
Home » Uncategorized (Page 132)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Using molecular techniques, researchers improved diagnosis and treatment of cancer

The ABC Medical Center, located in Mexico City, implemented various molecular diagnostic methods that can detect the genetic alterations in several types of cancer, so they can select a personalized therapy for each patient and direct it against the mutated genes that cause disease.
For this the Laboratory of Molecular Pathology was created, thanks to a cancer patient who decided to make a donation to the hospital to acquire the necessary equipment, sensitive to the existing need in Mexico for a place where timely diagnosis of the disease is made.
“While the techniques of molecular biology have been applied to clinical diagnosis for over 20 years, they had just been established in the country. It is estimated that only about five years ago in some of the National Institutes of Health and universities, where it was first used in the field of research, “says Dr. César Lara Torres, head of…
View original post 419 more words
Dovitinib in phase 3 for Cancer, bladder (urothelial carcinoma)

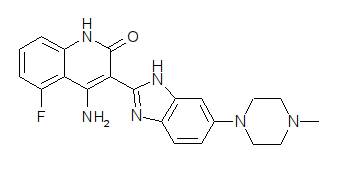
Dovitinib
(3E)-4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1,3-dihydrobenzimidazol-2-ylidene]quinolin-2-one, is one kind of white crystalline powder, odorless, little bitter taste.
4-Amino-5-fluoro-3-[6-(4-methyl-1-piperazinyl)-1H-benzimidazol-2-yl]-2(1H)-quinolinone 2-hydroxypropanoate
4-Amino-5-fluoro-3-[6-(4-methyl-1-piperazinyl)-1H-benzimidazol-2-yl]-2(1H)-quinolinone 2-hydroxypropanoate hydrate (1:1:1);
| CAS No. | 405169-16-6 (free base), 915769-50-5, 804551-71-1 of lactate | ||
| TKI-258; CHIR-258. | |||
| Formula | C21H21FN6O.C3H6O3.H2O | ||
| Molecular Weight | 500.53 |
for treatment of cancer
Novartis Ag, innovator
Dovitinib lactate is the orally bioavailable lactate salt of a benzimidazole-quinolinone compound with potential antineoplastic activity. Dovitinib strongly binds to fibroblast growth factor receptor 3 (FGFR3) and inhibits its phosphorylation, which may result in the inhibition of tumor cell proliferation and the induction of tumor cell death. In addition, this agent may inhibit other members of the RTK superfamily, including the vascular endothelial growth factor receptor; fibroblast growth factor receptor 1; platelet-derived growth factor receptor type 3; FMS-like tyrosine kinase 3; stem cell factor receptor (c-KIT); and colony-stimulating factor receptor 1; this may result in an additional reduction in cellular proliferation and angiogenesis, and the induction of tumor cell apoptosis. The activation of FGFR3 is associated with cell proliferation and survival in certain cancer cell types
Dovitinib (TKI258) is a highly potent, novel multitargeted growth factor receptor kinase inhibitor with IC50 of 1, 2, 10, 8, 27, 36 nM for FLT3, c-KIT, VEGFR1/2/3, PDGFRß and CSF-1R, respectively. It shows both antitumor and antiangiogenic activities in vivo. [1] It potently inhibits FGFR3 with an inhibitory concentration of 50% (IC50) of 5 nM in in vitro kinase assays and selectively inhibited the growth of B9 cells and human myeloma cell lines expressing wild-type (WT) or activated mutant FGFR3. Antiproliferative activity of Dovitinib (TKI258) against MV4;11 was ~24-fold greater compared with RS4;11, indicating more potent inhibition against cells with constitutively activated FLT3 ITD. [2][3]
References on Dovitinib (TKI258)
- [1] Clin Cancer Res 2005;11:3633-3641
- [2] Blood 2005;105: 2941-2948
- [3] Clin Cancer Res 2005;11:5281-5291
Dovitinib lactate is an angiogenesis inhibitor in phase III clinical trials at Novartis for the treatment of refractory advanced/metastatic renal cell cancer. Early clinical trials are also under way at the company for the oral treatment of several types of solid tumors, multiple myeloma and glioblastoma multiforme. Phase II trials are ongoing for the treatment of castration-resistant prostate cancer and for the treatment of Von-Hippel Lindau disease, for the treatment of non-small cell lung cancer (NSCLC) and for the treatment of colorectal cancer. Novartis and Seoul National University Hospital are conducting phase II clinical studies for the treatment of adenoid cystic carcinoma. Additional phase II clinical trials are ongoing at Asan Medical Center for the treatment of metastatic or advanced gastrointestinal stromal tumors (GIST). The University of Pennsylvania is conducting phase II clinical trials for the treatment of advanced malignant pheochromocytoma or paraganglioma. Phase II clinical studies are ongoing by Novartis for the treatment of advanced malignant pleural mesothelioma which has progressed following prior platinum-antifolate chemotherapy (DOVE-M) and for the oral treatment of hepatocellular carcinoma.
In 2009, Novartis discontinued development of dovitinib lactate for the treatment of acute myeloid leukemia (AML) based on the observation of time dependent drug accumulation. A phase I trial was also stopped for the same reason.
The drug candidate has been shown to inhibit multiple growth factor tyrosine kinases, including vascular endothelial growth factor receptor (VEGFR) tyrosine kinases VEGFR1 and VEGFR2, fibroblast growth factor receptor (FGFR) and platelet-derived growth factor receptor (PDGFR) tyrosine kinases. In previous studies, the benzimidazole-quinoline inhibited VEGF-mediated human microvascular endothelial cell (HMVEC) proliferation and demonstrated concentration-dependent antiangiogenic activity in in vitro assays, as well as potent antiproliferative activity against a subset of cancer cell lines.
In 2013, an orphan drug designation was assigned in the U.S. for the treatment of adenoid cystic carcinoma.
“Molecularly Targeted Agents for Renal Cell Carcinoma: The Next Generation”, C. Lance Cowey and Thomas E. Hutson -Clinical Advances in Hematology & Oncology, 2010, 8, 357.
Lee S. H.; Lopes de Menezes, D. Vora, J. Harris, A.; Ye, H. Nordahl, L.; Garrett, E.; Samara, E.; Aukerman, S. L.; Gelb, A. B. Heise, C. In Vivo Target Modulation and Biological Activity of CHIR-258, a Multitargeted Growth Factor Receptor Kinase Inhibitor, in Colon Cancer Models. Clin. Cancer Res. 2005, 11 (10), 3633–3641.
Lopes de Menezes, D. E.; Peng, J.; Garrett, E. N.; Louie, S. G.; Lee, S. H.; Wiesmann, M.; Tang, Y.; Shephard, L.; Goldbeck, C.; Oei, Y.; Ye, H.; Aukerman, S. L.; Heise, C. CHIR-258: A Potent Inhibitor of FLT3 Kinase in Experimental Tumor Xenograft Models of Human Acute Myelogenous Leukemia. Clin. Cancer Res. 2005, 11 (14), 5281–5291.
Trudel, S.; Li, Z. H.; Wei, E.; Wiesmann, M.; Chang, H.; Chen, C.; Reece, D.; Heise, C.; Stewart, A. K. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood 2005, 105 (7), 2941–2948.
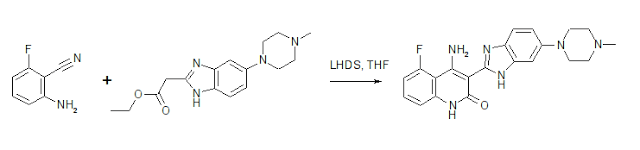
Synthesis of Dovitinib
Tetrahedron Letters 47 (2006) 657–660
LHMDS mediated tandem acylation–cyclization of 2-aminobenzenecarbonitriles with 2-benzymidazol-2-yl acetates: a short and efficient route to the synthesis of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones
William R. Antonios-McCrea, Kelly A. Frazier, Elisa M. Jazan, Timothy D. Machajewski, Christopher M. McBride, Sabina Pecchi, Paul A. Renhowe, Cynthia M. Shafer and Clarke Taylor


cas 852433-84-2
…………………………..
852433-84-2
…………………………..
WO 2002022598 or https://www.google.com/patents/EP1317442A1?cl=en


………………………………
WO 2003087095 or http://www.google.fm/patents/US20030028018?cl=un

…………………..
WO 2005046589 or http://www.google.com/patents/EP1692085A2?cl=en
lactate salt of the compound of
Structure I or the tautomer thereof is administered to the subject and/or is used to prepare the medicament. [0062] In some embodiments, the compound of Structure I has the following formula

……………………
WO 2006125130
http://www.google.com/patents/WO2006125130A1?cl=en
formula IHB

HIB
Scheme 1



Example 4
Synthesis of 4-Amino-5-fluoro-3-[6-(4-rnethyl-piperazin-1 -yl)-1 H-benzimidazol- 2-yl]-1 H-quinolin-2-one Procedure A

[0149] [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (250 g, 820 mmol) (dried with ethanol as described above) was dissolved in THF (3800 ml_) in a 5000 ml_ flask fitted with a condenser, mechanical stirrer, temperature probe, and purged with argon. 2-Amino-6-fluoro-benzonitrile (95.3 g, 700 mmol) was added to the solution, and the internal temperature was raised to 40°C. When all the solids had dissolved and the solution temperature had reached 4O0C, solid KHMDS (376.2 g, 1890 mmol) was added over a period of 5 minutes. When addition of the potassium base was complete, a heterogeneous yellow solution was obtained, and the internal temperature had risen to 62°C. After a period of 60 minutes, the internal temperature decreased back to 40°C, and the reaction was determined to be complete by HPLC (no starting material or uncyclized intermediate was present). The thick reaction mixture was then quenched by pouring it into H2O (6000 ml_) and stirring the resulting mixture until it had reached room temperature. The mixture was then filtered, and the filter pad was washed with water (1000 ml_ 2X). The bright yellow solid was placed in a drying tray and dried in a vacuum oven at 50°C overnight providing 155.3 g (47.9%) of the desired 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1 H-benzimidazol-2- yl]-1 H-quinolin-2-one.
Procedure B
[0150] A 5000 mL 4-neck jacketed flask was equipped with a distillation apparatus, a temperature probe, a N2 gas inlet, an addition funnel, and a mechanical stirrer. [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (173.0 g, 570 mmol) was charged into the reactor, and the reactor was purged with N2 for 15 minutes. Dry THF (2600 mL) was then charged into the flask with stirring. After all the solid had dissolved, solvent was removed by distillation (vacuum or atmospheric (the higher temperature helps to remove the water) using heat as necessary. After 1000 mL of solvent had been removed, distillation was stopped and the reaction was purged with N2. 1000 mL of dry THF was then added to the reaction vessel, and when all solid was dissolved, distillation (vacuum or atmospheric) was again conducted until another 1000 mL of solvent had been removed. This process of adding dry THF and solvent removal was repeated at least 4 times (on the 4thdistillation, 60% of the solvent is removed instead of just 40% as in the first 3 distillations) after which a 1 mL sample was removed for Karl Fischer analysis to determine water content. If the analysis showed that the sample contained less than 0.20% water, then reaction was continued as described in the next paragraph. However, if the analysis showed more than 0.20% water, then the drying process described above was continued until a water content of less than 0.20% was achieved.
[0151] After a water content of less than or about 0.20% was achieved using the procedure described in the previous paragraph, the distillation apparatus was replaced with a reflux condenser, and the reaction was charged with 2-amino-6- fluoro-benzonitrile (66.2 g, 470 mmol)( in some procedures 0.95 equivalents is used). The reaction was then heated to an internal temperature of 38-420C. When the internal temperature had reached 38-420C, KHMDS solution (1313 g, 1.32 mol, 20% KHMDS in THF) was added to the reaction via the addition funnel over a period of 5 minutes maintaining the internal temperature at about 38-50°C during the addition. When addition of the potassium base was complete, the reaction was stirred for 3.5 to 4.5 hours (in some examples it was stirred for 30 to 60 minutes and the reaction may be complete within that time) while maintaining the internal temperature at from 38-420C. A sample of the reaction was then removed and analyzed by HPLC. If the reaction was not complete, additional KHMDS solution was added to the flask over a period of 5 minutes and the reaction was stirred at 38-420C for 45-60 minutes (the amount of KHMDS solution added was determined by the following: If the IPC ratio is < 3.50, then 125 ml_ was added; if 10.0 >IPC ratio >3.50, then 56 mL was added; if 20.0 ≥IPC ratio >10, then 30 mL was added. The IPC ratio is equal to the area corresponding to 4-amino-5-fluoro-3-[6- (4-methyl-piperazin-1 -yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one) divided by the area corresponding to the uncyclized intermediate). Once the reaction was complete (IPC ratio > 20), the reactor was cooled to an internal temperature of 25- 300C, and water (350 mL) was charged into the reactor over a period of 15 minutes while maintaining the internal temperature at 25-35°C (in one alternative, the reaction is conducted at 400C and water is added within 5 minutes. The quicker quench reduces the amount of impurity that forms over time). The reflux condenser was then replaced with a distillation apparatus and solvent was removed by distillation (vacuum or atmospheric) using heat as required. After 1500 mL of solvent had been removed, distillation was discontinued and the reaction was purged with N2. Water (1660 mL) was then added to the reaction flask while maintaining the internal temperature at 20-300C. The reaction mixture was then stirred at 20-300C for 30 minutes before cooling it to an internal temperature of 5- 100C and then stirring for 1 hour. The resulting suspension was filtered, and the flask and filter cake were washed with water (3 x 650 mL). The solid thus obtained was dried to a constant weight under vacuum at 5O0C in a vacuum oven to provide 103.9 g (42.6% yield) of 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1H- benzimidazol-2-yl]-1H-quinolin-2-one as a yellow powder.
Procedure C

[0152] [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (608 g, 2.01 mol) (dried) and 2-amino-6-fluoro-benzonitrile (274 g, 2.01 mol) were charged into a 4-neck 12 L flask seated on a heating mantle and fitted with a condenser, mechanical stirrer, gas inlet, and temperature probe. The reaction vessel was purged with N2, and toluene (7.7 L) was charged into the reaction mixture while it was stirred. The reaction vessel was again purged with N2 and maintained under N2. The internal temperature of the mixture was raised until a temperature of 630C (+/- 3°C) was achieved. The internal temperature of the mixture was maintained at 63°C (+/- 30C) while approximately 2.6 L of toluene was distilled from the flask under reduced pressure (380 +/- 10 torr, distilling head t = 40°C (+/- 1O0C) (Karl Fischer analysis was used to check the water content in the mixture. If the water content was greater than 0.03%, then another 2.6 L of toluene was added and distillation was repeated. This process was repeated until a water content of less than 0.03% was achieved). After a water content of less than 0.03% was reached, heating was discontinued, and the reaction was cooled under N2 to an internal temperature of 17-19°C. Potassium t-butoxide in THF (20% in THF; 3.39 kg, 6.04 moles potassium t-butoxide) was then added to the reaction under N2 at a rate such that the internal temperature of the reaction was kept below 20°C. After addition of the potassium t-butoxide was complete, the reaction was stirred at an internal temperature of less than 2O0C for 30 minutes. The temperature was then raised to 25°C, and the reaction was stirred for at least 1 hour. The temperature was then raised to 30°C, and the reaction was stirred for at least 30 minutes. The reaction was then monitored for completion using HPLC to check for consumption of the starting materials (typically in 2-3 hours, both starting materials were consumed (less than 0.5% by area % HPLC)). If the reaction was not complete after 2 hours, another 0.05 equivalents of potassium t-butoxide was added at a time, and the process was completed until HPLC showed that the reaction was complete. After the reaction was complete, 650 mL of water was added to the stirred reaction mixture. The reaction was then warmed to an internal temperature of 50°C and the THF was distilled away (about 3 L by volume) under reduced pressure from the reaction mixture. Water (2.6 L) was then added drop wise to the reaction mixture using an addition funnel. The mixture was then cooled to room temperature and stirred for at least 1 hour. The mixture was then filtered, and the filter cake was washed with water (1.2 L), with 70% ethanol (1.2 L), and with 95% ethanol (1.2 L). The bright yellow solid was placed in a drying tray and dried in a vacuum oven at 50°C until a constant weight was obtained providing 674 g (85.4%) of the desired 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1 H- benzimidazol-2-yl]-1 H-quinolin-2-one.
Preparation of Lactic Acid salt of 4-Amino-5-fluoro-3-[6-(4-methyl-piperazin-1- yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one

D,L-Lactic Acid

[0154] A 3000 ml_ 4-necked jacketed flask was fitted with a condenser, a temperature probe, a N2 gas inlet, and a mechanical stirrer. The reaction vessel was purged with N2 for at least 15 minutes and then charged with 4-amino-5-fluoro- 3-[6-(4-methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-1 H-quinoiin-2-one (484 g, 1.23 mol). A solution of D,L-Lactic acid (243.3 g, 1.72 mol of monomer-see the following paragraph), water (339 mL), and ethanol (1211 mL) was prepared and then charged to the reaction flask. Stirring was initiated at a medium rate, and the reaction was heated to an internal temperature of 68-720C. The internal temperature of the reaction was maintained at 68-72°C for 15-45 minutes and then heating was discontinued. The resulting mixture was filtered through a 10-20 micron frit collecting the filtrate in a 12 L flask. The 12 L flask was equipped with an internal temperature probe, a reflux condenser, an addition funnel, a gas inlet an outlet, and an overhead stirrer. The filtrate was then stirred at a medium rate and heated to reflux (internal temperature of about 780C). While maintaining a gentle reflux, ethanol (3,596 mL) was charged to the flask over a period of about 20 minutes. The reaction flask was then cooled to an internal temperature ranging from about 64-700C within 15-25 minutes and this temperature was maintained for a period of about 30 minutes. The reactor was inspected for crystals. If no crystals were present, then crystals of the lactic acid salt of 4-amino-5-fluoro-3-[6-(4-methyl- piperazin-1-yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one (484 mg, 0.1 mole %) were added to the flask, and the reaction was stirred at 64-7O0C for 30 minutes before again inspecting the flask for crystals.
[0155] Once crystals were present, stirring was reduced to a low rate and the reaction was stirred at 64-700C for an additional 90 minutes. The reaction was then cooled to about 00C over a period of about 2 hours, and the resulting mixture was filtered through a 25-50 micron fritted filter. The reactor was washed with ethanol (484 ml_) and stirred until the internal temperature was about 00C. The cold ethanol was used to wash the filter cake, and this procedure was repeated 2 more times. The collected solid was dried to a constant weight at 500C under vacuum in a vacuum oven yielding 510.7 g (85.7%) of the crystalline yellow lactic acid salt of 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1 -yl)-1 H-benzimidazol-2-yl]-1 H-quinolin- 2-one. A rubber dam or inert conditions were typically used during the filtration process. While the dry solid did not appear to be very hygroscopic, the wet filter cake tends to pick up water and become sticky. Precautions were taken to avoid prolonged exposure of the wet filter cake to the atmosphere.
[0156] Commercial lactic acid generally contains about 8-12% w/w water, and contains dimers and trimers in addition to the monomeric lactic acid. The mole ratio of lactic acid dimer to monomer is generally about 1.0:4.7. Commercial grade lactic acid may be used in the process described in the preceding paragraph as the monolactate salt preferentially precipitates from the reaction mixture.
[0157] It should be understood that the organic compounds according to the invention may exhibit the phenomenon of tautomerism. As the chemical structures within this specification can only represent one of the possible tautomeric forms at a time, it should be understood that the invention encompasses any tautomeric form of the drawn structure. For example, the compound having the formula NIB is shown below with one tautomer, Tautomer INBa:

HIB

Tautomer HIBa
Other tautomers of the compound having the formula NIB, Tautomer INlBb and Tautomer IHBc, are shown below:

Tautomer IIIBb

Tautomer IIIBc
……………………….
WO 2006127926
……………………..
Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: A novel class of receptor tyrosine kinase inhibitors
J Med Chem 2009, 52(2): 278
………………………………
WO 2003087095
…………………..
WO 2005046589
……………………
WO 2006125130
……………………….
WO 2006127926
……………………..
Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: A novel class of receptor tyrosine kinase inhibitors
J Med Chem 2009, 52(2): 278
Ezatiostat……….designed to stimulate the production of blood cells in the bone marrow

Ezatiostat
168682-53-9 (Ezatiostat); 286942-97-0 (Ezatiostat HCl salt)
gamma-Glu-S-BzCys-PhGly diethyl ester

Ezatiostat hydrochloride
Target: glutathione S-transferase P1-1 (GSTP1-1) inhibitor
Pathway: hsa00480 Glutathione metabolism
Activity: Treatment of disorders of bone marrow cellular growth and differentiation
see http://www.ncbi.nlm.nih.gov/pubmed?term=TLK-199&cmd=search
| Telik, Inc. |
innovator
TLK199; TLK-199; TLK 199; Brand name: TELINTRA®。ethyl (2R)-[(4S)-4-amino-5-ethoxy-5-oxopentanoyl]-S-benzyl-L-cysteinyl-2- phenylglycinate.
ethyl (2S)-2-amino-5-[[(2R)-3-benzylsulfanyl-1-[[(1R)-2-ethoxy-2-oxo-1-phenylethyl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoate.
IUPAC/Chemical name:
(S)-ethyl 2-amino-5-(((R)-3-(benzylthio)-1-(((S)-2-ethoxy-2-oxo-1-phenylethyl)amino)-1-oxopropan-2-yl)amino)-5-oxopentanoate
C27H35N3O6S
Exact Mass: 529.2246
nmr.http://www.medkoo.com/Product-Data/Ezatiostat/ezatiostat-QC-CRB40225web.pdf
Telintra is a small molecule product candidate designed to stimulate the production of blood cells in the bone marrow. Many conditions are characterized by depleted bone marrow, including myelodysplastic syndrome, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the 3 major blood elements (white blood cells, red blood cells and platelets). A reduction in blood cell levels is also a common, toxic effect of many standard chemotherapeutic drugs.
Ezatiostat is a liposomal small-molecule glutathione analog inhibitor of glutathione S-transferase (GST) P1-1 with hematopoiesis-stimulating activity. After intracellular de-esterification, the active form of ezatiostat binds to and inhibits GST P1-1, thereby restoring Jun kinase and MAPK pathway activities and promoting MAPK-mediated cellular proliferation and differentiation pathways. This agent promotes the proliferation and maturation of hematopoietic precursor cells, granulocytes, monocytes, erythrocytes and platelets
Phase II trial myelodysplastic syndrome (MDS): Cancer. 2012 Apr 15;118(8):2138-47.
Phase I trial myelodysplastic syndrome (MDS): J Hematol Oncol. 2012 Apr 30;5:18. doi: 10.1186/1756-8722-5-18; Blood. 2009 Jun 25;113(26):6533-40; J Hematol Oncol. 2009 May 13;2:20.
Ezatiostat hydrochloride is the hydrochloride acid addition salt of ezatiostat. Ezatiostat, also known as TLK199 or TER 199, is a compound of the formula:

Ezatiostat has been shown to induce the differentiation of HL-60 promyelocyte leukemia cells in vitro, to potentiate the activity of cytotoxic agents both in vitro and in vivo, and to stimulate colony formation of all three lineages of hematopoietic progenitor cells in normal human peripheral blood.
In preclinical testing, ezatiostat has been shown to increase white blood cell production in normal animals, as well as in animals in which white blood cells were depleted by treatment with cisplatin or fluorouracil. Similar effects may provide a new approach to treating myelodysplastic syndrome (MDS).
Many conditions, including MDS, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the three major blood elements (white blood cells, red blood cells, and platelets), are characterized by depleted bone marrow. Myelosuppression, which is characterized by a reduction in blood cell levels and in a reduction of new blood cell generation in the bone marrow, is also a common, toxic effect of many standard chemotherapeutic drugs.
Ezatiostat hydrochloride in a liposomal injectable formulation was studied in a clinical trial for the treatment of MDS, and results from this trial, reported by Raza et al., J Hem. One, 2:20 (published online 13 May 2009), demonstrated that administration of TLK199 was well tolerated and resulted in multi-lineage hematologic improvement.
Ezatiostat hydrochloride in a tablet formulation has been evaluated in a clinical trial for the treatment of MDS, as reported by Raza et al., Blood, 113:6533-6540 (prepublished online 27 April 2009) and a single-patient report by Quddus et al., J Hem. One, 3:16 (published online 23 April 2010), and is currently being evaluated in clinical trials for the treatment of MDS and for severe chronic idiopathic neutropenia.
When used for treating humans, it is important that a crystalline therapeutic agent like ezatiostat hydrochloride retains its polymorphic and chemical stability, solubility, and other physicochemical properties over time and among various manufactured batches of the agent. If the physicochemical properties vary with time and among batches, the administration of a therapeutically effective dose becomes problematic and may lead to toxic side effects or to ineffective therapy, particularly if a given polymorph decomposes prior to use, to a less active, inactive, or toxic compound.
Therefore, it is important to choose a form of the crystalline agent that is stable, is manufactured reproducibly, and has physicochemical properties favorable for its use as a therapeutic agent.
Ezatiostat hydrochloride (USAN) has the molecular weight of 566.1, the trademark of Telintra®, and the CAS registry number of 286942-97-0. Ezatiostat hydrochloride has been evaluated for the treatment of myelodysplastic syndrome (MDS), in a Phase I-IIa study using a liposomal formulation (U.S. Pat. No. 7,029,695), as reported at the 2005 Annual Meeting of the American Society for Hematology (Abstract #2250) and by Raza et al. in Journal of Hematology & Oncology, 2:20 (published online on 13 May 2009); and in a Phase I study using a tablet formulation, as reported at the 2007 Annual Meeting of the American Society for Hematology (Abstract #1454) and by Raza et al. in Blood, 113:6533-6540 (prepublished online on 27 Apr. 2009), and in a single patient case report by Quddus et al. in Journal of Hematology & Oncology, 3:16 (published online on 23 Apr. 2010).
…………………………………………………………………
http://www.google.com/patents/US20110301376
Preparation of Ezatiostat Hydrochloride
In another aspect, this invention provides a process comprising the steps of contacting a compound of formula:

or a salt thereof with a compound of formula:

or a salt thereof and an activating agent under conditions which provide a compound of formula:

In one embodiment, the process further comprises deprotecting the compound of formula:

under conditions which provide a compound of formula:

or a salt thereof. In another embodiment, the compound provided is ezatiostat hydrochloride.
In another aspect, this invention provides a process comprising contacting a compound of formula:

or a salt thereof with an ethylating agent under conditions which provide a compound of formula:

In another embodiment, the process further comprises debenzylating the compound of formula:

under conditions which provide a compound of formula:

or a salt thereof.
In another aspect, this invention provides a process comprising the steps of contacting a compound of formula:

or a salt thereof having a t-butoxycarbonyl group with an activating agent and a compound of formula:

or a salt thereof under conditions which provide a compound of formula:

In another embodiment, the process further comprises deprotecting the tertiarybutyloxycarboyl (Boc) group under conditions to provide a compound of formula:

or a salt thereof.
Certain preferred embodiments of this invention are illustrated in the reaction scheme and described below. In the peptide coupling the amino acid reagents are used generally at a 1:1 molar ratio, and the activating reagent (isobutyl chloroformate) and the base (NMM) are used in slight excess over the amino acid reagents; while in the esterification of the N-BOC-L-glutamic acid γ-benzyl ester the esterifying agent (diethyl sulfate) and base are used in about 1.4-fold excess.

EXAMPLESAs relevant and unless otherwise noted, all operations were conducted under nitrogen purge and with stirring. Water was osmosis purified, and solvents were filtered. Unless otherwise stated, all temperatures are in degrees Celcius (° C.) and the following abbreviations have the following definitions:Et EthylHCl(g) HCl gasN-BOC or N-Boc N-tertiarybutyloxycarbonylL LiterKg KilogramNMM N-methylmorpholineMol Molew/w weight by weightExample 1Preparation of S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3)Without stirring, 45.1 Kg N-BOC-S-benzyl-L-cysteine (1) was added to a 600 L jacketed glass-lined reactor, followed by 45 L ethyl acetate. Stirring was started and the temperature was reduced to 13° C. NMM, 15.3 Kg, was added over 50 minutes, and rinsed in with 6 L ethyl acetate, and stirring stopped. Ethyl acetate, 315 L, was added to an 800 L cooled jacketed glass-lined reactor, followed by 20.7 Kg isobutyl chloroformate, rinsed in with 11 L ethyl acetate, and the mixture cooled to −10° C. The N-BOC-S-benzyl-L-cysteine NMM salt solution was added to the 800 L reactor over 5 hours, its reactor rinsed with 11 L ethyl acetate, and the rinse solution added to the 800 L reactor, while maintaining the temperature at (−10˜−7)° C. D-Phenylglycine ethyl ester hydrochloride, 31.2 Kg, was added in 8 portions over 50 minutes, followed by 15.3 Kg NMM in 8 portions over 1.3 hours, rinsed in with 2×5 L portions of ethyl acetate, allowing the mixture to warm to −1° C. by the end of the addition. The mixture was gradually warmed to 1° C. for 30 minutes, then to 20° C. over 2 hours, and maintained at (20˜25)° C. for 5 hours. The reaction mixture was washed twice with water: the first time adding 66 L water, stirring at room temperature for 40 minutes, allowing the phases to separate for 30 minutes, then removing the aqueous phase; the second time adding 68 L water, bringing the pH to 1.9 with the addition of 0.45 L 36% hydrochloric acid, stirring at room temperature for 35 minutes, allowing the phases to separate for 1 hour, then removing the aqueous phase. The organic phase was then heated to 38° C., and the pressure reduced to about 0.25 bar until no further gas was released, then to about (0.07-0.1) bar and solvents removed by distillation until 266 L of distillate had been removed. Four cycles of addition of 45 L ethyl acetate and removal of 45 L solvent by distillation were performed, and the water content of the remaining mixture was checked to ensure that it was below 0.1%. With the mixture at 36° C., 194 L heptanes was added, maintaining the temperature about 36° C., and held at that temperature for 2.3 hours. A further 194 L heptanes was added, allowing the temperature to cool to 30° C., and the temperature then reduced to −1° C. over 2.3 hours and then to −5° C. over 1 hour, and N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester recovered by filtration, washing twice with 30 L each of heptanes at −5° C., giving 85 Kg (63 Kg dry basis) N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester. Without stirring, the damp N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester was loaded into an 800 L jacketed glass-lined reactor, followed by 257 L ethyl acetate. Stirring was started and the temperature brought to 22° C., then the nitrogen purge stopped and 12.2 Kg hydrogen chloride gas was added through an immersion tube over 1.8 hours, allowing the temperature to increase to 38° C. The temperature was increased to 41° C., and the mixture held at that temperature for 9 hours. About 280 L of solvents were removed by distillation at that temperature and a pressure of (0.2˜0.1) bar over about 2 hours. Two cycles of addition of ethyl acetate and removal of solvent by distillation were performed, using 52 L in the first cycle and 77 L in the second cycle, and the viscous solution of S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3) in ethyl acetate, 148 Kg, was cooled to room temperature and filtered into a storage drum.Example 2Preparation of N-BOC-L-glutamic acid α-ethyl ester (6)Without stirring, 41 Kg N-BOC-L-glutamic acid γ-benzyl ester (4) was added to an 800 L jacketed glass-lined reactor, followed by 2.5 L water and 123 L ethyl acetate. The mixture was then stirred until the N-BOC-L-glutamic acid γ-benzyl ester completely dissolved, keeping the temperature below 15° C. Potassium carbonate fine powder, 23.4 Kg, was added in five batches, and the mixture then heated to 55° C. and maintained at that temperature for 40 minutes, giving a heterogeneous and completely fluid mixture. Diethyl sulfate, 26.2 Kg, was added over 2 hours, and rinsed in with 5 L ethyl acetate, with the temperature remaining at about 52° C. The nitrogen purge was stopped and a solution of 20 Kg ammonium chloride in 73 L water at room temperature added over 2 hours to the mixture, maintaining the temperature near 50° C., then rinsing in with 10 L water. Nitrogen purging was resumed, and the mixture was maintained at about 50° C. for 3 hours, then lowered to about 45° C., the stirring stopped, the phases allowed to separate for 30 minutes, and the lower, aqueous, phase removed. The organic phase, containing N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), was washed three times with water, each time adding 41 L water, stirring at room temperature for 30 minutes, allowing the phases to separate for 30 minutes, then removing the aqueous phase. The organic phase was heated to 35° C., and the pressure reduced, starting at about 0.2 bar and reducing as necessary until 82 Kg solvent had been removed by distillation, leaving about (70˜80) L of slightly opalescent solution. This solution was heated to 53° C., and 102 L heptanes was added, maintaining the same temperature. The solution was then filtered, rinsing with a further 13 L heptanes, then cooled to 32° C. to cause crystallization and maintained at that temperature for 1 hour. A further 66 L heptanes was added, and the mixture cooled to 22° C. and held for 1 hour, then cooled to −5° C. and held for another 1 hour. The mixture was then filtered to isolate the N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), which was washed twice, each time with 25 L heptanes cooled to (−5˜0)° C., and dried under vacuum at 40° C., giving 39.3 Kg N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5).A 4000 L hydrogenator was purged with nitrogen, then under nitrogen sweep and no stirring loaded with 39.2 Kg N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), 2.0 Kg 5% palladium on carbon, and 432 L ethyl acetate, and purged (3 bar) and decompressed (0.2 bar) twice with nitrogen and twice with hydrogen. Stirring was begun and the mixture heated to (37±2)° C., hydrogenated at that temperature under 2.8 bar hydrogen pressure until no further hydrogen absorption occurred, then held under 2.8 bar hydrogen pressure for 12 hours. Completion of hydrogenation was confirmed by thin-layer chromatography of a sample. The mixture was cooled to 28° C., the hydrogen purged from the hydrogenator, and the hydrogenator purged (2 bar) and decompressed (0.2 bar) twice with nitrogen. The mixture was filtered through a filter precoated with 10 Kg powdered cellulose in 200 L ethyl acetate, then the filter washed with the ethyl acetate used to form the precoat, giving a total of 626 Kg of a dilute ethyl acetate solution containing 29.5 Kg N-BOC-L-glutamic acid α-ethyl ester (6). This was distilled at (35˜40)° C. and (0.16˜0.18) bar to give 67 L of concentrated solution, then 29 L of ethyl acetate added and the solution redistilled to again give 67 L of concentrated solution.Example 3Preparation of Ezatiostat HydrochlorideThe concentrated solution of N-BOC-L-glutamic acid α-ethyl ester (6), 61.2 Kg (containing 27.8 Kg N-BOC-L-glutamic acid α-ethyl ester), was added to a 600 L jacketed glass-lined reactor, rinsed in with 5 L ethyl acetate, then cooled to 14° C. NMM, 10.8 Kg, was added over 50 minutes and rinsed in with 5 L ethyl acetate, then stirring stopped, giving an ethyl acetate solution of N-BOC-L-glutamic acid α-ethyl ester NMM salt. Ethyl acetate, 475 L, was added to a 1300 L cooled jacketed glass-lined reactor, followed by 14.5 Kg isobutyl chloroformate, rinsed in with 2×10 L ethyl acetate, and the mixture cooled to −11° C. The N-BOC-L-glutamic acid α-ethyl ester NMM salt solution was added to the 1300 L reactor over 1.3 hours, its reactor rinsed with 10 L ethyl acetate, and the rinse solution added to the 1300 L reactor, then stirred for an additional 30 minutes, while maintaining the temperature at about −13° C.S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3) in ethyl acetate, 112 Kg (containing 41.3 Kg S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride) was added in 4 portions over 45 minutes, and rinsed in with 5 L ethyl acetate, followed by 10.8 Kg NMM in 8 portions over 1.3 hours, rinsed in with 2×5 L portions of ethyl acetate, allowing the mixture to warm to −4° C. by the end of the addition. The mixture was gradually warmed to 30° C. over 2 hours, and maintained at (30˜35)° C. for 2 hours. The reaction mixture was washed twice with water: the first time adding 100 L water, heating to 41° C., allowing the phases to separate for 30 minutes, then removing the aqueous phase; the second time adding 100 L water, bringing the pH to 2.0 with the addition of 0.8 L 36% hydrochloric acid, stirring at 43° C. for 30 minutes, allowing the phases to separate for 1 hour, then removing the aqueous phase. The organic phase was then heated to 42° C., and the pressure reduced to about 0.25 bar until no further gas was released and solvents removed by distillation until 495 L of distillate had been removed. Four cycles of addition of 120 L ethyl acetate and removal of 120 L solvent by distillation were performed, and the water content of the remaining mixture was checked to ensure that it was below 0.1%. With the mixture at 42° C., 610 L of ethyl acetate was added, maintaining the temperature about 41° C., then heating to 58° C. to ensure dissolution. The solution was filtered, rinsing the filter with 18 L ethyl acetate, and the solution allowed to cool to 22° C. The nitrogen purge was stopped and 22.2 Kg hydrogen chloride gas was added through an immersion tube over 2 hours, then the mixture held at that temperature for 2 hours. The mixture was heated to 31° C. over 1.5 hours, and held at about that temperature for 15.5 hours. Solvents were removed by distillation at 33° C. and a pressure of about 0.13 bar over about 1.5 hours to give a volume of concentrated solution of about 630 L. Ethyl acetate, 100 L, was added, and the mixture cooled to 25° C. and held at that temperature for 30 minutes. The crude ezatiostat hydrochloride was recovered by filtration and washed with 30 L ethyl acetate, giving 113 Kg damp crude ezatiostat hydrochloride, which was dried at 40° C. under vacuum for 24 hours to give 52.8 Kg dry crude ezatiostat hydrochloride.Example 4Crystallization of Ezatiostat Hydrochloride to Form Pure Crystalline Ezatiostat Hydrochloride Ansolvate Form D61.5 Kg crude ezatiostat hydrochloride was added to a reactor at room temperature, followed by 399 liter (L) ethanol, and this mixture was heated to 68° C. to completely dissolve the ezatiostat hydrochloride, filtered, then allowed to cool to 65° C. and checked for clarity and the absence of crystallization. About 1.3 Kg of ezatiostat hydrochloride ansolvate form D was suspended in 9 L of ethyl acetate, and about one-half of this suspension was added to the ethanol solution. The mixture was cooled to 63° C. and the second half of the suspension added to the mixture. The resulting mixture was cooled gradually to 45° C., 928 L ethyl acetate was added, and the mixture was cooled to 26° C. and held at about that temperature for about 5 hours, then cooled to −2° C. The mixture, containing crystalline ezatiostat hydrochloride ansolvate, was filtered, and the residue washed twice with 65 L of chilled (0-5° C.) ethyl acetate. The crystalline ezatiostat hydrochloride ansolvate was dried at 30° C. for 48 hours, then cooled to room temperature and sieved. Analysis of the material by DSC and XRPD confirmed its identity as crystalline ezatiostat hydrochloride ansolvate, and Karl Fischer analysis showed a water content of 0.1%.Example 5Purifying Ezatiostat Hydrochloride Crystals to Form Pure Crystalline Ezatiostat Hydrochloride Ansolvate Form DCrude ezatiostat hydrochloride, 51.4 Kg, was added to a 600 L jacketed glass-lined reactor at room temperature, followed by 334 L of ethanol. The mixture was heated to 68° C. to completely dissolve the ezatiostat hydrochloride. The resulting solution was filtered into a 1300 L jacketed glass-lined reactor, and an additional 27 L ethanol warmed to 66° C. used to rinse the first reactor into the second reactor through the filter. The resulting solution in the second reactor was cooled to 63° C. and checked for complete dissolution; then 4 L of a seeding suspension of crystalline ezatiostat hydrochloride ansolvate in ethyl acetate was added, and the mixture cooled to 60° C. The remaining 4 L of the seeding suspension was added, and the mixture cooled to 47° C. over 2 hours. The solids in the mixture were shown by DSC to contain more than one form of ezatiostat hydrochloride, so the stages of heating to dissolution, cooling, and adding seeding suspension (this time 2×2 L), were repeated, then the mixture cooled to 41° C. This time the solids in the mixture were confirmed by DSC to be crystalline ezatiostat hydrochloride ansolvate. Ethyl acetate, 776 L, was added, and the mixture was cooled to 25° C. over 1.3 hours and further to 20° C. over an additional 5 hours, then cooled to −3° C. The mixture, containing crystalline ezatiostat hydrochloride ansolvate, was filtered and the solids washed twice with 54 L each of chilled (−5˜0)° C. ethyl acetate. The damp solids of crystalline ezatiostat hydrochloride ansolvate, 70 Kg, were dried in a vacuum oven at 25° C. for 16 hours, 35° C. for 7 hours, then at room temperature for 1 hour, then sieved. The crystalline ezatiostat hydrochloride ansolvate, 44.2 Kg, had a loss on drying at 40° C. under vacuum for 2 hours of 0.09%, and a water content by Karl Fischer analysis of 0.09%.

……………………………………………………………
U.S. Pat. No. 5,763,570
https://www.google.com/patents/US5763570
……………………………..
http://www.google.com/patents/WO2011156025A1?cl=en
Example 1. Preparation Of Ezatiostat Hydrochloride Ansolvate By Slurrying
[0082] Ezatiostat hydrochloride monohydrate was added to methyl tert-butyl ether at room temperature in excess, so that undissolved solids were present. The mixture was then agitated in a sealed vial at room temperature for 4 days, and the solids were then isolated by suction filtration. XRPD analysis of the solids established that the isolated solids were ezatiostat hydrochloride ansolvate.
[0083] Ezatiostat hydrochloride monohydrate was added to hexanes at 60 °C in excess, so that undissolved solids were present. The mixture was then agitated in a sealed vial at 60 °C for 4 days, and the solids were then isolated by suction filtration. XRPD analysis of the solids established that the isolated solids were ezatiostat hydrochloride ansolvate.
Example 2. Preparation Of Crystalline Ezatiostat Hydrochloride Ansolvate By Heating
[0084] DSC of crystalline ezatiostat hydrochloride monohydrate showed the pattern in FIG. 1, as discussed in paragraph above. Hot stage microscopy showed an initial melt followed by a recrystallization at 153 °C and a final melt at 166 °C. VT-XRPD, where XRPD patterns were obtained at 28 °C, 90 °C, and 160 °C during heating, and 28 °C after cooling of the formerly heated material, showed the presence of ezatiostat hydrochloride monohydrate at 28 °C and 90 °C during heating and of crystalline ezatiostat hydrochloride ansolvate at 160 °C and 28 °C after cooling of the formerly heated material. This confirmed that the transition at around 153/156 °C was a conversion of ezatiostat hydrochloride monohydrate form A to crystalline ezatiostat hydrochloride ansolvate form D and that the final DSC endothermic peak at about 177 °C (166 °C in the hot stage microscopy) was due to the melting of crystalline ezatiostat hydrochloride ansolvate. This was further confirmed by XRPD of the TG-IR material, where XRPD patterns obtained at room temperature both before and after heating to about 160 °C showed that the material before heating was form A and that the material after heating was form D ansolvate. DSC of crystalline ezatiostat hydrochloride ansolvate prepared by recrystallization showed the pattern in FIG. 5, with only the endothermic peak at about
177 °C followed by a broad endotherm at about (205 – 215) °C. Accordingly, the presence of the DSC endothermic peak at about 177 °C, for example at (177±2) °C, when measured under the conditions described above, is considered characteristic of crystalline ezatiostat hydrochloride ansolvate, and the substantial absence of thermal events at temperatures below this is considered indicative of the absence of other forms of ezatiostat hydrochloride
………………………………..
http://www.google.com/patents/WO2013082462A1?cl=en
Ezatiostat, also known as TLK199 or TER 199, is a compound of the formula:

[0003] Ezatiostat has been shown to induce the differentiation of HL-60 promyelocytic leukemia cells in vitro, to potentiate the activity of cytotoxic agents both in vitro and in vivo, and to stimulate colony formation of all three lineages of hematopoietic progenitor cells in normal human peripheral blood. In preclinical testing, ezatiostat has been shown to increase white blood cell production in normal animals, as well as in animals in which white blood cells were depleted by treatment with cisplatin or fluorouracil. Similar effects may provide a new approach to treating myelodysplasia syndrome (MDS).
[0004] Many conditions, including MDS, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the three major blood elements (white blood cells, red blood cells, and platelets), are characterized by depleted bone marrow.
Myelosuppression, which is characterized by a reduction in blood cell levels and in a reduction of new blood cell generation in the bone marrow, is also a common, toxic effect of many standard chemotherapeutic drugs.
[0005] Ezatiostat hydrochloride is the hydrochloride acid addition salt of ezatiostat.
Ezatiostat hydrochloride in a liposomal injectable formulation was studied in a clinical trial for the treatment of MDS, and results from this trial, reported by Raza et al., J. Hem. One, 2:20 (published online 13 May 2009), demonstrated that administration of TLK199 was well tolerated and resulted in multi-lineage hematologic improvement. Ezatiostat hydrochloride in a tablet formulation has been evaluated in a clinical trial for the treatment of MDS, as reported by Raza et al, Blood, 113:6533-6540 (prepublished online 27 April 2009) and a single-patient report by Quddus et al, J. Hem. One, 3:16 (published online 23 April 2010), and is currently being evaluated in clinical trials for the treatment of MDS and for severe chronic idiopathic neutropenia.
…………………………………..
http://www.google.com/patents/WO2013082462A1?cl=en
Example 1
[0048] 80 mg of crystalline ezatiostat hydrochloride was placed in a round bottom flask and dissolved in 25 mL of methanol. The solvent was then evaporated on a rotary evaporation apparatus under reduced pressure at 30 °C. After 30 minutes, the solid sample was removed from the round bottom flask and stored in a sealed vial at 2 °C in a refrigerator. Analysis of this sample was carried out within 24 hours of removing it from the rotary evaporation apparatus.
[0049] The resulting amorphous material was analyzed by 1H NMR, 13C NMR, DSC, and X-Ray powder diffraction experiments. The DSC conditions were 30 to 300 °C at 10 °C/ min using 7 mg of the amorphous material. The X-Ray powder diffraction was taken at 0-60 of 2theta. The crystalline ezatiostat hydrochloride was also analyzed.
Phase II trials: Ezatiostat is the first GSTP1-1 inhibitor shown to cause clinically significant and sustained reduction in RBC transfusions, transfusion independence, and multilineage responses in MDS patients. The tolerability and activity profile of ezatiostat may offer a new treatment option for patients with MDS. (source: Cancer. 2012 Apr 15;118(8):2138-47.)
|
References |
1: Galili N, Tamayo P, Botvinnik OB, Mesirov JP, Brooks MR, Brown G, Raza A. Prediction of response to therapy with ezatiostat in lower risk myelodysplastic syndrome. J Hematol Oncol. 2012 May 6;5:20. doi: 10.1186/1756-8722-5-20. PubMed PMID: 22559819; PubMed Central PMCID: PMC3407785.
2: Raza A, Galili N, Mulford D, Smith SE, Brown GL, Steensma DP, Lyons RM, Boccia R, Sekeres MA, Garcia-Manero G, Mesa RA. Phase 1 dose-ranging study of ezatiostat hydrochloride in combination with lenalidomide in patients with non-deletion (5q) low to intermediate-1 risk myelodysplastic syndrome (MDS). J Hematol Oncol. 2012 Apr 30;5:18. doi: 10.1186/1756-8722-5-18. PubMed PMID: 22546242; PubMed Central PMCID: PMC3416694.
3: Lyons RM, Wilks ST, Young S, Brown GL. Oral ezatiostat HCl (Telintra®, TLK199) and idiopathic chronic neutropenia (ICN): a case report of complete response of a patient with G-CSF resistant ICN following treatment with ezatiostat, a glutathione S-transferase P1-1 (GSTP1-1) inhibitor. J Hematol Oncol. 2011 Nov 2;4:43. doi: 10.1186/1756-8722-4-43. PubMed PMID: 22047626; PubMed Central PMCID: PMC3235963.
4: Raza A, Galili N, Smith SE, Godwin J, Boccia RV, Myint H, Mahadevan D, Mulford D, Rarick M, Brown GL, Schaar D, Faderl S, Komrokji RS, List AF, Sekeres M. A phase 2 randomized multicenter study of 2 extended dosing schedules of oral ezatiostat in low to intermediate-1 risk myelodysplastic syndrome. Cancer. 2012 Apr 15;118(8):2138-47. doi: 10.1002/cncr.26469. Epub 2011 Sep 1. PubMed PMID: 21887679.
5: Quddus F, Clima J, Seedham H, Sajjad G, Galili N, Raza A. Oral Ezatiostat HCl (TLK199) and Myelodysplastic syndrome: a case report of sustained hematologic response following an abbreviated exposure. J Hematol Oncol. 2010 Apr 23;3:16. doi: 10.1186/1756-8722-3-16. PubMed PMID: 20416051; PubMed Central PMCID: PMC2873355.
6: Steensma DP. Novel therapies for myelodysplastic syndromes. Hematol Oncol Clin North Am. 2010 Apr;24(2):423-41. doi: 10.1016/j.hoc.2010.02.010. Review. PubMed PMID: 20359635.
7: D’Alò F, Greco M, Criscuolo M, Voso MT. New treatments for myelodysplastic syndromes. Mediterr J Hematol Infect Dis. 2010 Aug 11;2(2):e2010021. doi: 10.4084/MJHID.2010.021. PubMed PMID: 21415972; PubMed Central PMCID: PMC3033133.
8: Raza A, Galili N, Callander N, Ochoa L, Piro L, Emanuel P, Williams S, Burris H 3rd, Faderl S, Estrov Z, Curtin P, Larson RA, Keck JG, Jones M, Meng L, Brown GL. Phase 1-2a multicenter dose-escalation study of ezatiostat hydrochloride liposomes for injection (Telintra, TLK199), a novel glutathione analog prodrug in patients with myelodysplastic syndrome. J Hematol Oncol. 2009 May 13;2:20. doi: 10.1186/1756-8722-2-20. PubMed PMID: 19439093; PubMed Central PMCID: PMC2694211.
9: Raza A, Galili N, Smith S, Godwin J, Lancet J, Melchert M, Jones M, Keck JG, Meng L, Brown GL, List A. Phase 1 multicenter dose-escalation study of ezatiostat hydrochloride (TLK199 tablets), a novel glutathione analog prodrug, in patients with myelodysplastic syndrome. Blood. 2009 Jun 25;113(26):6533-40. doi: 10.1182/blood-2009-01-176032. Epub 2009 Apr 27. PubMed PMID: 19398716.
| WO2013082462A1 * | Nov 30, 2012 | Jun 6, 2013 | Telik, Inc. | Amorphous ezatiostat ansolvate |
| US20120251496 * | Mar 20, 2012 | Oct 4, 2012 | Telik, Inc. | Ezatiostat for treating multiple myeloma |
……….

Known Yes1 kinase inhibitors, dasatinib and saracatinib.
| Compound name and NCGC ID | Structure | Clinical phase | Known targets | Yes1 IC50 (nM) |
|---|---|---|---|---|
| Dasatinib (1) NCGC00181129 |
 |
Approved | Lyn, PDGFR, KIT, Lck, BTK, Bcr–Abl, Fyn, Yes1, c-Src |
0.5 (<1.0)a |
| Saracatinib (2) NCGC00241099 |
 |
Phase II/III | c-Src, Bcr–Abl, Yes1, Lck | 6.2 (0.70)a |
| AEE-788 (3) NCGC00263149 |
 |
Phase I/II | EGFR, HER-2, VEGFR-2 | 17.5 (13.1)a |
| Dovitinib (4) NCGC00249685 |
|
Phase III | FGFR, EGFR, PDGFR, VEGFR-1,2 | 31 (1.4)a |
| DCC-2036 (5) NCGC00263172 |
 |
Phase I/II | Bcr–Abl, Tie-2, Lyn, FLT3, VEGFR-2 | 2.5 (1.5)a |
| SGI-1776 (6) NCGC00263186 |
 |
Discontinued | Pim-1, FLT3 | 2670 (240)a |
| AMG-Tie-2-1 (7) NCGC00263199 |
 |
Preclinical | Tie-2 | 8.7 (22.0)a |
| AZ-23 (8) NCGC00250381 |
 |
Preclinical | Trk | 39.1 (3.0)a |
| Dorsomorphin (9) NCGC00165869 |
 |
Preclinical | AMPK, BMPR, TGFβ Receptor | 195.9 (29.8)a |
| AZ-628 (10) NCGC00250380 |
 |
Preclinical | Raf Kinase B,C | 348.3 (51.2)a |
- http://www.sciencedirect.com/science/article/pii/S0960894X13006677
- Data in parentheses were gathered by Reaction Biology Corp. using a [γ-33P]-ATP radiolabeled enzyme activity assay at an ATP concentration of 10 μM (www.reactionbiology.com).
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
Very First Human Trials Using Cannbis To Treat Brain Cancer Are Under Way

The picture to your left is showing immunofluorescence of the human glioma cell line. (View more pictures here)
A European based pharmaceutical company called GW Pharmaceuticals is set to commence its first phase of clinical trials for the treatment of Glioblastoma Multiforme (GBM). It’s a bio-pharmaceutical company focused on discovering, developing and commercializing novel therapeutics from its proprietary cannabinoid product platform.
According to the New England Journal of Medicine, GBM accounts for approximately 50% of the 22,500 new cases of brain cancer diagnosed in the United States alone each year.(1) Treatment with regards to brain cancer are very limited which makes the study of cannabis and its effect on brain tumors crucial.
La cantera de los biológicos
Los medicamentos biotecnológicos están en plena ebullición, de forma continuada aparecen nuevas moléculas. Muchas de ellas se encuentran en fases preliminares de desarrollo y se espera que en los próximos años se vayan aprobando. ¿Se mantendrá este ritmo en el tiempo?
View original post 320 more words
New method sneaks drugs into cancer cells before triggering release

Biomedical engineering researchers have developed an anti-cancer drug delivery method that essentially smuggles the drug into a cancer cell before triggering its release. Credit: Ran Mo
Biomedical engineering…
View original post 456 more words
New technique uses ATP as trigger for targeted anti-cancer drug delivery

Biomedical engineering researchers from North Carolina State University and the University of North Carolina have developed a new technique that uses adenosine-5′-triphosphate (the so-called ‘energy molecule’) to trigger the release of anti-cancer drugs directly into cancer cells. The spherical nanoparticles are coated with a shell that incorporates hyaluronic acid, which interacts with proteins found on the surface of some cancer cells. The nanoparticle is filled with DNA molecules that release anti-cancer drug doxorubicin when they come into contact with the adenosine-5′-triphosphate inside a cancer cell. Credit: Ran Mo
Biomedical engineering researchers have developed a new technique that uses adenosine-5′-triphosphate (ATP), the so-called “energy molecule,” to trigger the release of anti-cancer drugs directly into cancer cells. Early laboratory tests show it increases the effectiveness of drugs targeting breast cancer. The technique was developed by researchers at North Carolina State University and the University of North Carolina at Chapel Hill.
“This is…
View original post 339 more words
Regadenoson……..Adenosine A2a receptor agonist, for Coronary artery disease; Sickle cell anemia

2-{4-[(methylamino)carbonyl]- 1H-pyrazol-1-yl}adenosine
(1-{9-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-6-aminopurin-2-yl}pyrazol-4-yl)-N-methylcarboxamide.

Regadenoson (INN, code named CVT-3146) is an A2A adenosine receptor agonist that is a coronary vasodilator. It produces hyperemia quickly and maintains it for a duration that is useful for radionuclide myocardial perfusion imaging.[1]
It was approved by the United States Food and Drug Administration on April 10, 2008 and is marketed by Astellas Pharma under the tradename Lexiscan.[2] It is approved for use in the European Union and under the name of Rapiscan. It is currently being marketed by GE Healthcare and is being sold in both the United Kingdom and Germany.
Regadenoson has a 2- to 3-minute biological half-life, as compared with adenosine‘s 30-second half-life. Regadenoson stress protocols using a single bolus have been developed, obviating the need for an intravenous line. Regadenoson stress tests are not affected by the presence of beta blockers, as regadenoson vasodilates but does not stimulate beta adrenergic receptors.


Regadenoson is an A2A adenosine receptor agonist that is a coronary vasodilator [see CLINICAL PHARMACOLOGY]. Regadenoson is chemically described as adenosine, 2-[4-[(methylamino)carbonyl]-1H-pyrazol-1-yl]-, monohydrate. Its structural formula is:
 |
The molecular formula for regadenoson is C15H18N8O5 • H2O and its molecular weight is 408.37. Lexiscan is a sterile, nonpyrogenic solution for intravenous injection. The solution is clear and colorless. Each 1 mL in the 5 mL pre-filled syringe contains 0.084 mg of regadenoson monohydrate, corresponding to 0.08 mg regadenoson on an anhydrous basis, 10.9 mg dibasic sodium phosphate dihydrate or 8.7 mg dibasic sodium phosphate anhydrous, 5.4 mg monobasic sodium phosphate monohydrate, 150 mg propylene glycol, 1 mg edetate disodium dihydrate, and Water for Injection, with pH between 6.3 and 7.7.
Regadenoson is also referred to in the literature as CVT- 3146 or (1-{9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-6- aminopurin-2-yl}pyrazol-4-yl)-N-methylcarboxamide and has the formula:

Methods for synthesizing regadenoson and related compounds are set forth in U.S. Patent No. 6,403,567, the specification of which is incorporated herein by reference in its entirety.
Regadenoson may be administered by pharmaceutical administration methods that are known in the art. It is preferred that regadenoson is dosed i.v. It is more preferred that regadenoson is administered in a single dose i.v. The term “single dose” refers generally to a single quickly administered dose of a therapeutic amount of regadenoson. The term “single dose” does not encompass a dose or doses administered over an extended period of time by, for example continuous i.v. infusion.
Regadenoson will typically be incorporated into a pharmaceutical composition prior to use. The term “pharmaceutical composition” refers to the combination of regadenoson with at least one liquid carrier that together form a solution or a suspension. Lyophilized powders including compositions of this invention fall within the scope of “pharmaceutical compositions” so long as the powders are intended to be reconstituted by the addition of a suitable liquid carrier prior to use. Examples of suitable liquid carriers include, but are not limited to water, distilled water, de-ionized water, saline, buffer solutions, normal isotonic saline solution, dextrose in water, and combinations thereof.
Regadenoson [(l-{9-[(4S, 2R, 3R, 5R)-3,4-dihydroxy-5-(hydroxymethyl)oxalan-2-yl]-6- aminopurin-2-yl}pyrazol-4-yl)-N-methylcarboxamine] is a selective A2A-adenosine receptor agonist that is a coronary vasodilator. It is currently marketed in the form of a monohydrate as a pharmacologic stress agent indicated for radionuclide myocardial perfusion imaging (MPI) in patients unable to undergo adequate exercise stress.
U.S. Patent No. 8,106,183 describes amorphous regadenoson, and three forms of regadenoson, referred to as Form A (a monohydrate), Form B and Form C.
The synthesis of regadenoson is described, for example, in U.S. Patent Nos. 6,403,567 and 7,183,264. The syntheses disclosed are multi-step processes that proceed via 2- hydrazinoadenosine, which is prepared from the corresponding iodo-derivative (2- iodoadenosine).


……………………………
http://www.google.com/patents/WO2012149196A1?cl=en
EXAMPLE 1
Synthesis of N-Methyl-4-carboxamide
20 g (143 mmol, 1 equiv) of ethyl pyrazole-4-carboxylate and 200 mL (2310 mmol, 16.2 equiv) of a 40 % aqueous solution of methylamine were added to a three-necked flask equipped with a condenser and a heating mantle. The mixture was stirred to aid dissolution, and heated to 65 °C for 2 hours. The reaction was monitored using HPLC at 220 nm with a C18 column. The reaction mixture was then concentrated in vacuo to obtain a syrup / solid. The crude product was co-evaporated with acetonitrile (3 x 200 mL). 100 mL of acetonitrile was then added to the solids and the mixture was stirred for several hours until the solids were well suspended. The solids were then isolated by filtration, washed with 100 mL acetonitrile, and dried in an oven at 40°C to afford 14.4 g (80 % yield) of N-methyl-4-carboxamide with a purity of 93.5% by HPLC.
EXAMPLE 2
Synthesis of IDAAR-Cu+2
This preparation has reported in the literature. See, e.g., Chinese Chemical Letters, (21(1), 51-54, 2010.
An Erlenmeyer flask was charged with 350 mL of water and 75 g of Chelex 100 resin. With stirring, an aqueous solution of copper sulfate pentahydrate (59 g in 350 mL of water) was slowly added over a period of 15 minutes. The resulting slurry was stirred for 2 hours, then filtered. The resulting solids were washed with 100 – 200 mL of water and dried in a vacuum oven at 50 °C for 16 hours to afford 18 g of IDAAR-Cu+2. The copper content of the product was determined to be 11 wt % using Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES).
EXAMPLE 3
Synthesis of Regadenoson Monohydrate
5 g (17.5 mmol, 1 equiv) of 2-fluoroadenosine, 3.07g (24.5 mmol, 1.4 equiv) of N- methylpyrazole-4-carboxamide, and 32 mL of dimethylsulfoxide were added under a nitrogen atmosphere to a dry 3-necked reaction flask equipped with a condenser and a heating mantle.. The mixture was stirred to afford a solution. 100 mL of acetonitrile was then added followed by the addition of 2.2 g of IDAAR-Cu2+ and 5.34 g (5.24 mL, 35.1 mmol, 2 equiv) of
diazabicycloundecene (DBU). The reaction mixture was heated to 70 – 80 °C overnight and monitored by HPLC at 260 nm with a C18 column until the reaction was complete. Then, the reaction mixture was evaporated in vacuo to remove most of the acetonitrile. The remaining dimethylsufoxide solution was purified by reverse phase chromatography using methanol and water. The product was dried in vacuo at a temperature that did not exceed 40° C to afford 3 g (44% yield) of regadenoson monohydrate.
EXAMPLE 4
Synthesis of 2-Hydazineadenosine
2-fluoroadenosine (4g, 14 mmol) was dissolved in 100 mL ethanol in a 300 mL three- necked flask. Hydrazine hydrate (4.1 mL, 6 equivalents, 84 mmol) was added and the mixture was heated to reflux for 1 hour. The reaction mixture was allowed to cool to room temperature and stirred overnight (16 hours). The resulting white precipitate was isolated by filtration and dried in oven at 40°C overnight to afford 2-hydrazinoadenosine (yield: 94%, 3.5g, 96% purity).
EXAMPLE 5
Synthesis of Regadenoson Form D
2-Fluoroadenosine (45 g, 0.158 moL, 1 eq.), 4-(N-methylcarboxamido)pyrazole (27.64 g, 0.221 moL, 1.4 eq.), dimethylsulfoxide (DMSO) (320 mL) and acetonitrile (960 mL) were added to a dry 3000 ml 3-neck reaction flask equipped with a condenser and heating mantle. After stirring for 10 minutes, IDAAR-Cu (20.07 g, 0.032 moL, 0.2 eq.) and DBU (48.0 g, 0.316 moL, 2 eq.) were added. The resulting mixture was then heated to 65°C overnight (18 hours).
The reaction mixture was then filtered and the filtrate was evaporated followed by 2 x 500 mL co-evaporation with xylene. The residue was diluted with 5 L acetonitrile, transferred to a 10 L flask and kept in a cold room (4°C) overnight. The resulting white precipitate was isolated by filtration and stirred in 1.8 L of water. The mixture was heated to 80° C for 2 hours, then allowed to cool in a cold room (4°C) overnight.
The white precipitate was isolated by filtration, then dissolved in 200 ml of 1 : 1 mixture of DMSO and methanol. The clear and slightly yellow solution was loaded to a reverse phase column (10 L) and eluted with water/methanol (gradient with a 5% increase of MeOH every 10 L).
The fractions with HPLC purity of more than 99.9% were combined and concentrated to a paste. The supernatant liquid was decanted and the flask heated in an oil-bath at 150° C under reduced pressure of 20mmHg for 6 hours to afford 6.2 g of Regadenoson Form D as white solid (99.94% HPLC, KF analysis 0.8%).
The fractions with HPLC purity between 50 and 99.8% (~ 23g of product as indicated by HPLC) were combined and subjected to a second purification stage.
………………………………
WO 0078779
https://www.google.com/patents/WO2000078779A2?cl=en
Example 5

(l-{9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-6-aminopurin-2- yl}pyrazol-4-yl)-N-methylcarboxamide (16)
Compound 12 (0.05 mg, 0.12 mmol) was added to 4 mL methylamine (40% sol. In water). The mixture heated at 65 °C in for 24 h. After concentration in vacuo, the residue was purified using prep. TLC (10% MeOH:DCM). ‘HNMR (CD3OD) 62.90 (s, 3 H), 3.78 (m, 1
H), 3.91 (m, 1 H), 4.13 (d, 1 H), 4.34 (d, 1 H), 4.64 (m, 1 H), 6.06 (d, 1 H), 8.11 (s, 1 H), 8.38
(s, 1 H), 9.05 (s, 1 H).
…………………..
https://www.google.com/patents/US6403567
U.S. Patent Nos. 6,403,567
Scheme 1.

Compound I can be prepared by reacting compound 1 with appropriately substituted 1,3 -dicarbonyl in a mixture of AcOH and MeOH at 80° C. (Holzer et al., J. Heterocycl. Chem. (1993) 30, 865). Compound II, which can be obtained by reacting compound I with 2,2-dimethoxypropane in the presence of an acid, can be oxidized to the carboxylic acid III, based on structurally similar compounds using potassium permanganate or pyridinium chlorochromate (M. Hudlicky, (1990) Oxidations in Organic Chemistry, ACS Monographs, American Chemical Society, Washington D.C.). Reaction of a primary or secondary amine having the formula HNR6R7, and compound III using DCC (M. Fujino et al., Chem. Pharm. Bull. (1974), 22, 1857), PyBOP (J. Martinez et al., J. Med. Chem. (1988) 28, 1874) or PyBrop (J. Caste et al. Tetrahedron, (1991), 32, 1967) coupling conditions can afford compound IV.

Compound V can be prepared as shown in Scheme 2. The Tri TBDMS derivative 4 can be obtained by treating compound 2 with TBDMSCl and imidazole in DMF followed by hydrolysis of the ethyl ester using NaOH. Reaction of a primary or secondary amine with the formula HNR6R7, and compound 4 using DCC (M. Fujino et al., Chem. Pharm. Bull. (1974), 22, 1857), PyBOP (J. Martinez et al., J. Med. Chem. (1988) 28, 1874) or PyBrop (J. Caste et al. Tetrahedron, (1991), 32, 1967) coupling conditions can afford compound V.


A specific synthesis of compound 11 is illustrated in Scheme 3. Commercially available guanosine 5 was converted to the triacetate 6 as previously described (M. J. Robins and B. Uznanski, Can. J. Chem. (1981), 59, 2601-2607). Compound 7, prepared by following the literature procedure of Cerster et al. (J. F. Cerster, A. F. Lewis, and R. K. Robins, Org. Synthesis, 242-243), was converted to compound 9 in two steps as previously described (V. Nair et al., J. Org. Chem., (1988), 53, 3051-3057). Compound 1 was obtained by reacting hydrazine hydrate with compound 9 in ethanol at 80° C. Condensation of compound 1 with ethoxycarbonylmalondialdehyde in a mixture of AcOH and MeOH at 80° C. produced compound 10. Heating compound 10 in excess methylamine afforded compound 11.

The synthesis of 1,3-dialdehyde VII is described in Scheme 4. Reaction of 3,3-diethoxypropionate or 3,3-diethoxypropionitrile or 1,1-diethoxy-2-nitroethane VI (R3=CO2R, CN or NO2) with ethyl or methyl formate in the presence of NaH can afford the dialdehyde VII (Y. Yamamoto et al., J. Org. Chem. (1989) 54, 4734).
EXAMPLE 5

(1-{9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-6-aminopurin-2 -yl}pyrazol-4N-methylcarboxamide which can also be identified as 2-(4-methylaminocarbonylpyrazol-1-yl)adenosine (16)
The mixture heated at 65° C. in for 24 h. After concentration in vacuo, the residue was purified using prep. TLC (10% MeOH:DCM). 1HNMR (CD3OD) δ2.90 (s, 3 H), 3.78 (m, 1 H), 3.91 (m, 1 H), 4.13 (d, 1 H), 4.34 (d, 1 H), 4.64 (m, 1 H), 6.06 (d, 1 H), 8.11 (s, 1 H), 8.38 (s, 1 H), 9.05 (s, 1 H).
………………………….
US 7,183,264
http://www.google.com/patents/US7183264
EXAMPLE 5

(1-{9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-6-aminopurin-2-yl}pyrazol-4-yl)-N-methylcarboxamide (16)
Compound 12 (0.05 mg, 0.12 mmol) was added to 4 mL methylamine (40% sol. In water). The mixture heated at 65° C. in for 24 h. After concentration in vacuo, the residue was purified using prep. TLC (10% MeOH:DCM). 1HNMR (CD3OD) δ2.90 (s, 3 H), 3.78 (m, 1 H), 3.91 (m, 1 H), 4.13 (d, 1 H), 4.34 (d, 1 H), 4.64 (m, 1 H), 6.06 (d, 1 H), 8.11 (s, 1 H), 8.38 (s, 1 H), 9.05 (s, 1 H).
References
- Cerqueira MD (July 2004). “The future of pharmacologic stress: selective A2A adenosine receptor agonists”. Am. J. Cardiol. 94 (2A): 33D–40D; discussion 40D–42D. doi:10.1016/j.amjcard.2004.04.017. PMID 15261132.
- CV Therapeutics and Astellas Announce FDA Approval for Lexiscan(TM)
|
12-28-2007
|
Use of A2A Adenosine Receptor Agonists in the Treatment of Ischemia
|
|
|
2-28-2007
|
N-pyrazole A2A receptor agonists
|
|
|
1-10-2007
|
Polymer coating for medical devices
|
|
|
5-5-2006
|
Polymer coating for medical devices
|
|
1-32-2012
|
USE OF A2A ADENOSINE RECEPTOR AGONISTS
|
|
|
1-32-2012
|
PROCESS FOR PREPARING AN A2A-ADENOSINE RECEPTOR AGONIST AND ITS POLYMORPHS
|
|
|
10-21-2011
|
PROCESS FOR PREPARING AN A2A-ADENOSINE RECEPTOR AGONIST AND ITS POLYMORPHS
|
|
|
6-8-2011
|
PROCESS FOR PREPARING AN A2A-ADENOSINE RECEPTOR AGONIST AND ITS POLYMORPHS
|
|
|
6-9-2010
|
Process for preparing an A2A-adenosine receptor agonist and its polymorphs
|
|
|
3-3-2010
|
PROCESS FOR PREPARING AN A2A-ADENOSINE RECEPTOR AGONIST AND ITS POLYMORPHS
|
|
|
2-3-2010
|
Use of A2A adenosine receptor agonists
|
|
|
9-3-2008
|
Polymer coating for medical devices
|
|
|
7-4-2008
|
POLYMER COATING FOR MEDICAL DEVICES
|
|
|
4-30-2008
|
Polymer coating for medical devices
|
| US6403567 | Jun 22, 1999 | Jun 11, 2002 | Cv Therapeutics, Inc. | To stimulate mammalian coronary vasodilatation and for imaging the heart; regadenoson |
| US7183264 | Aug 29, 2003 | Feb 27, 2007 | Cv Therapeutics, Inc. | Such as ethyl-1-(9-((4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxy-methyl)oxolan-2-yl)-6 -aminopurin-2-yl)pyrazole-4-carboxylate; adenosine receptors (A2A); for stimulating mammalian coronary vasodilatation for therapy and imaging the heart |
| US7732595 | Feb 2, 2007 | Jun 8, 2010 | Gilead Palo Alto, Inc. | Process for preparing an A2A-adenosine receptor agonist and its polymorphs |
| US8106183 | Apr 22, 2010 | Jan 31, 2012 | Gilead Sciences, Inc. | Process for preparing an A2A-adenosine receptor agonist and its polymorphs |
NEW PATENT
Novel process for the preparation of (1-{9-[(4S,2R,3R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl)-6-aminopurin-2-yl}pyrazole-4-yl)-N-methylcarboxamide
Biophore India Pharmaceuticals Pvt Ltd
Engineered Virus With Dual Protease Key System Opens to Release Gene Therapy

Viruses cause many diseases but can also serve as vectors for delivery of genetic cargo for therapeutic purposes. Rice University researchers have now modified the adeno-associated virus, commonly used to deliver gene therapy, to work like a lock box that opens itself up only in the presence of two different chemical “keys”.
The virus responds to proteases, enzymes that break down other proteins, opening up and releasing the cargo only when both of the markers are present. By selecting which proteases unlock the virus, a new form of therapy may develop that allows doctors to precisely tune where gene delivery happens.
More from Rice:
“We were looking for other types of biomarkers beyond cellular receptors present at disease sites,” [Junghae Suh, bioengineer at Rice] said. “In breast cancer, for example, it’s known the tumor cells oversecrete extracellular proteases, but perhaps more important are the infiltrating immune cells that migrate into the tumor…
View original post 262 more words
RO 5114436……..The chemokine receptor CCR5 is a clinically validated target for Human Immunodeficiency Virus (HIV) disease and a potentially interesting target for the inflammation therapy area
RO 5114436
1220514-67-9 CAS OF FREE BASE
1220514-58-8 0F HCL SALT
3-Furancarboxamide, N-[(1S)-3-[(3aR,6aS)-5-[(4,6-dimethyl-5-pyrimidinyl)carbonyl]hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]-1-(3-fluorophenyl)propyl]tetrahydro-, (3R)-
(R)-Tetrahydrofuran-3-carboxylic acid [(S)-3-[5-(4,6-dimethylpyrimidine-5-carbonyl)hexahydropyrrolo[3,4-c]pyrrol-2-yl]-1-(3-fluorophenyl)propyl]amide
The chemokine receptor CCR5 is a clinically validated target for Human Immunodeficiency Virus (HIV) disease and a potentially interesting target for the inflammation therapy area. The first small-molecule CCR5 antagonist on the market, maraviroc (Selzentry), was approved by the FDA for treatment of HIV-1 infection.(1) Medicinal chemistry research at Roche led to the discovery of a series of 3,7-diazabicyclo[3.3.0]octane compounds,(2) represented by RO5114436 (1), that are potent CCR5 antagonists. Compound 1 also showed high potency in functional assays for inflammation. The PK properties of 1 were superior to those of maraviroc in preclinical species, including rat, dog, and monkey.
octahydro-pyrrolo[3,4-c]pyrrole derivatives useful in the treatment of a variety of disorders, including those in which the modulation of CCR5 receptors is implicated. More particularly, the present invention relates to 3-(hexahydro- pyrrolo[3,4-c]pyτrol-2-yl)-l-phenyl-propylamine and [3-(hexahydro-pyrrolo[3,4- c]pyτrol-2-yl)-propyl]-phenyl-amine compounds and related derivatives, to compositions containing, to uses of such derivatives and to processes for preparing said compoundsz. Disorders that may be treated or prevented by the present derivatives include HIV and genetically related retroviral infections (and the resulting acquired immune deficiency syndrome, AIDS), diseases of the immune system and inflammatory diseases.
A-M. Vandamme et al. (Antiviral Chemistry & Chemotherapy, 1998 9:187-203) disclose current HAART clinical treatments of HIV- 1 infections in man including at least triple drug combinations. Highly active anti-retroviral therapy (HAART) has traditionally consisted of combination therapy with nucleoside reverse transcriptase inhibitors (NRTI), non-nucleoside reverse transcriptase inhibitors (NNRTI) and protease inhibitors (PI). These compounds inhibit biochemical processes required for viral replication. In compliant drug-naive patients, HAART is effective in reducing mortality and progression of HIV- 1 to AIDS. While HAART has dramatically altered the prognosis for HIV infected persons, there remain many drawbacks to the current therapy including highly complex dosing regimes and side effects which can be very severe (A. Carr and D. A. Cooper, Lancet 2000356(9239):1423-1430). Moreover, these multidrug therapies do not eliminate HIV-1 and long-term treatment usually results in multidrug resistance, thus limiting their utility in long term therapy. Development of new drug therapies to provide better HIV-1 treatment remains a priority. Compounds of the present invention modulate the activity of the chemokine CCR5 receptors. The chemokines are a large family of pro-inflammatory peptides that exert their pharmacological effect through G-protein-coupled receptors. The name “chemokine”, is a contraction of “chemotactic cytokines”. The chemokines are a family of leukocyte chemotactic proteins capable of attracting leukocytes to various tissues, which is an essential response to inflammation and infection. Human chemokines include approximately 50 small proteins of 50-120 amino acids that are structurally homologous. (M. Baggiolini etal, Annu. Rev. Immunol. 1997 15:675-705)
Modulators of the CCR5 receptor may be useful in the treatment of various inflammatory diseases and conditions, and in the treatment of infection by HIV-1 and genetically related retroviruses. As leukocyte chemotactic factors, chemokines play an indispensable role in the attraction of leukocytes to various tissues of the body, a process which is essential for both inflammation and the body’s response to infection. Because chemokines and their receptors are central to the pathophysiology of inflammatory and infectious diseases, agents which are active in modulating, preferably antagonizing, the activity of chemokines and their receptors, are useful in the therapeutic treatment of such inflammatory and infectious diseases. The chemokine receptor CCR5 is of particular importance in the context of treating inflammatory and infectious diseases. CCR5 is a receptor for chemokines, especially for the macrophage inflammatory proteins (MIP) designated MIP- la and MIP- lb, and for a protein which is regulated upon activation and is normal T-cell expressed and secreted (RANTES).
……………..
OPRD [PAPER

http://pubs.acs.org/doi/abs/10.1021/op100020z
A practical asymmetric synthesis of a 3,7-diazabicyclo[3.3.0]octane derivative (1), a representative of a new class of potent CCR5 receptor antagonists, is described. The benzylamine stereogenic center of 1 was introduced by a ruthenium-catalyzed asymmetric reductive amination using (R)-MeOBIPHEP as ligand. Aldehyde 4, prepared by Parikh−Doering oxidation, was used without workup in the reductive amination reaction, which not only simplified the process but also overcame the instability of 4. The 3,7-diazabicyclo[3.3.0]octane core was obtained by a [3 + 2] cycloaddition.
Example 14
(S)-4,4-Difluoro-cyclohexanecarboxylic acid [3- [5-(4,6-dimethyl-pyrimidine-5- carbonyl)-hexahydro-pyrrolo[3,4-c]pyrrol-2-yl]-l-(3-fluoro-phenyl)-propyl]-amide (I- 485)

56 step 2 I ^ 57a: R = Boc 57b: R = H

step 1 – To a solution of 56 (562 mg, 2.1 mmol, prepared as described in WO2004/018425) and 44 (518 mg, 2.1 mmol) in DCM (20mL) containing HOAc (0.31 mL) was added NaBH(OAc)3 (579 mg, 2.73 mmol) in 1 portion and the reaction mixture was stirred for 18 hrs at RT. The reaction was quenched by the addition of 10% K2CO3 (20 mL) and stirring continued for 30 min. The product was twice extracted with DCM (25 mL). The combined extracts were dried (MgSO ) and concentrated in vacuo. The crude product was purified by flash chromatography on silica eiuting with DCM/ 5% MeOH (containing 2% NH4OH) to afford 821 mg (79% theory) of 57a as a white foam: ms (ES+) m/z 498 (M+H)+. step 2 – A solution of 57a (821 mg, 1.65 mmol) dissolved in 10 M HCl in MeOH(40 mL) was heated at 65° C for 2 h. The MeOH was evaporated under reduced pressure and the residue cautiously partitioned between DCM (35 mL) and 20% K2CO3 solution. The aqueous layer was extracted with DCM (2 x 35 mL). The combined organic extracts were dried (Na2SO4) and concentrated in vacuo to afford 641 mg (98%) of 57b as a viscous liquid: ms (ES+) m/z 398 (M+H)+. step 3 – To a solution of 57b (98 mg, 0.25 mmol) in DCM (4 mL) at RT was added 4,4-difluorocyclohexanecarboxyιic acid (49 mg, 0.30mmol). To the resulting solution was added sequentially EDCI (61.4 mg, 0.32 mmol), HOBt (43 mg. 0.32 mmol) and DIPEA (0.13 mL, 0.74 mmol). The mixture was stirred for 4 h. The reaction mixture washed with brine and dried (Na2SO4), then concentrated in vacuo. The crude product was flash chromatographed on silica eiuting with DCM/ 7.5% MeOH (containing 2% NH OH) to afford 113 mg (84%) of 1-485 a white foam: ms (ES+) m/z 544 (M+H)+.
Example 13
Cyclopentanecarboxylic acid {3- [5-(4,6-dimethyl-pyrimidine-5-carbonyl)- hexahydro-pyrrolo[3,4-c]pyrrol-2-yl]-l-phenyl-propyl}-amide (1-29)

To a solution of 12 (0.24 g, 0.70 mmol) in DCM (10 mL) were added 4,6-dimethyl- pyrimidine-5-carboxylic acid (55, 0.12 g, 0.84 mmol) , EDCI (0.17 g, 0.91 mmol), HOBt (0.12 g, 0.91 mmol) and DIPEA (0.36 mL, 2.10 mmol). The mixture was stirred at RT for 3 h. The reaction mixture was washed with saturated NaHCO3 and the organic layer was dried (Na2SO4). The crude product was purified by SiO2 column chromatography eiuting with DCM:MeOH:NH4OH (150:10:1) to afford 0.27 g (81 %) of 1-29:: mp 48.0- 49.0 °C; ms (ES+) m/z 476 (M + H); Anal. (C28H37N5O2.0.2M CH2C12) C; calcd, 68.76; found, 68.61; H; calcd, 7.65; found, 7.51; N; calcd, 14.22; found, 14.28
-
1 Haycock-Lewandowski, S. J.; Wilder, A.; Åhman, J. Org. Process Res. Dev. 2008, 12, 1094– 1103(b) Åhman, J.; Birch, M.; Haycock-Lewandowski, S. J.; Long, J.; Wilder, A. Org. Process Res. Dev. 2008, 12, 1104– 1113
and references therein
-
2.Lee, E. K.; Melville, C. R.; Rotstein, D. M. Chem. Abstr. 2005, 144, 69821PCT Int. Publication Number WO/2005/121145 A2, 2005
