
Home » Uncategorized (Page 174)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
OTC Drug (Meclizine) to Treat Infectious Diseases and Cancer
Meclizine, an over-the-counter drug used for decades to treat nausea and motion sickness, has the potential for new uses to treat certain infectious diseases and some forms of cancer, according to Vishal M. Gohil, Texas A&M AgriLife Research biochemist.
The research on meclizine appears in the current online version of the Journal of Biological Chemistry.
FDA approves Gazyva for chronic lymphocytic leukemia
Drug is first with breakthrough therapy designation to receive FDA approval
The U.S. Food and Drug Administration today approved Gazyva (obinutuzumab) for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia (CLL).
read all at
http://www.pharmalive.com/fda-approves-roche-s-gazyva
my old article cut paste
Roche’s new leukaemia drug, Obinutuzumab, superior to Rituxan in clinical trial
JULY 25, 2013 12:52 AM / 6 COMMENTS / EDIT
 July 24 2013 | By Márcio Barra
July 24 2013 | By Márcio Barra
Roche has announced that its experimental leukemia drug GA101, or obinutuzumab, used in combination with chemotherapy, was better than Rituxan at helping people with chronic lymphocytic leukemia live longer without their disease worsening, according to the results from the second phase of the clinical trial. Both drugs were tested and compared in combination with chlorambucil.
Roche’s Phase III leukemia drug Obinutuzumab (GA101) yields positive results
- GA101 is the first glycoengineered, type II anti-CD20 mAb.

Roche’s Phase III leukemia drug Obinutuzumab (GA101) yields positive results
Obinutuzumab (GA101)
| FORMULA | C6512H10060N1712O2020S44 |
|---|
GA101 is the first glycoengineered, type II anti-CD20 monoclonal antibody (mAb) that has been designed for increased antibody-dependent cellular cytotoxicity (ADCC) and Direct CellDeath.1 This agent is being investigated in collaboration with Biogen Idec.
Swiss pharmaceutical company Roche has announced that its early Phase III trial of Leukemia drug obinutuzumab (GA101) demonstrated significantly improved progression-free survival in people with chronic lymphocytic leukemia (CLL).
The positive results yield from stage 1 of a three-arm study called CLL11, designed to investigate the efficacy and safety profile of obinutuzumab (GA101) plus chlorambucil, a chemotherapy, compared with chlorambucil alone in people with previously untreated chronic lymphocytic leukemia (CLL).
This phase of the study met its primary endpoint and an improvement in progression-free survival was achieved; obinutuzumab plus chlorambucil significantly reduced the risk of disease worsening or death compared to chlorambucil alone.
Roche chief medical officer and global product development head Hal Barron said; “the improvement in progression-free survival seen with GA101 is encouraging for people with CLL, a chronic illness of older people for which new treatment options are needed.”
“GA101 demonstrates our ongoing commitment to the research and development of new medicines for this disease.”
Obinutuzumab is Roche’s most advanced drug in development for the treatment of hematological malignancies.
It has been specifically designed as the first glycoengineered, type 2 anti-CD20 monoclonal antibody in development for B cell malignancies.
Afutuzumab is a monoclonal antibody being developed by Hoffmann-La Roche Inc. for the treatment of lymphoma.[1] It acts as an immunomodulator.[2][3] It was renamed obinutuzumab in 2009.[4]
References
- Robak, T (2009). “GA-101, a third-generation, humanized and glyco-engineered anti-CD20 mAb for the treatment of B-cell lymphoid malignancies”. Current opinion in investigational drugs (London, England : 2000) 10 (6): 588–96. PMID 19513948.
- Statement On A Nonproprietary Name Adopted By The Usan Council – Afutuzumab,American Medical Association.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
-
OBINUTUZUMAB ISMONOCLONAL ANTIBODY TYPE Whole antibody SOURCE Humanized (from mouse) TARGET CD20
Biosimilar drugs in Portugal
November 1 ,2013 | By Márcio Barra
What follows is a list of Biosimilar drugs available in Portugal. This data has been compiled from the INFOMED database, managed by the Portuguese National Competent Authrority on Medicines, INFARMED. The Portuguese Marketing approval date was also provided. In the Market Status, you may find “no data” on some drugs. This means that the drug in question has no information displayed on the INFOMED database, save for its name.
View original post 486 more words
Covidien sells Confluent product line for $235 million
October 30, 2013 | By Anabela Farrica
![]()
Covidien announced yesterday that it has reached an agreement with Integra LifeSciences Corporation to sell its Confluent Surgical product line for $235 million in up-front cash, plus an additional $30 million in milestones. The transaction depends on a number of regulatory approvals, but it is expected to be finished by March 31, 2014.
View original post 152 more words
Sales of Biogen Idec’s multiple sclerosis drug Tecfidera soar
October 29 ,2013 | By Márcio Barra

Tecfidera (dymethil Fumarate), Biogen Idec’s prized multiple sclerosis drug, is fast approaching blockbuster status according to the recently released third-quarter numbers of Biogen Idec’s, beating along the way analyst expectation and pushing Biogen Idec’s net profit up 22%.
For Q3, Tecfidera sales garnered $286.4 million, far higher than the $217.2 million that analysts expected. It is now expected that Tecfidera will reach $3.5 billion in annual revenue by 2016, a no doubt impressive number for a drug that, in its first quarter in the market (the drug was launched in March 2013 in the US), posted $192.1 million in sales. This revenue mark pushed Biogen’s net profit up 22% to $488 million, up from $398 million last year.
View original post 302 more words
Atomoxetine

Atomoxetine
Atomoxetine hydrochloride (CAS NO.: 82248-59-7)
(R)-(-)-N-Methyl-gamma-(2-methylphenoxy)benzenepropanamine hydrochloride
| Patent No | PatentExpiry Date | |
|---|---|---|
| 5658590 | Nov 26, 2016 | |
| 5658590*PED | May 26, 2017 |
nda 021411 app 2002-11-26
TREATMENT OF ATTENTION-DEFICIT HYPERACTIVITY DISORDER
label
|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 5658590 | 1997-05-26 | 2017-05-26 |
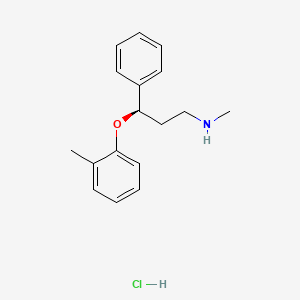
The HCl salt of atomoxetine , with the (R)-configuration], which is marketed under the trade name Strattera, is used for treating attention-deficit hyperactivity disorder (ADHD
Atomoxetine is a drug approved for the treatment of attention-deficit hyperactivity disorder(ADHD).[1] It is a selective norepinephrine reuptake inhibitor (NRI),[1] not to be confused with serotonin norepinephrine reuptake inhibitors (SNRIs) or selective serotonin reuptake inhibitors (SSRIs), both of which are currently the most prescribed form of antidepressants.
his compound is manufactured, marketed and sold in theUnited States under the brand name Strattera by Eli Lilly and Company as a hydrochloride salt (atomoxetine HCl), the original patent filing company, and current U.S. patent owner. Generics of atomoxetine are sold in all other countries; they are manufactured by Torrent Pharmaceuticals using the label Tomoxetin, Ranbaxy Laboratories (through its Division: Solus) using the label Attentin, Sun Pharmaceuticals(through its Division: Milmet Pharmaceuticals), and Intas Biopharmaceuticals There is currently no generic manufactured directly in the United States since it is under patent until 2017.[2]
On August 12, 2010, Lilly lost a lawsuit that challenged Lilly’s patent on Strattera, increasing the likelihood of an earlier entry of a generic into the US market.[3] On September 1, 2010, Sun Pharmaceuticals announced it would begin manufacturing a generic in the United States.[4] In a July 29, 2011 conference call, however, Sun Pharmaceutical’s Chairman stated “Lilly won that litigation on appeal so I think [generic Strattera]’s deferred.”[5]
Atomoxetine is designated chemically as (−)-N-methyl-3-phenyl-3-(o-tolyloxy)-propylamine hydrochloride, and has a molecular mass of 291.82.[1] It has a solubility of 27.8 mg/mL in water.[1] Atomoxetine is a white solid that exists as a granular powder inside the capsule, along with pre-gelatinized starch and dimethicone.[1] The capsule shells contain gelatin, sodium lauryl sulfate, FD&C Blue No. 2, synthetic yellow iron oxide, titanium dioxide, red iron oxide, edible black ink, and trace amounts of other inactive ingredients.[1]
The compound (-)-N-methyl-3-(2-methylphenoxy)-3-phenylpropylamine, or (-)-Λ/-methyl-3-phenyl-3-(o-tolyloxy)-propylamine hydrochloride, is usually known by its adopted name “atomoxetine hydrochloride.” It is represented as shown in Formula 1 and is a selective norepinephrine reuptake inhibitor. A commercialatomoxetine hydrochloride product is sold as STRATTERA™ in the form of capsules containing 10, 18, 25, 40, 60, 80, or 100 mg of atomoxetine, for treating attention-deficit/hyperactivity disorder.
- “STRATTERA® (atomoxetine hydrochloride) CAPSULES for Oral Use. Full Prescribing Information.” Eli Lilly and Company, 2002, 2013. Revised August 5, 2013. [1]
- “Patent and Exclusivity Search Results”. Electronic Orange Book. US Food and Drug Administration. Retrieved 26 April 2009.
- “Drugmaker Eli Lilly loses patent case over ADHD drug, lowers revenue outlook”. Chicago Tribune.
- “Sun Pharma receives USFDA approval for generic Strattera capsules”. International Business Times.
- “Sun Pharma Q1 2011-12 Earnings Call Transcript 10.00 am, July 29, 2011”.
- Strattera by Eli Lilly and Company
- RxList.com – Strattera
- Detailed Strattera Consumer Information: Uses, Precautions, Side Effects
- All disclosed Lilly trials
- MSDS for Atomoxetine HCl
- Strattera Related Published Studies
Synthesis
Also known as: Atomoxetine hydrochloride, Strattera, Atomoxetine HCL, (R)-Tomoxetine hydrochloride, TOMOXETINE HYDROCHLORIDE, Tomoxetine, 82248-59-7
First step appears to be a Mannich reaction between acetophenone, paraformaldehyde and dimethylamine, although not formally written in the scheme.

Foster, B. J.; Lavagnino, E. R.; European Patent, 1982, EP 0052492.

Eli Lilly’s Strattera capsules.


Atomoxetine, designated chemically as (-)-N-methyl-3-phenyl-3-(0-tolyloxy)- propylamine hydrochloride, is structurally represented by the compound of Formula-I and is indicated for the potential treatment of attention-deficit hyperactivity disorder (ADHD). This compound is manufactured, marketed and sold in the United States under the brand name Strattera.

Formula-I Atomoxetine was first disclosed in US Patent No 4314081. The said patent disclosed Atomoxetine, its pharmaceutically acceptable salts and composition containing them.
4-hydroxy Atomoxetine, chemically known as R-(-)-N-methyl-3-(2-methyl-4- hydroxyphenyl)oxy)-3 -phenyl- 1-aminopropane, structurally represented by Formula-II, is a metabolite of Atomoxetine.

Fomnula-ll
4-hydroxy Atomoxetine hydrochloride was first disclosed in US Patent No 7384983, wherein 4-hydroxy Atomoxetine free base was dissolved in ethylacetate, treated the solution with 0.1N HC1; followed by lyophilization yielded a yellow solid which was dissolved in methanol and passed through a short column of activated carbon; the solvent was removed and finally the hydrochloride salt was recrystallized from water to afford 4-hydroxy Atomoxetine hydrochloride. However, this patent does not mention about the nature of the polymorph obtained through this process.
The asymmetric epoxidation of (E)-3-phenyl-2-propen-1-ol (I) by means of titanium tetraisopropoxide, (+)-diethyl tartrate (+)-(DET) and tBu-OOH in dichloromethane gives the chiral epoxide (II), which is opened by means of bis(2-methoxyethoxy)aluminum hydride (Red-Al) in DME to yield the chiral diol (III). The regioselective reaction of (III) with Ms-Cl and TEA in ethyl ether affords the primary mesylate (IV), which is condensed with 2-methylphenol (V) by means of PPh3 and DEAD in ethyl ether to provide the adduct (VI). Finally this compound is treated with methylamine in hot aq. THF to give rise to the target (R)-tomoxetine.

The reduction of omega-chloropropiophenone (I) with NaBH4 in ethanol gives 3-chloro-1-phenyl-1-propanol (II), which is treated with butyric anhydride and pyridine in dichloromethane to yield the corresponding racemic ester (III). The optical resolution of (III) with immobilized lipase B from Candida antarctica (CALB) affords a mixture of unreacted (S)-ester and (R)-alcohol (IV) that are separated by column chromatography. Condensation of th (R)-alcohol (IV) with 2-methylphenol (V) by means of PPh3 and diethyl azodicarboxylate (DEAD) in THF gives the corresponding ether (VI), which is finally treated with methylamine in refluxing ethanol.
more info
U.S. Patent No. 4,314,081 describes 3-Aryloxy-3-phenyl polyamines, which possess central nervous system activity. Atomoxetine is a member of the above class of compounds, and is a useful drug for the treatment of depression.Atomoxetine was claimed in U. S. Patent No. 4,314,081 and the patent describes a process for the preparation of atomoxetine and related compounds in two different ways as depicted below as Scheme A and Scheme B, respectively.
Scheme A
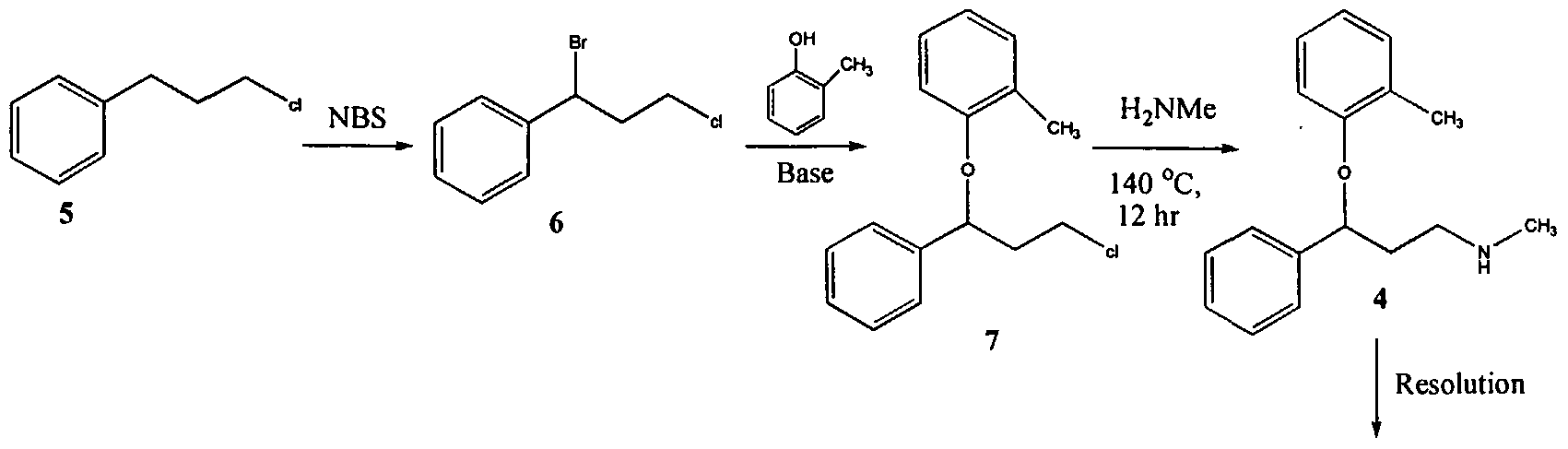
Atomoxetinc
Scheme B

The process illustrated in Scheme A involves the preparation of the atomoxetineusing 3-phenyl chloropropyl amine (Formula 5) as a starting material. The process involves bromination of said starting compound (Formula 5) by using N-bromosuccinimide. Further the bromo derivative is condensed with o-cresol to result in a compound of Formula 7, which is then subjected to amination using methylamine. Though the process looks very simple, it involves the following disadvantages: i) N-bromosuccinimide being a corrosive and sensitive chemical, its usage demands special care; ii) the workup of the compound formula 7 involves high vacuum (0.03 torr) distillation at 135-1450C, which is a tedious and cumbersome process to carry out at the plant level; and iii) the reaction conditions involved in some of the steps are harsh, for example the amination reaction is conducted at 14O0C. at pressures of 10 kg/cm2 for 12 hours in an autoclave.
All the above points make the process not viable for practicing on a commercial scale. Further, as described in U.S. Patent 4,314,081 , the free base compounds exist as high boiling oils, but form white crystalline salts.
On the other hand, Scheme B describes the preparation of atomoxetine using β-dimethylaminopropiophenone produced by a Mannich reaction; which is reduced to the hydroxy derivative having Formula 9 using diborane; further the hydroxy compound (Formula 9) is converted to the corresponding chloro derivative of Formula 10 using dry HCI gas and thionyl chloride and is followed by condensation with o-cresol.
The said reaction is carried out in methanol at reflux for a duration of five days to achieve the compound of formula 11 and is followed by demethylation using cyanogen bromide to end up with atomoxetine. As can be clearly understood the process is associated with the following problems: i) the use of costly reagents such as diborane makes the process uneconomical; ii) the passage of dry HCI gas followed by thionyl chloride addition is ^ very cumbersome and is not advisable in the plant; iii) this is a time-consuming process, involving a reaction which requires five days for its completion; and iv) use of cyanogen bromide, which is highly toxic, is not desirable.
All of the above-quoted drawbacks make the process unfriendly to practice in a production plant as well as to the environment.
Further, M. Srebnik et al., Journal of Organic Chemistry, Vol. 53, pages2916-2920 (1988); E. Corey et al., Tetrahedron Letters, Vol. 30, pages 5207-5210 (1989);
U.S. Patent No. 4,868,344; Y. Gao et al., Journal of Organic Chemistry, Vol. 53, pages 4081-4084 (1988); J. Deeter et al.,
Tetrahedron Letters, Vol. 31, pages 7101-7104 (1990);
and U.S. Patent No. 4,950,791 disclose stereospecific methods for the preparation of 3-aryloxy-3-phenylpropylamines; the enantiomers of 3-hydroxy-3-phenylpropylamines are prepared by the stereospecific reduction of the corresponding ketones. The thus obtained (S)-3-hydroxy-3-phenyl propylamines are subjected to condensation with aryl alcohols using the Mitsunobo reaction. As can be seen in Scheme C, the reaction involves two critical steps.
Scheme C
Dusopinocampheny) chloroborane


OH
,CH,
DEAD/ tn phenyl phosphine

The first critical step is an asymmetric reduction of the ketone to its corresponding alcohol. The second critical step involves the condensation of the obtained enantiomeric alcohol with the corresponding aryl alcohol. The process suffers from the following disadvantages:
1) the reagent used for the asymmetric reduction of the ketone is highly expensive;
2) the reagent diethyl azodicarboxylate (“DEAD”) is expensive;
3) the DEAD reagent is known to be highly carcinogenic, thus creating problems in handling; and
4) the reaction involves the use of triphenylphosphine and DEAD and the resulting byproducts formed in the reaction, phoshineoxide and a hydrazine derivative, are very difficult to remove.
Therefore, commercial applicability of the said process is limited owing to the above noted disadvantages.
International Patent Publication No. WO 00/58262 relates to a stereo- specific process for the preparation of atomoxetineusing nucleophilic aromatic displacement of an aromatic ring having a functional group, which can be converted to a methyl group. As can be seen, the process is very lengthy and involves many steps and is thus not commercially desirable.
U.S. Patent No. 5,847,214 describes the nucleophilic aromatic displacement reaction of 3-hydroxy-3-arylpropylamines with activated aryl halides, for example the reaction of N-methyl-3-phenyl-3-hydroxypropylamine with 4- triflouromethyl-1-cholro benzene has been reported; the success of this reaction is mainly due to electron withdrawing group on benzene ring of the aryl halides.
U.S. Patent No. 6,541 ,668 describes a process for the preparation of atomoxetine and its pharmaceutically acceptable addition salts which comprises reacting an alkoxide of N-methyl-3-phenyl-3-hydroxy propyl amine or an N protected derivative thereof, with 2-flouro toluene in the presence of 1 ,3-Dimethyl – 2-imidazolidinone (“DMI”) or N-Methyl-3-pyrrolidinone (“NMP”) as the solvent. The process disclosed in the said patent can be shown as Scheme D. Further, the process disclosed in the said patent restricts itself to the solvents DMI and NMP.
Scheme D

Nevertheless, a new crystalline form of N-methyl-3-phenyl-3-(o- tolyloxy)propylamine oxalate and an isolation technique of (±)-atmoxetine free base in a solid form, an intermediate useful in the synthesis of atomoxetine hydrochloride, is desirable.

http://www.sciencedirect.com/science/article/pii/S0040403906025068
There have been several methods reported for preparing (R)-(−)-N-methyl-3-(2-methylphenoxy)-3-phenylpropylamine (Atomoxetine®). For example, U.S. Pat. No. 4,868,344 discloses a process as shown in the following scheme:

In this example, 3-chloropropiophenone is used as the starting material to be asymmetrically reduced with (−)-diisopinocamphenylchloroborane ((−)-IPc2BC1) to give the corresponding chiral alcohol. The resulting chiral alcohol is then reacted with o-cresol via Mitsunobu reaction to form the chiral ether compound. Subsequently, amination of the chiral ether compound with methylamine provided atomoxetine. In this process, the materials such as chiral-borane ((−)-IPc2BC1) and diethyl azodicarboxylate (DEAD) are expensive, and result in high manufacturing cost.
Further, WO 2006/009884 discloses another method for preparing atomoxetine, including the step of reacting N-methyl-3-phenyl-3-hydroxypropylamine with 2-fluorotoluene which is followed by resolution of the resulting product to provide optically pure atomoxetine as shown in the following scheme:

This process involving a chiral resolution step is inefficient due to low product yield, complicated and long time process that renders this process economically less competitive.
………………………………………………………………………………………………

see below
B.-F. Chen and co-inventors describe a synthesis of 5 that avoids costly reagents. It includes the preparation of the chiral amino alcohol 3 as a key intermediate. The route for preparing 5 starts with a Mannich reaction between benzophenone, N,O-dimethylhydroxylamine, and paraformaldehyde to give compound 1, isolated in 87.6% yield.
Ketone 1 is asymmetrically reduced to form alcohol 2 by using the chiral ruthenium catalyst RuCl2-[(S)-DMSEGPHOS)][(S)-DAIPEN]. The hydrogen pressure is described as “predetermined”, but no value is given. Product 2 is recovered as an oily product in 98.7% yield with 98.8% purity and 99% ee. It appears that the catalyst is not removed before the next step in which the oil is hydrogenated over a Raney nickel catalyst to form amino alcohol 3.
Intermediate 3 is also isolated as an oil in 96.4% yield, 96.5% % purity, and 99% ee. After recrystallization from toluene–heptane, the solid product is recovered with 100% ee. In the last step, 3 is treated with fluorotoluene 4 in the presence of t-BuOK to form atomoxetine, isolated as an oil in 91% yield with 97% ee. The purification of 5 and its conversion to the HCl salt are not described.
The inventors provide basic 1H-NMR data for all compounds except 5. The example describing the preparation of 1 lists one of the reactants as 2-acetylthiophene, which is clearly incorrect; and another reagent is called “32% hydrochloride”. These errors should have been spotted by anyone with a fundamental knowledge of chemistry who was involved in writing the patent—perhaps none were. (Sci Pharmtech [Taiwan]. US Patent 8,299,305, Oct. 30, 2012; Keith Turner)
View the full-text patent here.
| Patent Number: | US 8299305 |
| Title: | Process for preparation of atomoxetine |
| Inventor(s): | Chen, Bo-Fong; Li, Yan-Wei; Yeh, Jinun-Ban; Wong, Wei-Chyun |
| Patent Assignee(s): | SCI Pharmtech, Inc., Taiwan |
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab

NIVOLUMAB
Anti-PD-1;BMS-936558; ONO-4538
PRONUNCIATION nye vol’ ue mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL DESCRIPTION
A fully human IgG4 antibody blocking the programmed cell death-1 receptor (Medarex/Ono Pharmaceuticals/Bristol-Myers Squibb)
MOLECULAR FORMULA C6362H9862N1712O1995S42
MOLECULAR WEIGHT 143.6 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION MDX-1106, BMS-936558
CAS REGISTRY NUMBER 946414-94-4
Bristol-Myers Squibb announced promising results from an expanded phase 1 dose-ranging study of its lung cancer drug nivolumab
Nivolumab (nye vol’ ue mab) is a fully human IgG4 monoclonal antibody designed for the treatment of cancer. Nivolumab was developed by Bristol-Myers Squibb and is also known as BMS-936558 and MDX1106.[1] Nivolumab acts as an immunomodulator by blocking ligand activation of the Programmed cell death 1 receptor.
A Phase 1 clinical trial [2] tested nivolumab at doses ranging from 0.1 to 10.0 mg per kilogram of body weight, every 2 weeks. Response was assessed after each 8-week treatment cycle, and were evaluable for 236 of 296 patients. Study authors concluded that:”Anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non–small-cell lung cancer, melanoma, or renal-cell cancer; the adverse-event profile does not appear to preclude its use.”[3]
Phase III clinical trials of nivolumab are recruiting in the US and EU.[4]
- Statement On A Nonproprietary Name Adopted By The USAN Council – Nivolumab, American Medical Association.
- A Phase 1b Study of MDX-1106 in Subjects With Advanced or Recurrent Malignancies (MDX1106-03), NIH.
- Topalian SL, et al. (June 2012). “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer”. New England Journal of Medicine 366. doi:10.1056/NEJMoa1200690. Lay summary – New York Times.
- Nivolumab at ClinicalTrials.gov, A service of the U.S. National Institutes of Health.
The PD-1 blocking antibody nivolumab continues to demonstrate sustained clinical activity in previously treated patients with advanced non-small cell lung cancer (NSCLC), according to updated long-term survival data from a phase I trial.
Survival rates at one year with nivolumab were 42% and reached 24% at two years, according to the median 20.3-month follow up. Additionally, the objective response rate (ORR) with nivolumab, defined as complete or partial responses by standard RECIST criteria, was 17% for patients with NSCLC. Results from the updated analysis will be presented during the 2013 World Conference on Lung Cancer on October 29.
“Lung cancer is very difficult to treat and there continues to be a high unmet medical need for these patients, especially those who have received multiple treatments,” David R. Spigel, MD, the program director of Lung Cancer Research at the Sarah Cannon Research Institute and one of the authors of the updated analysis, said in a statement.
“With nivolumab, we are investigating an approach to treating lung cancer that is designed to work with the body’s own immune system, and these are encouraging phase I results that support further investigation in larger scale trials.”
In the phase I trial, 306 patients received intravenous nivolumab at 0.1–10 mg/kg every-other-week for ≤12 cycles (4 doses/8 week cycle). In all, the trial enrolled patients with NSCLC, melanoma, renal cell carcinoma, colorectal cancer, and prostate cancer.
The long-term follow up focused specifically on the 129 patients with NSCLC. In this subgroup, patients treated with nivolumab showed encouraging clinical activity. The participants had a median age of 65 years and good performance status scores, and more than half had received three or more prior therapies. Across all doses of nivolumab, the median overall survival was 9.9 months, based on Kaplan-Meier estimates.
In a previous update of the full trial results presented at the 2013 ASCO Annual Meeting, drug-related adverse events of all grades occurred in 72% of patients and grade 3/4 events occurred in 15%. Grade 3/4 pneumonitis related to treatment with nivolumab emerged early in the trial, resulting in 3 deaths. As a result, a treatment algorithm for early detection and management was developed to prevent this serious side effect.
Nivolumab is a fully human monoclonal antibody that blocks the PD-1 receptor from binding to both of its known ligands, PD-L1 and PD-L2. This mechanism, along with early data, suggested an associated between PD-L1 expression and response to treatment.
In separate analysis presented at the 2013 World Conference on Lung Cancer, the association of tumor PD-L1 expression and clinical activity in patients with NSCLC treated with nivolumab was further explored. Of the 129 patients with NSCLC treated with nivolumab in the phase I trial, 63 with NSCLC were tested for PD-L1 expression by immunohistochemistry (29 squamous; 34 non-squamous).
Bristol-Myers Squibb announced promising results from phase 2b study of its rheumatoid arthritis drug clazakizumab
NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL
CLAZAKIZUMAB
PRONUNCIATION klaz” a kiz’ ue mab
THERAPEUTIC CLAIM Autoimmune diseases, rheumatoid arthritis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6) (human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 heavy chain), disulfide with human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 κ-chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 (B-cell stimulatory factor 2, CTL differentiation factor, hybridoma growth factor, interferon beta-2)); humanized rabbit monoclonal BMS-945429/ALD518 [300-alanine(CH2-N67>A67)]1 heavy chain (223-217′)-disulfide with humanized rabbit monoclonal BMS-945429/ALD518 light chain dimer (229-229”:232-232”)-bisdisulfide, O-glycosylated
MOLECULAR FORMULA C6426H9972N1724O2032S42
MOLECULAR WEIGHT 145.2 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION BMS-945429, ALD518
CAS REGISTRY NUMBER 1236278-28-6
Monoclonal antibody
Type Whole antibody
Source Humanized
Target IL6
CAS number 1236278-28-6
Clazakizumab is a humanized monoclonal antibody designed for the treatment of rheumatoid arthritis.[1]
Clazakizumab was developed by Alder Biopharmaceuticals and Bristol-Myers Squibb.
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH

FDA approves GE’s imaging drug Vizamyl for Alzheimer’s
October 28, 2013 | By Anabela Farrica

Last Friday, FDA approved Vizamyl (flutemetamol F 18), a radioactive agent to be used with PET to help evaluate the brain of patients for Alzheimer’s disease or dementia. Vyzamil works by binding to beta-amyloid plaques, which can be found in the brain of people with Alzheimer’s disease or other dementias, as well as in the brain of elderly people who do not have neurological problems.
View original post 206 more words

{kind=link}