
Home » Phase2 drugs (Page 27)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
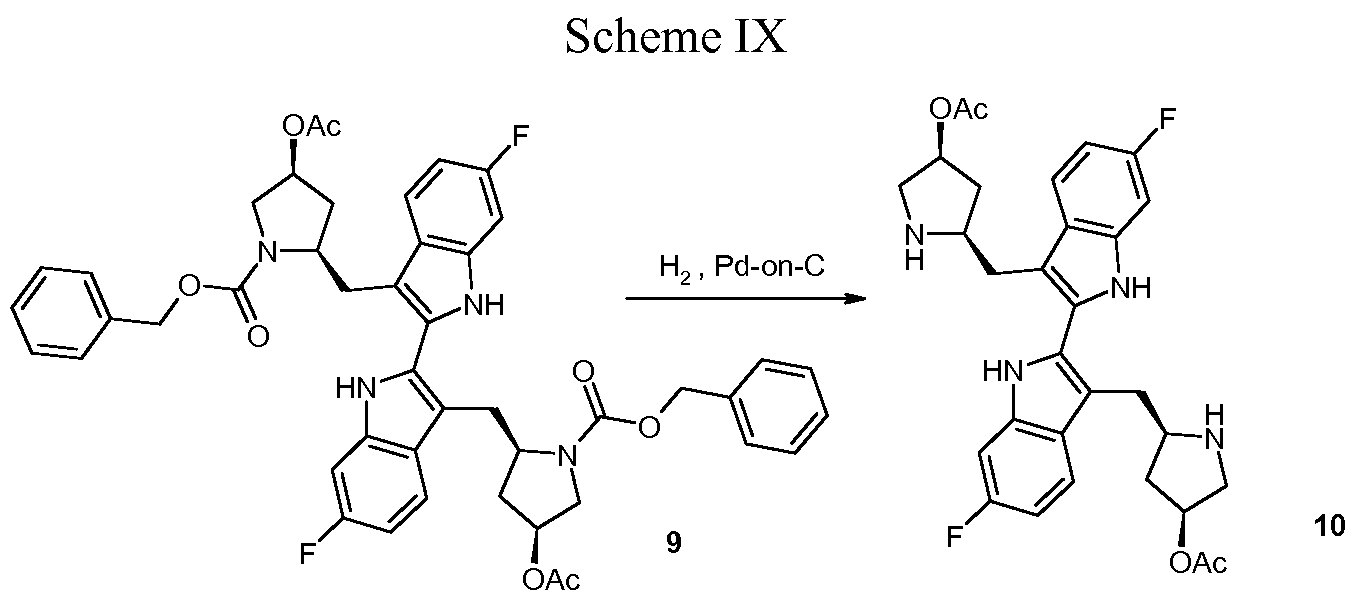
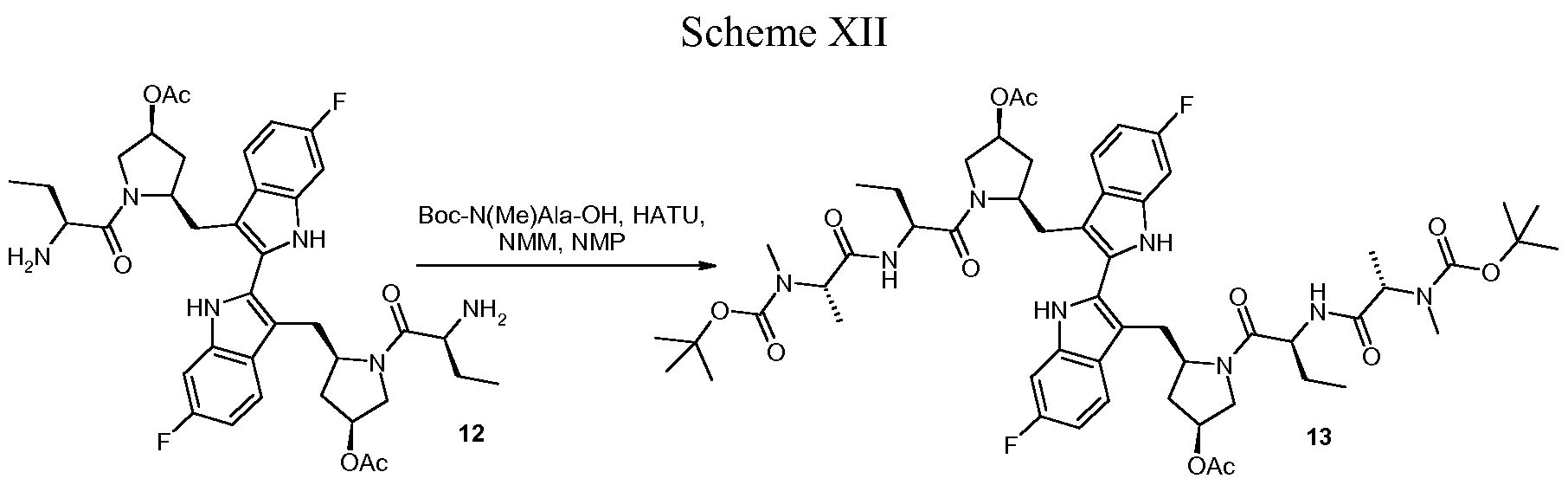
PHASE 2 -TetraLogic’s BIRINAPANT for treatment of acute myeloid leukemia, pancreatic cancer, or ovarian cancer
BIRINAPANT, Apoptosis inhibitor
(2S,2’S)-N,N’-((2S,2’S)-((3S,3’S,5R,5’R)-5,5′-((6,6′-difluoro-1H,1’H-[2,2′-biindole]-3,3′-diyl)bis(methylene))bis(3-hydroxypyrrolidine-5,1-diyl))bis(1-oxobutane-2,1-diyl))bis(2-(methylamino)propanamide)
1260251-31-7 cas no
Birinapant is an antagonist of XIAP and cIAP1 with Kd value of 45 nM and <1 nM, respectively.
US20110003877,WO 2013049350 A1
| Molecular Weight: | 806.94 |
| Birinapant Formula: | C42H56F2N8O6 |
Birinapant, also known as TL32711, is a synthetic small molecule and peptido mimetic of second mitochondrial-derived activator of caspases (SMAC) and inhibitor of IAP (Inhibitor of Apoptosis Protein) family proteins, with potential antineoplastic activity. As a SMAC mimetic and IAP antagonist, TL32711 binds to and inhibits the activity of IAPs, such as X chromosome-linked IAP (XIAP) and cellular IAPs 1 and 2. Since IAPs shield cancer cells from the apoptosis process, this agent may restore and promote the induction of apoptosis through apoptotic signaling pathways in cancer cells. IAPs are overexpressed by many cancer cell types and suppress apoptosis by binding and inhibiting active caspases-3, -7 and -9 via their baculoviral lAP repeat (BIR) domains
Birinapant is currently in Phase II clinical trials in patients with acute myeloid leukemia, pancreatic cancer, or ovarian cancer. Although these trials don’t have a control group, the emerging data are promising, TetraLogic chief executive officer John M. Gill told C&EN. (Early-stage cancer clinical trials are commonly run without placebo groups.) The birinapant trials show preliminary evidence both that the drug is having the desired effect and that this effect is associated with signs of clinical activity. Given these results, the company plans to launch randomized Phase II studies early in 2014
Birinapant, also called TL32711, is a potent antagonist for XIAP with Kd value of 45 nM and cIAP1 with Kd value <1 nM [1].
Birinapant not only binds to the isolated BIR3 domains of cIAP1, cIAP2, XIAP but the single BIR domain of ML-IAP with high affinity and degrades TRAF2-bound cIAP1 and cIAP2 rapidly accordingly inhibiting the activation of TNF-mediated NF- kB. Additionally, birinapantcan promote the formation of caspase-8: RIPK1 complex in response to TNF stimulation, which result in downstream caspasesactivation [4].
In the inorganic SUM149- and SUM190-derived cells, which with differential XIAP expression (SUM149 wtXIAP, SUM190 shXIAP) and other high cIAP1/2 but low XIAP binding affinity bivalent Smac mimetic GT13402, XIAP inhibition are needed for increasing TRAIL potency. Opposite, single agent efficacy of Birinapant is owing to pan-IAP antagonism. Rapid cIAP1 degradation was caused by birinapant, as well as NF-κB activation, PARP cleavage andcaspase activation. While combined withTNF-α, showing strong combination activity, the combination was more effective than individual. The response in spheroid models was conserved, whereas in vivo birinapant inhibited tumor growth without adding TNF-α in vitro to resistant cell lines. In a parental cell line, TNF-αcombined withbirinapantinhibited the growth of a melanoma cell line with acquired resistance to the same extent of BRAF inhibition [1, 2].
Drug treatment increased the mean [18F]ICMT-11 tumor uptake with a peak at 24 hours for CPA (40 mg/kg; AUC40-60: 8.04 ± 1.33 and 16.05 ± 3.35 %ID/mL × min at baseline and 24 hours, respectively) and 6 hours for birinapant (15 mg/kg; AUC40-60: 20.29 ± 0.82 and 31.07 ± 5.66 %ID/mL × min, at baseline and 6 hours, respectively). Voxel-based spatiotemporal analysis of tumor-intrinsic heterogeneity showed that [18F] ICMT-11 could detect the discrete pockets of caspase-3 activation. Caspase-3 activation that measured ex vivo associated with the increased tumor [18F] ICMT-11, and early radiotracer uptake predicted apoptosis, distinct from the glucose metabolism with [18F] fluorodeoxyglucose-PET, which depicted the continuous loss of cell viability [3].

 Guangyao Yu, Yijun Deng, Gurpreet Singh Kapoor, Condon, Susan Rippin, Martin Graham, Angeline Mufalli, Jennifer Burns, Martin Seipel, Eric Neiman, Thomas Haimowitz, Christopher Benetatos, Yasuhiro Mitsuuchi, Srinivas Chunduru Not pictured: Divya Goel")
Birinapant was designed to reinstate cancer cells’ ability to die. Many cancers that are resistant to conventional chemotherapy drugs have defects in the cell death pathway known as apoptosis. The human body uses apoptosis every day to clear away abnormal or unwanted cells.
Apoptosis is a tightly regulated process, Condon explains, with a network of proteins that activate or block the process. TetraLogic’s target is a family of proteins called the Inhibitor of Apoptosis proteins. As their name suggests, these proteins block apoptosis. They interfere with protease enzymes that carry out cellular dismantling.
TetraLogic’s aim is to lift that blockade to restart apoptosis in tumors. Many tumors have excesses of the apoptosis inhibitor proteins relative to normal cells, so targeting this process has the potential to be less toxic to normal cells compared with conventional chemotherapy.
It turns out nature has a way of negating the inhibitor proteins’ actions—a protein known as Smac. TetraLogic’s founders demonstrated that only a tiny portion of Smac is necessary to keep the inhibitor proteins at bay—the four amino acids at the protein’s N-terminus. “Once you can get a protein down to a tetrapeptide,” about the size of a small-molecule drug, “you start getting a lot of interest from the pharma community,” Condon told C&EN.
Peptides fall apart in the body too quickly to be drugs, so Condon’s team worked with molecular mimics of the Smac tetrapeptide. Their biggest advance was realizing that combining two copies of their tetrapeptide mimics into one molecule made their compounds highly effective at reinstating apoptosis in cancer cell lines. Many proteins in the apoptosis pathway function as dimers, so using these so-called bivalent mimics against them makes sense, Condon said.
However, several of the bivalent compounds were associated with pronounced body weight loss in mice. Condon’s team eventually learned that replacing a branched side chain on their peptide mimics and adding a hydroxyl group to a proline residue improved the tolerability for the animals without impacting the antitumor effect. With that, birinapant was born.
In mice, birinapant shrank tumors. The compound has been in clinical trials since 2009, both on its own and in combination with other chemotherapy drugs such as irinotecan and gemcitabine. On the basis of other biochemical work on the apoptosis pathway, TetraLogic thinks these drugs could act in synergy with birinapant to treat cancer, Condon said.
Birinapant is currently in Phase II clinical trials in patients with acute myeloid leukemia, pancreatic cancer, or ovarian cancer. Although these trials don’t have a control group, the emerging data are promising, TetraLogic chief executive officer John M. Gill told C&EN. (Early-stage cancer clinical trials are commonly run without placebo groups.) The birinapant trials show preliminary evidence both that the drug is having the desired effect and that this effect is associated with signs of clinical activity. Given these results, the company plans to launch randomized Phase II studies early in 2014.
1H NMR













spectral data
1H NMR (300 MHz, CDC13): 511.74 (s, 2H), 8.27 (d, J= 8.7 Hz, 2H), 7.71 (dd, J= 5.4, 8.4 Hz, 2H), 7.55 (dd, J =2.4, 9.6 Hz, 2H), 6.88 (ddd, J= 2.4, 9.3, 9.3 Hz, 2H), 4.62-4.78 (m, 4H), 4.43 (dd, J= 9.3, 9.9 Hz, 2H), 4.03 (dd, J= 4.8, 11.4 Hz, 2H), 3.80 (d, J = 11.4 Hz, 2H), 3.66 (dd, J= 2.7, 14.4 Hz, 2H), 3.53 (dd, J = 11.4, 14.4 Hz, 2H), 3.11 (q, J = 6.9 Hz, 2H), 2.56 (s, 6H), 2.45 (m, 2H), 2.19 (d, J= 14.4 Hz, 2H), 1.76-2.10 (m, 6H), 1.59 (br s, 2H), 1.39 (d, J= 6.9 Hz, 6H), 1.22-1.38 (m, 2H), 1.07 (t, J = 7.2 Hz, 6H) ppm;
13C NMR (75 MHz, d6– DMSO): 5175.2, 172.8, 161.6, 158.5, 137.3, 137.2, 128.4, 128.3, 126.4, 120.8, 120.6, 109.4, 108.7, 108.4, 98.4, 98.0, 70.8, 60.2, 59.9, 56.6, 51.8, 36.4, 35.3, 28.3, 25.6, 20.0, 10.6 ppm.
Mass spectrum (ESI), m/z 807.5 [(M)+; calcd for C42H56F2N806: 806.9].
Paper
Click to access op5b00390_si_001.pdf
Click to access op5b00390_si_001.pdf
Process Development and Synthesis of Birinapant: Large Scale Preparation and Acid-Mediated Dimerization of the Key Indole Intermediate

Birinapant/TL32711 (1) is a novel bivalent antagonist of the inhibitor of apoptosis (IAP) family of proteins which is currently in clinical development for the treatment of cancer and hepatitis B virus (HBV) infection. In this report, we present a detailed description of the 1 drug substance synthesis used to support our ongoing clinical studies. Key transformations in this process included the development of a scalable, high-yielding route to acyl indole 14 as well as a two-step dimerization/oxidation of indole 19 that afforded biindole 21 in excellent yield and purity (70% yield, 2 steps; >95 area% purity by HPLC analysis). In addition, partial defluorination of 21 was observed following hydrogen-mediated benzyloxycarbonyl (Cbz) protective group removal which was obviated by the use of HBr/HOAc for this transformation. The use of commercially available amino acid derivatives afforded related impurities which proved difficult to purge in subsequent steps. Thus, defining the impurity specification for these reagents was critical to providing 1 drug substance of >99 area% chemical purity. Using this process, we have successfully prepared 1 drug substance multiple times on >500-g-scale in support of our clinical development program.
References:
1.Allensworth JL, Sauer S, Lyerly HK, et al. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-a-independent mechanism. Breast Cancer Research, 2013, 137:359-371.
2.Krepler C, Chunduru SK, Halloran MB, et al. The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clinical Cancer Research, 2013, 19 (7): 1784-1794.
3.Nguyen QD, Lavdas I, Gubbins J, et al. Temporal and Spatial Evolution of Therapy-Induced Tumor Apoptosis Detected by Caspase-3–Selective Molecular Imaging. Clinical Cancer Research, 2013, 19 (14): 3914-3924.
4.Benetatos CA, Mitsuuchi Y, Burns JM, et al. Birinapant (TL32711), a Bivalent SMAC Mimetic, Targets TRAF2-Associated cIAPs, Abrogates TNF-Induced NF-kB Activation, and Is Active in Patient-Derived Xenograft Models. 2014, 13(4):867-879.

phase 2-LGX818, Novartis Research Foundation to treat melanoma with a specific mutation in B-RAF kinase V600E

LGX818
Methyl [(2S)-1-{[4-(3-{5-chloro-2-fluoro-3-[(methylsulfonyl)amino]phenyl}-1-isopropyl-1H-pyrazol-4-yl)-2-pyrimidinyl]amino}-2-propanyl]carbamate
Novartis Institutes for Biomedical Research and Genomics Institute of the Novartis Research Foundation to treat melanoma with a specific mutation in B-RAF kinase V600E, selective mutant B-RAF kinase inhibitor
LGX818 is currently in Phase Ib/II clinical trials. Patients with colon cancer or melanoma with the BRAF mutation, including patients resistant to other BRAF-targeted drugs, are receiving LGX818 pills alone or as part of drug cocktails to determine whether the drug is safe and efficacious
A phase Ib/II drug structure by Novartis disclosed at the spring 2013 American Chemical Society meeting in New Orleans to treat melanoma with a V600E mutation in the B-RAF kinase which it inhibits.[1][2][3]
Several clinical trials of LGX818 , either alone or in combinations with the MEK inhibitorMEK162[4], CDK4 inhibitor LEE011[5] are being run. The initial results are encouraging [6].
- C. Drahl, Liveblogging First-Time Disclosures of Drug Structures from #ACSNOLA, 2013, http://cenblog.org/the-haystack/2013/04/liveblogging-first-time-disclosures-of-drug-structures-from-acsnola/
- http://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011023773
- http://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011025927
- http://www.cancer.gov/clinicaltrials/search/view?cdrid=728588&version=HealthProfessional&protocolsearchid=11645832
- http://www.cancer.gov/clinicaltrials/search/view?cdrid=745927&version=HealthProfessional&protocolsearchid=11645832
- http://www.novartis.com/downloads/investors/event-calendar/2012/6-bridging-science-and-patients.pdf
Phase 2, AZD5423 BY ASTRAZENECA FOR COPD

AZD5423
The compound is now in a Phase II study in patients with COPD, where its efficacy and safety are being measured against that of a typical steroid or a placebo
A phase II drug structure by AstraZeneca disclosed at the spring 2013 American Chemical Society meeting in New Orleans to treat respiratory diseases and in particular the chronic obstructive pulmonary disease.[1][2][3][4]
- C. Drahl, Liveblogging First-Time Disclosures of Drug Structures from #ACSNOLA, 2013, http://cenblog.org/the-haystack/2013/04/liveblogging-first-time-disclosures-of-drug-structures-from-acsnola/
- GB2010051905 COMBINATIONS COMPRISING A GLUCOCORTICOID RECEPTOR MODULATOR FOR THE TREATMENT OF RESPIRATORY DISEASES
- SE2009050900 A COMBINATION OF (A) GLUCOCORTICOID RECEPTOR MODULATOR AND (B) A MUSCARINIC ANTAGONIST
- SE2009000264 COMBINATION OF (A) GLUCOCORTICOID RECEPTOR MODULATOR AND (B) A B2-AGONIST
Sorbent begins CLP-1001 Phase 2b trial in heart failure patients
APRIL 2013
Sorbent Therapeutics has begun Phase 2b trial evaluating the safety and efficacy of CLP-1001 in treating signs and symptoms of fluid overload in heart failure patients.
Non-absorbed oral polymer CLP-1001 binds to and removes excess sodium and fluid in the GI tract independently of the kidneys……………read original article at pharmaceutical business review
Phase 2 , Sarepta, shows eteplirsen for Duchenne muscular dystrophy
ETEPLIRSEN
27 MAR 2013
The clock is ticking for Sarepta Therapeutics , a multitude of biotech investors and the boys who suffer from Duchenne muscular dystrophy.
Armed with promising Phase IIb data from a small study of eteplirsen involving only 12 patients with the lethal disease, Sarepta CEO Chris Garabedian is completing one of the most closely-watched high wire acts in the industry. At a time most companies would be focused solely on organizing a pivotal late-stage study, there’s intense speculation that the biotech will shoot for an accelerated approval with the data in hand. And it all comes down to their sit-down with the FDA to review mid-stage data.
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
The drug is a Morpholino antisense oligomer which triggers excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
TheraVida Presents Positive Phase 2 Data for Tolenix (THVD-201) in Patients with Overactive Bladder
mar18, 2013
TheraVida, Inc., a clinical-stage biopharmaceutical company developing novel combination drug products, presented positive results from a Phase 2 clinical trial of its lead product candidate, Tolenix ™ (THVD-201), for the treatment of overactive bladder (OAB) and urge urinary incontinence (UUI) at the 28 th Annual Congress of the European Urological Association (EAU) in Milan, Italy.
Tolenix ™ is a twice-daily (BID) proprietary combination of tolterodine, to treat OAB and UUI, and pilocarpine, to reduce the significant dry mouth (xerostomia) caused by muscarinic antagonist medications such as tolterodine.

tolterodine

pilocarpine
The objectives of the randomized, double-blinded, multiple-crossover Phase 2 trial were to assess the safety and efficacy of Tolenix ™ in reducing the frequency of micturition (urination) and incontinence episodes per day, as compared to both placebo control and active control Detrol ® (tolterodine). In addition, common side effects of muscarinic antagonist therapies, such as dry mouth, were carefully assessed in the 138 patients enrolled in the trial. This international Phase 2 clinical trial was conducted in South Korea, Australia, and New Zealand.
Patients receiving Tolenix ™ (2mg tolterodine plus 9mg pilocarpine, administered BID) experienced statistically significant improvements in their OAB and UUI symptoms over placebo, with a reduction in daily micturitions of 0.88 (p<0.0001) and a reduction in daily incontinence episodes of 0.47 (p<0.0001). This efficacy was similar in magnitude to the maximum dose of active control Detrol ® (2mg tolterodine, administered BID).
TONIX Completes Pre-Phase 3 Meeting With U.S. Food and Drug Administration for TNX-102 SL in Fibromyalgia

Regulatory Acceptance of Design of Registrational Clinical Studies; Dosing in First Safety and Efficacy Trial to Commence in the Third Quarter of 2013
NEW YORK, NY
March 11, 2013) – Tonix Pharmaceuticals Holding Corp. , a specialty pharmaceutical company developing novel treatments for challenging disorders of the central nervous system, including fibromyalgia (“FM”) and post-traumatic stress disorder (“PTSD”), announced that it recently held an End-of-Phase 2/Pre-Phase 3 meeting with the U.S. Food and Drug Administration (“FDA”) to discuss its proposed New Drug Application (“NDA”) plan for the Company’s novel sublingual tablet formulation of cyclobenzaprine for bedtime use, TNX-102 SL, for the management of FM. Official FDA meeting minutes indicate FDA acceptance of the clinical program and provide clear direction to achieve a successful NDA filing of TNX-102 SL in FM.
The registrational clinical trials will consist of two randomized, double-blind, placebo-controlled 12-week safety and efficacy studies in FM patients who will take either a TNX-102 SL (cyclobenzaprine HCl 2.8 mg) tablet or placebo at bedtime. The primary endpoint of both trials will be the change in pain from baseline to Week 12 as measured by the Numeric Rating Scale. The Company plans to conduct these trials in sequence, and expects to begin dosing in the first trial in the third quarter of 2013. This trial will enroll 100 to 200 FM patients, and top-line data are anticipated to become available in the second half of 2014.
Following the completion of the double-blind randomized portion of these studies, patients may be eligible to enroll in open-label extension studies of TNX-102 SL. The FDA agreed that the safety database needed to support a 505(b)(2) NDA submission for TNX-102 SL would contain a total exposure of at least 300 FM patients, with at least 100 patients receiving TNX-102 SL for six months and at least 50 patients for one year.
Seth Lederman, M.D., Chief Executive Officer of TONIX, said, “We view our meeting with the FDA as a major milestone for TONIX. We are pleased to have concurrence from the FDA on the design and selection of efficacy endpoints of our registrational clinical studies in FM in addition to receiving clear guidance on the remaining requirements for the TNX-102 SL NDA program. We are also pleased with the FDA’s requirements on chronic exposure, which are appropriately less than those typically needed for a new drug to be approved for a chronic use indication. We look forward to advancing TNX-102 SL towards a successful NDA filing.”
About Tonix Pharmaceuticals Holding Corp.
TONIX is developing innovative prescription medications for challenging disorders of the central nervous system. The Company targets conditions characterized by significant unmet medical need, inadequate existing treatment options, and high dissatisfaction among both patients and physicians. TONIX’s core technology improves the quality of sleep in patients with chronic pain syndromes, which is believed to translate into reductions in pain and other symptoms. An Investigational New Drug Application (“IND”) has been filed for the Company’s lead product candidate, TNX-102 SL, a novel under-the-tongue tablet formulation of cyclobenzaprine, the active ingredient in two FDA-approved muscle relaxants. TONIX expects to begin a registrational clinical study of TNX-102 SL in FM in the third quarter of 2013. TONIX expects to file an IND for TNX-102 SL in PTSD in the third quarter of 2013, and to begin a Phase 2 trial in this indication in the fourth quarter of 2013. To learn more, please visit www.tonixpharma.com.
Certain statements in this press release are forward-looking within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be identified by the use of forward-looking words such as “anticipate,” “believe,” “forecast,” “estimated” and “intend,” among others. These forward-looking statements are based on TONIX’s current expectations and actual results could differ materially. There are a number of factors that could cause actual events to differ materially from those indicated by such forward-looking statements. These factors include, but are not limited to, substantial competition; our ability to continue as a going concern; our need for additional financing; uncertainties of patent protection and litigation; uncertainties of government or third party payer reimbursement; limited sales and marketing efforts and dependence upon third parties; and risks related to failure to obtain FDA clearances or approvals and noncompliance with FDA regulations. As with any pharmaceutical under development, there are significant risks in the development, regulatory approval and commercialization of new products. TONIX does not undertake an obligation to update or revise any forward-looking statement. Investors should read the risk factors set forth in the Annual Report on Form 10-K filed with the SEC on March 30, 2012 and future periodic reports filed with the Securities and Exchange Commission. All of the Company’s forward-looking statements are expressly qualified by all such risk factors and other cautionary statements. The information set forth herein speaks only as of the date hereof
Cyclobenzaprine, N,N-dimethyl-3-(dibenzo[a,d]cyclohepten-5-ylidene)propylamine, is synthesized by reacting 5H-dibenzo[a,d]cyclohepten-5-one with 3-dimethylaminopropylmagnesium chloride and subsequent dehydration of the resulting carbinol in acidic conditions into cyclobenzaprine. 
- H.La Roche, GB 858187 (1961).
- F.J. Villani, C.A. Ellis, C. Teihman, C. Biges, J. Med. Pharm. Chem., 5, 373 (1962).
- Winthrop, S. O.; Davis, M. A.; Myers, G. S.; Gavin, J. G.; Thomas, R.; Barber, R. (1962). “New Psychotropic Agents. Derivatives of Dibenzo[a,d]-1,4-cycloheptadiene”. The Journal of Organic Chemistry 27: 230. doi:10.1021/jo01048a057.
Phase 2 ORM-12741, Orion’s Experimental Alzheimer’s Drug Shows Promise, Study Finds
 ORM-12741 in WO 2003082866 ORM-12741 in WO 2003082866 |
The purpose of this study is to determine whether ORM-12741 is safe and effective in the treatment of Alzheimer’s disease.,
March 11, 2013
A small Finnish study is raising hopes for a new drug designed to help stave off memory loss among patients struggling with moderate Alzheimer’s disease.
Still in the preliminary stages of investigation, the drug — called ORM-12741 — showed promise during a three-month trial involving 100 such patients, half of whom were given the medication on top of their current drug treatment.
By the end of the study, memory scores plummeted by 33 percent among the 50 patients who were given a dummy pill (placebo) rather than the new drug, while patients who took the new drug showed a 4 percent improvement on the tests.
“The bottom line is that this was the first study investigating [effectiveness] of a drug with a novel mechanism of action in patients with Alzheimer’s disease,” said study lead author Dr. Juha Rouru, who heads the central nervous system therapy area at Orion Pharma in Turku, Finland.
“The results were clearly positive,” he said, adding they were seen particularly on important episodic memory, which involves remembering events and personal experiences. Orion, the maker of ORM-12741, funded the research.
Rouru and his colleagues are scheduled to present their work in San Diego at a meeting of the American Academy of Neurology, which starts Saturday.
By 2050, as the elderly population increases, an estimated 13.8 million Americans will have Alzheimer’s, a progressive brain disease that robs people of their memory and the ability to perform even simple everyday tasks. There is no cure for the disease, and drugs aimed at controlling the debilitating symptoms are only moderately effective, Rouru said.
With that in mind, the team set out to assess the potential of ORM-12741, the first drug to target a specific receptor in the brain, called alpha-2C. This receptor is thought to play a role in the brain’s “fight or flight” response to stress, and the authors noted that the new drug’s impact on alpha-2C had shown promise in prior animal studies.
All the patients in the study were already taking a cholinesterase drug. Some were also using memantine, another type of Alzheimer’s medication.
Fifty patients were then given a placebo on top of their current regimen, while 50 were given either a low-dose (30 to 60 milligrams) twice daily supply of ORM-12741 or a high-dose (100 to 200 milligrams) version.
Computerized memory tests highlighted an apparent memory benefit (without prompting severe side effects) among the ORM-12741 patients, and Rouru suggested that the new drug should be seen as just one more potentially effective tool in an ongoing battle to reign in “a devastating disease.”
“I am afraid that wonder drugs hardly exist,” he noted. “In the present study, our drug was used on top of existing Alzheimer’s medications. In that setting it showed clear effect, which suggests that it is giving additional clinically significant benefit for patients that are already using Alzheimer’s medications.”
Catherine Roe, an assistant professor of neurology at Washington University School of Medicine in St. Louis, described Rouru’s research as “impressive.”
“This is really a new approach, in terms of the biology that they’re targeting,” she noted. “And they showed significant results after only three months of treatment, which is exciting particularly because this drug combination was tested on people who had moderate Alzheimer’s disease.”
Many experts have thought moderate Alzheimer’s disease would be untreatable, she said. “By the time it’s that advanced, the nerves have already died and it would be too late to do anything about memory by this stage,” she explained.
Still, much more testing will need to be done, Roe cautioned. “And these results will have to be replicated with other groups of people,” she said. “But if they can do that, this would be awesome.”
The data and conclusions of research presented at medical meetings are typically considered preliminary until published in a peer-reviewed journal.
More information
For more on Alzheimer’s disease, visit the U.S. National Institutes of Health.
Phase 2, Sarepta Therapeutics, Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
s excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Clinical studies
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
References
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods.. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303.. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
Phase 2 Drug: Ustekinumab A monoclonal antibody against the p40 subunit of IL-12/23 Other Name: Stelara
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-12 and IL-23
|

Ustekinumab, CAS number 815610-63-0, is also known by it’s brand name Stelara, which is marketed by Janssen Biotech, Inc. Developed as a treatment for adults with moderate to severe plaque psoriasis
Rockefeller University, MAR 2013
http://clinicaltrials.gov/ct2/show/NCT01806662
Atopic dermatitis (AD) is a chronic disease associated with intense itching, which affects most aspects of everyday life in the majority of patients. Acute inflammation and extensor/facial involvement is common in infants, whereas chronic inflammation increases in prevalence with age, as do localization to flexures. AD has a complex background characterized by immune activation, increased epidermal thickness in chronic diseased skin, and defective barrier function. In normal, healthy skin, the outer layer of the epidermis, the stratum corneum is made up flattened dead cells called corneocytes held together by a mixture of lipids and proteins. The stratum corneum and, in particular, the lipid layer are vital in providing a natural barrier function that locks water inside the skin and keeps allergens and irritants out. In people with AD, the barrier function is defective, which leads to dry skin. As the skin dries out, it cracks allowing allergens and irritants to penetrate.
Mild AD can be controlled with emollients and topical medications. However, moderate to severe AD is extremely difficult to control and requires systemic treatment that is often unsatisfactory due to impracticality and lack of effectiveness. Only three therapeutic options exist for moderate to severe AD, including: 1) oral steroids 2) cyclosporine A (CsA), that is not widely used in the US as it is not FDA approved for AD and 3) ultraviolet phototherapy. Oral steroids and CsA treatments have major side effects and UV radiation therapy is highly inconvenient for patients. Several biologic medications, such as TNF-alpha inhibitors, are effective, convenient, and relatively safe therapies for psoriasis, but have thus far not shown efficacy in AD. Ustekinumab is a unique biologic medication that may specifically target AD.
The investigators study will determine whether there is a reversal of the skin thickness and the immune pathways involved in the disease during treatment with Ustekinimab and what specific immune cells are involved. The investigators are also interested to understand how the clinical reversal of the disease will correlate with tissue reversal of the disease.
Detailed Description:
In psoriasis, epidermal hyperplasia is driven by underlying immune activation, whether as a direct response to IL-20 family cytokines that induces hyperplasia and inhibits keratinocyte terminal differentiation or as an indirect response to immune-mediated injury to keratinocytes. The epidermal reaction in psoriasis is largely restored to normal with selective immune suppression. Hence, one might hypothesize that similar epidermal responses should occur in the presence of “generalized” cellular immune activation, in diseases with similar inflammatory infiltrate and epidermal hyperplasia, such as AD. In fact, psoriasis and AD share features of dense T-cells and dentritic cell infiltrates, as well as over-expression of IL-22 in skin lesions. These diseases also share similar epidermal hyperplasia in their chronic phases.
Work from the investigators group showed that IL-22 is a key cytokine in the pathogenesis of both AD and psoriasis. The investigators have demonstrated that in psoriasis, ustekinumab suppresses the production of IL-12, IL-23, and IL-22. Additionally, by RT-PCR the investigators demonstrated that the mRNA expression of p40 cytokine and the IL23R is up-regulated in AD as compared to both normal skin and psoriasis. The investigators therefore hypothesize that ustekinumab will suppress IL-22 and possibly also p40 production in AD lesions and reverse both the epidermal growth/differentiation defects and the underlying immune activation, and hence will suppress disease activity. Interestingly, p40 was also found to be significantly up-regulated in non-lesional AD skin as compared with normal skin.
Although AD is thought to be predominately a disease of Th2-type cells, in the chronic stage, there is large Th1 component. To date, the precise mechanism by which sequential activation of Th2 and Th1 cells in AD is achieved remains unknown. IL-12 induces the differentiation and maturation of human Th cells into Th1-type cells. Recent circumstantial evidence suggests that in AD patients IL-12 may facilitate a change from the Th2-type to a Th1 cytokine profile. IL-12 was recently shown to be highly elevated in pediatric AD and its levels were strongly associated with disease severity.
Expression of IL-12 p40 mRNA is significantly enhanced in lesional skin from AD, suggesting that the enhanced local production of IL-12 in dendritic cells and macrophages may be responsible for the increased production of IFN-γ in chronic lesions potentially suggesting that IL-12 may have a pivotal role in promoting inflammation in atopic dermatitis. Topical steroids which constitute a mainstay of therapy in AD are known to strongly down-regulate IL-12 expression, possibly also indicating that targeted anti IL-12 therapy might important role in treating AD.
Recently, the Th1/Th2 paradigm in autoimmunity and allergy has been revisited to include a role for a new population of IL-17-producing Th cells known as Th17. Th17 cells are characterized by the production of inflammatory cytokines such as IL-17A, IL-17F, IL-22, and IL-26. One of the key factors involved in naive Th-cell commitment to a Th17 phenotype is IL-23.
Patients with acute AD were found to have increased Th17 T-cells in peripheral blood by flow cytometry and intracellular cytokine staining 26 as well as by immunohistochemistry (IHC) in lesions. Since IL-23 is the major inducer of Th17 T-cells, as well as “T22” T-cells, neutralization of IL-23 could potentially result in both decreased Th17 signal in acute AD as well as decreased “T22/IL22″ signal. Therefore the investigators postulate that ustekinumab in AD will act both inhibiting the IL-12-dependent Th1 shift in chronic AD stage as well as the pathogenic IL-22/”T22” axis in this disease.
Ustekinumab [1] (INN, experimental name CNTO 1275, proprietary commercial name Stelara,[2] Centocor) is a human monoclonal antibody. It is directed against interleukin 12 and interleukin 23, naturally occurring proteins that regulate the immune system and immune-mediated inflammatory disorders.[3]
In two Phase III trials for moderate to severe psoriasis, the longest >76 weeks, ustekinumab was safe and effective.[4][5]
A third Phase III trial, ACCEPT, compared the efficacy and safety of ustekinumab with etanercept in the treatment of moderate to severe plaque psoriasis.[6] This trial found a significantly higher clinical response with ustekinumab over the 12-week study period compared to high-dose etanercept.[6] It also demonstrated the clinical benefit of ustekinumab among patients who failed to respond to etanercept.[6]
Ustekinumab is approved in Canada, Europe and the United States to treat moderate to severe plaque psoriasis.[7]
As of November 2009, the drug is being investigated for the treatment of psoriatic arthritis.[8][9] It has also been tested in Phase II studies for multiple sclerosis[10] and sarcoidosis, the latter versus golimumab (Simponi).[11]
- Cingoz, Oya (2009). “Ustekinumab”. MAbs 1 (3): 216–221. doi:10.4161/mabs.1.3.8593. PMC 2726595. PMID 20069753.
- ^ European Medicines Agency, 20 November 2008, http://www.emea.europa.eu/pdfs/human/opinion/Stelara_58227008en.pdf
- ^ Reddy M, Davis C, Wong J, Marsters P, Pendley C, Prabhakar U (May 2007). “Modulation of CLA, IL-12R, CD40L, and IL-2Ralpha expression and inhibition of IL-12- and IL-23-induced cytokine secretion by CNTO 1275”. Cell. Immunol. 247 (1): 1–11. doi:10.1016/j.cellimm.2007.06.006. PMID 17761156.
- ^ Leonardi CL, Kimball AB, Papp KA, et al. (May 2008). “Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1)”. Lancet 371 (9625): 1665–74. doi:10.1016/S0140-6736(08)60725-4. PMID 18486739.
- ^ Papp KA, Langley RG, Lebwohl M, et al. (May 2008). “Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2)”. Lancet 371 (9625): 1675–84. doi:10.1016/S0140-6736(08)60726-6. PMID 18486740.
- ^ a b c Griffiths C, Strober B, van de Kerkhof P et al. (2010). “Comparison of Ustekinumab and Etanercept for Moderate-to-Severe Psoriasis”. N Engl J Med 362 (2): 118–28. doi:10.1056/NEJMoa0810652. PMID 20071701.
- ^ Medarex to Receive Milestone Payment for Approval of STELARA(TM) (Ustekinumab) for the Treatment of Moderate to Severe Plaque Psoriasis
- ^ ClinicalTrials.gov NCT00267956 A Study of the Safety and Efficacy of CNTO 1275 in Patients With Active Psoriatic Arthritis
- ^ ClinicalTrials.gov NCT01009086 A Study of the Safety and Efficacy of Ustekinumab in Patients With Psoriatic Arthritis
- ^ ClinicalTrials.gov NCT00207727 A Safety and Efficacy Study of CNTO1275 in Patients With Multiple Sclerosis
- ^ ClinicalTrials.gov NCT00955279 A Study to Evaluate the Safety and Effectiveness of Ustekinumab or Golimumab Administered Subcutaneously (SC) in Patients With Sarcoidosis
- ^ http://www.empr.com/stelara-approved-for-moderate-to-severe-psoriasis/article/149760/
- ^ a b Centocor 12/19/08 Press Release, http://www.centocor.com/centocor/i/press_releases/FDA_ISSUES_COMPLETE_RESPONSE_LETTER_TO_CENTOCOR_FOR_USTEKINUMAB_BIOLOGIC_LICENSE_APPLICATION_
- ^ Johnson LL. “Study: Drug for serious psoriasis tops competition” The Associated Press. 18 Sept 2008.[dead link]
- ^ Wild, David (November 2011), “Novel IL-12/23 Antagonist Shows Potential in Severe Crohn’s”, Gastroenterology & Endoscopy News 62 (11), retrieved 2011-12-04
- ^ a b c Weber J, Keam SJ (2009). “Ustekinumab”. BioDrugs 23 (1): 53–61. doi:10.2165/00063030-200923010-00006. PMID 19344192.
- ^ Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH (September 2008). “Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study”. Lancet Neurol 7 (9): 796–804. doi:10.1016/S1474-4422(08)70173-X. PMID 18703004.
- ^ “Important Safety Information”. STELARA® (ustekinumab). Janssen Biotech.
External links
- Centocor Ortho Biotech official site
- CNTO 1275 research studies registered with U.S. National Institutes of Health:
- ClinicalTrials.gov NCT00207727 Phase II Study on Multiple Sclerosis
- ClinicalTrials.gov NCT00320216 Phase II Study on Psoriasis
- ClinicalTrials.gov NCT00267969 Phase III Study on Psoriasis
- ClinicalTrials.gov NCT00307437 Phase III Study on Psoriasis
- ClinicalTrials.gov NCT00267956 Phase II Study on Psoriatic Arthritis
- Sylvester, Bruce (2006-03-06). “CNTO 1275 Shows Efficacy for Psoriasis: Presented at AAD”. Doctor’s Guide Publishing. Retrieved 2007-01-25.

{kind=link}