




| Patent | Submitted | Granted |
|---|---|---|
| NORMALIZATION OF CULTURE OF CORNEAL ENDOTHELIAL CELLS [US2015044178] | 2012-12-27 | 2015-02-12 |
Home » PHASE1 (Page 9)
Category Archives: PHASE1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SEMAPIMOD
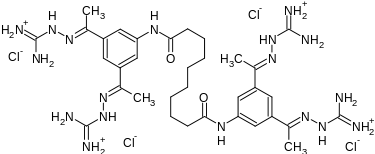

Semapimod Mesylate
CPSI-2364, AXD-455, CN-1493, CNI 1493
CAS No. 352513-83-8(Semapimod base)
Cas 164301-51-3 4x HCl
CAS 872830-80-3 (Semapimod mesylate)
MW 1129
CROHNS DISEASE, PHASE 1
N,N’-bis[3,5-bis[(E)-N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl]decanediamide
Decanediamide, N,N’-bis[3,5-bis[1-[(aminoiminomethyl)hydrazono]ethyl]phenyl]-, methanesulfonate
N,N’-Bis(3,5-bis(1-(carbamimidoylhydrazono)ethyl)phenyl)decanediamide
- N,N’-Bis(3-acetylphenyl)decane diamide tetrakis (amidinohydrazone)
- N,N’-bis(3,5-bis{(1E)-N-[amino(imino)methyl]ethanehydrazonoyl}phenyl)decanediamide
- N,N’-bis[3,5-bis[(E)-N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl]decanediamide
- N,N’-bis[3,5-bis[(E)-N-guanidino-C-methyl-carbonimidoyl]phenyl]decanediamide
A nitric oxide synthesis inhibitor and a p38 MAPK inhibitor potentially for the treatment of Crohn’s disease.

Crohn’s disease (CD) is a chronic inflammatory disease involving the upper and lower gastrointestinal tract and characterized by abdominal pain, weight loss, gastrointestinal bleeding and formation of fistulas between loops of bowel and from the bowel to the skin or other organs. Current therapy for active Crohn’s disease consists of symptomatic treatment, nutritional therapy, salicylates and immunosuppressants or surgical management.
Tumor necrosis factor a (TNF-a) plays a central role in the initiation and amplification of the granulomatous inflammatory reaction seen in CD (van Deventer, 1997). Increased TNF-a is present in gut mucosa as well as in stool of patients with active CD (Braegger et al, 1992). CNI-1493 is a synthetic guanylhydrazone compound that is an inhibitor of TNF-a synthesis. A monoclonal antibody to TNF, infliximab, is now approved for treatment of CD, but not all patients respond and many who do respond eventually become refractory to this treatment as well.
CNI-1493 is a synthetic compound which blocks the production of several inflammatory cytokines, including TNF. Because it blocks production of multiple inflammatory mediators, it may be more active than products targeted to a specific cytokine. In addition, as it is not a biologic, it should not cause hypersensitivity reactions or induce formation of antibodies.
The purpose of this trial is to determine if CNI-1493 is safe and effective in treating patients with moderate to severe Crohn’s Disease in a placebo controlled setting………https://clinicaltrials.gov/ct2/show/NCT00038766
Semapimod (INN), formerly known as CNI-1493, is an investigational new drug which has anti-inflammatory,[1] anti-cytokine,[2] immunomodulatory,[3] antiviral[4] and antimalarial[5] properties.
History
Semapimod was developed at the former Picower Institute for Medical Research, and is now licensed to Cytokine PharmaSciences. In 2000, Cytokine PharmaSciences licensed anti-infective applications of semapimod to Axxima Pharmaceuticals, but Axxima became insolvent in Dec. 2004 and its assets were acquired by GPC Biotech, which has recently merged into Agennix AG[1]. Although the disposition of Axxima’s partial rights to semapimod was not specified in these merger announcements, Cytokine PharmaSciences does not currently list any licensees for semapimod on its website.
Mechanism of action
Semapimod was first developed to inhibit nitric oxide synthesis by inflammatory macrophages, via inhibition of the uptake of arginine which macrophages require for nitric oxide synthesis.[1] Subsequently it was found that suppression of nitric oxide synthesis occurred even at semapimod concentrations 10-fold less than required for inhibition of arginine uptake, suggesting that this molecule was a more general inhibitor of inflammatory responses.[2] Further work revealed that semapimod suppressed the translation efficiency of tumor necrosis factor production.[6] Specifically, semapimod was found to be an inhibitor of p38 MAP kinase activation.[7] Surprisingly, however, the primary mode of action in vivo is now thought to be via stimulation of the vagus nerve, thereby down-regulating inflammatory pathways via the recently discovered cholinergic anti-inflammatory pathway.[8][9]
Pharmacology and clinical trials
In a preclinical study in rats, semapimod was found to suppress cytokine-storm induction by the anticancer cytokine interleukin-2 (IL-2) without decreasing its anticancer properties, allow larger doses of IL-2 to be administered.[10] A subsequent phase I trial in humans failed to show an increase in the tolerated dose of IL-2, although indications of pharmacological activity as an inhibitor of tumor necrosis factor production were observed.[11]
In a preliminary clinical trial of semapimod in patients with moderate to severe Crohn’s disease, positive clinical changes were observed, including endoscopic improvement, positive responses in some patients not responding to infliximab, healing of fistulae, and indications for tapering of steroids; no significant adverse effects were observed.[12]
In a small clinical trial against post-ERCP pancreatitis, significant suppression was not observed, although investigators observed a significant reduction of the incidence of hyperamylasemia and the levels of post-ERCP amylase.[13]
In the clinical trials above, semapimod tetrahydrochloride was administered by intravenous injection. This route has drawbacks such as dose-limiting phlebitis.[2] Recently Cytokine PharmaSciences has announced the development of novel salt forms of semapimod which are said to be orally absorbable; a phase I clinical trial of one of these salt forms, CPSI-2364, has been completed, and a phase II trial is planned for 2010.[3][4]
Chemistry
Semapimod is synthesized by reacting 3,5-diacetylaniline[14] with sebacoyl chloride in the presence of pyridine, followed by reaction of the resulting tetraketone with aminoguanidine hydrochloride.[1]
PATENT
-
N,N′-bis(3,5-diacetylphenyl) decanediamide tetrakis (amidinohydrazone) tetrahydrochloride (CNI-1493), which has the following structural formula:
-

SYNTHESIS
The reaction of decanedioyl dichloride (I) with 3,5-diacetylaniline (II) by means of pyridine in dichloromethane gives the corresponding diamide (III), which is condensed with aminoguanidine (IV) in refluxing aqueous ethanol to afford the target tetrakis amidinohydrazone. EP 0746312; EP 1160240; US 5599984; WO 9519767
http://www.google.com/patents/EP0746312A1?cl=en
References
- 1 Bianchi, M.; Ulrich, P.; Bloom, O.; Meistrell m, M. , I. I.; Zimmerman, G. A.; Schmidtmayerova, H.; Bukrinsky, M.; Donnelley, T.; Bucala, R.; Sherry, B.; Manogue, K. R.; Tortolani, A. J.; Cerami, A.; Tracey, K. J. (Mar 1995). “An inhibitor of macrophage arginine transport and nitric oxide production (CNI-1493) prevents acute inflammation and endotoxin lethality”. Molecular Medicine (Cambridge, Mass.) 1 (3): 254–266. ISSN 1076-1551. PMC 2229913. PMID 8529104.
- 2
- Bianchi, M.; Bloom, O.; Raabe, T.; Cohen, P. S.; Chesney, J.; Sherry, B.; Schmidtmayerova, H.; Calandra, T.; Zhang, X.; Bukrinsky, M.; Ulrich, P.; Cerami, A.; Tracey, K. J. (Mar 1996). “Suppression of proinflammatory cytokines in monocytes by a tetravalent guanylhydrazone”. The Journal of Experimental Medicine 183 (3): 927–936. doi:10.1084/jem.183.3.927. ISSN 0022-1007. PMC 2192362. PMID 8642296.
- 3
- Martiney, J.; Rajan, A. J.; Charles, P. C.; Cerami, A.; Ulrich, P. C.; MacPhail, S.; Tracey, K. J.; Brosnan, C. F. (Jun 1998). “Prevention and treatment of experimental autoimmune encephalomyelitis by CNI-1493, a macrophage-deactivating agent” (Free full text). Journal of immunology (Baltimore, Md. : 1950) 160 (11): 5588–5595. ISSN 0022-1767. PMID 9605164.
- 4
- Hauber, I.; Bevec, D.; Heukeshoven, J.; Krätzer, F.; Horn, F.; Choidas, A.; Harrer, T.; Hauber, J. (Jan 2005). “Identification of cellular deoxyhypusine synthase as a novel target for antiretroviral therapy”. The Journal of Clinical Investigation (Free full text) 115 (1): 76–85. doi:10.1172/JCI21949. ISSN 0021-9738. PMC 539192. PMID 15630446.
- 5
- Specht, S.; Sarite, R.; Hauber, I.; Hauber, J.; Görbig, F.; Meier, C.; Bevec, D.; Hoerauf, A.; Kaiser, A. (May 2008). “The guanylhydrazone CNI-1493: an inhibitor with dual activity against malaria-inhibition of host cell pro-inflammatory cytokine release and parasitic deoxyhypusine synthase”. Parasitology research 102 (6): 1177–1184. doi:10.1007/s00436-008-0891-x. ISSN 0932-0113. PMID 18256853.
- 6
- Cohen, P. S.; Nakshatri, H.; Dennis, J.; Caragine, T.; Bianchi, M.; Cerami, A.; Tracey, K. J. (Apr 1996). “CNI-1493 inhibits monocyte/macrophage tumor necrosis factor by suppression of translation efficiency”. Proceedings of the National Academy of Sciences of the United States of America 93 (9): 3967–3971. Bibcode:1996PNAS…93.3967C. doi:10.1073/pnas.93.9.3967. ISSN 0027-8424. PMC 39469. PMID 8632999.
- 7
- Cohen, P. S.; Schmidtmayerova, H.; Dennis, J.; Dubrovsky, L.; Sherry, B.; Wang, H.; Bukrinsky, M.; Tracey, K. J. (May 1997). “The critical role of p38 MAP kinase in T cell HIV-1 replication”. Molecular Medicine (Cambridge, Mass.) 3 (5): 339–346. ISSN 1076-1551. PMC 2230081. PMID 9205949.
- 8
- Tracey, J. (Feb 2007). “Physiology and immunology of the cholinergic antiinflammatory pathway”. The Journal of Clinical Investigation (Free full text) 117 (2): 289–296. doi:10.1172/JCI30555. ISSN 0021-9738. PMC 1783813. PMID 17273548.
- 9
- Oke, L.; Tracey, J. (Mar 2008). “From CNI-1493 to the immunological homunculus: physiology of the inflammatory reflex” (Free full text). Journal of Leukocyte Biology 83 (3): 512–517. doi:10.1189/jlb.0607363. ISSN 0741-5400. PMID 18065685.
- 10
- Kemeny, M. M.; Botchkina, G. I.; Ochani, M.; Bianchi, M.; Urmacher, C.; Tracey, K. J. (1998). “The tetravalent guanylhydrazone CNI-1493 blocks the toxic effects of interleukin-2 without diminishing antitumor efficacy”. Proceedings of the National Academy of Sciences of the United States of America 95 (8): 4561–4566. Bibcode:1998PNAS…95.4561K. doi:10.1073/pnas.95.8.4561. PMC 22529. PMID 9539777.
- 11
- Atkins, M. B.; Redman, B.; Mier, J.; Gollob, J.; Weber, J.; Sosman, J.; MacPherson, B. L.; Plasse, T. (2001). “A phase I study of CNI-1493, an inhibitor of cytokine release, in combination with high-dose interleukin-2 in patients with renal cancer and melanoma”. Clinical Cancer Research 7 (3): 486–492. PMID 11297238.
- 12
- Hommes, D.; Van Den Blink, B.; Plasse, T.; Bartelsman, J.; Xu, C.; MacPherson, B.; Tytgat, G.; Peppelenbosch, M.; Van Deventer, S. (2002). “Inhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn’s disease”. Gastroenterology 122 (1): 7–14. doi:10.1053/gast.2002.30770. PMID 11781274.
- 13 Vanwesterloo, D.; Rauws, E.; Hommes, D.; De Vos, A.; Van Der Poll, T.; Powers, B.; Fockens, P.; Dijkgraaf, M.; Bruno, M. (2008). “Pre-ERCP infusion of semapimod, a mitogen-activated protein kinases inhibitor, lowers post-ERCP hyperamylasemia but not pancreatitis incidence”. Gastrointestinal Endoscopy 68 (2): 246–254. doi:10.1016/j.gie.2008.01.034. PMID 18455169.
- 14 Ulrich, P.; Cerami, A. (Jan 1984). “Trypanocidal 1,3-arylene diketone bis(guanylhydrazone)s. Structure-activity relationships among substituted and heterocyclic analogues”. Journal of Medical Chemistry 27 (1): 35–40. doi:10.1021/jm00367a007. ISSN 0022-2623. PMID 6690682.
| Patent | Submitted | Granted |
|---|---|---|
| Neural tourniquet [US2005282906] | 2005-12-22 | |
| Guanylhydrazone Salts, Compositions, Processes of Making, and Methods of Using [US2008262090] | 2008-10-23 | |
| Protective role of semapimod in necrotizing enterocolitis [US7795314] | 2007-12-06 | 2010-09-14 |
| METHOD OF TREATING ILEUS BY PHARMACOLOGICAL ACTIVATION OF CHOLINERGIC RECEPTORS [US2011112128] | 2011-05-12 | |
| Method of treating ileus by pharmacological activation of cholinergic receptors [US2007213350] | 2007-09-13 | |
| Pharmaceutically active aromatic guanylhydrazones [US2005171176] | 2005-08-04 | |
| Guanylhydrazone salts, compositions, processes of making and methods of using [US7244765] | 2006-01-19 | 2007-07-17 |
| GUANYLHYDRAZONE SALTS, COMPOSITIONS, PROCESSES OF MAKING, AND METHODS OF USING [US8034840] | 2008-06-19 | 2011-10-11 |
| METHOD FOR TREATING GLIOBLASTOMAS AND OTHER TUMORS [US2014323576] | 2014-03-14 | 2014-10-30 |
| Methods of treatment of fatty liver disease by pharmacological activation of cholinergic pathways [US8865641] | 2012-06-14 | 2014-10-21 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N,N’-bis[3,5-bis[N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl] decanediamide tetrahydrochloride
|
|
| Identifiers | |
| CAS Number | 164301-51-3 352513-83-8 (base) |
| ATC code | None |
| PubChem | CID: 5745214 |
| UNII | 9SGW2H1K8P |
| ChEMBL | CHEMBL2107779 |
| Chemical data | |
| Formula | C34H56Cl4N18O2 |
| Molecular mass | 890.73984 g/mol |
see………http://worlddrugtracker.blogspot.in/2015/12/semapimod.html
/////////Semapimod Mesylate, CPSI-2364, AXD-455, CN-149, PHASE 1, FERRING, CNI 1493
CC(=NN=C(N)N)C1=CC(=CC(=C1)NC(=O)CCCCCCCCC(=O)NC2=CC(=CC(=C2)C(=NN=C(N)N)C)C(=NN=C(N)N)C)C(=NN=C(N)N)C
RQ 00000010 for the treatment of GERD, functional dyspepsia and chronic constipation.


RQ 00000010
CAS 907607-22-1
| Molecular Formula: | C22H27F3N2O6 |
|---|---|
| Molecular Weight: | 472.45479 g/mol |
HSMMHNBGQLGCBY-UHFFFAOYSA-N;
RaQualia Pharma Inc
PFIZER INNOVATOR
RQ-00000010; RQ-10
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid
ΦirΦfff^fΣ^^-TrifluoroethoxyVi.a-benzisoxazol-S-vnoxylmethvπpiperidin-i-vπmethylltetrahydro-2H-pyran-4-carboxylic acid
4-[[4-[[4-(2,2,2-trifluoroethoxy)-1,2-benzoxazol-3-yl]oxymethyl]piperidin-1-yl]methyl]oxane-4-carboxylic acid
PHASE 1 for the treatment of GERD, functional dyspepsia and chronic constipation.
Useful for treating diseases mediated by 5-HT4 receptor activity eg such as gastroesophageal reflux disease (GERD), gastric motility disorder, dyspepsia, constipation, esophagitis, diabetes, CNS and cardiovascular diseases.
RaQualia, following its spin-out from Pfizer, is developing RQ-00000010, a 5-HT4 receptor partial agonist, for the treatment of gastric motility disorders, including gastroparesis associated with Parkinson’s disease.
In November 2015, the drug was reported to be in phase 1 clinical development. RaQualia and licensee CJ CheilJedang are investigating the drug for the treatment of GERD, functional dyspepsia and chronic constipation.
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid is disclosed in PL1 as a 5-HT4 receptor agonist, which is useful in the treatment or alleviation of disease conditions mediated by 5-HT4 receptor activity; in particular 5-HT4 receptor agonistic activity, such as gastroesophageal reflux disease (GERD), gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia (FD), irritable bowel syndrome (IBS), constipation, dyspepsia, esophagitis, gastroesophageal disease, gastritis, nausea, central nervous system disease, Alzheimer’s disease, cognitive disorder, emesis, migraine, neurological disease, pain, cardiovascular disorders, cardiac failure, heart arrhythmia, diabetes, and apnea syndrome (See NPL 1 to 13 and PL 2 to 7).
Simply an white solid has been produced in the previously known methods of preparing 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid, described in PL 1. A generic disclosure of pharmaceutically-acceptable salts of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid of the instant application is disclosed, and the free base of the compound of the instant invention is disclosed and claimed, in PL 1 having an international filing date of December 6, 2006, assigned to the assignee hereof. Thus any salts of the compound have been neither pacifically described nor synthesized in prior art.
It has been found that HCl-salt, HBr-salt, pTSA-salt and EDSA-salt of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid shown below, can be isolated as a crystalline form which has advantageous properties such as ease of making a formulation, high solubility, and good stability. In addition the salts of the present invention are more easily purified than a non-crystalline form disclosed in PL 1 (WO2006/090224) and crystalline form disclosed in PL 3 (WO2012/157288).
Patent Literature
{PL 1} WO2006/090224.
{PL 2} US Patent No. 6,106,864.
{PL 3} WO2012/157288
{PL 4} WO00/35298.
{PL 5} WO91/11172.
{PL 6} WO94/02518.
{PL 7} WO98/55148.
Non Patent Literature
{NPL 1} Bockaert J. et al., TiPs 13; 141-145, 1992.
{NPL 2} Ford A. P et al., Med. Res. Rev. 13: 633-662, 1993.
{NPL 3} Gullikson G. W. et al., Drug Dev. Res. 26; 405-417, 1992.
{NPL 4} Richard M. Eglen et al., TiPs 16; 391-398, 1995.
{NPL 5} Bockaert J. et al., CNS Drugs 1; 6-15, 1994.
{NPL 6} Romanelli M. N. et al., Arzheim Forsch./Drug Res., 43; 913-918, 1993.
{NPL 7} Kaumann A. J. et al., Naunyn-Schmiedebergs Arch Pharmacol., 344; 150-159, 1991.
{NPL 8} Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
{NPL 9} Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
{NPL 10} Tablets, Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
{NPL 11} Pharmaceutical Technology On-line, 25(2), 1-14, by Verma et al. (2001).
{NPL 12} J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
{NPL 13} Evrard, B., et al., Journal of Controlled Release 96 (3), pp. 403-410, 2004.
{NPL 14} Byrn S. R. et al., Solid-State Chemistry of Drugs 2nd ed., pp 3-43 and 461-503, 1999, SSCI, Inc.

PATENT
WO2006090224
| PFIZER JAPAN INC. |
EXAMPLE 1 :
ΦirΦfff^fΣ^^-TrifluoroethoxyVi.a-benzisoxazol-S-vnoxylmethvπpiperidin-i-vπmethylltetrahydro-2H-pyran-4-carboxylic acid
Step 1. Methyl 2-hvdroxy-6-(2,2,2-trifluoroethoxy)benzoate
A mixture of 5-hydroxy-2,2-dimethyl-4tø-1 ,3-benzodioxin-4-one (123 g, 633 mmol, Synth. Commun.
1994, 24t 1025), potassium carbonate (262 g, 1.9 mol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (95.8 mL, 665 mmol) in Λ/,Λ/-dimethylformamide (600 mL) was stirred at 50 0C for 30 min. Then methanol (300 ml_) was added to the mixture, and stirring was continued for 5 h at that temperature. After cooling to room temperature, the mixture was diluted with water (500 ml_) and neutralized with 2Λ/ hydrochloric acid. Product was extracted with a mixture of ethyl acetate-hexane (5:1 , 500 mL x 3). Combined organic layers were washed with water (500 mL), dried over magnesium sulfate and concentrated under reduced pressure. The residual solid was recrystallized from methanol-water to afford 125 g (79%) of the desired product as colorless crystals.
1H-NMR (CDCI3) δ: 11.47 (1 H, s), 7.36 (1 H, t, J = 8.4 Hz), 6.72 (1 H, dd, J = 1.1 , 8.4 Hz), 6.38 (1 H, q, J = 8.1 Hz), 4.36 (2 H, q, J= 8.0 Hz), 3.96 (3 H, s).
MS (ESI) m/z: 251 (M+H) +, 249 (M-H) \
Step 2. 4-(2,2,2-Trifluoroethoxy)-1 ,2-benzisoxazol-3-ol
To a solution of hydroxylamine sulfate (120 g, 732 mmol) in water (360 mL) was added potassium carbonate (121 g, 875 mmol) at 0 0C. After 30 min of stirring, sodium sulfite (3.74 g, 29.7 mmol) and a methanolic solution of methyl 2-hydroxyl-6-(2,2,2-trifluoroethoxy)benzoate (36.4 g, 146 mmol, EXAMPLE 1 , step 1 , in 360 mL of methanol) were added to the mixture. Then the mixture was warmed to 50 °C and stirred for 30 h. After cooling to room temperature, reaction mixture was partially concentrated to approx. 2/3 volume and acidified with 2Λ/ hydrochloric acid. Product was extracted three times with ethyl acetate. Combined organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to afford the desired product as a crystalline solid. Crude product (36.3 g) was used for the next step without further purification.
The described above crude product (5.56 g, 22.14 mmol) was suspended in tetrahydrofuran (22.0 mL) and heated at 50 °C. 1 ,1 ‘-carbonyldiimidazole (7.54 g, 46.48 mmol) was added to the suspension at 50 °C. After addition, the mixture was stirred at 50 0C for 14 h, the mixture was cooled to room temperature. 2Λ/ hydrochloric acid was added to the mixture and extracted with ethyl acetate. The organic layer was extracted with 10% aq. potassium carbonate (100 mL x 5). The water layers were acidified with 2Λ/ hydrochloric acid and extracted with ethyl acetate (200 mL x 2). The extracts were combined and dried over sodium sulfate and concentrated in vacuo to give brown solid. The residual solid was recrystallized from ethyl acetate/hexane to give 3.21 g (61 %) of the title compound as colorless needles.
1H-NMR (CDCl3) δ: 7.53 (1 H1 1, J = 8.5 Hz), 7.14 (1 H, d, J= 8.5 Hz), 6.73 (1 H, d, J = 7.9 Hz), 4.63 (2 H, q, J= 8.0 Hz), 3.83 (1 H, br).
MS (ESI) m/z: 234 (M+H) +, 232 (M-H) “.
Step 3. rMethoxy(tetrahydro-4H-pyran-4-ylidene)methoxyKtrimethyl)silane
To a stirred solution of diisopropylamine (5.2 mL, 37 mmol) in tetrahydrofuran (15 mL) was added dropwise n-butyllithium (1.6 M in hexane, 21 mL, 34 mmol) at 0 0C and stirred for 20 min. A mixture of methyl tetrahydro-2W-pyran-4-carboxylate (4.5 g, 31 mmol) and trimethylsilyl chloride (4.3 mL, 34 mmol) was added to the mixture at -40 0C, then trimethylsilyl chloride (0.4 mL, 0.3 mmol) was added to the mixture. The mixture was stirred at room temperature for 2 h. The volatile components were removed by evaporation and the residual mixture was filtered through a pad of celite washing with hexane. The filtrate was evaporated to give 6.9 g (quant.) of the title compound as a clear yellow oil.
1H-NMR (CDCI3) δ: 3.64-3.59 (4 H, m), 3.52 (3 H, s), 2.24 (2 H, t, J = 5.6 Hz), 2.15 (2 H, t, J = 5.4 Hz), 0.22 (9 H, s).
Step 4. Methyl 4-{f4-(hvdroxymeth’vDpiperidin-1 -yllmethylltetrahvdro^rt-pyran^-carboxylate
To a stirred mixture of piperidin-4-ylmethanol (5.0 g, 43.4 mmol), f-butyldimethylsilylchloride (7.2 g, 47.8 mmol), and triethylamine (7.3 ml_, 52.1 mmol) in dichloromethane (50 mL) was added 4-dimethylaminopyridine (530 mg, 4.3 mmol) at 0 0C. After being stirred at 0 0C for 2 h, 50 mL of water was added to the mixture. The mixture was extracted with dichloromethane (50 mL x 3) and the extracts were combined, dried over sodium sulfate, and concentrated in vacuo to give 10.2 g of a crude oil. The residual oil was dissolved with 86 mL of ethanol, and potassium carbonate (7.2 g, 52.1 mmol) and paraformaldehyde (1.56 g, 52.1 mmol) were added to the solution. After being stirred at room temperature for 2 days, the mixture was filtered and the filtrate was concentrated in vacuo to give a yellow oil. The residual oil was dissolved with 45 mL of acetonitrile and magnesium chloride (414 mg, 4.3 mmol) was added to the solution. [methoxy(tetrahydro-4H-pyran-4-ylidene)methoxy](trimethyl)silane (11.3 g, 52.1 mmol, EXAMPLE 1 , step 3) was added to the mixture at 0 0C. After being stirred at 0 0C for 20 h, 100 mL of 2Λ/ hydrochloric acid was added to the mixture. The mixture was stirred for 30 min and washed with diethyl ether (100 mL x 2). The water layer was neutralized with aq. ammonia and extracted with ethyl acetate (100 mL x 2). The extracts were combined and dried over sodium sulfate and concentrated in vacuo to give a yellow oil. The residual oil was purified by silica gel column chromatography (dichloromethane/methanol/aq. ammonia 400: 10: 1 ) to give 6.8 g (41%) of the title compound as a colorless waxy solid.
1H-NMR (CDCI3) δ: 3.75-3.90 (2 H, m), 3.71 (3 H, s), 3.40-3.55 (4 H, m), 2.73 (2 H, m), 2.49 (2 H, m), 2.10-2.25 (2 H, m), 1.95-2.10 (2 H, m), 1.50-1.70 (4 H, m), 1.30-1.50 (2 H, m), 1.10-1.30 (2 H, m).
MS (ESI) m/z: 272 (M+H) +.
Step 5. Methyl 4-{r4-((r4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-vπoxy)methyl)piperidin-1 -yllmethyll-tetrahydro-2H-pyran-4-carboxylate
A mixture of 4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-ol (230 mg, 1 mmol, EXAMPLE 1 , step
2), methyl 4-{[4-(hydroxymethyl)piperidin-1 -yl]methyl}tetrahydro-2/-/-pyran-4-carboxylate (270 mg, 1 mmol, EXAMPLE 1 , step 4), and cyanomethyltributylphosphorane (400 mg, 1.5 mmol) in toluene (1.0 mL) was stirred at 100 0C for 16 h. After cooling, the mixture was concentrated in vacuo to give a dark brown oil. The residual oil was purified by silica gel column chromatography (hexane/ethyl acetate 2 : 1 ) to give 250 mg (51 %) of the title compound as a white solid.
1H-NMR (CDCl3) δ: 7.44 (1 H, dd, J= 7.9, 8.4 Hz), 7.12 (1 H, d, J= 8.4 Hz), 6.61 (1 H, d, J= 7.9 Hz), 4.49 (2 H, q, J= 8.1 Hz), 4.24 (2 H, d, J= 6.4 Hz), 3.88-3.78 (2 H, m), 3.72 (3 H, s), 3.54-3.41 (2 H, m), 2.83-2.71 (2 H, m), 2.52 (2 H, s), 2.35-1.29 (11 H, m).
MS (ESI) m/z: 487 (M+H) +.
Step 6. 4-(r4-(ir4-(2,2,2-Trifluoroethoxy)-1 ,2-benzisoxazol-3-vπoxy)methyl)piperidin-1 -ylimethylltetrahydro-2H-pyran-4-carboxylic acid
A mixture of methyl 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2/+pyran-4-carboxylate (89 mg, 0.18 mmol, EXAMPLE 1 , Step 5) in tetrahydrofuran (1 mL), methanol (1 ml_) and 2 Λ/ aq. sodium hydroxide (1 ml_) was stirred at 70 °C for 17 h. The mixture was neutralized with 2 N hydrochloric acid (1 mL) and formed precipitate was filtered.
The precipitate was triturated with diethylether to give 50 mg (58%) of the title compound as a white solid.
1H-NMR (DMSO-d6) δ: 7.59 (1 H1 dd, J= 8.1 , 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J= 8.7 Hz), 4.19 (2 H, d, J= 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H was not observed.
MS (ESI) m/z: 473 (M+H) +, 471 (M-H) “.
m.p.: 171.7 °C.
IR (KBr) v: 2950, 1617, 1527, 1188, 1113 cm”1.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.72; H, 5.78; N, 5.80.
PATENT
WO2015174098
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015174098
PATENT
WO2014080633
http://www.google.com/patents/WO2014080633A1?cl=en
PATENT
WO 2015178020
The present invention relates to novel salts of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid. More particularly, the invention relates to salt forms (HCl-salt, HBr-salt, p-toluenesulfonate salt and ethanedisulfonate salt), and to processes for the preparation of, compositions containing and to uses of, such salt forms.
EXAMPLE 1
Preparation of
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid according to the conventional process
A slurry of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]-methyl}tetrahydro-2H-pyran-4-carboxylic acid (1.326 kg, 2.807 mol, a white solid) in ethyl acetate (18.564 L) is dissolved at 70 oC. The solution is cooled to 64 oC during 35 min and 200 mg of seed crystal (0.423 mmol) is seeded to the mixture. The mixture is cooled to 40 oC over 5 h period and stirred at this temperature for 14.5 h. The slurry is gradually cooled to 19 oC during 6 h period and the mixture is stirred at this temperature for 46 h. The formed precipitate is collected by filtration and the filter cake is washed with 2.0 L of ethyl acetate. The filter cake is dried under reduced pressure at 50 oC to afford 1.140 kg of the desired crystalline form of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]-
methyl}tetrahydro-2H-pyran-4-carboxylic acid (86%).
1H-NMR (DMSO-d6) delta: 7.59 (1 H, dd, J = 8.1, 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J = 8.7 Hz), 4.19 (2 H, d, J = 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H is not observed.
m.p. (DSC onset): 169 oC.
The temperature has a margin of error of +/- 1 oC.
Crystallinity by PXRD: Crystal (Figure 1): Main peaks at 2-Theta: 5.9, 9.3, 9.8, 11.9, 13.7, 14.3, 15.0, 17.8, 18.2-19.3, 19.7, 22.6, 23.4-24.5 and 24.9 (o ). Each peak has a margin of error of +/- 0.2.
IR nu (diffuse reflection) (Figure 6): 4389-4383, 3426, 2943-2937, 2120, 1904, 1724, 1614, 1535, 1508, 1437, 1420, 1287, 1261, 1221, 1180, 1121, 1094, 1059, 1022, 991, 974, 957, 934, 918, 868, 827, 783, 746, 731, 654, 638, 615, 588, 554, 542 and 507 cm-1. Each peak has a margin of error of +/- 2 cm-1.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.76; H, 5.74; N, 5.85.
PATENT
WO2012/157288
http://www.google.co.in/patents/WO2012157288A1?cl=pt-PT
EXAMPLE 1
Preparation of
4-{[4-({[4-(2,2,2-Trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylic acid according to the conventional process
A mixture of methyl 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylate (89 mg, 0.18 mmol, PCT WO2006090224 EXAMPLE 1, Step 5) in tetrahydrofuran (1 mL), methanol (1 mL) and 2 N aq. sodium hydroxide (1 mL) is stirred at 70 oC for 17 h. The mixture is neutralized with 2 N hydrochloric acid (1 mL) and formed precipitate is filtered. The precipitate is triturated with diethylether to give 50 mg (58%) of the title compound as a white solid.
1H-NMR (DMSO-d6) delta: 7.59 (1 H, dd, J = 8.1, 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J = 8.7 Hz), 4.19 (2 H, d, J = 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H is not observed.
MS (ESI) m/z: 473 (M+H) +, 471 (M-H) –.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.72; H, 5.78; N, 5.80.
| Patent | Submitted | Granted |
|---|---|---|
| Benzisoxazole Derivatives [US2008207690] | 2008-08-28 | |
| 5-HT4 Receptor Agonist as a Prokinetic Agent [US2014051726] | 2012-03-23 | 2014-02-20 |
| Polymorph Form of 4-methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylic acid [US2014187583] | 2012-05-18 | 2014-07-03 |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
WO-2015178020-A1 |
2015-11-26 |
EN
|
|
||
|
2.
WO-2015174098-A1 |
2015-11-19 |
EN
|
|
||
|
3.
US-9187463-B2 |
2015-11-17 |
|
|||
|
4.
US-20150322055-A1 |
2015-11-12 |
|
|||
|
5.
EP-2922849-A1 |
2015-09-30 |
EN
|
|
||
|
6.
EP-2710002-A4 |
2014-10-01 |
EN
|
|
||
|
7.
US-8816090-B2 |
2014-08-26 |
|
|||
|
8.
EP-1856114-B1 |
2014-08-20 |
EN
|
|
||
|
9.
US-20140187583-A1 |
2014-07-03 |
|
|||
|
10.
WO-2014080633-A1 |
2014-05-30 |
EN
|
|
||
|
11.
EP-2710002-A1 |
2014-03-26 |
EN
|
|
||
|
12.
US-20140051726-A1 |
2014-02-20 |
|
|||
|
13.
EP-2688648-A1 |
2014-01-29 |
EN
|
|
||
|
14.
WO-2012157288-A1 |
2012-11-22 |
EN
|
|
||
|
15.
WO-2012127878-A1 |
2012-09-27 |
EN
|
|
||
|
16.
US-20080207690-A1 |
2008-08-28 |
|
|||
|
17.
EP-1856114-A1 |
2007-11-21 |
EN
|
|
||
|
18.
WO-2006090224-A1 |
2006-08-31 |
EN
|
|
see……….http://apisynthesisint.blogspot.in/2015/12/rq-00000010-for-treatment-of-gerd.html
/////c12c(cccc1onc2OCC3CCN(CC3)CC4(CCOCC4)C(=O)O)OCC(F)(F)F
C1CN(CCC1COC2=NOC3=C2C(=CC=C3)OCC(F)(F)F)CC4(CCOCC4)C(=O)O
Zidebactam, WCK 5107 in PHASE 1 FROM WOCKHARDT

Zidebactam, WCK 5107
![]()
Useful for treating bacterial infections
CAS 1436861-97-0, UNII: YPM97423DB, Wockhardt Biopharm
Molecular Formula, C13-H21-N5-O7-S
Molecular Weight, 391.4029
Disclosed in PCT International Patent Application No. PCT/IB2012/054290D
- 01 Aug 2015 Phase-I clinical trials in Bacterial infections (In volunteers, Combination therapy) in USA (IV) (NCT02532140)
trans- sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(2S, 5R)-sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(1R,2S,5R)-l,6-Diazabicyclo [3.2.1] octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-[2-[(3R)-3-piperidinylcarbonyl]hydrazide]
trans- sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(2S, 5R)-sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(lR,2S,5R)-l,6-Diazabicyclo [3.2.1] octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-[2-[(3R)-3 -piperidinylcarbonyl] hydrazide]
1,6-Diazabicyclo(3.2.1)octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-(2-((3R)-3-piperidinylcarbonyl)hydrazide), (1R,2S,5R)-

Zidebactam potassium
cas is 1706777-49-2
Zidebactam sodium ………..below

Cas 1706777-46-9
Sodium;[(2S,5R)-7-oxo-2-[[[(3R)-piperidine-3-carbonyl]amino]carbamoyl]-1,6-diazabicyclo[3.2.1]octan-6-yl] sulfate
UNII-NHY7N0Y9DG; NHY7N0Y9DG; Zidebactam sodium; Zidebactam sodium, (-)-; 1,6-Diazabicyclo(3.2.1)octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-(2-((3R)-3-piperidinylcarbonyl)hydrazide), sodium salt (1:1), (1R,2S,5R)-; 1706777-46-9;
| Molecular Formula: | C13H20N5NaO7S |
|---|---|
| Molecular Weight: | 413.381969 g/mol |
In September 2015, the drug was reported to be in phase I clinical trial.One of the family members US09132133, claims a combination of sulbactam and WCK-5107.
Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate.
Treatment of infections caused by resistant bacteria remains a key challenge for the clinician community. One example of such challenging pathogen is Acinetobacter baumannii (A. baumannii), which continues to be an increasingly important and demanding species in healthcare settings. The multidrug resistant nature of this pathogen and its unpredictable susceptibility patterns make empirical and therapeutic decisions more difficult. A. baumannii is associated with infections such as pneumonia, bacteremia, wound infections, urinary tract infections and meningitis.
Therefore, there is a need for development of newer ways to treat infections that are becoming resistant to known therapies and methods. Surprisingly, it has been found that a compositions comprising cefepime and certain nitrogen containing bicyclic compounds (disclosed in PCT/IB2012/054290) exhibit unexpectedly synergistic antibacterial activity, even against highly resistant bacterial strains.

PATENT
http://www.google.com/patents/WO2013030733A1?cl=en

Scheme-1

function with Boc group)
o ormua –
Scheme-2
Example-2 trans-sulfuric acid mono-r2-(N,-r(R)-piperidin-3-carbonyll-hvdrazinocarbonyl)-7-oxo-l,6- diaza-bicyclo Γ3.2.11 oct-6-νΠ ester
Step-1: Preparation of trans-3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-1 of Example- 1 above, and by using trans-6- benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylic acid (25 gm, 0.084 mol), N,N- dimethyl formamide (625 ml), EDC hydrochloride (24 gm, 0.126 mol), HOBt (16.96 gm, 0.126 mol), (R)-N-tert-butoxycarbonyl-piperidin-3-carboxylic acid hydrazide (21.40 gm , 0.088 mol) to provide the title compound in 17.0 gm quantity, 41% yield as a white solid.
Analysis: MS (ES+) CzsHasNsOe = 502.1 (M+l);
I^NMR (CDCI3) = 8.40 (br s, IH), 7.34-7.44 (m, 5H), 5.05 (d, IH), 4.90 (d, IH), 4.00 (br d, IH), 3.82 (br s, IH), 3.30 (br s, IH), 3.16-3.21 (m, IH), 3.06 (br d, IH), 2.42 (br s, IH), 2.29-2.34 (m, IH), 1.18-2.02 (m, 4H), 1.60-1.75 (m, 4H), 1.45-1.55 (m, 2H),1.44 (s, 9H).
Step-2: Preparation of trans-3-[N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-2 of Example- 1 above, and by using trans-3- [N ‘ -(6-benzyloxy-7-oxo- 1 ,6-diaza-bicyclo [3.2.1 ]octane-2-carbonyl)-hydrazinocarbonyl] -(R)- piperidin-l-carboxylic acid tert-butyl ester (16.5 gm , 0.033 mol), methanol (170 ml) and 10% palladium on carbon (3.5 gm) to provide the title compound in 13.5 gm quantity as a pale pink solid and it was used for the next reaction immediately.
Analysis: MS (ES+) CiglfeNsOe = 411.1 (M+l);
Step-3: Preparation of tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza- bicyclo [3.2.1] octane-2-carbonyl)-hydrazinocarbonyl] -(R)-piperidin- 1 -carboxylic acid tert- butyl ester:
By using the procedure described in Step-3 of Example- 1 above, and by using trans-3- [N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)- piperidin-1 -carboxylic acid tert-butyl ester (13.5 gm , 0.033 mol), pyridine (70 ml) and pyridine sulfur trioxide complex (26.11 gm, 0.164 mol), 0.5 N aqueous potassium dihydrogen phosphate solution (400 ml) and tetrabutylammonium sulphate (9.74 gm, 0.033 mol) to provide the title compound in 25 gm quantity as a yellowish solid, in quantitative yield.
Analysis: MS (ES-)
as a salt = 490.0 (M-l) as a free sulfonic acid;
Step-4: trans-sulfuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7- oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl]ester:
By using the procedure described in Step-4 of Example- 1 above, and by using tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester (24 gm , 0.032 mmol), dichloromethane (60 ml) and trifluoroacetic acid (60 ml) to provide the title compound in 10 gm quantity as a white solid, in 79% yield.
Analysis: MS (ES-)= C13H21N5O7S = 390.2 (M-l) as a free sulfonic acid;
HXNMR (DMSO-d6) = 9.97 (d, 2H), 8.32 (br s, 2H), 4.00 (br s, IH), 3.81 (d, IH), 3.10-3.22 (m, 3H), 2.97-3.02 (m, 2H), 2.86-2.91 (m, IH), 2.65-2.66 (m, IH), 1.97-2.03 (m, IH), 1.57-1.88 (m, 7H).
-32.6°, (c 0.5, water).
PATENT
http://www.google.com/patents/WO2015059643A1?cl=en

Both, cefepime and a compound of Formula (I) may be present in the composition in their free forms or in the form of their pharmaceutically acceptable derivatives (such as salts, pro-drugs, metabolites, esters, ethers, hydrates, polymorphs, solvates, complexes, or adducts).
Individual amounts of a compound of Formula (I) or a stereoisomer or a pharmaceutically acceptable derivative thereof, and cefepime or pharmaceutically acceptable derivative thereof in the composition may vary depending on clinical requirements. In some embodiments, a compound of Formula (I) or a stereoisomer or a pharmaceutically acceptable derivative thereof in the composition is present in an amount from about 0.01 gram to about 10 gram. In some other embodiments, cefepime or a pharmaceutically acceptable derivative thereof in the composition is present in an amount from about 0.01 gram to about 10 gram.
PATENT
http://www.google.com/patents/WO2015063653A1?cl=en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015110885
Formula (I)

(a) hydrogenolysis of a compound of Formula (II) to obtain a compound of Formula (III);

convertin a compound of Formula (III) to a compound of Formula (IV);



Example 1
Synthesis of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (25, 5R)-6-hydroxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III):
(25, 5i?)-6-benzyloxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazino-carbonyl] -l,6-diazabicyclo[3.2.1]octane (II) (130 g, 0.259 mol) was dissolved in methanol (1040 ml) to obtain a clear solution. To this solution, was added 10% palladium on carbon (13 g, 0.26 mol). The suspension was stirred under 230-250 psi hydrogen atmosphere at temperature of about 30 °C for about 2 hour. The catalyst was filtered over celite bed and catalyst containing bed was washed with additional methanol (400 ml). The methanolic solution was re-filtered through fresh celite bed and washed with methanol (100 ml). The filtrate was concentrated under vacuum at temperature of about 30°C to obtain the off white solid as product. The so obtained solid was stirred with cyclohexane (750 ml). The solid was then filtered and washed with cyclohexane (320 ml) and dried under suction to obtain 107 g of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo [3.2.1]octane (III).
Analysis:
Mass: 412.4 (M+l); for Molecular Formula of C18H29N5O6 and Molecular Weight of 411.5; and
Purity as determined by HPLC: 98.02%.
Step-2: Preparation of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1] octane (IV):
A solution of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) (106 g, 0.26 mol) in dichloromethane was charged with triethyl amine (110 ml, 0.78 mol) under stirring. To this clear solution was added pyridine sulfur trioxide complex (82.5 g, 0.53 mol) under nitrogen atmosphere and stirred at temperature of about 30°C for about 2 hour. The reaction mixture was diluted with 0.5 N aqueous potassium dihydrogen phosphate solution (2100 ml) followed by ethyl acetate (2100 ml). The turbid solution was stirred for 15 minute and then the layers were separated. The aqueous layer was washed with dichloromethane (530 ml) and then with ethyl acetate (1060 ml). Tetrabutyl ammonium sulfate (79 g, 0.23 mol) was added to the separated aqueous layer and stirred for 12 hour. The extraction of the product was done using dichloromethane as solvent (1150 ml x 2). The organic layer was dried over sodium sulfate and then evaporated under vacuum at temperature below 40°C to furnish 108 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo
[3.2.1] octane (IV).
Analysis:
Mass: 490.3 (M-l) as free sulfonic acid; for Molecular Formula of Ci8H28N509S.N(C4H9)4 and Molecular weight of 733.0; and
Purity as determined by HPLC: 86.50 %.
Step-3: Preparation of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) (88 g, 0.12 mol) was dissolved in dichloromethane (225 ml). The reaction mass was cooled to about -10°C and to this trifluoroacetic acid (225 ml) was added slowly. The reaction mixture was stirred for 1 hour at temperature of about -10°C. The solvent was removed under high vacuum at about 30°C. The residue (280 g) was stirred with diethyl ether (1320 ml) for 1 hour. The precipitated solid was filtered and the cake was washed with fresh diethyl ether (440 ml). This process was repeated with fresh diethyl ether (1320 ml + 440 ml). The obtained white solid was dried at temperature of about 30°C and suspended in acetone (1320 ml). The pH of the suspension was adjusted to 6.5-7.0 using 10% solution of sodium 2-ethyl hexanoate in acetone. The resulting suspension was filtered under suction and the wet cake was washed with acetone (440 ml) to provide the crude solid. The solid was further dried under vacuum at 40°C to yield 40 g of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I).
Analysis:
Mass: 392.2 (M+l); for Molecular formula of C13H21N5O7S and Molecular Weight of 391.4;
Purity as determined by HPLC: 92.87%; and
Melting point as determined by DSC: 274°C.
Example 2
Synthesis of Pure (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (25, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III):
The procedure for the synthesis of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) is same as given in Step- 1 of Example 1.
Step-2: Preparation of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1] octane (IV):
A solution of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) (106 g, 0.26 mol) in dichloromethane was charged with triethylamine (110 ml, 0.78 mol) under stirring to provide a clear solution. To this clear solution was added pyridine sulfur trioxide complex (82.5 g, 0.53 mol) under nitrogen atmosphere and stirred at temperature of about 30 °C for 2 hours. The reaction mixture was diluted with 0.5 N aqueous potassium dihydrogen phosphate solution (2100 ml) followed by ethyl acetate (2100 ml). The turbid solution was stirred for 15 minutes and then the layers were separated. The aqueous layer was washed with dichloromethane (530 ml) and then with ethyl acetate (1060 ml) respectively. Tetrabutyl ammonium sulfate (79 g, 0.23 mol) was added to the separated aqueous layer and stirred for 12 hours. The extraction of the product was done using dichloromethane as solvent (1150 ml x 2). Aliquot of the organic layer was dried over sodium sulfate for purity check. Considering the purity of the product as obtained above, silica gel (530 g) was added to the dichloromethane layer and stirred for 1 hour. This was filtered and again silica was taken in dichloromethane (3200 ml) and stirred for 45 minutes and filtered. Combined dichloromethane layer was filtered through the celite bed again and washed with additional 200 ml dichloromethane. The solvent was removed to obtain 88 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-!, 6-diaza-bicyclo[3.2.1]octane (IV) as white foam.
Analysis:
Mass: 490.3 (M-l) as a free sulfonic acid; for Molecular Formula of Ci8H28N509S.N(C4H9)4 and Molecular Weight of 733.0; and
Purity as determined by HPLC: 98.34%.
Step-3: Preparation of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
The above obtained tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) having purity of more than 98% (88 g, 0.12 mol) was dissolved in dichloromethane (225 ml). The reaction mass was cooled to temperature of about -10°C and to this trifluoroacetic acid (225 ml) was added slowly. The reaction mixture was stirred for 1 hour at about -10°C. The solvent was removed under high vacuum at temperature of about 30°C. The residue (280 g) was stirred with diethyl ether (1320 ml) for 1 hour. The precipitated solid was filtered and the cake was washed with fresh diethyl ether (440 ml). This process was repeated with fresh diethyl ether (1320 ml + 440 ml). The obtained white solid was dried at about 30°C and suspended in acetone (1320 ml). The pH of the suspension was adjusted to 6.5-7.0 using 10% solution of sodium 2-ethyl hexanoate in acetone. The resulting suspension was filtered under suction and the wet cake was washed with acetone (440 ml) to provide the crude solid. The solid was further dried under vacuum at 40°C to yield 40 g of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I).
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4; and
Purity as determined by HPLC: 98.7%.
Recovery of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1] octane (IV):
The silica recovered from the Step-2 was stirred with dichloromethane containing 2%
methanol (2000 ml) for one hour. Silica was filtered, washed with additional same composition of solvents (500 ml). Combined dichloromethane was filtered through the celite bed and washed with same composition of solvents (200 ml), evaporated to afford 1 1 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l , 6-diaza-bicyclo[3.2.1] octane (IV) as off white solid.
Repeating Step-3 with the above obtained tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2- [((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1 , 6-diaza-bicyclo [3.2.1] octane (IV) produced additional 7 g of compound of Formula (I).
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of CnH^NsOvS and Molecular Weight of 391.4;
Purity as determined by HPLC: 98.7%; and
Assay as determined by HPLC: 104% against reference standard of compound of Formula (I).
Example 3
Preparation of amorphous form of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1, 6-diaza-bicyclo[3.2. l]octane (I) :
Tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) (60 g, 0.081 mol), obtained in Step-2 of Example-2 was dissolved in dichloromethane (150 ml, 2.5 volume) to obtain a clear solution. Reaction mass was cooled to about -10°C and to it trifluoroacetic acid (150 ml) was slowly added. The reaction mixture was stirred for 1 hour at about – 10°C. The solvent was removed under high vacuum at about 30°C. Diethyl ether (600 ml x 3) was added to the residue ( 184 g) and stirred for 15 minute every time. The solvent was decanted off and the residue was washed with acetonitrile (600 ml x 3). This process was also repeated with dichloromethane (600 ml x 3). The off white solid was
isolated and dried under high vacuum at about 35 °C for 3 hour to obtain 33 g of amorphous form of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I). The XRD is shown in Figure 1.
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4;
HPLC purity: 92.26%; and
Melting point as determined by DSC: 210°C (loss of moisture below 100°C).
Example 4
Preparation of crystalline form of (25, 5R)-7-oxo-6-sulpho-oxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
The (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I) obtained as white solid (40 g) in Step-3 of Example 2 was dissolved in demineralised water (40 ml) to obtain a clear solution. To this isopropyl alcohol (280 ml) was added under stirring at room temperature. The obtained turbid solution became sticky initially then slowly started to convert into white solid, stirring continued for about 17 hours at temperature of about 30°C. The precipitated solid was filtered and washed with water: isopropyl alcohol mixture (20 ml: 140 ml). White solid was dried under high vacuum at temperature of about 45 °C for 5 hours to get 34 g of crystalline form of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1] octane (I).
Analysis:
Mass: 392.2 (M+l) for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4;
Purity as determined by HPLC: 98.7%;
Assay as determined by HPLC: 104% against reference standard of compound of Formula (I); and
Melting point as determined by DSC: 278°C (9% loss of moisture at 143-152°C).
X-ray powder diffraction pattern comprising a peak selected from the group consisting of 10.31 (± 0.2), 10.59 (± 0.2), 12.56 (± 0.2), 13.84 (± 0.2), 15.65 (± 0.2), 18.19 (± 0.2), 18.51(± 0.2), 20.38 (± 0.2), 20.65 (± 0.2), 24.30 (± 0.2), 24.85 (± 0.2) and 25.47 (± 0.2) degrees 2 theta.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014135931
Scheme 1.


Formula (I)
preparation of a compound of Formula (I), comprising:

Formula (I)
(a) reacting a compound of Formula (II) with a compound of Formula (III) to obtain a compound of Formula (IV);

Formula (II) Formula (III)

Formula (IV)
(b) hydrogenolysis of a compound of Formula (IV) to obtain a compound of Formula

X. Formula (V)
(c) sulfonating a compound of Formula (V) to obtain a compound of Formula (VI); and

Formula (VI)
(d) converting a compound of Formula (VI) into a compound of Formula (I).
Example -1
Preparation of (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (II):
Step-1: Preparation of (R)-Ethyl-N-Boc-piperidine-3-carboxylate (VIII)
To a solution of (R)-N-Boc-piperidine-3-carboxylic acid (1 kg. 4.36 mol) in N,N-dimethylacetamide (3 L) was charged potassium carbonate (0.664 kg, 4.80 mol) under mechanical stirring and the resulting suspension was stirred for 30 minutes at room temperature. To the reaction mass, ethyl iodide (0.75 kg, 4.80 mol) was charged via addition funnel and the reaction mass was stirred for 15 minutes at room temperature followed by at 50°C for 1 hour. The reaction was monitored using TLC (ethyl acetate: hexane 1:1). After the reaction was complete, the reaction mass was allowed to cool to room temperature and diluted with ethyl acetate (5 L). The suspension was filtered under suction and the wet cake was washed with ethyl acetate (5 L). The filtrate was stirred with 5% w/v sodium thio sulfate (15 L) and layers were separated. The aqueous layer was re-extracted with additional ethyl acetate (5 L). The combined organic layer was washed with water (5 L) and dried over sodium sulfate. The organic layer was evaporated under vacuum to provide semi-solid which solidifies upon standing as (R)-ethyl-N-Boc-piperidine-3-carboxylate in 1.1 kg quantity in 99.5% yield.
Analysis:
NMR: (CDC13): 4.63 (q, 2H), 3.90 (d, 1H), 2.87-2.95 (m, 2H), 2.73 (td, 1H), 2.32-2.39 (m, 1H), 1.66-2.01 (m, 2H), 1.52-1.68 (m, 2H), 1.39 (s, 9H), 1.19 (t, 3H).
Mass: (M+l): 258.1 for C13H23N04;
Step-2: Preparation of (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (II):
(R)-N-Boc-ethyl-piperidine-3-carboxylate (1.1 kg, 4.28 mol) was liquefied by warming and transferred to a round bottom flask (10 L), to this was charged hydrazine hydrate (0.470 kg, 9.41 mol) and stirring was started. The reaction mixture was stirred at about 120°C to 125°C for 5 hours. As the TLC showed (Chloroform: methanol 9:1) completion of reaction, the reaction mixture was cooled to room temperature and diluted with water (5.5 L) followed by dichloromethane (11 L) and was stirred for 20 minutes. The layers were separated and aqueous layer was extracted with additional dichloro methane (5.5 L). Combined organic layer was washed with water (2.75 L). The organic layer was dried over sodium sulfate and evaporated under vacuum to provide a thick gel which upon stirring and seeding in the presence of cyclohexane (5.5 L) provided white solid. The suspension was filtered and wet cake was washed with fresh cyclohexane (0.5 L). The cake was dried at 35°C under vacuum to provide (R)-N-Boc-piperidine-3-carboxylic acid hydrazide as a white solid in 0.90 kg quantity in 87% yield.
Analysis
NMR: (CDC13): 7.42 (br s, 1H), 3.92 (d, 1H), 3.88 (s, 2H), 3.54-3.65 (br s, 1H), 3.17 (br t, 1H), 2.98 (br s, 1H), 2.22-2.32 (br s, 1H), 1.82-1.90 (br m, 2H), 1.76 (s, 1H), 1.60-1.70 (m, 1H), 1.45 (s, 9H).
Mass (M+l): 244.1 for C11H21N303.
Specific rotation: [ ]25D = -53.5° (c 0.5, Methanol).
HPLC purity: 99%
Example 2
Preparation of (2S, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)- hydrazinocarbonyl] -l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (2S, 5R)- 6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1 ,6-diaza-bicyclo [3.2.1 ] octane(IV) :
Sodium (2S, 5R)-7-oxo-6-benzyloxy-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylate (III, 200 gm, 0.67 mol; prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) was dissolved in water (2.8 L) to obtain a clear solution under stirring at room temperature. To the clear solution was added successively, (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (171 gm, 0.70 mol), EDC hydrochloride (193 gm, 1.01 mol), and HOBt (90.6 gm, 0.67 mol) followed by water (0.56 L) under stirring at 35°C. The reaction mixture was stirred at 35°C for 20 hours. As maximum precipitation was reached, TLC (acetone: hexane 35:65) showed completion of reaction. The suspension was filtered under
suction and the wet cake was washed with additional water (2 L). The wet cake was suspended in warm water (10 L) and stirred for 5 hours. It was filtered under suction and dried under vacuum at 45°C to furnish (2S, 5R)-6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (IV) as a white powder in 270 gm quantity in 87% yield.
Analysis
NMR: (CDC13): 8.40 (br s, 1H), 7.34-7.44 (m, 5H), 5.05 (d, 1H), 4.90 (d, 1H), 4.00 (br d, 1H), 3.82 (br s, 1H), 3.30 (br s, 1H), 3.16-3.21 (m, 1H), 3.06 (br d, 1H), 2.42 (br s, 1H), 2.29-2.34 (m, 1H), 1.18-2.02 (m, 4H), 1.60-1.75 (m, 4H), 1.45-1.55 (m, 2H),1.44 (s, 9H).
Mass: (M+l) = 502.1 for C25H35N506
HPLC purity: 98.4%
Step-2: Preparation of (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2. l]octane (V):
(2S,5R)-6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino-carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (153 gm, 0.305 mol) was dissolved in methanol (1.23 L) to obtain a clear solution. To this solution, was added 10% Pd-C (15.3 gm, 50% wet) catalyst. The suspension was stirred for 3 hours under 100 psi hydrogen atmosphere at 35°C. As reaction showed completion on TLC (TLC system methanol: chloroform 10:90), the catalyst was filtered through celite under suction. The catalyst was washed with additional methanol (600 ml). The filtrate was evaporated under vacuum below 40°C to provide a crude residue. The residue was stirred with cyclohexane (1.23 L) for 1 hour. The solid was filtered at suction and the wet cake was washed with additional cyclohexane (0.25 L) to furnish (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (V) in 125 gm quantity as a solid in quantitative yield. The product being unstable was used immediately for the next reaction.
Analysis:
NMR: (CDC13): 9.0 (br s, 2H), 4.01 (br d, 2H), 3.80 (br s, 1H), 3.74 (br s, 1H), 3.48 (s, 1H), 3.13-3.26 (m, 3H), 2.96 (br s, 1H), 2.47 (br s, 1H), 2.28-2.32 ( br dd, 1H), 2.08 (br s, 1H), 1.90-2.0 (m, 3H),1.65-1.80 (m, 3H) 1.44 (s, 9H).
Mass: (M-l): 410.3 for C18H29N506
HPLC purity: 96.34%
Step-3: Preparation of Tetrabutyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]- 1 ,6-diaza-bicyclo[3.2.1 ] octane (VI) :
A solution of (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (113 gm, 0.274 mol), in dichloromethane (1.13 L) was charged with triethylamine (77 ml, 0.548 mol) under stirring to provide a clear solution. To the clear solution, was added pyridine sulfur trioxide complex (57 gm, 0.356 mol) under stirring at 35°C. The reaction mixture was stirred for 3 hours. The reaction mixture was worked up by adding 0.5 M aqueous potassium dihydrogen phosphate (1.13 L) followed by ethyl acetate (2.26 L) and the biphasic mixture was stirred for 15 minutes at 35°C. Layers were separated. Aqueous layer was re-extracted with dichloromethane ethyl acetate mixture (1:2 v/v, 2.26 L twice). Layers were separated. To the aqueous layer, was added solid tetrabutyl ammonium hydrogen sulfate (84 gm, 0.247 mol) and stirring was continued for 3 hours at room temperature. Dichloromethane (1.13 L) was added to the reaction mixture. Layers were separated. The aqueous layer was re-extracted with additional dichloromethane (0.565 L). Layers were separated. To the combined organic layer was added silica gel (226 gm) and the suspension was stirred for 1 hour. Suspension was filtered and silica gel was washed with dichloromethane (1 L). The combined filtrate was evaporated under vacuum to provide solid mass. To the solid mass was added cyclohexane (0.9 L) and stirred till complete solidification occurred (about 1 to 2 hours). The suspension was filtered under suction and the wet cake was dried under vacuum below 40°C to furnish tetrabutyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (VI) as a white solid in 122 gm quantity in 60% yield.
Analysis
NMR: (CDC13): 8.50 (br s, 2H), 4.32 (br s, 1H), 3.97 (d, 2H), 3.15-3.37 (m, 12H), 2.43 (br s, 1H), 2.33 (d, 1H), 2.10-2.2 (br m, 1H), 1.84-1.95 (m, 3H), 1.60-1.73 (m, 13H), 1.39-1.48 (m, 19H), 0.98 (t, 12H).
Mass: (M-l): 490.4 as a free sulfonic acid for C18H28N509S.N(C4H9)4;
HPLC purity: 96.3%
Step-4: Synthesis of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2. l]octane (I):
Tetra-butyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (113 gm, 0.154 mol) was dissolved in dichloromethane (280 ml) and to the clear solution was slowly added trifluoroacetic acid (280 ml) between 0 to 5°C. The reaction mixture was stirred between 0 to 5°C for 1 hour. The solvent and excess trifluoroacetic acid was evaporated under vacuum below 40°C to approximately 1/3 of it’s original volume to provide pale yellow oily residue. The oily residue was stirred with diethyl ether (2.25 L) for 1 hour to provide a suspension. The precipitate was filtered under suction and transferred to a round bottom flask, to it was added diethyl ether (1.1 L) under stirring. The suspension was stirred for 30 minutes and filtered under suction to provide a solid. The solid was charged in a round bottom flask and to it was added acetone (1.130 L). The pH of suspension was adjusted to 4.5 to 5.5 by adding 10% solution of sodium-2-ethyl hexanoate in acetone carefully. The resulting suspension was filtered under suction and the wet cake was washed with acetone (550 ml) to provide a crude solid. The obtained solid was dried under vacuum below 40°C to furnish 65 gm of a crude mass. The crude mass was dissolved in water (65 ml) under stirring and to the clear solution was added isopropyl alcohol (455 ml). The suspension was stirred for 24 hours and filtered under suction. The wet cake was washed with isopropyl alcohol (225 ml) and dried under vacuum below 40°C to provide a crystalline (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I) free from impurities in 48 gm quantity in 80% yield.
Analysis:
NMR: (DMSO-d6) = 9.97 (d, 2H), 8.32 (br s, 2H), 4.00 (br s, IH), 3.81 (d, IH), 3.10-3.22 (m, 3H), 2.97-3.02 (m, 2H), 2.86-2.91 (m, IH), 2.65-2.66 (m, IH), 1.97-2.03 (m, IH), 1.57-1.88 (m, 7H).
Mass: (M-l): 390.3 for C13H21N507S
HPLC purity: 95.78%
Specific rotation: [(X]25D: – 32.6° (c 0.5, water)
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 10.28 (+ 0.2), 10.57 (± 0.2), 12.53 (± 0.2), 13.82 (± 0.2), 15.62 (± 0.2), 18.16 (± 0.2), 18.49 (± 0.2), 20.35 (+ 0.2), 20.64 (± 0.2), 21.33 (+ 0.2), 22.99 (+ 0.2), 23.18 (+ 0.2), 24.27 (± 0.2), 24.81 (+ 0.2), 25.45 (± 0.2), 29.85 (+ 0.2), 30.45 (± 0.2), 32.39 (+ 0.2), 36.84 (± 0.2).
REFERENCES
Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of WCK-5107 Alone and in Combination With Cefepime (NCT02532140) https://clinicaltrials.gov/show/NCT02532140
ClinicalTrials.gov Web Site 2015, September 01, To evaluate the safety,tolerability and pharmacokinetics of single intravenous doses of WCK 5107 alone and in combination with cefepime in healthy adult human subjects.
| WO2013030733A1 * | Aug 24, 2012 | Mar 7, 2013 | Wockhardt Limited | 1,6- diazabicyclo [3,2,1] octan-7-one derivatives and their use in the treatment of bacterial infections |
| WO2014135931A1 * | Oct 12, 2013 | Sep 12, 2014 | Wockhardt Limited | A process for preparation of (2s, 5r)-7-oxo-6-sulphooxy-2-[((3r)-piperidine-3-carbonyl)-hydrazino carbonyl]-1,6-diaza-bicyclo [3.2.1]- octane |
| IB2012054290W | Title not available |


Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.

///////see………http://apisynthesisint.blogspot.in/2015/11/wck-5107-in-phase-1-from-wockhardt.html
SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
C1C[C@H](CNC1)C(=O)NNC(=O)[C@@H]2CC[C@@H]3C[N@]2C(=O)N3OS(=O)(=O)O
or
O=C(NNC(=O)[C@@H]2CC[C@@H]1CN2C(=O)N1OS(=O)(=O)O)[C@@H]3CCCNC3
C1CC(CNC1)C(=O)NNC(=O)C2CCC3CN2C(=O)N3OS(=O)(=O)[O-].[Na+]
SCYX 7158
SCYX-7158
[4-fluoro-N-(1-hydroxy-3,3-dimethyl-1,3-dihydro-benzo[c]oxaborol-6-yl-2-trifluoromethyl benzamide]
4-Fluoro-N-(1-hydroxy-3,3-diméthyl-1,3-dihydro-2,1-benzoxaborol-6-yl)-2-(trifluorométhyl)benzamide
Benzamide, N-(1,3-dihydro-1-hydroxy-3,3-dimethyl-2,1-benzoxaborol-6-yl)-4-fluoro-2-(trifluoromethyl)-
4-fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihydro- benzo[c][l,2]oxaborol-6-yl-2-trifluoromethyl benzamide
4-fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihydro- benzo[c][l,2]oxaborol-6-yl)-2-trifluoromethyl benzamide
SCYX-7158
1266084-51-8
UNII-2IOR2OO3GW
AN 5568
PHASE 1..Anacor Pharmaceuticals Drugs for Neglected Diseases Initiative, Trypanosomiasis, African (Sleeping sickness)
SEE……Future Medicinal Chemistry (2011), 3(10), 1259-1278.
- C17H14BF4NO3
- Average mass 367.103 Da
Human African trypanosomiasis (HAT) is an important public health problem in sub-Saharan Africa, affecting hundreds of thousands of individuals. An urgent need exists for the discovery and development of new, safe, and effective drugs to treat HAT, as existing therapies suffer from poor safety profiles, difficult treatment regimens, limited effectiveness, and a high cost of goods. We have discovered and optimized a novel class of small-molecule boron-containing compounds, benzoxaboroles, to identify SCYX-7158 as an effective, safe and orally active treatment for HAT.
The presence of a boron atom in the heterocyclic core structure has been found essential for trypanocidal activity of orally active series of benzoxaborole-6-carboxamides in murine models of human African trypanosomiasis. SCYX-7158 has been identified as an effective, safe and orally active treatment for human African trypanoso-miasis to enter preclinical studies, with expected progression to phase 1 clinical trials in 2011 ………http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3764666/
21. Jacobs RT, Plattner JJ, Nare B, Wring SA, Chen D, Freund Y, et al. Benzoxaboroles: a new class of potential drugs for human African trypanosomiasis. Future Med Chem. 2011;3:1259–1278. [PubMed]
22. Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl Trop Dis. 2011;5:e1151. [PMC free article]
A drug discovery project employing integrated biological screening, medicinal chemistry and pharmacokinetic characterization identified SCYX-7158 as an optimized analog, as it is active in vitro against relevant strains of Trypanosoma brucei, including T. b. rhodesiense and T. b. gambiense, is efficacious in both stage 1 and stage 2 murine HAT models and has physicochemical and in vitro absorption, distribution, metabolism, elimination and toxicology (ADMET) properties consistent with the compound being orally available, metabolically stable and CNS permeable.
In a murine stage 2 study,SCYX-7158 is effective orally at doses as low as 12.5 mg/kg (QD×7 days). In vivo pharmacokinetic characterization of SCYX-7158 demonstrates that the compound is highly bioavailable in rodents and non-human primates, has low intravenous plasma clearance and has a 24-h elimination half-life and a volume of distribution that indicate good tissue distribution.
Most importantly, in rodents brain exposure of SCYX-7158 is high, with Cmax >10 µg/mL and AUC0–24 hr >100 µg*h/mL following a 25 mg/kg oral dose. Furthermore, SCYX-7158 readily distributes into cerebrospinal fluid to achieve therapeutically relevant concentrations in this compartment.

Medicinal Chemistry Synthesis of SCYX-7158 SCHEME1
While the original route was eff ective for producing multi-gram quantities of the API, it was not amenable to scale-up. The route started with 2, a relatively expensive aryl boronic acid. This was protected as borocan 3 and halogen-lithium exchange followed by reaction with acetone and subsequent deprotection provided the oxaborole 4. This protection/alkylation/deprotection sequence added two steps to the overall synthesis and the metalation was not reliable. However, the biggest concern in the sequence was nitration of 4 to give 5. This was accomplished by adding a concentrated solution of 4 to cold fuming nitric acid. Besides the signifi cant safety considerations, the reaction did not scale well. Reduction of the nitro group to give aniline 6 was followed by amide formation to provide 1. While this end game was effi cient, the material produced was dark in color. The colored impurities were not removed by crystallization of 1 and furthermore a mixture of two polymorphs was formed under the original conditions.

Scheme 2 – Process Chemistry Synthesis of SCYX-7158
The process chemistry route to SCYX-7158 is shown in Scheme 2. When considering alternative routes to 1, the readily available and inexpensive methyl 2-bromobenzoate (8) was identifi ed as an attractive starting point. Gratifyingly, treatment of 8 with methylmagnesium bromide aff orded 2-bromocumyl alcohol (9) in high yield using simple operating conditions. Lithiumhalogen exchange followed by reaction with triisopropyl borate and acidic work-up provided benzoxaborole 4, along with cumyl alcohol (10). While this conversion was not completely atom-effi cient, it was easily scalable and several strategies are available to suppress the by-product in the future.
With benzoxaborole 4 in hand, attention turned to the introduction of a nitrogen-linked amide at the C(6) position. This was accomplished using the same nitration/reduction/acylation strategy used in Scheme 1. Yet signifi cant changes to the chemistry were required for safety and reliability reasons. The fi rst task was introduction of the nitrogen. Nitration was demonstrated using acetic anhydride/nitric acid. However, due to slow rates of nitration and potential for accumulation of a reactive intermediate, alternative conditions had to be identifi ed. These limitations were overcome by use of trifl uoroacetic anhydride/nitric acid, which provided a more reactive nitrating intermediate, thus improving the rate of nitration and aff ording a process in which nitric acid was slowly added until 4 was consumed. Full safety assessment of the nitration reaction, including extensive calorimetry studies, demonstrated the safety of this reaction. This process was used to prepare kilogram quantities of 5.
Following reduction of nitrobenzoxaborole 5 to aniline 6 under standard catalytic hydrogenation conditions, acylation with 7 provided the fi nal drug candidate in high chemical yield. Two challenges remained which needed to be addressed through further optimization of the process. The fi rst challenge was color and purity of the API, which derived from a highly colored impurity generated in the nitration reaction which carried through to fi nal product and was not removed by crystallization. The second challenge was to consistently obtain a single polymorph of the API. Both challenges were addressed by isolation of crystalline isopropyl boronate 11 which rejected colored impurities, followed by regeneration of 1 through addition of water and azeotropic removal of isopropanol. This crystallization provided the API as a single polymorph. The API was isolated in good yield, very high purity and was white in color.
PATENT
https://www.google.co.in/patents/WO2011019616A1?cl=en
N-(3,3-Dimethyl-l-phenyl-2,3-dihvdro-lH-benzotblborol-6-yl)-4-fluoro-2- trifluoromethylbenzatnide
HNO3

 This is not the compd, see precursor
This is not the compd, see precursorTo a suspension of 2-bromophenylboronic acid (75.Og, 373.4 mmol) in toluene (525 niL) was added JV-butyldiethanolamine (64.ImL, 392.1 mmol, 1.05 equiv.) via a syringe. The mixture was heated at 50 0C for two hours. After cooling to room temperature, the toluene was evaporated under reduced pressure and the remaining clear colorless oil was treated with heptanes (500 mL). The heptanes mixture was then sonicated for 5 min and the resulting suspension was allowed to stand at room temperature overnight. The solid that precipitated was collected by filtration, washed with heptanes, and dried in a vacuum oven overnight to yield 2-(2′- bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan as a white solid. Data: 1H NMR (400 MHz, CHLOROFORM-^) δ ppm 0.86 (t, J=7.4 Hz, 3 H) 1.14 – 1.25 (m, 2 H) 1.51 – 1.62 (m, 2 H) 2.61 – 2.70 (m, 2 H) 3.01 – 3.11 (m, 2 H) 3.26 – 3.37 (m, 2 H) 4.09 – 4.26 (m, 4 H) 7.10 (td, J=7.6, 2.0 Hz, 1 H) 7.24 (td, J=7.3, 1.1 Hz, 1 H) 7.51 (d, J=7.9 Hz, 1 H) 7.81 (dd, J=IA, 1.9 Hz, 1 H). Amount obtained, 123.7 g (98.6% yield).
To a solution of 2-(2′-bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan (30.0g, 89.2 mmol) in THF (740 mL) at -78 0C was added /?-BuLi (42.8 mL, 2.5M in hexane, 107.0 mmol, 1.2 equiv.) dropwise via a syringe over a period of 10 min while maintaining reaction temperature at -78 0C. After the addition the reaction solution was stirred for 20 min at -78 0C before acetone (7.5 mL, 124.8 mmol, 1.4 equiv.) was added dropwise via a syringe over a period of 10 min while maintaining the reaction temperature at -78 0C. The resulting mixture was allowed to stir for 20 min at -78 0C then warm to room temperature gradually. Once the reaction vessel reached room temperature, 6N HCl solution (150 mL) was added and the mixture was stirred for an additional 30 min. The mixture was extracted with EtOAc (3X). The EtOAc extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The light yellow oil was then subjected to flash chromatography (Isco Companion, 8Og SiO2 cartridge, solid loaded SiO2, neat heptanes to 20:80 EtOAc gradient at 60 ml/min for 90 min). 3,3-Dimethyl-3H-benzo[c][l,2]oxaborol-l-ol was recovered as clear colorless oil. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.44 (s, 6 H) 7.31 (d, J=Ll Hz, 1 H) 7.38 – 7.47 (m, 2 H) 7.66 (d, J=7.2 Hz, 1 H) 8.99 (s, 1 H). Amount obtained: 9.4O g (65.2 % yield).
To 60 mL fuming HNO3 at -45 0C was slowly added a solution of 3,3- dimethyl-3H-benzo[c][l,2]oxaborol-l-ol (9.4 g, 58.0 mmol) in 11.9 mL nitrobenzene via a syringe while maintaining the reaction temperature between -40 to -45 0C. Once the addition was complete the resulting solution was allowed to stir at -45 ° C for an additional 45 min before poured into crushed ice. The ice mixture was allowed to melt and the aqueous solution was extracted with DCM (3X). The combined DCM extracts were dried over Na2SO4 then evaporated. The crude oil remaining was mixed with one liter 1 : 1 DCM/heptanes. The volume of the solution was reduced under reduced pressure by half and the resulting solution was allowed to stand overnight in a -20 0C freezer. The precipitate formed was filtered out, washed with heptanes and vacuum dried to give 3,3-dimethyl-6-nitro-3H-benzo[c][1.2]oxaborol-l-ol as a white solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.46 (s, 6 H) 7.69 (d, J=8.4 Hz, 1 H) 8.28 (dd, J=8.4, 2.3 Hz, 1 H) 8.48 (d, J=2.2 Hz, 1 H) 9.41 (br. s., 1 H). Amount obtained: 7.31 g (60.4 % yield).
To a solution of 3,3-dimethyl-6-nitro-3H-benzo[c][l .2]oxaborol-l-ol (6.98 g, 33.3 mol) in THF ( 277 mL) was added 6N HC1( 16.6 mL, 100.2 mmol, 3.0 equiv.). The vessel was vacuum/N2 purged three times and 5% Pd/C (3.5 g) was added. The mixture was again vacuum/N2 purged three times then vacuum purged again. H2 was then introduced from a balloon and the reaction was allowed to stir at room
temperature over night. The reaction solution was filtered through a short pad of celite and the filtrate was evaporated to yield 6-amino-3, 3 -dimethyl -3H- benzo[c][l,2]oxaborol-l-ol HCl salt as a dark brown foamy solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). Amount obtained: 8.29 g (100% yield).
To a solution of 6-amino-3, 3 -dimethyl -3H-benzo[c][l,2]oxaborol-l-ol HCl salt (8.29 g, 33.3 mmol) in DCM (170 mL) was added Et3N (11.6 mL, 83.2 mmol, 2.5 equiv.). The mixture was cooled to 0 0C and 2-trifluoromethyl-4- fluorobenzoyl chloride (6.1 mL, 39.9 mmol, 1.2 equiv.) was added slowly via a syringe. The resulting solution was allowed to warm to room temperature gradually and stir for 2 hours. The reaction solution was diluted with DCM, washed with IN HCl, H2O, brine and then dried over Na2SO4, filtered and the filtrate was concentrated under reduced pressure to give an off- white solid. The solid was recrystallized from DCM/heptanes to give 4-fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihydro- benzo[c][l,2]oxaborol-6-yl-2-trifluoromethyl benzamide as a white solid. LCMS (M/Z) : 368 (M+H); 1H NMR (DMSO-d6) δ: 10.58 (s, IH), 9.11 (s, IH), 8.02 (d, J = 1.7 Hz, IH), 7.75 – 7.83 (m, 2H), 7.60 – 7.71 (m, 2H), 7.38 (d, J = 8.2 Hz, IH), 1.44 (s, 6H). Amount obtained: 11.7 g (96% yield)………IS SCYX 7158
BELOW NOT SCYX 7158
The title compound was prepared using a similar procedure to that of N-(I- phenyl- 1 ,3 -dihydrobenzo [c] [ 1 ,2]oxaborol-6-yl)-2-trifluoromethylbenzamide with phenyl magnesium bromide replacing p-to IyI magnesium bromide and 4-fluoro-iV-(l- hydroxy-3,3-dimethyl-2,3-dihydro-lH-benzo[b]borol-6-yl)-2-trifluoromethyl benzamide replacing N-(I -hydroxy-1 ,3-dihydrobenzo[c] [ 1 ,2]oxaborol-6-yl)-2- trifiuoromethylbenzamide. Data: LCMS m/e: 428 (M+H); 1H NMR (400 MHz, DMSO-J6) δ ppm 1.59 (s, 6 H) 7.46 – 7.62 (m, 4 H) 7.71 (td, J=8.5, 2.7 Hz,l H) 7.77 – 7.90 (m, 3 H) 8.00 – 8.09 (m, 2 H) 8.39 (d, J=2.0 Hz, 1 H) 10.66 (s, 1 H). 10 N-fl-p-Tolyl-lJ-dihydro-benzofcIflJIoxaborol-ό-vD-benzatnide
PATENT
82 4-Fluow-N-(l-hydwxy-3,3-dimethyl-l,3-dihydw-benzofcIfl,2Ioxabowl-6- yl-2-trifluoromethyl benzamide

To a suspension of 2-bromophenylboronic acid (10. Og, 49.7 mmol) in toluene (70 niL) was added N-butyldiethanolamine (8.5 mL, 52.2 mmol, 1.05 equiv.) via a syringe. The mixture was heated at 50 0C for two hours. After cooling to room temperature, the toluene was evaporated under reduced pressure and the remaining clear colorless crude oil was treated with heptanes (~ 500 mL). The heptanes mixture was then sonicated ~ 5 min and the resulting suspension was allowed to stand at room temperature overnight. The solid that precipitated was collected by filtration, washed with heptanes, and dried in a vacuum oven overnight to yield a white solid as the titled compound. 1U NMR (400 MHz, CHLOROFORM-J) δ ppm 0.86 (t, J=7.4 Hz, 3 H) 1.14 – 1.25 (m, 2 H) 1.51 – 1.62 (m, 2 H) 2.61 – 2.70 (m, 2 H) 3.01 – 3.11 (m, 2 H) 3.26 – 3.37 (m, 2 H) 4.09 – 4.26 (m, 4 H) 7.10 (td, J=7.6, 2.0 Hz, 1 H) 7.24 (td, J=7.3, 1.1 Hz, 1 H) 7.51 (d, J=7.9 Hz, 1 H) 7.81 (dd, J=IA, 1.9 Hz, 1 H). Amount obtained, 16.0 g, (98 % yield).
To a solution of 2-(2′-bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan (3.0g, 9.2 mmol) in THF (76 mL) at -78 0C was added /?-BuLi (4.4 mL, 2.5M in hexane, 11.0 mmol, 1.2 equiv.) dropwise via a syringe over a period of 10 min while maintaining reaction temperature at -78 0C. After the addition the reaction solution was stirred 20 min at -78 0C before acetone (946 μL, 12.8 mmol, 1.4 equiv.) was added dropwise via a syringe over a period of 10 min while maintaining the reaction temperature at -78 0C. The resulting mixture was allowed to stir for 20 min at -78 0C then warm to room temperature gradually. Once the reaction vessel reached room temperature, 6M HCl solution (30 mL) was added and the mixture was stirred for 30 min. The mixture was extracted with EtOAc (3X). The EtOAc extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The crude slightly yellow in color residual oil remaining was then subjected to flash chromatography (Isco Companion, 8Og SiO2 cartridge, solid loaded SiO2, neat heptane to 20:80 EtOAc gradient at 60 ml/min for 90 min). The product was recovered as clear colorless oil. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.44 (s, 6 H) 7.31 (d, J=Ll Hz, 1 H) 7.38 – 7.47 (m, 2 H) 7.66 (d, J=7.2 Hz, 1 H) 8.99 (s, 1 H). Amount obtained: 1.76 g (61%).
To 14.2 ml fuming HNO3 at -45 0C was added a solution of 3,3-dimethyl- 3H-benzo[c][l,2]oxaborol-l-ol (2.28 g, 14.1 mmol) in 3.0 ml nitrobenzene slowly via a syringe while maintaining the reaction temperature between -40 to -45 0C. Once the addition was complete the resulting solution was allowed to stir at -45 ° C for an additional 45 min before poured into crushed ice (600 g). The ice mixture was allowed to melt and the aqueous solution was extracted with dichloromethane. The combined dichloromethane extracts were dried over Na2SO4 then evaporated. The crude oil remaining was mixed with one liter 1 : 1 DCM:heptane. The volume of the solution was reduced on a rotovap by half and the resulting solution was allowed to stand overnight in a -20 0C freezer overnight. The precipitate formed was filtered out, washed with heptanes and vacuum dried to give the titled compound as a white solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.46 (s, 6 H) 7.69 (d, J=8.4 Hz, 1 H) 8.28 (dd, J=8.4, 2.3 Hz, 1 H) 8.48 (d, J=2.2 Hz, 1 H) 9.41 (br. s., 1 H). Amount obtained: 2.01 g (68%).
To a solution of 3,3-dimethyl-6-nitro-3H-benzo[c][1.2]oxaborol-l-ol (790 mg, 3.8 mmol) in THF ( 20 mL) was added HOAc (1.7 mL, 30 mmol). The vessel was vacuum/N2 purged three times and 5% Pd/C (200 mg) was added. The mixture was again vacuum/N2 purged three times then vacuum purged again. H2 was then introduced from a balloon and the reaction was allowed to stir for 2.5 hours. The reaction solution was filtered through a short pad of celite and the filtrate was evaporated to yield the title compound as a dark brown foamy solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). Amount obtained: 670 mg (89%). [0382] To a solution of 6-amino-3, 3 -dimethyl -3H-benzo[c][l,2]oxaborol-l-ol acetate salt (100 mg, 0.42 mmol) in DCM (2 niL) was added Et3N ( 117.3 μL, 0.84 mmol). The mixture was cooled to 0 0C and the 2-trifluoromethyl-4-fluorobenzoyl chloride (70.0 μL, 0.46 mmol) was added slowly via a syringe. The resulting solution was allowed to warm to room temperature gradually and stir for 2 hours. The reaction solution was diluted with DCM, washed with IN HCl, H2O and then dried over Na2SO4, filtered and the filtrate was concentrated under reduced pressure and the crude material was subjected to flash chromatography (Isco Companion, 4 g SiO2 cartridge, SiO2 solid load, neat heptanes to neat EtOAc gradient over 45 min, flow rate = 18 ml/min). The title compound was recovered as a white foam. LCMS (M/Z) : 368 (M+H); 1H NMR (DMSO-d6) δ: 10.58 (s, IH), 9.11 (s, IH), 8.02 (d, J = 1.7 Hz, IH), 7.75 – 7.83 (m, 2H), 7.60 – 7.71 (m, 2H), 7.38 (d, J = 8.2 Hz, IH), 1.44 (s, 6H). Amount obtained: 144.6 mg (93% yield).
Alternate Synthesis

82e
82b
A 500 mL round-bottomed-flask equipped with a magnetic stir bar and ice- H2O bath was charged with 82a (18.4g, 85.5 mmol) and anhydrous THF (200 mL). MeMgCl (68 mL, 3.0M in 2-methylTHF) was added dropwise through an additional funnel. The mixture was allowed to warm to rt. gradually and stirred overnight. After cooling back to 0 0C, the white milky suspension was carefully treated with HCl (3M) until the upper layer turned clear with white precipitate at the bottom of the flask (pH = 6). The upper clear solution was decanted into a separatory funnel. The precipitate was rinsed with methyl tert-butyl ether (MTBE) (100 mL) 3 times. Combined MTBE with the clear solution and the mixture was washed with H2O (100 mL) 3 times, brine (100 niL), dried over MgSO4, filtered and concentrated under reduced pressure to give 82b as a light yellow oil (20.2g, 100%).
82c
A 50 mL round-bottomed-flask equipped with a magnetic stir bar and ice- H2O bath was charged with 82b (860 mg, 4.0 mmol) and anhydrous THF (20 mL). MeMgBr (1.3 mL, 2.0 M in THF) was slowly added via a syringe. The mixture was stirred at 0 0C for 10 minute and the ice bath was replaced with a dry ice-acetone bath at -40 0C. BuLi (1.9 mL, 2.5 M in hexanes) was added dropwise via a syringe. The resulting mixture was stirred at -40 0C for another 2h before B(O-ipr)3 (1.4 mL, 4.8 mmol) was added dropwise. The mixture was allowed to warm up to rt gradually and stirred overnight. After carefully quenched the reaction with H2O (1 mL), HCl (3M, 10 mL) was added and the mixture was stirred at rt for Ih. The mixture was extracted with EtOAc (20 mL) 3 times. Combined extracts was washed with H2O (20 mL), brine (20 mL), dried over MgSO4, filtered and concentrated under reduced pressure to give a clear oil. The oil solidified overnight to give 82c as a pale yellow waxy solid (544mg, 82.4%).
82d
A 3 L round-bottomed-flask equipped with a mechanical stirrer, thermocouple and ice bath was charged with 82c (86.2 g of 58 wt%, 309 mmol) and trifluoroacetic acid (259 mL). Trifluoroacetic anhydride (129 mL, 926 mmol) was added in one portion. An exotherm of 18 0C was observed. The solution was again cooled to 0 0C and 90% nitric acid (18.0 mL, 386 mmol) was added via syringe pump over 2 h. After the addition was complete, the solution was aged for 1 h. Water (1.75 L) was added. Note: Initially the quench is quite exothermic. Add the water in 5 mL aliquots until the exotherm subsides. The resulting suspension was stirred for 16 h while warming to rt. The solids were collected on a frit, rinsed with water (2 x 500 mL), and air dried to constant weight to provide 50.3 g of crude 82d as a free-flowing orange solid. Note: the crude 82d can be carried forward without recrystallization. The solid was charged to a IL three-necked round-bottomed-flask equipped with a nitrogen inlet adapter, thermocouple, heating mantle and mechanical stirrer.
Isopropylacetate (IPAc, 75 mL) was added and the resulting slurry was warmed to 75 0C and heptanes (250 mL) was added over 15 min while maintaining an internal temp of > 65 0C. The slurry was allowed to cool to rt over night. The solids were collected on a frit and rinsed with 10% IP Ac/heptanes (100 mL) and then heptanes (100 rnL). The product was air dried to constant weight to provide a tan solid (31.7 g, 58%).
82e
A 500 mL round-bottomed-flask equipped with a magnetic stir bar, thermocouple and septum was charged with 82d (29.7 g, 192 mmol) and THF (150 mL, anhydrous stabilizer free). The vessel was inerted by cycling vacuum the nitrogen three times and 5% Pd/C (6.0 g, 50% wet, Degussa type NO/W) was added. The vessel was again inerted by cycling vacuum then nitrogen three times. A hydrogen filled balloon was attached via needle and the atmosphere was changed by cycling vacuum the hydrogen three times. The slurry was stirred vigorously for 16 h. The atmosphere was changed again to nitrogen by cycling vacuum then nitrogen three times. The mixture was filtered through a 1″ pad of celite and the cake was rinsed with THF (50 mL). Concentration in vacuo provided a light tan powder (26.82 g). In a 500 mL round bottomed-flask, the solids were slurried in IPAc (50 mL) and warmed in an 80 0C water bath. Heptanes (150 mL) were added over 10 min. The resulting slurry was allowed to cool to rt and stir for 16 h. The solids were collected on a frit, rinsed with heptanes (50 mL) and air dried to provide an off- white solid (24.39 g, 96%).
4-Fluoro-N-(l-hvdroxy-3,3-dimethyl-l,3-dihvdro-benzofcJfl,2Joxaborol-6-yl-2- triβuoromethyl benzatnide
A lL three-necked round-bottomed-flask equipped with a nitrogen inlet adapter, mechanical stirrer and thermocouple was charged with 82e (15.7g, 88.4 mmol), THF (160 mL, anhydrous, stabilizer free) and K2CO3 (14.7g, 106 mmol). The suspension was stirred at rt and 4-fluoro-2-(trifluoromethyl)benzoyl chloride (22.Og, 97.3 mmol) was added over 10 min. The resulting suspension was aged for 24 h at rt. Water (80 mL) and isopropyl acetate (160 mL) were added and the phases were partitioned. The organic phase was further extracted with water (80 mL) and then brine (50 mL). The organic phase was dried over MgSO4 (20 g) and concentrated in vacuo to provide a tan solid (34.26 g). The solid was dissolved with acetone (195 mL) and transferred to a mechanically stirred IL round-bottomed-flask. Distilled water (113 mL) was added in one portion and the mixture was stirred for 30 min to produce a seed bed and then additional distilled water (60 mL) was added over 30 min. The suspension was stirred at rt overnight and the solids were collected on a frit. The cake was rinsed with 1 : 1 acetone/water (100 rnL) and air dried to constant weight to provide an off-white solid (30.5 g, 94%).
Alternate Synthesis
HNO3 CF3CO2H (CF3CO)2O

to RT


1-1
A 72 L round-bottomed-flask was equipped with a cold bath, mechanical stirrer, nitrogen inlet adaptor, oxygen sensor, thermowell and 2 L dropping funnel. The flask was charged with methyl 2-bromobenzoate (2513 g, 11.7 mol) and the system was flushed with nitrogen to <0.1% O2. THF (18L, anhydrous, inhibitor free) was added and the cold bath was charged with ice and acetone. When the internal temp reached -4 0C, MeMgBr (11.6 L of a 3M solution in ether, 34.8 mol) was added via dropping funnel over 3 h. The internal temp was maintained below 15 0C throughout. At the end of addition, the cold bath was drained and the reaction was aged overnight at ambient temperature. The bath was again charged with ice and acetone and the suspension cooled to below 15 0C. HPLC indicated incomplete conversion (92:8 product, starting ester), so additional MeMgBr (2.3L of a 3M solution in ether) was added. After Ih, HPLC showed the conversion to be >99: 1. The reaction was quenched by slow addition of IN HCl (42 L) keeping the internal temp below 15 0C throughout. At the end of the quench, the pH was adjusted to 6 with IN HCl. The mixture was extracted with MTBE (10 L then 2x5L). The combined organic phases were dried over MgSO4, filtered and concentrated via rotary evaporation to provide 2482 g of 2-(2-bromophenyl)-propan-2-ol as a pale yellow oil. 1H NMR (CHLOPvOFORM-d) δ: 7.62 – 7.67 (m, IH), 7.53 – 7.58 (m, IH), 7.24 – 7.30 (m, IH), 7.03 – 7.10 (m, IH), 1.70 – 1.75 (m, 6H). 1-2
A 72L round-bottomed-flask was equipped with a mechanical stirrer, O2 sensor, thermowell, 2L dropping funnel, N2inlet adaptor, and cold bath. The vessel was inerted to 0.01% O2 and charged with THF (27L, anhydrous, inhibitor free). The resulting solution was cooled to -70 0C using dry ice and acetone and n-BuLi (8.2 L of a 2.5M solution in heptane, 20.5 mol) was added over Ih. 2-(2-Bromophenyl)- propan-2-ol (1994 g, 9.27 mol) was dissolved in THF (9L) and the solution was added to the BuLi via dropping funnel over 2h, keeping the internal temp below -70 0C. The resulting thin yellow suspension was aged for 30 min then B(OiPr)3 (244 Ig, 13.0 mol) was added rapidly via addition funnel. The cold bath was drained and the misture was allowed to warm to room temperature while aging over night. HPLC analysis shows an 81 :19 ratio of desired product: 2-phenyl-2-propanol. The mixture was cooled to -10 0C and 2N HCl (9.3 L) was added via dropping funnel over 30 min, keeping the reaction mixture below 10 0C. After 3 h, the pH was adjusted to 4 with additional HCl. The reaction mixture was extracted with MTBE (2 x 4L). The combined organic phases were concentrated to provide 2028 g of a heavy oil. The oil was dissolved in MTBE (14L) and extracted with IN NaOH (4.6, then 5, then 4L). The aqueous phases were combined and acidified with 2N HCl (6.8 L) to a pH of 4-5. The mixture was extracted with MTBE (5L). The organic phase was dried over MgSO4 (282 g) and concentrated to provide 1450 g (ca 60 wt%) of 3,3-dimethyl-3H- benzo[c][l,2]oxaborol-l-ol as a waxy white solid. LC/MS: m/z 163 (M+H)+; 1H NMR (DMSO-de) δ: 8.96 (br. s., IH), 7.62 (d, J = 7.2 Hz, IH), 7.33 – 7.45 (m, 2H), 7.25 – 7.30 (m, IH), 1.40 (s, 6H).
1-3
A 22 L round-bottomed-flask equipped with a mechanical stirrer, thermocouple, 2 L dropping funnel and cold bath was charged with 3,3-dimethyl-3H- benzo[c][l,2]oxaborol-l-ol (508 g, 300 g contained, 1.85 mol) and trifluoroacetic acid (1.54 L). The solution was cooled to 5 0C. Trifluoroacetic anhydride (722 mL, 5.56 mol, 3.00 eq) was added via dropping funnel over 15 min. After aging at 0 – 3 0C for 30 min, nitric acid (90% fuming, 108 mL, 2.31 mol, 1.5 eq) was added dropwise over 2h 50 min keeping the internal temp below 5 0C. After aging for 1 h, icewater (10.4L) was added over 50 min maintaining the reaction temp below 15 0C to provide a slurry. The slurry was aged at 0 0C overnight to provide an orange suspension. The solids were collected on a frit, rinsed with cold water (5L) and air dried under a stream of air to constant weight (ca 24h) to provide 364 g of 3,3- dimethyl-6-nitro-3H-benzo[c][l,2]oxaborol-l-ol as a 92.4 wt% pure solid (88%). LC/MS : m/z 208 (M+H)+; 1H NMR (DMSO-d6) δ: 8.52 (d, J = 2.2 Hz, IH), 8.32 (dd, J = 8.4, 2.2 Hz, IH), 7.74 (d, J = 8.4 Hz, IH), 1.50 (s, 6H)
1-4
A 2 gallon stirred pressure vessel was charged with 3,3-dimethyl-6-nitro- 3H-benzo[c][l,2]oxaborol-l-ol (966 g, 812 g corrected, 3.92 mol), 5% Pd/C (193 g, 50% wet, Degussa type 101 NO/W) and THF (4.83 L, inhibitor free). The vessel was sealed, the atmosphere was changed to H2 (5 psi) and the reaction was fun for 16 h. An exotherm to 30 0C was observed over about 30 min. The vessel was purged with N2, and completion of reaction was determined by HPLC. The reaction was vacuum filtered through a pad of celite (very slow filtration) and the filter cake was rinsed with THF (2L). The filtrate was concentrated via rotary evaporation to provide 982 g of a dark brown solid. This was transferred to a 22L round-bottomed-flask and warmed to 80 0C in iPAc (1.83 L) to provide a dark brown slurry. The slurry was cooled to 60 0C and heptanes (5.49L) were added over 2 h. The slurry was allowed to age with stirring over night while cooling to room temperature. The solids were collected on a frit, rinsed with heptanes (4L) and air dried to provide a dark brown solid (747 g).
The solids (747 g) were transferred to a 22L rbf and slurried in iPAc (3 L) at 70 0C. The batch was allowed to cool to 40 0C and heptanes (3L) were added over 5 h. The slurry was aged at room temperature over night and the solids were collected on a frit, rinsed with 1 : 1 iP Ac/heptanes (2L) then heptanes (IL) and air dried to provide 554 g of 6-amino-3,3-dimethyl-3H-benzo[c][l,2]oxaborol-l-ol as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). 4-Fluow-N-(l-hvdwxy-3,3-dimethyl-l,3-dihvdw-benzofcJfl,2Joxabowl-6-yl)-2- triβuoromethyl benzatnide
A 22L four-necked round-bottomed-flask equipped with a nitrogen inlet adapter, mechanical stirrer and thermocouple was charged with 6-amino-3,3- dimethyl-3H-benzo[c][l,2]oxaborol-l-ol (554g, 3.13 mol), THF (5.5 L, anhydrous, stabilizer free) and K2CO3 (865 g, 6.26 mol). The suspension was stirred at room temperature for 30 min and 4-fluoro-2-(trifluoromethyl)benzoyl chloride (780 g, 3.44 mol) was added over 30 min. The resulting suspension was aged for 24 h at room temperature. HPLC showed unreacted 6-amino-3,3-dimethyl-3H-benzo[c][l,2] oxaborol-1-ol so an additional 42 niL of the acid chloride was added. After 30 min, water (2.8 L) and isopropyl acetate (5.5 L) were added and the phases were partitioned. The organic phase was further extracted with water (2.8 L) and then brine (2.8 L). The organic phase was dried over MgSO4 and concentrated in vacuo to provide a tan solid. The solid was dissolved with acetone (3.0 L) and transferred to a mechanically stirred 5OL round-bottomed-flask. Distilled water (2.0 L) was added in one portion and the mixture was stirred for 30 min to produce a seed bed and then additional water (1.0 L) was added over 30 min. The suspension was stirred at room temperature overnight and the solids were collected on a frit. The cake was rinsed with 1 : 1 acetone/water (1.0 L) and air dried to constant weight to provide 4-fluoro-N- ( 1 -hydroxy-3 ,3 -dimethyl- 1 ,3 -dihydro-benzo [c] [ 1 ,2]oxaborol-6-yl)-2-trifluoromethyl benzamide as a dark tan solid (1.3 kg).
Recrystallization of4-Fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihvdro- benzotcl t 1,21 oxaborol-6-yl)-2-trifluoromethyl benzamide
A 22 L round-bottomed-flask was charged with the dark tan crude 4- fluoro-N-(l -hydroxy-3, 3 -dimethyl- 1 ,3-dihydro-benzo[c] [ 1 ,2]oxaborol-6-yl)-2- trifluoromethyl benzamide (1.3 kg), acetone (8L) and Darco G-60 (55 g, 400 mesh) and water (5.3L). The resulting suspension was stirred for 15 min, filtered through a pad of celite (ca 500 g) to provide a brown solution. The celite pad was washed with 60% acetone/water (8L). The combined filtrate and rinse were transferred to a 50 L round-bottomed-flask and water (2L) was added. The solution was seeded (5 g) to initiate crystallization and additional water (2.2 L) was added slowly via addition funnel. After aging at room temperature overnight, the solids were collected and the filter cake was rinsed with 30% acetone/water (4L). The solids were air dried for 24 h then dried in a room temperature vacuum oven for 5 days to constant weight to provide 969 g (72% recovery) of 4-fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihydro- benzo[c][l,2]oxaborol-6-yl)-2-trifluoromethyl benzamide as a light tan solid.
LC/MS: m/z 368 (M+H)+;
1H NMR (400 MHz, DMSO-d6) δ ppm 1.44 (s, 5 H) 1.49 (s, 2 H) 7.39 (d, J=8.2 Hz, 1 H) 7.61 – 7.76 (m, 2 H) 7.77 – 7.84 (m, 2 H) 7.86 – 7.90 (m, 0 H) 8.03 (d, J=I.7 Hz, 1 H) 9.09 (s, 1 H) 10.58 (s, 1 H).
POTASSIUM SALT
Formation of potassium salt

To a 50OmL three-neck flask fitted with a mechanical stirrer was charged KOH (1.51 g, 26.9 mmol, 1.0 eq.). Under a nitrogen atmosphere, anhydrous acetone (140 mL) and H2O (2.5 mL, 5 eq.) were added via syringe. A solution of 4-fluoro-N- (l-hydroxy-3,3-dimethyl-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl-2-trifluoromethyl benzamide (10.0 g, 27.2 mmol, 1.0 eq.) in anhydrous acetone (60 mL) was added to the flask with vigorous stirring. The resulting clear solution was stirred at room temperature. The potassium salt precipitated from the solution over ca. 4 hours to afford a thick suspension. The precipitate was collected by filtration, washed with acetone (200 mL) and dried in a vacuum oven overnight to afford a white solid (10.6g, 91.9% yield). 1H NMR (methanol-d4) δ: 7.70 – 7.76 (m, IH), 7.53 – 7.60 (m, 2H), 7.47 – 7.53 (m, IH), 7.33 – 7.36 (m, IH), 7.01 – 7.06 (m, IH), 1.46 (s, 6H); M.P. (range) 197 – 200 0C; Elemental analysis: Theory: C 48.25%, H 3.57%, N 3.31%, K 9.24%; Found: C 48.70%, H 3.41%, N 3.25%, K 9.19%.
REFERENCES
http://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0001151
- touguia J, Costa J (1999) Therapy of human African trypanosomiasis: current situation. Mem Inst Oswaldo Cruz 94: 221–224
- Barrett MP, Boykin DW, Brun R, Tidwell RR (2007) Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br J Pharmacol 152: 1155–1171.
- 1986. Epidemiology and control of African trypanosomiasis. Report of a WHO expert committee. World Health Organization. Geneva, Switzerland. Technical Report Series, No. 739. 126 pp.
- Benzoxaboroles: a new class of potential drugs for human African trypanosomiasis. Robert T Jacobs, Jacob J Plattner, Bakela Nare, Stephen A Wring, Daitao Chen, Yvonne Freund, Eric G Gaukel, Matthew D Orr, Joe B Perales, Matthew Jenks, Robert A Noe, Jessica M Sligar, Yong-Kang Zhang, Cyrus J Bacchi, Nigel Yarlett, and Robert Don. Future Medicinal Chemistry. August 2011. Vol. 3, No. 10. Pages 1259-1278.
http://www.swisstph.ch/fileadmin/user_upload/Pdfs/Events/2010_09_Jacobs.pdf ……….POWERPOINT
Lead optimization investigation of oxaboroles for the treatment of human African trypanosomiasis
238th Am Chem Soc (ACS) Natl Meet (August 16-20, Washington) 2009, Abst MEDI 345
LINK
https://www.acsmedchem.org/ama/orig/abstracts/mediabstractf2009.pdf
Robert Jacobs, bob.jacobs@scynexis.com
Daitao Chen1 , Matt Orr1 , Jessica Sligar1 , Matt. Jenks1 , Andy Noe1 , Bakela Nare2 , Luke T. Mercer2 , Tana S. Bowling2 , Cindy Rewerts1 , Stephen Wring1 , Cyrus Bacchi3 , Nigel Yarllet3 , Charles Ding4 , Yvonne Freund5 , Kurt Jarnagin5 , Jacobs Plattner5 , and Robert Don6 . (1) Scynexis Inc, Duhram, NC 27713, (2) SCYNEXIS, Inc, Research Triangle Park, NC 27709-2878, (3) Pace University, New York, NY, (4) Anacor Pharmaceuticals, Inc, Palo Alto, CA, (5) Anacor Pharmaceuticals, Inc, (6) Drugs for Neglected Diseases initiative, Geneva, Switzerland

///////////SCYX-7158
Mavatrep, JNJ 39439335,


Mavatrep; UNII-F197218T99; Mavatrep (USAN); JNJ-39439335; 956274-94-5;
2-(2-(2-(2-(4-trifluoromethylphenyl)vinyl)-1H-benzimidazol-5-yl)phenyl)propan-2-ol
(E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
(E)-2-(2-(2-(4-(Trifluoromethyl)styryl)-1H-benzo[d]imidazol-5-yl)phenyl)-propan-2-ol Hydrochloride
Phase I Musculoskeletal pain; Pain
- 01 Mar 2013 Janssen Research and Development completes a phase I trial in Japanese and Caucasian adult male volunteers in the US (NCT01631487)
- 01 Mar 2013 Janssen completes enrolment in its phase I trial for Pain (in volunteers) in the USA (NCT01631487)
- 05 Feb 2013 Janssen Research and Development initiates enrolment in a phase I trial for Pain (Japanese and Caucasian volunteers) in USA (NCT01631487)
- Originator Johnson & Johnson Pharmaceutical Research & Development
- Developer Janssen Research & Development
- Class Analgesics; Benzimidazoles; Small molecules
- Mechanism of Action TRPV1 receptor antagonists
| PHASE 1 Johnson & Johnson Pharmaceutical Research & Development, L.L.C. |
|
| Public title: | A Clinical Study to Investigate the Effect on Pain Relief of a Single Dose of JNJ-39439335 in Patients With Chronic Osteoarthritis Pain of the Knee |
http://clinicaltrials.gov/ct2/show/NCT01006304
http://apps.who.int/trialsearch/trial.aspx?trialid=NCT00933582
http://www.ama-assn.org/resources/doc/usan/mavatrep.pdf SEE STRUCTURE IN THIS FILE
MAVATREP IS JNJ-39439335
—
(E)-2-(2-(2-(4-(trifluoromethyl)styryl)-1H-benzo[d]imidazol-6-yl)phenyl)propan-2-ol hydrochloride
956282-89-6 CAS NO OF HCl SALT
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00271, http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00271

The process development of Mavatrep (1), a potent transient receptor potential vanilloid-1 (TRPV1) antagonist, is described. The two key synthetic transformations are the synthesis of (E)-6-bromo-2-(4-(trifluoromethyl)styryl)1H-benzo[d]imidazole (4) and the Suzuki coupling of 4 with 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol (5). Compound 1a was prepared in four chemical steps in 63% overall yield.
HCl SALT
1a as an off-white solid. 1H NMR (DMSO-d6, 500 MHz) δ 8.35 (d, J = 16.6 Hz, 1H), 7.96 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.80 (dd, J = 8.1, 1.3 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 1.4 Hz, 1H), 7.53 (d, J = 6.6 Hz, 1H), 7.43 (dd,J = 8.4, 1.5 Hz, 1H), 7.39 (ddd, J = 8.1, 7.4, 1.5 Hz, 1H), 7.27 (ddd, J = 7.4, 7.4, 1.3 Hz, 1H), 7.04 (dd, J = 7.5, 1.5, 1H), 1.29 (s, 6H); 13C NMR (DMSO-d6, 125 MHz) δ 147.7 (C), 147.4 (C), 142.3 (C), 140.3 (CH), 139.0 (C), 138.0 (C), 131.7 (CH), 131.1 (C), 130.5 (C), 130.2 (C), 128.7 (2CH), 128.3 (CH), 127.4 (CH), 126.2 (CH), 126.2 (CH), 125.8 (2CH), 124.0 (CF3), 114.3 (CH), 113.2 (CH), 112.4 (CH), 71.6 (C), 32.4 (2CH3); Anal. Calcd for C25H22F3ClN2O·1.2H2O: C, 62.49; H, 5.12; Cl, 7.38; F, 11.86; N, 5.83. Found: C, 62.34; H, 4.93; Cl, 7.24; F, 11.61; N, 5.78. Water wt % calcd, 4.50%; found, 4.52% (determined by KF analysis).
Patent




CLICK ON IMAGE FOR CLEAR VIEW
Example 10 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol(Cpd 18)
Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0278]A solution of 4-trifluoromethylbenzaldehyde (7.7 mL, 57.7 mmol), malonic acid (12.0 g, 115.4 mmol), 0.567 μL piperidine (5.75 mmol) in 30 mL of pyridine was stirred at 70° C. for 18 h. The reaction solution was cooled to room temperature. Water (300 mL) was added and the resulting mixture was acidified to pH 4 (litmus) using concentrated hydrochloric acid to give a precipitate. The solid was filtered, and washed with water until the filtrate was neutral. The solid product was dried in vacuo to give the title Compound 10a as a white powder (11.2 g, 90%). 1HNMR (400 MHz, DMSO-d6) δ (ppm): 12.60 (bs, 1H), 7.92 (d, 2H, J=8.2 Hz), 7.77 (d, 2H, J=8.2 Hz), 7.66 (d, 1H, J=16.0 Hz), 6.70 (d, 1H, J=16.0 Hz).
-
[0000]

Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0279]A solution of Compound 10a (20.6 g, 95.4 mmol) in anhydrous methylene chloride (200 mL) was treated with oxalyl chloride (16.6 mL, 190 mmol) and “3 drops” of anhydrous dimethylformamide. The resulting solution was stirred at room temperature under an argon atmosphere for 18 h. The solvent was concentrated to give 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b as a solid, which was used without further purification in the next step.
-
[0280]To a solution of 4-bromo-benzene-1,2-diamine (16.1 g, 86.7 mmol) in acetic acid (100 mL) was added dropwise a solution of Compound 10b (assumed 95.4 mmol) in acetic acid (100 mL). The reaction mixture was stirred at 100° C. for 18 h. The reaction mixture was cooled to room temperature, and a mixture of ethyl acetate and hexanes 3:7 (500 mL) was added. The mixture was triturated at room temperature for 3 h to give a precipitate. The solid was filtered, and dried in vacuo to give the title Compound 10c (23.2 g, 73%). 1H NMR (400 MHz, DMSO-d6/CDCl3) δ (ppm): 8.45 (d, 1H, J=16.7 Hz), 7.84-7.90 (m, 1H), 7.74 (d, 2H, J=8.3
-
[0281]Hz), 7.56-7.62 (m, 3H), 7.50-7.52 (m, 1H), 7.34 (d, 1H, 16.7 Hz).
-
[0000]


Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0282]To a solution of methyl 2-bromobenzoate (20.76 g, 96 mmol) in 120 mL of anhydrous ether under Argon at 0° C. was slowly added methylmagnesium bromide (77 mL, 3.26 M) at a rate that the internal temperature of the mixture was below 20° C. A white suspension resulted, and the mixture was stirred at room temperature for 2 h. The mixture was cooled in an ice-water bath. To the reaction mixture was very slowly added hydrochloric acid (400 mL, 0.5 M). The pH of the final mixture was adjusted to less than about 6 with few drops of 2M hydrochloric acid. The layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined and dried over magnesium sulfate. The organic fraction was filtered, and the filtrate was concentrated to yield the title compound as a pale yellow liquid, which was distilled under vacuum to afford the title Compound 10d as a colorless liquid (16.9 g, 82%, b.p. about 65-70° C./0.3 mmHg). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.67 (dd, 1H, J=1.7, 7.9 Hz), 7.58 (dd, 1H, J=1.3, 7.9 Hz), 7.30 (ddd, 1H, J=1.4, 7.4, 7.9 Hz), 7.10 (ddd, 1H, J=1.7, 7.4, 7.8 Hz), 2.77 (br s, 1H), 1.76 (s, 6H).
-
[0000]

Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0283]To a solution of n-BuLi (166 mL, 2.6 M, 432 mmol) in 200 mL of THF at −78° C. under argon was slowly added a solution of Compound 10d (42.2 g, 196 mmol) in 60 mL of THF at a rate that the internal temperature remained below −70° C. The mixture was stirred at −75° C. for 2 h. To the reaction mixture was then added triisopropylborate (59 mL, 255 mmol) in three portions. The mixture was allowed to warm slowly to room temperature overnight. The mixture was then cooled to 0° C., and was carefully quenched with dilute hydrochloric acid (250 mL, 2N). The mixture was then stirred at room temperature for 1 h. The pH of the mixture was checked and adjusted to acidic using additional 2N HCl if prophetic. The two layers were separated, and the aqueous layer was extracted twice with ether. The organic layers were combined, and dried with magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure to yield a pale yellow oil. The residue was then diluted with ethyl acetate (400 mL) and, washed with 1N sodium hydroxide solution (150 mL×3). The basic aqueous layers were combined and acidified with 2N HCl. The clear solution turned cloudy when the acid was added. The mixture was extracted with ether (150 mL×3). The organic layers were combined and dried with magnesium sulfate. The solution was filtered, and the filtrate was concentrated under reduced pressure to yield the title Compound 10e as a colorless oil (26.2 g, 82%) which was used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.00 (s, 1H), 7.66 (dm, 1H, J=7.3 Hz), 7.45 (dt, 1H, J=1.1, 7.7 Hz), 7.40 (dm, 1H, J=7.6 Hz), 7.31 (dt, 1H, J=1.2, 7.1 Hz), 1.44 (s, 6H).
-
[0000]

Step E. (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
[0284]To a mixture of Compound 10e (11.7 g, 71 mmol), Compound 10c (19.9 g, 54 mmol), sodium carbonate (46 g, 435 mmol) and PdCl2(dppf).CH2Cl2 (8.9 g, 11 mmol) in a 1 L round bottom flask equipped with water condenser was added 400 mL of anhydrous DME and 200 mL of water. The mixture was evacuated and filled with Argon three times. The mixture was heated to 100° C. for 20 h. The mixture was then cooled to room temperature. The biphasic system was transferred to a 1 L separatory funnel and the two layers were separated. The organic layer was washed with brine (2×300 mL). The aqueous layers were combined and extracted with ethyl acetate once (about 300 mL). The organic layers were combined, dried with sodium sulfate, and filtered. The volume of the filtrate was reduced to about 170 mL under reduced pressure. The mixture was then filtered through a pad of silica gel and the pad was washed with ethyl acetate until the filtrate did not contain any product. After concentration, a light pink/beige solid was obtained. The solid was triturated with 50 mL ethyl acetate, and the mixture was heated to 85° C. for 5 min. The mixture was slowly cooled to r.t., then cooled at 0° C. for 0.5 h. The mixture was filtered, and the solid was washed with cold ethyl acetate twice, and dried under vacuum at 40° C. to yield the title Compound 18 as a light beige solid (7.58 g, 33%). RP-HPLC 95% pure.
-
1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.73 (m, 1H,), 7.90 (d, 2H, J=8.2 Hz), 7.85 (dd, 1H, J=8.0, 0.6 Hz), 7.78 (d, 2H, J=8.4 Hz), 7.74 (d, 1H, J=16.8 Hz), 7.59-7.47 (m, 1H), 7.41 (s, 1H), 7.37-7.32 (m, 2H), 7.21 (dt, 1H, J=1.2, 7.4 Hz), 7.06 (s, 1H), 7.02 (d, 1H, J=7.4 Hz), 4.85 (s, 1H), 1.21 (s, 6H).
-
Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3.
-
m.p. (uncorr.) 250-251° C.
Example 10.1 Scale Up Preparation of (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol (Cpd 18) Step A. 3-(4-trifluoromethyl-phenyl)-acrylic acid
-
[0286]A 2-L 4-neck round bottom flask equipped with an air condenser/argon inlet, mechanical stirrer, thermocouple and a stopper was charged with 4-(trifluoromethyl)benzaldehyde (250 g, 196.2 mL, 1.44 mol), malonic acid (302.6 g, 2.87 mol), and pyridine (750 mL). An exotherm developed (about 38-40° C.), which was maintained for 30 min. Piperidine (14.202 mL, 143.58 mmol) was then added to the reaction and a second exotherm developed (Tmax about 42° C. after about 10 min.). The reaction was stirred for 30 min and then heated to 60° C. for 18 h (overnight). The reaction appeared to be complete by TLC, and was cooled to about 40° C., diluted into water (2 L; done to prevent reaction freezing), cooled to room temperature, and further diluted with water (4 L, 6 L total). The slurry was acidified to pH=2.0-3.0 with concentrated hydrochloric acid (about 675-700 mL). The material was stirred for 30 min., and a white solid was collected by filtration. The filter cake was washed with water until the filtrate was neutral (pH about 5.5-6, 2.5 L), air-dried in a Buchner funnel for 2 h, and then further dried in a vacuum oven at 60° C. overnight to provide 300.5 g (96%) of the title Compound 10a as a white solid.
Step B. (E)-5-bromo-2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazole
-
[0287]To a 5-L 4-neck round bottom flask equipped with a magnetic stirrer, argon inlet-argon outlet to a carbonate scrub, two stoppers, and a room temperature water bath was charged with 4-(trifluoromethyl)cinnamic acid (315 g, 1.46 mol) and dichloromethane (3.15 L) to give a slurry. To the slurry was added oxalyl chloride (151.71 mL, 1.75 mol) and DMF (1.13 mL, 14.57 mmol). Upon addition of DMF, gas evolution commenced, and the reaction was continued for about 3 h during which time a solution developed. When the reaction was complete (LC-MS), it was concentrated to dryness to give 342.4 g of 3-(4-trifluoromethyl-phenyl)-acryloyl chloride Compound 10b (>100%) as a yellow oily solid.
-
[0288]A 5-L 4-neck round bottom flask equipped with mechanical stirrer, thermocouple, air condenser with argon inlet, and a stopper was charged with 4-bromo-benzene-1,2-diamine (244 g, 1.27 mol) and acetic acid (2.13 L). To this solution was added a solution of Compound 10b (327 g, 1.39 mol) in toluene (237 mL). After this addition, the temperature spiked to 45° C. in about 30 seconds and then subsided. The reaction was then heated to 90° C. for 16 h (overnight). The reaction was cooled to 40° C., and poured into a mixed solution of EtOAc and heptane (about 1:3, 5.75 L) and a precipitate occurred. The resulting slurry was stirred for 3 h, and the solid was collected by filtration, washed with EtOAc:heptane (1:3, 3 L), and then dried in a vacuum oven (60° C.) to give 324.3 g (65%) of the title Compound 10c as a partial acetate salt.
Step C. 2-(2-bromo-phenyl)-propan-2-ol
-
[0289]A 12-Liter 4-neck flask equipped with a thermocouple, condenser, septum, addition funnel and overhead mechanical stirrer under argon was charged with methyl-2-bromobenzoate (226.5 g, 1.05 mol) and THF (1.6 L, 19.66 mol). The mixture was cooled to a temperature between 2 and 5° C. with stirring and held for 30 min. To the solution was slowly added methyl magnesium bromide in diethyl ether (3M, 1.05 L; 3.15 mol) via the addition funnel at a rate to maintain the reaction temperature below 15° C. An exotherm was observed during the addition, the reaction temperature warmed from 3 to 15° C. The addition of 1.05 L Grignard was complete in 4 h (approximate feed rate was 4.17 mL/min). The reaction mixture appeared to be off-white/yellow slurry. The reaction was allowed to warm to room temperature and stirred overnight (15 h). The reaction was sampled by HPLC/TLC and showed no starting material present. The ice bath was again applied to the reaction flask and a 0.5 M HCl solution (4.5 L; 2.25 mol) was slowly added over a period of 2 h. The temperature increased dramatically from 0 to 15° C. After the quench was complete, the reaction was stirred at room temperature for 30 min. Additional 2 N HCl (500 mL; 1.00 mol) was slowly added to maintain a pH less than 6. MTBE (1 L) was added to help with the phase split. The reaction was stirred at room temperature for 1 to 2 h to dissolve the solid material into the aqueous phase (most likely Mg(OH)2 which is very basic). The pH must be checked and adjusted with additional acid when necessary. The phases were separated and the aqueous layer was washed with an additional 1 L MTBE (2×500 mL). The organic phases were combined, washed with NaHCO3 solution (2×300 mL), dried over MgSO4, filtered and the filtrate was concentrated under vacuum to yield the title Compound 10d (220.83 g, 97.48% yield) as a clear yellow oil.
Step D. 3,3-dimethyl-3H-benzo[c][1,2]oxaborol-1-ol
-
[0290]A 12-Liter 4-neck round bottom flask equipped with a thermocouple, condenser, addition funnel and overhead mechanical stirrer under dry Argon was charged with anhydrous THF, (3 L) and chilled to −70 to −78° C. via a dry ice/acetone bath. n-Butyl lithium (2.5N in hexanes, 860 mL, 2.15 mol) was slowly added via addition funnel. An exotherm was observed as the temperature rose from −78 to −70° C. To the addition funnel was added a solution of Compound 10d (220 g, 979.97 mmol) in anhydrous THF (1 L). The 2-(2-bromophenyl)propan-2-ol solution was slowly added to the n-BuLi solution. The addition took 90 min in order to maintain a reaction temperature below −70° C. After the addition was complete, the reaction mixture was stirred at −70 to −75° C. for 30 min. The triethylborate (230 mL, 1.35 mol) was quickly added in 3 portions at −70° C. An exotherm was observed, the batch temperature rose from −70 to −64° C. The reaction was stirred at −70° C. and slowly warmed to room temperature over night. After the reaction was cooled to 0-5° C., the reaction was slowly quenched with 2 M HCl (1 L, 2.00 mol) added via the addition funnel while maintaining the batch temperature 0-5° C. The reaction mixture was stirred for 1 h. The aqueous phase pH was 9-10. The pH was then adjusted to acidic (4-5) with 2 M HCl (200 mL). The two phases were separated and the aqueous layer was extracted with MTBE (2×500 mL). The combined organic phases were dried with anhydrous magnesium sulfate. The solution was filtered and concentrated to yield a yellow oil. The yellow oil was diluted with MTBE (1.5 L) and washed with 1M NaOH (3×500 mL). The product containing basic aqueous phases were combined and acidified with 2 M HCl (800 mL) (the clear solution turns turbid with the addition of acid). After stirring the turbid solution for 15 min (pH=4-5) (Note 1), it was extracted with MTBE (2×500 mL). The organic phases were combined and dried over MgSO4. The solution was filtered and the filtrate was concentrated to yield the title Compound 10e as a clear yellow oil (121.78 grams, 77% yield).
Step E. (E)-2-(2-{2-[2-(4-Trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol
-
A 5-L 4-neck flask equipped with a thermocouple controller, condenser, overhead mechanical stirrer, Firestone Valve® and a nitrogen inlet/outlet was charged with dimethoxyethane (2 L), DI water (1 L) and sodium carbonate (230.9 g, 2.18 mol). The solution was degassed and purged with N2 three times. Compound 10e (71.7 g, 0.35 mol) and Compound 10c (100.0 g, 0.27 mol) were added to the degassed solution. The solution was degassed and purged with N2 three times. PdCl2(dppf) (44.48 g, 54.4 mmol) was added to the solution, and the solution was degassed and purged with N2 three times. The resulting two-phase suspension was heated to reflux for 18 h, and then cooled to room temperature. The reaction mixture was transferred to a 12-L separatory funnel, and the layers were separated. The organic layer was washed with brine (1 L). The two aqueous layers were combined and extracted with EtOAc (1 L). The combined organic layers were dried (Na2SO4), filtered, and the filtrate was concentrated to an oil. Two separate 100 g coupling reactions were combined and purified by chromatography in 10 successive chromatography runs on an ISCO preparative chromatography system (10×1.5 Kg SiO2, 5 column volumes of EtOAc, 250 mL/min flow rate). The combined fractions were transferred to two 22 L 4-neck round bottom flasks, and Silicycle Si-thiol functionalized silica gel (2 g) was added to each solution. The solutions were warmed to 40° C. and aged for 1 h. The solutions were filtered thru a medium glass funnel and washed with EtOAc (4 L) and combined. The filtrate was evaporated to a semi solid, which was transferred to a 2 L round bottom flask, to which EtOAc (0.4 L) was added. The resulting white precipitate slurry was cooled to −5° C. and stirred for 1 h. The slurry was filtered and washed twice with cold EtOAc (100 mL). The solids were dried in a vacuum oven at 40° C. for 40 h to afford 84.0 g (36.5% yield, 98.8 area % purity) of the title Compound 18 as a white solid. Anal. Calcd for C25H21N2OF3.0.04% H2O.0.15 mol MeOH: C, 70.48; H, 5.14: N, 6.42; F, 13.06 Found: C, 70.54; H, 4.83: N, 6.18; F, 13.33
Example 10.2 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol monosodium salt (Cpd 18)
-
A 5-L 4-neck flask equipped with a thermocouple controller, an overhead mechanical stirrer, and a nitrogen inlet/outlet was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (125.0 g, 0.510 mol) and MeOH (1.25 L). A solution of sodium methoxide in methanol (0.5 M, 592 mL, 0.3 mol) was added. The reaction was heated to 65° C. for 30 min and all solids dissolved. The solution was cooled and evaporated to dryness. The foam was collected by scraping it out of the flask. The solids were placed in vacuum oven for 24 h at 40° C. to afford 139 g (about 100% isolated yield) of the title Compound 18 monosodium salt as a yellowish solid. 1H NMR (400 MHz, DMSO-d6) δ 7.80-7.84 (m, 3H), 7.74 (d, 2H, J=8.59 Hz), 7.65 (d, 1H, J=16.4 Hz), 7.40-7.44 (m, 2H), 7.25-7.37 (m, 2H), 7.16-7.20 (m, 1H), 7.01-7.05 (m, 1H), 6.84-6.87 (m, 1H), 1.23 (s, 6H). Mass Spectrum (LCMS, APCI pos.) Calcd. For C25H21F3N2O: 423.2 (M+H). Found 423.3. m.p. (uncorr.) 258-259° C.
Example 10.3 (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol hydrochloride salt (Cpd 18)
-
A 250-mL separatory funnel was charged with (E)-2-(2-{2-[2-(4-trifluoromethyl-phenyl)-vinyl]-1H-benzimidazol-5-yl}-phenyl)-propan-2-ol. Compound 18 (1.0 g, 2.4 mmol) and EtOAc (20 mL). Aqueous HCl (1M, 20 mL) was added to the white slurry, and the separatory funnel was shaken. The solid product quickly dissolved, and a white precipitate started to form. The organic layer was transferred to a 100 mL round bottom flask equipped with a magnetic stir bar, and was stirred for 2 h. The thick slurry was filtered, rinsed with EtOAc (2×5 mL), and put into a vacuum oven at 40° C. for 36 h to afford 0.95 g (87.5%) of the title Compound 18 hydrochloride salt.
////////////Phase I, Musculoskeletal pain, Pain, Mavatrep, JNJ 39439335,
Altiratinib
Altiratinib
DCC-2701; DP-5164
CAS :1345847-93-9
N-[4-({2-[(cyclopropylcarbonyl)amino]pyridin-4-yl}oxy)-2,5-difluorophenyl]-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
N-(4-((2-(cyclopropanecarboxamido)pyridin-4-yl)oxy)-2,5-difluorophenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
Mechanism of Action:MET/TIE2/VEGFR2/TRK (A,B,C) kinase inhibitor
Indication:invasive solid tumors. The FDA has granted altiratinib Orphan Drug Designation for glioblastoma multiforme (GBM)
Development Stage:Phase I
Developer:Deciphera Pharmaceuticals, Llc

Altiratinib, also known as DCC-270, DP-5164, is an oral, selective and highly potent inhibitor of MET, TIE2, VEGFR2 and TRK kinases with potential anticancer activity. DCC-2701 effectively reduces tumor burden in vivo and blocks c-MET pTyr(1349)-mediated signaling, cell growth and migration as compared with a HGF antagonist in vitro. Importantly, DCC-2701’s anti-proliferative activity was dependent on c-MET activation induced by stromal human fibroblasts and to a lesser extent exogenous HGF. DCC-2701 may be superior to HGF antagonists that are in clinical trials and that pTyr(1349) levels might be a good indicator of c-MET activation and likely response to targeted therapy as a result of signals from the microenvironment. Inhibition of MET kinase blocks a key mechanism in tumor cells that causes cancer invasiveness and metastasis. (Oncogene. 2015 Jan 8;34(2):144-53.)
DCC-2701 is an angiogenesis inhibitor acting on Tie2 receptor, HGF receptor and VEGFR-2. The product is being evaluated in phase I clinical studies at Deciphera for the oral treatment of solid tumors.
Altiratinib(DCC-2701) is a novel c-MET/TIE-2/VEGFR inhibitor; effectively reduce tumor burden in vivo and block c-MET pTyr(1349)-mediated signaling, cell growth and migration as compared with a HGF antagonist in vitro.

![]()
SYNTHESIS
CLICK ON IMAGE TO ENLARGE
OR SEE
http://apisynthesisint.blogspot.in/2015/10/altiratinib-dcc-2701-dp-5164.html

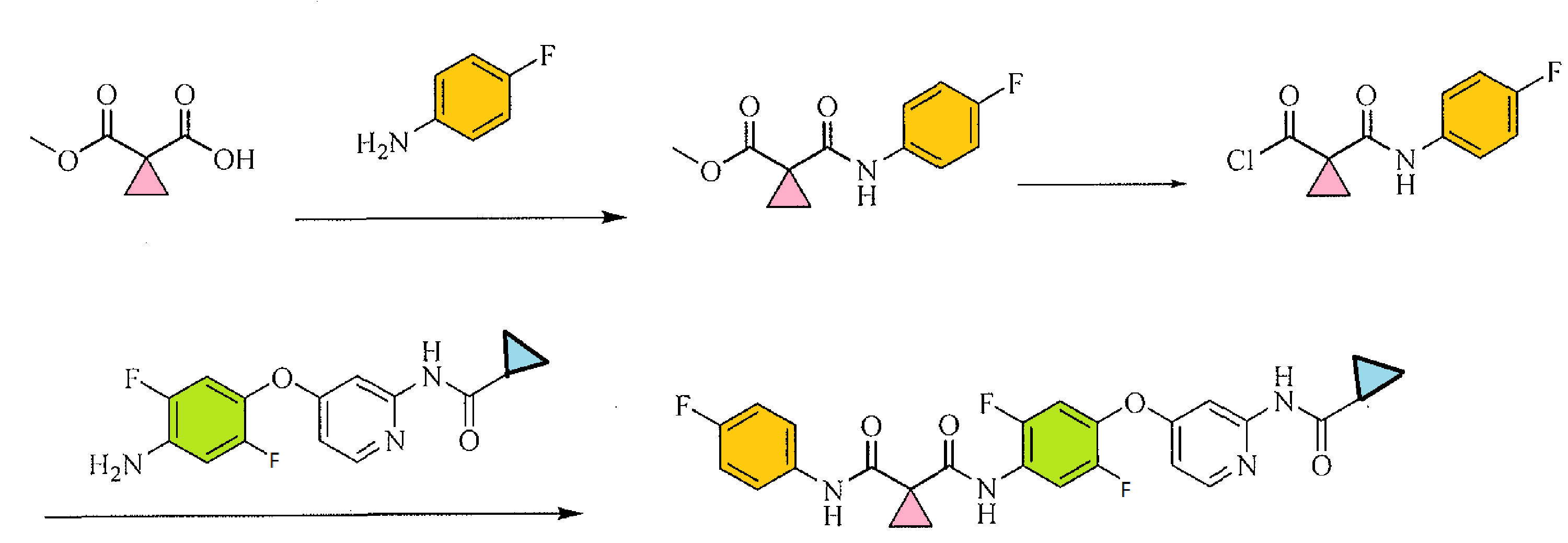
PATENT
https://www.google.co.in/patents/WO2011137342A1?cl=en

Scheme 1

BEAWARE THIS IS NOT THE COMPD
Scheme 11
INTERMEDIATES
Scheme 9
[00108] A non-limiting example of Scheme 9 is illustrated below for the synthesis of 36, a specific example of 26 wherein X is F, Y is CI, and Zl , Z2, and Z3 are CH (Scheme 10). Addition of l ,2,4-trifluoro-5-nitrobenzene (33) to a solution of 2-chloropyridin-4-ol (34) and sodium hydride in DMF at 0 °C yields the nitro intermediate 35. The nitro moiety of 35 is subsequently reduced at RT in the presence of zinc dust and ammonium chloride in solution of me hanol and THF to yield amine 36.

Scheme 10
[00109] A non-limiting example of Scheme 7 is illustrated in Scheme 11, beginning with intermediate 36, prepared in Scheme 10. Thus, 36 readily reacts with acid chloride 13 (see Scheme 3) in the presence of triethylamine to yield chloro-pyridine 37. Chloro-pyridine 37 is then converted to 38, a specific example of 1 wherein Rl is F, X is F, Zl , Z2, and Z3 are CH and R3 is -C(0)CH3, upon treatment with acetamide (an example of R3-NH2 27 where R3 is acetyl) and cesium carbonate in the presence of a catalytic amount of palladium acetate and xantphos.
NOTE 38 IS NOT THE FINAL PRODUCT
REFERENCES
Kwon Y, Smith BD, Zhou Y, Kaufman MD, Godwin AK. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene. 2015 Jan 8;34(2):144-53. doi: 10.1038/onc.2013.539. Epub 2013 Dec 23. PubMed PMID: 24362531; PubMed Central PMCID: PMC4067476.
////Altiratinib, DCC-2701, DP-5164, Phase I, Deciphera Pharmaceuticals
Pevonedistat

Millennium Pharmaceuticals, Inc. INNOVATOR
Millennium Pharmaceuticals, Inc., a subsidiary of Takeda Pharmaceutical Company Limited,
MLN4924, MLN 4924-003, TAK-924
905579-51-3 BASE
1160295-21-5 HcL
A potent and selective inhibitor of NAE. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. The ubiquitin-proteasome pathway mediates the destruction of unwanted proteins.
(((1S,2S,4R)-4-{4-[(S)-2,3-Dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride) (pevonedistat), a novel NEDD8-activating enzyme (NAE) inhibitor, has demonstrated in vitro cytotoxic activity against a variety of human malignancies and is currently being developed by Takeda Pharmaceuticals Company Limited as a clinical candidate for the treatment of cancer
In 2011, orphan drug designation was assigned to MLN-4924 for the treatment of MDS and for the treatment of acute myelogenous leukemia.
PHASE 1…….CANCER SOLID TUMOR

………………….
PATENT
http://www.google.com/patents/US20120330013

preparing a compound represented by the following formula 1 by reacting the compound of formula 11 with TFA (step 9):


The retrosynthetic analysis of MLN4924 (1), as the final desired nucleoside, is shown in the following.

MLN 4924 (1) can be synthesized by condensing cyclic sulfate 3 as the glycosyl donor with a purine base. The glycosyl donor 3 can be produced from diol 4, which in turn can be obtained from cyclopentanone 5 via a stereoselective reduction and a regioselective cleavage of the isopropylidene moiety. The cyclopentanone 5 can be synthesized from cyclopentenone 6 by stereoselective reduction. The intermediate cyclopentenone 6 can be easily derived from D-ribose according to our previously published procedure (Jeong, L. S. et al., J. Org. Chem. 2004, 69, 2634-2636).
The synthetic route for the glycosyl donor 3 is shown in the following scheme 1.

Example 1 Preparation of MLN4924 Step 1: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-one (Compound 5)

To a suspension of the compound 6 (20.0 g, 47.1 mmol) in methanol (400 ml) was added 10% palladium on activated carbon (1.0 g), and the mixture was stirred at room temperature overnight under H2 atmosphere. After filtration of the reaction mixture, the solvent was removed and the residue was dissolved in methylene chloride and then filtered through short pad silica gel. Then, the solvent was evaporated to give the compound 5 (20.1 g, 100%) as a colorless syrup.
[α]20 D −28.32 (c 1.49, MeOH); HR-MS (ESI): m/z calcd for C25H32NaO4Si [M+Na]+ 447.1968, Found 447.1956; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.40 (m, 6H), 4.84 (t, J=4.4 Hz, 1H), 4.22 (dd, J=1.2, 4.8 Hz, 1H), 3.96 (dd, J=8.0, 10.0 Hz, 1H), 3.82 (dd, J=6.8, 10.0 Hz, 1H), 2.37 (m, 1H), 2.30 (ddd, J=1.2, 8.4, and 18.4 Hz, 1H), 2.20 (ddd, J=1.2, 12.0, and 18.4 Hz, 1H), 1.37 (s, 3H), 1.35 (s, 3H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 112.6, 80.5, 77.6, 77.2, 76.9, 63.6, 38.1, 36.9, 27.1, 27.02, 27.01, 25.3, 19.5; Anal. Calcd for C25H32O4Si: C, 70.72; H, 7.60. Found: C, 70.79; H, 7.75.
Step 2: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-ol (Compound 7)

To a suspension of the compound 5 (20.1 g, 47.1 mmol) in methanol (500 ml) were added sodium borohydride (2.17 g, 57.4 mmol) and cerium (III) chloride heptahydrate (21.3 g, 57.2 mmol) at 0° C., and the mixture was stirred at room temperature for 30 min. After the solvent was removed, the residue was partitioned between ethyl acetate and water. The organic layer was then washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=5/1) to give the compound 7 (20.86 g, 98%) as a colorless syrup.
[α]20 D +34.55 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C25H34NaO4Si [M+Na]+: 449.2124; Found: 449.2110; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.39 (m, 6H), 4.62 (t, J=5.6 Hz, 1H), 4.44 (t, J=5.6 Hz, 1H), 3.89 (dd, J=6.0, 7.6 Hz, 1H), 3.84 (m, 1H), 3.68 (dd, J=6.4, 10.0 Hz, 1H), 1.91 (m, 2H), 1.26 (m, 1H), 1.42 (s, 3H), 1.33 (s, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 135.8, 134.2, 134.1, 129.8, 129.7, 127.8, 127.7, 110.6, 79.4, 78.9, 77.6, 77.2, 76.9, 72.5, 62.9, 41.6, 33.4, 27.0, 25.9, 27.0, 25.9, 24.4, 19.5; Anal. Calcd for C25H34O4Si: C, 70.38; H, 8.03. Found: C, 70.41; H, 8.08.
Step 3: Preparation of 3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentane-1,2-diol (Compound 4)

To a solution of the compound 7 (20.86 g, 47.12 mmol) in methylene chloride was added trimethylaluminum (2.0 M in toluene, 132.1 ml) at 0° C., and the mixture was stirred at room temperature for 2 days. The mixture was cooled to 0° C., slowly quenched with an aqueous saturated ammonium chloride solution, filtered, and evaporated. The residue was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 4 (13.42 g, 62%) as a colorless syrup.
[α]20 D +3.30 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C26H38NaO4Si [M+Na]+: 465.2437; Found: 465.2423; 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 4H), 7.41 (m, 6H), 4.05 (dd, J=4.4, 7.2 Hz, 1H), 3.93 (m, 1H), 3.72 (m, 2H), 3.59 (dd, J=3.6, 12.0 Hz, 2H), 2.70 (d, J=20.8 Hz, 1H), 2.10 (m, 2H), 1.60 (m, 1H), 1.20 (s, 9H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 133.5, 130.0, 129.9, 127.9, 127.9, 77.6, 77.2, 76.9, 74.9, 73.8, 72.7, 72.1, 63.3, 42.1, 34.0, 28.5, 27.0, 19.4; Anal. Calcd for C26H38O4Si: C, 70.55; H, 8.65. Found: C, 70.61; H, 8.70.
Step 4: Preparation of (4-tert-butoxy-2,2-dioxo-tetrahydro-2-yl-6-cyclopenta[1,3,2]-dioxathiol-5-ylmethoxy)-tert-butyl-diphenyl-silane (Compound 3)

To a solution of the compound 4 (13.42 g, 30.3 mmol) in methylene chloride were added triethyl amine (14.5 ml, 101.0 mmol) and thionyl chloride (3.7 ml, 47.4 mmol) at 0° C., and the reaction mixture was stirred at 0° C. for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the cyclic sulfite (14.37 g, 97%) as a white foam.
[α]20 D +20.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO5SSi [M+Na]+: 511.1950; Found: 511.1929; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.40 (m, 6H), 5.23 (m, 1H), 5.04 (dd, J=4.4, 6.0 Hz, 1H), 4.01 (t, J=4.8 Hz, 1H), 3.68 (dd, J=3.6, 10.4 Hz, 1H), 3.56 (dd, J=8.0, 10.4 Hz, 1H), 2.07 (m, 2H), 1.96 (m, 1H), 1.14 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.8, 135.7, 133.9, 133.8, 129.9, 129.9, 127.9, 127.8, 85.7, 83.2, 77.6, 77.2, 76.9, 75.0, 71.1, 62.7, 44.7, 31.4, 28.5, 27.1, 19.4; Anal. Calcd for C26H36O5SSi: C, 63.90; H, 7.42; S, 6.56. Found: C, 63.94; H, 7.45; S, 6.61.
To a solution of the cyclic sulfite obtained above (14.37 g, 29.4 mmol) in the mixture of carbon tetrachloride, acetonitrile and water (1:1:1.5, 210 ml) were added sodium metaperiodate (18.56 g, 56.4 mmol) and ruthenium chloride (1.72 g, 8.25 mmol), and the reaction mixture was stirred at room temperature for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=4/1) to give the compound 3 (13.36 g, 90%) as a white solid.
mp 101-104° C.; [α]20 D −80.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO6SSi [M+Na]+: 527.1900; Found: 527.1881; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.41 (m, 6H), 5.13 (m, 1H), 4.83 (dd, J=4.4, 6.8 Hz, 1H), 4.13 (t, J=4.0 Hz, 1H), 3.92 (dd, J=6.4, 10.4 Hz, 1H), 3.69 (dd, J=5.2, 10.4 Hz, 1H), 2.11 (m, 2H), 2.02 (m, 1H), 1.15 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.7, 135.0, 133.8, 133.7, 130.0, 128.0, 127.9, 83.5, 82.2, 77.6, 77.2, 76.9, 75.4, 70.4, 70.4, 62.2, 43.9, 31.3, 28.2, 27.1, 26.8, 19.4; Anal. Calcd for C26H36O6SSi: C, 61.87; H, 7.19; S, 6.35. Found: C, 61.91; H, 7.14; S, 6.30.
Step 5: Preparation of 2-tert-butoxy-3-(tert-butyl-diphenyl-silanyloxymethyl)-5-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 8)

A suspension of N6-indanyl-7-deazaadenine (8.80 g, 35.2 mmol), sodium hydride (1.38 g, 45.7 mmol) and 18-crown-6 (9.11 g, 45.7 mmol) in THF (200 ml) was stirred at 80° C. To the reaction mixture was added a solution for the compound 3 (13.36 g, 26.5 mmol) in THF (150 ml), and the stirring was continued at 80° C. overnight. The reaction mixture was cooled down to 0° C., and conc. HCl was added slowly until pH reaches 1-2. Then the reaction mixture was further stirred at 80° C. for 2 hours. After neutralized with saturated aqueous NaHCO3 solution, the reaction mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 8 (11.62 g, 65%) as a white foam.
UV (CH2Cl2) λmax 272.5 nm; [α]20 D −8.89 (c 0.45, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O3Si [M+H]+: 675.3730; Found: 675.3717; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.70 (m, 4H), 7.41 (m, 6H), 6.92 (d, J=3.6 Hz, 1H), 6.29 (d, J=3.2 Hz, 1H), 5.91 (dd, J=7.6, 14.8 Hz, 1H), 5.14 (br d, J=6.8 Hz, 1H), 4.77 (m, 1H), 4.36 (t, J=6.0 Hz, 1H), 4.22 (dd, J=5.2, 10.8 Hz, 1H), 3.84 (dd, J=5.6, 10.4 Hz, 1H), 3.73 (dd, J=8.4, 10.4 Hz, 1H), 3.37 (d, J=5.6 Hz, 1H), 3.06 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.75 (m, 1H), 2.58 (m, 1H), 2.38 (m, 1H), 2.15 (m, 1H), 1.98 (m, 1H), 1.65 (s, 1H), 1.55 (s, 1H), 1.16 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.8, 150.3, 144.1, 143.8, 135.9, 134.0, 129.9, 128.2, 127.9, 127.9, 127.0, 125.1, 124.4, 123.3, 103.8, 97.4, 77.8, 77.6, 77.2, 76.9, 74.9, 72.4, 63.5, 62.1, 56.3, 43.9, 34.9, 30.5, 30.5, 28.5, 27.2, 19.5; Anal. Calcd for C41H50N4O3Si: C, 72.96; H, 7.47; N, 8.30. Found: C, 73.01; H, 7.45; N, 8.36.
Step 6: Preparation of {7-[3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentyl]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}-indan-1-yl-amine (Compound 9)

To a solution of the compound 8 (11.62 g, 17.2 mmol) in methylene chloride (300 ml) were added N,N-dimethylaminopyridine (5.64 g, 51.6 mmol) and phenyl chlorothionocarbonate (4.3 ml, 34.4 mmol), and the reaction mixture was stirred at room temperature overnight. After the solvent was removed, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the thiocarbonate (13.82 g, 99%) as a white foam.
UV (MeOH) λmax 271.50 nm; [α]20 D +10.00 (c 0.15, MeOH); HR-MS (ESI): m/z calcd for C48H55N4O4SSi [M+H]+: 811.3713; Found: 811.3687; 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.61 (dd, J=1.6, 7.6 Hz, 4H), 7.34 (m, 5H), 7.26 (m, 4H), 7.18 (m, 6H), 6.86 (s, 1H), 6.25 (d, J=3.2 Hz, 1H), 6.00 (dd, J=3.2, 8.4 Hz, 1H), 5.83 (d, J=6.8 Hz, 1H), 5.19 (m, 1H), 5.07 (br s, 1H), 4.48 (t, J=3.6 Hz, 1H), 3.82 (dd, J=7.2, 10.4 Hz, 1H), 3.52 (dd, J=7.2, 10.0 Hz, 1H), 2.99 (m, 1H), 2.88 (m, 2H), 2.69 (m, 2H), 2.18 (dd, J=11.2, 13.6 Hz, 1H), 1.94 (m, 2H), 1.12 (s, 9H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 194.9, 153.5, 152.1, 143.9, 135.9, 135.8, 134.1, 129.9, 129.6, 128.3, 127.9, 127.0, 126.7, 125.1, 124.6, 123.2, 122.0, 87.9, 77.6, 77.2, 76.9, 74.6, 70.4, 63.5, 57.3, 42.8, 35.0, 30.7, 30.5, 29.9, 28.7, 27.1, 19.4; Anal. Calcd for C48H54N4O4SSi: C, 71.08; H, 6.71; N, 6.91; S, 3.95. Found: C, 71.14; H, 6.75; N, 6.95; S, 4.01.
To a solution of the thiocarbonate obtained above (13.82 g, 17.0 mmol) in toluene (200 ml) were added tri-n-butyltinhydride (9.4 ml, 34.1 mmol) and 2,2′-azo-bis-isobutyronitrile (4.32 g, 26.3 mmol), and the reaction mixture was stirred at 110° C. for 1 hour. After the mixture was cooled down, the solvent was removed. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=3/1) to give the compound 9 (9.21 g, 82%) as a white foam.
UV (MeOH) λmax 272.50 nm; [α]20 D −10.00 (c 0.20, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O2Si [M+H]+: 659.3781; Found: 659.3757; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.69 (m, 4H), 7.41 (m, 6H), 7.29 (m, 2H), 7.23 (m, 2H), 6.92 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.6 Hz, 1H), 5.90 (dd, J=7.2, 14.8 Hz, 1H), 5.38 (m, 1H), 5.15 (br s, 1H), 4.33 (dd, J=5.2, 8.4 Hz, 1H), 3.88 (dd, J=6.4, 10.0 Hz, 1H), 3.68 (dd, J=7.2, 10.4 Hz, 1H), 3.05 (m, 1H), 2.96 (dd, J=7.6, 15.6 Hz, 1H), 2.76 (m, 1H), 2.45 (d, J=5.2 Hz, 1H), 2.29 (m, 2H), 2.06 (m, 1H), 1.95 (m, 2H), 1.55 (s, 1H), 1.13 (s, 9H), 1.06 (s, 9H);13C NMR (100 MHz, CDCl3) δ 156.3, 151.9, 144.1, 143.9, 135.9, 135.8, 134.3, 129.8, 128.2, 127.8, 127.0, 125.1, 124.6, 121.8, 77.6, 77.2, 76.7, 73.5, 72.2, 63.6, 56.4, 52.8, 46.8, 42.8, 34.9, 34.5, 30.5, 28.6, 27.2, 28.7, 19.4; Anal. Calcd for C41H50N4O2Si: C, 74.73; H, 7.65; N, 8.30. Found: C, 74.79; H, 7.61; N, 8.25.
Step 7: Preparation of 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 10)

To a solution of the compound 9 (9.21 g, 13.97 mmol) in the mixture of THF and pyridine (1:1, 160 ml) was added dropwise pyridine hydrofluoride (18.42 ml, 190.0 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 1 hour. The mixture was neutralized with saturated aqueous NaHCO3 solution and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. Then, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/3) to give the compound 10 (5.63 g, 99%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −6.36 (c 1.10, MeOH); HR-MS (ESI): m/z calcd for C25H33N4O2 [M+H]+: 421.2604; Found: 421.2599; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.30 (d, J=7.6 Hz, 1H), 7.22 (d, J=7.2 Hz, 2H), 7.15 (t, J=6.8 Hz, 1H), 6.88 (d, J=3.2 Hz, 1H), 6.23 (d, J=3.6 Hz, 1H), 5.83 (dd, J=7.2, 15.2 Hz, 1H), 5.28 (m, 1H), 5.06 (m, 1H), 4.47 (dd, J=5.6, 10.4 Hz, 1H), 3.78 (m, 1H), 3.70 (m, 1H), 3.24 (t, J=5.2 Hz, 1H), 2.98 (m, 1H), 2.87 (m, 1H), 2.68 (m, 1H), 2.46 (m, 1H), 2.37 (m, 2H), 1.93 (m, 2H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.2, 151.8, 147.9, 143.9, 143.9, 128.3, 126.9, 125.1, 124.5, 121.9, 97.7, 77.6, 77.2, 76.9, 75.5, 74.9, 63.4, 56.4, 53.8, 44.2, 42.2, 34.9, 33.2, 30.5, 28.6; Anal. Calcd for C25H32N4O2: C, 71.40; H, 7.67; N, 13.32. Found: C, 71.46; H, 7.60; N, 13.35.
Step 8: Preparation of sulfamic acid 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 11)

Preparation of 2.0 M solution of chlorosulfonamide in acetonitrile: Formic acid (14.15 ml, 166.0 mmol) was added dropwise to chlorosulfonyl isocyanate (32.0 ml, 162.5 mmol) under nitrogen atmosphere at 0° C. When the addition was completed, the mixture was solidified. To the mixture was added acetonitrile (61.3 ml), and the resulting solution was left to stand under nitrogen source at room temperature overnight.
To a solution of the compound 10 (5.63 g, 13.83 mmol) and triethyl amine (9.7 ml, 0.74 mmol) in acetonitrile (278 ml) was added 2.0 M solution of chlorosulfonamide in acetonitrile (13.83 ml, 27.76 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 45 minutes. Additional 2.0 M chlorosulfonamide solution in acetonitrile (13.83 ml, 27.76 mmol) was added and the mixture was stirred at room temperature for 15 minutes. The reaction was quenched with methanol, and the solvent was removed. The residue was purified by silica gel column chromatography (methylene chloride/methanol=20/1) to give the compound 11 (6.37 g, 92%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −18.00 (c 0.50, MeOH); HR-MS (ESI): m/z calcd for C25H34N5O4S [M+H]+: 500.2332; Found: 500.2331; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.29 (d, J=7.2 Hz, 1H), 7.22 (m, 2H), 6.95 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.2 Hz, 1H), 5.89 (d, J=6.4 Hz, 1H), 5.10 (s, 2H), 4.41 (m, 2H), 4.26 (m, 1H), 3.05 (m, 1H), 2.94 (m, 1H), 2.76 (m, 2H), 2.27 (m, 3H), 2.06 (m, 1H), 1.97 (m, 1H), 1.76 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.9, 149.9, 143.9, 143.8, 128.3, 126.9, 125.1, 124.5, 121.9, 121.9, 103.5, 97.9, 77.4, 77.2, 76.9, 74.3, 71.9, 71.3, 56.4, 53.1, 49.0, 42.3, 34.9, 34.3, 30.5, 28.6; Anal. Calcd for C25H33N5O4S: C, 60.10; H, 6.66; N, 14.02; S, 6.42. Found: C, 60.15; H, 6.71; N, 13.98; S, 6.39.
Step 9: Preparation of sulfamic acid 2-hydroxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 1)

A solution of the compound 11 (6.37 g, 12.72 mmol) in 70% trifluoroacetic acid (149.24 ml) was stirred at room temperature for 2 hours. The solvent was removed and the residue was purified by silica gel column chromatography (hexane/ethylene acetate=1/2) to give the compound 1 (5.08 g, 90%) as a white foam. BASE
UV (MeOH) λmax 279.50 nm; [α]20 D −6.41 (c 2.34, MeOH);
HR-MS (ESI): m/z calcd for C21H26N5O4S [M+H]+: 444.1705; Found: 444.1706;
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J=1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J=3.6 Hz, 1H), 5.86 (t, J=7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J=2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1;
Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.
…………………….
http://www.google.com/patents/WO2010132110A1?cl=en
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (//) is described in Intl. App. Pub. No. WO 07/092213, U.S. App. Pub. No. 2007/0191293, and U.S. App. Pub. No. 2009/0036678. The potassium salt of ((lS,2S,4R)-4-{4-[( 1 S)-2,3-dihydro- 1 H-inden- 1 -ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl } -2-hydroxycyclopentyl)methyl sulfamate is disclosed in Intl. App. Pub. No. WO 07/092213 and U.S. App. Pub. No. 2007/0191293.

(H)
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH- inden-l-ylamino]-7H-pyπOlo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate (/):

Step 3: Synthesis of ((lS,2S.4R)-4-(4-r(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolor2.3-dlpyrimidin-7-yl}-2-hvdroxycvclopentyl)methyl sulfamate hydrochloride Form 1
[0158] A reactor was charged with ((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (13.4 Kg, 30.2 mol) and 200-proof ethanol (106.2 Kg). The mixture was heated to reflux to afford a clear solution. The mixture was cooled to 50 ± 5 0C and passed through a cartridge filter. 200 proof ethanol (8.9 Kg) was used to rinse the filter. 1.27M hydrogen chloride in ethanol (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then seeded with Form 1 (67 g). Further 1.27M HCl (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then stirred at 50 ± 5 0C for about 3 hours. The mixture was then cooled to 20 ± 5 0C over about 3 hours and then stirred for about 2.5 hours. The solid product was then isolated by filtration and washed with 200-proof ethanol (I x 20.4 Kg and 1 x 21.2 Kg). The solids were dried by aspiration on the filter until no supernatant was seen to be collected, and then further dried under reduced pressure at <30 0C to afford the title compound (12.2 Kg) as a white solid determined to be Form 1 by XRPD. IH NMR (300MHz, DMSO, δ): 9.83 (s, IH), 8.34 (s, IH), 7.62 (s, IH), 7.44 (s, 2H), 7.30 (m, 3H), 7.22 (t, IH), 7.07 (s, IH), 5.86 (dd, IH), 5.42 (m, IH), 4.32 (m, IH), 4.21 (dd, IH), 4.02 (dd, IH), 3.04 (m, IH), 2.88 (m, IH), 2.67 (m, 2H), 2.15 (m, 2H), 2.08 (m, 2H), 1.94 (m, IH). XRPD data for Form 1 is shown in FIGURE 1 and Table 1; DSC data is shown in FIGURE 2, and TGA data for Form 1 is shown in FIGURE 3.
…………..
http://www.google.com/patents/WO2007092213A2?cl=en
Example 70: Diastereoisomeric mixture of (lS/2R/4R)-4-{4-[(lS)-2/3-dihydro-lH-inden-l- ylaimnol-ZH-pyrrolop^-dlpyxirnidin-Z-ylJ^-hydroxycyclopentyl s ulf amate and (lRf2S/4S)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolo[2,3d]- pyrimidin-7-yl}-2-hydroxycyclopentyl sulfamate (Compounds 1-77 and 1-78)

Step a: Cyclopent-3-en-l-yl methanesulfonate
[0335] 3-Cydopentene-l-ol (0.500 g, 5.94 mmol) was stirred in DCM (95 mL).
Pyridine (2.40 mL), N,N-dimethylaminopyridine (0.10 g, 1.00 mmol) and methanesulfonyl chloride (0.690 mL, 8.92 mmol) were added, and the reaction mixture was stirred at 350C for 4 h. N,N-Dimethylarrιinopyridirιe (0.14 g, 1.2 mmol) and methanesulfonyl chloride (0.69 mL, 8.92 mmol) were added, and the reaction was stirred overnight. TLC indicated complete conversion. The reaction mixture was cooled and concentrated. The residue was purified by silica gel chromatography, eluting with DCM, to afford the title compound as a clear oil (0.660 g, 68%).
Step b: 7-Cyclopent-3-en-l-yl-N-r(lSV2,3-dihydro-lH-inden-l-yn-7H-pyrrolor2,3-rfl- pyrmτidin-4-arnine
[0336] N-[(lS)-2,3-DihydrcHlH-mden-l-yl]-7H-pyrrolo[2/3-d]p3αimidin-4-amine (1.32 g, 5.29 mmol) was azeotroped with toluene and placed under high vacuum for 30 min. N,N-Dimethylformamide (17.7 mL) was added, followed by cesium carbonate (1.99 g, 6.10 mmol). The mixture was stirred at 700C for 10 min. Cyclopent-3-en-l-yl methanesulfonate (0.660 g, 4.07 mmol) in N,N-dimethylformarnide (12.6 mL) was added dropwise. The reaction mixture was heated to 1100C for 1 h. The reaction mixture was cooled, quenched with brine and diluted with H2O. The aqueous layer was extracted with EtOAc (3x), washed with H2O and brine, dried (Na2SO4), filtered, and concentrated. The residue -was purified by via silica gel chromatography, eluting with a gradient of 0 to 5% MeOH in DCM followed by 25 to 50% EtOAc in hexanes, to afford the title compound as a pale brown solid (0.684 g, 53%). LC/MS: R1 = 1.38 min, ES+ 317 (FA standard). Step c: (lR,2S,45)-4-{4-r(lS)-2,3-dihydro-lH-inden-l-ylaininol-7H-pyrrolof2.3- rf1pyrimidin-7-yl}cyclopentane-l,2-diol
[0337] 7-Cyclopent-3-en-l-yl-N-[(lS)-2^-dihyrdo-lH-inden-l-yl]-7H-pyrrolo[2,3- d]pyτimidin-4-amine (0.312 g, 0.986 mmol) was stirred in tert-butyl alcohol (4.9 mL) and H2O (4.9 mL). AD-mix-α (Sigma- Aldrich, 1.4 g) was added, and the suspension was stirred at rt overnight. TLC indicated complete conversion. The reaction was quenched with sodium sulfite (1.48 g, 11.7 mmol), and the mixture was stirred for 5 h. The reaction mixture was diluted with EtOAc and H2O, and the aqueous layer was extracted with EtOAc (2x). The organic layer was dried (Na2SO4), filtered, and concentrated. The residue was purified via silica gel chromatography, eluting with EtOAc, to afford the title compound as a white solid (0.190 g, 55%).
Step d: Diastereoisomeric mixture of (lS,2R,4R)-4-{4-r(15)-23-dihydro-lH-inden-l- ylarninoi^jH-pyrrolofΣ^dlpyrirnidin-y-yll-l-hydroxycyclopentyl sulfamate and (lR,2S,4S)-4-{4-iαSV2,3-dihydro-lH-inden-l-ylarninol-7H-pyrrolor2,3- rf1pyrimidm-7-yl)-2-hydroxycyclopenryl sulfamate (Compounds 1-77 and 1-78)
[0338] (lR,2S,4S)-4-{4-[(lS)-2,3-Dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2/3- d]pyrimidin-7-ylJcyclopentane-l,2-diol (0.080 g, 0.23 mmol) was azeotroped with toluene and then was dissolved in anhydrous acetonitrile (2.3 mL). Pyridine (0.0369 mL, 0.458 mmol) was added. The reaction mixture was cooled to 00C, and a 2N solution of chlorosulfonamide in acetonitrile (0.144 mL) was added dropwise. The reaction was stirred for 1 h, and then additional 2N chlorosulfonamide in acetonitrile (0.028 mL) was added. After 30 min, additional 2N chlorosulfonamide in acetonitrile (0.0342 mL) was added, and the reaction mixture was stirred for 2 h. The reaction was quenched with methanol, and the mixture was concentrated in vacuo. The residue was purified by preparative thin layer chromatography using DCM:AcCN:MeOH (50:45:5). The relevant band was cut, washed with acetone, filtered, and concentrated to give a mixture of diastereomers as a white solid. (11 mg, 11%). 1H NMR (CDCl3, 400 NMR, δ): 8.36-8.27 (m, IH); 7.38-7.09 (m, 5H); 6.90-6.80 (m, IH); 6.36- 6.20 (m, IH); 5.95-5.76 (m, IH); 5.51-5.22 (m, 2H); 4.83-4.68 (m, IH); 3.87-3.72 (m, IH); 3.12- 2.83 (m, 2H); 2.75-2.53 (m, IH); 2.50-2.14 (m, 2H); 2.08-1.79 (m, 2H) ppm. LC/MS: R, = 1.16 min, ES+ 430 (FA standard).
…………
WO 2012061551
http://www.google.im/patents/WO2012061551A1?cl=en
The compound ((lS,2S,4R)-4-(4-((lS)-2,3-dihydro-lH-inden-l-ylamino)-7H-pyrrolo[2,3-d]- pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl sulfamate:

also known as MLN4924, is an inhibitor of NEDD8-activating enzyme (NAE). Inhibition of NAE has been shown to induce cancer cell death and inhibit the growth of tumors in xenograft models. See, e.g., T.A. Soucy et al., Nature, 2009, 458, 732-737; T.A. Soucy ei al., Clin. Cancer Res., 2009, 15 (12), 3912-3916; and J.E. Brownell et al., Mol. Cell., 2010, 37 (1), 102-111, each of which is hereby incorporated by reference herein in its entirety. MLN4924, pharmaceutical compositions of MLN4924, processes for its synthesis, and polymorphic forms have been described previously. See, e.g., US Patent Appl. Nos. 11/700,614 (Publ. No. 2007/0191293), 12/221,399 (Publ. No. 2009/0036678) and 12/779,331 (Publ. No. 2011/0021544),
……………
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00209, http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00209

A practical synthesis of a novel NEDD8-activating enzyme (NAE) inhibitor pevonedistat (MLN4924) is described. Key steps include an enantioselective synthesis of an amino-diol cyclopentane intermediate containing three chiral centers and a novel, regioselective sulfamoylation using N-(tert-butoxycarbonyl)-N-[(triethylenediammonium)sulfonyl]azanide. The linear process, involving six solid isolations, has been carried out in multiple cGMP productions on 15–30 kg scale to produce pevonedistat in 98% (a/a) chemical purity and 25% overall yield.

((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate (1)
The reaction yielded 1 (0.285 kg, 58.5%, 93.0% a/a) as an off-white solid.
HPLC retention time of 1 BASE(Method C): 22.6 min;
1H NMR (400 MHz, DMSO) δ 8.19 (s, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.45 (s, 2H), 7.31–7.26 (m, 2H), 7.22 (t, J = 6.6 Hz, 2H), 7.15 (t, J = 7.2 Hz, 1H), 6.66 (d, J = 3.5 Hz, 1H), 5.92 (q, J = 8.0 Hz, 1H), 5.39 (qd, J = 8.8, 5.7 Hz, 1H), 4.95 (d, J = 3.9 Hz, 1H), 4.42–4.31 (m, 1H), 4.25 (dd, J = 9.7, 7.0 Hz, 1H), 4.07 (dd, J = 9.6, 8.0 Hz, 1H), 3.01 (ddd, J = 15.7, 8.7, 3.0 Hz, 1H), 2.95–2.81 (m, 1H), 2.81–2.65 (m, 1H), 2.58–2.49 (m, 1H), 2.31–1.86 (m, 5H);
13C NMR (100 MHz, DMSO) δ 155.91, 151.18, 149.02, 144.66, 142.98, 127.30, 126.28, 124.49, 124.11, 121.68, 102.83, 98.86, 70.82, 69.37, 54.48, 52.15, 42.58, 42.25, 33.50, 33.26, 29.72;
m/z: 444.4 (M + H)+;
mp: 164–166 °C.
((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate·Hydrochloride (Pevonedistat)
Pevonedistat (14.0 g, 92.5%, 99.0% a/a) as a white solid.
HPLC retention time of pevonedistat (Method C): 22.6 min;
1H NMR (400 MHz, DMSO) δ 9.70 (s, 1H), 8.39 (s, 1H), 7.63 (s, 1H), 7.45 (s, 2H), 7.41–7.20 (m, 4H), 7.04 (s, 1H), 5.78 (s, 1H), 5.44 (s, 1H), 4.42–4.28 (m, 1H), 4.24 (dd, J = 9.7, 6.9 Hz, 1H), 4.05 (dd, J = 9.6, 8.0 Hz, 1H), 3.18–2.99 (m, 1H), 2.91 (dt, J = 15.6, 7.7 Hz, 1H), 2.81–2.57 (m, 2H), 2.24–1.86 (m, 6H).
13C NMR (100 MHz, DMSO) δ 149.12, 145.71, 143.23, 142.11, 141.30, 128.28, 126.64, 124.97, 124.82, 124.49, 102.57, 101.74, 70.67, 69.22, 57.38, 53.14, 42.52, 42.40, 33.57, 32.56, 29.80;
m/z: 444.4 (M + H)+;
mp: 155–157 °C.
……………..
J. Org. Chem., 2011, 76 (9), pp 3557–3561
DOI: 10.1021/jo2001897

MLN4924 (1), which is in clinical trials as an anticancer agent, was stereoselectively synthesized from d-ribose via a route involving stereoselective reduction, regioselective cleavage of an isopropylidene moiety, and selective displacement of a cyclic sulfate moiety as key steps.
Sulfamic Acid 2-Hydroxy-4-[4-(indan-1-ylamino)pyrrolo[2,3-d]pyrimidin-7-yl]cyclopentylmethyl Ester (1) BASE
purified by silica gel column chromatography (hexane/ethyl acetate = 1/2) to give 1 (5.08 g, 90%) as a white foam:
UV (MeOH) λmax 279.50 nm;
[α]20D −6.41 (c 2.34, MeOH);
HR-MS (ESI) m/z calcd for C21H26N5O4S [M + H]+ 444.1705, found 444.1706;
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J = 1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J = 3.6 Hz, 1H), 5.86 (t, J = 7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J = 2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1. Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.



NMR FROM CHEMIETEK
| WO2012061551A1 * | Nov 3, 2011 | May 10, 2012 | Millennium Pharmaceuticals, Inc. | Administration of nedd8-activating enzyme inhibitor |
| WO2013028832A2 * | Aug 23, 2012 | Feb 28, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| WO2013028832A3 * | Aug 23, 2012 | May 2, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| US8809356 | Aug 23, 2012 | Aug 19, 2014 | Millennium Pharmaceuticals, Inc. | Inhibitors of NEDD8-activating enzyme |
| Reference | ||
|---|---|---|
| 1 | * | Lee, et. al., Journal of Organic Chemistry (2011), 76(9), 3557-3561. |
1H NMR PREDICT

13 C NMR


//////////Pevonedistat, MLN4924, Millennium Pharmaceuticals, TAKEDA, TAK-924 , PHASE 1, orphan drug designation
Tazemetostat

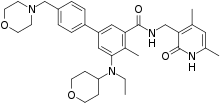
Tazemetostat
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
HBR 1467052-75-0
タゼメトスタット臭化水素酸塩
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
HBR
Chemical Formula: C34H44N4O4
Exact Mass: 572.33626
USFDA APPROVED 23/1/2020 AS HBR SALT, TAZVERIK, EPIZYME
N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[1,1′-biphenyl]-3-carboxamide
SIMLES: O=C(C1=CC(C2=CC=C(CN3CCOCC3)C=C2)=CC(N(CC)C4CCOCC4)=C1C)NCC5=C(C)C=C(C)NC5=O
(1,1′-Biphenyl)-3-carboxamide, N-((1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(4-morpholinylmethyl)-
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(oxan-4-yl)amino)-4-methyl-4′-((morpholin-4-yl)methyl)(1,1′-biphenyl)-3-carboxamide
UNII-Q40W93WPE1
Tazemetostat, sold under the brand name Tazverik, is a medication used for the treatment of adults and adolescents aged 16 years and older with metastatic (when cancer cells spread to other parts of the body) or locally advanced (when cancer has grown outside the organ it started in, but has not yet spread to distant parts of the body) epithelioid sarcoma not eligible for complete resection (surgically removing all of a tissue, structure, or organ).[1]
Tazemetostat is a cancer drug that acts as a potent selective EZH2 inhibitor.[2]
Tazemetostat blocks activity of the EZH2 methyltransferase, which may help keep the cancer cells from growing.[1] Most cases of epithelioid sarcoma begin in the soft tissue under the skin of an extremity, though it can start in other areas of the body.[1] Surgical removal is considered the main treatment when the cancer is localized to one area of the body.[1] Chemotherapy or radiation may also be given.[1] However, there is a high likelihood for local and regional spread of the disease even with treatment and approximately 50% of patients have metastatic disease at the time of diagnosis.[1] Metastatic disease is considered life-threatening to the patient.[1]
The most common side effects are pain, fatigue, nausea, decreased appetite, vomiting and constipation.[1] People taking tazemetostat are at increased risk of developing secondary malignancies including: T-cell lymphoblastic lymphoma (a type of blood cancer that affects the lymphatic system usually found in the lymph nodes), myelodysplastic syndrome (a disorder resulting from poorly formed or dysfunctional blood cells) and acute myeloid leukemia (a cancer of the blood and bone marrow).[1]
According to the NCI Drug Dictionary, “tazemetostat is an orally available, small molecule selective and S-adenosyl methionine (SAM) competitive inhibitor of histone methyl transferase EZH2, with potential antineoplastic activity. Upon oral administration, tazemetostat selectively inhibits the activity of both wild-type and mutated forms of EZH2. Inhibition of EZH2 specifically prevents the methylation of histone H3 lysine 27 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways and results in decreased tumor cell proliferation in EZH2 mutated cancer cells. EZH2, which belongs to the class of histone methyltransferases (HMTs), is overexpressed or mutated in a variety of cancer cells and plays a key role in tumor cell proliferation.”[3]
History
The U.S. Food and Drug Administration (FDA) approved tazemetostat in January 2020,[1] based on the results of a clinical trial (NCT02601950) enrolling 62 subjects with metastatic or locally advanced epithelioid sarcoma.[1][4] During the clinical trial, subjects received 800 milligrams (mg) of tazemetostat twice a day until the disease progressed or the subject reached an unacceptable level of toxicity.[1][4] Tumor response assessments were performed every eight weeks during the clinical trial.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] The overall response rate was 15%, with 1.6% of subjects having a complete response and 13% having a partial response.[1] Of the nine subjects that had a response, six (67%) subjects had a response lasting six months or longer.[1]
The trial was conducted at 22 sites in France, United Kingdom, Taiwan, Italy, Canada, Belgium, and the United States.[4]
The FDA granted the application for tazemetostat accelerated approval and orphan drug designation.[1] The FDA granted the approval of Tazverik to Epizyme Inc.[1]
PATENT
PRODUCT PAT
US 8410088 EXP 21/1/2034
WO 2012142504
US 9090562 EXP 13/4/32
SEE Proceedings of the National Academy of Sciences of the United States of America (2013), 110(19), 7922-7927, S7922/1-S7922/5….http://www.pnas.org/content/110/19/7922.abstract
http://www.epizyme.com/wp-content/uploads/2014/11/Ribrag-ENA-FINAL.pdf
Tazemetostat, also known as EPZ-6438, is a potent, selective, and orally bioavailable small-molecule inhibitor of EZH2 enzymatic activity. EPZ-6438 induces apoptosis and differentiation specifically in SMARCB1-deleted MRT cells.
Treatment of xenograft-bearing mice with EPZ-6438 leads to dose-dependent regression of MRTs with correlative diminution of intratumoral trimethylation levels of lysine 27 on histone H3, and prevention of tumor regrowth after dosing cessation.
These data demonstrate the dependency of SMARCB1 mutant MRTs on EZH2 enzymatic activity and portend the utility of EZH2-targeted drugs for the treatment of these genetically defined cancers. EPZ-6438 is currently in clinical trials.
Epizyme, Inc., Eisai R&D Management Co.Ltd.
Epizyme is developing tazemetostat, a lead from several small molecule EZH2 inhibitors, for treating cancer (phase 1 clinical, as of April 2015). Japanese licensee Eisai was developing the program for the potential oral treatment of cancers, including non-Hodgkin’s lymphoma; however, in March 2015, Epizyme regained worldwide, ex-Japan, rights to the program.
It appeared that Eisai was planning to investigate the program in Japan .
WO-2015057859 From, Eisai Research Institute; Epizyme Inc, indicates Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
see WO2013155317, claiming novel hydrobromide salt of tazemetostat.
PREDICT

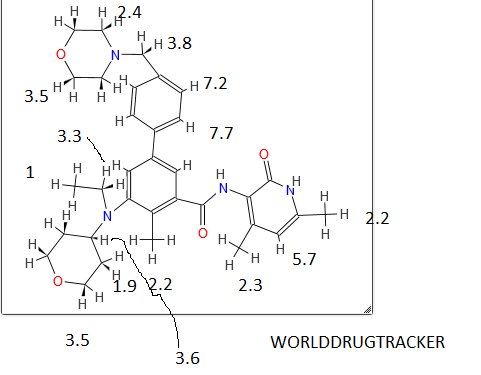
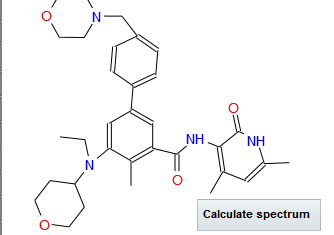
………………………………….
PATENT
WO 2012142504
http://www.google.com/patents/WO2012142504A1?cl=en

Example 44: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(moφholinomethyl)-[l , – biphenyl]-3-carboxamide

Compound 44
[Step 1 : Synthesis of 5-brom -2-methyl-3-nitrobenzoic acid

To stirred solution of 2-methyl-3-nitrobenzoic acid ( 100 g, 552 mmol) in cone. H2S04 (400 mL), 1 ,3-dibromo-5,5-dimethyl-2,4-imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid ( 140 g, 98%). The isolated compound was taken directly into the next step. Ή NMR (DMSO-4$, 400 MHz) δ 8.31 (s, 1 H), 8.17 (s, 1 H), 2.43 (s, 3H).
Step 2: Synthesis of methyl -bromo-2-methyl-3-nitrobenzoate

To a stirred solution of 5-bromo-2-methyl-3-nitrobenzoic acid (285 g, 1 105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate ( 1 L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).
Step 3: Synthesis of methyl 3-amino-5-bromo-2-methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-nitrobenzoate (290 g,
1058 mmol) in ethanol (1 .5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in-vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 7.37 (s, 1 H), 6.92 (s, 1 H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).
Step 4: Synthesis of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate

To a stirred solution of methyl 3-amino-5-bromo-2-methylbenzoate (15 g, 61 .5 mmol) and dihydro-2H-pyran-4(3)-one (9.2 g, 92 mmol) in dichloroethane (300 mL) was added acetic acid (22 g, 369 mmol) and the reaction mixture stirred at room temperature for 15 minutes, then the reaction mixture was cooled to 0°C and sodium triacetoxyborohydnde (39 g, 184 mmol) was added. The reaction mixture was stirred overnight at room temperature. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH of 7-8 was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a solid ( 14 g, 69%). ‘H NMR (DMSO-<fc, 400 MHz) δ 7.01 (s, 1 H), 6.98 (s, 1 H), 5.00 (d, 1 H, J=7.6 Hz), 3.84-3.87 (m, 2H), 3.79 (s, 31 1), 3.54-3.56 (mf 1 H), 3.43 (L 21 1, J 12 Hz), 2.14 (s. 31 1). 1 . 1 – 1 .84 (m: 211). 1 .47- 1 .55 (m, 2H).
Step 5: Synthesis of methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid ( 15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium
triacetoxyborohydnde (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). Ή NMR (DMSO-cfo 400 MHz) δ 7.62 (s, 1 H), 7.52 (s, 1 H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1 H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1 .37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).
Step 6: Synthesis of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3 -(ethyl (tetrahydro-2H-pyra -4-yl) amino)-2-methylbenzamide

To a stirred solution of 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate (14 g, 39.4 mmol) in ethanol ( 100 mL) was added aqueous NaOH (2.36 g, 59.2 mmol in 25mL water) and the resulting mixture was stirred at 60 °C for 1 h. Upon completion of the reaction as determined by TLC, the solvent was removed under reduced pressure and the residue obtained was acidified with IN HC1 until a pH 7 was obtained and then aqueous citric acid solution was added until a pH 5-6 was obtained. The aqueous layer was extracted with 10% MeOH in DCM (200mL X 3), the combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the respective acid (14 g, 100%).
The above acid (14 g, 40.9 mmol) was then dissolved in DMSO (70 mL) and 3- (amino methyl)-4, 6-dimethylpyridin-2( l H)-one ( 12.4 g, 81 .9 mmol) was added to it. The reaction mixture was stirred at room temperature for 15 minutes, then PYBOP (31.9 g, 61.4 mmol) was added and stirring was continued for overnight at room temperature. Upon completion of the reaction as determined by TLC, the reaction mixture was poured onto ice- cold water (700 mL), stirred for 30 minutes and the precipitated solid was collected by filtration, washed with water (500 mL) and air dried. The solid obtained was stirred with acetonitrile (75mL X 2), filtered and air dried. The solid obtained was again stirred with 5% MeOH in DCM ( l OOmL), filtered and dried completely under vacuum to afford the title compound as a solid ( 14 g, 74 %). Ή NMR (DMSO- 6, 400 MHz) δ 1 1.47 (s, 1 H), 8.23 (t, 1 H), 7.30 (s, 1 H), 7.08 (s, 1 H), 5.85 (s, 1 H), 4.23 (d, 2H, J=4.4 Hz), 3.81 (d, 2H, J=l 0.4 Hz), 3.20-3.26 (m, 2H), 3.00-3.07 (m, I H), 2.91 -2.96 (m, 2H), 2.18 (s, 3H), 2.14 (s, 3H), 2.10 (s, 3H), 1.58-1.60 (m, 2H), 1.45-1.50 (m, 2H), 0.78 (t, 3H, J=6.8 Hz).
Step 7: Synthesis of N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl) amino)-4-methyl-4′-(morpholinomethyl)-[l , l ‘-biphenyl]-3- carboxamide
 TITLE COMPD
TITLE COMPDTo a stirred solution of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzamide (14 g, 29.5 mmol) in dioxane/ water mixture (70 mL/ 14 mL) was added 4-(4-(4, 4, 5, 5-tetramethyl- l , 3, 2- dioxaborolan-2-yl) benzyl) morpholine (13.4 g, 44.2 mmol) followed by addition of Na2C03 (1 1 .2 g, 106.1 mmol). The solution was purged with argon for 15 minutes and then Pd (PPh3)4 (3.40 g, 2.94 mmol) was added and the solution was again purged with argon for a further 10 min. The reaction mixture was heated at 100°C for 4 h. After completion (monitored by TLC), the reaction mixture was diluted with water and extracted with 10% MeOH/DCM.
The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with methanol: DCM to the title compound as a solid (12 g, 71 %).
Analytical Data: LCMS: 573.35 (M + 1 )+; HPLC: 99.5% (@ 254 nm) (R,;3.999; Method: Column: YMC ODS-A 1 50 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol : 10 μΐ, Col. Temp.: 30 °C; Flow rate: 1 .4 mL/min.;
Gradient: 5% B to 95% B in 8 min, Hold for 1 .5 min, 9.51 -12 min 5% B);
Ή NMR (DMSO-i 6, 400 MHz) 5 1 1 .46 (s, I H), 8. 19 (t, 1 H), 7.57 (d, 2H, J=7.2 Hz), 7.36-7.39 (m, 3H), 7.21 (s, I H), 5.85 (s, I H), 4.28 (d, 2H, J=2.8 Hz), 3.82 (d, 2H, J=9.6 Hz), 3.57 (bs, 4H), 3.48 (s, 2H), 3.24 (t, 2H, J=10.8Hz), 3.07-3.09 (m, 2H), 3.01 (m, I H), 2.36 (m, 4H), 2.24 (s, 3H), 2.20 (s, 3H), 2.10 (s, 3H), 1 .64-1 .67 (m, 2H), 1 .51 – 1 .53 (m, 2H), 0.83 (t, 3H, J=6.4 Hz).
TRIHYDROCHLORIDE
Step 8: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3-yl)methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[ 1 , 1 ‘-biphenyl]-3- carboxamide trihydrochloride

N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5-(ethyl (tetrahydro- 21 l-pyran-4-yl) amino)-4-methyI-4′-(niorpholinornethyl)-[ 1 , 1 ‘-biphenyl]-3-carboxamide ( 12 g, 21.0 mmol) was dissolved in methanolic HC1 (200 mL) and stirred at room temperature for 3 h. After three hours of stirring, the reaction mixture was concentrated under reduced pressure. The solid obtained was stirred with ether ( l OOmL X 2) to afford the desired salt as a solid ( 1 1 g, 77 %).
Analytical Data of the tri-HCl salt: LCMS: 573.40 (M + 1 )+; HPLC: 99.1 % (@ 254 nm) (R,;3.961 ; Method: Column: YMC ODS-A 150 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol: 10 pL, Col. Temp.: 30 °C; Flow rate: 1.4 mL/min.; Gradient: 5% B to 95% B in 8 min, Hold for 1.5 min, 9.51 -12 min 5% B);
Ή NMR (D20 400 MHz) δ 7.92 (bs, I H,) 7.80 (s, I H), 7.77 (d, 2H, J=8 Hz), 7.63 (s, I H), 7.61 (s, I H), 6.30 (s, I H), 4.48 (s, 2H), 4.42 (s, 2H), 4.09-4.1 1 (m, 4H), 3.95-3.97 (m, 2H), 3.77 (t, 3H, J=10.4 Hz), 3.44-3.47 (m, 3H), 3.24-3.32 (m, 3H), 2.42 (s, 3H), 2.35 (s, 3H), 2.26 (s, 3H), 2.01 (m, 2H), 1 .76 (m, 2H), 1 .04 (t, 3H, J=6.8 Hz).
…………………………………………
PATENT
WO2013155317
http://www.google.com/patents/WO2013155317A1?cl=en
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl] -3-carboxamide hydrobromide:

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl
(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3- carboxamide hydrobromide:

As used herein, “Compound I” refers to N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxamide. The hydrobromide of Compound I can be used to inhibit the histone methyltransferase activity of EZH2, either in a subject or in vitro. The hydrobromide of Compound I can also be used to treat cancer in a subject in need thereof.
Scheme 1

……………………………………..Compound I Compound I – HBr
HPLC
HPLC was conducted on an Agilent 1200 HPLC quaternary pump, low pressure mixing, with an in-line degasser. Analytical method conditions: 8 μΐ^ sample (20 mg of ER-581982-06 diluted with 50 mL of a methanol to provide approximately 0.4 mg/mL solution) was injected onto a Agilent Zorbax Eclipse XDB-C18 (4.6 x 150 mm, 3.5 um), Chromatography conditions: mobile phase A, water with 5mM ammonium formate; mobile phase B, 5 mM ammonium formate in 50/45/5 acetonitrile/methanol/water; flow rate, 1.5 ml/min.; gradient: isocratic at 10% B from 0 to 3 min; linear increase to 70% B from 3 to 7 min; isocratic at 70% B from 7 to 12 min; linear increase to 100% B from 12 to 15 min isocratic at 100% B from 15 to 20 min;
column temperature, 35 °C; detection, UV 230 nm. Approximate retention time of Compound I = 10.7 min.
Synthesis of Polymorph A

5-bromo-2-methyl-3-nitrobenzoic acid stirred solution of 2-methyl-3-nitrobenzoic acid (100 g, 552 mmol) in cone. H2S04 (400 mL), l,3-dibromo-5,5-dimethyl-2,4- imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid (140 g, 98%). The isolated compound was taken directly into the next step. 1H NMR (DMSO-J6, 400 MHz) δ 8.31 (s, 1H), 8.17 (s, 1H), 2.43 (s, 3H).

Methyl 5-bromo-2-methyl-3-nitrobenzoate To a stirred solution of 5-bromo-2- methyl-3-nitrobenzoic acid (285 g, 1105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate (1L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. 1H NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).

Methyl 3-amino-5-bromo-2-methylbenzoate (1) To a stirred solution of methyl 5- bromo-2-methyl-3-nitrobenzoate (290 g, 1058 mmol) in ethanol (1.5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in- vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. 1H
NMR (CDCI3, 400 MHz) δ 7.37 (s, 1H), 6.92 (s, 1H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).

Methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (2) A reactor was charged with methyl 3-amino-5-bromo-2-methylbenzoate (455.8 g, 1.87 mol), 1,2- Dichloroethane (4.56 L), and acetic acid (535 ml, 9.34 mol). To the mixture were added dihydro-2H-pyran-4(3H)-one (280 g, 2.80 mol) and sodium triacetoxyborohydride (594 g, 2.80 mol) maintaining the internal temperature below 40 °C. The mixture was stirred at 25 °C for 2.5 h and then the reaction was quenched with a solution of sodium hydroxide (448 g, 11.20 mol) in water (5.61 L). After stirring for 20 minutes at ambient temperature, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (3.65 L). The organic layers were combined, washed with brine (1.5 L), and concentrated under vacuum.
The residue was treated with ethyl acetate (1.8 L) and heated to 65-70 °C. The mixture was stirred at 65-70 °C for 15 minutes to give a clear solution and then treated with n-heptane (7.3 L) maintaining the temperature between 60-70 °C. Once the heptane was completely added to the solution, the mixture was held at 65-70 °C for 15 minutes and then allowed to cool to 18- 22 °C over 3 h. The resulting suspension was stirred at 18-22 °C for 4 h, cooled to 0-5 °C over 1 h, and held at 0-5 °C for 2 h. The precipitate was filtered, washed twice with n-heptane (1.4 L), and dried under vacuum to give the title compound (540 g, 88%). The XRPD pattern of this compound is shown in Figure 17.

Methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzoate (3)
To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid (15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium triacetoxyborohydride (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). 1H NMR DMSO-d6, 400 MHz) δ 7.62 (s, 1H), 7.52 (s, 1H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1.37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).

Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-
[l,l’-biphenyl]-3-carboxylate (4): A mixture of methyl 5-bromo-3-(ethyl(tetrahydro-2H-pyran- 4-yl)amino)-2-methylbenzoate (580 g, 1.63 mol), 4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)benzyl)morpholine (592 g, 1.95 mol), 1,4-dioxane (3.86 L), sodium carbonate (618 g, 5.83 mol), and water (771 ml) was degassed by bubbling nitrogen through the mixture at 20 °C for 20 minutes and treated with tetrakis(triphenylphosphine)palladium(0) (14.11 g, 12.21 mmol). The resulting mixture was degassed for an additional 20 minutes and then heated to 87-89 °C for 17 h. After cooling to 20 °C, the mixture was diluted with ethyl acetate (5.80 L) and a solution of (R)-2-Amino-3-mercaptopropionic acid (232 g) in water (2.320 L). After stirring for 1 h at 20 °C, the organic layer was separated and washed again with a solution of (R)-2-Amino-3- mercaptopropionic acid (232 g) in water (2.320 L). The aqueous layers were combined and extracted with ethyl acetate (5.80 L). The organic layers were combined, washed with a solution of sodium hydroxide (93 g) in water (2.32 L), and concentrated under vacuum at 35 °C to give the title compound as an orange oil (1.21 kg, 164% yield).

5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxylic acid (5): Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylate (69.0 g, 152.5 mmol) (based on the theoretical yield from the previous step) was suspended in ethanol (380 mL) and treated with a solution of sodium hydroxide (24.84 g, 621.0 mmol) in water (207 mL). The mixture was stirred at 40°C for 18 h. After cooling to 0-5 °C, the mixture was neutralized to pH 6.5 with 1 N hydrochloric acid (580 mL) maintaining the temperature below 25 °C. Then, the mixture was extracted twice with a mixture of dichloromethane (690 mL) and methanol (69.0 mL). The organic layers were combined and concentrated under vacuum to give a crude product as a yellow solid (127g).
The crude product was dissolved in 2-methyltetrahydrofuran (656 mL) at 70 °C and then treated with IPA (828 mL). The mixture was allowed to cool to rt over 3-4 h and then stirred overnight at rt. The precipitate was filtered, washed twice with IPA (207 mL), and dried under vacuum to give the title compound as an off white solid (53.54 g, 80%). The XRPD pattern of this compound is shown in Figure 9.

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H- pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3-carboxamide
(Compound I): A mixture of 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylic acid (540 g, 1.23 mol) and 3-(aminomethyl)- 4,6-dimethyl-dihydro-pyridin-2(lH)-one hydrochloride (279 g, 1.48 mol) was suspended in DMSO (2.70 L) and treated with triethylamine (223 ml, 1.60 mol). The mixture was stirred at 25 °C for 30 min and treated with EDC-HC1 (354 g, 1.85 mol) and HOBT hydrate (283 g, 1.85 mol). The reaction mixture was stirred at rt for 16 h. After addition of triethylamine (292 ml, 2.09 mol), the mixture was cooled to 15 °C, diluted with water (10.1 L) maintaining the temperature below 30 °C, and stirred at 19-25 °C for 4 h. The resulting precipitate was filtered, washed twice with water (2.70 L), and dried under vacuum to give a crude product (695 g, wt-wt analysis = 78%).
For the further purification of the product, recrystallization was conducted. A crude product (20.00 g, 34.92 mmol) was suspended in a mixture of ethanol (190 ml) and water (10.00 ml) and heated to 75°C until a clear solution was obtained. The solution was allowed to cool to rt overnight. The precipitate was filtered, washed twice with a mixture of ethanol (30.0 ml) and water (30.0 ml), and dried under vacuum at 35 °C to give the title compound as an off white solid (14.0 g, 70% recovery from the crude and 90% yield based on wt-wt assay).

4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′- (ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4-yl)methyl)morpholin- 4-ium bromide (Polymorph A): A crude N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)am
biphenyl]-3-carboxamide (595 g, 464 g based on wt-wt assay, 810.3 mmol) was suspended in ethanol (3.33 L). After heating to 70 °C, the mixture was treated with 48% aqueous HBr (97 ml, 850.8 mmol) and stirred at 70 °C for 30 min. The resulting orange-red solution was treated with ethyl acetate (3.33 L) maintaining the temperature above 60 °C. The mixture was slowly cooled to rt over 18 h. The mixture was cooled to 0 °C over 1 h and stirred at that temperature for 5.5 h. The resulting precipitate was filtered, washed twice with ethyl acetate (1.39 L), and dried under vacuum to give the title compound as an off white solid (515 g, 97% yield).
Recrystallization of Polymorph A: 4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4- yl)methyl)morpholin-4-ium bromide (0.50 g, 0.77 mmol; 95.6% pure by HPLC) was suspended in ethanol (3.0 mL) and heated to 80 °C until a clear solution was obtained. To the solution was added MTBE (5.0 mL) slowly. The resulting solution was allowed to cool to 18-22 °C over 3 h and stirred at 18-22 °C for 15 h. The precipitate was filtered, washed twice with MTBE (2 mL) and dried under vacuum to give 0.45 g of the title compound (89% recovery, 96.6% pure by HPLC).
Compound I is protonated at the nitrogen of the morpholino substituent, providing a monohydrobromide of Compound I having the following structure:

This particular monohydrobromide can be referred to as “4-((3′-(((4,6-dimethyl-2-oxo- l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′- methyl-[l, -biphenyl]-4-yl)methyl)morpholin-4-ium bromide.” Figure 11 depicts the X-ray crystal structure of this particular salt form.
…………………………………………………………..
see
WO-2015057859
Eisai Research Institute; Epizyme Inc
Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
…………………
Synthesis
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| USA | Tazverik | Epizyme, 2020 |
Formulations
- oral tabs. and suspension
References
-
- Knutson, S. K. et al., Proc. Natl. Acad. Sci USA, (2013) 110(19), 7922-7927.
- WO 2012 142504 (Epizyme/Eisai Co; 18.10.2012; appl. 13.4.2012; USA-prior. 13.4.2011).
- WO 2013 155317 (Epizyme/Eisai Co; 17.10.2013; appl. 11.4.2013).
- WO 2015 057859 (Epizyme/Eisai Co; 23.4.2015; appl. 15.10.2014; USA-prior. 16.10.2013).
-
EZH2 inhibitors for treating lymphona:
- WO 2015 195848 (Epizyme; 23.12.2015; appl. 17.6.2015; USA-prior. 17.6.2014).
////////
PAPER
RSC Advances (2015), 5(33), 25967-25978
http://pubs.rsc.org/en/content/articlelanding/2015/ra/c5ra02365c#!divAbstract
RSC Adv., 2015,5, 25967-25978,
DOI: 10.1039/C5RA02365C
The histone lysine methyltransferase EZH2 has been implicated as a key component in cancer aggressiveness, metastasis and poor prognosis. This study discovered a new class of hexahydroisoquinolin derivatives as EZH2 inhibitors. A structure–activity relationship study showed that the steric hindrance was important to the activity for EZH2. A preliminary optimization study led to the discovery of several potent compounds with low nanomolar to sub-nanomolar potency for EZH2. Biological evaluation indicated that SKLB1049 was a highly potent with improved solubility compared to EPZ6438, SAM-competitive, and cell-active EZH2 inhibitor that decreased global H3K27me3 in SU-DHL-6 and Pfeiffer lymphoma cells in a concentration- and time-dependent manner. Further study indicated that SKLB1049 caused cell arrest in G0/G1 phase. These compounds would be useful as chemical tools to further explore the biology of EZH2 and provided us with a start point to develop new EZH2 inhibitors.

|
In vitro protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
In vivo protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
References |
1: Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, Copeland RA, Pollock RM, Kuntz KW, Raimondi A, Keilhack H. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS One. 2014 Dec 10;9(12):e111840. doi: 10.1371/journal.pone.0111840. eCollection 2014. PubMed PMID: 25493630; PubMed Central PMCID: PMC4262195.
2: Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014 Apr;13(4):842-54. doi: 10.1158/1535-7163.MCT-13-0773. Epub 2014 Feb 21. PubMed PMID: 24563539
3. Inhibitors of human histone methyltransferase EZH2, and methods of use thereof for treating cancer. By Kuntz, Kevin W.; Knutson, Sarah K.; Wigle, Timothy James Nelson . From U.S. Pat. Appl. Publ. (2013), US 20130040906 A1 20130214.
4. Aryl-or heteroaryl-substituted benzamide compounds as anticancer agents and their preparation By Kuntz, Kevin Wayne; Chesworth, Richard; Duncan, Kenneth William; Keilhack, Heike; Warholic, Natalie; Klaus, Christine; Zheng, Wanjun; Seki, Masashi; Shirotori, Syuji; Kawano, Satoshi From PCT Int. Appl. (2012), WO 2012142504 A1 20121018.
5: Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. doi: 10.1073/pnas.1303800110. Epub 2013 Apr 25. PubMed PMID: 23620515; PubMed Central PMCID: PMC3651445.
| WO2013155317A1 * | Apr 11, 2013 | Oct 17, 2013 | Epizyme, Inc. | Salt form of a human hi stone methyltransf erase ezh2 inhibitor |
| WO2013155464A1 * | Apr 12, 2013 | Oct 17, 2013 | Epizyme, Inc. | Combination therapy for treating cancer |
| WO2014049488A1 * | Sep 16, 2013 | Apr 3, 2014 | Pfizer Inc. | Benzamide and heterobenzamide compounds |
| WO2014062732A1 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014062733A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014172044A1 * | Mar 14, 2014 | Oct 23, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015004618A1 * | Jul 9, 2014 | Jan 15, 2015 | Glaxosmithkline Intellectual Property (No.2) Limited | Enhancer of zeste homolog 2 inhibitors |
| WO2015010049A1 * | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015010078A2 | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted 6,5-fused bicyclic heteroaryl compounds |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012142504A1 * | Apr 13, 2012 | Oct 18, 2012 | Eisai Co., Ltd. | Aryl-or heteroaryl-substituted benzene compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014062720A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Methods of treating cancer |
| WO2011140324A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indoles |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012005805A1 * | May 5, 2011 | Jan 12, 2012 | Glaxosmithkline Llc | Azaindazoles |
| US4522811 | Jul 8, 1982 | Jun 11, 1985 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US5763263 | Jul 24, 1996 | Jun 9, 1998 | Dehlinger; Peter J. | Method and apparatus for producing position addressable combinatorial libraries |
| US7563589 | May 27, 2005 | Jul 21, 2009 | The University Of North Carolina At Chapel Hill | Including EED, EZH2 and SUZ12 wherein the reconstituted complex has histone methyltransferase (HMTase) activity for lysine 27 of histone H3 (H3-K27); cancer |
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment option specifically for patients with epithelioid sarcoma, a rare soft tissue cancer”. U.S. Food and Drug Administration (FDA) (Press release). 23 January 2020. Retrieved 23 January 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Lue JK, Amengual JE (October 2018). “Emerging EZH2 Inhibitors and Their Application in Lymphoma”. Curr Hematol Malig Rep. 13 (5): 369–382. doi:10.1007/s11899-018-0466-6. PMID 30112706. S2CID 52010283.
- ^ “Tazemetostat”. NCI Drug Dictionary. National Cancer Institute.
- ^ Jump up to:a b c “Drug Trials Snapshots: Tazverik”. U.S. Food and Drug Administration (FDA). 23 January 2020. Retrieved 22 February 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Tazemetostat”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Tazverik |
| Other names | EPZ-6438 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620018 |
| License data |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C34H44N4O4 |
| Molar mass | 572.750 g·mol−1 |
| 3D model (JSmol) | |
About EPZ-‐6438 Epizyme is developing EPZ-‐6438, a small molecule inhibitor of EZH2 created with our
proprietary product platform, for the treatment of non-‐Hodgkin lymphoma patients and patients with INI1-‐deficient solid tumors. In many human cancers, misregulated EZH2 enzyme activity results in misregulation of genes that control cell proliferation—without these control mechanisms, cancer cells are free to grow
About Epizyme, Inc.
Epizyme, Inc. is a clinical stage biopharmaceutical company creating novel epigenetic therapeutics for cancer patients.
Epizyme has built a proprietary product platform that the company uses to create small molecule inhibitors of a 96 member class of enzymes known as histone methyltransferases, or HMTs. HMTs are part of the system of gene regulation, referred
to as epigenetics, that controls gene expression. Genetic alterations can result in changes to
the activity of HMTs, making them oncogenic (cancer -‐causing). By focusing on the genetic drivers of cancers, Epizyme’s targeted science seeks to match the right medicines with the right patients.
Epizyme®, Inc.
400 Technology Square, 4th Floor
Cambridge, MA 02139
Phone: (617) 229-5872
Fax: (617) 349-0707
contact@Epizyme.com
![]()

Victoria Richon, vice president of biological sciences, Epizyme Inc.


 Jason Rhodes (left) has been appointed to president of Epizyme Inc.,
Jason Rhodes (left) has been appointed to president of Epizyme Inc.,

100 Technology Square

Central Square – The square – Cambridge, MA, United States
/////////TAZVERIK, EPIZYME, FDA 2020, APPROVALS 2020, Tazemetostat, EPZ-6438, EPZ 6438
K 912, NC 6300, Epirubicin nano



Cancer Research UK

Irish Cancer Society
Macmillan Cancer Support
NHS Evidence
 Epirubicin – Substance Summary
Epirubicin – Substance Summary
PubChem
MedlinePlus
– See more at: http://www.cancerindex.org/Epirubicin#sthash.BBioCC6K.dpuf
PHASE 1 JAPAN SOLID TUMOURS
DNA/RNA Synthesis Inhibitor
WITH Nano Carrier Co.,Ltdhttp://pdf.irpocket.com/C4571/qnwX/eFou/vG1J.pdf
KOWA COMPANY LTD
CAS FREE FORM. 56420-45-2
Smiles
- O=C2c1c(O)c5c(c(O)c1C(=O)c3cccc(OC)c23)C[C@@](O)(C(=O)CO)C[C@@H]5O[C@@H]4O[C@H]([C@H](O)[C@@H](N)C4)C
- NMR
- http://file.selleckchem.com/downloads/nmr/S122302-Epirubicin-Hydrochloride-NMR-Selleck.pdf
- http://www.medkoo.com/Product-Data/Epirubicin/Epirubicin-QC-TZC20130604web.pdf
-
Epirubicin CAS No.: 56420-45-2 Synonyms: - 4′-Epi-DX;
- Epirubicina;
- WP 697;
- Pidorubicin;
- 4′-epiadriamycin;
- Adriblastin;
- epi-dx;
- Epiadriamycin;
- farmorubicin;
- imi28;
- Farmarubicin;
Formula: C27H29NO11 Exact Mass: 543.17400
NC-6300, an epirubicin-incorporating micelle, extends the antitumor effect and reduces the cardiotoxicity of epirubicin.
Epirubicin is widely used to treat various human tumors. However, it is difficult to achieve a sufficient antitumor effect because of dosage limitation to prevent cardiotoxicity. We hypothesized that epirubicin-incorporating micelle would reduce cardiotoxicity and improve the antitumor effect. NC-6300 comprises epirubicin covalently bound to PEG polyaspartate block copolymer through an acid-labile hydrazone bond. The conjugate forms a micellar structure of 40-80 nm in diameter in an aqueous milieu. NC-6300 (10, 15 mg/kg) and epirubicin (10 mg/kg) were given i.v. three times to mice bearing s.c. or liver xenograft of human hepatocellular carcinoma Hep3B cells. Cardiotoxicity was evaluated by echocardiography in C57BL/6 mice that were given NC-6300 (10 mg/kg) or epirubicin (10 mg/kg) in nine doses over 12 weeks. NC-6300 showed a significantly potent antitumor effect against Hep3B s.c. tumors compared with epirubicin. Moreover, NC-6300 also produced a significantly longer survival rate than epirubicin against the liver orthotopic tumor of Hep3B. With respect to cardiotoxicity, epirubicin-treated mice showed significant deteriorations in fractional shortening and ejection fraction. In contrast, cardiac functions of NC-6300 treated mice were no less well maintained than in control mice. This study warrants a clinical evaluation of NC-6300 in patients with hepatocellular carcinoma or other cancers.

K-912(NC-6300)の概要 K-912(NC-6300)は、世界的に幅広く使用されているアントラサイクリン系の抗が ん剤の一つであるエピルビシンを内包したミセル化ナノ粒子製剤で、その特性により、 エピルビシンの有する心毒性の軽減が期待できます。さらに、pH 応答性システムを採 用することで、腫瘍細胞内でのエピルビシンの放出量を高め、既存のエピルビシンに比 べより強力な抗腫瘍効果が期待できます。
Epirubicin is an anthracycline drug used for chemotherapy. It can be used in combination with other medications to treat breast cancer in patients who have had surgery to remove the tumor. It is marketed by Pfizer under the trade name Ellence in the US andPharmorubicin or Epirubicin Ebewe elsewhere.
Similarly to other anthracyclines, epirubicin acts by intercalating DNA strands. Intercalation results in complex formation which inhibits DNA and RNA synthesis. It also triggers DNA cleavage by topoisomerase II, resulting in mechanisms that lead to cell death. Binding to cell membranes and plasma proteins may be involved in the compound’s cytotoxic effects. Epirubicin also generates free radicalsthat cause cell and DNA damage.
Epirubicin is favoured over doxorubicin, the most popular anthracycline, in some chemotherapy regimens as it appears to cause fewer side-effects. Epirubicin has a different spatial orientation of the hydroxyl group at the 4′ carbon of the sugar – it has the opposite chirality – which may account for its faster elimination and reduced toxicity. Epirubicin is primarily used against breast and ovarian cancer, gastric cancer, lung cancer and lymphomas.
Development history
The first trial of epirubicin in humans was published in 1980.[1] Upjohn applied for approval by the U.S. Food and Drug Administration(FDA) in node-positive breast cancer in 1984, but was turned down because of lack of data.[2] It appears to have been licensed for use in Europe from around this time however.[3] In 1999 Pharmacia (who had by then merged with Upjohn) received FDA approval for the use of epirubicin as a component of adjuvant therapy in node-positive patients.
Patent protection for epirubicin expired in August 2007.
References
- Bonfante, V; Bonadonna, G; Villani, F; Martini, A (1980). “Preliminary clinical experience with 4-epidoxorubicin in advanced human neoplasia”. Recent results in cancer research 74: 192–9. PMID 6934564. PM6934564.
- “On Target”.
- According to the proprietary database iddb.com
External links
- http://www.bccancer.bc.ca/HPI/DrugDatabase/DrugIndexPro/Epirubicin.htm
- http://www.pfizerpro.com/page_not_found?rid=/wyeth_html/home/minisites/oncology/ellence/pi/description.html
1H NMR PREDICT

13C NMR PREDICT

COSY
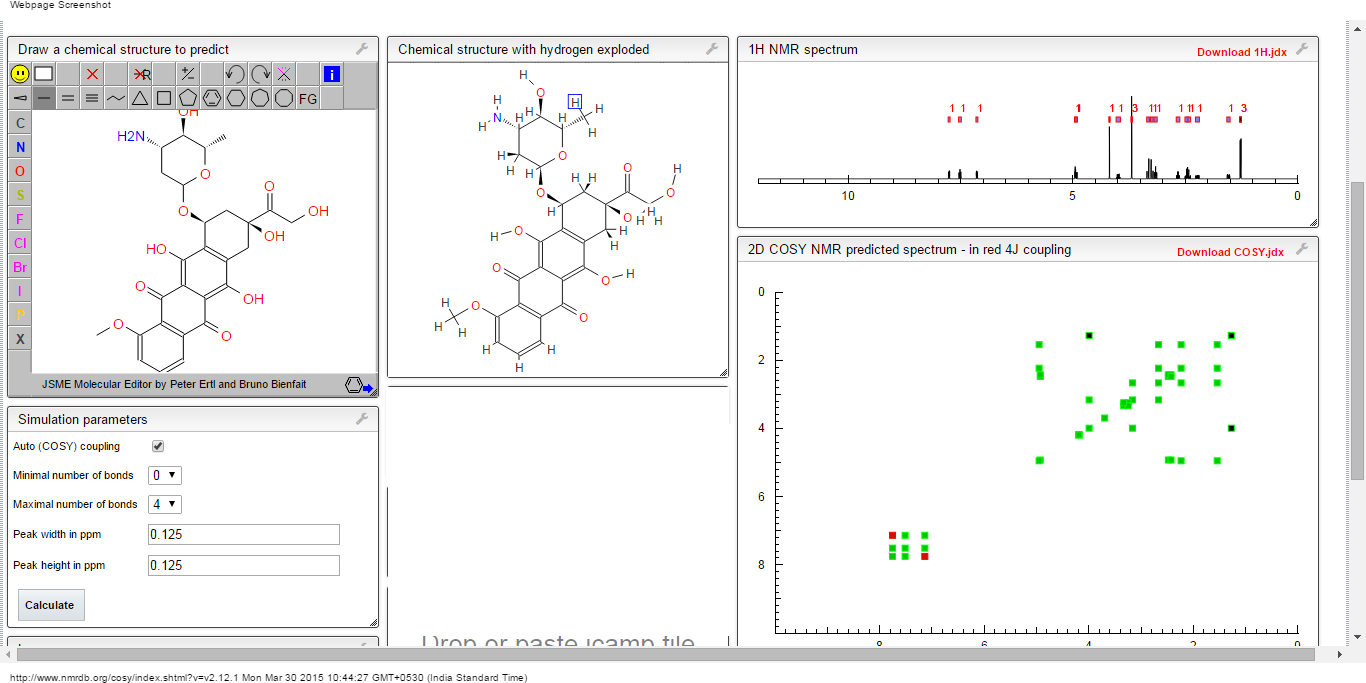
1H NMR

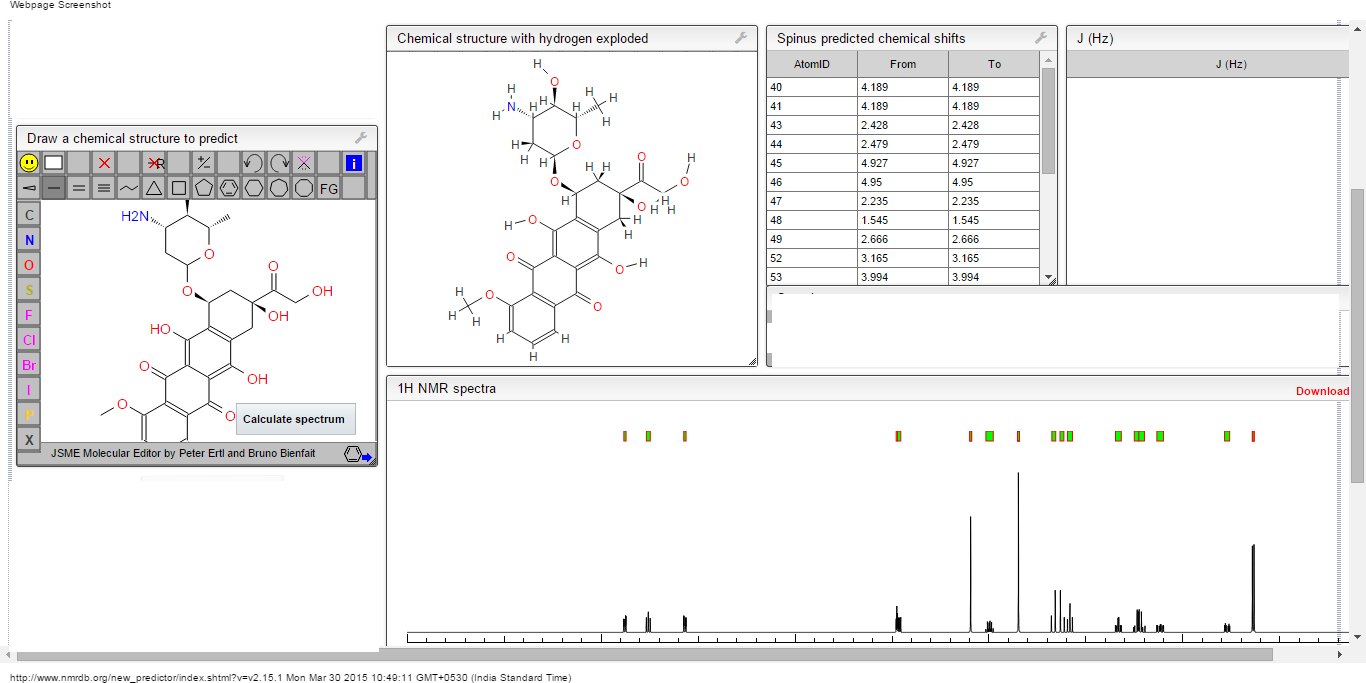
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (8R,10S)-10-((2S,4S,5R,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione | |
| Clinical data | |
| Trade names | Ellence |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a603003 |
| Intravenous | |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Protein binding | 77% |
| Metabolism | Hepatic glucuronidationand oxidation |
| Excretion | Biliary and renal |
| Identifiers | |
| 56420-45-2 |
|
| L01DB03 | |
| PubChem | CID 41867 |
| DrugBank | DB00445 |
| ChemSpider | 38201 |
| UNII | 3Z8479ZZ5X |
| KEGG | D07901 |
| ChEBI | CHEBI:47898 |
| ChEMBL | CHEMBL417 |
| Chemical data | |
| Formula | C27H29NO11 |
| 543.519 g/mol | |
KOWA COMPANY LTD


Nano Carrier Co


P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

GSK 923295, a CENP-E Inhibitor

GSK-923295A
1088965-37-0
Synonym: GSK-923295; GSK 923295; GSK923295.
CENP-E Inhibitor
IUPAC/Chemical name:
3-Chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}-4-[(1-methylethyl)oxy]benzamide
3-chloro-N-[(1S)-2-[[2-(dimethylamino)acetyl]amino]-1-[[4-[8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl]phenyl]methyl]ethyl]-4-(1-methylethoxy)- Benzamide,
3-Chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}-4-[(1-methylethyl)oxy]benzamide
3-Chloro-N-[(1S)-2-[(N,N-dimethylglycyl)amino]-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(1-methylethyl)oxy]benzamide
3-Chloro-N-[1-(N,N-dimethylglycinamido)-3-[4-[8-[1(S)-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl]phenyl]propan-2(S)-yl]-4-isopropoxybenzamide
C32H38ClN5O4
Exact Mass: 591.26123
Molecular Weight: 592.12822
Elemental Analysis: C, 64.91; H, 6.47; Cl, 5.99; N, 11.83; O, 10.81
Kinesin-like protein KIF11 inhibitor; Centromere protein E inhibitor
GSK-923295 is a novel antimitotic inhibitor of centromere-associated protein E (CENP-E) with potential anticancer activity. GSK923295A demonstrated significant antitumor activity against solid tumor models, inducing CRs in Ewing sarcoma, rhabdoid, and rhabdomyosarcoma xenografts.
GSK-923295, a small-molecule inhibitor of centromere associated protein (CENP), is in early clinical development at Cytokinetics for the treatment of refractory cancer. No recent development has been reported for early clinical research which had been ongoing at GlaxoSmithKline.
Clinical study showed that GSK923295 had dose-proportional pharmacokinetics and a low number of grade 3 or 4 adverse events. The observed incidence of myelosuppression and neuropathy was low. Further investigations may provide a more complete understanding of the potential for GSK923295 as an antiproliferative agent.
GSK923295 is a first-in-class, specific allosteric inhibitor of CENP-E kinesin motor ATPase with Ki of 3.2 nM, and less potent to mutant I182 and T183. Phase 1.
The compound potently inhibits CENP-E ATPase activity and exerts broad-spectrum antiproliferative activity against cancer cells and xenografts. GSK-923295 has demonstrated a broad spectrum of activity against a range of human tumor xenografts grown in nude mice, including models of colon, breast, ovarian, lung and other tumors.
Cytokinetics was developing GSK-923295, the lead from a series of small-molecule mitotic kinesin spindle protein inhibitors, for treating cancer including advanced solid tumors. However, since October 2014, the program was no longer listed on the Cytokinetics’ website
In 2001, a strategic alliance was established between Cytokinetics and GlaxoSmithKline to discover, develop and commercialize novel small-molecule therapeutics targeting mitotic kinesins for applications in the treatment of cancer and other diseases.

…………………….
PATENT
US8772507
http://www.google.com/patents/US8772507


1,1-Dimethylethyl [(1S)-2-(4-bromophenyl)-1-(hydroxymethyl)ethyl]carbamate

To a solution of 4-bromo-N-{[(1,1-dimethylethyl)oxy]carbonyl}-L-phenylalanine (72.6 mmol), in anhydrous diethyl ether (550 mL) at 0° C. was added slowly lithium aluminum hydride, 95% (108.9 mmol). The resulting solution was stirred for an additional 2 h at 0° C. The reaction was then carefully quenched with a saturated aqueous solution of sodium bicarbonate (73 mL) which stirred at RT for half an hour. Lithium aluminium salts crashed out of solution and were removed by filtration. The filtrate was concentrated and vacuum pumped for 24 h to afford the title product as a white solid (97%). ESMS [M+H]+: 331.2.
1,1-Dimethylethyl {(1S)-2-(4-bromophenyl)-1-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate

To a solution of 1,1-dimethylethyl [(1S)-2-(4-bromophenyl)-1-(hydroxymethyl)ethyl]carbamate (70.6 mmol), tripheylphosphine (84.7 mmol), and phthalimide (84.7 mmol) in anhydrous tetrahydrofuran (550 mL) at 0° C. was added dropwise diisopropyl azodicarboxylate (84.7 mmol) over 10 minutes. The reaction continued to stir allowing to warm to RT over 5 h. The reaction was then concentrated in vacuo and product was triturated out of solution using ethyl acetate (500 mL). The precipitate was filtered, washed with ethyl acetate (3×100 mL), and dried to afford the title product as a white solid (57%). ESMS [M+H]+: 460.4.
1,1-Dimethylethyl {(1S)-2-[4-(bromoacetyl)phenyl]-1-[(1,3-d oxo-1,3-dihydro-21′-isoindol-2-yl)methyl]ethyl}carbamate

A solution of 1,1-dimethylethyl {(1S)-2-(4-bromophenyl)-1-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (21.7 mmol), 1-ethoxyvinyltri-n-butylin (43.5 mmol), and trans-dichlorobis(triphenylphosphine)palladium(II) (5 mol %) were stirred in anhydrous dioxane (300 mL) at 100° C. for 3 h. The reaction was then concentrated in vacuo and redissolved in a solution of tetrahydrofuran and water (3:1, 400 mL). The mixture was treated with N-bromosuccinimide (108.8 mmol) and stirred at RT for half an hour. The reaction solution was then concentrated to dryness and redissolved in ethyl acetate (150 mL). Precipate formed upon addition of hexanes (350 mL) and was filtered and dried to afford the title product as yellow solid (71%). ESMS [M+H]+: 502.4.
1,1-Dimethylethyl [(1S)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]carbamate

A mixture of 1,1-dimethylethyl{(1S)-2-{4-(bromoacetyl)phenyl]-1-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (1.90 g, 3.79 mmol), 1-(2-amino-3-pyridinyl)ethanol (0.523 g, 3.79 mmol), and solid sodium bicarbonate (0.398 g, 4.72 mmol) in isopropanol (24 mL) was refluxed for 3.0 h. The mixture was concentrated in vacuo and the residue dissolved in ethyl acetate, washed with water and saturated sodium chloride, dried (Na2SO4), and concentrated to give the title compound (1.79 g, 87%) as a light pink solid. MS (ES+) m/e 541 [M+H]+.
3-Chloro-N-[(1S)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(1-methylethyl)oxy]benzamide

A mixture of 1,1-dimethylethyl [(1S)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]carbamate (1.79 g, 3.31 mmol) and 4 M HCl in 1,4-dioxane (20 mL, 80 mmol) was stirred at room temperature for 45 minutes. The reaction was concentrated to dryness and redissolved in DMF (30 mL). To this solution was added N,N-diisopropylethylamine (2.14 g, 16.55 mmol) and pentafluorophenyl 3-chloro-4 [(1-methylethyl)oxy]benzoate (1.36 g, 3.31 mmol). The mixture was stirred overnight at room temperature, diluted with water, and extracted into ethyl acetate. The extracts were washed with water, dried (Na2SO4), and concentrated in vacuo to give the title compound (2.10 g, 100%) as a tan solid. MS (ES+) m/e 637 [M+H]+.
N-[(1S)-2-Amino-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-3-chloro-4-[(1-methylethyl)oxy]benzamide

A mixture of 3-chloro-N-[(1S)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(1-methylethyl)oxy]benzamide (2.10 g, 3.30 mmol) and hydrazine monohydrate (0.83 g, 16.5 mmol) in ethanol (30 mL) was heated at 57° C. overnight. The reaction was cooled, diluted with ethanol, filtered, and concentrated to give the title compound (1.67 g, 100%) as a pale yellow powder. MS (ES+) m/e 507 [M+H]+.
3-Chloro-N-[(1S)-2-[(N,N-dimethylglycyl)amino]-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(1-methylethyl)oxy]benzamide

A mixture of N-[(1S)-2-amino-1-({4-[8-(1-hydroxyethyl)imidazo[1,2-a]pyridin-2-yl]phenyl}methyl)ethyl]-3-chloro-4-[(1-methylethyl)oxy]benzamide (0.912 g, 1.80 mmol), EDCI (0.69 g, 3.6 mmol), N,N-diisopropylethylamine (0.466 g, 3.6 mmol), and N,N-dimethylglycine (0.372 g, 3.6 mmol) in methylene chloride (17 mL) was stirred overnight at room temperature. The reaction was diluted with water, washed with brine, dried (Na2SO4), and concentrated. The residue was purified by flash chromatography on silica gel (8%-10% MeOH:CH2Cl2) to give the title compound (0.515 g, 48%) as a pale yellow solid. MS (ES+) ink 592 [M+H]+.
………………….
WO2005107762
https://www.google.im/patents/WO2005107762A2
Example 1

cheme E:
ide


NaHCOj, IPA 100 ‘C


1 , 1 -Dimethylethyl [( 1 S)-2-(4-bromophenyl)- 1 -(hydroxymethyl)ethyl]carbamate:

To a solution of 4-bromo-N-{[(l ,1 -dimethylethyl)oxy] carbonyl }-L- phenylalanine (72.6 mmol), in anhydrous diethyl ether (550 mL) at 0 °C was added slowly lithium aluminum hydride, 95% (108.9 mmol). The resulting solution was stiπed for an additional 2 h at 0 °C, The reaction was then carefully quenched with a saturated aqueous solution of sodium bicarbonate (73 mL) which stiπed at RT for half an hour. Lithium aluminium salts crashed out of solution which were removed by filtration. The filtrate was concentrated and vacuum pumped for 24 h to afford the title product as a white solid (97%).
ESMS [M+H]+: 331.2.
1,1 -Dimethylethyl {(lS)-2-(4-bromophenyl)-l-[(l,3-dioxo-l,3-dihydro-2H-isoindol-2- yl)methyl]ethyl}carbamate:

To a solution of 1 ,1 -dimethylethyl [(lS)-2-(4-bromophenyl)-l –
(hydroxymethyl)ethyl]carbamate (70.6 mmol), tripheylphosphine (84.7 mmol), and phthalimide (84.7 mmol) in anhydrous tetrahydrofuran (550 mL) at 0 °C was added dropwise diisopropyl azodi carboxyl ate (84.7 mmol) over 10 minutes. The reaction continued to stir allowing to wai to RT over 5h, The reaction was then concentrated in vacuo and product was tritarated out of solution usingl acetate (500 mL). The precipitate was filtered, washed with ethyl acetate (3 x 100 mL), and dried to afford the title product as a white solid (57%).
ESMS [M+H]+: 460.4.
1 ,1 -Dimethylethyl {(15)-2-[4-(bromoacetyl)phenyl]-l -[(l,3-dioxo-l ,3-dihydro-2H-isoindol- 2-yl)methyl]ethyl}carbamate:

A solution of 1,1 -dimethyl ethyl {(lS)-2-(4-bromophenyl)-l-[(l,3-dioxo-l,3- dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (21.7 mmol), 1-ethoxyvinyltri-n-butylin (43.5 mmol), and /ra/?s–dichlorobis(triphenylphospine)palladιum(II) (5 mol%) were stiπed in anhydrous dioxane (300 mL) at 100 °C for 3h. The reaction was then concentrated in vacuo and redissolved in a solution of tetrahydrofuran and water (3:1, 400mL) and treated with N- bromosuccinimide (108.8 mmol) and stined at RT for half an hour. The reaction solution was then concentrated to dryness and redissolved in ethyl acetate (150 mL) and precipate formed upon addition of hexanes (350 mL). The precipitate was filtered and dried to afford the title product as yellow solid (71%). ESMS [M+Η]+: 502.4. l,l-Dimethylethyl [(lS)-2-(l ,3-dioxo-l,3-dihydro-2H-isoindol-2-yl)-l-({4-[8-(l- hydroxyethyl)imidazo[l,2-β]pyridin-2-yl]phenyl}methyl)ethyl]carbamate:

A mixture of l!l-dimethylethyl{(lS)-2-{4-(biOinoacetyl)phenyl]-l-[(l,3- dioxo-l ,3-dihydro-2H-isoindol-2-yl)methyl]ethyl}carbamate (1.90 g, 3.79 mmol), l-(2- amino-3-pyτidinyl)ethanol (0.523 g, 3.79 mmol), and solid sodium bicarbonate (0.398 g, 4,72 mmol) in isopropanol (24 mL) was refluxed for 3.0 h. and concentrated in vacuo. The residue was dissolved in ethyl acetate, washed with water and saturated sodium chloride, dried (Na2S04), and concentrated to give the title compound (1.79 g, S7%) as a light pink solid. MS(ES+) m/e 541 [M+Η]+.
3-Chloro-N-[(lS)-2-(l,3-dioxo-l ,3-dihydro-2H-isoindol-2-yl)-l-({4-[8-(l- hydroxyethyl)imidazo[l,2-Λ]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(l – methylethyl)oxy]benzamide:

A mixture of 1,1 -dimethylethyl [(15)-2-(l,3-dioxo-l,3-dihydro-2H-isoindol-2- yl)-l-({4-[8-(l-hydroxyethyl)imidazo[l,2-fl]pyridin-2-yl]phenyl}methyl)ethyl]carbamate (1.79 g, 3.31 mmol) and 4M ΗC1 in 1,4-dioxane (20 mL, 80 mmol) was stirred at room temperature for 45 minutes. The reaction was concentrated to dryness ,redissolved in DMF (30 mL), and to this solution was added N,N-diisopropylethylamine (2.14 g, 16,55 mmol) and pentafluorophenyl 3-chloro-4 [(l-methylethyl)oxy]benzoate (1.36 g, 3.31 mmol). The mixture was stirred overnight at room temperature, diluted with water, and extracted into ethyl acetate. The extracts were washed with water, dried (Na SO ), and concentrated in vacuo to give the title compound (2.10 g, 100%) as a tan solid. MS(ES+) m/e 637 [M+H]+.
N-[(lS)-2-Amino-l-({4-[8-(l-hydroxyethyl)imidazo[l,2-α]p>tidin-2- yl]phenyl}methyl)eth)’l]-3-chloro-4-[(l-methylethyl)oxy]benzamide:

A mixture of 3-chloro-N-[(lS)-2-(l,3-dioxo-l ,3-dihydro-2N-isoindol-2-yl)-l-
({4-[8-(l -hydiOxyethyl)imidazo[l,2-β]pyridin-2-yl]phenyl}methyl)ethyl]-4-[(l- methylethyl)oxy]benzamide (2.10 g, 3.30 mmol) and hydrazine monohydrate (0.83 g, 16.5 mmol) in ethanol (30 mL) was heated at 57°C ovemight. The reaction was cooled, diluted with ethanol, filtered, and concentrated to give the title compound(1.67 g, 100%) as a pale yellow powder. MS(ES+) m/e 507 [M+H]+.
3-Chloro-N-[(15)-2-[(7VN-dimethylglycyl)amino]-l-({4-[8-(l-hydroxyethyl)imidazo[l ,2- «]pyitdin-2-yl]phenyl}methyl)ethyl]-4-[(l-methylethyl)oxy]benzamide:

A mixture ofN-[(lS)-2-amino-l-({4-[S-(l-hydroxyethyl)imidazo[l,2- α]pyridin-2-yl]phenyl)methyl)ethyl]-3-chloro-4-[(l-methylethyl)oxy]benzamide (0.912 g, 1 ,80 mmol), EDCI (0.69 g, 3,6 mmol), NN-diisopropylethylamine (0.466 g, 3,6 mmol), and N,N-dimethylglycine (0.372 g, 3.6 mmol) in methylene chloride (17 mL) was stirred overnight at room temperature. The reaction was diluted with water, washed with brine, dried (Νa2S0 ), and concentrated. The residue was purified by flash chromatography on silica gel (8%-10% MeOH:CH2Cl2) to give the title compound ( 0.515 g, 48%) as a pale yellow solid. MS(ES+) m/e 592 [M+H]+.
SEE
WO2008 / 138561
………………..
Organic Process Research & Development (2010), 14(5), 1254-1263
Org. Process Res. Dev., 2010, 14 (5), pp 1254–1263
DOI: 10.1021/op100186c
http://pubs.acs.org/doi/abs/10.1021/op100186c

The discovery and development of an efficient manufacturing route to the CENP-E inhibitor 3-chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}−4-[(1-methylethyl)oxy]benzamide (GSK923295A) is described. The existing route to GSK923295A was expensive, nonrobust, used nonideal reagents, and consistently struggled to deliver the API needed for clinical studies. The new synthesis commences from the readily available l-phenylalaninol, which is smoothly converted through to GSK923295A using key Friedel−Crafts acylation as well as selective acylation chemistries. Downstream chemistry to GSK923295A is both high yielding and robust, and the resulting process has been demonstrated first on the kilo scale and subsequently in the pilot plant where 55 kg was successfully prepared. The resulting process is simple, uses cheaper raw materials, is greener in that it avoids using aluminum, tin, and bromination chemistries, and obviates the need for chromatographic purification. Also discussed are the route derived impurities, how they were unambiguously prepared to confirm structure and processing amendments to control their formation, and enhancements to the new process to facilitate future processing.
1H NMR (400 MHz, CD3OD) δH 1.34 (6H, d, J = 6.0, (CH3)2), 1.59 (3H, d, J = 7.0, CH3CH), 2.21 (6H, s, N(CH3)2), 2.87−3.01 (4H, m, CH2Ph and CH2N(CH3)2), 3.49 (2H, m, CH2NPhthal), 4.50 (1H, m, CHNH), 4.70 (1H, m, (CH3)2CHO)), 5.49 (1H, q, J = 7.0, CHOH), 6.88 (1H, t, J = 7.0, H-j), 7.08 (1H, d, J = 7.5, H-b), 7.33−7.37 (3H, m, H-k and H-d), 7.63 (1H, dd, J = 7.5 and 2.0, H-c), 7.78 (1H, s, H-a), 7.83 (2H, d, J = 7.0, H-e), 8.09 (1H, m, H-h), 8.27 (1H, d, J = 8.0, H-i);
13C NMR (100 MHz, CD3OD) δC 22.2, 24.1, 39.3, 43.8, 46.1, 53.0, 63.7, 66.2, 73.0, 110.4, 113.8, 115.3, 121.2, 124.5, 126.1, 127.5, 128.4, 128.5, 130.6, 130.7, 133.3, 136.0, 139.4, 145.1, 146.1, 157.6, 168.5 and 173.6;
HRMS (ESI+) m/z calculated for [M+H]+ C32H39N5O4Cl 592.2691, found 592.2684.
…………………….
PREDICTIONS
http://orgspectroscopyint.blogspot.in/2015/03/gsk-923295.html

1H NMR PREDICT
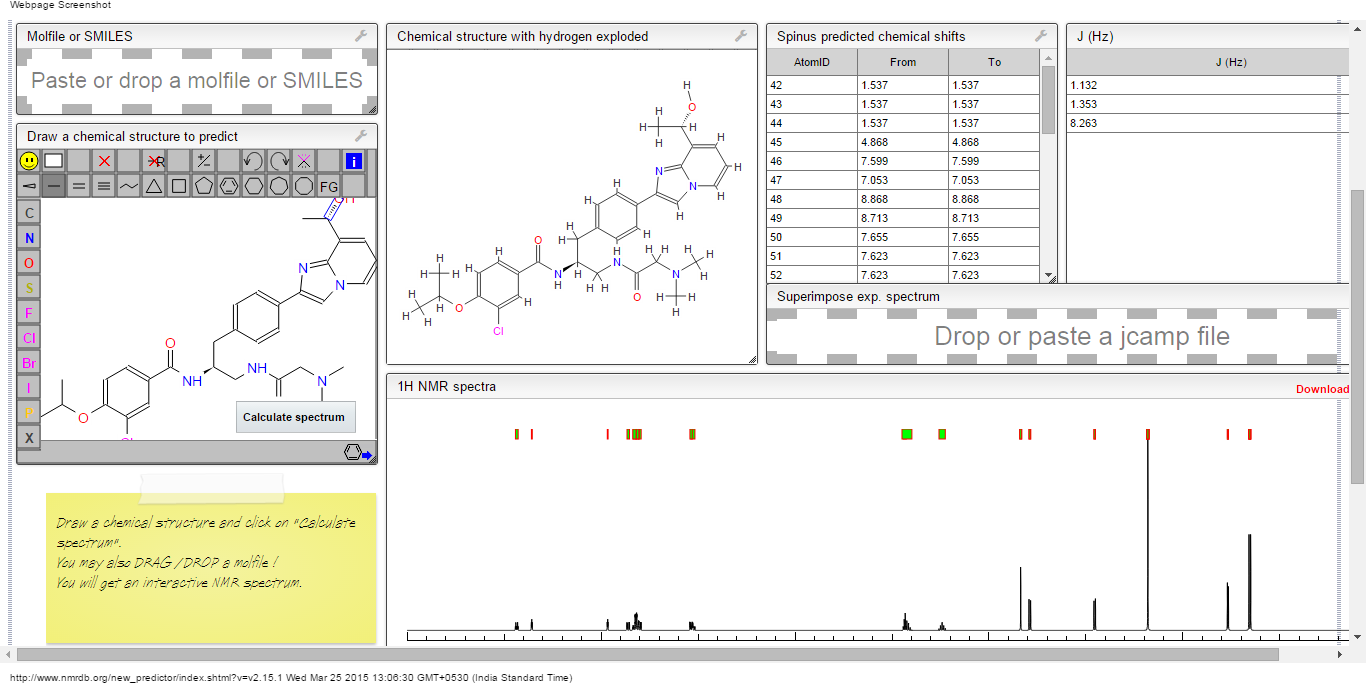

see http://orgspectroscopyint.blogspot.in/2015/03/gsk-923295.html
ACS Medicinal Chemistry Letters (2010), 1(1), 30-34
http://pubs.acs.org/doi/abs/10.1021/ml900018m

Inhibition of mitotic kinesins represents a novel approach for the discovery of a new generation of anti-mitotic cancer chemotherapeutics. We report here the discovery of the first potent and selective inhibitor of centromere-associated protein E (CENP-E) 3-chloro-N-{(1S)-2-[(N,N-dimethylglycyl)amino]-1-[(4-{8-[(1S)-1-hydroxyethyl]imidazo[1,2-a]pyridin-2-yl}phenyl)methyl]ethyl}-4-[(1-methylethyl)oxy]benzamide (GSK923295; 1), starting from a high-throughput screening hit, 3-chloro-4-isopropoxybenzoic acid 2. Compound 1 has demonstrated broad antitumor activity in vivo and is currently in human clinical trials.
SEE
WO-2015037460
Method for producing optically active 3-(biphenyl-4-yl)-2-[(t-butoxycarbonyl)amino]propan-1-ol
Process for preparing optically active 3-(biphenyl-4-yl)-2-[(t-butoxycarbonyl)amino]propan-1-ol, useful as an intermediate in the synthesis of pharmaceuticals described in WO2005107762 and WO2008138561 (such as GSK-923295 and tubulysin derivatives respectively). Appears to be a new area of interest to the assignee.
…………..
WO2010118207
|
References |
1: Mayes PA, Degenhardt YY, Wood A, Toporovskya Y, Diskin SJ, Haglund E, Moy C, Wooster R, Maris JM. Mitogen-activated protein kinase (MEK/ERK) inhibition sensitizes cancer cells to centromere-associated protein E inhibition. Int J Cancer. 2013 Feb 1;132(3):E149-57. doi: 10.1002/ijc.27781. Epub 2012 Sep 28. PubMed PMID: 22948716.
2: Chung V, Heath EI, Schelman WR, Johnson BM, Kirby LC, Lynch KM, Botbyl JD, Lampkin TA, Holen KD. First-time-in-human study of GSK923295, a novel antimitotic inhibitor of centromere-associated protein E (CENP-E), in patients with refractory cancer. Cancer Chemother Pharmacol. 2012 Mar;69(3):733-41. doi: 10.1007/s00280-011-1756-z. Epub 2011 Oct 22. PubMed PMID: 22020315.
3: Lock RB, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH, Maris JM, Wozniak AW, Gorlick R, Kolb EA, Houghton PJ, Smith MA. Initial testing of the CENP-E inhibitor GSK923295A by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012 Jun;58(6):916-23. doi: 10.1002/pbc.23176. Epub 2011 May 16. PubMed PMID: 21584937; PubMed Central PMCID: PMC3163687.
4: Balamuth NJ, Wood A, Wang Q, Jagannathan J, Mayes P, Zhang Z, Chen Z, Rappaport E, Courtright J, Pawel B, Weber B, Wooster R, Sekyere EO, Marshall GM, Maris JM. Serial transcriptome analysis and cross-species integration identifies centromere-associated protein E as a novel neuroblastoma target. Cancer Res. 2010 Apr 1;70(7):2749-58. doi: 10.1158/0008-5472.CAN-09-3844. Epub 2010 Mar 16. PubMed PMID: 20233875; PubMed Central PMCID: PMC2848992.
5: Wood KW, Lad L, Luo L, Qian X, Knight SD, Nevins N, Brejc K, Sutton D, Gilmartin AG, Chua PR, Desai R, Schauer SP, McNulty DE, Annan RS, Belmont LD, Garcia C, Lee Y, Diamond MA, Faucette LF, Giardiniere M, Zhang S, Sun CM, Vidal JD, Lichtsteiner S, Cornwell WD, Greshock JD, Wooster RF, Finer JT, Copeland RA, Huang PS, Morgans DJ Jr, Dhanak D, Bergnes G, Sakowicz R, Jackson JR. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc Natl Acad Sci U S A. 2010 Mar 30;107(13):5839-44. doi: 10.1073/pnas.0915068107. Epub 2010 Feb 18. PubMed PMID: 20167803; PubMed Central PMCID: PMC2851928.
