
Home » Uncategorized (Page 111)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
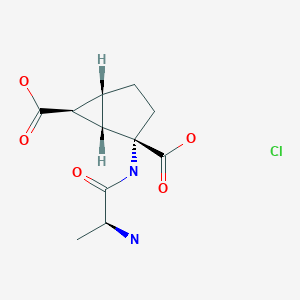
Talaglumetad hydrochloride
|
Talaglumetad hydrochloride
|
|
| Formula |
C11H16N2O5. HCl
|
|---|---|
| Exact mass |
292.0826
|
| Mol weight |
292.7161
|
(1S,2S,5R,6S)-2-[2(S)-Aminopropionamido]bicyclo[3.1.0]hexane-2,6-dicarboxylic acid hydrochloride
Treatment of anxiety and stress disorders [metabotropic glutamate [mGlu] agonist]
Talaglumetad hydrochloride, a prodrug of the type II metabotropic glutamate receptor agonist eglumetad, reached phase III clinical studies for the treatment of anxiety at Lilly.

-
In recent years, with the repeated cloning of glutamate receptor genes, it has become clear that there are surprisingly many subtypes of glutamate receptors. At present, glutamate receptors are broadly classified into two types: the “ionotropic type”, in which the receptor has an ion channel structure, and the “metabotropic type”, in which the receptor is coupled to G-proteins (Science, 258, 597-603, 1992). Ionotropic receptors are classified pharmacologically into three types: N-methyl-D-asparaginic acid (NMDA), α-amino-3-hydroxy-5-methyl isoxazole-4-propionate AMPA), and kynate (Science, 258, 597-603, 1992). Metabotropic receptors are classified into eight types, type 1 through type 8 (J. Neurosci., 13, 1372-1378, 1993; Neuropharmacol., 34, 1-26, 1995).
-
The metabotropic glutamate receptors are classified pharmacologically into three groups. Of these, group 2 (mGluR2/mGluR3) bind with adenylcyclase, and inhibit the accumulation of the Forskolin stimulation of cyclic adenosine monophosphate (cAMP) (Trends Pharmacol. Sci., 14, 13 (1993)), which suggests that compounds that act on group 2 metabotropic glutamate receptors should be useful for the treatment or prevention of acute and chronic psychiatric and neurological disorders. As a substance that acts on group 2 metabotropic glutamate receptors, (+)-(1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid has been disclosed in Japanese Unexamined Patent Publication, No. Hei 8-188561 [1996].
-
Fluorine atoms tend to be strongly electron-attractive and to confer high fat solubility, and compounds into which fluorine atoms are introduced greatly change their physical properties. Thus introducing fluorine atoms might greatly affect the absorbability, metabolic stability, and pharmacological effects of a compound. But it is by no means easy to introduce fluorine atoms. In fact, Japanese Unexamined Patent Publication No. Hei 8-188561 [1996] does not even discuss the introduction of fluorine atoms into (+)-(1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid.
………………………………………………………….
Process development of (1S,2S,5R,6S)-spiro[bicyclo[3.1.0]hexane-2′,5′-dioxo-2,4′-imidazolidine]-6-carboxylic acid, (R)-alpha-methylbenzenemethanamine salt (LSN344309)
Org Process Res Dev 2006, 10(1): 28
http://pubs.acs.org/doi/abs/10.1021/op049829e
LY544344 hydrochloride 6 is Talaglumetad

Process development and a pilot-plant process for the synthesis of 4 and its resolution to obtain (1S,2S,5R,6S)-spiro[bicyclo[3.1.0]hexane-2‘,5‘-dioxo-2,4‘-imidazolidine]-6-carboxylic acid, (R)-α-methylbenzenemethanamine salt (5) are described. Starting from the inexpensive raw 2-cyclopenten-1-one and sulfur ylide 1 the racemic bicyclo keto ester 2 was synthesized. Reaction of 2 with potassium cyanide and ammonium carbonate under Bücherer−Berg’s reaction conditions affords racemic 3 in 80% yield. Hydrolysis of 3 followed by the resolution with (R)-(+)-α-methylbenzylamine gave 4 in excellent yield and purity under optimized conditions. The improvement of the original discovery process to accommodate safety and environmental requirements for scale-up in manufacturing facilities is also discussed.
LY544344 hydrochloride 6 is a new chemical entity under investigation by Eli Lilly & Company as a potential treatment of neurological or psychiatric disorders related to the mammalian central nervous system (CNS)

Scheme 1. Original process for the synthesis of LSN344309 an intermediate of Talaglumetad
…………………………………………………….
Journal of Medicinal Chemistry (2005), 48(16), 5305-5320
http://pubs.acs.org/doi/full/10.1021/jm050235r

…………………………………………………….
WO 2002055485
OR;
http://www.google.im/patents/US20040138304?cl=un

………………………………………………………….
http://www.google.com/patents/EP1052246A1?cl=en

………………………………………………
REFERENCES
New approaches in the development of orally bioavailable selective group 2 metabotropic glutamate receptor agonists
Drugs Fut 2002, 27(Suppl. A): Abst C39
Utility of influx transporters to enhance oral bioavailability
241st ACS Natl Meet (March 27-30, Anaheim) 2011, Abst MEDI 163
The intestinal absorption of a prodrug of the mGlu2/3 receptor agonist LY354740 is mediated by PEPT1: In situ rat intestinal perfusion studies
J Pharm Sci 2010, 99(3): 1574
Dipeptides as effective prodrugs of the unnatural amino acid (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740), a selective group II metabotropic glutamate receptor agonist
J Med Chem 2005, 48(16): 5305
An efficient synthesis of LY544344.HCl: A prodrug of mGluR2 agonist LY354740
Tetrahedron Lett 2005, 46(43): 7299
Pharmacodynamics of a novel anxiolytic (LY544344)
24th CINP Congr (June 20-24, Paris) 2004, Abst P02.269
| WO2000004010A1 * | Jul 14, 1999 | Jan 27, 2000 | Stephen Richard Baker | Bicyclohexane derivatives |
| EP0696577A1 * | Aug 11, 1995 | Feb 14, 1996 | Eli Lilly And Company | Synthetic excitatory amino acids |
| EP1052246A1 * | Jan 27, 1999 | Nov 15, 2000 | Taisho Pharmaceutical Co. Ltd | Fluorine-containing amino acid derivatives |
Complaints and Recalls: new EU-GMP Chapter 8 published
DRUG REGULATORY AFFAIRS INTERNATIONAL
GMP News: Complaints and Recalls: new EU-GMP Chapter 8 published
|
View original post 450 more words
If a Facility stores Medicinal Products for more than 36 Hours GDP will apply
DRUG REGULATORY AFFAIRS INTERNATIONAL
GMP News: If a Facility stores Medicinal Products for more than 36 Hours GDP will apply
Since the EU Good Distribution Practice (GDP) Guide has been revised, a number of questions regarding its interpretation have been raised. One of these questions relates to storage facilities and so called distribution hubs. In the past, many facilities which have been involved in the supply chain were not managed under GDP and didn’t posses a licence for their activities.
The British Medicines Authority MHRA published a press release on 18 August 2014 to explain what they consider to be a facility which must be licensed and which needs to implement the GDP requirements. According to the MHRA: “The GDP Inspectorate is raising awareness of the impact of the new regulations to those parties that are either directly or indirectly affected and any freight consolidator or freight forwarder either in the air, sea or road transport sector…
View original post 59 more words
FDA publishes ICH Q4B – Annex 6 on Uniformity of Dosage Units
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
GMP News: FDA publishes ICH Q4B – Annex 6 on Uniformity of Dosage Units
On 16 June 2014, the FDA published the ICH harmonised Guideline entitled “Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions on Uniformity of Dosage Units General Chapter (Q4B Annex 6)”. This ICH Guideline thus came into force in the USA, too.
The objective of the ICH Q4B Working Group is to reach mutual recognition by regulatory authorities in the ICH regions for all testing methods listed in the ICH Q6A Guideline on Specifications. Through this, comparable testing laid down in the different pharmacopeias shouldn’t be performed separately when it has been assessed by the authorities that those are similar and interchangeable.
The Annex 6 states that the following official texts :
- Ph.Eur. 2.9.40 (Uniformity of Dosage Units
- JP 6.02 Uniformity of Dosage Units
- USP General Chapter <905> Uniformity of Dosage Units
View original post 91 more words
Acai counteracts oxidative stress, lengthens lifespan in fruit flies
24 AUG 2012
Bewildered by the array of antioxidant fruit juices on display in the supermarket and the promises they make? To sort out the antioxidant properties of fruits and berries, scientists at Emory University School of Medicine turned to fruit flies for help.
They found that a commercially available acai berry product can lengthen the lives of fruit flies, when the flies’ lives are made short through additional oxidative stress. Under certain conditions (a simple sugar diet) acai supplementation could triple flies’ lifespans, from eight to 24 days. Acai could also counteract the neurotoxic effects of the herbicide paraquat on the flies.
The results were recently published by the journal Experimental Gerontology, which awarded the paper its inaugural “Outstanding paper” prize. The lead author is Alysia Vrailas-Mortimer, a postdoctoral fellow in Emory University School of Medicine’s Department of Cell Biology.
Vrailas-Mortimer says she didn’t start out focusing on acai…
View original post 645 more words
AMIODARONE



In December 1985, amiodarone was approved by the FDA for the treatment of arrhythmias.[6] This makes amiodarone one of the few drugs approved by the FDA without rigorous randomized clinical trials.
Amiodarone is an antiarrhythmic agent used for various types of cardiac dysrhythmias, both ventricular and atrial. It was discovered in 1961. Despite relatively common side-effects, it is used in arrhythmias that are otherwise difficult to treat with medication.

A more recent synthesis of amiodarone reports the cyclisation of α-phenoxyhexanal 389 under acidic conditions to yield the substituted benzofuran 390 (Scheme 76). A Friedel–Crafts acylation next introduces the aryl ring at the 3-position. Demethylation, iodination and a final alkylation with a diethylaminoethane fragment yields amiodarone [115-117].
- 115 Witczak, M.; Kwiecień, H. Synth. Commun. 2005, 35, 2223–2230. doi:10.1080/00397910500182747
Return to citation in text: [1] - Wang, Z. J. Synthetic Process for 2-Butyl-3-(hydroxy-3,5-diiodobenzoyl)-benzofuran. Chin. Patent 1,858,042, Nov 8, 2006……….116
Return to citation in text: [1] - Ha, H. R.; Stieger, B.; Grassi, G.; Altorfer, H. R.; Follath, F. Eur. J. Clin. Pharmacol. 2000, 55, 807–814.doi:10.1007/s002280050701….117
![[1860-5397-7-57-i76]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i76.png?scale=2.0&max-width=1024&background=FFFFFF)
External links
- Siddoway LA (December 2003). “Amiodarone: guidelines for use and monitoring”. Am Fam Physician 68 (11): 2189–96. PMID 14677664.
- Amiodarone (MedicineNet.com)
- Amiodarone (FamilyPracticeNotebook.com)
- Amiodarone (The WorldWide Intensivist)
- U.S. National Library of Medicine: Drug Information Portal – Amiodarone
- Amiodarone (FDA MedWatch)
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2-{4-[(2-butyl-1-benzofuran-3-yl)carbonyl]-2,6-diiodophenoxy}ethyl)diethylamine | |
| Clinical data | |
| Trade names | Cordarone, Nexterone |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a687009 |
| Pregnancy cat. |
|
| Legal status |
|
| Routes | oral or intravenous |
| Pharmacokinetic data | |
| Bioavailability | 20–55% |
| Metabolism | Liver |
| Half-life | 58 days (range 15-142 days) |
| Excretion | Primarily Hepatic and Biliary |
| Identifiers | |
| CAS number | 1951-25-3 |
| ATC code | C01BD01 |
| PubChem | CID 2157 |
| IUPHAR ligand | 2566 |
| DrugBank | DB01118 |
| ChemSpider | 2072 |
| UNII | N3RQ532IUT |
| KEGG | D02910 |
| ChEBI | CHEBI:2663 |
| ChEMBL | CHEMBL633 |
| Chemical data | |
| Formula | C25H29I2NO3 |
| Mol. mass | 645,31 g/mol |
AMIODARONE
|
|
Title: Amiodarone
CAS Registry Number: 1951-25-3
CAS Name: (2-Butyl-3-benzofuranyl)[4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl]methanone
Additional Names: 2-butyl-3-benzofuranyl-4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl ketone; 2-butyl-3-[3,5-diiodo-4-(b-diethylaminoethoxy)benzoyl]benzofuran
Molecular Formula: C25H29I2NO3
Molecular Weight: 645.31
Percent Composition: C 46.53%, H 4.53%, I 39.33%, N 2.17%, O 7.44%
Literature References: Benzofuran derivative with multiple electrophysiological effects. Prepn: FR 1339389; R. Tondeur, F. Binon, US 3248401 (1963, 1966 to Soc. Belge l’Azote Prod. Chim. Marly). Physicochemical properties: M. Bonati et al., J. Pharm. Sci. 73, 829 (1984). HPLC determn in plasma: M. De Smet, D. L. Massart, J. Pharm. Biomed. Anal. 6, 277 (1988). Comprehensive description: T. A. Plomp, Anal. Profiles Drug Subs. 20, 1-120 (1991). Review of pharmacology, clinical efficacy and safety: M. Chow, Ann. Pharmacother. 30, 637-643 (1996); B. N. Singh, Clin. Cardiol. 20, 608-618 (1997). Clinical trial in cardiac resuscitation: P. J. Kudenchuk et al., N. Engl. J. Med. 341, 871 (1999); to prevent atrial fibrillation: D. Roy et al., ibid. 342, 913 (2000).
Derivative Type: Hydrochloride
CAS Registry Number: 19774-82-4
Manufacturers’ Codes: L-3428
Trademarks: Amiodar (Sanofi Winthrop); Ancaron (Taisho); Cordarex (Sanofi Winthrop); Cordarone (Wyeth); Ortacrone (Sanofi Winthrop); Pacerone (Upsher-Smith); Tachydaron (AWD); Trangorex (Sanofi Winthrop)
Molecular Formula: C25H29I2NO3.HCl
Molecular Weight: 681.77
Percent Composition: C 44.04%, H 4.44%, I 37.23%, N 2.05%, O 7.04%, Cl 5.20%
Properties: Crystalline powder, mp 156°. Also reported as crystals from acetone, mp 159 ±2° (Bonati). Soly at 25° (g/100ml): chloroform 44.51; methylene chloride 19.20; methanol 9.98; ethanol 1.28; benzene 0.65; tetrahydrofuran 0.60; acetonitrile 0.32; 1-octanol 0.30; ether 0.17; 1-propanol 0.13; water 0.07; hexane 0.03 petroleum ether 0.001. Sparingly sol in isopropanol; slightly sol in acetone, dioxane, and carbon tetrachloride. pH (5% soln) 3.4-3.9. pKa (25°C) 6.56 ±0.06. uv max (methanol): 208, 242 nm (E1%1cm 662 ±8, 623 ±10).
Melting point: mp 156°; mp 159 ±2° (Bonati)
pKa: pKa (25°C) 6.56 ±0.06
Absorption maximum: uv max (methanol): 208, 242 nm (E1%1cm 662 ±8, 623 ±10)
Therap-Cat: Antiarrhythmic (class III).
Keywords: Antiarrhythmic.
|
Amiodarone
-
- ATC:C01BD01
- Use:antiarrhythmic
- Chemical name:(2-butyl-3-benzofuranyl)[4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl]methanone
- Formula:C25H29I2NO3
- MW:645.32 g/mol
- CAS-RN:1951-25-3
- InChI Key:IYIKLHRQXLHMJQ-UHFFFAOYSA-N
- InChI:InChI=1S/C25H29I2NO3/c1-4-7-11-22-23(18-10-8-9-12-21(18)31-22)24(29)17-15-19(26)25(20(27)16-17)30-14-13-28(5-2)6-3/h8-10,12,15-16H,4-7,11,13-14H2,1-3H3
- EINECS:217-772-1
- LD50:178 mg/kg (M, i.v.); >4 g/kg (M, p.o.)
Derivatives
hydrochloride
- Formula:C25H29I2NO3 • HCl
- MW:681.78 g/mol
- CAS-RN:19774-82-4
Substance Classes
Synthesis Path
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Cordarex | Sanofi-Aventis | |
| Cornaron | TAD Pharma | ||
| F | Corbionax Ge | Winthrop/Sanofi-Aventis | |
| Cordarone | Sanofi-Aventis | ||
| GB | Cordaron | Sanofi-Aventis | |
| I | Amiodar | Sigma-Tau | |
| Cordarone | Sanofi-Aventis | ||
| J | Ancaron | Sanofi-Aventis | |
| USA | Cordarone | Wyeth-Ayerst | as hydrochloride |
Formulations
- inj. sol. 150 mg/3ml; tabl. 100 mg, 200 mg
References
-
- FR 1 339 389 (Labaz; appl. 22.11.1962).
- US 3 248 401 (Labaz; 26.4.1966; D-prior. 24.11.1961).
-
2-butylbenzofuran:
- Buu-Hoï, N.P. et al.: J. Chem. Soc. (JCSOA9) 1964, 173.
PATENT
CN109053652-PREPARATION METHOD OF AMIODARONE HYDROCHLORIDE INTERMITTENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN235615504&_cid=P11-KL0AU0-06410-1
| Amiodarone hydrochloride is currently the most widely used antiarrhythmic drug. In addition, amiodarone hydrochloride has become the first choice for long-term medication for patients with arrhythmia due to its stable curative effect and minor side effects. In today’s society, people’s work rhythm is constantly increasing, the pressure they face is increasing, the number of patients with cardiovascular diseases is increasing, the demand for anti-arrhythmic drugs is greatly increasing, and the market share of amiodarone hydrochloride is bound to continue to increase. |
| 2-Butyl-3-(4-hydroxybenzoyl)benzofuran is an important intermediate of amiodarone hydrochloride. Patent CN104262304 uses compounds 1 and 2 as starting materials, and the resulting compound 3 is reacted with sodium methoxide as a base and toluene as a solvent to form compound 5; compound 5 is reacted with compound 7 under Lewis acid conditions to form compound 8; compound 8 Hydrolyzed under Lewis acid conditions, the reaction produces 2-butyl-3-(4-hydroxybenzoyl)benzofuran 9. |
| |
| This synthetic route has the following shortcomings: Compound 3 can be directly obtained without hydrolysis under the condition of sodium methoxide as base and toluene as solvent. However, the difficulty of demethylation is much greater than that of decarboxylation, resulting in the formation of compound 5. The conversion rate of the cyclization reaction is low, the by-products are many, and the separation is difficult; resulting in low total yield and increased cost. |
| The synthetic route reported in patent CN107382925, except that the route for preparing compound 5 from compound 3 is different, other reaction routes are the same, but the reaction conditions are different. In this reaction route, the aldehyde group of compound 3 is first protected with trimethyl orthoformate to produce compound 0, and then compound 0 is hydrolyzed to produce compound 4. Compound 4 is cyclized under the catalysis of p-toluenesulfonyl chloride to obtain compound 5. |
| |
| This route has the following disadvantages: when compound 3 is prepared to compound 5, trimethyl orthoformate is added to protect the aldehyde group, then the ester group is first hydrolyzed to form an acid, and then the protective group of the aldehyde group is removed; although side reactions of the aldehyde group can be reduced, but The reaction steps are added, and the trimethyl orthoformate is highly flammable and has potential safety hazards. The yield of compound 4 to compound 5 catalyzed by p-toluenesulfonyl chloride is low, and the cost is also increased. |
| Example 1: |
| Add 15.00kg (122.8mol, 1eq) compound 1 and 30.82kg (147.4mol, 1.2eq) compound 2 to 120.00kg (8w/w) ethyl acetate, add 24.00kg (73.7mol, 0.6eq) cesium carbonate, 1.00 kg (2.5 mol, 0.02 eq) methyl trioctyl ammonium chloride, stirred and heated to 75-85°C, reacted for 1 to 2 hours. After the reaction, the filtrate was filtered with suction, and the filtrate was washed with 60.00kg (4w/w) purified water, and the organic phase was concentrated to dryness under reduced pressure at 40°C to obtain 30.21kg (120.7mol) of compound 3 with a yield of about 98.3%. |
| 30.00kg (120.0mol, 1eq) of compound 3 was added to 14.40kg (360.0mol, 3eq) of 150.00kg (5w/w) aqueous solution of sodium hydroxide, and stirred at 20-30°C. After the reaction, add 1N dilute hydrochloric acid to adjust the pH to 4, add 30.00kg (1w/w) ethyl acetate for extraction, add 30.00kg (1w/w) for the organic phase and wash once with purified water, and add 30.00kg (1w/w) for the organic phase Wash with saturated sodium chloride aqueous solution, add 1.50kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40° C. to obtain 27.30 kg (115.6 mol) of compound 4 with a yield of about 96.4%. |
| Add 27.00kg (114.3mol, 1eq) of compound 4 to 216.00kg (8.0w/w) of acetic anhydride, add 75.60kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 270.00kg (10w/w) purified water, stirred for 2 hours, and then extracted with 81.00kg (3w/w) ethyl acetate. The organic phase was washed twice with 27.00kg (1.0w/w) purified water, and the organic phase was washed with 27.00kg (1.0w/w) saturated aqueous sodium chloride solution, and dried with 2.70kg (0.1w/w) anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 14.26kg (81.8mol) of compound 5 was obtained, and the yield was about 71.6%. 1 HNMR(400MHz,d DMSO )δ:0.92~0.96(t,3H,-CH 3 ),1.37~1.42(m,2H,-CH 2 CH 3 ),1.68~1.72(m,2H,-CH 2 CH 2 CH 3 ),2.77~2.81(t,2H,Ar-CH 2 -CH 2 ), 6.59 (s, 1H, -ArH), 7.20 ~ 7.24 (m, 2H, ArH), 7.49 ~ 7.56 (m, 2 H, ArH). 13 CNMR (400 Hz, DMSO) δ: 159.76, 154.46, 129.07, 123.61, 122.95, 120.72, 111.03, 102.41, 29.71, 27.79, 22.12, 14.05 See attached drawings 1-2. |
| Add 17.00kg (111.7mol, 1eq) of compound 6 to 34.00kg (2w/w) toluene, add 33.31kg (280.0mol, 2.5eq) of thionyl chloride, heat to 75~85℃, keep warm for 2~4 hour. After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 18.99 kg (111.3 mol) of compound 7 as a colorless solution with a yield of about 97.4%. |
| Add 10.70kg (80.4mol, 1.0eq) of aluminum trichloride to 60.00kg of 1,2-dichloroethane at -20~-10℃, add 14.00kg (80.4mol, 1.0eq) of compound 5 under stirring, After stirring uniformly, 16.46kg (96.5mol, 1.2eq) of compound 7 is added at -20~-10°C, and the reaction is kept for 1~2 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 45.00kg purified water, the organic phase was washed with 45.00kg saturated sodium chloride aqueous solution, and 1.40kg was added. Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 55°C to obtain 20.11 kg (65.0 mol) of compound 8. The yield is about 81.2%. |
| Add 20.00kg (64.9mol, 1.0eq) of compound 8 to 60.00kg (3w/w) of toluene, add 9.52kg (71.4mol, 1.1eq) of aluminum trichloride, and raise it to 80~90℃ to react for 2~4 hours . After the reaction, transfer the reaction solution to 90.00kg (4.5w/w) purified water, adjust the pH to 2 with dilute hydrochloric acid, separate the organic phase and wash 2 times with 50.00kg (2.5w/w) purified water, and add 50.00 for the organic phase kg (2.5w/w) saturated sodium chloride aqueous solution wash, add 2.00kg (0.1w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. The wet product was filtered with suction and dried under vacuum at 80°C to obtain 16.60 kg (56.4 mol) of compound 9 as a white crystalline powder with a yield of about 86.9%. |
| 1 HNMR(400MHz,d DMSO )δ: 0.81~0.85(t,3H,-CH 3 ),1.24~1.29(m,2H,-CH 2 CH 3 ),1.68~1.70(m,2H,-CH 2 CH 2 CH 3 ),2.82~2.85(t,2H,Ar-CH 2 -CH 2 ), 6.91~7.72(m,8H,ArH), 10.46(m,1H,-OH). 13 CNMR(400 Hz,DMSO)δ:189.72,163.49,162.69,153.48,132.08,130.16,127.28,124.87,123.98 ,121.16,116.90,11 5.78,111.51,29.94,27.51,22.09,13.84. See attached drawings 3~4. |
| According to the operation of Example 1, the total yield was 46.6%. |
| Example 2: |
| Add 15.00kg (122.8mol, 1eq) of compound 1 and 30.82kg (147.4mol, 1.2eq) of compound 2 to 120.00kg (8w/w) ethyl acetate, add 10.19kg (73.7mol, 0.6eq) potassium carbonate, 1.00 kg (2.5 mol, 0.02 eq) methyl trioctyl ammonium chloride, stirred and heated to 75-85° C., reacted for 1 to 2 hours. After the reaction is completed, suction filtration, the filtrate is washed with 60.00kg (4w/w) purified water, and the organic phase is concentrated to dryness under reduced pressure at 40°C. Obtain 26.21kg (104.7mol) of compound 3, the yield is about 85.3%. |
| 26.00kg (103.9mol, 1eq) of compound 3 was added to 12.47kg (311.7mol, 3eq) of sodium hydroxide in 130.00kg (5w/w) methanol solution, and stirred at 20-30°C. After the reaction, it was concentrated to dryness under reduced pressure at 40°C. Add 130.00kg (5w/w) aqueous solution to dissolve, add 1N dilute hydrochloric acid to adjust the pH to 4, add 26.00kg (1w/w) ethyl acetate for extraction, add 26.00kg (1w/w) purified water for organic phase and wash once, organic phase Add 26.00kg (1w/w) saturated sodium chloride aqueous solution to wash, add 1.30kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40°C. 23.50 kg (99.5 mol) of compound 4 was obtained, and the yield was about 95.8%. |
| Add 23.00kg (97.4mol, 1eq) of compound 4 to 184.00kg (8.0w/w) of acetic anhydride, add 64.40kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 230.00kg (10w/w) purified water, stirred for 2 hours, and 69.00kg (3w/w) ethyl acetate was added for extraction. The organic phase was washed twice with 23.00kg (1.0w/w) purified water, the organic phase was washed with 23.00kg (1.0w/w) saturated sodium chloride aqueous solution, and 2.30kg (0.1w/w) was dried with anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 12.28kg (70.5mol) of compound 5 was obtained, and the yield was about 72.4%. |
| Add 15.00kg (98.6mol, 1eq) of compound 6 to 30.00kg (2w/w) of toluene, add 35.20kg (295.8mol, 3eq) of thionyl chloride, heat to 75~85℃, keep warm and react for 2~4 hours . After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 16.63 kg (97.5 mol) of compound 7 as a colorless solution, with a yield of about 98.9%. |
| Add 27.56kg (206.7mol, 3.0eq) of aluminum trichloride to 60.00kg of 1,2-dichloroethane at -20~-10℃, add 12.00kg (68.9mol, 1.0eq) of compound 5 with stirring, After stirring uniformly, 14.11kg (82.7mol, 1.2eq) of compound 7 is added at -20~-10℃, and the reaction is kept for 1~2 hours. After the reaction, the reaction solution was transferred to 80.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 40.00kg purified water, and the organic phase was washed with 40.00kg saturated sodium chloride aqueous solution, and 1.20kg was added. Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 55°C to obtain 17.17 kg (55.7 mol) of compound 8, with a yield of about 80.8%. |
| Add 17.00kg (55.1mol, 1.0eq) of compound 8 to 51.00kg (3w/w) of toluene, add 22.04kg (165.3mol, 3.0eq) of aluminum trichloride, raise to 80~90℃ and react for 2~4 hours . After the reaction, the reaction solution was transferred to 76.50kg (4.5w/w) purified water, adjusted to pH 2 with dilute hydrochloric acid, separated, the organic phase was washed twice with 42.50kg (2.5w/w) purified water, and the organic phase was added 42.50kg (2.5w/w) saturated sodium chloride aqueous solution was washed, and 1.70kg (0.1w/w) anhydrous sodium sulfate was added for drying. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. After suction filtration, the wet product was dried in vacuum at 80° C. to obtain 13.81 kg (46.9 mol) of compound 9 as a white crystalline powder with a yield of about 85.1%. |
| According to the operation of Example 2, the total yield was 40.2%. |
| Example 3: |
| Add 15.00kg (122.8mol, 1eq) compound 1 and 30.82kg (147.4mol, 1.2eq) compound 2 to 120.00kg (8w/w) toluene, add 24.00kg (73.7mol, 0.6eq) cesium carbonate, 1.00kg (2.5mol, 0.02eq) methyl trioctyl ammonium chloride, stir and raise the temperature to 75~85℃, and react for 1~2 hours. After the reaction is completed, suction filtration, the filtrate is washed with 60.00kg (4w/w) purified water, and the organic phase is concentrated to dryness under reduced pressure at 40°C. 30.36 kg (121.3 mol) of compound 3 was obtained, and the yield was about 98.8%. |
| 30.00kg (120.0mol, 1eq) of compound 3 was added to 20.20kg (360.0mol, 3eq) of 150.00kg (5w/w) aqueous solution of potassium hydroxide, and stirred at 20-30°C. After the reaction, add 1N dilute hydrochloric acid to adjust the pH to 4, add 30.00kg (1w/w) ethyl acetate for extraction, add 30.00kg (1w/w) for the organic phase and wash once with purified water, and add 30.00kg (1w/w) for the organic phase Wash with saturated sodium chloride aqueous solution, add 1.50kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40°C. 27.11 kg (114.7 mol) of compound 4 was obtained, and the yield was about 95.6%. |
| Add 27.00kg (114.3mol, 1eq) of compound 4 to 432.00kg (16.0w/w) of acetic anhydride, add 75.60kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 540.00kg (20w/w) purified water, stirred for 5 hours, and 81.00kg (3w/w) ethyl acetate was added for extraction. The organic phase was washed twice with 27.00kg (1.0w/w) purified water, and the organic phase was washed with 27.00kg (1.0w/w) saturated aqueous sodium chloride solution, and dried with 2.70kg (0.1w/w) anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 14.57kg (83.6mol) of compound 5 was obtained, and the yield was about 73.1%. |
| Add 17.00kg (111.7mol, 1eq) of compound 6 to 34.00kg (2w/w) of toluene, add 35.20kg (295.8mol, 3eq) of thionyl chloride, heat to 75~85℃, keep the temperature and react for 2~4 hours . After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 19.21 kg (112.6 mol) of compound 7 as a colorless solution, with a yield of about 100.0%. |
| Add 10.70kg (80.4mol, 1.0eq) of aluminum trichloride to 60.00kg of toluene at -20~-10℃, add 14.00kg (80.4mol, 1.0eq) of compound 5 with stirring, and after stirring, add at -20 16.46kg (96.5mol, 1.2eq) of compound 7 was added at ~-10°C, and the reaction was incubated for 1 to 2 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 45.00kg purified water, the organic phase was washed with 45.00kg saturated sodium chloride aqueous solution, and 1.40kg was added. Dry with water sodium sulfate. Add 10.72 kg (80.4 mol, 1.0 eq) of aluminum trichloride to the organic phase and raise it to 80-90° C. and react for 2 to 4 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 50.00kg purified water, the organic phase was washed with 50.00kg saturated sodium chloride aqueous solution, and 2.00kg Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. The wet product was filtered with suction and dried in vacuum at 80°C to obtain 14.47 kg (45.2 mol) of compound 9 as a white crystalline powder with a yield of 61.2%. |
| According to the operation of Example 3, the total yield was 42.3%. |
| Comparative Example 1 (CN104262304): |
| 700g (5.7mol, 1.0eq) of compound 1 was added to 2000g of ethyl acetate. After stirring, 1400g (6.7mol, 1.2eq) of compound 2 was slowly added. After the addition, 150g (0.46mol, 0.08eq) were added in sequence. Cesium carbonate and 50g (0.12mol, 0.02eq) methyl trioctyl ammonium chloride, and then slowly heated to 80 ℃, kept the reaction for 8 hours, at the end of the reaction, the ethyl acetate was evaporated under reduced pressure. After the evaporation, 2500g of toluene was added and controlled Warm to 20℃, continue to add 250g (4.6mol, 0.8eq) sodium methoxide, heat to reflux, keep the reaction for 5 hours; when the reaction is over, add dilute hydrochloric acid, adjust the pH to 7, stand still, separate the water phase; continue to add 600g Extract twice with water, discard the water phase, distill the organic phase under reduced pressure to remove the toluene, and collect the 135°C fraction. 383 g (2.2 mol) of compound 5 was obtained as a light yellow transparent liquid, and the yield was about 38.6%. |
| Add 360g (2.1mol, 1.0eq) of compound 5 to 2400g of toluene, heat to reflux, keep dehydration for 4 hours; after dehydration, cool to 15℃, add 420g (2.5mol, 1.2eq) of compound 7, 240g (1.8mol) , 0.86eq) zinc chloride, 60g (0.81mol, 0.4eq) N-nitrosodimethylamine, slowly increase the temperature to 80°C, keep the reaction for 10 hours; after the reaction is over, add dilute hydrochloric acid and adjust the pH to 1~2 , Continue to heat and stir for 2 hours, let stand, and discard the water phase; continue to add 450g of water to extract twice, discard the water phase, heat the organic phase to reflux, and keep dehydration for 4 hours; after dehydration, cool to 10°C, add 270g (2.1 mol, 1eq) Aluminum trichloride, slowly increase the temperature to 75°C, keep the reaction for 8 hours; after the reaction is over, add saturated sodium bicarbonate aqueous solution, adjust to pH 7, let stand, discard the water phase; add 360g saturated brine for extraction Three times, the water phase was discarded, the organic phase was distilled under reduced pressure, and it was steamed until a solid precipitated, and the temperature was reduced to 0°C, and the temperature was kept and stirred for 4 hours. After suction filtration, the wet product was vacuum dried at 80° C. for 8 hours to obtain 323 g (1.1 mol, 1 eq) of compound 9 as an off-white crystalline powder with a yield of about 52.4%. |
| According to the operation of Comparative Example 1, the total yield was 20.2%. |
| Comparative Example 2 (CN107382925): |
| 610g (5.0mol, 1.0eq) of compound 1 and 1728g (12.5mol, 2.5eq) of potassium carbonate were added to the mixture of 610kg (1w/w) DMF and 1830kg (3w/w) toluene, and heated to 60~ with stirring. Incubate at 70°C for half an hour, heat up to 80-100°C and add dropwise, 1098g (5.3mol, 1.05eq) of compound 2, react for 2 hours. After the reaction, 1830 g of water was added to wash twice, and the organic phase was concentrated to dryness under reduced pressure at 80°C. 1126 g (121.3 mol) of compound 3 was obtained, and the yield was about 90.1%. |
| 1100g (4.4mol, 1.0eq) of Compound 3 was added to 514g (4.8mol, 1.1eq) of trimethyl orthoformate and 7.6g (47mmol, 0.01eq) of p-toluenesulfonic acid in 11kg (10w/w) methanol solution, Stir at 20-30°C for 3 hours, add 2.4 g (44 mmol, 0.01 eq), and stir for 1 hour. Then it was concentrated to dryness under reduced pressure at 40°C. 1170 g (3.9 mol) of compound 0 was obtained, and the yield was about 89.7%. |
| Add 1100g (3.7mol, 1eq) of compound 0 to 2200g (2w/w) toluene, add 163g (4.1mol, 1.1eq) sodium hydroxide and 2200g (2w/w) water under stirring, 35~40 Stir and keep at ℃ for 4 hours. At the end of the reaction, 110g (0.1w/w) sodium chloride is added, and hydrochloric acid is added dropwise to adjust the pH to 1~2. The liquid was separated to obtain a toluene organic phase containing compound 4. Add 415g (4.1mol, 1.1eq) of triethylamine to the organic phase, raise it to 70~80℃ for reaction, add 782g (4.1mol, 1.1eq) of p-toluenesulfonyl chloride dropwise, and finish the reaction at 70~80℃ for 2 hours. A solution of 164g (4.1mol, 1.1eq) sodium hydroxide and 2200g (2w/w) water was added dropwise, and the reaction was kept at 70-80°C for 2 hours. Separate the liquids, wash the organic phase with 1100 g of 0.1M hydrochloric acid until neutral, and concentrate the organic phase to dryness at 80°C under reduced pressure. 453 g (2.6 mol) of compound 5 was obtained, and the yield was about 70.0%. |
| Add 409g (2.4mol, 1.0eq) compound 7, 336g (2.5mol, 1.05eq) aluminum trichloride to 60.00kg 1,2-dichloroethane at -20~-10℃, add 418g( 2.4 mol, 1.0 eq) Compound 5, incubated and reacted for 4 hours. Add 320g (2.4mol, 1.0eq) of aluminum trichloride to the organic phase, raise to reflux, and react under reflux for 6 hours. After the reaction, the reaction solution was transferred to 1000g purified water, the organic phase was separated and washed twice with 500g purified water, and the organic phase was purified by column chromatography to obtain 283g (1.0mol) of compound 9 as white crystalline powder with a yield of about 40.1%. . |
| According to the operation of Comparative Example 2, the total yield was 22.7%. |
| Description of the drawings |
| Figure 1 shows the proton nuclear magnetic resonance spectrum of compound 5; |
| Figure 2 shows the carbon nuclear magnetic resonance spectrum of compound 5; |
| Figure 3 shows the proton nuclear magnetic resonance spectrum of compound 9; |
| Figure 4 shows the carbon nuclear magnetic resonance spectrum of compound 9. |
PATENT
CN106946822
| Example 1: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione and 56.1g (0.5mol) of acrolein dimer in organic solvent tetrahydrofuran (2L). ), 126.9 g (0.5 mol) of elemental iodine and 3.4 g (25 mmol) of zinc chloride were added, and the above reaction solution was stirred and reacted in a reactor equipped with magnetic stirring at 40°C for 2 hours. After the reaction, the acidic reaction system was neutralized to neutrality with saturated sodium thiosulfate and saturated sodium bicarbonate respectively; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phase was combined and concentrated under reduced pressure; ethyl acetate and Diethyl ether was recrystallized and separated to obtain 98.6 g (0.32 mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 98.6g (0.32mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 2.1g (16mmol) of aluminum oxide was dissolved in 2L of acetonitrile; the mixture was stirred and reacted at 80°C for 5 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain 52.9 g (0.18 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 1 was 36%, wherein the yield of step (1) was 64%, and the yield of step (2) was 56%. |
| Example 2: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 58.5g (0.25mol) of 1-(4-methoxyphenyl)-1,3-heptanedione and 56.1g (0.5mol) of acrolein dimer in the organic solvent methylene chloride (2L), then add 165.8g (0.5mol) of carbon tetrabromide, BBr 3 6.26g (25mmol), the above reaction solution was stirred and reacted for 4 hours at 25°C in a reactor equipped with magnetic stirring. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with dichloromethane; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized to obtain 2-butane Benzyl-3-(4-methoxybenzoyl)benzofuran 49.3 g (0.16 mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 49.3g (0.16mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 1.1g (8mmol) of boron diethyl ether was dissolved in 2L of 1,2-dichloroethane; the mixture was stirred and reacted at 60℃ for 5 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution The aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain the 2-butyl-3-(4-hydroxybenzoyl) benzo Furan 32.37g (0.11mol). 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 2 was 44%, wherein the yield of step (1) was 64%, and the yield of step (2) was 68%. |
| Example 3: Preparation of 2-butyl-3-(4-methylbenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 56.1g (0.5mol) of acrolein dimer in organic solvent ethanol (2L) ), then add 66.8g (0.5mol) of N-chlorosuccinimide, ZrCl 4 5.8 g (25 mmol), the above reaction solution was stirred and reacted in a reactor equipped with magnetic stirring at 70°C for 8 hours. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane 104.7 g (0.34 mol) of phenyl-3-(4-methoxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 104.7g (0.34mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 6.5g (34mmol) of boron chloride was dissolved in 2L acetonitrile; the mixture was stirred and reacted at 0°C for 1 hour. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 79.4 g (0.27 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 3 was 54%, wherein the yield of step (1) was 68%, and the yield of step (2) was 79%. |
| Example 4: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 112.0g (1.0mol) of acrolein dimer in organic solvent toluene (2L) ), then add 142.9g (0.5mol) of dibromoglycine, AlCl 3 5.8 g (50 mmol), the above reaction liquid was stirred and reacted in a reactor equipped with magnetic stirring at 100°C for 1 hour. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane Group-3-(4-methoxybenzoyl)benzofuran 110.9g (0.36mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 110.9g (0.36mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 10.2g (72mmol) of boron diethyl ether was dissolved in 2L acetonitrile; the mixture was stirred and reacted at 80°C for 2 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by liquid extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain the 2-butyl-3-(4-hydroxybenzoyl)benzofuran 82.3g (0.28mol) . 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 4 was 56%, wherein the yield of step (1) was 72%, and the yield of step (2) was 78%. |
| Example 5: Preparation of 2-butyl-3-(4-methylbenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 28g (0.25mol) of acrolein dimer in the organic solvent dichloromethane ( 2L), then add 159.8g (0.5mol) of liquid bromine, ZnCl 2 6.8 g (50 mmol), the above reaction solution was stirred and reacted for 8 hours at 25°C in a reactor equipped with magnetic stirring. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane Group-3-(4-methoxybenzoyl)benzofuran 58.5g (0.19mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 58.5g (0.19mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 7.2g (38mmol) of sulfonic acid was dissolved in 2L of toluene; the mixture was stirred and reacted at 100°C for 4 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain 41.2 g (0.14 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 5 was 28%, wherein the yield of step (1) was 38%, and the yield of step (2) was 74% |
| In addition to the specific types of halogenated reagents, acid catalysts, and organic solvent raw materials used in the above embodiments, other halogenated reagents, acid catalysts, and organic solvents can also be used; wherein, the halogenated reagent in step (1) is preferably Liquid bromine, elemental iodine, N-iodosuccinimide, N-bromosuccinimide, N-chlorosuccinimide, 1,3-dichloro-5,5-dimethyl Any of hydantoin, dibromohydantoin, bromochlorohydantoin, carbon tetrabromide, and the acid catalyst is boron tribromide, boron trifluoride ether, aluminum chloride, hydrogen fluoride, zinc chloride, zirconium chloride At least one of the organic solvents is dichloromethane, 1,2-dichloroethane, acetonitrile, tetrahydrofuran, ethanol; the acid catalyst in step (2) is boron tribromide, trifluoride One of boron diethyl ether, aluminum chloride, hydrogen fluoride, sulfuric acid, and p-toluenesulfonic acid, and the organic solvent is any of methylene chloride, 1,2-dichloroethane, acetonitrile, tetrahydrofuran, ethanol, and toluene . The specific types of acid catalysts and organic solvents used in steps (1) and (2) of the present invention may be the same or different; in addition, the boiling point of the organic solvent used must be lower than the corresponding treatment temperature. |
| Unless otherwise specified, the various reaction raw materials in the present invention (eg, acrolein dimer, 1-(4-methoxyphenyl)-1,3-heptanedione, etc.) are commercially available The purity is preferably analytically pure. |
PATENT
CN109988131
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN248953969&_cid=P11-KL0BBI-08827-1
| Amiodarone (Amiodarone), also known as amiodarone, was first introduced as a coronary artery dilator, and Rosenbanm was the first to use it in the treatment of anti-arrhythmia in 1976. Amiodarone is extremely toxic. The lethal dose of intravenous injection is 10 times that of the therapeutic dose. The large oral lethal dose is negligible. Long-term larger doses are safe. Amiodarone hydrochloride (ADHC) is the hydrochloride of amiodarone, which was first marketed in Italy in 1984, and its structure is as follows: |
| |
| Amiodarone hydrochloride is a class III antiarrhythmic drug. It is mainly used clinically for supraventricular and ventricular tachyarrhythmias. It is also used for various organic heart diseases and acute coronary syndromes. It can be used as a symptomatic The first-line treatment of atrial fibrillation with left ventricular insufficiency or chronic heart failure. At present, it has become the drug of choice for the prevention of AMI with ventricular tachyarrhythmia, post-infarction ventricular arrhythmia, heart failure with arrhythmia, and sudden cardiac death. |
| In the existing synthetic methods, amiodarone hydrochloride is all obtained by etherification and salt formation with 2-butyl-(4-hydroxy-3,5-diiodobenzoyl)benzofuran as the key intermediate. The structure of this key intermediate is as follows: |
| |
| As a key intermediate of amiodarone hydrochloride, its synthesis is relatively mature. The traditional synthetic route has been applied to industrial production. The route is as follows: |
| |
| This process has the following problems: 1. Long route, complicated steps, and various operations; 2. Low yield and high cost; 3. Many wastes and high waste liquid treatment cost. Therefore, this route is no longer suitable for the current industrial and environmental protection requirements, and more efficient and green methods need to be developed. |
| Patent CN104262304 and CN1858042 improved the above synthesis method and developed a new synthesis method, the route is as follows: |
| |
| This method has been greatly improved compared with the earlier process, but the yield and purity of 2-butylbenzofuran prepared by the route method are low, which leads to a greatly reduced yield and purity of amiodarone hydrochloride. CN107382925 continues to improve the above process, and the purity and yield have increased, but column chromatography purification is required, which is not suitable for industrial large-scale production. |
| Example 1: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| |
| Put 5.5 g K 2 CO 3 , 076 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 60 mg of nickel catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 50°C and stirred for 18-22 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and twice with 40 ml of water. The filtrate was concentrated under reduced pressure to obtain 3.12 g of a dark yellow solid, namely 2-butylbenzofuran, with a yield of 89.7%. |
| 2. Synthesis of intermediate 2-butyl-(4-methoxybenzoyl)benzofuran: |
| |
| Add 5.50 g of aluminum trichloride and 24 ml of dichloromethane to a 100 ml reaction flask, stir and lower the temperature to 0°C, and add 7.0 g of p-methoxybenzoyl chloride dropwise to it within 5°C, and keep warm after dropping. Stir for 1 hour. Dissolve 6.00 g of 2-butylbenzofuran in 24 ml of dichloromethane and add dropwise to the above reaction solution within 5°C of temperature control. After dropping, slowly raise the temperature to 25°C, keep the temperature for 2 hours, and complete the reaction. After cooling to room temperature, it was poured into ice water for separation, the aqueous phase was extracted with dichloromethane, the organic layers were combined, washed twice with water, and the organic phase was concentrated under reduced pressure to obtain 10.60 g of oil. |
| 3. Synthesis of 2-butyl-(4-hydroxybenzoyl)benzofuran: |
| |
| Add 10.00 g of 2-butyl-(4-methoxybenzoyl) benzofuran, 4.50 g of aluminum trichloride and 40 ml of toluene into a 100 ml reaction flask. The temperature is raised to reflux, and the reaction is kept warm for 6 hours. The reaction is complete . Cool down to 0℃, pour into ice water, separate the liquids, extract the aqueous phase with toluene, combine the organic phases, add equal volumes of water, adjust the pH to above 12 with NaOH solution, separate the liquids, adjust the pH to less than 3 with hydrochloric acid for the aqueous phase , Filtered, and the filter cake was vacuum dried to obtain 8.16 g of light yellow solid. |
| 4. Synthesis of 2-butyl-(4-hydroxy-3,5-diiodobenzoyl)benzofuran: |
| |
| Add 8 g of 2-butyl-(4-hydroxybenzoyl) benzofuran, 15.18 g of iodine, 8.26 g of potassium carbonate and 48 ml of ethanol to a 100 ml reaction flask, and heat to reflux with stirring, and keep the reaction temperature for 2 hours. The reaction is over. The temperature was lowered to room temperature, filtered, the filtrate was added dropwise to the sodium metabisulfite aqueous solution, after dripping, the mixture was kept and stirred for 0.5 hours, filtered, the filter cake was washed twice with water, and the solid was vacuum dried to obtain 14.61 g of off-white solid. |
| 5. Synthesis of 2-butyl-[4-[2-(diethylamino)hydroxyethyl]-3,5-diiodobenzoyl]benzofuran: |
| |
| Add 12.00 g of 2-butyl-(4-hydroxy-3,5-diiodobenzoyl) benzofuran and 120 ml of toluene into a 250 ml reaction flask. The temperature is raised to 60°C. After the solid is dissolved, add to it 4.94 g of 2-diethylaminochloroethane hydrochloride, 5.65 g of potassium carbonate and 8.50 g of water were heated to reflux, stirred and reacted for 8 hours, and the reaction was completed. The reaction solution was washed 3 times with water, 0.60 g activated carbon was added to the organic phase, the temperature was raised to reflux and stirred for 1 hour, and then filtered with suction. The filtrate was concentrated under reduced pressure until solids began to precipitate, and the temperature was reduced to 0°C for crystallization. The cold toluene was washed twice, and the solid was vacuum-dried at 80°C to obtain 13.97 g of a finished white solid, which was the finished product of amiodarone hydrochloride. |
| Compared with the existing production process, the preparation method of amiodarone hydrochloride in this embodiment simplifies the operation and improves the convenience of operation and the stability of the product. Through the control of the catalyst and material ratio, the purity and yield of each intermediate are improved, and the product does not require column chromatography to purify, which saves costs and improves production efficiency, which provides convenience for industrial large-scale production. |
| Example 2: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 190 mg of ruthenium catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 50°C and stirred for 22-28 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.25 g of a dark yellow solid, which was 2-butylbenzofuran, with a yield of 93.4%. |
| The remaining steps are the same as in Example 1. |
| Example 3: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 89 mg of palladium catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 40°C under nitrogen and stirred for 26-30 hours. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.36 g of a dark yellow solid, which was 2-butylbenzofuran, with a yield of 96.6%. |
| The remaining steps are the same as in Example 1. |
| Example 4: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 100 mg of rhodium catalyst were added to it, and replaced with nitrogen 3 Next, the reaction was kept at 40°C under nitrogen protection and stirred for 20-24 hours. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 2.93 g of dark yellow solid, which is 2-butylbenzofuran, with a yield of 84.2%. |
| The remaining steps are the same as in Example 1. |
| Example 5: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 80 mg of gold catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 40°C and stirred for 20-28 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.07 g of dark yellow solid, which is 2-butylbenzofuran, with a yield of 88.2%. |
| The remaining steps are the same as in Example 1. |
?///////////
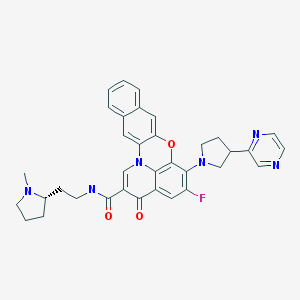
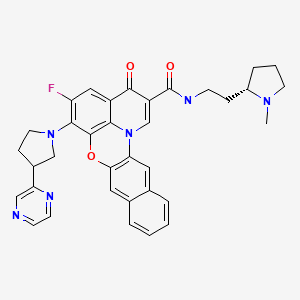
Quarfloxin, Itarnafloxin , CX-3543….Inhibits rRNA biogenesis.

Quarfloxin, Itarnafloxin
CAS: 865311-47-3.
Chemical Formula: C35H33FN6O3
Exact Mass: 604.25982
Molecular Weight: 604.67
Elemental Analysis: C, 69.52; H, 5.50; F, 3.14; N, 13.90; O, 7.94
Synonym: CX 3543; CX3543; CX-3543; QuarfloxacinTA1-1B
- CX 3543
- CX-3543
- Itarnafloxin
- Quarfloxacin
- Quarfloxin
- UNII-8M31J5031Q
IUPAC/Chemical name:
5-fluoro-N-(2-((S)-1-methylpyrrolidin-2-yl)ethyl)-3-oxo-6-((R)-3-(pyrazin-2-yl)pyrrolidin-1-yl)-3H-benzo[b]pyrido[3,2,1-kl]phenoxazine-2-carboxamide.
-
5-Fluoro-N-(2-((2S)-1-methylpyrrolidin-2-yl)ethyl)-3-oxo-6-(3-(pyrazin-2- yl)pyrrolidin-1-yl)-3H-benzo(b)pyrido(3,2,1-kl)phenoxazine-2-carboxamide
-
3H-Benzo(b)pyrido(3,2,1-kl)phenoxazine-2-carboxamide, 5-fluoro-N-(2-((2S)- 1-methyl-2-pyrrolidinyl)ethyl)-3-oxo-6-(3-pyrazinyl-1-pyrrolidinyl)-
Quarfloxin, also known as Quarfloxacin and CX-3543, is a fluoroquinolone derivative with antineoplastic activity. Quarfloxin disrupts the interaction between the nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis that is overexpressed in cancer cells; disruption of this G-quadruplex DNA:protein interaction in aberrant rRNA biogenesis may result in the inhibition of ribosome synthesis and tumor cell apoptosis.
CX-3543, developed at Cylene Pharmaceuticals, is a multi-targeting oncogene inhibitor evaluated in phase II clinical studies for the treatment of low or intermediate grade neuroendocrine carcinoma, including carcinoid and islet cell cancer. In 2008, a trial for the treatment of chronic lymphocytic leukemia (CLL) was withdrawn prior to patient enrollment. In 2010, phase I clinical studies for the treatment of solid tumors and for the treatment of lymphoma were terminated upon observation that the modified dose schedule presented no advantage over previously studies schedule solid tumors.
CX-3543 was developed using the company’s Quadruplex Targeting technology which is based on quadruplex motifs in genomic DNA that regulate the expression of clusters of key oncogenes but not normal cellular genes. In 2013, the product was licensed to TetraGene by Cylene Pharmaceuticals on an exclusive, worldwide basis for development for the treatment of cancer. Cylene ceased operations in 2013.
Current developer: Cylene Pharmaceuticals Inc. phase 2
Clinical trial news: Quarfloxin is a ground-breaking small-molecule targeted cancer therapeutic derived from the validated fluoroquinolone class of drugs. Rationally designed to selectively inhibit ribosomal RNA (rRNA) biogenesis in cancer cells, quarfloxin disrupts the interaction between the Nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis and one that is amplified in cancer cells. As a result, quarfloxin selectively induces apoptotic cell death in cancers. Many commercialized cancer therapeutics act indirectly on rRNA Biogenesis through upstream modulators, but quarfloxin is the first agent to directly target this cancer-specific aberrant cell function. According to news released on June 19, 2011, Cylene Pharmaceuticals announced the initiation of a Phase II clinical trial of quarfloxin (CX-3543) in patients with carcinoid/neuroendocrine tumors (C/NET), which are malignant cancers arising from neural crest cells.
Cylene Pharmaceuticals today announced the initiation of a Phase II clinical trial of quarfloxin (CX-3543) in patients with carcinoid/neuroendocrine tumors (C/NET), which are malignant cancers arising from neural crest cells.
“Quarfloxin (CX-3543) is a small molecule that disrupts a protein:rDNA complex that forms in the abnormal nucleoli of cancer cells, thereby selectively inducing apoptotic cell death in cancers,” said Dr. William Rice, President and Chief Executive Officer of Cylene Pharmaceuticals. “Many commercialized cancer therapeutics act on or through the nucleolus, but quarfloxin is the first agent designed to directly target a key function within the nucleolus. Quarfloxin has been well tolerated in humans and has demonstrated signs of biological benefit for patients with C/NET in Phase I clinical trials. Moreover, biodistribution studies revealed that quarfloxin accumulates in the tissues in which C/NET arise.”
In this open-label Phase II trial, quarfloxin will be administered to patients with low or intermediate grade C/NET, including those receiving concomitant treatment with a stable dose of octreotide. This multi-centered study will include an assessment of improvements in patients’ symptoms and biochemical markers, in addition to RECIST tumor response measurements. The first patient was enrolled and treated at Front Range Cancer Specialists in Fort Collins, CO under the care of Robert Marschke Jr., M.D. This study is expected to enroll up to 25 patients at several leading cancer centers.
“The initiation of this Phase II trial with quarfloxin is a major milestone for Cylene, but more importantly, we hope that quarfloxin will be an effective treatment for cancer patients with limited therapeutic alternatives,” added Dr. Daniel Von Hoff, Cylene’s Co-Founder and Vice President, Medical Affairs. “Quarfloxin has demonstrated potent in vivo efficacy against a broad range of tumors and a considerable therapeutic window in preclinical antitumor models, and has a unique profile of concentrating in neural crest tissues. For these reasons, we are enthusiastic about offering a Phase II clinical trial for patients with carcinoid/neuroendocrine tumors.”
About Quarfloxin (CX-3543), a Nucleolus Targeting Agent (NTA)
Quarfloxin is a ground-breaking small-molecule targeted cancer therapeutic derived from the validated fluoroquinolone class of drugs. Rationally designed to selectively inhibit ribosomal RNA (rRNA) biogenesis in cancer cells, quarfloxin disrupts the interaction between the Nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis and one that is amplified in cancer cells. As a result, quarfloxin selectively induces apoptotic cell death in cancers. Many commercialized cancer therapeutics act indirectly on rRNA Biogenesis through upstream modulators, but quarfloxin is the first agent to directly target this cancer-specific aberrant cell function.
About Cylene Pharmaceuticals, Inc.
Cylene Pharmaceuticals is a biotech pharmaceutical company dedicated to the discovery, development and commercialization of targeted small-molecule drugs to treat life-threatening cancers. Cylene has created a diverse portfolio of product candidates, including novel inhibitors of cancer-linked serine/threonine kinases, as well as innovative Nucleolus Targeting Agents (NTAs) that target the abnormal nucleolus functions of cancer cells and selectively kill cancer cells. More information can be found athttp://www.cylenepharma.com.
………………………………………………………..
http://www.google.com/patents/US20060029950
To a series of solutions of the fluoroacid (0.5 mmol) in NMP (3.6 mL) was added the amines NHR1R2 (0.5-2.0 mmol) at room temperature. The vessels were sealed and heated on a 90° C. hotplate with constant stirring for 1-2 hours until the reactions were determined to be complete by HPLC/MS analysis. The reaction mixtures were allowed to cool to room temperature and water was added (20 mL). The resulting precipitates were collected by vacuum filtration and dried under vacuum. In cases where 1.0 equivalent of amine was used, the resulting reaction mixtures were used in the next step “as is.” The resulting solids or solutions were treated with HBTU (2.5 eq.) and DIEA in 3.6 mL NMP and allowed to stir for 30 minutes at room temperature under an inert atmosphere. These solutions were added to a series of amines NHR3R4 (2.5 equivalents) in a 96 well format (Whatman Uniplate, 2 mL) and allowed to react for 2 hours. Methanol was then added (50-100 μL) and the plate was filtered (Whatman Unifilter Polypropylene). The resulting liquids were directly chromatographed on reverse HPLC (Waters Xterra 19×50 mm) with mass directed collection (Micromass ZQ, Waters FCII). The fractions were analyzed for purity (MS TIC, UV) and dried by vacuum evaporation (Savant) with an average yield of 5-10 mg). Examples of substituted quinobenzoxazines analogs are described in Table 1.
Example 48Synthesis of CX-3092 and CX-3543
One method for synthesizing CX-3543 is shown below. As shown in Scheme 2, CX-3543 is synthesized in a convergent manner, assembling the substructures 1, 1A and 2A in the final two synthetic steps (Scheme 2), to form CX-3543 having a 50:50 ratio of RS and SS isomers. CX-3092 may be synthesized in a similar manner using a non-chiral form of 1A.


In more detail, pyrazinopyrrolidine 1A is synthesized via a [3+2] cycloaddition chemistry. Conversion of L-proline 7 to cyano-1-aminopyrrolidine 8 without loss of stereochemistry, followed by reduction provides the chiral 2-aminoethyl-1-methylpyrrolidine 2A in high yield. CX-3543 was found to have a formulated solubility of approximately 20 mg/mL.
Example 70This example describes a method for preparing a substituted benzoxazine analog from reaction of the corresponding ester with an amine, and aluminum chloride.

To a solution of 2,3,4,5-tetrafluorobenzoic acid (100 g, 510 mmol), in methylene chloride (0.5 L) was added oxalyl chloride (68 g, 540 mmol) and DMF (ca 3 drops) and the reaction mixture was allowed to stir at room temperature overnight allowing for the produced gasses to escape. The solvent was removed in vacuo and the vessel was placed on high vacuum (ca 0.5 mm Hg) for 2 hours to afford the acid chloride as a viscous oil (105 g) and was used in the subsequent reaction without further purification.

To a suspension of potassium ethyl malonate (97 g, 570 mmol) and magnesium chloride (55 g, 570 mmol) in acetonitrile and the suspension was chilled to 0° C. To this suspension was added the crude 2,3,4,5-benzoyl chloride (105 g, 520 mmol) over 5 minutes. Triethylamine was slowly added at a rate sufficient to keep the reaction temperature below 10° C. and the mixture was allowed to warm to room temperature and was stirred overnight. The solvent was removed in vacuo and replaced with toluene (300 mL) and 1N HCl (500 mL) was added and the mixture was allowed to stir for 1 hour. The organic layer was separated and washed with 1N HCl (100 mL) and brine (100 mL) and dried over sodium sulfate, filtering over a pad of silica gel (50×100 mm), eluting with ethyl acetate. The solvent was removed in vacuo and the resulting oil was dissolved in ethanol/water (9:1) and was allowed to crystallize overnight. The resulting crystals were Isolated by filtration, washing with ethanol/water (8:2) to afford the ketoester (43.75 g, 166 mmol) as a white crystalline solid.

To a 250 mL round bottom flask was added the tetrafluoroketoester (10.0 g, 37.9 mmol), triethylorthoformate (8.6 mL, 56.8 mmol) and acetic anhydride (7.15 mL, 75.8 mmol) and the reaction mixture was heated to 145° C. for 2 hours. The reaction was allowed to cool to room temperature and placed on high vacuum (ca 0.5 mm Hg) for 1 hour. The resulting oil was dissolved in ethanol (100 mL) and 2-amino-1-naphthol (6.02 g, 37.9 mmol) was added at room temperature and the solution became briefly clear and then product began to precipitate. The reaction was allowed to stir for 2 hours and was then filtered and washed with ethanol (100 mL) to afford the enamine as a yellow solid (12.5 g, 28.9 mmol).

To a solution of the enamine (12.13 g, 27.95 mmol) in dry DMF (50 mL) was added potassium carbonate (4.24 g, 1.1 eq.) and the mixture was heated to 90° C., with constant stirring, for 2 hours. The mixture was allowed to cool to room temperature without stirring and was allowed to remain at room temperature for an additional hour. The crystalline solid was collected by filtration, washing with water. Recrystallization from THF afforded the difluoroester as a white crystalline solid (9.3 g, 23.6 mmol).

To a solution of the difluoroester (1.0 g, 2.5 mmol) in NMP (10 mL) was added N-Boc-3-(2-pyrazino)pyrrolidine (870 mg, 3.5 mmol) and the mixture was heated to reflux for 3 hours. The reaction mixture was then allowed to cool to room temperature and the product was collected by filtration. Crystallization from THF afforded the pyrazine ester as a yellow solid (910 mg, 1.74 mmol).

To a solution of the pyrazine ester (250 mg, 0.48 mmol) and 2-(2-aminoethyl)-1-methylpyrrolidine (80 mg, 0.63 mmol) in methylene chloride at room temperature was added aluminum chloride (83 mg, 0.63 mmol) and the reaction mixture was allowed to stir for 2 hours. The solvent was removed in vacuo and saturated L-tartaric acid was added (5 mL) and the mixture was allowed to stir for 1 hour. Methylene chloride (10 mL) was then added and the mixture was basified with 1N NaOH. The organic layer was separated and washed with a saturated solution of Rochelle’s salt, brine and dried over sodium sulfate. The solvent was removed in vacuo and the resulting solid was dissolved in THF and filtered and the solvent was removed again. The crude solid was recrystallized in ethyl acetate to afford the amide as a yellow solid (225 mg, 0.37 mmol, 98.5% pure).
Example 71
This example describes a method for preparing a substituted benzoxazine analog from reaction of the corresponding carboxylic acid with an amine, and aluminum chloride.

The pyrazinoester (2.0 g, 3.8 mmol) was dissolved in ethanol (100 mL) and conc HCl was added (20 mL) and the mixture was refluxed overnight. The mixture was allowed to cool to room temperature and the solid was collected by vacuum filtration, washing with ethanol to afford the pyrazinoacid as a light tan powder (1.6 g, 3.2 mmol).

To a mixture of the fluoroaminoacid (1.6 g, 3.2 mmol) and HBTU (2.0 g, 5.3 mmol) in NMP (20 mL) was added N,N-diisopropyl-N-ethylamine (1.0 mL, 6 mmol) and the mixture was allowed to stir at room temperature, under argon, for 1 hour (the solution became clear). (S)-2-(2-aminoethyl)-1-methylpyrrolidine (Mizuno, A.; Hamada, Y.; Shioiri, T., Synthesis, 1980, 12 1007)(1.0 mL, 6.9 mmol) was added and the mixture was allowed to stir for 30 minutes. Water (200 mL) was added and the resulting solid was collected by vacuum filtration, washing with water, and dried to afford the pyrazine as a yellow solid. The yellow solid was purified on silica gel (10% MeOH/CH2Cl2 first eluting off impurities followed by eluting with 5% NH4OH/15% MeOH/CH2Cl2. The combined fractions were evaporated to afford the compound as a yellow solid. (1.2 g, 2.0 mmol, 85% pure).
……………………………..
http://www.google.com/patents/WO2007137000A2?cl=en
The present disclosure provides an improved method of treating cancer using a combination of a G-quadruplex-interactive compound that binds to G- quadruplexes in rDNA to release the nucleolin already bound to these G- quadruplexes together with a PARP inhibitor. This results in an increase in apoptosis in cancer cells. The PARP inhibitor can be administered to a patient (human or animal) in need of cancer treatment simultaneously or from 0.1 to 24 hours prior to or 0.1 to 24 after the administration of the G-quadruplex-interactive agent that releases the nucleolin bound to the G-quadruplex and triggers enhanced apoptosis of cancer cells, or the PARP inhibitor and the enhancer of nucleolin binding can be administered simultaneously, with each agent being administered in an amount sufficient to inhibit the growth and/or cell division of cancer (neoplastic) cells, and preferably to cause cancer cell death. In the methods provided herein, the PARP inhibitor can be benzamide (as specifically exemplified) or it can be 3- benzamide, 3-methoxybenzamide, carba-NAD+, nicotinamide, a dihydroisoquinolinone, an isoquinolinone such as 5-methyl-dihydroisoquinolinone, a benzimidazole-4-carboxamide, a 2-aryl-benzimidazole-4-carboxamide, a benzoxazole-4-carboxamide, an N,N-dimethylaminomethyl, pyrrolidinomethyl or bis- benzamide derivative, for example 1 ,5-di(3- carbamoylphenyl)aminocarbonyloxy)pentane, a phthalazinone, a quinazolinone, an isoindolinone, a phenanthhdinone, among others. The G-quadruplex-interactive agent that releases nucleolin from the rDNA bound to the G-quadruplexes and triggers apoptosis of cancer cells is desirably a substituted quinobenzoxazine analog; in an embodiment of the invention, it is CX-3543

(see also US Patent Publication 2006-0029950, which is incorporated by reference herein). This combination chemotherapy can be administered in a single dose, or it can be administered at intervals chosen by a medical or veterinary practitioner.
[0003] The present disclosure further provides compositions comprising a PARP inhibitor and a G-quadruplex-interactive compound that triggers the release of nucleolin from the G-quadruplexes in the rDNA and triggers apoptosis of cancer cells. The compositions desirably further comprises a pharmaceutically acceptable excipient, especially one which is compatible with intravenous administration in human patients. In an embodiment of the invention, the composition comprises benzamide and CX-3543.
……………………………
pubs.acs.org/doi/abs/10.1021/jm3013486

Nowadays, it has been demonstrated that DNA G-quadruplex arrangements are involved in cellular aging and cancer, thus boosting the discovery of selective binders for these DNA secondary structures. By taking advantage of available structural and biological information on these structures, we performed a high throughput in silico screening of commercially available molecules databases by merging ligand- and structure-based approaches by means of docking experiments. Compounds selected by the virtual screening procedure were then tested for their ability to interact with the human telomeric G-quadruplex folding by circular dichroism, fluorescence spectroscopy, and photodynamic techniques. Interestingly, our screening succeeded in retrieving a new promising scaffold for G-quadruplex binders characterized by a psoralen moiety.
……………………………..
US 20070293485
http://www.google.com/patents/US20070293485
…………………………..
see
US 20130005720
http://www.google.com/patents/US20130005720
…………………………….
see
WO 2004091504 or http://www.google.com/patents/EP1610759A2?cl=en
|
References: |
1. Bayes, M.; Rabasseda, X.; Prous, J. R., Gateways to clinical trials. Methods Find Exp Clin Pharmacol 2007, 29, (1), 53-71.
2. Brennan, A. B.; Long, C. J.; Bagan, J. W.; Schumacher, J. F.; Spiecker, M. M. Surface topographies for non-toxic bioadhesion control. US20100226943A1.
3. Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C. B.; Proffitt, C.; Trent, K.; Whitten, J. P.; Lim, J. K. C.; Von, H. D.; Anderes, K.; Rice, W. G., Anticancer Activity of CX-3543: A Direct Inhibitor of rRNA Biogenesis. Cancer Res. 2009, 69, (19), 7653-7661.
4. Hurley, L. H.; Guzman, M. Combination cancer chemotherapy. WO2007137000A2, 2007.
5. Lim, J.; Whitten, J. P. Drug administration methods. WO2007143587A1, 2007.
6. Neidle, S., Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 277, (5), 1118-1125.
7. O’Brien, S.; Siddiqui-Jain, A. Targeting quadruplex sequences in human nucleic acids by identifying interacting quinoline and porphyrin derivatives. WO2007056113A2, 2007.
8. Ryckman, D. M.; Drygin, D.; Whitten, J. P.; Anderes, K.; Trent, K.; Darjania, L.; Haddach, M.; O’Brien, S.; Rice, W. G. Methods for treating aberrant cell proliferation disorders. US20080318938A1, 2008.
9. Tian, M.; Zhang, X.; Pan, R.; Zhao, C.; Tang, Y., Structure of G-quadruplex in the oncogene c-myc promoter and small ligands targeting the G-quadruplex. Huaxue Jinzhan 22, (5), 983-992.
10. Whitten, J. P.; O’Brien, S. Methods for treating ophthalmic disorders. US20080318939A1, 2008.
11. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. US20060074089A1, 2006.
12. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. WO2006034113A2, 2006.
13. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Process for the preparation of benzothiazole and phenoxazine compounds. US20060063761A1, 2006.
14. Whitten, J. P.; Schwaebe, M.; Siddiqui-Jain, A.; Moran, T. Preparation of substituted quinobenzoxazine analogs as antitumor agents. US20060029950A1, 2006.
| “CX-3543, 386705” ANNUAL DRUG DATA REPORT, PROUS, BARCELONA, ES, vol. 27, no. 4, 2005, page 379, XP009092663 ISSN: 0379-4121 | ||
| 2 | * | CEPEDA, V. ET AL.: “Poly(ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors in Cancer Chemotherapy” RECENT PATENTS ON ANTI-CANCER DRUG DISCOVERY, vol. 1, no. 1, January 2006 (2006-01), pages 39-53, XP007903584 ISSN: 1574-8928 |
| 3 | * | DATABASE INTEGRITY [Online] Prous science; DailyDrugNews.com (Daily Essentials) 22 July 2005 (2005-07-22), “CX-3543 begins phase I cancer trial” XP007903594 retrieved from INTEGRITY.PROUS.COM Database accession no. 386705 |
| 4 | * | JIN ET AL: “In vivo efficacy of CX-3543, a novel c-Myc oncogene inhibitor” PROCEEDINGS OF THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, NEW YORK, NY, US, vol. 45, 2004, page ABS. LB-243, XP001537665 ISSN: 0197-016X |
| 5 | * | RICE WILLIAM G ET AL: “Design of CX-3543, a novel multi-targeting antitumor agent” PROCEEDINGS OF THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, NEW YORK, NY, US, vol. 46, April 2005 (2005-04), pages 594-ABS. 2530, XP001536592 ISSN: 0197-016X |
| 6 | * | SHIOKAWA D ET AL: “Inhibitors of poly(ADP-ribose) polymerase suppress nuclear fragmentation and apoptotic-body formation during apoptosis in HL-60 cells” FEBS LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 413, no. 1, 11 August 1997 (1997-08-11), pages 99-103, XP004261237 ISSN: 0014-5793 |
| 7 | * | VALERIOTE F ET AL: “SYNERGISTIC INTERACTION OF ANTICANCER AGENTS: A CELLULAR PERSPECTIVE” CANCER CHEMOTHERAPY REPORTS, vol. 59, no. 5, September 1975 (1975-09), pages 895-900, XP009019750 |
| S7910600 | Aug 29, 2008 | Mar 22, 2011 | Cylene Pharmaceuticals, Inc. | Therapeutic kinase modulators |
| US7956064 | Aug 31, 2007 | Jun 7, 2011 | Cylene Pharmaceuticals, Inc. | Fused tricyclic compounds as serine-threonine protein kinase and PARP modulators |
| US8481529 | May 9, 2007 | Jul 9, 2013 | The Arizona Board Of Regents On Behalf Of The University Of Arizona | Combination cancer chemotherapy |
| EP2023935A1 * | Jun 1, 2007 | Feb 18, 2009 | Cylene Pharmaceuticals, Inc. | Drug administration methods |
| WO2007137000A2 * | May 9, 2007 | Nov 29, 2007 | Univ Arizona | Combination cancer chemotherapy |
| WO2007143587A1 * | Jun 1, 2007 | Dec 13, 2007 | Cylene Pharmaceuticals Inc | Drug administration methods |
GMP Question & Answer Guide
DRUG REGULATORY AFFAIRS INTERNATIONAL

GMP Question & Answer Guide
The requirements defined in the GMP Guidelines often leave room for interpretation. However, regulators worldwide (EMA, FDA, TGA etc) sometimes publish frequently asked questions on GMP. In a new ECA document these Q&As are summarized in a single source. The Q&As are structured in 4 main GMP Areas (General GMPs, GMP for APIs, GMP for Medicinal Products, GMP for IMPs). The document contains 150 pages of Q&As and is available at no cost on the ECA Webpage. A first set of ECA Q&As have also been included and additional GMP Q&As are planned for the future. Here you can access the GMP Questions and Answers Guide
http://www.gmp-compliance.org/eca_gmp-guide.html
| GMP Question and Answer Guide „GMP Advisor“ |
| http://www.gmp-compliance.org/eca_gmp-guide.html |
| Searching for concrete answers to GMP questions is a time-consuming activity. The document we now offer is intended to provide a single source of information. We have summarized GMP questions… |
View original post 334 more words
Still a GMP problem? Or already a criminal act? Do we need more stringent measures and enforcement in certain situations?
DRUG REGULATORY AFFAIRS INTERNATIONAL
Still a GMP problem? Or already a criminal act? Do we need more stringent measures and enforcement in certain situations?
Sometimes EU and FDA Inspectors discover serious GMP deviations and fraud during an inspection. What are the consequences and do we need to think about additional measures? Please read more in our GMP News.
When GMP issues are discussed, different interpretations are possible. Sometimes, the implementation of GMP regulations and expectations can be a challenge. However, everyone involved should do his/her best to make sure that GMP has been put in place and that patient safety is ultimately guaranteed.
Now and again, companies may receive GMP Non-Compliance Statements from EU Inspectors or Warning Letters from US FDA Inspectors because of non-compliance issues identified during inspections. This is a serious situation for the companies involved. Organisational problems and frequently also gross mismanagement can be the reasons for these deviations. In…
View original post 436 more words
Commentary Regarding new USP Chapters and for Particulate Matter Guidance
DRUG REGULATORY AFFAIRS INTERNATIONAL
Commentary Regarding new USP Chapters and for Particulate Matter Guidance
There are new chapters in the USP regarding testing of subvisible particles. Chapter Subvisible Particulate Matter in Therapeutic Protein Injections <787> became official August 1, 2014. The informational chapter <1787> was developed to support chapter <787> and will be published in USP 38 in November and become official on May 1, 2015. Read more.
During the current (2010-2015) USP Expert Committee cycle, the Dosage Forms Expert Committee has developed both new and revised general chapters that provide guidance on particulate matter content of injectable drug products. For visible particles, methods are based upon human detection sensitivity as described in Visible Particulates in Injections <790>, which applies to all sterile injectable dosage forms. For subvisible particle content, which is based upon instrumental determination, new particulate matter guidance has been established specifically for sterile injectable biotherapeutic products.
The new…
View original post 490 more words