
Home » Uncategorized (Page 148)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Green…Asymmetric hydrogentation of unfunctionalised olefins/enamines/imines
Asymmetric hydrogentation of unfunctionalised olefins/enamines/imines
The reaction survey found that the predominant strategy for the introduction of chirality was through classical chemical resolutions as opposed to introductions through biotransformation or transition metal or organometallic catalytic means.
Asymmetric hydrogenation provides an elegant methodology for the introduction of chirality, meeting many of the goals of green chemistry and is finding increasing application in API synthesis.47
The efficiency of this approach is elegantly exemplified by the Merck second generation synthesis of sitagliptin 5 (Scheme ), where an unprecedented final stage asymmetric hydrogenation of the unprotected enamide 6 resulted in an increase in overall yield of almost 50% and produced 100 kg less waste per kg sitagliptin48 when compared with the first generation approach.49
 |
||
| Scheme The synthesis of sitagliptin. | ||
There are challenging areas remaining within the field, for example, the hydrogenation of enamides and related substrates in the synthesis of amino acids has numerous examples50 but few examples exist for unsubstitued enamines41 and imines. Some classes of alkene offer additional challenges.51 For the pharmaceutical industry, the limited time for synthetic route identification is an issue and access to catalyst and ligand diversity is required to ensure the application of this approach.52
Some pharmaceutical companies have synthesised their own ligands and have found very effective catalysts.53 The majority of academic asymmetric hydrogenation approaches are based on homogeneous catalysis to overcome issues of activation and mass transfer. For pharmaceutical use, efficient catalyst and ligand recovery, and eliminating heavy metal contamination of the API are significant requirements for the industry.
These controls are often easier to achieve with heterogeneous methodology where there are less examples.50 The demonstration of organocatalytic hydride transfer offers the possibility of future access to metal free asymmetric hydrogenations.54
- 47………V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793. See also Asymmetric Catalysis on Industrial Scale Challenges, Approaches and Solutions, ed. H.-U. Blaser and E. Schmidt, Wiley-VCH, Weinheim, 2004 Search PubMed .
- 48………..http://www.epa.gov/greenchemistry/pubs/pgcc/winners/gspa06.html .
- 49……K. B. Hansen, J. Balsells, S. Dreher, Y. Hsiao, M. Kubryk, M. Palucki, N. Rivera, D. Steinhuebel, J. D. Armstrong III, D. Askin and E. J. J. Grabowski, Org. Process Res. Dev., 2005, 9, 634–639 Search PubMed .
- 50………..M. Studer, H.-U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS Search PubMed .
- 51……..X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS Search PubMed
- 52……….I. C. Lennon and C. J. Pilkington, Synthesis, 2003, 1639–1642 CrossRef CAS Search PubMed .
- 53………G. Hoge, H.-P. Wu, W. S. Kissel, D. A. Plum, D. J. Greene and J. Bao, J. Am. Chem. Soc., 2004, 126, 5966–5967 CrossRef CAS Search PubMed .
- 54……..H. Adolfsson, Angew. Chem., Int. Ed., 2005, 44, 3340–3342 CrossRef CAS Search PubMed .
Garcinia Cambogia Kills 89% of Pancreatic Cancer Cells and Synergizes with Curcumin

Garcinia Cambogia Extract Explained
The latest in innovation in weight loss supplements is Garcinia Cambogia. It is unparalleled in its ability to help boost your body’s weight loss potential, and help you achieve your perfect weight.
There’s no wonder it’s quickly gained a huge following, with endorsements from celebrities to health experts, with scientifically proven ability to help you increase your fat burning power.
As with all supplements like this, there are questions as to how it works, and just how it can benefit you, with your health and in losing weight. This site’s goal is to hopefully answer some of these questions, and to show you just how you can benefit from this amazing supplement.

What is Garcinia Cambogia?
Garcinia Cambogia is a fruit, that is grown all over Asia, but originating in Indonesia and grows particularly well grows best with tropical conditions. It rose to prominence after appearance on the massively lauded American health show, Doctor Oz. It had recently been subject to a medical trial where the study scientifically proved it was highly effective in increasing burning up fat and aiding in overall weight loss.
Can Garcinia Cambogia Extract Help Me Lose Weight?
Well Garcinia Cambogia contains a useful compound called Hydroxycitric Acid, which I’ll refer to as HCA for ease of reference. Garcinia Cambogia contains one of the highest known concentrations of HCA, and this was why it was noticed as a potential weight loss supplement. HCA has two main mechanisms in which it works to boost your fat burning potential:

Firstly it will reduce the ability for the body to convert carbohydrates into fat cells, meaning that even without a calorific controlled diet; you will be able to aid your body’s ability to burn of existing fat, while not gaining additional fat.
Secondly it will also suppress your appetite, meaning that it will not only help reduce the weight you can put on by stopping putting on additional fat, it will also massively reduce the cravings and hunger that usually lead to breaking a diet and weight loss routine. This means that your body will just be burning off the existing fat, helping you to achieve that perfect weight!
What About Side Effects form Garcinia Cambogia?
The most amazing thing about Garcinia Cambogia is that the side effects of the product are almost non-existent in the all-natural extract. By this I mean an extract that contains purely Garcinia Cambogia extract without any additional additives that some unrepeatable sellers will try to pass off as the quality product. Those extracts that contain additives can cause side effect in users of Garcinia Cambogia, which are related to the different additives and binding agents added.
The cost of Garcinia Cambogia from a supplier, whom ensures a high quality and natural product, will range from $40-50 a bottle. There is however introductory offers from some suppliers, such as Miracle Garcinia Cambogia currently offering a free bottle of Garcinia Cambogia with every order.
This means the overall cost per bottle of this amazing product can drop as low as $28.99. Most of these offers unfortunately do have a limited stock and therefore won’t be around forever.
Garcinia gummi-gutta is a tropical[2] species of Garcinia native to Indonesia. Common names include garcinia cambogia (a former scientific name), as well as gambooge, brindleberry,[3] brindall berry, Malabar tamarind,[2] assam fruit, vadakkan puli (northern tamarind) and kudam puli (pot tamarind).[4] This fruit looks like a small pumpkin and is green to pale yellow in color. It has recently received considerable media attention because of its purported effects on weight loss, although there is no clinical evidence to support this claim.

Cultivation


Ripe fruit
Garcinia gummi-gutta is grown for its fruit in southeast Asia, coastal Karnataka/Kerala, India, and west and central Africa. It thrives in most moist forests.
Garcinia gummi-gutta is one of several closely related Garcinia species from the plant family Guttiferae.[5] With thin skin and deep vertical lobes, the fruit of G. gummi-gutta and related species range from about the size of an orange to that of a grapefruit; G. gummi-gutta looks more like a small yellowish, greenish or sometimes reddish pumpkin.[6] The color can vary considerably. When the rinds are dried and cured in preparation for storage and extraction, they are dark brown or black in color.
Along the west coast of South India, G. gummi-gutta is popularly termed “Malabar tamarind,” and shares culinary uses with the tamarind (Tamarindus indica). The latter is a small and the former a quite large evergreen tree. G. gummi-gutta is also called “goraka” or, in some areas, simply “kattcha puli” (souring fruit).
Uses
Cooking
Garcinia gummi-gutta is used in cooking, including in the preparation of curries. The fruit rind and extracts of Garcinia species are called for in many traditional recipes,[7] and various species of Garcinia are used similarly in food preparation in Assam (India), Thailand, Malaysia, Burma and other Southeast Asian countries. In the Indian Ayurvedic medicine, “sour” flavors are said to activate digestion. The extract and rind of Garcinia gummi-gutta is a curry condiment in India. It is an essential souring ingredient in the Southern Thai variant of kaeng som, a sour curry.
Garcinia gummi-gutta is employed commercially in fish curing, especially in Sri Lanka (Colombo curing) and South India, which makes use of the antibacterial qualities of the fruit.
The trees can be found in forested areas and also are protected in plantations otherwise given over to pepper, spice, and coffee production.
Traditional medicine
Aside from its use in food preparation and preservation, extracts of G. gummi-gutta are sometimes used in traditional medicine aspurgatives. The fruit rind is also used to make medicine.
Weight loss
In late 2012, a United States television personality, Dr. Oz, promoted Garcinia cambogia extract as a “magic” weight-loss aid. Dr. Oz’s previous endorsements have often led to a substantial increase in consumer interest in the promoted products. However, a dearth of scientific evidence and clinical trials do not support claims that Garcinia cambogia is an effective weight-loss aid.[8][9] A meta-analysis found a possible small, short-term weight loss effect (under 1 kilogram).[10] However, side effects—namely hepatotoxicity (chemical-driven liver damage)—led to one preparation being withdrawn from the market.[11][12]
A 1998 randomized controlled trial looked at the effects of hydroxycitric acid, the purported active component in Garcinia gummi-gutta, as a potential antiobesity agent in 135 people. The conclusion from this trial was that “Garcinia cambogia failed to produce significant weight loss and fat mass loss beyond that observed with placebo”.[13]

When the fruit is sun dried for several days, it becomes black with a shrivelled body
References
- “Garcinia gummi-gutta (L.) Roxb.”. The Plant List. Royal Botanic Gardens, Kew and Missouri Botanical Garden. Retrieved 1 June 2013.
- “USDA GRIN Taxonomy”.
- “Potential treatments for insulin resistance in the horse: A comparative multi-species review”. Science Direct. Retrieved 6 October 2013.
- “Meals that heal – Soul curry”. The Hindu. Retrieved 3 October 2013.
- Publications & Information Directorate, Council of Scientific & Industrial Research (1986). G. cambogia Desr. The Useful Plants of India. (New Delhi: Publications & Information Directorate, 1986) 229.
- “Fruit yellowish or reddish, size of an orange having six or eight deep longitudinal grooves in its fleshy pericarp. Pulp acid of a pleasant flavor. It is dried among the Singalese who use it in curries.” Uphof, J.C. Th. (1968).
- “The acid rinds of the ripe fruit are eaten, and in Ceylon are dried, and eaten as a condiment in curries.” Drury, Heber (1873). “Garcinia gambogia(Desrous) N. 0. Clusiaceae”. The Useful Plants of India, second edition. London: William H. Allen & Co. p. 220.
- Belluz, Julia; Hoffman, Steven J. (1 January 2013). “Dr. Oz’s Miraculous Medical Advice; Pay no attention to that man behind the curtain”. Slate. The Slate Group. Retrieved 31 May 2013.
- Márquez F1, Babio N, Bulló M, Salas-Salvadó J (2012). “Evaluation of the safety and efficacy of hydroxycitric acid or Garcinia cambogia extracts in humans”. Crit Rev Food Sci Nutr 52 (7): 585–94. doi:10.1080/10408398.2010.500551. PMID 22530711.
- Hepatotoxicity (from hepatic toxicity) implies driven liver damage.
- Lobb, A. (2009). “Hepatoxicity associated with weight-loss supplements: A case for better post-marketing surveillance”. World Journal of Gastroenterology 15 (14): 1786–1787. doi:10.3748/wjg.15.1786. PMC 2668789. PMID 19360927.
- Kim YJ1, Choi MS, Park YB, Kim SR, Lee MK, Jung UJ (2013). “Garcinia Cambogia attenuates diet-induced adiposity but exacerbates hepatic collagen accumulation and inflammation”. World J Gastroenterol 19 (29): 4689–701. doi:10.3748/wjg.v19.i29.4689. PMID 23922466.
- Heymsfield, S. B.; Allison, D. B.; Vasselli, J. R.; Pietrobelli, A.; Greenfield, D.; Nunez, C. (1998). “Garcinia cambogia (Hydroxycitric Acid) as a Potential Antiobesity Agent: A Randomized Controlled Trial”. JAMA: the Journal of the American Medical Association 280 (18): 1596–1600.doi:10.1001/jama.280.18.1596. PMID 9820262.
BERAPROST….Stable prostacyclin analog.

BERAPROST
https://www.ama-assn.org/resources/doc/usan/beraprost.pdf
2,3,3a,8b-tetrahydro-2-hydroxy-1-(3-hydroxy-4-methyl-1-octen-6-ynyl)-1H-cyclopenta(b)benzofuran-5-butanoic acid
(±)-(IR*,2R*,3aS*,8bS*)-2,3,3a,8b-tetrahydro-2-hydroxy-1-[(E)-(3S*)-3-hydroxy-4-methyl-1-octene-6-inyl]-1H-cyclopenta[b]benzofuran-5-butyric acid
rac-4-{(1R,2R,3aS,8bS)-2-hydroxy-1-[(1E,3S,4RS)-3-hydroxy-4-methyloct-1-en-6-ynyl]-2,3,3a,8b-tetrahydro-1H-cyclopenta[b][1]benzofuran-5-yl}butanoic acid
- Beraprost
- Beraprostum
- Beraprostum [INN-Latin]
- MDL 201229
- MDL-201229
- ML 1229
- ML-1229
- UNII-35E3NJJ4O6
Beraprost is a synthetic analogue of prostacyclin, under clinical trials for the treatment of pulmonary hypertension. It is also being studied for use in avoiding reperfusion injury.
As an analogue of prostacyclin PGI2, beraprost effects vasodilation, which in turn lowers the blood pressure. Beraprost also inhibits plateletaggregation, though the role this phenomenon may play in relation to pulmonary hypertension has yet to be determined.
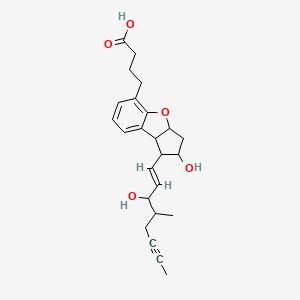
Beraprost …sodium salt
ML 1129; Procyclin; TRK 100 (CAS 88475-69-8)

| Synonyms |
|
|---|---|
| Formal Name | 2,3,3a,8b-tetrahydro-2-hydroxy-1-(3-hydroxy-4-methyl-1-octen-6-ynyl)-1H-cyclopenta[b]benzofuran-5-butanoic acid, monosodium salt |
| CAS Number | 88475-69-8 |
| Molecular Formula | C24H29O5 · Na |
| Formula Weight | 420.5 |
- Beraprost sodium is a prostacyclin analog and an NOS3 expression enhancer that was first launched in 1992 in Japan pursuant to a collaboration between Astellas Pharma and Toray for the oral treatment of peripheral vascular disease (PVD), including Raynaud’s syndrome and Buerger’s disease. In 2000, the drug was commercialized for the treatment of pulmonary hypertension. Development for the oral treatment of intermittent claudication associated with arteriosclerosis obliterans (ASO) was discontinued at Kaken and United Therapeutics after the product failed to demonstrate statistically significant results in a phase III efficacy trial.
- In terms of clinical development, beraprost sodium is currently in phase II clinical trials at Kaken for the treatment of lumbar spinal canal stenosis and at Astellas Pharma for the oral treatment of primary chronic renal failure. The company is also conducting phase III trials for the treatment of nephrosclerosis. The drug has also been studied through phase II clinical trials at Kaken for the oral treatment of diabetic neuropathy, but recent progress reports for this indication have not been made available.
- Beraprost is an oral form of prostacyclin, a member of the family of lipid molecules known as eicosanoids. Prostacyclin is produced in the endothelial cells from prostaglandin H2 by the action of the enzyme prostacyclin synthase. It has been shown to keep blood vessels dilated and free of platelet aggregation.
- Beraprost sodium was originally developed at Toray in Japan, and rights to the drug were subsequently acquired by Astellas Pharma. A 1972 alliance between Toray and Kaken Pharmaceutical to develop and commercialize prostaglandin led to a later collaboration agreement for the development of beraprost. In 1990, Toray granted the right to market the drug to Sanofi (formerly known as sanofi-aventis), a licensing agreement that was later expanded to include Canada, the U.S., South America, Africa, Southeast Asia, South Asia, Korea and China. In September 1996, Bristol-Myers Squibb entered into separate agreements with Sanofi and Toray to acquire all development and marketing rights to beraprost in the U.S. and Canada. In January 1999, United Therapeutics and Toray agreed to cooperatively test the drug in North America, and in July 2000, a new agreement was signed pursuant to which United Therapeutics gained exclusive North American rights to develop and commercialize sustained-release formulations of beraprost for all vascular and cardiovascular diseases. In 1999, orphan drug designation was received in the U.S. for the treatment of pulmonary arterial hypertension associated with any New York Heart Association classification (Class I, II, III, or IV). In 2011, orphan drug designation was assigned in the U.S. for the treatment of pulmonary arterial hypertension.
-
The compound name of beraprost which is used as an antimetastasis agent of malignant tumors according to the present invention is (±)-(IR*,2R*,3aS*,8bS*)-2,3,3a,8b-tetrahydro-2-hydroxy-1-[(E)-(3S*)-3-hydroxy-4-methyl-1-octene-6-inyl]-1H-cyclopenta[b]benzofuran-5-butyric acid. This compound has the following structure.

Beraprost is described in Japanese Laid-open Patent Application (Kokai) Nos. 58-32277, 57-144276 and 58-124778 and the like as a PGI₂ derivative having a structure in which the exoenol moiety characteristic to beraprost is converted to inter-m-phenylene structure. However, it is not known that beraprost has an activity to inhibit metastasis of malignant tumors.
-
The beraprost which is an effective ingredient of the agent of the present invention includes not only racemic body, but also d-body and l-body. Beraprost can be produced by, for example, the method described in the above-mentioned Japanese Laid-open Patent Application (Kokai) No. 58-124778. The salts of beraprost include any pharmaceutically acceptable salts including alkaline metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as magnesium salt and calcium salt; ammonium salt; primary, secondary and tertiary amine salts; and basic amino acid salts.

…………………..
EXAMPLE 6 Beraprost of the Formula (I)
0.246 g (0.6 mmol) of compound of the general formula (II) obtained in Example 5 is dissolved in 1 ml of methanol and 1 ml of 1 M aqueous sodium hydroxide solution is added dropwise slowly thereto. After stirring for an hour the methanol is distilled off from the reaction mixture in vacuum. The aqueous residue is diluted with 10 ml of water extracted with methyl-tert.butyl-ether and the combined organic phase is washed with saturated NaCl solution, dried on Na2SO4 and evaporated. The residue of evaporation is crystallized from ethylacetate-hexane mixture and the pure above mentioned title compound is obtained as colourless crystals.
Yield: 0.21 g (87%)
TLC-Rf (toluene-dioxan-acetic acid 20:10:1)=0.41
Melting point: 98–112° C.
1H NMR (400 MHz, CDCl3), δH (ppm): 1.00d, 1.03d [3H; J=6.8 Hz; 21-H3]; 1.79m [1H; 16-H]; 1.80t, 1.81t [3H, J=2.5,2.4 Hz; 20-H3]; 2.3–1.9m [5H, 3-H2, 10Hb, 17-H2]; 2.34t [1H; J=7.4 Hz; 2-H2]; 2.43m [1H; 12-H]; 2.64m [3H; 10-Ha, 4-H2]; 3.43t, 3.44t [1H, J=8.7,8.5 Hz; 8-H]; 3.92m [1H; 11-H]; 4.07t, 4.17t [1H, J=7.3,5.6 Hz; 15-H]; 4.3b [2H; OH]; 5.09m [1H, 9-H]; 5.58dd, 5.61dd [1H; J=15.3,6.5 Hz; 14-H]; 5.67dd, 5.68dd [1H; J=15.3,8.0 Hz; 13-H]; 6.77m [1H; 2′-H]; 6.95m [2H; 1′-H,3′-H]13C NMR (100 MHz, CDCl3), δC (ppm): 3.5, 3.6 [C-20]; 14.7, 15.8 [C-21]; 22.3, 22.6 [C-17]; 24.6 [C-2]; 29.1 [C-4]; 33.1 [C-3]; 38.2, 38.3 [C-16]; 41.2 [C-10]; 50.4 [C-8]; 58.8 [C-12]; 75.8, 76.3, 76.4 [C-11, C-15]; 77.2, 77.4 [C-18, C-19]; 84.5, 84.6 [C-9]; 120.6 [C-2′]; 121.9 [C-3′]; 123.2 [C-5]; 129.0 [C-1′]; 129.7 [C-7]; 132.3, 133.0, 133.8, 134.0 [C-13, C-14]; 157.2 [C-6]; 178.3 [C-1].
EXAMPLE 7 Beraprost Sodium Salt (The Sodium Salt of the Compound of Formula (I)
0.199 g of beraprost is dissolved in 2 ml of methanol, 0.5 ml of 1 M aqueous solution of sodium hydroxide is added thereto and after their mixing the solvent is evaporated in vacuum and thus the above title salt is obtained as colourless crystals.
Yield: 0.21 g (100%)
Melting point: >205° C.
1H NMR (400 MHz, DMSO-d6), δH (ppm): 0.90d, 0.92d [3H; J=6.7 Hz; 21-H3]; 1.75–1.55m [7H; 10Hb, 16-H, 3-H2, 20-H3]; 1.89t [2H, J=7.6 Hz; 2-H2]; 1.94m [1H; 17-Hb]; 2.16q [1H, J=8.5 Hz; 12-H]; 2.25m [1H; 17-Ha]; 2.44t [2H; J=7.5 Hz; 4-H2]; 2.50o [1H; 10-Ha]; 3.39t [1H, J=8.5 Hz; 8-H]; 3.72td [1H; J=8.5,6.1 Hz; 11-H]; 3.84t 3.96t [1H, J=6.5,6.0 Hz; 15-H]; 4.85b [2H, OH]; 5.01dt [1H, J=8.5,6.6 Hz; 9-H]; 5.46dd, 5.47dd [1H; J=15.4,6.5 Hz, J=15.4,6.0 Hz; 14-H]; 5.65dd, 5.66dd [1H; J=15.4,8.5 Hz; 13-H]; 6.71m [1H; 2′-H]; 6.92m [2H; 1′-H, 3′-H] During the above thin layer chromatography (TLC) procedures we used plates MERCK Kieselgel 60 F254, thickness of layer is 0.2 mm, length of plates is 5 cm.


…………….
-
Reaction Scheme A.




-
The starting material of bromocarboxylic acid, Compound 1, and the process for the preparation thereof are disclosed in Japanese Patent Application No. 29637/81.
-
Scheme B.
REACTION SCHEME B
-

- REACTION SCHEME C
-

 Org Lett 2012, 14(1): 299
Org Lett 2012, 14(1): 299
EP0024943A1 Sep 2, 1980 Mar 11, 1981 Toray Industries, Inc. 5,6,7-Trinor-4,8-inter-m-phenylene PGI2 derivatives and pharmaceutical compositions containing them EP0084856A1 Jan 19, 1983 Aug 3, 1983 Toray Industries, Inc. 5,6,7-Trinor-4, 8-inter-m-phenylene prostaglandin I2 derivatives JP3069909B Title not available
-
Sulfoaildenafil …. An analog of Sildenafil which has been used as an illegal adulterant in some dietary supplements

Sulfoaildenafil
An analog of Sildenafil which has been used as an illegal adulterant in some dietary supplements.
856190-47-1 cas no
5-(5-(((3R,5S)-3,5-Dimethylpiperazin-1-yl)sulfonyl)-2-ethoxyphenyl)-1-methyl-3-propyl-1H-pyrazolo[4,3-d]pyrimidine-7(4H)-thione
-
7H-Pyrazolo(4,3-d)pyrimidine-7-thione, 5-(5-(((3R,5S)-3,5-dimethyl-1-piperazinyl)sulfonyl)-2-ethoxyphenyl)-1,6-dihydro-1-methyl-3-propyl-, rel-
- Sildenafil thione
- Thioaildenafil
- UNII-33DX49E09G
-
-
C23-H32-N6-O3-S2
- 504.6768
-
Sulfoaildenafil (thioaildenafil) is a synthetic chemical compound that is a structural analog of sildenafil (Viagra).[1] It was first reported in 2005,[2] and it is not approved by any health regulation agency. Like sildenafil, sulfoaildenafil is a phosphodiesterase type 5 inhibitor.
Sulfoaildenafil has been found as an adulterant in a variety of supplements which are sold as “natural” or “herbal” sexual enhancement products.[3][4][5][6] A range of designer analogues of USA FDA-approved inhibitors of type-5 cGMP-specific phosphodiesterase (PDE5), such as sildenafil and vardenafil, have been detected in recent years as adulturants in over-the-counter herbal aphrodisiac products and dietary supplements,[7][8][9] in an apparent attempt to circumvent both the legal restrictions on sale of erectile dysfunction drugs, which are prescription-onlymedicines in most Western countries, and the patent protection which prevents sale of these drugs by competitors except under license to their inventors.

Figure 1. Biological pathway of penile erection
These compounds have been demonstrated to display PDE5 inhibitory activity in vitro and presumably have similar effects when consumed, but have undergone no formal testing in either humans or animals, and as such represent a significant health risk to consumers of these products due to their unknown safety profile.[10] Some attempts have been made to ban these drugs as unlicensed medicines, but progress has been slow so far, as even in those jurisdictions which have laws targeting designer drugs, the laws are drafted to ban analogues of illegal drugs of abuse, rather than analogues of prescription medicines. However at least one court case has resulted in a product being taken off the market.[11]

Figure 2. PDE5 domains

In December 2010, the United States Food and Drug Administration (FDA) issued a warning to consumers about such products stating, “The FDA has found many products marketed as dietary supplements for sexual enhancement during the past several years that can be harmful because they contain active ingredients in FDA-approved drugs or variations of these ingredients.”[12]

Figure 3. PDE5 Domains
Volume 50, Issue 2, 8 September 2009, Pages 228–231
Phosphodiesterase type 5 (PDE-5) inhibitors represent a class of drugs used primarily in the treatment of erectile dysfunction. Currently, three PDE-5 inhibitors have been approved by the U.S. Food and Drug Administration (FDA) for use in the United States: sildenafil citrate, tadalafil, and vardenafil hydrochloride trihydrate. A bulk material, labeled as an ingredient for a dietary supplement, was analyzed for the presence of PDE-5 inhibitors. The compound that was detected displayed structural similarities to sildenafil, and was characterized further using LC–MSn, FTICRMS, X-ray crystallography and NMR. The compound was given the name sulfoaildenafil. When compared to sildenafil, sulfoaildenafil contains a sulfur atom substitution for the oxygen atom in the pyrazolopyrimidine portion of the molecule, and a 3,5-dimethyl substitution on the piperazine ring, rather than the 4-methyl moiety. The X-ray crystallographic data indicate that the material in this sample is comprised of two polymorphs, which may affect the chemical and/or biological properties of any product formulated with this compound.
……………..

http://www.theresonance.com/2012/categories/pharmaceutical/adulterated-natural-products
……………….
Herbal Supplement for Erectile Dysfunction Found to Contain Thio Structural Analog of Sildenafil (Viagra)
A herbal supplement marketed to alleviate erectile dysfunction was recently submitted for testing in our laboratory because it was surprisingly effective considering it should only contain the traditional herbals utilized for this problem such as Oyster, 2-Deoxy-D Glucose, Barberry, Snow Lotus, Bombyx Mori L., Ginger Root, Salfron Crocus.
http://process-nmr.com/WordPress/?cat=5
References
- Gratz, SR; Zeller, M; Mincey, DW; Flurer, CL (2009). “Structural characterization of sulfoaildenafil, an analog of sildenafil”. Journal of pharmaceutical and biomedical analysis 50 (2): 228–31. doi:10.1016/j.jpba.2009.04.003. PMID 19427155.
- Li, Shuxin; Ren, Jianping; Zhao, Yanjin; Lv, Qiujun; Guo, Jinhua. Pyrazolopyrimidinethione Derivatives, Salts and Solvates thereof, Preparation Methods and Use thereof. WO 2005058899
- Gryniewicz, CM; Reepmeyer, JC; Kauffman, JF; Buhse, LF (2009). “Detection of undeclared erectile dysfunction drugs and analogues in dietary supplements by ion mobility spectrometry”. Journal of pharmaceutical and biomedical analysis 49 (3): 601–6. doi:10.1016/j.jpba.2008.12.002. PMID 19150190.
- FDA warns consumers to avoid sexual enhancement pills, Sanjay Gupta, CNN, December 13th, 2010
- Reepmeyer JC, d’Avignon DA (January 2009). “Structure elucidation of thioketone analogues of sildenafil detected as adulterants in herbal aphrodisiacs”. Journal of Pharmaceutical and Biomedical Analysis 49 (1): 145–50. doi:10.1016/j.jpba.2008.10.007. PMID 19042103.
- Balayssac S, Trefi S, Gilard V, Malet-Martino M, Martino R, Delsuc MA (November 2008). “2D and 3D DOSY (1)H NMR, a useful tool for analysis of complex mixtures: Application to herbal drugs or dietary supplements for erectile dysfunction”. Journal of Pharmaceutical and Biomedical Analysis 50 (4): 602–12. doi:10.1016/j.jpba.2008.10.034. PMID 19108978.
- Zou P, Oh SS, Hou P, Low MY, Koh HL (February 2006). “Simultaneous determination of synthetic phosphodiesterase-5 inhibitors found in a dietary supplement and pre-mixed bulk powders for dietary supplements using high-performance liquid chromatography with diode array detection and liquid chromatography-electrospray ionization tandem mass spectrometry”. J Chromatogr A 1104 (1-2): 113–22. doi:10.1016/j.chroma.2005.11.103. PMID 16364350.
- Gratz SR, Gamble BM, Flurer RA (2006). “Accurate mass measurement using Fourier transform ion cyclotron resonance mass spectrometry for structure elucidation of designer drug analogs of tadalafil, vardenafil and sildenafil in herbal and pharmaceutical matrices”. Rapid Commun. Mass Spectrom. 20 (15): 2317–27. doi:10.1002/rcm.2594. PMID 16817245.
- Hou P, Zou P, Low MY, Chan E, Koh HL (September 2006). “Structural identification of a new acetildenafil analogue from pre-mixed bulk powder intended as a dietary supplement”. Food Addit Contam 23 (9): 870–5. doi:10.1080/02652030600803856. PMID 16901855.
- Oh, SS; Zou, P; Low, MY; Koh, HL (2006). “Detection of sildenafil analogues in herbal products for erectile dysfunction.”. Journal of toxicology and environmental health. Part A 69 (21): 1951–8.doi:10.1080/15287390600751355. PMID 16982533.
- Venhuis, BJ; Blok-Tip, L; De Kaste, D (2008). “Designer drugs in herbal aphrodisiacs.”. Forensic Science International 177 (2–3): e25–7. doi:10.1016/j.forsciint.2007.11.007. PMID 18178354.
- FDA warns consumers to avoid Man Up Now capsules, United States Food and Drug Administration, Dec. 15, 2010
Advanced Nanoparticle System Kills Cancer Cells From Within

The latest cancer targeting nanoparticles being developed in labs around the world are getting ever more complex and are utilizing multiple mechanisms to find and strike their targets. Researchers at North Carolina State University and the University of North Carolina at Chapel Hill just published an article in Nature Communications describing a nanoparticle that delivers its killer payload only when inside cells by homing in on ATP (adenosine triphosphate).
ATP is the famous energy molecule that powers the activity inside of cells, and the new nanoparticle carries DNA strands bound to doxorubicin, an anti-cancer drug, than unfold when high levels of ATP are present. The nanoparticles themselves have a layer of hyaluronic acid (HA) that attracts some types of cancer cells, allowing the nanoparticles to enter and open up, releasing the folded DNA strands.
From study abstract in Nature Communications:
The half-maximal inhibitory concentration of ATP-responsive nanovehicles is 0.24 μM in MDA-MB-231 cells…
View original post 71 more words
Chemists devise a new way to manufacture peptide drugs, which hold promise for treating many diseases

MIT chemists have devised a way to rapidly combine amino acids into protein fragments known as peptides. Credit: Alexander Vinogradov
Small protein fragments, also called peptides, are promising as drugs because they can be designed for very specific functions inside living cells. Insulin and the HIV drug Fuzeon are some of the earliest successful examples, and peptide drugs are expected to become a $25 billion market by 2018.
However, a major bottleneck has prevented peptide drugs from reaching their full potential: Manufacturing the peptides takes several weeks, making it difficult to obtain large quantities, and to rapidly test their effectiveness.
That bottleneck may soon disappear: A team of MIT chemists and chemical engineers has designed a way to manufacture peptides in mere hours. The new system, described in the March 21st issue of journal ChemBioChem, could have a major impact on peptide drug development, says Bradley Pentelute, an assistant…
View original post 683 more words
100th approval … Pradaxa® (dabigatran etexilate) now approved in more than 100 countries for stroke prevention in atrial fibrillation

More than 100 countries have now approved Boehringer Ingelheim’s Pradaxa® for the prevention of stroke and systemic embolism for adult patients with the most common sustained heart rhythm condition (non-valvular atrial fibrillation, nvAF).
The 100th approvalwas announced by the Jordan Food and Drug Administration. Further regulatory approvals for Pradaxa® are expected to be received in the near future. The continuous flow of regulatory approvals from health authorities all over the world reaffirms the overarching benefits delivered to patients by the treatment and supports previous announcements by the U.S. Food and Drugs Administation (FDA) and the European Medicines Agency (EMA).Pradaxa®, in addition, offers the most robust clinical data set and the longest real-world experience for stroke prevention in atrial fibrillation (SPAF) compared to any of the novel oral anticoagulants, providing ongoing support for physician use of the novel treatment

Cabazitaxel

Cabazitaxel
For treatment of patients with hormone-refractory metastatic prostate cancer previously treated with a docetaxel-containing treatment regimen.
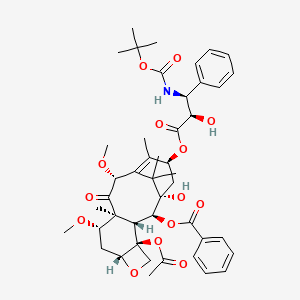
4-acetoxy-2α-benzoyloxy-5β,20-epoxy-1-hydroxy-7β,10β-dimethoxy-9-oxotax-11-en-13α-yl(2R,3S)-3-tert-butoxycarbonylamino-2-hydroxy-3-phenyl-propionate
(1S,2S,3R,4S,7R,9S,10S,12R,15S)-4-(Acetyloxy)-15-{[(2R,3S)-3-{[(tert-butoxy)carbonyl]amino}-2-hydroxy-3-phenylpropanoyl]oxy}-1-hydroxy-9,12-dimethoxy-10,14,17,17-tetramethyl-11-oxo-6-oxatetracyclo[11.3.1.03,10.04,7]heptadec-13-ene-2-yl benzoate
183133-96-2
Cabazitaxel is prepared by semi-synthesis from 10-deacetylbaccatin III (10-DAB) which is extracted from yew tree needles. The chemical name of cabazitaxel is (2α,5β,7β,10β,13α)-4-acetoxy-13-({(2R,3S)-3-[(tert-butoxycarbonyl)amino]-2-hydroxy-3-phenylpropanoyl}oxy)-1-hydroxy-7,10-dimethoxy-9-oxo-5,20-epoxy-tax-11-en-2-yl benzoate and is marketed as a 1:1 acetone solvate (propan-2-one),
Cabazitaxel is an anti-neoplastic used with the steroid medicine prednisone. Cabazitaxel is used to treat people with prostate cancer that has progressed despite treatment with docetaxel. Cabazitaxel is prepared by semi-synthesis with a precursor extracted from yew needles (10-deacetylbaccatin III). It was approved by the U.S. Food and Drug Administration (FDA) on June 17, 2010.
Cabazitaxel (previously XRP-6258, trade name Jevtana) is a semi-synthetic derivative of a natural taxoid.[1] It was developed by Sanofi-Aventis and was approved by the U.S. Food and Drug Administration (FDA) for the treatment of hormone-refractory prostate cancer on June 17, 2010. It is a microtubule inhibitor, and the fourth taxane to be approved as a cancer therapy.[2]
Nagesh Palepu, “CABAZITAXEL FORMULATIONS AND METHODS OF PREPARING THEREOF.” U.S. Patent US20120065255, issued March 15, 2012.

Cabazitaxel in combination with prednisone is a treatment option for hormone-refractory prostate cancer following docetaxel-based treatment.
Clinical trials
In a phase III trial with 755 men for the treatment of castration-resistant prostate cancer, median survival was 15.1 months for patients receiving cabazitaxel versus 12.7 months for patients receiving mitoxantrone. Cabazitaxel was associated with more grade 3–4 neutropenia (81.7%) than mitoxantrone (58%).[3]
| United States | 5438072 | 2010-06-17 | exp 2013-11-22 |
| United States | 5698582 | 2010-06-17 | 2012-07-03 |
| United States | 5847170 | 2010-06-17 | 2016-03-26 |
| United States | 6331635 | 2010-06-17 | 2016-03-26 |
| United States | 6372780 | 2010-06-17 | 2016-03-26 |
| United States | 6387946 | 2010-06-17 | 2016-03-26 |
| United States | 7241907 | 2010-06-17 | 2025-12-10 |
JEVTANA (cabazitaxel) is an antineoplastic agent belonging to the taxane class. It is prepared by semi-synthesis with a precursor extracted from yew needles.
The chemical name of cabazitaxel is (2α,5β,7β,10β,13α)-4-acetoxy-13-({(2R,3S)-3[(tertbutoxycarbonyl) amino]-2-hydroxy-3-phenylpropanoyl}oxy)-1-hydroxy-7,10-dimethoxy-9oxo-5,20-epoxytax-11-en-2-yl benzoate – propan-2-one(1:1).
Cabazitaxel has the following structural formula:
 |
Cabazitaxel is a white to almost-white powder with a molecular formula of C45H57NO14•C3H6O and a molecular weight of 894.01 (for the acetone solvate) / 835.93 (for the solvent free). It is lipophilic, practically insoluble in water and soluble in alcohol.
JEVTANA (cabazitaxel) Injection 60 mg/1.5 mL is a sterile, non-pyrogenic, clear yellow to brownish-yellow viscous solution and is available in single-use vials containing 60 mg cabazitaxel (anhydrous and solvent free) and 1.56 g polysorbate 80. Each mL contains 40 mg cabazitaxel (anhydrous) and 1.04 g polysorbate 80.
DILUENT for JEVTANA is a clear, colorless, sterile, and non-pyrogenic solution containing 13% (w/w) ethanol in water for injection, approximately 5.7 mL.
JEVTANA requires two dilutions prior to intravenous infusion. JEVTANA injection should be diluted only with the supplied DILUENT for JEVTANA, followed by dilution in either 0.9% sodium chloride solution or 5% dextrose solution.
The taxane family of terpenes has received much attention in the scientific and medical community, because members of this family have demonstrated broad spectrum of anti-leukemic and tumor-inhibitory activity. A well-known member of this family is paclitaxel (Taxol®).

Paclitaxel (Taxol) Paclitaxel was first isolated from the bark of the pacific yew tree (Taxus brevifolia) in 1971 , and has proved to be a potent natural anti-cancer agent. To date, paclitaxel has been found to have activity against different forms of leukemia and against solid tumors in the breast, ovary, brain, and lung in humans.
As will be appreciated, this beneficial activity has stimulated an intense research effort over recent years with a view to identifying other taxanes having similar or improved properties, and with a view to developing synthetic pathways for making these taxanes, such as paclitaxel.
This research effort led to the discovery of a synthetic analogue of paclitaxel, namely, docetaxel (also known as Taxotere®). As disclosed in U.S. Patent No. 4,814,470, docetaxel has been found to have a very good anti-tumour activity and better bioavailability than paclitaxel. Docetaxel is similar in structure to paclitaxel, having t- butoxycarbonyl instead of benzoyl on the amino group at the 3′ position, and a hydroxy group instead of the acetoxy group at the C-10 position.

As will be appreciated, taxanes are structurally complicated molecules, and the development of commercially viable synthetic methods to make taxanes has been a challenge. A number of semi-synthetic pathways have been developed over the years, which typically begin with the isolation and purification of a naturally occurring starting material, which can be converted to a specific taxane derivative of interest. Cabazitaxel (I) is an anti-tumor drug which belongs to the taxol family. It differs from docetaxel in that it has methoxy groups at positions 7 and 10 of the molecule, as opposed to the hydroxyl groups at equivalent positions in docetaxel. Cabazitaxel is obtained by semi-synthesis from an extract of Chinese yew (Taxus mairei). It is understood that cabazitaxel can be obtained via semi-synthesis from other taxus species including T.candensis, T.baccatta, T.chinensis, T. mairei etc.

Cabazitaxel is a semi-synthetic derivative of the natural taxoid 0-deacetylbaccatin III (10-DAB) with potentially unique antineoplastic activity for a variety of tumors.
Cabazitaxel binds to and stabilizes tubulin, resulting in the inhibition of microtubule depolymerization and cell division, cell cycle arrest in the G2/M phase, and the inhibition of tumor cell proliferation. This drug is a microtubule depolymerization inhibitor, which can penetrate blood brain barrier (BBB).
Cabazitaxel was recently approved by the US Federal Drug Administration (FDA) for the treatment of docetaxel resistant hormone refractory prostate cancer. It has been developed by Sanofi-Aventis under the trade name of Jevtana. The CAS number for the compound is 183133-96-2. A synonym is dimethoxydocetaxel. The compound is also known as RPR-1 16258A; XRP6258; TXD 258; and axoid XRP6258.
The free base form of cabazitaxel has the chemical name
(2aR,4S,4aS,6R,9S, 1 1 S,12S,12aR, 12bS)-12b-acetoxy-9-(((2R,3S)-3-((tert- butoxycarbonyl)amino)-2-hydroxy-3-phenylpropanoyl)oxy)-11-hydroxy-4,6-dimethoxy- 4a,8, 13, 13-tetramethyl-5-oxo-2a,3,4,4a,5,6,9, 10, 11 , 12, 12a, 12b-dodecahydro-1 H- 7, 1 1-methanocyclodeca[3,4]benzo[1 ,2-b]oxet-12-yl benzoate. In a first part of this description, taxel drugs including paclitaxel (taxol), docetaxel (taxotere) and cabazitaxel may be prepared starting from 10-deacetylbaccatin (known as 10-DAB) derived from Taxus plants, via semi-synthesis. Furthermore, the same inventive methodologies can be used to semi-synthesize cabazitaxel starting from 9- dihydro-13-acetylbaccatin III (9-DHB).
Patent numbers CN1213042C, CN152870, CN1179716 and CN1179775 disclose methods to prepare cabazitaxel from 10-DAB (herein compound II).

10-DAB (II)
A typical prior art synthesis route is as follows:


OCOCH3
OCOC6H5
The method above which synthesizes cabazitaxel has many synthetic steps, a very low overall yield and high price.
There is therefore a need in the art to develop new methods to synthesize cabazitaxel and its intermediates to improve the yield of cabazitaxel, simplify the methodology and optimize the synthetic technology.
Cabazitaxel, chemically known as 4-acetoxy-2α-benzoyloxy-5β,20-epoxy-1-hydroxy-7β,10β-dimethoxy-9-oxotax-11-en-13α-yl(2R,3S)-3-tert-butoxycarbonylamino-2-hydroxy-3-phenyl-propionate, is represented by formula (I).

It is a microtubule inhibitor, indicated in combination with prednisone for treatment of patients with hormone-refractory metastatic prostate cancer previously treated with a docetaxel-containing treatment regimen, under the trade name Jevtana®.
Cabazitaxel is known from U.S. Pat. No. 5,847,170. Process for preparation of Cabazitaxel as described in U.S. Pat. No. 5,847,170 involves column chromatography, which is cumbersome tedious and not commercially viable.
The acetone solvate of 4-acetoxy-2α-benzoyloxy-5β-20-epoxy-1-hydroxy-7β, 10β-dimethoxy-9-oxotan-11-en-13α-yl-(2R,3S)-3-tert-butoxycarbonylamino-2-hydroxy-3-phenylpropionate (Form A) is formed by crystallization by using acetone and is characterized by XRD in U.S. Pat. No. 7,241,907.
U.S. 20110144362 describes anhydrous crystalline Forms B to Form F, ethanolates Form B, D, E and F and mono and dihydrate Forms of Cabazitaxel. All the anhydrous crystalline forms are prepared either by acetone solvate or ethanol solvate. Mono and dihydrate forms are formed at ambient temperature in an atmosphere containing 10 and 60% relative humidity, respectively.
Cabazitaxel (also called dimethoxy docetaxel) is a dimethyl derivative of docetaxel, which itself is semi-synthetic, and was originally developed by Rhone-Poulenc Rorer and was approved by the U.S. Food and Drug Administration (FDA) for the treatment of hormone-refractory prostate cancer on Jun. 17, 2010. Cabazitaxel is a microtubule inhibitor. The acetone solvate crystalline form of cabazitaxel and a process for its preparation is disclosed in the U.S. Pat. No. 7,241,907.
U.S. Pat. No. 5,847,170 describes cabazitaxel and its preparation methods. One of the methods described in U.S. Pat. No. 5,847,170 includes a step-wise methylation of 10-DAB (the step-wise methylation method is shown in FIG. 1) to provide the key intermediate (2αR,4S,4αS,6R,9S,11S,12S,12αR,12βS)-12β-acetoxy-9,11-dihydroxy-4,6-dimethoxy-4α,8,13,13-tetramethyl-5-oxo-2α,3,4,4α,5,6,9,10,11,12,12α,12β-dodecahydro-1H-7,11-methanocyclodeca[3,4]benzo[1,2-b]oxet-12-yl benzoate, herein referred to as 7,10-di-O-methyl-10-DAB (XVa). The intermediate XVa is coupled with the 3-phenylisoserine side chain derivative VI to provide XVa′, which is followed by removal of the oxazolidine protecting group from the side chain of XVa′ to give cabazitaxel.
Another method described in U.S. Pat. No. 5,847,170 utilizes methylthiomethyl (MTM) ethers as shown in FIG. 2. MTM ethers can be prepared from alcohols using two common methods. One method comprises deprotonation of an alcohol with a strong base to form an alkoxide followed by alkylation of the alkoxide with a methylthiomethyl halide. This approach is only useful when the alcohol is stable to treatment with a strong base. 10-DAB and some of its derivatives in which C7-OH is not protected displays so instability in the presence of strong bases and epimerization of the C7-OH can occur upon contact of 10-DAB and some of its derivatives in which C7-OH is not protected with strong bases. Another method for the synthesis of MTM ethers from alcohols utilizes Ac2O and DMSO. One disadvantage of this method is that it can also lead to the oxidation of alcohols to aldehydes or ketones. For example when the synthesis of the 10-di-O-MTM derivative of 10-DAB without protecting groups at the C13 hydroxyl group is attempted undesired oxidation of the C13-OH to its corresponding ketone occurs.
U.S. Pat. No. 5,962,705 discloses a method for dialkylation of 10-DAB and its derivatives to furnish 7,10-di-O-alkyl derivatives, as shown in FIG. 3. This has been demonstrated as a one-step, one-pot reaction, however, provides the best isolated yield when potassium hydride is used at −30° C. From an industrial point of view, the use of low reaction temperature is less favorable than using ambient temperature. Furthermore the use of a strong base can cause some epimerization of the C7-OH chiral center with an associated loss of yield. Potassium hydride is a very reactive base and must be treated with great caution.
Accordingly, there is a need for an alternative processes for the preparation of cabazitaxel and its key intermediate, 7,10-di-O-methyl-10-DAB (XVa) that is short in number of synthetic steps and avoids the use of low temperatures and strong bases such as metal hydrides in the C7-O methyl ether formation step. Such a process would also be useful for the preparation of analogues of cabazitaxel wherein the C7-O and C10-O functional groups were substituted with other alkyl groups.
FIG. 1 shows the chemistry employed in the examples of U.S. Pat. No. 5,847,170.
FIG. 2 shows the chemistry employed in the examples of U.S. Pat. No. 5,847,170.
FIG. 3 shows the chemistry employed in the examples of U.S. Pat. No. 5,962,705.
FIG. 4 shows key steps of the general synthetic scheme as per Method A/A′ of the present invention for the synthesis of cabazitaxel and cabazitaxel analogues.
FIG. 5 shows key steps of the general synthetic scheme as per Method B/B′ of the present invention for the synthesis of cabazitaxel and cabazitaxel analogues.
FIG. 6 shows the general scheme for the hydrodesulfurization reaction.
FIG. 7 shows the complete synthetic route of Method A that can be used for conversion of 10-DAB to cabazitaxel.
FIG. 8 shows the complete synthetic route of Method B that can be used for conversion of 10-DAB to cabazitaxel.
FIG. 9 shows the complete synthetic route of Method A′ that can be used for conversion of 10-DAB, via XIV′, to cabazitaxel.
FIG. 10 shows the complete synthetic route of Method B′ that can be used for conversion of 10-DAB, via XVI′, to cabazitaxel.
FIG. 11 shows the synthetic relationship between two methods (A and B) used to convert 7,10-di-O-alkyl-10-DAB (XV) to cabazitaxel.
FIG. 12 shows the synthetic scheme for the preparation of XIVa.
FIG. 13 shows the synthetic scheme for the preparation of XIVb from 10-DAB.
FIG. 14 shows the synthetic scheme for the preparation of XIVb from XIVb′.
FIG. 15 shows the synthetic scheme for the preparation of XIVc.
FIG. 16 shows the synthetic scheme for the preparation of XIVa′ from XIVa.
FIG. 17 shows the synthetic scheme for the preparation of XIVa′ from XX.
FIG. 18 shows the synthetic scheme for the preparation of XIVb′.
FIG. 19 shows the synthetic scheme for the preparation of XIVc′.
FIG. 20 shows the synthetic scheme for the preparation of XVa from XIVa.
FIG. 21 shows the synthetic scheme for the preparation of XVa from XIVb.
FIG. 22 shows the synthetic scheme for the preparation of XVa from XIVc.
FIG. 23 shows the synthetic scheme for the preparation of XVa′ from XIVa.
FIG. 24 shows the synthetic scheme for the preparation of XVa′ from XIVa′.
FIG. 25 shows the synthetic scheme for the preparation of XVa′ from XIVb′.
FIG. 26 shows the synthetic scheme for the preparation of XVa′ from XIVc′.
FIG. 27 shows the synthetic scheme for the preparation of XVa′ from XVa.
FIG. 28 shows the synthesis of cabazitaxel.
FIG. 29 shows the synthesis of XVIIa.
FIG. 30 shows the synthesis of XVIIIa.
FIG. 31 shows the synthesis of XIXa.
FIG. 32 shows the synthesis of XVIa.
FIG. 33 shows the synthesis of XVa from XVIa.











see this at http://www.google.com/patents/US20130116444
………..
Detailed description
The invention provides a new method for the preparation of cabazitaxel, one embodiment of which can be summarized as follows, showing the preparation of a protected taxane intermediate and its deprotection to taxane compounds:



OH OCOCH3
OCOC6H5
This reaction is also depicted in Figure 1. The reaction of the invention reduces the number of steps and increases yield of cabazitaxel.
The deprotection methods of the invention can also be used for the preparation of paclitaxel (taxol):

The deprotection methods of the invention can also be applied to the preparation of docetaxel:

10-DAB synthetic routes
Example 12
Dissolve 100 g of 2′-THP-cabazitaxel in 1730 ml of HOAc/H20/THF (3:1 :1 ). under N2 atmosphere, increase temperature to 50 degrees C and stir 4 hrs. Then cool to room temperature. Add 2L of ethyl acetate, 2 L of H20, stir, separate layers, wash organic layer with saturated NaHC03 (3 L x 2), saturated NaCI (3 L), dry with Na2S04.
Concentrate to obtain white 77.8 g of cabazitaxel (yield 83%).
MS(m/z) :859(M+Na)„ jHNMR (500MHz) δ 1.21(611, d) , 1.36(911, s) , 1.59(lH, s) , 1.64(lH,s) , 1.79(lH,m) , 1.87 (3H, s) ,2.27 (2H, m) , 2.35(3H,m) ,2.69(lH,m) ,3.30 (3H, s) ,
3.45 (3H, s) , 3.85 (2H, m) , 4.16 (1H, d) , 4.29 (1H, d) , 4.62 (1H, bs) , 4.79 (1H, s) , 5.29 (1H, m),5.42(lH, d),5.62(lH, d),6.21 (1H, t),7.2 ~ 7.4(6H, m) , 7.48 (2H, t),7.59(lH, t) , 8.11 (2H, d) ,
References
- http://www.cancer.gov/drugdictionary/?CdrID=534131
- “Jevtana (cabazitaxel) Injection Approved by U.S. FDA After Priority Review” (Press release). sanofi-aventis. 2010-06-17. Retrieved June 17, 2010.
- “Cabazitaxel Effective for Hormone Refractory Prostate Cancer After Failure of Taxotere”.
- Cabazitaxel – Official web site of manufacturer.
- Cabazitaxel Prescribing Information – Official prescribing information.
- U.S. National Library of Medicine: Drug Information Portal – Cabazitaxel
Patents
Patent :
Patent Number : 5438072
Country : United States
Approved : 2010-06-17
Expires : 2013-11-22
Patent :
Patent Number : 5698582
Country : United States
Approved : 2010-06-17
Expires : 2012-07-03
Patent :
Patent Number : 5847170
Country : United States
Approved : 2010-06-17
Expires : 2016-03-26
Patent :
Patent Number : 6331635
Country : United States
Approved : 2010-06-17
Expires : 2016-03-26
Patent :
Patent Number : 6372780
Country : United States
Approved : 2010-06-17
Expires : 2016-03-26
Patent :
Patent Number : 6387946
Country : United States
Approved : 2010-06-17
Expires : 2016-03-26
Patent :
Patent Number : 7241907
Country : United States
Approved : 2010-06-17
Expires : 2025-12-10
|
3-13-2009
|
SELF-EMULSIFYING AND SELF-MICROEMULSIFYING FORMULATIONS FOR THE ORAL ADMINISTRATION OF TAXOIDS
|
|
|
3-32-2005
|
Semi-solid formulations for the oral administration of taxoids
|
|
|
2-4-2005
|
Self-emulsifying and self-microemulsifying formulations for the oral administration of taxoids
|
|
|
6-12-2002
|
Use of taxoid derivatives
|


Orphan Drugs: FDA Approval For Tropical Disease
The FDA announces on March 19th the approval of Impavido (Miltefosine), an oral medicine for the treatment of the tropical disease Leishmaniasis. Leishmaniasis is caused by a parasite, Leishmania, which is transmitted by sand fly bites to humans. It occurs mainly in people who live in the tropics and subtropics. The drug is already approved for sale in Europe, the Indian subcontinent, and Central and South America.
The FDA granted Impavido Fast Track Designation, Priority Review, and Orphan Drug Designation (ODD) (October 2006). Paladin Therapeutics, Impavido’s manufacturer, is awarded a FDA Tropical Disease Priority Review Voucher. This type of Priority Review Voucher (PRV) is awarded under a provision in the FDA Amendments Act of 2007 that encourages the development of new drugs and vaccines for neglected tropical diseases. “The PRV is transferable and can be sold and entitles the bearer to a priority review for any product. To…
View original post 115 more words
The U.S. FDA approved Impavido (miltefosine) to treat a tropical disease called leishmaniasis

MILTEFOSINE
2-(hexadecoxy-oxido-phosphoryl)oxyethyl-trimethyl-azanium
58066-85-6
March 19, 2014 — The U.S. Food and Drug Administration today approved Impavido (miltefosine) to treat a tropical disease called leishmaniasis.
Leishmaniasis is a disease caused by Leishmania, a parasite which is transmitted to humans through sand fly bites. The disease occurs primarily in people who live in the tropics and subtropics. Most U.S. patients acquire leishmaniasis overseas.
Impavido is an oral medicine approved to treat the three main types of leishmaniasis: visceral leishmaniasis (affects internal organs), cutaneous leishmaniasis (affects the skin) and mucosal leishmaniasis (affects the nose and throat). It is intended for patients 12 years of age and older. Impavido is the first FDA-approved drug to treat cutaneous or mucosal leishmaniasis.
“Today’s approval demonstrates the FDA’s commitment to making available therapeutic options to treat tropical diseases,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
The FDA granted Impavido fast track designation, priority review, and orphan product designation. These designations were granted because the drug demonstrated the potential to fill an unmet medical need in a serious disease or condition, the potential to be a significant improvement in safety or effectiveness in the treatment of a serious disease or condition, and is intended to treat a rare disease, respectively. With this approval, Impavido’s manufacturer, Paladin Therapeutics, is awarded a Tropical Disease Priority Review Voucher under a provision included in the Food and Drug Administration Amendments Act of 2007 that aims to encourage development of new drugs and biological products for the prevention and treatment of certain tropical diseases.
Impavido’s safety and efficacy were evaluated in four clinical trials. A total of 547 patients received Impavido and 183 patients received either a comparator drug or a placebo. Results from these trials demonstrated that Impavido is safe and effective in treating visceral, cutaneous and mucosal leishmaniasis.
The labeling for Impavido includes a boxed warning to alert patients and health care professionals that the drug can cause fetal harm and therefore should not be given to pregnant women. Health care professionals should advise women to use effective contraception during and for five months after Impavido therapy.
The most common side effects identified in clinical trials were nausea, vomiting, diarrhea, headache, decreased appetite, dizziness, abdominal pain, itching, drowsiness and elevated levels of liver enzymes (transaminases) and creatinine.
Paladin Therapeutics is based in Montreal, Canada
Miltefosine (INN, trade names Impavido and Miltex) is a phospholipid drug. Chemically it is a derivative of alkylphosphocholinecompounds discovered in the early 1980s. It was developed in the late 1980s as an anticancer drug by German scientists Hansjörg Eibl and Clemens Unger.[2] Simultaneously but independently it was found that the drug could kill Leishmania parasites, and since the mid-1990s successful clinical trials were conducted. The drug became the first (and still the only prescribed) oral drug in the treatment ofleishmaniasis. It is now known to be a broad-spectrum antimicrobial drug, active against pathogenic bacteria and fungi,[1][3] as well as human trematode Schistosoma mansoni and its vector host, the snail Biomphalaria alexandrina.[4] It can be administered orally and topically.
In the target cell, it acts as an Akt inhibitor. Therefore, it is also under investigation as a potential therapy against HIV infection.[5][6]
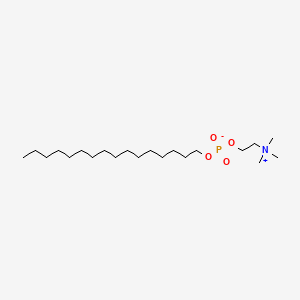
Phospholipid group alkylphosphocholine were known since the early 1980s, particularly in terms of their binding affinity with cobra venom.[7]In 1987 the phosholids were found to potent toxins on leukemic cell culture.[8] Initial in vivo investigation on the antineoplastic activity showed positive result, but then only at high dosage and at high toxicity.[9] At the same time in Germany, Hansjörg Eibl, at the Max Planck Institute for Biophysical Chemistry, and Clemens Unger, at the University of Göttingen, demonstrated that the antineoplastic activity of the phospholipid analogue miltefosine (at the time known as hexadecylphosphocholine) was indeed tumour-specific. It was highly effective against methylnitrosourea-induced mammary carcinoma, but less so on transplantable mammary carcinomas and autochthonous benzo(a)pyrene-induced sarcomas, and relatively inactive on Walker 256 carcinosarcoma and autochthonous acetoxymethylmethylnitrosamine-induced colonic tumors of rats.[10][11] It was subsequently found that miltefosine was strucrally unique among lipds having anticancer property in that it lacks the glycerol group, is highly selective on cell types and acts through different mechanism.[12][13]
In the same year as the discovery of the acticancer property, miltefosine was reported by S. L. Croft and his team at the London School of Hygiene and Tropical Medicine as having antileishmanial effect as well. The compound was effective against Leishmania donovani amastigotes in cultured mouse peritoneal macrophages at a dose of 12.8 mg/kg/day in a five-day course.[14] However priority was given to the development of the compound for cutaneous metastases of breast cancer. In 1992 a new research was reported in which the compound was highly effective in mouse against different life cycle stages of different Leishmania species, and in fect more potent than the conventional sodium stibogluconate therapy by a factor of more than 600.[15] Results of the first clinical trial in humans were reported from Indian patients with chronic leishmaniasis with high degree of success and safety.[16] This promising development promulgated a unique public–private partnership collaboration between ASTA Medica (later Zentaris GmbH), the WHO Special Programme for Research and Training in Tropical Diseases, and the Government of India. Eventually, several successful Phase II and III trials led to the approval of miltefosine in 2002 as the first and only oral drug for leishmaniasis.[1]
Miltefosine is registered and used by Zentaris GmbH in India, Colombia and Germany for the treatment of visceral and cutaneous leishmaniasis, and is undergoing clinical trials for this use in several other countries, such as Brazil[17] and Guatemala.[18]
Miltefosine is a phosphocholine analogue that was originally launched in 1993 by Baxter Oncology for the treatment of cancer. In 2003, Zentaris (formerly part of Asta Medica) launched the drug for the oral treatment of visceral leishmaniasis. Zentaris has also brought the product to market for the treatment of cutaneous leishmaniasis. Jado Technologies is conducting phase II clinical trials for the treatment of antihistamine resistant urticaria. Clinical trials had been ongoing for several indications, including the treatment of cutaneous mastocytosis or cutaneous involvement of systemic mastocytosis. Jado is investigating topical and oral versions of the compound in phase II trials in several allergy indications.
Miltefosine is effective against promastigotes and intracellular amastigotes, which survive and multiply in phagolysosomal compartments of macrophages and make up the two stages of the leishmania lifecycle. Although the exact mechanism of action of the drug has not been determined, it may exert its therapeutic effect through inhibition of phospholipid metabolism. Another theory suggests that miltefosine may interfere with leishmaniacal membrane signal transduction, lipid metabolism and glycosylphosphatidylinositol anchor biosynthesis. The drug is well absorbed in the gastrointestinal tract after a single oral administration and is widely distributed throughout the body.
Miltefosine was originally developed under a collaboration between the Indian government, the German biopharmaceutical company Zentaris, and the Tropical Disease Research (TDR) programme, co-sponsored by the World Health Organization and the United Nations Development Programme (UNDP). Subsequent to the product’s approval, Zentaris partnered with various organizations for its distribution. In February 2004, Roche and Zentaris entered into a marketing agreement, pursuant to which Roche agreed to support Zentaris in the registration process and to market miltefosine in Brazil.
Several medical agents have some efficacy against visceral or cutaneous leishmaniasis, however a 2005 survey concluded that Miltefosine is the only effective oral treatment for both forms of leishmaniasis.[19]
Miltefosine is being investigated by researchers interested in finding treatments for infections which have become resistant to existing drugs. Animal and in vitro studies suggest it may have broad anti-protozoal and anti-fungal properties:
- Animal studies suggest miltefosine may also be effective against Trypanosoma cruzi, the parasite responsible for Chagas’ disease.[20]
- Several studies have found the drug to be effective against Cryptococcus neoformans, Candida, Aspergillus and Fusarium.[21]
- An in vitro study found that miltefosine is effective against metronidazole-resistant variants of Trichomonas vaginalis, a sexually transmitted protozoal disease.[22]
- Hexadecyltrimethylammonium bromide, a compound structurally similar to miltefosine, was recently found to exhibit potent in vitro activity against Plasmodium falciparum.[23]
- Miltefosine is being made available in the United States through the CDC for emergency use under an expanded access IND protocol for treatment of free-living amoeba (FLA) infections:Primary amoebic meningoencephalitis caused by Naegleria fowleri and Granulomatous Amebic Encephalitis caused by Balamuthia mandrillaris, and Acanthamoeba species.[24][25]
Investigatory usage against HIV infection
Miltefosine targets HIV infected macrophages, which play a role in vivo as long-lived HIV-1 reservoirs. The HIV protein Tat activates pro-survival PI3K/Akt pathway in primary human macrophages. Miltefosine acts by inhibiting the PI3K/Akt pathway, thus removing the infected macrophages from circulation, without affecting healthy cells.[5] It significantly reduces replication of HIV-1 in cocultures of human dendritic cells (DCs) and CD4(+) T cells, which is due to a rapid secretion of soluble factors and is associated with induction of type-I interferon (IFN) in the human cells.[26]
In leishmanisis the recommended dose as oral monotherapy is 2.5 mg/kg/day for a total of 28 days. However, due to frequent commercial shortage of the 10 mg capsule, dosages are often altered. For example, the Indian government recommends 100 mg/day miltefosine for patients with a body weight ≥25 kg (corresponding to ∼1.7–4 mg/kg/day) and 50 mg/day for body weights <25 kg (corresponding to ∼2–5.5 mg/kg/day).[1] Even up to 150 mg/day for 28 days was found to be quite safe.[27]
The main side effects reported with miltefosine treatment are nausea and vomiting, which occur in 60% of patients. Adverse effect is more severe in women and young children. The overall effects are quite mild and easily reverse.[28] It is embryotoxic and fetotoxic in rats and rabbits, and teratogenic in rats but not in rabbits. It is therefore contraindicated for use during pregnancy, andcontraception is required beyond the end of treatment in women of child-bearing age.[29]
 |
miltefosine (1-hexadecylphosphoryl-choline, HePC); Calbiochem 475841 |
Compounds o f the general formula I belonging to the class of phospholipids (X is O and R2 is a group of formula II), e.g. alkyloxy phospholipids (Y is O) and the corresponding alkylthio derivatives (Y is S), can be prepared as described in the literature (Bittman, R.; J. Med. Chem. 1997, 40, 1391-1395; Reddy, K. C.; Tetrahedron Lett. 1994, 35, 2679-2682; Guivisdalsky, P. N.; J. Med. Chem. 1990, 33, 2614-2621 and references cited therein) or by standard variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO and SCO, respectively) can be accomplished by standard acylation of the hydroxy or thio precursor materials.
f the general formula I belonging to the class of phospholipids (X is O and R2 is a group of formula II), e.g. alkyloxy phospholipids (Y is O) and the corresponding alkylthio derivatives (Y is S), can be prepared as described in the literature (Bittman, R.; J. Med. Chem. 1997, 40, 1391-1395; Reddy, K. C.; Tetrahedron Lett. 1994, 35, 2679-2682; Guivisdalsky, P. N.; J. Med. Chem. 1990, 33, 2614-2621 and references cited therein) or by standard variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO and SCO, respectively) can be accomplished by standard acylation of the hydroxy or thio precursor materials.
Compounds of the general formula I belonging to the class of phosphonolipids (X is a direct bond and R2 is a group of formula II), e.g alkyloxy phosphonolipids (Y is O and R2 is a group of formula II) and the corresponding alkylthio derivatives (Y is S) can be prepared as published by Bittman et al. (Bittman, R.; J. Med. Chem. 1993, 36, 297-299; Bittman, R.; J. Med. Chem.1994, 37, 425-430 and references cited therein) or by synthetic variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO or SCO) can be accomplished by standard acylation of the hydroxy or thio precursor materials.
SEE
Antitumor ether lipids: An improved synthesis of ilmofosine and an enantioselective synthesis of an ilmofosine analog
Tetrahedron Lett 1994, 35(17): 2679
AND
Hexadecylphosphocholine, a new antineoplastic agent: Cytotoxic properties in leukaemic cells
J Cancer Res Clin Oncol 1986, 111: 24
References
- Dorlo, T. P. C.; Balasegaram, M.; Beijnen, J. H.; de Vries, P. J. (2012). “Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis”. Journal of Antimicrobial Chemotherapy 67 (11): 2576–2597. doi:10.1093/jac/dks275.PMID 22833634.
- Eibl, H; Unger, C (1990 Sep). “Hexadecylphosphocholine: a new and selective antitumor drug.”. Cancer Treatment Reviews 17 (2-3): 233–42. PMID 2272038.
- Almeida Pachioni, JD; Magalhães, JG; Cardoso Lima, EJ; Moura Bueno, LD; Barbosa, JF; Malta de Sá, M; Rangel-Yagui, CO (2013). “Alkylphospholipids – a promising class of chemotherapeutic agents with a broad pharmacological spectrum.”. Journal of Pharmacy & Pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques 16 (5): 742–59. PMID 24393556.
- Eissa, Maha M; El Bardicy, Samia; Tadros, Menerva (2011). “Bioactivity of miltefosine against aquatic stages of Schistosoma mansoni, Schistosoma haematobium and their snail hosts, supported by scanning electron microscopy”. Parasites & Vectors 4 (1): 73.doi:10.1186/1756-3305-4-73. PMC PMC3114006. PMID 21569375.
- ^ Jump up to:a b Chugh P, Bradel-Tretheway B, Monteiro-Filho CM, et al. (2008). “Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy”. Retrovirology 5 (1): 11. doi:10.1186/1742-4690-5-11. PMC 2265748. PMID 18237430.
- “Parasitic Drug Shows HIV-Fighting Promise”. AIDSmeds.com. 2008-02-01. Retrieved 2008-02-02.
- Teshima, K; Ikeda, K; Hamaguchi, K; Hayashi, K (1983). “Bindings of cobra venom phospholipases A2 to micelles of n-hexadecylphosphorylcholine.”. Journal of Biochemistry 94(1): 223–32. PMID 6619110.
- Fleer, EA; Unger, C; Kim, DJ; Eibl, H (1987). “Metabolism of ether phospholipids and analogs in neoplastic cells.”. Lipids 22 (11): 856–61. PMID 3444378.
- Berger, MR; Petru, E; Schmähl, D (1987). “Therapeutic ratio of mono or combination bacterial lipopolysaccharide therapy in methylnitrosourea-induced rat mammary carcinoma.”. Journal of Cancer Research and Clinical Oncology 113 (5): 437–45. PMID 3624299.
- Muschiol, C; Berger, MR; Schuler, B; Scherf, HR; Garzon, FT; Zeller, WJ; Unger, C; Eibl, HJ; Schmähl, D (1987). “Alkyl phosphocholines: toxicity and anticancer properties.”. Lipids 22 (11): 930–4. PMID 3444388.
- Berger, MR; Muschiol, C; Schmähl, D; Eibl, HJ (1987). “New cytostatics with experimentally different toxic profiles.”. Cancer treatment Reviews 14 (3-4): 307–17. PMID 3440252.
- Hilgard, P; Stekar, J; Voegeli, R; Engel, J; Schumacher, W; Eibl, H; Unger, C; Berger, MR (1988). “Characterization of the antitumor activity of hexadecylphosphocholine (D 18506).”.European Journal of Cancer & Clinical Oncology 24 (9): 1457–61. PMID 3141197.
- Eibl, H; Unger, C (1990 Sep). “Hexadecylphosphocholine: a new and selective antitumor drug.”. Cancer Treatment Reviews 17 (2-3): 233–42. PMID 2272038.
- Croft, S.L.; Neal, R.A.; Pendergast, W.; Chan, J.H. (1987). “The activity of alkyl phosphorylcholines and related derivatives against Leishmania donovani”. Biochemical Pharmacology 36 (16): 2633–2636. doi:10.1016/0006-2952(87)90543-0.
- Kuhlencord, A; Maniera, T; Eibl, H; Unger, C (1992). “Hexadecylphosphocholine: oral treatment of visceral leishmaniasis in mice.”. Antimicrobial Agents and Chemotherapy 36(8): 1630–1634. doi:10.1128/AAC.36.8.1630. PMC PMC192021. PMID 1329624.
- Sundar, Shyam; Rosenkaimer, Frank; Makharia, Manoj K; Goyal, Ashish K; Mandal, Ashim K; Voss, Andreas; Hilgard, Peter; Murray, Henry W (1998). “Trial of oral miltefosine for visceral leishmaniasis”. The Lancet 352 (9143): 1821–1823. doi:10.1016/S0140-6736(98)04367-0.PMID 9851383.
- Cristina, Márcia; Pedrosa, Robert (September 2005). “Hospital de Doenças Tropicais testa droga contra calazar”. Sapiência (in Portuguese) (Fundação de Amparo à Pesquisa do Estado do Piauí). Archived from the original on 2006-08-22. Retrieved 2006-09-01.
- Soto J, Berman J (2006). “Treatment of New World cutaneous leishmaniasis with miltefosine.”. Trans R Soc Trop Med Hyg 100: S34. doi:10.1016/j.trstmh.2006.02.022.PMID 16930649.
- Berman, J. (2005). “Clinical status of agents being developed for leishmaniasis”. Expert Opinion on Investigational Drugs 14 (11): 1337–1346. doi:10.1517/13543784.14.11.1337.PMID 16255674.
- Saraiva V, Gibaldi D, Previato J, Mendonça-Previato L, Bozza M, Freire-De-Lima C, Heise N (2002). “Proinflammatory and cytotoxic effects of hexadecylphosphocholine (miltefosine) against drug-resistant strains of Trypanosoma cruzi.”. Antimicrob Agents Chemother 46 (11): 3472–7. doi:10.1128/AAC.46.11.3472-3477.2002. PMC 128733. PMID 12384352.
- Widmer F, Wright L, Obando D, Handke R, Ganendren R, Ellis D, Sorrell T (2006).“Hexadecylphosphocholine (miltefosine) has broad-spectrum fungicidal activity and is efficacious in a mouse model of cryptococcosis.”. Antimicrob Agents Chemother 50 (2): 414–21. doi:10.1128/AAC.50.2.414-421.2006. PMC 1366877. PMID 16436691.
- Blaha C, Duchêne M, Aspöck H, Walochnik J (2006). “In vitro activity of hexadecylphosphocholine (miltefosine) against metronidazole-resistant and -susceptible strains of Trichomonas vaginalis”. J. Antimicrob. Chemother. 57 (2): 273–8.doi:10.1093/jac/dki417. PMID 16344287.
- Choubey V, Maity P, Guha M, et al. (February 2007). “Inhibition of Plasmodium falciparum choline kinase by hexadecyltrimethylammonium bromide: a possible antimalarial mechanism”. Antimicrob. Agents Chemother. 51 (2): 696–706. doi:10.1128/AAC.00919-06.PMC 1797733. PMID 17145794.
- Naegleria fowleri – Primary Amebic Meningoencephalitis (PAM)
- Brain-Eating Amoeba: How One Girl Survived
- Garg, Ravendra; Tremblay, Michel J. (October 2012). “Miltefosine represses HIV-1 replication in human dendritic cell/T-cell cocultures partially by inducing secretion of type-I interferon”.Virology 432 (2): 271–276. doi:10.1016/j.virol.2012.05.032. PMID 22704066.
- Sundar, Shyam; Jha, T.K.; Thakur, C.P.; Bhattacharya, S.K.; Rai, M. (2006). “Oral miltefosine for the treatment of Indian visceral leishmaniasis”. Transactions of the Royal Society of Tropical Medicine and Hygiene 100 (Suppl 1): S26–S33. doi:10.1016/j.trstmh.2006.02.011.PMID 16730038.
- S.D. Seth (2008). “Drug therapy of leishmaniasis”. In S.D. Seth. Textbook of Pharmacology. Elsevier India. p. 31. ISBN 9788131211588.
- Sindermann, H.; Engel, J. (December 2006). “Development of miltefosine as an oral treatment for leishmaniasis”. Transactions of the Royal Society of Tropical Medicine and Hygiene 100 (Suppl 1): S17–S20. doi:10.1016/j.trstmh.2006.02.010. PMID 16730362.
|
7-4-2012
|
LOCAL TREATMENT OF NEUROFIBROMAS
|
|
|
10-28-2011
|
METHODS FOR THE TREATMENT AND AMELIORATION OF ATOPIC DERMATITIS
|
|
|
8-17-2011
|
Methods for the treatment and amelioration of atopic dermatitis
|
|
|
11-16-2007
|
Mucosal formulation
|
|
|
7-20-2007
|
NOVEL ALKYL PHOSPHOLIPID DERIVATIVES WITH REDUCED CYTOTOXICITY AND USES THEREOF
|
 an animation to soothe ones eye
an animation to soothe ones eye