
Home » Uncategorized (Page 24)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BIFONAZOLE
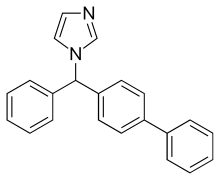
BIFONAZOLE
- Molecular FormulaC22H18N2
- Average mass310.392 Da
(±)-1-(p,a-Diphenylbenzyl)imidazole
(±)-Bifonazole
1-([1,1′-Biphenyl]-4-ylphenylmethyl)-1H-imidazole
1-(p,α-Diphenylbenzyl)imidazole
262-336-6[EINECS]
4887
60628-96-8[RN]
бифоназол
بيفونازول
联苯苄唑
- BAY H 4502
- BAY-H-4502
Bifonazole
CAS Registry Number: 60628-96-8
CAS Name: 1-([1,1¢-Biphenyl]-4-ylphenylmethyl)-1H-imidazole
Additional Names: (±)-1-(p,a-diphenylbenzyl)imidazole
Manufacturers’ Codes: Bay h 4502
Trademarks: Amycor (Lipha); Azolmen (Menarini); Bedriol (Andromaco); Mycospor (Bayer); Mycosporan (Bayer)
Molecular Formula: C22H18N2, Molecular Weight: 310.39
Percent Composition: C 85.13%, H 5.85%, N 9.03%
Literature References: Antimycotic deriv of imidazole. Prepn: E. Regel et al.,DE2461406; eidem,US4118487 (1976, 1978 both to Bayer). Series of articles on in vitro and in vivo antimycotic efficacy, microscopic studies, pharmacokinetics, efficacy in dermatomycoses and comparison with clotrimazole and miconazole, q.q.v.:Arzneim.-Forsch.33, 517-551, 745-754 (1983). Toxicology: G. Schlüter, ibid. 739.
Properties: Crystals from acetonitrile, mp 142°. Very lipophilic. Sol in alcohols, DMF, DMSO. Soly in water at pH 6: <0.1 mg/100 ml. Stable in aq soln at pH 1-12. LD50 in male mice, rats (mg/kg): 2629, 2854 orally (Schlüter).
Melting point: mp 142°
Toxicity data: LD50 in male mice, rats (mg/kg): 2629, 2854 orally (Schlüter)
Therap-Cat: Antifungal.
Keywords: Antifungal (Synthetic); Imidazoles.
BrandsAmycor (Merck) / Azolmen (Menarini) / Bayclear Plus (Bayer) / Bifonol (Mayado Seiyaku) / Canespor (Bayer) / Canesten (Bayer) / Mycospor (Bayer)
Bifonazole (trade name Canespor among others[1]) is an imidazole antifungal drug used in form of ointments.
It was patented in 1974 and approved for medical use in 1983.[2] There are also combinations with carbamide for the treatment of onychomycosis.
Bifonazole is an azole antifungal drug used to treat fungal skin infections, such as dermatomycosis.
- Synonyms:Bifonazolum
- ATC:D01AC10
- MW:310.40 g/mol
- CAS-RN:60628-96-8
- InChI Key:OCAPBUJLXMYKEJ-UHFFFAOYSA-N
- InChI:InChI=1S/C22H18N2/c1-3-7-18(8-4-1)19-11-13-21(14-12-19)22(24-16-15-23-17-24)20-9-5-2-6-10-20/h1-17,22H
- EINECS:262-336-6
- LD50:57 mg/kg (M, i.v.); 2629 mg/kg (M, p.o.);
63 mg/kg (R, i.v.); 1463 mg/kg (R, p.o.);
>500 mg/kg (dog, p.o.)
Derivatives
Monohydrochloride
- Formula:C22H18N2 • HCl
- MW:346.86 g/mol
- CAS-RN:60629-09-6
Sulfate
- Formula:C22H18N2 • xH2O4S
- MW:unspecified
- CAS-RN:60629-08-5
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 98-88-4 | C7H5ClO | benzoyl chloride | Benzoyl chloride |
| 92-52-4 | C12H10 | biphenyl | 1,1′-Biphenyl |
| 7515-73-3 | C19H15Cl | (±)-4-(chlorophenylmethyl)biphenyl | 1,1′-Biphenyl, 4-(chlorophenylmethyl)- |
| 288-32-4 | C3H4N2 | imidazole | 1H-Imidazole |
SYN
Synthesis Reference
Regal, E., Draber, W., Buchel, K.H.and Plempel, M.; U.S. Patent 4,118,487; October 3,1978; assigned to Bayer A.G.
SYN

SYN
(CAS NO.: ), with its systematic name of , 1-(alpha-(4-biphenylyl)benzyl)-, could be produced through many synthetic methods.
Following is one of the synthesis routes: (I) could be reduced with NaBH4 in ethanol to produce 4-phenylbenzhydrol (II), and the yielding product is then condensed with imidazole (III) in the presence of SOCl2 in acetonitrile.

PAT
https://patents.google.com/patent/DE10332684B3/en
- The The present invention relates to a process for the preparation of Bifonazole (1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole) by reacting 1-biphenyl-4-yl (phenyl) methanol with a chlorinating reagent in cyclohexane and subsequent coupling with imidazole.
- [0002]The compound bifonazole (1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole) is off DE-A 2 461 406 known and corresponds to the formula (I). Due to its antifungal activity, it can be used as an agent for the treatment of fungal diseases.
- [0003]Various methods for preparing this compound are known. So describes DE-A 2 461 406 the synthesis (process 1) of bifonazole (Example 1) starting from biphenyl-4-yl (phenyl) methanol by reaction with imidazole and thionyl chloride in acetonitrile with a yield of only 56% of theory. An alternative synthesis described therein (process 2) starting from 4- [chloro (phenyl) methyl] biphenyl, which is prepared from biphenyl-4-yl (phenyl) methanol by reaction with thionyl chloride in toluene, by reaction with trimethylsilylimidazole bifonazole provides only in a yield of 52% of theory.
- [0004]ES-A 2 024 363 describes also starting from 4- [chloro (phenyl) methyl] biphenyl, which is prepared from biphenyl-4-yl (phenyl) methanol by reaction with hydrogen chloride in acetonitrile, by reaction with imidazole in acetonitrile using a phase transfer catalyst, the synthesis (method 3) of bifonazole.
- [0005]AT-B 396 931 describes the preparation (method 4) of bifonazole by means of reductive amination of biphenyl-4-yl (phenyl) methanone with imidazole and formic acid. However, this requires high reaction temperatures (220 ° C.) and long reaction times. DE-A 3 538 873 describes a comparable process (process 5) with the additional use of p-toluenesulfonic acid, wherein the reaction temperature is 180 ° C.
- [0006]This in ES 539 345 described method (method 6) for the preparation of bifonazole involves a Gringard reaction between 4-biphenylmagnesium bromide and benzoylated imidazole. Finally, it is tosylated and reduced to bifonazole.
- [0007]ES 549 793 describes the synthesis (method 7) of bifonazole starting from a cyclocondensation between biphenyl-4-yl (phenyl) methylamine, 2-chloro-1-aminoethane and ethyl orthoacetate. The final dehydrogenation is carried out by reaction with 2,3-dichloro-5,6-dicyano-p-benzoquinone in benzene.
- [0008]All known processes have various disadvantages which are particularly unfavorable in the preparation of the compound of the formula (I) on an industrial scale. The solvents used in processes 1 and 2 acetonitrile and toluene are of concern to health. Their use should be avoided in the manufacture of active ingredients used in medicines. By using toluene in process 2, chlorination to give 4- [chloro (phenyl) methyl] biphenyl also produces a toluene-specific, undesired by-product which can only be removed incompletely and thus deteriorates the product quality. The yield is unsatisfactory in both processes. A significant disadvantage of method 3 is, in addition to the use of acetonitrile as solvent, the use of a phase transfer catalyst, which is difficult to separate from the product during work-up. Methods 4 and 5 both operate at very high temperatures and are therefore disadvantageous in a technical use due to the energy consumption and the potential hazard. In method 6, the use of the Gringard reagent is disadvantageous, since this must be produced under considerable safety expense and difficult to handle on an industrial scale. Disadvantage in process 7 is the use of the very toxic compounds 2,3-dichloro-5,6-dicyano-p-benzoquinone and benzene. Their use should be avoided especially in the production of active ingredients used in pharmaceuticals
- Embodiment:
- Synthesis of bifonazole (1- [Biphenyl-4-yl (phenyl) methyl] -1H-imidazole)
- 1st step: 4- [chloro (phenyl) methyl] biphenyl (III)
- [0038]140 g (0.54 mol) dry (water content <0.3%) biphenyl-4-yl (phenyl) methanol (II) are suspended in 1550 ml of cyclohexane and treated with 90 g (0.76 mol) thionyl chloride at a temperature of 50 to 55 ° C added. The reaction mixture is stirred for 0.5 h at a temperature of 50 to 55 ° C stirred. Subsequently, in the Vacuum (<100 mbar) Distilled off thionyl chloride and cyclohexane. A distillation bottoms containing 4- [chloro (phenyl) methyl] biphenyl remains.
- 2nd step: 1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole (Bifonazole)
- [0039]162 g (2.4 mol) of imidazole are suspended in 1350 ml of acetone and dissolved at 50 ° C. This solution is added to the distillation bottoms from step 1 containing 4- [chloro (phenyl) methyl] biphenyl (III). The reaction mixture is heated at reflux for 3 h. After cooling, the reaction solution is mixed with 2 g of activated carbon and 2 g of bleaching earth at a temperature of 50 to 55 ° C, stirred for 0.5 h and filtered. The filtrate is cooled to about 0 ° C. The title compound crystallizes by addition of seed crystals, is filtered off and washed with a mixture of acetone / water (1: 1). For recrystallization, the product is dissolved in 1250 ml of isopropanol, treated with 0.5 g of activated charcoal and 0.5 g of bleaching earth, heated to reflux and filtered hot. The filtrate is cooled to 10 ° C. The title compound crystallizes out by addition of seed crystals, is filtered off, washed with isopropanol and dried. The yield is 101 g (61.9% of theory). The purity of the product is 98.68% by weight.
Melting point: 142 ° C - Comparative method:
- [0040]In the comparative method, instead of cyclohexane, toluene is used as solvent in step 1 as in DE-A 2 461 406 described. Step 2 is performed as described above. 1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole (bifonazole) is obtained in a purity of 97.66% by weight.
///////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Adverse effects
The most common side effect is a burning sensation at the application site. Other reactions, such as itching, eczema or skin dryness, are rare.[3] Bifonazole is a potent aromatase inhibitor in vitro.[4][5]
Pharmacology
Mechanism of action
Bifonazole has a dual mode of action. It inhibits fungal ergosterol biosynthesis at two points, via transformation of 24-methylendihydrolanosterol to desmethylsterol, together with inhibition of HMG-CoA. This enables fungicidal properties against dermatophytes and distinguishes bifonazole from other antifungal drugs.[3][6]
Pharmacokinetics
Six hours after application, bifonazole concentrations range from 1000 µg/cm³ in the stratum corneum to 5 µg/cm³ in the papillary dermis.[3]
References
- ^ International Drug Names: Bifonazole.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 502. ISBN 9783527607495.
- ^ Jump up to:a b c Haberfeld H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Canesten Bifonazol-Creme.
- ^ Trösken ER, Fischer K, Völkel W, Lutz WK (February 2006). “Inhibition of human CYP19 by azoles used as antifungal agents and aromatase inhibitors, using a new LC-MS/MS method for the analysis of estradiol product formation”. Toxicology. 219 (1–3): 33–40. doi:10.1016/j.tox.2005.10.020. PMID 16330141.
- ^ Egbuta C, Lo J, Ghosh D (December 2014). “Mechanism of inhibition of estrogen biosynthesis by azole fungicides”. Endocrinology. 155 (12): 4622–8. doi:10.1210/en.2014-1561. PMC 4239419. PMID 25243857.
- ^ Berg D, Regel E, Harenberg HE, Plempel M (1984). “Bifonazole and clotrimazole. Their mode of action and the possible reason for the fungicidal behaviour of bifonazole”. Arzneimittel-Forschung. 34 (2): 139–46. PMID 6372801.
Further reading
- Lackner TE, Clissold SP (August 1989). “Bifonazole. A review of its antimicrobial activity and therapeutic use in superficial mycoses”. Drugs. 38 (2): 204–25. doi:10.2165/00003495-198938020-00004. PMID 2670516.
| Clinical data | |
|---|---|
| Trade names | Canespor, many others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Topical |
| ATC code | D01AC10 (WHO) |
| Legal status | |
| Legal status | In general: Over-the-counter (OTC) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 60628-96-8 |
| PubChem CID | 2378 |
| DrugBank | DB04794 |
| ChemSpider | 2287 |
| UNII | QYJ305Z91O |
| KEGG | D01775 |
| ChEBI | CHEBI:31286 |
| ChEMBL | ChEMBL277535 |
| CompTox Dashboard (EPA) | DTXSID9045631 |
| ECHA InfoCard | 100.056.651 |
| Chemical and physical data | |
| Formula | C22H18N2 |
| Molar mass | 310.400 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////BIFONAZOLE, бифоназол , بيفونازول , 联苯苄唑 , BAY H 4502, BAY-H-4502
C1=CN(C=N1)C(C1=CC=CC=C1)C1=CC=C(C=C1)C1=CC=CC=C1

NEW DRUG APPROVALS
ONE TIME
$10.00
Melitracen

Melitracen
- Molecular FormulaC21H25N
- Average mass291.430 Da
10563-70-9[RN]
1568
1-Propanamine, 3-(10,10-dimethyl-9(10H)-anthracenylidene)-N,N-dimethyl-
225-858-5[EINECS], 234-150-5[EINECS]
3-(10,10-Dimethyl-9(10H)-anthracenyliden)-N,N-dimethyl-1-propanamine
Q7T0Y1109Z
Thymeol
мелитрацен[Russian][INN]
ميليتراسان[Arabic][INN]
美利曲辛[Chinese][INN]
Melitracen
CAS Registry Number: 5118-29-6
CAS Name: 3-(10,10-Dimethyl-9(10H)-anthracenylidene)-N,N-dimethyl-1-propanamine
Additional Names:N,N,10,10-tetramethyl-D9(10H),g-anthracenepropylamine; 9,10-dihydro-10,10-dimethyl-9-(3-dimethylaminopropylidene)anthracene; 9-[3-(dimethylamino)propylidene]-10,10-dimethyl-9,10-dihydroanthracene; N,N-dimethyl-3-(10,10-dimethyl-9(10H)-anthrylidene)propylamine
Molecular Formula: C21H25N, Molecular Weight: 291.43
Percent Composition: C 86.55%, H 8.65%, N 4.81%
Literature References: Prepn of the hydrochloride: Holm, Acta Chem. Scand.17, 2437 (1963); idem,GB939856 corresp to US3177209 (1963, 1965, both to Kefalas A/S). Crystal structure: J. Lopez de Lerma et al.,Acta Crystallogr.B35, 1739 (1979). Toxicity data: P. V. Petersen et al.,Acta Pharmacol. Toxicol.24, 121 (1966).
Derivative Type: Hydrochloride
CAS Registry Number: 10563-70-9
Manufacturers’ Codes: U-24973A
Trademarks: Melixeran (Lusofarmaco); Trausabun (Promonta); Dixeran (Lundbeck)
Molecular Formula: C21H25N.HCl, Molecular Weight: 327.89
Percent Composition: C 76.92%, H 7.99%, N 4.27%, Cl 10.81%
Properties: Crystals from acetone, mp 245-248°. LD50 i.v. in mice: 52 mg/kg (Petersen).
Melting point: mp 245-248°
Toxicity data: LD50 i.v. in mice: 52 mg/kg (Petersen)
Therap-Cat: Antidepressant.
Keywords: Antidepressant; Tricyclics.
Melitracen (brand names Melixeran) is a tricyclic antidepressant (TCA), for the treatment of depression and anxiety.[1][2][3][4] In addition to single drug preparations, it is also available as Deanxit, marketed by Lundbeck, a combination product containing both melitracen and flupentixol.[5][6][7][8]
The pharmacology of melitracen has not been properly investigated and is largely unknown, but it is likely to act in a similar manner to other TCAs. Indeed, melitracen is reported to have imipramine and amitriptyline-like effects and efficacy against depression and anxiety, though with improved tolerability and a somewhat faster onset of action.[9][10]
- ATC:N06AA14
- MW:291.44 g/mol
- CAS-RN:5118-29-6
- InChI Key:GWWLWDURRGNSRS-UHFFFAOYSA-N
- InChI:InChI=1S/C21H25N/c1-21(2)19-13-7-5-10-17(19)16(12-9-15-22(3)4)18-11-6-8-14-20(18)21/h5-8,10-14H,9,15H2,1-4H3
- EINECS:225-858-5
- LD50:52 mg/kg (M, i.v.); 315 mg/kg (M, p.o.);
170 mg/kg (R, p.o.)
Derivatives
hydrochloride
- Formula:C21H25N • HCl
- MW:327.90 g/mol
- CAS-RN:10563-70-9
- EINECS:234-150-5
- LD50:52 mg/kg (M, i.v.); 315 mg/kg (M, p.o.);
170 mg/kg (R, p.o.)
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 90-44-8 | C14H10O | anthrone | 9(10H)-Anthracenone |
| 85118-29-2 | C21H27NO | 9-[3-(dimethylamino)propyl]-9,10-dihydro-10,10-dimethyl-9-anthracenol | 9-Anthracenol, 9-[3-(dimethylamino)propyl]-9,10-dihydro-10,10-dimethyl- |
| 19070-16-7 | C5H12ClMgN | 3-dimethylaminopropylmagnesium chloride | Magnesium, chloro[3-(dimethylamino)propyl]- |
| 5447-86-9 | C16H14O | 10,10-dimethylanthrone | 9(10H)-Anthracenone, 10,10-dimethyl- |
SYN

English: DOI number: 10.3891/acta.chem.scand.17-2437 GB 939856 corresp to US 3177209 (1963, 1965, both to Kefalas A/S).
SYN
https://pubs.rsc.org/en/content/articlehtml/2020/re/d0re00087f
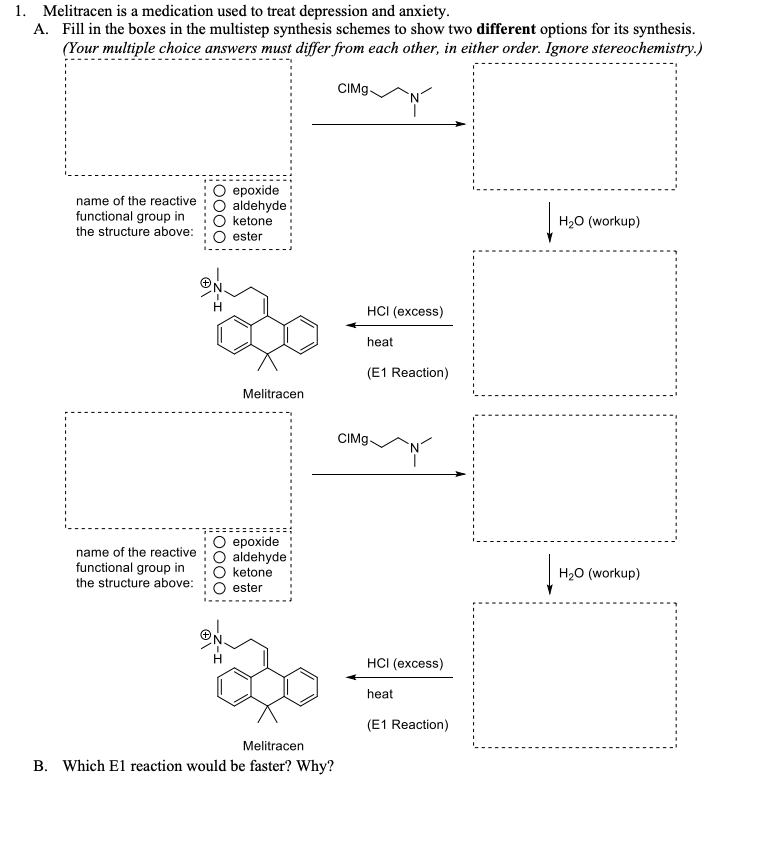
| Fig. 10 Synthesis of melitracen HCl-(36) by Kiil and co-workers making use of a one-flow system. Adapted with permission from Org. Process Res. Dev., 2018, 22, 228–235. Copyright 2018 American Chemical Society.35 |
Grignard reactions are commonly used for the construction of carbon–carbon bonds and show exothermic behaviour which can be dangerous in large-scale batch processes. The use of Grignard reagents in flow can be beneficial because of the high control of reaction conditions, facile heat transport and small effective reaction volume.6,34 A recent example was published by Kiil and co-workers, who synthesised melitracen (36) in a one-flow system.35 Kiil hypothesised that the seven unit operations required in batch could be decreased by combining a hydrolysis and dehydration step, and removing a phase separation (Fig. 10).
The investigation commenced with finding a suitable solvent for the Grignard reaction in which starting materials 34, 35 and intermediate products would dissolve. After having identified THF as the most suitable option, the next challenge was to find an acid that could induce both hydrolysis and dehydration in a single step. Hydrochloric acid was able to perform both transformations, however, precipitation was observed. Thus, hydrochloric acid molarities ranging from 1–12 M were tested. However, while even at the lowest molarity precipitation was observed, it also appeared that below 6 M the dehydration reaction did not proceed. Since the precipitation could not be prevented, a molarity of 12 M was eventually used. The individually optimised transformations were then combined in a one-flow continuous system. Most troublesome was that addition of HCl to the reaction mixture led to an exothermic reaction and boiling of the solvent. Therefore, a back-pressure regulator was employed so that melitracen (36) could be successfully synthesised as its HCl-salt in approximately 85% yield.
SYN
https://pubs.acs.org/doi/pdf/10.1021/acs.oprd.7b00368
A Grignard-based batch process, for the preparation of Melitracen HCl, has been redesigned to fit a continuous reactor system. The Grignard addition is carried out at room temperature, with subsequent hydrolysis of the magnesium alkoxide intermediate followed by dehydration of the resulting alcohol. The product undergoes further workup by simple gravimetric phase separation and then crystallization with 2 M HCl in diethyl ether to afford pure Melitracen HCl. All steps in the laboratory setup were concatenated, and the setup was proven capable of producing a significant portion of the commercial quantities of Melitracen HCl. The flow setup profits from a reduced footprint, lower energy consumption, fewer synthetic steps, and reduced raw material usage compared to the batch process.

As illustrated in Scheme 1, four synthetic steps are involved in the manufacturing of Melitracen HCl (6). The four steps are a classic Grignard addition to a ketone, a hydrolysis of a magnesium alkoxide, a dehydration of an alcohol and a salt precipitation to isolate the API. The Grignard addition is between 10,10-dimethylanthrone (10,10-DMA (1)) and 3-(N,N-dimethylamino)propylmagnesium chloride (DMPC-MgCl (2)), resulting in formation of the magnesium alkoxide 3. The magnesium alkoxide 3 is then hydrolyzed to the alcohol 4 and dehydrated to form product 5. The last step is a crystallization of the API as a salt, where HCl is added to obtain the Melitracen HCl (6)
Scheme 1: Syntheses of magnesium alkoxide 3, alcohol 4 and dehydrated product 5 in the manufacturing process of Melitracen HCl 6, from ketone 1 and Grignard reagent 2.

Current Batch Synthesis The current batch synthesis involves individual synthetic steps, as illustrated in Figure 1. DMPC-MgCl 2 is made in-house before it is used, due to its limited storage shelf life, in a toluene-THF solvent mixture. THF is present in trace amounts in order to stabilize the magnesium in the Grignard reagents.45 A solution of 10,10-DMA 1 is prepared in toluene and is slowly transferred to the DMPC-MgCl 2, maintaining a temperature of 50°C. DMPC-MgCl 2 is used in an equivalence of 1.6 compared to 10,10-DMA 1. The formed magnesium alkoxide 3 is hydrolyzed with water and acetic acid (80%). The aqueous phase is discarded and concentrated hydrochloric acid (37%) is used to dehydrate alcohol 4 to form dehydrated product 5. Toluene is replaced with ethanol by a solvent swap. Crystallization of the dehydrated product 5 from the ethanol phase is done with HCl gas to obtain the final Melitracen HCl (6), which is subsequently isolated by filtration.
Precipitation of Melitracen HCl from THF The dehydrated product 5 was crystallized as the final HCl salt in the THF in a batch experiment, in order to remove a solvent swap to ethanol. The crystallization was carried out with 2 M HCl in Et2O, as this was considered more suited for a later flow process and more easily implemented in the laboratory setup. An equivalence of 1.1 HCl was used and the requirement was an achievement of pH<2. The mixture was kept stirred during the crystallization and carried out at ambient temperature. After 10 minutes, fine white solids started to form, followed by a massive precipitation of Melitracen HCl 6. The Melitracen HCl 6 was filtered with a Büchner funnel and washed with THF. The isolated yield was 80% and within the specifications for the in-house analysis methods used in the routine production (CHN, TGA, UV-vis, HPLC, melting point). Figure 3 is a microscope picture of the isolated Melitracen HCl 6. For full-scale production, the HCl gas would still be more desirable for the crystallization and the 2 M HCl in Et2O merely serves as a proof of concept for the laboratory flow setup.
CLIP
PATENT
https://patents.google.com/patent/CN105418436B/en
Melitracen (Melitracen), is a kind of tricyclics, entitled 10, the 10- dimethyl -9- γ-two of chemistry Methylamino acrylic -9,10- dihydro-anthraquinone, Clinical practice is its hydrochloride.Melitracen can suppress in presynaptic membrane To the effect of the reuptake of norepinephrine and serotonin, and therefore improve containing for monoamine transmitterses in synaptic cleft Amount.
On the preparation method of melitracen, document report both domestic and external is seldom, existing as described below:
US3177209, GB939856, DK97400, are the compound patents of Lundbeck drugmaker of Denmark, it is mentioned that Synthetic method is that, with 10,10- dimethylanthracene -9- ketone and N, TMSDMA N dimethylamine base propyl group magnesium chloride is generated in the middle of melitracen Body, then by intermediate be dissolved under chloroform, reflux state lead to hydrogen chloride prepare melitracen crude product, then crystallized again with acetone Melitracen is obtained, this method needs to be passed through hydrogen chloride at reflux, there is substantial amounts of smog to produce, and reaction condition is not yet It is easy to control, it there is larger safety factor.
CN103877088A is Lundbeck drugmaker of Denmark in a kind of safe melitracen group disclosed in 2014 Compound, wherein the purity to melitracen in drug regimen proposes more strict requirements, especially to that may make in clinic Cause the impurity (formula I, formula II) of the adverse reactions such as anxiety, irritated and excitement in, even more propose:Formula I<0.1%, formula II< 0.1, I+formula of formula II<0.1% rigors.The melitracen of patent US3177209, GB939856, DK97400 method synthesis Impurity is more, and primary purification can not obtain satisfactory active pharmaceutical ingredient (API).
It is also mentioned that the preparation method of melitracen hydrochloride, this method is with 10,10- diformazans in patent CN103877088A The γ of base-9-dimethylaminopropyl-9- anthrols are raw material, add dichloromethane and hydrochloric acid, are heated to reflux, reaction system alkaline hydrolysis from The free alkali obtained afterwards, is re-dissolved in acetone and leads to hydrogen chloride into salt, obtain melitracen crude product, then isolated and purified with column chromatography Obtain the melitracen of high-purity.The melitracen yield that it is prepared into is low, and purifies and separates process needs column chromatography, it is impossible to meet The need for large-scale production.
Embodiment 1
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
10,10- dimethylanthracene -9- ketone carry out grignard reaction with 3- dimethylaminos-n-propyl chloride in the presence of initiator, obtain To melitracen intermediate, detailed process is as follows:
340g magnesium rods and 17.5L absolute ethers are added in 20L glass reaction kettles, stirring is warming up to 30~35 DEG C, addition 1.75kg 3- dimethylaminos-n-propyl chloride, finish insulated and stirred, add 1g iodine and 2mL 1,2- Bromofume as initiator, 9h is stirred at reflux, magnesium rod disappears completely, reaction system is cooled into 10~20 DEG C, 1.5kg 10,10- dimethyl is slowly added to Anthracene -9- ketone, then it is warming up to 30~35 DEG C, back flow reaction 1 hour;TLC monitoring reactions are complete, and reaction system is cooled into 10~20 DEG C, then add 5.5L water, ether layer is separated, anhydrous sodium sulfate is added and is concentrated under reduced pressure drying, obtain melitracen intermediate 2.03kg, receive The ﹪ of rate 97.2, purity 98.5%.
TLC monitoring methods:Add water and be quenched after sampling, take organic layer point plate;Solvent is petroleum ether:Ethyl acetate=2:1 (volume ratio);The Rf of 10,10- dimethylanthracene -9- ketone is 0.6, and the Rf of melitracen intermediate is 0.1.
(2) melitracen crude product is prepared
2kg melitracens intermediate, 10L chloroforms and 2.4L concentrated hydrochloric acids are put into 20L glass reaction kettles, stirred molten Solution, obtains pale yellow solution, and 60 DEG C of heating stirring reaction 2 hours, TLC monitoring reactions are complete, and separate aqueous layer, organic phase is concentrated under reduced pressure Dry, it is melitracen crude product 2.03kg, yield 95.7%, purity 99.41%, containing Formulas I to obtain white solid:0.20%, formula II:0.13%;Formulas I, II1HNMR spectrograms, melitracen crude product liquid phase spectrogram are shown in accompanying drawing 1,2,3 respectively;
TLC monitoring methods:Organic phase point plate is extracted reaction solution, solvent is dichloromethane:Methanol:Acetic acid=150:10:2 (volume ratio).
Formulas I:1H NMR(400MHz,DMSO)δ7.78-7.82(m,2H),δ7.50-7.53(m,2H),δ7.28-7.35 (m, 4H), δ 2.11 (S, 6H), δ 2.08 (d, J=6.8Hz, 2H), δ 1.96 (t, J=6.4Hz, 2H), δ 1.72 (s, 3H), δ 1.61(s,3H),δ1.26(brs,1H),δ1.02-1.09(m,2H)
Formula II:1H NMR(400MHz,DMSO)δ8.95(s,2H),δ7.47-7.63(m,4H),δ7.27-7.37(m, 4H), δ 6.06 (t, J=7.2Hz, 1H), δ 3.09 (t, J=7.2Hz, 2H), δ 2.91 (m, 2H), δ 2.54 (s, 3H), δ 1.53 (s,6H)
(3) purifying of melitracen crude product
Take 2.03kg melitracens crude product (purity 99.41%, Formulas I:0.20%, Formula II:0.13%) 4 times of amount (W/, are added V isopropanol), 20~25 DEG C of stirring 4h (mashing), is filtered, and is dried, is obtained product 2.0kg, yield is 98.5%, and purity is 99.61%, containing Formulas I:0.054%, without Formula II;Melitracen crude product is shown in accompanying drawing 4 through isopropanol mashing sample liquid chromatography(LC figure;
Product after 2kg is beaten is added in 30L glass reaction kettle, adds 16kg isopropanols, and backflow is dissolved, then Cool to 10 DEG C and stir crystallization and stay overnight, suction filtration is dried under reduced pressure, and obtains melitracen 1.89kg, and yield 94.5%, purity 99.98% contains Formulas I:0.0026%, without Formula II;See accompanying drawing 5 through isopropanol recrystallization liquid phase spectrogram.
Embodiment 2
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 1;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 1;
(3) purifying of melitracen crude product
Take 10g melitracens crude product (purity 99.41%, Formulas I:0.20%, Formula II:0.13%) 4 times of amounts (W/V), are added Ethanol, 20~25 DEG C stirring 4h (mashing), filtering, drying, obtain product 9.79g, yield is 97.9%, purity 99.69% contains Formula I 0.047%, containing formula II 0.005%;Melitracen crude product is shown in accompanying drawing 6 through ethanol mashing sample liquid chromatography(LC figure;
Product after 9.0g ethanol is beaten is added in 250mL round-bottomed flask, adds the dissolving of 230mL alcohol refluxs, Then 10 DEG C are cooled to stir crystallization and stay overnight, suction filtration is dried under reduced pressure, obtain melitracen 8.4g, yield 93.3%, purity 99.98%, Containing Formulas I:0.0041%, without Formula II;See accompanying drawing 7 through ethanol recrystallization liquid phase spectrogram.
Embodiment 3
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 1;
(2) melitracen crude product is prepared
100g melitracens intermediate, 500mL chloroforms and 120mL concentrated hydrochloric acids are put into 1L three-necked bottles, stirred molten Solution, obtains pale yellow solution, and 60 DEG C of heating stirring reaction 2 hours, TLC monitoring reactions are complete, and separate aqueous layer, organic phase is concentrated under reduced pressure Dry, it is melitracen crude product 104g, yield 98.3%, purity 99.38%, containing Formulas I to obtain white solid:0.22%, Formula II: 0.15%;Melitracen crude product liquid phase spectrogram is shown in accompanying drawing 8;
TLC monitoring methods:Organic phase point plate is extracted reaction solution, solvent is dichloromethane:Methanol:Acetic acid=150:10:2 (volume ratio).
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the methanol of 4 times of amounts (W/V) is added, 20~25 DEG C of stirring 4h (mashing) obtain product Weight is 18.48g, and yield is 92.4%, and purity is 99.66%, containing Formulas I:0.05%, Formula II:0.008%, see accompanying drawing 9.
Embodiment 4
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the n-butanol of 4 times of amounts (W/V) is added, 20~25 DEG C of stirring 5h (mashing) are produced Thing weight is 19.6g, and yield is 98%, and purity is 99.54%, containing Formulas I:0.05%, Formula II:0.009%, see accompanying drawing 10.
Embodiment 5
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 30~35 DEG C of stirring 5h (mashing) are produced Thing weight is 18.06g, and yield is 90.3%.
Embodiment 6
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 50 DEG C of stirring 3h (mashing) obtain product weight Measure as 14.2g, yield is 71%.
Embodiment 7
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 5-10 DEG C of stirring 5h (mashing) obtains product Weight is 19.7g, and yield is 98.5%, and purity is 99.53%, containing Formulas I:0.054%, Formula II:0.014%, see accompanying drawing 11.
Embodiment 8
With reference to CN103877088A, crystallized using acetone, that is, take 10g melitracens intermediate and 24mL dichloromethane, 6.7mL concentrated hydrochloric acids are heated to reflux 2h and are cooled to room temperature, and pH is to 8-9 for regulation, then are extracted with dichloromethane and product, are concentrated to give free Alkali cpd, acetone is dissolved in by the free alkali compound, concentrated hydrochloric acid is added dropwise to pH=0.1, stirring, cooling separate out solid 7.1g, This solid crystallizes to obtain sample 6.4g with acetone again, and total recovery is 60.9%, and purity is 99.64%, containing Formulas I:0.09%, Formula II: 0.04%.Melitracen is shown in accompanying drawing 12 only with acetone crystallization liquid chromatography(LC figure.
Repeat literature method crystallized only with acetone obtained by product in impurity Formulas I, Formula II impurity summation be 0.13%, The adverse reactions such as anxiety, irritated and excitement may be caused in Clinical practice.
In summary, the effect of mashing is to make melitracen crude product rapid dispersion, and the effect of methanol mashing is similar with ethanol, But it is good without isopropanol effect, but methanol mashing yield is decreased obviously trend;N-butanol mashing needs the extension time to reach To the effect same with ethanol, but be not as good as isopropanol effect, and because the viscosity of n-butanol is slightly larger, melitracen crude product is at it In disperse slightly worse, invention has the granular solids that not readily dissolve after filtering, and the removal effect to other impurities is also poor;Isopropanol Temperature is raised during mashing, yield is decreased obviously, and reduces temperature, yield has no raising, though to the removal effect of impurity Formula II It can so control in the range of conforming to quality requirements, but compared to being decreased obviously in embodiment 1.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Swiss Pharmaceutical Society (2000). Index Nominum 2000: International Drug Directory (Book with CD-ROM). Boca Raton: Medpharm Scientific Publishers. ISBN 3-88763-075-0.
- ^ Hall, Chapman and; Chemical Abstracts Service, American Chemical Society; Rhodes, P. H (1996). Dictionary of organic compounds. London: Chapman & Hall. ISBN 0-412-54090-8.
- ^ O’Neil, Maryadele J. (2001). The Merck index: an encyclopedia of chemicals, drugs, and biologicals. Rahway, NJ: Merck Research Laboratories. ISBN 0-911910-13-1.
- ^ José Miguel Vela; Helmut Buschmann; Jörg Holenz; Antonio Párraga; Antoni Torrens (2007). Antidepressants, Antipsychotics, Anxiolytics: From Chemistry and Pharmacology to Clinical Application. Weinheim: Wiley-VCH. ISBN 978-3-527-31058-6.
- ^ Muller, Niels F; Dessing, Rudolf P; Pharmacy, European Society of Clinical (1998). European Drug Index, 4th Edition. Boca Raton: CRC Press. ISBN 3-7692-2114-1.
- ^ Van Moffaert M, Dierick M, De Meulemeester F, Vereecken A (1983). “Treatment of depressive anxiety states associated with psychosomatic symptoms. A double-blind multicentre clinical study: mianserin versus melitracen-flupentixol”. Acta Psychiatrica Belgica. 83 (5): 525–39. PMID 6670581.
- ^ Bin Yaacob H (April 1985). “Flupenthixol and Melitracen in the management of trigeminal neuralgia”. Dental Journal of Malaysia. 8 (2): 37–8. PMID 3917005.
- ^ Hashash JG, Abdul-Baki H, Azar C, et al. (June 2008). “Clinical trial: a randomized controlled cross-over study of flupenthixol + melitracen in functional dyspepsia”. Alimentary Pharmacology & Therapeutics. 27 (11): 1148–55. doi:10.1111/j.1365-2036.2008.03677.x. PMID 18331614. S2CID 40714136.
- ^ Aronson, Jeffrey Kenneth (2008). Meyler’s Side Effects of Psychiatric Drugs (Meylers Side Effects). Amsterdam: Elsevier Science. ISBN 978-0-444-53266-4.
- ^ Author Unknown (1970). Ann Reports Medicinal Chem V5 (v. 5). Boston: Academic Press. ISBN 0-12-040505-9.
{{cite book}}:|author=has generic name (help)
| Clinical data | |
|---|---|
| Trade names | Adaptol, Dixeran, Melixeran, Thymeol, Trausabun |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral, intramuscular injection |
| ATC code | N06AA14 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 5118-29-6 |
| PubChem CID | 25382 |
| ChemSpider | 23697 |
| UNII | Q7T0Y1109Z |
| KEGG | D08171 |
| ChEMBL | ChEMBL110094 |
| CompTox Dashboard (EPA) | DTXSID4048274 |
| ECHA InfoCard | 100.023.507 |
| Chemical and physical data | |
| Formula | C21H25N |
| Molar mass | 291.438 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
//////////Melitracen, Q7T0Y1109Z, Thymeol, мелитрацен , ميليتراسان , 美利曲辛 , U 24973A, Antidepressant, Tricyclics,

NEW DRUG APPROVALS
ONE TIME
$10.00
VIP 152, BAY 1251152

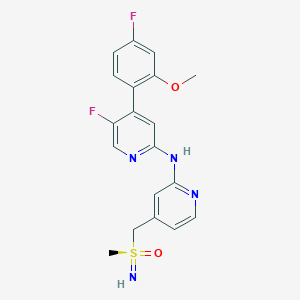
VIP 152, BAY 1251152
CAS RN.: 1610358-56-9
C19H18F2N4O2S
5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-[4-[(methylsulfonimidoyl)methyl]pyridin-2-yl]pyridin-2-amine
- 2-Pyridinamine, 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-[4-[(S-methylsulfonimidoyl)methyl]-2-pyridinyl]-, (+)-
(+)-BAY-1251152 is a CDK9 inhibitor extracted from patent WO 2014076091 A1, example 1.
RN: 1610408-97-3
UNII: 1255AT22ZJ
UNII-1255AT22ZJ
2-Pyridinamine, 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-[4-[[[S(S)]-S-methylsulfonimidoyl]methyl]-2-pyridinyl]-
Molecular Formula, C19-H18-F2-N4-O2-S, Molecular Weight, 404.4336
- OriginatorBayer
- DeveloperBayer; Vincerx Pharma
- ClassAntineoplastics; Fluorinated hydrocarbons; Organic sulfur compounds; Phenyl ethers; Pyridines; Small molecules
- Mechanism of ActionCyclin dependent kinase 9 inhibitors; Positive transcriptional elongation factor B inhibitors
- Orphan Drug StatusYes – Diffuse large B cell lymphoma
- Phase IChronic lymphocytic leukaemia; Haematological malignancies; Non-Hodgkin’s lymphoma; Richter’s syndrome; Solid tumours
- 17 Dec 2021Vincerx Pharma plans phase II trials for Cancer (IV, Infusion), in the second half of 2022
- 16 Dec 2021Phase-I clinical trials in Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (IV)
- 16 Dec 2021Phase-I clinical trials in Richter’s syndrome (Second-line therapy or greater) (IV) in USA

First-in-human dose escalation study of cyclin-dependent kinase-9 inhibitor VIP152 in patients with advanced malignancies shows early signs of clinical efficacyJennifer R. Diamond, Valentina Boni, Emerson Lim, Grzegorz Nowakowski, Raul Cordoba, Daniel Morillo, Ray Valencia, Isabelle Genvresse, Claudia Merz, Oliver Boix, Melanie M. Frigault, Joy M. Greer, Ahmed M. Hamdy, Xin Huang, Raquel Izumi, Harvey Wong and Victor Moreno
DOI: 10.1158/1078-0432.CCR-21-3617
Abstract
Purpose: To report on the first-in-human phase I study of VIP152 (NCT02635672), a potent and highly selective CDK9 inhibitor. Patients and Methods: Adults with solid tumors or aggressive non-Hodgkin lymphoma (NHL) who were refractory to or had exhausted all available therapies received VIP152 monotherapy as a 30-minute intravenous, once weekly infusion, as escalating doses (5, 10, 15, 22.5, or 30 mg in 21-day cycles) until the maximum tolerated dose (MTD) was determined. Results: Thirty-seven patients received {greater than or equal to} 1 VIP152 dose, with 30 mg identified as the MTD based on dose-limiting toxicity of grade 3/4 neutropenia. The most common adverse events were nausea and vomiting (75.7% and 56.8%, respectively), all of grade 1/2 severity. Of the most common events, Grade 3/4 events occurring in > 1 patient were neutropenia (22%), anemia (11%), abdominal pain (8%), increased alkaline phosphatase (8%), and hyponatremia (8%). Day 1 exposure for the MTD exceeded the predicted minimum therapeutic exposure and reproducibly achieved maximal pathway modulation; no accumulation occurred after multiple doses. Seven of 30 patients with solid tumors had stable disease (including 9.5 and 16.8 months in individual patients with pancreatic cancer and salivary gland cancer, respectively), and 2 of 7 patients with high-grade B-cell lymphoma with MYC and BCL2/BCL6 translocations (HGL) achieved durable complete metabolic remission (ongoing at study discontinuation, after 3.7 and 2.3 years of treatment). Conclusion: VIP152 monotherapy, administered intravenously once weekly, demonstrated a favorable safety profile and evidence of clinical benefit in patients with advanced HGL and solid tumors.
CLIP
Preclinical bioconjugation platform designed to overcome limitations of small–molecule and antibody–drug conjugates use to treat cancer
Vincera Pharma, Inc., a biopharmaceutical company aspiring to address the unmet medical needs of patients with cancer through paradigm-shifting therapeutics, today announced the signing of an exclusive license agreement with Bayer AG for the development and commercialization of an early development oncology portfolio. The license will become effective upon the closing of the transaction with LSAC (described below), and Vincera intends to use the funds it will receive upon closing of such transaction to initiate its clinical program.
Under the terms of the license agreement, Vincera will in-license VIP152 (formerly BAY 1251152 & CAS RN.: 1610358-56-9), a clinical-stage, highly selective, positive transcription elongation factor b (PTEFb)/cyclin-dependent kinase 9 (CDK9) inhibitor for the treatment of cancer. Additionally, Vincera will receive assets and license technology for a preclinical bioconjugation platform to address the limitations of small-molecule and antibody-drug conjugates in oncology. The preclinical assets include VIP236, a small molecule drug conjugate (SMDC) targeting advanced and metastatic cancer; as well as VIP943 (formerly BAY-943) and VIP924 (formerly BAY-924), two antibody-drug conjugates (ADC) targeting hematologic tumors; and VIP217, an oral PTEFb/CDK9 inhibitor in discovery. “This license agreement with Bayer creates the foundation of Vincera’s targeted clinical oncology pipeline, with a potentially best-in-class asset, while positioning us for long-term growth across two therapeutic platforms,” said Ahmed Hamdy M.D., Chief Executive Officer of Vincera. “Our lead asset, VIP152, is a small molecule PTEFb/CDK9 inhibitor with very encouraging data from monotherapy Phase 1 studies, including 2 of 7 patients with durable remissions of over 2 years in the very aggressive indication of relapsed/refractory double-hit DLBCL. In addition, preclinical data support our belief that VIP152 is the most selective CDK9 inhibitor in the clinic with on-target depletion of oncogenic MYC and MCL1 mRNA transcripts in patients. These results, combined with the acceptable safety profile seen to date, suggest that VIP152 could be an important new treatment option for patients with MYC- and MCL1-driven malignancies. Importantly, with proof-of-concept clinical data in hand, we are poised to execute on a strategic clinical development plan with the potential for multiple accelerated approvals in the U.S. Expansion of the current Phase 1b study to include these patient populations is expected to begin in 2021.”
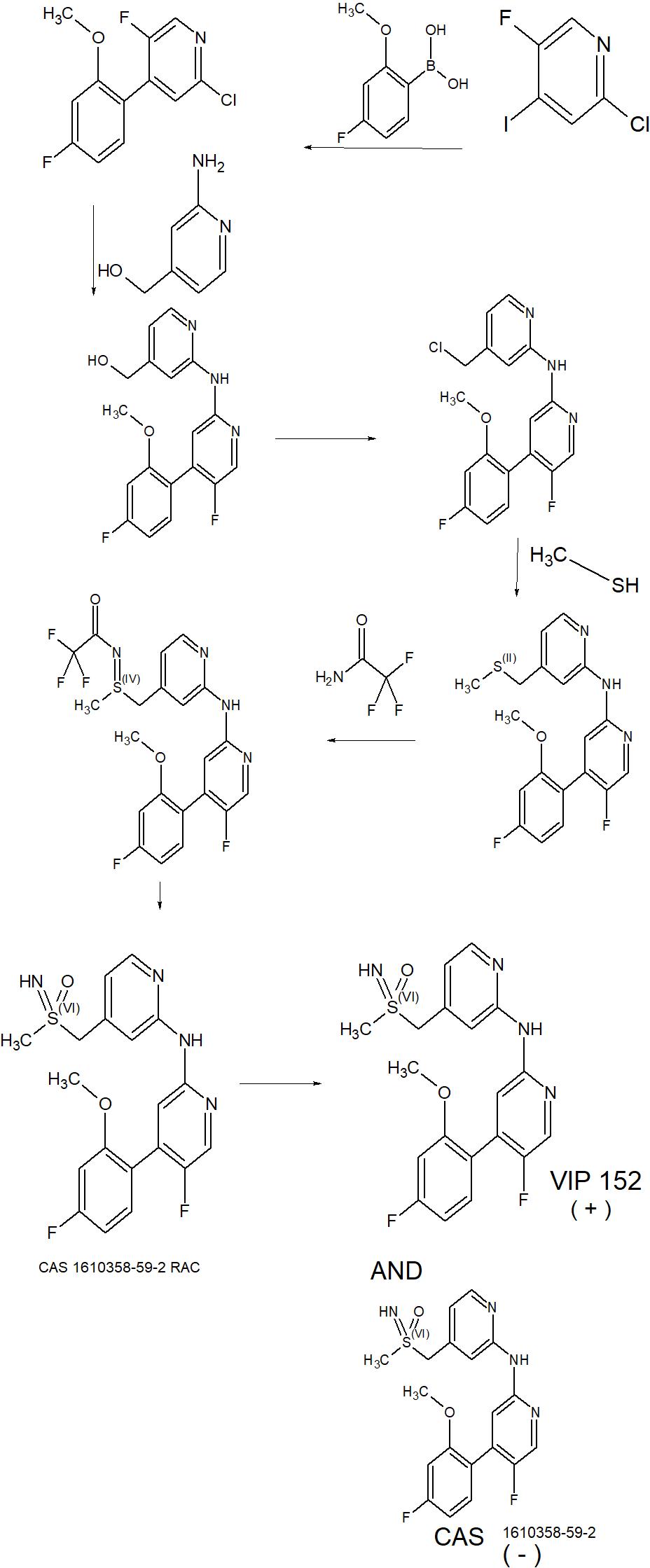
IP Information:
WO2014076091A1 (Product Patent)
Assignee: Bayer Pharma Aktiengesellschaft, Germany
Application Date: 2013-11-12
Family Equivalents:
AP3872A; AR093505A1; AU2013346939A1; AU2013346939B2; BR112015010707A2; BR112015010707A8; CA2891358A1; CL2015001304A1; CN105102444A; CN105102444B; CR20150256A; CU20150052A7; CY1118441T1; DK2928878T3; DOP2015000118A; EA027226B1; EA201590890A1; EP2928878B1; ES2612978T3; HK1213255A1; HRP20161547T1; HUE032868T2; IL238322A; JO3332B1; JP2015537015A; JP6263193B2; KR20150084968A; LT2928878T; MA38090A1; MA38090B1; ME02880B; MX2015006169A; NZ707084A; PE20151071A1; PH12015501003A1; PH12015501003B1; PL2928878T3; PT2928878T; RS55580B1; SG11201503079PA; SI2928878T1; SV2015004979A; TN2015000185A1; TW201420569A; TWI613193B; UA115254C2; US2015291528A1; US2017202815A1; US9650340B2; US9877954B2; UY35141A; WO2014076091A1
Title: 5-FLUORO-N-(PYRIDIN-2-YL)PYRIDIN-2-AMINE DERIVATIVES CONTAINING A SULFOXIMINE GROUP.
Abstract
The present invention relates to 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfoximine group of general formula (I) as described and defined herein, and methods for their preparation, their use for the treatment and/or prophylaxis of disorders, in particular of hyper-proliferative disorders and/or virally induced infectious diseases and/or of cardiovascular diseases. The invention further relates to intermediate compounds useful in the preparation of said compounds of general formula (I).
“CDK9 represents a validated target for malignancies such as CLL where other less selective CDK inhibitors have shown clinical activity in high-risk patients,” says Dr. John C. Byrd, Chair of the Scientific Advisory Board of Vincera. “VIP-152 represents an exciting new therapy for this disease, particularly those with prior resistance to ibrutinib and venetoclax where a true unmet need exists for new treatments.”
Dr. Hamdy continued, “In addition to our planned clinical program, we intend to advance, in parallel, the development of our preclinical bioconjugation platform. We believe our next-generation platform has the potential to generate first-in-class and best-in-class opportunities in oncology, improving the specificity of drug targeting and release through a modular platform with innovative warhead design and linker-payload technologies. We are thrilled that the Bayer license will allow us to pursue the commercial potential of this promising oncology portfolio and look forward to providing updates as we execute across our pipeline in the coming quarters.”
In exchange for this license, Vincera will pay Bayer an upfront license fee and development and commercial sales milestone payments. In further consideration of the rights granted, we will also pay an annual royalty on the commercial sale of licensed products in the single- to low-double-digit percentage range on net commercial sales of licensed products.
On September 29, 2020, Vincera announced that it has entered into a merger agreement with LifeSci Acquisition Corp. (“LSAC”), a publicly-traded blank check company targeting biopharma, medical technology, digital health, and healthcare services sectors. Following the completion of the merger, the combined company is expected to have approximately $60 million in cash to fund its preclinical and clinical pipeline. Additional information about the merger and related transactions, including a copy of the merger agreement, are included in a Current Report on Form 8-K filed by LSAC with the SEC on September 29, 2020, and available at www.sec.gov.
About Vincera Pharma, Inc.
Vincera is a recently formed clinical-stage life sciences company focused on leveraging its extensive development and oncology expertise to advance new therapies intended to address unmet medical needs for the treatment of cancer. Vincera’s executive team has assembled a management team of biopharmaceutical experts with extensive experience in building and operating organizations that develop and deliver innovative medicines to patients. Vincera’s current pipeline is derived from an exclusive license agreement with Bayer and includes (i) a clinical-stage and follow-on small molecule drug program and (ii) a preclinical stage bioconjugation/next-generation antibody-drug conjugate platform. The company intends to develop multiple products through clinical proof-of-concept and potentially through Accelerated Approval in the United States. For more information, please visit www.vincerapharma.com.
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Patent
US 20150291528
Example 1
(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 1.12-Chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.27 (m, 1H), 7.33 (m, 1H), 7.24 (m, 1H), 6.75 (m, 2H), 3.83 (s, 3H). |
Preparation of Intermediate 1.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.15 (m, 2H), 7.61 (m, 1H), 7.40 (s, 1H), 7.35 (br, 1H), 7.29 (m, 1H), 6.82 (m, 1H), 6.75 (m, 2H), 3.83 (s, 3H), 3.62 (s, 2H), 2.03 (s, 3H). |
Alternative Procedure for the Preparation of Intermediate 1.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 1.3(2-{[5-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methanol
Preparation of End Product (Alternative Preparation of Intermediate 1.2)
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.15 (m, 2H), 7.61 (m, 1H), 7.40 (br, 2H), 7.29 (m, 1H), 6.82 (m, 1H), 6.75 (m, 2H), 3.83 (s, 3H), 3.62 (s, 2H), 2.03 (s, 3H). |
Alternative Procedure for the Preparation of Example 1Preparation of Intermediate 1.4(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.29 (m, 1H), 8.18 (m, 1H), 7.83 (s. 1H), 7.50 (br, 1H), 7.32 (m, 1H), 7.28 (m, 1H), 6.79 (m, 3H), 4.52 (d, 1H), 4.21 (d, 1H), 3.85 (s, 3H), 2.71 (s, 3H). |
Alternative Preparation of End Product (Example 1)
Example 2 and 3
Enantiomers of 5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
| (rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine (3.47 g) was separated into the single enantiomers by preparative chiral HPLC. |
Example 2(+)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Example 3(−)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Example 4(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 4.15-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=7.95 (m, 1H), 7.20 (m, 1H), 6.72 (m, 2H), 6.46 (m, 1H), 4.33 (br, 2H), 3.61 (s, 3H). |
Preparation of Intermediate 4.2(2-Chloro-6-methylpyridin-4-yl)methanol
| 1H NMR (400 MHz, CDCl 3, 300K) δ=7.18 (s, 1H), 7.09 (s, 1H), 4.72 (d, 2H), 2.55 (s, 3H), 2.17 (tr, 1H). |
Preparation of Intermediate 4.32-Chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=7.12 (s, 1H), 7.05 (s, 1H), 3.58 (s, 2H), 2.54 (s, 3H), 2.03 (s, 3H). |
Preparation of Intermediate 4.45-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediates 4.5 and 4.6(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide and (rac)-N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Intermediate 4.5:
Intermediate 4.6:
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.18 (s, 1H), 7.84 (s, 1H), 7.33 (s, 1H), 7.29 (m, 1H), 7.23 (m, 1H), 6.78 (m, 2H), 4.77 (d, 1H), 4.36 (d, 1H), 3.86 (s, 3H), 2.80 (s, 3H), 2.63 (s, 3H). |
Preparation of End Product:
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.16 (s, 1H), 7.60 (s, 1H), 7.39 (m, 1H), 7.30 (m, 2H), 6.79 (m, 3H), 4.34 (d, 1H), 4.22 (d, 1H), 3.86 (s, 3H), 3.02 (s, 3H), 2.79 (br, 1H), 2.48 (s, 3H). |
Alternative Procedure for the Preparation of Example 4Preparation of Intermediate 4.15-Fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
| 1H-NMR (300 MHz, DMSO-d 6, 300 K): δ [ppm]=7.85 (d, 1H), 7.25 (tr, 1H), 7.08-7.00 (m, 1H), 6.91-6.81 (m, 1H), 6.35 (d, 1H), 5.84 (s, 2H). |
Preparation of Intermediate 4.32-Chloro-6-methyl-4-[(methylsulfanyl)methyl]pyridine
| 1H-NMR (300 MHz, DMSO-d 6, 300 K): δ [ppm]=7.24 (s, 1H), 7.20 (s, 1H), 3.66 (s, 2H), 2.42 (s, 3H), 1.95 (s, 3H). |
Preparation of Intermediate 4.45-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
| 1H-NMR (300 MHz, CDCl 3, 300 K): δ [ppm]=8.16 (d, 1H), 7.56 (d, 1H), 7.36-7.29 (m, 2H), 7.21 (s, 1H), 6.85-6.73 (m, 2H), 6.72 (s, 1H), 3.86 (s, 3H), 3.61 (s, 2H), 2.45 (s, 3H), 2.06 (s, 3H). |
Preparation of Intermediate 4.5(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Preparation of Intermediate 4.6(rac)-N-{[(3-bromo-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-2-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
Intermediate 4.5:
Intermediate 4.6(1H-NMR was Taken from a Different Batch)
Preparation of Intermediates 4.7 and 4.8
| 3.76 g of racemic 2,2,2-trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ 4-sulfanylidene}acetamide were separated by chiral HPLC: |
Intermediate 4.7(+)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Intermediate 4.8(−)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methylpyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
Alternative Preparation of End Product (Example 4)(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Example 5
(rac)-5-Bromo-N-[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]-6-methyl-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.14 (m, 1H), 7.80 (s, 1H), 7.32 (m, 2H), 7.29 (m, 1H), 6.78 (m, 2H), 4.87 (d, 1H), 4.59 (d, 1H), 3.85 (s, 3H), 3.07 (s, 3H), 2.99 (br, 1H), 2.62 (s, 3H). |
Example 6(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 6.1:2-Chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=6.92 (s, 1H), 6.61 (s, 1H), 3.96 (s, 3H), 3.56 (s, 2H), 2.03 (s, 3H). |
Preparation of Intermediate 6.25-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(methylsulfanyl)methyl]pyridin-2-yl}pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.15 (m, 1H), 7.91 (m, 1H), 7.29 (m, 1H), 7.21 (s, 1H), 6.77 (m, 3H), 6.28 (s, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.58 (s, 2H), 2.06 (s, 3H). |
Preparation of Intermediate 6.3(rac)-2,2,2-Trifluoro-N-{[(2-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}-6-methoxypyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}acetamide
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.18 (m, 1H), 7.56 (m, 1H), 7.29 (m, 2H), 7.12 (m, 1H), 6.78 (m, 2H), 6.25 (s, 1H), 4.52 (d, 1H), 4.07 (d, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 2.70 (s, 3H). |
Preparation of End Product:
Alternative Procedure for the Preparation of Example 6(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{6-methoxy-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Example 7(rac)-N-{6-Chloro-4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
Preparation of Intermediate 7.12-Chloro-6-methoxy-4-[(methylsulfanyl)methyl]pyridine
| 1H NMR (400 MHz, d 6-DMSO, 300K) δ=10.06 (s, 1H), 8.25 (m, 1H), 7.71 (m, 1H), 7.56 (m, 1H), 7.35 (m, 1H), 7.10 (m, 1H), 6.93 (m, 1H), 6.85 (m, 1H), 5.47 (tr, 1H), 4.49 (d, 2H), 3.81 (s, 3H). |
Preparation of Intermediate 7.2N-{6-Chloro-4-[(methylsulfanyl)methyl]pyridin-2-yl}-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-amine
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.17 (s, 1H), 7.50 (m, 3H), 7.32 (m, 1H), 6.90 (s, 1H), 6.79 (m, 2H), 3.87 (s, 3H), 3.62 (s, 2H), 2.07 (s, 3H). |
Preparation of Intermediate 7.3(rac)-N-{[(2-chloro-6-{[5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridin-2-yl]amino}pyridin-4-yl)methyl](methyl)-λ4-sulfanylidene}-2,2,2-trifluoroacetamide
| 1H NMR (400 MHz, CDCl 3, 300K) δ=8.18 (s, 1H), 8.12 (br, 1H), 7.84 (s, 1H), 7.37 (m, 1H), 7.31 (m, 1H), 6.80 (m, 3H), 4.46 (d, 1H), 4.24 (d, 1H), 3.87 (s, 3H), 2.75 (s, 3H). |
Preparation of End Product:
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014076091
Example 1:
(rac)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
Preparation of Intermediate 1.1:
2-Chloro-5-fluoro-4-(4-fluoro-2-methoxyphenyl)pyridine
A batch with 2-chloro-5-fluoro-4-iodopyridine (1000 mg; 3.88 mmol; APAC Pharmaceutical, LLC), (4-fluoro-2-methoxyphenyl)boronic acid (660 mg; 3.88 mmol; Aldrich Chemical Company Inc.) and tetrakis(triphenylphosphin)palladium(0) (449 mg; 0.38 mmol) in 1,2-dimethoxyethane (10.0 mL) and 2 M aqueous solution of potassium carbonate (5.8 mL) was degassed using argon. The batch was stirred under an atmosphere of argon for 4 hours at 100 °C. After cooling, the batch was diluted with ethyl
acetate and THF and washed with a saturated aqueous solution of sodium chloride. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by column chromatography (hexane to hexane / ethyl acetate 50%) to give the desired product (947 mg; 3.70 mmol).
1H NMR (400MHz, CDCl3, 300K) δ = 8.27 (m, 1H), 7.33 (m, 1H), 7.24 (m, 1H), 6.75 (m, 2H), 3.83 (s, 3H).
Example 2: (+)-5-Fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S- methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine
1H-NMR (300 MHz, DMSO-d6, 300 K): δ [ppm] = 9.80 (s, 1H), 8.20 (m, 1H), 8.16 (m, 1H), 7.78 (m, 1H), 7.59 (s, 1H), 7.34 (m, 1H), 7.09 (m, 1H), 6.90 (m, 2H), 4.37 (d, 1H), 4.33 (d, 1H), 3.79 (s, 3H), 3.72 (s, 1H), 2.87 (s, 3H).
////////////VIP 152, BAY 1251152
COC1=C(C=CC(=C1)F)C2=CC(=NC=C2F)NC3=NC=CC(=C3)CS(=N)(=O)C

NEW DRUG APPROVALS
ONE TIME
$10.00
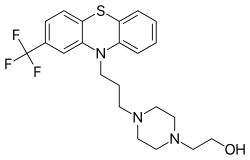
FLUPHENAZINE
Fluphenazine
- Molecular FormulaC22H26F3N3OS
- Average mass437.522 Da
- SQ 10733
- Squibb 16144
UNIIS79426A41Z
CAS number69-23-8
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Fluphenazine decanoate | FMU62K1L3C | 5002-47-1 | VIQCGTZFEYDQMR-UHFFFAOYSA-N |
| Fluphenazine enanthate | QSB34YF0W9 | 2746-81-8 | LRWSFOSWNAQHHW-UHFFFAOYSA-N |
| Fluphenazine hydrochloride | ZOU145W1XL | 146-56-5 | MBHNWCYEGXQEIT-UHFFFAOYSA-N |
2-(Trifluoromethyl)-10-[3-[1-(b-hydroxyethyl)-4-piperazinyl]propyl]phenothiazine
200-702-9[EINECS]
4-(3-(2-(trifluoromethyl)phenothiazin-10-yl)propyl)-1-Piperazineethanol
4-[3-[2-(Trifluoromethyl)-10H-phenothiazin-10-yl]propyl]-1-piperazineethanol
69-23-8[RN]
فلوفينازين[Arabic][INN]
氟奋乃静[Chinese][INN]
1-(2-Hydroxyethyl)-4-[3-(trifluoromethyl-10-phenothiazinyl)propyl]piperazine
10-[3′-[4”-(b-Hydroxyethyl)-1”-piperazinyl]propyl]-3-trifluoromethylphenothiazine
1-Piperazineethanol, 4-(3-(2-(trifluoromethyl)-10H-phenothiazin-10-yl)propyl)-
1-Piperazineethanol, 4-[3-[2-(trifluoromethyl)-10H-phenothiazin-10-yl]propyl]-
read https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/071413s019lbl.pdfFluphenazineCAS Registry Number: 69-23-8
CAS Name: 4-[3-[2-(Trifluoromethyl)-10H-phenothiazin-10-yl]propyl]-1-piperazineethanol
Additional Names: 1-(2-hydroxyethyl)-4-[3-(trifluoromethyl-10-phenothiazinyl)propyl]piperazine; 10-[3¢-[4¢¢-(b-hydroxyethyl)-1¢¢-piperazinyl]propyl]-3-trifluoromethylphenothiazine; 2-(trifluoromethyl)-10-[3-[1-(b-hydroxyethyl)-4-piperazinyl]propyl]phenothiazine
Manufacturers’ Codes: S-94; SQ-4918
Molecular Formula: C22H26F3N3OS, Molecular Weight: 437.52
Percent Composition: C 60.39%, H 5.99%, F 13.03%, N 9.60%, O 3.66%, S 7.33%
Literature References: Prepn: H. L. Yale, F. Sowinski, J. Am. Chem. Soc.82, 2039 (1960); GB829246; G. E. Ullyot, US3058979 (1960, 1962 both to SKF); GB833474 (1960 to Scherico), C.A.54, 21143e (1960); E. L. Anderson et al.,Arzneim.-Forsch.12, 937 (1962); H. L. Yale, R. C. Merrill, US3194733 (1965 to Olin Mathieson). Metabolism: J. Dreyfuss, A. J. Cohen, J. Pharm. Sci.60, 826 (1971). Comprehensive description of the enanthate ester: K. Florey, Anal. Profiles Drug Subs.2, 245-262 (1973); of the dihydrochloride: idem,ibid. 263-294; of the decanoate ester: G. Clarke, ibid.9, 275-294 (1980).
Properties: Dark brown viscous oil, bp0.5 268-274°; bp0.3 250-252°.
Boiling point: bp0.5 268-274°; bp0.3 250-252°
Derivative Type: Dihydrochloride
CAS Registry Number: 146-56-5
Trademarks: Anatensol (BMS); Dapotum (BMS); Lyogen (Promonta Lundbeck); Moditen (Sanofi Winthrop); Omca (BMS); Pacinol (Schering); Permitil (Schering); Prolixin (Apothecon); Siqualone (BMS); Tensofin (BMS); Valamina (Schering)
Molecular Formula: C22H26F3N3OS.2HCl, Molecular Weight: 510.44
Percent Composition: C 51.77%, H 5.53%, F 11.17%, N 8.23%, O 3.13%, S 6.28%, Cl 13.89%
Properties: Crystals from abs ethanol, mp 235-237°. Also reported as mp 224.5-226°.
Melting point: mp 235-237°; Also reported as mp 224.5-226°
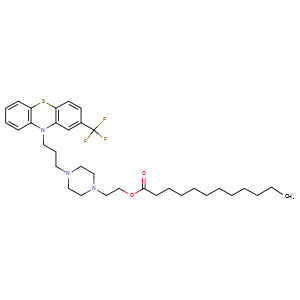
Derivative Type: Decanoate
CAS Registry Number: 5002-47-1
Manufacturers’ Codes: SQ-10733; QD-10733
Trademarks: Modecate (Sanofi Winthrop)
Molecular Formula: C32H44F3N3O2S, Molecular Weight: 591.77
Percent Composition: C 64.95%, H 7.49%, F 9.63%, N 7.10%, O 5.41%, S 5.42%
Properties: Pale yellow-orange, viscous liquid. Slowly crystallizes at room temp. mp 30-32°. Very sol in chloroform, ether, cyclohexane, methanol, ethanol. Insol in water.
Melting point: mp 30-32°
Derivative Type: Enanthate
CAS Registry Number: 2746-81-8
Manufacturers’ Codes: SQ-16144
Molecular Formula: C29H38F3N3O2S, Molecular Weight: 549.69Percent Composition: C 63.36%, H 6.97%, F 10.37%, N 7.64%, O 5.82%, S 5.83%
Properties: Pale yellow to yellow-orange viscous liquid or oily solid.
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Phenothiazines.
Fluphenazine is a phenothiazine used to treat patients requiring long-term neuroleptic therapy.
A phenothiazine used in the treatment of psychoses. Its properties and uses are generally similar to those of chlorpromazine.
Fluphenazine, sold under the brand names Prolixin among others, is a high-potency typical antipsychotic medication.[1] It is used in the treatment of chronic psychoses such as schizophrenia,[1][2] and appears to be about equal in effectiveness to low-potency antipsychotics like chlorpromazine.[3] It is given by mouth, injection into a muscle, or just under the skin.[1] There is also a long acting injectable version that may last for up to four weeks.[1] Fluphenazine decanoate, the depot injection form of fluphenazine, should not be used by people with severe depression.[4]
Common side effects include movement problems, sleepiness, depression and increased weight.[1] Serious side effects may include neuroleptic malignant syndrome, low white blood cell levels, and the potentially permanent movement disorder tardive dyskinesia.[1] In older people with psychosis as a result of dementia it may increase the risk of dying.[1] It may also increase prolactin levels which may result in milk production, enlarged breasts in males, impotence, and the absence of menstrual periods.[1] It is unclear if it is safe for use in pregnancy.[1]
Fluphenazine is a typical antipsychotic of the phenothiazine class.[1] Its mechanism of action is not entirely clear but believed to be related to its ability to block dopamine receptors.[1] In up to 40% of those on long term phenothiazines, liver function tests become mildly abnormal.[5]
Fluphenazine came into use in 1959.[6] The injectable form is on the World Health Organization’s List of Essential Medicines.[7] It is available as a generic medication.[1] It was discontinued in Australia around mid 2017.[8]
Synthesis Reference
Ullyot, G.E.; U.S. Patent 3,058,979; October 16, 1962; assigned to Smith Kline & French Laboratories.
syn
syn
Antipsychotics (Neuroleptics)
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Fluphenazine
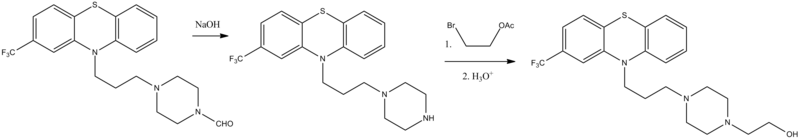
Fluphenazine, 4-[3-[2-(trifluoromethyl)phenothiazin-10-yl]propyl]-1-piperazineethanol (6.1.8), is synthesized by any of the methods described above [21–27]. Alkylation of 2-trifluoromethylphenothiazine using 4-formyl-1-piperazineylpropylchlo-ride in the presence of sodium amide synthesizes 2-trifluoromethyl-10-[3-(4-formyl-1-piperazinyl)propyl]phenothizine (6.1.6). Further alkaline hydrolysis removes the N-formyl group, giving 2-trifluoromethyl-10-[3-(1-piperazinyl)propyl]phenothiazine (6.1.7). This is alkylated by 2-bromethanol-1 acetate, which upon further acidic hydrolysis removes the protecting acetyl group, yielding fluphenazine (6.1.8) [27,28].

Fluphenazine is an extremely strong antipsychotic drug. A stimulatory effect accompanies the neuroleptic effect. It is used in psychiatry for treating various forms of schizophrenia and other mental illnesses. The most common synonyms are fluorphenazine, moditen, dapotum, motival, permitil, and others.SYN
Manufacturing Process
A suspension of 69.0 grams of 2-trifluoromethylphenothiazine in 1 liter of toluene with 10.9 grams of sodium amide is heated at reflux with high speed stirring for 15 minutes. A solution of 54.1 grams of 1-formyl-4-(3’chloropropyl)-piperazine, [prepared by formylating 1-(3′-hydroxypropyl)piperazine by refluxing in an excess of methyl formate, purifying the 1-formyl4-(3′-hydroxypropyl)-piperazine by vacuum distillation, reacting this compound with an excess of thionyl chloride at reflux and isolating the desired 1-formyl-4(3′-chloropropyl)-piperazine by neutralization with sodium carbonate solution followed by distillation] in 200 ml of toluene is added. The reflux period is continued for 4 hours. The cooled reaction mixture is treated with 200 ml of water. The organic layer is extracted twice with dilute hydrochloric acid. The acid extracts are made basic with ammonia and extracted with benzene. The volatiles are taken off in vacuo at the steam bath to leave a dark brown oil which is 10-[3′-(N-formylpiperazinyl)-propyl]-2trifluoromethylphenothiazine. It can be distilled at 260°C at 10 microns, or used directly without distillation if desired.
A solution of 103.5 grams of 10-[3′-(N-formylpiperazinyl)-propyl]-2trifluoromethylphenothiazine in 400 ml of ethanol and 218 ml of water containing 26 ml of 40% sodium hydroxide solution is heated at reflux for 2 hours. The alcohol is taken off in vacuo on the steam bath. The residue is swirled with benzene and water. The dried benzene layer is evaporated in vacuo. The residue is vacuum distilled to give a viscous, yellow oil, 10(3’piperazinylpropyl)-2-trifluoromethylphenothiazine, distilling at 210° to235°C at 0.5 to 0.6 mm.
A suspension of 14.0 grams of 10-(3′-piperazinylpropyl)-2trifluoromethylphenothiazine, 6.4 grams of β-bromoethyl acetate and 2.6 grams of potassium carbonate in 100 ml of toluene is stirred at reflux for 16 hours. Water (50 ml) is added to the cooled mixture. The organic layer is extracted into dilute hydrochloric acid. After neutralizing the extracts and taking the separated base up in benzene, a viscous, yellow residue is obtained by evaporating the organic solvent in vacuo. This oil is chromatographed on alumina. The purified fraction of 7.7 grams of 10-[3′-(Nacetoxyethylpiperazinyl)-propyl] -2-trifluoromethylphenothiazine is taken up in ethyl acetate and mixed with 25 ml of alcoholic hydrogen chloride. Concentration in vacuo separates white crystals of the dihydrochloride salt, MP 225° to 227°C.
A solution of 1.0 gram of 10-[3′-(N-acetoxyethylpiperazinyl)-propyl]-2trifluoromethylphenothiazine in 25 ml of 1 N hydrochloric acid is heated at reflux briefly. Neutralization with dilute sodium carbonate solution and extraction with benzene gives the oily base, 10-[3′-(N-βhydroxyethylpiperazinyl)-propyl]-2-trifluoromethylphenothiazine. The base is reacted with an excess of an alcoholic hydrogen chloride solution. Trituration with ether separates crystals of the dihydrochloride salt, MP 224° to 226°C, (from US Patent 3,058,979).
Chemical Synthesis
Fluphenazine, 4-[3-[2-(trifluoromethyl)phenothiazin-10-yl]propyl]-1- piperazineethanol (6.1.8), is synthesized by any of the methods described above [21–27]. Alkylation of 2-trifluoromethylphenothiazine using 4-formyl-1-piperazineylpropylchloride in the presence of sodium amide synthesizes 2-trifluoromethyl-10-[3-(4-formyl- 1-piperazinyl)propyl]phenothizine (6.1.6). Further alkaline hydrolysis removes the N-formyl group, giving 2-trifluoromethyl-10-[3-(1-piperazinyl)propyl]phenothiazine (6.1.7). This is alkylated by 2-bromethanol-1 acetate, which upon further acidic hydrolysis removes the protecting acetyl group, yielding fluphenazine (6.1.8) [27,28].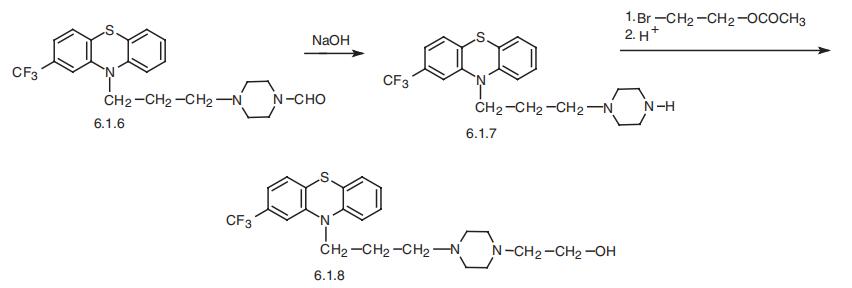
SYN
Indian Pat. Appl., 2014MU02033,
PATENT
CN 105153062
https://patents.google.com/patent/CN105153062A/en
Embodiment 1(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 100g(0.356mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 10g(0.178mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 67.5g, yield about 80%.By 3-trifluoromethyl pentanoic 60g(0.253mol), sublimed sulphur 8g(0.253mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 3g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 200g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 100g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 29g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 85%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 79g(0.5mol) 1,3-bromo-chloropropane, 320g toluene add in reaction flask, 130g(1.0mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 95g, yield about 92%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 28g(0.105mol), toluene 140g, granular sodium hydroxide 28g(0.7mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 50g toluene, the dropping process lasts about 1.5 hours of 26g (0.126mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 150g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 100g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 150g toluene.In aqueous phase, add toluene 140g, drip the sodium hydroxide solution of 20% of 62g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 15g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 33g fluorine and put forth energy to be nearly alkali, yield 72%.(4) preparation of fluophenazine hydrochloride: 32g alkali is dissolved in 128g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 50g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 36g, yield about 95%.embodiment 2.(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 500g(1.78mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 50g(0.89mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 346g, yield about 82%.By 3-trifluoromethyl pentanoic 300g(1.265mol), sublimed sulphur 40g(1.265mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 15g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 1000g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 1000g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 147g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 86%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 395g(2.5mol) 1,3-bromo-chloropropane, 1600g toluene add in reaction flask, 650g(5.0mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 470g, yield about 91%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 140g(0.525mol), toluene 700g, granular sodium hydroxide 140g(3.5mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 300g toluene, the dropping process lasts about 1.5 hours of 130g (0.63mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 750g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 500g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 750g toluene.In aqueous phase, add toluene 720g, drip the sodium hydroxide solution of 20% of 310g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 75g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 168g fluorine and put forth energy to be nearly alkali, yield 73%.(4) preparation of fluophenazine hydrochloride: 160g alkali is dissolved in 640g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 300g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 182g, yield about 96%.embodiment 3.(1) preparation of 2-trifluoromethyl thiodiphenylamine: by 1000g(3.56mol) Tecramine adds in reaction flask, be heated to 180-190 DEG C, open and stir, treat that it melts in backward reaction flask completely and add 100g(1.78mol) iron powder, stirring reaction about 2 hours at 180-190 DEG C of temperature, after reaction terminates, reaction solution is cooled to pour in beaker by reaction solution while hot when 100 DEG C, and iron powder stays (used water flushing) bottom reaction flask.Reaction solution is added underpressure distillation in clean reaction flask, collects 134-135 DEG C of (3mmHg) cut, obtain weak yellow liquid 3-trifluoromethyl pentanoic and be about 1029g, yield about 82%.By 3-trifluoromethyl pentanoic 600g(2.53mol), sublimed sulphur 80g(2.53mol) add in reaction flask, whipped state is warming up to about 130 DEG C, after the complete melting of sulphur, in reaction flask, add 30g elemental iodine, continue to be warming up to 185-190 DEG C, react about 1 hour at this temperature.There is hydrogen sulfide to release in reaction process, note tail gas absorption.After reaction terminates, reaction solution is cooled to about 100 DEG C, adds 2000g toluene in reaction flask, about raised temperature to 100 DEG C, in reaction flask, add 1000g water, stir layering while hot after 5 minutes, water layer discarded, toluene layer returns reaction flask, and whipped state borehole cooling, to 15-18 DEG C, filters, filtrate retains (to be recycled apply mechanically 3-trifluoromethyl pentanoic), filter cake adopts 60 DEG C, vacuum to dry 10 hours, and obtain 294g intermediate 2-trifluoromethyl thiodiphenylamine, yield is about 86%(and calculates by sulphur)(2) preparation of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine: by 790g(5mol) 1,3-bromo-chloropropane, 3200g toluene add in reaction flask, 1300g(10mol is dripped under control 32-35 DEG C condition) 1-(2-hydroxyethyl) piperazine, time for adding about 2 hours.After dropwising, 32-35 DEG C of stirring reaction 10 hours, after reaction terminates, passes into hydrogen chloride gas to reaction system, regulate PH=8, solids removed by filtration, filtrate decompression distillation and concentration removing toluene solvant and unreacted complete 1,3-bromo-chloropropane, obtains viscous liquid product 940g, yield about 91%.(3) fluorine puts forth energy to be the preparation of nearly alkali: by 2-trifluoromethyl thiodiphenylamine 280g(1.05mol), toluene 1400g, granular sodium hydroxide 280g(7mol) drop in reaction flask, whipped state is warming up to reflux state (110-112 DEG C), drip (the mixing solutions solution of 1-(3-chloropropyl)-4-(2-hydroxyethyl) piperazine and 500g toluene, the dropping process lasts about 1.5 hours of 260g (1.26mol) at reflux.After dropwising, reflux state reaction about 8 hours, whole reflux course notices that system moisture removes by timely water trap.After reaction terminates, be cooled to room temperature, solids removed by filtration insolubles, 1500g purifying moisture three washing organic phases.Add the 10% concentration aqueous hydrochloric acid of 1000g to organic phase, stir static layering after 10 minutes, discard upper toluene organic phase, retain lower floor’s aqueous phase, wash aqueous phase at twice with 1500g toluene.In aqueous phase, add toluene 1400g, drip the sodium hydroxide solution of 20% of 620g under whipped state, in process, hierarchy of control temperature is no more than 45 DEG C, after dropwising, stir 20 minutes, static layering, discard lower floor’s aqueous phase, retain upper organic phase, organic phase 150g anhydrous sodium sulfate drying, underpressure distillation removing toluene solvant, residue carries out underpressure distillation, collect 230 DEG C of (0.5mmHg) cuts, obtain 344g fluorine and put forth energy to be nearly alkali, yield 75%.(4) preparation of fluophenazine hydrochloride: 320g alkali is dissolved in 1280g dehydrated alcohol, stirring is dissolved backward system completely and is led to hydrogen chloride gas, process temperature is no more than 20 DEG C, logical hydrogen chloride gas is stopped as PH=2, stir after 30 minutes and filter, filter cake 500g absolute ethanol washing, product puts into vacuum drying oven, dry after 10 hours for 45 DEG C and obtain fluophenazine hydrochloride 364g, yield about 96%.
PATENT
WO 2015103587
https://patents.google.com/patent/WO2015103587A2/no
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical use
A 2018 Cochrane review found that fluphenazine was an imperfect treatment and other inexpensive drugs less associated with side effects may be an equally effective choice for people with schizophrenia.[9]
Side effects
Discontinuation
The British National Formulary recommends a gradual withdrawal when discontinuing antipsychotics to avoid acute withdrawal syndrome or rapid relapse.[10] Symptoms of withdrawal commonly include nausea, vomiting, and loss of appetite.[11] Other symptoms may include restlessness, increased sweating, and trouble sleeping.[11] Less commonly there may be a feeling of the world spinning, numbness, or muscle pains.[11] Symptoms generally resolve after a short period of time.[11]
There is tentative evidence that discontinuation of antipsychotics can result in psychosis.[12] It may also result in reoccurrence of the condition that is being treated.[13] Rarely tardive dyskinesia can occur when the medication is stopped.[11]
Pharmacology
Pharmacodynamics
See also: Antipsychotic § Pharmacodynamics, and Antipsychotic § Comparison of medications
Fluphenazine acts primarily by blocking post-synaptic D2 receptors in the basal ganglia, cortical and limbic system. It also blocks alpha-1 adrenergic receptors, muscarinic-1 receptors, and histamine-1 receptors.[14][15]
| Site | Ki (nM) | Action | Ref |
|---|---|---|---|
| 5-HT1A | 145-2829 | ND | [16] |
| 5-HT1B | 334 | ND | [16] |
| 5-HT1D | 334 | ND | [16] |
| 5-HT1E | 540 | ND | [16] |
| 5-HT2A | 3.8-98 | ND | [16] |
| 5-HT2B | ND | ND | [16] |
| 5-HT2C | 174–2,570 | ND | [16] |
| 5-HT3 | 4,265- > 10,000 | ND | [16] |
| 5-HT5A | 145 | ND | [16] |
| 5-HT6 | 7.9 – 38 | ND | [16] |
| 5-HT7 | 8 | ND | [16] |
| D1 | 14.45 | ND | [16] |
| D2 | 0.89 | ND | |
| D2L | ND | [16] | |
| D3 | 1.412 | ND | [16] |
| D4 | 89.12 | ND | [16] |
| D5 | 95–2,590 | ND | [16] |
| α1A | 6.4-9 | ND | [16] |
| α1B | 13 | ND | [16] |
| α2A | 304-314 | ND | [16] |
| α2B | 181.6-320 | ND | [16] |
| α2C | 28.8-122 | ND | [16] |
| β1 | > 10,000 | ND | [16] |
| β2 | > 10,000 | ND | [16] |
| H1 | 7.3-70 | ND | [16] |
| H2 | 560 | ND | [16] |
| H3 | 1,000 | ND | [16] |
| H4 | > 10,000 | ND | [16] |
| M1 | 1,095-3,235.93 | ND | [16] |
| M2 | 2,187.76-7,163 | ND | [16] |
| M3 | 1441–1445.4 | ND | [16] |
| M4 | 5,321 | ND | [16] |
| M5 | 357 | ND | [16] |
| SERT | ND | ND | [16] |
| NET | ND | ND | [16] |
| DAT | ND | ND | [16] |
| NMDA (PCP) | ND | ND | [16] |
| Values are Ki (nM). The smaller the value, the more strongly the drug binds to the site. All data are for human cloned proteins, except 5-HT3 (rat), D4 (human/rat), H3 (guinea pig), and NMDA/PCP (rat).[16] |
Pharmacokinetics
History
Fluphenazine came into use in 1959.[6]
Availability
The injectable form is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[7] It is available as a generic medication.[1] It was discontinued in Australia around mid 2017.[8]
Other animals
In horses, it is sometimes given by injection as an anxiety-relieving medication, though there are many negative common side effects and it is forbidden by many equestrian competition organizations.[27]
References
- ^ Jump up to:a b c d e f g h i j k l m n o “fluphenazine decanoate”. The American Society of Health-System Pharmacists. Archived from the original on 8 December 2015. Retrieved 1 December 2015.
- ^ “Product Information: Modecate (Fluphenazine Decanoate Oily Injection )” (PDF). TGA eBusiness Services. Bristol-Myers Squibb Australia Pty Ltd. 1 November 2012. Archived from the original on 2 August 2017. Retrieved 9 December 2013.
- ^ Tardy M, Huhn M, Engel RR, Leucht S (August 2014). “Fluphenazine versus low-potency first-generation antipsychotic drugs for schizophrenia”. The Cochrane Database of Systematic Reviews. 8 (8): CD009230. doi:10.1002/14651858.CD009230.pub2. PMID 25087165.
- ^ “Modecate Injection 25mg/ml – Patient Information Leaflet (PIL) – (eMC)”. http://www.medicines.org.uk. Retrieved 6 November 2017.
- ^ “Fluphenazine”. livertox.nih.gov. Retrieved 6 November 2017.
- ^ Jump up to:a b McPherson EM (2007). Pharmaceutical Manufacturing Encyclopedia (3rd ed.). Burlington: Elsevier. p. 1680. ISBN 9780815518563.
- ^ Jump up to:a b World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:a b Rossi S, ed. (July 2017). “Fluphenazine – Australian Medicines Handbook”. Australian Medicines Handbook. Adelaide, Australia: Australian Medicines Handbook Pty Ltd. Retrieved 8 August 2017.
- ^ Matar HE, Almerie MQ, Sampson SJ (June 2018). “Fluphenazine (oral) versus placebo for schizophrenia”. The Cochrane Database of Systematic Reviews. 6: CD006352. doi:10.1002/14651858.CD006352.pub3. PMC 6513420. PMID 29893410.
- ^ Joint Formulary Committee, BMJ, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISBN 978-0-85369-845-6.
Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.
- ^ Jump up to:a b c d e Haddad P, Haddad PM, Dursun S, Deakin B (2004). Adverse Syndromes and Psychiatric Drugs: A Clinical Guide. OUP Oxford. pp. 207–216. ISBN 9780198527480.
- ^ Moncrieff J (July 2006). “Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse”. Acta Psychiatrica Scandinavica. 114 (1): 3–13. doi:10.1111/j.1600-0447.2006.00787.x. PMID 16774655. S2CID 6267180.
- ^ Sacchetti E, Vita A, Siracusano A, Fleischhacker W (2013). Adherence to Antipsychotics in Schizophrenia. Springer Science & Business Media. p. 85. ISBN 9788847026797.
- ^ Siragusa S, Saadabadi A (2020). “Fluphenazine”. StatPearls. PMID 29083807.
- ^ PubChem. “Fluphenazine”. pubchem.ncbi.nlm.nih.gov. Retrieved 30 September 2019.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al Roth, BL; Driscol, J. “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
- ^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
- ^ Jump up to:a b Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID 6931472.
- ^ Jump up to:a b Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
- ^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID 6143748.
- ^ Jump up to:a b Curry SH, Whelpton R, de Schepper PJ, Vranckx S, Schiff AA (April 1979). “Kinetics of fluphenazine after fluphenazine dihydrochloride, enanthate and decanoate administration to man”. British Journal of Clinical Pharmacology. 7 (4): 325–31. doi:10.1111/j.1365-2125.1979.tb00941.x. PMC 1429660. PMID 444352.
- ^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
- ^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID 4992598.
- ^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID 3545764.
- ^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID 7185768.
- ^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
- ^ Loving NS (31 March 2012). “Effects of Behavior-Modifying Drug Investigated (AAEP 2011)”. The Horse Media Group. Archived from the original on 6 January 2017. Retrieved 13 December 2016.
External links
- “Fluphenazine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Prolixin, Modecate, Moditen others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682172 |
| License data | US DailyMed: Fluphenazine |
| Pregnancy category | AU: C |
| Routes of administration | By mouth, Intramuscular injection, depot injection (fluphenazine decanoate) |
| Drug class | Typical antipsychotic |
| ATC code | N05AB02 (WHO) |
| Legal status | |
| Legal status | AU: DiscontinuedCA: ℞-onlyUK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 2.7% (by mouth) |
| Metabolism | unclear[1] |
| Elimination half-life | IM 15 hours (HCL), 7–10 days (decanoate)[1] |
| Excretion | Urine, feces |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 69-23-8 |
| PubChem CID | 3372 |
| IUPHAR/BPS | 204 |
| DrugBank | DB00623 |
| ChemSpider | 3255 |
| UNII | S79426A41Z |
| KEGG | D07977 |
| ChEBI | CHEBI:5123 |
| ChEMBL | ChEMBL726 |
| CompTox Dashboard (EPA) | DTXSID2023068 |
| ECHA InfoCard | 100.000.639 |
| Chemical and physical data | |
| Formula | C22H26F3N3OS |
| Molar mass | 437.53 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
////////////Fluphenazine, فلوفينازين , 氟奋乃静 , SQ 10733, Squibb 16144
OCCN1CCN(CCCN2C3=CC=CC=C3SC3=C2C=C(C=C3)C(F)(F)F)CC1

NEW DRUG APPROVALS
ONE TIME
$10.00
TAUROLIDINE

TAUROLIDINE
- Molecular FormulaC7H16N4O4S2
- Average mass284.356 Da
19388-87-5[RN]
243-016-5[EINECS]
2H-1,2,4-Thiadiazine, 4,4′-methylenebis[tetrahydro-, 1,1,1′,1′-tetraoxide
4,4′-methanediylbis(1,2,4-thiadiazinane) 1,1,1′,1′-tetraoxide
UNII-8OBZ1M4V3V
тауролидин
توروليدين
牛磺利定
NMR https://www.apexbt.com/downloader/document/C4559/NMR-2.pdf
MS https://www.apexbt.com/downloader/document/C4559/MS-2.pdf
Taurolidine
CAS Registry Number: 19388-87-5
CAS Name: 4,4¢-Methylenebis(tetrahydro-1,2,4-thiadiazine) 1,1,1¢,1¢-tetraoxide
Additional Names: 4,4¢-methylenebis(perhydro-1,2,4-thiadiazine 1,1-dioxide); bis(1,1-dioxoperhydro-1,2,4-thiadiazin-4-yl)methane
Trademarks: Drainasept (Geistlich); Taurolin (HMR); Tauroflex (Geistlich)
Molecular Formula: C7H16N4O4S2, Molecular Weight: 284.36
Percent Composition: C 29.57%, H 5.67%, N 19.70%, O 22.51%, S 22.55%
Literature References: Broad spectrum, synthetic formaldehyde carrier formed by the condensation of two molecules of taurine and three molecules of formaldehyde. Prepn: FR1458701; R. W. Pfirrmann, US3423408 (1966, 1969 both to Ed. Geistlich Söhne). Antibacterial activity in mice: M. K. Browne et al.,J. Appl. Bacteriol.41, 363 (1976). Anti-endotoxin activity in lab animals: R. W. Pfirrmann, G. B. Leslie, ibid.46, 97 (1979). Mechanism of action: E. Myers et al.,ibid.48, 89 (1980). HPLC determn of metabolites in plasma: A. D. Woolfson et al.,Int. J. Pharm.49, 135 (1989). Pharmacokinetics: C. Steinbach-Lebbin et al.,Arzneim.-Forsch.32, 1542 (1982). Metabolism in humans: B. I. Knight et al.,Br. J. Clin. Pharmacol.12, 695 (1981). Clinical trials in peritonitis: M. K. Browne et al.,Surg. Gynecol. Obstet.146, 721 (1978); G. Wesch et al.,Fortschr. Med.101, 545 (1983); in wound sepsis: A. K. Halsall et al.,Pharmatherapeutica2, 673 (1981); in pleural infection: A. A. Conlan et al.,S. Afr. Med. J.64, 653 (1983).
Properties: White crystals, mp 154-158°. Sol in water.
Melting point: mp 154-158°
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic).
Taurolidine is an antimicrobial that is used to try to prevent infections in catheters.[1] Side effects and the induction of bacterial resistance is uncommon.[1] It is also being studied as a treatment for cancer.[2]
It is derived from the endogenous amino acid taurine. Taurolidine’s putative mechanism of action is based on a chemical reaction. During the metabolism of taurolidine to taurinamide and ultimately taurine and water, methylol groups are liberated that chemically react with the mureins in the bacterial cell wall and with the amino and hydroxyl groups of endotoxins and exotoxins. This results in denaturing of the complex polysaccharide and lipopolysaccharide components of the bacterial cell wall and of the endotoxin and in the inactivation of susceptible exotoxins.[3]
PATENT
https://patents.google.com/patent/WO2012070066A1/en
Taurolidine is an antibacterial drug and also has antiendotoxic substance, which is used as an antiseptic solution in surgery for washing out the abdominal cavity and it also prevents septic shock. It is commercially sold as Taurolidine (Formula I). The present invention relates to a process for the preparation of Taurolidine which provides significant advantages over the existing processes.

Formula I
The current process for the preparation of Taurolidine is depicted in Scheme 1

Formula II

Formula IVFormula IThe present inventors thus propose an industrially viable procedure for isolation of Taurolidine in substantially pure form.Taurolidine is dissolved in a suitable solvent to obtain a clear solution. The product starts to precipitate and an anti solvent is added optionally to maximize the precipitation procedure. The solvents employed for the purification are non -aqueous aprotic solvents comprising DMSO, DMAc, DMF, Acetonitirle, DMSO being the most preferred solvent. The antisolvents employed are toluene, ethyl acetate, dichloromethane, ether; toluene being the most preferred.Taurolidine obtained by the instant procedure has purity greater than or equal to 99.5 %. The process of the invention is illustrated by the following examples to obtain Taurolidine. Example ICbz-Taurine sodium salt (Formula II)To 1000ml of water in the RBF charge 192gm of (3.0 eq) of sodium hydroxide under cooling followed by 200gm of Taurine and dissolve it until clear solution is obtained. Cool to 0°C to 5°C, and Charge 50% CBZ-C1 in toluene at 0°C to 5°C. After completion of addition, maintain at room temperature for 14h. Separate the toluene layer and wash the aqueous layer with 2x200ml of ethyl acetate. Add slowly 27gm of sodium hydroxide in 60ml of water to the aqueous layer and adjust pH to 12- 14. Cool to 0°C to 5°C and a white solid separates from the solution. Filter the solid and dry the solid at 60 -70 °C. Weight of the solid: 320 gExample 2Cbz-Taurinamide (Formula III)To a clean dry flask charge 1500ml of toluene and charge 320gm of Formula II and cool to 0°C to 5°C. Charge 308 gm of PC15 slowly at 0°C to 5°C for 2hrs. Maintain at 0°C to 5°C up to completion of reaction. Quench the RM into another flask containing 2 ltr of water at 0°C to 5°C. Separate the organic layer, wash and extract the aqueous layer with toluene. Dry the organic layer with sodium sulphate and cool to 0°C to 5°C. Purge ammonia gas into the reaction mass till the reaction is complete. Filter the solid and dissolve the solid in 21tr of water and extract the aqueous layer with 2x600ml of ethyl acetate. Dry the organic layer with sodium sulphate and concentrate it under reduced pressure to obtain a white solid. Weight of the solid: 150 gExample 3Taurinamide Succinate (Formula IV)Take a suspension of 100 g of Cbz-Taurinamide in 1000 ml methanol, and 10% Pd/C (1 .0 g) and subject to hydrogenation at 45-50 psi. Upon completion of the reaction filter the catalyst and add succinic acid (1 .0 eq) to the solvent and distill off the solvent under vacuum to provide the title compound in about 90% yield as a white solid.Example 4Taurolidine (Formula I)To a solution of 100 g Taurinamide succinate in water is added sat sodium bicarbonate solution and pH adjusted to 7-8. To the solution was added formaldehyde (50 ml) and allowed to stir for 4 h. The solid obtained was filtered and washed with water to give Taurolidine. The title compound was obtained in about 70% yield and about 98% purity.Example 5Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. The solid is filtered and washed with toluene and dried to give a white solid in 40 % yield. The product obtained was >99.5% pure.Example 6Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. To the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70 % yield. The product obtained was >99.5% pure by HPLC and passed elemental analysis within 0.4% of the theoretical values.Example 7Purification of TaurolidineTaurolidine (100 g) was dissolved in DMAc (800 ml) and to the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022007713&_cid=P12-KYJIDL-52672-1Taurolidine (English name Taurolidine, chemical name is 4,4′-methylenebis[tetrahydro-2H-1,2,4-thiadiazine] 1,1,1′,1′-tetraoxide , the molecular formula is C 7 H 16 N 4 O 4 S 2 ) is a derivative of the amino acid taurine, and its structure is as follows:
Taurolidine has anti-endotoxin, anti-bacterial and anti-adherent properties. In terms of bacteria, taurolidine can chemically react with cell walls, endotoxins and exotoxins to inhibit microbial adhesion and play an antibacterial role. In addition, in terms of anti-tumor, taurolidine can induce cytotoxicity of tumor cells by inducing apoptosis, autophagy and necrosis. The extent to which these processes are involved may vary with the type of tumor cell. Until July 2020, there were more than 260 foreign literature searches on taurolidine research reports, most of which focused on the exploration of the effect of taurolidine on tumor-related signaling pathways, while the application of taurolidine in antiviral activity was not yet available. See research reports.
PATENThttps://patents.google.com/patent/CN101274921B/en
Taurolidine synthetic operation step:1. the preparation of tauryl villaumite hydrochlorate
In being housed, ventpipe, escape pipe, thermometer and churned mechanically 300ml four-necked bottle add Mercaptamine 25g, 200ml methylene dichloride and 32ml dehydrated alcohol, under ice-water bath (below the 10 ℃) mechanical stirring, feed dry appropriate chlorine, reaction begins and heat release immediately, the thick solid of adularescent generates, and temperature remains on below 50 ℃ and stirs, reaction 5h.The whole process HCl gas and the monochloroethane gas of alkali lye absorption reaction process.Stop logical chlorine after reacting end, get yellow mercury oxide, suction filtration is used washed with dichloromethane four times, and vacuum-drying gets white solid 50g, 152~154 ℃ of fusing points.2. the preparation of tauryl azide salt hydrochlorate
The reaction flask ice-water bath of containing 45ml water is cooled to-15 ℃, adds NaN 3(2g), after stirring is molten entirely, add slightly pinkiness of tauryl villaumite hydrochlorate (9g) solution in batches, the water-bath of 20 minutes recession deicings, room temperature continues stirring 60 minutes.3. the preparation of tauryl amine hydrochlorate
Above-mentioned reaction solution is joined in the 500ml autoclave, add 0.5g 5%Pd/C, feed hydrogen, pressure is 7Mpa, stirring at room 6h.Turn off hydrogen, pour out reaction solution, elimination Pd/C gets colourless reaction solution.The reaction solution that takes a morsel adds in the nuclear-magnetism pipe, adds deuterated reagent D 2O, with 1HNMR determines the transformation efficiency of hydrogenation reaction.Two kinds of CH of tauryl amine hydrochlorate 2D 1The HNMR peak is 3.29~3.31 and 3.40~3.42ppm place, and two kinds of CH of reactant tauryl azide salt hydrochlorate 2D 1The HNMR peak is 3.37~3.38 and 3.82~3.84ppm place.Determine that with the peak height ratio of two kinds of compounds the 4th step added the amount of formaldehyde.4. the preparation of taurolidine
With the above-mentioned reaction solution that removes by filter Pd/C, add 5g NaHCO 3, be stirred to molten entirely, frozen water cooling, stir slowly splash into down formaldehyde solution (37%, 2ml), have milky white precipitate to produce after 30 minutes, continue to stir 1h, suction filtration, filter cake is washed 3 times with frozen water.Vacuum-drying gets white powdery solid 2.3g, 170~174 ℃ of fusing points.Embodiment 2:Making with extra care of taurolidine:The above-mentioned taurolidine white powder 5~10g that obtains adds 50~200ml acetonitrile, and heating for dissolving removes by filter a small amount of insolubles, concentrates, and cooling below 10 ℃ gets white powder 5~10g, 172~174 ℃ of fusing points.Embodiment 3:Proton nmr spectra ( 1H-NMR) data are as follows:1HNMR(DMSO-D6,TMS7.26-7.28(t,2H,NH),4.09-4.10(d,4H,N-CH 2),3.53(s,2H,N-CH 2-N),3.28-3.29(t,4H,N-CH 2-CH 2),2.96-2.97(t,4H,S-CH 2-CH 2)。The infrared absorption spectrum data are as follows:IR (KBr compressing tablet cm -1): 3425,3263,1633,1450,1404,1317,1278,1228,1160,1134,1073,1026,993,958,924,830,757,667,532,511.See Fig. 3.The ultimate analysis analytical value:C, 29.04%, N, 18.55%, H, 5.85%; Calculated value: C, 29.57%, N, 19.71%, H, 5.67%Embodiment 4:Taurolidine formulation optimizing injection type of the present invention, as: infusion solution, injection liquid, freeze-dried powder injection or powder ampoule agent for injection etc., more preferably infusion solution.The preparation of infusion solution[prescription 1] taurolidine 10.0~30.0gPVP 40.0~80.0gNaCl 2~5gAdd water to 1000ml[method for making] takes by weighing taurolidine, is dissolved in water, and stirs, and adds the PVP dissolving, and adjust pH to 7.0 is crossed the moisture film of 0.22 μ m, packing, and 121 ℃ of sterilizations 20 minutes, promptly.[prescription 2] taurolidine 10.0~30.0gCitric acid 0.1~1.0gLemon enzyme sodium 10.0~20.0gAdd water to 1000ml[method for making] takes by weighing taurolidine, is dissolved in water, stir, and the dissolving of adding citric acid sodium, adjust pH to 7.0, the moisture film of mistake 0.22 μ m, packing was sterilized 20 minutes for 121 ℃, promptly.
PATENThttps://patents.google.com/patent/US8952148B2/en

Example ICbz-Taurine Sodium Salt (Formula II)To 1000 ml of water in the RBF charge 192 gm of (3.0 eq) of sodium hydroxide under cooling followed by 200 gm of Taurine and dissolve it until clear solution is obtained. Cool to 0° C. to 5° C., and Charge 50% CBZ-Cl in toluene at 0° C. to 5° C. After completion of addition, maintain at room temperature for 14 h. Separate the toluene layer and wash the aqueous layer with 2×200 ml of ethyl acetate. Add slowly 27 gm of sodium hydroxide in 60 ml of water to the aqueous layer and adjust pH to 12-14. Cool to 0° C. to 5° C. and a white solid separates from the solution. Filter the solid and dry the solid at 60-70° C. Weight of the solid: 320 g
Example 2Cbz-Taurinamide (Formula III)To a clean dry flask charge 1500 ml of toluene and charge 320 gm of Formula II and cool to 0° C. to 5° C. Charge 308 gm of PCl5 slowly at 0° C. to 5° C. for 2 hrs. Maintain at 0° C. to 5° C. up to completion of reaction. Quench the RM into another flask containing 2 ltr of water at 0° C. to 5° C. Separate the organic layer, wash and extract the aqueous layer with toluene. Dry the organic layer with sodium sulphate and cool to 0° C. to 5° C. Purge ammonia gas into the reaction mass till the reaction is complete. Filter the solid and dissolve the solid in 2 ltr of water and extract the aqueous layer with 2×600 ml of ethyl acetate. Dry the organic layer with sodium sulphate and concentrate it under reduced pressure to obtain a white solid. Weight of the solid: 150 g
Example 3Taurinamide Succinate (Formula IV)Take a suspension of 100 g of Cbz-Taurinamide in 1000 ml methanol, and 10% Pd/C (1.0 g) and subject to hydrogenation at 45-50 psi. Upon completion of the reaction filter the catalyst and add succinic acid (1.0 eq) to the solvent and distill off the solvent under vacuum to provide the title compound in about 90% yield as a white solid.
Example 4Taurolidine (Formula I)To a solution of 100 g Taurinamide succinate in water is added sat sodium bicarbonate solution and pH adjusted to 7-8. To the solution was added formaldehyde (50 ml) and allowed to stir for 4 h. The solid obtained was filtered and washed with water to give Taurolidine. The title compound was obtained in about 70% yield and about 98% purity.
Example 5Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. The solid is filtered and washed with toluene and dried to give a white solid in 40% yield. The product obtained was >99.5% pure.
Example 6Purification of TaurolidineTaurolidine (100 g) was dissolved in DMSO (400 ml) and a clear solution is obtained and a precipitate is obtained immediately. To the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield. The product obtained was >99.5% pure by HPLC and passed elemental analysis within 0.4% of the theoretical values.
Example 7Purification of TaurolidineTaurolidine (100 g) was dissolved in DMAc (800 ml) and to the solution, toluene (1000 ml) is added. The solid is filtered and washed with toluene and dried to give a white solid in 70% yield.

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Defencath, a product created by CorMedix, Inc., was granted FDA approval on November 15, 2023. Its purpose is to decrease the occur rence of catheter-related bloodstream infections (CRBSI) in a limited
population of adult individuals who have chronic renal failure and undergo hemodialysis (HD) using a central venous catheter (CVC) [11]. Defencath is the first and only FDA-approved antimicrobial catheter
locking solution (CLS) in the United States. It is composed of the anticoagulant Heparin and the broad-spectrum non-antibiotic antimicrobial and antifungal agent Taurolidine. Taurolidine has been proven to have activity against the following microorganisms: Staphylococcus aureus, Staphylococcus epidermidis, Escherichia coli, Enterococcus faecalis, Pseudomonas aeruginosa, Klebsiella pneumoniae, Serratia marcescens, Candida albicans, Candida glabrata [12,13].

The synthesis route of Taurolidine is showed in Scheme 4 [14].
Benzyl (2-sulfamoylethyl)carbamate (TAUR-001) was removed benzy
loxycarbonyl (Cbz) group under the action of palladium carbon to obtain
TAUR-002. After the reaction with formaldehyde, the product Taur
olidine was obtained.
[11] A.K. Agarwal, P. Roy-Chaudhury, P. Mounts, E. Hurlburt, A. Pfaffle, E.C. Poggio,
Taurolidine/Heparin lock solution and catheter-related bloodstream infection in
hemodialysis: a randomized, double-blind, active-control, phase 3 study, Clin. J.
Am. Soc. Nephrol. 18 (2023) 1446–1455.
[12] C.H. van den Bosch, B. Jeremiasse, J.T. van der Bruggen, F.N.J. Frakking, Y.G.
T. Loeffen, C.P. van de Ven, A.F.W. van der Steeg, M.F. Fiocco, M.D. van de
Wetering, M. Wijnen, The efficacy of taurolidine containing lock solutions for the
prevention of central-venous-catheter-related bloodstream infections: a
systematic review and meta-analysis, J. Hosp. Infect. 123 (2022) 143–155.
[13] Y. Wouters, G.R.H. Mennen, R.H.M. Te Morsche, H.M.J. Roelofs, G.J.A. Wanten,
The antiseptic and antineoplastic agent taurolidine modulates key leukocyte
functions, In Vivo 36 (2022) 2074–2082.
[14] R.M. Berrios, Synthesis of Taurolidine, Purity Profiles and Polymorphs, 2023.
US11738120B1
Medical uses
Taurolidine is an antimicrobial agent used in an effort to prevent catheter infections. It however is not approved for this use in the United States as of 2011.[4]
- Catheter lock solution in home parenteral nutrition (HPN) or total parenteral nutrition (TPN): catheter-related blood stream infections (CRBSI) remains the most common serious complication associated with long-term parenteral nutrition. The use of taurolidine as a catheter lock solution shows a reduction of CRBSI.[1][5] The overall quality of the evidence however is not strong enough to justify routine use.[1][5]
- Catheter lock solution: Taurolidine decreases the adherence of bacteria and fungi to host cells by destructing the fimbriae and flagella and thus prevent the biofilm formation.[6][7] Taurolidine is the active ingredient of antimicrobial catheter lock solutions for the prevention and treatment of catheter related bloodstream infections (CRBSIs) and is suitable for use in all catheter based vascular access devices.[8][1] Bacterial resistance against taurolidine has never been observed in various studies.[9][10]
- The use of a taurolidine lock solution may decrease the risk of catheter infection in children with cancer but the evidence is tentative.[11]
Side effects
No systemic side effects have been identified. The safety of taurolidine has also been confirmed in clinical studies with long-term intravenous administration of high doses (up to 20 grams daily). In the body, taurolidine is metabolized rapidly via the metabolites taurultam and methylol taurinamide, which also have a bactericidal action, to taurine, an endogenous aminosulphonic acid, carbon dioxide and water. Therefore, no toxic effects are known or expected in the event of accidental injection. Burning sensation while instilling, numbness, erythema, facial flushing, headache, epistaxis, and nausea have been reported.[12]
Toxicology
Taurolidine has a relatively low acute and subacute toxicity.[1] Intravenous injection of 5 grams taurolidine into humans over 0.5–2 hours produce only burning sensation while instilling, numbness, and erythema at the injection sites.[12] For treatment of peritonitis, taurolidine was administered by peritoneal lavage, intraperitoneal instillation or intravenous infusion, or by a combination thereof. The total daily dose ranged widely from 0.5 to 50 g. The total cumulative dose ranged from 0.5 to 721 g. In those patients who received intravenous taurolidine, the daily intravenous dose was usually 15 to 30 g but several patients received up to 40 g/day. Total daily doses of up to 40 g and total cumulative doses exceeding 300 g were safe and well tolerated.[12][13][14][15][16]
Pharmacology
- Metabolism: Taurolidine and taurultam are quickly metabolized to taurinamide, taurine, carbon dioxide and water. Taurolidine exists in equilibrium with taurultam and N-methylol-taurultam in aqueous solution.[17]
- Pharmacokinetic (elimination): The half-life of the terminal elimination phase of taurultam is about 1.5 hours, and of the taurinamide metabolite about 6 hours. 25% of the taurolidine dose applied is renally eliminated as taurinamide and/or taurine.[13][14][18]
Mechanism of action
Following administration of taurolidine, the antimicrobial and antiendotoxin activity of the taurolidine molecule is conferred by the release of three active methylol (hydroxymethyl) groups as taurolidine is rapidly metabolized by hydrolysis via methylol taurultam to methylol taurinamide and taurine. These labile N-methylol derivatives of taurultam and taurinamide react with the bacterial cell-wall resulting in lysis of the bacteria, and by inter- and intramolecular cross-linking of the lipopolysaccharide-protein complex, neutralization of the bacterial endotoxins which is enhanced by enzymatic activation. This mechanism of action is accelerated and maximised when taurolidine is pre-warmed to 37 °C (99 °F). Microbes are killed and the resulting toxins are inactivated; the destruction time in vitro is 30 minutes.[19]
The chemical mode of action of taurolidine via its reactive methylol groups confers greater potency in vivo than indicated by in vitro minimum inhibitory concentration (MIC) values, and also appears to preclude susceptibility to resistance mechanisms.[14]
Taurolidine binding to lipopolysaccharides (LPS) prevents microbial adherence to host epithelial cells, thereby prevents microbial invasion of uninfected host cells. Although the mechanism underlying its antineoplastic activity has not been fully elucidated, it may be related to this agent’s anti-adherence property.[6][7] Taurolidine has been shown to block interleukin 1 (IL-1) and tumour necrosis factor (TNF) in human peripheral blood mononuclear cells (PBMC).[20] In addition, taurolidine also promotes apoptosis by inducing various apoptotic factors and suppresses the production of vascular endothelial growth factor (VEGF), a protein that plays an important role in angiogenesis.[21]
Taurolidine is highly active against the common infecting pathogens associated with peritonitis and catheter sepsis, this activity extends across a wide-spectrum of aerobic and anaerobic bacteria and fungi (with no diminution of effect in the presence of biological fluids, e.g. blood, serum, pus).[15][16][22]
- Gram-positive bacteria (MIC of 1–2 mg/mL): Staphylococcus (including multiple-antibiotic resistant coagulase negative strains, methicillin-resistant S. aureus), Streptococcus, Enterococcus.
- Gram-negative bacteria (MIC of 0.5–5 mg/mL): Aerobacter species, Citrobacter species, Enterobacter species, Escherichia coli, Proteus species (indole negative), Proteus mirabilis, Pseudomonas species (including P. aeruginosa), Salmonella species, Serratia marcescans, Klebsiella species.
- Anaerobes (MIC 0.03–0.3 mg/mL): Bacteroides species (including B. fragilis), Fusobacteria, Clostridium species, Peptostreptococcus anaerobius.
- Fungi (MIC 0.3–5 mg/mL): Candida albicans, Cryptococcus neoformans, Aspergillus species, Trichophyton rubrum, Epidermophyton floccosum, Pityrosporum ovale.[10][17][22]
Chemical properties
The chemical name for taurolidine is 4,4′-Methylene-bis(1,2,4-thiadiazinane)-1,1,1’,1′-tetraoxide.
It is a white to off white odourless crystalline powder. It is practically insoluble in chloroform, slightly soluble in boiling acetone, ethanol, methanol, and ethyl acetate, sparingly soluble in water 8 at 20° and ethyl alcohol, soluble in dilute hydrochloric acid, and dilute sodium hydroxide, and freely soluble in N,N-dimethylformamide (at 60 °C).
History
Taurolidine was first synthesized in the laboratories of Geistlich Pharma AG, Switzerland in 1972. Clinical trials begun in 1975 in patients with severe peritonitis.
Research
Taurolidine demonstrates some anti-tumor properties, with positive results seen in early-stage clinical investigations using the drug to treat gastrointestinal malignancies and tumors of the central nervous system.[23] More recently, it has been found to exert antineoplastic activity. Taurolidine induces cancer cell death through a variety of mechanisms. Even now, all the antineoplastic pathways it employs are not completely elucidated. It has been shown to enhance apoptosis, inhibit angiogenesis, reduce tumor adherence, downregulate pro-inflammatory cytokine release, and stimulate anticancer immune regulation following surgical trauma. Apoptosis is activated through both a mitochondrial cytochrome-c-dependent mechanism and an extrinsic direct pathway. A lot of in vitro and animal data support taurolidine’s tumoricidal action.[24][25][26] Taurolidine has been used as an antimicrobial agent in the clinical setting since the 1970s and thus far appears nontoxic. The nontoxic nature of taurolidine makes it a favorable option compared with current chemotherapeutic regimens. Few published clinical studies exist evaluating the role of taurolidine as a chemotherapeutic agent. The literature lacks a gold-standard level 1 randomized clinical trial to evaluate taurolidine’s potential antineoplastic benefits. However, these trials are currently underway. Such randomized control studies are vital to clarify the role of taurolidine in modern cancer treatment.[21][2]
References
- ^ Jump up to:a b c d e f Liu Y, Zhang AQ, Cao L, Xia HT, Ma JJ (2013). “Taurolidine lock solutions for the prevention of catheter-related bloodstream infections: a systematic review and meta-analysis of randomized controlled trials”. PLOS ONE. 8 (11): e79417. Bibcode:2013PLoSO…879417L. doi:10.1371/journal.pone.0079417. PMC 3836857. PMID 24278133.
- ^ Jump up to:a b Neary PM, Hallihan P, Wang JH, Pfirrmann RW, Bouchier-Hayes DJ, Redmond HP (April 2010). “The evolving role of taurolidine in cancer therapy”. Annals of Surgical Oncology. 17 (4): 1135–43. doi:10.1245/s10434-009-0867-9. PMID 20039217. S2CID 23807182.
- ^ Waser PG, Sibler E (1986). “Taurolidine: A new concept in antimicrobial chemotherapy”. In Harms AF (ed.). Innovative Approaches in Drug Research. Elsevier Science Publishers. pp. 155–169.
- ^ O’Grady NP, Alexander M, Burns LA, Dellinger EP, Garland J, Heard SO, et al. (May 2011). “Guidelines for the prevention of intravascular catheter-related infections”. Clinical Infectious Diseases. 52 (9): e162-93. doi:10.1093/cid/cir257. PMC 3106269. PMID 21460264.
- ^ Jump up to:a b Bradshaw JH, Puntis JW (August 2008). “Taurolidine and catheter-related bloodstream infection: a systematic review of the literature”. Journal of Pediatric Gastroenterology and Nutrition. 47 (2): 179–86. doi:10.1097/MPG.0b013e318162c428. PMID 18664870. S2CID 19136945.
- ^ Jump up to:a b Gorman SP, McCafferty DF, Woolfson AD, Jones DS (April 1987). “Reduced adherence of micro-organisms to human mucosal epithelial cells following treatment with Taurolin, a novel antimicrobial agent”. The Journal of Applied Bacteriology. 62 (4): 315–20. doi:10.1111/j.1365-2672.1987.tb04926.x. PMID 3298185.
- ^ Jump up to:a b Blenkharn JI (July 1989). “Anti-adherence properties of taurolidine and noxythiolin”. Journal of Chemotherapy. 1 (4 Suppl): 233–4. PMID 16312382.
- ^ Liu H, Liu H, Deng J, Chen L, Yuan L, Wu Y (2014). “Preventing catheter-related bacteremia with taurolidine-citrate catheter locks: a systematic review and meta-analysis”. Blood Purification. 37 (3): 179–87. doi:10.1159/000360271. PMID 24777144.
- ^ Olthof ED, Rentenaar RJ, Rijs AJ, Wanten GJ (August 2013). “Absence of microbial adaptation to taurolidine in patients on home parenteral nutrition who develop catheter related bloodstream infections and use taurolidine locks”. Clinical Nutrition. 32 (4): 538–42. doi:10.1016/j.clnu.2012.11.014. PMID 23267744.
- ^ Jump up to:a b Torres-Viera C, Thauvin-Eliopoulos C, Souli M, DeGirolami P, Farris MG, Wennersten CB, et al. (June 2000). “Activities of taurolidine in vitro and in experimental enterococcal endocarditis”. Antimicrobial Agents and Chemotherapy. 44 (6): 1720–4. doi:10.1128/aac.44.6.1720-1724.2000. PMC 89943. PMID 10817739.
- ^ Simon A, Bode U, Beutel K (July 2006). “Diagnosis and treatment of catheter-related infections in paediatric oncology: an update”. Clinical Microbiology and Infection. 12 (7): 606–20. doi:10.1111/j.1469-0691.2006.01416.x. PMID 16774556.
- ^ Jump up to:a b c Gong L, Greenberg HE, Perhach JL, Waldman SA, Kraft WK (June 2007). “The pharmacokinetics of taurolidine metabolites in healthy volunteers”. Journal of Clinical Pharmacology. 47 (6): 697–703. doi:10.1177/0091270007299929. PMID 17395893. S2CID 31059736.
- ^ Jump up to:a b Knight BI, Skellern GG, Browne MK, Pfirrmann RW (November 1981). “Peritoneal absorption of the antibacterial and antiendotoxin taurolin in peritonitis”. British Journal of Clinical Pharmacology. 12 (5): 695–9. doi:10.1111/j.1365-2125.1981.tb01292.x. PMC 1401955. PMID 7332737.
- ^ Jump up to:a b c Stendel R, Scheurer L, Schlatterer K, Stalder U, Pfirrmann RW, Fiss I, et al. (2007). “Pharmacokinetics of taurolidine following repeated intravenous infusions measured by HPLC-ESI-MS/MS of the derivatives taurultame and taurinamide in glioblastoma patients”. Clinical Pharmacokinetics. 46 (6): 513–24. doi:10.2165/00003088-200746060-00005. PMID 17518510. S2CID 33321671.
- ^ Jump up to:a b Browne MK, MacKenzie M, Doyle PJ (May 1978). “C controlled trial of taurolin in established bacterial peritonitis”. Surgery, Gynecology & Obstetrics. 146 (5): 721–4. PMID 347606.
- ^ Jump up to:a b Browne MK (1981). “The treatment of peritonitis by an antiseptic – taurolin”. Pharmatherapeutica. 2 (8): 517–22. PMID 7255507.
- ^ Jump up to:a b Knight BI, Skellern GG, Browne MK, Pfirrmann RW (September 1981). “The characterisation and quantitation by high-performance liquid chromatography of the metabolites of taurolin”. British Journal of Clinical Pharmacology. 12 (3): 439–40. doi:10.1111/j.1365-2125.1981.tb01245.x. PMC 1401804. PMID 7295478.
- ^ Browne MK, Leslie GB, Pfirrmann RW (December 1976). “Taurolin, a new chemotherapeutic agent”. The Journal of Applied Bacteriology. 41 (3): 363–8. doi:10.1111/j.1365-2672.1976.tb00647.x. PMID 828157.
- ^ Braumann C, Pfirrman RW, et al. (2013). “Taurolidine, an Effective Multimodal Antimicrobial Drug Versus Traditional Antiseptics and Antibiotics”. In Willy C (ed.). Antiseptics in Surgery – Update 2013. Lindqvist Book Publishing. pp. 119–125.
- ^ Bedrosian I, Sofia RD, Wolff SM, Dinarello CA (November 1991). “Taurolidine, an analogue of the amino acid taurine, suppresses interleukin 1 and tumor necrosis factor synthesis in human peripheral blood mononuclear cells”. Cytokine. 3 (6): 568–75. doi:10.1016/1043-4666(91)90483-t. PMID 1790304.
- ^ Jump up to:a b Jacobi CA, Menenakos C, Braumann C (October 2005). “Taurolidine–a new drug with anti-tumor and anti-angiogenic effects”. Anti-Cancer Drugs. 16 (9): 917–21. doi:10.1097/01.cad.0000176502.40810.b0. PMID 16162968. S2CID 33876185.
- ^ Jump up to:a b Nösner K, Focht J (1994). “In-vitro Wirksamkeit von Taurolidin und 9 Antibiotika gegen klinische Isolate aus chirurgischem Einsendegut sowie gegen Pilze”. Chirurgische Gastroenterologie. 10 (Suppl 2): 10.
- ^ Stendel R, Picht T, Schilling A, Heidenreich J, Loddenkemper C, Jänisch W, Brock M (2004-04-01). “Treatment of glioblastoma with intravenous taurolidine. First clinical experience”. Anticancer Research. 24 (2C): 1143–7. PMID 15154639.
- ^ Calabresi P, Goulette FA, Darnowski JW (September 2001). “Taurolidine: cytotoxic and mechanistic evaluation of a novel antineoplastic agent”. Cancer Research. 61 (18): 6816–21. PMID 11559556.
- ^ Clarke NW, Wang JH, et al. (2005). “Taurolidine inhibits colorectal adenocarcinoma metastases in vivo and in vitro by inducing apoptosis”. Ir J Med Sci. 174 (Supplement 3): 1.
- ^ Stendel R, Scheurer L, Stoltenburg-Didinger G, Brock M, Möhler H (2003-06-01). “Enhancement of Fas-ligand-mediated programmed cell death by taurolidine”. Anticancer Research. 23 (3B): 2309–14. PMID 12894508.
| Clinical data | |
|---|---|
| ATC code | B05CA05 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 19388-87-5 |
| ChemSpider | 27486 |
| UNII | 8OBZ1M4V3V |
| CompTox Dashboard (EPA) | DTXSID00173001 |
| ECHA InfoCard | 100.039.090 |
| Chemical and physical data | |
| Formula | C7H16N4O4S2 |
| Molar mass | 284.35 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////////////TAUROLIDINE, UNII-8OBZ1M4V3V, тауролидин ,توروليدين , 牛磺利定 ,
C1CS(=O)(=O)NCN1CN2CCS(=O)(=O)NC2

NEW DRUG APPROVALS
ONE TIME
$10.00
Anthony crasto is now Consultant Glenmark Lifesciences at Glenmark Life Sciences!

I’m happy to share that I’m starting a new position as Consultant Glenmark Lifesciences at Glenmark Life Sciences!
17th Jan 2022, A new innings
I retired 16th Jan 2022 at 58 yrs from Glenmark . completed 16 yrs 2 months
30 plus years in the field of Process research
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
///////////////
DIPYRIDAMOLE
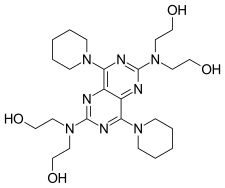
Dipyridamole
- Molecular FormulaC24H40N8O4
- Average mass504.626 Da
2,2′,2”,2”’-{[4,8-Di(piperidin-1-yl)pyrimido[5,4-d]pyrimidine-2,6-diyl]dinitrilo}tetraethanol
200-374-7[EINECS]
58-32-2[RN]
Ethanol, 2,2′,2”,2”’-[(4,8-di-1-piperidinylpyrimido[5,4-d]pyrimidine-2,6-diyl)dinitrilo]tetrakis-
дипиридамол [Russian] [INN]
ديبيريدامول [Arabic] [INN]
双嘧达莫 [Chinese] [INN]
0068373 [Beilstein]
DipyridamoleCAS Registry Number: 58-32-2
CAS Name: 2,2¢,2¢¢,2¢¢¢-[(4,8-Di-1-piperidinylpyrimido[5,4-d]pyrimidine-2,6-diyl)dinitrilo]tetrakisethanol
Additional Names: 2,6-bis(diethanolamino)-4,8-dipiperidinopyrimido-[5,4-d]pyrimidine
Manufacturers’ Codes: NSC-515776; RA-8
Trademarks: Anginal (Yamanouchi); Cardoxin (RAFA); Cleridium (Marcofina); Coridil (Delalande); Coronarine (NEGMA); Curantyl (Berlin-Chemie); Dipyridan (Hokuriku); Gulliostin (Taiyo); Natyl (Interdelta); Peridamol (Boehringer, Ing.); Persantine (Boehringer, Ing.); Piroan (Towa Yakuhin); Prandiol (Bottu); Protangix (Lefrancq)
Molecular Formula: C24H40N8O4
Molecular Weight: 504.63
Percent Composition: C 57.12%, H 7.99%, N 22.21%, O 12.68%
Literature References: Phosphodiesterase inhibitor that reduces platelet aggregation; also acts as a coronary vasodilator. Prepn: GB807826; F. G. Fischer, et al.,US3031450 (1959, 1962 both to Thomae). Activity studies: Saraf, Seth, Indian J. Physiol. Pharmacol.15, 135 (1971). Toxicological study: F. Takenaka et al.,Arzneim.-Forsch.22, 892 (1972). Symposium on pharmacology and clinical experience as antithrombotic: Thromb. Res.60, Suppl. 12, 1-99 (1990). Review of use as pharmacological stress agent in echocardiography: M. B. Buchalter et al.,Postgrad. Med. J.66, 531-535 (1990); in 201Tl cardiac imaging: S. G. Beer et al.,Am. J. Cardiol.67, Suppl., 18D-26D (1991).
Properties: Deep yellow needles from ethyl acetate, mp 163°. Bitter taste. Slightly sol in H2O; sol in dil acid having a pH of 3.3 or below; very sol in methanol, ethanol, chloroform; not too sol in acetone, benzene, ethyl acetate. Solns are yellow and show strong blue-green fluorescence. LD50 in rats: 8.4 g/kg orally; 208 mg/kg i.v. (Takenaka).Melting point: mp 163°
Toxicity data: LD50 in rats: 8.4 g/kg orally; 208 mg/kg i.v. (Takenaka)
Derivative Type: Combination with aspirin
Trademarks: Aggrenox (Boehringer, Ing.)
Literature References: Review of pharmacology and clinical efficacy in secondary prevention of stroke: P. S. Hervey, K. L. Goa, Drugs58, 469-475 (1999).
Therap-Cat: Antithrombotic; diagnostic aid (cardiac stress testing).
Keywords: Antithrombotic; Diagnostic Aid; Phosphodiesterase Inhibitor.
Dipyridamole (trademarked as Persantine and others) is a nucleoside transport inhibitor and a PDE3 inhibitor medication that inhibits blood clot formation[3] when given chronically and causes blood vessel dilation when given at high doses over a short time.
PATENT
https://patents.google.com/patent/WO2011151640A1/enDipyridamole, represented by structural formula (I), possesses platelet aggregation inhibiting, anti-thrombotic and vasodilator properties and it is marketed as an anti-platelet therapy for the treatment and prevention of disorders such as thrombo-embolisms.

A process for the preparation of dipyridamole, disclosed in patent US 3031450, involves the reaction of 2,6-dichloro-4,8-dipiperidino-pyrimido(5,4-d)pyrimidine with diethanolamine (see Scheme 1). The preparation of 2,6-dichloro-4,8-dipiperidino- pyrimido(5,4-d)pyrimidine is also reported in US 3031450 and is incorporated herein by reference. The reaction to prepare dipyridamole does not employ an additional reaction solvent and is a neat mixture of the two reactants carried out at a very high temperature of 190 to 195°C. The process also involves a cumbersome work-up to isolate dipyridamole, since the crude product obtained is a pasty mass which needs decantation of the mother liquor and further purification. This decantation process is not practical on commercial scale.

2,6-dichloro-4,8-dipiperidino- dipyridamole (Ί) pyflmido(5,4-d)pyrimidineScheme 1A similar process for the production of dipyridamole is described in patent DD 117456 wherein the reaction conditions exemplified are heating 2,6-dichloro-4,8-dipiperidino- pyrimido(5,4-d)pyrimidine and diethanolamine at 155 to 160°C under vacuum. However, this process again requires a high temperature which leads to the formation of impurities.A process for the preparation and purification of dipyridamole is disclosed in patent DE 1812918, wherein 2,6-dicMoro-4,8-dipiperidino-pyrimido(5,4-d)pyrimidine and diethanolamine are heated to 150 to 200°C. After completion of the reaction, the reaction mixture is dissolved in chloroform, which is further separated into an upper layer of diethanolamine and its hydrochloride and a chloroform solution. The chloroform solution obtained is separated and reduced to dryness after stirring with water. This process also requires a high temperature which can lead to the formation of impurities. In addition, the solvent used for the isolation of dipyridamole, chloroform, is inconvenient as it is a restricted solvent and its permitted limit in the final marketed dipyridamole is very low.A similar process, wherein dipyridamole is manufactured by the reaction of diethanolamine with 2,6-dichloro-4,8-dipiperidino-pyrimido(5,4-d)pyrimidine is disclosed in patent RO 104718. However, this process again requires high temperatures of 180 to 200°C which leads to the formation of impurities and, consequently, the yield of the final product is very low (58%) with a purity of less than 98%.A process is disclosed in patent DD 115670, wherein the purification of dipyridamole involves refluxing it in butyl acetate, AcOBu, for 2 hours in the presence of an equal amount of silica gel or column chromatography on silica gel at 60-100°C. However, purification by column chromatography is not economical and not feasible on industrial scale. Moreover, this purification process only removes one specific impurity, 2,4,6-tris- (diethanolamino) – 8 -pip eridino-pyrimido (5,4-d)pyrimidine .The processes described above to prepare dipyridamole do not employ an additional reaction solvent but involve neat mixtures of the two reactants, 2,6-dichloro-4,8- dipiperidino-pyrimido(5,4-d)pyrimidine and diethanolamine, which are heated at very high temperatures. The use of neat reaction mixtures and/ or high temperatures means that it is very difficult to control the levels of impurities formed.Another process for the preparation of dipyridamole, disclosed in patent application WO 2007/080463, involves reacting diethanolamine with 2,6-dichloro-4,8-dipiperidino- pyrimido(5,4-d)pyrimidine in a solvent selected from the group consisting of l-methyl-2- pyrrolidinone, sulpholane and polyethylene glycol. However, the exemplified reaction temperatures are very high at 190 to 200°C and the HPLC purity of the crude dipyridamole is reported to be only 90-94%. A purification method is disclosed using first a ketonic solvent and then an alcohol and water. Even though the process disclosed in this patent application uses a solvent in the reaction, the temperature of reaction is still very high and the purification in ketonic solvent is reported at high temperature (100 to 120°C). The HPLC purity after purification is reported as only 99.0-99.5%.As discussed above, all the processes disclosed in the prior art for the preparation of dipyridamole suffer from serious disadvantages with respect to commercial production. The prior art synthetic and purification processes employ high temperatures in the preparation of dipyridamole which leads to inefficiency and high processing costs. The high temperatures also lead to higher levels of impurities being formed during manufacture with the consequence that further cumbersome and expensive purification procedures are required.The high quality dipyridamole prepared by the processes according to the present invention can be used for the preparation of a pharmaceutical composition to use in the manufacture of a medicament for anti-platelet therapy. A preferred embodiment of the present invention, illustrated in Scheme 2, provides a process for the preparation of dipyridamole comprising reacting 2,6-dichloro-4,8- dipiperidino-pyrimido(5,4-d)pyrimidine with diethanolamine at 113-115°C. This reaction temperature is significantly lower than that used in the prior art processes to prepare dipyridamole.

Another preferred embodiment of the present invention, illustrated in Scheme 3, also provides a process for the preparation of dipyridamole by the reaction of 2,6-dichloro-4,8- dipiperidino-pyrimido(5,4-d)pyrimidine with diethanolamine in dimethylsulfoxide at 120- 125°C to afford the mono-substituted intermediate, 2-chloro-6-diethanolamino-4,8- dipiperidino-pyrimido(5,4-d)pyrimidine, which is isolated and then further converted to dipyridamole by heating in diethanolamine at 113-115°C.Although the solvent used in this preferred embodiment of the present invention is preferably dimethylsulfoxide (DMSO), other solvents can alternatively be used. Preferred alternative solvents are other polar aprotic solvents, such as dimethylformamide (DMF), dimethylacetamide (DMA) or N-methyl-2-pyrrolidinone (NMP). Alternatively, hydrocarbon solvents can be used. Preferred hydrocarbon solvents are aromatic hydrocarbon solvents such as toluene or xylene.


Example 1Preparation of crude dipyridamoleDiethanolamine (10 vol) and 2,6-dichloro-4,8-dipiperidino-pyrimido(5,4-d)pyrimidine (1 eq) were mixed at 25-30°C, stirred for 10 minutes and then heated at 113-115°C for 45-48 hours. After completion of the reaction, the mixture was cooled to 75-80°C. Ethanol (5 vol) was added at 75-80°C and the mixture was stirred at 75-80°C for 10 minutes. Toluene (10 vol) was added at 70-75°C and the mixture was stirred at 70-75°C for 15 minutes. Purified water (15 vol) was added at 70-75°C and the mixture was stirred at 60-65°C for 30 minutes. The mixture was then cooled and stirred at 25-30°C for 30 minutes. The precipitated solid was filtered and washed with purified water (2 x 5 vol) before drying at 75-80°C under reduced pressure afforded crude dipyridamole as a yellow crystalline solid. Yield (w/w) = 80-85%Yield (molar) = 58-62%HPLC purity > 98%Example 2Stage 1: Preparation of 2-chloro-6-diemanolamino-4,8-dipiperidino-pyrimido(5,4-d) pyrimidineDiethanolamine (3 eq) and 2,6-dichloro-4,8-dipiperidino-pyrimido(5,4-d)pyrimidine (1 eq) were added to dimefhylsulfoxide (10 vol) at 25-30°C, stirred for 10 minutes and then heated at 120-125°C for 4-5 hours. After completion of the reaction, the reaction mixture was cooled to 55-60°C. Acetone (5 vol) was added at 55-60°C and the mixture was stirred at 55-60°C for 10 minutes. Purified water (15 vol) was added at 55-60°C and the mixture was stirred at 50-55°C for 15 minutes. The mixture was cooled to 25-30°C and stirred at 25-30°C for 30 minutes. The precipitated solid was filtered, washed with purified water (2 x 5 vol) and dried at 75-80°C under reduced pressure to afford crude 2-chloro-6- diethanolamino-4,8-dipiperidino-pyrimido(5,4-d)pyrimidine as a yellow crystalline solid. Yield (w/w) = 110-120%Yield (molar) = 93-100%HPLC purity > 96%Stage 2: Preparation of crude dipyridamoleDiethanolamine (10 vol) and 2-chloro-6-diemanolamino-4,8-dipiperidino-pyrimido(5,4-d) pyrimidine (1 eq) were mixed at 25-30°C, stirred for 10 minutes and then heated at 113- 115°C for 45-48 hours. After completion of the reaction, the mixture was cooled to 75- 80°C. Ethanol (5 vol) was added and the mixture was stirred at 75-80°C for 10 minutes. Toluene (10 vol) was added and the mixture was stirred at 70-75°C for 15 minutes. Purified water (15 vol) was added and the mixture was stirred at 60-65°C for 30 minutes. The mixture was then cooled to 25-30°C and stirred for 30 minutes. The precipitated solid was filtered, washed with purified water (2 x 5 vol) and dried at 75-80°C under reduced pressure to afford crude dipyridamole as a yellow crystalline solid.Yield (w/w) = 95-97%Yield (molar) = 82-84%HPLC purity > 98%Example 3Crystallization of crude dipyridamoleCrude dipyridamole (1 eq) and diefhanolamine (8 vol) were stirred together at 25-30°C for 10 minutes and then heated to about 80°C for 10 minutes. The clear solution was cooled to 75-80°C, ethanol (5 vol) was added and the mixture was stirred at 75-80°C for 10 minutes. Toluene (10 vol) was added and the mixture was stirred at 70-75°C for 15 minutes. The mixture was cooled to 25-30°C, stirred at 25-30°C for 10 minutes and filtered. The filtrate was heated to 70-75°C for 10 minutes, purified water (15 vol) was added and the mixture was stirred at 60-65°C for 30 minutes before cooling to 25-30°C with stirring for 30 minutes. The precipitated solid was filtered, washed with purified water (2 x 5 vol) and dried at 75-80°C under reduced pressure to afford dipyridamole as a yellow crystalline solid.Yield (w/w and molar) = 90-95%HPLC purity > 99.9%
PATENT
https://patents.google.com/patent/WO2007080463A1/enDipyridamole which is chemically known as 4,8-Bis(piperidino)-N,N,N’,N’- tetra(2-hydroxyethyl)pyrimido[5,4-d]pyrimidine-2,6-diamine is a platelet adhesion inhibitor. It is useful in anti-platelet therapy and it is marketed as Persantin ® by Boehringer Ingelheim.The platelet aggregation inhibiting properties, anti-thrombotic and vasodilator properties of Dipyridamole is reported in US patent 3031450 which also describe a process for its preparation by reacting 2-chloro-6-diethanolamino-4, 8-dipiperidyl- pyrimido-pyrimidine with diethanolamine.German patent 117456 describes the process for the production of Dipyridamole from 2,6-dichloro-4,8-dipiperidinopyrimido[5,4-d]pyrimidine and diethanolamine at 130 to 200° C under vacuum. German patent 1812918 describes the process for the preparation and purification of Dipyridamole. According to this patent 2,6-dichloro-4,8,- dipiperidinopyrimido[5,4-d]pyrimidine and diethanolamine are heated to 150 to 2000C to obtain Dipyridamole. This is characterized by the fact that after the completion of the reaction, the reaction mixture is dissolved in chloroform, which is further separated into the upper layer of diethanolamine and its hydrochloride and the chloroform solution. Thus obtained chloroform solution is reduced to dryness after stirring with water.RO 104718 Bl describes a process where Dipyridamole is manufactured and purified by reaction of diethanolamine with 2,6-dichloro-4,8-dipiperidinopyrimido[5,4- d]pyrimidine. In this process the yield is very low (58%) and purity is only 98%.Another patent DDl 15670 Z describes a process for the purification of Dipyridamole by refluxing it in AcOBu for 2 h in the presence of an equal amount of silica gel or by column chromatography on silica gel at 60-1000C. The purification by column chromatography is not economical and feasible at industrial scale.All the above mentioned prior art are neat reaction in which it is difficult to control the impurity and is not easy to scale up. In the prior art process the obtained product is pasty which needs decanting the mother liquor and further purification.We focused our research to develop an improved and efficient process for the preparation and purification of Dipyridamole of formula (I) which will overcome the above mentioned prior art problems and will produce Dipyridamole in substantially good yield, high purity and with no mixture of solvents.Objectives of the InventionThe main objective of the present invention is to provide an improved process for the preparation and purification of compound of formula (I), which gives better purity and high yield of the product. Another objective of the present invention is to provide a process for the preparation and purification of compound of formula (I), which would be easy to scale up and implement at industrial level.Yet another objective of the present invention is to provide a process for the preparation and purification of compound of formula (I), which avoids, use of hazardous gas (SO2) and corrosive chemicals like HCl, H2SO4, acetic acid, NaOH, NH3 etc.Summary of the InventionAccordingly, the present invention provides a process for the preparation ofDipyridamole of formula (I) comprising reacting 2,6-Dichloro-4,8- dipiperidinopyrimido- (5,4-d)pyrimidine (DDH) of formula (II) with Diethanolamine (DEA) of formula (III) using a solvent. This process can be represented by the scheme given below:
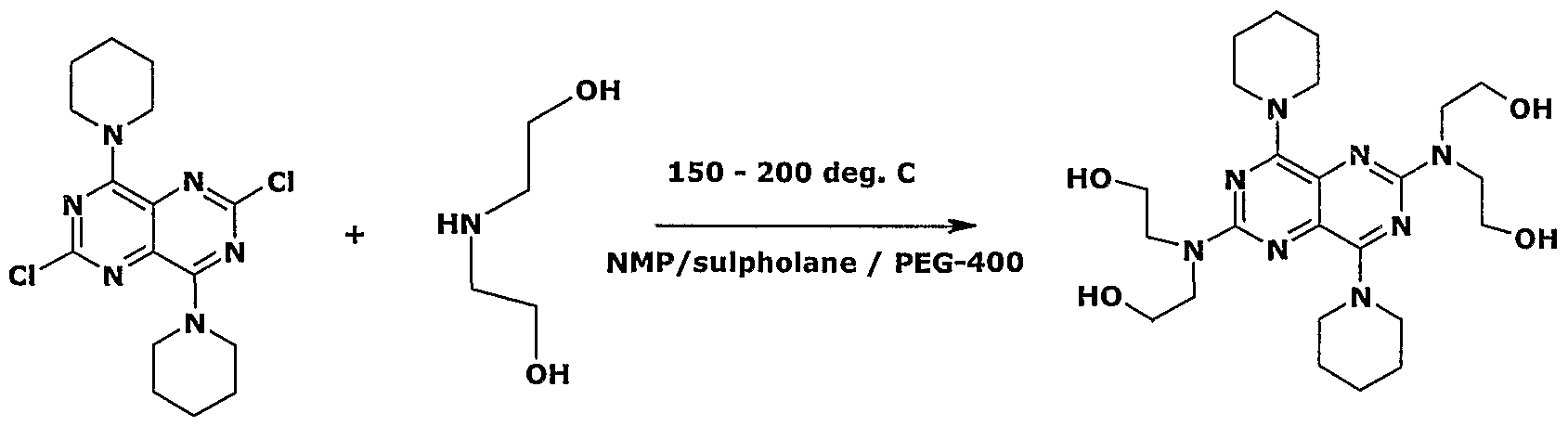
(II) (III) (I)The obtained wet or optionally dried crude Dipyridamole is purified by using ketonic solvent and aqueous alcoholic solvent or mixture thereof.Description of the InventionIn an embodiment of the present invention the solvent is selected from the group consisting of l-Methyl-2-pyrrolidinone, Sulpholane and Polyethylene glycol, preferably l-Methyl-2-pyrrolidinone (NMP). In another embodiment of the present invention, the polyethylene glycol used is PEG-20Q or PEG-400, preferably PEG-400.In yet another embodiment of the present invention, the reaction is carried out at a temperature of about 25° C to reflux temperature, preferably at a temperature of about 150° C to 2000C.In still another embodiment of the present invention the starting material of this invention is prepared according to the literature available in the prior art.In yet another embodiment the ketonic solvent is acetone, methyl ethyl ketone, methyl vinyl ketone or methyl isobutyl ketone (MIBK), preferably MIBK.In yet another embodiment the alcoholic solvent is selected from the group having Ci to C4 alkanol preferably isopropyl alcohol (IPA) or methanol.The present invention is illustrated with the following examples, which should not be construed for limiting the scope of the invention.Example 1Preparation of Dipyridamole (Crude)250 mL of l-Methyl-2-pyrrolidinone (NMP), 50g of 2,6-Dichloro-4,8 dipiperidinopyrimido(5,4-d)pyrimidine (DDH) and 136g of Diethanolamine (DEA) were charged into a 2.0 L four-necked RBF at 25-35 0C. The reaction mass was heated to 190 – 2000C and maintained for 1.5 to 2.5 hrs under stirring. The reaction mass was cooled to 25-350C and 450 mL of purified water was charged slowly into it and stirred for lhr. The solid reaction mass was filtered and washed with 500ml-purified water and dried the solid under vacuum at 50-550C for 8 to 10 hrs to get 55-60 g of crude Dipyridamole of 90-94% HPLC purity.Purification Of DipyridamoleMethyl isobutyl ketone (750 ml) and 50 g of Dipyridamole (crude) were charged into a clean 2.0 L four-necked RBF at 25-3O0C and heated to 100-1200C. It was stirred to dissolve and cooled to 25-350C and stirred for 30-60 min. The solid was Filtered and washed with 100 ml MIBK. The product was dried at 45-50 0C under vacuum. The obtained material was further purified as follows:Isopropyl alcohol (200 mL) and 40-45 g of Dipyridamole were charged into a clean 1.0 lit four-necked RBF at 25-350C. It was heated to 60-650C. Carbon (Ig) was added at 30-350C and filtered through celite and washed with 50 mL IPA. Water (500 mL) was charged slowly and stirred for 30 min. The solid was filtered and washed with a mixture of IPA : Water (1 :2) and dried the product at 45-50 0C under vacuum to obtain 43-5Og of Dipyridamole having HPLC purity 99.0 – 99.5%Example 2Sulpholane (15 mL), 5.0g of 2,6-Dichloro-4,8-dipiperidinopyrimido(5,4-d) pyrimidine (DDH) and 8.6g Diethanolamine (DEA) were charged into 100 mL four- necked RBF at 25-35°C. It was heated to 190-2000C and stirred for 2-3 hrs. The reaction mass was cooled to 25-35°C and 45 mL of water was added into it. The reaction mass was stirred. The solid was filtered and washed with water. The solid was purified with MIBK,IPA-Water as given in example 1.Example 3Polyethylene glycol-400 (15 mL), 5g of 2,6-Dichloro-4,8-dipiperidinopyrimido (5,4-d) pyrimidine (DDH) and 8.6 gm Diethanolamine (DEA) were charged into 10OmL four necked RBF at 25-35°C. The reaction mass was heated to 190-2000C and maintained for 2-3 hrs. The mixture was cooled to 25-350C. Water (45 mL) was added to the reaction mixture and stirred. The solid was filtered and washed with water. The solid was purified with MIBK,IPA- Water as given in example 1.Example 4 (Azeotrophic removal of water in MIBK purification)Preparation of Dipyridamole (Crude*) l-Methyl-2-pyrrolidinone (150 mL), 50g of 2,6-Dichloro-4,8-dipiperidinopyrimido(5,4-d)pyrimidine (DDH) and 136g Diethanolamine (DEA) was charged into a 2.0 L four-necked RBF at 25-35 0C. The reaction mass was stirred & heated to 190 – 2000C and stirring was continued for 1.5 to 2.5 hrs. The reaction mass was cooled to 25-350C and 450 mL of purified water was charged slowly into it and stirred for lhr. The solid reaction mass was filtered and washed with 500ml-purified water to obtain 110-13Og of wet crude Dipyridamole.Purification Of DipyridamoleMethyl isobutyl ketone (750 ml) and 65 g of Dipyridamole (wet crude) were charged into a clean 2.0 L four-necked RBF at 25-3O0C and heated to 100-1200C followed by azeotrophic separation of water. It was cooled to 25-350C and stirred for30-60 min. The solid was filtered and washed with 100 ml MIBK. The product was dried at 45-50 0C under vacuum. The obtained material was further purified as follows:Isopropyl alcohol (200 mL) and 45-48 g of Dipyridamole were charged into a clean 1.0 L four-necked RBF at 25-350C. It was heated to 60-650C and stirred to dissolve. Carbon (Ig) was added at 30-350C and filtered through celite and washed with 50 mL IPA. Water (500 mL) was charged slowly and stirred for 30 min. The solid was filtered and washed with a mixture of IPA : Water (1:2) and dried the product at 45-50 0C under vacuum to obtain 43-5Og of Dipyridamole having HPLC purity 99.0-99.5%Purification with Methaanol-waterMethanol (200 mL) and 45-48 g of Dipyridamole were charged into a clean 1.0 L four-necked RBF at 25-350C. It was heated to 60-650C and stirred to dissolve. Carbon (Ig) was added at 30-350C and filtered through celite and washed with 50 mL methanol. Water (500 mL) was charged slowly and stirred for 30 min. The solid was filtered and washed with a mixture of methanol : Water (1 :2) and dried the product at 45-50 0C under vacuum to obtain 45g of Dipyridamole having HPLC purity 99%.
PATENThttps://patents.google.com/patent/CN108069972A/enEmbodiment 1Weigh urea 120g(2mol), ethylene glycol 62g(1mol)120 DEG C are heated in a kettle, in Catalyzed by p-Toluenesulfonic Acid Effect is lower to carry out step(1)Reaction, generate compound 3;Then 2,3- diamino succinic acid 37g is weighed(0.25mol)With step The compound 3 generated in rapid 1 carries out the reaction generation compound 5 of step 2,220 DEG C, reaction time 3h of reaction temperature, catalyst For the nickel-base catalyst of support type, catalyst is filtered out after the completion of reaction;Exist in phosphorus oxychloride, phosphorus trichloride and lead to chlorine In the case of, compound 5 carries out chlorination reaction generation compound 6,110 DEG C of reaction temperature, reaction time 30h;Weigh piperidines 85g (1mol)Step is carried out with compound(4)Reaction, after reaction liquid hydrolysis, cooling filtering, be dried to obtain compound 9;Claim Take 105g(1mol)Diethanol amine and compound 9 carry out step(6)Reaction, 220 DEG C of reaction temperature, reaction time 3h fills 1.5 MPa of Hydrogen Vapor Pressure, catalyst are the crude product of the nickel-base catalyst, after reaction cold filtration of support type, and crude product passes through It is refining to obtain Dipyridamole finished product, quality 62.10g, purity 99.21%, product yield 48.74%(Yield is with 2,3- diamino fourths Diacid is calculating benchmark).Embodiment 2Weigh urea 120g(2mol), ethylene glycol 62g(1mol)120 DEG C are heated in a kettle, in Catalyzed by p-Toluenesulfonic Acid Effect is lower to carry out step(1)Reaction, generate compound 3;Then 2,3- diamino succinic acid 30g is weighed(0.2mol)With step The compound 3 generated in rapid 1 carries out the reaction generation compound 5 of step 2,220 DEG C, reaction time 3h of reaction temperature, catalyst For the nickel-base catalyst of support type, catalyst is filtered out after the completion of reaction;Exist in phosphorus oxychloride, phosphorus trichloride and lead to chlorine In the case of, compound 5 carries out chlorination reaction generation compound 6,110 DEG C of reaction temperature, reaction time 30h;Weigh piperidines 85g (1mol)Step is carried out with compound(4)Reaction, after reaction liquid hydrolysis, cooling filtering, be dried to obtain compound 9;Claim Take 105g(1mol)Diethanol amine and compound 9 carry out step(6)Reaction, 220 DEG C of reaction temperature, reaction time 3h fills 1.5 MPa of Hydrogen Vapor Pressure, catalyst are the crude product of the nickel-base catalyst, after reaction cold filtration of support type, and crude product passes through It is refining to obtain Dipyridamole finished product, quality 50.34g, purity 99.3%, product yield 49.53%(Yield is with 2,3- diamino fourth two Acid is calculating benchmark).Embodiment 3Other steps are the same as embodiment 2, step(6)In reaction temperature for 240 DEG C, product yield 50.31%.Comparative example 1Weigh urea 36g(0.6mol), ethyl acetoacetate 26g(0.2mol), add in ethanol-hydrogen chloride liquid(30% hydrochloric acid:95% second Alcohol=1:4)Then 200ml, drying and dehydrating after stirring add in sodium hydroxide solution and are warming up to 95 DEG C, then cool to 75 DEG C, add Hydrochloric acid adjusts PH=1, cold filtration, the 6- methyluracils of washing filtering;Nitric acid is added in reaction pot, is cooled to less than 10 DEG C, Stirring adds in 6- methyluracils, be warming up to 30 DEG C of heat preservations 1 it is small when the nitro whey liquid that filters;Take a policy powder in water, stirring After dissolving plus nitro whey liquid, temperature control keep the temperature 30min at 35 DEG C, add the static 3h of hydrochloric acid, stir 2h, filtration drying obtains amino breast Clear liquid;Again weigh urea 36g and add in reaction kettle with amino whey liquid, stirring is warming up to 100 DEG C of heat preservation 20min, cools to 90 DEG C add in 2mol/L sodium hydroxide solution, be warming up to 100 DEG C heat preservation dissolving 1h.Cool to 40 DEG C, filter tetrahydroxy pyrimidine- [4,5d] and pyrimidine sodium salt, adds water, 60 DEG C of heat preservation 30min add hydrochloric acid to adjust PH=4, is cooled to 15 DEG C of filterings, washing, dries 2,4,6,8- tetrahydroxys pyrimidine-[4,5d] and pyrimidine;Tetrahydroxy object, phosphorus oxychloride, phosphorus trichloride are added in reaction kettle, is stirred, 10 DEG C of similarly hereinafter chlorine are warming up to 110 DEG C of reflux for 24 hours, be cooled to 15 DEG C of filterings, washing, dry 2,4,6,8- tetrachloro-pyrimidines- [4,5d] and pyrimidine;Acetone, tetrachloride are sequentially added, piperidines-acetone mixture, 30 DEG C of heat preservations are added dropwise in 20 DEG C of heat preservation 30min 1h, adds water to stir 1h, and filtration drying obtains 2,6- bis- chloro- 4,8 ,-two piperidines-pyrimidine(Dichloride);Weigh diethanol amine 63g with Dichloride is mixed, and is warming up to 200 DEG C of heat preservation 15min, is cooled to less than 25 DEG C plus acetone and stirs 30min, then 30 DEG C 4h is kept the temperature, filtration drying obtains Dipyridamole crude product, then carries out refined Dipyridamole finished product 13.53, purity 98.5%, yield 13.21%(Using ethyl acetoacetate as calculating standard).It is found by being compared with comparative example:The method of the production Dipyridamole of the present invention is short with synthetic route, into The characteristics of product high income, production cost is low.
SYN
GB 807826 U.S. Patent 3,031,450

SYN
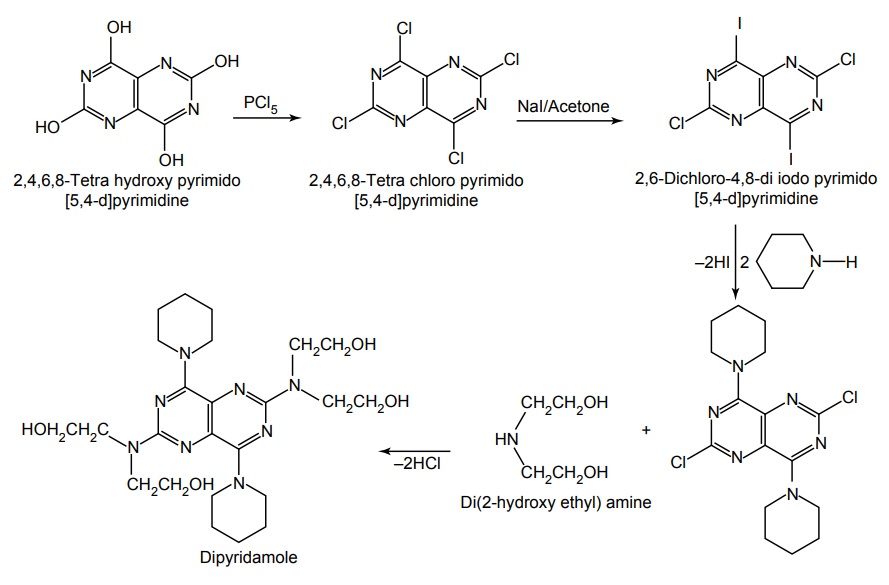
SYN
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Dipyridamole
Dipyridamole, 2,2′,2″,2′″-[(4,8-dipiperidinopirimido[5,4-d]pirimidin-2,6-diyl)-diimino]-tetraethanol (19.4.13), is easily synthesized from 5-nitroorotic acid (19.4.8), easily obtained, in turn, by nitrating of 2,4-dihydroxy-6-methylpyrimidine, which is usually synthesized by the condensation of urea with acetoacetic ether. Reduction of the nitro group in 5-nitroorotic acid by various reducing agents gives 5-aminoorotic acid (19.4.9), which is reacted with urea or with potassium cyanide to give 2,4,6,8-tetrahydroxypyrimido[5,4-d]pyrimidine (19.4.10). This undergoes a reaction with a mixture of phosphorous oxychloride and phosphorous pentachloride, which forms 2,4,6,8- tetra-chloropyrimido[5,4-d]pyrimidine (19.4.11). Reacting the resulting tetrachloride with piperidine replaces the chlorine atoms at C4 and C8 of the heterocyclic system with piperidine, giving 2,6-dichloropyrimido-4,8-dipiperidino[5,4-d]pyrimidine (19.4.12). Reacting the resulting product with diethanolamine gives dipyridamole (19.4.13) [32,33].

Dipyridamole increases coronary blood circulation, increases oxygen flow to the myocardium, potentiates adenosine activity, and impedes its metabolization. It inhibits aggregation of thrombocytes, blocks phosphodiesterase, increases microcirculation, and inhibits the formation of thrombocytes.
It is used for chronic coronary insufficiency, as well as for preventing and treating thrombosis. Synonyms of this drug are anginal, curantyl, stenocor, thrompresantin, and many others.SYN
Chemical Synthesis
Dipyridamole, 2,2′,2”,2”’-[(4,8-dipiperidinopirimido[5,4-d]pirimidin-2,6- diyl)-diimino]-tetraethanol (19.4.13), is easily synthesized from 5-nitroorotic acid (19.4.8), easily obtained, in turn, by nitrating of 2,4-dihydroxy-6-methylpyrimidine, which is usually synthesized by the condensation of urea with acetoacetic ether. Reduction of the nitro group in 5-nitroorotic acid by various reducing agents gives 5-aminoorotic acid (19.4.9), which is reacted with urea or with potassium cyanide to give 2,4,6,8- tetrahydroxypyrimido[5,4-d]pyrimidine (19.4.10). This undergoes a reaction with a mixture of phosphorous oxychloride and phosphorous pentachloride, which forms 2,4,6,8- tetrachloropyrimido[ 5,4-d]pyrimidine (19.4.11). Reacting the resulting tetrachloride with piperidine replaces the chlorine atoms at C4 and C8 of the heterocyclic system with piperidine, giving 2,6-dichloropyrimido-4,8-dipiperidino[5,4-d]pyrimidine (19.4.12). Reacting the resulting product with diethanolamine gives dipyridamole (19.4.13).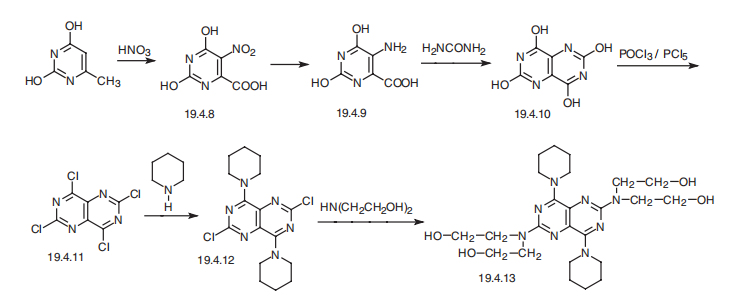
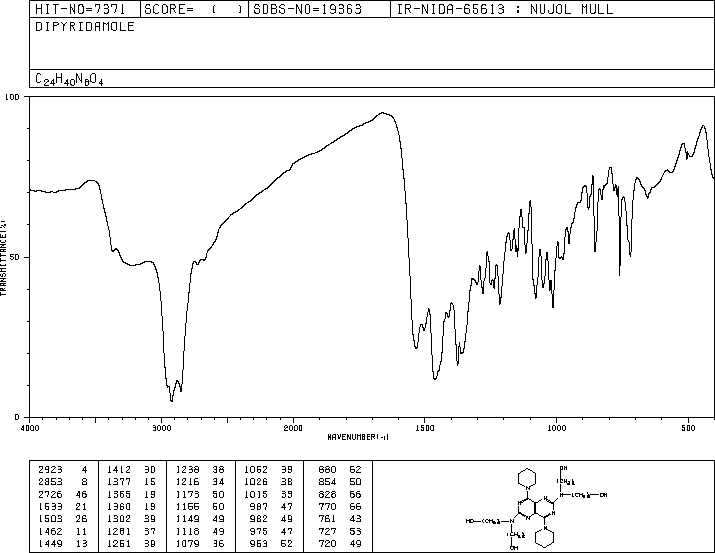
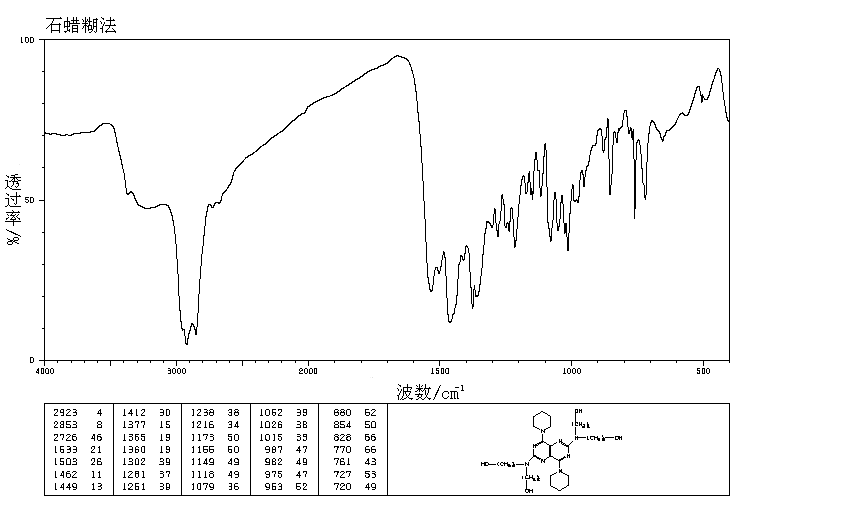
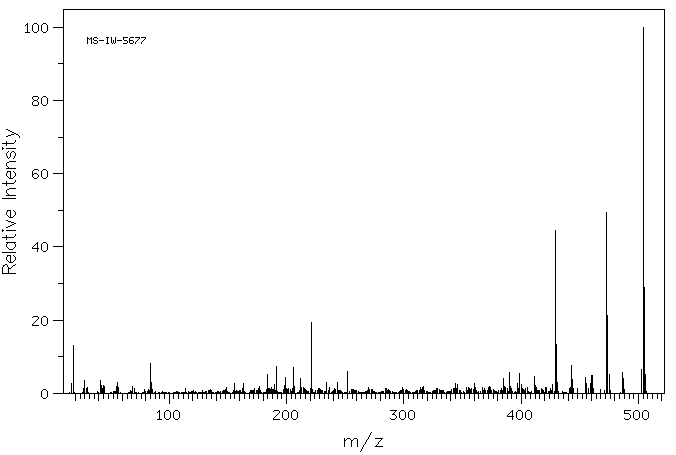
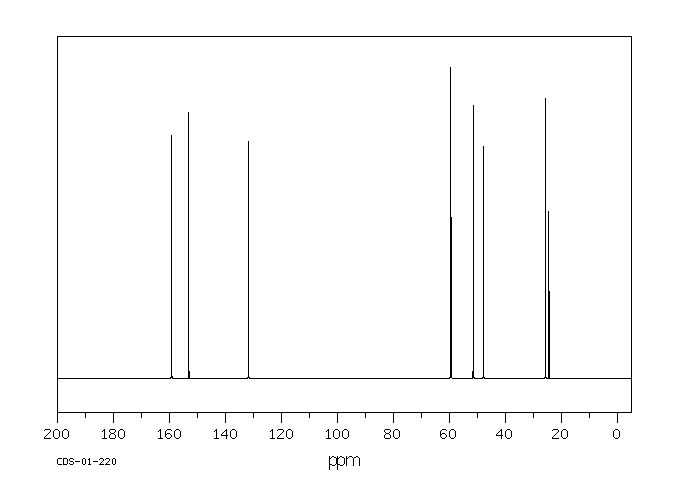
clipA general outline of the procedure for synthesizing dipyridamole is shown in Scheme 1. Reaction of the pyrimidino pyrimidine-2,4,6,8- tetraol (1) with a mixture of phosphorous oxychloride and phosphorous pentachloride gives the tetrachloro derivative (2). The halogens at the peri positions 4 and 8 are more reactive to substitution than are the remaining halogen pairs 2 and 6, which are in effect the two positions of the pyrimidines. Thus, reaction with piperidine at ambient temperature gives the 4, 8 diamine (3). Subsequent reaction with bis-2-hydroxy ethylamine under more strenuous conditions gives dipyridamole (4) [12, 13].12. F.G. Fischer, J. Roch, and A. Kottler, U.S. Patent 3, 031, 450 (1962). 13. D. Lednicer and L.A. Mitscher, The Organic Chemistry of Drug Synthessis Volume 1, John Wiley and Sons, New York, p. 428 (1977).
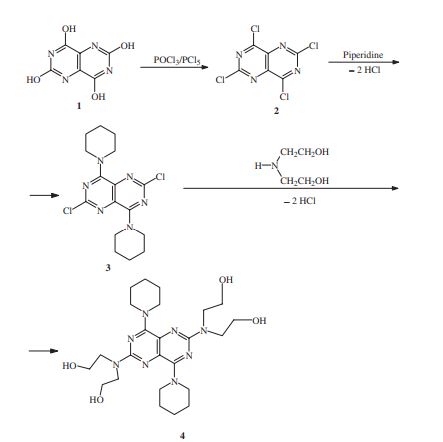
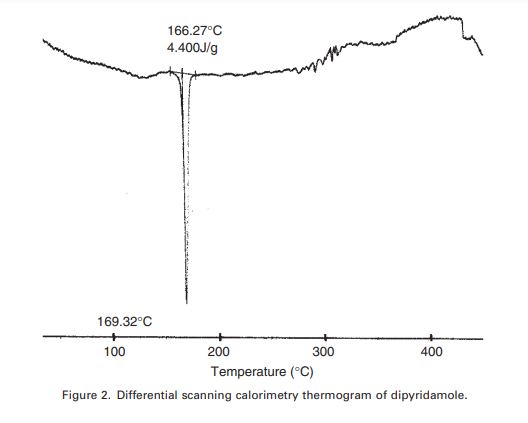
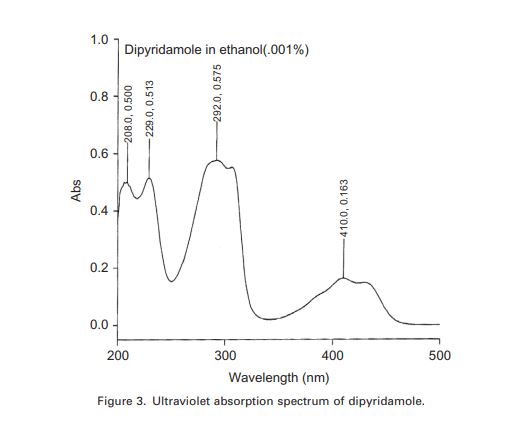
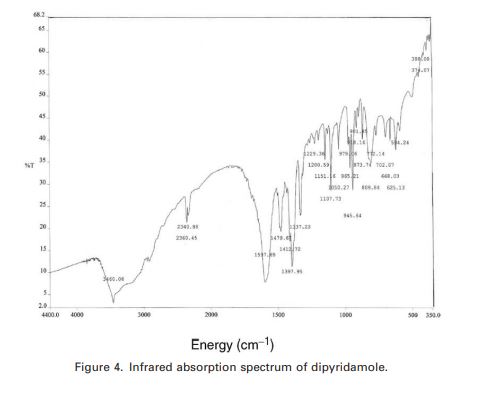
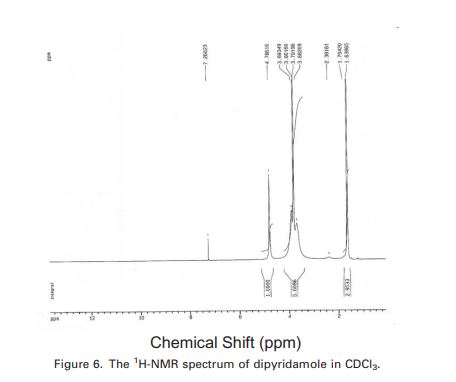
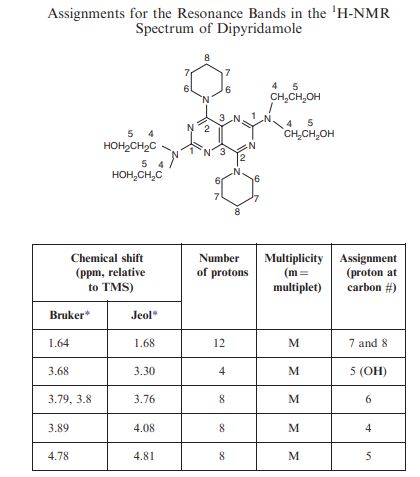
set 2
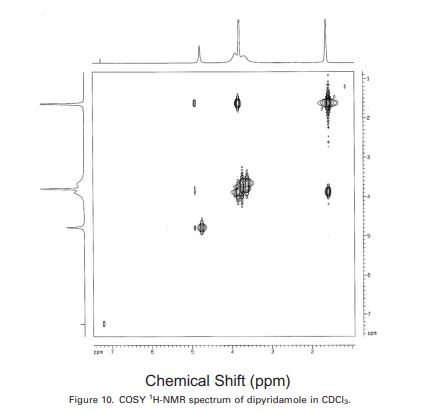
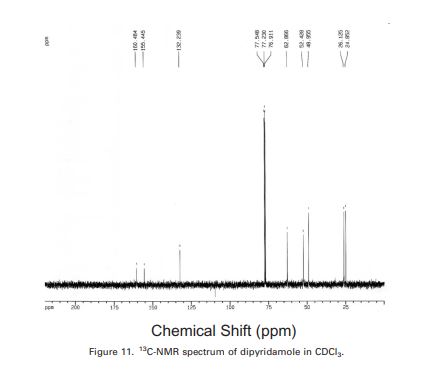
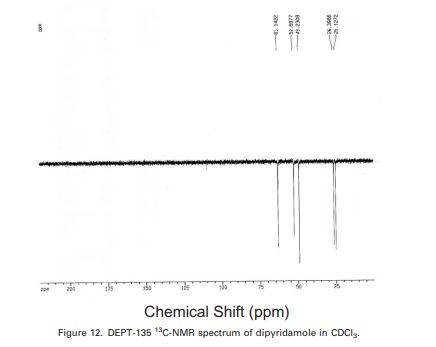
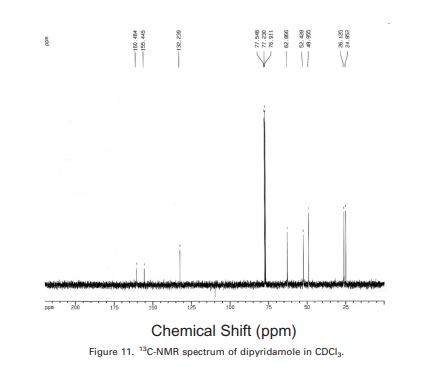
set 3
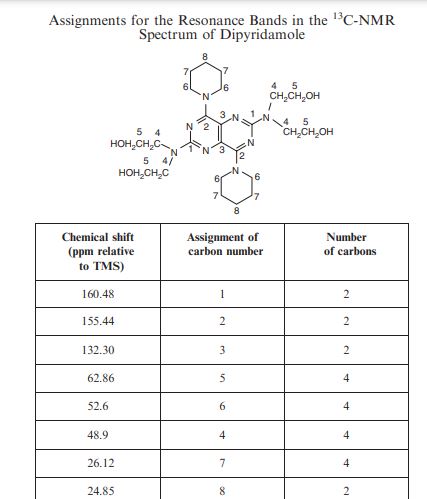
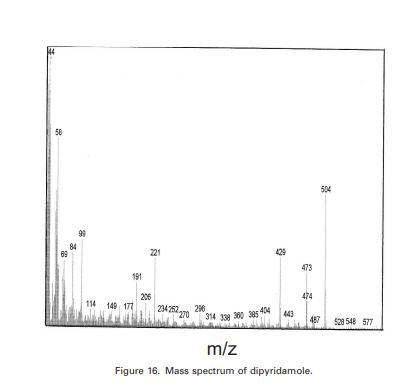

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
- Dipyridamole is used to dilate blood vessels in people with peripheral arterial disease and coronary artery disease[4]
- Dipyridamole has been shown to lower pulmonary hypertension without significant drop of systemic blood pressure
- It inhibits formation of pro-inflammatory cytokines (MCP-1, MMP-9) in vitro and results in reduction of hsCRP[clarification needed] in patients.
- It inhibits proliferation of smooth muscle cells in vivo and modestly increases unassisted patency of synthetic arteriovenous hemodialysis grafts.[5]
- It increases the release of tissue plasminogen activator from brain microvascular endothelial cells.
- It results in an increase of 13-hydroxyoctadecadienoic acid and decrease of 12-hydroxyeicosatetraenoic acid in the subendothelial matrix and reduced thrombogenicity of the subendothelial matrix.
- Pretreatment it reduced reperfusion injury in volunteers.
- It has been shown to increase myocardial perfusion and left ventricular function in patients with ischemic cardiomyopathy.
- It results in a reduction of the number of thrombin and PECAM-1 receptors on platelets in stroke patients.
- Cyclic adenosine monophosphate impairs platelet aggregation and also causes arteriolar smooth muscle relaxation. Chronic therapy did not show significant drop of systemic blood pressure.
- It inhibits the replication of mengovirus RNA.[6]
- It can be used for myocardial stress testing as an alternative to exercise-induced stress methods such as treadmills.
Stroke
A combination of dipyridamole and aspirin (acetylsalicylic acid/dipyridamole) is FDA-approved for the secondary prevention of stroke and has a bleeding risk equal to that of aspirin use alone.[4] Dipyridamole absorption is pH-dependent and concomitant treatment with gastric acid suppressors (such as a proton pump inhibitor) will inhibit the absorption of liquid and plain tablets.[7][8] Modified release preparations are buffered and absorption is not affected.[9][10]
However, it is not licensed as monotherapy for stroke prophylaxis, although a Cochrane review suggested that dipyridamole may reduce the risk of further vascular events in patients presenting after cerebral ischemia.[11]
A triple therapy of aspirin, clopidogrel, and dipyridamole has been investigated, but this combination led to an increase in adverse bleeding events.[12]
- Vasodilation occurs in healthy arteries, whereas stenosed arteries remain narrowed. This creates a “steal” phenomenon where the coronary blood supply will increase to the dilated healthy vessels compared to the stenosed arteries which can then be detected by clinical symptoms of chest pain, electrocardiogram and echocardiography when it causes ischemia.
- Flow heterogeneity (a necessary precursor to ischemia) can be detected with gamma cameras and SPECT using nuclear imaging agents such as Thallium-201, Tc99m–Tetrofosmin and Tc99m–Sestamibi. However, relative differences in perfusion do not necessarily imply any absolute decrease in blood supply in the tissue supplied by a stenosed artery.
Other uses
Dipyridamole also has non-medicinal uses in a laboratory context, such as the inhibition of cardiovirus growth in cell culture.[citation needed]
Drug interactions
Due to its action as a phosphodiesterase inhibitor, dipyridamole is likely to potentiate the effects of adenosine. This occurs by blocking the nucleoside transporter (ENT1) through which adenosine enters erythrocyte and endothelial cells.[13]
According to Association of Anaesthetists of Great Britain and Ireland 2016 guidelines, dipyridamole is considered to not cause risk of bleeding when receiving neuroaxial anaesthesia and deep nerve blocks. It does not therefore require cessation prior to anaesthesia with these techniques, and can continue to be taken with nerve block catheters in place.[14]
Overdose
Dipyridamole overdose can be treated with aminophylline[2]: 6 or caffeine which reverses its dilating effect on the blood vessels. Symptomatic treatment is recommended, possibly including a vasopressor drug. Gastric lavage should be considered. Since dipyridamole is highly protein bound, dialysis is not likely to be of benefit.
Mechanisms of action
Dipyridamole has two known effects, acting via different mechanisms of action:
- Dipyridamole inhibits the phosphodiesterase enzymes that normally break down cAMP (increasing cellular cAMP levels and blocking the platelet aggregation, response[4] to ADP) and/or cGMP.
- Dipyridamole inhibits the cellular reuptake of adenosine into platelets, red blood cells, and endothelial cells, leading to increased extracellular concentrations of adenosine.
Experimental studies[
Dipyridamole is currently undergoing repurposing for treatment of ocular surface disorders. These include pterygium and dry eye disease. The first report of topical dipyridamole’s benefit in treating pterygium was published in 2014.[15] A subsequent report of outcomes in 25 patients using topical dipyridamole was presented in 2016.[16]
See also
References
- ^ Nielsen-Kudsk, F; Pedersen, AK (May 1979). “Pharmacokinetics of Dipyridamole”. Acta Pharmacologica et Toxicologica. 44 (5): 391–9. doi:10.1111/j.1600-0773.1979.tb02350.x. PMID 474151.
- ^ Jump up to:a b “Aggrenox (aspirin/extended-release dipyridamole) Capsules. Full Prescribing Information” (PDF). Boehringer Ingelheim Pharmaceuticals, Inc. Retrieved 1 December 2016.
- ^ “Dipyridamole” at Dorland’s Medical Dictionary
- ^ Jump up to:a b c Brown DG, Wilkerson EC, Love WE (March 2015). “A review of traditional and novel oral anticoagulant and antiplatelet therapy for dermatologists and dermatologic surgeons”. Journal of the American Academy of Dermatology. 72 (3): 524–34. doi:10.1016/j.jaad.2014.10.027. PMID 25486915.
- ^ Dixon BS, Beck GJ, Vazquez MA, et al. (2009). “Effect of dipyridamole plus aspirin on hemodialysis graft patency”. N Engl J Med. 360 (21): 2191–2201. doi:10.1056/nejmoa0805840. PMC 3929400. PMID 19458364.
- ^ Dipyridamole in the laboratory: Fata-Hartley, Cori L.; Ann C. Palmenberg (2005). “Dipyridamole reversibly inhibits mengovirus RNA replication”. Journal of Virology. 79 (17): 11062–11070. doi:10.1128/JVI.79.17.11062-11070.2005. PMC 1193570. PMID 16103157.
- ^ Russell TL, Berardi RR, Barnett JL, O’Sullivan TL, Wagner JG, Dressman JB. pH-related changes in the absorption of “dipyridamole” in the elderly. Pharm Res (1994) 11 136–43.
- ^ Derendorf H, VanderMaelen CP, Brickl R-S, MacGregor TR, Eisert W. “Dipyridamole” bioavailability in subjects with reduced gastric acidity. J Clin Pharmacol (2005) 45, 845–50.
- ^ “Archived copy”. Archived from the original on 2009-07-05. Retrieved 2010-02-06.
- ^ Stockley, Ivan (2009). Stockley’s Drug Interactions. The Pharmaceutical Press. ISBN 978-0-85369-424-3.
- ^ De Schryver EL, Algra A, van Gijn J (2007). Algra A (ed.). “Dipyridamole for preventing stroke and other vascular events in patients with vascular disease”. Cochrane Database of Systematic Reviews (2): CD001820. doi:10.1002/14651858.CD001820.pub3. PMID 17636684.
- ^ Sprigg N, Gray LJ, England T, et al. (2008). Berger JS (ed.). “A randomised controlled trial of triple antiplatelet therapy (aspirin, clopidogrel and dipyridamole) in the secondary prevention of stroke: safety, tolerability and feasibility”. PLOS ONE. 3 (8): e2852. Bibcode:2008PLoSO…3.2852S. doi:10.1371/journal.pone.0002852. PMC 2481397. PMID 18682741.
- ^ Gamboa A, Abraham R, Diedrich A, Shibao C, Paranjape SY, Farley G, et al. Role of adenosine and nitric oxide on the mechanisms of action of dipyridamole. Stroke. 2005;36(10):2170-2175.
- ^ AAGBI Guidelines Neuraxial and Coagulation June 2016
- ^ Carlock, Beth H.; Bienstock, Carol A.; Rogosnitzky, Moshe (2014-03-25). “Pterygium: Nonsurgical Treatment Using Topical Dipyridamole – A Case Report”. Case Reports in Ophthalmology. 5 (1): 98–103. doi:10.1159/000362113. ISSN 1663-2699. PMC 3995373. PMID 24761148.
- ^ “Topical Dipyridamole for Treatment of Pterygium and Associated Dry Eye Symptoms: Analysis of User-Reported Outcomes”. ResearchGate. Retrieved 2019-05-19.
| Clinical data | |
|---|---|
| Trade names | Persantine, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682830 |
| Pregnancy category | B |
| Routes of administration | By mouth, IV |
| ATC code | B01AC07 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 37–66%[1] |
| Protein binding | ~99% |
| Metabolism | Liver (glucuronidation)[2] |
| Elimination half-life | α phase: 40 min, β phase: 10 hours |
| Excretion | Biliary (95%), urine (negligible) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 58-32-2 |
| PubChem CID | 3108 |
| IUPHAR/BPS | 4807 |
| DrugBank | DB00975 |
| ChemSpider | 2997 |
| UNII | 64ALC7F90C |
| KEGG | D00302 |
| ChEBI | CHEBI:4653 |
| ChEMBL | ChEMBL932 |
| CompTox Dashboard (EPA) | DTXSID6040668 |
| ECHA InfoCard | 100.000.340 |
| Chemical and physical data | |
| Formula | C24H40N8O4 |
| Molar mass | 504.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESOCCN(CCO)C(N=C1N2CCCCC2)=NC3=C1N=C(N(CCO)CCO)N=C3N4CCCCC4 | |
| showInChI | |
| (verify) |
Patent
Publication numberPriority datePublication dateAssigneeTitleUS3031450A1959-04-301962-04-24Thomae Gmbh Dr KSubstituted pyrimido-[5, 4-d]-pyrimidinesDE1812918A11968-04-251969-11-06Dresden Arzneimittel2,6-Bis (diethanolamino-4,8-dipiperidino-pyrimido (5,4-d)-pyrimidine – purification by simple procedure giving good yieldsDD115670A11974-02-191975-10-12DD117456A11975-02-131976-01-12DE2927539A1 *1979-07-071981-01-08Margineanu Dan Axente Dipl IngBis:di:ethanol-amino-di:piperidino-pyrimido-pyrimidine prepn. – from methyl acetoacetate and urea via amino-orotic acidRO104718B11989-08-091994-09-30Medicamente DePRODUCTION METHOD OF PURE 2,6-bis-(DIETHANOL AMIDE)-4,8-DI- PIPERIDINE-PYRIMIDO-(5,4-d)-PYRIMIDINEWO2007080463A12006-01-122007-07-19Orchid Chemicals & Pharmaceuticals LimitedAn improved process for the preparation of dipyridamoleFamily To Family CitationsDE115670C *JPS5191295A *1975-02-051976-08-10Jipiridamooruno kairyoseizohoJPS5757038B2 *1977-09-301982-12-02Yamanouchi Pharma Co LtdJPS57209291A *1981-06-171982-12-22Kyowa Hakko Kogyo Co LtdPurification of dipyridamoleUS6232312B1 *1995-06-072001-05-15Cell Pathways, Inc.Method for treating patient having precancerous lesions with a combination of pyrimidopyrimidine derivatives and esters and amides of substituted indenyl acetic acidesCN1425461A *2003-01-032003-06-25贵州益佰制药股份有限公司Injection preparation for resisting platelet aggregation and its producing methodCN1634085A *2004-11-242005-07-06崔晓廷Injectio of aspirin and dipyridamole and its preparing process
Non-Patent
TitleCURTIN, NICOLA J. ET AL: “Resistance-Modifying Agents of Pyrimido[5,4-d]pyrimidine Modulators of Antitumor Drug Activity. Synthesis and Structure-Activity Relationships for Nucleoside Transport Inhibition and Binding to .alpha.1-Acid Glycoprotein”, JOURNAL OF MEDICINAL CHEMISTRY , 47(20), 4905-4922 CODEN: JMCMAR; ISSN: 0022-2623, 26 August 2004 (2004-08-26), XP002651697 *
CN104710431B *2015-03-182017-03-01常州康普药业有限公司A kind of purifying process of dipyridamoleCN107782805B *2016-08-252021-02-02亚宝药业集团股份有限公司HPLC analysis method for key intermediate impurity synthesized by dipyridamoleCN106380471B *2016-08-312018-11-06广州市桐晖药业有限公司A kind of preparation method of DipyridamoleCN108069972A *2016-11-162018-05-25湖南尔康制药股份有限公司A kind of production method of Dipyridamole bulk pharmaceutical chemicalsCN106946887B *2017-03-242019-05-28大连万福制药有限公司A kind of preparation method introducing catalyst optimization synthesis Dipyridamole
/////////////////Dipyridamole, дипиридамол , ديبيريدامول , 双嘧达莫 , 0068373 , NSC-515776, RA-8
OCCN(CCO)C(N=C1N2CCCCC2)=NC3=C1N=C(N(CCO)CCO)N=C3N4CCCCC4

NEW DRUG APPROVALS
ONE TIME
$10.00
Racecadotril


Racecadotril
- Molecular FormulaC21H23NO4S
- Average mass385.477 Da
(±)-Acetorphan
(RS)-Benzyl N-[3-(acetylthio)-2-benzylpropanoyl]glycinate
2-{[2-[(acetylthio)methyl]-1-oxo-3-phenylpropyl]amino}acetic acid (phenylmethyl) ester7378
76K53XP4TO
81110-73-8[RN]
Benzyl N-[3-(acetylsulfanyl)-2-benzylpropanoyl]glycinate [ACD/IUPAC Name]
Cadotril
Dexecadotril[INN]
Glycine, N-[3-(acetylthio)-1-oxo-2-(phenylmethyl)propyl]-, phenylmethyl ester
Hidrasec [Trade name]
рацекадотрил[Russian][INN]
راسيكادوتريل[Arabic][INN]
消旋卡多曲[Chinese][INN]
RacecadotrilCAS Registry Number: 81110-73-8
CAS Name:N-[2-[(Acetylthio)methyl]-1-oxo-3-phenylpropyl]glycine phenylmethyl ester
Additional Names:N-[(R,S)-3-acetylthio-2-benzylpropanoyl]glycine benzyl ester; acetorphan
Trademarks: Hidrasec (GSK); Tiorfan (Bioprojet)
Molecular Formula: C21H23NO4S, Molecular Weight: 385.48
Percent Composition: C 65.43%, H 6.01%, N 3.63%, O 16.60%, S 8.32%
Literature References: Antisecretory enkephalinase inhibitor. Prepn: B. Roques et al.,EP38758 (1981); eidem,US4513009 (1985 to Bioprojet). Pharmacology: J.-M. Lecomte et al.,J. Pharmacol. Exp. Ther.237, 937 (1986). Effect on intestinal transit: J. F. Bergmann et al.,Aliment. Pharmacol. Ther.6, 305 (1992). Clinical trial in acute diarrhea: P. Baumer et al.,Gut33, 753 (1992); in children: E. Salazar-Lindo et al.,N. Engl. J. Med.343, 463 (2000). Symposium on pharmacology and clinical experience: Aliment. Pharmacol. Ther.13, Suppl. 6, 1-32 (1999). Review of clinical development: J.-C. Schwartz, Int. J. Antimicrob. Agents14, 75-79 (2000); J. M. Lecomte, ibid. 81-87.
Properties: White crystals from ether mp 89°., Melting point: mp 89°
Derivative Type: (S)-Form
CAS Registry Number: 112573-73-6
Additional Names: Ecadotril; sinorphan
Manufacturers’ Codes: Bay-y-7432
Molecular Formula: C21H23NO4S, Molecular Weight: 385.48
Percent Composition: C 65.43%, H 6.01%, N 3.63%, O 16.60%, S 8.32%
Literature References: Prepn: P. Duhamel et al.,EP318377; eidem,US5208255 (1989, 1993 both to Bioprojet); and pharmacology: B. Giros et al.,J. Pharmacol. Exp. Ther.243, 666 (1987). Clinical effect on plasma ANP levels in CHF: J. C. Kahn et al.,Lancet335, 118 (1990); on renal function: F. Schmitt et al.,Am. J. Physiol.267, F20 (1994). Clinical trial in heart failure: C. M. O’Connor et al.,Am. Heart J.138, 1140 (1999); J. G. F. Cleland, K. Swedberg, Lancet351, 1657 (1998).
Properties: mp 71°. [a]D25 -24.1° (c = 1.3 in methanol). LD50 i.v. in mice: >100 mg/kg (Duhamel, 1993).
Melting point: mp 71°
Optical Rotation: [a]D25 -24.1° (c = 1.3 in methanol)
Toxicity data: LD50 i.v. in mice: >100 mg/kg (Duhamel, 1993)
Therap-Cat: Antidiarrheal.
Keywords: Antidiarrheal; Neutral Endopeptidase Inhibitor.
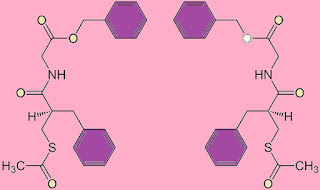
Racecadotril is an anti-secretory enkephalinase inhibitor useful in the treatment of diarrhea.Racecadotril has been investigated for the basic science and treatment of Diarrhea, Acute Diarrhea, and Acute Gastroenteritis.
Racecadotril, also known as acetorphan, is an antidiarrheal medication which acts as a peripheral enkephalinase inhibitor.[3] Unlike other opioid medications used to treat diarrhea, which reduce intestinal motility, racecadotril has an antisecretory effect — it reduces the secretion of water and electrolytes into the intestine.[3] It is available in France (where it was first introduced in ~1990) and other European countries (including Germany, Italy, the United Kingdom, Spain, Portugal, Poland, Finland, Russia and the Czech Republic) as well as most of South America and some South East Asian countries (including China, India and Thailand), but not in the United States. It is sold under the tradename Hidrasec, among others.[4] Thiorphan is the active metabolite of racecadotril, which exerts the bulk of its inhibitory actions on enkephalinases.[5]
Medical uses
Racecadotril is used for the treatment of acute diarrhea in children and adults and has better tolerability than loperamide, as it causes less constipation and flatulence.[6][7] Several guidelines have recommended racecadotril use in addition to oral rehydration treatment in children with acute diarrhea.[8]
Contraindications
Racecadotril has no contraindications apart from known hypersensitivity to the substance.[9][10]
There is insufficient data for the therapy of chronic diarrhea, for patients with renal or hepatic failure, and for children under three months. Additional contraindications for the children’s formulation are hereditary fructose intolerance, glucose-galactose malabsorption and saccharase deficiency, as it contains sugar.[7][9]
Racecadotril (CAS NO.: 81110-73-8), with its systematic name of Glycine, N-(2-((acetylthio)methyl)-1-oxo-3-phenylpropyl)-, phenylmethyl ester, (+-)-, could be produced through many synthetic methods.
Following is one of the synthesis routes: 2-Benzylacrylic acid (I) reacts with SOCl2 in hot toluene to afford the acyl chloride (II), which is condensed with N-tosylglycine benzyl ester (III) in the presence of TEA in toluene to yield the corresponding amide (IV). Finally, this compound is condensed with thioacetic acid by heating at 80 °C to afford the target acylthio compound.

{kind=link}
{kind=link}
{kind=link}
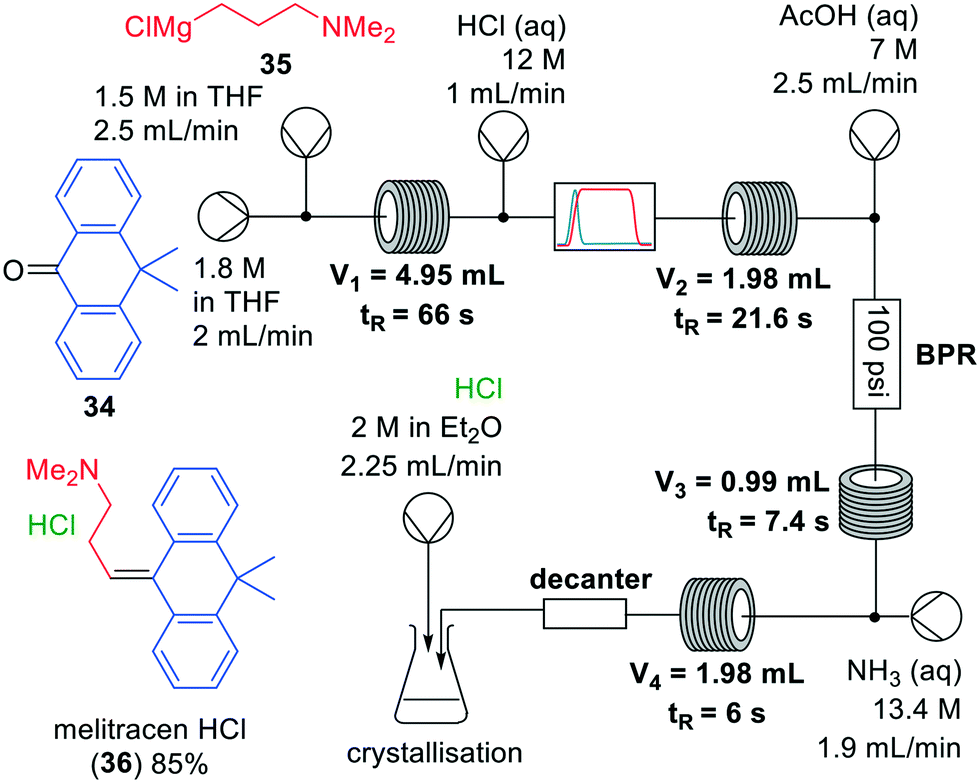{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Racecadotril is a neutral endopeptidase inhibitor used as antidiarrheal in the treatment of chronic cardiac insufficiency and is available under the brand names Hidrasec and Tiorfan. Racecadotril is chemically known as N-[2-[(acetylthio) methyl]- l-oxo-3-phenylpropyl] glycine phenyl methyl ester, (herein after referred by its generic name racecadotril) and represented by the formula (I).

U.S. Patent No. US 4,513,009 describes amino acid derivatives including racecadotril, a pharmaceutical composition and a method of treatment.
The US’009 patent also discloses a process for the preparation of racecadotril which is illustrated by below scheme:

U.S. Patent No. US 6,835,851 B2 discloses a process for the preparation of racecadotril which is illustrated by scheme below:
European Patent No. EP 0501870B 1 discloses a process for the preparation of racec

Racecadotril
The use of coupling agents like hydroxyl benzotriazole (HOBT) and dicyclohexyl amine carbodiimide (DCC) generally induces the formation of side products such as dicyclohexylurea. These side products do lead to major problems, wherein purification by chromatography may be contemplated, but the side products are extremely difficult to remove on an industrial scale.
Consequently, efforts have been made to replace the peptidic coupling step
so as to avoid the formation of side products associated with the use of the coupling agents. Thus, it appears that, even if the preparation of N-(mercaptoacyl)amino acid derivatives from .alpha.-substituted acrylic acids by Michael addition of a thio acid and conversion of acid to acid chloride by using thionyl chloride and then coupling of an amino ester may be advantageous on a laboratory scale, such reactions are difficult to adapt on an industrial use.
The aforementioned processes described above involves expensive reagents such as hydroxyl benzotriazole (HOBT) and dicyclohexyl amine carbodiimide (DCC) and hazardous reagent like thionyl chloride thus rendering the processes expensive and not feasible on industrial scale.
SYNTHESIS BY WORLDDRUGTRACKER
Patent
https://www.google.com/patents/WO2013098826A1?cl=en
EXAMPLES
Example-1: Preparation of Racecadotril (I):
Step A) Preparation of 2-acetyIsulfanyI methyI-3-phenyI propionic acid (IV)
16.2 g of 2-benzylacrylic acid and 12.3 ml of thioacetic acid were were charged into a clean and dry R.B.flask and stirred at about 30°C for about 1 hour. The reaction mixture was heated to about 60°C and stirred for about 4 hours.The excess of thioacetic acid was distilled off completely to afford the title compound as residue. Yield: 23.8 g. Step B) Preparation of Racecadotril crude (la)
23.8 g. of 2-acetyl sulfanylmethyl-3-phenyl-propionic acid (IV), 200ml of methylene chloride and 16.7 ml of triethylamine were charged into a clean and dry R.B.flask. 10. 5 ml of ethylchloroformate was added at about -5°C. The resultant reaction mixture was stirred at about 0°C for about 30 min. 33.7 g of glycine benzyl ester p-tosyalte (II), 14 ml of triethylamine and 100ml of methylene chloride was added as a mixture to the reaction mass at about 0°C. Then the resultant reaction mixture was stirred at about 0°C for about 1 hr. followed by at about 30°C for about 30 min. After completion of the reaction as determined by TLC, the reaction mass was washed with 65 ml of distilled water, 65 ml 4% sodium bicarbonate solution and followed by 65 ml distilled water. The organic and aqueous phases were separated and the solvent was distilled completely, 2 x 50 ml Isopropyl alcohol was charged and again distilled off the solvent completely to give residue. The residue
obtained was triturated with a mixture of isopropyl alcohol 4 ml) and n-hexane (94 ml) at about 5°C to the title compound as crude. Yield: 34 g.
ExampIe-2: Purification of Racecadotril (Crude):
34 g. of crude Racecadotril and 35 ml of 20 % v/v aqueous .methanol were charged in a clean and dry R.B.flask and heated to about 65°C. 3g. of SP.carbon was charged and stirred at about 65°C for about 10 min. The reaction suspension was stirred at about 65°C for about 10 min. The reaction suspension was filtered on hyflow bed (diatomous earth) and washed the hyflow bed with 30 ml of aqueous methanol. The filtrate obtained was cooled to about 0°C for about 30 min. The solid separated was filtered and the solid obtained washed with 60 ml of precooled aqueous methanol to afford the pure racecadotril (I).
Yield: 29 g.; Purity by HPLC: 99.5 area %; The overall yield is 75.3%.
PATENT
https://www.google.com/patents/CN104356036A?cl=en
Example 1
The 40. 0g Racecadotril dissolved in 200ml of absolute ethanol and water bath heated to 40 ° C, and stir until the whole solution, stirring was stopped, the solution was placed in 15 ° C water bath was allowed to stand, when starting When there is precipitation of crystals, and then placed under the 0 ° C crystallization, after filtration, to 45 ° C under hot air drying cycle 6 hours to obtain 29. 2g, purity 99.6% of Racecadotril a polymorph crystals.
reflection angle X-ray powder diffraction pattern 20 at 4.3 °, 8.7 °, 13.2 °, 16.8 °, 17.8 ° and 20.0 ° at the show X-ray powder diffraction peaks. In 1135. 19CHT1,1551. 46CHT1,1644. 73CHT1,1687. 57CHT1, 1731. 35CHT1 and 3289. 20CHT1 displayed at an infrared absorption peak.
Clips
US 20020055645
PATENT
Racecadotril, chemical name N_ [(R, S) -3- acetyl-mercapto-2-benzyl-propionyl)] glycine benzyl ester, is a neprilysin inhibitor, selectively, reversible inhibition of neprilysin, so that the inner protection from degradation of endogenous enkephalins, prolong the physiological activity of endogenous enkephalins in the digestive tract, mainly used in clinical treatment of children and adults with acute diarrhea. Its structural formula is as follows:

Racecadotril as enkephalinase inhibitors, developed in France in 1993 Bioprojet listed acute diarrhea treatment, trade name Tiorfan.
In W02011116490A1, US5945548 and CN101768095A and other documents, documented racecadotril the synthesis process, but did not report the crystal form; therefore the present inventors have not reported Racecadotril crystalline polymorph conduct further
Example 1
[0032] The 40. 0g Racecadotril dissolved in 200ml of absolute ethanol and water bath heated to 40 ° C, and stir until the whole solution, stirring was stopped, the solution was placed in 15 ° C water bath was allowed to stand, when starting When there is precipitation of crystals, and then placed under the 0 ° C crystallization, after filtration, to 45 ° C under hot air drying cycle 6 hours to obtain 29. 2g, purity 99.6% of Racecadotril a polymorph crystals.
[0033] reflection angle X-ray powder diffraction pattern 20 at 4.3 °, 8.7 °, 13.2 °, 16.8 °, 17.8 ° and 20.0 ° at the show X-ray powder diffraction peaks. In 1135. 19CHT1,1551. 46CHT1,1644. 73CHT1,1687. 57CHT1, 1731. 35CHT1 and 3289. 20CHT1 displayed at an infrared absorption peak.
SYN
EP 0038758
Alternatively, the condensation of dimethyl malonate (VI) with benzaldehyde (VII) by means of piperidine in refluxing toluene gives dimethyl benzylidenemalonate (VIII), which is reduced with H2 over Pd/C in toluene to yield the corresponding benzyl derivative (IX). The hydrolysis of (IX) with NaOH in water affords the benzylmalonic acid (X). Alternatively, intermediate (X) can also be obtained starting from diethyl malonate (XI), which is condensed with with benzaldehyde (VII) by means of piperidine in refluxing toluene to give diethyl benzylidenemalonate (XII). Reduction of (XII) with H2 over Pd/C in toluene yields the corresponding benzyl derivative (XIII), which is then hydrolized with NaOH in water. The monodecarboxylation of (X) and its condensation with paraformaldehyde and diethylamine in refluxing ethyl acetate provides 2-benzylacrylic acid (XIV), which is condensed with thioacetic acid (V) by heating at 70 C to afford 2-(acetylsulfanylmethyl)-3-phenylpropionic acid (XV). Finally, this compound is condensed with N-tosylglycine benzyl ester (XVI) by means of HOBt, DCC and TEA in THF.
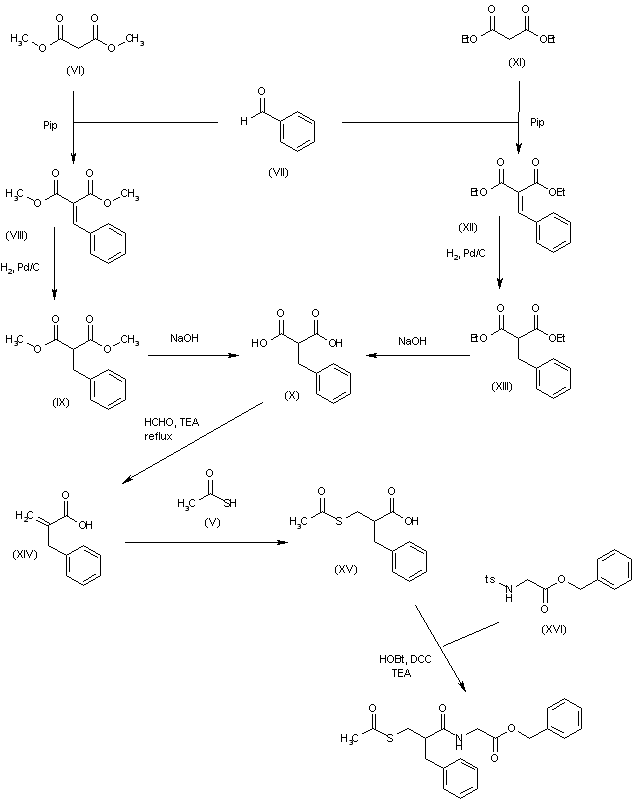
SYN
| EP 0729936 |
Reaction of benzaldehyde (I) with dimethyl malonate (II) in refluxing toluene in the presence of piperidine and HOAc provides dimethyl benzylidene malonate (III), which is then hydrogenated over Pd/C to afford dimethyl benzyl malonate (IV). Reduction of (IV) with LiAlH4 in refluxing THF furnishes 2-benzyl-1,3-propanediol (V), which is then subjected to reaction with vinyl acetate (VI) by means of Novozym 435 enzyme to yield diacetate (VII). Enantioselective removal of one acetyl group from (VII) by treatment with Pseudomonas fluorescens Lipase in acetone/phosphate buffer (pH = 7) at 30 C gives 3-acetoxy-2(S)-benzyl-propanol (S)-(VIII), which is then oxidized by means of Jones reagent in acetone/isopropanol to provide carboxylic acid (R)-(IX). The hydrolysis of (IX) with LiOH in THF/H2O gives 2(R)-benzyl-3-hydroxypropanoic acid (R)-(X). Alternatively, intermediate (X) can also be synthesized as follows: Condensation of benzaldehyde (I) with methyl acrylate (XV) by means of diaza-1,4-bicyclo[2.2.2.]octane affords methyl beta-hydroxy-alpha-methylene-benzenepropanoate (XVI), which is then subjected to hydrolysis with KOH in MeOH/H2O to yield carboxylic acid (XVII). Treatment of (XVII) with p-toluenesulfonic acid in refluxing HOAc gives (E)-2-(acetoxymethyl)-3-phenylpropionic acid (XVIII), which is finally converted into (X) by enantioselective hydrogenation in the presence of S-Binap and ruthenium catalyst [CodRu(all)2]. Derivative (R)-(X) is then converted into 3-(acetylsulfanyl)-2(S)-benzylpropionic acid (XI) by means of a Mitsunobu reaction with thioacetic acid, diisopropyl azodicarboxylate (DIAD) and triphenylphosphine (PPh3). Compound (XI) is then subjected to optical purification by formation and isolation of the corresponding salt with (-)-ephedrine and subsequent hydrolysis with HCl to furnish enantiomerically pure (S)-(XII). Finally, carboxylic acid (S)-(XII) is converted into ecadotril by its coupling with benzyl glycinate (XIV), either by means of Et3N, DCC and HOBt in CHCl3, or by first reaction with thionyl chloride to give acid chloride (S)-(XIII) and subsequent coupling with glycinate (XIV) by means of Et3N in CH2Cl2.
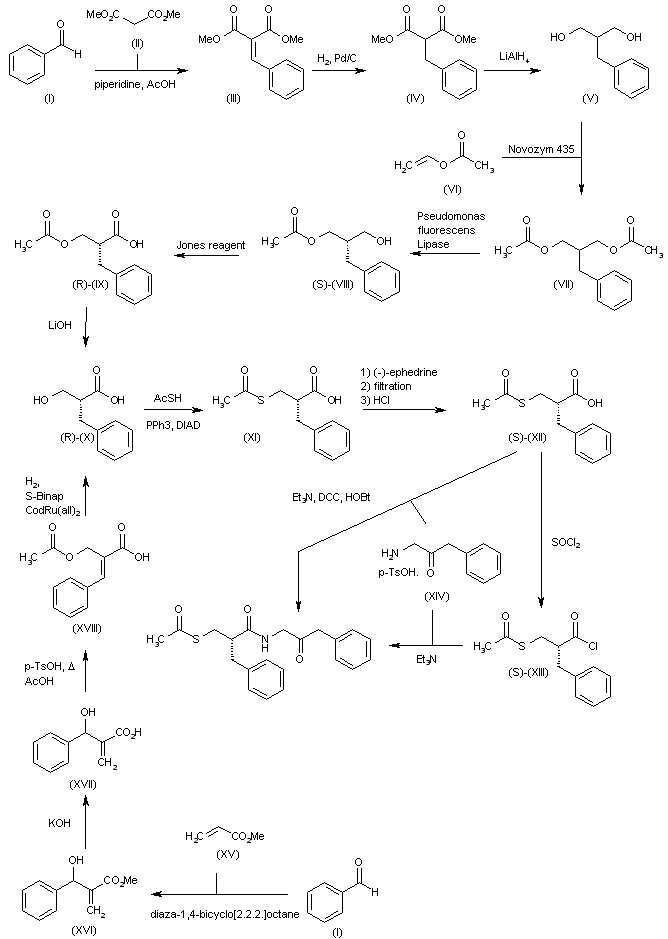
SYN
The reaction of 2-benzylacrylic acid (I) with SOCl2 in hot toluene gives the acyl chloride (II), which is condensed with N-tosylglycine benzyl ester (III) by means of TEA in toluene to yield the corresponding amide (IV). Finally, this compound is condensed with thioacetic acid by heating at 80 C to afford the target acylthio compound.
| FR 2816309; US 2002055645 |
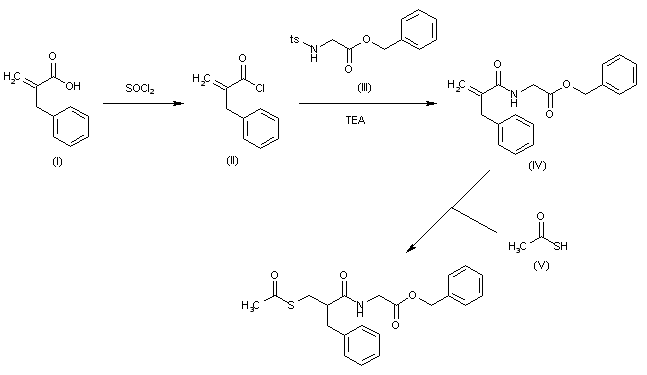
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Side effects
The most common adverse effect is headache, which occurs in 1–2% of patients.[7] Rashes occur in fewer than 1% of patients. Other described skin reactions include itching, urticaria, angioedema, erythema multiforme, and erythema nodosum.[9][10]
Overdose
No cases of overdose are known. Adults have tolerated 20-fold therapeutic doses without ill effects.[10]
Interactions
No interactions in humans have been described. Combining racecadotril with an ACE inhibitor can theoretically increase the risk for angioedema.[9][10]
Racecadotril and its main metabolites neither inhibit nor induce the liver enzymes CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. They also do not induce UGT enzymes.[10] This means that racecadotril has a low potential for pharmacokinetic interactions.
Pharmacology
Mechanism of action
Enkephalins are peptides produced by the body that act on opioid receptors with preference for the δ subtype.[11] Activation of δ receptors inhibits the enzyme adenylyl cyclase, decreasing intracellular levels of the messenger molecule cAMP.[7]
The active metabolite of racecadotril, thiorphan, inhibits enkephalinase enzymes in the intestinal epithelium with an IC50 of 6.1 nM, protecting enkephalins from being broken down by these enzymes. (Racecadotril itself is much less potent at 4500 nM.)[7][8] This reduces diarrhea related hypersecretion in the small intestine without influencing basal secretion. Racecadotril also has no influence on the time substances, bacteria or virus particles stay in the intestine.[10]
Pharmacokinetics

Some metabolites of racecadotril.
top left: precursor to the active metabolite
top right: active metabolite
bottom row: inactive metabolites
Racecadotril is rapidly absorbed after oral administration and reaches Cmax within 60 minutes. Food delays Cmax by 60 to 90 minutes but does not affect the overall bioavailability. Racecadotril is rapidly and effectively metabolized to the moderately active S-acetylthiorphan the main active metabolite thiorphan, of which 90% are bound to blood plasma proteins. In therapeutic doses, racecadotril does not pass the blood–brain barrier. Inhibition of enkephalinases starts 30 minutes after administration, reaches its maximum (75–90% inhibition with a therapeutic dose) two hours after administration, and lasts for eight hours. The elimination half-life, measured from enkephalinase inhibition, is three hours.[7][8][9]
Thiorphan is further metabolized to inactive metabolites such as the methyl thioether and the methyl sulfoxide. Both active and inactive metabolites are excreted, mostly via the kidney (81.4%), and to a lesser extent via the feces (8%).[10]
Society and culture
Brand names
In both France and Portugal it is sold as Tiorfan and in Italy as Tiorfix. In India it is available as Redotril and Enuff.[4]
See also
- Ecadotril, the (S)-enantiomer of racecadotril
- D/DL-Phenylalanine
- RB-101
References
- ^ https://www.ema.europa.eu/documents/psusa/racecadotril-list-nationally-authorised-medicinal-products-psusa/00002602/202003_en.pdf
- ^ Jump up to:a b c d “SPC-DOC_PL 39418-0003.PDF” (PDF). Medicines and Healthcare Products Regulatory Agency. Bioprojet Europe Ltd. 26 December 2012. Retrieved 7 May 2014.
- ^ Jump up to:a b Matheson AJ, Noble S (April 2000). “Racecadotril”. Drugs. 59 (4): 829–35, discussion 836–7. doi:10.2165/00003495-200059040-00010. PMID 10804038.
- ^ Jump up to:a b Brayfield, A, ed. (13 December 2013). “Racecadotril”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 6 May 2014.
- ^ Spillantini MG, Geppetti P, Fanciullacci M, Michelacci S, Lecomte JM, Sicuteri F (June 1986). “In vivo ‘enkephalinase’ inhibition by acetorphan in human plasma and CSF”. European Journal of Pharmacology. 125 (1): 147–50. doi:10.1016/0014-2999(86)90094-4. PMID 3015640.
- ^ Fischbach, Wolfgang; Andresen, Viola; Eberlin, Marion; Mueck, Tobias; Layer, Peter (2016). “A Comprehensive Comparison of the Efficacy and Tolerability of Racecadotril with Other Treatments of Acute Diarrhea in Adults”. Frontiers in Medicine. 3: 44. doi:10.3389/fmed.2016.00044. ISSN 2296-858X. PMC 5064048. PMID 27790616.
- ^ Jump up to:a b c d e f Dinnendahl, V; Fricke, U, eds. (1982). Arzneistoff-Profile (in German). Eschborn, Germany: Govi Pharmazeutischer Verlag. ISBN 978-3-7741-9846-3.
- ^ Jump up to:a b c Eberlin, Marion; Mück, Thomas; Michel, Martin C. (2012). “A Comprehensive Review of the Pharmacodynamics, Pharmacokinetics, and Clinical Effects of the Neutral Endopeptidase Inhibitor Racecadotril”. Frontiers in Pharmacology. 3: 93. doi:10.3389/fphar.2012.00093. ISSN 1663-9812. PMC 3362754. PMID 22661949.
- ^ Jump up to:a b c d e Mediq.ch: racecadotril. Accessed 2019-12-30.
- ^ Jump up to:a b c d e f g Haberfeld, H, ed. (2019). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Hidrasec 100 mg-Hartkapseln.
- ^ Cumming, P (2019). “A Survey of Molecular Imaging of Opioid Receptors”. Molecules. 24 (22): 4190. doi:10.3390/molecules24224190. PMC 6891617. PMID 31752279.
External links
- “Racecadotril”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Hidrasec, Tiorfan, Zedott, others |
| Other names | Benzyl 2-[3-(acetylthio)-2-benzylpropanamido]acetate |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | By mouth |
| ATC code | A07XA04 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)EU: Rx-only [1] |
| Pharmacokinetic data | |
| Protein binding | 90% (active metabolite thiorphan)[2] |
| Metabolism | Liver-mediated[2] |
| Onset of action | 30 min |
| Elimination half-life | 3 hours[2] |
| Excretion | Urine (81.4%), feces (8%)[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 81110-73-8 |
| PubChem CID | 107751 |
| DrugBank | DB11696 |
| ChemSpider | 96913 |
| UNII | 76K53XP4TO |
| KEGG | D08464 |
| ChEMBL | ChEMBL2103772 |
| CompTox Dashboard (EPA) | DTXSID8045513 |
| ECHA InfoCard | 100.214.352 |
| Chemical and physical data | |
| Formula | C21H23NO4S |
| Molar mass | 385.48 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| Melting point | 89 °C (192 °F) |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
{kind=link}

| CN101103960A * | Jul 14, 2006 | Jan 16, 2008 | 海南盛科生命科学研究院 | Dry mixed suspension containing racecadotril and preparation method thereof |
| CN101768095A * | Dec 26, 2008 | Jul 7, 2010 | 山东齐都药业有限公司 | Preparation method of racecadotril |
| WO2001097803A1 * | Jun 20, 2001 | Dec 27, 2001 | Laboratoire Glaxosmithkline | Pharmaceutical preparations comprising racecadotril (acetorphan) |
| WO2013098826A1 * | Dec 26, 2011 | Jul 4, 2013 | Symed Labs Limited | “a process for the preparation of n-[2-[(acetylthio) methyl]-1-oxo-3-phenylpropyl] glycine phenyl methyl ester and intermediates thereof” |
| Reference |
|---|
| Reference | ||||
|---|---|---|---|---|
| CN101103960A * | Jul 14, 2006 | Jan 16, 2008 | 海南盛科生命科学研究院 | Dry mixed suspension containing racecadotril and preparation method thereof |
| CN101768095A * | Dec 26, 2008 | Jul 7, 2010 | 山东齐都药业有限公司 | Preparation method of racecadotril |
| WO2001097803A1 * | Jun 20, 2001 | Dec 27, 2001 | Laboratoire Glaxosmithkline | Pharmaceutical preparations comprising racecadotril (acetorphan) |
| WO2013098826A1 * | Dec 26, 2011 | Jul 4, 2013 | Symed Labs Limited | “a process for the preparation of n-[2-[(acetylthio) methyl]-1-oxo-3-phenylpropyl] glycine phenyl methyl ester and intermediates thereof” |
| 1 | * | 金庆平 等: “神经内肽酶抑制剂消旋卡多曲(Racecadotril)的合成工艺研究“, 《中国现代应用药学杂志》, vol. 20, no. 7, 31 August 2003 (2003-08-31) |
| Reference | ||||
|---|---|---|---|---|
| Citing Patent | Filing date | Publication date | Applicant | Title |
| US6013829 * | Feb 4, 1997 | Jan 11, 2000 | Societe Civile Bioprojet | Process for the asymmetric synthesis of S-acyl derivatives of 2-mercaptomethyl -3- phenyl propanoic acid, application to the synthesis of N-(mercaptoacyl) amino acid derivatives |
| US20040009956 * | Apr 29, 2003 | Jan 15, 2004 | Dehua Pei | Inhibition of protein tyrosine phosphatases and SH2 domains by a neutral phosphotyrosine mimetic |
| 1 | * | MOHAMED A.O. ET AL.: ‘Stability-indicating methods for the determination of racecadotril in the presence of its degradation products‘ BIOSCIENCE TRENDS vol. 3, no. 6, 2009, pages 247 – 252, XP055074337 | ||
| CN104356036A * | Nov 7, 2014 | Feb 18, 2015 | 山东齐都药业有限公司 | Alpha crystal form of racecadotril and preparation method of alpha crystal form |
///////////Racecadotril, рацекадотрил , راسيكادوتريل , 消旋卡多曲 , Antidiarrheal, Neutral Endopeptidase Inhibitor, Cadotril, Dexecadotril ,
CC(=O)SCC(CC1=CC=CC=C1)C(=O)NCC(=O)OCC1=CC=CC=C1

NEW DRUG APPROVALS
ONE TIME
$10.00
CEFADROXIL

CEFADROXIL
- Molecular FormulaC16H17N3O5S
- Average mass363.388 Da
(6R,7R)-7-{[(2R)-2-Amino-2-(4-hydroxyphenyl)acetyl]amino}-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
256-555-6[EINECS]
50370-12-2[RN]
5-Thia-1-azabicyclo(4.2.0)oct-2-ene-2-carboxylic acid, 7-(((2R)-amino(4-hydroxyphenyl)acetyl)amino)-3-methyl-8-oxo-, (6R,7R)-
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[(2R)-2-amino-2-(4-hydroxyphenyl)acetyl]amino]-3-methyl-8-oxo-, (6R,7R)-
цефадроксил [Russian] [INN]
سيفادروكسيل [Arabic] [INN]
头孢羟氨苄 [Chinese] [INN]
Cephos
- Molecular FormulaC16H19N3O6S
- Average mass381.404 Da
5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[(2R)-2-amino-2-(4-hydroxyphenyl)acetyl]amino]-3-methyl-8-oxo-, (6R,7R)-, monohydrate
66592-87-8[RN]
(6R,7R)-7-{[(2R)-2-amino-2-(4-hydroxyphenyl)acetyl]amino}-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid hydrate
(6R,7R)-7-{[(2R)-2-Amino-2-(4-hydroxyphenyl)acetyl]amino}-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid hydrate (1:1)
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Cefadroxil hemihydrate | J9CMF6461M | 119922-85-9 | AJAMDISMDZXITN-QXBGZBSVSA-N |
| Cefadroxil monohydrate | 280111G160 | 66592-87-8 | NBFNMSULHIODTC-CYJZLJNKSA-N |
| Cefadroxil sodium | SSZ6380I0I | 42284-83-3 | GQOVFIUWRATNJC-CYJZLJNKSA-M |
CefadroxilCAS Registry Number: 66592-87-8
CAS Name: (6R,7R)-7-[[(2R)-Amino-(4-hydroxyphenyl)acetyl]amino]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid monohydrate
Additional Names: 7-[D-(-)-a-amino-a-(4-hydroxyphenyl)acetamido]-3-methyl-3-cephem-4-carboxylic acid monohydrate; p-hydroxycephalexine monohydrate
Manufacturers’ Codes: BL-S578; MJF-11567-3
Trademarks: Baxan (BMS); Bidocef (BMS); Cefa-Drops (Fort Dodge); Cefamox (BMS); Ceforal (Farmoffer); Cephos (CT); Duracef (BMS); Duricef (BMS); Kefroxil (Torre); Oracéfal (BMS); Sedral (BMS); Ultracef (BMS)
Molecular Formula: C16H17N3O5S.H2O
Molecular Weight: 381.40
Percent Composition: C 50.39%, H 5.02%, N 11.02%, O 25.17%, S 8.41%
Literature References: Semi-synthetic cephalosporin antibiotic. Prepn: NL6812382; L. B. Crast, Jr., US3489752 (1969, 1970 both to Bristol-Myers); T. Takahashi et al.,DE2216113; eidem,US3816253 (1972, 1974, both to Takeda). Prepn of crystalline monohydrate: D. Bouzard et al.,US4504657 (1985 to Bristol-Myers). Antimicrobial activity: R. E. Buck, K. E. Price, Antimicrob. Agents Chemother.11, 324 (1977). Pharmacology: M. Pfeffer et al.,ibid. 331; A. I. Hartstein et al.,ibid.12, 93 (1977). Review:J. Antimicrob. Chemother.10, Suppl. B, 1-162 (1982). Series of articles on clinical trials in respiratory tract infections: Drugs32, Suppl. 3, 1-56 (1986).Properties: White crystals, mp 197° (dec).
Melting point: mp 197° (dec)
Therap-Cat: Antibacterial.
Therap-Cat-Vet: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Cephalosporins.
Cefadroxil is a cephalosporin antibiotic used in the treatment of various bacterial infections, such as urinary tract infections, skin and skin structure infections, and tonsillitis.
Cefadroxil (formerly trademarked as Duricef) is a broad-spectrum antibiotic of the cephalosporin type, effective in Gram-positive and Gram-negative bacterial infections. It is a bactericidal antibiotic.
It was patented in 1967 and approved for medical use in 1978.[1]
DURICEF (cefadroxil) is a semisynthetic cephalosporin antibiotic intended for oral administration. It is a white to yellowish-white crystalline powder. It is soluble in water and it is acid- stable. It is chemically designated as 5-Thia-l-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[amino(4-hydroxyphenyl)acetyl]amino]-3-methyl-8-oxo-, monohydrate[6R- [6α,7β(R*)]]-. It has the formula C16H17N3O5S•H20 and the molecular weight of 381.40. It has the following structural formula:
 |
DURICEF (cefadroxil) film-coated tablets, 1 g, contain the following inactive ingredients: microcrystalline cellulose, hydroxypropyl methylcellulose, magnesium stearate, polyethylene glycol, polysorbate 80, simethicone emulsion, and titanium dioxide.
DURICEF (cefadroxil) for Oral Suspension contains the following inactive ingredients: FD&C Yellow No. 6, flavors (natural and artificial), polysorbate 80, sodium benzoate, sucrose, and xanthan gum.
DURICEF (cefadroxil) capsules contain the following inactive ingredients: D&C Red No. 28, FD&C Blue No. 1, FD&C Red No. 40, gelatin, magnesium stearate, and titanium dioxide.
SYN

IR spectrum of pure cefadroxil drug.
Synthesis Reference
Leonardo Marsili, “Substantially anhydrous crystalline cefadroxil and method for producing it.” U.S. Patent US5329001, issued April, 1978.
SYN
Antibiotics
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Cefadroxil
Cefadroxil, [6R-[6α,7β(R)]]-3-methyl-8-oxo-7-[[amino(4-hydroxyphenyl) acetyl]amino]-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid (32.1.2.14), is an analog of cephalexin and differs only in the presence of a hydroxyl group in the fourth position of the phenyl ring of phenylglycine, and is synthesized by a scheme analogous to the scheme of cephradin synthesis [90–96].

Cefadroxil has a broad spectrum of antimicrobial action; it is active with respect to Gram-positive and Gram-negative microorganisms. Like all of the other drugs described above, it acts as a bactericide by disrupting the process of restoring the membranes of bacteria. Synonyms of this drug are bidocef, cefadril, duracef, ultracef, and others.SYN
Cefadroxil
- ATC:J01DA09
- MW:363.39 g/mol
- CAS-RN:50370-12-2
- InChI Key:BOEGTKLJZSQCCD-UEKVPHQBSA-N
- InChI:InChI=1S/C16H17N3O5S/c1-7-6-25-15-11(14(22)19(15)12(7)16(23)24)18-13(21)10(17)8-2-4-9(20)5-3-8/h2-5,10-11,15,20H,6,17H2,1H3,(H,18,21)(H,23,24)/t10-,11-,15-/m1/s1
- EINECS:256-555-6
- LD50:>1.5 g/kg (M, i.v.); >10 g/kg (M, p.o.);
>1 g/kg (R, i.v.); >10 g/kg (R, p.o.);
>2 g/kg (dog, p.o.)
Synthesis
Substances
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 22252-43-3 | C8H10N2O3S | 7-amino-3-deacetoxycephalosporanic acid | 5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-amino-3-methyl-8-oxo-, (6R-trans)- |
| 53487-89-1 | C13H15NO5 | d(–)-4-hydroxy-N-(2-methoxycarbonyl-1-methylethenyl)phenylglycine | Benzeneacetic acid, 4-hydroxy-α-[(3-methoxy-1-methyl-3-oxo-1-propenyl)amino]-, (R)- |
PATENT
https://patents.google.com/patent/WO2005042543A1/enOral cephalosporin antibiotics, including cefprozil, cefatrizine, and cefadroxii, commonly have a 4-hydroxyphenylglycine group, as represented by the following formula:

The compound of the above formula is cefprozil when A is -C=CH-CH3, cefatrizine when A is 1 H-1 ,2,3-triazole-4-yl-thiomethyl, and cefadroxii when A is -CH3. Conventionally, there have been known various processes for preparing oral cephalosporin antibiotics, such as cefprozil, cefatrizine, and cefadroxii, by reacting reactive derivatives of 4-hydroxyphenylglycine with 3-cephem compounds. For example, U.S. Patent No. 3,985,741 discloses a process for preparing a cefadroxii, which includes reacting 4-hydroxyphenylglycine and ethylchloroformate inN-methylmorpholine to obtain an anhydride, followed by reaction with7-amino-deacetoxy-cephalosporanic acid (7-ADCA). However, the yield and quality of the product are poor. U.S. Patent Nos. 4,520,022, 4,591 ,641 , and 4,661 ,590 disclose a condensation reaction between 4-hydroxyphenylglycine with a protected amino group and a cephem compound in the presence of Λ/.Λ/’-dicyclohexylcarbodimide. However,Λ/,Λ/’-dicyclohexylurea produced after the condensation reaction is not easily removed, which restricts industrial applications. U.S. Patent No. 4,336,376 discloses a process for preparing a cefadroxii, which includes reacting a 4-hydroxyphenylglycine salt having a protected amino group with trimethylsilyl-2-oxazolidinone to protect a 4-hydroxyl group followed by reaction with acylchloride to obtain a 4-hydroxyphenylglycine anhydride and then reaction with 7-ADCA. However, silylation is prerequisite and these reactions are annoying, and thus, this process is not suitable for industrial application. U.S. Patent No. 4,708,825 discloses a technique of reacting4-hydroxyphenylglycine having a substituted amino group with thionyl chloride using a gaseous hydrogen chloride to obtain a 4-hydroxyphenylglycyl chloride hydrochloride followed by reaction with a cephem compound. However, handling property of the thionyl chloride and the gaseous hydrogen chloride is poor, and thus, this technique is not suitable for industrial application. U.S. Patent Nos. 3,925,418, 4,243,819, and 4,464,307 disclose a process for producing 4-hydroxyphenylglycine using excess phosgene. However, difficulty in handling of highly toxic phosgene, removal of excess residual phosgene, and control of reaction conditions renders mass production difficult. As a process for preparing a reactive anhydride of 4-hydroxyphenylglycine, there are reported a method for the preparation of acid chloride using phosphorus pentachloride, phosphorus oxychloride, or thionyl chloride, and a method for the preparation of active ester using imidazole, mercaptobenzothiazole, or hydroxybenzotriazole. However, an acid chloride of 4-hydroxyphenylglycine has poor reactivity due to a hydroxyl group and an active ester of 4-hydroxyphenylglycine has poor reactivity and involves a side reaction. In addition, Korean Patent Laid-Open Publication Nos. 2002-69431 , 2002-69432, 2002-69437, and 2002-69440 disclose a process for preparing a pivaloyl or succinimide derivative of 4-hydroxyphenylglycine and a process for preparing a cephem compound such as cefprozil using the pivaloyl or succinimide derivative of 4-hydroxyphenylglycine. Meanwhile, there have been known various preparation processes for 3-(Z)-propenyl cephem derivative which is a compound useful as an intermediate for preparation of cefprozil which is an oral cephalosporin antibiotic. WO93/16084 discloses a process of selectively separating a 3-(Z)-propenyl cephem compound by means of a hydrochloride, metal, or tertiary amine salt of7-amino-3-(1-propen-1-yl)-3-cephem-carboxylic acid or by adsorption chromatography. However, there is a disadvantage in that separation and purification are cost-ineffective. U.K. Patent No. 2,135,305 discloses a process for preparing cefprozil from a4-hydroxyphenylglycine compound with a t-butoxycarbonyl-protected amino group and a cephem compound with a benzhydryl-protected carboxyl group. However, incorporation of a 3-propenyl group after acylation lowers reaction efficiency and high-performance liquid chromatography is required for isomer separation, which render industrial application difficult. U.S. Patent No. 4,727,070 discloses a technique of removing an E-isomer cefprozil from a mixture of 27E cefprozil, which includes incorporating an active group such as sodium imidazolidinone into the mixture of 2VE cefprozil by reaction of the mixture of 27E cefprozil with acetone followed by deprotection. However, purification by chromatography incurs enormous costs. In view of the above problems, Korean Patent Laid-Open Publication No.2002-80838 discloses a process for preparing a 3-(Z)-propenyl cephem compound by reacting a phosphoranylidene cephem compound with acetaldehyde in a mixed solvent essentially consisting of ether in the presence of a base. According to a disclosure in this patent document, ether is essentially used. In this respect, in the case of using methylenechloride or tetrahydrofuran, even when other reaction conditions, for example, reaction temperature, reaction duration, base, catalyst, and the like are adjusted, it is very difficult to adjust the content of the Z-isomer to more than 83%.DETAILED DESCRIPTION OF THE INVENTION Technical Goal of the Invention The present invention provides a process for simply preparing a cephalosporin antibiotic in high yield and purity using a novel reactive intermediate, i.e., a 4-hydroxyphenylglycine derivative. The present invention also provides a novel reactive intermediate, i.e., a 4-hydroxyphenylglycine derivative which is used in simply preparing a cephalosphorin antibiotic in high yield and purity, and a preparation process thereof. While searching for a process for stereospecifically preparing a novel3-(Z)-propenyl cephem derivative, the present inventors found that use of a mixed solvent including methylenechloride, isopropylalcohol, and water in a predetermined ratio can stereospecifically and efficiently produce the 3-(Z)-propenyl cephem derivative, which is in contrary to the disclosure in Korean Patent Laid-Open Publication No. 2002-80838. Therefore, the present invention also provides a process for stereospecifically preparing a 3-(Z)-propenyl cephem derivative using a mixed solvent including methylenechloride, isopropylalcohol, and water in a predetermined ratio.



Example 9 Preparation of7-r2-amino-2-(4-hvdroxyphenyl)acetamido1-3-methyl-3-cephem-4-carboxylic acid(cefadroxii) The reaction solution obtained in step A of Example 1 was cooled to -40 °C and a solution obtained by dissolving 6.21 g (0.029mol) of 7-amino-3-methyl-3-cephem-4-carboxylic acid in 40 ml of methylenechloride, 10 ml of water, and 6.5 g of triethylamine was gradually dropwise added thereto for 1 hour. Then, the reaction mixture was incubated at the same temperature for 2 hours and cooled to 0°C to obtain an insoluble solid. The insoluble solid was filtered. A filtrate was sent to a reactor and then stirred for 1 hour after addition of 20 ml of 6N HCI. The reaction solution was adjusted to pH of 3.2 by addition of 10% NaOH, stirred at0°C for 2 hours, and filtered to give 9.1g (83%) of the titled compound as a white solid. H-NMR( δ , D20-d2) : 1.79(3H, d, 8.6Hz, -CH3), 3.22(1 H, d, 18Hz, 2-H),3.55(1 H. d. 18Hz, 2-H), 5.15(1 H, d, 4.6Hz, 6-H), 5.66(1 H, d, 4.6Hz, 7-H), 6.91 (2H, d,8.0Hz, phenyl-H), 7.38(2H, d, 8.0Hz, phenyl-H)
PAPER
Deshmukh, J. H.; Asian Journal of Chemistry 2010, V22(3), P1760-1768
Journal of the Chinese Chemical Society (Weinheim, Germany), 66(12), 1649-1657; 2019
Journal of the Indian Chemical Society, 93(6), 593-598; 2016
Biotechnology Letters, 34(9), 1719-1724; 2012
PATENT
WO 2011113486
By Gupta, Niranjan Lal et alFrom Indian, 184842, 30 Sep 2000
PAPER
European Journal of Organic Chemistry, (10), 1817-1820; 2001
PAPER
Organic Letters, 2(18), 2829-2831; 2000
The cephalosporin antibiotic Cefadroxil can be epimerized at the α-carbon of its amino acid side chain using pyridoxal as the mediator. By clathration with 2,7-dihydroxynaphthalene, the desired diastereomer can be selectively withdrawn from the equilibrating mixture of epimers. In this way, an asymmetric transformation of Cefadroxil can be accomplished. This opens the possibility of the production of Cefadroxil starting from racemic p-hydroxyphenylglycine, in contrast to the current industrial synthesis that employs the d-amino acid in enantiopure form.

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical use
Cefadroxil is a first-generation cephalosporin antibacterial drug that is the para-hydroxy derivative of cephalexin, and is used similarly in the treatment of mild to moderate susceptible infections such as the bacterium Streptococcus pyogenes, causing the disease popularly called strep throat or streptococcal tonsillitis, urinary tract infection, reproductive tract infection, and skin infections.
Cefadroxil is used as an antibiotic prophylaxis before dental procedures, for patients allergic to penicillins.
Spectrum of bacterial resistance and susceptibility
Cefadroxil has a broad spectrum of activity and has been effective in treating bacteria responsible for causing tonsillitis, and infections of the skin and urinary tract. The following represents MIC susceptibility data for a few medically significant microorganisms.[2]
- Escherichia coli: 8 μg/ml
- Staphylococcus aureus: 1 – 2 μg/ml
- Streptococcus pneumoniae: ≤1 – >16 μg/ml
Side effects
The most common side effects of cefadroxil are diarrhea (which, less commonly, may be bloody), nausea, upset stomach, and vomiting. Other side effects include[3] rashes, hives, and itching.
Pharmacokinetics
Cefadroxil is almost completely absorbed from the gastrointestinal tract. After doses of 500 mg and 1 g by mouth, peak plasma concentrations of about 16 and 30 micrograms/ml, respectively, are obtained after 1.5 to 2.0 hours. Although peak concentrations are similar to those of cefalexin, plasma concentrations are more sustained. Dosage with food does not appear to affect the absorption of cefadroxil. About 20% of cefadroxil is reported to be bound to plasma proteins. Its plasma half-life is about 1.5 hours and is prolonged in patients with renal impairment.
Cefadroxil is widely distributed to body tissues and fluids. It crosses the placenta and appears in breast milk. More than 90% of a dose of cefadroxil may be excreted unchanged in the urine within 24 hours by glomerular filtration and tubular secretion; peak urinary concentrations of 1.8 mg/ml have been reported after a dose of 500 mg. Cefadroxil is removed by haemodialysis.
Dosage
Cefadroxil is given by mouth, and doses are expressed in terms of the anhydrous substance; 1.04 g of cefadroxil monohydrate is equivalent to about 1 g of anhydrous cefadroxil.
Veterinary use
It can be used for treating infected wounds on animals. Usually in powder form mixed with water, it has a color and smell similar to Tang. Given orally to animals, the amount is dependent on their weight and severity of infection.
References
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 493. ISBN 9783527607495.
- ^ “Cefadroxil, Free Acid Susceptibility and Minimum Inhibitory Concentration (MIC) Data” (PDF).
- ^ “Cefadroxil side effects”. Drugs.
| Clinical data | |
|---|---|
| Trade names | Duricef |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682730 |
| Routes of administration | Oral |
| ATC code | J01DB05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | plasma protein |
| Metabolism | unknown |
| Elimination half-life | 1.5 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 66592-87-8 |
| PubChem CID | 47964 |
| DrugBank | DB01140 |
| ChemSpider | 43629 |
| UNII | 280111G160 |
| KEGG | D02353 |
| ChEBI | CHEBI:53667 |
| ChEMBL | ChEMBL1644 |
| CompTox Dashboard (EPA) | DTXSID8022749 |
| ECHA InfoCard | 100.051.397 |
| Chemical and physical data | |
| Formula | C16H17N3O5S |
| Molar mass | 363.39 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
{kind=link}
////////////CEFADROXIL, цефадроксил , سيفادروكسيل , 头孢羟氨苄 , BL-S578; MJF-11567-3, BL S578, MJF 11567-3
[H][C@]12SCC(C)=C(N1C(=O)[C@H]2NC(=O)[C@H](N)C1=CC=C(O)C=C1)C(O)=O

NEW DRUG APPROVALS
ONE TIME
$10.00
MY NEW DRUG APPROVALS BLOG HAS CLOCKED 35 LAKH VIEWS IN 7 CONTINENTS, 226 COUNTRIES

MY NEW DRUG APPROVALS BLOG HAS CLOCKED 35 LAKH VIEWS IN 7 CONTINENTS, 226 COUNTRIES
Service to education is service to humanity, Taking free education to the world, Every difficulty an opportunity,In the dark but yet in light,proud of my disabilty, Taking industry experience to students, See the ability not the disabilty, The only disability in life is a bad attitude !!!, Dreamt a billion when hit by million, Mentor to the world
![]() All my awards, https://lnkd.in/fGxx9VF
All my awards, https://lnkd.in/fGxx9VF
Click on above links