
Home » PHASE 1 (Page 9)
Category Archives: PHASE 1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BMS 986120

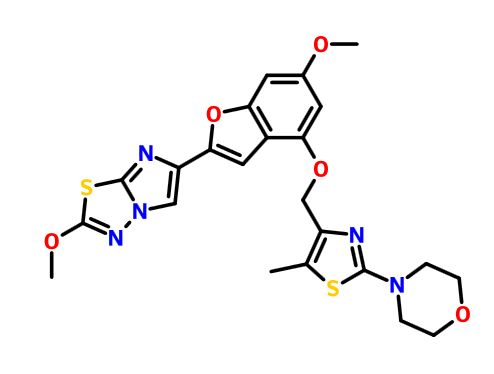
 .
.
Picture credit….Bethany Halford
BMS 986120
Originator Bristol-Myers Squibb
Bristol-Myers Squibb Company, Université de Montréal
| Molecular Formula: | C23H23N5O5S2 |
|---|---|
| Molecular Weight: | 513.58922 g/mol |
4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]morpholine
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine
Imidazo[2,1-b] -1,3,4-thiadiazole, 2-methoxy-6-[6-methoxy-4-[[5-methyl-2-(4-morpholinyl)-4- thiazolyl]methoxy]-2-benzofuranyl]-
CAS 1478712-37-6
Phase I Thrombosis
- 02 Apr 2015 Bristol-Myers Squibb plans a phase I trial in Thrombosis (In volunteers) in United Kingdom (NCT02439190)
- 01 Aug 2014 Preclinical trials in Thrombosis in USA (PO)
https://clinicaltrials.gov/ct2/show/NCT02208882
https://clinicaltrials.gov/ct2/show/NCT02439190
Class Imidazoles; Small molecules; Thiadiazoles
$BMY antithrombic compound
PATENT
http://www.google.com/patents/WO2013163279A1?cl=en
Thromboembolic diseases remain the leading cause of death in developed countries despite the availability of anticoagulants such as warfarin (COUMADIN®), heparin, low molecular weight heparins (LMWH), synthetic pentasaccharides, and antiplatelet agents such as aspirin and clopidogrel (PLAVIX®).
Current anti-platelet therapies have limitations including increased risk of bleeding as well as partial efficacy (relative cardiovascular risk reduction in the 20 to
30% range). Thus, discovering and developing safe and efficacious oral or parenteral antithrombotics for the prevention and treatment of a wide range of thromboembolic disorders remains an important goal.
Alpha-thrombin is the most potent known activator of platelet aggregation and degranulation. Activation of platelets is causally involved in atherothrombotic vascular occlusions. Thrombin activates platelets by cleaving G-protein coupled receptors termed protease activated receptors (PARs). PARs provide their own cryptic ligand present in the N-terminal extracellular domain that is unmasked by proteolytic cleavage, with subsequent intramolecular binding to the receptor to induce signaling (tethered ligand mechanism; Coughlin, S.R., Nature, 407:258-264 (2000)). Synthetic peptides that mimic the sequence of the newly formed N-terminus upon proteolytic activation can induce signaling independent of receptor cleavage. Platelets are a key player in atherothrombotic events. Human platelets express at least two thrombin receptors, commonly referred to as PARI and PAR4. Inhibitors of PARI have been investigated extensively, and several compounds, including vorapaxar and atopaxar have advanced into late stage clinical trials. Recently, in the TRACER phase III trial in ACS patients, vorapaxar did not significantly reduce cardiovascular events, but significantly increased the risk of major bleeding (Tricoci, P. et al, N. Eng. J. Med., 366(l):20-33 (2012). Thus, there remains a need to discover new antiplatelet agents with increased efficacy and reduced bleeding side effects.
There are several early reports of preclinical studies of PAR4 inhibitors. Lee, F-Y. et al., “Synthesis of l-Benzyl-3-(5′-hydroxymethyl-2′-furyl)indazole Analogues as Novel Antiplatelet Agents”, J. Med. Chem., 44(22):3746-3749 (2001) discloses in the abstract that the compound

58
“was found to be a selective and potent inhibitor or protease-activated receptor type 4 (PAR4)-dependent platelet activation. ”
Compound 58 is also referred to as YD-3 in Wu, C-C. et al, “Selective Inhibition of Protease-activated Receptor 4-dependent Platelet Activation by YD-3”, Thromb. Haemost., 87: 1026-1033 (2002). Also, see Chen, H.S. et al, “Synthesis and platelet activity”, J. Bioorg. Med. Chem., 16: 1262-1278 (2008).
EP1166785 Al and EP0667345 disclose various pyrazole derivatives which are useful as inhibitors of platelet aggregation.\
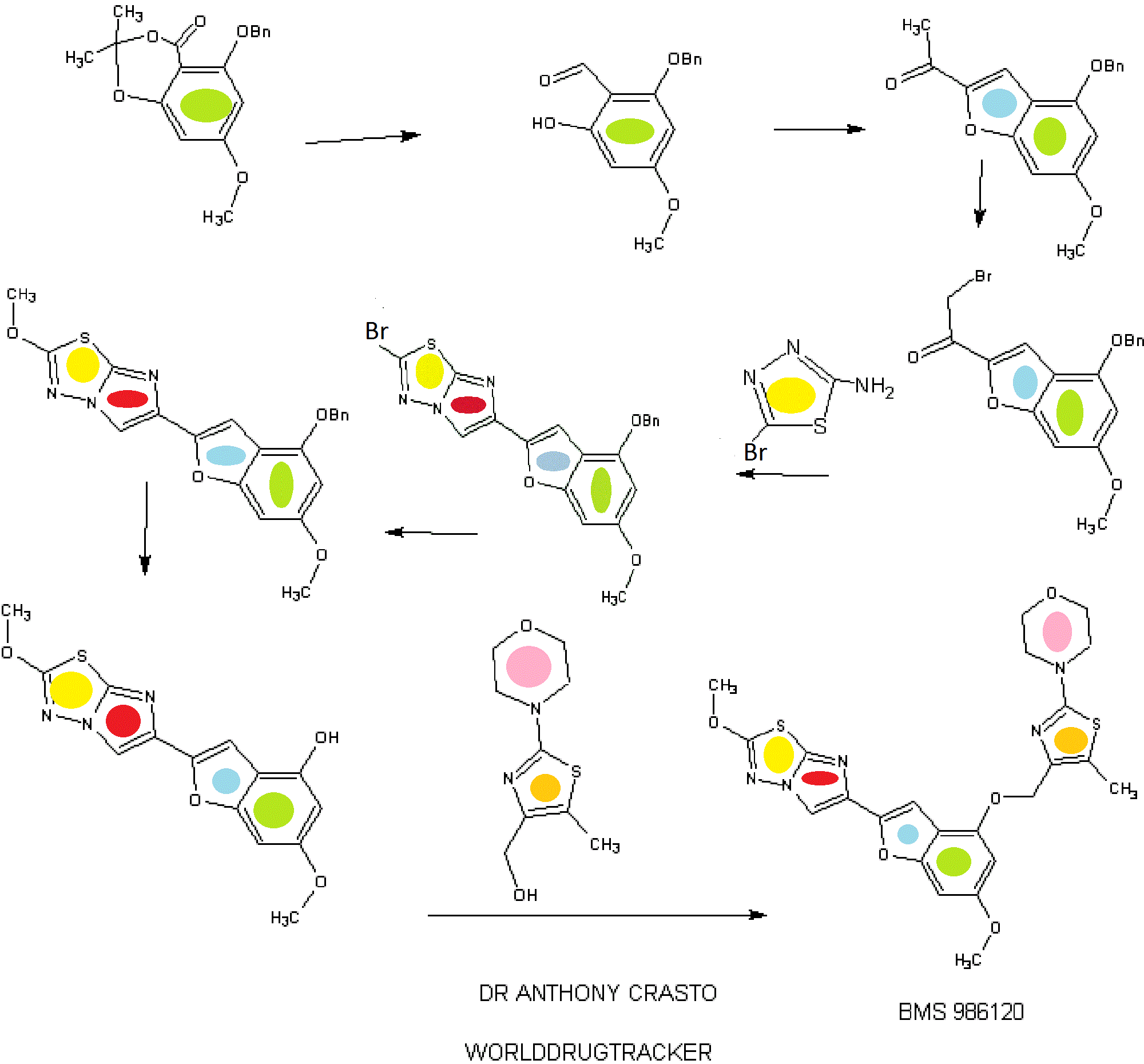
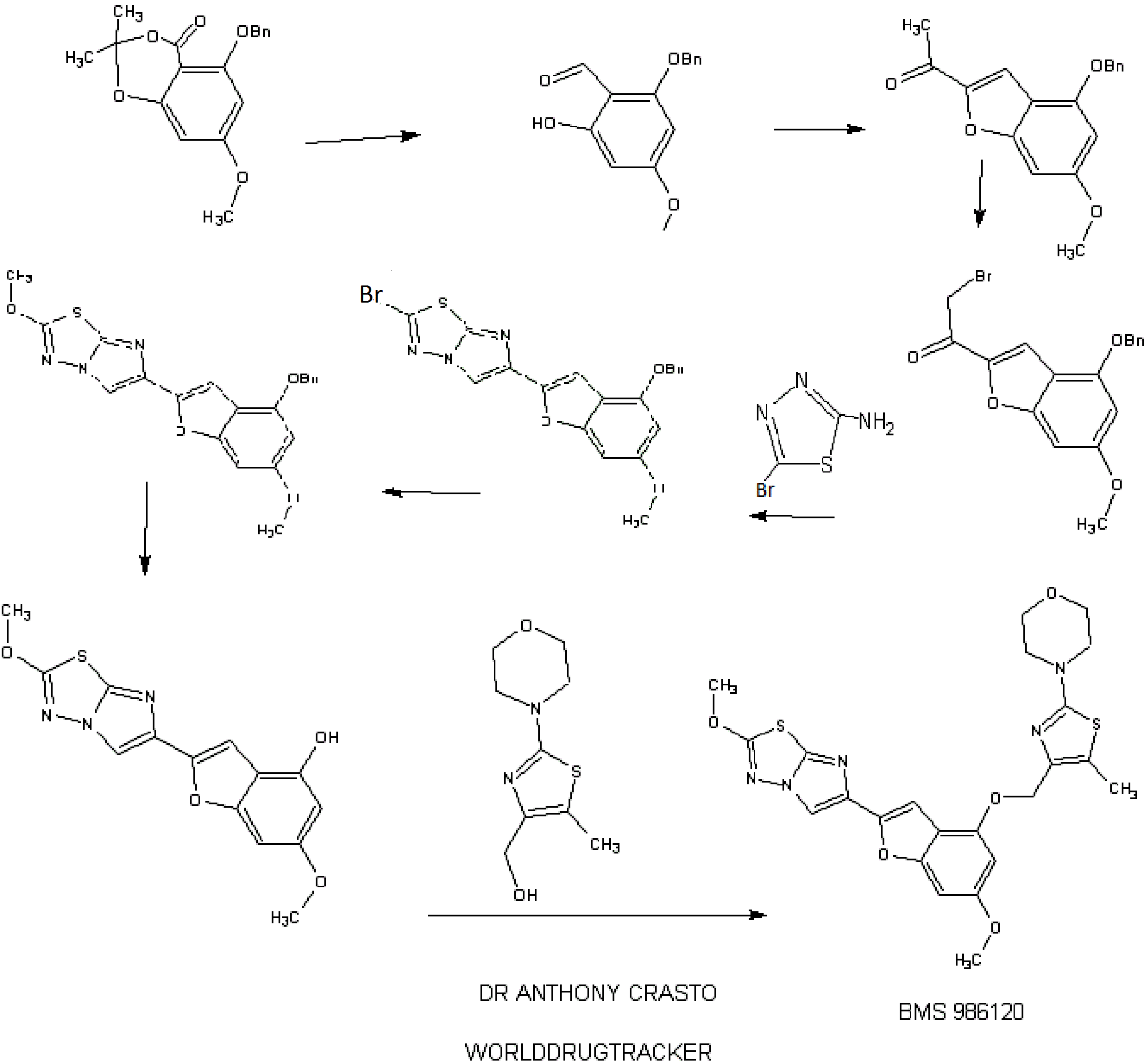
IB. 5-(Benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4-one

A solution of 5-hydroxy-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4- one (30.00 g, 0.134 mol, see Kamisuki, S. et al, Tetrahedron, 60:5695-5700 (2004) for preparation) in N,N-dimethylformamide (400 mL) was treated with powdered anhydrous potassium carbonate (19.41 g, 0.14 mol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with benzyl bromide (24.03 g, 0.14 mol) added dropwise over 15 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting material left by tic). The solid was filtered and washed with N,N- dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (500 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Crystallization form ethyl acetate (50 mL) and hexane (150 mL) gave 35.17 g of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H- benzo[d][l ,3]dioxin-4-one as large colorless prisms. Chromatography of the mother liquors on silica gel (4 x 13 cm, elution toluene – ethyl acetate 0-5%) gave 6.64 g of additional material to afford a total yield of 41.81 g (99%). HRMS(ESI) calcd for
Ci8Hi905 [M+H]+ m/z 315.1227, found 315.1386. 1H NMR (CDC13, 600 MHz) δ 1.68 (s, 6H), 3.77 (s, 3H), 5.19 (s, 2H), 5.19 (s, 2H), 6.04 (d, J = 2.03 Hz, 1H), 6.15 (d, J = 2.03 Hz, 1H), 7.27 (broad t, 1H), 7.36 (broad t, 2H), 7.52 (broad d, 2H).
1 C. 2-(Benzyloxy)-6-hydroxy-4-methoxybenzaldehyde

A solution of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l ,3]dioxin- 4-one (Example IB, 6.76 g, 21.5 mmol) in dichloromethane (120 mL) was cooled to -78 °C and treated with 43 mL (64.5 mmol) of a 1.5 M solution of diisobutylaluminum hydride in toluene added dropwise over 20 min. The resulting mixture was then stirred at -78 °C for 3 h. The reaction mixture was quenched by the careful addition of methanol (5 mL) added dropwise over 15 min, followed by IN hydrochloric acid (50 mL) added dropwise over 15 min. The cooling bath was then removed and an additional 150 mL of IN hydrochloric acid was added over 20 min. The mixture was then stirred at 22 °C for 2 h and diluted with dichloromethane (400 mL). The organic phase was collected and the aqueous phase (pH ~1) was extracted with dichloromethane (3 x 50 mL). The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residual oil was diluted with tetrahydrofuran (70 mL), treated with 10 mL of 0.1N hydrochloric acid and stirred at 20 °C for 2 h. The reaction mixture was diluted with ethyl acetate (300 mL), washed with brine, dried over anhydrous magnesium sulfate, evaporated in vacuo to give a clear oil. Chromatography on silica gel (4 x 13 cm, elution toluene) gave 4.08 g (73% yield) of the title aldehyde as a clear oil which solidified on standing. LC (Method C): 2.237 min. HRMS(ESI) calcd for Ci5Hi504 [M+H]+ m/z 259.0965, found 259.1153. 1H NMR (CDC13, 600 MHz) δ 3.80 (s, 3H), 5.07 (s, 2H), 5.97 (d, J= 2.1 Hz, 1H), 6.01 (d, J= 2.1 Hz, 1H), 7.3 – 7.4 (m, 5 H), 10.15 (s, 1H), 12.49 (s, 1H).
ID. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)ethanone

A solution of 2-(benzyloxy)-6-hydroxy-4-methoxybenzaldehyde (Example 1C, 3.46 g, 13.4 mmol) in N,N-dimethylformamide (50 mL) was treated with powdered anhydrous cesium carbonate (4.58 g, 14.05 mmol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with chloroacetone (1.74 g, 18.7 mmol) added dropwise over 5 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting aldehyde left by tic and formation of the intermediate alkylated aldehyde). The solid was filtered and washed with N,N-dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (300 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. This syrup was diluted with tetrahydrofuran (50 mL) and ethyl acetate (50 mL), treated p- toluenesulfonic acid monohydrate (0.2 g) and stirred at 20 °C for 1 h (tic indicated complete cyclization of the intermediate alkylated aldehyde to the benzofuran). The reaction mixture was diluted with ethyl acetate (300 mL), washed with saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 2-4%) gave 3.51 g (88% yield) of the title benzofuran as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave the title material as large yellow prisms (3.15 g). LC (Method D): 2.148 min. HRMS(ESI) calcd for Ci8Hiv04 [M+H]+ m/z 297.1121, found 297.1092. 1H NMR (CDC13, 600 MHz) δ 2.51 (s, 3H), 3.82 (s, 3H), 5.13 (s, 2H), 6.37 (d, J= 1.77 Hz, 1H), 6.63 (broad s, 1H), 7.34 (broad t, 1H), 7.39 (broad t, 2H), 7.44 (broad d, 2H), 7.55 (d, J = 0.7 Ηζ,ΙΗ). IE. l-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone

A 250-mL, three-necked flask is equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with anhydrous tetrahydrofuran (25 mL) followed by 9.3 mL (9.3 mmol) of a 1M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran. The mixture was cooled to -78 °C and treated with a solution of l-(4- (benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 2.40 g, 8.1 mmole) in tetrahydrofuran (20 mL) added dropwise over 10 min. The resulting mixture was then stirred at -78 °C for 45 min. Then chlorotrimethylsilane (1.18 mL, 9.31 mmol) was added dropwise over 5 min and the resulting solution was stirred at -78 °C for another 20 min. The cooling bath was then removed and the mixture is allowed to warm to room temperature over 30 min. The reaction mixture was then quenched by addition to a cold solution of ethyl acetate (200 mL), saturated sodium bicarbonate (30 mL) and ice. The organic phase was rapidly dried over anhydrous magnesium sulfate (magnetic stirring) and evaporated in vacuo to give the silyl enol ether as an oil which is co-evaporated with toluene (20 mL). The silyl enol ether was then dissolved in dry tetrahydrofuran (40 mL), cooled to -20 °C and treated with solid sodium bicarbonate (0.10 g) followed by N- bromosuccinimide (1.44 g, 8.1 mmol) added in small portions over 15 min. The reaction mixture was allowed to warm to 0 °C over 2h and then quenched by addition of ethyl acetate (300 mL) and saturated sodium bicarbonate. The organic phase was washed with brine, dried over anhydrous magnesium sulfate and evaporated to give an orange oil. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 0-5%) gave 2.62 g (86% yield) of the title bromomethylketone as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave yellow prisms (2.30 g). LC (Method E): 1.977 min. HRMS(ESI) calcd for Ci8Hi6Br04 [M+H]+ m/z 375.0226, found 375.0277. 1H NMR (CDCls, 600 MHz) δ 3.84 (s, 3H), 4.33 (s, 2H), 5.14 (s, 2H), 6.38 (d, J = 1.76 Hz, 1H), 6.64 (broad s, 1H), 7.35 (broad t, 1H), 7.40 (broad t, 2H), 7.44 (broad d, 2H), 7.70 (s, 1H). 1 EE. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone

Benzyltrimethylammonium dichloroiodate (117 g, 169 mmol) was added to a solution of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 50 g, 170 mmol) in THF (500 mL) in a 1 L multineck round bottom flask under nitrogen atmosphere. The reaction mixture was stirred at RT for 6 h, cooled to 0 °C and quenched with 10% NaHCC”3 solution. The organic layer was washed with 1 M sodium thiosulphate solution, water, and brine, dried over Na2S04, and concentrated in vacuo (bath temperature <45 °C). The residue was triturated with 5% EtOAc in pet. ether and dried to obtain the title chloromethylketone as a pale yellow solid (48 g, 130 mmol, 78%). 1H NMR (300 MHz, DMSO-d6) δ 3.84-3.82 (d, J =4.5Hz, 3H) 4.98 (s, 2H), 5.27(s, 2H), 6.62 -6.61 (d, J = 1.8Hz, 1H), 6.92-6.93 (m, 1H), 7.54-7.36 (m, 5H), 8.10-8.09 (d, J = 3Hz, 1H); MS m/z: [M+H]+ 331.0. IF. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoimidazo[2, 1 – b] [ 1 ,3 ,4]thiadiazole

A mixture of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone (Example IE, 3.00 g, 8.0 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (1.65 g, 9.16 mmol) in isopropanol (100 mL) was heated in a pressure flask equipped with a magnetic stirring bar at 78-80 °C for 18 h (homogeneous after 20 min and then formation of a precipitate after 2 h). The cooled mixture is then transferred into five 20 mL microwave vials and then heated in a microwave apparatus to 150 °C for 30 min. Each vial was then diluted with dichloromethane (250 mL) washed with saturated sodium bicarbonate (25 mL) and brine (25 mL), dried over anhydrous magnesium sulfate. The fractions were combined and concentrated in vacuo. Chromatography of the orange-brown residual solid on silica gel (4 x 10 cm, slow elution with dichloromethane due to poor solubility) gave 2.96 g of the title imidazothiadiazole contaminated with some l-(4-(benzyloxy)-6- methoxybenzofuran-2-yl)ethanone. The solid material was triturated with ethyl acetate (20 mL), filtered, washed with ethyl acetate (10 ml) and dried in vacuo to give 2.34 g (64% yield) of pure title imidazothiadiazole as an off white solid which is used as such for the next step. LC (Method E): 2.188 min. HRMS(ESI) calcd for C2oHi5BrN303S [M+H]+ m/z 456.00175, found 456.00397. 1H NMR (CDC13, 600 MHz) δ 3.82 (s, 3H), 5.16 (s, 2H), 6.38 (d, J= 1.67 Hz, 1H), 6.66 (broad s, 1H), 7.15 (s, 1H), 7.31 (broad t, 1H), 7.38 (broad t, 2H), 7.45 (broad d, 2H), 8.02 (s, 1H).
Alternatively, Example IF, 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole, was prepared as follows:
A 1000-mL, three-necked flask equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with dry NMP (200 mL) followed by 1- (4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone (Example 1EE, 50 g, 150 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (27.2 g, 151 mmol). The resulting mixture was stirred at 80 °C for 8h. TLC (8:2 dichloromethane/pet. ether) and LC/MS showed intermediate uncyclized material (m/z 476) and the reaction mixture was stirred at 120 °C for 3h. The reaction mixture was cooled to RT, quenched with water and extracted with EtOAc (3X). The combined organic layers were washed with brine, dried over Na2S04, and concentrated in vacuo. The thick brown residue was purified by silica gel chromatography (0 to 100% dichloromethane in pet. ether) to give a brown solid. This material was triturated with EtOAc and dried to obtain the title imidazothiadiazole (24 g, 50 mmol, 33%>) as a light brown solid. (See the procedure set forth above for analytical data).
1 G. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-methoxyimidazo[2, 1 – b][l,3,4]thiadiazole

A solution of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole (Example IF, 2.30 g, 5.04 mmol) in a mixture of dichloromethane (180 mL) and methanol (45 mL) was treated at 22 °C with 4.2 mL of a 25 wt.% solution of sodium methoxide in methanol (0.2 mmol) added in one portion. More methanol (45 mL) was added and the mixture was stirred for 1 h. The reaction mixture was quenched by the addition of 25 mL of IN hydrochloric acid followed by 20 ml of saturated sodium bicarbonate. The solvent was evaporated under reduced pressure and the residue was diluted with dichloromethane (400 mL), washed with brine, dried over anhydrous magnesium sulfate and evaporated in vacuo. Chromatography of the residue on silica gel (3 x 10 cm, elution with dichloromethane – ethyl acetate 0-4%) gave 1.70 g (83% yield) of the title compound as a white solid. This material was recrystallized from ethyl acetate (30 mL per gram, 80% recovery) to give white needles. LC (Method
D): 2.293 min. HRMS(ESI) calcd for C21H18N3O4S [M+H]+ m/z 408.1013, found 408.1024. 1H NMR (CDC13, 600 MHz) δ 3.81 (s, 3H), 4.18 (s, 3H), 5.16 (s, 2H), 6.37 (d, J = 1.75 Hz, 1H), 6.67 (broad s, 1H), 7.07 (s, 1H), 7.31 (broad t, 1H), 7.37 (broad t, 2H), 7.45 (broad d, 2H), 7.81 (s, 1H).
1H. 6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-ol

A mixture of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- methoxyimidazo[2,l-b][l,3,4]thiadiazole (Example 1G, 1.250 g, 3.06 mmol) and pentamethylbenzene (3.17 g, 21.4 mmol) in dichloromethane (200 mL) was cooled to -78 °C under a nitrogen atmosphere and then treated immediately (to avoid crystallization) with 8 mL (8 mmol) of a 1 M solution of boron trichloride in dichloromethane added dropwise over 3 min. The resulting mixture was stirred at -78 °C for 1 h. The reaction mixture was then quenched by the addition of a solution of sodium bicarbonate (6 g) in water (100 mL) added in one portion. The cooling bath was removed and the resulting mixture was stirred at room temperature for 1 h. The solid formed was filtered, washed successively with water (50 m) and dichloromethane (50 mL). The filter cake was allowed to soak with anhydrous ethanol (15 ml) and then sucked dry. The white solid obtained was then dried under vacuum for 24 h to give 0.788 g (80%> yield) of pure title material (> 95% by hplc). The combined filtrate and washings were diluted with dichloromethane (600 mL) and stirred in a warm water bath till the organic phase was clear with no apparent solid in suspension. The organic phase was collected, dried over anhydrous magnesium sulfate and rapidly filtered while still warm. The filtrate was evaporated and the residue (product and pentamethylbenzene) was triturated with toluene (20 mL), the solid collected and washed with toluene (20 mL) to give 0.186 g (19% yield, 99% combined yield) of title material as a tan solid (> 95% by hplc). LC (Method E): 1.444 min. HRMS(ESI) calcd for C14H12N3O4S [M+H]+ m/z 318.0543, found 318.0578. 1H NMR (DMSO-de, 600 MHz) 5 3.71 (s, 3H), 4.16 (s, 3H), 6.21 (d, J = 1.87 Hz, 1H), 6.61 (broad s, 1H), 6.95 (s, 1H), 8.29 (s, 1H), 9.96 (s, 1H).
Example 94
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine

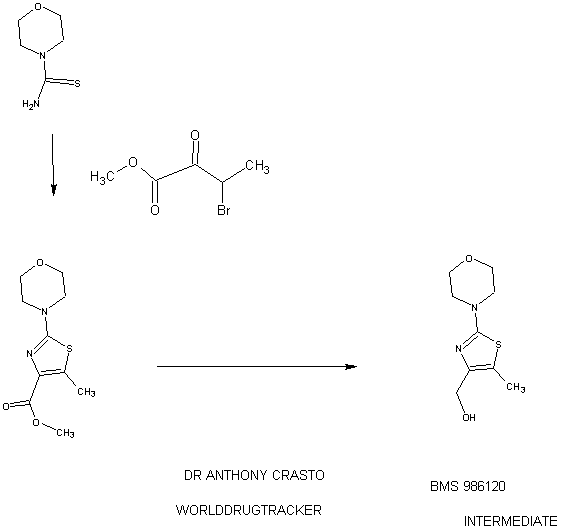
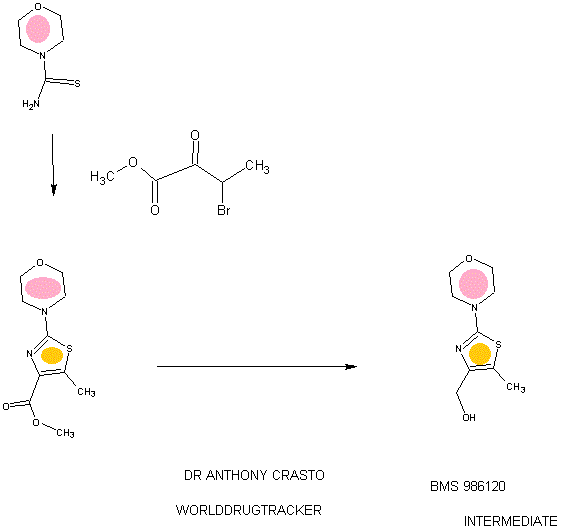
94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate [00258] A solution of methyl 2-bromo-5-methylthiazole-4-carboxylate (2.80 g, 11.86 mmol) and morpholine (4.5 mL, 51.7 mmol) in THF (10 mL) was heated at reflux under nitrogen for 18 h. The volatiles were then removed under reduced pressure and the crude product was purified on the ISCO using a REDISEP® 40 g column (0 to 40% EtOAc- DCM), to give the title compound (2.20 g, 77%) as a yellow solid. LCMS (APCI): calcd for CioHisNzOsS [M+H]+ m/z 243.07, found 243.1. 1H NMR (CDC13, 400 MHz) δ ppm: 3.89 (s, 3H), 3.77-3.83 (m, 4H), 3.41-3.47 (m, 4H), 2.64 (s, 3H). [00259] Alternatively, Example 94A, methyl 5-methyl-2-morpholinothiazole-4- carboxylate, was prepared as follows:
94AA. Methyl 3-bromo-2-oxobutanoate

A 5L 4-neck round bottom flask equipped with a mechanical stirrer, temperature thermocouple, condenser and a 1L addition funnel, was charged copper(II) bromide (962 g, 4310 mmol) and ethyl acetate (2 L). A solution of methyl 2-ketobutyrate (250 g, 2150 mmol) in CHC13 (828 mL) was added dropwise. A scrubber (400 mL 1 N NaOH) was connected and the reaction mixture was heated to reflux (75 °C). The reaction started as a dark green color and as heating progressed, it became a light green with a white precipitate forming. NMR after one hour at reflux indicated that the reaction was complete. The reaction was cooled to RT and filtered through a pad of CELITE®. The filtrate was concentrated to an oil, dissolved in methylene chloride (500 mL) and filtered again through CELITE®. The filtrate was then passed through a pad of silica gel and eluted with ethyl acetate. Concentration of the filtrate provided the title bromoketoester (399 g, 2040 mmol, 95%) as a yellow oil. 1H NMR (400MHz, CDC13) δ 5.18 (q, J = 6.7 Hz, 1H), 3.94 (s, 3H), 1.83 (d, J = 6.8 Hz, 3H). 94AAA. Morpholine-4-carbothioamide

To a solution of morpholine (199 g, 2280 mmol) in CHC13 (1 L) was added isothiocyanatotrimethylsilane (150 g, 1140 mmol) dropwise. A white precipitate formed almost immediately, and the reaction was stirred for 1 h at RT. The reaction was then filtered and the resulting solid was washed with additional CHC13 and dried in vacuo to give the title thiourea as a white solid. (137 g, 937 mmol, 82%). 1H NMR (400MHz, DMSO-de) δ 3.81 – 3.71 (m, 2H), 3.17 – 3.08 (m, 2H).
94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate

To a solution of morpholine-4-carbothioamide (Example 94 AAA, 175 g, 1200 mmol) in methanol (500 mL) was charged methyl 3-bromo-2-oxobutanoate (Example 94AA, 233 g, 1200 mmol). The reaction was then heated to reflux for 1 hour, cooled to RT, and filtered. The filtrate was concentrated and the crude product was purified on by silica gel chromatography. The title thiazole (206g, 850 mmol, 71%) was isolated as a yellow oil. (See the procedure set forth above for analytical data).
(5-Methyl-2-morpholinothiaz l-4-yl)methanol

The compound was prepared according to the protocol described for Example 92B. The crude product was purified on the ISCO using a REDISEP® Gold 24 g column (0 to 50% EtOAc-DCM) to give the title compound as a white solid (0.086 g, 51%). LCMS (APCI): calcd for C9Hi5N202S [M+H]+ m/z 215.08, found 215.1. 1H NMR (CDCI3, 400 MHz) δ ppm: 4.48 (d, J= 4.7 Hz, 2H), 3.77-3.83 (m, 4H), 3.37-3.43 (m, 4H), 2.30 (t, J= 4.7 Hz, 1H), 2.28 (s, 3H).
Example 94. 4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2, 1 -b] [ 1 ,3,4]thiadiazol-6-yl) benzofuran-4-yl)oxy)methyl)-5 -methylthiazol-2-yl)morpholine

The title compound was prepared according to the protocol described for Example 86. The crude product was purified on the ISCO using a REDISEP® 4 g column (0 to 40% EtOAc-DCM) and the obtained solid was suspended in MeOH, sonicated, filtered and dried to give the title compound as an off-white solid (0.094 g, 53%). LC (Method C): 2.314 min. HRMS(ESI): calcd for C23H24N505S2 [M+H]+ m/z 514.122, found 514.126. 1H NMR (CDC13, 400 MHz) δ ppm: 7.83 (s, 1H), 7.06 (d, J = 0.8 Hz, 1H), 6.69 (d, J= 0.8 Hz, 1H), 6.50 (d, J= 2.0 Hz, 1H), 5.05 (s, 2H), 4.21 (s, 3H), 3.85 (s, 3H), 3.78- 3.84 (m, 4H), 3.39- 3.46 (m, 4H), 2.37 (s, 3H).
ABSTRACT
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 263


| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094297 | 2015-04-02 | IMIDAZOTHIADIAZOLE AND IMIDAZOPYRAZINE DERIVATIVES AS PROTEASE ACTIVATED RECEPTOR 4 (PAR4) INHIBITORS FOR TREATING PLATELET AGGREGATION |
////////BMS 986120, phase 1, Bristol-Myers Squibb , Imidazoles, Small molecules, Thiadiazoles, 1478712-37-6
c1(sc2nc(cn2n1)c3cc4c(cc(cc4o3)OC)OCc5nc(sc5C)N6CCOCC6)OC
CC1=C(N=C(S1)N2CCOCC2)COC3=C4C=C(OC4=CC(=C3)OC)C5=CN6C(=N5)SC(=N6)OC
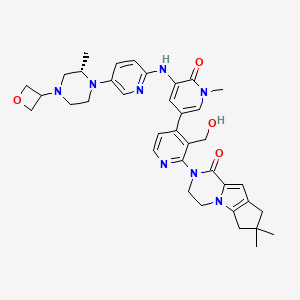
GDC 0853, Fenebrutinib
 .
.
Picture credit….Bethany Halford
GDC 0853, Fenebrutinib
GDC-0853; RG 7845
| Molecular Formula: | C37H44N8O4 |
|---|---|
| Molecular Weight: | 664.79646 g/mol |
2-[3-(hydroxymethyl)-4-[1-methyl-5-[(7-methyl-6,8-dihydro-5H-[1,2,4]triazolo[1,5-a]pyrazin-2-yl)amino]-6-oxo-3-pyridyl]-2-pyridyl]-3,4,6,7,8,9-hexahydropyrazino[1,2-a]indol-1-one
3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
2H-Cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one, 2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S) -2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino] -6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7- dimethyl-
s ISoMER 1434048-34-6 desired
r iSoMER 1434048-57-3 undesired
Phase 1
Patients with Patients with Resistant B-Cell Lymphoma or Chronic Lymphocytic Leukemia..
@genentech‘s Btk inhibitor
https://clinicaltrials.gov/ct2/show/NCT01991184
Bruton tyrosine kinase inhibitor
- 01 Sep 2015 Phase-I clinical trials in Autoimmune disorders (In volunteers) in USA (PO, Capsule and Tablet) (NCT02699710)
- 16 Oct 2014 Discontinued – Phase-I for Non-Hodgkin’s lymphoma (Second-line therapy or greater) in USA (unspecified route)
- 16 Oct 2014 Discontinued – Phase-I for Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (unspecified route)

GDC-0853; RG 7845; RO 7010939
2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S)-2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino]-6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7-dimethyl-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one
GDC-0853 is an orally bioavailable, selective, and reversible Bruton’s tyrosine kinase (BTK) inhibitor with IC50s ranging from 2-9 nM for basophil activation, B cell receptor activation, and constitutive p-BTK activity in whole blood lysates.1,2 In rats, treatment for longer than 7 days leads to pancreatic toxicity but it does not occur in mice or dogs, even at higher doses.3 Formulations containing GDC-0853 were well-tolerated in Phase I clinical trials and are in additional clinical trials for rheumatoid arthritis, lupus erythematosus, lymphoma, and leukemia.
- Originator Genentech
- Class Antineoplastics; Antirheumatics; Piperazines; Pyrazines; Pyridines
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
- Phase II Rheumatoid arthritis; Systemic lupus erythematosus; Urticaria
- Phase I Autoimmune disorders
- Discontinued Chronic lymphocytic leukaemia; Non-Hodgkin’s lymphoma
Most Recent Events
- 01 Jun 2018 Chemical structure information added
- 07 Nov 2017 Genentech initiates enrolment in a phase II extension trial for Systemic Lupus Erythematosus in Spain (EudraCT2017-001764-37)
- 13 Sep 2017 Genentech initiates enrolment in a phase I trial in Healthy volunteers in United Kingdom (NCT03290703)
BTK inhibitor GDC-0853 An orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, GDC-0853 inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
SCHEME

MAIN

Patent
WO 2013067274
https://www.google.co.in/patents/WO2013067274A1?cl=en
part
Example 271a (S)-tert-Butyl 4-(6-(5-Chloro-2-methoxypyridin-3-ylamino)pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271a
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (40 mL), (S)-tert-butyl 4-(6-amino pyridin-3-yl)-3-methylpiperazine-1-carboxylate 101h (2.04 g, 7.0 mmol), 3-bromo-5-chloro-2-methoxypyridine (2.8 g, 12.6 mmol), Pd2(dba)3 (640 mg, 0.70 mmol), XantPhos (404.6 mg, 0.70 mmol), and cesium carbonate (4.56 g, 14.0 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 °C for 4 h. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 1:3 ethyl acetate/petroleum ether to afford 271a (1.7 g, 57%) as a yellow solid. MS-ESI: [M+H]+ 434.2
Example 271btert-Butyl (3S)-4-(6-{[5-(2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}-3-(hydroxymethyl)pyridin-4-yl)-2-methoxypyridin-3-yl] amino}pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271b
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 271a (650 mg, 1.50 mmol), {3-[(acetyloxy)methyl]-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridin-4-yl}boronic acid 199e (1.79 g, 4.5 mmol), Pd2(dba)3 (137.2 mg, 0.15 mmol), P(cy)3(167.4 mg, 0.60 mmol), Cs2CO3 (978 mg, 3.0 mmol), dioxane (20 mL), and water (0.5 mL). After three cycles of vacuum/argon flush, the mixture was heated at 110°C for 16 h. After this time the reaction was cooled to room temperature. Lithium hydroxide monohydrate (1.89 g, 45 mmol) and water (2.0 mL) were added. The resulting mixture was stirred at 45°C for 4 h. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 ethyl acetate/petroleum ether to afford 271b (290 mg, 27%) as a yellow solid. MS-ESI: [M+H]+ 709.3
Example 271c 10-[3-(Hydroxymethyl)-4-[5-({5-[(2S)-2-methylpiperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridin-2-yl]-4,4-dimethyl-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-9-one 271c
A solution of 271b (286.6 mg, 0.40 mmol) in dioxane/HCl (30 mL) was stirred at 50 °C for 2 h. It was evaporated under reduced pressure to afford 271c (450 mg, crude) as a black solid. MS-ESI: [M+H]+ 595.3
Example 271 3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one 271
To a solution of 271c (450 mg, 0.75 mmol) in methanol (10 mL) was added oxetan-3-one (162 mg, 2.25 mmol), NaBH3CN (141.8 mg, 2.25 mmol), and ZnCl2 (306 mg, 2.25 mmol). The reaction was stirred at room temperature for 3 h. The mixture was evaporated under reduced pressure and the residue was diluted with water (5 mL). It was then extracted with dichloromethane (3 X 10 mL) and the combined dichloromethane extract was concentrated under reduced pressure. The residue was purified by reverse-phase prep-HPLC to afford 271 (23.0 mg, 8.8%, over two steps) as a yellow solid. MS-ESI: [M+H]+651.3. 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 8.74 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 5.0 Hz, 1H), 7.99 (d, J = 3.0 Hz, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.41 (d, J = 4.5 Hz, 1H), 7.35 (dd, J = 2.5 Hz, 8.5 Hz, 1H), 6.87 (s, 1H), 6.85 (d, J = 9.0 Hz, 1H), 5.16-5.13 (m, 1H), 4.72-4.69 (m, 5H), 4.54-4.53 (m, 1H), 4.36-4.35 (m, 1H), 4.19-4.17 (m, 2H), 3.89-3.87 (m, 1H), 3.56-3.49 (m, 2H), 3.11-3.09 (m, 2H), 2.60-2.48 (m, overlap, 7H), 2.24-2.21 (m, 1H), 1.29 (s, 6H), 1.02 (d, J = 6.0 Hz, 3H)
271
………………………..
syn of 191 j
is intermediatenot product, is acid
To a mixture of 4-chloro-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridine-3-carbaldehyde 108a (500 mg, 1.46 mmol), tert-butyl alcohol (20 mL), and dichloromethane (5 mL) was added 2-methyl-2-butene (3066 mg, 43.8 mmol). An aqueous solution (8 mL) of NaClO2 (263 mg, 2.92 mmol) and NaH2PO4·2water (683 mg, 4.38 mmol) was added dropwise at -10°C and the reaction mixture was stirred at -10 °C for overnight. It was concentrated under reduced pressure and the residue was extracted with ethyl acetate (4 × 20 mL). The combined organic extract was dried over MgSO4 and concentrated. The residue was purified with reverse-phase prep-HPLC to afford 210a (315 mg, 60%) as a pale yellow solid. MS-ESI: [M+H]+ 360.1
Example 210b 2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl} -4-[1-methyl-5-({5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridine-3-carboxylic Acid 210b
A 25-mL round-bottomed flask equipped with a reflux condenser was charged with 210a (400 mg, 1.1 mmol), (S)-1-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-ylamino)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2(1H)-one 191j (536 mg, 1.1 mmol), PdCl2(dppf) (81 mg, 0.11 mmol), K3PO4 (466 mg, 2.2 mmol), sodium acetate (216 mg, 2.2 mmol), acetonitrile (10 mL), and water (0.2 mL). After three cycles of vacuum/argon flush, the mixture was heated at 100°C for 3 h. It was then filtered and the filtrate was evaporated in vacuo. The residue was purified by silica-gel column chromatography eluting with 1:3 petroleum/ethyl acetate to afford 210b as a yellow solid (306 mg, 41%). MS-ESI: [M+H]+ 679.3
construction, use your discretion
Example 130a (3S)-tert- utyl 3-methyl-4-(6-nitropyridin-3-yl)piperazine-l-carboxylate 130a

130a
Following the procedures as described for compound lOlg, reaction of 5-bromo-2-nitropyridine (10.5 g, 50 mmol), and (JS)-tert-butyl-3 -methylpiperazine- 1 -carboxylate (10.0 g, 50 mmol) afforded 130a as a yellow solid (8.05 g, 50%). LCMS: [M+H]+ 323
Example 130b (3 S)-tert-butyl-4-(6-aminopyridin-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130b

130b
Following the procedures as described for compound lOlh, hydrogenation of 130a (5.8 g) afforded 130bas a brown solid (4.9 g, 96%). LCMS: [M+H]+ 293
Example 130c (3 S)-tert-Butyl-4-(6-(5 -bromo- 1 -methyl -2 -oxo- 1,2-dihydropyridin-3 -yl amino) pyridine-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130c
N

Following the procedures as described for compound lOli, reaction of 130b (4.0 g) and 3,5-dibromo-l-methylpyridin-2(lH)-one (5.5 g) afforded 130c as a yellow solid (5.4 g, 83%). LCMS: [M+H]+ 478
Example 130d (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- 2-ylamino)pyridine-2(lH)-one 130d

Following the procedures as described for compound lOlj, acidic hydrolysis of the Boc group of 130c (3.1 g) afforded 130d as a yellow solid (2.3 g, 95%). LCMS: [M+H]+ 380.
Example 130e (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2 -methyl-4-(ox etan-3-yl)piperazin-l-yl) pyridine -2-ylamino)pyridin-2(lH)-one 130e

Following the procedures as described for compound 101k, reductive amination of 130d (2.35 g) with oxetan-3-one (0.4 mL) afforded 130e as a yellow solid (2.6 g, 98%). LCMS: [M+H]+ 434.
Example 13 Of (3S)-l-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)pyridin-2-ylamino) -5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 130f
 check pyridine ring position
check pyridine ring position
A 100 mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 130e (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), AcOK (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90 °C for 4 hrs, then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3: 1 PE/EA (80 mL) to afford 130f as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
check pyridine ring position, use your discretion
Example 191h ( 3S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- -ylamino)pyridine-2(lH)-one 191h

Following the procedure described for compound lOlj and starting with (3S)-tert-butyl 4-(6-(5 -bromo- 1 -methyl-2-oxo- 1 ,2-dihydropyridin-3 -ylamino)pyridine-3 -yl)-3 -methyl-piperazine-l-carboxylate 191g (3.1 g, 6.5 mmol) afforded 191h as a yellow solid (2.3 g, 94%). MS-ESI: [M+H]+ 378.
Example 1 1 i (S)-5 -Bromo- 1 -methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin- 1 -yl)pyridin-2-ylamino)pyridin-2(lH)-one 191i
A mixture of (5)-5-bromo-l-methyl-3-(5-(2-methylpiperazin-l-yl)pyridin-2-ylamino)pyridin-2(lH)-one 191h (40.0 g, 106 mmol), oxetan-3-one (1 1.4 g, 159 mmol), NaBH3CN (10.0 g, 159 mmol), and zinc chloride (21.3 g, 159 mmol) in methanol (700 mL) was stirred at 50°C for 5 hours. The mixture was added to water (100 mL) and concentrated under reduced pressure. The residue was extracted with dichloromethane (200 mL x 3). The combined organic layer was concentrated under reduced pressure and the residue was purified by silica-gel column chromatography eluting with 40: 1 dichloromethane /methanol to afford 191i (35 g, 73%). MS: [M+H]+ 434.
Example 191j (J5)-l-Methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)-pyridin- -ylamino) -5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 191j

191 i 191j
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with (5)-tert-butyl-4-(6-(5-bromo-l-methyl-2-oxo-l ,2-dihydropyridin-3-ylamino)pyridine-3-yl)-3-methylpiperazine-l-carboxylate 191i (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), potassium acetate (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90°C for 4 h. It was then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3 : 1 petroleum ether/ethyl acetate (80 mL) to afford 191j as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
pipeline
http://www.gene.com/medical-professionals/pipeline

Pictrelisib, GDC-0941, RG7321 and GNE0941
| Patent ID | Date | Patent Title |
|---|---|---|
| US8921353 | 2014-12-30 | Heteroaryl pyridone and aza-pyridone compounds |
| US2014378432 | 2014-12-25 | HETEROARYL PYRIDONE AND AZA-PYRIDONE COMPOUNDS |
| US8716274 | 2014-05-06 | Heteroaryl pyridone and aza-pyridone compounds |
Development of an Efficient Manufacturing Process for Reversible Bruton’s Tyrosine Kinase Inhibitor GDC-0853
 , Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin† 
Efforts toward the process development of reversible Bruton’s tyrosine kinase (BTK) inhibitor GDC-0853 (1) are described. A practical synthesis of GDC-0853 was accomplished via a key highly regioselective Pd-catalyzed C–N coupling of tricyclic lactam 5 with 2,4-dichloronicotinaldehyde (6) to afford the C–N coupling product 3, a Suzuki–Miyaura cross-coupling of intermediate 3 with boronic ester 4 derived from a Pd-catalyzed borylation of tetracyclic bromide 7, to generate penultimate aldehyde intermediate 2 and subsequent aldehyde reduction and recrystallization. Process development of starting materials 5, 6, and 7 is also discussed.
(S)-2-(3′-(Hydroxymethyl)-1-methyl-5-((5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino)-6-oxo-1,6-dihydro-[3,4′-bipyridin]-2′-yl)-7,7-dimethyl-2,3,4,6,7,8-hexahydro-1H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1-one (crude GDC-0853, 1)
GDC-0853 (1, 196 kg, 81% yield, >99 A%, Pd < 10 ppm): mp 271 °C (DSC);
FTIR (cm–1, neat) 3430, 3313, 2945, 2865, 1606, 1573;
1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 2.2 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 2.3 Hz, 2H), 7.36 (d, J = 5.1 Hz, 1H), 7.31 (dd, J = 8.9, 2.8 Hz, 1H), 6.87–6.76 (m, 2H), 5.18–4.98 (m, 1H), 4.77–4.58 (m, 5H), 4.50 (m, 1H), 4.33 (m, 1H), 4.16 (m, 2H), 3.86 (m, 1H), 3.71 (s, 3H), 3.61–3.38 (m, 2H), 3.07 (m, 2H), 2.67–2.39 (m, 7H), 2.20 (dd, J = 10.8, 6.3 Hz, 1H), 1.27 (s, 6H), 0.98 (d, J = 6.3 Hz, 3H);
13C NMR (101 MHz, CDCl3) δ 161.7, 157.6, 154.3, 150.3, 148.4, 141.9, 140.0, 131.4, 131.1, 129.7, 128.8, 127.7, 125.8, 123.9, 117.2, 116.3, 112.4, 111.3, 75.5, 75.5, 59.4, 59.1, 56.3, 52.9, 50.0, 49.2, 48.2, 45.9, 42.7, 40.9, 39.6, 38.5, 30.3, 15.3.
HRMS (ESI+) calcd for C37H45N8O4 ([M + H]+), 665.3564; found, 665.3588.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00134/suppl_file/op8b00134_si_001.pdf


/////////////
O=C1N(C)C=C(C2=CC=NC(N3CCN4C(C3=O)=CC5=C4CC(C)(C)C5)=C2CO)C=C1NC(N=C6)=CC=C6N7CCN(C8COC8)C[C@@H]7C
//////GDC 0853, genentech, Btk inhibitor, phase 1, Patients with Resistant B-Cell Lymphoma, Chronic Lymphocytic Leukemia, Bruton tyrosine kinase inhibitor, GDC-0853, RG 7845, 1434048-34-6, Fenebrutinib
N1(CCN(CC1C)C2COC2)c3cnc(cc3)NC=4C(N(\C=C(/C=4)c5c(c(ncc5)N6CCn7c(C6=O)cc8CC(Cc78)(C)C)CO)C)=O
CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
SIDE CHAIN
MAIN
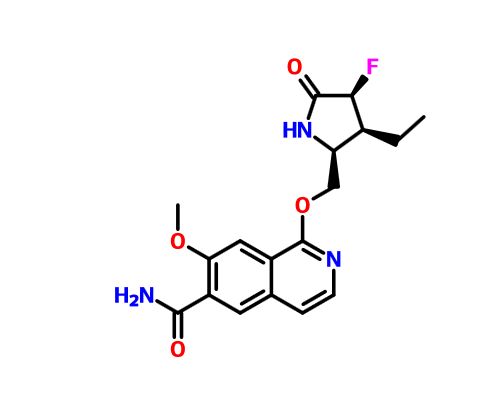
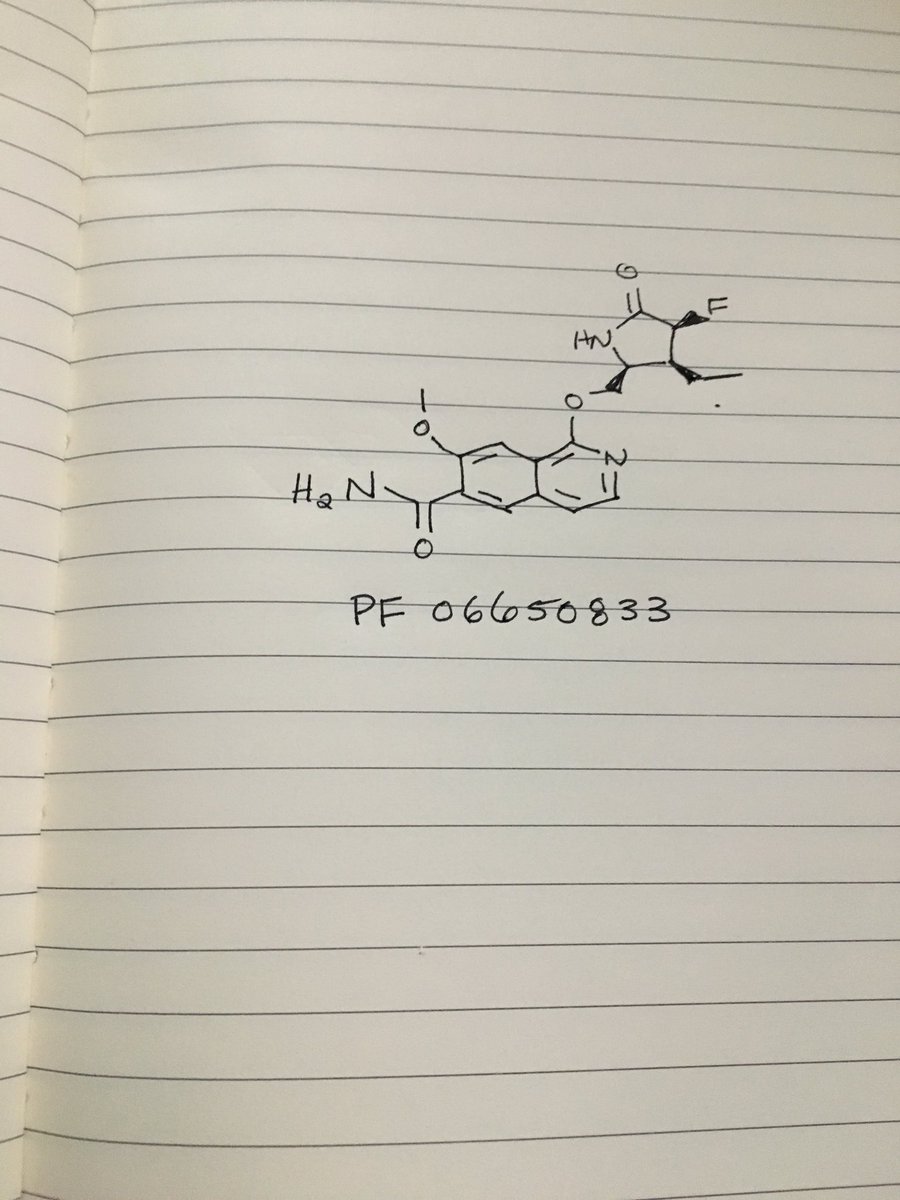
PF 06650833
 .
.
Picture credit….Bethany Halford
PF 06650833
MFC18H20FN3O4, MW361.37
1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide
6-Isoquinolinecarboxamide, 1-[[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxo-2-pyrrolidinyl]methoxy]-7-methoxy-
CAS 1817626-54-2
WO 2015150995
1st disclosures is @pfizer‘s on inflammatory disease treatment targeting IRAK4
$PFE IRAK4 inhibitor
Phase I Lupus vulgaris
- 01 Feb 2016 Pfizer completes a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Nov 2015 Pfizer initiates a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Jun 2015 Pfizer completes a phase I trial for Lupus (In volunteers) in USA (PO) (NCT02224651)
Compounds useful for the treatment of autoimmune and inflammatory diseases associated with lnterleukin-1 Receptor Associated Kinase (IRAK) and more particularly compounds that modulate the function of IRAK4.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified in tyrosine and serine/threonine kinases. Inappropriate activity arising from dysregulation of certain kinases by a variety of mechanisms is believed to underlie the causes of many diseases, including but not limited to, cancer, cardiovascular diseases, allergies, asthma, respiratory diseases, autoimmune diseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative diseases. As such, potent and selective inhibitors of kinases are sought as potential treatments for a variety of human diseases.
There is considerable interest in targeting the innate immune system in the treatment of autoimmune diseases and sterile inflammation. Receptors of the innate immune system provide the first line of defense against bacterial and viral insults. These receptors recognize bacterial and viral products as well as pro-inflammatory cytokines and thereby initiate a signaling cascade that ultimately results in the up-regulation of inflammatory cytokines such as TNFa, IL6, and interferons. Recently it has become apparent that self-generated ligands such as nucleic acids and products of inflammation such as high-mobility group protein B1 (HMGB1) and Advanced Glycated End-products (AGE) are ligands for Toll-like receptors (TLRs) which are key receptors of the innate immune system (O’Neill 2003, Kanzler et al 2007, Wagner 2006). This demonstrates the role of TLRs in the initiation and perpetuation of inflammation due to autoimmunity.
lnterleukin-1 receptor associated kinase 4 (I RAK4) is a ubiquitously expressed serine/threonine kinase involved in the regulation of innate immunity (Suzuki & Saito 2006). IRAK4 is responsible for initiating signaling from TLRs and members of the I L- 1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice were reported to cause reductions in TLR and IL-1 induced pro-inflammatory cytokines (Kawagoe et al 2007; Fraczek et al. 2008; Kim et al. 2007). IRAK4 kinase-dead knock-in mice have also been shown to be resistant to induced joint inflammation in the antigen-induced-arthritis (AIA) and serum transfer-induced (K/BxN) arthritis models (Koziczak-Holbro 2009). Likewise, humans deficient in IRAK4 also appear to display the inability to respond to challenge by Toll ligands and IL-1 (Hernandez & Bastian 2006). However, the immunodeficient phenotype of IRAK4-null individuals is narrowly restricted to challenge by gram positive bacteria, but not gram negative bacteria, viruses or fungi. This gram positive sensitivity also lessens with age, implying redundant or compensating mechanisms for innate immunity in the absence of IRAK4 (Lavine et al 2007).
These data indicate that inhibitors of IRAK4 kinase activity should have therapeutic value in treating cytokine driven autoimmune diseases while having minimal immunosuppressive side effects. Additional recent studies suggest that targeting IRAK4 may be useful in other inflammatory pathologies such as atherosclerosis and diffuse large B-cell lymphoma (Rekhter et al 2008; Ngo et al 2011). Therefore, inhibitors of IRAK4 kinase activity are potential therapeutics for a wide variety of diseases including but not limited to autoimmunity, inflammation, cardiovascular diseases, cancer, and metabolic diseases. See the following references for additional information: N. Suzuki and T. Saito, Trends in Immunology, 2006, 27, 566. T. Kawagoe, S. Sato, A. Jung, M. Yamamoto, K. Matsui, H. Kato, S. Uematsu, O. Takeuchi and S. Akira, Journal of Experimental Medicine, 2007, 204, 1013. J. Fraczek, T. W. Kim, H. Xiao, J. Yao, Q. Wen, Y. Li, J.-L. Casanova, J. Pryjma and X. Li, Journal of Biological Chemistry, 2008, 283, 31697. T. W. Kim, K. Staschke, K. Bulek, J. Yao, K. Peters, K.-H. Oh, Y. Vandenburg, H. Xiao, W. Qian, T. Hamilton, B. Min, G. Sen, R. Gilmour and X. Li, Journal of Experimental Medicine, 2007, 204, 1025. M. Koziczak-Holbro, A. Littlewood- Evans,
B. Pollinger, J. Kovarik, J. Dawson, G. Zenke, C. Burkhart, M. Muller and H. Gram, Arthritis & Rheumatism, 2009, 60, 1661. M. Hernandez and J. F. Bastian, Current Allergy and Asthma Reports, 2006, 6, 468. E. Lavine, R. Somech, J. Y. Zhang, A. Puel, X. Bossuyt, C. Picard, J. L. Casanova and C. M. Roifman, Journal of Allergy and Clinical Immunology, 2007, 120, 948. M. Rekhter, K. Staschke, T. Estridge, P. Rutherford, N. Jackson, D. Gifford-Moore, P. Foxworthy,
C. Reidy, X.-d. Huang, M. Kalbfleisch, K. Hui, M.S. Kuo, R. Gilmour and C. J. Vlahos, Biochemical and Biophysical Research Communications, 2008, 367, 642. O’Neill, L. A. (2003). “Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases.” Curr Opin Pharmacol 3(4): 396. Kanzler, H et al. (2007) “Therapeutic targeting of innate immunity with toll-like receptor agonists and antagonists.” Nature Medicine 13:552. Wagner, H. (2006) “Endogenous TLR ligands and autoimmunity” /Advances in Immunol 91 : 159. Ngo, V. N. et al. (2011) “Oncogenically active MyD88 mutations in human lymphoma” Nature 470: 115.
PATENT
Preparation 1 : 1-chloro-7-methoxyisoquinoline-6-carbonitrile (P1) Step 1. Synthesis of methyl 4-iodo-3-methoxybenzoate (CAS 35387-92-9. CD.
To a solution of 3-hydroxy-4-iodobenzoic acid (CAS 58123-77-6, C12) (10800 g, 40.9 moles) in DMF (65 L) was added K2C03 (25398 g, 184 moles), followed by the slow addition of dimethyl sulfate (11352 g, 90 moles). This mixture was heated to about 50 °C for over night. The reaction mixture was cooled to about 25 °C, diluted with EtOAc (50 L) and filtered through a plug of Celite®. The solid was thoroughly washed with EtOAc (10 L X 3). The combined EtOAc filtrates were poured into water. After stirring for about 30 min, the EtOAc layer was separated and it was further washed sequentially with water, 1 M NaOH and brine. The EtOAc layer was separated, dried over Na2S04, filtered and concentrated to provide the title compound C1. Yield: 11750 g (98%).
Step 2. Synthesis of (4-iodo-3-methoxyphenyl)methanol (CAS 244257-61-2, C2).
To a solution of compound C1 (11750 g, 40.2 moles) in THF (35 L) was added NaBH4 (7645 g, 201.09 moles) and refluxed. While refluxing, MeOH (25 L) was slowly added into the reaction mixture at a rate of about 1 L per hour. After completion of the reaction, it was poured into a solution of cold dilute HCI. Once the excess of NaBH4was quenched, the solution was filtered and extracted with EtOAc (2.5 L X 3). The combined EtOAc extracts were washed sequentially with water, brine and dried over Na2S04. The solvent was evaporated under reduced pressure and the resulting crude material was treated with MTBE. The resulting solid was filtered and filtrate was washed with water, brine, dried over Na2S0 , and filtered. The solvent was evaporated under reduced pressure to provide the title compound C2. Yield: 9900 g (93%).
Step 3. Synthesis of 4-iodo-3-methoxybenzaldehyde (CAS 121404-83-9, C3).
To a solution of compound C2 (9900 g, 34.5 moles) in CHCI3 (186 L), was added manganese dioxide (18000 g, 207 moles) and the resulting mixture was refluxed for about 16 h. The mixture was cooled to about 25 °C and filtered through a Celite pad, which was then washed thoroughly with CHCI3. The CHCI3 was evaporated under reduced pressure to provide the title compound C3. Yield: 9330 g (95%). 1 H NMR (400 MHz, CDCI3): δ 9.95 (s, 1 H), 7.99 (d, 1 H), 7.14 (dd, 1 H), 3.95 (s, 3 H).
Step 3. Synthesis of 6-iodo-7-methoxyisoquinoline (CAS 244257-63-4. C4).
To a solution of compound C3 (9300 g, 35 moles) in toluene (60 L) was added amino acetaldehyde dimethyl acetal (5590 g, 53 moles) and the mixture was refluxed for about 4 h, while removing the liberated water by the use of a Dean – Stark water separator. The reaction mixture was cooled to about 0 °C, after which trifluoroacetic anhydride (22305 g, 106 moles) followed by BF3-Et20 (15080 g, 106 moles) were added, keeping internal temperature below 5 °C. The reaction mixture was stirred at about 25 °C for about 16 h and quenched by pouring into a mixture of ice and ammonium hydroxide. The product was extracted with EtOAc (10 L X 3), and the combined EtOAc extracts were washed sequentially with water and brine. The combined EtOAc extracts were dried over Na2S04, filtered, and concentrated to afford a dark tan colored residue. This was treated with a mixture of MTBE and hexane (1 :1 v/v, 30 L), followed by 6 M HCI (9 L), with stirring. The precipitated solid was filtered and washed with MTBE. The solid was suspended in EtOAc (5 L) and made alkaline with ammonium hydroxide. The EtOAc layer was separated, washed with brine, dried over Na2S04, filtered, and concentrated to afford crude compound C4 as a brown solid. HPLC (230 nm) showed it to be about 83% pure.
The crude material (1000 g) was taken in AcOH (2.5 L) and stirred for about 90 min at about 25 °C. The solid was filtered and washed with AcOH (500 ml_). The filtrate was neutralized with saturated aqueous Na2C03 solution. The resulting precipitated solid was filtered, washed with water (4 L), and oven dried at about 70 – 75 °C for about 5 h to afford about 780 g of pure C4. Similarly, the remaining crude C4 (4 kg) was purified to provide the title compound C4. Yield: 4300 g (42%). 1H NMR (400 MHz, CDCI3): δ 9.15 (s, 1 H), 8.45 (d, 1 H), 8.35 (s, 1 H), 7.45 (d, 1 H), 7.15 (s, 1 H) 4.00 (s, 3 H).
Step 4. Synthesis of 7-methoxyisoquinoline-6-carbonitrile (C5).
To a solution of compound C4 (4300 g , 15 moles) in DMSO (39 L) was added copper(l) cyanide (2954 g, 33 moles) and the mixture was heated to about 120 °C for about 3 h. The reaction mixture was quenched by pouring into a mixture of ice and ammonium hydroxide (40 L) and filtered. The filtrate was extracted with EtOAc (10 L X 2). While stirring, the solid residue was again treated with ammonium hydroxide solution (10 L) and EtOAc (10 L). After filtration, the precipitated material was repeatedly washed with a mixture of MeOH and CHCI3 (1 :9, v/v) several times and the combined extracts were washed with brine. The extracts were dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting crude material was triturated with hexane to provide the title compound C5. Yield: 2250 g (87%). 1H NMR (400 MHz, CDCI3): δ 9.25 (br. s, 1 H), 8.55 (br. s, 1 H), 8.15 (s, 1 H), 7.60 (d, 1 H), 7.30 (s, 1 H), 4.05 (s, 3 H).

A solution of a reactant such as 1-(((2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl)methoxy)-7-methoxyisoquinoline-6-carbonitrile (200 mg, 0.5 mmol) in concentrated H2SO4 (1.5 ml.) was warmed to about 55 °C for about two hours, then cooled to about 20 °C. The reaction mixture was added dropwise with vigorous stirring to 7.3 ml_ of ice cold concentrated ammonium hydroxide with cooling in ice. The precipitated solid was filtered and washed with water, heptane, ether, and dried under vacuum. The residue may be used directly for subsequent work, or it may be purified by chromatography or HPLC.
ABSTRACTS
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 261




//////////PF 06650833, IRAK4 inhibitor, inflammatory disease treatment , PFIZER, 1817626-54-2
N1C([C@H](C([C@H]1COc3c2cc(c(cc2ccn3)C(=O)N)OC)CC)F)=O
NC(=O)c2cc3ccnc(OC[C@H]1NC(=O)[C@@H](F)[C@H]1CC)c3cc2OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
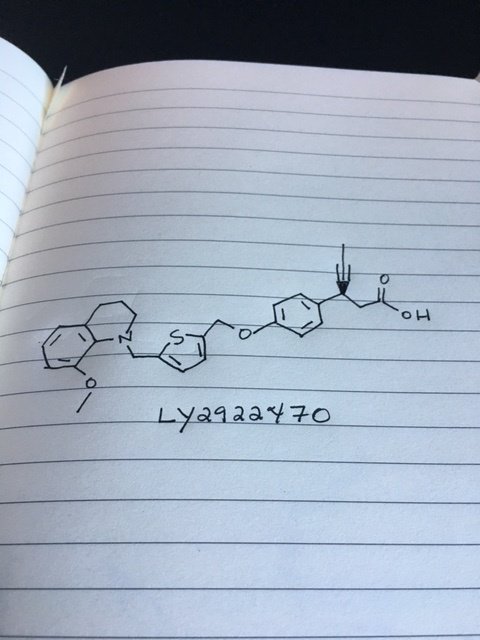
LY 2922470

LY 2922470

LY 2922470
Picture credit….Bethany Halford
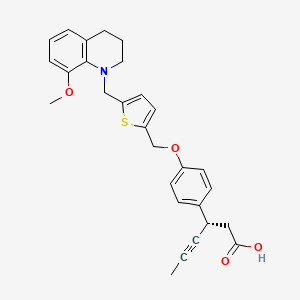
(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophen-2-yl]methoxy]phenyl]hex-4-ynoic acid
Benzenepropanoic acid, 4-[[5-[(3,4-dihydro-8-methoxy-1(2H)-quinolinyl)methyl]-2-thienyl]methoxy]-β-1-propyn-1-yl-, (βS)-
Glucose Lowering Agents, Signal Transduction Modulators
| CAS | 1423018-12-5 |
|---|---|
| Molecular Formula: | C28H29NO4S |
| Molecular Weight: | 475.59916 g/mol |
|---|
https://clinicaltrials.gov/ct2/show/NCT01867216
- Phase I Type 2 diabetes mellitus
Eli Lilly
Antihyperglycaemics
- 28 Jan 2014 Eli Lilly completes a phase I trial in Type-2 diabetes mellitus in USA (NCT01867216)
- 30 Jun 2013 Phase-I clinical trials in Type-2 diabetes mellitus in USA (PO)
- 14 Jun 2013 Eli Lilly plans a phase I trial for Type-2 diabetes mellitus in USA (NCT01867216)
PATENT
WO 2013025424
https://www.google.com/patents/US20130045990?cl=de
| Also published as | CA2843474A1, CA2843474C, CN103687856A, CN103687856B, EP2744806A1, US8431706, WO2013025424A1, Less « |
| Inventors | Chafiq Hamdouchi |
| Original Assignee | Eli Lilly And Company |



Preparation 18-Methoxyquinoline
Add potassium hydroxide (435 g, 7.76 mol) to a solution of 8-hydroxy quinoline (250 g, 1.724 mol) in THF (10 L) at ambient temperature and stir. Add methyl iodide (435 g, 2.58 mol) dropwise and stir overnight. Filter the reaction mixture and wash the solid with THF (2 L). Concentrate the solution to dryness; add water; extract with dichloromethane (2×3 L); combine the organic layers; and wash with brine. Collect the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a red oil, which solidifies on standing, to give the title compound (281 g, 102%), which can be used without further purification. ESI (m/z) 160(M+H).
Preparation 2
8-Methoxy-1,2,3,4-tetrahydroquinoline
Add sodium cyanoborohydride (505 g, 8.11 mol) in EtOH (1 L) to a solution of 8-methoxy quinoline (425 g, 2.673 mol) in EtOH (9 L), and stir. Cool the reaction mixture to an internal temperature of 0° C. and add HCl (35%, 1.12 L, 10.962 mol) dropwise over 60 min so that the internal temperature did not rise above 20° C. Allow the reaction mixture to warm to ambient temperature and then heat to reflux for 2.5 hours. Cool to ambient temperature and stir overnight. Add ammonium hydroxide (25%, 1 L); dilute with water (15 L); and extract the mixture with dichloromethane (3×10 L). Combine the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography, eluting with ethyl acetate: hexane (1:10) to give the title compound (357 g, 82%). ESI (m/z) 164(M+H).
Preparation 3
Methyl-5-methylthiophene-2-carboxylate
Add thionyl chloride (153 ml, 2.1 mol) dropwise over 20 min to a solution of 5-methylthiophene-2-carboxylic acid (100 g, 0.703 mol) in MeOH (1 L) at 0° C. and stir. After the addition is complete, heat the reaction mixture to reflux for 3.5 hours. Cool and concentrate in vacuo to give a thick oil. Dilute the oil with EtOAc (500 ml) and sequentially wash with water (300 ml) then brine (300 ml). Dry the organic layer over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give the title compound (106 g, 97%), which is used without further purification. ESI (m/z) 156(M+H).
Preparation 4
Methyl 5-(bromomethyl)thiophene-2-carboxylate
Add freshly recrystallised NBS (323.8 g, 1.81 mol) to a solution of methyl-5-methylthiophene-2-carboxylate (258 g, 1.65 mol) in chloroform (2.6 L) at room temperature, and stir. Add benzoyl peroxide (3.99 g, 0.016 mol) and heat the reaction mixture to reflux for 7 hours. Cool the reaction mixture to ambient temperature and filter through diatomaceous earth. Wash the filter cake with chloroform (250 ml). Collect the organic layers and remove the solvent to give the title compound (388 g, 100%), which is used without further purification. ESI (m/z) 236(M+H).
Preparation 5
Methyl-5-[8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophene-2-carboxylate
Add methyl-5-(bromoethyl)thiophene-2-carboxylate (432.5 g, 1.84 mol) in EtOH (500 ml) to a solution of 8-methoxy-1,2,3,4-tetrahydroquinoline (300 g 1.84 mol) in EtOH (1 L) and stir. Add DIPEA (641 ml, 3.67 mol) dropwise and stir at room temperature overnight. After completion of the reaction, remove the EtOH in vacuo, and add water (5 L). Extract the aqueous with EtOAc (3×3 L); combine the organic layers; and dry over sodium sulfate. Filter the solution and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography eluting with ethyl acetate: hexane (6:94) to give the title compound (325 g, 56%). ESI (m/z) 318(M+H).
Preparation 6
[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methanol
Add DIBAL-H (1 M in toluene 2.7 L, 2.66 mol) slowly via a cannula over a period of 1.5 h to a stirred solution of methyl-5-(8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-carboxylate (281 g, 0.886 mol) in THF (4 L) at −70° C. Monitor the reaction via thin layer chromatography (TLC) for completion. After completion of the reaction, allow the reaction mixture to warm to 20° C. and add a saturated solution of ammonium chloride. Add a solution of sodium potassium tartrate (1.3 Kg in 5 L of water), and stir overnight. Separate the organic layer; extract the aqueous phase with EtOAc (2×5 L); then combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration. Remove the solvent from the filtrate under reduced pressure to give the title compound as a white solid (252 g, 98%). ESI (m/z) 290(M+H).
Preparation 7
Ethyl(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoate
Add tributylphosphine (50% solution in EtOAc, 543 ml, 1.34 mol) to a solution of ADDP (282.5 g, 1.5 eq) in THF (3 L) and cool the mixture to an internal temperature of 0° C., then stir for 15 minutes. Add (S)-ethyl 3-(4-hydroxyphenyl)hex-4-ynoate (173.5 g, 0.747 mol) in THF (3 L) dropwise over 15 min; then add 5-((8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-yl)methanol (216 g, 0747 mol) in THF (5 L) dropwise. Allow the reaction mixture to warm to ambient temperature and stir overnight. Filter the reaction mixture through diatomaceous earth and wash the filter cake with ethyl acetate (2 L). Concentrate the organic filtrate to dryness. Add water (4 L); extract with ethyl acetate (3×5 L); combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration and concentrate under reduced pressure to give an oil. Purify the residue by silica gel flash chromatography by eluting with ethyl acetate: hexane (6:94) to give the title compound (167 g, 44%). ESI (m/z) 504(M+H).
Example 1
(3S)-3-[4-[[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoic acid

Add a solution of potassium hydroxide (49.76 g, 0.88 mol) in water (372 ml) to a solution of (S)-ethyl-3-(4-((5-8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophen-2-yl)methoxy) phenyl)hex-4-ynoate (149 g, 0.296 mol) in EtOH (1.49 L) at room temperature and stir overnight. Concentrate the reaction mixture to dryness and add water (1.3 L). Extract the resulting solution with EtOAc (2×300 ml) and separate. Adjust the pH of the aqueous layer to pH=6 with 2 N HCl. Collect the resulting solids. Recrystallise the solids from hot MeOH (298 ml, 2 vol) to give the title compound (91 g, 65%). ESI (m/z) 476(M+H).
Abstract
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260

Presenter

Chafiq Hamdouchi
Senior Research Advisor at Eli Lilly and Company
https://www.linkedin.com/in/chafiq-hamdouchi-4988126
Summary
Dr. Hamdouchi earned his bachelor’s degree and doctorate in organic chemistry from Louis Pasteur University, Strasbourg-France.
Following two postdoctoral fellowships, sponsored by the National Science Foundation-USA and Ministerio de Educación y Ciencia-Spain, he joined Eli Lilly and Company in 1995.
Throughout his 20 years of career at Lilly, he has contributed to a sustainable drug discovery portfolio from preclinical hypothesis to clinical proof-of-concept that spans the oncology, neuroscience and endocrinology therapeutic areas. He has led multidisciplinary (chemistry, pharmacology, ADMET, PK, medical) scientific teams in USA, Europe and Asia to deliver a number of compounds that achieved first human dose.
He is a co-inventor of six innovative molecules being pursued in clinical development for the treatment of Diabetes, Cancer and Neurodegenerative Diseases.
He has an extensive patent and publication record and deep experience in conducting drug discovery and development in Asia through effective partnership and mentorship.
SEE AT…………ONE ORGANIC CHEMIST ONE DAY BLOG
LINK……http://oneorganichemistoneday.blogspot.in/2016/03/chafiq-hamdouchi-senior-research.html
| Patent ID | Date | Patent Title |
|---|---|---|
| US8431706 | 2013-04-30 | 1,2,3,4-tetrahydroqinoline derivative useful for the treatment of diabetes |
References
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260
//////Phase 1, LY2922470, LY 2922470, Eli Lilly, Type 2 diabetes mellitus, 1423018-12-5, Chafiq Hamdouchi
CC#CC(CC(=O)O)C1=CC=C(C=C1)OCC2=CC=C(S2)CN3CCCC4=C3C(=CC=C4)OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
SUVN-G3031, from Suven Life Sciences Ltd

 .2HCl
.2HCl
SUVN-G3031
N-[4-(1-cyclobutyl piperidin-4-yloxy)-phenyl]-2-(morpholin-4-yl) acet amide dihydrochloride
N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl]-2-(morpholin-4-yl) acetamide dihydrochloride

SUVN-G3031
Base
Cas 1394808-82-2
SUVN-G3031 (in phase I)
Suven Life Sciences Limited, IN 2011CH00520
- Phase I Cognition disorders associated with Alzheimer disease patients.
https://clinicaltrials.gov/ct2/show/NCT02342041
Useful for treating cognitive disorders, dementia, attention deficit hyperactivity disorder, epilepsy, sleep disorders, obesity, schizophrenia, eating disorders and pain.
Histamine H3 receptor antagonists
Neuropsychotherapeutics; Nootropics
Suven Life Sciences is developing, Histamine H3 receptor antagonists, SUVN-G3031 (in phase I)

- 13 Jul 2015Suven Life Sciences has patent protection for SUVN G3031 in China and South Africa
- 16 Mar 2015SUVN G3031 is available for licensing as of 16 Mar 2015. http://www.suven.com/
- 16 Mar 2015Suven Life Sciences receives patents for SUVN G3031 in USA and New Zealand

H 3 receptors play a critical role as neuromodulators through their widespread distribution in the central nervous system. Blockade of this receptor augments the pre-synaptic release of both histamine and other neurotransmitters including acetylcholine from cholinergic neurons. Currently, several H 3 receptor antagonists/inverse agonists are in different stages of clinical trials for the potential treatment of narcolepsy, cognitive impairments associated with Alzheimer’s disease, Parkinson’s disease, schizophrenia and attention deficit hyperactivity disorder.
Histamine H3 receptor is a G-protein coupled receptor (GPCR) and one out of the four receptors of Histamine family. Histamine H3 receptor is identified in 1983 and its cloning and characterization were done in 1999. Histamine H3 receptor is expressed to a larger extent in central nervous system and lesser extent in the peripheral nervous system.
Literature evidence suggests that Histamine H3 receptor ligands can be used in treatment of cognitive disorders (British Journal of Pharmacology, 2008, 154(6), 1 166-1181), dementia (Drug News Perspective, 2010, 23(2), 99-103), attention deficit hyperactivity disorder, obesity (Indian Journal of Pharmacology, 2001, 33, 17-28), schizophrenia (Biochemical Pharmacology, 2007, 73(8), 1215-1224) and pain (Journal of Pharmacology and Experimental Therapeutics, 2011, 336(1), 30-37).
Patent publications WO 2007/137955, US 2009/0170869, US 2010/0029608, US 2010/0048580, WO 2009/100120, WO 2009/121812 and WO 2009/135842 disclosed series of compounds as ligands at Histamine H3 receptors. While some Histamine H3 receptor ligands have been disclosed, no compound till date is launched in market in this area of research, and there still exists a need and scope to discover new drugs with novel chemical structures for treatment of disorders affected by Histamine H3 receptors.
Suven Life completes Phase 1 studies for SUVN- G3031 for Schizophrenia – Cognitive Impairment
Drugmaker Suven Life Science, which is mostly into researching for new molecules used for ailments of the central nervous system, has completed the single ascending dose (SAD) studies for SUVN- G3031, which is likely to be used for cognitive dysfunction associated with Alzheimer’s and schizophrenia.
The phase-1 study was said to be designed to evaluate safety, tolerability and pharmacokinetics of SUVN-G3031 in healthy volunteers. It was found that the tolerability of SUVN-G3031 up to the highest dose administered in SAD study was ‘excellent’ with ‘no serious adverse events’. The drug candidate was demonstrated for one-day dosing.
OLD CLIPS
SUVN-G3031 for Cognition in Alzheimer’s Disease commenced Phase 1 Clinical Trial in USA under US-IND 123179
HYDERABAD, INDIA (Nov 03, 2014) – Suven Life Sciences today informed that their NCE SUVN-3031 has commenced Phase 1 clinical trial in USA. SUVN-G3031 – A potent, selective, brain penetrant and orally active Histamine H3 antagonist for the treatment of cognitive dysfunction associated with Alzheimer’s Disease / Schizophrenia has completed all the pre-clinical, safety and early toxicological studies, GLP toxicological studies and was submitted forInvestigational New Drug Application {IND) to conduct Phase 1 clinical trial with the indication for Cognition in Alzheimer’s Disease under 505(1) of the Federal Food, Drug and Cosmetic Act (FDCA) which was assigned an IND number 123179.
Based on the IND “A Single Center, Double-blind, Placebo-controlled, Randomized, Phase 1 Study to Evaluate the safety, Tolerability, and Pharmacokinetics of SUVN-G3031 after Single Ascending Doses and Multiple Ascending Doses in Healthy Male Subjects” for Cognition in Alzheimer’s Disease is underway in USA
“We are very pleased that the second compound from our pipeline of molecules in CNS has moved into clinical trial that is being developed for cognitive disorders in Alzheimer’s and Schizophrenia with high unmet medical need which has huge market potential globally” says Venkat Jasti, CEO of Suven.
Suven Life Science is a biopharmaceutical company focused on discovering, developing and commercializing novel pharmaceutical products, which are first in class or best in class CNS therapies through the use of GPCR targets. The Company has eleven (11) internally-discovered therapeutic drug candidates currently in pre-clinical stage of development targeting conditions such as ADHD, dementia, major depressive disorder (MDD), Huntington’s disease, Parkinson’s disease and obesity in addition to this Phase 1 developmental candidate SUVN-G301 and Phase 2 a (PoC) ready SUVN-502 for Alzheimer’s disease and Schizophrenia.
SYNTHESIS

PATENT
WO2012114348
OR SEE
https://www.google.com/patents/US20140135304?cl=en22
PATENT
Scheme I as shown below.

PATENT
process for large scale production of N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl]-2-(morpholin-4-yl) acetamide dihydrochloride of formula (I).
N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl]-2-(moφholin-4-yl) acetamide dihydrochloride, is a promising pharmaceutical agent, which is potent and selective Histamine ¾ receptor ligand intended for the symptomatic treatment of cognitive disorders, dementia, attention deficit hyperactivity disorder, epilepsy, sleep disorders, sleep apnea, obesity, schizophrenia, eating disorders and pain. N-[4-(l-Cyclobutyl piperidin-4-yloxy) phehyl]-2-(morpholin-4-yl) acetamide dihydrochloride and its synthesis is disclosed by Ramakrishna et al. in WO20121 14348.
Currently N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl] -2-(morpholin-4-yl) acetamide dihydrochloride has completed preclinical studies and is ready to enter human clinical trials. The demand for N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl]-2-(morpholin-4-yl) acetamide dihydrochloride as a drug substance has increased substantially with the advent of its clinical testing. The future need for much larger amounts is projected due to the intended commercialization of N-[4-( 1 -Cyclobutyl piperidin-4-yloxy) phenyl]-2-(morpholin-4-yl) acetamide dihydrochloride.
For the person skilled in art, it is a well known fact that various parameters will change during the manufacture of a compound on a large scale when compared to the synthetic procedures followed in laboratory. Therefore, there is a need to establish and optimize large scale manufacturing process. The process for the preparation of N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl]-2-(morpholin-4-yl) acetamide dihydrochloride disclosed in WO20121 14348 was proved to be unsatisfactory for adaptation to the large scale manufacturing. Hence it is highly desirable to establish optimized manufacturing process of N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl] -2 -(morpholin-4-yl) acetamide dihydrochloride of formula (I), which is amenable to the large scale manufacturing of the compound.
Example 1: Preparation of N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl]-2-(raorpholin-4-yl) acetamide dihydrochloride
Step (i): Preparation of l-cycIobutylpiperidin-4-ol
Ethylene dichloride (235 L) was charged into the reactor at 20-25 °C followed by 4-hydroxy piperidine (9.5 Kg, 93.92 M). The mass was stirred for ~ 15 minutes to obtain a clear, solution. Then cyclobutanone (7.9 Kg, 1 12.71 M) was charged into the reactor at 20-25 °C and stirred the mass for 90 minutes at the same temperature. The mass was cooled to 15-20 °C and started lot wise addition of sodium triacetoxy borohydride (39.9 Kg, 188.26 M) maintaining the mass temperature below 25 °C in ~ 110 minutes. After completion of addition, the mass was stirred for 30 minutes at ~ 20 °C. The mass temperature was raised to 25-30 °C and maintained at the same temperature for ~ 13.1 hours, while monitoring the progress of the reaction by Thin Layer Chromatography (TLC). After completion of the reaction, water (1 12 L) was charged into the reactor at 25-30 °C. The mass was then cooled to 15-20 °C and pH of the reaction mass was adjusted to 13.0-13.5 with a solution of aqueous sodium hydroxide (24.6 Kg of sodium hydroxide dissolved in 106 L of demineralised water (DM water) maintaining the mass
temperature below 20 °C in about 1 hour 20 minutes. In the meanwhile, nutsche filter with hyflow bed (using 4.75 Kg hyflow and 47.5 L DM water) was made ready for filtration of dirt and sodium acetate salt, for the purpose of clean layer separations during extraction of the product. The reaction mass was filtered through nutsche and the nutsche was washed with 23.75 L of ethylene dichloride. The filtrate containing the product was collected into clean and dedicated containers. The combined filtrate and washings were transferred to a reactor, stirred 15 minutes and settled for 15 minutes at 25-30 °C. The bottom organic layer (containing the product) was collected in dedicated containers and the mass was dried over anhydrous sodium sulfate (9.5 Kg). The supernatant, clean, dry organic layer was taken in a reactor and solvent was removed by distillation under vacuum maintaining mass temperature below 50 °C. The residual crude mass was cooled to 25-30 °C.
2nd extraction of the aqueous layer: The aqueous layer separated as above was taken in a reactor and charged dichloromethane (DCM) (56 L) at 25-30 °C. The mass was stirred 15 minutes and settled for 15 minutes. The bottom organic layer (containing product) was separated into dedicated containers. The aqueous layer was collected and taken for 3 rd extraction.
3 rd extraction of the aqueous layer: The aqueous layer separated as above was takenin a reactor and charged DCM (56 L) at 25-30 °C. The mass was stirred 15 minutes and settled for 15 minutes. The bottom organic layer (containing product) was separated into dedicated containers. The aqueous layer was collected and taken for 4th extraction.
4th extraction of the aqueous layer: The aqueous layer separated as above was taken in a reactor and charged DCM (56 L) at 25-30 °C. The mass was stirred 15 minutes and settled for 15 minutes. The bottom organic layer (containing product) was separated into dedicated containers. The aqueous layer was collected and taken for 5th extraction.
5th extraction of the aqueous layer: The aqueous layer separated as above was taken in a reactor and charged dichloromethane (56 L) at 25-30 °C. The mass was stirred 15 minutes and settled for 15 minutes. The bottom organic layer
(containing product) was separated into dedicated containers. The aqueous layer was collected in dedicated containers and kept aside.
The organic layer obtained from second extraction to fifth extraction was combined and dried over anhydrous sodium sulfate (13.5 Kg). The supernatant, clean, dry organic layer was taken in the reactor, containing the crude product obtained from first extraction, and solvent was removed by distillation under reduced pressure (>500 mm Hg) maintaining mass temperature below 50 °C. The residual mass was cooled to 25-30 °C and collected the technical product (14.36 Kg).
Yield: 98.49 %;
Ή-NMR (δ ppm, CDC13): 1.55 – 1.69 (5H, m), 1.83 – 2.02 (8H, m), 2.65 – 2.69 (3H, m), 3.66 – 3.70 (1H, m);
Mass (m/z): 156.2 (M+H)+.
Step (ii): Preparation of 4-(l-cyclobutylpiperidin-4-yIoxy)-l-nitrobenzene
Tetrahydrofuran (THF) (43.2 L) was charged into a Stainless steel reactor (SS reactor) at 25-30 °C under nitrogen atmosphere followed by addition of sodium hydride (5.22 Kg) maintaining mass temperature at 25-30 °C under nitrogen atmosphere. The contents were stirred for 15 minutes at 25-30 °C. The temperature of the reaction mass was raised to 35-40 °C.
THF (56.7 L) was charged into another SS reactor at 25-30 °C under nitrogen atmosphere by the addition of above obtained step (i) material (13.5 Kg, 86.96 M). The mass was stirred for 15 minutes at 25-30 °C to obtain a clear solution. The resulting solution was added to the above reactor containing sodium hydride in THF, maintaining the mass temperature of the main reactor at 35-40 °C over a period of ~ 45 minutes under nitrogen atmosphere. The resulting mass was further stirred for 90 minutes at 35-40 °C.
In the meanwhile THF (35.8 L) was charged into another SS reactor at 25-30 °C under nitrogen atmosphere, followed by the addition of 4-fluoro-l-nitrobenzene (14.72 Kg, 104.32 M). The contents of the reactor were stirred for 15 minutes at 25-30 °C to obtain a clear solution. The clear solution, thus obtained, was slowly transferred to the main reactor in ~ 45 minutes maintaining the mass temperature of the main reactor at 35-40 °C. The temperature of the reaction mass was further maintained at 35-40 °C for 5 hours under stirring and under nitrogen atmosphere, while monitoring the progress of the reaction by TLC. After completion of the reaction, the reaction mass was cooled to 15-20 °C.
. Charged water (675 L) into another SS reactor under nitrogen atmosphere. The contents of the reactor were cooled to 5-10 °C. Then the reaction mass from the main reactor was transferred carefully to this reactor containing water, maintaining the mass temperature below 20 °C in ~ 45 minutes. The resulting mass was further stirred for 30 minutes maintaining the temperature at 15-20 °C. The solid mass was centrifuged and the mother liquors were collected in dedicated containers. The cake on the centrifuge was washed with water (2 x 135 L) and spin dried to obtain technical product (19.80 Kg).
Purity: 99.5 %.
Purification: Dissolved the technical product obtained as above (19.80 Kg) in ~ 200 L of 10 % aqueous acetic acid solution (~ 20.59 Kg acetic acid diluted with 180 L with water) at 25-30 °C.
1st toluene extraction: Stirred 15 minutes and then charged toluene (33 L) at 25-30 °C. Stirred 15 minutes and settled for 15 minutes and layers separated, The top organic layer containing the impurities was kept aside in a dedicated container.
2nd toluene extraction: The lower aqueous product layer was taken into the reactor again and charged toluene (33 L) at 25-30 °C. Stirred 15 minutes and settled for 15 minutes and layers separated. The top organic layer containing the impurities was kept aside in the dedicated container.
3rd toluene extraction: The lower aqueous product layer was taken again into the reactor and charged toluene (25 L) at 25-30 °C. Stirred 15 minutes and settled for 15 minutes and layers separated. The top organic layer containing the impurities was kept aside in the dedicated container.
The aqueous product layer was charged into the reactor at 25-30 °C. The mass was cooled to 10 – 15 °C. pH of the reaction mass was adjusted to 1 1.5 -12.0; with 20 % w/v aqueous sodium hydroxide solution (prepared by dissolving 15.44 Kg sodium hydroxide flakes in 69.3 L of DM water) while maintaining mass temperature at 10-15 °C for 1.45 hours. The resulting mass was stirred for 15 minutes at 25-30 °C at pH 11.55. The solids that separated were centrifuged. The cake was washed with (40 L x 2) DM water and the product was spin dried (19.9 Kg), Yield: 53.56 %
Purity: 99.52 %.
Ή-NMR (δ ppm, CDC13): 1.58 – 1.73 (2H, m), 1.84 – 1.93 (4H, m), 2.02 – 2.06 (4H, m), 2.19 (2H, s), 2.62 (2H, s), 2.71 – 2.76 (1H, m), 4.45 (1H, s), 6.93 – 6.95 (2H, d, J = 9.07 Hz), 8.18 – 8.20 (2H, d, J = 9.02 Hz);
Mass (m/z): 277.2 (M+H)+.
The aqueous layer (obtained after eentrifuging and washing the product) was collected in dedicated containers for isolation of the second crop.
Step (iii): Preparation of 4-(l-cyclobutylpiperidin-4-yloxy) aniline
The reaction was done in a SS reactor under nitrogen blanket. DM Water
(33.59 L) was charged into a SS reactor at 25-30 °C followed by iron powder (10.43 Kg, 186.75 M, 1 :4 ratio) under stirring. Then ammonium chloride (11.5 Kg, 215 M) was charged at 25-30 °C and stirred the contents for 15 minutes at 25-30 °C. The mass temperature was raised slowly to 95- 100 °C and maintained at that temperature (95-100 °C).for.^.90 minutes. The mass was cooled to 75-80 °C.
In the meanwhile, ethyl alcohol (128.7 L) was charged into another reactor at 25-30 °C, followed by addition above obtained compound (19.9 Kg). The contents were stirred for 15 minutes and then raised the mass temperature to 50-55 °C, where by a clear solution was obtained. The mass was slowly transferred to the main reactor, containing the activated iron powder at 78-80 °C over a period of ~ 70 minutes. The mass was further stirred for 3 hours, while maintaining the mass temperature at 75-80 °C. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mass was cooled to 25-30 °C and filtered through nutsche, containing hyflow bed. The filtrate was collected into dedicated containers. The bed was washed with 3 x 32.18 L of ethyl alcohol and collected the washings into dedicated containers. The combined filtrate was charged into a clean SS reactor at 25-30 °C. All the volatiles are distilled off under reduced pressure (> 500 mm Hg) maintaining the mass temperature below 55 °C. The residual mass was cooled to 25-30 °C and charged DM water (32.18 L). The pH of the reaction mass was adjusted to 9.0 – 10.0 with 91 L of sodium carbonate solution (prepared by dissolving 21.5 Kg of sodium carbonate in 80 L of DM water), while maintaining the mass temperature at 25-30 °C. Final pH is 9.14. The solid mass, separated in the reactor, was cehtrifuged and collected the filtrate in dedicated containers. The product was spin dried (20.34 Kg).
Ethylacetate (EtOAc) (80 L) was charged into a clean SS reactor at 25-30 °C followed by the wet cake (20.34 Kg) obtained above. The mass was stirred for 15 minutes at 25-30 °C. Then added DM water (32 L) and further stirred the mass for 15 minutes and settled for 15 minutes. The aqueous layer was separated and collected in dedicated containers.
The organic layer containing the product was filtered through nutsche filter through hyflow bed (formed with 5.15 Kg hyflow and 26 L water) and filtrate was collected in dedicated containers. The bed was washed with EtOAc (13 L). The combined organic layer and EtOAc washings were charged into a clean SS reactor. Charged 20 L DM water, stirred for 15 minutes and settled for 15 minutes at 25-30 °C. The aqueous layer is separated and the organic layer was dried over anhydrous sodium sulfate (20 Kg).
The clean, dried organic layer was charged into a reactor at 25-30 °C. Solvent was distilled off under reduced pressure (> 500 mm Hg) below 50 °C (Solvent recovered: 70 L). The residual product was cooled to 25-30 °C and unloaded into dedicated containers (12.30 Kg) and sent for complete analysis. Weight of the product: 12.3 Kg (wet with solvent EtOAc: 9.1 %),
Yield (on dry basis): 9.7.5 %;
Purity: 97.79 %;
IR (cm-‘): 3424, 3345, 2943, 1627, 1509, 1229, 1 168, 1044, 821 ;
1H-NMR (5 ppm, DMSO): 1.49 – 1.61 (4H, m), 1.71 – 1.83 (4H, m), 1.92 – 1.97 (5H, m), 2.52 – 2.53 (2H, m), 3.99 – 4.04 (1 H, m), 4.59 (2H, bs), 6.46 – 6.48 (2H, d, J = 8.60 Hz), 6.61 – 6.63 (2H, d, J = 8.66 Hz);
Mass (m/z): 247.4 (M+H)+.
Step (iv): Preparation of 2-chloro-N-[4-(l-cycIobutyI piperidin-4-yloxy).
phenyl] acetamide
The reaction was done in a SS reactor under nitrogen blanket. THF (89.6
L) was charged into a Glass reactor (GLR) at 25-30 °C followed by addition of above obtained material (1 1.2 Kg on dry basis, 45.46 M). The contents were stirred 15 minutes. Then charged anhydrous potassium carbonate (K2C03) powder (12.54 Kg, 90.73 M) into the reactor and stirred the mass for 15 minutes at 25-30 °C. The reaction mass was cooled to -10 to -5 °C by circulating brine in the jacket. Then a solution of chloroacetylchloride (6.72 Kg, 59.5 M) dissolved in THF (44.8 L) was slowly introduced into the reactor through a holding tank, under nitrogen atmosphere, in ~ 2.5 hours maintaining the mass temperature at -10 to -5 °C. The reaction mass was further maintained under stirring at -10 to -5 °C for another 2 hours while monitoring the progress of the reaction by TLC.
After completion of the reaction, slow addition of chilled DM water (186 L) through the addition funnel started at -10 to -5 °C. Towards the end of addition of DM water (addition time 45 minutes), it was so adjusted that the mass temperature reached 10-15 °C. After completion of addition of DM water the mass temperature was raised to 25-30 °C.
1st extraction: Ethyl acetate (1 12 L) charged into the reactor at 25-30 °C. The mass was stirred 30 minutes and settled for 30 minutes. Layers separated and the organic product layer was collected in dedicated containers.
2nd extraction: The aqueous layer obtained as above was charged into the reactor followed by EtOAc (1 12 L) at 25-30 °C. The mass was stirred 30 minutes and settled for 30 minutes. Layers separated and the organic product layer and the aqueous layer were collected in dedicated containers.
The combined organic layer, obtained from the above extractions, was charged into a clean GLR followed by the addition of 116 L of brine solution (prepared by dissolving 33.6 Kg sodium chloride in 1 12 L DM water) at 25-30 °C. The mass was stirred for 30 minutes and settled for 30 minutes at 25-30 °C. The aqueous layer was separated and collected in dedicated containers. The organic product layer was dried over anhydrous sodium sulfate (22.4 Kg). The volume of the organic layer was 360 L. The organic layer obtained as above was charged into a clean GLR at 25-30 °C. Solvent was distilled off under reduced pressure (> 500 mm Hg) maintaining mass temperature below 55 °C (volume of recovered solvent; 178 L). The mass was cooled to 25-30 °C. Solid mass separated in the reactor.
Recrystallization
Isopropanol (72.8 L) was charged into the reactor containing the solids (~ 13.5 Kg) at 25-30 °C, followed by methanol (~ 58.2 L) at 25-30 °C. Stirred the reaction mass at 25-30 °C for 30 minutes. The mass temperature was raised slowly to reflux temperature and maintained at reflux till a clear solution is obtained (~ 30 minutes). Then the mass was cooled to 25-30 °C and stirred the mass for 60 minutes. The mass was further cooled to -12 -15 °C, stirred for 30 minutes and centrifuged the material. The cake on the centrifuge was washed with 2 x 7 L isopropanol (25-30 °C) and spin dried thoroughly.
The wet cake (1 1.2 Kg) was dried in a vacuum tray drier (VTD) for ~ 4 hours at 40-50 °C to obtain crystallized product (9.7 Kg).
Yield: 66.12 %;
Purity (by HPLC): 99.56 %; – IR (cm-1): 3307, 3278, 2951, 1670.43, 1612, 1554.69, 1508.4/1240.28, 1 171.81 , 1047.39, 953.84, 832.32;
1H-NMR (δ ppm, DMSO): 1.53 – 1.61 (4H, m), 1.72 – 1.74 (2H, m), 1.87 – 1.99 (6H, m), 2.49 – 2.53 (2H, m), 2.64 – 2.68 (1H, m), 4.19 (2H, s), 4.24 – 4.29 (1H, m), 6.88 – 6.90 (2H, d, J = 8.96 Hz), 7.44 – 7.46 (2H, d, J = 8.96 Hz), 10.12 (1H, s); …. . . .. ■÷. “
Mass (m/z): 323.3, 325.2 (M+H)+.
Mother liquor obtained, after recrystallization and centrifuging the product, was processed for isolating second crop.
Step (v): Preparation of N-[4-(l-cycIoburyl piperidin-4-yIoxy) phenyI]-2-(morphoIin-4-yl) acetamide
Acetonitrile (1.41 L) was charged into the GLR at 25-30 °C under nitrogen atmosphere, followed by addition of the above obtained material (9.4 Kg, 29.11 M). Then, charged anhydrous K2C03 granules (6.0 Kg, 43.41 M) into the reactor at 25-30 °C. Stirred the reaction mass in the reactor for 10 minutes and charged morpholine (3.3 Kg, 37.88 M). The contents of the reactor were stirred for 15 minutes at 25-30 °C. The temperature of the reaction mass was raised slowly to reflux (80-82 °C) and maintained at reflux for 4 hours while monitoring the progress of the reaction every two hours by HPLC.
Analysis of the sample by HPLC after 4 hours reflux: 89.61 % product and 8.83 % starting material (SM).
Charged morpholine (253 grams) and K2C03 (400 grams) and further refiuxed. Analysis by of the sample at 7.5 hours: 92.8 % product and 5.63 % SM. So charged morpholine (506 grams), K2C03 (810 grams) and acetonitrile (30 L) and heated the mass at reflux for another five hours. Analysis of the sample at 12.5 hours: 96.78 % product and 2.06 % SM. Again charged K2C03 (820 grams), morpholine (255 gm) and acetonitrile (40 L) and maintained the mass under reflux. Analysis of the sample at 19.5 hours: 97.52 % product and 0.9 % SM. The reaction mass was cooled to 30-35 °C and filtered solids through nutsche at 30-35 °C. The cake on the nutsche was washed with 15 L acetonitrile; Mother liquors (~ 210 L filtrate) were taken back into the main reactor (GLR) and kept under stirring at 30 – 35 °C, while workup of the solid cake (22.4 Kg), containing the product along with salts, was going on in another reactor.
Wet weight of cake: 22.4 Kg (contained ~ 23 % product).
Charged 30 L water into another reactor followed by the wet cake obtained after nutsche filtration (22.4 Kg). Stirred the mass for 30 minutes and charged EtOAc (47 L). The mass was stirred 15 minutes and settled for 15 minutes. The organic layer containing the product was collected in dedicated containers. pH of the aqueous mother liquors was found to be 10.05 on pH meter.
2nd extraction: Charged the above obtained aqueous layer into the reactor followed by EtOAc (47 L). The mass was stirred 15 minutes and settled for 15 minutes and layers separated. The organic layer containing the product was collected in dedicated containers.
3nd extraction: Charged the above obtained aqueous layer into the reactor followed by EtOAc (40 L). The mass was stirred 15 minutes and settled for 15 minutes and layers separated. The organic layer containing the product was collected in dedicated containers.
The combined organic layer was dried over sodium sulfate (9.4 Kg) and the clean organic layer was taken for distillation under reduced pressure (> 500 mm Hg) at 50-55 °C. The mass was cooled to 25-30 °C. Added 23.5 L of acetonitrile and stirred well.
Part of the reaction mass (65 L of acetonitrile solution) from GLR was unloaded and charged into the above reaction mass at 25-30 °C and stirred 30 minutes, whereby a clear solution was obtained. The mass was transferred to the main reactor. Washing was given to this reactor with 20 L fresh acetonitrile at 40-45 °C and again transferred to the main reactor and stirred 15 minutes before sampling.
The final, uniformly mixed reaction mass was sampled from the main GLR and analyzed. HPLC: 99.09 % product and 0.31 % SM. So charged morpholine (510 grams) and K2C03 (825 grams) and the mass was heated to reflux and further maintained the mass at reflux temperature for 2 hours. A sample was analyzed after 2 hours reflux. Starting material was absent (product purity: 99.24 %).
The reflux was further continued for another 2 hours and then cooled the mass temperature to 30-35 °C. Solvent was distilled off under reduced pressure (> 500 mm Hg), maintaining mass temperature below 55 °C.
1st Extraction: Charged DM water (23.5 L) to the residual mass at 25-30 °C. Stirred the mass for 15 minutes and charged ethyl acetate (80 L). A clear solution was obtained. Stirred the mass for 15 minutes and settled the mass for 15 minutes. Layers separated and the product organic layer collected in dedicated containers. 2ndExtraction: The aqueous layer obtained as above (pH was found to be 9.9 on meter) was charged into the reactor followed by ethyl acetate (40 L). Stirred the mass for 15 minutes and settled the mass for 15 minutes. Layers separated and the product organic layer collected in dedicated containers.
3nd Extraction: The aqueous layer obtained as above was once again charged into the reactor followed by ethyl acetate (40 L). Stirred the mass for 15 minutes and settled the mass for 15 minutes. Layers separated and the product organic layer collected in dedicated containers.
Brine washing: The combined organic layer was taken in the reactor and charged
~ 35 L brine solution (prepared by dissolving 9.4 Kg sodium chloride in 28.2 L DM water). The mass was stirred for 15 minutes and settled for 30 minutes.
Layers separated and collected aqueous layer in dedicated containers.
The organic product layer was dried over anhydrous sodium sulfate (18.8
Kg). Total volume of the organic layer was 185 L. The solvent was distilled off under reduced pressure (> 500 mm Hg) maintaining mass temperature below 55 °C. Solid mass (Step-5 material) separated in reactor.
Yield: Quantitative; 5
Purity: 99.51 %;
1H-NMR (CDC13, δ ppm): 1.65 – 2.04 (12H, m), 2.61 – 2.63 (6H, m), 2.69 – 2.77 (1H, m), 3.12 (2H, s), 3.76 – 3.78 (4H, m), 4.26 – 4.27 (1H, m), 6.87 – 6.89 (2H, d, J = 8.82 Hz), 7.43 – 7.45 (2H, d, J – 8.80 Hz), 8.91 (1H, s);
Mass (m/z): 374.4 (M+H)+.
Step (vi): Preparation of N-[4-(l-CyclobutyI piperidin-4 yloxy) phenyl]-2-(morphoIin-4-yl) acetamide dihydrochloride
Charged isopropyl alcohol (75 L) into the reactor containing step (v) product. The reaction mass temperature was raised to 50-55 °C and stirred for 30 minutes to obtain a clear solution. The mass was cooled to 25 °C before starting the addition of isopropanolic hydrochloride (Isopropanolic HC1).
Isopropanolic HC1 (16.2 L, 16.1 % w/v) was diluted with isopropanol (8 L) and charged into a holding tank. Isopropanolic HC1 in the holding tank was transferred slowly into the reactor in 90 minutes, maintaining mass temperature ~ 22 – 28 °C (now and then giving jerks with brine in the reactor jacket). The resulting mass was further stirred under maintenance at 25-30 °C for 6 hours. The mass was centrifuged; the cake on the centrifuge was washed with fresh isopropanol, 16 L (for slurry wash) + 5.5 L (for spray wash) and spin dried to obtain 20.26 Kg of wet product. Purity: 99.37 %. The material was unloaded into trays and dried in a VTD at 50 – 60 °C for 16 hours.
Final weight: 12.62 Kg;
Yield: 97 %;
Ή-NMR (δ ppm, DMSO): 1.65 – 2.0 (4H, m), 2.13 – 2.19 (4H, m), 2.33 – 2.48 (2H, m), 2.8 – 3.42 (6H, m), 3.67 – 3.92 (6H, m), 4.16 (2H, s), 4.49 – 4.70 (2H, m), 6.97 – 7.03 (2H, m), 7.51 – 7.54 (2H, m), 10.54 (1H, bs), 10.73 (1H, bs), 1 1.01 (lH, bs);
Mass (m/z): 374.4 (M+H)+.
Step (vii): Recrystallization of N-[4-(l-CycIobutyl piperidin-4-yloxy) phenyl]-2-(morphoIin-4-yl) acetamide dihydrochloride
The reaction was done in a GLR reactor under nitrogen blanket. Methanol (24.8 L) was charged into a GLR followed by addition of above obtained technical material (6.2 Kg, 13.89 M) at 25-30 °C. The mass was stirred for 30 minutes to obtain a clear solution. Filtered the mass through nutsche and washed the nutsche with methanol (6.2 L). The filtrate and washing were charged into a clean GLR at 25-30 °C.
The contents of the reactor were heated to 62-63 °C, where a gentle reflux of methanol started. Addition of isopropanol (31 L) through the addition tank started at this temperature of ~ 62 °C. Addition of isopropanol was completed in one hour, while maintaining mass temperature at 62-63 °C. The mass was allowed to cool on its own to room temperature by applying air in the jacket. Solids were separated in the reactor at 48 °C in 3 hours. The mass was allowed to cool to ~ 35 °C on its own. The mass was further cooled to ~ 15 – 20 °C in 2 hours (brine jerks given to the reactor jacket) and the temperature was maintained at ~ 15 – 20 °C for 15 minutes.
The mass was centrifuged. The wet cake on the filter was washed with isopropanol (slurry wash) using 9 L isopropanol at 25-30 °C. The mass was spin dried in the centrifuge for 1 hour, unloaded (wet weight: 5.0 Kg) taken to vacuum tray drier and dried at 50-60 °C for 12 hours.
Weight of the product: 4.20 Kg;
Yield: 67.7 %;
HPLC purity (gradient): 99.71 %;
Any other impurity: < 0.1 %;
Salt content (di HC1): 16.16 %;
Melting Range: 247.0 – 249.5 °C;
DSC (2 °C / min, onset): 246.41 °C
TGA (5 °C / min): 0.45 %
Chemical Assay (% w/w): 101.53 %;
IR (cm“1): 3280, 3085, 2935, 2498, 1689, 1604, 1552, 1505, 1235, 1 120 and 830. Ή-NMR (δ ppm, DMSO): 1.62 – 2.0 (4H, m), 2.12 – 2.16 (4H, m), 2.37 – 2.42
(2H, m), 2.78 – 2.91 (2H, m), 3.16 – 3.60 (6H, m), 3.66 – 3.91 (5H, m), 4.17 (2H, s), 4.47 – 4.70 (1 H, m), 6.96 – 7.03 (2H, m), 7.52 – 7.56 (2H, m), 10.69 (1H, bs),
10.86 – 10.89 (1H, bd), 1 1.36 – 1 1.37 (1 H, bd);
Mass (m/z): 374.4 (M+H)+.
13C-NMR (DMSO, δ ppm): 13.48, 13.61, 24.94, 25.10, 25.98, 27.89, 43.85, 47.06,
52.00, 57.08, 58.16, 63.38, 67.29, 71.20, 1 16.33, 1 17.07, 121.36, 132.02, 132.24,
153.03, 153.37, 162.43.
SCHEME 1
Step (i): coupling of 4-hydroxy piperidine of formula (1) with cyclobutanone of formula (2) in presence of sodium triacetoxy borohydride in a suitable solvent to obtain l-cyclobutylpiperidin-4-ol of formula (3). The solvent used in the reaction can be selected from halohydrocarbons, preferably ethylene dichloride. This reaction is carried out at a temperature of 20 °C to 30 °C, preferably 25 °C to 30 °C. The duration of the reaction may range from 12 hours to 14 hours, preferably from a period of 13 hours to 13.5 hours.
Step (ii): coupling of 1 -cyclobutylpiperidin-4-ol of formula (3) with 4-fluoro-l-nitrobenzene of formula (4) in a suitable solvent and base to obtain 4-(l-cyclobutylpiperidin-4-yloxy)-l -nitrobenzene of formula (5). The solvent used in the reaction can be selected from ethers, preferably tetrahydrofuran. The base used in the reaction can be selected from alkali metal hydrides, preferably sodium hydride. This reaction is carried out at temperature of 30 °C to 45 °C, preferably 35 °C to 40 °C. The duration of the reaction may range from 5 hours to 6 hours, preferably from a period of 5.5 hours to 6 hours.
Step (iii): reduction of 4-(l-cyclobutylpiperidin-4-yloxy)-l -nitrobenzene of formula (5) using ammonium chloride and iron powder, in a suitable solvent to obtain 4-(l-cyclobutylpiperidin-4-yloxy) aniline of formula (6). The solvent used in the reaction can be selected from aqueous alcohols, preferably aqueous ethyl alcohol. This reaction is carried out at temperature of 70 °C to 85 °C, preferably 75 °C to 80 °C. The duration of the reaction may range from 3 hours to 5 hours, preferably for a period of 4 hours.
Step (iv): reaction of 4-(l-cyclobutylpiperidin-4-yloxy) aniline of formula (6) with chloroacetylchloride of formula (7) in a suitable solvent and base to obtain 2-chloro-N-[4-(l-cyclobutyl piperidin-4-yloxy)phenyl]acetamide of formula (8). The solvent used in reaction can be selected from ethers, preferably tetrahydrofuran. The base used in reaction can be selected from alkali metal carbonates, preferably potassium carbonate. This reaction is carried out at a temperature of -10 °C to 0 °C, preferably -10 °C to -5 °C. The duration of the reaction may range from 4.5 to 5.5 hours, preferably for a period of 5 hours.
Step (v): reaction of 2-chloro-N-[4-(l -cyclobutyl piperidin-4-yloxy)phenyl]acetamide of formula (8) with morpholine of formula (9) in a suitable solvent and base to obtain N-[4-(l-cyclobutyl piperidin^-yloxy) phenyl]-2-(morpholin-4-yl) acetamide of formula (10). The solvent used in the reaction can be selected from nitrile solvents, preferably acetonitrile. The base used in the reaction can be selected from alkalimetal carbonates, preferably potassium carbonate. This reaction is carried out at temperature of 75 °C to 85 °C, preferably 80 °C to 82 °C. The duration of the reaction may range from 20 hours to 30 hours, preferably for a period of 24 hours to 26 hours.
Step (vi): converting N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl]-2-(morpholin-4-yl) acetamide of formula (10) in presence of isopropanolic hydrochloride and isopropanol to N-[4-(l-cyclobutyl piperidin-4-yloxy) phenyl]-2-(morpholin-4-yl) acetamide dihydrochloride of formula (11). This reaction is carried out at a temperature of 20 °C to 30 °C, preferably 25 °C to 30 °C. The duration of the reaction may range from 7 hours to 8.5 hours, preferably from a period of 7.5 hours to 8 hours.
Step (vii): recrystallization of N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl]-2-(morpholin-4-yl) acetamide dihydrochloride of formula (11) in presence of isopropanol and methanol to obtain N-[4-(l-Cyclobutyl piperidin-4-yloxy) phenyl] -2-(morpholin-4-yl) acetamide dihydrochloride of formula (I). This reaction is carried out at a temperature of 58 °C to 63 °C, preferably 62 °C to 63 °C. The duration of the reaction may range from 4 hours to 5 hours, preferably for a period of 4.5 hours.
![]()
SUVEN Life Sciences Ltd
REFERENCES
http://www.alzheimersanddementia.com/article/S1552-5260(14)01286-2/abstract
http://suven.com/news_Apr2015_13.htm
///////SUVN-G3031, HISTAMINE H3 RECEPTOR ANTAGONIST, TREATMENT OF COGNITIVE DEFICITS, SUVN G3031, PHASE 1, SUVEN
O=C(CN1CCOCC1)Nc4ccc(OC2CCN(CC2)C3CCC3)cc4
SUVN-D4010 from Suven Life Sciences Ltd
1H-Indazole, 3-[5-[1-(3-methoxypropyl)-4-piperidinyl]-1,3,4-oxadiazol-2-yl]-1-(1-methylethyl)-
CAS BASE 1428862-32-1, C21 H29 N5 O2, 383.49

SUVN-D4010

C21 H29 N5 O2 . C2 H2 O4
1H-Indazole, 3-[5-[1-(3-methoxypropyl)-4-piperidinyl]-1,3,4-oxadiazol-2-yl]-1-(1-methylethyl)-, ethanedioate (1:1)
1-isopropyl-3-{5-[1-(3-methoxypropyl)-piperidin-4-yl]-[1,3,4]oxadiazol-2-yl}-1H-indazole oxalate
l-isopropyl-3-{5-[l-(3-methoxy propyl) piperidin-4-yl]- [l,3>4]oxadiazol-2-yl}-lH-indazole oxalate salt
SUVN-1004028; SUVN-D-1208045; SUVN-D1003019; SUVN-D1104010; SUVN-D1108121;
l-ISOPROPYL-3-{5-[l-(3-METHOXYPROPYL) PIPERIDIN-4-YL]-[l,3,4]OXADIAZOL-2-YL}-1H-INDAZOLE OXALATE
OXALATE CAS 1428862-33-2
IN 2011CH03203, WO2013042135, WO 2015092804,
In phase I, for treating cognitive dysfunction associated with Alzheimer’s disease, schizophrenia and neurological diseases.
Suven Life Sciences Limited, Phase I Alzheimer’s disease; Schizophrenia
https://www.clinicaltrials.gov/ct2/show/NCT02575482
- Class Antidementias
- Mechanism of Action Serotonin 4 receptor agonists
Used as 5-HT4 receptor agonist for treating Alzheimer’s disease, cognitive disorders, Attention deficit hyperactivity disorder, Parkinson’s and schizophrenia
- 05 Jan 2016Suven Life Sciences has patent protection for chemical entities targeting serotonin receptors for the treatment of neurodegenerative disorders in Canada, Africa and South Korea
- 11 Dec 2015Suven Life Sciences receives patent allowance for chemical entities targeting serotonin receptors in Eurasia, Europe, Israel and Macau
- 02 Nov 2015SUVN D4010 is available for licensing as of 02 Nov 2015. http://www.suven.com
SUVN-D4010 for Cognition in Alzheimer’s disease commenced Phase 1 Clinical Trial in USA under US-IND 126099
HYDERABAD, INDIA (Sept 02, 2015) – Suven Life Sciences today informed that their NCE SUVN-D4010 has commenced Phase 1 clinical trial in USA. SUVN-D4010 is a potent, selective, brain penetrant and orally active 5-HT4 receptor partial agonist for the treatment of cognitive dysfunction associated with Alzheimer’s disease and other dementias. Suven submitted Investigational New Drug Application (IND) to US FDA to conduct Phase-1 clinical trial for Cognition in Alzheimer’s Disease, under 505(1) of the Federal Food, Drug and Cosmetic Act (FDCA) which was assigned an IND number 126099.
Based on the IND# 126099, “A Single Center, Double-blind, Placebo-controlled, Randomized, Phase 1 Study to Evaluate the safety, Tolerability, and Pharmacokinetics of SUVN-D4010 after Single Ascending Doses and Multiple Ascending Doses in Healthy Male Subjects” for Cognition in Alzheimer’s Disease is underway in USA
“We are very pleased that the third compound from our pipeline of molecules in CNS has moved into clinical trial that is being developed for cognitive disorders in Alzheimer’s and Schizophrenia, a high unmet medical need which has huge market potential globally” says Venkat Jasti, CEO of Suven.
Suven Life Science is a biopharmaceutical company focused on discovering, developing and commercializing novel pharmaceutical products, which are first in class or best in class CNS therapies through the use of GPCR targets.Suven has 3 clinical stage compounds, a Phase 2 initiated candidate SUVN-502, Phase 1 completed candidate SUVN-G3031 and Phase 1 initiated candidate SUVN-D4010 for Alzheimer’s disease and Schizophrenia. In addition to that the Company has ten (10) internally-discovered therapeutic drug candidates currently in pre-clinical stage of development targeting conditions such as ADHD, dementia, depression, Huntington’s disease, Parkinson’s disease and pain
![]() SUVEN Life Sciences Ltd
SUVEN Life Sciences Ltd
Alzheimer’s disease (AD) is a neurodegenerative disorder of advanced age characterized by loss of memory, accumulation of amyloid beta protein (Αβ) deposits and decreased levels of the neurotransmitter acetylcholine. Approximately forty percent of AD patients suffer from significant depression. 5-HT4 receptor partial agonists may be of benefit for both the symptomatic and disease-modifying treatment for AD and may offer improved clinical efficacy and/or tolerability relative to acetylcholine esterase inhibitors. 5-HT4 receptor agonists also have antidepressant like properties (Expert Review of Neurotherapeutics, 2007, 7, 1357-1374; Experimental Neurology, 2007, 203(1), 274- 278; Neuroscience & Medicine, 201 1 , 2, 87 – 92; Schizophrenia Bulletin, 2007, 33 (5), 1 100 – 1 1 19).
1 -Isopropyl-3 – { 5 – [ 1 -(3 -methoxypropyl) piperidin-4-yl] – [ 1 ,3 ,4]oxadiazol-2-y 1 } -1 H-indazole oxalate of formula (I) is a promising pharmaceutical agent, which is a potent, selective and orally bioavailable 5-HT4 receptor partial agonist intended for both disease modifying and symptomatic treatment of Alzheimer’s disease and other disorders of memory and cognition like Attention deficient hyperactivity,
Parkinson’s and Schizophrenia. . In addition to the pro-cognitive effects, the compound also demonstrated dose dependent antidepressant like effects in the mouse forced swim test. l-Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yl]-[l,3,4]oxadiazol-2-yl}-lH-indazole oxalate and its synthesis is disclosed by Ramakrishna et al. in WO2013042135.
At present, l-Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yl]-[l,3,4] oxadiazol-2-yl}-l H-indazole oxalate of formula (I) has completed preclinical studies and is ready to enter human clinical trials. The demand for l-Isopropyl-3-{ 5- [ 1 -(3 -methoxypropyl) piperidin-4-yl]- [ 1 ,3 ,4]oxadiazol-2-yl } – 1 H-indazole oxalate of formula (I) as a drug substance would be increased substantially with the advent of its human clinical trials. The future need for much larger amounts is projected due to the intended commercialization of l-Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yl]-[l ,3,4]oxadiazol-2-yl}-lH-indazole oxalate of formula (I).
For the person skilled in art, it is a well known fact that various parameters will change during the manufacturing of a compound on a large scale when compared to the synthetic procedures followed in laboratory. Therefore, there is a need to establish and optimize large scale manufacturing process. The process for the preparation of l -Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yl]-[l ,3,4] oxadiazol-2-yl}-l H-indazole oxalate of formula (I) which was disclosed in WO2013042135 had been proved to be unsatisfactory for the large scale synthesis. Eventually, it is highly desirable to establish optimized manufacturing process for l-Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yl]-[l ,3,4] oxadiazol-2-yl}-l H-indazole oxalate of formula (I) which is amenable to the large scale preparation.
PATENT
WO2013042135
http://www.google.com/patents/WO2013042135A1?cl=en
Example 3: Preparation of l-isopropyl-3-{5-[l-(3-methoxy propyl) piperidin-4-yl]- [l,3>4]oxadiazol-2-yl}-lH-indazole oxalate salt
Step (i): Preparation of l-isopropyI-3-{5-[l-(3-methoxy propyl) piperidin-4-yI]- [l,3,4]oxadiazol-2-yl}-lH-indazo!e
To the mixture of l-isopropyl-lH-indazole-3-carboxylic acid hydrazide (15.0 grams, 68.8 mmol) and l-(3-Methoxy propyl)-piperidine-4-carboxylic acid hydrochloride (20.9 grams, 88.2 mmol, obtained in preparation 7) cooled at 0 °C was added phosphoryl chloride (130 mL). The reaction temperature was gradually raised to 100 °C and stirred was 2 hours. Upon completion of the reaction, it was cooled to 0 °C and triturated with hexanes (3 x 250 mL). The crude product was basified with aqueous sodium hydroxide solution and extracted with 5% methanol in dichloromethane. The combined organic layer was dried over anhydrous sodium sulphate and the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography to obtain l-isopropyl-3-{5-[l-(3-methoxy propyl) piperidin-4-yl]- [l,3,4]oxadiazol-2-yl}-lH-indazole (15.78 grams)
Yield: 59 %.
Ή – NMR (CDCb): δ 8.35 (d, J = 8.1 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.47 (t, J *= 7.0 Hz, 1H), 7.33 (t, J = 7.4 Hz, 1H), 5.05-4.90 (m, 1H), 3.44 (t, J = 6.4 Hz, 2H), 3.35 (s, 3H), 3.15-2.97 (m, 3H), 2.48 (t, J = 7.3 Hz, 2H), 2.26-2.02 (m, 6H), 1.88-1.75 (m, 2H), 1.67 (d, J = 6.7 Hz, 6H);
Mass (m/z): 384.5 (M+H)+.
Step (ii): Preparation of l-Isopropyl-3-{5-[l-(3-methoxy-propyl)-piperidin-4-yl]- [l,3,4]oxadiazoI-2-yl}-lH-indazole oxalate salt
To a stirred solution of l-isopropyl-3-{5-[l-(3-methoxy propyl) piperidin-4-yl]- [l,3,4]oxadiazol-2-yl}-lH-indazole (12.55 grams, 32.7 mmol, obtained in the above step) in 2-propanol (200 mL), oxalic acid (4.12 grams, 32.7 mmol) was added. After stirring at room temperature for 1 hour the reaction was further diluted with 2-propanol and refluxed for 2 hours. The crystalline product which was precipitated after cooling the reaction mixture to room temperature was filtered, dried under vacuum to obtain 1- isopropyl-3-{5-[l-(3-methoxy propyl) piperidin-4-yl]-[l,3,4]oxadiazol-2-yl}-lH- indazole oxalate salt (16.4 grams)
Yield: 88 %
Ή – NMR (DMSO-d6): δ 8.18 (d, J = 8.1 Hz, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.54 (t, J = 7.4 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 5.23 – 5.10 (m, 1H), 3.50 – 3.40 (m, 3H), 3.37 (t, J = 5.9 Hz, 2H), 3.23 (s, 3H), 3.10 -2.96 (m, 4H), 2.35 – 2.25 (m, 2H), 2.18-2.02 (m, 2H), 1.94 – 1.85 (m, 2H), 1.53 (d, J = 6.6 Hz, 6H);
Mass (m/z): 384.3 (M+H)+.
Patent
The large scale manufacturing process for preparation of l-Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yl]-[l ,3,4]oxadiazol-2-yl}-lH-indazole oxalate of

Scheme-1
Preparation 1: Preparation of l-Isopropyl-lH-indazoIe-3-carboxylic acid

To a stirred solution of dimethylformamide (DMF) (50 L) at 25 °C to 30 °C under nitrogen atmosphere, sodium tert-butoxide (6.0 Kg, 62.43 mols) was added over a period of 15 minutes. The reaction mixture was stirred for 10 minutes after which it was cooled to 0 °C to 5 °C. A solution of indazole-3-carboxylic acid (4.0 Kg, 24.67 mols) in DMF (50 L) was added slowly into the reactor over a period of 45 minutes, maintaining the reaction mass temperature at 0 °C to 5 °C. The cooling was removed and the reaction temperature was gradually raised to 25 °C to 30 °C over a period of 30 minutes. After stirring at this temperature for 1 hour the reaction mixture was cooled to 0 °C and isopropyl iodide (6.32 Kg, 37.18 mo!s) was added over a period of 30 minutes. The cooling was removed and the reaction temperature was allowed to rise to 25 °C to 30 °C. After 17 hours of stirring, the HPLC analysis of the reaction mixture revealed <10 % of indazole-7-carboxylic acid remaining. The reaction mass was diluted cautiously with water (200 L) and washed with ethylacetate (2 x 100 L). The resultant aqueous layer was acidified to 4.0 – 4.5 pH with aqueous hydrochloride solution (6.0 N, 21.5 L) and extracted with ethylacetate (2 x 144 L). The combined organic layer was washed with water (2 x 100 L), brine solution (200 L) and dried over anhydrous sodium sulfate (4.0 Kg). The filtered organic layer was subjected to solvent removal under reduced pressure (> 500 mm of Mercury) at 50 °C to 60 °C to obtain a crude mass. The obtained crude mass was diluted with dichloromethane (DCM) (28.0 L) and was stirred for 15 minutes. The solids precipitated (un-reacted indazole-7-carboxylic acid) were filtered through nutsche filter and the filter bed was washed once with DCM (8.0 L). The combined filtrate was distilled under reduced pressure (> 500 mm of Mercury) at 45 °C to 55 °C to obtain a crude mass which was stirred with ether (7.0 L) for 30 minutes and filtered through nutsche filter to obtain the wet solid which was dried further in vacuum oven under reduced pressure (> 500 mm of Mercury) at 45 °C to 55 °C to obtain above titled compound (3.0 Kg) as an off-white crystalline powder.
Yield: 59.5 %;
Purity: 99.86 %;
IR (cm-‘): 2980, 1729, 1682, 1487, 1287, 1203, 1 170, 1 127, 1085, 754;
Ή-NMR (δ ppm, CDC13): 8.27 (d, J= 8.1 Hz, 1H), 7.55 (d, J= 8.4 Hz, 1H), .7.46 (t, J = 7.6 Hz, 1H), 7.34 (t, J = 7.4 Hz, 1H), 5.01 – 4.95 (m, 1H), 1 .68 (d, J = 6.65 Hz, 6H);
Mass (m/z): 205.1 (M+H)+.
Preparation 2: Preparation of l-(3-Methoxypropyl) piperidine-4-carboxyIic acid hydrazide

Step (i): Preparation of Ethyl 1 -(3-methoxj propyl) piperidine-4-carboxylate

To a stirred solution of acetonitrile (97.5 L) under nitrogen atmosphere at 25 °C to 30 °C, ethyl isonipecotate (6.5 Kg, 41.35 mols) was added. The contents were stirred for 10 minutes after which potassium carbonate powder (7.35 Kg, 53.2 mols) and l-Bromo-3-methoxy propane (6.89 Kg, 45.0 mols) were sequentially added. The reaction mixture was gradually heated to reflux (82 °C – 85 °C) over a period of 30 minutes and was maintained at this temperature for 7 hours. At this time, the TLC revealed complete consumption of ethylisonipecotate. The volatiles were distilled off under reduced pressure (> 500 mm of Mercury) at 50 °C to 60 °C. The crude mass was cooled to 25 °C to 30 °C and was diluted with water (71.5 L) and DCM (136.5 L). After stirring the contents the two layers were separated. The organic layer was washed with water (71.5 L), dried over anhydrous sodium sulfate (6.5 Kg) and the volatiles were removed under reduced pressure (> 500 mm of Mercury) at 50 °C to 55 °C to obtain the desired product (9.3 Kg) as pale yellow colored liquid.
Yield: 98 %;
Purity: 98.8 %;
IR (cm“‘): 2949, 1732, 1449, 1376, 1 179, 11 19, 1048;
Ή-NMR (6 ppm, CDC13): 4.06 (q, J = 7.1 Hz, 2H), 3.37 – 3.34 (t, J – 6.4 Hz, 2H), 3.27 (s, 3H), 2.83 – 2.80 (m, 2H), 2.34 (t, J = 7.5 Hz, 2H), 2.22 – 2.18 (m, 1H), 1.96 – 1.94 (m, 2H), 1.85 – 1.82 (m, 2H), 1.74 -1.68 (m, 4H), 1.19 (t, J= 7.04 Hz, 3H);
Mass (m/z): 230.4 (M+H)+.
Step (ii): Preparation of l-(3-Methoxypropyl) piperidine-4-carboxylic acid hydrazide

To a stirred solution of methanol (38 L) under nitrogen atmosphere at 25 °C to 30 °C, ethyl l-(3-methoxypropyl) piperidine-4-carboxylate (5.0 Kg, 21.8 mols, obtained in above step) was added. After stirring the reaction mixture for 15 minutes, hydrazine hydrate (80 % w/v, 4.1 Kg, 65.4 mols) was added over a period of 15 minutes. The reaction mixture was gradually heated to reflux (70 °C) over 30 minutes and continued stirring for 4 hours. Additional amount of hydrazine hydrate (80 % w/v, 4.1 Kg, 65.4 mols) was added and the stirring continued for another 4 hours. Another installment of hydrazine hydrate (80 % w/v, 4.1 Kg, 65.4 mols) was added and the stirring was continued for 16 hours at 70 °C, upon which the Thin Layer Chromatography (TLC) reveals < 5 % of ester. The volatiles were distilled off under reduced pressure (> 500 mm of Mercury) at 60 °C until syrupy mass appeared. After cooling syrypy mass to room temperature (25 °C – 30 °C), it was diluted with DCM (38.0 L) and was stirred for 15 minutes. The observed two layers were then separated. The organic layer was dried over anhydrous sodium sulfate (5.0 Kg) and the solvent was evaporated under reduced pressure (> 500 mm of Mercury) at 55 °C until dryness. The solid product which was separated was cooled to 25 °C to 30 °C, diluted with hexanes (15.0 L) and the resultant slurry was filtered at nutsche filter. The filter bed was washed once with hexanes (15.0 L) and ethylacetate (2 x 10.0 L). The product cake was vacuum dried and the solid material thus separated was further dried in vacuum oven under reduced pressure (> 500 mm of Mercury) at 50 °C for 6 hours to obtain the above titled compound (4.1 Kg) as an off-white crystalline powder.
Yield: 87 %;
Purity: 99.79 %;
IR (cm-‘): 3290, 3212, 2948, 2930, 1637, 1530, 1378, 1 124, 1 1 13, 986, 948, 789, 693;
Ή-NMR (δ ppm, CDC13): 6.83 (s, 1H), 3.86 (bs, 2H), 3.41 (t, J = 6.4 Hz, 2H), 3.32 (s, 3H), 2.99 – 2.96 (m, 2H), 2.42 (t, J= 7.44 Hz, 2H), 2.1 1 – 1.96 (m, 3H), 1.82 – 1.73 (m, 6H);
Mass (m/z): 216.3 (M+H)+.
Example 1: Preparation of l-Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yI]-[l,3,4]oxadiazol-2-yl}-lH-indazole oxalate
Step (i): Preparation of N-[l-(3-Methoxypropyl) piperidine-4-carbonyI] ‘-(l-isopropyI-lH-indazole-3-carbonyl) hydrazine

To a stirred solution of 1 ,2-dichloroethane (19.8 L) under nitrogen atmosphere at 25 °C to 30 °C, l -isopropyl-lH-indazole-3-carboxylic acid (3.0 Kg, 14.69 moles, obtained in preparation 1 ) was added and the reaction mixture was stirred for 15 minutes for complete dissolution. Thionyl chloride (3.6 Kg, 30.25 mols) was then added to the reaction mixture by maintaining its temperature below 30 °C over a period of 15 minutes. The reaction temperature was then gradually raised to 75 °C over a period of 30 minutes and was stirred for 2 hours at that temperature. The TLC revealed complete conversion of acid to acid chloride. The solvent 1,2-dichloroethane and excess thionyl chloride was removed under reduced pressure (> 500 mm of Mercury) below 60 °C temperature. The obtained residual mass was cooled to 25 °C to 30 °C, and diluted with DCM (15.6 L). The contents were further cooled to 0 °C to 5 °C. A solution of l-(3-Methoxypropyl) piperidine-4-carboxylic acid hydrazide (3.0 Kg, 1 3.94 mols, obtained in the preparation 2) in DCM (18.0 L) was added to the reaction mass over a period of 30 minutes. The reaction temperature was then gradually raised to 25 °C to 30 °C and the reaction mixture was stirred for 2 hours. The progress of the reaction was monitored by TLC which showed absence of hydrazide (< 1.0 %). The reaction mixture was then diluted with water (30.0 L), stirred for 15 minutes and the two layers were separated. The aqueous layer was washed with DCM (1 x 30.0 L), cooled to 0 °C to 5 °C and cautiously basified to pH 7.6 with aqueous sodium bicarbonate solution (10 % w/v, 46.5 L). The basified aqueous layer was then extracted with DCM (2 x 30.0 L). The combined organic layer was dried over anhydrous sodium sulfate (6.0 Kg) and the solvent was removed under reduced pressure (> 500 mm of Mercury) below 55 °C. The residue was then cooled to 25 °C – 30 °C and diluted with solvent hexane (9.0 L). The slurry, thus obtained, was centrifuged at room temperature under nitrogen atmosphere and the wet product cake was washed with hexanes (6.0 L). The wet product was then dried in oven at 55 °C -60 °C until loss on drying was < 1.0 % to obtain the above titled compound (4.4 Kg) as an off white crystalline powder.
Yield: 74.5 %;
Purity: 98.75 %;
IR (cm-1): 3506, 3233, 2943, 1703, 1637, 1523, 1487, 1 195, 1 1 16, 750;
Ή-NMR (δ ppm, CDC13): 9.35 (bs, 1H), 8.70 (bs, 1H), 8.30 (d, J = 8.1 Hz, 1H), 7.48 (d, J = 8.4 Hz, 1H), 7.42 (t, J = 8.2 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 4.90 -4.85 (m, 1H), 3.40 (t, J = 6.4 Hz, 2H), 3.33 (s, 3H), 2.94 – 2.85 (m, 2H), 2.39 -2.31 (m, 3H), 1.92 – 1.88 (m, 4H), 1.76 – 1.65 (m, 4H), 1.59 (d, J = 6.6 Hz, 6H); Mass (m/z): 402.2 (M+H)+.
Step (ii): Preparation of l-Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yl]-[l,3»4]oxadiazol-2-yl}-lH-indazole

To a stirred solution of 1 ,2-dichloroethane (60 L) under nitrogen atmosphere at 25 °C to 30 °C, N-[l-(3-methoxypropyl) piperidine-4-carbonyl] N’-(l -isopropyl-1 H-indazole-3-carbonyl) hydrazine (3.0 Kg, 7.47 mols, obtainted in above step) was added and the contents were stirred for 15 minutes afterwhich, thionyl chloride (1.77 Kg, 15.0 mols) was added over 15 minutes time. The reaction mixture temperature was then gradually raised to 79 °C – 83 °C over a period of 30 minutes at which the reaction mixture starts refluxing. Upon completion of 9 hours, the reaction mass showed complete consumption of starting material when checked by TLC. The excess thionyl chloride and solvent 1,2-dichloroethane were distilled off under reduced pressure (> 500 mm of Mercury) below 60 °C. The reaction mass was cooled to 25 °C – 30 °C, diluted with water (39.0 L) and solvent ether (19.5 L). The resulting mass was stirred for 15 minutes and the two layers were separated. The pH of the aqueous layer was adjusted to 9 – 10 by adding an aqueous solution of sodium hydroxide (2.5N, 3.0 L). The basified aqueous layer was then extracted with DCM (2 x 54.0 L). The combined organic layer was washed with cold (5 °C – 10 °C) aqueous sodium hydroxide solution (0.6 N, 54.0 L), dried over anhydrous sodium sulfate (6.0 Kg) and the solvent was removed under reduced pressure (> 500 mm of Mercury) below 55 °C, which yielded above titled compound (2.6 Kg) as brown colored syrupy mass.
Yield: 90.5 %;
Purity: 99.3 %;
IR (cm“1): 3054, 2946, 2808, 1599, 1563, 1462, 1389, 121 1, 1 120, 1069, 999, 749; Ή-NMR (6 ppm, CDC13): 8.34 (d, J = 8.12 Hz, 1H), 7.53 (d, J – 8.44 Hz, 1H), 7.45 (t, J = 7.58 Hz, 1H), 7.32 (t, J = 7.44 Hz, 1H), 4.98 – 4.93 (m, 1H), 3.44 (t, J = 6.44 Hz, 2H), 3.03 – 3.00 (m, 3H), 3.34 (s, 3H), 2.46 (t, J = 7.54 Hz, 2H), 2.20 -2.02 (m, 6H), 1.80 (t, J= 7.27 Hz, 2H), 1.66 (d, J= 6.72 Hz, 6H);
Mass (m/z): 384.3 (M+H)+.
Step (iii): Purification of l-Isopropyl-3-{5-[l-(3-methoxypropyI) piperidin-4-yl]-[l,3.4]oxadiazoI-2-yl}-lH-indazole
The above obtained crude step (ii) product was dissolved in a stirring aqueous acetic acid solution (10 % w/v, 26.0 L) and washed with ethylacetate (2 x 26.0 L). The resultant aqueous layer pH was adjusted to 9.0 – 10.0 by adding an aqueous sodium hydroxide solution (0.5N, 52.0 L). The basified aqueous layer was extracted with solvent ether (2 x 26.0 L) and the combined organic layer was dried over anhydrous sodium sulfate (3.0 Kg). The volatiles were removed under reduced pressure (> 500 mm of Mercury) below 55 °C to obtain a brown colored syrupy mass (2.19 Kg).
Yield: 84 %;
Purity: 99.72 %;
IR (cm“1): 3054, 2978, 2946, 2808, 2772, 1599, 1563, 1462, 1389, 1 194, 1 177, 1 120, 1069, 999, 749;
Ή-NMR (δ ppm, CDC13): 8.34 (d, J = 8.12 Hz, 1H), 7.53 (d, J = 8.44 Hz, 1H), 7.45 (t, J = 7.58 Hz, 1H), 7.32 (t, J = 7.44 Hz, l H), 4.98 – 4.93 (m, 1H), 3.44 (t, J = 6.44 Hz, 2H), 3.03 – 3.00 (m, 3H), 3.34 (s, 3H), 2.46 (t, J = 7.54 Hz, 2H), 2.20 -2.02 (m, 6H), 1.80 (t, J= 7.27 Hz, 2H), 1.66 (d, J = 6.72 Hz, 6H);
Mass (m/z): 384.4 (M+H)+.
Step (iv): Preparation of l-Isopropyl-3-{5-[l-(3-methoxypropyl) piperidin-4-yI]-[l,3,4]oxadiazol-2-yi}-lH-indazole oxalate
To a stirred solution of isopropanol (60.8 L) under nitrogen atmosphere at 25 °C -30 °C, l-isopropyl-3-{5-[l -(3-methoxypropyl) piperidin-4-yl]-[l,3,4]oxadiazol-2-yl}-lH-indazole (6.08 Kg, 15.86 mols, obtained in step (iii) was added, followed by oxalic acid (1.46 Kg, 16.2 mols) addition. The reaction mixture was stirred for 2 hours and solid product that is precipitated was filtered through nutsche filter under nitrogen atmosphere. The wet product bed was washed with isopropanol (10.0 L) and solvent ether (60.8 L) to obtain a technical grade product.
IR (cm“1): 3437, 2975, 2932, 2890, 1703, 1604, 1564, 1458, 1391, 1281, 1217, 1 192, 1 1 14, 992, 750;
Ή-NMR (δ ppm, DMSO-d6): 10.72, (bs, 2H), 8.16 (d, J = 8.1 Hz, 1H), 7.85 (d, J = 8.5 Hz, 1H), 7.51 (t, J = 7.4 Hz, 1 H), 7.35 (t, J = 7.7 Hz, 1H), 5.20 – 5.07 (m, 1H), 3.55 – 3.43 (m, 3H), 3.36 (t, J = 5.9 Hz, 2H), 3.21 (s, 3H), 3.1 8 – 2.98 (m, 4H), 2.40 – 2.30 (m, 2H), 2.26-2.12 (m, 2H), 1.96 – 1.85 (m, 2H), 1.53 (d, J = 6.6 Hz, 6H);
Mass (m/z): 384.4 (M+H)+.
Step (v): Recrystallization of l-Isopropyl-3-{5-[l-(3-methoxypropyI) piperidin-4-yl]-[l,3,4]oxadiazol-2-yl}-lH-indazole oxalate
The above obtained product was suspended in a mixture of isopropanol (35.26 L) and water (7.3 L) and refluxed (76 °C) for 4 hours until complete dissolution. The homogenous solution thus obtained was gradually cooled to 25 °C – 30 °C and maintained at this temperature under slow stirring for 16 hours. The precipitated oxalate salt was centrifuged under nitrogen atmosphere. The product cake was washed with isopropanol (15.0 L) and ether (60.8 L). The suction dried product was then dried in vacuum oven at 25 °C – 30 °C for 2 hours and at 65 °C for 1 hour to obtain above titled compound (4.24 Kg) as light cream colored crystalline material.
Yield: 60 %;
Purity: 99.92 %;
Salt content (oxalate salt): 20.37 %;
Heavy metals: < 20 ppm;
IR (cm-1): 3437, 2975, 2932, 2890, 1703, 1604, 1564, 1458, 1391, 1281, 1217, 1 192, 1 1 14, 992, 750;
1H-NMR (δ ppm, DMSO-d6): 10.72, (bs, 2H), 8.16 (d, J- 8.1 Hz, 1H), 7.85 (d, J = 8.5 Hz, 1H), 7.51 (t, J = 7.4 Hz, 1H), 7.35 (t, J = 7.7 Hz, 1H), 5.20 – 5.07 (m, 1H), 3.55 – 3.43 (m, 3H), 3.36 (t, J = 5.9 Hz, 2H), 3.21 (s, 3H), 3.18 – 2.98 (m, 4H), 2.40 – 2.30 (m, 2H), 2.26-2.12 (m, 2H), 1.96 – 1.85 (m, 2H), 1.53 (d, J= 6.6 Hz, 6H);
Mass (m/z): 384.4 (M+H)+.
REFERENCES
http://www.sciencedirect.com/science/article/pii/S1552526014012874
http://www.suven.com/news_Sep2015_02.htm
SUVN-D4010: Novel 5-HT4 receptor partial agonist for the treatment of Alzheimer’s disease
45th Annu Meet Soc Neurosci (October 17-21, Chicago) 2015, Abst 54.08
SEE BELOW
Characterization of SUVN-D1104010: A potent, selective and orallyactive 5-HT4 receptor partial agonist
Alzheimer’s Assoc Int Conf (AAIC) (July 14-19, Vancouver) 2012, Abst P2-392
SUVN-D1104010 displayed IC50 values > 45 and > 10 mcM for cytochrome P450 3A4 and 2D6, respectively. In dog, rat and human liver microsome preparations, it showed respective stabilities of 64, 26 and 26%. It displayed rat brain, rat plasma and human plasma protein binding values of 94, 89 and 93%, respectively. For parmacokinetic studies, the agent was administered to male Wistar rats (1 mg/kg i.v.; 3 mg/kg p.o.) and male Beagle dogs (1 mg/kg i.v. and p.o.). Following intravenous administration, the rats showed AUC(0-24 h), t1/2, MRT Last, Cl and Vdss values of 245 ng·h/mL, 1.1 hours, 1.1 hours, 67 mL/min/kg and 5.3 L/kg, respectively. Following intravenous administration to dogs, these respective values were 951 ng·h/mL, 6 hours, 3.9 hours, 18 mL/min/kg and 5.1 L/kg. Following oral administration to rats, the respective values were 136 ng·h/mL, 0.42 hours, 222 hours, 1.4 mL/min/kg and 1.4 L/kg. For dogs, these respective values were 179 ng·h/mL, 0.58 hours, 711 hours, 4.6 mL/min/kg and 4.0 L/kg. Oral bioavailabilty values in rats and dogs were 30 and 72%, respectively. The brain penetration profile was studied 1 hour after the administration of 1, 3 and 10 mg/kg p.o. in rats. Plasma, cerebrospinal fluid (CSF), whole brain samples were collected and drug concentrations were analyzed by liquid chromatography – mass spectrometry. Dosing at 1, 3 and 10 mg/kg p.o. was associated with respective plasma concentrations of 42, 136 and 537 nM; respective brain concentrations of 120, 352 and 1674 nM; respective CSF concentrations of 7, 18 and 90 nM; ratios of CSF concentrations over Ki values of 0.3, 0.8 and 3.8; ratios of brain concentrations over Ki values of 5, 5 and 70; and ratios of brain over plasma concentrations of 2.8, 2.5 and 3. Further studies included in vivo receptor occupancy (brain 5-HT4 receptor) analysis. The drug showed dose-dependent occupancy in the rat striatum and gained ready access to the brain. An ED50 of 2.75 mg/kg p.o. was noted. Brain cortical soluble amyloid precursor protein alpha (sAPPalpha) levels were assessed in male C57BL6 mice injected with 1-10 mg/kg s.c. and sacrificed 30/60 minutes later. Results were compared to vehicle-treated mice. At 3 and 10 mg/kg doses, significant increases in sAPPalpha levels were noted (P values < 0.05 and < 0.01, respectively) using ELISA. To study changes in CSF beta-amyloid levels, Wistar rats were administered the drug orally at 0.03-3 mg/kg and 2 hours later, CSF was collected and analyzed for beta-amyloid protein 42 (Abeta42) and 40 (Abeta40) by ELISA. The drug induced a decrease of 19-35% in Abeta42 levels and a decrease of 20-38% in Abeta40 levels in rat CSF at a dose of 0.1 mg/kg (P < 0.01). Toxicity studies are currently under way.
March 16, 2015
Drug firm Suven Life Sciences has been granted a patent each by the US and New Zealand for a drug used in the treatment of neuro-degenerative diseases.
The patents are valid until 2030 and 2031, respectively, Suven Life Sciences said in a filing to the BSE.
Commenting on the development, Suven Life CEO Venkat Jasti said: “We are very pleased by the grant of these patents to Suven for our pipeline of molecules in CNS arena that are being developed for cognitive disorders with high unmet medical need with huge market potential globally.”
SUVEN, Chief executive and chairman Venkat Jasti

The company has “secured patents in USA and New Zealand to one of their new chemical entity (NCE) for CNS therapy through new mechanism of action – H3 Inverse agonist…,” Suven Life Sciences said.
With these new patents, Suven has a total of 20 granted patents from US and 23 granted patents from New Zealand.
“These granted patents are exclusive intellectual property of Suven and are achieved through the internal discovery research efforts.
“Products out of these inventions may be out-licensed at various phases of clinical development like at Phase-I or Phase-II,” Suven said.
http://www.bseindia.com/xml-data/corpfiling/AttachLive/suven_life_sciences_ltd_160315.pdf

Suven Life Sciences secures 2 (two) Product Patents for their NCE’s through New mechanism of action – H3 Inverse Agonist in USA & New Zealand HYDERABAD, INDIA (March 16, 2015) – Suven Life Sciences Ltd (Suven) announced today that they secured patents in USA (us 8912179) and New Zealand (614567) to one of their New Chemical Entity (NCE) for CNS therapy through new mechanism of action – H3 Inverse agonist and these patents are valid until 2030 and 2031 respectively. The granted claims of the patent include the class of selective H3 ligands discovered by Suven and are being developed as therapeutic agents and are useful in the treatment of cognitive impairment associated with neurodegenerative disorders



Suven Life Sciences Ltd.
6th Floor, SDE Serene Chambers,
Avenue – 7, Road No. 5, Banjara Hills,
Hyderabad-500 034, Telangana, INDIA
Phone : +91-40-2354-1142, 2354-3311
Fax : +91~40~2354-1152
Email id: info@suven.com
INDIAN PATENT
- Nirogi, Ramakrishna; Shinde, Anil Karbhari; Kambhampati, Ramasastri; Namala, Rambabu; Dwarampudi, Adi Reddy; Kota, Laxman; Gampa, Murlimohan; Kodru, Padmavathi; Tiriveedhi, Taraka Naga Vinaykumar; Kandikere, Vishwottam Nagaraj; et al
- From Indian Pat. Appl. (2012), IN 2010CH02551
PATENT
http://www.google.com/patents/US8912179
The present invention relates to heterocyclyl compounds of formula (I) and their pharmaceutically acceptable salts, its process of preparation and compositions containing them, for the treatment of various disorders that are related to Histamine H3 receptors.

1-Cyclobutyl-piperidin-4-ol (1.6 grams, 10 mmol) in tetrahydrofuran (20 mL) was treated with cooled and stirred suspension of sodium hydride (0.9 grams, 18 mmol) in tetrahydrofuran (20 mL) slowly over a period of 30 minutes; the reaction mixture was stirred for 1 hour. A solution of 2-Bromo-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester (3 grams, 9 mmol, obtained in preparation 1) in tetrahydrofuran (30 mL) was added drop wise over a period of 15 minutes and refluxed the reaction for 6 hours. Reaction mass was quenched with ice cold water and the product was extracted with ethyl acetate (3×50 mL). Combined organics were washed with water followed by brine and dried over anhydrous sodium sulphate. Organic volatiles were evaporated under vacuum. The residue was purified by flash chromatography (ethylacetate/n-hexane, 1/1) to obtain the title compound (2.0 grams).
1H-NMR (δ ppm): 1.48 (9H, s), 1.65-1.72 (2H, m), 1.85-1.92 (4H, m), 2.01-2.07 (4H, m), 2.18-2.19 (2H, m), 2.57 (2H, m), 2.62-2.66 (2H, m), 2.71-2.75 (1H, m), 3.70 (2H, m), 4.43 (2H, m), 4.93 (1H, m);
Mass (m/z): 394.2 (M+H)+.
Step (ii): Preparation of 2-(1-Cyclobutyl-piperidin-4-yloxy)-4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridineA solution of 2-(1-Cyclobutyl-piperidin-4-yloxy)-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester (2.0 grams, 5 mmol, obtained in above step) in dichloromethane (30 mL) was treated with trifluroacetic acid (5.0 mL, 50 mmol) at 0° C. Reaction mass was stirred for 4 hours. After completion of reaction, the reaction mass was quenched into ice cold water and adjust pH to 10, by using 40% aqueous sodium hydroxide solution. The product was extracted with dichloromethane (3×50 mL), combined organics were washed with water followed by brine and dried over anhydrous sodium sulphate. Organic volatiles were evaporated under vacuum to obtain the title compound (1.3 grams).
1H-NMR (δ ppm): 1.68-1.74 (2H, m), 1.85-1.93 (4H, m), 2.06 (4H, m), 2.19 (2H, m), 2.60-2.61 (4H, m), 2.73-2.80 (1H, m), 2.90-3.10 (1H, m), 3.13-3.16 (2H, m), 3.85 (2H, s), 4.90-4.93 (1H, m);
Mass (m/z): 294.2 (M+H)+.
Step (iii): Preparation of 1-[2-(1-Cyclobutyl-piperidin-4-yloxy)-6,7-dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-propan-1-oneA solution of 2-(1-Cyclobutyl-piperidin-4-yloxy)-4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridine (1.3 grams, 4 mmol, obtained in above step) and triethylamine (1.9 mL, 13 mmol) in dichloromethane (30 mL) was cooled to 0° C. Propionylchloride (0.4 mL, 5 mmol) in dichloromethane (5 mL) was added drop wise over a period of 15 minutes and stirred the reaction for 30 minutes. Reaction mass was poured onto ice cold water and the product was extracted with ethyl acetate (3×50 mL). Combined organics were washed with water followed by brine and dried over anhydrous sodium sulphate. Organic volatiles were evaporated under vacuum. The residue was purified by flash chromatography (methanol/chloroform, 2/98) to obtain the title compound (1.0 gram).
1H-NMR (δ ppm): 1.17-1.21 (3H, m), 1.65-1.72 (5H, m), 1.87-1.91 (4H, m), 2.01-2.07 (4H, m), 2.22 (1H, m), 2.38-2.45 (2H, m), 2.45 (1H, m), 2.68-2.76 (3H, m), 3.72-3.74 (1H, m), 4.47-4.62 (2H, m), 4.92-4.94 (1H, m).
Mass (m/z): 350.4 (M+H)+.
Step (iv): Preparation of 1-[2-(1-Cyclobutyl-piperidin-4-yloxy)-6,7-dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-propan-1-one tartrateA solution of 1-[2-(1-Cyclobutyl-piperidin-4-yloxy)-6,7-dihydro-4H-thiazolo[5,4-c]pyridin-5-yl]-propan-1-one (0.8 grams, 2.3 mmol, obtained in above step) in methanol (10 mL) was treated with L(+)-Tartaric acid (0.34 grams, 2.3 mmol) at 0° C. Stirred the reaction mass for about 1 hour and the solvent was evaporated under vacuum to dryness. The solids were washed with diethyl ether and dried under vacuum to obtain the title compound (1.1 grams).
1H-NMR (δ ppm): 1.12-1.20 (3H, m), 1.82-1.87 (2H, m), 2.16-2.32 (7H, m), 2.45-2.55 (2H, m), 2.63-2.66 (3H, m), 2.72 (1H, m), 3.20 (2H, m), 3.47-3.50 (1H, m), 3.66-3.70 (1H, m), 3.81-3.88 (2H, m), 4.45 (2H, s), 4.60 (2H, s), 5.18 (5H, m);
Mass (m/z): 350.4 (M+H)+.
| Publication number | US8912179 B2 |
| Publication type | Grant |
| Application number | US 13/818,152 |
| PCT number | PCT/IN2010/000740 |
| Publication date | Dec 16, 2014 |
| Filing date | Nov 15, 2010 |
| Priority date | Sep 2, 2010 |
| Also published as | CA2812970A1, 4 More » |
| Inventors | Ramakrishna Nirogi, Anil Karbhari Shinde,Ramasastri Kambhampati, Rambabu Namala,Adi Reddy Dwarampudi, Laxman Kota,Murlimohan Gampa, Padmavathi Kodru,Taraka Naga Vinaykumar Tiriveedhi,Vishwottam Nagaraj Kandikere, Nageshwara Rao Muddana, Ramanatha Shrikantha Saralaya, Pradeep Jayarajan, Dhanalakshmi Shanmuganathan, Ishtiyaque Ahmad,Venkateswarlu Jasti, Less « |
| Original Assignee | Suven Life Sciences Limited |
| Export Citation | BiBTeX, EndNote, RefMan |
| Patent Citations (12), Non-Patent Citations (10), Classifications (16),Legal Events (1) | |
| External Links: USPTO, USPTO Assignment, Espacenet | |
……………….
Banjara Hills,Hyderabad
 TAJ KRISHNA
TAJ KRISHNA

//////
CC(C)n4nc(c1nnc(o1)C2CCN(CCCOC)CC2)c3ccccc34
PF 04995274, a 5-HT4Partial Agonist
PF-04995274,
(R)-4-((4-(((4-(Tetrahydrofuran-3-yloxy)-1,2-benzisoxazol-3-yl)oxy)methyl)piperidin-1-yl)methyl)tetrahydro-2H-pyran-4-ol
4-(4-{4-[(R)-(Tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-yloxymethyl}-piperidin-1-ylmethyl)-tetrahydro-pyran-4-ol
CAS 1331782-27-4
UNII: XI179PG9LV
MF C23-H32-N2-O6
MW 432.5138
a 5-HT4Partial Agonist
PHASE 1 Alzheimer’s type dementia.
Pfizer Inc. INNOVATOR
5-HT4 agonists have attracted attention for therapeutic value in the treatment of Alzheimer’s Disease (AD) and cognitive impairment.Acting to increase levels of acetylcholine and soluble APP alpha, 5-HT4 agonists have the potential to demonstrate both ameliorative and disease modifying effects
(R)-4-((4-((4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidin-1-yl)methyl)tetrahydro-2/-/-pyran-4-ol and pharmaceutically acceptable salts thereof. This invention also is directed, in part, to a method for treating a 5-HT4 mediated disorder in a mammal. Such disorders include acute neurological and psychiatric disorders, stroke, cerebral ischemia, spinal cord trauma, head trauma, perinatal hypoxia, cardiac arrest, hypoglycemic neuronal damage, dementia, Alzheimer’s disease, Huntington’s Chorea, amyotrophic lateral sclerosis, ocular damage, retinopathy, cognitive disorders, idiopathic and drug- induced Parkinson’s disease, muscular spasms and disorders associated with muscular spasticity including tremors, depression, epilepsy, convulsions, migraine, urinary incontinence, substance tolerance, substance withdrawal, psychosis, schizophrenia, anxiety, mood disorders, trigeminal neuralgia, hearing loss, tinnitus, macular degeneration of the eye, gastroesophageal reflux disease, gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia, irritable bowel syndrome, constipation, dyspepsia, esophagitis, gastroesophageral disease, nausea, emesis, brain edema, pain, tardive dyskinesia, sleep disorders, attention deficit/hyperactivity disorder, attention deficit disorder, disorders that comprise as a symptom a deficiency in attention and/or cognition, and conduct disorder

a(a) SOCl2, DMAP, acetone, DME, RT, 81%;
(b) DEAD, PPh3, THF, RT, 65%;
(c) K2CO3, MeOH, RT, 92%;
(d) K2CO3, water, MeOH, 50 °C, 76%;
(e) CDI, THF, 50 °C, 43%;
(f) DEAD, PPh3, THF, reflux, 51%;
(g) HCl, Et2O, RT, 81%;
(h) TEA, MeOH, reflux, 50%.

PAPER
Journal of Medicinal Chemistry (2012), 55(21), 9240-9254
http://pubs.acs.org/doi/abs/10.1021/jm300953p

The cognitive impairments observed in Alzheimer’s disease (AD) are in part a consequence of reduced acetylcholine (ACh) levels resulting from a loss of cholinergic neurons. Preclinically, serotonin 4 receptor (5-HT4) agonists are reported to modulate cholinergic function and therefore may provide a new mechanistic approach for treating cognitive deficits associated with AD. Herein we communicate the design and synthesis of potent, selective, and brain penetrant 5-HT4 agonists. The overall goal of the medicinal chemistry strategy was identification of structurally diverse clinical candidates with varying intrinsic activities. The exposure–response relationships between binding affinity, intrinsic activity, receptor occupancy, drug exposure, and pharmacodynamic activity in relevant preclinical models of AD were utilized as key selection criteria for advancing compounds. On the basis of their excellent balance of pharmacokinetic attributes and safety, two lead 5-HT4 partial agonist candidates 2d and 3 were chosen for clinical development.
PATENT
https://www.google.co.in/patents/WO2011101774A1?cl=en
(R)-4-((4-((4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidin-1-yl)methyl)tetrahydro-2H-pyran-4-ol , hereinafter referred to as “Compound X,” and having the following structure:

Compound X
Example 1 : Synthesis of iR)-4-ii4-i(4-itetrahvdrofuran-3-yloxy)benzord1isoxazol-3-yloxy)methyl)piperidin-1 -yl)methyl)tetrahvdro- 2 -pyran-4-ol

Methyl 2-fluoro-6-hydroxybenzoate (2): To a 20L jacketed reactor were charged 2-fluoro-6-hydroxybenzoic acid (Oakwood Products; 0.972 kg, 6.31 mol), methanol (7.60 L) and sulfuric acid (0.710 kg, 7.24 mol, 1 .15 eq). The jacket temperature was heated to 60°C and the reaction mixture was stirred for 45 h. The reaction mixture was concentrated under vacuum and approximately 7.5 L of methanol distillates were collected. The resulting thin oil was cooled to 20°C. Water (7.60 L) and ethyl acetate (7.60 L) were charged to the reactor, and the product extracted into the organic layer. The EtOAc solution was washed with a solution of sodium bicarbonate (1.52 Kg) in water (6.92 L) followed by a brine solution of sodium chloride (1.74 kg) in water (4.08 L). The resulting EtOAc solution was concentrated to dryness. A light orange oil was isolated; the oil slowly crystallized upon standing to give the title compound (2) (0.952 Kg, 5.60 mol, 89% yield). 1 H NMR (400 MHz, CDCI3) δ ppm 3.97 (s, 3H), 6.59 (ddd, J=10.9, 8.2,1 .2, 1 H), 6.76 (dt, J=8.2, 1 .1 , 1 H), 7.35 (td, J=8.6, 6.3, 1 H), 1 1.24 (s, 1 H); 13C NMR (400 MHz, CDCI3) δ ppm 52.65, 102.56 (d, J=13), 106.90 (d, J=23), 1 13.31 (d, J=3.1 ), 135.34 (d, J=1 1 .5), 161 .02, 163.31 (d, J=62.2), 169.87 (d, 3.8); MS 171.045 (m+1 ). 2-Fluoro-N,6-dihydroxybenzamide (3): To a 50L reactor was charged water (4.47 L) and hydroxylamine sulfate (6.430 kg, 39.17 mol), the mixture was stirred at 25°C. A solution of potassium carbonate (3.87 Kg, 27.98 mol) in water (5.05 L) was slowly added to the reaction mixture to form a thick white mixture that was stirred at 20°C. A solution of methyl 2-fluoro-6-hydroxybenzoate (2) (0.952 Kg, 5.60 mol) in methanol (9.52 L) was slowly added to the reactor resulting in mild off gassing. The reaction mixture was then heated to 35°C and stirred for 20 h. The reaction mixture was cooled to 15°C and stirred for 1 h. The mixture was filtered to remove inorganic material. The reactor was rinsed with methanol (2.86 L) and the tank rinse was used to wash the inorganic cake.
Analysis of the cake indicated that it contained product. To a 20L reactor was charged methanol (10 L) and the inorganic cake and the mixture was stirred at 25°C for 30 min. The mixture was filtered and the cake washed with methanol (3 L).
The combined filtrates were charged back into the reactor and concentrated under vacuum with the jacket temperature set at 40°C until approximately 10 L remained. The mixture was held at 25°C and cone. HCI (5.51 L) was added. The reactor was cooled to 15°C and stirred for 2 h. The white slurry was filtered and the resulting product cake was washed with water (4.76L), blown dry with nitrogen and then dried in a vacuum oven at 40°C for 12 h. The desired product (3) (747 g, 4.36 mol), was isolated in 78% yield. 1 H NMR (400 MHz, CD3OD) δ ppm 4.91 (s, 3H), 6.63 (ddd, J=10.9, 8.5, 0.8, 1 H), 6.72 (dt, J=8.2, 0.8, 1 H), 7.31 (td, J=8.2, 6.6, 1 H); MS 172.040 (m+1 ).
4-Fluorobenzo[d]isoxazol-3-ol (4): To a 20L jacketed reactor were charged tetrahydrofuran (2.23 L) and 1 ,1 ‘-carbonyldiimidazole (0.910 Kg, 5.64 mol). The resulting mixture was stirred at 20°C. Then a solution of 2-fluoro-N,6-dihydroxybenzamide (3) (744 g, 4.34 mol) in tetrahydrofuran (4.45 L) was slowly charged to the reactor maintaining the temperature below 30°C and stirred at 25°C for 30 min during which some off gassing was observed. The reaction mixture was heated to 60°C over 30 min and stirred for 6 h. The reactor was cooled to 20°C followed by the addition of 1 N aqueous hydrogen chloride (7.48L) over 15 min to adjust the pH to 1. The jacket temperature was set to 35°C and the reaction mixture concentrated under vacuum to remove approximately 6.68L of THF. The reactor was cooled to 15°C and stirred for 1 h. The resulting white slurry was filtered, the cake was washed with water (3.71 L) and dried in a vacuum oven at 40°C for 12 h. The desired product, (4) (597 g, 3.90 mol), was isolated in 90% yield. 1 H NMR (400 MHz, CD3OD) δ ppm 4.93 (b, 1 H), 6.95 (dd, J=10.1 , 8.6, 1 H), (d, J=8.6, 1 H), 7.52-7.57 (m, 1 H); LRMS 154.029 (m+1 ).
Tert-butyl 4-(tosyloxymethyl)piperidine-1-carboxylate (5): To a 20L jacketed reactor were charged dichloromethane (8 L), N-boc-4-piperdine methanol (0.982 Kg, 4.56 mol) and p-toluenesulfonyl chloride (0.970 Kg, 5.09 mol) and the resulting mixture was stirred at 20°C for 5 min. Triethylamine (0.94 Kg, 9.29 mol) was added to the reactor via an addition funnel and the resulting deep red solution was stirred at 25°C for 16 h. A solution of sodium carbonate (0.96 Kg, 9.06 mol) in water (7.04 L) was charged to the reaction mixture and stirred for 1 h at 20°C. The phases were split and the organic layer washed with brine (6 L) and concentrated at 40°C to a low stir volume. Dimethylacetamide (2 L) was charged to the reactor and concentration continued under full vacuum at 40°C for 1 h. The solution of tert-butyl 4-(tosyloxymethyl)piperidine-l -carboxylate (5) in dimethyl acetamide was held for further processing. Yield was assumed to be 100% with approximately
90% potency. A sample was pulled and concentrated to dryness for purity analysis. 1 H NMR (400 MHz, CDCI3) δ ppm 1 .02-1 .12 (m, 2H), 1.14 (s, 9H), 1 .59-1.64 (m, 2H), 1.75-1.87 (m, 1 H), 2.43 (s, 3H), 2.55-2.75 (m, 2H), 3.83 (d, J=6.7, 2H), 3.95-4.20 (b, 2H), 7.33 (d, 8.6, 2H), 7.76 (d, 8.2, 2H); 13C NMR (400 MHz, CDCI3) δ ppm 21 .64, 28.15, 28.39, 35.74, 73.97, 79.50, 126.99, 127.84, 129.86, 132.84, 144.84, 154.63; LRMS 739.329 (2m+1 ).
Tert-butyl 4-((4-fluorobenzo[d]isoxazol-3-yloxy)methyl)piperidine-1-carboxylate (6): To a 20L jacketed reactor were charged dimethylacetamide (4.28 L), tert-butyl 4-(tosyloxymethyl)piperidine-1 -carboxylate (5) (1.68 Kg, 4.56 mol), 4-fluorobenzo[d]isoxazol-3-ol (4) (540 g, 3.51 mol), and potassium carbonate (960 g, 6.98 mol) resulting in a thick beige slurry. The reaction mixture was heated to 50°C and stirred for 20 h and then cooled to 20°C, followed by the addition of water (7.5 L) and ethyl acetate (5.37 L). After mixing for 15 min, the phases were settled and split. The organic layer was washed with water (5.37 L), sending the aqueous wash to waste. The organic mixture was distilled under vacuum with a maximum jacket temperature of 40°C until approximately 5 L remained in the reactor. Methanol (2.68 L) was added and the resulting solution concentrated under vacuum to about 3 L of a yellow oil. Methanol (2.68 L) was charged to the reactor and the resulting solution was stirred at 25°C for 15 min. Water (0.54 L) was added over 15 min resulting in a white slurry. The mixture was cooled to 15°C, stirred for 1 h and then filtered. The filter cake was washed with a solution of water (0.54 L) in methanol (2.14 L), then air dried for 30 min, transferred to a vacuum oven and dried at 40°C for 12 h. The desired product, (6) (746 g, 2.13 mol), was isolated in 61 % yield. 1 H NMR (400 MHz, CDCI3) δ ppm 1.23-1 .37 (m, 2H), 1 .45 (s, 9H), 1 .78-1 .88 (m, 2H), 2.04-2.17 (m, 1 H), 2.67-2.83 (m, 2H), 4.02-4.26 (m, 2H), 4.28 (d, 6.6, 2H), 6.89 (dd, J=8.6, 7.5, 1 H), 7.21 (d, J=9, 1 H), (td, 8.6, 4.9); LRMS 351.171 (m+1 ).
(R)-Tert-butyl 4-((4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidine-1-carboxylate (8): To a 20 L glass reactor with the jacket set to 20°C were charged (R)-tetrahydrofuran-3-ol (7) (297 g, 3.37 mol) and dimethylacetamide (5.1 L). 2.0 M sodium bis(trimethylsilyl)amide in THF (1.37 L, 2.74 mol) was slowly added via an addition funnel while maintaining a pot temperature less than 30°C. The resulting orange/red solution was stirred at 25°C for 30 min. Then, tert-butyl 4-((4-fluorobenzo[d]isoxazol-3-yloxy)methyl)piperidine-1 -carboxylate (6) (640.15 g, 1.83 mol) was charged and the reaction mixture was stirred at 25°C for 16 h. The reaction mixture was cooled to 20°C and water (6.4 L) was slowly added over 45 min maintaining a pot temperature of less than 35°C. Ethyl acetate (6 L) was added and the biphasic mixture was stirred for 15 min and then separated. The aqueous layer was back extracted with additional ethyl acetate (4 L). The combined organics were then washed with water (5 L) and a 20% brine solution (5 L). The organic mixture was concentrated under vacuum with the jacket temperature set to 40°C to approximately 3 L and held for further processing. Quantitative yield of the desired product, (8) (0.76 Kg, 1 .82 mol), in ethyl acetate was assumed. A sample was pulled and concentrated to dryness for purity analysis. 1 H NMR (400 MHz, CDCI3) δ ppm 1 .25-1.38 (m, 2H), 1 .44 (s, 9H), 1.76-1 .84 (m, 2H), 1 .89-1.97 (b, 1 H), 1 .99-2.12 (m, 1 H), 2.14-2.28 (m, 2H), 2.63-2.84 (m, 2H), 3.90-4.21 (m, 6H), 4.24 (d, J=6.3, 2H), 5.00-5.05 (m, 1 H), 6.48 (d, J=8.2, 1 H), 6.98 (d, J=8.6, 1 H), 7.37 (t, J=8.2, 1 H); LRMS 419.216 (m+1 ).
(R)-3-(Piperidin-4-ylmethoxy)-4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazole 4-methylbenzenesulfonate (9): To a 20L jacketed reactor charged ethyl acetate (6.1 L), (R)-tert-butyl 4-((4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidine-1 -carboxylate (8) (0.76 kg, 1 .82 mol) and p-toluenesulfonic acid monohydrate (0.413 kg, 2.17 mol) and stirred at 20°C for 30 min. The reactor jacket was heated from 20 to 65°C over
1 h and then held at 65°C for 16 h. The reactor was cooled to 15°C over 1 h and granulated for 2 h. The resulting slurry was filtered, the cake was washed with EtOAc (3 L) and then air dried on the filter for 30 min. The cake was transferred to a vacuum oven and dried at 40°C for 12 h. The desired product, (9) (854 g, 1.74 mol), was isolated in 96% yield (two steps). 1 H NMR (400
MHz, CD3OD) δ ppm 1.54-1 .67 (m, 2H), 2.04-2.18 (m, 3H), 2.19-2.36 (m, 2H), 2.33 (s, 3H), 3.01 -3.12 (m, 2H), 3.41-3.50 (m, 2H), 3.86-4.01 (m, 4H), 4.26 (d, J=6.3, 2H), 4.90 (s, 2H), 5.14-5.19 (m, 1 H), 6.72 (d, J=8.2, 1 H), 7.02 (d, J=8.6, 1 H), 7.21 (d, J=7.8, 2H), 7.48 (t, J=8.6, 1 H), 7.70 (d, J=8.2, 2H); LRMS 319.165 (m+1 ).
(R)-4-((4-((4-(Tetrahydrofuran-3-yloxy)benzo[d]isoxazol-3-yloxy)methyl)piperidin-1-yl)methyl)tetrahydro-2H-pyran-4-ol (11): To a
20L jacketed reactor were charged water (7.5 L) and sodium carbonate (0.98 kg); the mixture was stirred at 20°C until all solids had dissolved. Then (R)-3-(piperidin-4-ylmethoxy)-4-(tetrahydrofuran-3-yloxy)benzo[d]isoxazole 4-methylbenzenesulfonate (9) (750 g, 1 .53 mol) and ethyl acetate (6.0 L) were added to the reactor and stirred at 20°C for 30 min. The phases were split and the lower aqueous layer was back extracted twice with ethyl acetate (6.0 L and then 3.75 L). The organic layers were combined in the 20L reactor and washed twice with brine (3.0 L). The ethyl acetate solution was concentrated to under vacuum at 45°C to a low stir volume. Isopropyl alcohol (3.75 L) was added and concentration continued until 2 L remained in the reactor.
Additional isopropyl alcohol (2.75 L) was added and the mixture cooled to 25°C. To the reactor was charged 1 ,6-dioxaspiro[2.5]octane (10) (260 g, 2.29 mol) and the resulting solution heated to 50°C and stirred for 16 h. The reaction mixture was cooled to 30°C and water (15 L) was added over 60 min. Product crystallized from solution and the resulting slurry was cooled to 15°C over 1 h and then granulated for 4 h. The product was filtered and washed with water (3.75 L). The cake was blown dry with nitrogen for 30 min and then transferred to a vacuum oven and dried at 40°C for 12 h. The desired product, (11 ) (588 g, 1 .36 mol), was isolated in 89% yield.
1 H NMR (400 MHz, CDCI3) δ ppm 1 .41-1 .63 (m, 6H), 1.71 -1.81 (m, 2H), 1.81 -1.94 (m, 1 H), 2.17-2.26 (m, 2H), 2.33 (s, 2H), 2.4 (td, J=1 1.7, 2.3, 2H), 2.92 (d, J=1 1 .8, 2H), 3.46 (s, 1 H), 3.71-3.84 (m, 4H), 3.91 -4.10 (m, 4H), 4.24 (d, J=5.9, 2H), 5.03-5.08 (m, 1 H), 6.50 (d, J=8.2, 1 H), 7.00 (d, J=8.2, 1 H), 7.38 (t, J=8.2, 1 H);
13C NMR (400 MHz, CDCI3) δ ppm 29.1 1 , 33.10, 35.20, 36.92, 36.96, 56.15, 63.93, 67.14, 67.46, 68.27, 72.94, 74.06, 78.37, 103.17, 105.15, 131.71 , 152.71 , 166.02, 166.28;
LRMS 433.232 (m+1 ).
Example 2: Synthesis of iR)-4-ii4-i(4-itetrahvdrofuran-3-yloxy)benzord1isoxazol-3-yloxy)methyl)piperidin-1 -yl)methyl)tetrahvdro- 2H-pyran-4-ol

5-Hydroxy-2,2-dimethyl-benzo[1,3]dioxin-4-one: Thionyl chloride (83.8 g, 0.71 mol) was slowly added to a solution of 2,6-dihydroxy-benzoic acid (77 g, 0.5 mol), acetone (37.7 g, 0.65 mol) and DMAP (3.1 g, 0.025 mol) in dimethoxyethane (375 mL). The mixture was stirred at RT for 7 h. The residue obtained after concentration under reduced pressure was dissolved in ethyl
acetate and washed with water and aqueous saturated sodium bicarbonate solution. The organic layer was dried (Na2S04) and concentrated to afford 79 g desired product as a red solid (81 % yield). 1 H NMR (400 MHz, CDCI3) δ ppm 1 .68 (s, 6H), 6.37 (dd, J=8, 0.8, 11-1) 6.56 (dd, J=8, 0.8, 1 H), 7.34 (t, J=8, 1 H), 10.27( brs, 1 H).
2,2-Dimethyl-5-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[1,3]dioxin-4-one:
Diethyl azodicarboxylate (130.5 g, 0.75 mol) was added in a dropwise fashion to a mixture of 5-hydroxy-2,2-dimethyl-benzo[1 ,3]dioxin-4-one (100 g, 0.51 mol), triphenylphosphine (196.5 g, 0.75 mol), and (S)-tetrahydro-furan-3-ol (44 g, 0.5 mol) in 600 ml. of anhydrous THF. The resulting mixture was stirred at RT for 18 h. The solvent was removed under reduced pressure and the crude material was purified on a silica gel flash column, eluting with petroleum ether/ ethyl acetate (15:1 -> 3:1 ). 86 g (65% yield) of product was isolated as a colorless oil. 1 H NMR (400 MHz, CDCI3) δ ppm 1.67 (s, 6H), 2.30 (m, 2H), 4.2 (m, 4H) 4.97 (m, 1 H), 6.49 (d, J=8.4, 1 H) 6.51 (d, J=8.4, 1 H), 7.39 (t,
J=8.4, 1 H).
2-Hydroxy-6-[(R)-(tetrahydro-furan-3-yl)oxy]-benzoic acid methyl ester: Potassium carbonate (134.8 g, 0.98 mol) was added to a solution of 2,2-dimethyl-5-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[1 ,3]dioxin-4-one (86 g, 0.33 mol) in 1 L methanol. The mixture was stirred at RT for 2 h, then concentrated in vacuo. The residue was dissolved in ethyl acetate and washed with aqueous ammonium chloride solution. The organic layer was dried (Na2S04) and concentrated to afford 72 g of the product as a yellow solid (92% yield). 1 H NMR (400 MHz, CDCI3) δ ppm 2.20 (m, 2H), 3.99 (s, 3H), 4.80(m, 4H). 4.94 (m, 1 H), 6.31 (dd, J=8.4, 0.8, 1 H), 6.59 (dd, J=8.4, 0.8, 1 H), 7.30 (t, J=8.4, 1 H).
2,N-Dihydroxy-6-[(R)-(tetrahydro-furan-3-yl)oxy]-benzamide: Potassium carbonate (121 g. 0.867mmol) was added portionwise to a solution of hydroxylamine sulfate (120 g, 0.732 mol) in 360 ml. of water at 0°C. After stirring for 30 min, sodium sulfite (3.74 g, 0.029 mol) and a solution of 2-hydroxy-6-[(R)-(tetrahydro-furan-3-yl)oxy]-benzoic acid methyl ester (35 g, 0.146 mol) in 360 ml. of methanol were added and the mixture was stirred at 50°C for 30 h. Methanol was removed from the cooled reaction mixture under reduced pressure and the resulting aqueous layer was acidified with 2N HCI. The aqueous layer was extracted with ethyl acetate and the organic layer was dried (Na2S04) and concentrated to afford 25 g (76% yield ) of the product as a yellow solid. 1 H NMR (400 MHz, CDCI3) δ ppm 2.00 (m, 1 H), 2.15 (m, 1 H), 3.80 (m, 4H), 5.05 (m, 1 H), 6.48 (d, J=8, 1 H), 6.49 (d, J=8, 1 H), 7.19 (t, J=8, 1 H), 10.41 (brs, 1 H), 1 1.49 (brs, 1 H); LRMS m/z 239 (m+1 ).
4-[(R)-(Tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-ol: A solution of 2, N-dihydroxy-6-[(R)-(tetrahydro-furan-3-yl)oxy]-benzamide (25 g, 0.105 mol) in 250 ml. of THF was heated to 50°C. Carbonyl diimidazole was added portionwise and the resulting mixture was stirred at 50°C for 14 h. After cooling to RT, 100 ml. of 2N HCI was added and the aqueous layer was extracted with ethyl acetate. The combined organic layers were then extracted three times with 10% aqueous potassium carbonate. The potassium carbonate aqueous extracts were washed with ethyl acetate and then acidified to pH 2 – 3 with 2N HCI. The acidified aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were washed with brine, dried (Na2S04) and concentrated to afford 20 g of product as a yellow solid (43% yield). 1 H NMR (400 MHz, CDCI3) δ ppm 2.20 (m, 2H), 3.89 (m, 1 H), 4.01 (m, 3H), 5.05 (m, 1 H), 6.48 (d, J=7.6, 1 H). 6.92 (d, J=7.6, 1 H), 7.37 (t, J=7.6, 1 H); LRMS m/z 222 (m+1 ).
4-{4-[(R)-(Tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-yloxymethyl}-piperidine-1-carboxylic acid tert-butyl ester: Diethyl azodicarboxylate (15.6 g, 0.09 mol) was added to a mixture of 4-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-ol (10 g, 0.045 mol), 4-hydroxymethyl-piperidine-1 -carboxylic acid tert-butyl ester (1 1.6 g, 0.054 mol) and triphenylphosphine (23.5 g, 0.09 mol) in 300 mL THF. After the addition was complete the mixture was heated at reflux for 18 h. After concentration in vacuo, the crude product was purified on a silica gel flash column, eluting with petroleum ether/ ethyl acetate (15:1 -» 5:1 ) to afford 22 g of the product as an oil (51 % yield). 1 H NMR (400 MHz, CDCI3) δ ppm 1.25 (m, 2H), 1.39 (s, 9H), 1.76 (m, 2H), 1.99 (m, 1 H). 2.15 (m, 2H), 2.70 (bt, J=1 1.6, 2H), 3.95 (m, 4H). 4.13 (m, 2H). 4.34 (d J=6.4, 2H), 4.98 (m, 1 H), 6.43 (d, J=8, 1 H), 6.93 (d, J=8, 1 H), 7.31 (t, J=8, 1 H).
3-(Piperidin-4-ylmethoxy)-4-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazole: A 0°C solution of 4-{4-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-yloxymethyl}-piperidine-1 -carboxylic acid tert-butyl ester in 500 mL ether was treated with a saturated solution of HCI (g) in 200 mL ether. After addition was complete, the mixture was warmed to RT and stirred for 16 h. The reaction mixture was filtered. The white solid was washed with ethyl acetate followed by ether and dried to yield 15 g (81 % yield) of the desired product as a white solid. 1 H NMR (400 MHz, CD3OD) 5 ppm 1 .51 – 1.69 (m, 2 H) 2.04 – 2.19 (m, 3 H) 2.22 – 2.37 (m, 2 H) 2.99 – 3.14 (m, 2 H) 3.40 – 3.51 (m, 2 H) 3.85 – 4.02 (m, 4 H) 4.25 – 4.31 (m, 2 H) 5.17 (td, J= >1^ , 1 .56 Hz, 1 H) 6.72 (d, J=8.00 Hz, 1 H) 7.01 (d, J=8.59 Hz, 1 H) 7.47 (t, J=8.20 Hz, 1 H); LRMS m/z 319 (m+1 ).
4-(4-{4-[(R)-(Tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazol-3-yloxymethyl}-piperidin-1-ylmethyl)-tetrahydro-pyran-4-ol: 1 ,6-Dioxa-spiro[2.5]octane (Focus Synthesis; 9.7 g, 0.084 mol) and triethylamine (8.6 g, 0.084 mol) were added to a solution of 3-(piperidin-4-ylmethoxy)-4-[(R)-(tetrahydro-furan-3-yl)oxy]-benzo[d]isoxazole (15 g, 0.042 mol) in 200 mL methanol. The resulting solution was heated at reflux for 18 h. The cooled mixture was concentrated and ethyl acetate and water were added to the residue. The layers were separated and the organic extracts were washed with brine, dried (Na2S04) and concentrated to provide 17 g crude product as a yellow oil. The crude material was purified by prep HPLC to afford 10 g of the desired product as a white solid. (50% yield).
1 H NMR (400 MHz, CDCI3) δ ppm 1.41 -1.63 (m, 6H), 1.71-1.81 (m, 2H), 1 .81 -1 .94 (m, 1 H), 2.17-2.26 (m, 2H), 2.33 (s, 2H), 2.4 (td, J=1 1 .7, 2.3, 2H), 2.92 (d, J=1 1.8, 2H), 3.46 (s, 1 H), 3.71-3.84 (m, 4H), 3.91-4.10 (m, 4H), 4.24 (d, J=5.9, 2H), 5.03-5.08 (m, 1 H), 6.50 (d, J=8.2, 1 H), 7.00 (d, J=8.2, 1 H), 7.38 (t, J=8.2, 1 H);
13C NMR (101 MHz, CDCI3) δ ppm 29.1 1 , 33.10, 35.20, 36.92, 36.96, 56.15, 63.93, 67.14, 67.46, 68.27, 72.94, 74.06, 78.37, 103.17, 105.15, 131.71 , 152.71 , 166.02, 166.28.
PAPER
Two Routes to 4-Fluorobenzisoxazol-3-one in the Synthesis of a 5-HT4Partial Agonist
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00389

A potent 5-HT4 partial agonist, 1 (PF-04995274), targeted for the treatment of Alzheimer’s disease and cognitive impairment, has been prepared on a multi-kilogram scale. The initial synthetic route, that proceeded through a 4-substituted 3-hydroxybenzisoxazole core, gave an undesired benzoxazolinone through a Lossen-type rearrangement. Route scouting led to two new robust routes to the desired 4-substituted core. Process development led to the efficient assembly of the API on a pilot plant scale under process-friendly conditions with enhanced throughput. In addition, crystallization of a hemicitrate salt of the API with pharmaceutically beneficial properties was developed to enable progression of clinical studies.
REFERNCES
Noguchi, H.; Waizumi, N. Preparation of benzisoxazole derivatives for treatment of 5-HT4 mediated disorders. PCT Int. Appl. WO/2011/101774 A1, 20110825
////////PF-04995274, PF 04995274, PFIZER, Alzheimer’s type dementia, PHASE 1
c1cc2c(c(c1)O[C@@H]3CCOC3)c(no2)OCC4CCN(CC4)CC5(CCOCC5)O
Fresolimumab

Fresolimumab
GC 1008, GC1008
UNII-375142VBIA
cas 948564-73-6
Structure
- immunoglobulin G4, anti-(human transforming growth factors beta-1, beta-2 (G-TSF or cetermin) and beta-3), human monoclonal GC-1008 γ4 heavy chain (134-215′)-disulfide with human monoclonal GC-1008 κ light chain, dimer (226-226”:229-229”)-bisdisulfide
- immunoglobulin G4, anti-(transforming growth factor β) (human monoclonal GC-1008 heavy chain), disulfide with human monoclonal GC-1008 light chain, dimer
For Idiopathic Pulmonary Fibrosis, Focal Segmental Glomerulosclerosis,and Cancer
An anti-TGF-beta antibody in phase I clinical trials (2011) for treatment-resistant primary focal segmental glomerulosclerosis.
A pan-specific, recombinant, fully human monoclonal antibody directed against human transforming growth factor (TGF) -beta 1, 2 and 3 with potential antineoplastic activity. Fresolimumab binds to and inhibits the activity of all isoforms of TGF-beta, which may result in the inhibition of tumor cell growth, angiogenesis, and migration. TGF-beta, a cytokine often over-expressed in various malignancies, may play an important role in promoting the growth, progression, and migration of tumor cells.

Fresolimumab (GC1008) is a human monoclonal antibody[1] and an immunomodulator. It is intended for the treatment of idiopathic pulmonary fibrosis (IPF), focal segmental glomerulosclerosis, and cancer[2][3] (kidney cancer and melanoma).
It binds to and inhibits all isoforms of the protein transforming growth factor beta (TGF-β).[2]
History
Fresolimumab was discovered by Cambridge Antibody Technology (CAT) scientists[4] and was one of a pair of candidate drugs that were identified for the treatment of the fatal condition scleroderma. CAT chose to co-develop the two drugs metelimumab (CAT-192) and fresolimumab with Genzyme. During early development, around 2004, CAT decided to drop development of metelimumab in favour of fresolimumab.[5]
In February 2011 Sanofi-Aventis agreed to buy Genzyme for US$ 20.1 billion.[6]
As of June 2011 the drug was being tested in humans (clinical trials) against IPF, renal disease, and cancer.[7][8] On 13 August 2012, Genzyme applied to begin a Phase 2 clinical trial in primary focal segmental glomerulosclerosis[9] comparing fresolimumab versus placebo.
As of July 2014, Sanofi-Aventis continue to list fresolimumab in their research and development portfolio under Phase II development.[10]

References
2 National Cancer Institute: Fresolimumab
- 3 Statement On A Nonproprietary Name Adopted By The USAN Council – Fresolimumab
- 4 Grütter, Christian; Wilkinson, Trevor; Turner, Richard; Podichetty, Sadhana; Finch, Donna; McCourt, Matthew; Loning, Scott; Jermutus, Lutz; Grütter, Markus G. (2008-12-23). “A cytokine-neutralizing antibody as a structural mimetic of 2 receptor interactions”. Proceedings of the National Academy of Sciences 105 (51): 20251–20256. doi:10.1073/pnas.0807200106. ISSN 0027-8424. PMC 2600578. PMID 19073914.
- 5 http://www.independent.co.uk/news/business/news/cat-may-abandon-skin-drug-after-trial-results-disappoint-569445.html
- 6 http://www.bbc.co.uk/news/business-12477750
- 7 http://www.genengnews.com/gen-news-highlights/scientists-trigger-white-fat-to-become-brown-fat-like-to-treat-obesty-and-type-2-diabetes/81245389/
- 8 Clinicaltrials.gov for Fresolimumab
- 9 http://clinicaltrials.gov/show/NCT01665391
- 10 http://en.sanofi.com/rd/rd_portfolio/rd_portfolio.aspx
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | TGF beta 1, 2 and 3 |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Number | 948564-73-6 |
| ATC code | None |
| ChemSpider | none |
| KEGG | D09620 |
| Chemical data | |
| Formula | C6392H9926N1698O2026S44 |
| Molar mass | 144.4 kDa |
////////////
What was the drug in Clinical Trial Tragedy In France Jan 2016
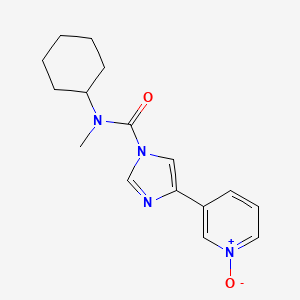
BIA 10-2474
cas 1233855-46-3
3-(1-(cyclohexyl(methyl)carbamoyl)-1H-imidazol-4-yl)pyridine 1-oxide
1H-Imidazole-1-carboxamide, N-cyclohexyl-N-methyl-4-(1-oxido-3-pyridinyl)-
C16 H20 N4 O2, 300.36
| Bial-Portela & Ca. S.A. |
BIA 10-2474 is an experimental fatty acid amide hydrolase inhibitor[1] developed by the Portuguese pharmaceutical company Bial-Portela & Ca. SA. The drug was developed to relieve pain,[2][3] to ease mood and anxiety problems, and to improve movement coordination linked to neurodegenerative illnesses.[4] It interacts with the human endocannabinoid system.[5][6] It has been linked to severe adverse events affecting 5 patients in a drug trial in Rennes, France, and at least one death, in January 2016.[7]

Synthesis
WO 2014017938
Example 5. 3-(l-(cyclohexyl(methyl)carbamoyl-lfl-imidazol-4-yl)pyridine l-oxide (compound A)

C16H20N4O C16H20N4O2
MW 284,36 MW 300,36
To a solution of N-cyclohexyl-N-methyl-4-(pyridm-3-yl)-lH-imidazole-l-carboxamide in dichioromethane at 25°C was added peracetic acid (38%; the concentration is not critical, and may be varied) in a single portion. The reaction mixture was then maintained at 25°C for at least 20 h, whereupon the reaction was washed four times with water (in some embodiments, the water for the extraction step may be supplemented with a small amount (e.g. 1%) of acetic acid, which helps to promote product solubility in the DCM). The dichioromethane solution was then filtered prior to diluting with 2-propanol. Dichioromethane (50%) was then distilled off under atmospheric pressure, whereupon, 2-propanol was charged at the same rate as the distillate was collected. The distillation was continued until >90% of the dichioromethane was collected. The resulting suspension was then cooled to 20°C and aged for at least 30 min. prior to cooling to 0°C and aging for a further 60 min. The reaction mixture was then filtered and the product washed with additional 2-propanol, before drying at 50°C under vacuum to afford the title compound as an off-white crystalline solid.
The purity of the product was ascertained by HPLC, with identity confirmable by NMR. The yield was consistently >80% in several production runs.
PATENT
Example 1. Preparation of N-cyclohexyl-N-methyl-4-(pyridin-3yl)-lH-imidazole-l-carboxamide

C8H7N3 C15H1 1N302 C16H20N4O
MW 145,16 MW 265,27 MW 284,36
To a suspension of 3-(l/ -imidazol-4-yl)pyridine in tetrahydrofuran (THF) containing pyridine at 25°C was slowly added a solution of phenyl chloroformate in THF over 60 to 90 min. The resulting fine white suspension was then maintained at 25°C for at least 60 min. before the addition of N-methyl- -cyclohexylamine in a single portion, causing the suspension to thin and become yellow in colour. The reaction mixture was then stirred for 90 min. before filtering and washing the filter cake with additional THF. The mother liquors were then maintained at 25°C for at least 18 h, whereupon 65% of the volume of THF was distilled off under atmospheric pressure. The resulting solution was then diluted with 2-propanol and maintained at > 50°C for 10 min. prior to cooling down to 20°C. The resulting suspension was aged at 20°C for 15 min. prior to cooling to 0°C and aging for a further 60 min. The reaction mixture was then filtered and the product was washed with additional 2-propanol, before drying at 50°C under vacuum to afford the title compound as an off-white crystalline solid.
The purity of the product was ascertained by HPLC, with identity confirmable by NMR. The yield was consistently around 50% in several production runs.
Example 2. 3-(l-(cyclohexyl(methyl)carbamoyl-l//-imidazol-4-yl)pyridine 1 -oxide (compound A)

C16H20N4O Ci6H2oN402
MW 284,36 MW 300,36
To a solution of N-cyclohexyl-N-methyl-4-(pyridin-3-yl)-lH-imidazole-l-carboxamide in dichloromethane at 25°C was added peracetic acid (38%; the concentration is not critical, and may be varied) in a single portion. The reaction mixture was then maintained at 25°C for at least 20 h, whereupon the reaction was washed four times with water. The dichloromethane solution was then filtered prior to diluting with 2-propanol. Dichloromethane (50%) was then distilled off under atmospheric pressure, whereupon, 2-propanol was charged at the same rate as the distillate was collected. The distillation was continued until >90% of the dichloromethane was collected. The resulting suspension was then cooled to 20°C and aged for at least 30 min. prior to cooling to 0°C and aging for a further 60 min. The reaction mixture was then filtered and the product washed with additional 2-propanol, before drying at 50°C under vacuum to afford the title compound as an off-white crystalline solid.
The purity of the product was ascertained by HPLC, with identity confirmable by NMR. The yield was consistently >80% in several production runs. It will be appreciated that this gives an overall yield of compound A many times greater than that achieved in the prior art.
In a further run of this synthesis, in a 2L reactor to a mixture of N-cyclohexyl-N-methyl-4-(pyridin-3-yl)-l H- imidazole-l-carboxamide (90 g, 317 mmol) and dichloromethane (1350 ml) was added peracetic acid (84 ml, 475 mmol). The reaction mixture was stirred at 25°C. Completion of the reaction was monitored by HPLC for the disappearance of N-cyclohexyl-N-methyl-4-(pyridin-3-yl)-lH- imidazole- 1-carboxamide. After reaction completion a solution of sodium metabisulfite (60.2 g, 317 mmol) in water (270ml) was added to the reaction mixture maintaining the temperature below 30°C. After phase separation the organic phase was washed with water. After phase separation the organic phase was concentrated at atmospheric pressure until 5 vol. Then solvent was swapped to isopropanol (1350 ml) and the suspension was cooled to 0°C during 4 hours and stirred at that temperature for 1 hour. The resulting solid was collected by filtration and was rinsed with water (270 ml) and isopropanol (270 ml) to afford a white crystalline solid in 84.8g (89%).
PATENT
Preparation of compound 362 a) N-cyclohexyl-N-methyl-4-(pyridin-3-yl)- 1 H-imidazole- 1 -carboxamide

To a stirred suspension of 3-( 1 H-imidazol-4-yl)pyridine dihydrochloride (1.745 g, 8 mmol) in a mixture of tetrahydrofuran (29 mL) and DMF (2.90 mL) was added potassium 2-methylpropan-2-olate (1.795 g, 16.0 mmol) and the mixture was refluxed for 30 minutes. The resulting brown suspension was cooled to room temperature and treated with pyridine (0.979 mL, 12 mmol) and N,N-dimethylpyridin-4-amine (0.098 g, 0.8 mmol), followed by the addition of cyclohexyl(methyl)carbamic chloride (1.476 g, 8.4 mmol). The reaction was heated to 90 0C overnight, whereupon the mixture was diluted with water and extracted with ethyl acetate. The organic phase was dried (MgSO^) and filtered. After evaporation, the crude product was chromatographed over silica gel using a dichloromethane/methanol (9:1) mixture. Homogenous fractions were pooled and evaporated to leave a white powder, (160 mg, 7 %).
b) 3-( 1 -(cyclohexyl(methyl)carbamoyl)- 1 H-imidazol-4-yl)pyridine 1 -oxide

To a stirred solution of N-cyclohexyl-N-methyl-4-(pyridin-3-yl)-l H-imidazole- 1 -carboxamide (90 mg, 0.317 mmol) in chloroform (5 mL) was added 3-chlorobenzoρeroxoic acid (149 mg, 0.475 mmol) in one portion. The reaction was allowed to stir at room temperature for 20 h. TLC showed the reaction to be complete and the mixture was evaporated to dryness. The residue was triturated with ether and the resulting white crystals were filtered off and dried in air. Recrystallisation from hot isopropanol gave a white powder (46 mg, 46 %).
Structure and action
French newspaper Le Figaro has obtained Bial study protocol documents listing the the chemical name of BIA-10-2474 as 3-(1-(cyclohexyl(methyl)carbamoyl)-1H-imidazol-4-yl)pyridine 1-oxide.[8] A Bial news release described BIA-10-2474 as “a long-acting inhibitor of FAAH”.[9]
Fatty acid amide hydrolase (FAAH) is an enzyme which degrades endocannabinoid neurotransmitters like anandamide,[10] which relieves pain and can affect eating and sleep patterns.[11][12] FAAH inhibitors have been proposed for a range of nervous-system disorders including anxiety, alcoholism, pain and nausea.
The Portuguese pharmaceutical company Bial holds several patents on FAAH enzyme inhibitors.[12][13][14][15]
No details of the preclinical testing of this molecule have been made public by the manufacturer Bial. However, the French newspaper Le Figaro has obtained and published an apparently legitimate copy of the full clinical trial protocol (BIA-102474-101).[8] The protocol presents a summary of what appears to be a full package of pharmacodynamic, pharmacokinetic and toxicological studies that might be expected to support a first-in-man study, including safety pharmacology studies in two species (rat, dog) and repeated dose toxicity studies in four species (13 week sub-chronic studies in mouse, rat, dog and monkey). The summary presented however includes no assessment of the relevance of the animal species selected for study (that is, in terms of physiological and genetic similarities with humans and the mechanism of action of the study drug).
Of note, few adverse events were observed in any of the studies, with the 13-week oral No Observed Adverse Effect Level (NOAEL) varying between 10 mg/kg/day in mice to 75 mg/kg/day in monkeys. The authors suggest that these were the maximum doses tested in these studies, though it is not clear. The authors also report no effects of significance in the animal models used for the CNS safety pharmacology studies, which studied a dose of up to 300 mg/kg/day.[8]
Notably absent from the protocol are calculations of receptor occupancy; predictions of in vivo ligand binding saturation levels; measures of target affinity; or assessment of the molecule’s activity in non-target tissues or non-target binding interactions as suggested by the European guidance for Phase I studies,[16] assuming BIA 10-2474 could be considered ‘high risk’).[8]
The trial protocol makes no reference to chimpanzee studies (only monkeys) which contradicts a previous statement to the media in which the French Health Minister stated that the drug had been tested on animals including chimpanzees.[4][17] [18] Some experts had remarked that drug testing in chimpanzees was unlikely.[19]
These findings provide no explanation for the type and severity of events observed in Rennes. In describing the rationale for the starting dose, the authors conclude that:
No target organ was identified during toxicology studies and few adverse clinical findings were observed at the highest dose tested. For the single ascending dose part [of the clinical trial], a starting dose of 0.25 mg was judged to be safe for a first-in-human administration. [8]
The protocol defines no starting dose for the multi-dose treatment groups, noting that this will be based on the outcome of the single dose portion of the trial (an approach known as adaptive trial design). The authors note that nonetheless, the starting dose will not exceed 33% of the maximum tolerated dose (MTD) identified in the single dose groups (or 33% of the maximum administered dose if the MTD is not reached).[8]
Death and serious adverse events during phase I clinical trial
In July 2015 Biotrial, a contract research organization, began testing the drug in a human phase one clinical trial for the manufacturer. The study was approved by French regulatory authority, the Agence Nationale de Sécurité du Médicament (ANSM), on June 26, 2015, and by the Brest regional ethics committee on July 3, 2015.[20] The trial commenced on July 9, 2015,[21] in the city of Rennes, and recruited 128 healthy volunteers, both men and women aged 18 to 55. According to French authorities, the study employed a three-stage design with 90 of the volunteers having received the drug during the first two stages of the trial, with no serious adverse events being reported .[17][20] Participants of the study were to receive €1,900 and, in turn, asked to stay at Biotrial’s facility for two weeks during which time they would take the drug for ten days and undergo tests.[22]
In the third stage of the trial evaluating multiple doses, six male volunteers received doses by mouth, starting on 7 January 2016. The first volunteer was hospitalized at the Rennes University Hospital on January 10, became brain dead,[17][23][24][25] and died on January 17.[26] The other five men in the same dosage group were also hospitalized, in the period of January 10 through January 13[27] four of them suffering injuries including deep hemorrhagic and necrotic lesions seen on brain MRI.[7] The six men who were hospitalised were the group which received the highest dose.[26] A neurologist at the University of Rennes Hospital Center, Professor Pierre-Gilles Edan, stated in a press conference with the French Minister for Health, that 3 of the 4 men who were displaying neurological symptoms “already have a severe enough clinical picture to fear that even in the best situation there will be an irreversible handicap” and were being given corticosteroids to control the inflammation.[27] The sixth man from the group was not showing adverse effects but had been hospitalized for observation.[25][28][29] Biotrial stopped the experiment on January 11, 2016.[4]
No details of the trial have been made public by the manufacturer Bial. The study does not appear in searches of any of the key clinical trial registries, including EudraCT and ClinicalTrials.gov which would normally contain details of approved clinical studies.[30][31][32][33] The trial protocol published by Le Figaro provides extensive detail on what was planned for the study, but many details of the key multi-dose part are not included and were to have been finalised at the conclusion of the single-dose part of the trial.[8]
The French health minister Marisol Touraine called the event “an accident of exceptional gravity” and promised to investigate the matter.[4] On January 18 it was reported authorities were investigating if a manufacturing or transport error might be involved.[34]
Le Figaro posted a 96-page clinical study protocol for BIA 10-2474 that the French newspaper procured from an unnamed source.
According to the document, BIA 10-2474 is 3-(1-(cyclohexyl(methyl)carbamoyl)-1H-imidazol-4-yl)pyridine 1-oxide.
BIA 10-2474 “is designed to act as a long-active and reversible inhibitor of brain and peripheral FAAH,” notes the protocol. The compound “increases anandamide levels in the central nervous system and in peripheral tissues.”
The clinical trial protocol also notes that the company tested BIA 10-2474 on mice, rats, dogs, and monkeys for effects on the heart, kidneys, and gastrointestinal tract, among other pharmacological and toxicological evaluations.

One man is dead and five men were hospitalized after participating in a Phase I clinical trial in Rennes, France
The clinical trial, conducted by the company Biotrial on behalf of the Portuguese pharmaceutical firm Bial, was evaluating a pain relief drug candidate called BIA 10-2474 that inhibits fatty acid amide hydrolase (FAAH) enzymes. Blocking these enzymes prevents them from breaking down cannabinoids in the brain, a family of compounds that includes the euphoria-inducing neurotransmitter anandamide and Δ9-tetrahydrocannabinol, the major psychoactive component of marijuana.
Phase I clinical trials are conducted to check a drug candidate’s safety profile in healthy, paid volunteers. In this case, the drug caused hemorrhagic and necrotic brain lesions in five out of six men in a group who received the highest doses of the drug, said Gilles Edan, a neurologist at the University Hospital Center of Rennes.

The most severely affected man was pronounced brain-dead after hospitalization and then died on Jan. 17. Four men remain in the hospital in stable condition. The only man in the high-dose group who had no adverse symptoms has been released from the hospital.
Clinical trials are an essential part of the drug development process. In order to get life-improving and life-saving medicines to patients, they first have to go through an extensive series of tests. Even before a drug makes it to Phase 1 testing, where its safety, dosage amount, and side effects are tested in a small group of humans, it will undergo testing in animals. As a result, it is not common for a medicine undergoing clinical tests to have a very serious adverse effect on a human. This makes you wonder what happened to a group of patients involved in a clinical study in Rennes, France.
According to news reports, a drug undergoing testing in a French clinic has left one person dead, two others with what may be permanent brain damage, and and two others critically ill. The drug has thus far been unnamed, but it appears to have been produced by the Portuguese company Bial. The French health minister has stated the drug acted on natural receptors found in the body known as endocannibinoids, which regulate mood and appetite. It did not contain cannabis or anything derived from it, as was originally reported. All six trial participants were administered the doses simultaneously.
The trial was being performed at Biotrial, a French-based firm that was formed in 1989 and has conducted thousands of trials. A message on the company’s website stated that they are working with health authorities to understand the cause of the accident, while extending thoughts to the patients and their families. Bial has disclosed the drug was a FAAH (fatty acid amide hydrolase) inhibitor, which is an enzyme produced in the brain and elsewhere that breaks down neurotransmitters called endocannabinoids. Two scientists from the Nottingham Medical School who have worked with FAAH tried over the weekend to try and identify the drug by examining a list of drugs Bial currently has in its pipeline. They believe the culprit is one identified by the codename BIA 10-2474. That same codename appeared on a recruitment form that was given to a volunteer, which was published in a French newspaper. Little more is known about it, and there does not appear to be any entry for it in clinical trial registries.
The French health ministry is reporting the six patients were all in good health prior to taking the oral medicine, which was administered to 90 volunteers. The trial recruited 128 individuals, and the remaining participants received a placebo. Health minister Marisol Touraine, describing the situation as a very serious accident, noted the patients were taking part in a trial in Brittany, Rennes involving a medicine developed by a “European laboratory”, refusing to comment further until additional information became available. She has also asked the Inspector General of Social Affairs to lead an investigation into the circumstances around the trial, which has obviously been suspended. She notes the drug had been tested on animals, including chimpanzees. France’s National Agency for Medicine and Health Products Safety approved the trial on in June 2015.
One thing we do know is that the trial was a Phase 1 clinical study that included 90 healthy volunteers. Regulations that oversee all clinical trials in Europe do attempt to minimize the risk associated with trials, but there is always a risk involved with administering an unapproved medicine to humans. At this time the chief neuroscientist at the hospital where the patients are being treated has said there is no known antidote for the drug.
The drug, administered to men between the ages of 28 and 49, was intended to treat mood disorders such as anxiety. While the men were administered varying doses, the patients who are hospitalized were taking the drug “regularly”.

Old 2006 case
While safety issues like this are rare, they are not unheard of. In 2006, a clinical trial in London left six men ill. All were taking part in a study testing a drug designed to fight auto-immune disease and leukemia. Within hours of taking the drug TGN1412, all experienced a serious reaction, were admitted to intensive care, and had to be treated for organ failure. Two became critically ill, with one eventually losing all of his fingers and toes. All were told they would have a higher risk of developing cancers or auto-immune diseases.
This of course led many to wonder about the future of trials, and whether the situation could happen again. The Duff Report, written in response to the TGN1412 trial, noted the medicine should have been tested in one person at a time. It also helped to put additional safety measures in place. The Medicines and Health Products Regulatory Agency (MHRA) now requires committees to look at pre-clinical data to determine the proper initial dose, and rules are in place to stop the trial if unintended reactions occur.
However, since patients can fall ill immediately after being administered a medication, certain risks will still exist.
The company that manufactured TGN1412, TeGenero Immuno Therapeutics, later went bankrupt. However the drug was later purchased by a Russian investor and renamed TABO8. TheraMAB, a Russian biotech company, then conducted a new trial of the drug in a much lower dose. A later Phase 2 study was started in patients with Rheumatoid Arthritis.
Other pharmaceutical companies, including Merck, Pfizer, Johnson & Johnson, Sanofi and Vernalis, have previously taken other FAAH inhibitors into clinical trials without experiencing such adverse events (e.g. respectively, MK-4409,[35][36] PF-04457845, JNJ-42165279,[37] SSR411298 and V158866.[38][39] Related enzyme inhibitor compounds such as URB-597 and LY-2183240 have been sold illicitly as designer drugs,[40][41] all without reports of this type of toxicity emerging, so the mechanism of the toxicity observed with BIA 10-2474 remains poorly understood.
Following the events in Rennes, Janssen announced that it was temporarily suspending dosing in two Phase II clinical trials with its own FAAH inhibitor JNJ-42165279, headlining the decision as “precautionary measure follows safety issue with different drug in class”. Janssen was emphatic that no serious adverse events had been reported in any of the clinical trials with JNJ-42165279 to date. The suspension is to remain in effect until more information is available about the BIA 10-2474 study.[42]
References
- 1 “BRIEF-Bial says firmly committed to ensure wellbeing of test participants”. Reuters. 15 January 2016.
- 2 “BIA 10-2474”. Biocentury.com. Retrieved 2016-01-17.
- 3 “BIA 102474”. Adisinsight/springer.com. Retrieved 2016-01-17.
- 4 Adamson B (January 15, 2016). “Botched Drug Trial Leaves 1 Brain Dead, 5 in Hospital”. ABC, AP. Retrieved January 16, 2016.
- 5 Angeline Benoit,Makiko Kitamura (15 January 2016). “France Ties Brain-Dead Person to Tests of Bial-Portela Drug”. Bloomberg.com.
- 6 “France/Monde – Essai thérapeutique : 90 personnes ont pris la molécule”. Ledauphine.com. Retrieved 2016-01-17.
- 7 Enserink, Martin (2016). “More Details Emerge on Fateful French Drug Trial” (online). Science (January 16). Retrieved 16 January 2016.
- 8 “Drame de Rennes : le protocole de l’essai clinique en accusation”. sante.lefigaro.fr. Retrieved 2016-01-21.
- 9 “News Release – Phase I Clinical Trial Rennes”. http://www.bial.com. Retrieved 2016-01-21.
- 10 Debora Mackenzie. “Six in hospital after French pain relief drug trial goes wrong”. New Scientist.
- 11 “The Discovery and Development of Inhibitors of Fatty Acid Amide Hydrolase (FAAH)”. PubMed Central.
- 12 “Patent WO2015012708A1 – Imidazolecarboxamides and their use as faah inhibitors – Google Patents”. Google.com. Retrieved 2016-01-17.
- 13 “Patent WO2015016729A1 – Urea compounds and their use as faah enzyme inhibitors”. Google.com. Retrieved 2016-01-17.
- 14 “PT2014000052 UREA COMPOUNDS AND THEIR USE AS FAAH ENZYME INHIBITORS”. Patentscope.wipo.int. Retrieved 2016-01-17.
- 15 WO application 2015016729, Laszlo Erno KISS, Rita GUSMÃO DE NORONHA, Carla Patrícia ROSA DA COSTA PEREIRA, Rui PINTO, “Urea compounds and their use as faah enzyme inhibitors”, published Feb 5, 2015, assigned to BIAL
- 16″Strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products (CHMP/SWP/28367/07)” (PDF). European Medicines Agency. 1 September 2007. Retrieved 22 January 2016.
- 17 “Accident grave dans le cadre d’un essai clinique – Intervention de Marisol Touraine à Rennes”. Ministère des Affaires Sociales, de la Santé et des Droits des Femmes, France. 15 January 2016.
- 18Barbara Casassus (23 January 2016). “France investigates drug trial disaster” (PDF). The Lancet 387 (10016): 326. doi:10.1016/S0140-6736(16)00154-9.
- 19
- “Expert reaction to French drug trial – reports of one patient dying and five others in hospital and of the Paris prosecutor’s office having opened an investigation into what happened”. Science Media Centre, London. 16 January 2016. Retrieved 21 January 2016.
- 20
- “La survenue d’effets graves ayant entraîné l’hospitalisation de 6 patients, dont un en état de mort cérébrale, a conduit à l’arrêt prématuré d’un essai clinique du laboratoire BIAL – Point d’information”. Agence Nationale de Sécurité du Médicament, France (ANSM). 15 January 2016.
- 21
- “Six hospitalized in Bial clinical trial in France”. BioWorld.com. Retrieved 2016-01-17.
- 22
- Enserink M (January 16, 2016). “More details emerge on fateful French drug trial”. Science Magazine. Retrieved January 18, 2016.
- 23
- “France clinical trial: 90 given drug, one man brain-dead”. BBC. January 15, 2016. Retrieved January 16, 2016.
- 24
- “Ce que l’on sait de l’accident survenu lors d’un essai clinique à Rennes”, Le Monde, 15 January 2016
- 25
- Blamont M (January 15, 2016). “French drug trial disaster leaves one brain dead, five injured”. Reuters. Retrieved January 16, 2016.
- 26
- Clarisse Lucas, AFP (January 17, 2016). “Man dies after being left brain-dead in French drug trial”. Yahoo. Retrieved January 17, 2016.
- 27
- “Accident “inédit” lors d’un essai clinique: un homme en état de mort cérébrale, cinq hospitalisés”. La Depeche. January 15, 2016. Retrieved January 18, 2016.
- 28
- “France clinical trial: ‘No known antidote’ to drug”. BBC News. 15 January 2016. Retrieved 18 January 2016.
- 29
- Enserink, Martin (2016). “What We Know So Far About the Clinical Trial Disaster in France” (online). Science (January 15). Retrieved 16 January 2016.
- 30
- “EU Clinical Trials Register”. European Medicines Agency. Retrieved 20 January 2016.
- 31
- “WHO International Clinical Trials Registry Platform”. World Health Organization. Retrieved 20 January 2016.
- 32
- “ANSM – Répertoire public des essais cliniques de médicaments”. Agence Nationale de Sécurité du Médicament et des Produits de Santé, France. Retrieved 20 January 2016.
- 33
- “ClinicalTrials.gov Registry”. U.S. National Institutes of Health. Retrieved 20 January 2016.
- 34
- “Drug trial volunteer dies as ‘manufacturing error’ theory investigated”. The Independent. January 18, 2016. Retrieved January 18, 2016.
- 35
- Chobanian; et al. (10 April 2014). “Discovery of MK-4409, a Novel Oxazole FAAH Inhibitor for the Treatment of Inflammatory and Neuropathic Pain”. ACS Med. Chem. Lett.
- 36
- Merck (15 October 2009). “Merck Pipeline, Oct 2009” (PDF). Merck.
- 37
- “Seven studies found for: jnj-42165279”. Clinicaltrials.gov. Retrieved 2016-01-19.
- 38
- “2 studies found for: V158866”. Clinicaltrials.gov. Retrieved 2016-01-19.
- 39
- Bisogno T, Maccarrone M. Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin Drug Discov. 2013 May;8(5):509-22. PMID 23488865 doi: 10.1517/17460441.2013.780021.
- 40
- Shanks KG, Behonick GS, Dahn T, Terrell A. Identification of novel third-generation synthetic cannabinoids in products by ultra-performance liquid chromatography and time-of-flight mass spectrometry. J Anal Toxicol. 2013 Oct;37(8):517-25. PMID 23946450 doi: 10.1093/jat/bkt062.
- 41
- Uchiyama N, Matsuda S, Kawamura M, Shimokawa Y, Kikura-Hanajiri R, Aritake K, Urade Y, Goda Y. Characterization of four new designer drugs, 5-chloro-NNEI, NNEI indazole analog, α-PHPP and α-POP, with 11 newly distributed designer drugs in illegal products. Forensic Sci Int. 2014 Oct;243:1-13. PMID 24769262 doi: 10.1016/j.forsciint.2014.03.013.
- “Janssen Research & Development, LLC Voluntarily Suspends Dosing in Phase 2 Clinical Trials of Experimental Treatment for Mood Disorders”. Janssen.com. 17 January 2016. Retrieved 21 January 2016.
External links
| WO2005073199A1 * | Jan 15, 2005 | Aug 11, 2005 | Aventis Pharma Gmbh | Indazole derivatives as inhibitors of hormone-sensitive lipases |
| WO2010074588A2 | Dec 23, 2009 | Jul 1, 2010 | BIAL – PORTELA & Cª, S.A. | Pharmaceutical compounds |
| WO2012015324A1 | Jul 28, 2011 | Feb 2, 2012 | Bial – Portela & Ca, S.A. | Process for the synthesis of substituted urea compounds |
| US4051252 * | Nov 24, 1975 | Sep 27, 1977 | Bayer Aktiengesellschaft | 3-aminoindazole-1 and 2-carboxylic acid derivatives |
| US4331678 * | Jan 14, 1980 | May 25, 1982 | Fbc Limited | Carbamoyl pyrazole compounds and their pesticidal application |
| US4973588 * | Feb 10, 1989 | Nov 27, 1990 | Mitsui Petrochemical Industries, Ltd. | Imidazole derivatives having anti-hypoxia properties |
| US5578627 * | Oct 27, 1993 | Nov 26, 1996 | Toyama Chemical Co., Ltd. | 1,2-benzoisoxazole derivative or its salt and brain-protecting agent comprising the same |
 |
|||||
| Systematic (IUPAC) name | |||||
|---|---|---|---|---|---|
|
3-(1-(cyclohexyl(methyl)carbamoyl)-1H-imidazol-4-yl)pyridine 1-oxide
|
|||||
| Clinical data | |||||
| Legal status |
|
||||
| Routes of administration |
Oral | ||||
| Identifiers | |||||
| PubChem | CID: 46831476 | ||||
| Chemical data | |||||
| Formula | C16H20N4O2
|
||||
/////////
C1C(CCCC1)N(C)C(=O)n2cc(nc2)c3ccc[n+](c3)O
SB 1578

SB1578
ONX 0805
(9E)-15-(2-(Pyrrolidin-1-yl)ethoxy)-7,12,25-trioxa-19,21,24-triaza-tetracyclo[18.3.1.1(2,5).1(14,18)]hexacosa-1(24),2,4,9,14(26), 15,17,20,22-nonaene
7,12,26-Trioxa-19,21,24-triazatetracyclo[18.3.1.12,5.114,18]hexacosa-1(24),2,4,9,14,16,18(25),20,22-nonaene, 15-[2-(1-pyrrolidinyl)ethoxy]-, (9E)-
Phase 1 clinical trials
C26 H30 N4 O4
CAS 937273-04-6
CITRATE 1262279-15-1
HCL 1262279-16-2
S*Bio Pte Ltd INNOVATOR
SB1578 (disclosed in WO2007058627 and in WO2011008172 as the citrate salt) is in ongoing phase I studies for the treatment of rheumatoid arthritis. SB 1578 is shown below.

SB1578, also known as ONX-0805, is a novel, orally bioavailable JAK2 inhibitor with specificity for JAK2 within the JAK family and also potent activity against FLT3 and c-Fms. SB1578 blocks the activation of these kinases and their downstream signaling in pertinent cells, leading to inhibition of pathological cellular responses. The biochemical and cellular activities of SB1578 translate into its high efficacy in two rodent models of arthritis. SB1578 not only prevents the onset of arthritis but is also potent in treating established disease in collagen-induced arthritis mice with beneficial effects on histopathological parameters of bone resorption and cartilage damage. SB1578 abrogates the inflammatory response and prevents the infiltration of macrophages and neutrophils into affected joints. It also leads to inhibition of Ag-presenting dendritic cells and inhibits the autoimmune component of the disease. In summary, SB1578 has a unique kinase spectrum, and its pharmacological profile provides a strong rationale for the ongoing clinical development in autoimmune diseases. ( J Immunol. 2012 Oct 15;189(8):4123-34)
Synonym: ONX 0805; ONX0805; ONX0805; SB1578; SB1578; SB 1578.

PATENT
WO 2011008172
http://www.google.im/patents/WO2011008172A1?cl=en
The compound 9E-15-(2-pyrrolidin-1-yl-ethoxy)-7,12,25-trioxa-19,21 ,24-triaza-tetracyclo[18.3.1.1(2,5).1(14,18)]hexacosa-1 (24),2,4,9,14,16l18(26)l20,22-nonaene (Compound I) was first described in PCT/SG2006/000352 and shows significant promise as a pharmaceutically active agent for the treatment of a number of medical conditions. Pharmaceutical development of this compound is underway based on the activity profiles demonstrated by the compound.

Compound I
In the development of a drug suitable for mass production and ultimately commercial use acceptable levels of drug activity against the target of interest is only one of the important variables that must be considered. For example, in the formulation of pharmaceutical compositions it is imperative that the pharmaceutically active substance be in a form that can be reliably reproduced in a commercial
manufacturing process and which is robust enough to withstand the conditions to which the pharmaceutically active substance is exposed.
From a manufacturing perspective, it is important that the commercial manufacturing process of a pharmaceutically active substance is such that the same material is produced when the same manufacturing conditions are used. In addition, it is desirable that the pharmaceutically active substance exists in a solid form where minor changes to the manufacturing conditions do not lead to major changes in the solid form of the pharmaceutically active substance produced. For example, it is important that the manufacturing process produces material having the same crystalline properties on a reliable basis, and also that the process produces material having the same level of hydration.
In addition, it is important that the pharmaceutically active substance be stable to degradation, hygroscopicity and subsequent changes to its solid form. This is important to facilitate the incorporation of the pharmaceutically active ingredient into pharmaceutical formulations. If the pharmaceutically active substance is hygroscopic (“sticky”) in the sense that it absorbs water over time it is almost impossible to reliably formulate the pharmaceutically active substance into a drug as the amount of substance to be added to provide the same dosage will vary greatly depending upon the degree of hydration. Furthermore, variations in hydration or solid form (“polymorphism”) can lead to changes in physico-chemical properties, such as solubility or dissolution rate, which can in turn lead to inconsistent oral absorption in a patient.
Accordingly, chemical stability, solid state stability, and “shelf life” of the pharmaceutically active agent are very important factors. In an ideal situation the pharmaceutically active agent and any compositions containing it, should be capable of being effectively stored over appreciable periods of time without exhibiting a significant change in the physico-chemical characteristics of the active component such as its activity, moisture content, solubility characteristics, solid form and the like.
In relation to 9E-15-(2-pyrrolidin-1-yl-ethoxy)-7,12,25-trioxa-19,21 ,24-triaza-tetracyclo[18.3.1.1 (2,5).1(14,18)]hexacosa-1(24),2,4,9,14,16,18(26),20,22-nonaene
initial studies were carried out on the hydrochloride salt and indicated that polymorphism was prevalent, with the compound being found to adopt more than one crystalline form depending upon the manufacturing conditions. In addition it was observed that the ratio of the polymorphs varied from batch to batch even when the manufacturing conditions remained constant. These batch-to-batch inconsistencies made the hydrochloride salt less desirable from a commercial viewpoint.
Accordingly it would be desirable to develop salts of 9E-15-(2-pyrrolidin-1-yl-ethoxy)-7, 12,25-trioxa-i 9,21 ,24-triaza-tetracyclo[18.3.1.1 (2,5).1 (14,18)]hexacosa-1(24)l2,4,9,14,16,18(26),20,22-nonaene which overcome or ameliorate one or more of the above identified problems.
Figure 22 shows a 1H NMR spectrum for Batch 4 in d6-DMSO.
Figure 23 shows a 1H NMR spectrum for Batch 4 in D2O.
List of hydrochloride and citrate salt batches used for comparative studies

Example 4 – Formation of the Citrate salt (Batch 4) in THF as solvent:
The free base of compound 1 (0.30Og, 0.648mmoles, 1.eq) was added to 12mL of THF. The solution was heated to reflux until complete dissolution was observed and maintained for 1h. A solution of citric acid (0.149g, 0.778mmoles, 1.2eq) dissolved in 12mL THF was then added slowly at reflux conditions. The mixture was refluxed for a further 15min then cooled. Crystallization was observed on gradual cooling. The crystals were stirred at room temperature for 12h and filtered under vacuum. The product was dried under vacuum to afford 250mg.
PATENT
http://www.google.im/patents/WO2007058627A1?cl=en
Representative procedure for the synthesis of compounds type (XVIIIf)
5-(2-Chloro-pyrimidin-4-yl)-furan-2-carbaldehyde (XIIIfI) 
(XIIfI) (XIIIH) .
Compound (XIIIfI) was obtained using the same procedure described for compound (XIIIeI); LC-MS (ESI positive mode) /τVz 209 ([M+H]+)
[5-(2-Chloro-pyrimidin-4-yl)-furan-2-yl]-methanol (Xlllf2)

Compound (Xlllf2) was obtained using the same procedure described for compound (XXIb); LC-MS (ESI positive mode) m/z 211 ([M+H]+).
4-(5-Allyloxymethyl-furan-2-yl)-2-chloro-pyrimidine (XVfI)

Compound (XVfI) was obtained using the same procedure described for compound (XXIIb); LC-MS (ESI positive mode) m/z 251 ([M+H]+).
^-(S-Allyloxymethyl-furan-Σ-yO-pyrimidin^-yll-IS-allyloxymethyl^^-pyrrolidin-i-yl- ethoxy)-phenyl]-amine (XVIIfI)


(XVIb2) (XVIIfI)
Compound (XVIIfI) was obtained using the same procedure described for compound (XVIIbI); LC-MS (ESI positive mode) m/z 491.
Macrocycle Example 6 (Compound 38)

(XVIIfI)

Compound (38) was obtained using the same procedure described for compound (1) HPLC purity at 254nm: 99%; LC-MS (ESI positive mode) m/z 463 ([M+H]+); 1H NMR (MeOD-d4) δ 8.90 (d, 1 H), 8.33 (d, 1 H), 7.37 (d, 1 H), 7.17 (d, 1 H), 7.14-7.11 (m, 1 H)1 7.04 (d, 1 H), 6.67 (d, 1 H), 6.04 (dt, 1 H, CH, J = 5.2Hz, Jtrans = 15.8Hz), 5.96 (dt, 1 H, CH, J = 5.0Hz, Jtrans = 15.8Hz), 4.65 (s, 2H), 4.62 (s, 2H), 4.37 (t, 2H), 4.14 (d, 2H), 4.09 (d, 2H), 3.81 (br s, 2H), 3.66 (t, 2H), 3.33 (s, 2H), 2.21-1.98 (m, 4H).

PAPER
Discovery of the Macrocycle (9E)-15-(2-(Pyrrolidin-1-yl)ethoxy)-7,12,25-trioxa-19,21,24-triaza-tetracyclo[18.3.1.1(2,5).1(14,18)]hexacosa-1(24),2,4,9,14(26),15,17,20,22-nonaene (SB1578), a Potent Inhibitor of Janus Kinase 2/Fms-LikeTyrosine Kinase-3 (JAK2/FLT3) for the Treatment of Rheumatoid Arthritis
http://pubs.acs.org/doi/abs/10.1021/jm201454n
Herein, we describe the synthesis and SAR of a series of small molecule macrocycles that selectively inhibit JAK2 kinase within the JAK family and FLT3 kinase. Following a multiparameter optimization of a key aryl ring of the previously described SB1518 (pacritinib), the highly soluble 14l was selected as the optimal compound. Oral efficacy in the murine collagen-induced arthritis (CIA) model for rheumatoid arthritis (RA) supported 14l as a potential treatment for autoimmune diseases and inflammatory disorders such as psoriasis and RA. Compound 14l (SB1578) was progressed into development and is currently undergoing phase 1 clinical trials in healthy volunteers.
(9E)-15-(2-(Pyrrolidin-1-yl)ethoxy)-7,12,25-trioxa-19,21,24-triaza-tetracyclo[18.3.1.1(2,5).1(14,18)]hexacosa-1(24),2,4,9,14(26), 15,17,20,22-nonaene (14l)
REF
Madan B, Goh KC, Hart S, William AD, Jayaraman R, Ethirajulu K, Dymock BW, Wood JM. SB1578, a novel inhibitor of JAK2, FLT3, and c-Fms for the treatment of rheumatoid arthritis. J Immunol. 2012 Oct 15;189(8):4123-34. doi: 10.4049/jimmunol.1200675. Epub 2012 Sep 7. PubMed PMID: 22962687.
2: Poulsen A, William A, Blanchard S, Lee A, Nagaraj H, Wang H, Teo E, Tan E, Goh KC, Dymock B. Structure-based design of oxygen-linked macrocyclic kinase inhibitors: discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3). J Comput Aided Mol Des. 2012 Apr;26(4):437-50. doi: 10.1007/s10822-012-9572-z. Epub 2012 Apr 22. PubMed PMID: 22527961.
3: William AD, Lee AC, Poulsen A, Goh KC, Madan B, Hart S, Tan E, Wang H, Nagaraj H, Chen D, Lee CP, Sun ET, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW. Discovery of the macrocycle (9E)-15-(2-(pyrrolidin-1-yl)ethoxy)-7,12,25-trioxa-19,21,24-triaza-tetracyclo[18. 3.1.1(2,5).1(14,18)]hexacosa-1(24),2,4,9,14(26),15,17,20,22-nonaene (SB1578), a potent inhibitor of janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) for the treatment of rheumatoid arthritis. J Med Chem. 2012 Mar 22;55(6):2623-40. doi: 10.1021/jm201454n. Epub 2012 Mar 6. PubMed PMID: 22339472.
| WO2007058627A1 * | 15 Nov 2006 | 24 May 2007 | S Bio Pte Ltd | Oxygen linked pyrimidine derivatives |
| SG2006000352W | Title not available |






AUTHOR’S
• Principle lead and inventor of 3 clinical stage candidates,
1) SB1518 (Pacritinib)-A selective JAK2 inhibitor for myleofibrosis into phase 2,
2) SB1317 (TG02)-A mutikinase inhibitor CDK, JAK2, FLT3, and ERK5 into phase 1 and
3) SB1578-A more selective JAK2 inhibitor than pracritinib for autoimmune diseases such as Rheumatoid Arthritis (RA) and Psoriasis into phase 1
NEXT………..

Babita Madan
DUKE NUS Graduate Medical School
Experience
Asst. Professor
Duke NUS Graduate Medical Centre
December 2011 – Present (4 years 2 months)Singapore
Scientist
S*BIO Pte Ltd
January 2010 – October 2011 (1 year 10 months)Singapore
Senior Research Fellow
University Clinics Ulm, Germany
November 2002 – December 2008 (6 years 2 months)
Education

Dr. Babita Madan,
Scientist,
S*BIO Pte Ltd,
Singapore.

SEE……..http://apisynthesisint.blogspot.in/2016/01/sb1578-onx-0805.html
///////
N3=C1NC(=CC=N1)c2oc(cc2)COCC=CCOCc5cc3ccc5OCCN4CCCC4
OR
C1(C2=CC=C(O2)COC/C=C/COCC3=CC(N4)=CC=C3OCCN5CCCC5)=NC4=NC=C1

{kind=link}