61).[16] However, the 40 mg once-daily group, 60 mg once-daily group, and 30 mg twice-daily group did not show statistically significant differences compared to the placebo group (p-values: 40 mg group: 0.282, 60 mg group: 0.189, 30 mg twice-daily group: 0.090).[16]
Market reaction to phase II clinical trial
With a PANSS improvement of 7.5, NBI-111758 lagged behind xanomeline/trospium (KarXT) (Karuna Therapeutics) with 8.4 and emraclidine (Cerevel Therapeutics) with 12.7, both of which were in clinical trials at the same time. Moreover, the lack of dose-dependency led to disappointment in the stock market.[17] Neurocrine Biosciences’ share price dropped 19% on the day following the announcement of the Phase II clinical trial results.[18]
References
- “Delving into the Latest Updates on Direclidine with Synapse”. Synapse. 30 June 2025. Retrieved 27 July 2025.
- https://cdn.who.int/media/docs/default-source/international-nonproprietary-names-(inn)/pl133.pdf [bare URL PDF]
- Ye N, Wang Q, Li Y, Zhen X (March 2025). “Current emerging therapeutic targets and clinical investigational agents for schizophrenia: Challenges and opportunities”. Medicinal Research Reviews. 45 (2): 755–787. doi:10.1002/med.22086. PMID 39300769.
- “HTL 0016878”. AdisInsight. 2 September 2024. Retrieved 21 October 2024.
- “ニューロクライン社との統合失調症およびその他の精神神経疾患を対象とした新規ムスカリン受容体作動薬に関するライセンス契約締結のお知らせ” [Announcement of License Agreement with Neurocrine for Novel Muscarinic Receptor Agonist for Schizophrenia and Other Neuropsychiatric Disorders] (in Japanese). PR TIMES. 2021-11-22. Retrieved 2024-09-17.
- “ネクセラファーマ株価6.5%高 薬候補で毒性試験成功 – 日本経済新聞” [NexThera Pharma shares rise 6.5% as drug candidate passes toxicity test – Nikkei]. 日本経済新聞 電子版 (Nikkei Electronic Edition) (in Japanese). 日本経済新聞社 (Nikkei Inc.). 2024-04-17. Retrieved 2024-09-17.
- “当社提携先のニューロクライン社が、統合失調症を対象にしたNBI-1117568の第II相臨床試験の開始を発表[そーせいグループ] | NIKKEI COMPASS – 日本経済新聞” [Our partner Neurocrine, Inc. announces initiation of Phase II clinical trial of NBI-1117568 for schizophrenia [Sosei Group] | NIKKEI COMPASS – Nikkei Newspaper]. 日経コンパス (Nikkei Compass) (in Japanese). 日本経済新聞社 (Nikkei Inc.). 2022-10-28. Retrieved 2024-09-17.
- “アルツハイマー病等の主要症状の治療薬として開発中の新薬候補、 選択的ムスカリンM4受容体作動薬の第I相臨床試験で最初の被験者への投与を実施” [First subjects dosed in Phase I clinical trial of selective muscarinic M4 receptor agonist, a potential new drug candidate for treating major symptoms of Alzheimer’s disease] (PDF) (in Japanese). ネクセラファーマ(旧そーせいグループ株式会社). 2017-09-01. Retrieved 2024-09-17.
- “ムスカリン作動薬プログラムのグローバルな研究開発権・販売権が返還” [Global R&D and commercial rights to muscarinic agonist program returned]. プレスリリース・ニュースリリース配信シェアNo.1|PR TIMES (in Japanese). PR Times. 2021-01-05. Retrieved 2024-09-17.
- 日経バイオテクONLINE (2024-09-02). “ネクセラファーマ、統合失調症治療薬候補 NBI-1117568の第II相臨床試験の良好な結果によりニューロクライン社より35百万米ドルのマイルストンを受領” [Nexella Pharma Receives $35 Million Milestone Payment from Neurocrine Following Positive Results of Phase II Clinical Trial of NBI-1117568, a Potential Treatment for Schizophrenia]. 日経バイオテクONLINE (in Japanese). 日本経済新聞社. Retrieved 2024-09-17.
- “ネクセラ—大幅反落、ニューロクラインの株価下落に追随売り | 個別株 – 株探ニュース” [Nexella – Sharp decline, selling follows fall in Neurocrine stock price | Individual stocks – Kabutan News]. kabutan.jp (in Japanese). MINKABU THE INFONOID, Inc. 2024-08-29. Retrieved 2024-09-17.
- “当社提携先のニューロクライン社が、統合失調症を対象にしたNBI-1117568の第Ⅱ相臨床試験の開始を発表” [Our Partner Neurocrine Announces Initiation of Phase 2 Clinical Trial of NBI-1117568 for Schizophrenia]. プレスリリース・ニュースリリース配信シェアNo.1|PR TIMES (in Japanese). PR TIMES. 2022-10-28. Retrieved 2024-09-17.
- 日経バイオテク (Nikkei Biotech) ONLINE (2024-09-02). “ネクセラファーマ、統合失調症治療薬候補 NBI-1117568の第II相臨床試験の良好な結果によりニューロクライン社より35百万米ドルのマイルストンを受領” [Nexella Pharma Receives $35 Million Milestone Payment from Neurocrine Following Positive Results of Phase II Clinical Trial of NBI-1117568, a Potential Treatment for Schizophrenia]. 日経バイオテクONLINE (in Japanese). 日本経済新聞社. Retrieved 2024-09-17.
- “ネクセラファーマの統合失調症薬、最終治験を開始”. 日本経済新聞. 日本経済新聞社. 2025-05-01. Retrieved 2025-05-02.
- “Neurocrine Biosciences Initiates Phase 2 Clinical Study Evaluating NBI-1117568 in Adults with Schizophrenia”. ニューロクライン. 2022-10-27. Retrieved 2024-09-17.
- “ネクセラファーマ[4565]:ニューロクライン社との提携プログラムである統合失調症治療薬候補NBI-1117568の第2相臨床試験で良好な結果 2024年8月28日(適時開示) :日経会社情報DIGITAL:日本経済新聞” [Nexella Pharma [4565]: Positive results in Phase 2 clinical trial of NBI-1117568, a candidate for the treatment of schizophrenia, a collaboration program with Neurocrine, Inc. August 28, 2024 (timely disclosure) : Nikkei Company Information DIGITAL: Nikkei Shimbun]. 日本経済新聞 電子版 (in Japanese). 日本経済新聞社. Retrieved 2024-09-17.
- “Nxera Pharma Official IR Blog 「ニューロクライン社から35百万米ドルのマイルストン受領」” [Received $35 million milestone payment from Neurocrine]. soseiheptares.blogspot.com. ネクセラファーマ. 2024-09-02. Retrieved 2024-09-19.
- “Neurocrine Stock Down 19% on Mixed Schizophrenia Study Results”. Zacks Investment Research. Zacks Investment. 2024-08-29. Retrieved 2024-09-17.
| Clinical data | |
|---|---|
| Other names | HTL-0016878; NBI-1117568; NBI-568[1] |
| Routes of administration | Oral |
| Drug class | Muscarinic acetylcholine M4 receptor agonist |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1803346-98-6 |
| PubChem CID | 118295270 |
| ChemSpider | 133319625 |
| UNII | TXB4V44U24 |
| KEGG | D13221 |
| Chemical and physical data | |
| Formula | C19H30N4O2 |
| Molar mass | 346.475 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
NEXT
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com/
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.

Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com
/////direclidine, ANAX, BLUE JET, ADVECT, muscarinic M4 receptor positive allosteric modulator, TXB4V44U24, NBI-1117568, NBI 1117568
Brexanolone caprilcerbate



Brexanolone caprilcerbate
CAS 2681264-65-1
MFC48H78O12 MW 847.1 g/mol
1-O-[[(3R,5S,8R,9S,10S,13S,14S,17S)-17-acetyl-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl]oxycarbonyloxymethyl] 5-O-[1,3-di(octanoyloxy)propan-2-yl] 3-methylpentanedioate
1-[1,3-bis(octanoyloxy)propan-2-yl] 5-[({[(20-oxo-5α-pregnan3α-yl)oxy]carbonyl}oxy)methyl] 3-methylpentanedioate
GABAA receptor positive allosteric modulator, K3KLQ9T6WM, PHASE 2,
Brexanolone caprilcerbate (INNTooltip International Nonproprietary Name; developmental code names LYT-300, SPT-300) is an orally active prodrug of brexanolone (allopregnanolone) which is under development for the treatment of anxiety disorders.[1][2][3][4] It is a absorbed via the lymphatic system with oral administration.[5] The drug is being developed by Seaport Therapeutics and PureTech Health.[1][2] As of January 2025, it is in phase 2 clinical trials.[1]

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US335021515&_cid=P22-MKAJEO-33027-1
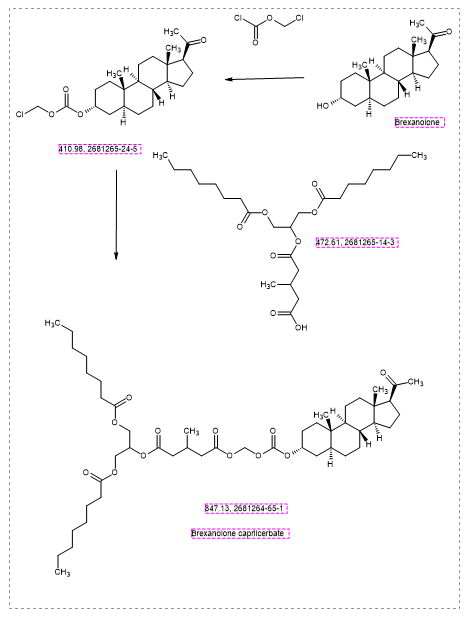
PAT
- Lipid prodrugs of neurosteroidsPublication Number: WO-2021159021-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2023338552-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2022395513-A1Priority Date: 2020-02-05
- Lipid prodrugs of neurosteroidsPublication Number: US-2021268115-A1Priority Date: 2020-02-05
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Allopregnanolone prodrug”. AdisInsight. 28 January 2025. Retrieved 26 February 2025.
- “Delving into the Latest Updates on Brexanolone caprilcerbate with Synapse”. Synapse. 15 February 2025. Retrieved 26 February 2025.
- “Proposed INN: List 131 International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 38 (2): 270. 2024.
brexanolonum caprilcerbas brexanolone caprilcerbate 1-[1,3-bis(octanoyloxy)propan-2-yl] 5-[({[(20-oxo-5α-pregnan3α-yl)oxy]carbonyl}oxy)methyl] 3-methylpentanedioate GABAA receptor positive allosteric modulator C48H78O12 2681264-65-1
- Carlini SV, Osborne LM, Deligiannidis KM (December 2023). “Current pharmacotherapy approaches and novel GABAergic antidepressant development in postpartum depression”. Dialogues in Clinical Neuroscience. 25 (1): 92–100. doi:10.1080/19585969.2023.2262464. PMC 10557560. PMID 37796239.
- Alashal N, Hussain N (2025). “Approach to the use of rescue medications in children for prolonged epileptic seizures in the community”. Paediatrics and Child Health. 35 (4): 113–117. doi:10.1016/j.paed.2025.01.004.
| Clinical data | |
|---|---|
| Other names | LYT-300; LYT300; SPT-300; SPT300; Allopregnanolone 3-O-caprilcerbate |
| Routes of administration | Oral[1] |
| Drug class | GABAA receptor positive allosteric modulator; Neurosteroid |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2681264-65-1 |
| PubChem CID | 158098654 |
| UNII | K3KLQ9T6WM |
| Chemical and physical data | |
| Formula | C48H76O12 |
| Molar mass | 845.124 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Brexanolone caprilcerbate, GABAA receptor positive allosteric modulator, K3KLQ9T6WM, PHASE 2,
Balinatunfib



Balinatunfib
CAS 2248726-53-4
MF C27H24F2N6O2, 502.5 g/mol
(1R,11R)-5-[2-(1-aminocyclobutyl)pyrimidin-5-yl]-18-(difluoromethoxy)-12-methyl-2,9,12-triazapentacyclo[9.8.1.02,10.03,8.014,19]icosa-3(8),4,6,9,14(19),15,17-heptaen-13-one
(7R,14R)-11-[2-(1-Aminocyclobutyl)pyrimidin-5-yl]-1-(difluoromethoxy)-6-methyl-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
(7R,14R)-11-[2-(1-aminocyclobutyl)pyrimidin-5-yl]-1-(difluoromethoxy)-6-methyl-6,7-dihydro-7,14-methano[1,3]benzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
tumor necrosis factor (TNF) signaling inhibitor, SAR441566, SAR 441566, PLY98MAN4C
- OriginatorSanofi
- ClassAmines; Anti-inflammatories; Antipsoriatics; Antirheumatics; Azabicyclo compounds; Benzimidazoles; Cyclobutanes; Fluorinated hydrocarbons; Heterocyclic compounds with 4 or more rings; Ketones; Phenyl ethers; Pyrimidines; Small molecules
- Mechanism of ActionTumour necrosis factor alpha inhibitors
- Phase IICrohn’s disease; Psoriasis; Rheumatoid arthritis; Ulcerative colitis
- No development reportedInflammation
- 09 Dec 2025Sanofi plans a phase-I trial (In volunteers) in December 2025 (PO, Tablet), (NCT07272629)
- 29 Oct 2025Sanofi plans a phase II SPECIFI-IBD-LTS trial for Crohn’s Disease or Ulcerative Colitis ( Treatment-experienced) in unknown location (PO, Tablet) in December 2025 (NCT07222189)
- 16 Sep 2025Chemical structure information added.
- You need to be a logged in or subscribed to view this c
Balinatunfib (SAR441566) is an experimental drug which acts as a potent small molecule inhibitor of TNF. Rather than blocking TNF receptors, balinatunfib inactivates TNF directly by stabilising an inactive form of the TNF trimer which fails to bind to its target receptors. It is in early stage clinical trials for rheumatoid arthritis and other chronic autoimmune diseases.[1][2]
SYN



PAT
(WO 2016/050975,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016050975&_cid=P22-MK3F7M-67505-1
Intermediate 40

(1R,3R)-1-[2-bromo-6-(difluoromethoxy)phenyl]-7-chloro-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-amine
Intermediate 38 (5 g, 11.64 mmol) was suspended in toluene (22 mL) and cooled to 0°C before addition of diphenylphosphoryl azide (3.4 mL, 15 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (2.5 mL, 16 mmol). The mixture was allowed to warm up to r.t and stirred for 2 hours and subsequently at 45°C overnight. The reaction mixture was diluted with EtOAc (150 mL) and the organic phase washed with a saturated aqueous solution of ammonium chloride (50 mL) then a saturated solution of aqueous sodium bicarbonate (50 mL), and concentrated in vacuo. The crude residue thus obtained was solubilized in THF (100 mL) and water (10 mL), trimethylphosphine (17.46 mL, 17.46 mmol) was added and the reaction mixture stirred overnight. The mixture was concentrated in vacuo, partitioned between EtOAc (200 mL) and water (150 mL). The organic layer was extracted with 0.2M HCl aq (3 x 200 mL). The combined acid layer was stirred in an ice bath, whilst 10% NaOH solution was added with stirring until pH increased to 10. The stirred was continued for further 15 minutes to complete precipitation. The precipitate was filtered, rinsed with water (20 mL), then dried under suction for 10 minutes before drying under high vacuum overnight to afford 3.92 g (78%) of the title compound as an off white solid. LCMS basic: RT 1.96 min. (ES+) 428/430 (M+H)+
EXAMPLE 11

(7R, 14R)-11-chloro-1-(difluoromethoxy)-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
Intermediate 40 (3.7 g, 8.6 mmol), activated molecular sieve 4A powder (1.2 g), potassium carbonate (1.5 equiv., 13 mmol) followed by dichloro[9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene]palladium(II) (0.04 equiv., 0.35 mmol) were poured into the center of the 100 mL Glass Parr reaction vessel. 3 cycles of vacuum (~20 mmHg) followed by Argon were applied to the closed reactor.
Anhydrous dimethyl sulfoxide (35 mL) was added, followed by phenol 5M in DMSO (1.1 equiv., 9.5 mmol). The solution was degassed by 3 vacuum (~20 mmHg) / argon cycles followed by 3 cycles of vacuum / CO resulting in a final CO pressure of 1 bar.
The mixture was stirred and heated overnight at 100 °C under the CO atmosphere . The reaction was cooled to 30°C, the reactor vessel was opened and EtOAc (40 mL) was added. The resulting mixture was filtered on a pad of Celite, evaporated in vacuo to yield a green oil.
The residue thus obtained was taken up in EtOAc (100 mL) and the organic layer was washed with water, K2CO3 (saturated aqueous solution) and brine (saturated aqueous solution). The aqueous layer was then re-extracted with EtOAc (1 x 50 mL). The combined organic layers were dried over MgSO4, filtered and evaporated to dryness. The obtained green solid (3.65 g), was taken up in EtOAc, the insoluble material was filtered and rinsed with Et2O to afford 1.06 g (33.1%) of the title compound as a grey solid.
The filtrate can be purified by flash chromatography to provide additional product if required:
LCMS basic: MH+ m/z = 376, RT 1.90 minutes.
1H NMR (300 MHz, DMSO) δ 9.12 (d, 1 H, J = 6.7 Hz), 8.23 (dd, 1 H, J = 7.0, 2.4 Hz), 7.60 (m, 5 H), 7.20 (dd, 1 H, J = 8.7, 2.1 Hz), 6.29 (d, 1 H, J = 7.1 Hz), 4.87 (dd, 1 H, J = 6.7 Hz, 6.7 Hz), 3.46 (m, 1 H), 2.72 (d, 1 H, J = 13.4 Hz).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US283322316&_cid=P22-MK3EWF-57090-1
Intermediate 3

(7R,14R)-11-Chloro-1-(difluoromethoxy)-6-methyl-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
Intermediate 17

tert-Butyl (1-{5-[(7R,14R)-1-(difluoromethoxy)-6-methyl-5-oxo-5,6,7,14-tetrahydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-11-yl]pyrimidin-2-yl}cyclobutyl)-carbamate
EXAMPLE 6

(7R,14R)-11-[2-(1-Aminocyclobutyl)pyrimidin-5-yl]-1-(difluoromethoxy)-6-methyl-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
To a solution of Intermediate 17 (18.0 g, 29.9 mmol) in 1,4-dioxane (25 mL) was added 4M hydrochloric acid in 1,4-dioxane (40 mL). The resulting mixture was stirred at room temperature for 1 h, then concentrated in vacuo. The residue was dissolved in water (500 mL) and washed with EtOAc (2×300 mL). The aqueous layer was basified to pH 9 with 2N aqueous sodium hydroxide solution, which resulted in precipitation of a solid. EtOAc (500 mL) was added and the mixture was stirred until all solids had dissolved. The residue was partitioned, then the aqueous layer was further extracted with EtOAc (500 mL). The combined organic layers were dried over Na 2SO 4 and filtered, then concentrated in vacuo and dried overnight under high vacuum. The foamy residue was suspended in a mixture of diethyl ether and hexane (150 mL), then stirred and shaken vigorously, before being concentrated in vacuo, to afford the title compound (12.4 g, 83%) as a white amorphous solid. δ H (400 MHz, DMSO-d 6) 9.05 (s, 2H), 8.32-8.22 (m, 1H), 7.91-7.66 (m, 3H), 7.62 (dd, J8.5, 1.8 Hz, 1H), 7.53-7.46 (m, 2H), 6.31 (d, J7.1 Hz, 1H), 5.26 (d, J 7.2 Hz, 1H), 3.52 (dt, J 14.2, 7.3 Hz, 1H), 3.36 (s, 3H), 2.84 (d, J 13.8 Hz, 1H), 2.63 (dtd, J11.5, 5.6, 2.5 Hz, 2H), 2.38 (s, 2H), 2.16-2.05 (m, 2H), 2.04-1.91 (m, 1H), 1.87-1.73 (m, 1H). LCMS (ES+APCI) [M-NH 2] − 486.0, RT 1.66 minutes (Method 2). LCMS (ES+) [M+H] + 503.0, RT 1.71 minutes (Method 1).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025008402&_cid=P22-MK3EWF-57090-1

(7R,14R)-1 l-[2-(l-aminocyclobutyl)pyrimidin-5-yl]-l-(difhroromethoxy)-6-methyl-6,7-dihydro-7, 14-methanobenzimidazo[l,2-b][2,5]benzodiazocin-5(14H)-one.
PAT
- Fused Pentacyclic Imidazole Derivatives as Modulators of TNF ActivityPublication Number: US-2021252012-A1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of TNF activityPublication Number: KR-102565132-B1Priority Date: 2017-04-25Grant Date: 2023-08-08
- Fused Pentacyclic Imidazole Derivatives as Modulators of TNF ActivityPublication Number: US-2025127795-A1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of TNF activityPublication Number: CN-110582495-BPriority Date: 2017-04-25Grant Date: 2022-04-01
- Fused Pentacyclic Imidazole DerivativesPublication Number: US-2017305932-A1Priority Date: 2014-10-03
- Fused pentacyclic imidazole derivatives as modulators of TNF activityPublication Number: CN-110582495-APriority Date: 2017-04-25
- Fused Pentacyclic Imidazole Derivatives as Modulators of TNF ActivityPublication Number: US-2023250105-A1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of TNF activityPublication Number: US-10980814-B2Priority Date: 2017-04-25Grant Date: 2021-04-20
- Fused pentacyclic imidazole derivatives as modulators of tnf activityPublication Number: EP-3939980-A1Priority Date: 2017-04-25
- Process for preparing fused pentacyclic imidazole derivatives and uses thereof as modulators of tnf activityPublication Number: EP-3939980-B1Priority Date: 2017-04-25Grant Date: 2023-07-26
- Preparation of bridged pentacyclic imidazole derivatives as modulators of tnf activity, intermeditates and their preparationPublication Number: WO-2025068505-A1Priority Date: 2023-09-29
- DERIVATIVES OF COMBINED PENTACYCLIC IMIDAZOLES AS MODULATORS OF TNF ACTIVITYPublication Number: HR-P20211927-T1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of tnf activityPublication Number: CA-3058980-A1Priority Date: 2017-04-25
- Fused pentacyclic imidazole derivatives as modulators of tnf activityPublication Number: EP-3615534-B1Priority Date: 2017-04-25Grant Date: 2021-09-15
- Fused Pentacyclic Imidazole Derivatives as Modulators of TNF ActivityPublication Number: US-2020046723-A1Priority Date: 2017-04-25
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Vugler A, O’Connell J, Nguyen MA, Weitz D, Leeuw T, Hickford E, et al. (2022). “An orally available small molecule that targets soluble TNF to deliver anti-TNF biologic-like efficacy in rheumatoid arthritis”. Frontiers in Pharmacology. 13 1037983. doi:10.3389/fphar.2022.1037983. PMC 9709720. PMID 36467083.
- Li Y, Ye R, Dai H, Lin J, Cheng Y, Zhou Y, et al. (January 2025). “Exploring TNFR1: from discovery to targeted therapy development”. Journal of Translational Medicine. 23 (1): 71. doi:10.1186/s12967-025-06122-0. PMC 11734553. PMID 39815286.
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2248726-53-4 |
| PubChem CID | 132042903 |
| IUPHAR/BPS | 13583 |
| ChemSpider | 129738176 |
| Chemical and physical data | |
| Formula | C27H24F2N6O2 |
| Molar mass | 502.526 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////Balinatunfib, tumor necrosis factor (TNF) signaling inhibitor, SAR441566, SAR 441566, PLY98MAN4C
Amogammadex



Amogammadex
CAS 1309580-40-2
MF C88H136N8O56S8 MW2458.56
(2R)-2-acetamido-3-[[(1S,3S,5S,6S,8S,10S,11S,13S,15S,16S,18S,20S,21S,23S,25S,26S,28S,30S,31S,33S,35S,36S,38S,40S,41R,42R,43R,44R,45R,46R,47R,48R,49R,50R,51R,52R,53R,54R,55R,56R)-10,15,20,25,30,35,40-heptakis[[(2R)-2-acetamido-2-carboxyethyl]sulfanylmethyl]-41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56-hexadecahydroxy-2,4,7,9,12,14,17,19,22,24,27,29,32,34,37,39-hexadecaoxanonacyclo[36.2.2.23,6.28,11.213,16.218,21.223,26.228,31.233,36]hexapentacontan-5-yl]methylsulfanyl]propanoic acid
L-CYSTEINE, S,S’,S”,S”’,S””,S”””,S”””,S”””’-(6A,6B,6C,6D,6E,6F,6G,6H-OCTADEOXY-.GAMMA.-CYCLODEXTRIN-6A,6B,6C,6D,6E,6F,6G,6H-OCTAYL)OCTAKIS(N-ACETYL-
AMOGAMMADEX [INN]
CYCLOOCTAKIS-(1->4)-(6-S-((2R)-2-ACETAMIDO-2-CARBOXYETHYL)-6-THIO-.ALPHA.-D-GLUCOPYRANOSYL)
cyclooctakis-(1→4)-{6-S-[(2R)-2-acetamido-2-carboxyethyl]-6-thio-α-Dglucopyranosyl}
rocuronium and vecuronium reversal agent, L-CYSTEINE, S,S’,S”,S”’,S””,S”””,S”””,S”””’-(6A,6B,6C,6D,6E,6F,6G,6H-OCTADEOXY-.GAMMA.-CYCLODEXTRIN-6A,6B,6C,6D,6E,6F,6G,6H-OCTAYL)OCTAKIS(N-ACETYL-
AMOGAMMADEX [INN]
CYCLOOCTAKIS-(1->4)-(6-S-((2R)-2-ACETAMIDO-2-CARBOXYETHYL)-6-THIO-.ALPHA.-D-GLUCOPYRANOSYL)
Pat
WO2012068981
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012068981&_cid=P21-MJW9RG-10499-1

CD-8
Weigh 23.7 g (0.088 mol) of N-acetylcysteine and measure 160 ml of dry DMF. Add both to a dry three-necked flask and stir until completely dissolved. Cool the reaction solution to approximately -10°C in a constant temperature bath. Slowly add 8.81 g of sodium hydride (60%) in portions under argon protection and mechanical stirring, maintaining the temperature below -5°C. After the addition is complete, continue stirring until no more bubbles emerge, then transfer the solution to approximately 5°C and react until no more bubbles emerge (approximately 2-3 hours).
With the temperature controlled at approximately 5°C in an ice bath, add 8.38 g (3.85 mmol) of DMF solution of 6-per-deoxy-6-per-iodo-γ-cyclodextrin to the reaction solution of the fully reacted N-acetylcysteine sodium salt. Under argon protection, mechanically stir to ensure homogeneity and continue stirring for 30 min. Gradually raise the temperature of the reaction solution to 70°C and react for 12 h. Then cool the reaction solution to room temperature, filter, wash the filter cake twice with DMF, and then wash with acetone until triphenylphosphine and triphenyloxyphosphine are removed. Dry under reduced pressure to obtain crude sodium salt. Dissolve the crude sodium salt in glacial acetic acid, and then pass dry hydrogen chloride gas into the solution under ice bath cooling. A white solid precipitates after 20 min. Filter after no more white solid precipitates (approximately 1 h). Dry acetone was gradually added to the filtrate, and a solid precipitated out. The mixture was filtered, and the filter cake was washed with acetone until there was no sour taste. The cake was dried under reduced pressure to obtain 6-per-deoxy-6-per-(N-acetylglycine methyl)thioether-γ-cyclodextrin (CD-8) with a yield of 48%.
Ή NMR spectra of CD-8 in heavy water (D2O ) : 52.02 (CH3,m,3H), 2.69,2.44 (CH2,m,2H), 3.02 (CH,m,H), 3.06,2.81 (CH2,m,2H), 3.73 (2CH,m,2H), 4.19 (CH,m,H), 4.74 (CH,m,H), 5.03 (CH,s,H) ppm.
PAT
CN102060941
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84636898&_cid=P21-MJW9XY-15988-1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////amogammadex
Aleniglipron



Aleniglipron
CAS 2685823-26-9
MF C49H55FN9O6P MW916.0 g/mol
3-[(1S,2S)-1-[2-[(4S)-3-[3-[4-diethylphosphoryl-3-(methylamino)phenyl]-2-oxoimidazol-1-yl]-2-(4-fluoro-3,5-dimethylphenyl)-4-methyl-6,7-dihydro-4H-pyrazolo[4,3-c]pyridine-5-carbonyl]-5-(oxan-4-yl)indol-1-yl]-2-methylcyclopropyl]-4H-1,2,4-oxadiazol-5-one

glucagon-like peptide 1 (GLP-1) receptor agonist, GSBR-1290, GSBR 1290, Z6XCL6R9SX
Aleniglipron (development code GSBR-1290) is a small-molecule GLP-1 agonist developed by Structure Therapeutics.[1] It is delivered orally and is in a Phase II trial as of 2023.[2][3][4] In June 2024, Structure Therapeutics reported positive topline data from a Phase 2a obesity study in which GSBR-1290 demonstrated clinically meaningful and statistically significant placebo-adjusted mean weight loss and generally favorable safety and tolerability results.[5]
- Aleniglipron Phase 2 Body Composition StudyCTID: NCT07169942Phase: Phase 2Status: Active, not recruitingDate: 2025-10-31
- A Dose-Range Study of Aleniglipron (GSBR-1290) in Participants Living With Obesity or Overweight With at Least One Weight-related ComorbidityCTID: NCT06703021Phase: Phase 2Status: Active, not recruitingDate: 2025-09-15
- A Phase 2b, Dose-range Finding Study of the Efficacy and Safety of Multiple Doses of Aleniglipron (GSBR-1290) in Participants Living With Obesity or Overweight With at Least One Weight-related ComorbidityCTID: NCT06693843Phase: Phase 2Status: Active, not recruitingDate: 2025-08-26
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US367934715&_cid=P10-MJRZ0C-74156-1


Example 2: Synthesis of
3-((1S,2S)-1-(2-((S)-3-(3-(4-(diethylphosphoryl)-3-(methylamino)phenyl)-2-oxo-2,3-dihydro-1H-imidazol-1-yl)-2-(4-fluoro-3,5-dimethylphenyl)-4-methyl-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridine-5-carbonyl)-5-(tetrahydro-2H-pyran-4-yl)-1H-indol-1-yl)-2-methylcyclopropyl)-1,2,4-oxadiazol-5(4H)-one (Compound 121a)


Step A: (4-bromo-2-fluorophenyl)diethylphosphine oxide
| 1H NMR (400 MHz, DMSO-d 6) δ 7.63-7.73 (m, 3H), 1.95-2.08 (m, 2H), 1.80-1.92 (m, 2H), 0.80-1.10 (m, 6H). |
Step B: (4-bromo-2-(methylamino)phenyl)diethylphosphine oxide
| 1H NMR (600 MHz, DMSO-d 6) δ 7.75-7.76 (m, 1H), 7.11 (dd, J=13.2, 8.4 Hz, 1H), 6.63-6.80 (m, 2H), 2.71 (d, J=5.4 Hz, 3H), 1.88-1.94 (m, 4H), 0.90-1.05 (m, 6H). |
Step C: tert-butyl (S)-3-(3-(4-(diethylphosphoryl)-3-(methylamino)phenyl)-2-oxo-2,3-dihydro-1H-imidazol-1-yl)-2-(4-fluoro-3,5-dimethylphenyl)-4-methyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxylate
Step D: (5)-1-(4-(diethylphosphoryl)-3-(methylamino)phenyl)-3-(2-(4-fluoro-3,5-dimethylphenyl)-4-methyl-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridin-3-yl)-1,3-dihydro-2H-imidazol-2-one hydrochloride
Step E: 3-((1S,2S)-1-(2-((S)-3-(3-(4-(diethylphosphoryl)-3-(methylamino) phenyl)-2-oxo-2,3-dihydro-1H-imidazol-1-yl)-2-(4-fluoro-3,5-dimethylphenyl)-4-methyl-4,5,6,7-tetrahydro-2H-pyrazolo[4,3-c]pyridine-5-carbonyl)-5-(tetrahydro-2H-pyran-4-yl)-1H-indol-1-yl)-2-methylcyclopropyl)-1,2,4-oxadiazol-5(4H)-one
PAT
- Heterocyclic glp-1 agonistsPublication Number: EP-4097099-A1Priority Date: 2020-02-07
- Heterocyclic GLP-1 agonistsPublication Number: CN-115698003-APriority Date: 2020-02-07
- Heterocyclic glp-1 agonistsPublication Number: EP-4097099-B9Priority Date: 2020-02-07Grant Date: 2025-04-30
- Heterocyclic glp-1 agonistsPublication Number: EP-4458834-A2Priority Date: 2020-02-07
- Heterocyclic GLP-1 agonistsPublication Number: US-11926643-B2Priority Date: 2020-02-07Grant Date: 2024-03-12
- Heterocyclic GLP-1 agonistsPublication Number: CN-119823184-APriority Date: 2020-02-07
- Heterocyclic GLP-1 agonistsPublication Number: CN-119841865-APriority Date: 2020-02-07
- Heterocyclic GLP-1 agonistsPublication Number: CN-119874775-APriority Date: 2020-02-07
- Heterocyclic GLP-1 agonistsPublication Number: US-11492365-B2Priority Date: 2020-02-07Grant Date: 2022-11-08
- Heterocyclic GLP-1 agonistsPublication Number: CN-115698003-BPriority Date: 2020-02-07Grant Date: 2024-10-11
- Heterocyclic glp-1 agonistsPublication Number: US-2022213130-A1Priority Date: 2020-02-07
- Heterocyclic glp-1 agonistsPublication Number: EP-4097099-B1Priority Date: 2020-02-07Grant Date: 2024-06-26
- Heterocyclic glp-1 agonistsPublication Number: US-2023174565-A1Priority Date: 2020-02-07
- Salts and solid forms of a compound having glp-1 agonist activityPublication Number: WO-2024125602-A1Priority Date: 2022-12-15
- Salts and solid forms of a compound having glp-1 agonist activityPublication Number: EP-4634180-A1Priority Date: 2022-12-15
- Heterocyclic glp-1 agonistsPublication Number: US-2024366639-A1Priority Date: 2021-08-12
- Heterocyclic glp-1 agonistsPublication Number: WO-2023016546-A1Priority Date: 2021-08-12
- Heterocyclic glp-1 agonistsPublication Number: WO-2021155841-A1Priority Date: 2020-02-07
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Mao, Ting; Meng, Qinghua; Zhang, Haizhen; Zhang, Jinqiang J.; Shi, Songting; Guan, Zhibo; Jiang, Xinglong; Zhang, Fang; Lei, Hui; Lin, Xichen (20 June 2023). “760-P: Discovery of GSBR-1290, a Highly Potent, Orally Available, Novel Small Molecule GLP-1 Receptor Agonist”. Diabetes. 72 (Supplement_1) 760-P. doi:10.2337/db23-760-P. S2CID 259430363.
- “Structure Therapeutics Initiates Phase 2a Study of Oral GLP-1 agonist GSBR-1290 for the Treatment of Type 2 Diabetes and Obesity”. BioSpace. 25 May 2023. Retrieved 4 November 2023.
- “Structure announces positive results from oral GLP-1 receptor agonist gsbr-1290”. Bariatric News. 2 October 2023. Retrieved 4 November 2023.
- Satija, Bhanvi (29 September 2023). “Structure Therapeutics surges as early data from obesity pill tops expectations”. Reuters. Retrieved 4 November 2023.
- “Structure Therapeutics Reports Positive Topline Data from its Phase 2a Obesity Study and Capsule to Tablet PK Study for its Oral Non-Peptide Small Molecule GLP-1 Receptor Agonist GSBR-1290”. BioSpace. 2024-06-03. Retrieved 2024-10-24.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2685823-26-9 |
| PubChem CID | 164809721 |
| DrugBank | DB18551 |
| UNII | Z6XCL6R9SX |
| Chemical and physical data | |
| Formula | C49H55FN9O6P |
| Molar mass | 916.008 g·mol−1 |
| InChI | |
//////////Aleniglipron, glucagon-like peptide 1 (GLP-1) receptor agonist, GSBR-1290, GSBR 1290, Z6XCL6R9SX
Zoracopan




Zoracopan
CAS 2243483-63-6
MF C31H31BrN6O3 MW 615.52
2-Azabicyclo[3.1.0]hexane-3-carboxamide, 2-[2-[3-acetyl-7-methyl-5-(2-methyl-5-pyrimidinyl)-1H-indol-1-yl]acetyl]-N-(6-bromo-3-methyl-2-pyridinyl)-5-methyl-, (1R,3S,5R)-
(1R,3S,5R)-2-{[3-acetyl-7-methyl-5-(2-methylpyrimidin5-yl)-1H-indol-1-yl]acetyl}-N-(6-bromo-3-methylpyridin2-yl)-5-methyl-2-azabicyclo[3.1.0]hexane-3-carboxamide
complement factor D inhibitor, ALXN-2080, ALXN 2080, E7799Y8LXY
Zoracopan is a selective complement factor D (CFD) inhibitor. When administered systemically (orally or intravenously), Zoracopan accumulates and is sustained-released in ocular tissues, primarily in the choroid-retinal pigment epithelium (C-RPE) and/or iridociliary body (I-CB).
Zoracopan is a small molecule drug. The usage of the INN stem ‘-copan’ in the name indicates that Zoracopan is a complement receptor antagonist/complement inhibitor. Zoracopan is under investigation in clinical trial NCT06173596 (A Study to Evaluate Potential Drug Interactions Between ALXN2080 and Itraconazole, Fluconazole & Carbamazepine in Healthy Adults). Zoracopan has a monoisotopic molecular weight of 614.16 Da.
- Safety and Tolerability, PK, and PD Study of Single and Multiple ALXN2080 Doses in Healthy ParticipantsCTID: NCT05428696Phase: Phase 1Status: CompletedDate: 2024-06-07
- A Study to Evaluate Potential Drug Interactions Between ALXN2080 and Itraconazole, Fluconazole & Carbamazepine in Healthy AdultsCTID: NCT06173596Phase: Phase 1Status: CompletedDate: 2024-06-20
- A Study to Investigate the Potential Drug Interactions Between ALXN2080 and Rosuvastatin and Metformin in Healthy Adult ParticipantsCTID: NCT06160414Phase: Phase 1Status: CompletedDate: 2025-04-24
WO2024259085
WO2024137329
SYN
426
https://patentscope.wipo.int/search/en/detail.jsf?docId=US289331902&_cid=P10-MJJEYB-31207-1
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018160889&_cid=P10-MJJEK1-12570-1

PAT
- Aryl, heteroaryl and heterocyclic pharmaceutical compounds for the treatment of medical disordersPublication Number: JP-7390449-B2Priority Date: 2017-03-01Grant Date: 2023-12-01
- Aryl, heteroaryl, and heterocyclic pharmaceutical compounds for treatment of medical disordersPublication Number: US-11708351-B2Priority Date: 2017-03-01Grant Date: 2023-07-25
- Aryl, heteroaryl and heterocyclic pharmaceutical compounds for the treatment of medical disordersPublication Number: JP-2022174122-APriority Date: 2017-03-01
- Aryl, heteroaryl, and heterocyclic pharmaceutical compounds for the treatment of medical disordersPublication Number: KR-20190126831-APriority Date: 2017-03-01
- Aryl, heteroaryl, and heterocyclic pharmaceutical compounds for treatment of medical disordersPublication Number: US-11084800-B2Priority Date: 2017-03-01Grant Date: 2021-08-10
- Aryl, heteroary, and heterocyclic pharmaceutical compounds for treatment of medical disordersPublication Number: EP-3985002-A1Priority Date: 2017-03-01
- Aryl, heteroaryl, and heterocyclic pharmaceutical compounds for the treatment of medical disordersPublication Number: KR-102632860-B1Priority Date: 2017-03-01Grant Date: 2024-02-02
- Aryi, Heteroaryl, and Heterocyclic Pharmaceutical Compounds for Treatment of Medical DisordersPublication Number: US-2020071301-A1Priority Date: 2017-03-01
- Aryl, Heteroaryl and Heterocyclic Pharmaceutical Compounds for the Treatment of Medical DisordersPublication Number: CN-110603252-APriority Date: 2017-03-01
- Aryl, heteroaryl, and heterocyclic pharmaceutical compounds for treatment of medical disordersPublication Number: US-2022396563-A1Priority Date: 2017-03-01
- Ocular drug depot for complement-mediated disordersPublication Number: US-2023126447-A1Priority Date: 2020-03-10
- Ocular drug depot for complement-mediated disordersPublication Number: WO-2021183555-A1Priority Date: 2020-03-10
- Aryl, heteroaryl, and heterocyclic pharmaceutical compounds for treatment of medical disordersPublication Number: US-12162860-B2Priority Date: 2017-03-01Grant Date: 2024-12-10
- Aryl, heteroaryl, and heterocyclic pharmaceutical compounds for treatment of medical disordersPublication Number: US-2023357199-A1Priority Date: 2017-03-01
- Aryl, heteroary, and heterocyclic pharmaceutical compounds for treatment of medical disordersPublication Number: WO-2018160889-A1Priority Date: 2017-03-01
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////zoracopan, complement factor D inhibitor, ALXN-2080, ALXN 2080, E7799Y8LXY

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}