
Home » Uncategorized (Page 18)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Canlitinib
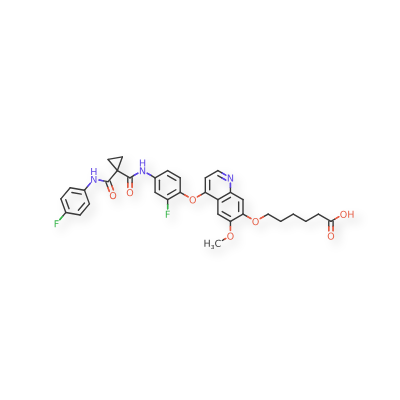


Canlitinib
Cas 2222730-78-9
| Molecular Weight | 619.61 |
|---|---|
| Formula | C33H31F2N3O7 |
6-[4-[2-fluoro-4-[[1-[(4-fluorophenyl)carbamoyl]cyclopropanecarbonyl]amino]phenoxy]-6-methoxyquinolin-7-yl]oxyhexanoic acid
CANLITINIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
Canlitinib is a tyrosine kinase inhibitor, extracted from patent WO2018072614 (IV-2). Canlitinib has the potential for cancer study.
Kanitinib is a tyrosine kinase inhibitor targeting the oncoprotein c-Met (hepatocyte growth factor receptor; HGFR; MET) and vascular endothelial growth factor receptor 2 (VEGFR2), with potential anti-angiogenic and antineoplastic activities. Upon oral administration, kanitinib targets and binds to c-Met and VEGFR2, thereby disrupting c-Met- and VEGFR2-dependent signal transduction pathways. This may induce cell death in tumor cells overexpressing c-Met and/or VEGFR2 protein. c-Met and VEGFR2 are both overexpressed in many tumor cell types and play key roles in tumor cell proliferation, survival, invasion, metastasis, and tumor angiogenesis
SCHEME

INT

PATENT
WO2020216188
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020216188&_cid=P20-MA3XXD-35471-1
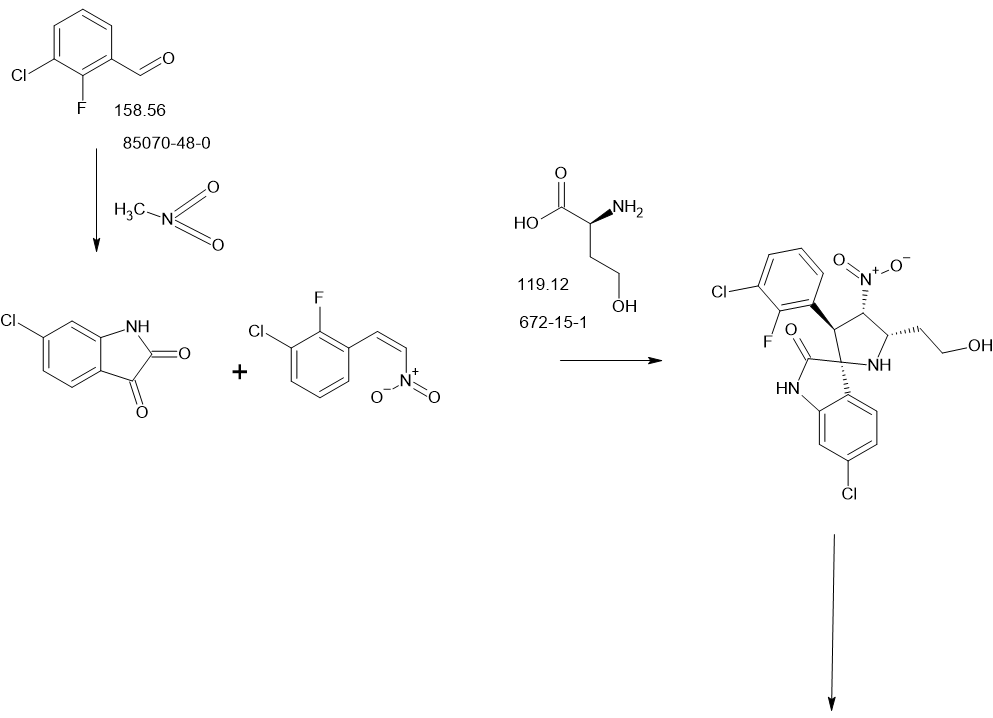
[0064]The preparation method of compound 1 is shown in Example 9 of compound patent WO 2018/072614 A1. Specifically, the preparation method of compound 1 is as follows.
[0065]
[0066]Under stirring, NaOH (4.4 g, 110 mmol) was added dropwise to a solution of methyl 6-[[4-[2-fluoro-4-[[1-[(4-fluorophenyl)carbamoyl]cyclopropanecarbonyl]amino]phenoxy]-6-methoxy-7-quinolyl]oxy]hexanoate (IV-1, 35.0 g, 55.2 mmol, prepared according to the method described in WO2013/040801A1) in ethanol (350 mL). After the addition was complete, water (50 mL) was added. The resulting mixture was stirred at 20-25°C for 18 h, the reaction solution was diluted with water (100 mL), stirred for 20 min, and the pH was adjusted to 3-4 with 1N HCl. The reaction mixture was concentrated under reduced pressure to distill off about 300 mL of ethanol. The solid product was collected by filtration to give 28.4 g of crude product, which was purified by silica gel column chromatography (eluent: ethyl acetate:methanol = 1:1, v/v) to give 6-[[4-[2-fluoro-4-[[1-[(4-fluorophenyl)carbamoyl]cyclopropanecarbonyl]amino]phenoxy]-6-methoxy-7-quinolyl]oxy]hexanoic acid (Compound 1), 9.6 g (yield: 28.1%).
[0067]Analytical data of compound 1: molecular weight 619.61; NMR hydrogen spectrum is shown in Figure 1, and NMR hydrogen spectrum data are as follows:
[0068]
1H-NMR(δ,DMSO-d6,400MHz):12.03(s,1H,OH),10.40(s,1H,NH),10.02(s,1H,NH),8.47~8.46(d,J=4,1H,CH),7.89-7.92(d,J=12,1H,CH),7.63-7.67(d,J=16,2H,2CH),7.51-7.52(d,J=4,2H2CH),7.39-7.43(t,2H,2CH),7.13-7.17(t,2H,2CH),6.41-6.42(d,J=4,1H,CH),4.12-4.15(t,2H,CH 2),3.95(s,3H,CH 3),2.24-2.28(t,2H,CH 2),1.78-1.85(m,2H,CH 2),1.57-1.64(m,2H,CH 2),1.43-1.51(m,6H,3CH 2)。
PATENT
CN111825609
PATENT
WO2018072614
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018072614&_cid=P20-MA3XZQ-37082-1
[0438]Preparation of 6-[[4-[2-fluoro-4-[[1-[(4-fluorophenyl)carbamoyl]cyclopropanecarbonyl]amino]phenoxy]-6-methoxy-7-quinolyl]oxy]hexanoic acid (IV-2), the reaction formula is as follows:
[0439]
[0440]Under stirring, NaOH (4.4 g, 110 mmol) was added dropwise to a solution of methyl 6-[[4-[2-fluoro-4-[[1-[(4-fluorophenyl)carbamoyl]cyclopropanecarbonyl]amino]phenoxy]-6-methoxy-7-quinolyl]oxy]hexanoate (IV-1, 35.0 g, 55.2 mmol, prepared according to the method described in WO2013/040801A1) in ethanol (350 mL). After the addition was complete, water (50 mL) was added. The resulting mixture was stirred at 20-25°C for 18 h, the reaction solution was diluted with water (100 mL), stirred for 20 min, and the pH was adjusted to 3-4 with 1N HCl. The reaction mixture was concentrated under reduced pressure to distill off about 300 mL of ethanol. The solid product was collected by filtration to give 28.4 g of crude product, which was purified by silica gel column chromatography (eluent: ethyl acetate:methanol = 1:1, v/v) to give 6-[[4-[2-fluoro-4-[[1-[(4-fluorophenyl)carbamoyl]cyclopropanecarbonyl]amino]phenoxy]-6-methoxy-7-quinolyl]oxy]hexanoic acid (IV-2), 9.6 g (yield: 28.1%). Analytical data:
1 H-NMR (400 MHz, DMSO-d
6 ): δ=8.17 (d, J=8.0 Hz, 1H), 7.81 (dd, J=2.8, 13.4 Hz, 1H) 7.62 (m, 2H), 7.51 (m, 4H), 7.39 (t, J=2.4 Hz, 2H), 6.44 (d, J=20.0 Hz, 1H), 4.13 (t, J=8.5 Hz, 2H), 3.85 (s, 3H), 2.27 (t, J=4.0 Hz, 2H), 1.83 (m, 2H), 1.68-1.46 (m, 8H). Mass spectrum (ESI) m/z: 620.2 [M+H]
+ .
PATENT
WO2013/040801
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013040801&_cid=P20-MA3Y3E-39505-1
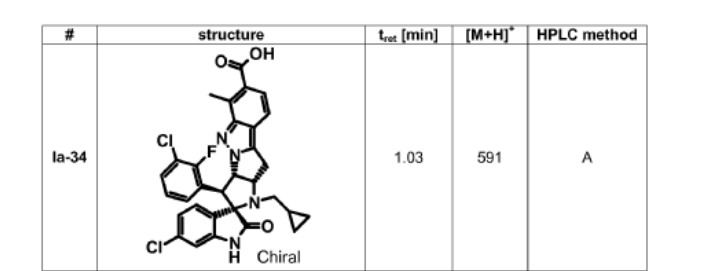
BRIGIMADLIN
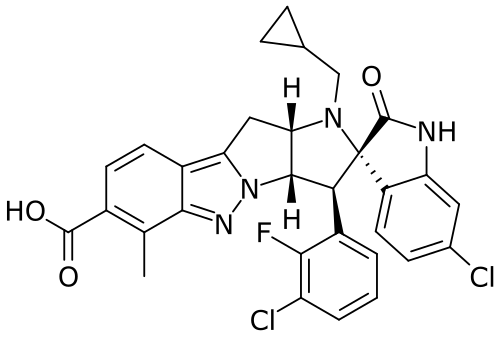
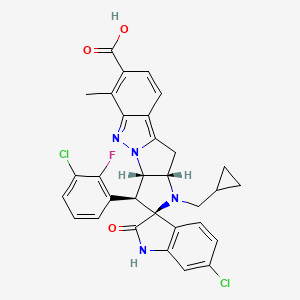
BRIGIMADLIN
Cas 2095116-40-6
WeightAverage: 591.46
Monoisotopic: 590.1287742
Chemical FormulaC31H25Cl2FN4O3
Spiro[3H-indole-3,2′(1′H)-pyrrolo[2′,3′:4,5]pyrrolo[1,2-b]indazole]-7′-carboxylic acid, 6-chloro-3′-(3-chloro-2-fluorophenyl)-1′-(cyclopropylmethyl)-1,2,3′,3′a,10′,10′a-hexahydro-6′-methyl-2-oxo-, (2′S,3′S,3′aS,10′aS)-
(2′S,3′S,3′aS,10′aS)-6-Chloro-3′-(3-chloro-2-fluorophenyl)-1′-(cyclopropylmethyl)-1,2,3′,3′a,10′,10′a-hexahydro-6′-methyl-2-oxospiro[3H-indole-3,2′(1′H)-pyrrolo[2′,3′:4,5]pyrrolo[1,2-b]indazole]-7′-carboxylic acid (
- (3S,10’S,11’S,14’S)-6-chloro-11′-(3-chloro-2-fluorophenyl)-13′-(cyclopropylmethyl)-6′-methyl-2-oxospiro[1H-indole-3,12′-8,9,13-triazatetracyclo[7.6.0.02,7.010,14]pentadeca-1,3,5,7-tetraene]-5′-carboxylic acid
- (3S,3’S,3a’S,10a’S)-6-chloro-3′-(3-chloro-2-fluorophenyl)-1′-(cyclopropylmethyl)-6′-methyl-2-oxo-1,2,3′,3a’,10′,10a’-hexahydro- 1’H-spiro[indole-3,2′-pyrrolo[2′,3′:4,5]pyrrolo[1,2-b]indazole]-7′- carboxylic acid
- (3S,3’S,3a’S,10a’S)-6-Chloro-3′-(3-chloro-2-fluorophenyl)-1′-(cyclopropylmethyl)-6′-methyl-2-oxo-1,2,3′,3a’,10′,10a’-hexahydro1’H-spiro[indole-3,2′-pyrrolo[2′,3′:4,5]pyrrolo[1,2-b]indazole]-7′-carboxylic acid
- Spiro[3H-indole-3,2′(1’H)-pyrrolo[2′,3′:4,5]pyrrolo[1,2-b]indazole]-7′-carboxylic acid, 6-chloro-3′-(3-chloro-2-fluorophenyl)-1′-(cyclopropylmethyl)-1,2,3′,3’a,10′,10’a-hexahydro-6′-methyl-2-oxo-, (2’S,3’S,3’aS,10’aS)-
Brigimadlin (BI-907828) is a small molecule MDM2–TP53 inhibitor developed for liposarcoma.[2][3][4][5][6]
Brigimadlin is an orally available inhibitor of murine double minute 2 (MDM2), with potential antineoplastic activity. Upon oral administration, brigimadlin binds to MDM2 protein and prevents its binding to the transcriptional activation domain of the tumor suppressor protein p53. By preventing MDM2-p53 interaction, the transcriptional activity of p53 is restored. This leads to p53-mediated induction of tumor cell apoptosis. Compared to currently available MDM2 inhibitors, the pharmacokinetic properties of BI 907828 allow for more optimal dosing and dose schedules that may reduce myelosuppression, an on-target, dose-limiting toxicity for this class of inhibitors.
SCHEME
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US231206177&_cid=P10-MA0ULZ-04263-1
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017060431&_cid=P10-MA0TY5-76812-1
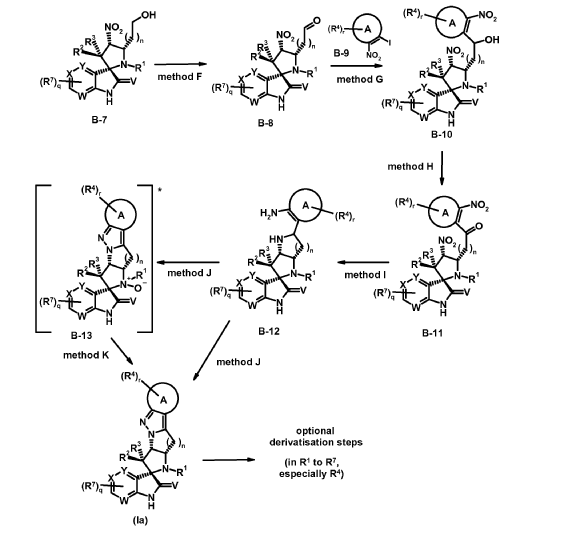
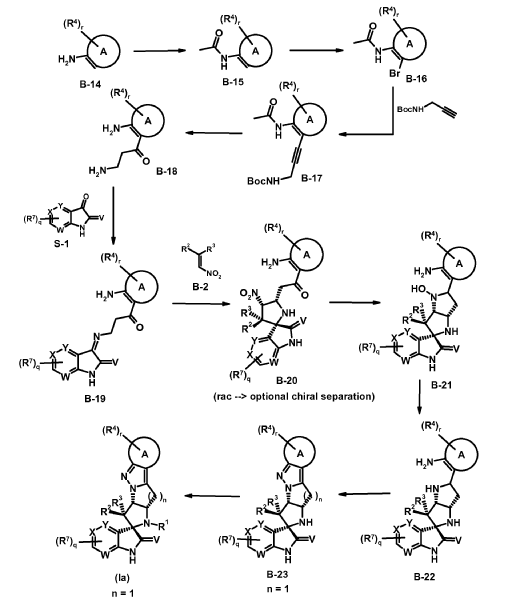
intermediates B-7
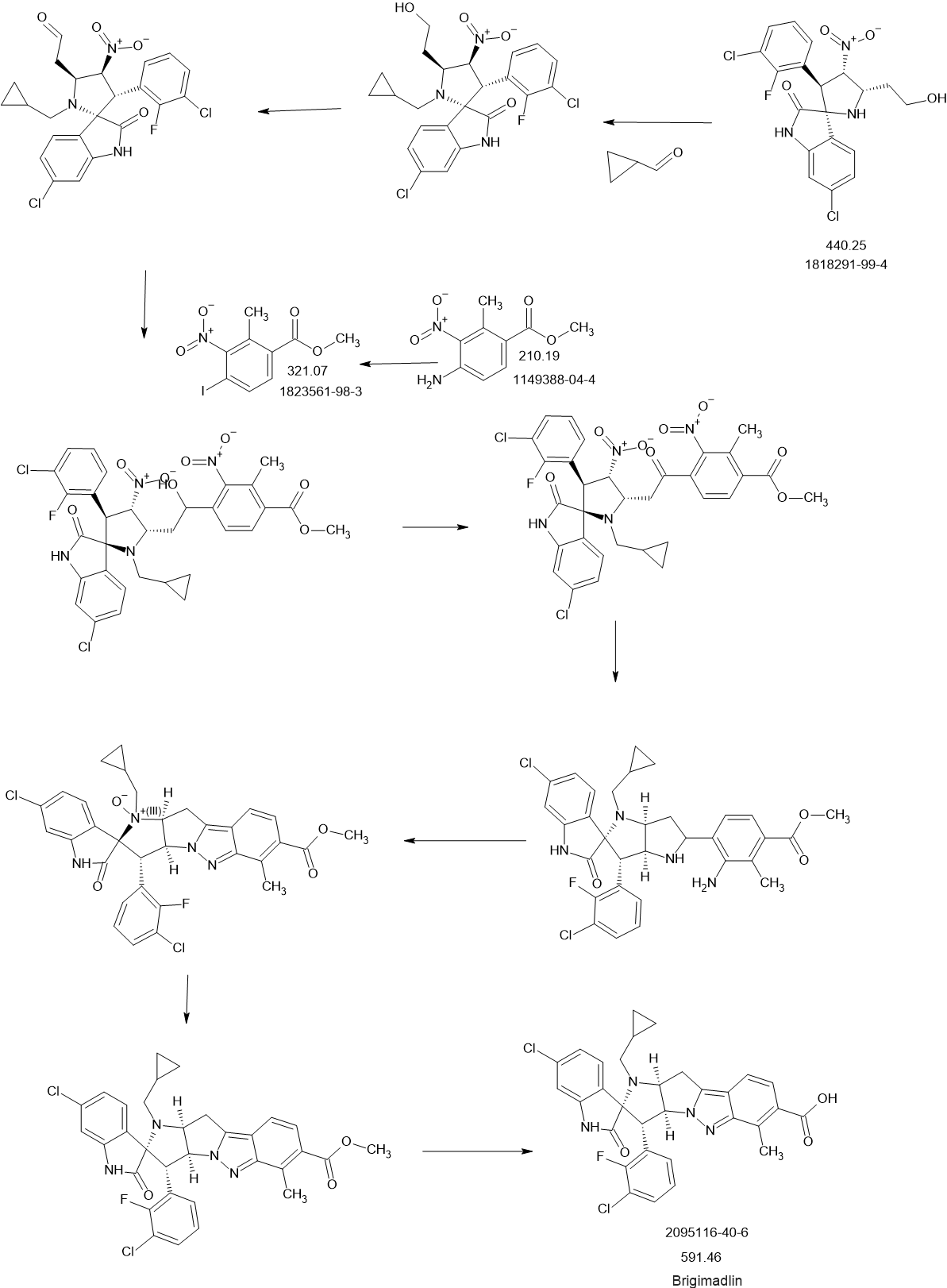
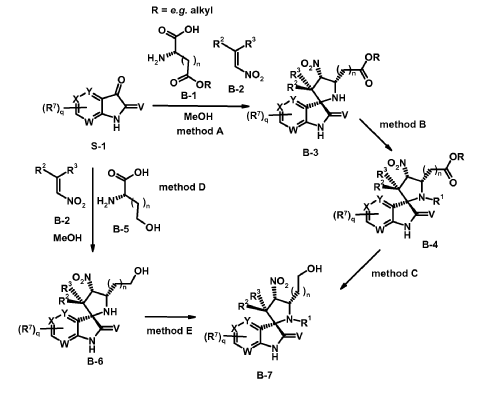
Experimental procedure for the synthesis of B-7 a (method E)
To a solution of cyclopropanecarbaldehyde (1.7 mL, 22.7 mmol) in AcOH (19.5 mL) is added intermediate B-6a (1.60 g, 3.8 mmol) and the reaction mixture is stirred for 15 min. Sodium triacetoxyborohydride (1.34 g, 6.3 mmol) is added and the reaction mixture is stirred overnight. Water is added to the reaction mixture and it is extracted with EtOAc. The combined organic layer is dried (MgSO4), filtered, concentrated in vacuo and the crude product B-7a is purified by chromatography if necessary.
Experimental procedure for the synthesis of B-3a (method A)
6-Chloroisatin S-1a (5 g, 27,0 mmol), 1-(3-chloro-2-fluoro-phenyl)-2-nitroethene B-2a (5.5 g, 27.0 mmol) and amino acid B-1a (4.4 g, 27.0 mmol) are refluxed in MeOH for 4 h. The reaction mixture is concentrated in vacuo and purified by crystallization or chromatography if necessary.
Synthesis of compounds (la) according to the invention
Experimental procedure for the synthesis of la-1 (method J)
To a solution of intermediate B-12a (329 mg, 0.65 mmol) in DCM (7 mL) is added a solution of Oxone® (793 mg, 1.29 mmol) in H2O (7 mL) at 0 °C dropwise. The biphasic reaction mixture is stirred vigorously for 20 min at 0 °C and for additional 2 h at rt. The reaction mixture is diluted with H2O and is extracted with DCM. The combined organic layer is dried (MgSO4), filtered, concentrated in vacuo and the crude product is purified by chromatography which gives compound la-1.
Experimental procedure for the synthesis of la-20 (method J + method K)
* The location of overoxidation/N-oxid formation is not entirely clear. B-13a as depicted seems to be probable.
To a solution of intermediate B-12j (417 mg, 0.68 mmol) in DCM (10 mL) is added a solution of Oxone® (841 mg, 1.37 mmol) in H2O (7 mL) at 0 °C dropwise. The biphasic reaction mixture is stirred vigorously for 20 min at 0 °C and for additional 6 h at rt. The reaction mixture is diluted with H2O and extracted with DCM. The combined organic layer is dried (MgSO4), filtered, concentrated in vacuo which gives a crude mixture of la-20 and an oxidized form B-13a (M+H = 621). This mixture is dissolved in MeCN (4.2 mL) and bis(pinacolato)diborone (326 mg, 1.28 mmol) is added. The reaction mixture is heated under microwave irradiation to 100 °C for 30 min. The reaction mixture is diluted with H2O and extracted with DCM. The combined organic layer is dried (MgSO4), filtered, concentrated in vacuo and the crude product is purified by chromatography which gives compound la-20.
References
^ “Brigimadlin”. pubchem.ncbi.nlm.nih.gov.
- ^ Rinnenthal, Joerg; Rudolph, Dorothea; Blake, Sophia; Gollner, Andreas; Wernitznig, Andreas; Weyer-Czernilofsky, Ulrike; Haslinger, Christian; Garin-Chesa, Pilar; Moll, Jürgen; Kraut, Norbert; McConnell, Darryl; Quant, Jens (1 July 2018). “Abstract 4865: BI 907828: A highly potent MDM2 inhibitor with low human dose estimation, designed for high-dose intermittent schedules in the clinic”. Cancer Research. 78 (13_Supplement): 4865. doi:10.1158/1538-7445.AM2018-4865. S2CID 56768874.
- ^ Rudolph, Dorothea; Reschke, Markus; Blake, Sophia; Rinnenthal, Jörg; Wernitznig, Andreas; Weyer-Czernilofsky, Ulrike; Gollner, Andreas; Haslinger, Christian; Garin-Chesa, Pilar; Quant, Jens; McConnell, Darryl B.; Norbert, Kraut; Moll, Jürgen (1 July 2018). “Abstract 4866: BI 907828: A novel, potent MDM2 inhibitor that induces antitumor immunologic memory and acts synergistically with an anti-PD-1 antibody in syngeneic mouse models of cancer”. Cancer Research. 78 (13_Supplement): 4866. doi:10.1158/1538-7445.AM2018-4866. S2CID 80770832.
- ^ Cornillie, J.; Wozniak, A.; Li, H.; Gebreyohannes, Y. K.; Wellens, J.; Hompes, D.; Debiec-Rychter, M.; Sciot, R.; Schöffski, P. (April 2020). “Anti-tumor activity of the MDM2-TP53 inhibitor BI-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring MDM2 amplification”. Clinical and Translational Oncology. 22 (4): 546–554. doi:10.1007/s12094-019-02158-z. PMID 31201607. S2CID 189862528.
- ^ Schöffski, Patrick; Lahmar, Mehdi; Lucarelli, Anthony; Maki, Robert G (March 2023). “Brightline-1: phase II/III trial of the MDM2–p53 antagonist BI 907828 versus doxorubicin in patients with advanced DDLPS”. Future Oncology. 19 (9): 621–629. doi:10.2217/fon-2022-1291. PMID 36987836. S2CID 257802972.
- ^ Schoeffski, P.; Lorusso, P.; Yamamoto, N.; Lugowska, I.; Moreno Garcia, V.; Lauer, U.; Hu, C.; Jayadeva, G.; Lahmar, M.; Gounder, M. (October 2023). “673P A phase I dose-escalation and expansion study evaluating the safety and efficacy of the MDM2–p53 antagonist brigimadlin (BI 907828) in patients (pts) with solid tumours”. Annals of Oncology. 34: S472 – S473. doi:10.1016/j.annonc.2023.09.1859. S2CID 264392338.
| Names | |
|---|---|
| IUPAC name(3S,10’S,11’S,14’S)-6-chloro-11′-(3-chloro-2-fluorophenyl)-13′-(cyclopropylmethyl)-6′-methyl-2-oxospiro[1H-indole-3,12′-8,9,13-triazatetracyclo[7.6.0.02,7.010,14]pentadeca-1,3,5,7-tetraene]-5′-carboxylic acid | |
| Identifiers | |
| CAS Number | 2095116-40-6 |
| 3D model (JSmol) | Interactive image |
| ChemSpider | 128922236 |
| DrugBank | DB18578 |
| EC Number | 826-645-5 |
| KEGG | D12842 |
| PubChem CID | 129264140 |
| UNII | 9A934ZAN94 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C31H25Cl2FN4O3 |
| Molar mass | 591.46 g·mol−1 |
| Hazards | |
| GHS labelling:[1] | |
| Pictograms | |
| Signal word | Danger |
| Hazard statements | H300, H360Df, H372, H413 |
| Precautionary statements | P203, P260, P264, P270, P273, P280, P301+P316, P318, P319, P321, P330, P405, P501 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
{kind=link}
{kind=link}
{kind=link}
/////////////BRIGIMADLIN, BI-907828, BI 907828, 9A934ZAN94
Bocodepsin



Bocodepsin, OKI-179
CAS 1834513-65-3
S-((3E)-4-((6S,9S)-12,12-DIMETHYL-4,8,11,14-TETRAOXO-9-(PROPAN-2-YL)-7-OXA -3,10,13-TRIAZA-1(2,4)-(1,3)THIAZOLACYCLOTETRADECAPHAN-6-YL)BUT-3-EN-1-YL) (2S)-2-AMINO-3-METHYLBUTANETHIOATE
S-(4-((7S,10S)-4,4-DIMETHYL-2,5,8,12-TETRAOXO-7-(PROPAN-2-YL)-9-OXA-16-THIA- 3,6,13,18-TETRAAZABICYCLO(13.2.1)OCTADECA-15(18),17-DIEN-10-YL)BUT-3-EN-1-YL) (2S)-2-AMINO-3-METHYLBUTANETHIOATE
| Molecular Weight | 581.75 |
|---|---|
| Formula | C26H39N5O6S2 |
- L-Valine, S-L-valyl-(3S,4E)-3-hydroxy-7-mercapto-4-heptenonyl-2-(aminomethyl)-4-thiazolecarbonyl-2-methylalanyl-, (5–>2)-lactone
- S-{(3E)-4-[(6S,9S)-12,12-dimethyl-4,8,11,14-tetraoxo-9-(propan-2-yl)-7-oxa-3,10,13-triaza-1(2,4)-[1,3]thiazolacyclotetradecaphan-6-yl]but-3-en-1-yl} (2S)-2-amino-3-methylbutanethioate
- S-{4-[(7S,10S)-4,4-dimethyl-2,5,8,12-tetraoxo-7-(propan-2-yl)-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-15(18),17-dien-10-yl]but-3-en-1-yl} (2S)-2-amino-3-methylbutanethioate
- Originator OnKure Therapeutics
- Class Antineoplastics; Small molecules
- Mechanism of ActionHDAC1 protein inhibitors
Phase I/II Malignant melanoma; Solid tumours
- No development reportedHaematological malignancies
29 Jan 2025 OnKure Therapeutics completes the phase-I/II Nautilus trial in Malignant melanoma (Late-stage disease, Metastatic disease, Second-line therapy or greater, Combination therapy) in USA (PO) (NCT05340621),
- 11 Oct 2023Pharmacodynamics data from a preclinical studies in Solid tumours presented at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics 2023 (AACR-NCI-EORTC-2023 2023)
- 11 Oct 2023Initial efficacy and adverse events data from a phase Ib/II NAUTILUS trial in Melanoma presented at the International Conference on Molecular Targets and Cancer Therapeutics 2023 (AACR-NCI-EORTC-2023)
Bocodepsin (OKI-179) is an orally active and selective HDAC inhibitor, with antitumor activity. Bocodepsin can be used for suppression on solid tumor and hematologic malignancies.
SCHEME


PATENT
WO2017201278
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017201278&_cid=P12-M9WKU5-87067-1

Examples
[00127] The following examples are provided for illustrative purposes only and are not intended to limit the scope of the invention.
[00128] Example 1: Preparation of (R)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12- tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10- yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride.
Step 1 : Preparation of (R)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12- tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10- yl)but-3-en-l-yl) 2-((tert-butoxycarbonyl)amino)-3-methylbutanethioate. (7S,10S)-10-((E)- 4- chlorobut-l-en-l-yl)-7-isopropyl-4,4-dimethyl-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-diene-2,5,8,12-tetraone (15 g, 0.03 mol), (R)-2- ((tert-butoxycarbonyl)amino)-3-methylbutanethioic S-acid (12.5 g, 0.06 mol), K2CO3 (11.2 g, 0.09 mol), and KI (0.89 g, 0.006 mol) were dissolved in 150mL of acetonitrile and the resulting mixture was warmed to 60-65°C and stirred under nitrogen. After 16 hours, the mixture was cooled to 20°C, 300 mL of water was added, and the resulting suspension was extracted with ethyl acetate (2X200 mL). The combined organic phases were dried with anhydrous sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (elution with ethyl acetate/petroleum ether = 1/1 to 4/1) to give (R)- 5- ((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[13.2.1] octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-((tert- butoxycarbonyl)amino)-3-methylbutanethioate (17.0 g, 80% yield).
Step 2: Preparation of (R)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12- tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10- yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride. (R)-S-((E)-4-((7S,10S)-7- isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[l 3.2.1] octadeca- 1 ( 17), 15 (18)-dien- 10-y l)but-3-en- 1 -y 1) 2-((tert-butoxy carbony l)amino)-3-methylbutanethioate (1.7 g, 0.025 mol) was dissolved in 150 mL of dichloromethane and trifluoroacetic acid (22.5 mL) was added at 10°C. After stirring at 10°C for 4 hours under nitrogen, the mixture was concentrated to dryness and the residue was dissolved in 100 mL of ethyl acetate and treated with 10 mL of 4M HCl/ethyl acetate solution. The mixture was then treated with petroleum ether (100 mL) and the resulting white solid was collected by filtration and dried to give (R)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[13.2.1] octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride (0.40 g, 26% yield). Mass Spec(m/z): 582.8 (M+l).
129] Example 2: Preparation of (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride.
Step 1 : (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-((tert-butoxy carbony l)amino)-3 -methy lbutanethioate. (7S,10S)-10-((E)-4-chlorobut- 1 -en- 1 -yl)-7-isopropyl-4,4-dimethyl-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-diene-2,5,8,12-tetraone (40 g, 0.0825 mol), (S)-2-((tert-butoxycarbonyl)amino)-3-methylbutanethioic S-acid (38.5 g, 0.165 mol), K2C03 (34.1 g, 0.247 mol), and KI (2.7 g, 0.0163 mol) were dissolved in 400 mL of acetonitrile and stirred at 60-65°C under nitrogen for 20 hours. The mixture was cooled to 20°C, water (300 mL) was added and the resulting suspension was extracted with ethyl acetate (2X200 mL). The organic phases were combined, dried with anhydrous sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (elution with ethyl acetate/petroleum ether = 1/1 to 4/1) to give (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-((tert-butoxycarbonyl)amino)-3-methylbutanethioate (49.8 g, 89% yield).
Step 2: (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate hydrochloride. (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-1 ( 17), 15( 18)-dien- 10-y l)but-3-en- 1 -y 1) 2-((tert-butoxy carbony l)amino)-3 -methylbutanethioate (47.8 g, 0.07 mol) was dissolved in dichloromethane (400 mL) and trifluoroacetic acid (65 mL) was added dropwise at 10 to 20°C while stirring under nitrogen. After the addition, the mixture was stirred at 15 to 20°C for 3 hours at which time an additional aliquot of trifluoroacetic acid (20 mL) was added and stirring at 15 to 20°C was continued for an additional 1.5 hours. The solution was then concentrated under vacuum to near dryness and the residue dissolved in ethyl acetate (250 mL). 20 mL of 4M HCl/ethyl acetate solution was then added while stirring at a temperature between 10 to 15°C resulting in the formation of a slurry. 250 mL n-heptane was then added and the solids were filtered, rinsed with n-heptane and dried in vacuo to give (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18- tetraazabicyclo[13.2.1] octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3 -methylbutanethioate hydrochloride as a white solid which contained some residual heptane. (49.0 g, 100% yield). Mass Spec(m/z): 582.8 (M+l)
130] Example 3: Preparation of (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3 -methylbutanethioate benzenesulfonate.
The product of Example 2, step 1 ((S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-((tert-butoxycarbonyl)amino)-3-methylbutanethioate) (1 eq.) was dissolved in acetonitrile (10 vol) at 20-25°C and the mixture was treated with
benzenesulfonic acid (3 eq.). After stirring at room temperature for 5 hours, the solvent was removed by decanting, the residual oil was treated with THF (5vol), and the resulting mixture was stirred over night at room temperature. The resulting white solid was collected by filtration and dried in vacuo to give (S)-S-((E)-4-((7S,10S)-7-isopropyl-4,4-dimethyl-2,5,8,12-tetraoxo-9-oxa-16-thia-3,6,13,18-tetraazabicyclo[13.2.1]octadeca-l(17),15(18)-dien-10-yl)but-3-en-l-yl) 2-amino-3-methylbutanethioate benzenesulfonate (90% yield; 98% purity). 1HNMR (d6-DMSO) δ: 0.56 to 0.57 (m, 3H), 0.76 to 0.78 (m, 3H), 0.92 to 0.94 (m, 3H), 0.96 to 0.98 (m, 3H), 1.45 to 1.48 (m, 3H), 1.70 to 1.72 (m, 3H), 2.07 to 2.16 (m, 2H), 2.27 to 2.28 (m, 2H), 2.93 to 2.95 (m, 1H), 2.94 to 2.95 (m, 1H), 2.97 to 3.1 (m, 1H), 4.13 to 4.15 (m, 1H), 4.28 to 4.33 (1H), 4.92 to 5.0 (m, 1H), 5.61 to 5.64 (m, 3H), 7.29 to 7.32 (m, 3H), 7.57 to 7.60 (m, 2H), 7.88 to 7.92 (m, 1H), 8.17 (s, 1H), 8.32 (s, 3H), 8.48 to 8.50 (m, 1H).
[1]. Diamond JR, et al. Preclinical Development of the Class-I-Selective Histone Deacetylase Inhibitor OKI-179 for the Treatment of Solid Tumors. Mol Cancer Ther. 2022 Mar 1;21(3):397-406. [Content Brief]
///////////Bocodepsin, K5D067O1SW, OKI-179, Malignant melanoma, Solid tumours, OnKure Therapeutics, OKI 006
Tegeprotafib



Tegeprotafib
CAS 2407610-46-0
| Molecular Weight | 326.30 |
|---|---|
| Formula | C13H11FN2O5S |
PTPN2/1-IN-1, YGY4WEM0NZ
5-(1-fluoro-3-hydroxy-7-methoxynaphthalen-2-yl)-1,1-dioxo-1,2,5-thiadiazolidin-3-one
Tegeprotafib (PTPN2/1-IN-1) (Compound 124) is an orally active PTPN1 and PTPN2 inhibitor with IC50s of 4.4 nM and 1-10 nM against PTPN2 and PTP1B, respectively.
| Cancer immunotherapy regimens targeting immune evasion mechanisms including checkpoint blockade (e.g., PD-1/PD-L1 and CTLA-4 blocking antibodies) have been shown to be effective in treating in a variety of cancers, dramatically improving outcomes in some populations refractory to conventional therapies. However, incomplete clinical responses and the development of intrinsic or acquired resistance will continue to limit the patient populations who could benefit from checkpoint blockade. |
SCHEME

PATENT
Calico Life Sciences LLC; AbbVie Inc. , WO2021127499
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021127499&_cid=P21-M9UYU6-17583-1


Example 25: 5-(1-fluoro-3-hydroxy-7-methoxynaphthalen-2-yl)-1λ6,2,5-thiadiazolidine-1,1,3-trione (Compound 124)
Example 25A: benzyl 3-(benzyloxy)-7-methoxynaphthalene-2-carboxylate
A mixture of 3-hydroxy-7-methoxy-2-naphthoic acid (75 g, 344 mmol) and cesium carbonate (336 g, 1031 mmol) in N,N-dimethylformamide (687 mL) was rapidly stirred for 5 minutes at 23 °C. Thereafter, benzyl bromide (84 mL, 705 mmol) was added. After 90 minutes, the mixture was poured into H2O (1 L) and extracted with ethyl acetate (4 × 300 mL). The combined organic layers were washed with saturated aqueous ammonium chloride (3 × 100 mL), dried over sodium sulfate, filtered, and concentrated in vacuo to afford a brown solid. The crude solid was collected by filtration, slurried with tert-butyl methyl ether/heptanes (1:2, 3 × 100 mL), then dried in vacuo (12 mbar) at 40 °C to afford the title compound (122.5 g, 307 mmol, 89% yield) as a beige solid. MS (APCI+) m/z 399 [M+H]+.
Example 25B: 3-(benzyloxy)-7-methoxynaphthalene-2-carboxylic acid
To a suspension of the product of Example 25A (122.5 g, 307 mmol) in methanol (780 mL) was added 6 M aqueous sodium hydroxide (154 mL, 922 mmol). The heterogeneous, brown slurry was agitated with an overhead mechanical stirrer and heated to an internal temperature of 68 °C. After 15 minutes, the mixture was cooled to room temperature in an ice bath, and 6 M HCl (250 mL) was added over 5 minutes. The off-white solid was collected by filtration, washed with H2O (3 × 500 mL), and dried to constant weight in vacuo at 65 °C to afford the title compound (84.1 g, 273 mmol, 89% yield) as a white solid. MS (APCI+) m/z 309 [M+H]+.
Example 25C: 3-(benzyloxy)-7-methoxynaphthalen-2-amine
To a suspension of the product of Example 25B (84.1 g, 273 mmol), in toluene (766 mL) and tert-butanol (766 mL) was added triethylamine (40.3 mL, 289 mmol). The homogeneous black solution was heated to an internal temperature of 80 °C under nitrogen, and diphenyl phosphorazidate (62.2 mL, 289 mmol) was added dropwise over 90 minutes with the entire
reaction behind a blast shield. After 5 hours, the reaction was cooled to room temperature, diluted with H2O (1.5 L), and extracted with ethyl acetate (3 × 150 mL). The combined organic layers were washed with brine (2 × 100 mL), dried over sodium sulfate, filtered and concentrated to give 180.1 g of a dark brown solid. The solid was carried forward to hydrolysis without further purification.
To the crude intermediate was added diethylenetriamine (475 mL, 4.40 mol). The heterogeneous suspension was heated to an internal temperature of 130 °C under nitrogen, at which time a homogeneous dark orange solution formed. After 16 hours, the mixture was cooled to room temperature in an ice bath, and H2O (1.5 L) was added slowly over 3 minutes, resulting in precipitation of a yellow solid and a concomitant exotherm to an internal temperature of 62 °C. Once the heterogeneous suspension had cooled to room temperature, the crude solid was dissolved in CH2Cl2 (1.5 L), and the layers were separated. The aqueous layer was back-extracted with CH2Cl2 (3 × 150 mL), and the combined organic layers were washed with brine (3 × 100 mL), dried over sodium sulfate, filtered, and concentrated in vacuo to afford 78.8 g of an orange solid. The solid was slurried with isopropanol (50 mL), collected via filtration, re-slurried with isopropanol (1 × 50 mL), and dried in vacuo (15 mbar) at 35 °C to afford the title compound (60.12 g, 215 mmol, 79% yield over two steps) as a yellow solid. MS (APCI+) m/z 280 [M+H]+.
Example 25D: methyl {[3-(benzyloxy)-7-methoxynaphthalen-2-yl]amino}acetate
To a mixture of the product of Example 25C (59.2 g, 212 mmol) and potassium carbonate (58.6 g, 424 mmol) in dimethylformamide (363 mL) and H2O (1.91 mL, 106 mmol) was added methyl 2-bromoacetate (30.1 mL, 318 mmol). The suspension was vigorously stirred at room temperature for 5 minutes and then heated to an internal temperature of 60 °C. After 70 minutes, the suspension was cooled to room temperature and diluted with H2O (600 mL) and ethyl acetate (500 mL). The aqueous layer was extracted with ethyl acetate (2 × 300 mL), and the combined organic layers were washed with saturated aqueous ammonium chloride (3 × 60 mL), dried over sodium sulfate, filtered, and concentrated to afford 104.3 g of a pale beige solid. The solid was triturated with heptanes (200 mL). The resulting beige solid was collected via filtration, washed with additional heptanes (2 × 30 mL), and dried in vacuo (15 mbar) at 35 °C to afford the title compound (72.27 g, 206 mmol, 97% yield) as an off-white solid. MS (APCI+) m/z 352 [M+H]+.
Example 25E: methyl {[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl]amino}acetate To a mixture of the product of Example 25D (30.0 g, 85 mmol) and N-fluorobenzenesulfonimide (26.9 g, 85 mmol) was added tetrahydrofuran (THF) (854 mL), and
the resulting homogeneous yellow solution was stirred at room temperature. After 90 minutes, residual oxidant was quenched by adding a solution of sodium thiosulfate pentahydrate (10.59 g, 42.7 mmol) in water (150 mL), and the mixture was stirred at room temperature for 30 minutes. Thereafter, ethyl acetate (600 mL) was added, the aqueous layer was separated, and the organic layer was washed with a solution of sodium carbonate (18.10 g, 171 mmol) in water (30 mL), followed by water:brine (1:1, 1 × 20 mL). The organic fraction was dried over sodium sulfate, filtered, and the concentrated in vacuo to afford a bright yellow/orange solid. The solids were triturated with tert-butyl methyl ether (300 mL), collected via filtration, and the filter cake (N-(phenylsulfonyl)benzenesulfonamide) was washed with tert-butyl methyl ether (2 × 100 mL). The filtrate was concentrated to afford 34.6 g of a dark red oil that was purified by flash chromatography (750 g SiO2, heptanes to 20% ethyl acetate/heptanes) to afford the title compound (16.07 g, 43.5 mmol, 51% yield) as a yellow solid. MS (APCI+) m/z 370 [M+H]+. Example 25F: methyl {[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl](sulfamoyl)amino}acetate
To a solution of chlorosulfonyl isocyanate (5.13 mL, 59.1 mmol) in dichloromethane (83 mL) at 0 °C was added tert-butanol (5.65 mL, 59.1 mmol) slowly so that the internal temperature remained less than 10 °C. After stirring for 30 minutes at 0 °C, a preformed solution of the product of Example 25E (14.55 g, 39.4 mmol) and triethylamine (10.98 mL, 79 mmol) in dichloromethane (68.9 mL) was added slowly via addition funnel so that the internal temperature remained below 10 °C. Upon complete addition, the addition funnel was rinsed with dichloromethane (23 mL). The resulting solution was stirred for 30 minutes at 0 °C, and then the reaction mixture was quenched with H2O (20 mL). The layers were separated, and the aqueous layer was extracted with dichloromethane (2 × 30 mL). The combined organic layers were washed with brine (1 × 30 mL), dried over sodium sulfate, filtered and concentrated in vacuo to give an orange oil. The residue was dissolved in ethyl acetate (200 mL) and washed with water:brine (1:1, 2 × 50 mL) to remove residual triethylamine hydrochloride. The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo to give methyl {[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl][(tert-butoxycarbonyl)sulfamoyl]amino}acetate which was used without purification.
To a solution of methyl {[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl][(tert-butoxycarbonyl)sulfamoyl]amino}acetate in dichloromethane (98 mL) was added trifluoroacetic acid (45.5 mL, 591 mmol), and the resulting dark solution was stirred at room temperature. After 20 minutes, the reaction was quenched by slow addition of saturated aqueous sodium bicarbonate (691 mL) via an addition funnel. The layers were separated, and the aqueous layer was extracted with dichloromethane (2 × 50 mL). The combined organic layers were concentrated to give a dark red oil; upon addition of tert-butyl methyl ether (60 mL), a yellow solid precipitated that was collected via filtration, washed with tert-butyl methyl ether (2 × 30 mL) and dried in vacuo (15 mbar) at 35 °C to give the title compound (13.23 g, 29.5 mmol, 75% yield over two steps) as a light yellow solid. MS (ESI+) m/z 449 [M+H]+.
Example 25G: 5-(1-fluoro-3-hydroxy-7-methoxynaphthalen-2-yl)-1λ6,2,5-thiadiazolidine-1,1,3-trione
To a solution of the product of Example 25F (13.23 g, 29.5 mmol) in tetrahydrofuran (THF) (355 mL) at room temperature was added solid potassium tert-butoxide (3.31 g, 29.5 mmol), and the resulting solution was stirred at room temperature. After 10 minutes, the reaction was quenched with 1 M hydrochloric acid (90 mL) and diluted with ethyl acetate (400 mL). The layers were separated, and the aqueous layer was extracted with ethyl acetate (2 × 120 mL). The combined organic layers were washed with brine (3 × 50 mL), then dried over sodium sulfate, filtered and concentrated. The crude 5-[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl]-1λ6,2,5-thiadiazolidine-1,1,3-trione was used in the subsequent reaction without further purification.
A mixture of crude intermediate, 5-[3-(benzyloxy)-1-fluoro-7-methoxynaphthalen-2-yl]-1λ6,2,5-thiadiazolidine-1,1,3-trione (12.28 g, 29.5 mmol) and pentamethylbenzene (13.11 g, 88 mmol) in dichloromethane (147 mL) was cooled to an internal temperature of –76 °C under an atmosphere of dry nitrogen. Subsequently, a 1 M solution of boron trichloride (59.0 mL, 59.0 mmol) in CH2Cl2 was added dropwise over 15 minutes, so as not to raise the internal temperature past –72 °C. Over the course of the addition, the reaction turned dark brown and became homogeneous. Incomplete conversion was observed, and additional boron trichloride (2 × 5.90 mL, 2 × 5.90 mmol) was added, resulting in full conversion. The reaction was quenched at –75 °C with CH2Cl2/methanol (10:1, 140 mL) via cannula transfer under nitrogen over 15 minutes, then slowly warmed to room temperature over 20 minutes under nitrogen. The volatiles were removed in vacuo to afford a brown/tan solid, which was collected by filtration, and slurried with heptanes (5 × 40 mL) and CH2Cl2 (3 × 40 mL). The crude solid was suspended in isopropanol (75 mL), warmed until the material dissolved, then allowed to cool slowly to room temperature over 1 hour. The solid was collected by filtration, washed with heptanes (2 × 30 mL), and dried in vacuo (15 mbar) at 60 °C to afford 5.11 g of a white solid. The mother liquor was concentrated, and the process was repeated to give an additional 1.96 g of a white solid. The batches were combined to obtain the title compound (7.07 g, 21.67 mmol, 73.5% yield over two steps). 1H NMR (CD3OD) δ ppm 7.60 (dd, J = 9.1, 1.5 Hz, 1H), 7.25 (d, J = 2.6, 1H), 7.16 (dd, J = 9.1, 2.6 Hz, 1H), 7.04 (s, 1 H), 4.56 (s, 2H), 3.89 (s, 3 H); MS (ESI–) m/z 325 [M–H]–.
PATENT
WO2020186199
WO2019246513
PATENT
compound 124 [US20230019236A1]
https://patentscope.wipo.int/search/en/detail.jsf?docId=US389737555&_cid=P21-M9UYQD-14144-1
///////Tegeprotafib, PTPN2/1-IN-1, YGY4WEM0NZ
Probenecid



Probenecid
- 57-66-9
- 4-(Dipropylsulfamoyl)benzoic acid
- Probenecid acid
- Benemid
4-(dipropylsulfamoyl)benzoic acid
C13H19NO4S, 285.359
HC 5006- NSC-18786
FDA APPROVED, 10/25/2024, sulopenem etzadroxil, probenecid, Orlynvah, To treat uncomplicated urinary tract infections (uUTI)
Drug Trial Snapshot
Probenecid, also sold under the brand name Probalan, is a medication that increases uric acid excretion in the urine. It is primarily used in treating gout and hyperuricemia.
Probenecid was developed as an alternative to caronamide[1] to competitively inhibit renal excretion of some drugs, thereby increasing their plasma concentration and prolonging their effects.
Experimental Properties
| Property | Value | Source |
|---|---|---|
| melting point (°C) | 195 °C | PhysProp |
| water solubility | 27.1 mg/L | Not Available |
| logP | 3.21 | HANSCH,C ET AL. (1995) |
| pKa | 3.4 | SANGSTER (1994) |
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US12109197 | No | 2024-10-08 | 2039-04-01 |  |
| US11554112 | No | 2023-01-17 | 2039-04-01 | |
| US11478428 | No | 2022-10-25 | 2039-12-23 | |
| US7795243 | No | 2010-09-14 | 2029-06-03 | |

PATENT
https://patents.google.com/patent/CN103613521A/en
At present, the production technique of probenecid mainly contains two kinds:
(1) p-methyl benzenesulfonic acid-dipropyl amine method
Take p-methyl benzenesulfonic acid as raw material, through potassium bichromate or potassium permanganate oxidation, then react generation with chlorsulfonic acid generation sulfonating chlorinating to carboxyl benzene sulfonyl chloride, amidate action occurs then in organic solvent and obtain the finished product probenecid.Reaction process route is as follows:

This technique in a large number with an organic solvent, seriously polluted; Heavy metal recovery and treatment cost are high; Chlorsulfonic acid transportation, storage and use are dangerous large, and acid mist is obvious.Along with the increasing of environmental protection pressure, people increase severely day by day to the concern of environment, and this route is substantially in end-of-life state.
(2) to methyl benzenesulfonamide-Halopropane method
To methyl benzenesulfonamide, through potassium bichromate or potassium permanganate oxidation, be P―Carboxybenzenesulfonamide, under the effect of alkali, with Halopropane generation alkylated reaction, after acidifying, obtain probenecid.Reaction process route is as follows:

This process using sodium dichromate 99 or potassium permanganate oxidation are to methyl benzenesulfonamide, and yield is on the low side (lower than 50%).In addition, the waste water that contains chromium or manganese is difficult to dispose, and these have all seriously restricted further developing of this technique.
Reaction scheme of the present invention is as follows:

embodiment 1
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 127.4ml hydrochloric acid (31%, 1.25mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to 0-5 ℃, drip sodium nitrite solution (34.5g Sodium Nitrite, 0.5mol, be dissolved in 190g water), control temperature at 10-20 ℃, it is 4 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 765ml hydrochloric acid (31%, 7.5mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at-3–1 ℃, when passing into 64g sulfurous gas (1mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 5 hours, is warming up to gradually 5-10 ℃, continues reaction 8 hours at this temperature; Filtration obtains 121g to carboxyl benzene sulfonyl chloride.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 152g dipropyl amine (1.5mol), open and stir, when temperature is greater than 15 ℃, start to divide gradually 40 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 3 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filter, obtain 135g probenecid crude product, put in 500ml pure water, agitator treating 1 hour, heavy 122.8g after filtering, being dried, yield 86.2%(is in para-amino benzoic acid), purity 98.2%.
embodiment 2
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 152.9ml hydrochloric acid (31%, 1.5mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to 0-5 ℃, drip sodium nitrite solution (36.0g Sodium Nitrite, 0.52mol, be dissolved in 190g water), control temperature at 0-10 ℃, it is 3 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 887ml hydrochloric acid (31%, 8.7mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at 0-5 ℃, when passing into 112g sulfurous gas (1.75mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 4 hours, is warming up to gradually 5-15 ℃, continues reaction 5 hours at this temperature; Filtration obtains 150g to carboxyl benzene sulfonyl chloride.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 192g dipropyl amine (1.9mol), open and stir, when temperature is greater than 15 ℃, start to divide gradually 35 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 2 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filter, obtain 155.4g probenecid crude product, put in 500ml pure water, agitator treating 1 hour, heavy 129.5g after filtering, being dried, yield 90.9%(is in para-amino benzoic acid), purity 98.7%.
embodiment 3
(1) diazotization reaction
Get 68.6g para-amino benzoic acid (0.5mol), 250g water and 203.9ml hydrochloric acid (31%, 2mol) join in 2000ml there-necked flask, in ice-water bath, stir, be cooled to-10–5 ℃, drip sodium nitrite solution (38.0g Sodium Nitrite, 0.55mol, be dissolved in 190g water), control temperature at 0-10 ℃, it is 5 hours that time for adding is controlled, after dropping finishes, at this temperature, continue reaction 1 hour, obtain diazotization reaction liquid.
(2) sulfonating chlorinating reaction
In 5000ml there-necked flask, add 250g water, 968ml hydrochloric acid (31%, 9.5mol), in ice-water bath, stir, be cooled to-5 ℃, start to pass into liquid sulfur dioxide, control temperature at 5-10 ℃, when passing into 160g sulfurous gas (2.5mol), sulfurous gas absorbs complete, obtains sulfonating chlorinating reagent.
In sulfonating chlorinating reagent, add diazotization reaction liquid, adding the time control of diazotization reaction liquid is 3 hours, is warming up to gradually 10-15 ℃, continues reaction 20 hours at this temperature; Filtration obtains 146.7g to carboxyl benzene sulfonyl chloride, needn’t be dried, and directly enters next step reaction.
(3) synthetic probenecid reaction
In 1000ml there-necked flask, add 350g water, 202g dipropyl amine (2mol), open to stir, when temperature is greater than 30 ℃, start to divide gradually 30 batches add step (2) gained to carboxyl benzene sulfonyl chloride, temperature control 40-50 ℃, adds and at this temperature, stirs 4 hours continuing after carboxyl benzene sulfonyl chloride.Drip hydrochloric acid (31%), regulate pH value to 2-3, continue to stir 1 hour.Filtration obtains 151.7g probenecid crude product, puts in 500ml pure water, and agitator treating 1 hour, heavy 128.5g after filtering, being dried, yield 90.2%(is in para-amino benzoic acid), purity 98.8%.Medical uses
Probenecid is primarily used to treat gout and hyperuricemia.
Probenecid is sometimes used to increase the concentration of some antibiotics and to protect the kidneys when given with cidofovir. Specifically, a small amount of evidence supports the use of intravenous cefazolin once rather than three times a day when it is combined with probenecid.[2]
It has also found use as a masking agent,[3] potentially helping athletes using performance-enhancing substances to avoid detection by drug tests.
Adverse effects
Mild symptoms such as nausea, loss of appetite, dizziness, vomiting, headache, sore gums, or frequent urination are common with this medication. Life-threatening side effects such as thrombocytopenia, hemolytic anemia, leukemia and encephalopathy are extremely rare.[4] Theoretically probenecid can increase the risk of uric acid kidney stones.
Drug interactions
Some of the important clinical interactions of probenecid include those with captopril, indomethacin, ketoprofen, ketorolac, naproxen, cephalosporins, quinolones, penicillins, methotrexate, zidovudine, ganciclovir, lorazepam, and acyclovir. In all these interactions, the excretion of these drugs is reduced due to probenecid, which in turn can lead to increased concentrations of these.[5]
Pharmacology
Pharmacodynamics
In gout, probenecid competitively inhibits the reabsorption of uric acid through the organic anion transporter (OAT) at the proximal tubules. This leads to preferential reabsorption of probenecid back into plasma and excretion of uric acid in urine,[6] thus reducing blood uric acid levels and reducing its deposition in various tissues.
Probenecid also inhibits pannexin 1.[7] Pannexin 1 is involved in the activation of inflammasomes and subsequent release of interleukin-1β causing inflammation. Inhibition of pannexin 1 thus reduces inflammation, which is the core pathology of gout.[7]
Pharmacokinetics
In the kidneys, probenecid is filtered at the glomerulus, secreted in the proximal tubule and reabsorbed in the distal tubule. Probenicid lowers the concentration of certain drugs in urine drug screens by reducing renal excretion of these drugs.
Historically, probenecid has been used to increase the duration of action of drugs such as penicillin and other beta-lactam antibiotics. Penicillins are excreted in the urine at proximal and distal convoluted tubules through the same organic anion transporter (OAT) as seen in gout. Probenecid competes with penicillin for excretion at the OAT, which in turn increases the plasma concentration of penicillin.[8]
History
During World War II, probenecid was used to extend limited supplies of penicillin. This use exploited probenecid’s interference with drug elimination (via urinary excretion) in the kidneys and allowed lower doses of penicillin to be used.[9]
Probenecid was added to the International Olympic Committee‘s list of banned substances in January 1988, due to its use as a masking agent.[10]
References
- ^ Mason RM (June 1954). “Studies on the effect of probenecid (benemid) in gout”. Annals of the Rheumatic Diseases. 13 (2): 120–130. doi:10.1136/ard.13.2.120. PMC 1030399. PMID 13171805.
- ^ Cox VC, Zed PJ (March 2004). “Once-daily cefazolin and probenecid for skin and soft tissue infections”. The Annals of Pharmacotherapy. 38 (3): 458–463. doi:10.1345/aph.1d251. PMID 14970368. S2CID 11449580.
- ^ Morra V, Davit P, Capra P, Vincenti M, Di Stilo A, Botrè F (December 2006). “Fast gas chromatographic/mass spectrometric determination of diuretics and masking agents in human urine: Development and validation of a productive screening protocol for antidoping analysis”. Journal of Chromatography A. 1135 (2): 219–229. doi:10.1016/j.chroma.2006.09.034. hdl:2318/40201. PMID 17027009. S2CID 20282106.
- ^ Kydd AS, Seth R, Buchbinder R, Edwards CJ, Bombardier C (November 2014). “Uricosuric medications for chronic gout”. The Cochrane Database of Systematic Reviews (11): CD010457. doi:10.1002/14651858.CD010457.pub2. PMC 11262558. PMID 25392987.
- ^ Cunningham RF, Israili ZH, Dayton PG (March–April 1981). “Clinical pharmacokinetics of probenecid”. Clinical Pharmacokinetics. 6 (2): 135–151. doi:10.2165/00003088-198106020-00004. PMID 7011657. S2CID 24497865.
- ^ “Probenecid”. PubChem. U.S. National Library of Medicine. Retrieved 2022-06-12.
- ^ Jump up to:a b Silverman W, Locovei S, Dahl G (September 2008). “Probenecid, a gout remedy, inhibits pannexin 1 channels”. American Journal of Physiology. Cell Physiology. 295 (3): C761 – C767. doi:10.1152/ajpcell.00227.2008. PMC 2544448. PMID 18596212.
- ^ Ho RH (January 2010). “4.25 – Uptake Transporters”. In McQueen CA, Kim RB (eds.). Comprehensive Toxicology (Second ed.). Oxford: Elsevier. pp. 519–556. doi:10.1016/B978-0-08-046884-6.00425-5. ISBN 978-0-08-046884-6.
- ^ Butler D (November 2005). “Wartime tactic doubles power of scarce bird-flu drug”. Nature. 438 (7064): 6. Bibcode:2005Natur.438….6B. doi:10.1038/438006a. PMID 16267514.
- ^ Wilson W, Derse E, eds. (2001). Doping in Elite Sport: The Politics of Drugs in the Olympic Movement. Human Kinetics. p. 86. ISBN 0-7360-0329-0.
| Clinical data | |
|---|---|
| Trade names | Probalan |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682395 |
| Routes of administration | By mouth |
| ATC code | M04AB01 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 75-95% |
| Elimination half-life | 2-6 hours (dose: 0.5-1 g) |
| Excretion | kidney (77-88%) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 57-66-9 |
| PubChem CID | 4911 |
| IUPHAR/BPS | 4357 |
| DrugBank | DB01032 |
| ChemSpider | 4742 |
| UNII | PO572Z7917 |
| KEGG | D00475 |
| ChEMBL | ChEMBL897 |
| CompTox Dashboard (EPA) | DTXSID9021188 |
| ECHA InfoCard | 100.000.313 |
| Chemical and physical data | |
| Formula | C13H19NO4S |
| Molar mass | 285.36 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
{kind=link}
{kind=link}
/////////probenecid, APPROVALS 2024, FDA 2024, Orlynvah, HC 5006, NSC-18786
#probenecid, #APPROVALS 2024, #FDA 2024, #Orlynvah, #HC 5006, #NSC-18786
Bleximenib



Bleximenib
CAS 2654081-35-1
WeightAverage: 599.796
Monoisotopic: 599.395916661
Chemical FormulaC32H50FN7O3
- CS-0636752
- DA-55335
- HY-148669
- PHASE 3
- JNJ-75276617; Menin-MLL inhibitor 24
- Benzamide, N-ethyl-5-fluoro-2-[[5-[2-[(1R)-4-[(2-methoxyethyl)methylamino]-1-(1-methylethyl)butyl]-2,6-diazaspiro[3.4]oct-6-yl]-1,2,4-triazin-6-yl]oxy]-N-(1-methylethyl)-
- N-ethyl-5-fluoro-2-{[5-(2-{(3R)-6-[(2-methoxyethyl)(methyl)amino]-2-methylhexan-3-yl}-2,6-diazaspiro[3.4]octan-6-yl)-1,2,4-triazin-6-yl]oxy}-N-(propan-2-yl)benzamide

2866179-95-3 (oxalate)
(R)-N-ethyl-5-fluoro-N-isopropyl-2-((5-(2-(6-((2-methoxyethyl)(methyl)amino)-2-methylhexan-3-yl)-2,6-diazaspiro[3.4]octan-6-yl)-1,2,4-triazin-6-yl)oxy)benzamide oxalate
Chemical Formula: C34H52FN7O7
Exact Mass: 689.39
Molecular Weight: 689.830
Elemental Analysis: C, 59.20; H, 7.60; F, 2.75; N, 14.21; O, 16.23
\Bleximenib is under investigation in clinical trials NCT04811560 (A Phase 1/2 Study of Bleximenib in Participants With Acute Leukemia) and NCT05453903 (A Study of Bleximenib in Combination With Acute Myeloid Leukemia (AML) Directed Therapies)
Bleximenib (JNJ-75276617) is an orally active and selective menin-KMT2A inhibitor, with IC50 values of 0.1 nM, 0.045 nM, and ≤0.066 nM for humans, mice, and dogs, respectively. Bleximenib can inhibit the proliferation and induce apoptosis and differentiation of tumor cells. Bleximenib can be used in the research of tumors such as leukemia.
Bleximenib is an orally bioavailable protein-protein interaction (PPI) inhibitor of the menin-mixed lineage leukemia (MLL; mixed-lineage leukemia 1; MLL1; myeloid/lymphoid leukemia; histone-lysine N-methyltransferase 2A; KMT2A) proteins, with potential antineoplastic activity. Upon oral administration, bleximenib inhibits the interaction between the two proteins menin and MLL and the formation of the menin-MLL complex. This reduces the expression of downstream target genes and results in an inhibition of the proliferation of leukemic cells with either KMT2A alterations such as gene rearrangements (KMT2A-r), duplications, and amplification, or nucleophosmin 1 gene (NPM1) alterations. The menin-MLL complex plays a key role in the survival, growth, transformation and proliferation of certain kinds of leukemia cells.
SCHEME

SIDECHAIN

PATENTS
Janssen Pharmaceutica NV; Johnson & Johnson (China) Investment Ltd.
WO2021121327
WO2022237719
PATENT
WO2022237720

PATENTS
PATENT
Compound A—(R)-N-ethyl-5-fluoro-N-isopropyl-2-((5-(2-(6-((2-methoxyethyl) (methyl)amino)-2-methylhexan-3-yl)-2,6-diazaspiro[3.4]octan-6-yl)-1,2,4-triazin-6-yl)oxy) benzamide
PATENT
WO2022262796
The present invention is directed to (R) -N-ethyl-5-fluoro-N-isopropyl-2- ( (5- (2- (6- ( (2-methoxyethyl) (methyl) amino) -2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) benzamide besylate salt (benzenesulfonate salt) :
[0011]
[0140]
tert-butyl (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate
[0141]
[0142]
The mixture 2- ( (5- (2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) -N-ethyl-5-fluoro-N-isopropylbenzamide (intermediate 3) (1.0 g, 2.4 mmol) , tert-butyl (5-methyl-4-oxohexyl) carbamate (intermediate 1) (830 mg, 3.62 mmol) and ZnCl 2(660 mg, 4.84 mmol) in MeOH (15 mL) was stirred at 80 ℃ for 0.5 h. Then NaBH 3CN (310 mg, 4.93 mmol) was added and the resulting mixture was stirred at 80 ℃ for 6 h. After cooled to RT, the mixture was concentrated under reduced pressure to give the crude product, which was further purified by preparative HPLC using a Waters Xbridge Prep OBD (column: C18 150×40 mm 10 um; eluent: ACN/H 2O (0.05%ammonia) from 45%to 75%v/v) to afford the title compound (700 mg, 46%yield) as colorless oil.
reparation of Compounds 62 and 63
[0144]
tert-butyl (R) – (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate
[0145]
tert-butyl (S) – (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate
[0146]
[0147]
tert-butyl (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate (Compound 61) (200 mg, 0.319 mmol) was purified by SFC over DAICEL CHIRALPAK IG (column: 250×30 mm 10 um; isocratic elution: EtOH (containing 0.1%of 25%ammonia) : supercritical CO 2, 40%: 60% (v/v) ) to afford the title compounds (Compound 62) (85 mg, 42%yield) and (Compound 63) (80 mg, 40%yield) both as light yellow oil.
[0148]
[0149]
(R) -2- ( (5- (2- (6-amino-2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) -N-ethyl-5-fluoro-N-isopropylbenzamide
[0150]
[0151]
To the solution of tert-butyl (R) – (4- (6- (6- (2- (ethyl (isopropyl) carbamoyl) -4-fluorophenoxy) -1, 2, 4-triazin-5-yl) -2, 6-diazaspiro [3.4] octan-2-yl) -5-methylhexyl) carbamate (Compound 62) (550 mg, 0.876 mmol) in DCM (4 mL) was slowly added TFA (4 mL) , and the resulting mixture was stirred at 25 ℃ for 1 h. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was diluted in DCM (40 mL) and the pH value was adjusted to around 12 by aq. NaOH (2 M, 16 mL) solution. The aqueous layer was extracted with DCM (10 mL x 2) . The combined organic layers were dried over anhydrous Na 2SO 4, filtered and concentrated in vacuo to afford the title compound (460 mg, crude) as yellow solid, which was used directly in next step without further purification.
[0152]
[0153]
(R) -N-ethyl-5-fluoro-N-isopropyl-2- ( (5- (2- (6- ( (2-methoxyethyl) amino) -2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) benzamide
[0154]
[0155]
The mixture of (R) -2- ( (5- (2- (6-amino-2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) -N-ethyl-5-fluoro-N-isopropylbenzamide (Compound 64) (120 mg, crude) , 1-bromo-2-methoxyethane (32 mg, 0.23 mmol) , Cs 2CO 3(222 mg, 0.681 mmol) , NaI (102 mg, 0.680 mmol) in DMF (1 mL) was stirred at 80 ℃ via microwave irradiation for 1 h. After cooling to RT, the mixture was diluted with H 2O (10 mL) and extracted with EtOAc (3 x 10 mL) . The combined organic layers were washed with H 2O (10 mL) , dried over Na 2SO 4, filtered and concentrated under reduced pressure to afford the crude product which was further purified by HPLC over a Phenomenex Gemini-NX (column: 150×30 mm 5 μm; eluent: ACN/H 2O (10mM NH 4HCO 3) from 51%to 71% (v/v) ) and further purified by SFC over DAICEL CHIRALCEL OD-H (column: 250×30 mm 5 um; eluent: supercritical CO 2in EtOH (0.1%v/v ammonia) 25/25, v/v) to afford the title compound (5.13 mg, 96%purity) as yellow solid.
[0156]
LC-MS (ESI) (Method 1) : R t= 2.997 min, m/z found 586.3 [M+H] +.
[0157]
[0158]
(R) -N-ethyl-5-fluoro-N-isopropyl-2- ( (5- (2- (6- ( (2-methoxyethyl) (methyl) amino) -2-methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) benzamide
[0159]
[0160]
The mixture of (R) -N-ethyl-5-fluoro-N-isopropyl-2- ( (5- (2- (6- ( (2-methoxyethyl) amino) -2- methylhexan-3-yl) -2, 6-diazaspiro [3.4] octan-6-yl) -1, 2, 4-triazin-6-yl) oxy) benzamide (Compound 11) (40.0 mg, 0.068 mmol) , formaldehyde (55.4 mg, 0.683 mol, 37%in water) and AcOH (8.2 mg, 0.137 mmol) in anhydrous MeOH (2 mL) was stirred at 45 ℃ for 1 h. Then, NaBH 3CN (8.6 mg, 0.137 mmol) was added to the mixture and the resulting mixture was stirred at 45 ℃ for another 1 h. After cooling to RT, the reaction mixture was treated with sat. aq. NaHCO 3(40 mL) to adjust the pH value to about 8 and further extracted with DCM (20 mL x 3) . The combined organic layers were dried over anhydrous Na 2SO 4, filtered and concentrated under reduced pressure to give the crude which was purified by preparative HPLC over Boston Prime (column: C18 150x30mm 5um, Mobile Phase A: H 2O (0.04%ammonia+10mM NH 4HCO 3) , Mobile Phase B: ACN, Flow rate: 25 mL/min, gradient condition B/A from 50%to 80% (50%B to 80%B) ) to afford the title compound (9.62 mg, 99.10%purity, 23.3%yield) as yellow oil.
- [1]. Kwon MC, et al. Preclinical efficacy of the potent, selective menin-KMT2A inhibitor JNJ-75276617 (bleximenib) in KMT2A- and NPM1-altered leukemias. Blood. 2024 Sep 12;144(11):1206-1220. [Content Brief][2]. Hogeling SM, et al. Bleximenib, the novel menin-KMT2A inhibitor JNJ-75276617, impairs long-term proliferation and immune evasion in acute myeloid leukemia. Haematologica. 2024 Dec 19. [Content Brief]
////////Bleximenib, CS-0636752, DA-55335, HY-148669, JNJ-75276617, Menin-MLL inhibitor 24
Tegomil fumarate


Tegomil fumarate
cas 1817769-42-8
dimethyl (2E,19E)-4,18-dioxo-5,8,11,14,17-pentaoxahenicosa-2,19-diene-1,21-dioate
4-O-[2-[2-[2-[2-[(E)-4-methoxy-4-oxobut-2-enoyl]oxyethoxy]ethoxy]ethoxy]ethyl] 1-O-methyl (E)-but-2-enedioate
- 1,21-Dimethyl (2E,19E)-4,18-dioxo-5,8,11,14,17-pentaoxaheneicosa-2,19-dienedioate
- 5,8,11,14,17-Pentaoxaheneicosa-2,19-dienedioic acid, 4,18-dioxo-, 1,21-dimethyl ester, (2E,19E)-
Chemical Formula: C18H26O11
Exact Mass: 418.15
Molecular Weight: 418.395
Elemental Analysis: C, 51.67; H, 6.26; O, 42.06
SCHEME

Patent
- Method for producing monomethyl fumarate compoundsPublication Number: WO-2017108960-A1Priority Date: 2015-12-22
- Mmf-derivatives of ethyleneglycolsPublication Number: EP-3131633-A1Priority Date: 2014-04-17
- Mmf-derivatives of ethyleneglycolsPublication Number: EP-3131633-B1Priority Date: 2014-04-17Grant Date: 2020-03-04
- Mmf-derivatives of ethyleneglycolsPublication Number: US-2017029357-A1Priority Date: 2014-04-17
- MMF-derivatives of ethyleneglycolsPublication Number: US-9969674-B2Priority Date: 2014-04-17Grant Date: 2018-05-15
Ratiopharm GmbH, WO2015158817
PATENT
WO2017108960
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017108960
Scheme 2: Synthesis of (E)-But-2-enedioic acid 2-(2-{2-[2-((E)-3-methoxycarbonyl- acryloyloxy)-ethoxy]-ethoxy}-ethoxy)-ethyl ester methyl ester
Step 1: Synthesis of (Z)-But-2-enedioic acid mono-[2-(2-{2-[2-((Z)-3- carboxy-acryloyloxy)-ethoxy]-ethoxy}-ethoxy)-ethyl] ester



///////////Tegomil fumarate, MXD6KMG2ZP
Bivamelagon



Bivamelagon
CAS 2641595-54-0
NO1Y8WRA8N, 629.3 g/mol, C35H53ClN4O4
MC-4R Agonist 2
- N-[(3S,5S)-1-[[(3S,4R)-4-(4-Chlorophenyl)-1-(1,1-dimethylethyl)-3-pyrrolidinyl]carbonyl]-5-(4-morpholinylcarbonyl)-3-pyrrolidinyl]-2-methyl-N-(cis-4-methylcyclohexyl)propanamide
- N-((3S,5S)-1-((3S,4R)-1-(tert-Butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-5-(morpholine-4-carbonyl)pyrrolidin-3-yl)-N-(cis-4-methylcyclohexyl)isobutyramide
- N-[(3S,5S)-1-[(3S,4R)-1-tert-butyl-4-(4-chlorophenyl)pyrrolidine-3-carbonyl]-5-(morpholine-4-carbonyl)pyrrolidin-3-yl]-2-methyl-N-(4-methylcyclohexyl)propanamide
LB54640; LB-54640; LR-19021; LR19021
MC-4R Agonist 2 (Example 1) is a MC4R agonist. MC-4R Agonist 2 can be used in the study of obesity, diabetes, inflammation, and erectile dysfunction[1].
SCHEME

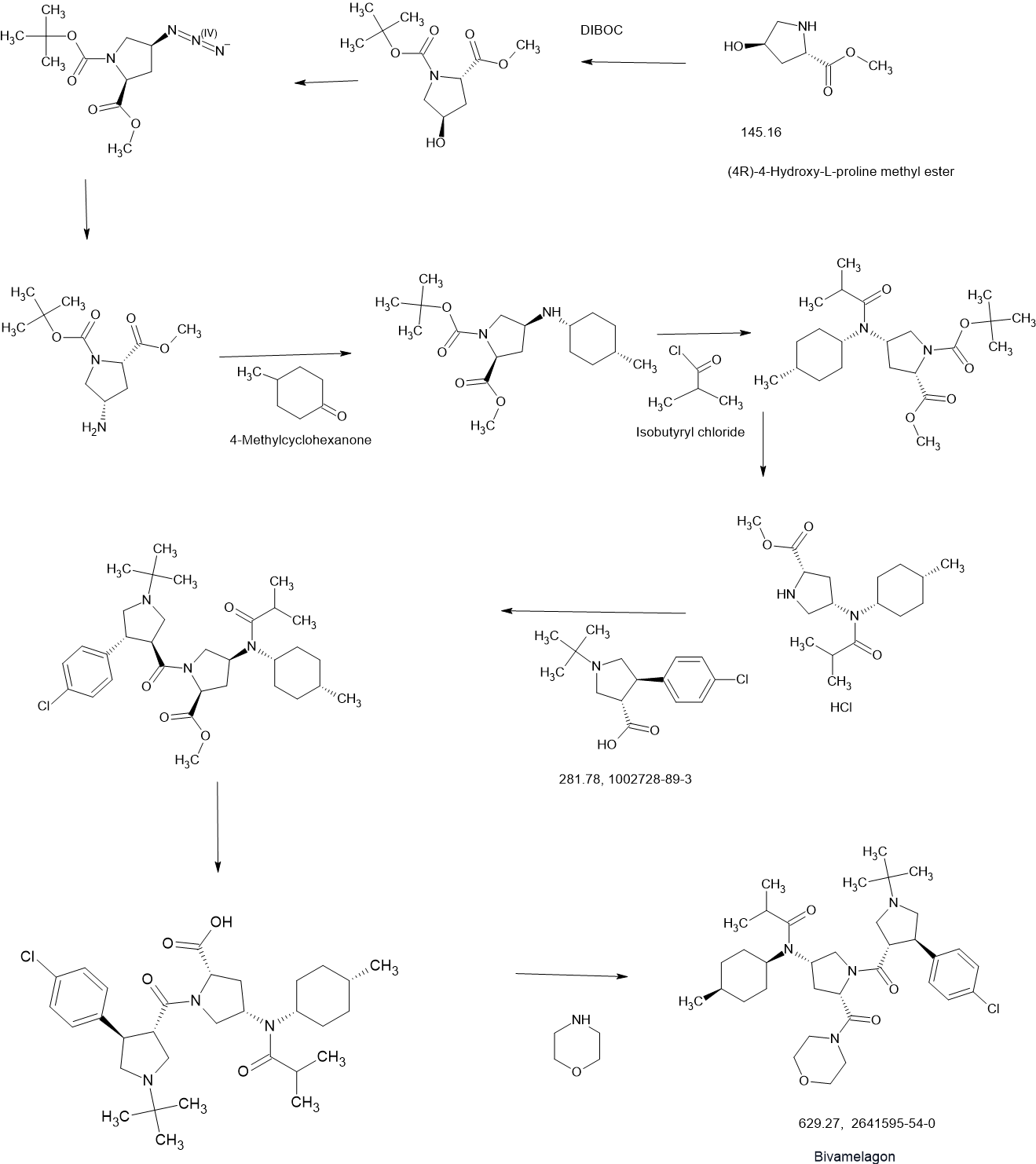
PATENT
WO2021091283A1.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021091283&_cid=P21-M9NZN0-90342-1
Step D: Preparation of N -((3 S ,5 S )-1-((3 S ,4 R )-1-( tert -butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-5-(morpholine-4-carbonyl)pyrrolidin-3-yl)- N -((1 s ,4 R )-4-methylcyclohexyl)isobutyramide hydrochloride
[173]
N -((3 S ,5 S )-1-((3 S ,4 R )-1-( tert -butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-5-(morpholine-4-carbonyl)pyrrolidin-3-yl)- N -((1 s ,4 R )-4-methylcyclohexyl)isobutyramide (5.0 g, 7.95 mmol) obtained in Step C was dissolved in ethyl acetate (50 ml), and a 2N hydrochloric acid ethyl acetate solution (3.97 ml, 15.89 mmol) was slowly added. After stirring at room temperature for 30 minutes, the reaction solvent was concentrated under reduced pressure. The resulting crude solid was purified by trituration using hexane and diethyl ether to obtain the title compound (5.23 g, 99%).
[174]
[175]
1H NMR (500 MHz, CD 3OD) δ 7.49-7.44 (m, 4H), 4.83 (m, 1H), 4.23-4.20 (m, 1H), 3.95-3.91 (m, 2H), 3.79-3.47 (m, 14H), 3.03-3.00 (m, 1H), 2.86-2.82 (m, 1H), 2.73-2.67 (m, 1H), 2.20-2.14 (m, 1H), 1.97 (m, 1H), 1.80-1.62 (m, 5H), 1.50 (s, 9H), 1.44-1.27 (m, 3H), 1.06-1.04 (m, 9H)
PATENT
WO2022235103
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022235103&_cid=P21-M9NZHZ-87240-1
Preparation of cyclohexyl-3-carbonyl-l)-5-(morpholine-4-carbonyl)pyrrolidin-3-yl)-N-((1s,4R)-4-methylcyclohexyl)isobutyramide (MC70)
[141]
[142]The title compound was obtained through the following steps A, B, and C.
[143]
Step A: Preparation of methyl (2S,4S)-1-((3S,4R)-1-(tert-butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-4-(N-((1s,4R)-4-methylcyclohexyl)isobutyramido)pyrrolidine-2-carboxylate
[144]Methyl (2S,4S)-4-(N-((1s,4R)-4-methylcyclohexyl)isobutyramido)pyrrolidine-2-carboxylate hydrochloride (28.7 g, 82.73 mmol) obtained in Manufacturing Example 1, (3S,4R)-1-(tert-butyl)-4-(4-chlorophenyl)pyrrolidine-3-carboxylic acid (24.5 g, 86.87 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (22.2 g, 115.83 mmol) and 1-hydroxybenzotriazole hydrate (15.7 g, 115.83 mmol) obtained in Manufacturing Example 2 were dissolved in N,N’-dimethylformamide (400 ml) and N,N’-diisopropylethylamine (72.0 ml, 413.66 mmol) was slowly added. The mixture was stirred at room temperature for 16 hours and the reaction solvent was concentrated under reduced pressure. A 0.5 N aqueous sodium hydroxide solution was added, and extraction was performed twice with ethyl acetate. The organic layer was washed twice with an aqueous sodium chloride solution and water, dried over anhydrous magnesium sulfate, and filtered. The filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography to obtain methyl (2S,4S)-1-((3S,4R)-1-(tert-butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-4-(N-((1s,4R)-4-methylcyclohexyl)isobutyramido)pyrrolidine-2-carboxylate (41.19 g, 87%).
[145]
[146]
Step B: Preparation of (2S,4S)-1-((3S,4R)-1-(tert-butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-4-(N-((1s,4R)-4-methylcyclohexyl)isobutyramido)pyrrolidine-2-carboxylic acid
[147]Methyl (2S,4S)-1-((3S,4R)-1-(tert-butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-4-(N-((1s,4R)-4-methylcyclohexyl)isobutyramido)pyrrolidine-2-carboxylate (39.4 g, 68.62 mmol) obtained in the above step A was dissolved in methanol (450 ml), and 6N sodium hydroxide aqueous solution (57.2 ml, 343.09 mmol) was added. The mixture was stirred at room temperature for 16 hours, and the pH was adjusted to about 5 with 6N hydrochloric acid aqueous solution, and then the reaction solution was concentrated under reduced pressure. The concentrate was dissolved in dichloromethane, and the insoluble solid was filtered through a paper filter. The filtrate was concentrated under reduced pressure to obtain the crude title compound (38.4 g, 99%), which was used in the next step without purification.
[148]
[149]
Step C: Preparation of N-((3S,5S)-1-((3S,4R)-1-(tert-butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-5-(morpholine-4-carbonyl)pyrrolidin-3-yl)-N-((1s,4R)-4-methylcyclohexyl)isobutyramide
[150](2S,4S)-1-((3S,4R)-1-(tert-butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-4-(N-((1s,4R)-4-methylcyclohexyl)isobutyramido)pyrrolidine-2-carboxylic acid (38.4 g, 68.60 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (18.4 g, 96.04 mmol) and 1-hydroxybenzotriazole hydrate (13.0 g, 96.04 mmol) obtained in the above step B were dissolved in N,N’-dimethylformamide (200 ml), and then morpholine (5.9 ml, 68.80 mmol) and N,N’-diisopropylethylamine were sequentially added. (59.7 ml, 343.02 mmol) was slowly added. The mixture was stirred at room temperature for 16 hours and the reaction solution was concentrated under reduced pressure, 0.5 N aqueous sodium hydroxide solution was added, and extraction was performed twice with ethyl acetate. The organic layer was washed twice with aqueous sodium chloride solution and water, dried over anhydrous magnesium sulfate, and filtered. The filtrate was concentrated under reduced pressure and purified by column chromatography to obtain N-((3S,5S)-1-((3S,4R)-1-(tert-butyl)-4-(4-chlorophenyl)pyrrolidine-3-carbonyl)-5-(morpholine-4-carbonyl)pyrrolidin-3-yl)-N-((1s,4R)-4-methylcyclohexyl)isobutyramide (37.05 g, 86%,
[151]
MS [M+H] = 630 (M+1)
Bivamelagon (INNTooltip International Nonproprietary Name; developmental code names LB54640, LR-19021) is a small-molecule melanocortin MC4 receptor agonist under development by LG Chem Life Sciences for the treatment of hypothalamic obesity, .[1][2] Unlike the older drug with the same mechanism of action, setmelanotide, it can be taken orally.[3][4][5] As of March 2024, it is in phase 2 clinical trials.[1]
References
- ^ Jump up to:a b “Rhythm Pharmaceuticals”. AdisInsight. 13 March 2024. Retrieved 25 February 2025.
- ^ “Delving into the Latest Updates on Bivamelagon with Synapse”. Synapse. 23 January 2025. Retrieved 25 February 2025.
- ^ Aronne, Sarah R. Barenbaum, Louis J. (2023). “Antiobesity Medications on the Horizon”. Handbook of Obesity – Volume 2 (5 ed.). CRC Press. pp. 394–401. doi:10.1201/9781003432807-42. ISBN 978-1-003-43280-7.
- ^ First-in-Human Study of Safety, Pharmacodynamics of LB54640, An Oral Melanocortin-4 Receptor Agonist Mirza, Victoria, MD, MPH; Lee, Jisoo, MD; Gwak, Heemin; Yang, Yunjeong; Kim, Mina. Obesity; Silver Spring Vol. 30, (Nov 2022): 145-146.
- ^ Piper, Noah B.C.; Whitfield, Emily A.; Stewart, Gregory D.; Xu, Xiaomeng; Furness, Sebastian G.B. (August 2022). “Targeting appetite and satiety in diabetes and obesity, via G protein-coupled receptors”. Biochemical Pharmacology. 202: 115115. doi:10.1016/j.bcp.2022.115115. PMID 35671790. S2CID 249452717.
| Clinical data | |
|---|---|
| Other names | LB54640; LB-54640; LR-19021; LR19021 |
| Routes of administration | Oral |
| Drug class | Melanocortin MC4 receptor agonist |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2641595-54-0 |
| PubChem CID | 165152355 |
| DrugBank | DB18331 |
| ChemSpider | 129440355 |
| UNII | NO1Y8WRA8N |
| Chemical and physical data | |
| Formula | C35H53ClN4O4 |
| Molar mass | 629.28 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
{kind=link}
[1]. Seung Wan Kang, et al. Melanocortin-4 receptor agonists. Patent WO2021091283A1.
////////Bivamelagon, LB54640, LB-54640, LR-19021, LR19021, NO1Y8WRA8N
Tisolagiline


Tisolagiline
CAS 1894207-44-3
PCH79KLX33
(2S)-2-[[4-[4-(trifluoromethyl)phenyl]phenyl]methylamino]propanamide
322.32 g/mol
SCHEME
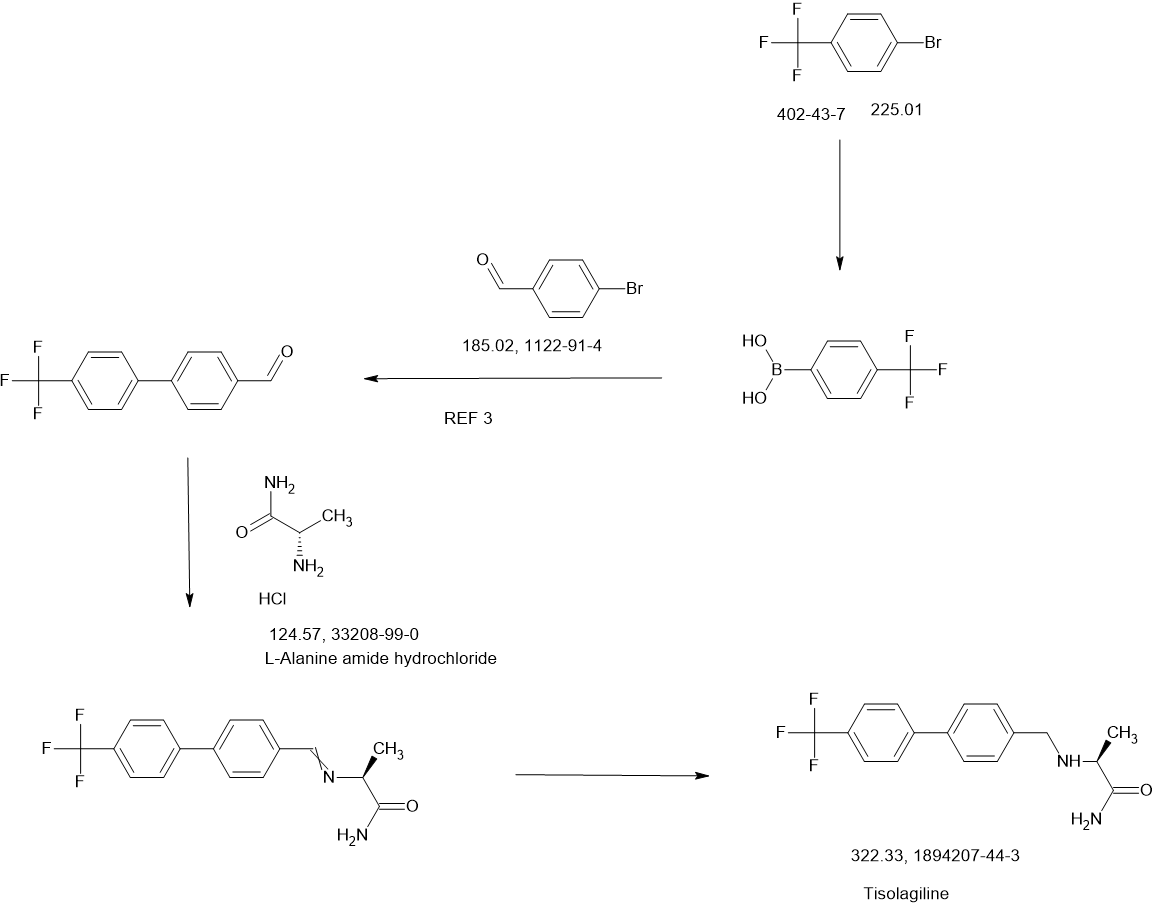
Tisolagiline (INNTooltip International Nonproprietary Name; developmental code names KDS-2010, SeReMABI) is a potent, highly selective, and reversible monoamine oxidase B (MAO-B) inhibitor which is under development for the treatment of Alzheimer’s disease and obesity.[1][2][3][4] It is taken by mouth.[1] Tisolagiline is being developed by NEUROBiOGEN and Scilex Bio.[1][2] As of December 2024, it is in phase 2 clinical trials for Alzheimer’s disease and obesity.[1][2]
Parkinson’s disease is a progressive disease that ranks second among degenerative neurological diseases, and the incidence rate is estimated to be about 6.3 million patients worldwide, and about 1 in 1,000 people develop Parkinson’s disease. The incidence rate is usually higher in the elderly, but it is now developing in young people as well. Parkinson’s disease is not easy to distinguish from other diseases because the symptoms progress slowly, and it is difficult to detect in the early stages. Clinical characteristics include tremors, rigidity, bradykinesia, postural instability, stooped posture, freezing of gait, depression, sleep disorders, urination disorders, and dementia.
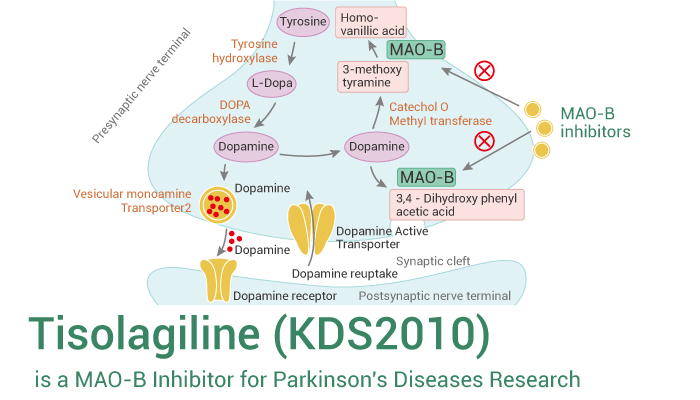
[3]Parkinson’s disease has an unknown cause, but it is known to be a disease that occurs when nerve cells that secrete the neurotransmitter dopamine in the brain are destroyed, resulting in a lack of dopamine. The most widely developed and used drug is levodopa therapy, which is generally administered by administering levodopa, which is converted into dopamine in the body. Levodopa is the most effective treatment for Parkinson’s disease, but there are cases where the drug-related effects decrease or various movement disorders occur during the treatment process. Other drugs used include COMT inhibitors and MAO-B inhibitors, which suppress dopamine metabolism and maintain the concentration of dopamine in the brain.
[4]MAO-B is known to play an important role in dopamine metabolism in the brain and to suppress damage to brain neurons. Although there is no clear evidence that MAO-B inhibitors actually slow down the progression of Parkinson’s disease, it is known that inhibiting MAO-B has an effect of suppressing degeneration or death of dopamine neurons, as it plays an important role in the development of Parkinson’s disease caused by MPTP or similar environmental toxicants. In addition, evidence from animal and clinical trials suggests that MAO-B inhibitors have a brain protective effect, unlike other drugs.
[5]The most representative MAO-B inhibitor approved is selegiline, which is prescribed as a treatment for Parkinson’s disease, but when taken, it is metabolized into amphetamine in the body, causing liver toxicity, and as an irreversible inhibitor, it has various side effects. Azilect, which contains rasagiline, was first marketed in Israel in 2005 and has recently been released in about 50 countries including Europe and the United States. Azilect does not have amphetamine side effects in the body when taken and is said to be more effective than other dopaminergic drugs. However, rasagiline, like selegiline, is an irreversible MAO-B inhibitor, so although it has an excellent MAO-B inhibition effect, it has the disadvantage of safety issues. Therefore, recently, drugs that are effective and can reversibly inhibit activity are being developed as alternatives to complement these shortcomings, but no notable reversible inhibitors have been prescribed to date.
[6]Meanwhile, obesity is a medical condition in which excessive fat accumulates in the body to the extent that it has a negative impact on health. Excessive weight can appear in combination with various diseases as the remaining energy is accumulated excessively due to the difference between energy consumed and energy used.
[7]Previous studies on the hypothalamus in relation to food regulation have focused on neurons that make up a portion of the brain, which has limited our understanding of the brain’s function in controlling food and obesity. Therefore, in order to comprehensively understand brain function, studies on glial cells, which make up the majority, must also be conducted in parallel. In addition, astrocytes, which are the most numerous among glial cells, have recently emerged as cells that can activate or inhibit surrounding neurons by secreting various signaling substances such as GABA (gamma-aminobutyric acid), glutamate, D-serine, and ATP. Astrocytes in the hypothalamus also interact closely with POMC (pro-opiomelanocortin) neurons and express leptin receptors, which can contribute to leptin signaling.
[8]There are two groups of POMC neurons in the hypothalamus: those that induce appetite reduction and those that induce energy consumption. Under normal circumstances, astrocytes help activate nearby POMC neurons that induce energy consumption. However, in obese states, unlike normal astrocytes, they are transformed into reactive astrocytes due to excessive leptin signals, and putrescine is converted into GABA by MAO-B (mono-aminoxidase B) and secreted. In addition, POMC neurons that induce energy consumption express GABAa receptors outside the synapse containing a4, a5, and a6 subunits due to excessive leptin signals, and are affected by persistent GABA secreted from anti-responsive astrocytes. As a result, POMC neurons are inhibited, energy consumption is reduced, and fat accumulation occurs.
[9]At this time, if MAOBI, the causal enzyme of GABA production, is inhibited, GABA production and secretion are inhibited, the inhibition of POMC neurons is relieved, and they are reactivated to promote energy consumption. However, POMC neurons that induce appetite reduction do not express GABAa receptors outside the synapse, so they are not continuously affected by GABA. Therefore, MAOBI inhibitors selectively act on POMC neurons that induce energy consumption and exhibit the effect of obesity treatment. However, most of the existing MAOBI inhibitors are irreversible inhibitors, and there is a problem that they are accompanied by various side effects. Accordingly, drugs that can reversibly inhibit MAOBI are being researched and developed, but no notable reversible MAOBI inhibitor that can effectively act on obesity has been prescribed to date.
REF
Regulatory Toxicology and Pharmacology (2020), 117, 104733
Toxicological Research (Cham, Switzerland) (2023), 39(4), 693-709
Combinatorial Chemistry & High Throughput Screening (2020), 23(9), 836-841
KR2023027416,
WO2023022256
WO2023022256
WO2016052928
PATENT
WO2016052928
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016052928&_cid=P20-M8XX0L-81795-1


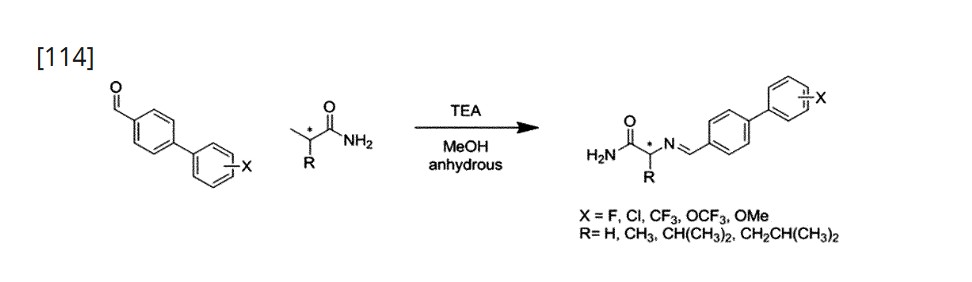
Using L-Alaninamide hydrochloride or D-Alaninamide hydrochloride, a reductive amination reaction was performed with the compound of step (a) to obtain an imine compound (step b, reaction scheme 1b), which was then reduced with sodium cyanoborohydride to obtain an amine compound (step c, reaction scheme 1c).
[112]Add 1.2 equivalents of Glycinamide hydrochloride or L-Alaninamide hydrochloride or D-Alaninamide hydrochloride or L-Valinamide hydrochloride or L-Leucinamide hydrochloride to anhydrous methanol at a concentration of 0.92 molar, and then add 1.5 equivalents of triethylamine. When the solution becomes transparent, add 1.0 equivalent of the aldehyde synthesized in step (a). After two hours, wash with ethyl acetate and distilled water. Dry the organic layer with sodium sulfate and concentrate in vacuo. Dissolve the concentrated reaction solution in anhydrous methanol at a concentration of 1.0 molar, and add 4.0 equivalents of sodium cyanoborohydride at 0 ℃. Then, react at room temperature for 18 hours, and after completion of the reaction, wash the reaction solution with ethyl acetate and distilled water. The organic layer was dried over sodium sulfate, concentrated in vacuo, and separated and purified using silica gel column chromatography.
References
- ^ Jump up to:a b c d “KDS 2010”. AdisInsight. 6 February 2025. Retrieved 24 February 2025.
- ^ Jump up to:a b c “Delving into the Latest Updates on KDS-2010 with Synapse”. Synapse. 23 January 2025. Retrieved 24 February 2025.
- ^ Nam MH, Sa M, Ju YH, Park MG, Lee CJ (April 2022). “Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease”. International Journal of Molecular Sciences. 23 (8): 4453. doi:10.3390/ijms23084453. PMC 9028367. PMID 35457272.
4.4. KDS2010 A recently developed KDS2010, which is ~12,500-fold more selective to MAOB than MAOA, differentiates the role of MAOB from MAOA and reports that MAOB does not contribute to DA degradation [39]. KDS2010 is a potent (IC50 = 7.6 nM), and selective MAOB inhibitor named shows no known off-target effect (no other enzymes or channels causing >40% inhibition) or toxicity for 4 weeks of repeated dosing in non-human primates [16,41]. KDS2010 was turned out to be highly effective for alleviating the PD-related motor symptoms and PD-like pathology, including reactive astrogliosis, excessive astrocytic GABA, and nigrostriatal DAergic neuronal loss in multiple rodent models of PD [41]. Its clinical efficacy is still waiting to be tested in future studies.
- ^ Duarte P, Cuadrado A, León R (2021). “Monoamine Oxidase Inhibitors: From Classic to New Clinical Approaches”. Handbook of Experimental Pharmacology. 264: 229–259. doi:10.1007/164_2020_384. ISBN 978-3-030-68509-6. PMID 32852645.
KDS2010 is a novel compound highly potent and selective reversible MAO-B inhibitor (Fig. 2). It has demonstrated learning and memory improvements, promotion of synaptic transmission, and reduction of astrogliosis and astrocytic GABA levels in APP/presenilin 1 mice (Park et al. 2019).
| Clinical data | |
|---|---|
| Other names | KDS-2010; KDS2010; SeReMABI |
| Drug class | Reversible monoamine oxidase B (MAO-B) inhibitor |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1894207-44-3 |
| PubChem CID | 132023446 |
| ChemSpider | 128942408 |
| UNII | PCH79KLX33 |
| ChEMBL | ChEMBL5314546 |
| Chemical and physical data | |
| Formula | C17H17F3N2O |
| Molar mass | 322.331 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
{kind=link}
///////////Tisolagiline, PCH79KLX33
Bemfivastatin, PPD 10558, RBx 10558



Bemfivastatin, PPD 10558, RBx 10558
cas 805241-79-6
| Molecular Weight | 588.67 |
|---|---|
| Formula | C34H37FN2O6 |


- PPD-10558 calcium salt
- Ppd-10558(calcium salt)
- 805241-64-9
- ppd-10558 calcium
- 3I8G750MW3
- calcium;(3R,5R)-7-[2-(4-fluorophenyl)-4-[[4-(hydroxymethyl)phenyl]carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate
- C68H72CaF2N4O12
- 1H-PYRROLE-1-HEPTANOIC ACID, 2-(4-FLUOROPHENYL)-.BETA.,.DELTA.-DIHYDROXY-4-(((4-(HYDROXYMETHYL)PHENYL)AMINO)CARBONYL)-5-(1-METHYLETHYL)-3-PHENYL-, CALCIUM SALT (2:1), (.BETA.R,.DELTA.R)-
- calcium;(3R,5R)-7-[2-(4-fluorophenyl)-4-[[4-(hydroxymethyl)phenyl]carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate
- UNII-3I8G750MW3
- 1H-Pyrrole-1-heptanoic acid, 2-(4-fluorophenyl)-beta,delta-dihydroxy-4-(((4-(hydroxymethyl)phenyl)amino)carbonyl)-5-(1-methylethyl)-3-phenyl-, calcium salt (2:1), (betaR,deltaR)-
- Bemfivastatin calcium
- Bemfivastatin hemicalcium
Bemfivastatin (PPD 10558) is an orally active, HMG-CoA Reductase (HMGCR) inhibitor, also known as Statin. Bemfivastatin enhances the activity of liver extraction. Bemfivastatin exhibits little developmental toxicity effects in pregnant rats and rabbits via daily oral doses during organogenesis period. The no observed adverse effect level (NOAEL) are ≥320 mg/kg/day for rats developmental toxicity, 12.5 mg/kg/day for rabbits maternal toxicity, and 25 mg/kg/day for rabbits developmental toxicity, respectively. Bemfivastatin can be used for research on Statin-related hypercholesterolemic myalgia with inability to tolerate statins.
Korean Patent No. 10-1329113 describes a method for preparing (3R,5R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-[(4-hydroxymethylphenylamino)carbonyl]-pyrrol-1-yl]-3,5-dihydroxy-heptanoic acid hemicalcium salt, as shown in the following reaction scheme.




SCHEME

MAIN

PATENT
WO2020040614
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020040614&_cid=P11-M8VDBE-14315-1
Step 3: Preparation of tert-butyl (3R,5R)-7-(2-(4-fluorophenyl)-4-((4-(hydroxymethyl)phenyl)carbamoyl)-5-isopropyl-3-phenyl-1H-pyrrol-1-yl)-3,5-dihydroxyheptanoate
[499]In step 2, tert-butyl 2-((4R,6R)-6-(2-(3-((4-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)carbamoyl)-5-(4-fluorophenyl)-2-isopropyl-4-phenyl-1H-pyrrol-1-yl)ethyl)-2,2-dimethyl-1,3-dioxan-4-yl)acetate (5 g) was dissolved in methanol (37 ml) and THF (37 ml), 1 N HCl aqueous solution (37 ml) was added, and the mixture was stirred at room temperature for 2 hours. EA was added to the reaction solution, diluted, and washed several times with distilled water and brine. The extracted organic layer was dried over Na
2 SO
4 and filtered under reduced pressure. The filtrate was concentrated under reduced pressure, EA and hexane were added, and the mixture was purified by recrystallization to obtain the title compound.
[500]White solid 4.6 g (yield quantitative);
[501]
1H NMR (500 MHz, CDCl 3): 7.24-7.14 (m, 9H), 7.06 (d, J = 8.5 Hz, 2H), 6.99 (t, J = 8.5 Hz, 2H), 6.87 (br s, 1H), 4.57 (s, 2H), 4.45-4.08 (m, 2H), 3.96-3.90 (m, 1H), 3.75-3.71 (m, 1H), 3.58 (sep, J = 7.0 Hz, 1H), 2.32 (d, J = 6.5 Hz, 2H), 1.73-1.65 (m, 1H), 1.64-1.58 (m, 1H), 1.54 (d, J = 7.0 Hz, 6H), 1.45 (s, 9H), 1.27-1.22 (m, 2H), MH+ 645.
Step 4: Preparation of (3R,5R)-7-(2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-((4-hydroxymethylphenylamino)carbonyl)-pyrrol-1-yl)-3,5-dihydroxyheptanoic acid hemicalcium salt
[503]In step 3, tert-butyl (3R,5R)-7-(2-(4-fluorophenyl)-4-((4-(hydroxymethyl)phenyl)carbamoyl)-5-isopropyl-3-phenyl-1H-pyrrol-1-yl)-3,5-dihydroxyheptanoate (4.19 g) obtained was dissolved in MeOH (65 ml) and THF (65 ml), and stirred in an ice bath. NaOH pellets (5 eq, 1.3 g) were added, and the mixture was stirred for 1 more hour at room temperature. After concentrating the reaction solution under reduced pressure, distilled water (44 ml) was added until the formed solid was completely dissolved. After concentrating the reaction solution under reduced pressure, distilled water (430 ml) was added until the solid was completely dissolved. 1 M Ca(OAc)
2 aqueous solution (3.6 ml) was slowly added dropwise, and the mixture was stirred for 15.5 hours at room temperature. After the generated solid was filtered under reduced pressure, it was washed several times with distilled water and the filtered solid was dried in an oven.
[504]2.98 g of white solid (yield 76%);
[505]
1H NMR (500 MHz, DMSO-d 6) δ 9.78 (br s, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.26-7.23 (m, 2H), 7.19 (t, J = 9.0 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 7.09-7.05 (m, 4H), 7.02-6.98 (m, 1H), 6.41 (br s, 1H), 5.04 (t, J = 5.5 Hz, 1H), 4.75 (br s, 1H), 4.39 (d, J = 5.5 Hz, 2H), 3.98-3.91 (m, 1H), 3.79-3.69 (m, 2H), 3.55-3.50 (m, 1H), 3.22 (sep, J = 7.0 Hz, 1H), 2.03 (dd, J = 15.0 Hz, 4.0 Hz, 1H), 1.90 (dd, J = 15.0 Hz, 8.0 Hz, 1H), 1.63-1.57 (m, 1H), 1.54-1.47 (m, 1H), 1.41-1.36 (m, 1H), 1.37 (d, J = 7.0 Hz, 6H), 1.23-1.16 (m, 1H), MH+ (acid+1) 589.
Step 5: Preparation of (3R,5R)-7-(2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-((4-hydroxymethylphenylamino)carbonyl)-pyrrol-1-yl)-3,5-dihydroxyheptanoic acid hemicalcium salt
[540]The title compound was prepared in the same manner as in step 4 of Example 15.
[541]
1H NMR (500 MHz, DMSO-d 6) δ 9.78 (br s, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.26-7.23 (m, 2H), 7.19 (t, J = 9.0 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 7.09-7.05 (m, 4H), 7.02-6.98 (m, 1H), 6.41 (br s, 1H), 5.04 (t, J = 5.5 Hz, 1H), 4.75 (br s, 1H), 4.39 (d, J = 5.5 Hz, 2H), 3.98-3.91 (m, 1H), 3.79-3.69 (m, 2H), 3.55-3.50 (m, 1H), 3.22 (sep, J = 7.0 Hz, 1H), 2.03 (dd, J = 15.0 Hz, 4.0 Hz, 1H), 1.90 (dd, J = 15.0 Hz, 8.0 Hz, 1H), 1.63-1.57 (m, 1H), 1.54-1.47 (m, 1H), 1.41-1.36 (m, 1H), 1.37 (d, J = 7.0 Hz, 6H), 1.23-1.16 (m, 1H), MH+ (acid+1) 589.
KR2001835 63%
KR2016103248
- [1]. Faqi AS, et al. Developmental toxicity of the HMG-CoA reductase inhibitor (PPD10558) in rats and rabbits. Birth Defects Res B Dev Reprod Toxicol. 2012 Feb;95(1):23-37. [Content Brief][2]. Wierzbicki A, et al. Drugs in development for the management of dyslipidaemia[J]. Future Prescriber, 2012, 13(2): 12-15.
/////////Bemfivastatin, PPD 10558, PPD-10558, RBx-10558; PPD10558, RBx10558, PPD 10558, RBx 10558, bemfivastatin CA, RBx 10558