
Home » Uncategorized (Page 104)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
(S)-Atenolol

(S)-atenolol
- Description
Selective β1 adrenoceptor antagonist
- Biological descriptionSelective β1 adrenoceptor antagonist. Orally active. Limited ability to cross the blood-brain barrier. Antihypertensive activity in vivo.
Properties
- Chemical name(S)-(-)-4-[2-Hydroxy-3-[(1-methylethyl)amino]propoxy]benzeneacetamide
- Molecular Weight 266.34
- Molecular formula C14H22N2O3
- CAS Number 93379-54-5
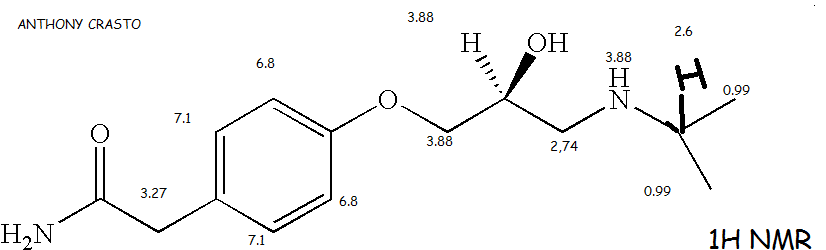
1H NMR (DMSO-d6): δ 0.99 (d, J=7 Hz, 6H, 2×CH3), 2.60 (m, 1H, CH), 2.74 (m, 2H, CH2), 3.27 (s, 2H, CH2), 3.88 (m, 4H, CH2, CH, NH), 6.83 (d, J=8 Hz, 2H, Ar—H), 7.14 (d, J=8 Hz, 2H, Ar—H), 7.40 (bs, 1H).
22.01, 22.09,
41.26, 48.39, 49.38, 67.73, 70.58, 114.16, 128.41, 129.93, 157.17, 172.59 ppm.

The compound (R,S)-atenolol (4-[2-hydroxy-3-[(1-methylethyl)amino]propoxy]-benzeneacetamide) is useful as a β-adrenegic blocker for the treatment of angina pectoris, arrhythmia and hypertension. It is known that atenolol is a 1-aryloxy-3-aminopropane-2-ol derivative wherein the hydroxy bearing carbon is an asymmetric carbon and hence exists as R- and S-isomers. It is also known that the S-isomer is particularly useful as a β-adrenegic blocker in view of its superior pharmacological activities. It is reported that S-atenolol has hypotensive activity and activity on brachycardia (A. A. Pearson, T. E. Gaffney, T. Walle, P. J. Privitera; J. Pharmacol. Exp. Ther., 250(3), 759, 1989).
In prior art, the optical resolution of racemic atenolol has been studied to obtain the desired optically active atenolol, however, any practical method has not been reported so far. It is also reported that the diastereomers of atenolol having high purity is obtained from racemic mixture by using (R,R)-O,O-di-toluoyltartaric acid anhydride (M. J. Wilson et al., J. Chromatogr. (NLD) 431 (1), 222–227, 1988). However, this method is not suitable for large scale production of optically active atenolol as it requires a large volume of solvent and further it is technically very troublesome to recycle (R,R)-O,O-di-toluoyltartaric acid anhydride.
Another method of preparing optically active atenolol has been proposed in JP-A-50-77331 and DE-A-2453324:

Wherein Z is halogen atom or sulphonyloxy group, and * means asymmetric carbon.
However, this process has some disadvantages as this process requires several steps for obtaining optically active S-atenolol stating from D-manitol; moreover the yield of S-atenolol by this process is less than 50% and the optical purity is just about 80% ee.
Another method for the preparation of S-atenolol has been reported in U.S. Pat. No. 5,223,646 which consists of reacting sodium salt of 4-carbamoylmethylphenol with R-epichlorohydrin at 0° to 35° C. to obtain an intermediate—an optically active glycidyl ether and then reacting the optically active intermediate glycidyl ether with isopropylamine to obtain S-atenolol (see also EP-435068 A2; EP-605384; JP 03077856 A2).
It has also been reported that the above procedure gives optically active glycidyl ether and atenolol of 90–96% ee optical purity. According to this report, the optical purity of atenolol may be enhanced to 98% or higher, if the intermediate optically active glycidyl ether is repeatedly recrystallised from a suitable solvent.
It has also been reported that the optically active atenolol in an optical purity of 98% or higher can be produced from atenolol of lower optical purity by converting it to its salt with Bronsted’s acid (K. Kazuhiro; T. Yosikazu; F. Yoshiro; Y. Hiroshi; O. Junzo, Chem. Pharm. Bull., 46(3), 505–507, 1998).
The separation of the atenolol salt having higher optical purity (>98% ee) is carried out by dissolving the atenolol salt having lower optical purity in a solvent, precipitating solid materials having a high content of racemic atenolol salt, and then isolating the desired atenolol salt having higher optical purity (>98% ee) by solid-liquid separation method. The optically active salt having high optical purity is then subjected to removal of acid moiety to isolate the desired optically active atenolol in free form. Though this process yields atenolol of higher optical purity, it involves salt formation and tedious separation of racemic salt from an optically active salt, which leads to the lower yields of desired optically active atenolol. Further, the salt has to be converted to free atenolol either by neutralisation or using ion exchange resins. Thus, this process gives lower overall yield of the desired optically active atenolol is low.
There is therefore a need to provide a process whereby S-atenolol may be obtained in high yield and high optical purity.
Emcure Pharmaceuticals Limited
http://www.google.com/patents/US6982349
Satish Ramanlal Mehta, Baburao Manikroa Bhawal, Vishnu Hari Deshpande, Mukund Keshav Gurjar
Accordingly, the present invention provides a process for the preparation of (S)-atenolol (1), which comprises the steps of:
-
- a) reacting a phenol of formula 2:

with an (R)-epichlorohydrin of formula (3):

in presence of an alkali metal hydroxide and a quaternary ammonium salt as phase transfer catalyst (PTC) in an aqueous solution at a temperature in a range of −10° C. to 0° C. to obtain optically active intermediate glycidyl ether of formula 4:

- b) reacting the optically active intermediate glycidyl ether (4) with isopropylamine at 10° to 40° C. to obtain (S)-atenolol of the formula 1:

in good chemical yield and high optical purity (>99 ee).
- a) reacting a phenol of formula 2:




One major advantage of this process is that S-atenolol may be obtained directly without going through the cumbersome step of recrystallization or additional salt formation step, as in the prior art.
The aqueous alkali metal hydroxide used in the process is selected from sodium hydroxide or potassium hydroxide and is used as aqueous solution in 1 to 1.5 moles to 1 mole of the phenol 2. The (R)-epichlorohydrin (3) used in the process is preferably of high optical purity and used in an amount of 1 to 3 moles, more preferably 1 to 1.6 moles, to 1 mole of phenol (2).
The quaternary ammonium salt has the formula:
R1R2R3R4N+X−
Wherein R1, R2, R3 and R4 are same or different, each an alkyl group having 1 to 16 carbon atoms (e.g. methyl, ethyl, propyl butyl etc), phenyl or benzyl, X is chlorine, bromine, iodine, hydrogen sulphate or hydroxyl group. The amount of quaternary ammonium salt used is 0.001 to 2% by weight of phenol (2).
The Applicant studied the reaction temperature extensively and found that it plays an important role in deciding optical purity of (S)-atenolol (1) formed via optically active glycidyl ether. When the reaction of phenol (2) and (R)-epichlorohydrin is carried out at 5° C. or at any other higher temperature, (S)-atenolol (1) of a lower optical purity was obtained via optically active glycidyl ether, as for example in EP 435068.
The Applicant, after studying the prior art processes found that during the course of these reactions, the phenoxide (or phenol) attacks the C-1 carbon atom of (R)-epichlorohydrin with the expulsion of chloride to yield (R)-glycidyl ether, which on reaction with isopropyl amine gives (R)-atenolol. The original epoxide ring remains unchanged in the reaction.

Thus, the reaction of phenol (2) at carbon centre C-1 of (R)-epichlorohydrin by nucleophilic displacement of chlorine leads to the formation of undesired (R)-atenolol via optically active (R)-glycidyl ether as a side product, which accounts for the low yield of optically active S-atenolol in the prior art.
The Applicant then conducted this reaction at a lower temperature and found to their surprise that S-atenolol could be obtained in high yield. The reason is that during the course of reaction, the phenoxide (or phenol) ion attacks the C-3 carbon atom of (R)-epichlorohydrin and opens the epoxide ring. The new epoxide ring formation takes place by the attack of O− on C-3 carbon with expulsion of chloride to give (S)-glycidyl ether, which on reaction with isopropyl amine gives (S)-atenolol. Thus, the reaction of phenol (2) at carbon centre C-3 of (R)-epichlorohydrin leads to the formation desired (S)-atenolol (1) as a major product via optically active glycidyl ether (4).

The lower optical purity in (S)-atenolol formation in the prior art may therefore be on account of the slow reaction rate at carbon atom 1 and the high yield of S-atenolol obtained by the process of the present invention may be due to the reaction at carbon atom 3 of (R)-epichlorohydrin (3). Both these reactions occurring on different atoms are shown as path ‘a’ and path ‘b’ in the following scheme herebelow.
Path ‘a’ is the process of the present invention whereas path ‘b’ is the process of the prior art.

EXAMPLE 1A mixture of (R)-epichlorohydrin ([α]D 25: −35.1 (neat), 138.75 g, 1.5 mole) and water (82 ml) was cooled to −7° C. and to this cold reaction mixture is added a solution of 4-hydroxyphenyl acetamide of formula 1 (151.00 g, 1 mole) and benzyltrimethylammonium chloride (1.3 g) in sodium hydroxide [40 g, 1 mole; dissolved in water (670 ml)] with stirring over a period of 3 hrs. maintaining the temperature at −7° C. to −5° C. The reaction mixture is then stirred further at −7° C. to −5° C. for 50 hrs. The precipitated solid is filtered, washed with water and dried at 60° C. to give 176 g of a mixture of S-glycidyl ether of formula 4 and S-chlorohydrin of formula 5 in about 3:2 ratio. m.p. 159–161° C.
EXAMPLE 2A mixture of isopropylamine (1.1 kg) and water (200 ml) is cooled to 10° C. and a mixture of S-glycidyl ether of formula 4 and S-chlorohydrin of formula 5 obtained in Example 1 (176 g) is added to it in lots maintaining temperature between 10 to 15° C. over a period of 3 hrs. The reaction is then stirred further for another 10 hr. The excess of isopropylamine is removed by distillation and the residue was treated with the water. The slurry so obtained is acidified with 5N HCl to pH 2.0. The resulting solution is then filtered, washed with water. The filtrate is basified with 2N NaOH to pH 11.7 and precipitated solid is filtered washed with water and dried to get (S)-atenolol (206 g, 91%) in 99.1% ee when analysed by using Chiracel OD column.
m.p. 152–153° C.
[α]D 25: −17.2 (c=1.0, 1N HCl).
IR: νmax 3352, 3168, 1635, 1242 cm−1.
1H NMR (DMSO-d6): δ 0.99 (d, J=7 Hz, 6H, 2×CH3), 2.60 (m, 1H, CH), 2.74 (m, 2H, CH2), 3.27 (s, 2H, CH2), 3.88 (m, 4H, CH2, CH, NH), 6.83 (d, J=8 Hz, 2H, Ar—H), 7.14 (d, J=8 Hz, 2H, Ar—H), 7.40 (bs, 1H).
13C NMR (DMSO-d6): 22.01, 22.09, 41.26, 48.39, 49.38, 67.73, 70.58, 114.16, 128.41, 129.93, 157.17, 172.59 ppm.
OTHER INFO
Despite both optical isomers being bioactive, as briefly mentioned in the Physical section, recent studies have shown that the S-ATENOLOL isomer was found to avoid the occasional side effect of an excessively lowered heart rate sometimes encountered with the racemate. The following steps are involved with the isolation of each enantiomer from 1-[p-[ (butoxy-carbonyl)methyl]phenoxy]-3-chloropropan-2-ol (7) using Lipase Catalysis.
The subsequent step highlights the production of S-ATENOLOL via (S-)1-[p-[ (butoxy-
carbonyl)methyl]phenoxy]-3-chloropropan-2-ol (8). This is fundamentally achieved using lipase from Pseudomonas Cepacia in a mixture of acetic anhydride and DIPE.
STEP 4 (b)

Whereas lipase from Pseudomonas Cepacia was used to isolate the S-ATENOLOL ISOMER, lipase from Candida Cylindracea is utilised in the production of the R + ATENOLOL ISOMER. The same principle applies in each isomeric scenario as demonstated below.
STEP 5 (a)

Acetic anhydride may also be used as a substitute for 1-butanol however, its inherent toxicity led to one opting for 1-butanol. Even though 1-butanol is harmful in its own right, on a relative scale is was the most suitable and effective alternative evolving an approximate 94 %conversion.
STEP 5 (b)

Yet again, greater than 95 % conversion is achieved after purifying the precipitate by treating(R) 1-[p-[(butoxy-carbonyl)methyl]phenoxy]-3-chloropropan-2-ol (10) with 2-methyl-ethanamine. This is then followed by addition of ammonium hydroxide in methanol and finally single recrystallisation in ethyl acetate.
References for (S)-(-)-Atenolol (ab120856)
This product has been referenced in:
- Agon P et al. Permeability of the blood-brain barrier for atenolol studied by positron emission tomography. J Pharm Pharmacol 43:597-600 (1991). Read more (PubMed: 1681079) »
- Tsuchihashi H et al. Characteristics of 125I-iodocyanopindolol binding to beta-adrenergic and serotonin-1B receptors of rat brain: selectivity of beta-adrenergic agents. Jpn J Pharmacol 52:195-200 (1990). Read more (PubMed: 1968985) »
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4085136 | Jun 9, 1976 | Apr 18, 1978 | Imperial Chemical Industries Limited | Adrenergic blocking agents |
| US5223646 | Apr 21, 1992 | Jun 29, 1993 | Daiso Company, Ltd. | Process for producing optically active atenolol and intermediate thereof |
| JPH0377856A | Title not available | |||
| JPH01102072A * | Title not available | |||
| JPH04198175A * | Title not available |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4085136 | Jun 9, 1976 | Apr 18, 1978 | Imperial Chemical Industries Limited | Adrenergic blocking agents |
| US5223646 | Apr 21, 1992 | Jun 29, 1993 | Daiso Company, Ltd. | Process for producing optically active atenolol and intermediate thereof |
| JPH0377856A | Title not available | |||
| JPH01102072A * | Title not available | |||
| JPH04198175A * | Title not available |
Nicox stock leaps on positive Ph III glaucoma drug data , 英文名称

- 4- (nitrooxy) butyl (5Z) -7 – {(1R, 2R, 3R, 5S) -3,5-dihydroxy-2 – [(3R) -3-hydroxy-5-phenylpentyl] cyclopentyl} hept-5- enoate
- CAS No.860005-21-6
- Formula C 27 H 41 NO 8
The firms have published top-line results from the pivotal Phase 3 studies conducted with Vesneo (latanoprostene bunod) for the reduction of intraocular pressure in patients with glaucoma or ocular hypertension. The drug is a nitric oxide-donating prostaglandin F2-alpha analog licensed by Nicox to Bausch + Lomb.
Read more at: http://www.pharmatimes.com/Article/14-09-25/Nicox_stock_leaps_on_positive_Ph_III_glaucoma_drug_data.aspx#ixzz3ETxo7SBd
prostaglandin nitrooxyderivatives, pharmaceutical compositions containing them and their use as drugs for treating glaucoma and ocular hypertension. Glaucoma is optic nerve damage, often associated with increased intraocular pressure (IOP), that leads to progressive, irreversible loss of vision. . Almost 3 million people in the United States and 14 million people worldwide have glaucoma; this is the third leading cause of blindness worldwide. Glaucoma occurs when an imbalance in production and drainage of fluid in the eye (aqueous humor) increases eye pressure to unhealthy levels. It is known that elevated IOP can be at least partially controlled by administering drugs which either‘ reduce the production of aqueous humor within the eye or increase the fluid drainage, such as beta-blockers, α- agonists, ■ ‘ cholinergic agents, carbonic anhydrase inhibitors, or prostaglandin analogs. . Several side effects are associated with the drugs conventionally used to treat glaucoma. . ■ Topical beta-blockers show serious pulmonary side effects, depression, fatigue,’ confusion, impotence, hair loss, heart failure and bradycardia. Topical -agonists have a fairly high incidence of allergic, .or toxic reactions; topical cholinergic agents (miotics) can cause visual side effects. The side effects associated with oral carbonic anhydrase inhibitors include fatigue, anorexia, depression, paresthesias and serum■ electrolyte abnormalities (The Merck Manual of Diagnosis and Therapy, Seventeenth Edition, M. H. Beers and R. Berkow Editors, Sec. 8, Ch. 100) . Finally, the topical prostaglandin analogs (bimatoprost, latanoprost, travoprost and unoprostone) ‘ used in the treatment of glaucoma, can produce ocular side effects, such as increased pigmentation of the iris, ocular irritation, conjunctival hyperaemia, iritis, uveitis and macular oedema (Martindale, Thirty-third edition, p. 1.445) U.S. Pat. No. 3,922,293 describes monocarboxyacylates of prostaglandins F-type and their 15β isomers, at the C-9 position, and processes for preparing them; U.S. Pat. No. 6,417,228 discloses 13-aza prostaglandins having functional PGF2α receptor agonist activity and their use in treating glaucoma and ocular hypertension. WO 90/02553 • discloses the use ‘ of prostaglandins derivatives of PGA, PGB, PGE and PGF, in which the omega chain contains a ring structure, for the treatment of glaucoma or ocular hypertension. WO 00/51978 describes novel nitrosated and/or nitrosylated prostaglandins, ‘ • in ‘ particular novel derivatives of PGEi, novel compositions and their use for treating sexual dysfunctions. • : U.S.- Pat. No. 5,625,083 • discloses” ‘diriitroglycerol esters of prostaglandins which may‘ be used as vasodilators, antihypertensive cardiovascular agents- or bronchodilators . U.S. Pat. No. 6,211,233 discloses compounds of the general formula A-Xι-N02,‘ wherein A contains ‘a ■■ – prostaglandin residue, .in ‘particular .‘PGEi, and Xi • is a bivalent connecting bridge; .’and their use fo ‘ treating impotence. It is an object of the present invention to provide new derivatives of prostaglandins able not only to eliminate or at least reduce the side ■ effects associated with these compounds, but also to possess an improved pharmacological activity. It has been surprisingly found that prostaglandin nitroderivatives have a significantly improved overall profile as compared to native, prostaglandins both in terms of -wider pharmacological .activity and enhanced tolerability. In particular, it has been recognized that the prostaglandin nitroderivatives of the present invention can be employed for treating glaucoma and ocular hypertension. The compounds of the present invention are indicated for the reduction of intraocular pressure in patients with open-angle glaucoma or with chronic angle- closure glaucoma who underwent peripheral iridotomy or laser iridoplasty.
Latanoprostene bunod
 Currently in Phase 3 clinical development with Nicox’s partner Bausch + Lomb
Currently in Phase 3 clinical development with Nicox’s partner Bausch + Lomb
Latanoprostene bunod is a nitric oxide-donating prostaglandin F2-alpha analog in Phase 3 clinical development for the reduction of intraocular pressure in patients with glaucoma and ocular hypertension. It was licensed to Bausch + Lomb by Nicox in March 2010
Bausch + Lomb initiated a global Phase 3 program for latanoprostene bunod (previously known as BOL-303259-X and NCX 116) in January 2013. This pivotal Phase 3 program includes two separate randomized, multicentre, double-masked, parallel-group clinical studies, APOLLO andLUNAR, designed to compare the efficacy and safety of latanoprostene bunod administered once daily (QD) with timolol maleate 0.5% administered twice daily (BID) in lowering intraocular pressure (IOP) in patients with open-angle glaucoma or ocular hypertension.
The primary endpoint of both studies, which will include a combined total of approximately 800 patients, is the reduction in mean IOP measured at specified time points during three months of treatment. The Phase 3 studies are pivotal for U.S. registration and will be conducted in North America and Europe.
In July 2013, Bausch + Lomb initiated two additional studies in Japan: JUPITER (Phase 3) and KRONUS (Phase 1). A confirmatory efficacy study is expected to be required for the Japanese registration of latanoprostene bunod.
Phase 2b top-line results
A phase 2b study conducted by Bausch + Lomb with latanoprostene bunod met its primary efficacy endpoint and showed positive results on a number of secondary endpoints, including responder rate.
No new class of drugs has come to market for treating glaucoma since 1996, when the FDA approved the first prostaglandin analogue, latanoprost (Xalatan). That could change soon: Experts who follow drug development are hopeful that we’re on the brink of reaping the benefits of years of research.
“It’s been a decade and a half and counting since we’ve had new class of drugs to treat glaucoma. We’ve had formulary improvements and fixed combinations, but no novel agents,” said Louis B. Cantor, MD, at Indiana University. “We’ve gone through a long dry spell but are just beginning to see, in the last couple of years, exploration by pharma of some new types of drugs.” But, he added, “We don t know how well those will pan out.
The uncertainty about “panning out” involves both drug efficacy and marketplace issues. As Dr. Cantor said, “Prostaglandin analogues are pretty effective. For a company to go into the investment of developing a new class of drugs for glaucoma, they have to be better than prostaglandin analogues.
Andrew G. Iwach, MD, at the University of California, San Francisco, agreed: “This is a unique time period for glaucoma medications in that we have very good drugs, usually well tolerated. And they’ve gone generic. That’s important, because having such strong generic contenders out there makes it harder for drug companies to try to introduce new molecules into this arena. Specifically, the prostaglandin analogues have set a high bar. It’s hard to compete with them.
Given this barrier, what are the marketplace incentives for development? Sheer numbers, for a start: Ten thousand people a day turn 65, and this rate will continue for 18 years, Dr. Cantor said. “The number of people who are going to need treatment for glaucoma has already begun to increase substantially.
Even more important, “Despite all the advances, our medical therapy fails not only for compliance reasons, but just fails,” Dr. Cantor said. “We need to continue to have new alternatives for treatment that are more effective, that last longer, and that have simple dosing requirements.
Thus, any new drug that makes it from the bench to the clinic will be a welcome addition. “Obviously, we want new and better therapies. We still have no cure for glaucoma. And while half of all patients are treatable with one drug, half are not. So we still need additional therapies to treat glaucoma,” said Gary D. Novack, PhD, president of Pharmalogic Development.
……………………….
http://www.google.com/patents/EP1704141A1?cl=en
EXAMPLE 1 Synthesis of [1R- [l (Z) , 2α (R*) , 3α, 5α] ] -7- [3, 5-dihydroxy-2- (3-hydroxy-5-phenylpentyl) cyclopentyl] -5-heptenoic acid 4- (nitrooxy) butyl ester (compound 1)
I Synthetic Pathway ONO,
MW 72.11 MW 153.02 MW 198.02

MW 390.51 MW 507.62
II EXPERIMENTAL II.1 Preparation of 4-bromobutanol
Tetrahydrofuran (12.5 g – 173 mmol) was charged under nitrogen in a reactor cooled to 5-10 °C. Hydrogen bromide (7.0 g. – 86.5 mmol) was then added slowly and the reaction ■medium was stirred over a period of 4.5 hours at 5-10°C. The mixture was diluted with 22.5 g of cold water and the pH of this solution was adjusted to pH=5-7 by adding 27.65% sodium hydroxide (2.0 g) keeping the temperature at 5-10 °C. The solution was then extracted twice with dichloromethane (13.25 g) . The combined organic phases were washed with -25% brine (7.5 g) , adjusted to pH=6-7 with 27.65% sodium hydroxide and dried over magnesium sulfate. Dichloromethane was distilled off and crude 4-bromobutanol (10.3 g – 66.9 mmol) was obtained in a yield of about 77%. II.2 Preparation of 4-bromobutyl nitrate
In reactor cooled to -5 to 5°C, nitric acid fuming (8.5 g – 135 mmol) was slowly added to a solution of 98% sulfuric acid (13.0 g – 130 mmol) in dichloromethane (18.0 g – 212 mmol). 4-bromobutanol (10.2 g – 66.6 mmol) was then added to this mixture and the reaction medium was stirred at -5 to 5°C over a period of 2-5 hours. The mixture was poured into cold water (110 g) keeping the temperature between -5 °C and 3°C. After decantation, the upper aqueous phase was extracted with dichloromethane and the combined organic phases were washed with water, adjusted to pH=6-7 by addition of 27.65% sodium hydroxide, washed with brine and dried over magnesium sulfate. Dichloromethane was distilled off under vacuum and crude 4-bromobutyl nitrate (12.7 g – 64.1 mmol) was recovered in a yield of about 96%.
II.3 Preparation of [1R- [lα-(Z) , 2β (R*) , 3α, 5α] ] -7- [3, 5- dihydroxy-2- (3-hydroxy-5-phenylpentyl) cyclopentyl] -5- heptenoic acid 4- (nitrooxy) butyl ester
Latanoprost acid (97.7%, S-isomer <1%) (213mg, 0.54 mmol) was dis.solved in 5.0 g anhydrous DMF. K2C03 (206′ mg, 1.49 mmol), KI (77 mg, 0.46 mmol)‘ and ‘4-bromobutylnitrate (805 mg, .25% w/w in methylene chloride, 1.02 mmol) were added. The reaction mixture was heated and stirred on a rotary evaporator at 45-50°C. fter 1.5. hour, TLC (Si, ■ CH2Cl2-MeOH, 5%) showed -no – starting acid. . . .. The reaction mixture was diluted with 100 ml ethyl acetate, washed with brine (3 x 50 ml), dried over MgS04 and evaporated to give yellowish oil (420 mg) .
5 1H NMR/13C NMR showed target molecule as a major product together with some starting 4-bromobutylnitrate and DMF. HPLC showed no starting acid. Residual solvent, 4- bromobutylnitrate and target ester were the main peaks. Butylnitrate ester showed similar UV spectrum as0 latanoprost and relative retention time was as expected.
Instrument: Bruker 300 MHz Solvent : CDC13 -5 H-NMR (CDC13) δ: 7.29-7.19 (5H, m, Ar) ; 5.45 (IH, m. CH=CH) ; 5.38 (IH, m, CH=CH) ;. 4.48 (2H, t, CH2-ON02) ; 4.18 (IH, m, CH-OH); 4.10 (2H, t, C00CH2) ; 3.95 (IH, m, CH-OH); 3.68 (IH, m, CH-OH); 2.87-2.60 (2H, ) ; 2.35 (2H, t) ; 2.25 (2H,m) ; 2.13 (2H,m) ; 1.90-1.35 (16H, m) .0 13C-NMR (CDCI3) ppm: 173.94 (C=0) ; 142.14; 129.55 (C5); 129.50 (C6) ; 128.50; 125.93 78.80 (Cu) ; 74.50 (C9) ; 72.70 (C-0N02) ; 71.39 (Ci5) ; 63.57; 52.99 (C12) 51.99 (C8); 41.30 (C10) ; 39.16 (Ci6) ; 33.66; 32.21; 29.73; 27.04; 26.70;5 25.04; 24.91; 23.72; 15.37.
 |
|
| Trabecular meshwork structure. The colors in this drawing delineate the layers of the TM. |
 |
|
| Hyperemia. A side effect that emerged in trials of ROCK inhibitors is hyperemia; researchers are exploring different strategies to reduce it. |
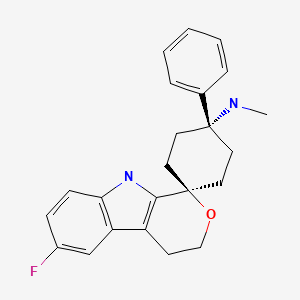
LEXANOPADOL, For Treatment of acute and chronic pain requiring opioid analgesia
LEXANOPADOL
trans-6′-Fluoro-N-methyl-4-phenyl-4′,9′-dihydro-3’H-spiro(cyclohexane-1,1′-pyrano(3,4-b)indol)-4-amine
PRONUNCIATION lex” an oh’ pa dol
THERAPEUTIC CLAIM Treatment of acute and chronic pain requiring opioid analgesia
CHEMICAL NAMES
1. Spiro[cyclohexane-1,1′(3’H)-pyrano[3,4-b]indol]-4-amine, 6′-fluoro-4′,9′-dihydro-N-methyl-4-phenyl-, trans-
2. Trans-6′-fluoro-N-methyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4-b]indol]-4-amine
3. Trans -6’-fluoro-4’,9’-dihydro-N-methyl-4-phenyl-spiro[cyclohexane-1,1’(3’H)-pyrano[3,4-b]indol]-4-amine
MOLECULAR FORMULA C23H25FN2O
MOLECULAR WEIGHT 364.5
SPONSOR Grűnenthal GmbH
CODE DESIGNATIONS GRT6006, GRT13106G
CAS REGISTRY NUMBER 1357348-09-4
UNIIDZ4NDW1LZX
WHO NUMBER 9765
gbk
The heptadecapeptide nociceptin is an endogenous ligand of the ORL1 (opioid receptor-like) receptor (Meunier et al., Nature 377, 1995, p. 532-535), which belongs to the family of opioid receptors and is to be found in many regions of the brain and spinal cord, and has a high affinity for the ORL1 receptor. The ORL1 receptor is homologous to the μ, κ and δ opioid receptors and the amino acid sequence of the nociceptin peptide has a marked similarity to those of the known opioid peptides. The receptor activation induced by nociceptin leads, via coupling with Gi/o proteins, to an inhibition of adenylate cyclase (Meunier et al., Nature 377, 1995, p. 532-535).
The nociceptin peptide shows a pronociceptive and hyperalgesic activity after intercerebroventicular administration in various animal models (Reinscheid et al., Science 270, 1995, p. 792-794). These findings can be explained as an inhibition of stress-induced analgesia (Mogil et al., Neuroscience 75, 1996, p. 333-337). In this connection, it has also been possible to demonstrate an anxiolytic activity of nociceptin (Jenck et al., Proc. Natl. Acad. Sci. USA 94, 1997, 14854-14858).
On the other hand, it has also been possible to demonstrate an antinociceptive effect of nociceptin in various animal models, in particular after intrathecal administration. Nociceptin has an antinociceptive action in various pain models, for example in the tail flick test in the mouse (King et al., Neurosci. Lett., 223, 1997, 113-116. It has likewise been possible to demonstrate an antinociceptive action of nociceptin in models for neuropathic pain, which is of particular interest inasmuch as the activity of nociceptin increases after axotomy of spinal nerves. This is in contrast to conventional opioids, the activity of which decreases under these conditions (Abdulla and Smith, J. Neurosci., 18, 1998, p. 9685-9694).
The ORL1 receptor is moreover also involved in regulation of further physiological and pathophysiological processes. These include, inter alia, learning and memory development (Manabe et al., Nature, 394, 1997, p. 577-581), audition (Nishi et al., EMBO J., 16, 1997, p. 1858-1864) and numerous further processes. A review article by Cabo et al. (Br. J. Pharmacol., 129, 2000, 1261-1283) gives an overview of the indications or biological processes in which the ORL1 receptor plays a role or with high probability could play a role. This mentions, inter alia: analgesia, stimulation and regulation of food intake, influence on μ-agonists, such as morphine, treatment of withdrawal symptoms, reduction in the addiction potential of opioids, anxiolysis, modulation of motor activity, impaired memory, epilepsy; modulation of neurotransmitter secretion, in particular glutamate, serotonin and dopamine, and therefore neurodegenerative diseases; influencing of the cardiovascular system, initiation of an erection, diuresis, anti-natriuresis, electrolyte balance, arterial blood pressure, water retention diseases, intestinal motility (diarrhea), relaxing effects on the respiratory tract, micturation reflex (urinary incontinence). The use of agonists and antagonists as anoretics, analgesics (also in co-administration with opioids) or nootropics is furthermore discussed.
The possible uses of compounds which bind to the ORL1 receptor and activate or inhibit this are correspondingly diverse. Alongside this, however, opioid receptors, such as the μ-receptor, but also the other sub-types of these opioid receptors, namely δ and κ, play a large role precisely in the area of pain therapy, but also in that of other indications of those mentioned. Accordingly, it is favourable if the compound also show an action on these opioid receptors.
SYN
http://www.google.com/patents/US20110319440

SYNTHESIS ……………..ON THE WAY ….. WATCH OUT
The dimethyl analogue is

Jirkovsky et al., J. Heterocycl. Chem., 12, 1975, 937-940;
Campaigne et al., J. Heterocycl. Chem., 2, 1965, 231-235;
Efange et al., J. Med. Chem., 41, 1998, 4486-4491;
Ellingboe et al., J. Med. Chem., 35, 1992, 1176-1183;
Pearson et al., Aust. J. Chem., 44, 1991, 907-917;
Yokohama et al., Chem. Pharm. Bull., 40, 1992, 2391-2398;
Beck et al., J. Chem. Soc. Perkin 1, 1992, 813-822;
Shinada et al., Tetrahedron Lett., 39, 1996, 7099-7102;
Garden et al., Tetrahedron, 58, 2002, 8399-8412;
Lednicer et al., J. Med. Chem., 23, 1980, 424-430.
|
2-10-2012
|
Pharmaceutical dosage forms comprising 6′-fluoro-(N-methyl- or N,N-dimethyl-)-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine
|
|
|
11-9-2011
|
Compositions containing spirocyclic cyclohexane compounds
|
|
|
10-12-2011
|
Spirocyclic Cyclohexane Compounds Useful To Treat Substance Dependency
|
|
|
5-32-2011
|
Spirocyclic Cyclohexane Compounds
|
|
|
1-21-2011
|
MIXED ORL1/MU-AGONISTS FOR THE TREATMENT OF PAIN
|
|
|
9-22-2010
|
SPIROCYCLIC CYCLOHEXANE COMPOUNDS
|
|
|
6-17-2009
|
Spirocyclic cyclohexane compounds
|
|
|
9-12-2008
|
Spirocyclic Cyclohexane Compounds Useful To Treat Substance Dependency
|
|
|
5-30-2008
|
Mixed ORL1/mu-agonists for the treatment of pain
|
| US8614245 | Jan 8, 2013 | Dec 24, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US8618156 * | Jul 6, 2012 | Dec 31, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US8765800 | Mar 15, 2013 | Jul 1, 2014 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US20130231381 * | Mar 15, 2013 | Sep 5, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US5356896 * | Dec 22, 1992 | Oct 18, 1994 | Sandoz Ltd. | Alkaline stabiling medium |
| US20060004034 * | May 11, 2005 | Jan 5, 2006 | Gruenenthal Gmbh | Treating conditions associated with the nociceptin/ORL1 receptor system, e.g. pain, drug withdrawal, anxiety, muscle relaxants, anxiolytic agents; e.g. 1,1-[3-dimethylamino-3-(pyridin-2-yl)pentamethylene]-3,4-dihydro-1H-2,9-diazafluorene |

MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
amcrasto@gmail.com 
keep watching for synthesis update on this drug
QP Declaration: EMA publishes Comments
DRUG REGULATORY AFFAIRS INTERNATIONAL
QP Declaration: EMA publishes Comments
More than three years ago, the EMA has published two draft documents for a template for the QP’s declaration concerning GMP compliance of the API used as starting material and verification of its supply chain called “The QP declaration template“:
1. The draft template for the Qualified Person’s declaration
and
2. the respective draft Q&A on the template for the Qualified Person’s declaration
The QP Declaration should be provided in support of an application for a new marketing authorisation, variation or renewal of a medicinal product(s) authorised in the Community, using EU or national procedures within the scope of the respective Directives.
The consultation for the template ended on 30 April 2011. In June 2014, the final version was published together with a template guidance. Now, three months after publication of the final document, the comments from 2011 have been published.
The…
View original post 559 more words
WHO publishes New Version of the Draft on “Hold-Time” Studies
DRUG REGULATORY AFFAIRS INTERNATIONAL
WHO publishes New Version of the Draft on “Hold-Time” Studies
The 2nd revision for comment was published already in February this year (we reported). Now, a 3rd version is available – also for comment. The document describes the design of hold-time studies for the determination of time limits which have to be determined according to the generally applicable intermediate and bulk products. This should avoid that the storage of intermediate or bulk products from having any negative influence on their quality or the quality of a finished before processing to the next stage.
Chapter 2 which defines what intermediate and bulk products are has been added. It is now explicitly pointed out that hold-time investigations are part of the process validation. In turn, the reference to retrospective observation has been removed from the current version as well as – fortunately – the incomprehensible paragraph on the ‚most probable /…
View original post 113 more words
How to identify Out-of-Trend Results in Stability Studies?
DRUG REGULATORY AFFAIRS INTERNATIONAL

How to identify Out-of-Trend Results in Stability Studies?
http://www.gmp-compliance.org/enews_4522_How-to-identify-Out-of-Trend-Results-in-Stability-Studies_8360,8348,8430,Z-QCM_n.html
An article in PharmTech from June 2013 (by Trajkovic-Jolevska et. al) deals with the methods to identify Out-of-Trend (OOT) results in ongoing stability studies.
With regard to stability studies, it is important to make the difference between Out-of-Specification (OOS) and Out-of-Trend (OOT). Both the pharmaceutical industry and authorities often misuse these two terms.
The article defines OOT results as those results which don’t follow the expected trend, either in comparison with other stability batches or compared to previous results collected during a stability study. OOT results aren’t necessarily OOS, but they don’t look like a typical data point.
Although OOT results are a serious problem, neither the scientific literature nor regulatory guidelines fully address them.
The aim of the study described in this Pharmtech article by Trajkovic-Jolevska et. al was to perform a statistical evaluation of the statistical methods used in…
View original post 112 more words
Pirenperone, R 47465

ON THE LEFT OR ABOVE
IS
3-(2- {4-[(4-fluorophenyl)carbonyl]piperidin- 1 -yl} ethyl)-2-methyl-4H-pyrido[ 1 ,2- α]pyrimidin-4-one
3-(2-{4-[(4-fluorophenyl)carbonyl]piperidin-1-yl}ethyl)-2-methyl-4H-pyrido[1,2-α]pyrimidin-4-one
3-[2-[4-(4-fluorobenzoyl)-1-piperidinyl]ethyl]-2-methyl-4H-pyrido[1,2-a]pyri- midin-4-one
pirenperone CAS : 75444-65-4
- C23 H24 F N3 O2
- 4H-Pyrido[1,2-a]pyrimidin-4-one, 3-[2-[4-(4-fluorobenzoyl)-1-piperidinyl]ethyl]-2-methyl-
- R 47465
Cardiovascular disease; Inflammatory disease; Neoplasm; Pain
Calcium channel modulator T-type
……………………………..
http://www.google.co.in/patents/US4342870
http://www.google.co.in/patents/EP0037265A1
Example XXIV
-
A solution of 2 parts of 3-[2-[4-(4-fluorobenzoyl)-1 -piperidinyl]ethyl]-2 -methyl-4H -pyricio[1, 2 -a]pyrimidin-4-one in 64 parts of 2-propanol is warm acidified with 2-propanol saturated with hydrogen chloride. The formed hydrochloride salt is allowed to crystallize. It is filtered off and dried, yielding 2 parts (85. 5%) of 3-[2-[4-(4-fluorobenzoyl)-1-piperidinyl]ethyl]-2-methyl-4H-pyrido[1,2-a]pyri- midin-4-one dihydrochloride; mp. + 300°C.
-
In a similar manner there are also prepared:
- 3-[2 -[4-(4-fluorobenzoyl)-1-piperidiny]yethyl7-2 -methyl-4H-pyrido-[1, 2 -a]pyrimidin-4-one sulfate (1 : 2); mp. 254. 7°C; and
- 3-[2-[4-(4-fluorobenzoyl)-1 -piperidinyl]ethyl]-2-methyl-4H-pyrido-[1, 2-a]pyrimidin-4-one phosphate (1 : 2) ; mp. 243.8°C.
WO-2014143915
http://www.google.com/patents/WO2014143915A1?cl=en
Novelcrystalline polymorphic forms of pirenperone, useful for treating disorders associated with T-type calcium ion channels such as pain syndrome, neoplasm, cardiovascular disease or inflammation. VM Discovery, from which VM Therapeutics was spun out, was investigating the VMD-C300 series of compounds which act as T-type calcium channel modulators, including VMD-3816 and VMD-3222, for treating cancer, pain, neurological diseases and cardiovascular diseases; but as of September 2014, this program was assumed to be discontinued. See WO2009108798, (by the inventor, assigned to VM Discovery) claiming use of the same compound for treating same indications.
It was first disclosed in the now-expired US Patent No. 4,342,870 (Claim 5), and intended to be used as potential anti-anxiety drug. However, the early human clinical studies has shown that the compound did not show any dose-related anti-anxiety effects as hoped, but otherwise the compound was safe in human (ref. Ansseau M, Doumo t A, Thlry D, Gelders Y. “Pilot study of a specific serotonergic antagonist, pirenperone, in the treatment of anxiety disorders”, Acta Psychiatr Belg, 1983 Sep-Oct;83(5):517-24). in the US Patent No. 4,342,870, there is no crystalline polymorphic form disclosed, nor disclosure of potential uses for management of pain and treatment of other related diseases or disorders.
It was further disclosed in the PCT patent application WO/2009/108798 as “Compound 10 (pirenperone)‘” to be used for novel T-type calcium ion channel antagonist for management of pain and treatment of other diseases or disorders associated to the T-type calcium ion channels.
Surprisingly, we have found that there are many crystalline polymorphic forms of this compound which may affect the compound’s pharmaceutical safety and pharmacology properties.
MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/
A CASE OF ICHCHTHYOSIS ; A TYPICAL SKIN DISORDER ; E.T.G AYURVEDASCAN TEST EVALUATION ; “इक्थियासिस” जैसे लाइलाज चर्म रोग का ई०टी०जी० आयुर्वेदास्कैन आधारित आन्कलन
चर्म रोग ICHCHTHYOSIS या इख्तोयासिस एक तरह की ऐसी तकलीफ है जो त्वचा के टिश्यूज से जुड़ी हुयी बीमारी है / इस बीमारी मे त्वचा का रन्ग काला पड़ जाता है और त्वचा मोटी हो जाती है / इसके अलावा त्वचा का रन्ग काला और वर्ण cracks यानी फटी हुयी और आकार मछली की खाल जैसा हो जाता है /
जिस मरीज का नीचे दिया गया चित्र है उसे लगभग चार साल से यह तकलीफ रही है / अन्ग्रेजी और देशी और होम्योपैथी का इलाज कराने के बाद इसे आराम नही मिला / हमारे यहा से इलाज करा चुके एक मरीज द्वारा हमारे सन्स्थान मे इलाज कराने के लिये प्रोत्साहित किये जाने के बाद यह मरीज इलाज के लिये हमारे यहां आया है /
मरीज के दोनो पैरो और शरीर के लगभग सभी हिस्सो मे इसी तरह के scabs मौजूद है /.
नीचे दिये गये चित्र मे यह चर्म रोग…
View original post 339 more words
Leflunomide

Leflunomide
RS-34821, SU-101, HWA-486, Arava,75706-12-6,

- Arava
- HSDB 7289
- HWA 486
- HWA-486
- Leflunomida
- Leflunomida [INN-Spanish]
- Leflunomide
- Leflunomidum
- Leflunomidum [INN-Latin]
- SU 101 (pharmaceutical)
- SU101
- UNII-G162GK9U4W
Leflunomide (brand names: Arabloc, Arava, Lunava, Repso) is an immunosuppressive disease-modifying antirheumatic drug (DMARD),[2] used in active moderate to severe rheumatoid arthritis and psoriatic arthritis. It is a pyrimidine synthesis inhibitor.[3]

Bottle of Leflunomide (Arava) and tablet
Medical use
Rheumatoid arthritis and psoriatic arthritis are the only indications that have received regulatory approval.[1][4] Clinical studies regarding the following diseases have been conducted:[5]
- Polyoma BK Virus Nephropathy[6]
- Kimura’s disease[7]
- Systemic lupus erythematosus[8]
- Felty’s syndrome [9]
- Takayasu arteritis[10]
- Wegener’s granulomatosis[9]
- Ankylosing spondylitis[11]
- Crohn’s disease[12][13]
- Sarcoidosis[14]
- Uveitis[15]
- Still’s disease[16]
- Prostate cancer[17]
- Pemphigoid[18]
- Prevention of organ transplant rejection[19]
Side effects
Its principle dose-limiting side effects are liver damage, lung disease and immunosuppression.[19] The most common side effects (occurring in >1% of those treated with it) are, in approximately descending order of frequency:[1][4][20][21][22][23][24] diarrhoea, respiratory tract infections, hair loss, high blood pressure, rash, nausea, bronchitis, headache, abdominal pain, abnormal liver function tests, back pain, indigestion, urinary tract infection, dizziness, infection, joint disorder, itchiness, weight loss, loss of appetite, cough, gastroenteritis, pharyngitis, stomatitis, tenosynovitis, vomiting, weakness, allergic reaction, chest pain, dry skin, eczema,paraesthesia, pneumonia, rhinitis, synovitis, cholelithiasis and shortness of breath. Whereas uncommon side effects (occurring in 0.1-1% of those treated with the drug) include:[4] constipation, oral thrush, stomatitis, taste disturbance, thrombocytopenia and hives. Rarely (in 0.1% of those treated with it) it can cause:[4] anaphylaxis, angiooedema, anaemia, agranulocytosis, eosinophilia,leucopenia, pancytopenia, vasculitis, toxic epidermal necrolysis, Stevens-Johnson syndrome, cutaneous lupus erythematosus, severe infection, interstitial lung disease, cirrhosis and liver failure.
Contraindications
Contraindications include:[1]
- Pregnancy, women of childbearing potential (unless contraception used)
- Liver disease, hepatitis B/Cseropositive
- Active serious infections
- Hypersensitivity
Interactions
Other immunomodulatory treatments should be avoided due to the potential for additive immunosuppressant effects, or in the case of immunostimulants like echinacea or astragalus, reduced therapeutic effects.[1] Likewise live vaccines (like haemophilus influenzae type b vaccine and yellow fever vaccines) should be avoided due to the potential for severe infection due to the immunosuppressive nature of the treatment.[1]
The concomitant use of methotrexate, in particular, may lead to severe or even fatal liver- or hepatotoxicity. Seventy-five percent of all cases of severe liver damage reported until early 2001 were seen under combined drug therapy leflunomide plus methotrexate.[25]However, some studies have shown that the combination of methotrexate and leflunomide in patients with rheumatoid arthritis gave better results than either drug alone.[25]
Mechanism of action
Leflunomide is an immunomodulatory drug that achieves its effects by inhibiting the mitochondrial enzyme dihydroorotate dehydrogenase(an enzyme involved in de novo pyrimidine synthesis) (abbreviation DHODH), which plays a key role in the de novo (from scratch) synthesis of the uridine monophosphate (rUMP), which is required for the synthesis of DNA and RNA, hence leflunomide inhibits the reproduction of rapidly dividing cells, especially lymphocytes.[19] The inhibition of human DHODH by teriflunomide, the active metabolite of leflunomide, occurs at levels (approximately 600 nM) that are achieved during treatment of rheumatoid arthritis (RA).[26] Teriflunomide also inhibits several tyrosine kinases.[19] Teriflunomide prevents the expansion of activated and autoimmune lymphocytes by interfering with their cell cycle progression while nonlymphoid cells are able to use another pathway to make their ribonucleotides by use of salvage pyrimidine pathway, which makes them less dependent on de novo synthesis.[26] Teriflunomide also has antiviral effects against numerous viruses including CMV, HSV1 and the BK virus, which it achieves by inhibiting viral replication by interfering with nucleocapsid tegumentation and hence virion assembly.[19]
Pharmacokinetics
It has an oral bioavailability of 80%, protein binding of >99%, metabolism sites of the GI mucosa and liver, volume of distribution (Vd) of 0.13 L/kg, elimination half-life of 14-18 days and excretion routes of faeces (48%) and urine (43%).[19][1][20]
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 5-methyl-N-[4-(trifluoromethyl) phenyl]-isoxazole-4-carboxamide | |
| Clinical data | |
| Trade names | Arabloc, Arava, Lunava, Repso |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a600032 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | |
| Legal status | |
| Routes | Oral (tablets) |
| Pharmacokinetic data | |
| Bioavailability | 80%[1] |
| Protein binding | >99%[1] |
| Metabolism | GI mucosa and liver[1] |
| Half-life | 14-18 days[1] |
| Excretion | Faeces (48%), urine (43%)[1] |
| Identifiers | |
| CAS number | 75706-12-6 |
| ATC code | L04AA13 |
| PubChem | CID 3899 |
| DrugBank | DB01097 |
| ChemSpider | 3762 |
| UNII | G162GK9U4W |
| KEGG | D00749 |
| ChEBI | CHEBI:6402 |
| ChEMBL | CHEMBL960 |
| Chemical data | |
| Formula | C12H9F3N2O2 |
| Mol. mass | 270.207 g/mol |
……………………………

…………………………..
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S86
Leflunomide is a pyrimidine synthase inhibitor of the DMARD-type (disease-modifying anti-rheumatic drug) marketed by Sanofi-Aventis. Unlike NSAIDs, which only deal with symptoms of rheumatoid arthritis, DMARDs target the cause of it. DMARDs are not necessarily structurally or mechanistically related. The effect of leflunomide is possibly due to its regulation of the immune system via affecting lymphocytes. Its synthesis [134] is relatively straightforward starting with a Knoevenagel condensation of ethyl acetoacetate (39) and triethyl orthoformate in the presence of acetic anhydride. The resulting ethyl ethoxymethylene acetoacetate (448) is next condensed with hydroxylamine hydrate in methanol to yield ethyl 5-methylisoxazole-4-carboxylate (449). The ethyl ester is hydrolysed under acidic conditions and the carboxylic acid activated with thionyl chloride in DMF for amide formation with 4-trifluoromethylaniline (450) (Scheme 86).
![[1860-5397-7-57-i86]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i86.png?scale=2.0&max-width=1024&background=FFFFFF)
US patent 5,494,911 discloses process for preparation of Teriflunomide in Example- 4 as shown in given below scheme-I.

References
- “Arava (leflunomide) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 11 March 2014.
- Dougados M, Emery P, Lemmel EM, Zerbini CA, Brin S, van Riel P (January 2005).“When a DMARD fails, should patients switch to sulfasalazine or add sulfasalazine to continuing leflunomide?”. Annals of the rheumatic diseases 64 (1): 44–51.doi:10.1136/ard.2003.016709. PMC 1755199. PMID 15271770.
- Pinto P, Dougados M (2006). “Leflunomide in clinical practice”. Acta reumatológica portuguesa 31 (3): 215–24. PMID 17094333.
- ^ Jump up to:a b c d Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- Jump up^ http://clinicaltrials.gov/ct2/results?term=Leflunomide
- Jump up^ Blanckaert, K; De Vriese, AS (23 September 2006). “Current recommendations for diagnosis and management of polyoma BK virus nephropathy in renal transplant recipients” (PDF). Nephrology Dialysis Transplantation 21 (12): 3364–3367.doi:10.1093/ndt/gfl404.
- Jump up^ Dai, L; Wei, XN; Zheng, DH; Mo, YQ; Pessler, F; Zhang, BY (June 2011). “Effective treatment of Kimura’s disease with leflunomide in combination with glucocorticoids.”.Clinical Rheumatology 30 (6): 859–65. doi:10.1007/s10067-011-1689-2.PMID 21286771.
- Jump up^ Wu, GC; Xu, XD; Huang, Q; Wu, H (February 2013). “Leflunomide: friend or foe for systemic lupus erythematosus?”. Rheumatology International 33 (2): 273–6.doi:10.1007/s00296-012-2508-z. PMID 22961090.
- ^ Jump up to:a b Sanders, S; Harisdangkul, V (2002). “Leflunomide for the treatment of rheumatoid arthritis and autoimmunity”. American Journal of Medical Sciences 323 (4): 190–3.doi:10.1097/00000441-200204000-00004. PMID 12003373.
- Jump up^ Unizony, S; Stone, JH; Stone, JR (January 2013). “New treatment strategies in large-vessel vasculitis.”. Current Opinion in Rheumatology 25 (1): 3–9.doi:10.1097/BOR.0b013e32835b133a. PMID 23114585.
- Jump up^ Haibel, H; Rudwaleit, M; Braun, J; Sieper, J (January 2005). “Six months open label trial of leflunomide in active ankylosing spondylitis.” (PDF). Annals of the Rheumatic Diseases 64 (1): 124–6. doi:10.1136/ard.2003.019174. PMC 1755172.PMID 15608310.
- Jump up^ Prajapati, DN; Knox, JF; Emmons, J; Saeian, K; Csuka, ME; Binion, DG (August 2003). “Leflunomide treatment of Crohn’s disease patients intolerant to standard immunomodulator therapy.”. Journal of Clinical Gastroenterology 37 (2): 125–8.doi:10.1097/00004836-200308000-00006. PMID 12869881.
- Jump up^ Holtmann, MH; Gerts, AL; Weinman, A; Galle, PR; Neurath, MF (April 2008). “Treatment of Crohn’s disease with leflunomide as second-line immunosuppression : a phase 1 open-label trial on efficacy, tolerability and safety.”. Digestive Diseases and Sciences 53 (4): 1025–32. doi:10.1007/s10620-007-9953-7. PMID 17934840.
- Jump up^ Panselinas, E; Judson, MA (October 2012). “Acute pulmonary exacerbations of sarcoidosis.” (PDF). Chest 142 (4): 827–36. doi:10.1378/chest.12-1060.PMID 23032450.
- Jump up^ Roy, M (August 2007). “Early clinical experience with leflunomide in uveitis.”. Canadian Journal of Ophthalmology 42 (4): 634. doi:10.3129/canjophthalmol.i07-085.PMID 17641721.
- Jump up^ Pirildar, T (May 2003). “Treatment of adult-onset Still’s disease with leflunomide and chloroquine combination in two patients.”. Clinical Rheumatology 22 (2): 157.doi:10.1007/s10067-002-0667-0. PMID 12740686.
- Jump up^ “Mitoxantrone and Prednisone With or Without Leflunomide in Treating Patients With Stage IV Prostate Cancer”. ClinicalTrials.gov. National Institute of Health. September 2012. Retrieved 11 March 2014.
- Jump up^ “Leflunomide Associated With Topical Corticosteroids for Bullous Pemphigoid (ARABUL)”. ClinicalTrials.gov. National Institute of Health. December 2008. Retrieved 11 March 2014.
- ^ Jump up to:a b c d e f Teschner, S; Burst, V (September 2010). “Leflunomide: a drug with a potential beyond rheumatology.”. Immunotherapy 2 (5): 637–50. doi:10.2217/imt.10.52.PMID 20874647.
- ^ Jump up to:a b “PRODUCT INFORMATION ARAVA®” (PDF). TGA eBusiness Services. sanofi-aventis australia pty ltd. 7 August 2012. Retrieved 11 March 2014.
- Jump up^ “Arava : EPAR – Product Information” (PDF). European Medicines Agency. Sanofi-Aventis Deutschland GmbH. 21 November 2013. Retrieved 11 March 2014.
- Jump up^ “Data Sheet Arava®” (PDF). Medsafe. sanofi-aventis new zealand limited. 29 June 2012. Retrieved 11 March 2014.
- Jump up^ “ARAVA (leflunomide) tablet, film coated [sanofi-aventis U.S. LLC]”. DailyMed. sanofi-aventis U.S. LLC. November 2012. Retrieved 11 March 2014.
- Jump up^ “Arava 100mg Tablets – Summary of Product Characteristic”. electronic Medicines Compendium. SANOFI. 21 February 2014. Retrieved 11 March 2014.
- ^ Jump up to:a b Lee, S.; Park, Y.; Park, J.; Kang, Y.; Nam, E.; Kim, S.; Lee, J.; Yoo, W.; Lee, S. (2009). “Combination treatment with leflunomide and methotrexate for patients with active rheumatoid arthritis”. Scandinavian journal of rheumatology 38 (1): 11–14.doi:10.1080/03009740802360632. PMID 19191187.
- ^ Jump up to:a b Fox, RI; Herrmann, ML; Frangou, CG; Wahl, GM; Morris, RE; Strand, V; Kirschbaum, BJ (December 1999). “Mechanism of action for leflunomide in rheumatoid arthritis.”. Clinical Immunology 93 (3): 198–208. doi:10.1006/clim.1999.4777.PMID 10600330.
External links
- National Rheumatoid Arthritis Society (NRAS) Information about Disease Modifying drugs such as Leflunomide
- http://www.arava.com/professional/home.do (full prescribing information)
- http://www.rheuma-online.de/medikamente/leflunomid-arava/studien-zu-leflunomid-arava/gibt-es-untersuchungen-zu-leflunomid-in-weiteren-einsatzgebieten.html (in German, regarding potential indications)
- http://www.arznei-telegramm.de/register/0204507.pdf (in German, regarding discontinuation of the drug)
- http://www.emea.europa.eu/pdfs/human/press/pus/561101en.pdf (warning as of 2001 regarding hepatotoxicity) (URL DEAD 16 Oct 2010)
- The safety of leflunomide Australian Prescriber
Dosages/Routes/Forms
by SANOFI AVENTIS US
| Strength | Form/Route | Marketing Status | RLD | TE Code |
|---|---|---|---|---|
| 10MG | TABLET;ORAL | 1 | 0 | AB |
| 20MG | TABLET;ORAL | 1 | 1 | AB |
| 100MG | TABLET;ORAL | 1 | 1 |
Approval History

Spectra
UV – spectrum
 |
||||
| Conditions : Concentration – 1 mg / 100 ml | ||||
| Solvent designation schedule | Methanol |
Water |
0.1 M HCl |
0.1M NaOH |
|---|---|---|---|---|
| The absorption maximum | 261 nm | 259 nm | 259 nm | Observed decay |
 |
732 | 610 | 610 | – |
| ε | 19800 | 16500 | 16500 | – |
IR – spectrum
| Wavelength (μm) |
|---|
 |
| Wavenumber (cm -1 ) |
Links
-
UV and IR Spectra. H.-W. Dibbern, RM Muller, E. Wirbitzki, 2002 ECV
-
NIST / EPA / NIH Mass Spectral Library 2008
-
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman.Academic Press, 2000.
-
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
LEFLUNOMIDE IMPURITY C [EP
5-Methyl-N-(3-(trifluoromethyl)phenyl)isoxazole-4-carboxamide
LEFLUNOMIDE IMPURITY D [EP]
5-Methylisoxazole-4-carboxylic acid
LEFLUNOMIDE IMPURITY E [EP]
3-Methyl-N-(4-(trifluoromethyl)phenyl)isoxazole-4-carboxamide
LEFLUNOMIDE IMPURITY F [EP]
5-Methyl-N-(2-(trifluoromethyl)phenyl)isoxazole-4-carboxamide
LEFLUNOMIDE IMPURITY G [EP]
5-Methyl-N-(4-methylphenyl)isoxazole-4-carboxamide
LEFLUNOMIDE IMPURITY H [EP]
2-Cyano-N-(4-(trifluoromethyl)phenyl)acetamide
