
Home » Uncategorized (Page 165)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

Sanfilippo Syndrome: Gene Therapy Developments
Gene therapy for rare genetic diseases continues to top the news in biotechnology. Biotechnology companies that develop and commercialize gene therapy is a rapidly growing field of medicine, especially for rare diseases. This is the tenth in a series of Blog Posts on the topic of recent business developments for gene therapy for rare diseases.
Sanfilippo (SF) Syndrome or Mucopolysaccharidosis III (MPS III) is a rare genetic metabolism disorder that prohibits the proper breakdown of the body’s sugar molecules. There are 4 types of MPS III (MPS III A, MPS III B, MPS III C, and MPS III D), each with a deficiency in one of four lysosomal enzymes:
• Heparin N-sulfatase for MPS III A
• N-acetyl-alpha-D-glucoasaminidase for MPS III B
• Acetyl-CoA:alpha-glucosaminidase for MPS III C
• N-acetylglucoasamine-G-sulfate sulfatase for MPS III D.
The disease first affects the central nervous system, causing severe brain damage, and typically results in hearing loss, vision loss, organ damage, bone…
View original post 378 more words
VANIPREVIR, MK 7009

| Molecular formula | C38H53N5O9S |
| Molar mass | 755.92 g mol−1 |
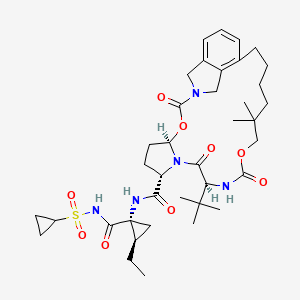
Vaniprevir (MK-7009) is a macrocyclic Hepatitis C virus (HCV) NS3/4a protease inhibitor, developed by Merck & Co., which is currently in clinical testing.[1]
- McCauley JA, McIntyre CJ, Rudd MT, Nguyen KT, Romano JJ, Butcher JW, Gilbert KF, Bush KJ, Holloway MK, Swestock J, Wan BL, Carroll SS, DiMuzio JM, Graham DJ, Ludmerer SW, Mao SS, Stahlhut MW, Fandozzi CM, Trainor N, Olsen DB, Vacca JP, Liverton NJ (March 2010). “Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor”. J. Med. Chem. 53 (6): 2443–63.doi:10.1021/jm9015526. PMID 20163176.
Abstract
Development of a practical synthesis of MK-7009, a 20-membered [corrected] macrocycle, is described. A variety of ring-closing strategies were evaluated, including ring-closing metathesis, intermolecular palladium-catalyzed cross-couplings, and macrolactamization. Ring closure via macrolactamization was found to give the highest yields under relatively high reaction concentrations. Optimization of the ring formation step and the synthesis of key intermediates en route to MK-7009 are reported
Synthesis of the HCV Protease Inhibitor Vaniprevir (MK-7009) Using Ring-Closing Metathesis Strategy.J. Org. Chem. 2012; 77: 3820-3828

2,6-Dichloro-1,4-benzoquinone was added to suppress isomerization of the allyl alkene in the isoindoline unit in C and consequent competing formation of a 19-membered ring by-product. An important contributor to the success of the RCM reaction was the high purity of crystalline B

nmr
Synthesis of the HCV protease inhibitor vaniprevir (MK-7009) using ring-closing metathesis strategy
J Org Chem 2012, 77(8): 3820
Song, Z.G.J.; Tellers, D.M.; Journet, M.; et al.
Synthesis of vaniprevir (MK-7009): Lactamization to prepare a 22-membered macrocycle
J Org Chem 2011, 76(19): 9553
PATENTS
WO 2013106631
WO 2013101550
WO 2007015787
WO 2007015855
WO 2013066753
WO 2012082672
WO 2011025849
WO 2003099274
WO 2007016441
…………………………………………………………………………….
EXAMPLE 46 (5R,7S,10S)-10-tert-Butyl-N-((1R,2R)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-ethylcyclopropyl)-15,15-dimethyl-3,9,12-trioxo-6,7,9,10,11,12,14,15,16,17,18,19-dodecahydro-1H,5H-2,23-ethano-5,8-methano-4,13,2,8,11-benzodioxatriazacyclohenicosine-7-carboxamide (III-231)

Step 1: 8-Hydroxy-1,2,3,4-tetrahydroisoquinoline hydrobromide

A mixture of 8-methoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride [Tetrahedron Letters, 1991, 32(17), 1965.] (3.0 g 15.0 mmol) and 45 mL of 48% aqueous HBr was heated for 18 h at 120° C. The resulting brown suspension was filtered and dried to provide 8-hydroxy-1,2,3,4-tetrahydroisoquinoline hydrobromide (2.8 g, 81% yield). LRMS (ESI) m/z 150.1 [(M+H)+; calcd for C9H1NO: 150.2].
Step 2: 1-tert-Butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate:

Carbonyldiimidazole (0.176 g, 1.086 mmol) was added to a stirred, room temperature solution of DMF (5 mL) and N-Boc-trans-4-hydroxy-L-proline methyl ester (0.21 g, 0.87 mmol) and the mixture was stirred 45 min. 8-Hydroxy-1,2,3,4-tetrahydroisoquinoline (0.20 g, 0.87 mmol) and Et3N (0.18 g, 1.74 mmol) were added and the resulting solution was heated at 50° C. for 2 h. The reaction mixture was poured into aqueous saturated NH4Cl and extracted with EtOAc, dried over Na2SO4and concentrated to an oil. The residue was purified by column chromatography on silica gel (gradient elution, 10 to 80% EtOAc in hexanes) to give 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (0.25 g, 0.60 mmol, 69% yield) as a colorless foam after evaporation of solvent. LRMS (ESI) m/z 321.3 [((M-Boc)+H)+; calcd for C16H21N2O5: 321.4].
Step 3: 1-tert-Butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoromethyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate

Trifluoromethanesulfonic anhydride (1.76 g, 6.24 mmol) was added to a stirred, 0° C. mixture of 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (1.81 g, 4.30 mmol) and Et3N (1.31 g, 12.90 mmol) in DCM (20 mL) and stirred for 18 h. The resulting mixture was poured into saturated aqueous NaHCO3 and extracted into dichloromethane. The organic layer was washed with 10% citric acid solution, dried over Na2SO4and concentrated to red oil. The oil was purified by column chromatography on silica gel (gradient elution, 10 to 70% EtOAc in hexanes) to give a yellow oil, 1-tert-butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoro methyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate (1.65 g, 69.4% yield). LRMS (ESI) m/z 453.2 [((M-Boc)+H)+; calcd for C17H20F3N2O7S: 453.4].
Step 4: 1-tert-Butyl 2-methyl (2S,4R)-4-{[(8-vinyl-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate

A solution of 1-tert-butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoromethyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate (1.74 g, 3.15 mmol), tri-n-butyl vinyl tin (1.10 g, 1.46 mmol) and lithium chloride (0.40 g, 9.45 mmol) in 25 mL DMF was purged with nitrogen for 10 min. Then bis(triphenylphosphine)palladium (II) chloride (0.22 g, 0.32 mmol) was added, and the mixture stirred at 25° C. under nitrogen for 18 h. The mixture was partitioned between EtOAc and saturated NaHCO3, the organic layer separated and washed with water then brine, dried over anhydrous sodium sulfate and concentrated to an oil. The oil was purified by column chromatography on silica gel (gradient elution, 10 to 65% EtOAc in hexanes) to give a colorless oil, 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-vinyl-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (1.00 g, 74% yield). LRMS (ESI) m/z 453.2 [(M+Na)+; calcd for C23H30N2O6Na: 453.5].
Step 5: (5R,7S,10S)-10-tert-Butyl-N-((1R,2R)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-ethylcyclopropyl)-15,15-dimethyl-3,9,12-trioxo-6,7,9,10,11,12,14,15,16,17,18,19-dodecahydro-1H,5H-2,23-ethano-5.8-methano-4,13,2,8,11-benzodioxatriazacyclohenicosine-7-carboxamide (III-231)


Synthesis of the HCV protease inhibitor Vaniprevir (MK-7009) using ring-closing metathesis strategy.
Kong J, Chen CY, Balsells-Padros J, Cao Y, Dunn RF, Dolman SJ, Janey J, Li H, Zacuto MJ.
J Org Chem. 2012 Apr 20;77(8):3820-8. doi: 10.1021/jo3001595. Epub 2012 Apr 10.
Synthesis of vaniprevir (MK-7009): lactamization to prepare a 20-membered [corrected] macrocycle.
Song ZJ, Tellers DM, Journet M, Kuethe JT, Lieberman D, Humphrey G, Zhang F, Peng Z, Waters MS, Zewge D, Nolting A, Zhao D, Reamer RA, Dormer PG, Belyk KM, Davies IW, Devine PN, Tschaen DM.
J Org Chem. 2011 Oct 7;76(19):7804-15. doi: 10.1021/jo2011494. Epub 2011 Aug 31. Erratum in: J Org Chem. 2011 Nov 18;76(22):9553.
Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor.
McCauley JA, McIntyre CJ, Rudd MT, Nguyen KT, Romano JJ, Butcher JW, Gilbert KF, Bush KJ, Holloway MK, Swestock J, Wan BL, Carroll SS, DiMuzio JM, Graham DJ, Ludmerer SW, Mao SS, Stahlhut MW, Fandozzi CM, Trainor N, Olsen DB, Vacca JP, Liverton NJ.
J Med Chem. 2010 Mar 25;53(6):2443-63. doi: 10.1021/jm9015526.
Pompei M, Di Francesco ME, Pesci S, Koch U, Vignetti SE, Veneziano M, Pace P, Summa V.
Bioorg Med Chem Lett. 2010 Jan 1;20(1):168-74. doi: 10.1016/j.bmcl.2009.11.005. Epub 2009 Nov 10.
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
Anakinra licensed in UK to treat CAPS in infants and adults

The Medicines and Healthcare Products Regulatory Agency (MHRA) in the UK has granted a licence to an interleukin-1 (IL-1) inhibitor Anakinra (Kineret) for the treatment of cryopyrin-associated periodic syndromes (CAPS) in adults and children as young as eight months.
Anakinra (brand name Kineret) is a drug used to treat rheumatoid arthritis.[1] It is aninterleukin-1 (IL-1) receptor antagonist.
Anakinra is an interleukin-1 (IL-1) receptor antagonist.Anakinra blocks the biologic activity of naturally occurring IL-1, including inflammation and cartilage degradation associated with rheumatoid arthritis, by competitively inhibiting the binding of IL-1 to the Interleukin-1 type receptor, which is expressed in many tissues and organs. IL-1 is produced in response to inflammatory stimuli and mediates various physiologic responses, including inflammatory and immunologic reactions. IL-1 additionally stimulates bone resorption and induces tissue damage like cartilage degradation as a result of loss ofproteoglycans. In patients with rheumatoid arthritis the natural IL-1 receptor antagonist is not found in effective concentrations in synovium and synovial fluid to counteract the elevated IL-1 concentrations in these patients.
Anakinra is not considered a ‘Disease-modifying antirheumatic drug‘ (DMARD) but rather a ‘Biological Response Modifier’ (BRM) because its able to selectively target the pathologic element of the disease.

Baxter seeks FDA approval of Rixubis in paediatric hemophilia B patients

Baxter International has filed an application to the US Food and Drug Administration (FDA) for a paediatric indication for Rixubis, Coagulation Factor IX (Recombinant), for the treatment of hemophilia B.
old article
Rixubis [Coagulation Factor IX (Recombinant)]
June 27, 2013 — The U.S. Food and Drug Administration yesterday approved Rixubis [Coagulation Factor IX (Recombinant)] for use in people with hemophilia B who are 16 years of age and older. Rixubis is indicated for the control and prevention of bleeding episodes, perioperative (period extending from the time of hospitalization for surgery to the time of discharge) management, and routine use to prevent or reduce the frequency of bleeding episodes (prophylaxis).
read all at


Rilpivirine

Rilpivirine
500287-72-9 cas no
4-{[4-({4-[(E)-2-cyanovinyl]-2,6-dimethylphenyl}amino)pyrimidin-2-yl]amino}benzonitrile

Rilpivirine (TMC278, trade name Edurant) is a pharmaceutical drug, developed byTibotec, for the treatment of HIV infection.[1][2] It is a second-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) with higher potency, longer half-life and reducedside-effect profile compared with older NNRTIs, such as efavirenz.[3][4]
Rilpivirine entered phase III clinical trials in April 2008,[5][6] and was approved for use in the United States in May 2011.[7] A fixed-dose drug combining rilpivirine with emtricitabine andtenofovir, was approved by the U.S. Food and Drug Administration in August 2011 under the brand name Complera.[8]
Like etravirine, a second-generation NNRTI approved in 2008, rilpivirine is a diarylpyrimidine(DAPY). Rilpivirine in combination with emtricitabine and tenofovir has been shown to have higher rates of virologic failure than Atripla in patients with baseline HIV viral loads greater than 100,000 copies.
- Rilpivirine bound to proteins in the PDB
- “TMC278 – A new NNRTI”. Tibotec. Retrieved 2010-03-07.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”.Eur. J. Med. Res. 12 (9): 483–95. PMID 17933730.
- Goebel F, Yakovlev A, Pozniak AL, Vinogradova E, Boogaerts G, Hoetelmans R, de Béthune MP, Peeters M, Woodfall B (2006). “Short-term antiviral activity of TMC278–a novel NNRTI–in treatment-naive HIV-1-infected subjects”. AIDS 20 (13): 1721–6.doi:10.1097/01.aids.0000242818.65215.bd. PMID 16931936.
- Pozniak A, Morales-Ramirez J, Mohap L et al. 48-Week Primary Analysis of Trial TMC278-C204: TMC278 Demonstrates Potent and Sustained Efficacy in ART-naïve Patients. Oral abstract 144LB.
- ClinicalTrials.gov A Clinical Trial in Treatment naïve HIV-1 Patients Comparing TMC278 to Efavirenz in Combination With Tenofovir + Emtricitabine
- ClinicalTrials.gov A Clinical Trial in Treatment naïve HIV-Subjects Patients Comparing TMC278 to Efavirenz in Combination With 2 Nucleoside/Nucleotide Reverse Transcriptase Inhibitors
- “FDA approves new HIV treatment”. FDA. Retrieved 2011-05-20.
- “Approval of Complera: emtricitabine/rilpivirine/tenofovir DF fixed dose combination”. FDA. August 10, 2011.
FORMULATION
EDURANT (rilpivirine, Janssen Therapeutics) is a non-nucleoside reverse transcriptase inhibitor (NNRTI) of human immunodeficiency virus type 1 (HIV-1). EDURANT is available as a white to off-white, film-coated, round, biconvex, 6.4 mm tablet for oral administration. Each tablet contains 27.5 mg of rilpivirine hydrochloride, which is equivalent to 25 mg of rilpivirine.
The chemical name for rilpivirine hydrochloride is 4-[[4-[[4-[(E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]2-pyrimidinyl]amino]benzonitrile monohydrochloride. Its molecular formula is C22H18N6 • HCl and its molecular weight is 402.88. Rilpivirine hydrochloride has the following structural formula:
 |
Rilpivirine hydrochloride is a white to almost white powder. Rilpivirine hydrochloride is practically insoluble in water over a wide pH range.
Each EDURANT tablet also contains the inactive ingredients croscarmellose sodium, lactose monohydrate, magnesium stearate, polysorbate 20, povidone K30 and silicified microcrystalline cellulose. The tablet coating contains hypromellose 2910 6 mPa.s, lactose monohydrate, PEG 3000, titanium dioxide and triacetin.
…………………………….
papers
Sun, et al.: J. Med. Chem., 41, 4648 (1998),
Kashiwada, et al.: Bioorg. Med. Chem. Lett., 11, 183 (2001)
Journal of Medicinal Chemistry, 2005 , vol. 48, 6 , pg. 2072 – 2079
………………………………………………
patents
WO201356003, WO200635067,
WO2013038425
The following PCT Publications describe the synthesis of Rilpivirine:
WO03016306, WO2005021001, WO2006024667, WO2006024668, W02994916581, WO2009007441, WO2006125809, and WO2005123662. [0006] Crystalline Rilpivirine base Forms I and II are described in the US Patent
Publication: US2010189796. Crystalline Rilpivirine HC1, Forms A, B, C, and D, are described in the US Patent Publications: US2009/012108, and US2011/0008434. Rilpivirine fumarate and a synthesis thereof are disclosed in WO2006024667.
country……………….patent……………approved……………expiry
| United States | 6838464 | 2011-05-20 | 2021-02-26 |
| United States | 7067522 | 2011-05-20 | 2019-12-20 |
| United States | 7125879 | 2011-05-20 | 2014-04-14 |
| United States | 7638522 | 2011-05-20 | 2014-04-14 |
| United States | 8080551 | 2011-05-20 | 2023-04-11 |
| United States | 8101629 | 2011-05-20 | 2022-08-09 |
Rilpivirine and its hydrochloride salt were disclosed in U.S. patent no. 7,125,879.Process for the preparation of rilpivirine was disclosed in U.S. patent no. 7,399,856 (‘856 patent). According to the ‘856 patent, rilpivirine can be prepared by reacting the (E)-3-(4-amino-3,5-dimethylphenyI)acrylonitrile hydrochloride of formula II with 4-(4-chloropyrimidin-2-ylamino)benzonitrile of formula III-a in the presence of potassium carbonate and acetonitrile under reflux for 69 hours. The synthetic procedure is illustrated in scheme I, below:
 Scheme 1 Process for the preparation of rilpivirine was disclosed in U.S. patent no. 7,705,148 (Ί48 patent). According to the Ί48 patent, rilpivirine can be prepared by reacting the 4-[[4-[[4-bromo-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile with acrylonitrile in the presence of palladium acetate, Ν,Ν-diethylethanamine and tris(2-methylphenyl)phosphine in acetonitrile. According to the Ί48 patent, rilpivirine can be prepared by reacting the compound of formula IV with 4-(4-chloropyrimidin-2-ylamino)benzonitrile formula Ill-a in the presence of hydrochloric acid and n-propanol to obtain a compound of formula Vll, and then the compound was treated with acetonitrile and potassium carbonate under reflux for 69 hours. The synthetic procedure is illustrated in scheme II, below:  Rilpivirine Scheme II U.S. patent no. 7,563,922 disclosed a process for the preparation of (E)-3-(4- amino-3,5-dimethylphenyl)acrylonitrile hydrochloride. According to the patent, (E)-3-(4- amino-3,5-dimethylphenyl)acrylonitrile hydrochloride can be prepared by reacting the 4- iodo-2,6-dimethyl-benzenamine in Ν,Ν-dimethylacetamide with acrylonitrile in the presence of sodium acetate and toluene, and then the solid thus obtained was reacted with hydrochloric acid in 2-propanol in the presence of ethanol and diisopropyl ether. U.S. patent no. 7,956,063 described a polymorphic Form A, Form B, Form C and Form D of rilpivirine hydrochloride. An unpublished application, IN 1415/CHE/201 1 assigned to Hetero Research Foundation discloses a process for the preparation of rilpivirine. According to the application, rilpivirine can be prepared by reacting the 4-(4-chloropyrimidin-2- ylamino)benzonitrile with (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride in the presence of p-toluene sulfonic acid monohydrate and 1 ,4-dioxane. It has been found that the rilpivirine produced according to the prior art procedures results in low yields.
|
The synthesis is as follows:
………………
more info………………………..
Rilpivirine, which is chemically known as 4-{[4-({4-[(lE)-2-cyanoethenyl]-2,6- dimethylphenyl} amino) pyrimidin-2-yl]amino}benzonitrile, is a non-nucleoside reverse transcriptase inhibitor (NNRTI) and exhibits human immunodeficiency virus (HIV) replication inhibiting properties. Rilpivirine is used as its hydrochloride salt in the anti-HIV formulations.

Conventionally, various processes followed for the synthesis of Rilpivirine hydrochloride (I), generally involve preparation of the key intermediate, (E)-4-(2- cyanoemenyl)-2,6-dimethylphenylamine hydrochloride of formula (II).

(E)-4-(2-cyanoethenyl)-2,6-dimethylphenylamine hydrochloride (II)
WO 03/016306 first disclosed the synthesis of Rilpivirine involving different routes for synthesis of 4-(2-cyanoethenyl)-2,6-dimethylphenylamine. The first route involved protection of the amino group of 4-bromo-2,6-dimemylphenylarnine by converting to Ν,Ν-dimethylmethanimidamide, followed by formylation involving n- butyl lithium and dimethylformamide. The resulting formyl derivative was treated with diethyl(cyanomethyl) phosphonate to give the cyanoethenyl compound which was deprotected using zinc chloride to yield the cyanoethenylphenylamine intermediate having an undisclosed E/Z ratio. This route involved an elaborate sequence of synthesis comprising protection of amine by its conversion into imide, use of a highly moisture sensitive and pyrophoric base such as butyl lithium and a low yielding formylation reaction. All these factors made the process highly unviable on industrial scale.
The second route disclosed in WO 03/016306 employed 4-iodo-2,6- dimethylphenylamine as a starting material for synthesis of cyanoemenylphenylamine intermediate, which involved reaction of the dimethylphenylamine derivative with acrylonitrile for atleast 12 hours at 130 C in presence of sodium acetate and a heterogeneous catalyst such as palladium on carbon. Isolation of the desired compound involved solvent treatment with multiple solvents followed by evaporation. This route also does not give any details of the E/Z ratio of the unsaturated intermediate product. Although this route avoids use of phosphine ligands but lengthy reaction time and problem of availability of pure halo-phenylamine derivatives coupled with moderate yields hampers the commercial usefulness of this route.
The third route disclosed in WO 03/016306 involved reaction of 4-bromo-2,6- dimethylphenylamine with acrylamide in presence of palladium acetate, tris(2- methylphenyl)phosphine and N,N-diethylethanamine. The resulting amide was dehydrated using phosphoryl chloride to give 4-(2-cyanoethenyi)-2,6- dimethylphenylamine in a moderate yield of 67% without mentioning the E/Z ratio. Although the E/Z isomer ratio for the cyanoethenyl derivative obtained from these routes is not specifically disclosed in the patent, however, reproducibility of the abovementioned reactions were found to provide an E/Z ratio between 70/30 and 80/20. Various other methods have also been reported in the literature for introduction of the ‘ cyanoethenyl group in Rilpivirine. The Journal of Medicinal Chemistry (2005), 48, 2072-79 discloses Wittig or Wadsworth-Emmons reaction of the corresponding aldehyde with cyanomethyl triphenylphosphonium chloride to provide a product having an E/Z isomer ratio of 80/20. An alternate method of Heck reaction comprising reaction of aryl bromide with acrylonitrile in presence of tri-o- tolylphosphine and palladium acetate gave the same compound with a higher E/Z isomer ratio of 90/10. The method required further purification in view of the presence of a significant proportion of the Z isomer in the unsaturated intermediate. A similar method was disclosed in Organic Process Research and Development (2008), 12, 530-536. However, the E/Z ratio of 4-(2-cyanoethenyl)-2,6- dimethylphenylamine was found to be 80/20, which was found to improve to 98/2 (E/Z) after the compound was converted to its hydrochloride salt utilizing ethanol and isopropanol mixture.
It would be evident from the foregoing that prior art methods are associated with the following drawbacks:
a) High proportion of Z isomer, which requires elaborate purification by utilizing column chromatographic techniques, crystallization, or successive treatment with multiple solvents, which decreases the overall yield,
b) Introduction of cyanoethenyl group to the formylated benzenamine derivatives involves a moisture sensitive reagent like n-butyl lithium, which is not preferred on industrial scale. Further, the method utilizes cyanomethyl phosphonate esters and is silent about the proportion of the Z isomer and the higher percentage of impurities which requires elaborate purification and ultimately lowers the yield,
c) Prior art routes involve use of phosphine ligands which are expensive, environmentally toxic for large scale operations,
d) Prior art methods utilize phase transfer catalysts such as tetrabutyl ammonium bromide in stoichiometric amounts and the reactions are carried out at very high temperatures of upto 140-150°C.
Thus, there is a need to develop an improved, convenient and cost effective process for preparation of (E)-4-(2-cyanoethenyl)-2,6-dimethylphenylamine hydrochloride of formula (II) having Z-isomer less than 0.5%, without involving any purification and does not involve use of phosphine reagent and which subsequently provides Rilpivirine hydrochloride (I) conforming to regulatory specifications.
……………………………..
http://www.google.com/patents/EP2643294A2?cl=en
The present inventors have developed a process for stereoselective synthesis of the key Rilpivirine intermediate, (E)-4-(2-cyanoethenyl)-2,6-dimemylphenylarnine hydrochloride (II), comprising diazotization of 2,6-dimethyl-4-amino-l- carboxybenzyl phenylamine followed by treatment with alkali tetrafluoroborate to provide the tetrafluoroborate salt of the diazonium ion which is followed by reaction with acrylonitrile in presence of palladium (II) acetate and subsequent deprotection of the amino group with an acid followed by treatment with hydrochloric acid to give the desired E isomer of compound (II) having Z isomer content less than 0.5% and with a yield of 75-80%. The compound (II) was subsequently converted to Rilpivirine hydrochloride of formula (I) with Z isomer content less than 0.1%.



……………………………………

Chemical structures of selected NNRTIs
…………………………….
http://pubs.acs.org/doi/full/10.1021/jm040840e
Purslane – The Gourmet Weed

Health benefits of Purslane
- This wonderful green leafy vegetable is very low in calories (just 16 kcal/100g) and fats; nonetheless, it is rich in dietary fiber, vitamins, and minerals.
- Fresh leaves contain surprisingly more omega-3 fatty acids (α-linolenic acid) than any other leafy vegetable plant. 100 grams of fresh purslane leaves provide about 350 mg of α-linolenic acid. Research studies show that consumption of foods rich in ω-3 fatty acids may reduce the risk of coronary heart disease, stroke, and help prevent the development of ADHD, autism, and other developmental differences in children.
- It is an excellent source of Vitamin A, (1320 IU/100 g, provides 44% of RDA) one of the highest among green leafy vegetables. Vitamin A is a known powerful natural antioxidant and is essential for vision. This vitamin is also required to maintain healthy mucus membranes and skin. Consumption of natural vegetables and fruits rich in vitamin A is known to help to protect from lung and oral cavity cancers.
- Purslane is also a rich source of vitamin C, and some B-complex vitamins like riboflavin, niacin, pyridoxine and carotenoids, as well as dietary minerals, such as iron, magnesium, calcium, potassium, and manganese.
- Furthermore, present in purslane are two types of betalain alkaloid pigments, the reddish beta-cyaninsand the yellow beta-xanthins. Both pigment types are potent anti-oxidants and have been found to have anti-mutagenic properties in laboratory studies. [Proc. West. Pharmacol. Soc. 45: 101-103 (2002)].
Portulaca oleracea (common purslane, also known as verdolaga, pigweed, little hogweed, or pursley, and moss rose) is an annual succulent in the family Portulacaceae, which may reach 40 cm in height.

Greek salad with Purslane
Approximately forty varieties currently are cultivated.[1] It has an extensive Old World distribution extending from North Africa through the Middle East and the Indian Subcontinentto Malesia and Australasia. The species status in the New World is uncertain: in general, it is considered an exotic weed, however, there is evidence that the species was in Crawford Lake deposits (Ontario) in 1430-89 AD, suggesting that it reached North America in the pre-Columbian era.[2] It is naturalised elsewhere and in some regions is considered an invasiveweed. It has smooth, reddish, mostly prostrate stems and alternate leaves clustered at stem joints and ends. The yellow flowers have five regular parts and are up to 6 mm wide. Depending upon rainfall, the flowers appear at anytime during the year. The flowers open singly at the center of the leaf cluster for only a few hours on sunny mornings. Seeds are formed in a tiny pod, which opens when the seeds are mature. Purslane has a taproot with fibrous secondary roots and is able to tolerate poor, compacted soils and drought.

A Purslane cultivar grown as a vegetable
Although purslane is considered a weed in the United States, it may be eaten as a leaf vegetable. It has a slightly sour and salty taste and is eaten throughout much of Europe, the middle east, Asia, and Mexico.[1][3] The stems, leaves and flower buds are all edible. Purslane may be used fresh as a salad, stir-fried, or cooked as spinach is, and because of its mucilaginous quality it also is suitable for soups and stews. Australian Aborigines use the seeds to make seedcakes. Greeks, who call it andrakla (αντράκλα) or glystrida (γλυστρίδα), fry the leaves and the stems with feta cheese, tomato, onion, garlic, oregano, and olive oil, add it in salads, boil it or add to casseroled chicken. In Turkey, besides being used in salads and in baked pastries, it is cooked as a vegetable similar to spinach. InAlbania it is called burdullak, and also is used as a vegetable similar to spinach, mostly simmered and served in olive oil dressing, or mixed with other ingredients as a filling for dough layers of byrek. In the south of Portugal (Alentejo), “baldroegas” are used as a soup ingredient.
Purslane contains more omega-3 fatty acids (alpha-linolenic acid in particular[4]) than any other leafy vegetable plant. Studies have found that Purslane has 0.01 mg/g ofeicosapentaenoic acid (EPA). This is an extraordinary amount of EPA for a land-based vegetable source. EPA is an Omega-3 fatty acid found mostly in fish, some algae, and flax seeds.[5] It also contains vitamins (mainly vitamin A, vitamin C, Vitamin E (alpha-tocopherol)[6] and some vitamin B and carotenoids), as well as dietary minerals, such asmagnesium, calcium, potassium, and iron. Also present are two types of betalain alkaloid pigments, the reddish betacyanins (visible in the coloration of the stems) and the yellow betaxanthins (noticeable in the flowers and in the slight yellowish cast of the leaves). Both of these pigment types are potent antioxidants and have been found to have antimutagenic properties in laboratory studies.[7]
100 Grams of fresh purslane leaves (about 1 cup) contain 300 to 400 mg of alpha-linolenic acid.[8] One cup of cooked leaves contains 90 mg of calcium, 561 mg of potassium, and more than 2,000 IUs of vitamin A. A half-cup of purslane leaves contains as much as 910 mg of oxalate, a compound implicated in the formation of kidney stones; however, many common vegetables, such as spinach, also can contain high concentrations of oxalates. Cooking purslane reduces overall soluble oxalate content by 27%, which is important considering its suggested nutritional benefits of being part of a healthy diet.[9]
When stressed by low availability of water, purslane, which has evolved in hot and dry environments, switches to photosynthesis usingCrassulacean acid metabolism (the CAM pathway): At night its leaves trap carbon dioxide, which is converted into malic acid (the souring principle of apples), and, in the day, the malic acid is converted into glucose. When harvested in the early morning, the leaves have ten times the malic acid content as when harvested in the late afternoon, and thus have a significantly more tangy taste.

Portulaca oleracea showing blooms

Seed pods, closed and open, revealing the seeds
Known as Ma Chi Xian (pinyin: translates as “horse tooth amaranth”) in traditional Chinese medicine, its active constituents include: noradrenaline, calcium salts, dopamine,DOPA, malic acid, citric acid, glutamic acid, asparagic acid, nicotinic acid, alanine, glucose, fructose, and sucrose.[10] Betacyanins isolated from Portulaca oleracea improved cognition deficits in aged mice.[11] A rare subclass of Homoisoflavonoids, from the plant, showed in vitro cytotoxic activities towards four human cancer cell lines.[12]Use is contraindicated during pregnancy and for those with cold and weak digestion.[10]Purslane is a clinically effective treatment for oral lichen planus,[13] and its leaves are used to treat insect or snake bites on the skin,[14] boils, sores, pain from bee stings, bacillary dysentery, diarrhea, hemorrhoids, postpartum bleeding, and intestinal bleeding.[10]
Portulaca oleracea efficiently removes bisphenol A, an endocrine-disrupting chemical, from a hydroponic solution. How this happens is unclear.[15]
Purslane, also known as Khulpha, Khursa in Hindi or Ghol in Marathi, is a water-retaining plant that can reach a height of 6″ – 12”. It’s smooth, reddish, thick leaves are wedge shaped. The leaves are alternately clustered at stem joints and are greenish on top and purplish on the underside.
The very tiny yellow flowers are around 6 mm wide and depending upon rainfall, the flowers appear at anytime during the year. Purslane has a taproot with fibrous secondary roots and is able to tolerate poor, compacted soils and drought.
It’s smooth, reddish, thick leaves are wedge shaped. The leaves are alternately clustered at stem joints and are greenish on top and purplish on the underside.
All that purslane needs to grow is part to full sun and clear ground. They are not picky about soil type or nutrition. If you decide to plant purslane seeds, simply scatter the seeds over the area that you plan on growing the purslane. Do not cover the seeds with soil. Purslane seeds need light to germinate, so they must stay on the surface of the soil. If you are using Purslane cuttings, lay them on the ground where you plan on growing purslane. Water the stems and they should take root in the soil in a few days.
 About a month after the seeds are planted, the first flowers will begin to appear. Once the flowers open, the seeds will begin to set within about a week to ten days. Since the Purslane is an invasive plant, it is difficult to get rid of. This is because the plant has stored enough energy for the seeds to continue to mature even after you pull the plant. Therefore, if you are trying to get rid of purslane, don’t try to compost it. If the compost pile is not hot enough to destroy the seeds, you will end up with more plants you don’t want.
About a month after the seeds are planted, the first flowers will begin to appear. Once the flowers open, the seeds will begin to set within about a week to ten days. Since the Purslane is an invasive plant, it is difficult to get rid of. This is because the plant has stored enough energy for the seeds to continue to mature even after you pull the plant. Therefore, if you are trying to get rid of purslane, don’t try to compost it. If the compost pile is not hot enough to destroy the seeds, you will end up with more plants you don’t want.
Purslane is ready to harvest in about 2 months from the time the seeds are sown. Make sure to harvest it regularly and be aware that it can become invasive. Harvesting before it develops flowers will help cut down on its spreading. Generally, you can harvest two or three times before the plants are exhausted.

The erect, tangy and succulent stems are high in Vitamin C. The leaves contain the highest concentration of Omega-3 fatty acids found in land plants. This is 5 times more than Spinach and 10 times more than any Lettuce or Mustard. It also contains Vitamin A, Vitamin C, and some Vitamin B and carotenoids as well as dietary minerals such as Magnesium, Calcium, Potassium and Iron.
100 Grams of fresh purslane leaves contain 300 to 400 mg of essential fatty acids (EFAs). One cup of cooked leaves contains 90 mg of Calcium, 561 mg of Potassium, and more than 2,000 IUs of Vitamin A.
As a companion plant, Purslane provides ground cover to create a humid microclimate for nearby plants, stabilizing ground moisture. Its deep roots bring up moisture and nutrients that those plants can use, and some, including corn, will “follow” purslane roots down through harder soil that they cannot penetrate on their own.
As a companion plant, Purslane provides ground cover to create a humid microclimate for nearby plants, stabilizing ground moisture. Its deep roots bring up moisture and nutrients that those plants can use, and some, including corn, will “follow” purslane roots down through harder soil that they cannot penetrate on their own (ecological facilitation). It is known as a beneficial weed in places that do not already grow it as a crop in its own right.
Widely used in East Mediterranean countries, archaeobotanical finds are common at manyprehistoric sites. In historic contexts, seeds have been retrieved from a protogeometric layer in Kastanas, as well as from the Samian Heraion dating to seventh century B.C. In the fourth century B.C., Theophrastus names purslane, andrákhne (ἀνδράχνη), as one of the several summer pot herbs that must be sown in April (H.P 7.12).[16] As portulaca it figures in the long list of comestibles enjoyed by the Milanese given by Bonvesin de la Riva in his “Marvels of Milan” (1288).[17]
In antiquity, its healing properties were thought so reliable that Pliny advised wearing the plant as an amulet to expel all evil (Natural History 20.120).[16]
A common plant in parts of India, purslane is known as Sanhti, Punarva, or Kulfa.
- The name verdolaga, associated with the plant that grows in South America is a nickname for Football clubs with green-white schemes in their uniforms, such as Colombia‘s Atletico Nacional and Argentina‘s Ferrocarril Oeste.
- Marlena Spieler (July 5, 2006). “Something Tasty? Just Look Down”. The New York Times.
- Byrne, R. and McAndrews, J. H. (1975). “Pre-Columbian puslane (Portulaca oleracea L.) in the New World”. Nature 253(5494): 726–727. doi:10.1038/253726a0.
- Pests in Landscapes and Gardens: Common Purslane. Pest Notes University of California Agriculture and Natural Resources Publication 7461. October 2003
- Jump up^ David Beaulieu. “Edible Landscaping With Purslane”. About.com.
- ARTEMIS P SIMOPOULOS Omega-3 Fatty Acids and Antioxidants in Edible Wild Plants. 2004. Biol Res 37: 263-277, 2004
- Simopoulos AP, Norman HA, Gillaspy JE, Duke JA. Common purslane: a source of omega-3 fatty acids and antioxidants. J Am Coll Nutr. 1992;11(4):374-82.
- Evaluation of the Antimutagenic Activity of Different Vegetable Extracts Using an In Vitro Screening Test
- A. P. Simopoulos, H. A. Norman, J. E. Gillaspy, and J. A. Duke. Common purslane: a source of omega-3 fatty acids and antioxidants. Journal of the American College of Nutrition, Vol 11, Issue 4 374-382, Copyright © 1992
- http://world-food.net/oxalate-content-of-raw-and-cooked-purslane/
- Tierra, C.A., N.D., Michael (1988). Planetary Herbology. Lotus Press. p. 199.
- Wang CQ. Yang GQ., “Betacyanins from Portulaca oleracea L. ameliorate cognition deficits and attenuate oxidative damage induced by D-galactose in the brains of senescent mice.,Phytomedicine. 17(7):527-32, 2010 Jun.
- Yan J, Sun LR, Zhou ZY, Chen YC, Zhang WM, Dai HF, Tan JW “Homoisoflavonoids from the medicinal plant Portulaca oleracea.” Phytochemistry. 2012 Aug;80:37-41
- Agha-Hosseini F, Borhan-Mojabi K, Monsef-Esfahani HR, Mirzaii-Dizgah I, Etemad-Moghadam S, Karagah A (Feb 2010). “Efficacy of purslane in the treatment of oral lichen planus”.Phytother Res. 24 (2): 240–4. doi:10.1002/ptr.2919.PMID 19585472.
- Bensky, Dan, et al. Chinese Herbal Medicine, Materia Medica. China: Eastland Press Inc., 2004.
- Watanabe I. Harada K. Matsui T. Miyasaka H. Okuhata H. Tanaka S. Nakayama H. Kato K. Bamba T. Hirata K.”Characterization of bisphenol A metabolites produced by Portulaca oleracea cv. by liquid chromatography coupled with tandem mass spectrometry.” , Biotechnology & Biochemistry. 76(5):1015-7, 2012.
- Megaloudi Fragiska (2005). “Wild and Cultivated Vegetables, Herbs and Spices in Greek Antiquity”.Environmental Archaeology 10 (1): 73–82.Noted by John Dickie, Delizia! The Epic History of Italians and Their Food (New York, 2008), p. 37.
- Noted by John Dickie, Delizia! The Epic History of Italians and Their Food (New York, 2008), p. 37.
“Portulaca oleracea”. FloraBase. Department of Environment and Conservation, Government of Western Australia.- Online Field guide to Common Saltmarsh Plants of Queensland
- Portulaca oleracea in West African plants – A Photo Guide.
- Purslane Recipes, Prairieland Community Supported Agriculture
| Nutritional value per 100 g (3.5 oz) | |
|---|---|
| Energy | 84 kJ (20 kcal) |
| Carbohydrates | 3.39 g |
| Fat | 0.36 g |
| Protein | 2.03 g |
| Water | 92.86 g |
| Vitamin A | 1320 IU |
| Thiamine (vit. B1) | 0.047 mg (4%) |
| Riboflavin (vit. B2) | 0.112 mg (9%) |
| Niacin (vit. B3) | 0.48 mg (3%) |
| Vitamin B6 | 0.073 mg (6%) |
| Folate (vit. B9) | 12 μg (3%) |
| Vitamin C | 21 mg (25%) |
| Vitamin E | 12.2 mg (81%) |
| Calcium | 65 mg (7%) |
| Iron | 1.99 mg (15%) |
| Magnesium | 68 mg (19%) |
| Manganese | 0.303 mg (14%) |
| Phosphorus | 44 mg (6%) |
| Potassium | 494 mg (11%) |
| Zinc | 0.17 mg (2%) |
| Link to USDA Database entry Percentages are roughly approximated using US recommendations for adults. Source: USDA Nutrient Database |
|
Preparation and serving methods
The stems and flower buds are also edible. Trim the tough stems near roots using a sharp knife. Cook under low temperature for a shorter period in order to preserve the majority of nutrients. Although antioxidant properties are significantly decreased on frying and boiling, its minerals, carotenes and flavonoids may remain intact with steam cooking.
India gift to the world
In fact, among the many names given to purslane around the world, there are some like the old Arabic baqla hamqa or the Spanish verdilacas or yerba orate that mean crazy plant. It is a reference not just to its appearance, but to the madly unrestrained way it grows, spreading rapidly in all directions at ground level in a mesh of stems, roots and leaves, which is one reason why for many gardeners purslane is one of the most annoying weeds.
Added to this is its remarkable resilience — it stores water it in its succulent stems and leaves, allowing it to tolerate hot, dry conditions, and can produce over 240,000 tiny seeds per plant, making it really hard to remove. It’s no surprise that purslane has spread remarkably widely, growing in different forms in most parts of the world and known by a wide variety of names such as portulaca or little door, from the way its seed pod opens, or the Hebrew regelah or foot, since that’s near where it grows, though the most unusual must be the term from Malawi that translates as ‘the buttocks of the chief’s wife”, an apparent reference to the fleshy rounded leaves of some forms.
Despite this wide range, most botanical studies give India as the origin for purslane, and some writers, like the American expert on wild food, Euell Gibbons, have even labelled it “India’s gift to the world.” But it is a gift that we have largely forgotten about, since few people here eat purslane these days, or even know that this weed is edible. It is rarely cultivated, but gathered from the wild and only rarely appears in places like Bhaji Gully because few know its value, other than old people or poor migrants from rural areas who have some memory of eating it.
One who did know the value of luni was Mahatma Gandhi, and while it’s a bit of a stretch to describe purslane as his favourite food, as some of its enthusiasts abroad have done, he did recommend it to several people and, in an article in his magazine Harijan, he wrote about “the nourishing properties of the innumerable leaves that are to be found hidden among the grasses that grow wild in India.” He had discovered these while living in Wardha and following a diet of uncooked food that required what he felt was an unreasonable amount of purchases from the local market. So he was delighted when an ashram resident “brought to me a leaf that was growing wild among the Ashram grasses. It was luni. I tried it, and it agreed with me.” It soon was a regular part of his diet.
Gandhi’s recommendations, of course, are no guide to taste, since he didn’t believe in enjoying food for its own sake. But luni has a pleasant lightly acid taste when raw, though with a slightly grassy, earthy undertone that does take some getting used to. It is probably never going to be one of those foods you have to try-before-you-die, but it is not bad at all to eat, either raw in a salad, or cooked. I find that the version we get here, which is rather less fleshy than purslane I’ve seen abroad, is worth stir-frying or adding to a dal, which brings out a nice, slightly peanutty taste. Another interesting way to cook it is in the Persian style, first sautéing it with onions and then cooking with eggs to make a firm omelette that has a nicely herbal taste when cut up and eaten cold.
The real reason for valuing purslane though is not taste, but health. It has always had a reputation for medicinal properties, with physicians over the centuries, from India to the Middle East to Europe, recommending it for everything from reducing fever, removing worms and soothing urinary infections. But modern science has made clear why it is of such value: apart from providing significant amounts of vitamins A, B and C. and decent amounts of protein, purslane probably contains more omega-3 fatty acids than any other commonly available vegetable source.
These fatty acids are essential for reducing cholesterol and heart diseases, but their most easily accessible source is oily fish, which makes it hard for vegetarians to get them. Some health conscious ones do force themselves to swallow fish oil capsules, or eat alsi (flax seeds) which are also a decent source of omega-3 acids. But purslane is probably a better source, and can be cooked and eaten as part of one’s meal. (The only caution is for people prone to kidney stones, since it also contains high levels of the oxalates which cause them). Luni may seem like a crazy thing to eat, but when people around the world are realising the value of this Indian plant, it is the way we are letting it become forgotten that may be what is really loony.
Loxiglumide

Loxiglumide, CR-1505
molecular formula :C21H30Cl2N2O5
molecular weight 461.3793
CAS NO:107097-80-3
WO 1987003869
Rottapharm (Originator)
Cholecystokinin (CCK) belongs to the group of substances known as brain-gut peptides and function as a neuropeptide and as a gut hormone. (Noble et al., Pharmacol. Rev. 1999, 51(4):745-781; Crawley et al., Peptides 1994, 15(4):731-755). It is now evident that at least two different receptors, namely CCK1 (formerly CCKA or alimentary) and CCK2 (formerly CCKB or brain) receptors, mediate CCK biological actions. (Noble et al., Pharmacol. Rev., 1999, 51(4):745-781; Woodruff and Hughes, Ann. Rev. Pharmacol. 1991, 31:469-501). CCK1 receptors are found in peripheral tissues, including the GI tract.
CCK is secreted primarily in response to meals and plays a well-recognized role in regulating gallbladder contraction and pancreatic enzyme secretion. Over the last decade, considerable evidence has emerged to support the concept that CCK plays an equally important role in the regulation of motor and sensory functions at various levels of the human upper GI tract. Specifically, the native peptide delays gastric emptying, modulates gastric sensory function (especially in response to fat), increases the rate of meal-induced, transient lower esophageal sphincter relaxations (TLESRs) and affects small bowel and colonic transit.
The CCK1 antagonists loxiglumide and dexloxiglumide have demonstrated the ability to reverse the physiologic effects of CCK on gastric emptying and to decrease dyspeptic symptoms induced by air distension and fat infusion. By example,loxiglumide reduced both exogenous and endogenous CCK-induced delay in gastric emptying of liquids and solids in healthy subjects (Borovicka et al., Am J Physiol. 1996, 271:448-453; Schwizer et al., Gut. 1997, 41(4):500-504). Dexloxiglumide reversed the diminished tolerance to water volume that occurred from CCK release in response to duodenal lipid infusion; the effect was due to reduction of intragastric volume, primarily due to accelerated gastric emptying (Lal et al., Am J Physiol Gastrointest Liver Physiol. 2004, 287(1):72-79). When proximal gastric relaxation was produced in healthy subjects by duodenal infusion of lipid, a potent stimulus of CCK release, the relaxation was reversed by loxiglumide (Feinle et al., Gastroenterology 1996, 110(5):1379-1385). Also, loxiglumide modulated antro-pyloroduodenal dysmotility, which is postulated to play a role in generation of dyspeptic symptoms, after it was experimentally induced in healthy subjects by intraduodenal infusion of a mixed liquid meal (Katschinski et al., Eur J Clin Invest. 1996, 26(7):574-583). Loxiglumide was also able to reverse the lowering of intragastric pressure of healthy subjects after duodenal infusion of lipids induced sensations such as fullness and nausea (See Feinle et al., 1996).
In patients with nonulcer dyspepsia and delayed gastric emptying, loxiglumide was shown to accelerate gastric emptying by comparison to placebo (Chua AS, Bekkering M, et al., 1994). Loxiglumide significantly improved dyspeptic symptoms in patients with non-ulcer dyspepsia in an 8-week study (Chua et al., Ann N Y Acad. Sci. 1994, 713:298-299). In another study in patients with functional dyspepsia, aggravation of nausea, fullness, discomfort, bloating and pain was produced by duodenal infusion of lipid with or without balloon distension; dexloxiglumide significantly improved dyspepsia symptom scores compared to placebo (Feinle et al., Gut. 2001, 48(3): 347-355).
Pharmaceutical compositions comprising CCKB antagonists and a proton pump inhibitor to control gastric acid secretion in gastrointestinal disorders have been described in the literature. (See WO 04/098610, WO 04/101533, WO 04/098609, WO 03/041714, WO 01/90078, WO 01/85724, WO 01/85723, WO 01/85704, WO 01/85167, and WO 93/12817) CCK-B receptors mediate CCK biological actions in the brain and are one of several regulators of gastric acid secretion. It is the CCK1 receptors, however, that mediate the CCK biological actions in peripheral tissues including gastric emptying and esophageal sphincter effects.
In addition, combination therapy of a PPI and a second agent, e.g., loxiglumide, to improve impaired esophageal motility has been disclosed as a possible treatment to gastroesophageal reflux disease. (Tonini et al., Drugs 2004, 64(4): 347-361). International Application Nos. PCT/EP2004/050936 and PCT/EP2005/050336 also disclose pharmaceutical combinations of a proton pump inhibitor and a compound that modifies gastrointestinal motility. Both international applications disclose that dexloxiglumide may be useful for therapy of irritable bowel syndrome (IBS) or GERD and may be used to modify gastrointestinal motility.
D,l-4-(3,4-dichlorobenzoylamino)-5-(N-3-methoxypropyl-pentylamino)-5-oxopentanoic acid (CR 1505; loxiglumide) is a newly developed analog of proglumide.

N-(3,4-dichlorobenzoyl)-glutamic acid anhydride (I) is condensed with N-(3-methoxypropyl)-N-pentylamine (II) in water at 5 °C to produce Loxiglumide.


FDA Advisory Committee Recommends Approval of Takeda’s Investigational Biologic Vedolizumab

Deerfield, Ill., December 9, 2013 and Osaka, Japan, December 10, 2013 — Takeda Pharmaceutical Company Limited (“Takeda”) and its wholly-owned subsidiary, Takeda Pharmaceuticals U.S.A., Inc., today announced that a joint panel of members from the Gastrointestinal Drugs and Drug Safety and Risk Management Advisory Committees of the United States (U.S.) Food and Drug Administration (FDA) voted to recommend approval of Takeda’s vedolizumab for the treatment of adults with moderately to severely active ulcerative colitis (UC) and Crohn’s disease (CD). All 21 committee members voted that based on currently available efficacy and safety data, the benefits outweigh the potential risks of vedolizumab to support approval for UC. Specifically, 13 committee members supported approval for UC patients who have failed steroids or immunosuppressants or TNF-α antagonists, while eight committee members supported approval for UC patients who have failed immunosuppressants or TNF-α antagonists (the indicated population would not include patients that failed steroids only). Twenty of the 21 committee members voted to support approval for CD. Specifically, 14 committee members supported approval for CD patients who have failed steroids or immunosuppressants or TNF-α antagonists while six supported approval for CD patients who have failed immunosuppressants or TNF-α antagonists (the indicated population would not include patients that failed steroids only).
read at
About Crohn’s disease and ulcerative colitis
Crohn’s disease (CD) and ulcerative colitis (UC) are the two most common forms of inflammatory bowel disease (IBD), which is marked by inflammation in the lining of the GI tract. CD can impact any part of the digestive tract, and common symptoms may include abdominal pain, diarrhea, rectal bleeding, weight loss, and/or fever. UC impacts the large intestine only, which includes the colon and the rectum. The most common symptoms of UC include abdominal discomfort and blood or pus in diarrhea. There is no known cause for CD or UC, although many researchers believe that the interaction of an outside agent, such as a virus or bacteria, with the body’s immune system may trigger them. No cure exists for CD or UC; the aim of IBD treatments is to induce and maintain remission, or achieve extended periods of time when patients do not experience symptoms.
About vedolizumab
Vedolizumab was developed for the treatment of CD and UC, as a gut-selective, humanized monoclonal antibody that specifically antagonizes the alpha4beta7 (α4β7) integrin, which is expressed on a subset of circulating white blood cells. These cells have been shown to play a role in mediating the inflammatory process in CD and UC. α4β7 binds with a specific adhesion molecule primarily expressed in the intestinal tract. Therefore, vedolizumab, by preventing this interaction, has a gut selective effect.
About Takeda Pharmaceutical Company Limited
Located in Osaka, Japan, Takeda is a research-based global company with its main focus on pharmaceuticals. As the largest pharmaceutical company in Japan and one of the global leaders of the industry, Takeda is committed to strive towards better health for patients worldwide through leading innovation in medicine. Additional information about Takeda is available through its corporate website, http://www.takeda.com.
Vedolizumab is a monoclonal antibody being developed by Millennium Pharmaceuticals, Inc. for the treatment of ulcerative colitis and Crohn’s disease.It binds to integrin α4β7(LPAM-1, lymphocyte Peyer’s patch adhesion molecule 1).[1][2]
The molecule was first identified by Dr. Andrew Lazarovits [1][2] as the murine MLN0002 homologue. His discovery of the mouse equivalent of this antibody—originally applied to anti-rejection strategies in kidney transplantation—was published in the journal Nature in 1996. The drug was then licensed to Millennium Pharmaceuticals of Boston for further development.
As of October 2009, vedolizumab is undergoing Phase III trials.[3] Clinical trials indicate that Vedolizumab was found safe and highly effective for inducing and maintaining clinical remission in patients with moderate to severe ulcerative colitis [3]. Dr. Brian Faegan, head researcher, reported an absence of any instances of progressive multifocal leukoencephalopathy (PML), which is a particularly important finding [4]. It looks like it will be an effective abiologic agent without some of the toxicity issues previously seen with anti-TNF drugs .
It is widely believed now that “vedolizumab can be used either as a first-line treatment or in case of anti-TNF failure”
- Statement On A Nonproprietary Name Adopted By The USAN Council – Vedolizumab, American Medical Association.
- Soler, D; Chapman, T; Yang, LL; Wyant, T; Egan, R; Fedyk, ER (2009). “The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases”. The Journal of Pharmacology and Experimental Therapeutics 330 (3): 864–75. doi:10.1124/jpet.109.153973. PMID 19509315.
- ClinicalTrials.gov NCT00790933 Study of Vedolizumab (MLN0002) in Patients With Moderate to Severe Crohn’s Disease (GEMINI II)

