








Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.
Home » Phase2 drugs (Page 16)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
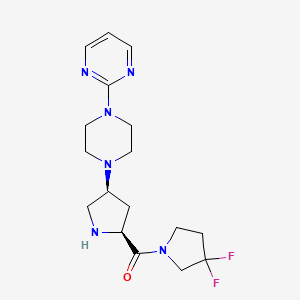
GOSOGLIPTIN
GOSOGLIPTIN
CAS 869490-23-3 FREE BASE
DIHYDROCHLORIDE..869490-47-1
GOSOGLIPTIN; UNII-GI718UO477; PF-00734200; PF-734200;
(3,3-difluoropyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-ylpiperazin-1-yl)pyrrolidin-2-yl]methanone
| Molecular Formula: | C17H24F2N6O |
|---|---|
| Molecular Weight: | 366.408866 g/mol |
| Company | Pfizer Inc. |
| Description | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Molecular Target | Dipeptidyl peptidase-4 (DPP-4) (CD26) |
| Mechanism of Action | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Latest Stage of Development | Phase II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
Type 2 diabetes mellitus is a chronic disorder characterized by hyperglycemia coupled with a gradual decline in insulin sensitivity and insulin secretion. The incretin hormone glucagon-like peptide-1 (GLP-1), which is released post-prandially from the L-cells of the intestine, stimulates the release of insulin from pancreatic β-cells. However, GLP-1 is rapidly degraded in vivo by peptidases, including dipeptidyl peptidase IV (DPP-4), which is a widely distributed serine protease that specifically cleaves N-terminal dipeptides from polypeptides with proline or alanine at the penultimate position.
In vivo administration of DPP-4 inhibitors to human subjects results in higher circulating concentrations of endogenous GLP-1 and subsequent decrease in plasma glucose. Long term treatment with a DPP-4 inhibitor leads to a reduction in circulating HbA1c (glycosylated hemoglobin). DPP-4 inhibition also offers the potential to improve the insulin producing function of the pancreas through either β-cell preservation or regeneration. Therefore, DPP-4 inhibition has emerged as a promising new treatment of Type 2 diabetes
PF-734200 is a potent, selective, orally active dipeptidyl peptidase IV inhibitor. It had been in phase II clinical development at Pfizer for the treatment of type 2 diabetes; however, in 2010 the company discontinued these trials. In 2012, the product was licensed to SatRx, a spin-off of the ChemRar High Tech Center, by Pfizer on an exclusive worldwide basis (with the exception of China) for the development and commercialization as monotherapy or in combination with other therapies for the treatment of type 2 diabetes. SatRx is conducting phase II clinical trials for the treatment of type 2 diabetes.
……………………….
PAPER
New synthetic route to a dipeptidyl peptidase-4 inhibitor
Org Process Res Dev 2012, 16(3): 409
http://pubs.acs.org/doi/abs/10.1021/op200309z

A new synthetic route to a dipeptidyl peptidase-4 (DPP4) inhibitor was developed and demonstrated on a multigram scale. This approach takes advantage of the cheap and readily available Boc-trans-4-hydroxy-l-proline methyl ester as starting material which was derivatized through an SN2 reaction. Several leaving groups were studied, and the nosylate group showed superiority over other derivatives. Formation of an amide using the most costly starting material, 3,3-difluoropyrrolidine, was performed late in the synthesis to minimize its economical impact on the overall cost of the API.
(3,3-Difluoropyrrolidin-1-yl)-(2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)methanone.FREE BASE
Mp 149 °C (decomp).
[α]d = −31.1 (T = 24 °C, c = 1, CHCl3). Specific rotation of product 4 prepared using the initial route: [α]d = −31.5 (T = 24 °C, c = 1, CHCl3).
1H NMR (400 MHz; CDCl3) δ 8.30 (d, J = 4 Hz, 2H), 6.48 (t, J = 4 Hz, 1H), 3.95–3.6 (m, 9H), 3.25–2.85 (m, 4H), 2.6–2.25 (m, 7H), 1.75–1.6 (m, 1H).
13C NMR (100 MHz; CDCl3) δ 172.28; 161.55; 157.70; 127.22 (t, 1J C–F = 248 Hz), 126.22 (t, 1J C–F = 246 Hz), 109.95; 66.54; 58.87; 57.99; 52.71 (t, 2 J C–F = 32 Hz); 52.00; 50.41; 43.03; 34.46, 34.37, 34.25; 19F NMR (377 MHz, CDCl3) δ −102.1 (m, 2F).
IR (neat): 2951w, 2864w, 2799w, 2759w, 1630s, 1585vs, 1547m, 1449m, 1172m, 1254m, 1129m, 982w, 923m, 796m, 638w.
HRMS (ES, N2) Calcd for C17H24F2N6O: 367.20524, found: 367.20592.
……………………….
PAPER
(3,3-difluoro-pyrrolidin-1-yl)-((2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl)-methanone: A potent, selective, orally active dipeptidyl peptidase IV inhibitor
Bioorg Med Chem Lett 2009, 19(7): 1991
http://www.sciencedirect.com/science/article/pii/S0960894X09001966?np=y
- Pfizer Global Research & Development, Groton/New London Laboratories, Pfizer Inc, Groton, CT 06340, United States
A series of 4-substituted proline amides was evaluated as inhibitors of dipeptidyl pepdidase IV for the treatment of type 2 diabetes. (3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone (5) emerged as a potent (IC50 = 13 nM) and selective compound, with high oral bioavailability in preclinical species.
………………….
PATENT
WO 2005116014
http://www.google.co.in/patents/WO2005116014A1?cl=en
Example 113 (3.3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1 – (S)-2-(3.3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1 -carboxylic acid tert-butyl ester
(S)-4-Oxo-pyrrolidine-1 ,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0°C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0°C to 5°C for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0°C to 5°C. The reaction mixture turned back to aslurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0°C to 5°C.
3,3-Difluoro- pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at – 10°C to 0°C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at -10°C to 0°C. Upon completion of triethylamine addition, the mixture was stirred for 1h at 0 to 5°C. The reaction was complete by HPLC assay (-1% starting material). The reaction was quenched with water (10 volumes) at 0°C to 5 °C. The mixture was heated to 20°C to 25 °C. The layers were separated, and the organic layer was washed with 0.5 M HCI (5 volumes). The organic layer was again washed with combined 5% NaHC03 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to -6 volumes. The mixture was held at 60°C to 65 °C for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20°C to 25 °C for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25°C to 35°C for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65°C to 70°C. The mixture was cooled to 60°C to 65 °C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20°C to 25 °C over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35°C to 45 °C yielded 435 grams of product. HPLC purity: 96.4%.
Step 2 – (2S.4S)-2-(3.3-Dif luoro-pyrrolidine-1 -carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidine-1 – carboxylic acid tert-butyl ester A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20°C to 25°C until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60°C to 70°C until a steady temperature of 66.9°C was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to -3°C to 7°C and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at -5°C to 15°C. The reaction mixture was heated to 20°C to 25°C and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30°C to 60°C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20°C to 25CC. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45°C to 50°C. To the slurry was added heptane (10 volumes) at 45°C to 50°C over 30 minutes. The mixture was cooled to 20°C to 25°C and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35°C to 45°C yield 5.35 kg (91.3%) of the product. Step 3 – (3.3-Dif luoro-pyrrolidin-1 -yl)-f(2S.4S)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidin-2-yll- methanone Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg,
1.0 equivalent). To the slurry was added concentrated HCI (37 wt% in water, 19.1 liters, 2 volumes) slowly at 20°C to 30°C over 4 hours. The slurry went into solution after 12 liters of HCI was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5°C to 15°C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20°C to 25°C and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution.
The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50°C to 60°C. The mixture was cooled to 20°C to 25°C over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1 :1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44°C for 12 hours.
Yield: 5.7 kg, 75.9%.
m.p. 156°C. MS m/z 367 (MH+).
FREE BASE
1H NMR (400 MHz, D20): δ 8.15 (d, 2H, J = 5.0 Hz, CH of pyrimidine), 6.55 (t, 1 H, J = 4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1 H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N-CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N-CH2 of piperazine), 2.97 (dd, 1 H, J = 10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1 H, H4b of proline), 2.69 (dd, 1 H, J = 10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N-CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1 H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
………………
US 2005/0256310
http://www.google.com/patents/US20050256310

This approach begins with N–t-Boc-4-oxo-l-proline (1) that undergoes a mixed anhydride activation with pivaloyl chloride at 0 °C, followed by amidation with 3,3-difluoropyrrolidine to yield the intermediate 2. Reductive amination with 1-(2-pyrimidyl)piperazine using sodium triacetoxyborohydride in THF/AcOH provided the desired stereoisomer 3 in high yield and selectivity, the undesired diastereomer being completely removed by crystallization. Deprotection of 3 with 6 N HCl, followed by neutralization with 50% NaOH and extraction provided PF-734200 (4) in good yield.
EXAMPLE 113 (3,3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1—(S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1-carboxylic acid tert-butyl
(S)-4-Oxo-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0° C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0° C. to 5° C. for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0° C. to 5° C. The reaction mixture turned back to a slurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0° C. to 5° C. 3,3-Difluoro-pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at −10° C. to 0° C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at −10° C. to 0° C. Upon completion of triethylamine addition, the mixture was stirred for 1 h at 0 to 5° C. The reaction was complete by HPLC assay (˜1% starting material). The reaction was quenched with water (10 volumes) at 0° C. to 5 ° C. The mixture was heated to 20° C. to 25 ° C. The layers were separated, organic layer was washed with 0.5 M HCl (5 volumes). The organic layer was again washed with combined 5% NaHCO3 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to ˜6 volumes. The mixture was held at 60° C. to 65° C. for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20° C. to 25 ° C. for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25° C. to 35° C. for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65° C. to 70° C. The mixture was cooled to 60° C. to 65° C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20° C. to 25° C. over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yielded 435 grams of product. HPLC purity: 96.4%.
Step 2—(2S,4S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester
A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20° C. to 25° C. until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60° C. to 70° C. until a steady temperature of 66.9° C. was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to −3° C. to 7° C. and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at −5° C. to 15° C. The reaction mixture was heated to 20° C. to 25° C. and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30° C. to 60° C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20° C. to 25° C. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45° C. to 50° C. To the slurry was added heptane (10 volumes) at 45° C. to 50° C. over 30 minutes. The mixture was cooled to 20° C. to 25° C. and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yield 5.35 kg (91.3%) of the product.
Step 3—(3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone
Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg, 1.0 equivalent). To the slurry was added concentrated HCl (37 wt % in water, 19.1 liters, 2 volumes) slowly at 20° C. to 30° C. over 4 hours. The slurry went into solution after 12 liters of HCl was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5° C. to 15° C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20° C. to 25° C. and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution. The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50° C. to 60° C. The mixture was cooled to 20° C. to 25° C. over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1:1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44° C. for 12 hours.
Yield: 5.7 kg, 75.9%. m.p. 156° C. MS m/z 367 (MH+).
1H NMR (400 MHz, D2O): δ 8.15 (d, 2H, J=5.0 Hz, CH of pyrimidine), 6.55 (t, 1H, J=4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N—CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N—CH2 of piperazine), 2.97 (dd, 1H, J=10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1H, H4b of proline), 2.69 (dd, 1H, J=10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N—CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
……………..
PAPER

Scheme 1.
Reagents and conditions: (a) 3,3-difluoropyrrolidine hydrochloride, EDC, HOBt, TEA, DCM, rt; (b) NaBH4, MeOH, (c) (1) trifluoromethane-sulphonyl chloride, DIPEA, DCM; (2) 2-(1-piperazinyl)pyrimidine, DCM, −10 °C; (d) 4 N HCl in dioxane, rt; (e) 2-(1-piperazinyl)pyrimidine, NaBH(OAc)3, AcOH, DCE; (f) R1R2NH hydrochloride, EDC, HOBt TEA, DCM, 0–rt; (g) N-heterocyclic piperazine, NaBH(OAc)3, AcOH, DCE.
……………………….
if image is not clear see at………..http://www.allfordrugs.com/2015/07/03/gosogliptin/
| Patent | Submitted | Granted |
|---|---|---|
| Therapeutic compounds [US7291618] | 2005-11-17 | 2007-11-06 |
| (2S,4S)-4-(piperazin-1-yl)pyrrolidine-2-methanone derivatives [US7465732] | 2007-05-03 | 2008-12-16 |
| THERAPEUTIC COMPOUNDS [US2007161664] | 2007-07-12 | |
| Therapeutic compounds [US2006079498] | 2006-04-13 |
//////////
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html
BEZ 235 (NVP-BEZ235), Dactolisib
BEZ235 (NVP-BEZ235)Dactolisib
4-[2,3-dihydro-3-methyl-2-oxo-8-(3-quinolinyl)-1H-imidazo[4, 5-c]quinolin-1-yl]-α,α-dimethyl-benzeneacetonitrile
2-methyl-2-{4-[3-methyl-2-oxo-8-(quinolin-3-yl)-1H,2H,3H-imidazo[4,5-c]quinolin-1-yl]phenyl}propanenitrile
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]-propionitrile
Chemical Formula: C30H23N5O
CAS Number: 915019-65-7
Molecular Weight: 469.54
PHASE 2, NOVARTIS
CANCER, BLADDER
NVP-BEZ235 is a dual inhibitor of phosphatidylinositol 3-kinase (P13K)and the downstream mammalian target of rapamycin (mTOR) by binding to the ATP-binding cleft of these enzymes. It specifically blocks the dysfunctional activation of the P13K pathway and induce G(1) arrest. NPV-BEZ235 has been shown to inhibit VEGF induced cell proliferation and survival in vitro and VEGF induced angiogenesis in vivo. It has also been shown to inhibit the growth of human cancer in animal models.
BEZ-235 is an orally active phosphatidylinositol 3-kinase (PI3K) inhibitor in early clinical trials at Novartis for the treatment of advanced breast cancer, renal cell carcinoma, solid tumors and castration-resistant prostate cancer. Phase I clinical trials were also under way at the company for the treatment of glioma, however, no developments in this indication has been reported. Phase II clinical trials are ongoing at Johann Wolfgang Goethe Universität for the treatment of relapsed or refractory acute leukemia.
PI3Ks perform various functions, promoting cell growth, proliferation, differentiation, motility, survival and intracellular trafficking. Mutations leading to increased activity of PI3Ks, including faulty production or action of PI3K antagonists, have been found in many cancers.

……………………………..
WO 2006122806
http://www.google.com/patents/WO2006122806A2?cl=en
![]()
…………………………..
WO 2008064093
2-methyl-2-[4-(3-methyl- 2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile of formula I (compound I),

Example 1
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]-propionitrile

In a suitable lab glass reactor are placed 45.0 g of starting 2[4-(8-bromo-3-methyl-2-oxo-2,3- dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]2-methyl-propionitrile together with 2.25 g of bistriphenylphosphine’palladium dichloride in 445 ml N,N-dimethylformamide. This mixture is heated to 95 0C and then a solution of 22.2 g of 3-quinoline boronic acid in a mixture of 225 ml DMF, 300 ml H2O and 60 g of KHCO3 is added. This mixture is heated for 2 h at 95 0C. Then 1080 ml H2O are added. The product 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl- 2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile precipitates. The mixture is cooled within 1.5 h to 0 – 5 °C. After stirring at that temperature for 2 h the crude product is filtered and washed with 300 ml H2O. This product is dried in vacuo at 60 0C for 18 h, to yield crude product.
40 g of this crude product is dissolved in 200 ml formic acid at 60 0C. 8 g of active charcoal and Smopex 234 are added. The mixture is stirred at 60 0C for 1 h, the charcoal is filtered, the residue washed with 80 ml formic acid and then 175 ml formic acid are distilled off in vacuo. Then 320 ml methanol are added and the mixture is heated at reflux for 3 h. The purified product precipitates from the reaction mixture. The mixture is cooled to 0 – 5 0C within 1 h, then stirred 2 h at that temperature is finally filtered and washed with 80 ml cold methanol. This recrystallisation procedure is repeated again. Finally the twice recrystallised material is dried in vacuo at 60 0C to yield purified 2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin- 3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile.
Example 1a 5-Bromo-2-(2-nitro-vinylamino)-benzoic acid

A suspension of 25 g (16 mmol) of 2-amino-5-bromo-benzoic acid (Fluka, Buchs, Switzerland) in H2O-HCI (37%) (10:1) is stirred for 8 h and then filtered (solution A). 8.17 g (255 mmol) of nitromethane (Fluka, Buchs, Switzerland) are added over 10 min to an ice- bath cooled mixture of 35 g of ice and 15.3 g (382 mmol) of NaOH. After stirring for 1 h at 0 0C and 1 h at rt, the solution is added at 0 0C to 28 g of ice and 42 ml of HCI (37%) (solution B). Solutions A and B are combined and the reaction mixture is stirred for 18 h at rt. The yellow precipitate is filtered off, washed with H2O and dried in vacuo at 400C to give the title compound. ES-MS: 287, 289 (M + H)+, Br pattern; 1H NMR (DMSO-d6): δ 13.7-14.6/br s (1 H), 12.94/d (1 H), 8.07/d (1 H), 8.03/dd (1 H), 7.83/dd (1 H), 7.71/d (1 H), 6.76/d (1 H).
Example 1b 6-Bromo-3-nitro-quinolin-4-ol

29 g (101 mmol) of 5-bromo-2-(2-nitro-vinylamino)-benzoic acid (Example 1a) and 11.9 g (121 mmol) of potassium acetate in 129 ml (152 mmol) of acetic anhydride are stirred for 1.5 h at 120 0C. The precipitate is filtered off and washed with acetic acid until the filtrate is colorless, then is washed with H2O and dried in vacuo to give the title compound. ES-MS: 269, 271 (M + H)+, Br pattern; analytical HPLC: W= 2.70 min (Grad 1).
Example 1c 6-Bromo-4-chloro-3-nitro-quinoline

20 g (74.3 mmol) of 6-bromo-3-nitro-quinolin-4-ol (Example 1b) in 150 ml (1.63 mol) of POCI3 are stirred for 45 min at 120 °C. The mixture is cooled to rt and poured slowly into ice- water. The precipitate is filtered off, washed with ice-cold water, and dissolved in CH2CI2. The organic phase is washed with cold brine, and the aqueous phase is discarded. After drying over MgSO4, the organic solvent is evaporated to dryness to provide the title compound. 1H NMR (CDCI3): J9.20/S (1H), 8.54/d (1H), 8.04/d (1H), 7.96/dd (1H); analytical HPLC: W= 4.32 min (Grad 1).
Example 1d 2-Methyl-2-(4-nitro-phenyl)-propionitrile
O .

To 15 g (92.5 mmol) of (4-nitro-phenyl)-acetonitrile (Fluka, Buchs, Switzerland), 1.64 mg (5.09 mmol) of tetrabutylammonium bromide (Fluka, Buchs, Switzerland) and 43.3 g (305 mmol) of iodomethane in 125 mL of CH2CI2 are added 1O g (250 mmol) of NaOH in 125 ml of water. The reaction mixture is stirred for 20 h at RT. After this time, the organic layer is separated, dried over MgSO4, and evaporated to dryness. The residue is dissolved in diethylether and treated with black charcoal for 30 min, filtered over Celite and evaporated in vacuo to give the title compound as a pale yellow solid. Analytical HPLC: tret= 3.60 minutes (Grad 1).Example 1e (2-(4-Amino-phenyl)-2-methyl-propionitrile

16 g (84.1 mmol) of 2-methyl-2-(4-nitro-phenyl)-propionitrile (Example 1d) and 4.16 g of Raney-Ni are shacked in 160 ml of THF-MeOH (1:1) under 1.1 bar of H2 for 12 h at rt. After completion of the reaction, the catalyst is filtered-off and the filtrate is evaporated to dryness. The residue is purified by flash chromatography on silica gel (hexane-EtOAc 3:1 to 1:2) to provide the title compound as an oil. ES-MS: 161 (M + H)+; analytical HPLC: tret= 2.13 minutes (Grad 1).
Example 1f 2-[4-(6-Bromo-3-nitro-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile

18 g (62.6 mmol) of 6-bromo-4-chloro-3-nitro-quinoline (Example 1c) and 11 g (68.9 mmol) of (2-(4-amino-phenyl)-2-methyl-propionitrile (Example 1e) are dissolved in 350 ml of acetic acid and stirred for 2 h. After this time, water is added and the yellow precipitate is filtered off and washed with H2O. The solid is dissolved in EtOAc-THF (1 :1), washed with sat. aqueous NaHCO3 and dried over MgSO4. The organic phase is evaporated to dryness to give the title compound as a yellow solid. ES-MS: 411 , 413 (M + H)+, Br pattern; analytical HPLC: tret= 3.69 min (Grad 1).
Example 1q 2-[4-(3-Amino-6-bromo-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile

24 g (58.4 mmol) of 2-[4-(6-bromo-3-nitro-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile (Example 1e) is shacked in 300 ml of MeOH-THF (1:1) under 1.1 bar of H2 in the presence of 8.35 g of Raney-Ni for 1 h. After completion of the reaction, the catalyst is filtered off and the filtrate is evaporated to dryness to give the title compound as a yellow foam. ES-MS: 381 , 383 (M + H)+, Br pattern; analytical HPLC: W= 3.21 min (Grad 1).
Example 1h
2-[4-(8-Bromo-2-oxo-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-2-methyl- propionitrile

A solution of 5 g (13.1 mmol) of 2-[4-(3-amino-6-bromo-quinolin-4-ylamino)-phenyl]-2- methyl-propionitrile (Example 1g) and 1.59 g (15.7 mmol) of triethylamine in 120 ml CH2CI2 is added over 40 min to a solution of 2.85 g (14.4 mmol) of trichloromethyl chloroformate (Fluka, Buchs, Switzerland) in 80 ml of CH2CI2 at 00C with an ice-bath. The reaction mixture is stirred for 20 min at this temperature then is quenched with sat. aqueous NaHCO3, stirred for 5 min and extracted with CH2CI2. The organic layer is dried over Na2SO4, filtered and evaporated in vacuo to give crude title compound as a brownish solid. ES-MS: 407, 409 (M + H)+, Br pattern; analytical HPLC: tret= 3.05 min (Grad 1). Example 1i
2-[4-(8-Bromo-3-methyl-2-oxo-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-2- methyl-propionitrile

To a solution of 3.45 g (8.47 mmol) of 2-[4-(8-bromo-2-oxo-2,3-dihydro-imidazo[4,5- c]quinolin-1-yl)-phenyl]-2-methyl-propionitrile (Example 1h), 1.8 g (12.7 mmol) of iodomethane (Fluka, Buchs, Switzerland) and 273 mg (0.847 mmol) of tetrabutylammonium bromide (Fluka, Buchs, Switzerland) in 170 ml of CH2CI2 is added a solution of 508 mg (12.7 mmol) of NaOH (Fluka, Buchs, Switzerland) in 85 ml of H2O. The reaction mixture is stirred for 2 days and 900 mg (6.35 mmol) of iodomethane and 254 mg (6.35 mmol) of NaOH in 5 ml of H2O are added. The reaction mixture is stirred for 1 day at rt . After this time, the reaction is quenched with H2O and extracted with CH2CI2 (2*). The organic layer is washed with brine, dried over Na2SO4, filtered and evaporated in vacuo to give the title compound as a beige solid. ES-MS: 421 , 423 (M + H)+, Br pattern; analytical HPLC: tret= 3.15 min (Grad 1).
Example 2
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]propionitrile p-toluenesulfonate salt
26.5 g of 2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1- yl)-phenyl]propionitrile are placed together with 55 ml formic acid into a glass reactor. This mixture is heated to 60 0C to get a clear solution. This solution is clearfiltered and washed with 36 ml formic acid. Then formic acid is distilled off until the volume of the residual solution is 55 ml. Then a solution of 11.3 g of p-toluenesulfonic acid in 228 ml acetone is added at 50 0C, followed by further addition of 822 ml acetone within 30 minutes. The salt precipitates from the reaction mixture. The mixture is cooled to 0 0C within 2 h, stirred at that temperature for 3 h, is then filtered and washed with 84 ml acetone. The product‘ is dried at 60 0C in vacuo for 18 h to yield 29.8 g (82.4 %) of the 2-Methyl-2-[4-(3-methyl-2-oxo-8- quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile p-toluenesulfonate salt (crystalline form A). The crystalline forms of the present invention are synthesized in accordance with the following examples which are illustrative without limiting the scope of the present invention.
Example 3:
Preparation of form A of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro- imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile
Form A of compound I can be manufactured in the following way: 241 g of free base are dissolved 2.4 I acetic acid at 50 0C. The solution is clearfiltered, washed with 250 ml acetic acid and then at 50 0C 7.2 I of water are added. The free base starts precipitating. The mixture is cooled within 1 h to 25 0C, is then filtered and washed with 10 I H2O. The free base is then dried in vacuo at 50 0C over night to yield 204 g of free base.
References
| WO2005054237A1 | 19 Nov 2004 | 16 Jun 2005 | Hans-Georg Capraro | 1h-imidazoquinoline derivatives as protein kinase inhibitors |
| WO2006122806A2 | 18 May 2006 | 23 Nov 2006 | Novartis Ag | 1,3-dihydro-imidazo [4,5-c] quinolin-2-ones as lipid kinase inhibitors |
| CL11872006A | Title not available |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009118324A1 * | 24 Mar 2009 | 1 Oct 2009 | Novartis Ag | 5imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| WO2013049300A1 * | 27 Sep 2012 | 4 Apr 2013 | Dana-Farber Cancer Institute, Inc. | Method of treating mucoepidermoid carcinoma |
| WO2013152717A1 | 9 Apr 2013 | 17 Oct 2013 | Shanghai Yunyi Healthcare Management Co., Ltd. | Fused pyrimidine compound, and preparation method, intermediate, composition, and uses thereof |
| EP2474323A2 * | 24 Mar 2009 | 11 Jul 2012 | Novartis AG | Imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| US8476294 | 2 Jun 2010 | 2 Jul 2013 | Novartis Ag | 1H-imidazo[4,5-c]quinolinone derivatives |
VX 787, PIMODIVIR, for Avian influenza

(2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
(2S,3S)-3-((5-Fluoro-2-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic Acid
399.394
C20 H19 F2 N5 O2
JNJ-872
VRT-0928787
VX-787
VRT-0928787
VX-787
vx 787
| Vertex Pharmaceuticals |
Janssen Pharmaceuticals, under license from Vertex Pharmaceuticals, is developing VX-787 and its back-up compound VX-353, an influenza A viral replication inhibitor, for treating influenza A virus infection, including pandemic and avian influenza strains. In May 2015, VX-787 was in phase II clinical trial.

Useful for treating influenza virus infection. For concurrent filing see WO2015073476 (claiming the polymorphic forms of VX-787) and WO2015073491 (claiming the composition comprising the hydrochloride salt of VX-787).
Polymorphic forms of hydrochloride (A,F and D) and tosylate salts (form A) of VX-787 are claimed. , useful for treating influenza virus infection. For concurrent filing see WO2015073481 (claiming the processes for the synthesis of VX-787 ) and WO2015073491 (claiming the composition comprising the hydrochloride salt of VX-787).
WO2010148197
http://www.google.com/patents/WO2010148197A1?cl=en
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
Compound 1070 was made in a similar fashion as described above for compounds 946 and 947.
………………….
WO 2013019828
http://www.google.com/patents/WO2013019828A1?cl=en
WO 2012083122
http://www.google.co.in/patents/WO2012083122A1?cl=en
Synthetic Scheme 1

(a) CHC13; (b) NaOMe, MeOH; (c) DPPA, Et3N, BnOH; (d) H2, Pd/C;
Synthetic Scheme 2

(a) Et3N, CH3CN; (b) cone. H2S04; (c) 9M H2S04; (d) Ag2C03, HOAc, DMSO, 100 °C; (e) X- phos, Pd2(dba)3, K3PO4, 2-methyl THF, H20, 120 °C (f) LiOH, THF, MeOH, 70 °C
Synthetic Scheme 3

(a) Et3N, THF; (b) chiral SFC separation; (c) 5-fluoro- l -(p-tolylsulfonyl)-3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-
………………………
See new patents
WO2015073491
……………………………..
Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2
J. Med. Chem., 2014, 57 (15), pp 6668–6678
DOI: 10.1021/jm5007275
http://pubs.acs.org/doi/abs/10.1021/jm5007275
Vertex Pharmaceuticals Inc

1H NMR (300 MHz, DMSO-d6) δ 12.71 (br s, 1H), 8.58 (s, 1H), 8.47 (dd, J = 9.6, 2.8 Hz, 1H), 8.41 (d, J = 4.8 Hz, 1H), 8.39–8.34 (m, 1H), 4.89–4.76 (m, 1H), 2.94 (d, J = 6.9 Hz, 1H), 2.05 (br s, 1H), 1.96 (br s, 1H), 1.68 (complex m, 7H);
13C NMR (300 MHz, DMSO-d6) δ 174.96, 157.00, 155.07, 153.34, 152.97, 145.61, 142.67, 140.65, 134.24, 133.00, 118.02, 114.71, 51.62, 46.73, 28.44, 28.00, 24.90, 23.78, 20.88, 18.98;
LCMS gradient 10–90%, 0.1% formic acid, 5 min, C18/ACN, tR = 2.24 min, (M + H) 400.14;
HRMS (ESI) of C20H20F2N5O2 [M + H] calcd, 400.157 95; found, 400.157 56.
Article
June 18, 2014
Vertex Licenses VX-787 to Janssen Pharmaceuticals for the Treatment of Influenza
Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced that it has entered into a licensing agreement with Janssen Pharmaceuticals, Inc. for the worldwide development and commercialization of VX-787, a novel medicine discovered by Vertex for the treatment of influenza. As part of the agreement, Vertex will receive an up-front payment of $30 million from Janssen and has the potential to receive additional development and commercial milestone payments as well as royalties on future product sales. Vertex completed a Phase 2a study of VX-787 in 2013 that showed statistically significant improvements in viral and clinical measurements of influenza infection. VX-787 is designed to directly inhibit replication of the influenza virus.
“With a deep history in developing new medicines for viral infections and diseases, Janssen is well-positioned to advance the global development of VX-787 for the treatment of influenza,” said Jeffrey Leiden, M.D., Ph.D., Chairman, President and Chief Executive Officer of Vertex. “This collaboration provides important support for the continued development of VX-787 in influenza and contributes to our financial strength to enable continued investment in our key development programs for cystic fibrosis and in research aimed at discovering new medicines.”
About the Collaboration
Under the terms of the collaboration, Janssen will have full global development and commercialization rights to VX-787. Vertex will receive a $30 million up-front payment from Janssen and could receive additional development and commercial milestone payments as well as royalties on future product sales. The collaboration, and the related $30 million up-front payment, is subject to the expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act.
About VX-787
VX-787 is an investigational medicine that is designed to directly inhibit replication of influenza A, including recent H1 (pandemic) and H5 (avian) influenza strains, based on in-vitro data. VX-787’s mechanism represents a new class of potential medicines for the treatment of influenza, distinct from neuraminidase inhibitors, the current standard of care for the treatment of influenza. VX-787 is intended to provide a rapid onset of action and an expanded treatment window.
In a Phase 2a influenza challenge study, statistically significant improvements in viral and clinical measurements of influenza infection were observed after treatment with VX-787. The study met its primary endpoint and showed a statistically significant decrease in the amount of virus in nasal secretions (viral shedding) over the seven-day study period. In addition, at the highest dosing regimen evaluated in the study, there was a statistically significant reduction in the severity and duration of influenza-like symptoms. In this study, VX-787 was generally well-tolerated, with no adverse events leading to discontinuation. Those who took part in the study volunteered to be experimentally exposed to an attenuated form of live H3N2 influenza A virus. H3N2 is a common type of influenza virus and was the most common type observed in the 2012/2013 influenza season in the United States.
VX-787 was discovered by Vertex scientists.
About Influenza
Often called “the flu,” seasonal influenza is caused by influenza viruses, which infect the respiratory tract.1 The flu can result in seasonal epidemics2 and can produce severe disease and high mortality in certain populations, such as the elderly.3 Each year, on average 5 to 20 percent of the U.S. population gets the flu4 resulting in more than 200,000 flu-related hospitalizations and 36,000 deaths.5 The overall national economic burden of influenza-attributable illness for adults is $83.3 billion.5 Direct medical costs for influenza in adults totaled $8.7 billion including $4.5 billion for adult hospitalizations resulting from influenza-attributable illness.5 The treatment of the flu consists of antiviral medications that have been shown in clinical studies to shorten the disease and reduce the severity of symptoms if taken within two days of infection.6 There is a significant need for new medicines targeting flu that provide a wider treatment window, greater efficacy and faster onset of action.
About Vertex
Vertex is a global biotechnology company that aims to discover, develop and commercialize innovative medicines so people with serious diseases can lead better lives. In addition to our clinical development programs focused on cystic fibrosis, Vertex has more than a dozen ongoing research programs aimed at other serious and life-threatening diseases.
Founded in 1989 in Cambridge, Mass., Vertex today has research and development sites and commercial offices in the United States, Europe, Canada and Australia. For four years in a row, Science magazine has named Vertex one of its Top Employers in the life sciences. For additional information and the latest updates from the company, please visit www.vrtx.com.
Vertex’s press releases are available at www.vrtx.com.
| WO2002024705A1 | 13 Sep 2001 | 28 Mar 2002 | Charles Jackson Barnett | Stereoselective process for preparing cyclohexyl amine derivatives |
| WO2003015798A1 | 13 Aug 2002 | 27 Feb 2003 | Toyama Chemical Co Ltd | Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 | 30 Mar 2005 | 13 Oct 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006069258A1 * | 20 Dec 2005 | 29 Jun 2006 | Amgen Inc | Substituted heterocyclic compounds and methods of use |
| WO2007084557A2 | 17 Jan 2007 | 26 Jul 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2008079346A1 | 21 Dec 2007 | 3 Jul 2008 | Vertex Pharma | 5-cyan0-4- (pyrrolo [2, 3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| WO2009073300A1 | 31 Oct 2008 | 11 Jun 2009 | Vertex Pharma | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * | 17 Jun 2010 | 23 Dec 2010 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| WO2011008915A1 * | 15 Jul 2010 | 20 Jan 2011 | Abbott Laboratories | Pyrrolopyridine inhibitors of kinases |
| US20100038988 | 12 Aug 2008 | 18 Feb 2010 | Gannon Ramy | Stator and Method of Making the Same |
| WO2003015798A1 | Aug 13, 2002 | Feb 27, 2003 | Toyama Chemical Co Ltd | Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 | Mar 30, 2005 | Oct 13, 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 | Jan 17, 2007 | Jul 26, 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2009073300A1 | Oct 31, 2008 | Jun 11, 2009 | Vertex Pharma | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * | Jun 17, 2010 | Dec 23, 2010 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US20100038988 | Aug 12, 2008 | Feb 18, 2010 | Gannon Ramy | Stator and Method of Making the Same |
……
.

Vertex Pharmaceuticals’ Boston Campus, United States of America
Lynette Hopkinson VP Commercial Regulatory Affairs, Global Regulatory Affairs Vertex Pharmaceuticals Incorporated, United States

swati Patel, a lead analyst, shared a toast with Mir Hussain, a systems engineer, at Vertex Pharmaceuticals during the Friday beer hour, which features beer and chips for employees.
On Fridays around 5 o’clock, after a hard week of work, Frank Holland likes to unwind with a beer. And he doesn’t have to leave work to get one.
Holland is a research scientist at Vertex Pharmaceuticals, which every Friday rings in “beer hour,” offering free adult beverages and munchies to its 1,300 Boston employees.
For Holland, the weekly ritual is a chance to escape the bubble of his chemistry lab and bump into colleagues from other departments — as well as Vertex’s top executives, who regularly attend. For those who prefer grapes to hops, there is also wine.
“Some of the other companies I worked at, you really had to go out of your way to meet people,” said Holland, 32. “At Vertex all you have to do is show up in the cafeteria on a Friday afternoon.”
Sure, free beer is common at hip tech offices; some even have their own bars. But Vertex, best known for its treatment for cystic fibrosis, was doing this way before it was cool. The beer-hour tradition goes back to the company’s founding days, in 1989. Back then, it was just two dozen people in a small office in Cambridge. Someone went to a corner store, bought a case of beer and some chips, and beer hour was born.

Virginia Carden Carnahan
Vice President, New Product Planning and Strategy, Vertex Pharmaceuticals

A scientist works in the lab at Boston-based Vertex Pharmaceuticals.
Vertex Pharmaceuticals Headquarters Lobby
…………
Lascufloxacin, KRP-AM1977, by Kyorin

Lascufloxacin
CAS 848416-07-9
Kyorin Pharmaceutical Co., Ltd., 杏林製薬株式会社
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-
7-((3S,4S)-3-((Cyclopropylamino)methyl)-4-fluoropyrrolidin-1-yl)-6-fluoro-1-(2-fluoroethyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
{(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
(KRP-AM1977X)
-
C21-H24-F3-N3-O4
- 439.4316
- SMILES……COc1c2c(cc(c1N3C[C@H](C(C3)CNC4CC4)F)F)c(=O)c(cn2CCF)C(=O)O
![]()
…………………………
Lascufloxacin hydrochloride
-
C21-H24-F3-N3-O4.Cl-H
- 475.8925
- CAS 1433857-09-0
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, hydrochloride (1:1)
……………….
Lascufloxacin mesylate
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, methanesulfonate (1:1)
-
C21-H24-F3-N3-O4.C-H4-O3-S
- 535.5372
- CAS 1433857-41-0
The other non-fluorinated quinolone under clinical development is KRP-AM1977, by Kyorin, which is in Phase I of clinical trials. The oral formulation of the compound (KRP-AM1977X) is being tested for treatment of respiratory infections and the I.V. formulation is under development for treatment of MRSA infections [1,2].
………………………………..
PATENT
WO 2013069297
http://www.google.co.in/patents/WO2013069297A1?cl=en
The present invention is represented by Formula (1) – {(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (hereinafter, compound (1) crystals of a salt also referred to), and a method for their preparation.
Typically, the pharmaceutical, in addition to the therapeutic effects on diseases, such as safety and quality are required. Therefore, the compound is the active ingredient of drugs, a variety of conditions and that is excellent in storage stability in the (light, temperature, humidity etc. influence the compound) are determined. Also, if the medicament is a dosage form such as oral preparations and injections, it is preferred that higher solubility in active ingredients of the water contained.
Compound (1) is safe, not only exhibit a strong antimicrobial action, conventional hard Gram-positive bacteria antimicrobial agents shown efficacy, particularly MRSA, PRSP, to VRE such resistant strains, to exhibit strong antibacterial activity It is known (for example, Patent Document 1).
WO 2005/026147
Patent Document 1, as the physicochemical characteristics of the compound (1) only has been shown to be a light brown free crystals. Also, Patent Document 1, the solubility in water of Compound (1), stability, no disclosure whatsoever information including characteristics of the crystal.
The present invention aims to provide a technique capable of improving the solubility and storage stability in water of the compound (1).
(Reference Example 4)
Bis (acetato -O) – [6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylate -O 3, O 4] boron Under a nitrogen atmosphere, boric acid (catalyst preparation) 86.4 g (1.40mol) was added acetic anhydride 17.9 L (190mol), and was heated and stirred for 30 minutes at 70.0 ~ 77.7 ℃. It was then cooling the mixture to an internal temperature of 24.7 ℃ (hot water set temperature 23.0 ℃). Subsequently, it was added portionwise boric acid to 4 times to the mixture. Specifically, the addition of boric acid (1 time) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.7 ~ 27.4 ℃. The addition of boric acid (second) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.3 ~ 26.3 ℃. In addition boric acid (third time) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 24.3 ~ 26.8 ℃. In addition boric acid (4 th) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 25.1 ~ 28.3 ℃. The mixture was stirred for 30 minutes at 50.0 ~ 54.9 ℃, was with boric acid triacetate adjusted solution.
In the boric acid triacetate adjusted solution, 6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylic acid ethyl ester 4.60kg (14. In a reaction preparation solution are added 0mol), and stirred for 3 hours at 53.7 ~ 56.9 ℃. The reaction preparation was cooled to 30.0 ℃, and allowed to stand overnight at room temperature. The reaction preparation was allowed to dissolve with heating to precipitate up to 55.0 ℃, acetone 13.8L was added and the reaction solution (1).
Separately, under nitrogen atmosphere, it is mixed Tsunemizu 161L and aqueous ammonia (28%) 28.2L (464mol), and cooled the mixture to 1.6 ℃. To the mixture, it was added the reaction solution of the above (1), to obtain a crude crystal acquisition solution crowded washed with acetone 9.20L. After cooling the crude crystal acquisition solution to 15.0 ℃, it was stirred for 1 hour at 6.2 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with Tsunemizu 46.0L, to give 9.07kg of wet crude crystals. Set temperature 65.0 to about 16 hours and dried under reduced pressure at ℃, the crude crystals were obtained 5.89kg.
Under a nitrogen atmosphere, it is mixed acetone and 29.5L crude crystal, the resulting mixture was heated and dissolved (melting temperature 52.6 ℃). When heated, it was dropped until the crystallization of diisopropyl ether 58.9L in a mixture (dropping amount 10.0L; 52.8 → 48.7 ℃; crystallization temperature 49.0 ℃). After crystallization confirmation, stirred for 15 minutes the mixture at 49.0 ~ 50.1 ℃, it was dropped the rest of diisopropyl ether to the mixture (50.1 → 46.4 ℃), 46.7 ~ 51.7 It was stirred for 15 minutes mixture at ℃. After cooling the mixture to 15 ℃, it was stirred for 30 minutes at 8.1 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with acetone and diisopropyl ether 5.89L 11.8L, to obtain 6.19kg of wet crystals. For about 20 hours drying under reduced pressure at warm water set temperature 65.0 ℃, bis (acetato -O) – [6,7-difluoro-1- (2-fluoroethyl) -8-methoxy-4-oxo-1,4- dihydro-3-carboxylate -O 3, O 4] was obtained 5.42kg boron (90.4% yield).
Melting point: 183 ~ 185 ℃ (dec).
Elemental analysis (%): calculated as C 17 H 15 BF 3 NO 8: C, 47.58; H, 3.52; N, 3.26.
Measured value: C, 47.91; H, 3.44; N, 3.04.
1 H-NMR (CDCl 3, 400 MHz) δ: 2.04 (6H, s), 4.22 (3H, d, J = 2.4Hz), 4.88 (2H, dt, J = 47.0 , 4.4Hz), 5.21 (2H, dt, J = 24.9,4.4Hz), 8.17 (1H, t, J = 8.8Hz), 9.11 (1H, s).
ESI MS (positive) m / z: 430 (M + H) +.
IR (KBr) cm -1: 3080,1703.
………………………………………….
WO 2005026147
http://www.google.com/patents/EP1666477A1?cl=en
KEY INTERMEDIATE

604798-54-1
3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-
| Chemical Name:3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-CAS: 604798-54-1Molecular Formula: C8H15FN2Molecular Weight: 158.2165032 |
………………………….
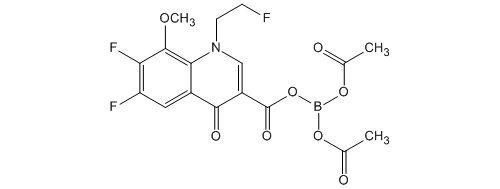
KEY INTERMEDIATE
CAS 848498-67-9
Boron, bis(acetato-κO)[6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-(oxo-κO)-3-quinolinecarboxylato-κO]-, (T-4)-
Coordination Compound
ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル)
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
| 化学物質名 | ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル) -8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ キシ]ボラン |
|---|---|
| 構造別分類コード番号 | F60622212422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
|
| 安衛法官報通し番号 | 21534 |
| 安衛法官報公示整理番号 | 8-(1)-3764 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 848498-67-9 |
| 出典 | 厚生労働省 |
……………………………….
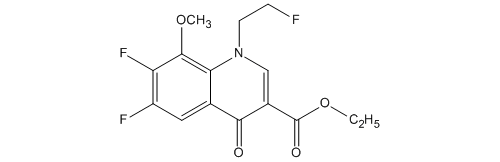
KEY INTERMEDIATE
3-Quinolinecarboxylic acid, 6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, ethyl ester
114214-60-7
C15H14F3NO4
6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
| 化学物質名 | 6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル |
|---|---|
| 構造別分類コード番号 | F60622322422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
 |
| 安衛法官報通し番号 | 21467 |
| 安衛法官報公示整理番号 | 8-(1)-3758 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 114214-60-7 |
| 出典 | 厚生労働省 |
| WO2003076428A1 * | 8 Mar 2002 | 18 Sep 2003 | Toshifumi Akiba | Quinolonecarboxylic acid derivative |
| WO2005026147A1 | 8 Sep 2004 | 24 Mar 2005 | Yoshikazu Asahina | 7-(4-substituted 3- cyclopropylaminomethyl-1 pyrrolidinyl) quinolonecarboxylic acid derivative |
| WO2007082471A1 * | 18 Jan 2007 | 26 Jul 2007 | Guangzhou Baiyunshan Pharmaceu | Anti-infective compound, preparation method thereof and use thereof |
| CN1158846A * | 9 May 1995 | 10 Sep 1997 | 昆山市康壮达兽药厂 | Synthesis technology of norfluxacini hydrochloride |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014174846A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174847A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174848A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Tablet |
- Kyorin. Kyorin—Main R&D Activities-1 (4 February 2013 Release). Available online: http://www.kyorin-pharm.co.jp/en/business/pdf/main_rd_activities_20130204_en.pdf (accessed on 4 February 2013).
- Kyorin. Drug discovery, development, and lcm with medical professionals and patients in mind. Available online: http://www.kyorin-gr.co.jp/en/business/gensen/r_and_d.shtml (accessed on 11 April 2013).
-
……….
![]()


Ochyanomizu Sola City 16F,
Kanda Surugadai 4-6, Chiyoda-ku,
Tokyo 101-8311 Japan
TEL: 03-3525-4711
Access
One-minute walk from the Hijiribashi exit of Ochanomizu station on JR Chuo and Sobu lines
One-minute walk from the B2 exit of Shin-Ochanomizu station on Tokyo Metro Chiyoda line
Four-minutes walk from the No.1 exit of Ochanomizu station on Tokyo Metro Marunouchi line
Six-minutes walk from the B3 exit of Ogawamachi station on Toei Subway Shinjuku line
| Trade Name | KYORIN Pharmaceutical Co.,Ltd. |
|---|---|
| Business | Manufacture and sales of prescription medicines |
| Head Office | Ochyanomizu Sola City 16F, Kanda Surugadai 4-6, Chiyoda-ku, Tokyo 101-8311 Japan (Access Map) |
| Telephone | 03-3525-4711 |
| Foundation | 1923 |
| Establishment | 1940 |
Shimotsuga-gun, Tochigi

 Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park –
Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park – .
.MARKET
Ochanomizu station


Motesanib (AMG-706)

Motesanib (AMG-706)
Amgen Inc.

Motesanib (AMG 706) is an experimental drug candidate originally developed by Amgen[1] but is now being investigated by theTakeda Pharmaceutical Company. It is an orally administered small molecule belonging to angiokinase inhibitor class which acts as an antagonist of VEGF receptors, platelet-derived growth factor receptors, and stem cell factor receptors.[2] It is used as thephosphatesalt motesanib diphosphate.
Motesanib, also known as AMG-706, is an orally administered multikinase inhibitor that selectively targets VEGF receptors, platelet-derived growth factor receptors, and Kit receptors.
Clinical trials
Motesanib was originally investigated for effectiveness against advanced nonsquamous non-small-cell lung cancer (NSCLC), withPhase II trials indicating an effectiveness comparable to bevacizumab when they were both used in combination withpaclitaxel/carboplatin.[3] However a later and more detailed Phase III trial failed to show any benefit for the treatment of NSCLC.[2][4]A second Phase III trial was started in 2012,[5] which focused on patients from Asian backgrounds (performed on the bases ofsubgroup analysis)[6] however this also failed to meet its primary endpoint.[7]
The drug has undergone a Phase II evaluation as first-line therapy for breast cancer[2] however this study found no evidence to support further investigation.[8] Phase II testing against persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas was also unsuccessful.[9]
There have also been 2 separate Phase II clinical trials for thyroid cancer which have both shown promising results.[10][11][12]
Developed at Amgen, the compound is also being evaluated as both monotherapy and in combination with other agents in the treatment of breast, colorectal, lung, thyroid and ovarian cancers. Clinical trials for the treatment of bladder cancer have been terminated.
The National Cancer Institute had been evaluating the potential of the drug in patients with low-grade neuroendocrine tumors; however, no recent development has been reported for this research. The FDA awarded fast track status to motesanib in 2004. In 2008, the compound was licensed to Takeda in Japan.

AMG-706 is synthesized as follows: 1-Acetyl-3,3-dimethyl-6-nitroindoline (I) is reduced by catalytic hydrogenation over Pd/C, giving the aminoindoline (II), which is then coupled with 2-chloronicotinoyl chloride (III) in the presence of DIEA to yield the corresponding nicotinamide (IV). Subsequent condensation of (IV) with neat 4-(aminomethyl)pyridine (V) at 120 °C affords the 2-aminonicotinamide derivative (VI). The N-acetyl group of (VI) is finally removed by acidic hydrolysis to furnish the title compound (1,2).
,………………………………………
US 2003125339
http://www.google.com/patents/US20030125339
………………………………………………….
US 2003225106
https://www.google.com/patents/US20030225106
EXAMPLE 133
[2295]

N-(3,3-Dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
Step A—Preparation of 1-acetyl-6-amino-3,3-dimethylindoline
1-Acetyl-3,3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL), the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc:CH2Cl2 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O—204.27.
Step B—Preparation of N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from 1-acetyl-6-amino-3,3-dimethylindoline (Step A) by the method described in Example 82.
Step C—Preparation of N-(3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N5O—373.45.
…………………….
http://www.google.com/patents/WO2012063085A3?cl=en
Example 133

N- (3, 3-Dimethy1indolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) }carboxamide Step A – Preparation of l-acetyl-6-amino-3 , 3- dimethylindoline l-Acetyl-3 , 3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL) , the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc :CH2C12 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O-204.27.
Step B – Preparation of N-(l-acetyl- 3 , 3-dimethylindolin-6- yl) (2-[ (4-pyridylmethyl) amino] (3-pyridyl) } carboxamide The titled compound was prepared from l-acetyl-6- amino-3 , 3-dimethylindoline (Step A) by the method described in Example 82.
Step C – Preparation of N- (3 , 3-dimethylindolin-6-yl) {2- [ (4- pyridylmethyl) amino] (3-pyridyl) }carboxamide
The titled compound was prepared from N-(l-acetyl- 3 , 3-dimethylindolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) } carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N50-373.45.
References
- Stafford, edited by Rongshi Li, Jeffrey A. (2009). “Chapter 5. Discovery of Motesanib”. Kinase inhibitor drugs. Hoboken, N.J.: Wiley. pp. 113–130. ISBN 978-0-470-27829-1.
- “Amgen and Takeda’s NSCLC Drug Fails in Phase III Study”. 30 Mar 2011.
- Blumenschein Jr, G. R.; Kabbinavar, F.; Menon, H.; Mok, T. S. K.; Stephenson, J.; Beck, J. T.; Lakshmaiah, K.; Reckamp, K.; Hei, Y.- J.; Kracht, K.; Sun, Y.- N.; Sikorski, R.; Schwartzberg, L. (14 February 2011). “A phase II, multicenter, open-label randomized study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced nonsquamous non-small-cell lung cancer”. Annals of Oncology 22 (9): 2057–2067. doi:10.1093/annonc/mdq731.
- Jump up^ Scagliotti, G. V.; Vynnychenko, I.; Park, K.; Ichinose, Y.; Kubota, K.; Blackhall, F.; Pirker, R.; Galiulin, R.; Ciuleanu, T.-E.; Sydorenko, O.; Dediu, M.; Papai-Szekely, Z.; Banaclocha, N. M.; McCoy, S.; Yao, B.; Hei, Y.-j.; Galimi, F.; Spigel, D. R. (2 July 2012). “International, Randomized, Placebo-Controlled, Double-Blind Phase III Study of Motesanib Plus Carboplatin/Paclitaxel in Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer: MONET1”. Journal of Clinical Oncology 30 (23): 2829–2836. doi:10.1200/JCO.2011.41.4987. PMID 22753922.
- “Takeda Initiates Phase 3 Trial of Motesanib in Japan and Additional Asian Countries”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J. H.; Shinkai, T.; Takeda, K.; Kim, S.- W.; Hsia, T.- C.; Li, R. K.; Tiangco, B. J.; Yau, S.; Lim, W.- T.; Yao, B.; Hei, Y.- J.; Park, K. (13 January 2014). “Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis”.Annals of Oncology 25 (2): 529–536. doi:10.1093/annonc/mdt552.
- Jump up^ “Takeda Announces Phase 3 MONET-A Study Evaluating Motesanib (AMG 706) in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer Does Not Meet Primary Endpoint”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Martin, Miguel; Roche, Henri; Pinter, Tamas; Crown, John; Kennedy, M John; Provencher, Louise; Priou, Frank; Eiermann, Wolfgang; Adrover, Encarna; Lang, Istvan; Ramos, Manuel; Latreille, Jean; Jagiełło-Gruszfeld, Agnieszka; Pienkowski, Tadeusz; Alba, Emilio; Snyder, Raymond; Almel, Sachin; Rolski, Janusz; Munoz, Montserrat; Moroose, Rebecca; Hurvitz, Sara; Baños, Ana; Adewoye, Henry; Hei, Yong-Jiang; Lindsay, Mary-Ann; Rupin, Matthieu; Cabaribere, David; Lemmerick, Yasmin; Mackey, John R (April 2011). “Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study”. The Lancet Oncology 12 (4): 369–376. doi:10.1016/S1470-2045(11)70037-7. PMID 21429799.
- Schilder, R.J.; Sill, M.W.; Lankes, H.A.; Gold, M.A.; Mannel, R.S.; Modesitt, S.C.; Hanjani, P.; Bonebrake, A.J.; Sood, A.K.; Godwin, A.K.; Hu, W.; Alpaugh, R.K. (April 2013). “A phase II evaluation of motesanib (AMG 706) in the treatment of persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas: A Gynecologic Oncology Group study”. Gynecologic Oncology 129 (1): 86–91. doi:10.1016/j.ygyno.2013.01.006. PMID 23321064.
- Motesanib Diphosphate Provides Anticancer Activity Among Patients with Progressive Thyroid Cancer, CancerConnect.com
- Jump up^ Schlumberger, M. J.; Elisei, R.; Bastholt, L.; Wirth, L. J.; Martins, R. G.; Locati, L. D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.-P.; Eschenberg, M. J.; Sun, Y.-N.; Juan, T.; Stepan, D. E.; Sherman, S. I. (29 June 2009). “Phase II Study of Safety and Efficacy of Motesanib in Patients With Progressive or Symptomatic, Advanced or Metastatic Medullary Thyroid Cancer”.Journal of Clinical Oncology 27 (23): 3794–3801. doi:10.1200/JCO.2008.18.7815. PMID 19564535.
- Sherman, Steven I.; Wirth, Lori J.; Droz, Jean-Pierre; Hofmann, Michael; Bastholt, Lars; Martins, Renato G.; Licitra, Lisa; Eschenberg, Michael J.; Sun, Yu-Nien; Juan, Todd; Stepan, Daniel E.; Schlumberger, Martin J. (3 July 2008). “Motesanib Diphosphate in Progressive Differentiated Thyroid Cancer”. New England Journal of Medicine 359 (1): 31–42.doi:10.1056/NEJMoa075853. PMID 18596272.
External links

Motesanib Diphosphate (AMG-706)
857876-30-3 diphosphate
453562-69-1 (free base)
N-(2,3-Dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide diphosphate
3-Pyridinecarboxamide, N-(2,3-dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-, phosphate (1:2)
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide diphosphate
| 569.4 | |
| Formula | C22H23N5O.2H3PO4 |
|---|
 |
|
| Names | |
|---|---|
| IUPAC name
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-[(pyridin-4-ylmethyl)amino]pyridine-3-carboxamide
|
|
| Other names
AMG 706
|
|
| Identifiers | |
| 453562-69-1 |
|
| ChEMBL | ChEMBL572881 |
| ChemSpider | 9842625 |
| Jmol-3D images | Image |
| PubChem | 11667893 |
| Properties | |
| C22H23N5O | |
| Molar mass | 373.45 g·mol−1 |
Stats today
6.8 lakh views on this blog
…………………..
TAKEDA, JAPAN
![]()

TOKYO HO

Takeda Pharmaceutical CEO Yasuchika Hasegawa
 Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.
Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.

Christophe Weber (L), the new president of Takeda Pharmaceutical Co., and CEO Yasuchika Hasegawa pose

 Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
 OSAKA
OSAKA

Dotonbori, Osaka, Japan
 OSAKA
OSAKA

Allisartan isoproxil

Allisartan isoproxil
CAS: 947331-05-7
553.01, C27 H29 Cl N6 O5
An angiotensin II receptor antagonist used to treat mild to moderate essential hypertension.
Approved china, cfda July 1 2012
![]()
Shanghai Allist Pharmaceutical, Inc.
Allist Shanghai Pharmaceutical Co., Ltd.

2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carboxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester,
2-Butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)biphenyl-4-ylmethyl]-1H-imidazole-5-carboxylic acid isopropoxycarbonyloxymethyl ester
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester
Allisartan is an orally-available angiotensin AT1 antagonist in phase II clinical trials at Shanghai Allist Pharmaceutical for the treatment of mild to moderate essential hypertension.
Shanghai Allist Pharmaceutical PHASE 2 for Hypertension

The prior art discloses Arleigh medoxomil illiquid, low bulk density, electrostatic phenomena evident. Chinese patent discloses a CN200710094131.0 Alicante medoxomil polymorph and method of preparation. Allie medoxomil based crystal prepared by the method has high stability characteristics, but relatively small bulk density of the crystal clear after the electrostatic phenomenon and poor liquidity dried, crushed and used for easy dispensing generate dust, operating the site clean and labor protection inconvenience, on the other hand also for accurate weighing and packaging products inconvenience.
CN200710094021.4 and CN201110289695.6 disclose the preparation of Alicante medoxomil, the inventor repeated, the proceeds of crystal and Chinese patent CN200710094131.0 consistent disclosed.

Allisartan isoproxil
Angiotensin II AT-1 receptor antagonist
Essential hypertension
Amorphous form of allisartan isoproxil is claimed in WO 2015062498. Useful for treating hypertension. Shenzhen Salubris Pharmaceuticals, in collaboration with Allist, has developed and launched allisartan isoproxil. In October 2012, Shenzhen Salubris signed a strategic cooperation framework agreement with Allist Pharmaceutical for the production and marketing of allisartan isoproxil. Family members of the product case of allisartanWO2007095789, expire in the EU and in the US in 2026. For a prior filing see WO2009049495 (assigned to Allist Pharmaceuticals), claiming the crystalline form of allisartan and its method of preparation.
The compound of formula (I) is an Ang II receptor antagonist. Its chemical name is 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carb-oxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester. Chinese Patent CN101024643A describes the structure, and its use as antihypertensive drugs.

As regards to the solid physical properties of the compound of formula (I), the patent document of CN101024643A discloses that it is a white solid, and its melting point is 134.5-136° C. However, CN101024643A dose not disclose the crystalline structure of the compound of formula (I).

 CHINA
CHINA
NEW PATENT
WO-2015062498
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015062498
2-butyl-4-chloro -1- [2 ‘- (1H- tetrazol-5-yl) -1,1’-biphenyl- – methyl] – imidazole-5-carboxylic acid, 1 – [(isopropoxy) – oxy] -, methyl ester, is a novel angiotensin Ⅱ receptor antagonist. China Patent CN200680000397.8 disclosed structural formula Alicante medoxomil compound. Allie medoxomil toxicity, blood pressure better than the same type of products (such as losartan), which by generating active metabolite (EXP3174) in vivo metabolism, and thus play its antihypertensive effect.
The prior art discloses Arleigh medoxomil illiquid, low bulk density, electrostatic phenomena evident. Chinese patent discloses a CN200710094131.0 Alicante medoxomil polymorph and method of preparation. Allie medoxomil based crystal prepared by the method has high stability characteristics, but relatively small bulk density of the crystal clear after the electrostatic phenomenon and poor liquidity dried, crushed and used for easy dispensing generate dust, operating the site clean and labor protection inconvenience, on the other hand also for accurate weighing and packaging products inconvenience.
CN200710094021.4 and CN201110289695.6 disclose the preparation of Alicante medoxomil, the inventor repeated, the proceeds of crystal and Chinese patent CN200710094131.0 consistent disclosed.
……………………..
PATENT
http://www.google.com/patents/CN103965171A?cl=en
Hypertension is a major disease threat to human health, looking for efficiency, low toxicity anti-hypertensive drugs can help relieve social pressures and family responsibilities, with good social and economic benefits.
Angiotensin II (Ang II) is the renin – angiotensin – aldosterone system (RAAS) main vasoconstrictor hormone, which plays an important role in the pathobiology of many chronic diseases, particularly its the role of blood pressure regulation is particularly prominent, and therefore Ang II receptor is believed to be a good target for the development of anti-hypertensive drugs.
EP0253310 discloses a series of imidazole derivatives, DuPont declared and obtained by the study of losartan potassium-listed in 1994, was the first non-peptide Ang II receptor antagonist anti-hypertensive drugs. Thereafter, he listed a series of losartan antihypertensive drugs: candesartan cilexetil, valsartan, irbesartan, telmisartan and olmesartan medoxomil, etc. (EP0253310, W02005049587, GB2419592, EP1719766, US5196444) .
The losartan potassium in the body, the active metabolite EXP3174 has a stronger antihypertensive effect than losartan potassium, but EXP3174 polar molecular structure, is difficult to form passive absorption by diffusion through the cell membrane. US5298915 discloses five carboxyl ester group transformation EXP3174 is a series of derivatives, focusing on the compound HN-65021, and discloses hypotensive test results HN-65021 administered by the oral route, its hypotensive activity with chlorine Similar losartan potassium (BritishJouurnal ofClinical Pharmacology, 40,1995,591).
CN200680000397.8 _5_ discloses a class of imidazole carboxylic acid derivatives, namely Alicante medoxomil compound 8 has a good blood pressure lowering effect, the structure of formula I, the preparation method disclosed in this patent document follows the route A, losartan potassium by oxidation, the protecting group into an ester, deprotected to give a compound of formula I, the route step oxidation process of hydroxyl to carboxyl groups, will be reduced to very fine granular potassium permanganate, manganese dioxide, filtration This manganese mud time-consuming, inefficient, polluting; the second step conversion was about 70%, and post-processing cumbersome; byproducts and produced the first two steps more. This makes the high cost of the entire route, not suitable for the production of amplification.

CN200710094021.4 discloses another method for preparing the compounds of formula I, the following route B, the starting material by nucleophilic substitution, oxidation, an ester, a tetrazole ring to obtain a compound of formula I, the first step of the method nucleophilic substitution easy to generate an imidazole ring -3 para isomer impurities difficult to remove; the last step into the ring to use sodium azide, operating dangerous.

CN201210020174.5 disclosed a series of anti-hypertensive compound and preparation method, the following line C, the temperature control in the first step of its preparation O ~ 5 ° C, a mixed solution of acetone and water, with a 5% aqueous solution of sodium hypochlorite oxidation, yield 70%, the second step use of potassium permanganate, manganese dioxide will produce the same, and a yield of only 40%, the first two steps total yield of 28%, is very low, and the post-treatment methods are by column separation, the first two steps are used are organic and inorganic mixed solvent is not conducive to recovery, not suitable for scale-up.






Example 8 2-Butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil crude)

To a 20L reactor 9800ml of methanol, stirring was started, the rotational speed is added at 200r / min 1225.3g solid compound of formula II, and heated to reflux. The reaction 8-10h evacuation HPLC detection, the formula II compound residue <1.0% seen as a response endpoint. After reaching the end of the reaction the heating was stopped, continued stirring speed of 180r / min. About 3_4h fell 20_25 ° C, colorless transparent crystalline solid precipitated. The reaction mixture was cooled to continue to 15-20 ° C, to maintain 15-20 ° C with stirring 3h, the reaction mixture was filtered to give a pale yellow clear filtrate. The filtrate was concentrated under reduced pressure to move 20L flask, vacuum degree of 0.075MPa, 40_45 ° C methanol distilled off under until no distillate. 800ml of absolute ethanol was added, a vacuum degree of 0.075MPa, 40-45 ° C under distillation until no distillate.
900ml of absolute ethanol was added, heated to reflux. N-heptane was added slowly 1100ml, reflux 15min, to -10 ° c / h speed cooled to 15 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration lh, was Allie medoxomil crude (800.lg, yield 93.8%).Purification was used directly in the next step without drying.
Example 9 2-butyl-4-chloro-_1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole-5-carboxylic acid, 1 – [(isopropylamino oxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

850ml of absolute ethanol was added to the 3L reaction vessel was charged with crude Alicante medoxomil (800.lg, 1.45mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1300ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration, the purified Alicante medoxomil (780.9g, 97.6% yield).
Example 10 2-butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

950ml of absolute ethanol was added to the 5L reaction vessel was charged with crude Alicante medoxomil (549.9g, 1.72mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1200ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = cake was washed with a mixture of 1/3, and dried under reduced pressure after filtration to obtain a purified Alicante medoxomil (540.0g, 98.2% yield).
……………….
PATENT
Example 122-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester (compound 8)

To a 100 ml of one-necked flask, 0.523 g of material, 0.124 g of potassium carbonate, 5 ml of N,N-dimethylacetamide were added in turn. The solution was stirred at room temperature for 20 minutes. Then 0.562 g of 1-chloromethyl isopropyl carbonate was added and the mixture was reacted at 45-50° C. for 16 hours. After the reaction was completed, the mixture solution was filtered, and 30 ml of water was added into the filtrate. The resulting mixture was extracted with 30 ml of ethyl acetate twice. The organic phase was dried and concentrated to give 1.724 g of oil, which was directly used in the next reaction without purification.
10 ml of dioxane and 5 ml of 4 mol/L HCl were added, and the resulting mixture was reacted at room temperature for 16 hours. The reaction was stopped and the solution was adjusted to pH 6-7 using aqueous sodium bicarbonate solution. The solution went turbid, and was extracted with ethyl acetate. The organic phase was washed with saturated brine, dried, concentrated to give 0.436 g of 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester.
In addition, the following reaction condition can be used to deprotect the protecting group. To 1.7 g of oily product, 5 ml absolute methanol was added and the mixture was heated slowly to reflux and stirred for 8 hours. When the insoluble solid disappeared totally, the mixture was discontinued to heating and cooled to 5° C. The white solid precipitated, and was separated by filtration, and the filter cake was washed with a small quantity of methanol. The combined filtrate was concentrated to dryness to give 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester with the yield of 70%.
1H-NMR (CDCl3) δ H (ppm): 0.89 (t, 3H, J=14.6), 1.24 (d, 6H, J=6.3), 0.37 (m, 2H, J=22.1), 1.69 (m, 2H, J=30.5), 2.64 (t, 2H, J=15.5), 4.81 (m, 1H, J=12.4), 5.54 (s, 2H), 5.86 (s, 2H), 6.95-7.64 (8H), 8.08 (d, 1H, J=7.42)
ESI(+) m/z: 552.7
Mp: 134.5-136° C.

USE CTRLand + Key, or click on picture
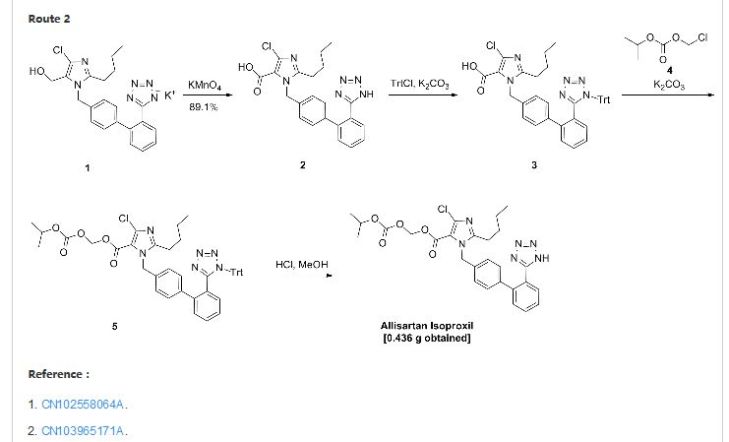



| WO2005011646A2 * | 20 Jul 2004 | 10 Feb 2005 | Nicoletta Almirante | Nitrooxy derivatives of losartan, valsatan, candesartan, telmisartan, eprosartan and olmesartan as angiotensin-ii receptor blockers for the treatment of cardiovascular diseases |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8455526 * | 6 Jun 2008 | 4 Jun 2013 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| US20100168193 * | 6 Jun 2008 | 1 Jul 2010 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| USRE44873 | 31 Jul 2006 | 29 Apr 2014 | Salubris Asset Management Co., Ltd. | Imidazole-5-carboxylic acid derivatives, the preparation method therefor and the uses thereof |
| CN101024643A | 20 Feb 2006 | 29 Aug 2007 | 上海艾力斯医药科技有限公司 | Imidazo-5-carboxylic-acid derivatives, its preparing method and use |
| US5298519 * | 24 Sep 1992 | 29 Mar 1994 | Chemish Pharmazeutische Forschungsgesellschaft M.B.H. | Acylals of imidazole-5-carboxylic acid derivatives, and their use as angiotensin (II) inhibitors |
……………….
update……………..
Example 1
Weigh 25g 2- butyl-4-chloro-1- [2 ‘- (1-trityl–1H- tetrazol-5-yl) -1,1’-biphenyl – methyl] – imidazole 5-carboxylic acid, 1 – [(isopropoxy) – carbonyloxy] -, methyl ester, was added to a 500ml three-necked flask, methanol was added 200ml, refluxed for 9h, methanol was distilled off under reduced pressure to give crude Alicante medoxomil .
To the residue (i.e., medoxomil crude Alicante) were added 33ml of isopropanol and 66ml of n-heptane, heated to 76 ℃ stirred for 2h. After cooling to 60 ℃ stirring for 1h, and then the system was slowly cooled to 0 ℃, stirring was continued for 3h. Filtered, the filter cake was washed with n-heptane. At 40 ℃ 8 hours and dried in vacuo to give 15.3g Alicante medoxomil (purity 99.3%) as a XRD spectrum as shown in Figure, the main peak of the diffraction peaks as shown in the following table, the DSC spectrum shown in figure II . Compared with the published crystal, the crystal obtained by the absence of significant electrostatic phenomena.
Shanghai , CHINA







RG-1577, EVT 302, Sembragiline, RO-4602522
RG-1577, EVT 302, Sembragiline, RO-4602522
CAS 676479-06-4, MW 342.36
- C19 H19 F N2 O3
- Acetamide, N-[(3S)-1-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-3-pyrrolidinyl]-
UNII-K3W9672PNJ
RG-1577, a selective and reversible monoamine oxidase B inhibitor, for treating AD (phase 2 clinical, as of May 2015).
Family members of the product case for RG-1577 (WO2004026825) hold protection in EU until 2023 and expire in US in 2024 with US154 extension. Follows on from WO2006097197, claiming a process for preparing RG-1577.
Alzheimer‘s Disease is a brain disease that slowly destroys memory and thinking skills, up to loss of the ability to carry out the simplest tasks. It is the most common cause of dementia among older people. Mild Alzheimer‘s Disease manifests itself in memory loss and small changes in other cognitive abilities, e.g getting lost, trouble handling money and managing daily tasks, having some mood and personality changes, etc.
In the stage of Moderate Alzheimer‘s Disease, the control of language, reasoning, sensory processing, and conscious thought are impacted. Memory loss and con usion grow worse, e.g patients have problems recognizing family and friends and become unable to learn new things, etc. hallucinations, delusions, and paranoia may occur. .Severe Alzheimer‘s Disease is the final stage. Patients cannot communicate anymore and are completely dependent.
N-[(3S)-l-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide has previously been described in the art. 1 WO 2006/097197 2 and WO 2006/0972703 relate to methods for preparing enantiomerically pure 4-pyrrolidinophenylbenzyl ether derivatives.
![]()
The processes of the prior art hamper from several drawbacks (e.g. long reaction sequence, low overall yield also due to loss of half of the product in the classical resolution step, the need for a chromatographic purification to remove by-products formed in the Mitsunobu reaction) and are therefore less suitable for the preparation of N-[(3S)-l-[4-[(3-fluorophenyl) methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide on large scale.
Most Recent Events
- 01 Aug 2014Roche completes a phase I trial in volunteers in USA (NCT02104648)
- 14 May 2014Roche completes enrolment in the MAyflOwer RoAD trial for Alzheimer’s disease (combination therapy, adjunctive treatment) in Australia, Canada, Czech Republic, France, Germany, Italy, Poland, South Korea, Spain, Sweden the United Kingdom and the USA (NCT01677754)
- 01 Apr 2014Roche initiates enrolment in a phase I trial in healthy volunteers in USA (NCT02104648)
http://www.evotec.com/uploads/media_library/10/2012-09_Evotec_Company_presentation_September_e.pdf

……………………..
WO2004026825
http://www.google.com/patents/WO2004026825A1?cl=en
………………….
WO2006097197
http://www.google.com/patents/WO2006097197A1?cl=en
……………………………………………..
PATENT
WO 2015063001
Novel, crystalline polymorphic forms A and B of a pyrrolidone derivative ie RG-1577, useful for treating Alzheimer’s disease (AD). Roche and its Japanese subsidiary Chugai, under license from Evotec, which previously licensed the drug from Roche, are developing RG 1577
formula 1 via the following routes

In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route A

1
In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route B

In a certain embodiment, present invention relates to a crystalline polymorph of a compound of formula 1.

synthesize a compound of formula 1 from a compound of formula 7

compound of formula 6 to a compound of formula 7

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 6 via the intermediate 6a to a compound of formula 7

further comprising reacting a compound of formula 3 with a compound of formula 5 to a compound of formula 6

comprising reacting a compound of formula 2 to a compound of formula 3

2 3
In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 10 to a compound of formula 6

eacting a compound of formula 9 with a compound of formula 5 to a compound of formula 10

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 8 to a compound of formula 9

(lS’)-N-[l-[4-(3-fluoro-benzyloxy)-phenyl]-5-oxo-pyrrolidin-3-yl-]acetamide (1)
To a suspension of chloride (7) (37.9 g, 100 mmol) in 2-methyltetrahydrofurane (600 ml) was added under vigorous stirring at 0°C 1.65 M potassium ie/t-butoxide in THF (75.5 ml, 125 mmol, ACROS) over 2.5 h. After additional stirring at 0°C for 1 h, the cold suspension was hydrolyzed with 0.1 M HCl (600 ml) and the reaction mixture was stirred at 30°C for 0.5 h. The organic layer was washed with water (300 ml), dried (Na2S04) and filtered. Removal of the solvent by rotary evaporation (50°C/>10 mbar) afforded 32.1 g crystalline residue, which was dissolved in 2-butanone (400 ml) at ca. 95°C and hot filtered. Crystallization, which was induced by seeding and cooling to room temperature and 0°C (4 h) afforded 25.4 g (74.2%) of the titled compound (1) as an off-white, crystalline powder,
Mp. 162-164°C (polymorph B).
Ee >99.8%, [cc]D20 = – 17.8 (DMF; c = 1).
1H NMR (400 MHz, DMSO- 6) δ ppm 1.82 (s, 3H), 2.34 (dd, J1=n. l, J2=3.9, 1H), 2.84 (dd, J/=17.1, J2=8.2, 1H), 3.55 (dd, J/=10.2, J2=3.2, 1H), 4.07 (dd, J/=10.2, J2=6.7, 1H), 4.32-4.41 (m, 1H), 5.13 (s, 2H), 7.02 & 7.55 (d, J=9.1, each 1H), 7.11-7.19 (m, 1H), 7.24-7.31 (m, 1H), 7.40-7.47 (m, 1H), 8.40 (d, J=6.4, 1H).
ESI-MS (m/z) 343 [M+H]+, 365 [M+Na]\. Anal.Calcd for Ci9H19FN203 (342.37): Calcd. C, 66.66; H, 5.59; N, 8.18; F, 5.02; O, 14.02. Found C, 66.76; H, 5.48; N, 8.13; F, 5.03; O, 13.99.
Crystallized (1) form previous step (9.5 g, 0.028 mol) was dissolved in 2-butanone (290 mL) upon heating. The hot solution was filtered over charcoal. The solution was concentrated by removal of 2-butanone (200 mL) by distillation prior to seeded cooling crystallization. Filtration, washing with chilled 2-butanone and drying at 50°C/25 mbar/16h afforded 9.18 g (93.9% corrected yield) of the title compound (1) as a crystalline powder of polymorphic form B with an assay of 100.4 %(w/w) and a purity of 99.97 %(area) (by HPLC).
Alternatively, to a stirred suspension of hydroxyamide (6) (30.0 g, 0.083 mol) in toluene (500 ml) was added at 50°C within 45 minutes thionyl chloride (10.40 g, 0.087 mol) and the resulting mixture was stirred for 3h at 50°C. The mixture was then heated up to 92°C and subsequently stirred at this temperature for 15 h. The Suspension was then cooled to 50°C and toluene was removed by distillation under reduced pressure. The distillation residue was cooled to ambient temperature and treated with N-methylpyrrolidone (210 ml) to obtain an almost clear solution. This solution was then cooled to -10°C and subsequently treated at this temperature within 2h with a solution of potassium iert-butoxide (12.40 g, 0.111 mol) in THF (60 g). The resulting mixture was stirred for another 60 minutes at -10°C, then warmed up to room temperature within 60 minutes and subsequently stirred at room temperature for 6 h. The reaction mixture was quenched with water (150 g) and the pH was adjusted with acetic acid (approx. 1.8 g) to pH 7-8. The mixture was then heated to 30-45°C and THF and toluene were distilled off under reduced pressure (<200 mbar) to obtain a clear NMP/water mixture (400 ml). This mixture was heated to 45°C and 260 mg of seed crystals were added. Water (320 ml) was then added within 3 h whereby the product crystallized. The resulting suspension was cooled to room temperature within 3 h and subsequently stirred at this temperature for 2 h. Filtration and washing of the filter cake with a mixture of water (100 ml) and N-methylpyrrolidone (20 ml) and subsequently only with water (150 ml) afforded after drying (70°C/10 mbar/20 h) 26.2 g (92%) of the title compound (1) as a crystalline powder with an assay of 99.6 %(w/w) and a purity of 99.7 %(area) (by HPLC).

HPLC
Purity (HPLC): Column: XSelect Phenyl Hexyl x2, 150 x 4.6mm, 3.5um. Starting
Pressure: 226 bar; temp.: 50°C. Inj. vol.: 2.0 μΐ^ + wash. Flow: 1.0 ml/min. Det: 204 nm. A: Water + 5% ACN, 77-2% in 7 min., hold for 1 min.; B: 0.1% HCOOH, 18% isocratic; C: MeOH, 5-80% in 7 min., hold for 1 min. Sample prep.: 2 mg/ml ACN. Retention times: β-acid 5.93 min., diacid 6.18 min., cc-acid 6.89 min., diester 6.96 min.
ee determination(HPLC): Column: Chiralpak IA-3 100 x 4.6mm, 3um; 91 bar, 2ml/min; temp.: 30°C. Inj. vol.: 10.0 μL· Det.: 206 nm. A: n-heptane, 80%; B: EtOH, 20%. Sample prep.: 4 mg/ml EtOH. Retention times: D-enantiomer 2.21 min., L-enantiomer 2.71 min
………………….
US 20050065204
EXAMPLE 11
Preparation of (S)-1-(4-Hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic Acid
8.00 g Polyethyleneglycol 6000 was dissolved in 150 mL (100 mM) magnesium acetate buffer pH 6.0 under stirring, and the solution added to a stirred suspension of 10.00 g (42.51 mmol) (RS)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid methyl ester (99.7%) in 40 mL methylcyclohexane. The mixture was heated to 28° C. and the pH readjusted to 6.0 with 2 M NaOH. The reaction was started by adding 33.2 mg Candida cylindraceae cholesterase (16.88 kU/g), and the pH was maintained at 6.0 by the controlled addition of 1.0 M NaOH solution under stirring. After a total consumption of 20.35 mL (20.35 mmol) 1.0 M sodium hydroxide solution (after 17.1 h; 47.9% conversion) the reaction mixture was passed through a sintered glass filter. The filtrate spontaneously separated into an aqueous and an organic phase.The aqueous phase was washed with 2×200 mL ethyl acetate to remove uncleaved ester. The aqueous phase was set to pH 4.0 with 25% sulfuric acid and concentrated in vacuo to a volume of ca. 80 mL (bath 60° C.). The solution was cooled to 1° C. (formation of white precipitate/crystals) and the pH set to 1.5 with 25% sulfuric acid. The precipitate/crystals were stirred overnight at 1° C., filtered off on a sintered glass filter (washed with a minimum amount of water) and dried overnight on high vacuum (RT, 6×10−2 mbar) to give 4.32 g (19.53 mmol; 45.9%) (S)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid. Analysis: HPLC (area A226nm): 99.3%, 0.7% ester. 98.9%ee. The product contains 5.3% water (according to Karl Fischer determination) and 2.1% (w/w) PEG (according to NMR).
| Company | Evotec AG |
| Description | Small molecule monoamine oxidase B (MAO-B) inhibitor |
| Molecular Target | Monoamine oxidase B (MAO-B) |
| Mechanism of Action | Monoamine oxidase B (MAO-B) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Alzheimer’s disease (AD) |
| Indication Details | Treat Alzheimer’s disease (AD) |
| Regulatory Designation | |
| Partner |
//////////
Chūō, japan




A Chūō Line (Rapid) E233 series (right) and A Chūō-Sōbu Line E231 series (June 2007)
 Chuo Dori street on a weekend afternoon
Chuo Dori street on a weekend afternoon

Zydus-Cadila is developing ZYH-7, a PPAR alpha modulator for the potential treatment of dyslipidemia

ZYH-7
Prediction of ZYH 7 below……..If it does not match then ZYH 7 will be a very close structure

1014989-63-9
Acetic acid, 2-[2-methyl-4-[1-[[[4-methyl-2-[4-(trifluoromethyl)phenyl]- 5-thiazolyl]methoxy]imino]ethyl]phenoxy]-
Zydus-Cadila is developing ZYH-7, a PPAR alpha modulator for the potential treatment of dyslipidemia .
By January 2012, phase II trials had begun ; in January 2014, the drug was still listed as being in phase II development
By January 2012 phase II trials had begun for Diabetes type 2 Lipoprotein disorders
Obesity
In August 2007, an IND was filed , and by March 2008, a phase I trial was underway ; by April 2011, the trial had been completed
| Zydus Cadila has filed an Investigational New Drug (NID) application for seeking DCGI’s permission for conducting clinical trials for its New Molecular Entity (NME) ZYH7. |
 |
| According to a company release, it claims that ZYH7 is a novel drug candidate for treating dyslipidemia and metabolic disorders. The company inform that ZYH7 had been conceptualised and developed by its scientists from Zydus Research Centre. |
| The company has its in-house research centre and it had recently concluded pre-clinical studies on ZYH7, which have reported interesting and encouraging finding which indicate a novel molecule to treat dyslipidemia and associated metabolic disorders. |
| Commenting on the new development, Pankaj Patel, chairman and managing director, Zydus Cadila said, “We have been building a promising pipeline of new molecular entities at the Zydus Research Centre and ZYH7 is an important step in this direction”. |
| Starting with its first IND filing in 2005, Zydus today has four INDs in various stages of clinical trials. NME – ZYH1 for treating dyslipidemia and ZYI1 for treating pain and inflammation are undergoing Phase II trials. ZYH2 for treating diabetes and the novel CB-1 antagonist, ZYO1 for treating obesity, are undergoing Phase I trials. |
| Diabetes, a worldwide health problem, affects more than 150 million people, a number expected to double to 300 million by 2025. People with diabetes are at especially high risk for dyslipidemia, particularly high triglyceride levels and low HDL levels. |
| Dyslipidemia is also a key independent risk factor for cardiovascular disease (CVD), which is the largest therapeutic segment in the world pharmaceutical market. |
| With an increasing correlation between several endocrine and metabolic disorders, there has been considerable emphasis in recent times on metabolic syndrome. The metabolic components of cardiovascular disease, diabetes and obesity, are linked in numerous ways with each having an impact on the other. |
| For instance, it is also well known that patients with Type 2 diabetes have a two to four-fold excess risk of coronary heart disease and that these patients very often have increased cardiovascular risk factors even before the onset of their diabetes. |
Dyslipidemia is an abnormal amount of lipids (e.g. cholesterol and/or fat) in the blood. In developed countries, most dyslipidemias are hyperlipidemias; that is, an elevation of lipids in the blood. This is often due to diet and lifestyle. Prolonged elevation of insulin levels can also lead to dyslipidemia. Likewise, increased levels of O-GlcNAc transferase (OGT) may cause dyslipidemia.
| Dyslipidemia | |
|---|---|
| Classification and external resources | |
| ICD–10 | E78 |
| ICD–9 | 272 |
| DiseasesDB | 33452 |
| MeSH | D050171 |
Classification
Physicians and basic researchers classify dyslipidemias in two distinct ways:
- Phenotype, or the presentation in the body (including the specific type of lipid that is increased)
- Etiology, or the reason for the condition (genetic, or secondary to another condition). This classification can be problematic, because most conditions involve the intersection of genetics and lifestyle issues. However, there are a few well-defined genetic conditions that are usually easy to identify.
Fredrickson Classification:[1]
For more a detailed version, see Hyperlipidemia#Classification.
| Phenotype | I | IIa | IIb | III | IV | V |
|---|---|---|---|---|---|---|
| Elevated Lipoprotein | Chylomicron | LDL | LDL and VLDL | IDL | VLDL | VLDL and chylomicrons |
WO 2008035359
https://www.google.com/patents/WO2008035359A2?cl=en
Scheme 1 below which comprises:

Scheme 2 below which comprises

| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009021740A2 | Aug 14, 2008 | Feb 19, 2009 | Sanofis Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010049946A2 * | Oct 22, 2009 | May 6, 2010 | Cadila Healthcare Limited | Thyroid receptor ligands |
| WO2010084512A1 * | Dec 22, 2009 | Jul 29, 2010 | Cadila Healthcare Limited | Novel oxime derivatives |
| WO2010110479A1 * | Mar 24, 2010 | Sep 30, 2010 | Nippon Chemiphar Co., Ltd. | Activator for peroxisome proliferator-activated receptor |
| WO2011157827A1 | Jun 17, 2011 | Dec 22, 2011 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2013037390A1 | Sep 12, 2011 | Mar 21, 2013 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014192023A1 * | May 20, 2014 | Dec 4, 2014 | Cadila Healthcare Limited | Novel compounds suitable for the treatment of dyslipidemia |
| EP2567959A1 | Sep 12, 2011 | Mar 13, 2013 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8742117 | Dec 22, 2009 | Jun 3, 2014 | Cadila Healthcare Limited | Oxime derivatives |
| US8822414 * | Dec 26, 2011 | Sep 2, 2014 | Cadila Healthcare Limited | Heterocyclic compounds suitable for the treatment of dyslipidemia |
………….
PARIS

Firategrast, T-0047

 Japan
Japan
Firategrast, 402567-16-2;
Firategrast, MS, Alpha4beta1 integrin
PHASE 2 GSK
Mitsubishi Tanabe Pharma INNOVATOR
Glaxo Group Limited, Mitsubishi Tanabe Pharma Corporation
SB 683699, SB-683699, UNII-OJY3SK9H5F
Firategrast; UNII-OJY3SK9H5F; SB-683699; Firategrast (USAN); 402567-16-2; SB683699; T-0047
Molecular Formula: C27H27F2NO6
Molecular Weight: 499.503186 g/mol
SYSTEMATIC NAME:
1,1′-Biphenyl)-4-propanoic acid, alpha-((2,6-difluorobenzoyl)amino)-4′-(ethoxymethyl)-2′,6′-dimethoxy-, (alphaS)-
N-(2,6-Difluorobenzoyl)-4-[4-(ethoxymethyl)-2,6-dimethoxyphenyl]-L-phenylalanine
N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
2S)-2-((2,6-Difluorobenzoyl)amino)-3-(4′-(ethoxymethyl)-2′,6′-dimethoxybiphenyl-4- yl)propanoic acid
(2S)-2-{[(2,6- difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4- biphenylyl]propanoic acid
(2S)-2-[[2,6-bis(fluoranyl)phenyl]carbonylamino]-3-[4-[4-(ethoxymethyl)-2,6-dimethoxy-phenyl]phenyl]propanoic acid
Pharmacological half-life is 2.5 – 4.5 hours, compared to 11 days for natalizumab, a drug in the same class
Orally bioavailable small molecule α4-integrin antagonist
see
http://www.msdiscovery.org/node/1377#node-biblio-1338
http://multiple-sclerosis-research.blogspot.com/2012/01/research-oral-tysabri-analogue.html
SB683699 is an alpha4 integrin antagonist that had been studied in phase II trials at GlaxoSmithKline under a license from Mitsubishi Tanabe Pharma for the oral treatment of multiple sclerosis (MS) in Europe. GlaxoSmithKline and Tanabe Seiyaku (now Mitsubishi Tanabe Pharma) had been studying the drug candidate for the treatment of asthma, rheumatoid arthritis (RA) and Crohn’s disease
MECHANISMS/EFFECTS
HUMAN:
Similar mechanism of action to natalizumab (α4-integrin blocker), but its faster elimination could improve safety profile

Firategrast
SYNTHESIS
………………….
PATENT
Scheme 1

Scheme 2

In a further aspect the present invention provides for a process for the preparation of compound of formula (II) which comprises coupling the compound of formula (V)

Suitable coupling conditions for the compound of formula (V) and the compound of formula (VI) include those shown in Scheme 2. In a further aspect of the invention there is provided the compound of formula (V):

1H NMR characterisation data for the compound of formula (V) were generated on an isolated and purified batch. 1H-NMR spectra were recorded on a Bruker Avance 400 at 400MHz, using TMS as an internal reference.1H NMR (400 MHz, DMSO-D6) δ ppm 1.17 (t, J=7.09 Hz, 3 H) 2.96 (dd, J=13.82, 9.90 Hz, 1 H) 3.1 1 (dd, J=13.82, 5.26 Hz, 1 H) 4.12 (q, J=7.09 Hz, 2 H) 4.63 (ddd, J=9.78, 7.82, 5.38 Hz, 1 H) 7.15 (t, J=7.95 Hz, 2 H) 7.25 (d, J=8.31 Hz, 2 H) 7.47 – 7.55 (m, 3 H) 9.23 (d, J=7.83 Hz, 1 H).
The present invention provides a process for the preparation of the compound of formula

which process comprises the steps: a) hydrolysis of an ester of formula (I la):

Recrvstallisation of (2S)-2-{r(2,6-difluorophenyl)carbonyllamino)-3-r4′-r(ethyloxy)methyll- 2′,6′-bis(methyloxy)-4-biphenylyllpropanoic acid
(2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4′-[(ethyloxy)methyl]-2′,6′-bis(methyloxy)- 4-biphenylyl]propanoic acid (9.38Kg) was charged into a clean reactor, followed by ethyl acetate (46.9L). The solution was heated to 50°C and filtered into the pre-warmed (35°C) crystallizing vessel. A line-wash with ethyl acetate (9.4L) was carried out. The combined ethyl acetate solutions were heated to 50°C, stirred to ensure complete dissolution. Filtered heptane (9.4L) was added maintaining the temperature at 50°C then the solution cooled to 30°C and seeded with (2S)-2-{[(2,6-difluorophenyl)carbonyl]amino}-3-[4 – [(ethyloxy)methyl]-2′,6′-bis(methyloxy)-4-biphenylyl]propanoic acid (47g) slurried in 1 :9 ethyl acetate:heptane (0.47L). The slurry was aged for 2 hours at 30°C. Filtered heptane (75L) was added over 3 hours. The slurry was then cooled to 0°C over 1 hour. The mixture was aged at 0°C for 1 hour then the solid was filtered off, washed with isopropyl ether (29.6L and dried under vacuum at 50±3°C to give the product (8.55Kg, 91 %). Characterised by having an infrared absorption spectrum with significant absorption bands at about 754, 768, 800, 820, 849, 866, 1006, 1 100, 1 122, 1 157, 1 188, 1225, 1242, 1268, 1292, 1317, 1352, 1417, 1466, 1530, 1580, 1624, 1650, 1662, 171 1 , 1728, 2938, 3302cm“
…………………………………..
PATENT
Example 10: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine ethyl ester.
(1) The product obtained in Example l-(4) (2.1 g) was acylated with 2 , 6-difluorobenzoyl chloride in a similar manner as described in Example 1 -(5) to give N- (2, 6-difluorobenzoyl) – 4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L-phenylalanine ethyl ester (2.75 g) . mp . 70-72 °C; IR (Nujol) 3400, 3263, 1735, 1654, 1624 cm“1; MS (APCI) m/z 500 (M+H) . (2) To a solution of the product obtained above (1.72 g) in DMSO (20 ml) were added Et3N (4.8 ml) and S03«pyridine (5.6 g) successively at room temperature. The whole mixture was stirred at room temperature for 25 minutes. The reaction mixture was poured into ice-water, and then the mixture was extracted with EtOAc. The organic layer was sequentially washed with 5% aqueous HCl, H20 and brine, dried (Na2S04) and then evaporated. The residue was purified by column chromatography (silica gel; eluent: n-hexane/EtOAc 5:1 to 1:1) to yield N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-formylphenyl) -L- phenylalanine ethyl ester (1.54 g) . mp. 114-116°C; IR (Nujol)
3332, 1735, 1695, 1657, 1644, 1623 cm“1; MS (APCI) m/z 498 (M+H) .
(3) The product obtained above (716 mg) was converted into the title compound (428 mg) in a similar manner as described in Example 1- (7) . mp . 87-89°C; IR (Neat+CHC13) 3300, 1739, 1668 cm“ 1; MS (APCI) m/z 528 (M+H) .
Example 11: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl ) -L-phenylalanine methyl ester.
(1) The product obtained in Example 2- (4) (1.00 g) was acylated with 2 , 6-difluorobenzoyl chloride to give N-(2,6- difluorobenzoyl) -4- (2 , 6-dimethoxy-4-hydroxymethylphenyl) -L- phenylalanine methyl ester (873 mg) in a similar manner as described in Example l-(5). IR (Nujol) 3257, 1743, 1655, 1624 cm“ 1; MS (APCI +Q1MS) m/z 503 (M+NH4) , 486 (M+H) . (2) The product obtained above (860 mg) was converted into the title compound (220 mg) in a similar manner as described in Example 2- (6) and (7).
Example 12: N- (2 , 6-Difluorobenzoyl) -4- (2 , 6-dimethoxy-4- ethoxymethylphenyl) -L-phenylalanine .
The product obtained in Example 10 (200 mg) was hydrolyzed in a similar manner as described in Example 3 to give the title compound (160 mg) . The product obtained in Example 11 (220 mg) was also hydrolyzed in a similar manner as described in Example 3 to give the title compound (167 mg) . mp. 156-158°C; IR (Nujol) 1735, 1655 cm“1; MS (ESI) m/z 498 (M-H) .
…………………….
PATENT
https://www.google.com/patents/WO2003072536A1?cl=en
OUT LINE
phenylalanine derivative of the formula (I) :

wherein X1 is a halogen atom, X2 is a halogen atom, Q is a group of the formula -CH2– or -(CH2)2– and Y is a lower alkyl group, or a pharmaceutically acceptable salt thereof, which has excellent inhibitory activity against α4 integrin-mediated cell adhesion.
Thus, the present invention relates to a process for preparing a compound of the formula (I) :

wherein the symbols are the same as defined above, or a pharmaceutically acceptable salt thereof, comprising : (1) coupling a compound of the formula (VI) :

wherein Z is a leaving group, R1NH is a protected amino group and C02R is a protected carboxyl group with a compound of the formula (V) :

wherein the symbols are the same as defined above, removing the protecting group from the protected amino group, and if necessary, converting the resulting compound into a salt, to yield a compound of the formula (IV) :

wherein the symbols are the same as defined above, or a salt thereof,
(2) condensing the compound (IV) or a salt thereof with a compound of the formula (III) :

wherein the symbols are the same as defined above, a salt or a reactive derivative thereof to yield a compound of the formula (II) :

Ethyl (ocS) – – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4- hydroxybenzene propionate and ethyl (otS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzene propionate are described in J. Med. Chem. , 33: 1620 (1990) and JP-A-7- 157472, respectively. 4-Bromo-3, 5-dimethoxybenzyl alcohol is described in, for example, J. Med. Chem. , 20: 299 (1977), and can also be prepared according to the following process.

Firstly, 4-bromo-3, 5-dihydroxybenzoic acid is methylated to give methyl 4-bromo-3, 5-dimethoxybenzoate, which is then reduced to yield 4-bromo-3, 5-dimethoxy benzyl alcohol. The methylation can be carried out by reacting with dimethyl sulfate in the presence of a base in a suitable solvent (e.g., ethyl acetate). The reduction can be carried out by reacting with an reducing agent (e.g., lithium alminium hydride, sodium borohydride and calcium borohydride) in a suitable solvent (e.g., tetrahydrofuran) .
EXAMPLES
The following Examples are provided to further illustrate the process of preparation according to the present invention. In the following examples, some compounds may be referred to by different compound name depending on the nomenclature, as illustrated below.
Ethyl (αS) -α-amino-4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate
Another name: ethyl (2S) -2-amino-3- [4- (4-ethoxymethyl- 2, 6-dimethoxyphenyl) phenyl]propanoate
Ethyl (αS) – [ [1, 1-dimethylethoxy] carbonyl] amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate
Another name 1: ethyl (2S) -2- [ (t-butoxycarbonyl) – amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) – phenyl]propanoate
Another name 2: Ethyl N- (t-butoxycarbonyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
Ethyl (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionate Another name 1: Ethyl (2S) -2- [ (2, 6- difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6- di ethoxyphenyl) phenyl] propanoate
Another name 2: Ethyl N- [2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
(ocS) – – [ (2, 6-Difluorobenzoyl) amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionic acid
Another name 1: (2S) -2- [ (2, 6-difluorobenzoyl) amino] -3- [4- (4-ethoxymethyl-2, 6-dimethoxyphenyl) phenyl]propanoic acid
Another name 2: N- [ 2 , 6-difluorobenzoyl) -4- (4- ethoxymethyl-2, 6-dimethoxyphenyl) -L-phenylalanine
EXAMPLE 1 (1) Under nitrogen atmosphere, pyridine (130.3 g) and trifluoromethanesulfonic anhydride (170.4 g) were added dropwise to a solution of ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-hydroxybenzenepropionate
(170.0 g) in dichloromethane (1.7 L) at 10 ° C or below. After stirring for 1 hour at the same temperature, water
(850 ml) was added dropwise to the mixture and the mixture was stirred for 2 hours at the same temperature. The organic layer was washed with 10 % aqueous citric acid solution and aqueous saturated sodium hydrogen carbonate solution, and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [ [ (1, 1- dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy)benzenepropionate (242.5 g) as oil . MS (m/z) : 441 (M+) (2) Under nitrogen atmosphere, to a mixture of ethyl (αS)- – [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4-
(trifluoromethanesulfonyloxy) benzenepropionate (66.2g), 4- ethoxymethyl-2, 6-dimethoxyphenylboric acid (54.0 g) , triphenylphosphine (9.83 g) and N-methylpyrrolidone (330 ml) were added palladium acetate (1.68 g) and diisopropylamine (24.9 g ), and the mixture was heated at 90 °C. After stirring for 1 hour at the same temperature, the mixture was cooled and toluene and water were added. The organic layers were washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield ethyl (αS) -α- [[ (1, 1-dimethylethoxy) carbonyl] amino] – 4′ -ethoxymethyl-2′ , 6′ -dimethox (1,1′ -biphenyl) -4-propionate (90.1 g) as oil.
The product was dissolved in ethanol (330 ml) , and after addition of p-toluenesulfonic acid monohydrate (28.5 g) , the mixture was stirred for 2 hours at 75 °C. After cooling to room temperature, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was dissolved in ethyl acetate with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS)-α- amino-4′ -ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4- propionate p-toluenesulfonate (63.4 g) .
MS (m/z) : 387 (M+-p-toluenesulfonic acid), M.p. 127-129°C
(3) To a mixture of ethyl (αS) -α-amino-4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate p- toluenesulfonate (29.0 g) , sodium hydrogen carbonate (15. 2 g) , water (290 ml) and ethyl acetate (290 ml) was added dropwise 2, 6-difluorobenzoyl chloride (9. 6 g) at 15 °C or below and the mixture was stirred for 30 minutes at the same temperature. The ethyl acetate layer was washed with saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo. The residue was recrystallized from isopropanol-water to yield ethyl (αS) -oi- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (26.4 g) . MS (m/z) : 527 (M+) , M.p. 87-89°C (4) To a solution of sodium hydroxide (2.9 g) in water- tetrahydrofuran (317 ml-159 ml) was added ethyl (oιS)-α- [ (2, 6-difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionate (31.7 g) at 15°C and the mixture was stirred for 4 hours at the same temperature. After neutralizing with IN HC1, the organic solvent was removed in vacuo. The aqueous layer was cooled, the crystalline precipitates were collected by filtration and recrystallized from ethanol-water to yield (αS) -a- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethoxy (1, 1′ -biphenyl) -4-propionic acid (28.8 g) . MS (m/z): 499 (M+) , M.p. 154-155°C
EXAMPLE 2 (1) Under nitrogen atmosphere, a mixture of ethyl (oιS)-o:- [[ (1, 1-dimethylethoxy) carbonyl] amino] -4-bromobenzene propanoate (11.17 g) , 4-ethoxymethyl-2, 6- dimethoxyphenylboronic acid (10.80 g ), palladium acetate (0.34 g), triphenylphosphine (1.57 g) , anhydrous potassium carbonate (12.44 g) , iV-methylpyrrolidone (56 ml) and water (11 ml) was stirred for 50 minutes at 80 °C. After completion of the reaction, the mixture was cooled to room temperature and extracted with ethyl acetate and water. The organic layer was washed with 10% aqueous citric acid solution and saturated aqueous NaCl solution, dried over magnesium sulfate and filtrated. The filtrate was concentrated under reduced pressure to yield ethyl (αS)-α- [ [ (1, 1-dimethylethoxy) carbonyl] amino] -4′ -ethoxymethyl- 2′ , 6′ -dimethox (1, 1′ -biphenyl) -4-propionate (20.4 g) as oil. The product was dissolved in ethanol (100 ml) , and after addition of p-toluenesulfonic acid monohydrate (5.7 g) , the mixture was stirred for 1.5 hours at 75 °C. After cooling, the mixture was filtrated over charcoal and the filtrate was concentrated under reduced pressure. The residue was suspended in toluene with heating. After cooling, the crystalline precipitates were collected by filtration and dried to yield ethyl (αS) – -amino-4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionate p- toluenesulfonate (13.80 g) . (2) The compound obtained in the above step (1) was treated in the same manner as described in Example 1 (2) to (4) to yield (αS) -a- [ [2 , 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1, 1′ -biphenyl) -4-propionic acid. The physicochemical data were the same as that obtained in Example 1.
EXAMPLE 3
To a solution of ethyl (αS) -α- [ (2, 6- difluorobenzoyl) amino] -4′ -ethoxymethyl-2′ , 6′ – dimethox (1, 1′ -biphenyl) -4-propionate (500 g ) in water (12.6 ml) and dioxane (50 ml) was added hydrochloric acid (12.4 g) and the mixture was stirred for 60 hours at 60 “C. The organic solvent was removed in vacuo and the aqueous layer was cooled. The crystalline precipitates were collected by filtration and recrystallized from ethanol- water to yield (αS) – – [ (2, 6-difluorobenzoyl) amino] -4′ – ethoxymethyl-2′ , 6′ -dimethoxy (1,1′ -biphenyl) -4-propionic acid (426 mg) . The physicochemical data were the same as that obtained in Example 1.
REFERENCE EXAMPLE 1
(1) To a mixture of 4-bromo-3, 5-dimethoxybenzylalcohol (44.5 g) , triethylammonium benzyl chloride (2.05 g) and 20% aqueous sodium hydroxide solution (288 g) was added diethyl sulfate (41.7 g) under ice-cooling, and the mixture was stirred overnight at 25-30 °C. After stirring for 1 hour at 70 °C, the mixture was cooled and extracted with toluene. The toluene layer was washed with water and saturated aqueous NaCl solution and dried over magnesium sulfate. The solvent was removed in vacuo to yield 4-bromo-3, 5- dimethoxybenzyl ethyl ether (49.5 g) as colorless oil. MS (m/z): 276 (M++2) , 274 (M+)
(2) Under nitrogen atmosphere, to a solution of 4-bromo- 3, 5-dimethoxybenzyl ethyl ether (440.0 g) in tetrahydrofuran (4.0 L) was added dropwise n-butyl lithium (1.6 M n-hexane solution, 1.1 L) at -60°C. After stirring for 15 minutes at the same temperature, trimethyl borate (249.3 g) was added. The temperature of the mixture was gradually elevated, followed by stirring for 1 hour under ice-cooling. To the mixture was added dropwise 10% aqueous sulfuric acid solution (835 g ) . The mixture was extracted with ethyl acetate and the organic layer was washed with water and saturated aqueous NaCl solution. After drying over magnesium sulfate, the solvent was removed in vacuo. The residue was dissolved in isopropyl ether with heating and cooled. The crystalline precipitates were collected by filtration and dried to yield 4-ethyoxymethyl-2, 6- dimetoxyphenylboronic acid (312.9 g) . M.p. 59-61°C
REFERENCE EXAMPLE 2
(1) To a suspension of 4-bromo-3, 5-dihydroxybenzoic acid (95.0 kg) in ethyl acetate (950 L) were added anhydrous potassium carbonate (270.8 kg) and dimethyl sulfate (174.7 kg) . The mixture was heated at 50-80 ‘C for about 4 hours and partitioned by adding water. The organic layer was washed with water and saturated aqueous NaCl solution and concentrated under reduced pressure. The residue was suspended into methanol, stirred under heating and cooled. The crystalline precipitates were collected by filtration and dried to yield methyl 4-bromo-3, 5-dimethoxybenzoate (98.8 kg) as pale yellow crystals. MS (m/z): 277 (M++2) , 275 (M+) , M.p. 120-122°C
(2) To a solution of calcium chloride (46.5 kg) in ethanol (336 L) were added tetrahydrofuran (672 L) and methyl 4- bromo-3, 5-dimethoxybenzoate (96.0 kg) to obtain a suspension. To the suspension was added sodium borohydride
(31.7 kg) by portions at room temperature, and the mixture was stirred for about 9 hours at temperature of room temperature to 45 °C. The reaction mixture was added dropwise to aqueous HC1 solution and stirred for about 16 hours at room temperature. Organic solvent was removed in vacuo, and water (1440 L) was added to the residue and stirred for 1 hour at 50 °C. After cooling, the crystalline precipitates were collected by filtration and dried to yield 4-bromo-3, 5-dimethoxybenzyl alcohol (83.3 kg) as colorless crystals. MS (m/z): 249 (M++2), 247 (M+) , M.p. 100-102°C.
INDUSTRIAL APPLICABILITY The process for preparation of the present invention makes it possible to afford a compound of the formula (I) or a pharmaceutically acceptable salt thereof with high- purity, in a high yield and inexpensively, and, therefore, the process of the present invention is industrially very useful.
References
5. Firategrast
| WO2002018320A2 | 27 Ago 2001 | 7 Mar 2002 | Tanabe Seiyaku Co | INHIBITORS OF α4 MEDIATED CELL ADHESION |
| WO2003072536A1 | 27 Fev 2003 | 4 Set 2003 | Tanabe Seiyaku Co | A process for preparing a phenylalanine derivative and intermediates thereof |
| WO2003072537A2 | 6 Fev 2003 | 4 Set 2003 | Abbott Lab | Selective protein tyrosine phosphatatase inhibitors |
Mitsubishi Tanabe Pharma Corporation


Mitsubishi Tanabe Pharma Corporation
Pharmacological research building

 |
||
| ■Mitsubishi Tanabe Pharma Corporation Pharmacological research building |

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Flupirtine Revisited

Flupirtine, D 9998
2-amino-6-(4-fluoro-benzylamino)- pyridin-3-yl)-carbamic acid ethyl ester, is unique as a non-opioid, non-NSAID, non-steroidal analgesic with a favorable tolerability. It first became available in Europe in 1984, and was sold mainly under the names Katadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor
PHASE 2
MS
- Neuronal potassium channels (7)
- Membrane resting potential (6)
- NMDA receptor channels (indirectly)(14)
- Originally developed by Asta Medica (1) (4)
- Being developed and commercialized to treat fibromyalgia by Synthetic Biologics (1)
Flupirtine

75507-68-5 maleate
56995-20-1 (free base)
56995-20-1 (free base)
| LAUNCHED | 1986 NEUROPATHIC PAIN |
Flupirtine maleate is the INN for 2-amino-3-ethylcarbamato-6- (4-fluoro-benzylamino) maleate, CAS: 75507-68-5, molar mass 420.40 g / mol, molecular formula C1 5 H17FN4O2 • C4H4O4, and corresponds to the structure of formula I.

Flupirtine maleate is used, for example, under the trade name Katadolon® as an analgesic.
56995-20-1
CAS Name: [2-Amino-6-[[(4-fluorophenyl)methyl]amino]-3-pyridinyl]carbamic acid ethyl ester
Additional Names: 2-amino-6-[(p-fluorobenzyl)amino]-3-pyridinecarbamic acid ethyl ester
Trademarks: D-9998
Molecular Formula: C15H17FN4O2
Molecular Weight: 304.32
Percent Composition: C 59.20%, H 5.63%, F 6.24%, N 18.41%, O 10.51%
Properties: Crystals from isopropanol, mp 115-116°. 5% ethanol soln is colorless, turns green on exposure to air for 20 hours. LD50 orally in mice, rats: 617, 1660 mg/kg (Jakovlev).
Melting point: mp 115-116°
Toxicity data: LD50 orally in mice, rats: 617, 1660 mg/kg (Jakovlev)
Derivative Type: Hydrochloride
Molecular Formula: C15H17FN4O2.HCl
Molecular Weight: 340.78
Percent Composition: C 52.87%, H 5.32%, F 5.57%, N 16.44%, O 9.39%, Cl 10.40%
Properties: Crystals from water, mp 214-215°. When prepd industrially contains intensely blue by-product.
Melting point: mp 214-215°
Derivative Type: Maleate
CAS Registry Number: 75507-68-5
Trademarks: Katadolon (AWD)
Molecular Formula: C15H17FN4O2.C4H4O4
Molecular Weight: 420.39
Percent Composition: C 54.28%, H 5.04%, F 4.52%, N 13.33%, O 22.84%
Properties: Colorless crystals from isopropanol, mp 175.5-176°. Formed as mixture of two crystalline forms A and B; mixtures containing 60-90% A are preferred.
Melting point: mp 175.5-176°
Therap-Cat: Analgesic.
TARGET:
Neuronal potassium channels
Membrane resting potential
NMDA receptor channels (indirectly)
STATUS FOR MS:
Phase II
COMMERCIAL:
Originally developed by Asta Medica
Being developed and commercialized to treat fibromyalgia by Synthetic Biologics
Marketed for pain indications in various European countries by Meda
TRADE NAME:
Effirma (US)
Katadolon (Brazil, Germany, Latvia, Estonia, Slovakia, Lithiania, Russian Federation)
SYNONYMS:
EINECS 260-503-8,UNII-MOH3ET196H, Effirma (US), Katadolon (Brazil, Germany, Latvia, Estonia, Slovakia, Lithiania, Russian Federation)
SYSTEMATIC NAME:
Carbamic acid, (2-amino-6-(((4-fluorophenyl)methyl)amino)-3-pyridinyl)-, ethyl ester
PROPERTIES:
Molecular weight: 304
MECHANISMS/EFFECTS
HUMAN:
Stabilizes membrane resting potential by activating neuronal Kv7 potassium channels
Indirectly antagonizes NMDA receptors
Reduces muscle spasticity in humans
Prevents apoptosis and reduced formation of reactive oxygen species by in cultured human retinal pigment epithelial cells

Structures of flupirtine, D13223, and retigabine.
Regulatory and Commercial Status
STATUS FOR MS:
Phase II
HIGHEST STATUS ACHIEVED (FOR ANY CONDITION):
Approved in Europe
ADMINISTRATION:
Oral
COMMERCIAL:
Originally developed by Asta Medica
Being developed and commercialized to treat fibromyalgia by Synthetic Biologics
Marketed for pain indications in various European countries by Meda
Flupirtine is an aminopyridine that functions as a centrally acting non-opioid analgesic. It first became available in Europe in 1984, and is sold mainly under the names Katadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor.[5] Flupirtine is sold by Intas Pharma under the brand name Pruf in India. Like nefopam, it is unique among analgesics in that it is a non-opioid, non-NSAID, non-steroidal centrally acting analgesic. In 2010 the chemically related drug (the difference being that the pyridine group in flupirtine is replaced with a phenyl group) retigabine (INN; ezogabine [USAN]) was approved by the FDA as an anticonvulsant for the treatment of refractory partial-onset seizures in treatment-experienced patients.[6] Retigabine also works by opening the neuronal KCNQ/Kv7 potassium channel, just like flupirtine.
History
Flupirtine was originally developed by Asta Medica, with the synthesis of the compound and the development of the drug described in patents from the 1970s to the 2000s.[7][8][9][10][11][12]
It was approved for the treatment of pain in 1984 in Europe. However, it has never been introduced to the United States market for any indication. In 2008, Adeona Pharmaceuticals, Inc. (now called Synthetic Biologics, Inc.) obtained an option to license issued and patent pending applications relating to flupirtine’s use in the treatment of ophthalmic indications, particularly retinitis pigmentosa.[13]
Mechanism of Action
Flupirtine is a selective neuronal potassium channel opener that also has NMDA receptor antagonist and GABAA receptor modulatory properties.[14]
Uses
Flupirtine is used as an analgesic for acute and chronic pain, in moderate-to-severe cases.[15] Its muscle relaxant properties make it popular for back pain and other orthopedic uses, but it is also used for migraines, in oncology, postoperative care, and gynecology.
Flupirtine has been noted for its neuroprotective properties, and it is being investigated for possible use in Creutzfeldt–Jakob disease, Alzheimer’s disease, and multiple sclerosis.[16][17] It has also been proposed as a possible treatment for Batten disease.[18]
Flupirtine underwent a clinical trial as a treatment for multiple sclerosis[19] and fibromyalgia.[20] Flupirtine showed promise for fibromyalgia due to its different action than the three approved by U.S. FDA drugs: Lyrica (pregabalin), Savella (milnacipran), and Cymbalta (duloxetine).[21] Additionally, there are case reports regarding flupirtine as a treatment for fibromyalgia.[22] Adeona Pharmaceuticals (now called Synthetic Biologics) sub-licensed its patents for using flupirtine for fibromyalgia to Meda AB in May 2010.[21]
Side Effects
The most serious side effect is frequent hepatotoxicity which prompted regulatory agencies to issue several warnings and restrictions.[23][24]
Flupirtine is devoid of negative psychological or motor function effects, or effects on reproductive function.[25][26]
Abuse and Dependence
Although some studies have reported flupirtine has no addictive properties,[27][28] there was suggestion that it may possess some abuse potential and liability.[29] There were at least two registered cases of flupirtine abuse.[30] Drug tolerance does not develop in most cases; however, tolerance may develop in single cases.[30]
Flupirtine is 2-amino-3-carbethoxyamino-6-(p-fluorobenzylamino) pyridine; CAS No: 56995-20-1 , an aminopyridine that functions as a centrally acting non-opioid analgesic. It first became available in Europe in 1984, and is sold mainly under the names atadolon, Trancolong, Awegal, Efiret, Trancopal Dolo, and Metanor. It is unique as a non- opioid, non-NSAID, non-steroidal analgesic. Flupirtine is used as an analgesic for acute and chronic pain, in moderate to severe cases. Its muscle relaxant properties make it popular for back pain and other orthopaedic uses, but it is also used for migraines, in oncology, postoperative care, and gynaecology. Flupirtine has been noted for its neuro-protective properties, as well as its possible uses for Creutzfeld- Jakob disease, Alzheimer’s disease, and multiple sclerosis are being investigated. It has also been proposed as a possible treatment for Batten disease. Flupirtine also acts as an antioxidant and prevent free radical- mediated structural damage.
US3481943 (hereinafter referred as ‘943) discloses the process for the preparation of flupirtine hydrochloride of formula (T) wherein p- fluorobenzylamine (formula R) is reacted with 2-amino-3-nitro-6- chloropyridine (Q) in n-propanol using potassium carbonate to prepare 2-amino-3-nitro-6-p-fluorobenzylamino-pyridine of formula (S) which is hydrogenated in dioxane using raney nickel at 50 C under a gauge pressure of 30 atmospheres. Solution is filtered off to remove the catalyst and then reacted with chloroformic acid ethyl ester (ethyl chloroformate) while stirring. The product is filtered off and recrystallized from water to give flupirtine hydrochloride salt of formula (T). The process disclosed therein in ‘943 is depicted as given below

Drawbacks associated with the process disclosed in ‘943 are:
1) The yield of 2-amino-3-nitro-6-p-fluorobenzylamino-pyridine of formula S obtained is around 40% only. ‘943 does not disclose the preparation of maleate salt of flupirtine.
2) During the preparation hydrochloride salt of flupirtine on an industrial scale, intensely blue colored by products are formed which are either difficult to remove or can not be removed completely.
3) Use of n-propanol as reaction solvent is expensive. reaction mass thereby hindering the progress of the reaction. Another most probable reason attributed for getting poor yield of 40% in the said process could be masking of hydrochlorides of both the reactants of formulae (Q’) and (R’) (as both reactants are amino compounds and form hydrochlorides) over potassium carbonate making it unavailable for further reaction posing problem towards the completion of reaction thereby adversely affecting the yield.

DE3133519 (US4481205) discloses the preparation of flupirtine maleate of formula (IA), wherein 2-amino-3-nitro-6-chloro-pyridine of formula (S) is prepared by taking 2,6-dichloro-3-nitropyridine of formula (P) (90%, water wet) in isopropanol at 20°-30°C and purging ammonia gas (or dropping liquid ammonia) into the said reaction mixture and then resulting 2-amino-3-nitro-6-chloro-pyridine of formula (Q) is reacted with p-fluorobenzylamine (R) in isopropanol using triethyl amine as a base ; the reaction mixture is refluxed for 6 hours. Thereupon after addition of a large volume of water the compound 2-amino-3-nitro-6-(p- fluorobenzylamino)-pyridine of formula (S) precipitates.
2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine of formula (S) is then hydrogenated in the presence of raney nickel at 5 bar at 60°C to give 2,3- diamino-6-(4-fluorobenzylamino) pyridine using 2-methoxy ethanol as hydrogenating solvent. This is followed by acylation with ethyl chloroformate using triethylamine as a base under inert gas atmosphere to obtain flupirtine base of formula (I). The catalyst is filtered off and filtrate containing dissolved triethyi amine hydrochloride is directly added to solution of maleic acid in isopropanol resulting into formation of crude flupirtine maleate (IA). It also discloses the importance of the exclusion of atmospheric oxygen by an intensive supply of inert gas and closed reactor system to avoid development of troublesome coloured complexes.
The purification of crude flupirtine maleate is carried out by converting crude flupirtine maleate into crude flupirtine base by contacting with ammonia or sodium hydroxide solution. Then the crude flupirtine base is recrystallized from isopropanol and, after contacting with activated carbon/kieselguh’r, it is reacted with a solution of maleic acid in isopropanol to give flupirtine maleate of formula (IA). The reaction scheme of DE3133519 is depicted herein below.

Drawbacks associated with the process disclosed in DE3133519 (US4481205) are:
1) Use of gaseous ammonia or liquid ammonia for the preparation of 2-amino-3-nitro-6-chloro-pyridine of formula (Q) starting from 2, 6- dichloro-3-nitropyridine of formula (P) contributes towards increased level of impurities of formulae X and Y as the gaseous ammonia and liquid ammonia as sources of ammonia are in concentrated forms and it is not easy to control the purging or addition in appropriate quantities and as a consequence it results in the formation of higher amounts of impurities and poor yield of the desired compound.

Another disadvantage of using ammonia gas is that it is classified as a hazardous material and is subject to strict regulations and risk management procedures for transport, storage, and handling. These requirements result in additional costs and may generate local community concerns over transporting and storing hazardous materials. While aqueous ammonia used by the inventors requires minimal special handling, social and regulatory requirements.
2) Preparation of 2-amino-3-nitro-6-(p-fluorobenzylamino)-pyridine of formula (S comprises reaction between 2-amino-3-nitro-6-chloro- pyridine of formula (Q) and p-fluorobenzylamine of formula (R) using isopropanol as solvent and triethyl amine as base. To induce separation of 2-amino-3-nitro-6-(p-fluorobenzylamino)-pyridine of formula (S from the reaction mixture in IP A a large volume of water is required which makes reaction mass highly voluminous therefore, not preferred at industrial scale. 3) Basification of crude flupirtine maleate comprising the process of liberating free flupirtine base using ammonia or sodium hydroxide produces an ammonium or sodium salt which pollutes the water.
4) Use of activated charcoal and kieselgulir during the purification of flupirtine base (that contains three amino groups known for their light and colour sensitive nature) takes prolonged time for filtration through filtering bed thereby exposing to environment producing high coloration.
5) The crude flupirtine maleate remains trapped with triethyl amine hydrochloride.
US59591 15A (hereinafter referred as Ί 15) discloses a process for the preparation of flupirtine maleate (IA) as discussed under DE3133519 (US4481205). It also discloses crystalline form “A” of flupirtine maleate by the use of water soluble alcohols (such as ethanol or isopropanol) during synthesis and/or purification. There are three proposed variants in Ί 15 as shown below: process variant:

A: ANFP (S)→hydrogenation→acylation→crude flupirtine base.
B: crude flupirtine base→maleic acid→crude flupirtine maleate
C-E (as shown in scheme-II): not applicable F: crude maleate→pure maleate.
1 s process variant comprises synthesis of oxygen sensitive crude base in situ in process step A and it was converted by a “very rapid” suction filtration process into an aqueous maleic acid solution from which coloured crude flupirtine maleate (IA) is obtained, which is to be purified by recrystallization from isopropanol-water.
2″ process variant:

A: ANFP (S)→hydrogenation→acylation→crude flupirtine base.
B: flupirtine base→maleic acid→crude flupirtine maleate.
C-F (as shown in scheme-II): Not applicable.
G: without isolation of the crude maleate→pure maleate.
As compared to the process step F in 1st variant, process step G in 2nd variant represents substantially shorter alternative process in which the precipitation of crude flupirtine maleate from the flupirtine base formed in situ in isopropanol is effected by Alteration with suction into an aqueous maleic acid solution at 50-60°C and, after that without isolation of the crude maleate, colourless pure material is obtained.
3rd process variant:

A: ANFP (S)→hydrogenation→acylation→cmde flupirtine base (isolated)
B: pure flupirtine base→maleic acid→pure flupirtine maleate.
Herein, after acylation, the flupirtine base (I) is precipitated preferably in ethanol or water and is purified by recrystallization and than treated with maleic acid to prepare pure flupirtine maleate (IA).
Ί 15 disclose hydrogenation of ANFP (S), acylation and precipitation in water-soluble alcohols, such as ethanol or isopropanol.
1) In 1st process variant “very rapid” suction filtration process is a great limitation at plant scale.
2) 2nd process variant also does not produce colorless pure maleate.
3) In 3 process variant, after acylation, the flupirtine base is precipitated preferably in ethanol or water and is purified by recrystallization and than treated with maleic acid to prepare pure flupirtine maleate salt (IA).

It also discloses that although the treatment of final product with activated carbon and recrystallization is known as a reasonably successful procedure to remove impurities. This approach is reluctantly accepted because of the losses in overall yield as it is applied in the last production step of a drug and particularly in the case of flupirtine, it is not a preferred/desirable procedure as it may result into the formation of colored impurities.
US47851 10A discloses a process for the preparation of 2-amino-3-nitro- 6-fluorobenzylamino pyridine of formula (S) comprising reaction of 2- amino-3-nitro-6-methoxypyridine of formula (T) (1 mole) with 4-fluoro- benzylamine of formula R (2-4 mole) optionally as a mineral acid salt in water at a temperature between 70°C and 150°C; preferably between 90° and 120°C. The said condensation is also performed in autoclave as the temperature is above 100°C.It also discloses the necessity of using basic material suitably as an aqueous solution in case when acid addition salts of 4-fluoro-benzylamine of formula (R) is used to liberate the free base for the reaction. It also discloses subsequent reduction of nitro group of 2-amino-3-nitro-6-methoxypyridine by various modes with preference to catalytic hydrogenation optionally in the presence of carriers selected from barium sulphate, calcium sulphate, magnesium sulphate, sodium sulphate etc.

The drawbacks associated with the process described in US47851 10A are: 1) As per the experimental section of the said process of condensation for the preparation of 2-amino-3-nitro-6-fluorobenzylamino pyridine of formula (S) discloses heating at boiling for ten hours. The temperature would be around 100°C as water is used as solvent. However, inventors of the subject invention disclose herein the same process comprising using 6-chlorpyridine instead of 6-methoxy pyridine and water as solvent’, wherein the reaction is carried out at temperature much below boiling point of water and reaction gets completed in 3 hrs compare to 10 hrs at temperature of boiling water as in’ 1 10. Furthermore, the said reaction disclosed herein in the present invention does not require autoclave. There is no teaching or anticipation on this aspect from Ί 10.
2) Excessive use of 2-4 moles of 4-fluoro-benzylamine of formula ( ) for the preparation of 2-amino-3-nitro-6-fluorobenzylamino pyridine of the formula (S) comprising the reaction of 2-amino-3-nitro-6- methoxypyridine of formula (T)with 4-fluoro-benzylamine of formula (R).Unreacted 4-fluoro-benzylamine is then removed by steam distillation which is not only time and energy consuming but also increase in an extra unit operation.
3) In case when acid addition salts of 4-fluoro-benzylamine are used that requires another additional operation of basification to liberate free base to enable 4-fluoro-benzylamine to be available to react further with 2- amino-3-nitro-6-methoxypyridine forming 2-amino-3-nitro-6- fluorobenzylamino pyridine of the formula (S)
DE 31 33 519 describes a process for the preparation of flupirtine maleate as a mixture of polymorphic forms A and B, wherein A is present in a proportion> 60%. The key reaction steps are the hydrogenation of 2-amino-6- (4-fluorobenzylamino) -3-nitropyridine (Formula II) shown in Figure 1, hereinafter also referred to as ANFP, by means of Ra-Nickel for 2,3-diamino- 6- (4-fluoro-benzylamino) -pyridine (Formula III) and subsequent regioselective acylation with chloroformate for free flupirtine base. By precipitation as maleate to blue contaminants that are incurred in the production of HCl salt, are eliminated. Purification of flupirtine maleate is obtained as maleate by releasing the base from the maleate, treatment with activated carbon and reprecipitation. Despite this lengthy and economically expensive purification strategy traces of colored impurities can be difficult to remove.
In WO 98/47872 a process for the preparation of flupirtine maleate is described, in which, in water-soluble alcohols (IPA) is carried out. There are three proposed variants. Option 1 includes an implementation of ANFP to Ra-nickel in the IPA is directly attached to the acylation and the precipitation of a product by Rohmaleat called “very fast” extraction process in an aqueous solution of maleic acid. It falls on a colored Rohmaleat which is to be purified by recrystallization from isopropanol / water. However, the enactment of this variant in the laboratory showed a colored product. In variant 2 should already be colorless an image obtained by aspiration in 50 to 60 0 C warm maleic Rohmaleat. This also could not be confirmed. According to the third variant, the Flupirtinbase formed after acylation is not converted in situ but precipitated in ethanol or water and recrystallized before further reaction with maleic acid. Even with the procedure referred to in this document is a pure white flupirtine maleate is not readily available.
………………….
PATENT
http://www.google.com.tr/patents/WO2010136113A1?cl=en&hl=tr
Example 3 Preparation of flupirtine maleate
50 g of 2-amino-6- (4-fluorobenzylamino) -3-nitropyridine, 2.5 g of palladium on activated carbon and 267 g of isopropanol were hydrogenated with hydrogen at 4.5 bar and 70 0 C. After completion of the reaction was additionally hydrogenated for 8 hours at 70 0 C. Then 20.2 g of ethyl chloroformate, 24.8 g of triethylamine and 4.96 g of ethyl chloroformate at 20 0 C was added. Thereafter, the reaction mixture was stirred for 1.5 h at 55 0 C. It was then filtered at room temperature. The filtrate was then added to a solution of 35.6 g of maleic acid in 1000 g of water at room temperature slowly. The resulting suspension was stirred for 1 h at room temperature. The precipitate was filtered off and washed with water and isopropanol. Dried filter cake (HPLC purity 91.5%) was dissolved in 828 g of isopropanol / water mixture (mass ratio 5.3: 1), and heated to 70 0 C. The resulting clear solution was cooled to room temperature and stirred at room temperature. The precipitate was filtered off and washed with isopropanol / water mixture. The filter cake was dried at 50 0 C. 43 g flupirtine maleate (HPLC purity 97.8%) was obtained as a white-gray solid. The yield was 55%.
………………
PATENT
http://www.google.com/patents/WO2013080215A1?cl=en
The invention relates to an improved process for the preparation of flupirtine of formula (I) and its pharmaceutically acceptable salts, particularly flupirtine maleate of formula (IA) preferably pure crystal modification A of flupirtine maleate.
A process for the preparation of the compound of formula (I)

and pharmaceutically acceptable acid addition salts thereof comprising the steps of:
(a) contacting 2, 6-dichloro-3-nitro pyridine of formula (P) with aqueous ammonia solution in a compatible solvent to produce 2-amino-3-nitro-6- chloro-pyridine of formula (Q);

(b) contacting said compound of formula (Q) with p-fluorobenzylamine taking water as a solvent in presence of a base to produce 2-amino-3- nitro-6-p-fluorobenzylamino-pyridine of formula (S);

(c) reducing nitro group of 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine of formula (S) in a solvent base combination as solvent system in the presence of a catalyst;

(d) contacting 2,3-diamino-6-p-fluorobenzyl amino pyridine produced in step c with an ethyl chloroformate in presence of a base optionally insitu without isolation to produce flupritine base of formula (I);

(e) contacting the said flupritine base of formula (I) with acid solution to produce corresponding acid addition salt.



Scheme (I):


EXAMPLE 1 : Preparation of 2-amino-3-nitro-6-chloro-pyridine.
A solution of 100 gm. 2, 6-dichloro-3-nitro-pyridine in 800 ml isopropyl alcohol is taken in round bottom flask. 300 ml of aqueous ammonia solution (20-25%) is added at 20-25°C. The reaction mass is stirred for 20-24 hours at 20-25°C. After completion of the reaction
The solid is filtered and washed with 100 ml isopropyl alcohol then dried to obtain 70-75 gm 2-amino-3-nitro-6-chloro-pyridine.
EXAMPLE 2: Preparation of 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine.
100 gm of 2-amino-3-nitro-6-chloro-pyridine is taken in 800 ml of water. 90 gm of p-fluorobenzylamine is added dropwise into the reaction mixture at 20-25°C. Then 87 gm triethylamine is also added dropwise into the reaction mixture at 20-25°C. After complete addition, the reaction mass is stirred at 40-45°C for half an hour again the reaction mass is heated to 80-85°C and stirred at this temperature for 3-4 hours. After completion of the reaction, the reaction mass is cooled to 20-25°C and stirred at this temperature for 2-3 hours and then stirred at 15-20°C for 3-4 hours. The solid mass is filtered and then washed with 200 ml of water and 100 ml isopropyl alcohol and then dried in air oven till constant weight to get 140-150 gm. of 2-amino-3-nitro-6-p- fluorobenzylamino-py ridine .
EXAMPLE 3: Preparation of flupirtine maleate.
In an autoclave, 100 gm. 2-amino-3-nitro-6-p-fluorobenzylamino- pyridine is taken in 500 ml. 1, 4-dioxane and 20 ml aqueous ammonia solution. 10 gm of raney nickel is added under nitrogen atmosphere and hydrogenated at 75-80°C for 2-3 hours under 4-5 kg pressure. After completion of the reaction, the reaction mass is cooled and filtered at 40- 45°Cthen in filtrate 45 ml of ethyl chloroformate is added slowly at 5- 10°C. The temperature is raised to 25-30°C and 80 ml triethyl amine is added under nitrogen atmosphere. The reaction mass, is heated at 55- 60°C under stirring for 3-4 hours. After completion of the reaction, the reaction mass is distilled up to 70-80% under vacuum. This concentrated reaction mass is added into aqueous solution of maleic acid (72 gm in 2000 ml DM water at 65-70°C and maintained at 65-70°C for 2 hours under nitrogen to get crude Flupirtine Maleate as a solid. The reaction mass is cooled to 25-30°C in 5-6 hours and maintained at this temperature for next 2-3 hours then filtered. The wet cake is washed with 200 ml water and dried to get 145 gm of flupirtine maleate.
EXAMPLE 4: Preparation of pure flupirtine maleate crystalline modification A.
1 15 gm crude Flupirtine maleate obtained in example 3 is taken in 1 150 ml methanol and 58 ml water. This mixture is heated to reflux and 58 ml water is added slowly to get a clear solution and refluxed for about half an hour. The reaction mixture is cooled slowly to 60°C and seeded with crystals of modification A. Then it is cooled slowly to 20-25°C and maintained at this temperature for 2 hours. The crystalline mass is filtered and washed with 100 ml chilled methanol and dried to give 92 gm. flupirtine maleate crystalline modification A.
………………….
PATENT
http://www.google.com.tr/patents/WO1998047872A1?cl=en
1. Example
Preparation of flupirtine maleate
75 g (0.286 mol) ANFP be in a suspension of 12.5 g of Raney nickel in 400 ml of isopropanol was hydrogenated at 65 ° C and 5 bar hydrogen pressure. After hydrogenation, the solution is then mixed with 26.4 ml of ethyl chloroformate and 50.6 ml of triethylamine. After adding a further 6.3 ml of ethyl chloroformate the reaction solution is stirred at 60 ° C. for 1 hour. Then sucks the hot solution with stirring in a 50 – 60 ° C heated solution of 53.3 g of maleic acid in 1, 5 IH 2 O and washed the catalyst with little isopropanol.
The flupirtine maleate is precipitated in colorless crystal suspension is cooled with further stirring at 20 ° C and maintained at this temperature for 20 minutes. It is suctioned off, washed with 500 ml of water and dried flupirtine maleate in vacuo at 35 ° C.
Yield: 107.55 g (89.6% of theory, based on ANFP.) Example 2
Preparation of flupirtine maleate
18.5 g (0.07 mol) ANFP be analogous to Example 1 in a suspension of 2.0 g of Raney nickel in 140 ml of ethanol 60 – 70 ° C and 5 bar hydrogen pressure After hydrogenation, the further reaction takes place at 40 – 50 ° C with 9.3 g of ethyl chloroformate (0.86 mol) of triethylamine and 9.2 g (0.91 mol) The separated from the catalyst reaction solution is added with stirring to 540 ml of water After 2 hours of stirring at room temperature suctioned the failed base off and washed with water and isopropanol and crystallized in the 3.7-fold amount of isopropanol to yield 18.4 g (86.0% of theory)
The precipitation and modification of pure flupirtine maleate is carried out according to the Examples 7 and 8
………………….
PATENT
CN104086481 (A) – Synthesis method of flupirtine maleate
http://worldwide.espacenet.com/publicationDetails/biblio?CC=CN&NR=104086481A&KC=A&FT=D
The invention provides a synthesis method of flupirtine maleate. Recrystallization by use of methanol is carried out in the refining step of the crude product of the flupirtine maleate so that the product is white in appearance and high in purity, and the crystal form of the product is pure A crystal and same as the crystal form of the commercial products. The optimal reaction solvent, reaction time and reaction temperature are explored and found out by use of a simplified process flow, and a method for preparing the flupirtine maleate in the pure A crystal form, which is high in yield, low in cost and simple to operate, uses easily available raw materials and is applicable to the industrial production is found.
………………
PATENT
http://www.google.com/patents/CN103333103A?cl=en
The preparation of a comprehensive literature about the ratio of maleic acid fluoride Jie Ting to 2_-amino-3-nitro-6-chloro-Jie ratio 唳 as a starting material, by condensation, reduction, acylation, salt and other processes for The most common route, however, due to the reduction, acylation, salt formation method of a three-step operation is different, not only the yield of the synthesis varies widely, and also on the flupirtine maleate product quality. This is mainly because of the intermediate 2,3-diamino-6-fluoro-benzyl amino pyridine and flupirtine multi-aminopyridine derivative, is easy to oxidative deterioration. So the use of continuous operation, not only simple steps, and can improve product quality and yield.
Chinese patent CN102241626 reported to 2,6_ dichloro _3_ nitropyridine as raw material by selective ammonia solution to give 2-amino-3-nitro-6-chloro-approved Li, then with amine fluoride Festival to afford a yellow solid 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine. After vacuum drying, the use of hydrogenation, and then under nitrogen and ethyl chloroformate acylation catalyst is filtered off and then a salt with maleic acid to give a pale green crude product yield was about 37% (2-amino-3-nitro-6-chloro-pyridin-meter).
Patent No. CN102838534 reported 2-amino-3-nitro-6-chloro-pyridine as starting material, the use of sub-step processing method, in a first reactor, and a condensation-fluorobenzyl amine, and dried in vacuo to give the intermediate 2-amino-3-nitro-6-p-fluorobenzyl-aminopyridine, in a second reactor to Raney nickel as the catalyst, the catalytic hydrogenation of hydrazine hydrate, after filtration the solvent was evaporated to give the intermediate 2,3-solid – diamino-6-p-fluorobenzyl-aminopyridine, in a third reactor with ethyl chloroformate acylated intermediate distillation under reduced pressure to give solid form of flupirtine with an aqueous solution of a salt of maleic acid, after purification, the total Yield 25% ~ 30%.
Patent W02012004391 discloses a method for preparing a high yield of flupirtine maleate method. In 2_-amino-3-nitro-6-chloro-fluoro-section batch Li and amines as raw material for condensation to give 2-amino-3-nitro-6-fluoro-section based on the amino pyridine granted, then using high-pressure hydrogenation the reduction, acylation step in a high pressure hydrogenation reactor concentrated completed, after the catalyst was filtered off and then the salt, the crude yield of greater than 70%. The preparation method using high-pressure hydrogenation apparatus, there are security risks, and takes too long, is not suitable for industrial production.
Patent No. CN102260209 discloses a 2_ amino _3_ _6_ fluorobenzyl nitro-pyridine as starting material, the reduction, acylation and salt-forming step of the continuous operation, the synthetic yield was improved to 58% so, no mention of product purity. Since the acylation step taken ethyl chloroformate, while an organic base is added, so that an increase in a side reaction, the product yield decreases; the same time, 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine as the raw material, the production cost high.
In the present invention, we consider the key intermediate 2,3-diamino-6-p-fluorobenzyl-aminopyridine and chemical properties of flupirtine, condensation, reduction, acylation, salt formation reaction is concentrated to the same conventional the reactor is completed, each step without intermediate separation, simplifying the process route and operations, improve efficiency, reduce costs, improve the overall yield of the crude by 40 percent following the step by step operation for more than 70% crude purity of more than 99% suitable for industrial scale production.

Example 4:
The 4Kg2_ amino-3-nitro-6-chloropyridine, 4.5Kg triethylamine, 40L of isopropanol into the reactor, stirred and heated to reflux for turn; the 4.4Kg of benzylamine was added to the fluorine reactor, the reaction under reflux conditions for 3 hours. After heating was stopped, the reaction solution was added to 40L of purified water, a lot of yellow solid was precipitated was filtered and the resulting wet product remains in the reaction vessel. To the reaction kettle was added 1.8Kg Raney nickel, 40L of isopropanol, stirred and heated to reflux for open, 7Kg80% hydrazine hydrate was added dropwise, the reaction was refluxed for 3 hours, after completion of the reaction down to room temperature in a nitrogen atmosphere, was added rapidly 3.6Kg chloro carboxylic acid ethyl ester, the reaction at room temperature for 3 hours. 3Kg of triethylamine was added, free 2 hours, filtered and the filtrate was added to 5Kg / 100L of maleic acid in isopropanol, cooling crystallization to give an off-white solid, 50 ° C blast drying, weight 7.8Kg, the yield was 80.5 %, purity 99.6%.
A sub-step treatment process research and data [0034] Comparative Example
The method according to Chinese patent CN102838534 disclosed flupirtine maleate was prepared, and a number of specific steps
………….
PATENT
FIG. 1 is flupirtine maleate 1H NMR.
[0021] FIG. 2 is flupirtine maleate A crystal X-ray diffraction pattern



Example 3
2-Amino-3-nitro-6-chloropyridine 246g, and 254g of triethylamine were added to 800ml of ethanol-necked flask and stirred under heating to reflux, fluorine was slowly added dropwise benzylamine 80g, reaction of 6 hours, the reaction was completed After the dropwise addition of purified water 500ml, cooled slowly with stirring to room temperature, filtered, dried to give 2-amino-3-nitro-6-p-fluoro-benzylamino-pyridine.
[0033] The ferric chloride hexahydrate was dissolved in purified water 41g 200ml, adding activated charcoal 20g, heated to 50 ° C, a saturated solution of sodium hydroxide was added 45g (24g of sodium hydroxide dissolved in 21ml water), 60 ° C with stirring I hours, cooled to room temperature, filtered, and dried to give ferric hydroxide / activated carbon catalyst.
[0034] A mixture of 2-amino-3-nitro-6-p-fluorobenzyl-aminopyridine 104.Sg, ferric hydroxide / activated carbon catalyst was added to 20g 2L reaction flask was added 95% ethanol 1200ml, heated with stirring to 90 ° C. Insulation 60% hydrazine hydrate was added dropwise 250g. Drops Bi insulation response to 3h. Completion of the reaction, the reaction solution is filtered hot with concentrated hydrochloric acid to 240ml and 95% ethanol IOOOml reaction flask. (TlO ° C crystallization I h, filtered, dried to give 2,3-amino-6-fluoro-benzyl-aminopyridine on
Hydrochloride.
[0035] A mixture of 2,3-diamino-6-p-fluoro-benzylamino-pyridine hydrochloride 132g, 800ml of isopropanol was added to a 2L reaction flask, the temperature control to 28 至 30 ° C, was slowly added dropwise acetic acid ester 39g. Stirred for 0.5 hours, was slowly added dropwise triethylamine 70g, after stirring for 0.5 hours, complement ethyl chloroformate 5g, stirred for 15 minutes, additional triethylamine remaining 10g. Continue stirring for I hour. The reaction solution was concentrated under reduced pressure to about 800ml of distillate was distilled out. The remaining reaction solution was poured into an aqueous solution of maleic acid with a good (39g of maleic acid was dissolved in purified water IlOOml), stirred for 30 minutes at room temperature, (T5 ° C was stirred for 5 ~ 8 hours, filtered, dried to give the maleic acid flupirtine crude.
[0036] The crude flupirtine maleate product 100g, 2000ml of ethanol into the reaction flask and heated to 70~80 ° C, was added 5g of activated carbon and dissolved, and incubated I hour, filtered hot, O~5 ° C CRYSTALLIZATION 3 hours, filtered and dried to give crude I. The crude product I 90g, 450ml of ethanol into the reaction flask and heated 20h, and then slowly cooled to room temperature, O~5 ° C for 2 hours, filtered, and dried to give crystal form A of flupirtine maleate product.
…………………..
PATENT
http://www.google.com/patents/CN102838534A?cl=en
flupirtine maleate is a non-opioid analgesic effects on the central nervous system drugs, which is a selective neuronal potassium channel opener (Selective Neuronal Potassium Channel Opener, SNEPCO), has analgesic, muscle relaxant and neuroprotective triple effect. Acute pain treatment is mainly used for various types of moderate, such as surgery, trauma-induced pain and headache / migraine and abdominal spasms.
flupirtine maleate English name: Flupirtine Maleate, chemical name: 2_ amino-6 – [((4-fluorophenyl) methyl) amino] pyridine-3-carboxylic acid ethyl ester maleic salt; Chemical Abstracts (CAS) number = 75507-68-5; formula = C15H17FN4O2 · C4H4O4; molecular weight: 420.39; its structural formula is:

From a structural perspective, flupirtine maleate molecular compounds, the derivatives of benzene and pyridine derivatives synthetically produced flupirtine, flupirtine and then forming an organic salt with maleic acid. Comprehensive literature, synthetic routes flupirtine maleate there are two major, now its main synthetic steps described below.
Route 1 (W0 98 / 47872Α1): The route to 2,6_ dichloro _3_ nitropyridine as raw material substitution, ammoniated, high-pressure hydrogenation, acylation, a process salt, refined and so on. The reaction formula is as follows:

Route 2 (US5959115A) to 2_ amino _3_ nitro _6_ methoxypyrido as the starting material, and on fluorobenzylamine substitution reaction to produce 2-amino-3-nitro–6 – fluorobenzyl amine of pyridine, the yield was 95.2%, and the high-pressure hydrogenation, the catalyst was filtered off, and then the occurrence of an acylation reaction with ethyl chloroformate to give the hydrochloride salt of flupirtine, three-step total yield of 53.3%. The reaction formula is as follows:

Route 1 starting material is different, but relatively speaking, the route I easily controlled reaction conditions, and 2-amino-3-nitro-6-chloro-pyridine is a common chemical raw materials, easy to buy on the market, This can shorten the reaction route. Route 2 two-step reaction process route is short, but the starting 2-amino-3-nitro-6-methoxy-approved Li expensive, hydrogenation, acidification two steps yield only 56.0%.
Chinese Patent Application Publication No. CN102241626A are disclosed and CN102260209A flupirtine maleate preparation method, but the application of these two methods for the preparation of a laboratory scale, for the industrial mass production were not optimized.
The method for purifying of flupirtine maleate in the final product are as follows:
650C ± 5 ° C under the flupirtine maleate crude and ethanol mass ratio of 1: 30-40 mixed, crude completely dissolved, then add 680g of activated carbon and stirred for 15–30 minutes, and hot filtration, the filtrate, stirring down to room temperature, and then cooled to 0 ° C crystallization 5–10 hours, filtered and the filter cake to take the filter cake can be dried.
BELOW AS FREE BASE
| Ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate | |
| CAS No.: | 56995-20-1 |
|---|---|
| Synonyms: |
|
| Formula: | C15H17FN4O2 |
| Exact Mass: | 304.13400 |
1H NMR INTERPRETATIONS/PREDICTIONS
![ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate NMR spectra analysis, Chemical CAS NO. 56995-20-1 NMR spectral analysis, ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-18/000/006/679/56995-20-1-1h.png)
13C NMR INTERPRETATIONS/PREDICTIONS
![ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate NMR spectra analysis, Chemical CAS NO. 56995-20-1 NMR spectral analysis, ethyl N-[2-amino-6-[(4-fluorophenyl)methylamino]pyridin-3-yl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-18/000/006/679/56995-20-1-13c.png)
…….
PAPER
Helvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
 AND
AND  GIVES PRODUCT
GIVES PRODUCT
 ALSO AN INTHelvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
ALSO AN INTHelvetica Chimica Acta, , vol. 77, # 8 p. 2175 – 2190
…………..
HPLC
Instrumentation An HPLC system (Agilent HPLC Model-1200) equipped with a C18 (Agilent BDS, 250 mm x 4.6 mm, 5µ) column, binary pump, rheodyne loop injector with 20 μL, and a photodiode array detector was used. The software used for HPLC data acquisition was EZChrome Elite. A flash chromatograph equipped with silica gel as the column material, and VWD-UV detection (using the software Analogix IF 280 V 5.10) was used for the isolation and purification of degradation products. 1 H-NMR was recorded on the Varian Unity Inova at 400 MHz (using TMS as internal standard and DMSO-d6 as solvent), 13C-NMR (Mercury Plus at (abundance 100 MHz), using DMSO-d6 as solvent), and mass spectral studies were performed on the API 3000 ABPCIES instrument.
Method Development and Optimization of the Chromatographic Conditions In preliminary experiments, the drug was subjected to the reversed-phase mode using a C18 column (Agilent, 250 x 4.6 mm, 5µ) and mobile phases consisting of water (pH 3.0 adjusted with orthophosphoric acid) and methanol by varying the % aqueous phase from 10% to 30%. The drug was retained on the column, but the peak shape was not good. It was noted that increasing the % aqueous phase in the mobile phase composition increases the retention time of flupiritine maleate. Based on the suitable retention time for SIAM, the 20% aqueous phase was optimized. To reduce the tailing effect, 0.2% triethylamine (TEA) was added and the pH was adjusted to 3.0 with orthophosphoric acid and the corresponding retention of FLU was 10.3 ± 0.3 min. Finally, the mobile phase of 0.2% v/v TEA (pH-adjusted to 3.0 with OPA) and methanol in the ratio of 20:80% v/v was optimized. The flow rate was 1.0 mLmin−1. The injection volume was 20 µL and the PDA detection wavelength was at 254 nm. The chromatogram obtained in the optimized condition is shown in Fig. 2. It was observed that eight degradation products were formed with retention times 3.9 ± 0.2 min (D1), 4.8 ± 0.2 min (D2), 6.4 ± 0.1 min (D3), 6.8 ± 0.2 min (D4), 8.2 ± 0.2 min(D5), 12.0 ± 0.2 min (D6), 14.1 ± 0.1 min (D7), and 15.0 ± 0.1 min (D8), respectively. The chromatographic resolution among all of the peaks was more than 2. The % degradation was about 5–30% depending on stress conditions.
………………..
paper
J Pharm Biomed Anal. 2014 Mar;90:27-34. doi: 10.1016/j.jpba.2013.11.015. Epub 2013 Nov 27.
Flupirtine maleate is a centrally acting, non-opioid, nonsteroidal antiinflammatory analgesic. During the manufacturing of flupirtine maleate, two unknown impurities present in the laboratory batches in the range of 0.05-1.0% along with the known impurities in HPLC analysis. These unknown impurities were obtained from the enriched mother liquor by column chromatography. Based on the complete spectral analysis (MS, (1)H, (13)C, 2D NMR and IR) and knowledge of the synthetic scheme of flupirtine maleate, these two new impurities were designated as diethyl 5-((4-fluorobenzyl)amino)-2-oxo-1H-imidazo[4,5-b]pyridine-1,3(2H)-dicarboxylate (impurity-I) and diethyl(6-((4-fluorobenzyl)amino)pyridine-2,3-diyl)dicarbamate (impurity-II). Impurity isolation, identification, structure elucidation and the formation of impurities were also discussed. Preparation and structure elucidation of impurity-III were also first reported in this paper.
…………………
journal of pharmaceutical and biomedical analysis, 90, 2014, 27-34
References: Substituted pyridine with central analgesic properties. Prepn: K. Thiele, W. von Bebenburg, ZA 6902364(1970 to Degussa); W. von Bebenburg et al., Chem. Ztg. 103, 387 (1979); eidem, ibid. 105, 217 (1981).
Prepn of maleate: W. von Bebenburg, S. Pauluhn, BE 890331; eidem, US 4481205 (1980, 1984 both to Degussa).
Comparison of pharmacology with other analgesics: V. Jakovlev et al., Arzneim.-Forsch. 35, 30 (1985).
Pharmacokinetic studies: K. Obermeier et al., ibid. 60.
Effect on driving ability: B. Biehl, ibid. 77.
Clinical trials in treatment of cancer pain: W. Scheef, D. Wolf-Gruber, ibid. 75.
Efficacy in treatment of pain after hysterectomy: R. A. Moore et al., Br. J. Anaesth. 55, 429 (1983).
Symposium on pharmacology and clinical efficacy: Postgrad. Med. J. 63, Suppl. 3, 1-113 (1987).
References
1
- Abrams, SML; L.R.I. Baker, P. Crome, A.S.T. White, A. Johnston, S.I. Ankier, S.J. Warrington, P. Turner, G. Niebch (1988). “Pharmacokinetics of flupirtine in elderly volunteers and in patients with moderate renal impairment”. The Fellowship of Postgraduate Medicine 64 (751): 361–363. doi:10.1136/pgmj.64.751.361. PMC 2428663. PMID 3200777.
- 2
- Narang, PK; Tourville JF; Chatterji DC; Gallelli JF (1984). “Quantitation of flupirtine and its active acetylated metabolite by reversed-phase high-performance liquid chromatography using fluorometric detection”. Journal of Chromatography 305 (1): 135–143. doi:10.1016/S0378-4347(00)83321-6. PMID 6707137.
- 3
- Methling, K; Reszka P; Lalk M; Vrana O; Scheuch E; Siegmund W; Terhaag B; Bednarski PJ (2008). “Investigation of the in Vitro Metabolism of the Analgesic Flupirtine”. Drug Metabolism and Disposition 37: 479–493. doi:10.1124/dmd.108.024364.
- 4
- Blackburn-Munro, G; W. Dalby-Brown; N. R. Mirza; J. D. Mikkelsen; R. E. Blackburn-Munro (2005). “Retigabine: Chemical Synthesis to Clinical Application”. CNS Drug Reviews 11 (1): 1–20. doi:10.1111/j.1527-3458.2005.tb00033.x. PMID 15867950.
- 5
- Flupirtine Drugs.com. Accessed 20 September 2011.
- 6
- “POTIGA® (ezogabine) Tablets, CV. Full Prescribing Information”. GlaxoSmithKline and Valeant Pharmaceuticals. Revised: September, 2013. Initial U.S. Approval: 2011. Retrieved 2 June 2014. Check date values in:
|date=(help) - 7
- http://www.freepatentsonline.com/5721258.html Primary and secondary neuroprotective effect of flupirtine in neurodegenerative diseases The synthesis of flupirtine and its pharmaceutically acceptable salts is described in EP 160 865 and 199 951. EP0199951 December, 1986 Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of 2-amino-3-carbethoxyamino-6-(4-fluorobenzylamino) pyridine.
- 8
- http://patent.ipexl.com/EP/EP0199951.html#reference Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of EPO Patent EP0199951 1986 German.
- 9
- http://www.patentfish.com/2-amino-3-carbethoxyamino-6-4-fluoro-benzylamino Process for the preparation of 2-amino-3-nitro-6-(4-fluorobenzylamino) pyridine and of 2-amino-3-carbethoxyamino-6-(4-fluorobenzylamino) pyridine EP 0199951 B1 1986. English.
- 10
- http://patent.ipexl.com/US/4481205.html 2-Amino-3-carbethoxyamino-6-(p-fluoro-benzylamino)-pyridine-maleate United States Patent 4481205. 1981
- 11
- http://www.freepatentsonline.com/3998834.html Novel N-(4-piperidinyl)-N-phenylamides and -carbamates having very potent analgesic activity, methods of preparing same and useful intermediates therefor. Patent 3998834. 1976.
- 12
- http://www.faqs.org/patents/app/20090306150 CARBOXYLIC ACID SALTS OF 2-AMINO-3-CARBETHOXYAMINO-6-(4-FLUORO-BENZYLAMINO)-PYRIDINE patent 20090306150. 2009. Flupirtine is commonly used in the form of pharmaceutically acceptable acid addition salts. Commercially, flupirtine is available as its maleate addition salt under the trademark Katadolon®. There are two known polymorphs of flupirtine maleate, designated in the art as flupirtine maleate A and B. European patentEP 0 977 736 discloses pure flupirtine maleate crystalline form A and a process for its preparation. Flupirtine and mixtures of flupirtine maleate polymorphs A and B can be synthesised according to EP 0 199 951.
- 13
- “Adeona Pharmaceuticals and National Neurovision Research Institute (NNRI) Collaborate to Test Flupirtine for Retinitis Pigmentosa”. Ann Arbor, MI and Owings Mills, MD: Synthetic Biologics, Inc. December 2, 2008. Retrieved 2 June 2014.
- 14
- Kornhuber, J.; Bleich, S.; Wiltfang, J.; Maler, M.; Parsons, C. G. (1999). “Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication”. Journal of neural transmission (Vienna, Austria : 1996) 106 (9–10): 857–867. doi:10.1007/s007020050206. PMID 10599868.
- 15
- McMahon, FG; Arndt WF Jr, Newton JJ, Montgomery PA, Perhach JL. (1987). “Clinical experience with flupirtine in the U.S”. Postgraduate Medical Journal 63 (3): 81–85. PMID 3328854.
- 16
- Klawe, C; Maschke, M (2009). “Flupirtine: pharmacology and clinical applications of a nonopioid analgesic and potentially neuroprotective compound”. Expert opinion on pharmacotherapy 10 (9): 1495–500. doi:10.1517/14656560902988528. PMID 19505216.
- 17
- Swedberg MD, Shannon HE, Nickel B, Goldberg SR (September 1988). “Pharmacological mechanisms of action of flupirtine: a novel, centrally acting, nonopioid analgesic evaluated by its discriminative effects in the rat”. J. Pharmacol. Exp. Ther. 246 (3): 1067–74. PMID 2901483.
- 18
- Dhar S, Bitting RL, Rylova SN et al. (April 2002). “Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons”. Ann. Neurol. 51 (4): 448–66. doi:10.1002/ana.10143. PMID 11921051.
- 19
- Flupirtine as Oral Treatment in Multiple Sclerosis (FLORIMS) Clinical Trials.gov Accessed 20 September 2011.
- 20
- Pipex Pharmaceuticals (PPXP)’ Oral Flupirtine Receives IND With FDA for Phase II Clinical Trial for Fibromyalgia 4/21/2008
- 21
- “Partnered Program. Effirma™ for Fibromyalgia”. Synthetic Biologics, Inc. Retrieved 2 June 2014.
- 22
- Stoll AL, Belmont MA. (2000) “Fibromyalgia Symptoms Relieved by Flupirtine: An Open-Label Case Series” Psychosomatics 41:371-372. Accessed 20 September 2011.
- 23
- EMA information about flupirtine
- 24
- article in Deutsches Ärzteblatt
- 25
- Singal, Rikki; Parveen Gupta; Nidhi Jain; Samita Gupta (2012). “Role of Flupirtine in the Treatment of Pain – Chemistry and its Effects”. Mædica — a Journal of Clinical Medicine 7 (2): 163–166. PMID 23401726.
- 26
- “DRUGDEX® Evaluations – Flupirtine”. Retrieved 24 March 2013.
- 27
- Preston, KL; Funderburk, FR; Liebson, IA; Bigelow, GE (Mar 1991). “Evaluation of the Abuse Potential of the Novel Analgesic Flupirtine Maleate”. Drug and Alcohol Dependence 27 (2): 101–113. doi:10.1016/0376-8716(91)90027-v. PMID 2055157.
- 28
- Sofia, RD; Diamantis, W; Gordon, R (1987). “Abuse Potential and Physical Dependence Liability Studies with Flupirtine Maleate in Laboratory Animals”. Postgraduate Medical Journal. 63 Suppl 3: 35–40. PMID 3447127.
- 29
- Gahr, M; Freudenmann, RW; Connemann, BJ; Hiemke, C; Schönfeldt–Lecuona, C (Dec 2013). “Abuse Liability of Flupirtine Revisited: Implications of Spontaneous Reports of Adverse Drug Reactions”. Journal of Clinical Pharmacology 53 (12): 1328–1333. doi:10.1002/jcph.164. PMID 24037995.
- 30
Stoessel, C; Heberlein, A; Hillemacher, T; Bleich, S; Kornhuber, J (Aug 16, 2010). “Positive Reinforcing Effects of Flupirtine—Two Case Reports”. Progress in Neuro-psychopharmacology & Biological Psychiatry 34 (6): 1120–1121. doi:10.1016/j.pnpbp.2010.03.031. PMID 20362025. Retrieved 2 June 2014.
References
2. Flupirtine
Note: Compound name must be entered under “Substance Identification” and then “Names and Synonyms” selected to view synonyms.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| ethyl {2-amino-6-[(4-fluorobenzyl)amino]pyridin-3-yl}carbamate | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Pharmacokinetic data | |
| Bioavailability | 90% (oral), 70% (rectal)[1] |
| Metabolism | Hepatic to 2-amino-3-acetylamino-6-(para-fluorobenzylamino) pyridine (which has 20-30% the analgesic potential of its parent compound), para-fluorohippuric acid[2] and a mercapturic acid metabolite, presumably formed from a glutathione adduct[3] |
| Half-life | 6.5 hrs (average), 11.2-16.8 hrs (average 14 hrs) (elderly), 8.7-10.9 hrs (average 9.8 hrs) (in those with moderate-level renal impairment)[1] |
| Excretion | 72% of flupirtine and its metabolites appear in urine and 18% appear in feces[4] |
| Identifiers | |
| 56995-20-1 |
|
| N02BG07 | |
| PubChem | CID 53276 |
| IUPHAR ligand | 2598 |
| ChemSpider | 48119 |
| UNII | MOH3ET196H |
| KEGG | D07978 |
| ChEMBL | CHEMBL255044 |
| Chemical data | |
| Formula | C15H17FN4O2 |
| 304.32 g/mol | |
