








| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 5387603 | 1993-12-01 | 2013-12-01 |
| United States | 5403847 | 1992-11-13 | 2012-11-13 |
Home » Uncategorized (Page 100)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
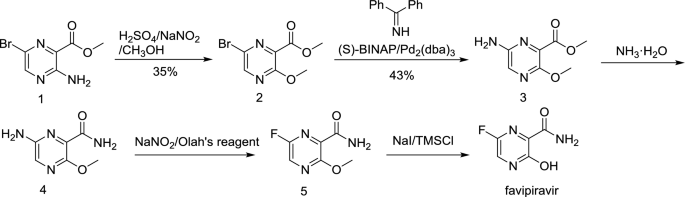
FAVIPIRAVIR, ファビピラビル

FAVIPIRAVIR
Toyama (Originator)
RNA-Directed RNA Polymerase (NS5B) Inhibitors
| Chemical Formula: | C5H4FN3O2 | |
| CAS #: | 259793-96-9 | |
| Molecular Weight: | 157.1 | |
ANTI-INFLUENZA COMPOUND |
||
| clinical trials | http://clinicaltrials.gov/search/intervention=Favipiravir | |
| Chemical Name: | 6-fluoro-3-hydroxy-2-pyrazinecarboxamide | |
| Synonyms: | T-705, T705, Favipiravir |
- Molecular FormulaC5H4FN3O2
- Average mass157.103 Da
259793-96-9 [RN]
2-Pyrazinecarboxamide, 6-fluoro-3,4-dihydro-3-oxo-
6-Fluoro-3-hydroxypyrazine-2-carboxamide
6-Fluoro-3-oxo-3,4-dihydro-2-pyrazinecarboxamide
8916
Avigan
ファビピラビル
Favipiravir
6-Fluoro-3-hydroxypyrazine-2-carboxamide
C5H4FN3O2 : 157.1
[259793-96-9]
The drug substance is a white to light yellow powder. It is sparingly soluble in acetonitrile and in methanol, and slightly soluble in water and in ethanol (99.5). It is slightly soluble at pH 2.0 to 5.5 and sparingly soluble at pH 5.5 to 6.1. The drug substance is not hygroscopic at 25°C/51% to 93%RH. The melting point is 187°C to 193°C, and the dissociation constant (pKa) is 5.1 due to the hydroxyl group of favipiravir. Measurement results on the partition ratio of favipiravir in water/octanol at 25°C indicate that favipiravir tends to be distributed in the 1-octanol phase at pH 2 to 4 and in the water phase at pH 5 to 13.
Any batch manufactured by the current manufacturing process is in Form A. The stability study does not show any change in crystal form over time; and a change from Form A to Form B is unlikely.
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 187℃ to 193℃ | https://www.pmda.go.jp/files/000210319.pdf |
| water solubility | slightly soluble in water | https://www.pmda.go.jp/files/000210319.pdf |
| pKa | 5.1 | https://www.pmda.go.jp/files/000210319.pdf |
T-705 is an RNA-directed RNA polymerase (NS5B) inhibitor which has been filed for approval in Japan for the oral treatment of influenza A (including avian and H1N1 infections) and for the treatment of influenza B infection.
The compound is a unique viral RNA polymerase inhibitor, acting on viral genetic copying to prevent its reproduction, discovered by Toyama Chemical. In 2005, Utah State University carried out various studies under its contract with the National Institute of Allergy and Infectious Diseases (NIAID) and demonstrated that T-705 has exceptionally potent activity in mouse infection models of H5N1 avian influenza.
T-705 (Favipiravir) is an antiviral pyrazinecarboxamide-based, inhibitor of of the influenza virus with an EC90 of 1.3 to 7.7 uM (influenza A, H5N1). EC90 ranges for other influenza A subtypes are 0.19-1.3 uM, 0.063-1.9 uM, and 0.5-3.1 uM for H1N1, H2N2, and H3N2, respectively. T-705 also exhibits activity against type B and C viruses, with EC90s of 0.25-0.57 uM and 0.19-0.36 uM, respectively. (1) Additionally, T-705 has broad activity against arenavirus, bunyavirus, foot-and-mouth disease virus, and West Nile virus with EC50s ranging from 5 to 300 uM.
Studies show that T-705 ribofuranosyl triphosphate is the active form of T-705 and acts like purines or purine nucleosides in cells and does not inhibit DNA synthesis
In 2012, MediVector was awarded a contract from the U.S. Department of Defense’s (DOD) Joint Project Manager Transformational Medical Technologies (JPM-TMT) to further develop T-705 (favipiravir), a broad-spectrum therapeutic against multiple influenza viruses.
Several novel anti-influenza compounds are in various phases of clinical development. One of these, T-705 (favipiravir), has a mechanism of action that is not fully understood but is suggested to target influenza virus RNA-dependent RNA polymerase. We investigated the mechanism of T-705 activity against influenza A (H1N1) viruses by applying selective drug pressure over multiple sequential passages in MDCK cells. We found that T-705 treatment did not select specific mutations in potential target proteins, including PB1, PB2, PA, and NP. Phenotypic assays based on cell viability confirmed that no T-705-resistant variants were selected. In the presence of T-705, titers of infectious virus decreased significantly (P < 0.0001) during serial passage in MDCK cells inoculated with seasonal influenza A (H1N1) viruses at a low multiplicity of infection (MOI; 0.0001 PFU/cell) or with 2009 pandemic H1N1 viruses at a high MOI (10 PFU/cell). There was no corresponding decrease in the number of viral RNA copies; therefore, specific virus infectivity (the ratio of infectious virus yield to viral RNA copy number) was reduced. Sequence analysis showed enrichment of G→A and C→T transversion mutations, increased mutation frequency, and a shift of the nucleotide profiles of individual NP gene clones under drug selection pressure. Our results demonstrate that T-705 induces a high rate of mutation that generates a nonviable viral phenotype and that lethal mutagenesis is a key antiviral mechanism of T-705. Our findings also explain the broad spectrum of activity of T-705 against viruses of multiple families.
Favipiravir, also known as T-705, Avigan, or favilavir is an antiviral drug being developed by Toyama Chemical (Fujifilm group) of Japan with activity against many RNA viruses. Like certain other experimental antiviral drugs (T-1105 and T-1106), it is a pyrazinecarboxamide derivative. In experiments conducted in animals Favipiravir has shown activity against influenza viruses, West Nile virus, yellow fever virus, foot-and-mouth disease virus as well as other flaviviruses, arenaviruses, bunyaviruses and alphaviruses.[1]Activity against enteroviruses[2] and Rift Valley fever virus has also been demonstrated.[3] Favipiravir has showed limited efficacy against Zika virus in animal studies, but was less effective than other antivirals such as MK-608.[4] The agent has also shown some efficacy against rabies,[5] and has been used experimentally in some humans infected with the virus.[6]
In February 2020 Favipiravir was being studied in China for experimental treatment of the emergent COVID-19 (novel coronavirus)disease.[7][8] On March 17 Chinese officials suggested the drug had been effective in treating COVID in Wuhan and Shenzhen.[9][10]
Discovered by Toyama Chemical Co., Ltd. in Japan, favipiravir is a modified pyrazine analog that was initially approved for therapeutic use in resistant cases of influenza.7,9 The antiviral targets RNA-dependent RNA polymerase (RdRp) enzymes, which are necessary for the transcription and replication of viral genomes.7,12,13
Not only does favipiravir inhibit replication of influenza A and B, but the drug shows promise in the treatment of influenza strains that are resistant to neuramidase inhibitors, as well as avian influenza.9,19 Favipiravir has been investigated for the treatment of life-threatening pathogens such as Ebola virus, Lassa virus, and now COVID-19.10,14,15
Mechanism of action
The mechanism of its actions is thought to be related to the selective inhibition of viral RNA-dependent RNA polymerase.[11] Other research suggests that favipiravir induces lethal RNA transversion mutations, producing a nonviable viral phenotype.[12] Favipiravir is a prodrug that is metabolized to its active form, favipiravir-ribofuranosyl-5′-triphosphate (favipiravir-RTP), available in both oral and intravenous formulations.[13][14] Human hypoxanthine guanine phosphoribosyltransferase (HGPRT) is believed to play a key role in this activation process.[15] Favipiravir does not inhibit RNA or DNA synthesis in mammalian cells and is not toxic to them.[1] In 2014, favipiravir was approved in Japan for stockpiling against influenza pandemics.[16] However, favipiravir has not been shown to be effective in primary human airway cells, casting doubt on its efficacy in influenza treatment.[17]
Approval status
In 2014, Japan approved Favipiravir for treating viral strains unresponsive to current antivirals.[18]
In March 2015, the US Food and Drug Administration completed a Phase III clinical trial studying the safety and efficacy of Favipiravir in the treatment of influenza.[19]
Ebola virus trials
Some research has been done suggesting that in mouse models Favipiravir may have efficacy against Ebola. Its efficacy against Ebola in humans is unproven.[20][21][22] During the 2014 West Africa Ebola virus outbreak, it was reported that a French nurse who contracted Ebola while volunteering for MSF in Liberia recovered after receiving a course of favipiravir.[23] A clinical trial investigating the use of favipiravir against Ebola virus disease was started in Guéckédou, Guinea, during December 2014.[24] Preliminary results showed a decrease in mortality rate in patients with low-to-moderate levels of Ebola virus in the blood, but no effect on patients with high levels of the virus, a group at a higher risk of death.[25] The trial design has been criticised by Scott Hammer and others for using only historical controls.[26] The results of this clinical trial were presented in February 2016 at the annual Conference on Retroviruses and Opportunistic Infections (CROI) by Daouda Sissoko[27] and published on March 1, 2016 in PLOS Medicine.[28]
SARS-CoV-2 virus disease
In March 2020, Chinese officials suggested Favipiravir may be effective in treating COVID-19.[29]
SYN
https://link.springer.com/article/10.1007/s11696-018-0654-9

Electronic supplementary material
Ref
https://pdfs.semanticscholar.org/be8e/cb882b99204983d2f60077c7ab8b53f4d62c.pdf
Drug Discoveries & Therapeutics. 2014; 8(3):117-120.
As a RNA polymerase inhibitor, 6-fluoro-3-hydroxypyrazine-2-carboxamide commercially named favipiravir has been proved to have potent inhibitory activity against RNA viruses in vitro and in vivo. A four-step synthesis of the compound is described in this article, amidation, nitrification, reduction and fluorination with an overall yield of about 8%. In addition, we reported the crystal structure of the title compound. The molecule is almost planar and the intramolecular O−H•••O hydrogen bond makes a 6-member ring. In the crystal, molecules are packing governed by both hydrogen bonds and stacking interactions.


2.2.1. Preparation of 3-hydroxypyrazine-2-carboxamide To a suspension of 3-hydroxypyrazine-2-carboxylic acid (1.4 g, 10 mmol) in 150 mL MeOH, SOCl2 was added dropwise at 40°C with magnetic stirring for 6 h resulting in a bright yellow solution. The reaction was then concentrated to dryness. The residue was dissolved in 50 mL 25% aqueous ammonia and stirred overnight to get a suspension. The precipitate was collected and dried. The solid yellow-brown crude product was recrystallization with 50 mL water to get the product as pale yellow crystals (1.1 g, 78%). mp = 263-265°C. 1 H-NMR (300 MHz, DMSO): δ 13.34 (brs, 1H, OH), 8.69 (s, 1H, pyrazine H), 7.93-8.11 (m, 3H, pyrazine H, CONH2). HRMS (ESI): m/z [M + H]+ calcd for C5H6N3O2 + : 140.0460; found: 140.0457.
2.2.2. Preparation of 3-hydroxy-6-nitropyrazine-2- carboxamide In the solution of 3-hydroxypyrazine-2-carboxamide (1.0 g, 7 mmol) in 6 mL concentrate sulfuric acid under ice-cooling, potassium nitrate (1.4 g, 14 mmol) was added. After stirring at 40°C for 4 h, the reaction mixture was poured into 60 mL water. The product was collected by fi ltration as yellow solid (0.62 g, 48%). mp = 250-252°C. 1 H-NMR (600 MHz, DMSO): δ 12.00- 15.00 (br, 1H, OH), 8.97 (s, 1H, pyrazine H), 8.32 (s, 1H, CONH2), 8.06 (s, 1H, CONH2). 13C-NMR (75 MHz, DMSO): δ 163.12, 156.49, 142.47, 138.20, 133.81. HRMS (ESI): m/z [M + H]+ calcd for C5H5N4O4 + : 185.0311; found: 185.0304.
2.2.3. Preparation of 6-amino-3-hydroxypyrazine-2- carboxamide 3-Hydroxy-6-nitropyrazine-2-carboxamide (0.6 g, 3.3 mmol) and a catalytic amount of raney nickel were suspended in MeOH, then hydrazine hydrate was added dropwise. The resulting solution was refl uxed 2 h, cooled, filtered with diatomite, and then MeOH is evaporated in vacuo to get the crude product as dark brown solid without further purification (0.4 g, 77%). HRMS (ESI): m/z [M + H]+ calcd for C5H7N4O2 + : 155.0569; found:155.0509.
2.2.4. Preparation of 6-fluoro-3-hydroxypyrazine-2- carboxamide To a solution of 6-amino-3-hydroxypyrazine-2- carboxamide (0.4 g, 2.6 mmol) in 3 mL 70% HFpyridine aqueous at -20°C under nitrogen atmosphere, sodium nitrate (0.35 g, 5.2 mmol) was added. After stirring 20 min, the solution was warmed to room temperature for another one hour. Then 20 mL ethyl acetate/water (1:1) were added, after separation of the upper layer, the aqueous phase is extracted with four 20 mL portions of ethyl acetate. The combined extracts are dried with anhydrous magnesium sulfate and concentrated to dryness to get crude product as oil. The crude product was purified by chromatography column as white solid (0.12 g, 30%). mp = 178-180°C. 1 H-NMR (600 MHz, DMSO): δ 12.34 (brs, 1H, OH), 8.31 (d, 1H, pyrazine H, J = 8.0 Hz), 7.44 (s, 1H, CONH2), 5.92 (s, 1H, CONH2). 13C-NMR (75 MHz, DMSO): δ 168.66, 159.69, 153.98, 150.76, 135.68. HRMS (ESI): m/z [M + H]+ calcd for C5H5FN3O2 + : 158.0366; found: 158.0360.
SEE
Chemical Papers (2019), 73(5), 1043-1051.
PAPER
Medicinal chemistry (Shariqah (United Arab Emirates)) (2018), 14(6), 595-603
http://www.eurekaselect.com/158990/article
PATENT
CN 107641106
PAPER
Chemical Papers (2017), 71(11), 2153-2158.
https://link.springer.com/article/10.1007%2Fs11696-017-0208-6

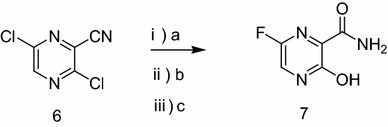
Below is the link to the electronic supplementary material.
References
- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D. F.; Barnard, D. L.; Gowen, B. B.; Julander, J. G.; Morrey, J. D. (2009). “T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections”. Antiviral Research 82 (3): 95–102. doi:10.1016/j.antiviral.2009.02.198. PMID 19428599. edit

- WO 2000010569
- WO 2008099874
- WO 201009504
- WO 2010104170
- WO 2012063931




CLIP
Influenza virus is a central virus of the cold syndrome, which has attacked human being periodically to cause many deaths amounting to tens millions. Although the number of deaths shows a tendency of decrease in the recent years owing to the improvement in hygienic and nutritive conditions, the prevalence of influenza is repeated every year, and it is apprehended that a new virus may appear to cause a wider prevalence.
For prevention of influenza virus, vaccine is used widely, in addition to which low molecular weight substances such as Amantadine and Ribavirin are also used
CLIP
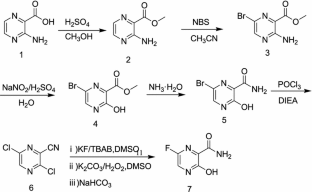
Synthesis of Favipiravir
ZHANG Tao1, KONG Lingjin1, LI Zongtao1,YUAN Hongyu1, XU Wenfang2*
(1. Shandong Qidu PharmaceuticalCo., Ltd., Linzi 255400; 2. School of Pharmacy, Shandong University, Jinan250012)
ABSTRACT: Favipiravir was synthesized from3-amino-2-pyrazinecarboxylic acid by esterification, bromination with NBS,diazotization and amination to give 6-bromo-3-hydroxypyrazine-2-carboxamide,which was subjected to chlorination with POCl3, fluorination with KF, andhydrolysis with an overall yield of about 22%.
PATENT
US6787544

| subs G1 | G2 | G3 | G4 | R2 |
| compd 32 N | CH | C—CF3 | N | H |
…………………
EP2192117
Example 1-1

To a 17.5 ml N,N-dimethylformamide solution of 5.0 g of 3,6-difluoro-2-pyrazinecarbonitrile, a 3.8 ml water solution of 7.83 g of potassium acetate was added dropwise at 25 to 35° C., and the solution was stirred at the same temperature for 2 hours. 0.38 ml of ammonia water was added to the reaction mixture, and then 15 ml of water and 0.38 g of active carbon were added. The insolubles were filtered off and the filter cake was washed with 11 ml of water. The filtrate and the washing were joined, the pH of this solution was adjusted to 9.4 with ammonia water, and 15 ml of acetone and 7.5 ml of toluene were added. Then 7.71 g of dicyclohexylamine was added dropwise and the solution was stirred at 20 to 30° C. for 45 minutes. Then 15 ml of water was added dropwise, the solution was cooled to 10° C., and the precipitate was filtered and collected to give 9.44 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyradinecarbonitrile as a lightly yellowish white solid product.
1H-NMR (DMSO-d6) δ values: 1.00-1.36 (10H, m), 1.56-1.67 (2H, m), 1.67-1.81 (4H, m), 1.91-2.07 (4H, m), 3.01-3.18 (2H, m), 8.03-8.06 (1H, m), 8.18-8.89 (1H, broad)
Example 1-2
4.11 ml of acetic acid was added at 5 to 15° C. to a 17.5 ml N,N-dimethylformamide solution of 5.0 g of 3,6-difluoro-2-pyrazinecarbonitrile. Then 7.27 g of triethylamine was added dropwise and the solution was stirred for 2 hours. 3.8 ml of water and 0.38 ml of ammonia water were added to the reaction mixture, and then 15 ml of water and 0.38 g of active carbon were added. The insolubles were filtered off and the filter cake was washed with 11 ml of water. The filtrate and the washing were joined, the pH of the joined solution was adjusted to 9.2 with ammonia water, and 15 ml of acetone and 7.5 ml of toluene were added to the solution, followed by dropwise addition of 7.71 g of dicyclohexylamine. Then 15 ml of water was added dropwise, the solution was cooled to 5° C., and the precipitate was filtered and collected to give 9.68 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile as a slightly yellowish white solid product.
Examples 2 to 5
The compounds shown in Table 1 were obtained in the same way as in Example 1-1.
| TABLE 1 | |||
 |
|||
| Example No. | Organic amine | Example No. | Organic amine |
| 2 | Dipropylamine | 4 | Dibenzylamine |
| 3 | Dibutylamine | 5 | N-benzylmethylamine |
Dipropylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) 6 values: 0.39 (6H, t, J=7.5 Hz), 1.10 (4H, sex, J=7.5 Hz), 2.30-2.38 (4H, m), 7.54 (1H, d, J=8.3 Hz)
Dibutylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) 6 values: 0.36 (6H, t, J=7.3 Hz), 0.81 (4H, sex, J=7.3 Hz), 0.99-1.10 (4H, m), 2.32-2.41 (4H, m), 7.53 (1H, d, J=8.3 Hz)
Dibenzylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) δ values: 4.17 (4H, s), 7.34-7.56 (10H, m), 8.07 (1H, d, J=8.3 Hz)
N-benzylmethylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) δ values: 2.57 (3H, s), 4.14 (2H, s), 7.37-7.53 (5H, m), 8.02-8.08 (1H, m)
Preparation Example 1

300 ml of toluene was added to a 600 ml water solution of 37.5 g of sodium hydroxide. Then 150 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile was added at 15 to 25° C. and the solution was stirred at the same temperature for 30 minutes. The water layer was separated and washed with toluene, and then 150 ml of water was added, followed by dropwise addition of 106 g of a 30% hydrogen peroxide solution at 15 to 30° C. and one-hour stirring at 20 to 30° C. Then 39 ml of hydrochloric acid was added, the seed crystals were added at 40 to 50° C., and 39 ml of hydrochloric acid was further added dropwise at the same temperature. The solution was cooled to 10° C. the precipitate was filtered and collected to give 65.6 g of 6-fluoro-3-hydroxy-2-pyrazinecarboxamide as a slightly yellowish white solid.
1H-NMR (DMSO-d6) δ values: 8.50 (1H, s), 8.51 (1H, d, J=7.8 Hz), 8.75 (1H, s), 13.41 (1H, s)
CLIP
jan 2014
Investigational flu treatment drug has broad-spectrum potential to fight multiple viruses
First patient enrolled in the North American Phase 3 clinical trials for investigational flu treatment drug
First patient enrolled in the North American Phase 3 clinical trials for investigational flu treatment drug
BioDefense Therapeutics (BD Tx)—a Joint Product Management office within the U.S. Department of Defense (DoD)—announced the first patient enrolled in the North American Phase 3 clinical trials for favipiravir (T-705a). The drug is an investigational flu treatment candidate with broad-spectrum potential being developed by BD Tx through a contract with Boston-based MediVector, Inc.
Favipiravir is a novel, antiviral compound that works differently than anti-flu drugs currently on the market. The novelty lies in the drug’s selective disruption of the viralRNA replication and transcription process within the infected cell to stop the infection cycle.
“Favipiravir has proven safe and well tolerated in previous studies,” said LTC Eric G. Midboe, Joint Product Manager for BD Tx. “This first patient signifies the start of an important phase in favipiravir’s path to U.S. Food and Drug Administration (FDA) approval for flu and lays the groundwork for future testing against other viruses of interest to the DoD.”
In providing therapeutic solutions to counter traditional, emerging, and engineered biological threats, BD Tx chose favipiravir not only because of its potential effectiveness against flu viruses, but also because of its demonstrated broad-spectrum potential against multiple viruses. In addition to testing favipiravir in the ongoing influenzaprogram, BD Tx is testing the drug’s efficacy against the Ebola virus and other viruses considered threats to service members. In laboratory testing, favipiravir was found to be effective against a wide variety of RNA viruses in infected cells and animals.
“FDA-approved, broad-spectrum therapeutics offer the fastest way to respond to dangerous and potentially lethal viruses,” said Dr. Tyler Bennett, Assistant Product Manager for BD Tx.
MediVector is overseeing the clinical trials required by the FDA to obtain drug licensure. The process requires safety data from at least 1,500 patients treated for flu at the dose and duration proposed for marketing of the drug. Currently, 150 trial sites are planned throughout the U.S.
SOURCE BioDefense Therapeutics
Malpani Y, Achary R, Kim SY, Jeong HC, Kim P, Han SB, Kim M, Lee CK, Kim JN, Jung YS.
Eur J Med Chem. 2013 Apr;62:534-44. doi: 10.1016/j.ejmech.2013.01.015. Epub 2013 Jan 29.
Eur J Med Chem. 2013 Apr;62:534-44. doi: 10.1016/j.ejmech.2013.01.015. Epub 2013 Jan 29.
| US3631036 * | Nov 4, 1969 | Dec 28, 1971 | American Home Prod | 5-amino-2 6-substituted-7h-pyrrolo(2 3-d) pyrimidines and related compounds |
| US3745161 * | Apr 20, 1970 | Jul 10, 1973 | Merck & Co Inc | Phenyl-hydroxy-pyrazine carboxylic acids and derivatives |
| US4404203 * | May 14, 1981 | Sep 13, 1983 | Warner-Lambert Company | Substituted 6-phenyl-3(2H)-pyridazinones useful as cardiotonic agents |
| US4545810 * | Mar 25, 1983 | Oct 8, 1985 | Sds Biotech Corporation | Herbicidal and plant growth regulant diphenylpyridazinones |
| US4565814 * | Jan 18, 1984 | Jan 21, 1986 | Sanofi | Pyridazine derivatives having a psychotropic action and compositions |
| US4661145 * | Sep 20, 1984 | Apr 28, 1987 | Rohm And Haas Company | Plant growth regulating 1-aryl-1,4-dihydro-4-oxo(thio)-pyridazines |
| US5420130 | May 16, 1994 | May 30, 1995 | Synthelabo | 2-aminopyrazine-5-carboxamide derivatives, their preparation and their application in therapeutics |
| US5459142 * | Aug 23, 1993 | Oct 17, 1995 | Otsuka Pharmaceutical Co., Ltd. | Pyrazinyl and piperazinyl substituted pyrazine compounds |
| US5597823 | Jun 5, 1995 | Jan 28, 1997 | Abbott Laboratories | Tricyclic substituted hexahydrobenz [e]isoindole alpha-1 adrenergic antagonists |
| US6159980 * | Sep 15, 1997 | Dec 12, 2000 | Dupont Pharmaceuticals Company | Pyrazinones and triazinones and their derivatives thereof |
| EP0023358A1 * | Jul 28, 1980 | Feb 4, 1981 | Rohm And Haas Company | Process for the preparation of pyridazine derivatives |
| GB1198688A | Title not available | |||
| HU9401512A | Title not available | |||
| JPH09216883A * | Title not available | |||
| JPS5620576A | Title not available |
- ^ Jump up to:a b Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD (June 2009). “T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections”. Antiviral Research. 82 (3): 95–102. doi:10.1016/j.antiviral.2009.02.198. PMID 19428599.
- ^ Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL (November 2013). “Favipiravir (T-705), a novel viral RNA polymerase inhibitor”. Antiviral Research. 100 (2): 446–54. doi:10.1016/j.antiviral.2013.09.015. PMC 3880838. PMID 24084488.
- ^ Caroline AL, Powell DS, Bethel LM, Oury TD, Reed DS, Hartman AL (April 2014). “Broad spectrum antiviral activity of favipiravir (T-705): protection from highly lethal inhalational Rift Valley Fever”. PLoS Neglected Tropical Diseases. 8 (4): e2790. doi:10.1371/journal.pntd.0002790. PMC 3983105. PMID 24722586.
- ^ Mumtaz N, van Kampen JJ, Reusken CB, Boucher CA, Koopmans MP (2016). “Zika Virus: Where Is the Treatment?”. Current Treatment Options in Infectious Diseases. 8 (3): 208–211. doi:10.1007/s40506-016-0083-7. PMC 4969322. PMID 27547128.
- ^ Yamada K, Noguchi K, Komeno T, Furuta Y, Nishizono A (April 2016). “Efficacy of Favipiravir (T-705) in Rabies Postexposure Prophylaxis”. The Journal of Infectious Diseases. 213 (8): 1253–61. doi:10.1093/infdis/jiv586. PMC 4799667. PMID 26655300.
- ^ Murphy J, Sifri CD, Pruitt R, Hornberger M, Bonds D, Blanton J, Ellison J, Cagnina RE, Enfield KB, Shiferaw M, Gigante C, Condori E, Gruszynski K, Wallace RM (January 2019). “Human Rabies – Virginia, 2017”. MMWR. Morbidity and Mortality Weekly Report. 67(5152): 1410–1414. doi:10.15585/mmwr.mm675152a2. PMC 6334827. PMID 30605446.
- ^ Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nature Reviews Drug Discovery 2020 Feb doi:10.1038/d41573-020-00016-0
- ^ BRIEF-Corrected-Zhejiang Hisun Pharma gets approval for clinical trial to test flu drug Favipiravir for pneumonia caused by new coronavirus. Reuters Healthcare, February 16, 2020.
- ^ [1]NHK World News ‘China: Avigan effective in tackling coronavirus’
- ^ Huaxia. Favipiravir shows good clinical efficacy in treating COVID-19: official. Xinhuanet.com, 17 March 2020
- ^ Jin Z, Smith LK, Rajwanshi VK, Kim B, Deval J (2013). “The ambiguous base-pairing and high substrate efficiency of T-705 (Favipiravir) Ribofuranosyl 5′-triphosphate towards influenza A virus polymerase”. PLOS ONE. 8 (7): e68347. Bibcode:2013PLoSO…868347J. doi:10.1371/journal.pone.0068347. PMC 3707847. PMID 23874596.
- ^ Baranovich T, Wong SS, Armstrong J, Marjuki H, Webby RJ, Webster RG, Govorkova EA (April 2013). “T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro”. Journal of Virology. 87 (7): 3741–51. doi:10.1128/JVI.02346-12. PMC 3624194. PMID 23325689.
- ^ Guedj J, Piorkowski G, Jacquot F, Madelain V, Nguyen TH, Rodallec A, et al. (March 2018). “Antiviral efficacy of favipiravir against Ebola virus: A translational study in cynomolgus macaques”. PLoS Medicine. 15 (3): e1002535. doi:10.1371/journal.pmed.1002535. PMC 5870946. PMID 29584730.
- ^ Smee DF, Hurst BL, Egawa H, Takahashi K, Kadota T, Furuta Y (October 2009). “Intracellular metabolism of favipiravir (T-705) in uninfected and influenza A (H5N1) virus-infected cells”. The Journal of Antimicrobial Chemotherapy. 64 (4): 741–6. doi:10.1093/jac/dkp274. PMC 2740635. PMID 19643775.
- ^ Naesens L, Guddat LW, Keough DT, van Kuilenburg AB, Meijer J, Vande Voorde J, Balzarini J (October 2013). “Role of human hypoxanthine guanine phosphoribosyltransferase in activation of the antiviral agent T-705 (favipiravir)”. Molecular Pharmacology. 84 (4): 615–29. doi:10.1124/mol.113.087247. PMID 23907213.
- ^ Koons C (7 August 2014). “Ebola Drug From Japan May Emerge Among Key Candidates”. Bloomberg.com.
- ^ Yoon JJ, Toots M, Lee S, Lee ME, Ludeke B, Luczo JM, et al. (August 2018). “Orally Efficacious Broad-Spectrum Ribonucleoside Analog Inhibitor of Influenza and Respiratory Syncytial Viruses”. Antimicrobial Agents and Chemotherapy. 62 (8): e00766–18. doi:10.1128/AAC.00766-18. PMC 6105843. PMID 29891600.
- ^ Hayden, Frederick. “Influenza virus polymerase inhibitors in clinical development”. Current Opinion in Infectious Diseases. doi:10.1097/QCO.0000000000000532.
- ^ “Phase 3 Efficacy and Safety Study of Favipiravir for Treatment of Uncomplicated Influenza in Adults – T705US316”. FDA. Retrieved 17 March 2020.
- ^ Gatherer D (August 2014). “The 2014 Ebola virus disease outbreak in West Africa”. The Journal of General Virology. 95 (Pt 8): 1619–24. doi:10.1099/vir.0.067199-0. PMID 24795448.
- ^ Oestereich L, Lüdtke A, Wurr S, Rieger T, Muñoz-Fontela C, Günther S (May 2014). “Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model”. Antiviral Research. 105: 17–21. doi:10.1016/j.antiviral.2014.02.014. PMID 24583123.
- ^ Smither SJ, Eastaugh LS, Steward JA, Nelson M, Lenk RP, Lever MS (April 2014). “Post-exposure efficacy of oral T-705 (Favipiravir) against inhalational Ebola virus infection in a mouse model”. Antiviral Research. 104: 153–5. doi:10.1016/j.antiviral.2014.01.012. PMID 24462697.
- ^ “First French Ebola patient leaves hospital”. Reuters. 4 October 2016.
- ^ “Guinea: Clinical Trial for Potential Ebola Treatment Started in MSF Clinic in Guinea”. AllAfrica – All the Time. Retrieved 28 December 2014.
- ^ Fink S (4 February 2015). “Ebola Drug Aids Some in a Study in West Africa”. The New York Times.
- ^ Cohen J (26 February 2015). “Results from encouraging Ebola trial scrutinized”. Science. doi:10.1126/science.aaa7912. Retrieved 21 January 2016.
- ^ “Favipiravir in Patients with Ebola Virus Disease: Early Results of the JIKI trial in Guinea | CROI Conference”. croiconference.org. Retrieved 2016-03-17.
- ^ Sissoko D, Laouenan C, Folkesson E, M’Lebing AB, Beavogui AH, Baize S, et al. (March 2016). “Experimental Treatment with Favipiravir for Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-Concept Trial in Guinea”. PLoS Medicine. 13(3): e1001967. doi:10.1371/journal.pmed.1001967. PMC 4773183. PMID 26930627.
- ^ “Japanese flu drug ‘clearly effective’ in treating coronavirus, says China”. The Guardian. 2020-03-18. Retrieved 2020-03-18.\
- Beigel J, Bray M: Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res. 2008 Apr;78(1):91-102. doi: 10.1016/j.antiviral.2008.01.003. Epub 2008 Feb 4. [PubMed:18328578]
- Hsieh HP, Hsu JT: Strategies of development of antiviral agents directed against influenza virus replication. Curr Pharm Des. 2007;13(34):3531-42. [PubMed:18220789]
- Gowen BB, Wong MH, Jung KH, Sanders AB, Mendenhall M, Bailey KW, Furuta Y, Sidwell RW: In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob Agents Chemother. 2007 Sep;51(9):3168-76. Epub 2007 Jul 2. [PubMed:17606691]
- Sidwell RW, Barnard DL, Day CW, Smee DF, Bailey KW, Wong MH, Morrey JD, Furuta Y: Efficacy of orally administered T-705 on lethal avian influenza A (H5N1) virus infections in mice. Antimicrob Agents Chemother. 2007 Mar;51(3):845-51. Epub 2006 Dec 28. [PubMed:17194832]
- Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K: Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother. 2005 Mar;49(3):981-6. [PubMed:15728892]
- Furuta Y, Takahashi K, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Watanabe Y, Narita H, Shiraki K: In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob Agents Chemother. 2002 Apr;46(4):977-81. [PubMed:11897578]
- Furuta Y, Komeno T, Nakamura T: Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(7):449-463. doi: 10.2183/pjab.93.027. [PubMed:28769016]
- Venkataraman S, Prasad BVLS, Selvarajan R: RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses. 2018 Feb 10;10(2). pii: v10020076. doi: 10.3390/v10020076. [PubMed:29439438]
- Hayden FG, Shindo N: Influenza virus polymerase inhibitors in clinical development. Curr Opin Infect Dis. 2019 Apr;32(2):176-186. doi: 10.1097/QCO.0000000000000532. [PubMed:30724789]
- Madelain V, Nguyen TH, Olivo A, de Lamballerie X, Guedj J, Taburet AM, Mentre F: Ebola Virus Infection: Review of the Pharmacokinetic and Pharmacodynamic Properties of Drugs Considered for Testing in Human Efficacy Trials. Clin Pharmacokinet. 2016 Aug;55(8):907-23. doi: 10.1007/s40262-015-0364-1. [PubMed:26798032]
- Nguyen TH, Guedj J, Anglaret X, Laouenan C, Madelain V, Taburet AM, Baize S, Sissoko D, Pastorino B, Rodallec A, Piorkowski G, Carazo S, Conde MN, Gala JL, Bore JA, Carbonnelle C, Jacquot F, Raoul H, Malvy D, de Lamballerie X, Mentre F: Favipiravir pharmacokinetics in Ebola-Infected patients of the JIKI trial reveals concentrations lower than targeted. PLoS Negl Trop Dis. 2017 Feb 23;11(2):e0005389. doi: 10.1371/journal.pntd.0005389. eCollection 2017 Feb. [PubMed:28231247]
- de Farias ST, Dos Santos Junior AP, Rego TG, Jose MV: Origin and Evolution of RNA-Dependent RNA Polymerase. Front Genet. 2017 Sep 20;8:125. doi: 10.3389/fgene.2017.00125. eCollection 2017. [PubMed:28979293]
- Shu B, Gong P: Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation. Proc Natl Acad Sci U S A. 2016 Jul 12;113(28):E4005-14. doi: 10.1073/pnas.1602591113. Epub 2016 Jun 23. [PubMed:27339134]
- Nagata T, Lefor AK, Hasegawa M, Ishii M: Favipiravir: a new medication for the Ebola virus disease pandemic. Disaster Med Public Health Prep. 2015 Feb;9(1):79-81. doi: 10.1017/dmp.2014.151. Epub 2014 Dec 29. [PubMed:25544306]
- Rosenke K, Feldmann H, Westover JB, Hanley PW, Martellaro C, Feldmann F, Saturday G, Lovaglio J, Scott DP, Furuta Y, Komeno T, Gowen BB, Safronetz D: Use of Favipiravir to Treat Lassa Virus Infection in Macaques. Emerg Infect Dis. 2018 Sep;24(9):1696-1699. doi: 10.3201/eid2409.180233. Epub 2018 Sep 17. [PubMed:29882740]
- Delang L, Abdelnabi R, Neyts J: Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antiviral Res. 2018 May;153:85-94. doi: 10.1016/j.antiviral.2018.03.003. Epub 2018 Mar 7. [PubMed:29524445]
- Nature Biotechnology: Coronavirus puts drug repurposing on the fast track [Link]
- Pharmaceuticals and Medical Devices Agency: Avigan (favipiravir) Review Report [Link]
- World Health Organization: Influenza (Avian and other zoonotic) [Link]
 |
|
| Names | |
|---|---|
| IUPAC name
5-Fluoro-2-hydroxypyrazine-3-carboxamide
|
|
| Other names
T-705; Avigan; favilavir
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C5H4FN3O2 | |
| Molar mass | 157.104 g·mol−1 |
| Pharmacology | |
| J05AX27 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
////////////

Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
WORLD DRUG TRACKER
one time
$10.00
PALINAVIR
PALINAVIR, BILA-2011-BS
UNII-632S1WU9Z2, 154612-39-2, n-[(1s)-1-[[(1s,2r)-1-benzyl-3-[(2s,4r)-2-(tert-butylcarbamoyl)-4-(4-pyridylmethoxy)piperidino]-2-hydroxypropyl]carbamoyl]-2-methylpropyl]quinaldamide,
N-[(2S)-1-[[(2S,3R)-4-[(2S,4R)-2-(tert-butylcarbamoyl)-4-(pyridin-4-ylmethoxy)piperidin-1-yl]-3-hydroxy-1-phenylbutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]quinoline-2-carboxamide
Molecular Formula:C41H52N6O5
Molecular Weight:708.88878 g/mol
| Patent | Submitted | Granted |
|---|---|---|
| Substituted pipecolinic acid derivatives as HIV protease inhibitors [US5614533] | 1997-03-25 | |
| Substituted pipecolinic acid derivatives as HIV protease inhibitors. [EP0560268] | 1993-09-15 | 1995-01-04 |

……………………….
PATENT
http://www.google.com/patents/WO2013105118A1?cl=en
Scheme 5: Synthesis of Palinavir (6):

The organic solvent mentioned according to the invention is selected from the group consisting of organic solvents, wherein the organic solvents are polar aprotic such as DCM, THF, Ethyl acetate, acetone, DMF, acetonitrile, DMSO ; polar protic solvents such as lower alcohol particularly (C1-C6) alkyl alcohol, water, acetic acid ; non-polar solvents such as hexane, benzene, toluene, chloroform, pet. ether, 1,4-dioxane, heptane either alone or mixtures thereof . Additionally the purification or separation of crude product can be accomplished by known techniques viz. extraction, column chromatography in a suitable organic solvent with the aid of instruments such as TLC, HPLC, GC, mass spectroscopy, or distillation, crystallization, derivatization.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
………………………….
J Org Chem 1997,62(11),3440

The reaction of tert-butoxycarbonyl-L-phenylalanine (I) with isobutyl chloroformate in THF gives the expected mixed anhydride which is treated with diazomethane and HCl yielding the corresponding chloromethyl ketone (II). The reduction of (II) with NaBH4 in THF affords the (S)-chlorohydrin (IV), which is treated with KOH in ethanol to obtain the chiral epoxide (V)(1,2). Ring opening of (V) with (?(cis)-N-tert-butyl-4-(4-pyridylmethoxy)piperidine-2-carboxamide (VI) by a treatment with LiCl in refluxing ethanol gives a mixture of diastereomers that is separated by chromatography giving the pure isomer (VII). The reaction of (VII) with tert-butoxycarbonyl-L-valine (VIII) by treatment first with trifluoroacetic acid (TFA), and condesation by means of BOP ((benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate) and NMM (N-methylmorpholine) affords the expected condensation product (IX). Finally, this compound is condensed with quinoline-2-carboxylic acid (X) by means of BOP and NMM as before. 2) The piperidine (VI) has been obtained by condensation of (?(cis)-N-(tert-butoxycarbonyl)-4-hydroxypiperidine-2-carboxamide (XI) with 4-(chloromethyl)pyridine (XII) by means of NaH in DMS, followed by hydrolysis with HCl.

Palinavir can also be obtained as follows: The controlled oxidation of 2(S)-(dibenzylamino)-3-phenyl-1-propanol (XIII) with pyridine-SO3 complex in DMSO gives the corresponding aldehyde (XIV), which is condensed with bromochloromethane (XV) by means of Li in THF followed by hydrolysis with HCl yielding regioselectively the 1-chloro-2-butanol (XVI). The debenzylation of (XVI) by hydrogenation over Pd/C affords the free amine (XVII), which is treated with tert-butoxycarbonyl anhydride/triethylamine and dehydrochlorinated with KOH in methanol to give the desired chiral epoxide (V).

The chiral piperidine (2S,4R)(VI) has been obtained as follows: The cyclization of 3-buten-1-ol (XXII) with (S)-1-phenylethylamine (XXIII) and glyoxylic acid (XXIV) by means of tosyl chloride in THF gives a mixture of the (2S,4R) and (2R,4S) lactones (XXV), which is resolved by fractional crystallyzation of their salts with the chiral camphorsulfonic acid (XXVI), followed by elimination of the acid with ammonia to afford (2S,4R)(XXVII). The reaction of lactone (XXVII) with isopropylmagnesium chloride and tert-butylamine in THF gives (2S,4R)-N-tert-butyl-4-hydroxy-1-(1(S)-phenylethyl)piperidine-2-carboxamide (XXVIII), which is debenzylated by hydrogenation and protected with tert-butoxycarbonyl anhydride yielding (2S,4R)-N-(tert-butoxycarbonyl)-4-hydroxypiperidine-2-carboxamide (2S,4R)(XI), which is finally condensed with 4-(chloromethyl)pyridine (XII) as before to obtain the chiral piperidine (2S,4R)(VI), already reported.

The condendsation of epoxide (V) with (2S,4R)(VI) by means of basic alumina in THF, followed by elimination of the protecting group with HCl and NaOH yields directly the condensation product (XVIII) as a pure diastereomer and with a free amino group. Finally, this compound is condensed with N-(2-quinolylcarbonyl)-L-valine (XIX) through its activation compound with isobutyl chloroformate (the 4(S)-isopropyl-2-(2-quinolyl)oxazol-5(4H)-one (XX)). The N-acyl-L-valine (XIX) has been obtained by acylation of L-valine (XXI) with quinoline-2-carboxylic acid (X) through its acyl chloride obtained with SOCl2.
………………………..

Palinavir is an inhibitor with five chiral centers. It contains the amino acid valine and pipecolinin acid. The previous way to create this drug faced three major obstacles. First, the reaction from 2 to 3 used diazomethane. Therefore, is is difficult, if not impossible, to produce large quantities. Secondly, the steps included in going from 4 to 5 gave way to racemers which is very inefficient. Finally, chromatography is needed at two separate times.

Four issues were addresses in route to product 1. First, because of the number of chiral centers, stereochemical control was a concern. high chemical yields were a second concern. Also, multi step procedures were advantageous to cut down on purification steps. Finally, the synthesis tried to restrict the use of hazardous reagents. The following retrosynthesis reaction was conceived and three target molecules were identified as seen in figure 1.

Molecule 3 uses a diaseteroselective addition of in situ (chloromethyl)lithium to N,N-dibenzylphenylalaninol and is derived from a four step process.

Recrystallization of 13 is required. Molecule 14 was not reached because it posed a problem later in the reaction. The N-benzyl protection group could not be removed to react with 9.

8 is a derivative of naturally occurring pipocolic acid, 16, named 3-buten-1-ol. Selective crystallization of diastereomeric salts can lead to 17a, but a more efficient way is by having a 60:40 mixture of lactones 17a,b. This leads to 18a,b using a Brodroux process. Crystallization of 18a,b lead to a poor overall yield. Instead, 18a,b undergoes salt crystallization with (-)-camphorsulfonic acid. Finally, 18a underwent hydrolysis and then addition of di-tert butyl dicarbonate leads to 8.
8 was then transformed to 5 in a three step process.
 8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid.
8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid.
 Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system.
Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system.
 In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a “viscous syrup”. 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.
In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a “viscous syrup”. 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.

Coupling of 10 and 9 is the final step in the synthesis , although there are still some purification steps left.

Two recrystallizations were required for the final 99.6% purity.

………………………..
J. Org. Chem., 1997, 62 (11), pp 3440–3448
DOI: 10.1021/jo9702655

Palinavir is a potent peptidomimetic-based HIV protease inhibitor. We have developed a highly convergent and stereoselective synthesis which is amenable to the preparation of multikilogram quantities of this compound. The synthetic sequence proceeds in 24 distinct chemical steps (with several integrated, multistep operations) from commercially available starting materials. No chromatographies are required throughout the process, and the final product is purified by crystallization of its dihydrochloride salt to >99% homogeneity.
crude palinavir (1) as a thick brown oil (yield not determined). HPLC analysis (Supelcosil LZ-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): 1, tR 17.80 min (84.1%); 24, tR 18.47 min (2.0%); 25, tR 19.97 min (1.45%).
palinavir dihydrochloride (1750 g, 51% yield) containing 0.25% w/w isopropanol (by 1H NMR):
mp 175−185 °C.
[α]25D −13.0° (c 1, MeOH). [α]25Hg365 +44.9° (c 1, MeOH).
IR (KBr) ν 3700−2300, 1660, 1555, 1520 cm-1.
1H NMR (DMSO-d6) δ 10.00 (broad s, 1H), 8.88 (d, J = 6.3 Hz, 2H), 8.61 (d, J = 8.4 Hz, 1H), 8.60 (s, 1H), 8.51 (d, J = 9.6 Hz, 1H), 8.35 (d, J = 8.7 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 6.0 Hz, 2H), 7.89 (t, J = 7.6 Hz, 1H), 7.74 (t, J = 7.5 Hz, 1H), 7.19 (d, J = 7.2 Hz, 2H), 7.08 (t, J = 7.5 Hz, 2H), 6.91 (t, J = 7.3 Hz, 1H), 4.86 (AB quartet, 2H), 4.37 (broad t, J = 7.8 Hz, 1H), 4.21 (d, J = 11.4 Hz, 1H), 4.11 (broad m, 1H), 3.96 (broad m, 1H), 3.80−3.65 (m, 2H), 3.26 (t, J = 7.4 Hz, 1H), 3.15−3.01 (m, 2H), 2.94 (broad d, J = 12.0 Hz, 1H), 2.62 (dd, J = 13.6, 10.6 Hz, 1H), 2.56 ((broad d, J = 12.0 Hz, 1H), 2.20−2.05 (m, 2H), 1.86 (m, 1H), 1.69 (q, J = 11.7 Hz, 1H), 1.31 (s, 9H), 0.81 (d, J = 6.3 Hz, 3H), 0.80 (d, J = 6.6 Hz, 3H).
13C NMR (DMSO-d6) δ 170.4, 166.4, 163.3, 158.3, 149.5, 145.9, 141.9, 138.6, 138.2, 130.7, 129.3, 129.1, 129.0, 128.3, 128.2, 128.0, 125.9, 124.1, 118.6, 72.3, 68.8, 67.2, 64.8, 58.0, 57.8, 54.4, 51.3, 51.1, 35.4, 34.1, 31.1, 28.2, 19.5, 17.9.
FAB-MS m/z 709 (MH+ of free base). Anal. Calcd for C41H54Cl2N6O5 (corrected for 8% water content as determined by Karl Fisher analysis and 0.25% w/w isopropanol as determined by 1H NMR): C, 58.31; H, 7.29; N, 9.93. Found: C, 57.76; H, 7.25; N, 9.89. Titration of HCl content using NaOH: 2.09 ± 0.03 mol HCl. HPLC homogeneity (Supelcosil LC-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): palinavir dihydrochloride, tR 18.24 min (99.51%); 25 tR 20.39 min (0.33%). HPLC homogeneity (Nova-Pak C8, 20−80% MeCN/50 mM NaH2PO4 in 25 min, 1 mL/min flow rate): palinavir dihydrochloride, tR 15.52 min (99.67%); 25 tR 13.52 min (0.33%).
PURE palinavir (1) as a white amorphous powder (1902 g, 84% yield):
mp 100−107 °C. [α]25D −11.5° (c 1, MeOH).
IR (KBr) ν 3700−3100, 1660, 1520, 1495 cm-1.
1H NMR (CDCl3) δ 8.54 (d, J = 5.7 Hz, 2H), 8.48 (d, J = 8.6 Hz, 1H), 8.31 (d, J = 8.6 Hz, 1H, part of AB), 8.22 (d, J = 8.3 Hz, 1H, part of AB), 8.13 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.80 (t, J = 7.6 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.25 (d, J = 5.4 Hz, 2H), 7.13 (d, J = 7.3 Hz, 2H), 7.07 (t, J = 7.5 Hz, 1H), 6.92 (t, J = 7.3 Hz, 1H), 6.59 (d, J = 8.3 Hz, 1H), 6.57 (s, 1H), 4.61 (d, J = 13.4 Hz, 1H, part of AB), 4.51 (d, J = 13.4 Hz, 1H, part of AB), 4.32 (dd, J = 8.6, 6.4 Hz, 1H), 4.22 (m, 1H), 3.97 (m, 1), 3.47−3.33 (m, 2H), 2.94 (dd, J = 14.3, 4.1 Hz, 1H), 2.89 (d, J= 8.6 Hz, 1H), 2.79−2.72 (m, 1H), 2.77 (dd, J = 14.3, 10.8 Hz, 1H), 2.43 (dd, J = 13.4, 8.3 Hz, 1H), 2.40−2.25 (m, 3H), 1.95 (broad d, J = 12.4 Hz, 1H), 1.65 (q J = 11.8 Hz, 2H), 1.32 (s, 9H), 0.95 (d, J = 7.0 Hz, 3H), 0.83 (d, J = 6.7 Hz, 3H).
13C NMR (CDCl3) δ 171.6, 171.2, 165.0, 149.8, 148.8, 147.9, 146.5, 137.6, 137.5, 130.3, 129.9, 129.5, 129.4, 129.0, 128.8, 128.5, 128.2, 127.7, 126.4, 121.7, 118.8, 75.0, 71.9, 68.1, 66.7, 59.4, 56.9, 54.6, 50.9, 50.2, 34.8, 33.3, 29.8, 29.7, 28.7, 19.6, 17.5.
FAB-MS m/z 709 (MH+). Anal. Calcd for C41H52N6O5(corrected for 0.7% water content as determined by Karl Fisher analysis): C, 68.98; H, 7.42; N, 11.77. Found: C, 68.71; H, 7.47; N, 11.71. HPLC homogeneity (Supelcosil LC-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): palinavir (1), tR 17.83 min (99.59%); 25 tR20.00 min (0.41%). HPLC homogeneity (Nova-Pak C8, 10−80% MeCN/50 mM NaH2PO4 in 25 min, 1 mL/min flow rate): palinavir (1), tR 17.37 min (99.51%); 25 tR 15.87 min (0.49%).
| Reference | ||
|---|---|---|
| 1 | * | ARUN K. GHOSH ET AL: “The Development of Cyclic Sulfolanes as Novel and High-Affinity P2 Ligands for HIV-1 Protease Inhibitors“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 37, no. 8, 1 April 1994 (1994-04-01), pages 1177-1188, XP055057710, ISSN: 0022-2623, DOI: 10.1021/jm00034a016 |
| 2 | * | KAY BRICKMANN ET AL: “Synthesis of Conformationally Restricted Mimetics of [gamma]-Turns and Incorporation into Desmopressin, an Analogue of the Peptide Hormone Vasopressin“, CHEMISTRY – A EUROPEAN JOURNAL, vol. 5, no. 8, 2 August 1999 (1999-08-02), pages 2241-2253, XP055057517, ISSN: 0947-6539, DOI: 10.1002/(SICI)1521-3765(19990802)5:8<2241: :AID-CHEM2241>3.0.CO;2-L |
| 3 | * | KIRAN I N C ET AL: “A concise enantioselective synthesis of (+)-goniodiol and (+)-8-methoxygoniodiol via Co-catalyzed HKR of anti-(2SR, 3RS)-3-methoxy-3-phenyl-1, 2-epoxypropane“, TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 52, no. 3, 19 January 2011 (2011-01-19), pages 438-440, XP027558447, ISSN: 0040-4039 [retrieved on 2010-12-14] |
| 4 | * | M. TOKUNAGA: “Asymmetric Catalysis with Water: Efficient Kinetic Resolution of Terminal Epoxides by Means of Catalytic Hydrolysis“, SCIENCE, vol. 277, no. 5328, 15 August 1997 (1997-08-15), pages 936-938, XP055057541, ISSN: 0036-8075, DOI: 10.1126/science.277.5328.936 |
| 5 | * | PARKES K E B ET AL: “STUDIES TOWARD THE LARGE-SCALE SYNTHESIS OF THE HIV PROTEINASE INHIBITOR RO 31-8959“, JOURNAL OF ORGANIC CHEMISTRY, ACS, US, vol. 59, no. 13/16, 1 January 1994 (1994-01-01), pages 3656-3664, XP002011975, ISSN: 0022-3263, DOI: 10.1021/JO00092A026 |
| 6 | * | R. SANTHOSH REDDY ET AL: “Co(iii)(salen)-catalyzed HKR of two stereocentered alkoxy- and azido epoxides: a concise enantioselective synthesis of (S,S)-reboxetine and (+)-epi-cytoxazone“, CHEMICAL COMMUNICATIONS, vol. 46, no. 27, 1 January 2010 (2010-01-01), page 5012, XP055057537, ISSN: 1359-7345, DOI: 10.1039/c0cc00650e |
| 7 | * | SHINJI NAGUMO ET AL: “Intramolecular Friedel-Crafts type reaction of vinyloxiranes linked to an ester group“, TETRAHEDRON, vol. 65, no. 47, 1 November 2009 (2009-11-01), pages 9884-9896, XP055057655, ISSN: 0040-4020, DOI: 10.1016/j.tet.2009.09.037 |
| 8 | * | SUNITA K. GADAKH ET AL: “Enantioselective synthesis of HIV protease inhibitor amprenavir via Co-catalyzed HKR of 2-(1-azido-2-phenylethyl)oxirane“, TETRAHEDRON: ASYMMETRY, vol. 23, no. 11-12, 1 June 2012 (2012-06-01), pages 898-903, XP055057475, ISSN: 0957-4166, DOI: 10.1016/j.tetasy.2012.06.003
Want to know everything on vir series click http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html AND http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html |
Sitasentan TBC 11251

Sitasentan,TBC 11251
210421-64-0
N-(4-chloro-3-methyl-1,2-oxazol-5-yl)-2-[2-(6-methyl-1,3-benzodioxol-5-yl)acetyl]thiophene-3-sulfonamide
Sitaxentan sodium (TBC-11251) is a medication for the treatment of pulmonary arterial hypertension (PAH).[1] It was marketed as Thelin by Encysive Pharmaceuticals until Pfizer purchased Encysive in February 2008. In 2010, Pfizer voluntarily removed sitaxentan from the market due to concerns about liver toxicity.[2]
Sitaxentan belongs to a class of drugs known as endothelin receptor antagonists (ERAs). Patients with PAH have elevated levels of endothelin, a potent blood vessel constrictor, in their plasma and lung tissue. Sitaxentan blocks the binding of endothelin to its receptors, thereby negating endothelin’s deleterious effects.
Mechanism of action
Sitaxentan is a small molecule that blocks the action of endothelin (ET) on the endothelin-A (ETA) receptor selectively (by a factor of 6000 compared to the ETB).[3] It is a sulfonamide class endothelin receptor antagonist (ERA) and is undergoing Food and Drug Administration (FDA) review for treating pulmonary hypertension. The rationale for benefit compared to bosentan, a nonselective ET blocker, is negligible inhibition of the beneficial effects of ETB stimulation, such as nitric oxide production and clearance of ET from circulation. In clinical trials, the efficacy of sitaxentan has been much the same as bosentan, but the hepatotoxicity of sitaxentan outweighs its benefits. Dosing is once daily, as opposed to twice daily for bosentan.
Regulatory status
On December 10, 2010 Pfizer announced it would be withdrawing sitaxentan worldwide (both from marketing and from all clinical study use), citing that it is a cause of fatal liver damage.[2]
Sitaxentan was approved for marketing in the European Union in 2006, in Canada in 2006[4] and in Australia in 2007. By February 2008 it had been launched commercially in Germany, Austria, The Netherlands, the United Kingdom, Ireland, France, Spain and Italy.
In March 2006, the FDA recommended an approvable status to sitaxentan but said it would not yet approve the product. In July 2006, sitaxentan received a second approvable letter stating that efficacy outcome issues raised in the context of the STRIDE-2 study were still unresolved. In July 2007, Encysive commenced a formal dispute resolution process in a preliminary meeting with the FDA. In September 2007 the company announced that it was making preparations for another phase III clinical trial (intended to be named STRIDE-5) to overcome the FDA’s concerns.[5] The takeover by Pfizer resulted in a reconfiguration and extension of these plans, to include combination therapy with sildenafil. The Sitaxentan Efficacy and Safety Trial With a Randomized Prospective Assessment of Adding Sildenafil (SR-PAAS) was an ongoing program of three clinical trials conducted in the United States (ClinicalTtrials.gov identifiers: NCT00795639, NCT00796666 and NCT00796510) with anticipated completion dates between June 2010 and January 2014.

N-(4-Chloro-3-methyl-5-isoxazolyl)-2-[2-(6-methyl-1,3-benzodioxol-5-yl)acetyl]-3-thiophenesulfonamide sodium salt, Sitaxsentan sodium salt, TBC-11251 sodium salt, Thelin
- CAS Number 210421-74-2
- Empirical Formula C18H14ClN2NaO6S2
- Molecular Weight 476.89
Adverse effects
Adverse effects observed with sitaxentan are class effects of endothelin receptor antagonists, and include :
- liver enzyme abnormalities (increased ALT and AST)
- headache
- edema
- constipation
- nasal congestion
- upper respiratory tract infection
- dizziness
- insomnia
- flushing
Because sitaxentan inhibits metabolism of warfarin, a decreased dose of warfarin is needed when co-administered with sitaxentan. This is because warfarin acts to prevent blood from clotting, and if it remains unmetabolized, it can continue to thin the blood.
http://www.google.com/patents/WO2007149568A2?cl=en
As used herein “sitaxsentan” refers to N-(4-chloro-3-methyl-5-isoxazolyl)-2-[2- methyl-4,5-(methylenedioxy)phenylacetyl]-thiophene-3-sulfonamide. Sitaxsentan is also known as TBCl 1251. Other chemical names for sitaxsentan include 4-chloro-3-methyl-5-(2- (2-(6-methylbenzo[d][l ,3]dioxol-5-yl)acetyl)-3-thienylsulfonamido)isoxazole and N-(4- chloro-3-methyl-5-isoxazolyl)-2-[3,4-(methylenedioxy)-6-methylphenylacetyl]-thiophene-3- sulfonamide.
The chemical name for sitaxsentan is N-(4-chloro-3-methyl-5-isoxazolyl)-2-[2- methyl-4,5-(methylenedioxy)phenylacetyl]-thiophene-3-sulfonamide, and its structural formula is as follows:

Sitaxsentan
Sitaxsentan is a potent endothelin receptor antagonist that has oral bioavailability in several species, a long duration of action, and high specificity for ETA receptors.
EXAMPLE 1
Preparation of 4-chloro-3-methyl-5-(2-(2-(6-methylbenzo[d] [l,3|dioxol-5-yl)aeetyl)-3- thienylsulfonamido)isoxazole, or N-(4-chloro-3-methyl-5-isoxazolyl)-2-[2-methy 1-4,5- (methylenedioxy)phenylacetyl]-thiophene-3-sulfonamide, or N-(4-chIoro-3-methyl-5- isoxazolyl)-2-[3,4-(methylenedioxy)-6-methylphenylacetyl]-thiophene-3-sulfonamide.
A. Preparation of (4-chIoro-3-methyl-5-(2-(2-(6-methylbenzo[d] [l,3]dioxol-5-yl)acetyl)- 3-thienylsuIfonamido)isoxazole 1. Preparation of 5-chloromethyI-6-methylbenzo[d][l,3]dioxole
To a mixture of methylene chloride (130 L), concentrated HCl (130 L), and tetrabuylammonium bromide (1.61 Kg) was added 5-methylbenzo[d][l,3]dioxole (10 Kg) followed by the slow addition of formaldehyde (14 L, 37 wt% in water). The mixture was stirred overnight. The organic layer was separated, dried with magnesium sulfate and concentrated to an oil. Hexane (180 L) was added and the mixture heated to boiling. The hot hexane solution was decanted from a heavy oily residue and evaporated to give almost pure 5-chloromethyl-6-methylbenzo[d][l,3]dioxole as a white solid. Recrystallization from hexane (50 L) gave 5-chloromethyl-6-methylbenzo[d][l,3]dioxole (80% recovery after recrystallization). 2. Formation of (4-chloro-3-methyl-5-(2-(2-(2-methyIbenzo[d][l,3]dioxol-5-yl) acetyl)-3-thienylsulfonamido)isoxazole
A portion of a solution of 5-chloromemyl-6-methylbenzo[d][l,3]di-oxole (16.8 g, 0.09 mol) in tetrahydrofuran (THF)(120 mL) was added to a well stirred slurry of magnesium powder, (3.3 g, 0.136 g-atom, Alfa, or Johnson-Mathey, -20 +100 mesh) in THF (120 mL) at room temperature. The resulting reaction admixture was warmed up to about 40-450C for about 2-3 min, causing the reaction to start. Once the heating activated the magnesium, and the reaction began, the mixture was cooled and maintained at a temperature below about 8 0C. The magnesium can be activated with dibromoethane in place of heat.
A flask containing the reaction mixture was cooled and the remaining solution of 5- chloromethlybenzo[d][l,3]dioxole added dropwise during 1.5 hours while maintaining an internal temperature below 8 0C. Temperature control is important: if the Grignard is generated and kept below 8 0C5 Wurtz coupling is suppressed. Longer times at higher temperatures promote the Wurtz coupling pathway. Wurtz coupling can be avoided by using high quality Mg and by keeping the temperature of the Grignard below about 8 0C and stirring vigorously. The reaction works fine at -20 0C, so any temperature below 8 0C is acceptable at which the Grignard will form. The color of the reaction mixture turns greenish.
The reaction mixture was stirred for an additional 5 min at 0 0C, while N2-methoxy- N2-methyl-3-(4-chloro-3-methyl-5-isoazolylsulfamoyl)-2-thiophenecarboxamide (6.6 g, 0.018 mol) in anhydrous THF (90 mL) was charged into the addition funnel. The reaction mixture was degassed two times then the solution of N2-methoxy-N2-methyl-3-(4-chloro-3- methyl-5-isoxazolylsulfamoyl)-2-thiophenecarboxamide was added at 0 0C over 5 min. TLC of the reaction mixture (Silica, 12% MeOHZCH2Cl2) taken immediately after the addition shows no N2-methoxy-N2-methyl-3-(4-chloro-3-methyl-5-isoxazolysulfamoyl)-2-thio- phenecarboxamide. The reaction mixture was transferred into a flask containing IN HCl (400 mL, 0.4 mol
HCl, ice-bath stirred), and the mixture stirred for 2 to 4 min, transferred into a separatory funnel and diluted with ethyl acetate (300 mL). The layers were separated after shaking. The water layer was extracted with additional ethyl acetate (150 mL) and the combined organics washed with half-brine. Following separation, THF was removed by drying the organic layer over sodium sulfate and concentrating under reduced pressure at about 39 0C to obtain the title compound. EXAMPLE 2
1.0 g Sitaxentan was dissolved in 10 ml ethyl acetate and 5 ml hexanes were added. The formed suspension was heated until a clear solution was obtained. Upon cooling light yellow plates were formed. After filtration and drying under vacuum 515 mg of sitaxentan polymorph A was obtained as light yellow plates in very high purity.
EXAMPLE 3
Preparation of 4-chloro-3-methyl-5-(2-(2-(6-methyIbenzo[dJ [l,3]dioxol-5-yl)acetyl)-3- thienylsulfonamido)isoxazole, Sodium Salt
The crystalline sitaxsentan from Example 2 is dissolved in ethyl acetate and washed with saturated NaHCO3 (5 x 10 mL). The solution is washed with brine, dried over Na2SO4 and concentrated in vacuo to obtain a solid residue. 10 mL OfCH2Cl2 is added and the mixture is stirred under nitrogen for 5 to 10 minutes. Ether (15 mL) is added and the mixture stirred for about 10 min. The product is isolated by filtration, washed with a mixture of CH2Cl2 /ether (1 :2) (10 mL) then with ether (10 mL) and dried under reduced pressure to obtain 4-Chloro-3-methyl-5-(2-(2-(6-methyIbenzo[d][l ,3]dioxol-5-yl)acetyl)-3- thienylsulfonamido)isoxazole, sodium salt.
………………………..
J. Med. Chem., 1997, 40 (11), pp 1690–1697
DOI: 10.1021/jm9700068
15q.Yellowpowder;
1HNMR(CDCl3):88.88(brs,1H),7.59(s,2H),6.72(s,1H),6.69(s,111),5.94(s,2H),4.22(s,2H),2.22(s,311),2.21(s,3H);
IR(KBrpellet):3455,3233,
3109,2899,1674,1632,1505,1487,1395,1373cm-1;
HRMS:[M+H]*455.0137
………………………..
see
Current Opinion in Investigational Drugs (PharmaPress Ltd.) (2001), 2(4), 531-536.
…………….
Synthesis of Sitaxsentan sodium
Yingyong Huaxue (2007), 24, (11), 1310-1313. Publisher: (Kexue Chubanshe, ) CODEN:YIHUED ISSN:1000-0518.
Yingyong Huaxue (2007), 24, (11), 1310-1313. Publisher: (Kexue Chubanshe, ) CODEN:YIHUED ISSN:1000-0518.
………………………………………
Table 1: Sitaxsentan Sodium Lyophilized Formulation

References
1Barst RJ, Langleben D, Frost A et al. (2004). “Sitaxsentan therapy for pulmonary arterial hypertension”. American Journal of Respiratory Critical Care Medicine 169 (4): 441–447. doi:10.1164/rccm.200307-957OC. PMID 14630619.
- 2
- Citing liver damage, Pfizer withdraws Thelin, Associated Press, December 12, 2010
- 3
- Girgis, RE; Frost, AE; Hill, NS; Horn, EM; Langleben, D; McLaughlin, VV; Oudiz, RJ; Robbins, IM et al. (2007). “Selective endothelinA receptor antagonism with sitaxsentan for pulmonary arterial hypertension associated with connective tissue disease”. Annals of the rheumatic diseases 66 (11): 1467–72. doi:10.1136/ard.2007.069609. PMC 2111639. PMID 17472992.
- 4
- “UPDATE 1-Encysive gets Canadian approval for hypertension drug”. Reuters. 30 May 2007. Retrieved 2007-07-08.
- 5
- “Encysive Pharmaceuticals to Conduct Phase III Study With Thelin (Sitaxsentan Sodium) in Pulmonary Arterial Hypertension”. PrimeNewswire via COMTEX News Network. 29 September 2007. Retrieved 2007-12-12.

External links
- http://www.drugs.com/nda/thelin_050714.html
- Mucke HAM: Sitaxsentan for the Oral Treatment of Pulmonary Arterial Hypertension: Benefits from Endothelin Receptor Subtype Selectivity? Clinical Medicine: Therapeutics 2009:1 111–121.
- ATS 2005. The International Conference of the American Thoracic Society. 20–25 May 2005. San Diego, CA.
- Robyn J. Barst, MD; Stuart Rich, MD, FCCP; Allison Widlitz, MS, PA; Evelyn M. Horn, MD; Vallerie McLaughlin, MD and Joyce McFarlin, RN : Clinical Efficacy of Sitaxsentan, an Endothelin-A Receptor Antagonist, in Patients With Pulmonary Arterial Hypertension Chest. 2002;121:1860-1868.
- American Heart Association. Primary or Unexplained Pulmonary Hypertension
| US20010021714 * | Apr 4, 1996 | Sep 13, 2001 | Ming Fai Chan | Compounds such as n-(4-bromo-3-methyl-5-isoxazolyl)-2-n-benzylbenzo(b)thiophene-3-sufonamide administered as endothelin peptide receptor antagonists |
| Reference | ||
|---|---|---|
| 1 | * | WU C ET AL: “Discovery of TBC11251, a Potent, Long Acting, Orally Active Endothelin Receptor-A Selective Antagonist” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 40, no. 11, 23 May 1997 (1997-05-23), pages 1690-1697, XP002164198 ISSN: 0022-2623 |
| Patent | Submitted | Granted |
|---|---|---|
| ANTIHYPERTENSIVE THERAPY METHOD [US2007293552] | 2007-12-20 | |
| Crystalline N-(4-chloro-3-methyl-5-isoxazolyl)-2-[2-methyl-4.5-(methylenedioxy)phenylacetyl]-thiophene-3-sulfonamide [US2008026061] | 2008-01-31 | |
| Gnrh agonist combination drugs [US2005215528] | 2005-09-29 | |
| THIENYL-, FURYL-, PYRROLYL- AND BIPHENYLSULFONAMIDES AND DERIVATIVES THEREOF THAT MODULATE THE ACTIVITY OF ENDOTHELIN [WO9631492] | 1996-10-10 | |
| SULFONAMIDES FOR TREATMENT OF ENDOTHELIN-MEDIATED DISORDERS [WO9849162] | 1998-11-05 |
| Patent | Submitted | Granted |
|---|---|---|
| Respiratory Drug Condensation Aerosols and Methods of Making and Using Them [US2009258075] | 2009-10-15 | |
| Method and Composition for Treating Alzheimer’s Disease and Dementias of Vascular Origin [US2010173872] | 2010-07-08 | |
| Method and Composition for Treating Alzheimer’s Disease and Dementias of Vascular Origin [US2010184725] | 2010-07-22 | |
| Formulations of sitaxsentan sodium [US2008076812] | 2008-03-27 | |
| Methods and compositions for treatment of sleep apnea [US2008085313] | 2008-04-10 | |
| Processes for the preparation of 4-chloro-3-methyl-5-(2-(2-(6-methylbenzo[d][1,3]dioxol-5-yl)acetyl)-3-thienylsulfonamido)isoxazole [US2008086010] | 2008-04-10 | |
| Method and composition for treating alzheimer’s disease and dementias of vascular origin [US2004092427] | 2004-05-13 | |
| Method for preventing or treating pulmonary inflammation by administering an endothelin antagonist [US2003004199] | 2003-01-02 | |
| Methods and Compositions for Treatment of an Interstitial Lung Disease [US2009004268] | 2009-01-01 | |
| Methods and compositions for treatment of diastolic heart failure [US2007232671] | 2007-10-04 | |
| Patent | Submitted | Granted |
|---|---|---|
| Isoxazolyl endothelin antagonists [US6043265] | 2000-03-28 | |
| Aminoguanidine hydrazone derivatives, process for producing the same and drugs thereof [US6350749] | 2002-02-26 | |
| Method for preventing or treating pain by administering an endothelin antagonist [US6573285] | 2002-06-27 | 2003-06-03 |
| Method for preventing or treating erectile dysfunction by administering an endothelin antagonist [US6268388] | 2001-07-31 | |
| Method and composition for potentiating the antipyretic action of a nonopioid analgesic [US7351692] | 2003-12-25 | 2008-04-01 |
| Method and Composition for Potentiating an Opiate Analgesic [US8114896] | 2010-05-06 | 2012-02-14 |
| SUBSTITUTED THIOPHENES [US7863308] | 2008-10-16 | 2011-01-04 |
| Respiratory drug condensation aerosols and methods of making and using them [US7550133] | 2004-06-03 | 2009-06-23 |
| SUBSTITUTED THIOPHENES [US2010280086] | 2010-11-04 | |
| Method and Composition for Potentiating an Opiate Analgesic [US2010311665] | 2010-12-09 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-(4-chloro-3-methyl-1,2-oxazol-5-yl)-2-[2-(6-methyl-2H-1,3-benzodioxol-5-yl)acetyl]thiophene-3-sulfonamide | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Licence data | EMA:Link |
| Legal status | |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 70 to 100% |
| Protein binding | >99% |
| Metabolism | Hepatic (CYP2C9– and CYP3A4-mediated) |
| Half-life | 10 hours |
| Excretion | Renal (50 to 60%) Fecal (40 to 50%) |
| Identifiers | |
| CAS number | 184036-34-8 210421-64-0 (sodium salt) |
| ATC code | C02KX03 |
| PubChem | CID 216235 |
| IUPHAR ligand | 3950 |
| DrugBank | DB06268 |
| ChemSpider | 21106381 |
| UNII | J9QH779MEM |
| KEGG | D07171 |
| ChEMBL | CHEMBL282724 |
| Synonyms | Sitaxsentan; TBC-11251 |
| Chemical data | |
| Formula | C18H15ClN2O6S2 |
| Molecular mass | 454.906 g/mol |

Structures and observed activities of the ETA receptor antagonists for the HipHop training set
 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HEREJoin me on Linkedin
 amcrasto@gmail.com
amcrasto@gmail.com
TAKE A TOUR
KISUMU, KENYA

Kisumu – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Kisumu
Kisumu is a port city in Kisumu County, Kenya 1,131 m (3,711 ft), with a population of 409,928 (2009 census). It is the third largest city in Kenya, the principal city …
Kisumu CountyKisumu County is one of the new devolved counties of Kenya. Its …
|
Kisumu International AirportKisumu International Airport is an airport in Kisumu, Kenya (IATA …
|

Boat riding in Kisumu

Local inhabitants near Kisumu, 1911


Clockwise: Lake Victoria Panorama, Kisumu Panorama, sunset at Oginga Odinga street, Downtown, Kiboko Point, Nighttime in Kisumu and Jomo Kenyatta Stadium.
|
|

Kisumu
|
|
Coordinates:  0°6′S 34°45′E 0°6′S 34°45′E |
|
| Country | |
|---|---|
| County | Kisumu County |

Kisumu panorama, viewed from Lake Victoria

Jomo Kenyatta Stadium

Kisumu Harbour. The green vegetation is water hyacinth

Nairobi University Kisumu Campus



Your visit to Kisumu is not complete if you do not visit this amazing beach, which happens to be a favourite spot fishing spot for the fishermen.


///////
WANT TO KNOW ON SENTAN SERIES
MEDICINAL CHEMISTRY AT ITS BEST, Tracks information on drugs on worldwide basis by Dr Anthony Melvin Crasto, worlddrugtracker, helping millions with websites, 6 million hits on google, one lakh connections worldwide, email amcrasto@gmail.com, call +91 9323115463 India
READ MORE ON SENTAN SERIES……http://medcheminternational.blogspot.in/p/sentan-series.html
| Antagonists of Endothelin type A receptor ETA | |
| Name | Structure |
| BQ-123 |  |
| Bosentan |  |
| Atrasentan |  |
| Tezosentan |  |
| Sitaxsentan | |
| Darusentan |  |
| Clazosentan |  |
| ZD-4054 (Zibotentan) |  |
| Ambrisentan |  |
| Tak-044 |  |
| Avosentan |  |
MAHABALIPURAM, INDIA
Mahabalipuram – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Mahabalipuram
Mahabalipuram, also known as Mamallapuram is a town in Kancheepuram district in the Indian state of Tamil Nadu. It is around 60 km south from the city of …Shore Temple – Seven Pagodas – Pancha Rathas –
.
.



Krishna’s Butter Ball in Mahabalipuram, India. The surface below the rock is …




http://www.weather-forecast.com/locations/Mamallapuram
 Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
 Moonraikers Restaurant, Mamallapuram
Moonraikers Restaurant, Mamallapuram


 Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
 .
.
A carving at the Varaha Temple, Mahabalipuram

/////////////
SWINE FLU ; AYURVEDA SUCCESSFUL TREATMENT ; स्वाइन प्लू का सुरक्षित आयुर्वेदिक इलाज
स्वाइन प्लू के लक्षणो पर आधारित सभी रोगियो का आयुर्वेदिक इलाज करने के बाद यह अनुभव मे आया है कि महामारी की तरह फैल रही बीमारी का बहुत सटीक और अचूक इलाज आयुर्वेद मे है /
वायरल / अथवा स्वाइन फ्लू के रोगियो के इलाज मे मैने निम्न दवाये दी है उन्हे मै सार्वजनिक तौर पर देश के सभी नागरिको के लिये यहा बता रहा हू /
स्वाइन फ्लू या इस जैसी बीमारी के इलाज के लिये मेरा नुस्खा इस तरह है /
महामृत्युन्जय रस दो गोली
कफ कुठार रस चार गोली
सुदर्शन घन वटी दो गोली
सप्त पर्ण घन वटी दो गोली
वयस्क व्यक्ति के लिये यह एक खुराक है /
सभी ऊपर लिखी गयी दवओ की गोलियो को गुन्गुने पानी से तीन तीन घन्टे के अन्तर से खिलाना चाहिये three hourly a with lukwarm water
कम उम्र के किशोरो को ऊपर लिखी दवाओ की एक एक गोली…
View original post 646 more words
Detailed Requirements concerning the DOE in the Regulatory Submission Dossier: EMA’s and FDA’s Recommendations
DRUG REGULATORY AFFAIRS INTERNATIONAL

The EMA has published together with the FDA a new question & answer (Q&A) paper at the end of 2014. This document answers questions on detailed requirements in connection with the documents concerning regulatory submissions. Among others it contains the answer to the question “What level of detail should be considered for design of experiments (DOEs) in a regulatory submission?”
GMP News
25/02/2015
In our News dated 18 February we reported on a question & answer (Q&A) paper which was published by EMA and FDA together at the end of 2014. This document answers questions on detailed requirements in connection with the documents concerning regulatory submissions. It also answers a question on the topic design of experiments (DOE).

The document answers the question “What level of detail should be considered for design of experiments (DOEs) in a regulatory submission?” as follows:
The level of detail should be commensurate…
View original post 165 more words
SILODOSIN………For treatment of benign prostatic hypertophy

SILODOSIN
Urief, 160970-54-7, Rapaflo, KMD 3213, Silodyx, KAD 3213, KMD-3213
Molecular Formula: C25H32F3N3O4
Molecular Weight: 495.53449 g/mol
Alpha 1A adrenoceptor antagonist
Prostate hyperplasia
Kissei Pharmaceutical Co Ltd INOVATOR
CAS 160970-54-7
2,3-Dihydro-1-(3-hydroxypropyl)-5-[(2R)-2-[[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl]amino]propyl]-1H-indole-7-carboxamide
160970-64-9 (racemate)
169107-04-4 (diHBr)
Properties: [a]D25 -14.0° (c = 1.01 in methanol).
Optical Rotation: [a]D25 -14.0° (c = 1.01 in methanol)
Therap-Cat: In treatment of benign prostatic hypertophy.
a-Adrenergic Blocker.
In February 2008, the FDA accepted for review an NDA for silodosin for the treatment of dysuria associated with BPH . In October 2008, the FDA approved the drug . In April 2009, Actavis launched silodosin for the treatment of the signs and symptoms of BPH .

1-(3-hydroxypropyl)-5-[(2R)-2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino]propyl]-2,3-dihydroindole-7-carboxamide
Kissei Pharmaceutical, Daiichi Sankyo (formerly Daiichi Seiyaku), Actavis (formerly Watson) and Recordati have developed and launched silodosin (Urief; Trupass; Rapaflo; Thrupas; Silodyx; Urorec; KMD-3213; Youlifu), an oral alpha 1A adrenoceptor antagonist selective for prostatic receptors . The product is comarketed in Europe by several licensees. The drug is indicated for the treatment of the signs and symptoms of benign prostatic hyperplasia (BPH).
Silodosin, a highly selective alpha1A-adrenoceptor antagonist, was launched in May 2006 in Japan for the oral treatment of urinary disturbance associated with benign prostatic hyperplasia (BPH). The product was launched in the U.S. for the treatment of signs and symptoms of benign prostatic hyperplasia in 2009. In 2009, a positive opinion was received in the E.U. for this indication and final approval was obtained in 2010. Launch in the E.U. took place the same year.
In May 2006, silodosin was launched as a capsule formulation in Japan. Actavis launched the drug in the US in April 2009. In June 2010, EU launched began, initially with Germany ; in November 2010, the drug was launched in France; by December 2010, the drug was launched in Spain.
In 2001, Kissei established an agreement with Daiichi Pharmaceutical to codevelop and comarket silodosin. An oral, once-daily formulation of silodosin filed in the U.S. by Watson (now Actavis) was approved in 2008. Watson (now Actavis) obtained exclusive rights in 2004 to develop and market the drug in the U.S.
PRODUCT Was developed and launched byKissei Pharmaceutical, Daiichi Sankyo, Actavis and Recordati. Family members of the product case EP0600675 have SPC protection in most EU states until 2018; while its Orange Book listed equivalent, US5387603, expire in the US in 2018 with US156 extension.
Silodosin (trade names Rapaflo (USA), Silodyx (Europe and South Africa), Rapilif (India), Silodal (India), Urief (Japan), Urorec (Russia)) is a medication for the symptomatic treatment of benign prostatic hyperplasia. It acts as an α1–adrenoceptor antagonist with high uroselectivity (selectivity for the prostate).
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-(3-hydroxypropyl)-5-[(2R)-({2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl}amino)propyl]indoline-7-carboxamide | |
| Clinical data | |
|
|
| Legal status |
|
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 32% |
| Protein binding | 97% |
| Metabolism | Hepatic glucuronidation (UGT2B7-mediated); also minor CYP3A4 involvement |
| Half-life | 13±8 hours |
| Excretion | Renal and fecal |
| Identifiers | |
| CAS number | 160970-54-7 |
| ATC code | G04CA04 |
| PubChem | CID 5312125 |
| IUPHAR ligand | 493 |
| ChemSpider | 4471557 |
| UNII | CUZ39LUY82 |
| ChEMBL | CHEMBL24778 |
| Synonyms | KAD-3213, KMD-3213 |
| Chemical data | |
| Formula | C25H32F3N3O4 |
| Molecular mass | 495.534 g/mol |
History
Silodosin received its first marketing approval in Japan in May 2006 under the tradename Urief, which is jointly marketed by Kissei Pharmaceutical Co., Ltd. and Daiichi Sankyo Pharmaceutical Co., Ltd.
Kissei licensed the US, Canadian, and Mexican rights for silodosin to Watson Pharmaceuticals, Inc. in 2004.
On February 12, 2008, Watson announced that the New Drug Application submitted to the United States Food and Drug Administration for silodosin has been accepted for filing. FDA approved this drug on October 9, 2008.[1] Silodosin is marketed under the trade names Rapaflo in the US and Silodyx in Europe.[2] and Rapilif in India (Ipca Urosciences)
Pharmacology
Since silodosin has high affinity for the α1A adrenergic receptor, it causes practically no orthostatic hypotension (in contrast to other α1 blockers). On the other side, the high selectivity seems to cause more problems with ejaculation.[3]
As α1A adrenoceptor antagonists are being investigated as a means to male birth control due to their ability to inhibit ejaculation but not orgasm, a trial with 15 male volunteers was conducted. While silodosin was completely efficacious in preventing the release of semen in all subjects, 12 out of the 15 patients reported mild discomfort upon orgasm. The men also reported the psychosexual side effect of being strongly dissatisfied by their lack of ejaculation.[4]

///////////////////////////////
CN 103848772
http://www.google.com/patents/CN103848772A?cl=en
silodosin (Silodosin) is 〃 2 Japanese orange Johnson invented – receptor antagonist, for the treatment of benign prostatic hyperplasia or hypertrophy, and other related symptoms. Clinical trials showed that 25% of patients with benign prostatic hyperplasia need for drugs or surgery. Although prostatectomy is better, the mortality rate is not high, but patients bring varying degrees of damage. So look for an effective and safe non-surgical treatment, not only can control the further development of the disease, while relieving the symptoms of the patient.
benign prostatic hyperplasia in older male patients have a higher prevalence, and clinical alternative drugs rarely, so the development of a benign prostatic hyperplasia drug treatment, not only has good social benefits, but also to bring good economic benefits. The study confirmed that silodosin is the treatment of benign prostatic hyperplasia in an important class of drugs.

Currently, the research reported in the published literature on the preparation of compounds of silodosin, are:


Early 1995, Kitazawa M et al patent US5387603, the reporter silodosin total synthesis method, but the method reaction step is long, the yield is not too high, not suitable for our industrial production.

In 2009, 翟富民 et al patent CN102115455A, which reported a method for preparing Sailuoduoxin key intermediates. The appropriate method for improving existing methods, although shorter than the previous method step in the step, but low synthesis yield of the process, we can not meet the needs of industrial production.
In summary, the compounds prepared silodosin more synthetic methods are constantly improved, but there are still a lot of flaws. Therefore, there is need for further research on the preparation of compounds of silodosin to get simple process, product yield, product easy separation of the new preparation. SUMMARY
The present invention is to overcome the above problems of the prior art, there is provided a method for preparing important intermediates silodosin, the present invention is simple process, high yield, easy separation of the product, the method suitable for industrial production .
To achieve the above technical object, to achieve the above technical result, the present invention is realized by the following technical scheme:
One kind of silodosin preparation of important intermediate, comprising the steps of:
Step I) in a flask, 282g of raw materials 1-acetyl-5- (2-bromo-propyl) indoline, 222g phthalimide potassium salt and 700mL DMF, was heated at 110 ° C for 2h; After completion of the reaction, to which was added the right amount of water to wash away the excess solvent DMF and salt extraction desolventizing after EA, was 296g crude;
Step 2) In a flask was added 296g crude product obtained in step I, dissolved 800mL ethanol, was added 165mL of hydrazine hydrate, 50 ° C is heated to precipitate a white solid; After completion of the reaction, cooling suction filtered, the filter cake washed with ethanol, and then the mother liquor removing solvent under reduced pressure; After dissolving EA, washed with water to wash away the excess hydrazine, and finally the organic phase the solvent was removed to give 165g intermediate, i.e. 1-acetyl-5- (2-aminopropyl) indoline;
Step 3) In the three-necked flask, 165g of Intermediate 1-acetyl-5- (2-aminopropyl) indoline, dissolved 600mL methanol, stirred at room temperature, and thereto was slowly added dropwise bromine; the addition was complete After stirring at room temperature 5-6h; After completion of the reaction, slowly poured into saturated NaHSO3, and wash away excess bromine; extracted with ethyl acetate, washed with water and saturated brine, dried over anhydrous sodium sulfate; After filtration, the solvent was removed in vacuo to give the crude product recrystallized from toluene to give 177g pure product;
Step 4) In a flask was added 177g of pure product obtained in Step 3 and 65g CuCN, after use 700mL DMF, was heated at 110 ° C for 3 to 5h; After completion of the reaction, the amount of water was added thereto, washing off excess solvent DMF and salt, EA desolventizing crude extract, after recrystallization 121g pure 1-acetyl-5- (2-bromo-propyl) -7-cyano-indoline that silodosin important intermediates;
Step (1), (2), (3), (4) synthesis reaction is:

Further, the step I) to step 4) by TLC plate tracking point detection reaction.
The beneficial effects of the present invention are:
Preparation silodosin important intermediate of the present invention, mention of the method is simple, high reaction yield, product easily separated, suitable for industrial production and so on.
Preparation Method A silodosin important intermediate, comprising the following steps: Step I) in a flask, 282g of raw materials 1-acetyl-5- (2-bromo-propyl) indoline, 222g o phthalimide potassium and 700mL DMF, heated at 110 ° C for 2h; After completion of the reaction, to which was added the right amount of water to wash away the excess solvent DMF and salt extraction desolventizing after EA, was 296g crude;
Step 2) In a flask was added 296g crude product obtained in step I, dissolved 800mL ethanol, was added 165mL of hydrazine hydrate, 50 ° C is heated to precipitate a white solid; After completion of the reaction, cooling suction filtered, the filter cake washed with ethanol, and then the mother liquor removing solvent under reduced pressure; After dissolving EA, washed with water to wash away the excess hydrazine, and finally the organic phase the solvent was removed to give 165g intermediate, i.e. 1-acetyl-5- (2-aminopropyl) indoline;
Step 3) In the three-necked flask, 165g of Intermediate 1-acetyl-5- (2-aminopropyl) indoline, dissolved 600mL methanol, stirred at room temperature, and thereto was slowly added dropwise bromine; the addition was complete After stirring at room temperature 5-6h; After completion of the reaction, slowly poured into saturated NaHSO3, and wash away excess bromine; extracted with ethyl acetate, washed with water and saturated brine, dried over anhydrous sodium sulfate; After filtration, the solvent was removed in vacuo to give the crude product recrystallized from toluene to give 177g pure product;
Step 4) In a flask was added 177g of pure product obtained in Step 3 and 65g CuCN, after use 700mL DMF, was heated at 110 ° C for 3 to 5h; After completion of the reaction, the amount of water was added thereto, washing off excess solvent DMF and salt, EA desolventizing crude extract, after recrystallization 121g pure 1-acetyl-5- (2-bromo-propyl) -7-cyano-indoline that silodosin important intermediates;
Step (1), (2), (3), (4) synthesis reaction is:

Further, the step I) to step 4) by TLC plate tracking point detection reaction.
…………………………………………………
WO2013056842
http://www.google.com/patents/WO2013056842A1?cl=en
Silodosin is commercially available under the tradenames RAPAFLO® or
UROPvEC as a capsule formulation for oral use containing 4 mg or 8 mg of the drug. The capsules are to be taken orally once daily for the treatment of the signs and symptoms of benign prostatic hyperplasia. US 5,387,603 and EP 0 600 675 disclose silodosin as a therapeutic agent for the treatment for dysurea associated with benign prostatic hyperplasia. The molecular structure of silodosin (XXV) is shown below.
(XXV)

The synthesis of silodosin is relatively complex and requires a sequence of multiple steps. A key intermediate compound in the synthesis of silodosin is the optically active amine compound represented by the general formula R-Y:

1
wherein, R represents a protecting group and R represents a cyano (CN) or carbamoyl (CONH2) group. The intermediate compound R-Y bears the asymmetric carbon atom that imparts the optical activity to silodosin. Therefore, it is important to obtain the compound R-Y with high optical purity, because according to the methods reported in the state of the art the optical purity of the compound R-Y determines the optical purity of the final product silodosin.
JP 2001-199956 discloses a process for the preparation of a compound of formula R-Y, wherein l-(3-benzoyloxypropyl)-7-cyano-5-(2-oxopropyl)-2,3- dihydroindole or the corresponding 7-carbamoyl derivative is reacted with an optically active amine, namely L-2-phenylglycinol or L-l-phenylethanamine, to afford an imine compound of formula III as depicted in the below scheme 1. Scheme l . JP 2001-199956

R1 = COPh; R2 = CN or CONH2; R3 = H or OH a = 1. cat. deprotection
2. frational crystallization with L-tartaric acid
b = 1. chromatographic separation
2. cat. deprotection
The optically active imine III is subjected to catalytic hydrogenation using platinum(IV) oxide as a catalyst affording the diastereomers IV in a ratio of 3.8:1. The chiral auxiliary II is subsequently removed by catalytic hydrogenation using 10% palladium on carbon, i. e. under the typical conditions which lead to the cleavage and removal of benzylic protecting groups from nitrogen or oxygen atoms. The catalytic deprotection reaction affords the desired intermediate compound R-Y with an optical purity corresponding to the ratio of the diasteromers obtained in the previous step, i. e. the ratio of compound R-Y to S-Y is approximately 3.8: 1, which corresponds to an optical purity of approximately 58.3% enantiomeric excess (e.e.).
In order to increase the optical purity of the intermediate R-Y JP 2001-199956 suggests to conduct a fractional crystallization of the desired enantiomer with L-tartaric acid. After a series of fractional crystallizations the compound R-Y is obtained with an optical purity of 97.6% enantiomeric excess. Alternatively, the diastereomers of the compound of formula IV are separated using chromatographic techniques as column chromatography on silicagel. The pure diastereomer R-TV affords the desired enantiomer R-Y with an optical purity of 100% e.e. after removal of the chiral auxiliary II with hydrogen using 10% palladium on carbon as catalyst.
Another approach for the synthesis of the key intermediate compound R-Y is reported in JP 2002-265444. The route of synthesis disclosed in said document is depicted in the below scheme 2.
Scheme 2. JP 2002-265444


R1 = CH2Ph (Bn); R2 = CN The process involves the reaction of an enantiomeric mixture of the compound of formula VI with (I S, 2R)-2-benzylaminocyclohexane methanol (VII) to obtain a diastereomeric mixture containing the salt VIII. After a series of crystallizations the diastereomer VIII was obtained with an optical purity of 92.8% diastereomeric excess (d.e.). Subsequently, the salt VIII was treated with an acidic aqueous solution to release the acid R-Vl from the salt. After extraction from the aqueous solution with ethyl acetate the acid R-Vl is converted into its amide IX. The compound IX is finally subjected to a Hofmann type rearrangement reaction to obtain the desired intermediate compound R-V.
WO 201 1/030356 discloses a process for the preparation of the intermediate compound R-V, which avoids the resolution of the enantiomers of specific intermediate compounds using chiral auxiliaries or optically active bases. The route of synthesis described in WO 201 1/030356 starts from L-alanine (X), which is a naturally occurring optically active amino acid. The process described in
WO 2011/030356 is depicted in the below scheme 3.

R1 = trimethylsilyl (TMS), tert-butyl dimethylsilyl (TBDMS), allyl, benzyl, propargyl R2 = CN or CONH2 The amino acid is protected by the addition of ethyl chloroformate and subsequently activated by the addition of oxalyl chloride to afford i?-(N-ethoxycarbonyl)alanine as an acyl chloride (XI). Said acyl chloride is reacted with hydroxy protected l-(3- hydroxypropyl)-7-cyano-2,3-dihydroindole of formula XII in a Friedel-Crafts acylation reaction, which gives a compound of formula XIII. The oxo group in compound XIII is reduced to afford a compound of formula XIV that is subsequently subjected to a hydrolysis reaction to yield the key intermediate compound R-Y. It is an object of the present invention to provide a process for preparing silodosin or a pharmaceutically acceptable salt thereof, which process affords the drug with high optical purity and with better yield compared to the prior art processes. This object is solved by the subject matter as defined in the claims.
Scheme 5. Conversion of com ound V to silodosin

R = protecting group
R2 = CN or CONH2
X = leaving group
Example 11. Silodosin (XXV)
A. The compound XXIV (18.0 g) was dissolved in methanol (150 ml) and 5% aqueous sodium hydroxide solution (50 ml). The reaction mixture was stirred at room temperature for 2 h. The deprotected compound XXIV, i. e. a compound of formula XXIV with R = hydrogen and R = cyano, was extracted with toluene. Subsequently, a 10% lactic acid solution (25 ml) was added to the toluene phase in order to extract the product in the aqueous phase. The aqueous solution was separated and then basified. The deprotected product was finally extracted with ethyl acetate. Removal of the solvent gives the deprotected compound to XXIV (R1 = H and R2 = CN; 1 1.0 g) as an oily mass.
B. A mixture of compound XXIV (R1 = H and R2 – CN; 10.0 g), DMSO (80 ml) and 5N NaOH solution (9.0 ml) was stirred for 15 min. at room temperature. An aqueous H202 (30%) solution (1 1.0 ml) was added to the reaction mixture, which was stirred at room temperature for additional 2 h after completion of the addition. Water was added to the reaction mixture, the product was extracted with ethyl acetate, and the solvent was subsequently evaporated to afford 9.0 g crude silodosin.
Example 12. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 85.0% e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 97.5% e.e.
Example 13. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 98.5% e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25 °C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 99.9% e.e.
Example 14. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 90.0 %e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 97.0% e.e.
Example 15. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 92.0% e.e.) was dissolved in isopropyl acetate (160 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 8.2 g of XXV with an optical purity of 98.0% e.e. Example 16. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 98.0% e.e.) was dissolved in isopropyl acetate (160 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 8.0 g of XXV with an optical purity of 99.5% e.e.
………………………………
EP2475634
http://www.google.com/patents/EP2475634A2?cl=en
Scheme- 1.

Scheme-2.

Scheme-3.




Scheme-4.

Scheme-5.

Example-14
Preparation of Preparation of l-(3-Hydroxy-propyl)-5-(2(R)-{2-[2-(2, 2, 2-trifluoro- ethoxy)-phenoxy]-ethyIamino}-propyl)-2,3-dihydro-lH-indol-7-carboxylic acid amide (I)(Silodosin)
To a solution of Benzoic acid 3-[5(R)-(2-amino-propyl)-7-cyano-2, 3-dihydro-indol-l- yl]-propyl ester (XV) (3.5 g, 10 mmole) in Dimethyl sulphoxide (60 ml), charged Hydrogen peroxide (10% w/w) (11 ml). Then added 5 N sodium hydroxide solution (12.3 ml) and reaction mass was stirred for 2 hours. After completion of reaction water was added and extracted the product in ethyl acetate. Organic layer was washed with brine and dried over sodium sulphate. The solvent was evaporated below 40°C under reduced pressure and added methanol (25 ml). To this solution charged glacial acetic acid (0.25 g, 4mmole) and [2-(2, 2, 2-Trifluoro-ethoxy)-phenoxy]-acetaldehyde (VIII) (3 g, 0.0125 mole). Reaction mixture was stirred at 25-30°C for 1 hour. Then reacted with sodium cyanoborohydride (0.15 g, 2.8 mmoles) and heated at 40-45°C for 2 hours. After the completion of reaction solvent was distilled off below 40°C under reduced pressure and added water to the residue. Reaction mass was then acidified with aqueous mineral acid. The aqueous layer was then basified and product was extracted in ethyl acetate. Organic layer was washed with water and dried over sodium sulphate. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel using a mixture of ethyl acetate and hexane (5/95) as eluent to give 0.8g of (I) as yellow solid. Purity (by HPLC) = 98%
Example 15
Preparation of l-(3-hydroxypropyl)-5-[(2R)-({2-[2-(2, 2, 2-trifIuoroethoxy) phenoxy]-ethyl} amino) propyl]-2, 3-dihydro-lH-indole-7-carbonitriIe (XVII) A mixture of 3-[7-Cyano-5 (R)-[-2-{2-[2-(2,2,2-trifluoroethoxy)-phenoxy] ethyl} amino) propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (XVI) (6.0 g , 0.010 mole), methanol (30 ml) and aqueous solution of Sodium hydroxide ( 1.6 g in 8 ml of water) was stirred at ambient temperature for 6 hours. To the reaction mixture water (90ml) was added and product was extracted with ethyl acetate (90 ml). The organic layer was washed with saturated sodium bicarbonate solution followed by brine wash and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to give 3.85 g of (XVII). Example 16
Preparation of l-(3-Hydroxy-propyl)-5(R)-(2-{2-[2-(2, 2, 2-trifluoro-ethoxy)- phenoxy]-ethylamino}-propyl)-2, 3-dihydro-lH-indol-7-carboxylic acid amide (I) (Silodosin)
To a solution of l-(3-hydroxypropyl)-5(R)-[2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy]- ethyl}amino)propyl]-2,3-dihydro-lH-indole-7-carbonitrile (XVII) (6.0 g , 0.013 mole) in dimethylsulfoxide (75 ml) was added 5 N sodium hydroxide solution (4.5 ml). To this reaction mixture, 30 % hydrogen peroxide (2.63 ml) was added slowly below 25°C. Reaction mixture was stirred at ambient temperature for 6 hours. Aqueous solution of sodium sulfite (2.1 in 150 ml water) was added to the reaction mixture. The reaction mixture was extracted with ethyl acetate. The combined ethyl acetate layer was extracted 2N hydrochloric acid. The aqueous layer was neutralized with sodium bicarbonate and extracted the product in ethyl acetate. The organic layer was washed with saturated sodium bicarbonate solution followed by brine wash and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate. The resulting solution was cooled to 5°C and filtered to get 4.51 g of (I) as solid.
…………………………………………………
WO2012147019
http://www.google.com/patents/WO2012147019A1?cl=en
The present invention provides a process for the preparation of Silodosin of formula (I). More particularly, the present invention provides the process for preparation of tartrate salt of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy ] ethyl } amino)propyl] -2, 3 -dihydro- 1 H-indol- 1 -y 1 } propyl benzoate of formula (IV), which is a precursor in the preparation of Silodosin.

Background of the Invention:
A compound of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy) phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (IV) is a key intermediate for preparation of Silodosin. The chemical name of Silodosin is l-(3- hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl} amino) propyl]-2,3-dihydro-lH-indole-7-carboxamide and structurally represented as
(IV)

(I)
U.S.Pat. No. 5,387,603 discloses Silodosin as therapeutic agents for the treatment of dysuria, urinary disturbance associated with benign prostatic hyperplasia.
U.S.Pat. No. 6,310,086 discloses a process for preparing a Silodosin analogue compound from reaction of (R)-3-{5-(2-aminopropyl)-7-cyano-2,3- dihydro- 1 H-indol- 1 -yl jpropylbenzoate with 2-(2-Ethoxyphenoxy)ethyl methane sulfonate and finally isolated as residue and purified by column chromatography on silicagel. The said literature process has certain drawbacks like use of column chromatography.
U.S.Pat. No. 7,834,193 (IN 3178/DELNP/2007) discloses the process for preparation of monooxalate salt of 3-{7-cyano-5-[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy ] ethyl } amino)propyl] -2, 3 -dihydro- 1 H-indol- 1 -y 1 } propyl benzoate (IV). This patent specifically discloses the preparation of monooxalate salt of formula (IV) helps to remove N,N-dialkyl impurity to certain extend. CN 101993405 A discloses the reaction of (R)-5-(2-aminopropyl)-l-(3-(4- fluorobenzoyloxy)propyl)-7-cyanoindoline with 2-(2-(2,2,2-trifluoroethoxy) phenoxy)ethyl methane sulfonate followed by oxalic acid salt preparation.
The main drawback in the prior art process, the formation of N,N-dialkyl impurity compound of formula (VI), as disclosed in detailed description, in the preparation of Silodosin, during condensation of compound of formula (II) with compound of formula (III), the impurity which is not removable by crystallization method or precipitation technique and column chromatography purification is not suitable for commercial purpose. So considering the commercial importance of Silodosin, the present invention focus on the preparation of pure Silodosin, and surprisingly found that the isolation of formula (IV) as tartrate salt helps to prepare Silodosin having less than 0.2 % of N,N dialkyl impurity and with good yield. None of the prior arts teaches or motivates isolation of tartaric acid addition salt of formula (IV). The preparation of Silodosin from tartrate salt of 3-{7-cyano-5-[(2R)- 2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH- indol-l-yl} propyl benzoate (IV) or its freebase of the present invention has purity of greater than 99.6 %.

Example 3
Preparation of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy) phenoxy] ethyl-} amino) propyl]-2,3-dihydro-lH-indole-7-carboxamide (Silodosin)
Method A: The compound of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy] ethyl- } amino)propyl] -2,3 -dihydro- 1 H-indole-7- carbonitrile of formula (V) in dimethylsulfoxide was treated with 48% hydrogen peroxide and 20% sodium hydroxide solution and stirred at room temperature till completion of reaction. After completion of reaction, reaction mass quenched with 5% sodium bisulphite solution and ethylacetate was added over it. The ethylacetate layer was separated and treated with 20 % aqueous hydrochloric acid. The aqueous layer separated, neutralized with sodium bicarbonate solution and extracted with ethylacetate. The separated organic layer was washed with 10% sodium bicarbonate solution, brine solution and dried under vacuum. The organic layer distilled upto residue under vacuum at 50-55°C. The obtained residue was crystallized in ethylacetate.
Method B: To the tartrate salt of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy) phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (IV) (100 grams) in methanol, aqueous potassium hydroxide solution (38.38 grams) was added and stirred at room temperature till reaction completion. After completion of reaction, DM water and dichloromethane was added over it under stirring. Organic layer separated, washed with brine solution distilled under vacuum upto less than 1 volume. To the solution, dimethyl sulphoxide, 20% sodium hydroxide and hydrogen peroxide was added and stirred till completion of reaction. After completion of reaction, water containing sodium bisulfite was added to the reaction mass. The pH of the reaction mixture adjusted to about 8.5 using 10% sodium hydroxide and extracted in dichloromethane twice, washed with water, dried and concentrated upto 1-2 volume under vacuum. To the obtained solution, toluene was added over it at room temperature under stirring. The reaction mixture maintained for complete solid formation, filtered and dried under vacuum. Yield 58 grams. Example 4
Purification of Silodosin:
Method A: To the mixture of toluene and acetonitrile solvent, Silodosin was added over it and heated to 50° – 55 °C for complete dissolution. The reaction mass gradually cooled to room temperature and maintained for completion of solid formation. The obtained solid is filtered, washed with toluene and dried under vacuum. Method B: To the mixture of ethyl acetate and toluene solvent, Silodosin was added over it and heated to 60° – 65 °C for complete dissolution. The reaction mass gradually cooled to room temperature and maintained for completion of solid formation. The obtained solid is filtered, washed with toluene and dried under vacuum.
//////////////////////////////////////////
CN101993407
http://www.google.com/patents/CN101993407B?cl=en
silodosin for selective inhibition of urethral smooth muscle contraction and reduce the pressure within the urethra, but no significant impact on blood pressure, for the treatment of benign prostatic hyperplasia. At present, the method of synthesis Silodosin many reports, but the lack of high yield method for industrial production.
JP200199956 reported that benzoic acid as a starting material, 1_ (3_ benzoyloxy-propyl) indoline hydrochloride (structural formula (1), R is a hydrogen atom) in 60% yield, then through the multi-step reaction was further prepared silodosin intermediate 1- (3-benzoyloxy-propyl) -5- (2-nitro-propyl) -7-cyano-indoline (structural formula VIII ), the total yield is low, and only 20 percent. Compound (VIII) with potassium carbonate, the reaction of hydrogen peroxide to yield compound (IX), impurities, and purified by column chromatography to be not suitable for industrial production. Compound (IX) under catalysis of molybdenum oxide, and L- (S) – benzyl glycyl alcohol asymmetric reactions, protecting groups may be due to steric hindrance is small, low chiral induction, is 3.8: I.




Silodosin Preparation: 12 Example
Example 11 to give 8 g solid, dissolved in DMSO 100ml, was added 5mol / L NaOH 12ml, 18 ~ 20 ° C was added dropwise slowly with 30% H2027 grams, then 30 ° C, the reaction ended 4h. Extracted with ethyl acetate, the combined organic layer was washed 2N HCl and then the organic layer, the aqueous layer was neutralized with sodium hydroxide, and then extracted with ethyl acetate, washed with saturated sodium bicarbonate, dried over anhydrous sodium sulfate, and evaporated concentrated and then dissolved in ethyl acetate, natural cooling crystallization, filtration, drying 5 g (87%), purity> 99%.
Mp 105 ~ 108 ° C
[a] 20d = -16.2 C = I, MeOH
1NMR spectrum (DMS0-d6): δ ppm 0.9-1.0 (3H, d), 1.5-1.6 (1H, s), 1.6-1.7 (2H, m),
2.3-2.4 (1H, dd), 2.6-2.7 (1H, dd), 2.8-3.0 (5H, m), 3.1-3.2 (2H, m), 3.3-3.4 (2H, m),
3.4-3.5 (2H, t), 4.0-4.1 (2H, t), 4.2-4.3 (1H, s), 4.6-4.8 (2H, t), 6.9-7.15 (6H, m),
7.2-7.3 (1H, s), 7.5-7.6 (1H, s)
…………………………………………………..
see
Silodosin is (I) (formula 1, claim 1, page 31).
Process for the prepartion of pure polymorphic gamma form of silodosin – comprising dissolving any polymorphic form of silodosin in a solvent and seeding gamma form of silodosin.
Crude (I) (50 g) was dissolved in methanol, filtered and solvent was distilled under vacuum. The residue was dissolved in isopropanol at 50 degreeC, cooled and seed of (I) gamma form was added and further cooled and cyclohexane (500 mL) was added, solid was filtered, washed and dried to obtain pure polymorphic form gamma of (I) having a toluene content of 12 ppm (example 10, pages 29-30).
A process for the preparation of silodosin and/or its salt is claimed, comprising the reaction of 3-[5-((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-1H-indol-1-yl]propyl benzoate(2R,3R)-monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate to form a cyano benzyloxy intermediate, followed by hydrolysis to form a cyano hydroxy intermediate, which is then reacted with tartaric acid and hydrolyzed in the presence of an oxidizing agent to obtain the product. An alternate method of preparation of silodosin comprising the hydrolyses of tartrate salt of cyano hydroxy intermediate in the presence of an oxidizing agent, pure polymorphic form gamma of silodosin, and the cyano hydroxy intermediate are also claimed. Further processes for the prepartion of the pure polymorphic form gamma of silodosin are claimed, wherein the process involves the dissolution of of any polymorphic form of silodosin in a solvent by heating at 30-100 degree C, cooling before and after seeding with gamma form of silodosin, adding an antisolven, isolating the polymorph and optionally micronizing.
The present invention provides an improved and efficient process for the preparation of

It acts as an selective ai -adrenoceptor antagonist and is useful in the symptomatic treatment of benign prostatic hyperplasia (BPH). Chemically it is known as l-(3-hydroxypropyl)-5-[(2R)- ( { 2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino) propyl] indoline-7-carboxamide.
Silodosin and its pharmaceutically acceptable salts are first disclosed in US patent 5,387,603. Synthetic approach for the production of silodosin, is described in patent ‘603 can be represented as shown below in scheme 1.
l

Scheme 1
As represented in scheme 1, silodosin is prepared by the reaction of l-acetyl-5-(2r aminopropyl)indoline-7-carbonitrile with 2-[2-(2,2,2-trifiuoroethoxy)phenoxy] ethyl methanesulfonate in the presence of sodium bicarbonate in ethanol to give l-acetyl-5-[2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino]propyl]indoline-7-carbonitrile, which upon reaction with di-tert-butyldicarbonate in methylene chloride produces protected acetyl indoline carbonitrile compound. Further deacetylation with sodium hydroxide in ethanol followed by treatment with acetic acid provides protected indoline carbonitrile compound, which upon hydrolysis using dimethyl sulfoxide, 30% hydrogen peroxide, sodium hydroxide and acetic acid gives protected indoline carboxamide, which upon further reaction with 2-tert-butyldimethylsiloxy)ethyl-4-nitrobenzene sulfonate in the presence of cis-dicyclohexano-18 crown-6 and potassium carbonate in dioxane gives protected (tert-butyl-dimethylsiloxy) ethyl indoline carbonitrile. Further treatment with tetrabutylammonium fluoride in tetrahydrofuran produces N-boc protected hydroxy deprotected propyl indoline carbonitrile, which under goes facile deprotection of boc group upon treatment with trifluoroacetic acid, in methylene chloride to yield silodosin. The complete process is very complex, make use of pyrophoric reagents
which are very difficult to handle in large scale and have many extra steps involving protection and depfotection. Further in US patent ‘603, concrete detail of preparation and purification of silodosin have not been reported. Furthermore, isolated silodosin is characterized using IR, NMR and specific rotation but the patent is silent on product appearance and crystalline nature. There are several processes known for the preparation of silodosin and its intermediates viz; in JP 4634560; JP 4921646; JP-2006- 188470; WO2011/124704 and WO2011/101864. In most of the inventions, silodosin is prepared by following reaction as shown in scheme 2. Major disadvantages of these processes are the formation of N,N dialkyl impurity, and other impurities which forms during the condensation of 3-[5-((2/?)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]propyl benzoate or its salts like monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methanesulfonate. N,N dialkyl impurity forms in about 12-15% and may form due to reaction of one molecule of benzoate compound with two molecules of methanesulfonate compound. Removal of this impurity is not possible by simple purification

wherein R is benzoyl, benzyl, tetrahydropyranyl, 2-trimethylsilylethyl, dinitrophenyl, diphenyl methyl and the like
Scheme 2
US patent 7,834,193 discloses a process for preparation of silodosin with similar condensation of 3-[5-((2R)-2-arriinopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]pfopyl benzoate or its salts like monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate, but 3-{7-cyano-5-[(2R)-2-({2-(2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propylbenzoate is purified by preparing monooxalate salt as shown below in
scheme 3. This patent specifically prepares monooxalate salt of 3- {7-cyano-5-[(2R)-2-({ 2- (2,2,2-trifluoroethoxy)-phenoxy]ethyl }amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propyl benzoate to remove N,N÷dialkyl impurity, but impurity has not been removed completely, only a certain % of it, has been removed.

Scheme 3
In PCT publication WO2012/131710, preparation of silodosin is described wherein improved processes for preparation of 3-[5-((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l- yl]propyl benzoate have been disclosed which is then converted to silodosin by condensation with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate. In exemplified process, 3-[5- ((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]propyl benzoate is condensed with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate and the resulting benzoate compound is hydrolyzed to give l-(3-hydroxy propyl)-5-[(2R)-2-({ 2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyr]-2,3-dihydro-lH-indol-7-carbonitrile.The carbonitrile compound is treated with oxalic acid to prepare its oxalate salt having purity greater than 99%, which is then hydrolyzed using a base to prepare free carbonitrile compound having purity greater than 99%, but this patent is silent about N, N- dialkyl impurity or its removal.
In PCT publication WO2012/147019, preparation of silodosin using 3-{ 7-cyano-5-[(2R)-2-({2- (2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propyl benzoate tartrate salt has been described as shown below in scheme 4.

Scheme 4
One other PCT publication WO2012/147107 describes preparation of silodosin by preparing hydrochloride and acetic acid salts of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyl]-2,3-dihydro-lH-indol-7-carbonitrile to remove N,N dialkyl impurity. It has been observed that in exemplified process, wherein hydroxy compound namely l-(3-hydroxy propyl)-5-[(2R)-2-({2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyl]-2,3-dihydro-lH-indol-7-carbonitrile is purified by preparing its acetate salt to, remove the impurities but still N, N-dialkyl impurity remains in an amount of 0.6%, which is difficult to remove in next stage or require extra purifications.
Beside to use highly pure silodosin, use of a pure polymorphic form of API is an essential requirement of drug formulation, these both aspects when address jointly, and obtained silodosin can be converted to pure polymorph then only a complete solution of prior art problems can be achieved. Apart from above mentioned process patents/publications which aimed to prepare the pure silodosin, there are exist some polymorph patents/publications which also aims to prepare pure polymorphic form of silodosin.
Polymorphism is considered as one of the- most important solid-state property of drug substance, since different polymorph have different physiochemical and biological properties and in pharmaceutical chemistry it is often desired to obtain one particular form that is biologically active and also offers ease of handling during formulation. The available literature references related to polymorph of silodosin are incorporated herein.
Japanese patent 3331048 (publication No.H07-330726), discloses a process for purification of silodosin wherein silodosin is dissolved in ethyl acetate, dried over anhydrous magnesium sulfate, solvent is distilled off and again dissolved in ethyl acetate at 70°C and crystallizes below room temperature. The resulting product is characterized by melting point, IR, NMR and specific rotation. Here also disclosure is silent about polymorphic form of product.
US patent publication US2006/0142374A1 (equivalent European patent EP1541554B 1) discloses polymorphic forms of silodosin including three crystalline polymorphic form of silodosin which are named as alpha (a), beta (β) and gamma (γ) and one amorphous form. These polymorphic forms have been characterized by X-ray powder diffraction pattern. In the patent publication, processes for the preparation of all these three crystalline forms have been disclosed. In. a given process, form alpha is prepared by dissolving crude silodosin in appropriate amount of ethyl acetate, ethyl formate, acetone, methyl ethyl ketone, acetonitrile, tetrahydrofuran or mixture of acetone and acetonitrile (1: 1), preferably ethyl acetate under heating, allowing to stand at room temperature to precipitate the crystal gradually. Similarly, form beta is prepared by dissolving crude silodosin in appropriate amount of methanol under heating, adding petroleum ether as a anti-solvent, crystal precipitation is ensured using vigorous stirring.
In a second process, to prepare the form beta, crude silodosin is dissolved in ethanol or 1-propanol and the reaction mass is cooled quickly. The crystalline form gamma is prepared by dissolving crude silodosin in appropriate amount of toluene or a mixture of acetonitrile and toluene (1:4) or ethyl acetate and toluene (1: 19), preferably in toluene, under heating, cooling to room temperature and allowing to precipitate gradually upon standing. In a second process to prepare form gamma, crude silodosin is dissolved in 2-propanol and the crystals are precipitated by adding an appropriate amount of toluene. In spite of disclosing three crystalline polymorphic forms, the patent publication prefers preparation and use of form alpha by highlighting the problems faced for preparation and use of other forms. It is disclosed that crystal form beta has manufacturing difficulties at industrial scale since precipitation occurs only when the nonpolar antisolvent is added to warm solution which leads to inconsistency in quality of crystals.
With the second process for preparation of form beta, desired level of yield and purity has not been achieved. Further, according to this publication, preparation of gamma form involves use of toluene which can not be removed completely from final product, because of its high boiling point and raises the problem of residual solvent. In the case of toluene, a class 2 solvent, its limits should not be more than 890 ppm. In the exemplified process, toluene content has not been disclosed, which clearly reflects that product was not suitable for pharmaceutical composition having problem of high residual content of toluene. Furthermore patent publication also states that all the three crystal forms donot have any difference in hygroscopicity and stabilities.
Thereafter, several patents/publications disclose preparation of polymorphic forms alpha and beta. For example a PCT publication WO2012/147107 discloses a process for preparation of beta form using isopropyl acetate and methyl isobutylketone. In another PCT publication WO2012/077138, preparation of alpha and beta forms are disclosed using various solvent , system. Similarly, in a Chinese patent CN102010359, crystalline form beta is prepared by dissolving the crude silodosin in alcoholic solvent by heating and the product is crystallized by cooling or by adding an antisolvent such as ketone or ether.
European patent EP2474529 discloses new polymorphic forms delta (δ) and eta (ε) of silodosin by using a solvent (tetrahydrofuran) and antisolvent (n-heptane, n-hexane, cyclohexane, tert butylmethyl ether).Further it discloses conversion of delta form to beta form by just heating the delta form at a particular temperature. The form delta can also be transformed into form eta by. slurrying in aqueous methanol. One new crystalline form designated as delta has also been disclosed in a Chinese patent publication CN102229558. An Indian patent application 478/MUM/2010, also discloses a new polymorphic form Zy-S which is prepared by using solvent such as esters, aromatic hydrocarbons, ketones, and alcohols.
All the above disclosures are silent about the preparation of gamma form of silodosin and only available disclosure reports that gamma form have problem of residual solvent, as impurity and is not suitable for pharmaceutical compositions.
Method C: l-(3-HydroxypropyI)-5-[(2R)-2-({2-[2,2,2-trifIuoroethoxy)phenoxy] ethyl} amino) propyl]-2,3-dihydro-lH-indol-7-carbonitrile tartrate (lOg) dissolved in dimethylsulfoxide (120 ml) and to this solution, was added 5 mol/L aqueous sodium hydroxide solution (15ml). To the reaction mixture, 30% hydrogen peroxide (5ml) was added and keeping the temperature below 25°C. The reaction mixture was stirred at 20-25°C, for 5 hours. To the reaction mixture, sodium sulfite (5g) dissolved in water (100ml) was added slowly. The reaction mixture was extracted with ethyl acetate (1x200ml) and ethyl acetate layer was concentrated under reduced pressure. The resulting product was dissolved in methanol and clear solution was filtered through micron filter paper of size 0.22 micron two times and filtrate was concentrated.The resulting compound was dissolved in toliiene (70ml) and isopropyl alcohol (7ml) at 50-55°C and the solution was cooled to 20-25°C, cyclohexane was added and stirred for further 4 hours, filtered and dried to give title compound having purity 99.86% and N,N-dialkyl impurity not detected by HPLC. Example 5: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin (15g) having toluene content 1872 ppm, was micronized under air pressure. The micronized product was dried under vacuum at 55°C-60°C for 23.0 hours to afford pure polymorphic form gamma of silodosin having toluene content 460 ppm.
Example 6: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin [having toluene content 1327 ppm] was micronized under air pressure. The micronized product was dried under vacuum at 55°C-60°C for 16 hours to afford pure polymorphic form gamma of silodosin having toluene content 350 ppm.
Example 7: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin crude (3.0g) was dissolved in isopropanol (12ml) at 50°C and reaction mass was cooled to 35°C and seed of silodosin gamma form (O.lg) was added. Thereafter reaction mass was again cooled to 15-20°C and cyclohexane (30ml) was added to the reaction mass and stirred for further 0.5 hour. The resulting solid, thus obtained, was filtered, washed with cyclohexane and dried to afford pure polymorphic form gamma of silodosin having toluene content 34 ppm.
References
- “Drugs.com, Watson Announces Silodosin NDA Accepted for Filing by FDA for the Treatment of Benign Prostatic Hyperplasia”. Retrieved 2008-02-13.
- European Medicines Agency: Assessment report for Silodyx
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Kobayashi K, Masumori N, Kato R, Hisasue S, Furuya R, Tsukamoto T. (December 2009). “Orgasm is preserved regardless of ejaculatory dysfunction with selective alpha1A-blocker administration.”. Int J Impot Res. 21 (5): 306–10. doi:10.1038/ijir.2009.27. PMC 2834370. PMID 19536124.
External links
a1a-Adrenoceptor antagonist. Prepn: M. Kitazawa et al., EP 600675; eidem, US 5387603 (1994, 1995 both to Kissei).PRODUCT PATENT
Adrenoceptor binding study: K. Shibata et al., Mol. Pharmacol. 48, 250 (1995); and tissue selectivity: S. Murata et al., J. Urol. 164, 578 (2000).
Pharmacology: K. Akiyama et al., Pharmacology 64, 140 (2002).
Series of articles on pharmacology, pharmacokinetcs and toxicology: Yakugaku Zasshi 126, 187-263 (2006).
Review of development and therapeutic potential: F. Kamali, Curr. Opin. Cent. Peripher. Nerv. Syst. Invest. Drugs 1, 248-252 (1999)
| CN101993405A * | Aug 27, 2009 | Mar 30, 2011 | 浙江华海药业股份有限公司;上海医药工业研究院 | Indoline derivative as well as preparation method and application thereof |
| JP2006188470A * | Title not available | |||
| US7834193 * | Apr 16, 2007 | Nov 16, 2010 | Kissei Pharmaceutical Co., Ltd. | industrial production of silodosin (for treating dysuria associated with benign prostatic hyperplasia) via mixing 3-{7-cyano-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino]propyl]-2,3-dihydro-1H-indol-1-yl}-propyl benzoate and oxalic acid, nitrilizing, hydrolyzing |
| WO2011030356A2 * | Sep 13, 2010 | Mar 17, 2011 | Sandoz Ag | Process for the preparation of indoline derivatives and their intermediates thereof |
| WO2011124704A1 * | Apr 8, 2011 | Oct 13, 2011 | Ratiopharm Gmbh | Process for preparing an intermediate for silodosin |
| WO2012131710A2 * | Mar 27, 2012 | Oct 4, 2012 | Panacea Biotec Ltd | Novel process for the synthesis of indoline derivatives |
| JP2006188470A * | Title not available |
| Patent | Submitted | Granted |
|---|---|---|
| Combination therapy for the treatment of benign prostatic hyperplasia [US6410554] | 2002-06-25 | |
| Indoline compound and process for producing the same [US7834193] | 2007-08-23 | 2010-11-16 |
| Agents and crystals for improving excretory potency of urinary bladder [US8252814] | 2009-10-22 | 2012-08-28 |
| METHODS FOR TREATING BENIGN PROSTATIC HYPERPLASIA [US2011319464] | 2011-12-29 | |
| PREVENTIVE AND/OR THERAPEUTIC AGENT FOR URINE COLLECTION DISORDER ACCOMPANYING LOWER URINARY TRACT OBSTRUCTION [US2009227651] | 2009-09-10 | |
| PREVENTIVE AND/OR THERAPEUTIC AGENT FOR URINE COLLECTION DISORDER ACCOMPANYING LOWER URINARY TRACT OBSTRUCTION [US2010137399] | 2010-06-03 | |
| Agents for improving excretory potency of urinary bladder [US2004116457] | 2004-06-17 | |
| Medicinal Composition for Prevention of Transition to Operative Treatment for Prostatic Hypertrophy [US2008090893] | 2008-04-17 | |
| METHODS FOR TREATING BENIGN PROSTATIC HYPERPLASIA [US2008242717] | 2008-10-02 | |
| Agents and crystals for improving excretory potency of urinary bladder [US2006281725] | 2006-12-14 |
Sex has another benefit: It makes humans less prone to disease over time
Public Release: 16-Feb-2015
Mixing our genes through sex helps purge us of disease mutations
University of Montreal
For decades, theories on the genetic advantage of sexual reproduction had been put forward, but none had ever been proven in humans, until now. Researchers at the University of Montreal and the Sainte-Justine University Hospital Research Centre in Montreal, Canada have just shown how humanity’s predispositions to disease gradually decrease the more we mix our genetic material together. This discovery was finally made possible by the availability in recent years of repositories of biological samples and genetic data from different populations around the globe.
What we already knew
As humans procreate, generation after generation, the exchange of genetic material between man and woman causes our species to evolve little by little. Chromosomes from the mother and the father recombine to create the chromosomes of their child (chromosomes are the larger building blocks of…
View original post 600 more words
FLUDABARINE

FLUDABARINE
CAS : 21679-14-1
9-b-D-Arabinofuranosyl-2-fluoro-9H-purin-6-amine
Additional Names: 9-b-D-arabinofuranosyl-2-fluoroadenine; 2-fluorovidarabine; 2-fluoro-9-b-D-arabinofuranosyladenine; 2-F-araA
Manufacturers’ Codes: NSC-118218; NSC-118218-H
Molecular Formula: C10H12FN5O4
Molecular Weight: 285.23
Percent Composition: C 42.11%, H 4.24%, F 6.66%, N 24.55%, O 22.44%
Properties: Crystals from ethanol + water, mp 260°. [a]D25 +17 ±2.5° (c = 0.1 in ethanol). uv max (pH 1, pH 7, pH 13): 262, 261, 262 nm (e ´ 10-3 13.2, 14.8, 15.0). Sparingly sol in water, organic solvents.
Melting point: mp 260°
Optical Rotation: [a]D25 +17 ±2.5° (c = 0.1 in ethanol)
Absorption maximum: uv max (pH 1, pH 7, pH 13): 262, 261, 262 nm (e ´ 10-3 13.2, 14.8, 15.0)

Derivative Type: 5¢-Monophosphate
CAS : 75607-67-9
Additional Names: 2-F-ara-AMP
Manufacturers’ Codes: NSC-328002; NSC-312887
Trademarks: Fludara (Schering AG)
Molecular Formula: C10H13FN5O7P
Molecular Weight: 365.21
Percent Composition: C 32.89%, H 3.59%, F 5.20%, N 19.18%, O 30.67%, P 8.48%
Properties: Sol in water.
Therap-Cat: Phosphate as antineoplastic.
| Systematic (IUPAC) name | |
|---|---|
| [(2R,3R,4S,5R)-5-(6-amino-2-fluoro-purin-9-yl)- 3,4-dihydroxy-oxolan-2-yl]methoxyphosphonic acid | |
| Clinical data | |
| Trade names | Fludara |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a692003 |
|
|
| Legal status | |
| Routes | Intravenous, oral |
| Pharmacokinetic data | |
| Bioavailability | 55% |
| Protein binding | 19 to 29% |
| Half-life | 20 hours |
| Excretion | Renal |
| Identifiers | |
| CAS number | 75607-67-9 |
| ATC code | L01BB05 |
| PubChem | CID 657237 |
| DrugBank | DB01073 |
| ChemSpider | 571392 |
| UNII | P2K93U8740 |
| KEGG | D01907 |
| ChEBI | CHEBI:63599 |
| ChEMBL | CHEMBL1568 |
| Chemical data | |
| Formula | C10H13FN5O7P |
| Molecular mass | 365.212 g/mol |
……………….
Fludarabine or fludarabine phosphate (Fludara) is a chemotherapy drug used in the treatment of hematological malignancies(cancers of blood cells such as leukemias and lymphomas). It is a purine analog, which interferes with DNA synthesis.


Indications
Fludarabine is highly effective in the treatment of chronic lymphocytic leukemia, producing higher response rates than alkylating agents such as chlorambucil alone.[1] Fludarabine is used in various combinations with cyclophosphamide, mitoxantrone,dexamethasone and rituximab in the treatment of indolent non-Hodgkins lymphomas. As part of the FLAG regimen, fludarabine is used together with cytarabine and granulocyte colony-stimulating factor in the treatment of acute myeloid leukaemia. Because of its immunosuppressive effects, fludarabine is also used in some conditioning regimens prior to allogeneic stem cell transplant.
Pharmacology
Fludarabine is a purine analog, and can be given both orally and intravenously. Fludarabine inhibits DNA synthesis by interfering withribonucleotide reductase and DNA polymerase. It is active against both dividing and resting cells. Being phosphorylated, fludarabine is ionized at physiologic pH and is effectually trapped in blood. This provides some level of specificity for blood cells, both cancerous and healthy.
Side effects
Fludarabine is associated with profound lymphopenia, and as a consequence, increases the risk of opportunistic infectionssignificantly. Patients who have been treated with fludarabine will usually be asked to take co-trimoxazole or to use monthly nebulised pentamidine to prevent Pneumocystis jiroveci pneumonia. The profound lymphopenia caused by fludarabine renders patients susceptible to transfusion-associated graft versus host disease, an oftentimes fatal complication of blood transfusion. For this reason, all patients who have ever received fludarabine should only be given irradiated blood components.
Fludarabine causes anemia, thrombocytopenia and neutropenia, requiring regular blood count monitoring. Some patients require blood and platelet transfusion, or G-CSF injections to boost neutrophil counts.
Fludarabine is associated with the development of severe autoimmune hemolytic anemia in a proportion of patients.[2]
Difficulties are often encountered when harvesting peripheral blood stem cells from patients previously treated with fludarabine.[3]
History
Fludarabine was produced by John Montgomery and Kathleen Hewson of the Southern Research Institute in 1968.[4] Their previous work involved 2-fluoroadenosine, which was unsafe for use in humans; the change to this arabinose analogue was inspired by the success of vidarabine.[4]

-
Fludarabine (9-β-D-arabinofuranosyl-2-fluoroadenine) (II) is a purine nucleoside antimetabolite resistant to adenosine deaminase, employed for the treatment of leukemia.

-
Fludarabine is usually administered as a pro-drug, fludarabine phosphate, which is also the natural metabolite. Fludarabine was firstly synthesised by Montgomery (US 4,188,378 and US 4,210,745) starting from 2-aminoadenine. The method comprised acetylation of 2-aminoadenine, reaction with a benzyl-protected chlorosugar, deacetylation of the amino groups, diazotization and fluorination of the 2-amino group followed by deprotection of the sugar residue.
-
Fludarabine phosphate can be obtained according to conventional phosphorylation methods, typically by treatment with trimethylphosphate and phosphoryl chloride. Recently, a method for preparing highly pure fludarabine, fludarabine phosphate and salts thereof has been disclosed by Tilstam et al. (US 6,46,322).
-
Enzymatic synthesis has been regarded as a valid alternative to conventional methods for the synthesis of nucleosides and nucleotides derivatives. EP 0 867 516 discloses a method for the preparation of sugar nucleotides from sugar 1-phosphates and nucleosides monophosphates by use of yeast cells having nucleoside diphosphate-sugar pyrophosphorylase activity. EP 0721 511 B1 discloses the synthesis of vidarabine phosphate and fludarabine phosphate by reacting an arabinonucleotide with an arylphosphate in the presence of a microorganism able to catalyse the phosphorylation of nucleosides. This method is particularly convenient in that it does not require purified enzymes, but it does not allow to synthesise vidarabine and fludarabine.

………………………………………………………………
paper
Simple Modification To Obtain High Quality Fludarabine
API R & D Centre, Emcure Pharmaceuticals Ltd, I.TBT Park, Phase-II, M.IDC Hinjewadi, Pune-411057, India
Org. Process Res. Dev., 2012, 16 (5), pp 840–842
DOI: 10.1021/op3000509
http://pubs.acs.org/doi/abs/10.1021/op3000509

A simple and improved debenzylation process is described to obtain fludarabine in greater than 99.8% purity and 90–95% yield.
……………………………………………………………………….
Patents
http://www.google.com/patents/EP1464708A1?cl=en

-
The present invention relates to a process for the preparation of fludarabine phosphate (I) illustrated in the scheme and comprising the following steps:
- a) reaction of 2-fluoroadenine with 9-β-D-arabinofuranosyl-uracil in the presence of Enterobacter aerogenes to give crude fludarabine (II);
- b) treatment of crude fludarabine with acetic anhydride to 2′,3′,5′-tri-O-acetyl-9-β-D-arabinofuranosyl-2-fluoroadenine (III);
- c) hydrolysis and recrystallisation of intermediate (III) to give pure fludarabine;
- d) phosphorylation of fludarabine to give fludarabine phosphate (I).
-
Step a) is carried out in a 0.03 – 0.05 M KH2PO4 solution, heated to a temperature comprised between 50 and 70°C, preferably to 60°C, adjusted to pH 7 with KOH pellets and added with 2-fluoroadenine, Ara-U and EBA. The concentration of 2-fluoroadenine in the solution ranges from 0.02 to 0.03 M, while 9-β-D-arabinofuranosyl-uracil is used in a strong excess; preferably, the molar ratio between 9-β-D-arabinofuranosyl-uracil and 2-fluoroadenine ranges from 5:1 to 7:1, more preferably from 5.5:1 to 6.5:1. 2 – 2.5 1 of cell culture per 1 of KH2PO4 solution are used. The mixture is stirred at 60°C, adjusting the pH to 7 with a 25% KOH solution and the reaction is monitored by HPLC. Once the reaction is complete (about 24-26 hours), the cell material is separated by conventional dialysis and the permeated solutions are recovered and kept cool overnight. Crystallised fludarabine contains 10% 9-β-D-arabinofuranosyl adenine, which can be conveniently removed by means of steps b) and c).
-
In step b) crude fludarabine from step a) is dissolved in 9-11 volumes of acetic anhydride, preferably 10 volumes and reacted at 90 – 100°C under stirring, until completion of the reaction (about 10 – 12 h). Acetic anhydride is co-evaporated with acetone and the product is suspended in water.
-
The hydrolysis of step c) is carried out with methanol and ammonium hydroxide. Typically, compound (III) from step b) is suspended in 9-11 volumes of methanol and 2.5 – 3.5 volumes of 25% NH4OH and stirred at room temperature until complete hydrolysis (about 20 hours; the completion of the reaction can be promoted by mildly warming up the mixture to 30-32°C). Fludarabine precipitates by cooling the mixture to 10°C and is further hot-crystallised with water, preferably with 50 – 70 ml of water per gram of fludarabine or with a water/ethanol mixture (1/1 v/v) using 30 – 40 ml of mixture per gram of fludarabine. Fludarabine is recovered as the monohydrate and has a HPLC purity higher than 99%.
-
Even though the conversion of fludarabine into fludarabine phosphate (step d) can be carried out according to any conventional technique, for example as disclosed in US 4,357,324, we have found that an accurate control of the reaction and crystallisation temperature allows to minimise product decomposition and significantly improves the yield. According to a preferred embodiment of the invention, the reaction between phosphorus oxychloride, triethylphosphate and fludarabine is carried out at -10°C, and fludarabine phosphate is precipitated from water at 0°C.
-
In summary, the present invention allows to obtain the following advantages: fludarabine is prepared by enzymatic synthesis without the use of pure enzymes and is therefore particularly suitable for industrial scale; fludarabine is easily recovered and purified from 9-β-D-arabinofuranosyl adenine by acetylation without the need of chromatographic purification, since the triacetyl-derivative precipitates from water with high purity and yield; fludarabine phosphate can be obtained in high yield by controlling the reaction and crystallisation temperature in the phosphorylation step.
-
The following examples illustrate the invention in more detail.

EXAMPLES
Example 1 – Crude 9-β-D-arabinofuranosyl-2-fluoroadenine (II)
-
A solution of KH2PO4 (123 g, 0,9 moles) in water (13 l) was heated to 60°C under stirring and the pH adjusted to 7 with KOH pellets (130 g, 2.32 moles), then added with Ara-U (1451 g, 5.94 moles), 2-fluoroadenine (150 g, 0.98 moles) and EBA (ATCC® n° 13048) cell culture (30 l).
-
The mixture was stirred at 60°C for 24-26 hours, adjusting the pH to 7 with a 25% KOH solution and monitoring the reaction by HPLC.
-
After 24-26 hours the cell material was separated by dialysis at 50°-55°C, diluting the mixture with water. The permeated yellow clear solutions were collected, pooled (50 l) and left to stand at 0°-5°C overnight. The resulting crystalline precipitate was filtered and washed with cold water (2 l).
-
The product was dried at 45°C under vacuum for 16 hours to give 110 g of the crude compound (II) which was shown by HPLC to be a mixture of (I) (90%) and 9-β-D-arabinofuranosyl adenine (10%).
Example 2
Pure 9-β-D-arabinofuranosyl-2-fluoroadenine (II)
-
9-β-D-arabinofuranosyl-2-fluoroadenine (II) (30 g, 0,095 moles) was suspended in acetic anhydride (300 ml) and heated to 95°C under stirring.
-
After 7 hours a clear solution was obtained and left to react at 95°C for further 2-3 hours until the acetylation was completed.
-
The resulting yellow solution was then concentrated under vacuum at 45°C and the residue was co-evaporated with acetone (2 x 50 ml) and suspended in water (600 ml). The water suspension was cooled to room temperature and left under stirring for 1 hour.
-
The product was collected by filtration and washed with water (2 x 100 ml) to give 34 g of wet 2′,3′,5′-tri-O-acetyl-9-β-D-arabinofuranosyl-2-fluoroadenine (III).
-
Wet compound (III) was suspended in methanol (300 ml) and added with 25% NH4OH (100 ml). The mixture was left to stand at room temperature overnight and after 19 hours was warmed to 30°-32°C for 3 hours, until no starting material was detected by HPLC.
-
The suspension was cooled to 10°C for 1 hour, then the product was collected by filtration and washed with a methanol-water mixture (2 x 25 ml, 3:1 v/v). The product was dried under vacuum at 45°C overnight to give 17.5 g of fludarabine (II) (98.4% HPLC purity).
Method A
-
Re-crystallisation of compound (II) (17.5 g, 0.061 moles) was also carried out by suspending the product in water (875 ml) and heating to 95°C until a clear solution was obtained. The solution was allowed to cool spontaneously to room temperature and the crystalline product was filtered, washed with cold water (2 x 50 ml) and dried under vacuum at 45°C overnight, to give 15.5 g of pure fludarabine (II) as the monohydrate (99.3% HPLC purity).
-
The monohydrate was further dried under vacuum at 90°C for 24 hours to give pure anhydrous fludarabine (II).
Method B
-
Fludarabine (II) (35 g, 0.123 moles) was also re-crystallized by suspending the product in a water/ethanol mixture (1/1, v/v) (1050 ml) and heating to 80°C until a clear solution was obtained. The solution was allowed to cool spontaneously to room temperature and the crystalline product was filtered, washed with a water/ethanol mixture (2 x 50 ml) and dried under vacuum at 45°C overnight, to give 32 g of pure fludarabine (II) as the monohydrate ( 99% HPLC purity ).
-
The monohydrate was further dried under vacuum at 90°C for 24 hours to give pure anhydrous fludarabine (II).
Example 3 – 9-β-D-arabinofuranosyl-2-fluoroadenine-5′-phosphate (I)Method A
-
Phosphorous oxychloride (5 g, 3 ml, 0.033 moles) was added to cold (0°C, ice-bath) triethylphosphate (50 ml) and the solution was kept at 0°C for 1 hour, thereafter added with anhydrous fludarabine (II) (5 g, 0.018 moles) under stirring.
-
After about 3 hours, the reaction mixture became homogeneous and turned light-yellow and was kept at 0°C overnight. Once the phosphorylation was completed (about 23 hours) the mixture was added with water (10 ml) and the solution was stirred for 3 hours at 0°C. The mixture was then poured into cold (0°C) methylene chloride (400 ml) and kept at 0°C under stirring until a clear methylene chloride phase was obtained (at least 1 hours).
-
The methylene chloride phase was removed by decantation and the residual yellowish oil was dissolved in warm (50°C) water (30 ml). The solution was allowed to cool spontaneously to room temperature overnight and the resulting crystalline product was collected by filtration and washed with water (10 ml) and ethanol (2 x 10 ml).
-
The product was dried at room temperature under vacuum for 24 hours to give 4 g of compound (I).
-
Compound (I) was re-crystallised as follows: compound (I) (4 g) was dissolved in 60 ml of preheated deionized water (73°-75°C) and the solution was stirred and rapidly cooled to 50°C to minimize product decomposition. The solution was then allowed to cool spontaneously to room temperature: the precipitation started at 40°C. The resulting precipitate was collected by filtration and washed with water (10 ml) and ethanol (2 × 10 ml). The product was dried at room temperature under vacuum for 24 hours to give 2.5 g of compound (I).
Method B
-
Phosphorous oxychloride (5 g, 3 ml, 0.033 mol) was added to cold (-10°C) triethylphosphate (50 ml) and the solution was kept at -10°C for 1 hour, thereafter anhydrous fludarabine (II) (5 g, 0,018 mol) was added with stirring at -10°C.
-
After about 6 hours the reaction mixture turned light-yellow and became homogeneous. The mixture was kept at -10°C overnight and after 23 hours the phosphorylation was completed. After addition of 40 ml of cold water (2°C) the solution was stirred for 1 hour at 0°C and extracted with cold (0°C) methylene chloride (100 ml and two 50-ml portions).
-
The aqueous solution was kept under vacuum at room temperature for 1 hour and allowed to stand at 0°C for 24 hours. The resulting crystalline product (I) was collected by filtration and washed with ethanol (2 x 20 ml).
-
The product was dried at 40°C under vacuum for 24 hours (Yield: 5 g).
-
A final crystallization was carried out as follows. Compound (I) (5 g) was dissolved in 75 ml of preheated deionized water (73°-75°C) and the solution was stirred and rapidly cooled to 50°C to minimize decomposition. The solution was then allowed to cool spontaneously to room temperature (the precipitation started at 40°C). The resulting precipitate was collected by filtration and washed with water (10 ml ) and ethanol (2 x 10 ml). The product was dried at 40°C under vacuum for 24 hours (Yield: 4 g).
……………………………………………….
http://www.google.com/patents/US20100290990

References
- Rai KR et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med 2000;343:1750-7. doi:10.1056/NEJM200012143432402 PMID 11114313
- Gonzalez H et al. Severe autoimmune hemolytic anemia in eight patients treated with fludarabine. Hematol Cell Ther. 1998;40:113-8. PMID 9698219
- Tournilhac O et al. Impact of frontline fludarabine and cyclophosphamide combined treatment on peripheral blood stem cell mobilization in B-cell chronic lymphocytic leukemia. Blood 2004;103:363-5. PMID 12969985
- Sneader, Walter (2005). Drug discovery: a history. New York: Wiley. p. 258. ISBN 0-471-89979-8.
Literature References:
Adenosine deaminase-resistant purine nucleoside antimetabolite. Prepn and in vitro cytotoxicity: J. A. Montgomery, K. Hewson, J. Med. Chem. 12, 498 (1969). Improved prepn: J. A. Montgomery et al., J. Heterocycl. Chem. 16, 157 (1979); J. A. Montgomery, US 4210745 (1980 to U.S. Dept. Health, Education and Welfare).
Inhibition of DNA synthesis and in vivo antileukemic activity: R. W. Brockman et al., Biochem. Pharmacol. 26, 2193 (1977). Metabolized to 5¢-monophosphate: R. W. Brockman et al., Cancer Res. 40, 3610 (1980).
HPLC determn in human leukemia cells: V. Gandhi et al., J. Chromatogr. 413,293 (1987). Prepn of 5¢-monophosphate: J. A. Montgomery, A. T. Shortnacy, US 4357324 (1982 to U.S. Dept. of Health and Human Services).
Pharmacokinetics in humans: M. R. Hersh et al., Cancer Chemother. Pharmacol. 17, 277 (1986).
Evaluation of therapeutic efficacy and CNS toxicity in acute refractory leukemia: R. P. Warrell, Jr., E. Berman, J. Clin. Oncol. 4, 74 (1986); H. G. Chun et al., Cancer Treat. Rep. 70, 1225 (1986). Series of articles on pharmacology and therapeutic use: Semin. Oncol. 17,Suppl. 8, 1-78 (1990).
External links
 COCK WILL TEACH YOU
COCK WILL TEACH YOU

DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin
Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com

Location of Khajuraho Group of Monuments in India.
 Location in Madhya Pradesh
Location in Madhya Pradesh
-
Khajuraho Group of Monuments – Wikipedia, the free …
en.wikipedia.org/wiki/Khajuraho_Group_of_MonumentsThe Khajuraho Group of Monuments are a group of Hindu and Jain temples in Madhya Pradesh, India. About 620 kilometres (385 mi) southeast of New Delhi, …








Hotel Chandela – A Taj Leisure Hotel




