

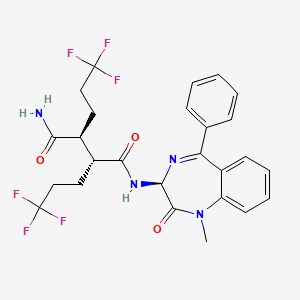





















Crystallization-Induced Dynamic Resolution toward the Synthesis of (S)-7-Amino-5H,7H-dibenzo[b,d]-azepin-6-one: An Important Scaffold for γ-Secretase Inhibitors
Home » PHASE 1 (Page 7)
Category Archives: PHASE 1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BMS 906024


BMS 906024
cas 1401066-79-2
- MF C26H26F6N4O3
- MW 556.500
(2R,3S)-N-[(3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)succinamide
Butanediamide, N1-((3S)-2,3-dihydro-1-methyl-2-oxo-5-phenyl-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluorophenyl)-, (2R,3S)-
(2R,35)-N-((35)-l-Methyl-2-oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-3- (2,2,2-trifluoroethyl)-2-(3,3,3-trifluoropropyl)succinamide
| Claude Quesnelle, Soong-Hoon Kim, Francis Lee, Ashvinikumar Gavai | |
| Applicant | Bristol-Myers Squibb Company |


Claude Quesnelle
Senior Research Investigator/Chemist at Bristol-Myers Squibb

RICHARD LEE
BMS-906024 is a novel, potent Notch receptor inhibitor . Cancers have a tendency to relapse or to become resistant to treatments that once worked. A family of proteins called Notch is implicated in that resistance and in cancer progression more generally. BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.
New Phase I drug structure by Bristol-Myers Squibb disclosed at the spring 2013 American Chemical Society meeting in New Orleans to treat breast, lung, and colon cancers and leukemia.[1] The drug works as an pan-Notch inhibitor. The structure is one of a set patented in 2012,[2] and it currently being studied in clinical trials.[3][4]
useful for the treatment of conditions related to the Notch pathway, such as cancer and other proliferative diseases.
Notch signaling has been implicated in a variety of cellular processes, such as cell fate specification, differentiation, proliferation, apoptosis, and angiogenesis. (Bray, Nature Reviews Molecular Cell Biology, 7:678-689 (2006); Fortini, Developmental Cell 16:633-647 (2009)). The Notch proteins are single-pass heterodimeric transmembrane molecules. The Notch family includes 4 receptors, NOTCH 1-4, which become activated upon binding to ligands from the DSL family (Delta-like 1, 3, 4 and Jagged 1 and 2).
The activation and maturation of NOTCH requires a series of processing steps, including a proteolytic cleavage step mediated by gamma secretase, a multiprotein complex containing Presenilin 1 or Presenilin 2, nicastrin, APH1, and PEN2. Once NOTCH is cleaved, NOTCH intracellular domain (NICD) is released from the membrane. The released NICD translocates to the nucleus, where it functions as a transcriptional activator in concert with CSL family members (RBPSUH, “suppressor of hairless”, and LAG1). NOTCH target genes include HES family members, such as HES- 1. HES- 1 functions as transcriptional repressors of genes such as HERP 1 (also known as HEY2), HERP2 (also known as HEY1), and HATH1 (also known as ATOH1).
The aberrant activation of the Notch pathway contributes to tumorigenesis. Activation of Notch signaling has been implicated in the pathogenesis of various solid tumors including ovarian, pancreatic, as well as breast cancer and hematologic tumors such as leukemias, lymphomas, and multiple myeloma. The role of Notch inhibition and its utility in the treatment of various solid and hematological tumors are described in Miele, L. et al, Current Cancer Drug Targets, 6:313-323 (2006); Bolos, V. et al, Endocrine Reviews, 28:339-363 (2007); Shih, I.-M. et al, Cancer Research, 67: 1879- 1882 (2007); Yamaguchi, N. et al., Cancer Research, 68: 1881-1888 (2008); Miele, L., Expert Review Anti-cancer Therapy, 8: 1 197-1201 (2008); Purow, B., Current Pharmaceutical Biotechnology, 10: 154-160 (2009); Nefedova, Y. et al, Drug Resistance Updates, 1 1 :210-218 (2008); Dufraine, J. et al, Oncogene, 27:5132-5137 (2008); and Jun, H.T. et al, Drug Development Research, 69:319-328 (2008).
There remains a need for compounds that are useful as Notch inhibitors and that have sufficient metabolic stability to provide efficacious levels of drug exposure. Further, there remains a need for compounds useful as Notch inhibitors that can be orally or intravenously administered to a patient.
U.S. Patent No. 7,053,084 Bl discloses succinoylamino benzodiazepine compounds useful for treating neurological disorders such as Alzheimer’s Disease. The reference discloses that these succinoylamino benzodiazepine compounds inhibit gamma secretase activity and the processing of amyloid precursor protein linked to the formation of neurological deposits of amyloid protein. The reference does not disclose the use of these compounds in the treatment of proliferative diseases such as cancer.
Applicants have found potent compounds that have activity as Notch inhibitors and have sufficient metabolic stability to provide efficacious levels of drug exposure upon intravenous or oral administration. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
![]()

PAPER

Structure–activity relationships in a series of (2-oxo-1,4-benzodiazepin-3-yl)-succinamides identified highly potent inhibitors of γ-secretase mediated signaling of Notch1/2/3/4 receptors. On the basis of its robust in vivo efficacy at tolerated doses in Notch driven leukemia and solid tumor xenograft models, 12 (BMS-906024) was selected as a candidate for clinical evaluation.
Discovery of Clinical Candidate BMS-906024: A Potent Pan-Notch Inhibitor for the Treatment of Leukemia and Solid Tumors
†Bristol-Myers Squibb Research and Development, Princeton, New Jersey 08543, United States
‡Bristol-Myers Squibb Research and Development, 5 Research Parkway, Wallingford, Connecticut 06492, United States
§ Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037,United States
ACS Med. Chem. Lett., 2015, 6 (5), pp 523–527
DOI: 10.1021/acsmedchemlett.5b00001, http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00001
*Phone: 609-252-5091. E-mail: ashvinikumar.gavai@bms.com.
(2R,3S)-N-((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4- benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
colorless solid: HPLC: RT = 9.60 min (HPLC Method D). Chiral LC/Analytical SFC conditions: Column: LuxCellulose-2 (0.46 x 25cm), Mobile phase: 10% methanol in CO2, Flow rate: 3 mL/min, wavelength: 220 nm; Temp.: 35C. RT = 9.21 min, Purity = 99.95%.
MS (ES): m/z = 557 [M+H]+ ;
1H NMR (400 MHz, DMSO-d6) 9.54 (1H, d, J = 7.28 Hz), 7.71 – 7.80 (1H, m), 7.68 (2H, d, J = 8.78 Hz), 7.50 – 7.62 (3H, m), 7.45 (2H, t, J = 7.28 Hz), 7.29 – 7.40 (2H, m), 7.15 (1H, s), 5.30 (1H, d, J = 7.28 Hz), 3.39 (3H, s), 2.74 – 2.86 (1H, m), 2.02 -2.32 (3H, m), 1.45 – 1.79 (4H, m);
[]D = -107.0° (5.73 mg/mL, DMSO).
Elemental analysis: Theoretical: C: 54.11%; H: 4.70%; N: 10.06%; Actual: C: 54.06%; H: 4.90%; N: 10.08%.
Karl Fisher Moisture: 0.48.
HPLC Method D: Sunfire C18 3.5um, 3.0x150mm column, solvent A: 5% acetonitrile – 95% water – 0.05% TFA, solvent B: 95% acetonitrile – 5% water – 0.05% TFA, flow=0.5 mL/min, gradient from 10%B to 100%B over 15min, 254 nm detector.

Patent
http://www.google.co.in/patents/WO2012129353A1?cl=en
Example 1
(2R,35)-N-((35′)-l-Methyl-2-oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-2,3- b -trifluoropropy l)succinamide
Preparation 1A: tert-Butyl 5, -trifluoropentanoate

[00219] To a stirred solution of 5,5,5-trifluoropentanoic acid (5 g, 32.0 mmol) in THF (30 mL) and hexane (30 mL) at 0 °C, was added tert-butyl 2,2,2-trichloroacetimidate (11.46 mL, 64.1 mmol). The mixture was stirred for 15 min at 0 °C. Boron trifluoride etherate (0.406 mL, 3.20 mmol) was added and the reaction mixture was allowed to warm to room temperature overnight. To the clear reaction mixture was added solid aHC03 (5 g) and stirred for 30 min. The mixture was filtered through MgS04 and washed with hexanes (200 mL). The solution was allowed to rest for 45 min, and the resulting solid material was removed by filtering on the same MgS04 filter again, washed with hexanes (100 mL) and concentrated under reduced pressure without heat. The volume was reduced to about 30 mL, filtered through a clean fritted funnel, washed with hexane (5 mL), and then concentrated under reduced pressure without heat. The resulting neat oil was filtered through a 0.45μηι nylon membrane filter disk to provide tert-butyl 5,5,5- trifluoropentanoate (6.6 g, 31.4 mmol 98% yield) as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 1.38 (s, 9 H) 1.74-1.83 (m, 2 H) 2.00-2.13 (m, 2 H) 2.24 (t, J=7.28 Hz, 2 H).
Preparation IB: (45)-4-(Propan-2- l)-3-(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00220] To a stirred solution of 5,5,5-trifluoropentanoic acid (5.04 g, 32.3 mmol) in DCM (50 mL) and DMF (3 drops) was added oxalyl chloride (3.4 mL, 38.8 mmol) dropwise over 5 min and the solution was stirred until all bubbling subsided. The reaction mixture was concentrated under reduced pressure to give pale yellow oil. To a separate flask charged with a solution of (45)-4-(propan-2-yl)-l,3-oxazolidin-2-one (4.18 g, 32.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (2.5M in hexane) (13.0 mL, 32.5 mmol) dropwise via syringe over 5 min. After stirring for 10 min, the above acid chloride dissolved in THF (20 mL) was added via cannula over 15 min. The reaction mixture was warmed to 0 °C, and was allowed to warm to room temperature as the bath warmed and stirred overnight. To the reaction mixture was added saturated NH4CI, and then extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The crude material was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 60% solvent A/B=hexanes/EtOAc, REDISEP® S1O2 120g). Concentration of appropriate fractions provided Preparation IB (7.39 g, 86%) as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 4.44 (1 H, dt, J=8.31, 3.53 Hz), 4.30 (1 H, t, J=8.69 Hz), 4.23 (1 H, dd, J=9.06, 3.02 Hz), 2.98-3.08 (2 H, m), 2.32-2.44 (1 H, m, J=13.91, 7.02, 7.02, 4.03 Hz), 2.13-2.25 (2 H, m), 1.88-2.00 (2 H, m), 0.93 (3 H, d, J=7.05 Hz), 0.88 (3 H, d, J=6.80 Hz). Preparation 1C: (25′,3R)-tert-Butyl 6,6,6-trifluoro-3-((5)-4-isopropyl-2-oxooxazolidine- 3 -carbonyl)-2-(3 ,3,3 -trifluoropropyl)hexanoate, and
Preparation ID: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((5)-4-isopropyl-2-oxooxazolidine- 3 -carbonyl)- -(3 ,3 ,3 -trifluoropropyl)hexanoate

(1 C) (1 D)
[00221] To a cold (-78 °C), stirred solution of diisopropylamine (5.3 mL, 37.2 mmol) in THF (59 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexane) (14.7 mL, 36.8 mmol), then warmed to 0 °C to give a 0.5M solution of LDA. A separate vessel was charged with Preparation IB (2.45 g, 9.17 mmol), the material was azeotroped twice with benzene (the RotoVap air inlet was fitted with nitrogen inlet to completely exclude humidity) then toluene (15.3 mL) was added. This solution was added to a flask containing dry lithium chloride (1.96 g, 46.2 mmol). To the resultant mixture, cooled to -78 °C, was added LDA solution (21.0 mL, 10.5 mmol) and stirred at -78 °C for 10 min, warmed to 0 °C for 10 min then recooled to -78 °C. To a separate reaction vessel containing Preparation 1A (3.41 g, 16.07 mmol), also azeotroped twice with benzene, was added toluene (15.3 mL), cooled to -78 °C and LDA (37.0 mL, 18.5 mmol) was added, the resulting solution was stirred at -78° for 25 min. At this time the enolate derived from the ester was transferred via cannula into the solution of the oxazolidinone enolate, stirred at -78 °C for an additional 5 min at which time the septum was removed and solid powdered bis(2-ethylhexanoyloxy)copper (9.02 g, 25.8 mmol) was rapidly added to the reaction vessel and the septum replaced. The vessel was immediately removed from the cold bath and immersed into a warm water bath (40 °C) with rapid swirling with a concomitant color change from the initial turquoise to brown. The reaction mixture was stirred for 20 min, was poured into 5% aqueous NH4OH (360 mL) and extracted with EtOAc (2x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 60% solvent A/B=hexanes/EtOAc, REDISEP® S1O2 120g). Concentration of appropriate fractions provided Preparation 1C (2.87 g, 66%) as pale yellow viscous oil. XH NMR showed the product was a 1.6: 1 mixture of diastereoisomers 1C: 1D as determined by the integration of the multiplets at 2.74 & 2.84 ppm: XH NMR (400 MHz, CDC13) δ ppm 4.43-4.54 (2 H, m), 4.23-4.35 (5 H, m), 4.01 (1 H, ddd, J=9.54, 6.27, 3.51 Hz), 2.84 (1 H, ddd, J=9.41, 7.28, 3.64 Hz), 2.74 (1 H, ddd, J=10.29, 6.27, 4.02 Hz), 2.37-2.48 (2 H, m, J=10.38, 6.98, 6.98, 3.51, 3.51 Hz), 2.20-2.37 (3 H, m), 1.92-2.20 (8 H, m), 1.64-1.91 (5 H, m), 1.47 (18 H, s), 0.88-0.98 (12 H, m). Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00222] To a cool (0 °C), stirred solution of Preparation 1C and ID (4.54 g, 9.51 mmol) in THF (140 mL) and water (42 mL) was sequentially added hydrogen peroxide (30% in water) (10.3 g, 91 mmol) and LiOH (685.3 mg, 28.6 mmol) and the mixture was stirred for 1 hr. At this time the reaction vessel was removed from the cold bath and then stirred for 1.5 hr. The reaction was judged complete by HPLC. To the reaction mixture was added saturated NaHC03 (45 mL) and saturated a2S03(15 mL), and then partially concentrated under reduced pressure. The resulting crude solution was extracted with DCM (3x). The aqueous phase was acidified to pH~l-2 with IN HC1, extracted with DCM (3x) and EtOAc (lx). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure to provide a mixture of Preparation IE and IF (3.00 g, 86%) as colorless oil: XH NMR (400 MHz, CDC13) δ ppm 2.76-2.84 (1 H, m, diastereoisomer 2), 2.64-2.76 (3 H, m), 2.04-2.35 (8 H, m), 1.88-2.00 (4 H, m), 1.71-1.83 (4 H, m), 1.48 (9 H, s, diastereoisomer 1), 1.46 (9 H, s, diastereoisomer 2); XH NMR showed a 1.7: 1 mixture of 1E: 1F by integration of the peaks for the ?-butyl groups.
Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00223] To a cold (-78 °C), stirred solution of diisopropylamine (1.7 mL, 11.93 mmol) in THF (19 mL) under nitrogen atmosphere was added n-BuLi (2.5M in hexanes) (4.8 mL, 12.00 mmol). The mixture was stirred for 5 min and then warmed to 0 °C. In a separate vessel, to a cold (-78 °C) stirred solution of the mixture of Preparation IE and IF (1.99 g, 5.43 mmol) in THF (18 mL) was added the LDA solution prepared above via cannula slowly over 25 min. The mixture was stirred for 15 min, then warmed to room temperature (placed in a 24 °C water bath) for 15 min, and then again cooled to -78 °C for 15 min. To the reaction mixture was added Et2AlCl (1M in hexane) (11.4 mL, 1 1.40 mmol) via syringe, stirred for 10 min, warmed to room temperature for 15 min and then cooled back to -78 °C for 15 min. Methanol (25 mL) was rapidly added, swirled vigorously while warming to room temperature, then concentrated to ~l/4 original volume. The mixture was dissolved in EtOAc and washed with IN HCl (50 mL) and ice (75 g). The aqueous phase was separated, extracted with EtOAc (2x). The combined organics were washed with a mixture of KF (2.85g in 75 mL water) and IN HCl (13 mL) [resulting solution pH 3-4], then with brine, dried (Na2S04), filtered and concentrated under reduced pressure to give a 9: 1 (IE: IF) enriched diastereoisomeric mixture (as determined by XH NMR) of Preparation IE and Preparation IF (2.13 g, >99%) as a pale yellow viscous oil: XH NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s). Preparation 1 G: (35)-3 -Amino- 1 -methyl-5-phenyl- 1 ,3 -dihydro-2H- 1 ,4-benzodiazepin-2- one, and
Preparation 1H: (3R)-3 -Amino- 1 -methyl-5-phenyl- 1 ,3-dihydro-2H- 1 ,4-benzodiazepin-2- one

(1G) (1 H)
[00224] Racemic 3-amino-l-methyl-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2- one (Rittle, K.E. et al, Tetrahedron Letters, 28(5):521-522 (1987)) was prepared according to the literature procedure. The enantiomers were separated under chiral-SFC conditions using the following method: CHIRALPAK® AS-H 5×25; Mobile phase: 30% MeOH+ 0.1% DEA in C02; Flow rate: 280 mL/min; Pressure: 100 bar; Temperature: 35 °C.
[00225] Obtained the S-enantiomer (Preparation 1G): HPLC: RT=1.75 min (30% MeOH + 0.1% DEA in C02 on CHIRALPAK® AS-H 4.6×250 mm, 3 mL/min, 35 °C, 100 bar, 230 nm, ΙΟμΙ injection); ¾ NMR (400 MHz, CDC13) δ ppm 7.58-7.63 (2 H, m), 7.55 (1 H, ddd, J=8.50, 7.1 1, 1.76 Hz), 7.40-7.47 (1 H, m), 7.34-7.40 (3 H, m), 7.31 (1 H, dd, J=7.81, 1.51 Hz), 7.14-7.22 (1 H, m), 4.46 (1 H, s), 3.44 (3 H, s), 3.42 (2 H, s); [a]D= -155° (c=1.9, MeOH) (Lit. Rittle, K.E. et al, Tetrahedron Letters, 28(5):521-522 (1987): [a]D=-236°).
[00226] Also obtained the R-enantiomer (Preparation 1H): HPLC: RT=1.71 min; [a]D=+165° (c=2.1, MeOH) (Lit [a]D= +227°).
Alternate procedure to make Preparation 1 G:
Preparation 1G»CSA salt: (35)-3-Amino-l-methyl-5-phenyl-l,3-dihydro-2H-l,4- benzodiazepin-2-one, (15)-(+)-10-camphorsulfonic acid salt

[00227] Preparation lG’CSA was prepared from racemic 3-amino-l-methyl-5-phenyl- l,3-dihydro-2H-l,4-benzodiazepin-2-one (9.98g, 37.6 mmol) (prepared according to the literature as shown above) according to the literature procedure (Reider, P.J. et al, J. Org. Chem., 52:955-957 (1987)). Preparation lG’CSA (16.91g, 99%) was obtained as a colorless solid: Optical Rotation: [a]D = -26.99° (c=l, H20) (Lit. [a]D = -27.8° (c=l,
H20))
Preparation II: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate, and Preparation 1J: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3-trifluoropropyl)hexanoate

(11) (U)
[00228] To a stirred solution of Preparation 1G (1.45 g, 5.47 mmol) and a 9: 1 mixture of Preparation IE and IF (1.989 g, 5.43 mmol) in DMF (19 mL) was added O- benzotriazol-l-yl-N,N,N’,N’-tetra-methyluronium tetrafluoroborate (1.79 g, 5.57 mmol) and triethylamine (3.0 mL, 21.52 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (125 mL) and the precipitated solid was collected by filtration, washed with water and air dried to provide an 8: 1 mixture of Preparation II and Preparation 1J (2.95 g, 89%) as a cream solid: MS (ES): m/z= 614 [M+H]+;XH NMR (400 MHz, CDC13) δ ppm 7.55-7.65 (3 H, m), 7.44- 7.52 (2 H, m), 7.35-7.45 (4 H, m), 5.52 (1 H, d, J=8.03 Hz), 3.48 (3 H, s), 2.63 (2 H, ddd, J=9.35, 3.95, 3.76 Hz), 2.14-2.25 (4 H, m), 1.90-2.03 (3 H, m), 1.69-1.82 (1 H, m), 1.51 (9 H, s).
Preparation IK: (25,,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- lH-l,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1L: (2R,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-

(1 K) (1 L)
[00229] To a cool (0 °C), stirred solution of the above mixture of Preparation II and Preparation 1 J (2.95 g, 4.81 mmol) in DCM (20 mL) was added TFA (20 mL, 260 mmol). The reaction mixture was stirred for lhr, then allowed to warm to room temperature and stirred for 2.5 hr. The reaction was judged complete by LCMS. The reaction mixture was diluted with toluene (50 mL) and concentrated under reduced pressure. The residue mixture was redissolved in toluene (50 mL) and concentrated under reduced pressure then dried under high vacuum. The crude product was dissolved in DCM, S1O2 (15g) was added, concentrated, then was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 0% to 45% solvent A/B=DCM/EtOAc, REDISEP® S1O2 80g). Concentration of appropriate fractions provided a mixture of Preparation IK and Preparation 1L (2.00 g, 75%) as a cream solid: HPLC: RT=2.770 min
(CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.1% TFA, 4 mL/min, monitoring at 254 nm); MS (ES): m/z= 558 [M+H]+; XH NMR (400 MHz, CDC13) δ ppm 8.32 (1 H, d, J=8.03 Hz), 7.65-7.71 (1 H, m), 7.50-7.60 (3 H, m), 7.41-7.49 (2 H, m), 7.39 (1 H, dd, J=7.91, 1.63 Hz), 7.23-7.35 (2 H, m), 5.59 (1 H, d, J=8.03 Hz), 3.51 (3 H, s), 2.81 (1 H, ddd, J=10.54, 6.90, 3.64 Hz), 2.67-2.76 (1 H, m), 2.22-2.33 (3 H, m), 1.99-2.12 (3 H, m), 1.85-1.94 (1 H, m), 1.79 (1 H, ddd, J=13.87, 7.84, 3.64 Hz). Example 1 :
[00230] To a stirred solution of an 8: 1 mixture of Preparation IK and Preparation 1L (3.46 g, 6.21 mmol) in DMF (25 mL) under nitrogen atmosphere was added ammonium chloride (3.32 g, 62.1 mmol), EDC (3.55 g, 18.52 mmol), HOBT (2.85 g, 18.61 mmol), and triethyl amine (16 mL, 1 15 mmol) and stirred overnight. The reaction was judged complete by LCMS. The reaction mixture was poured into water (200 mL) with vigorous swirling and then allowed to sit. The solid was collected by filtration, washed with water, allowed to dry to afford 3.6 g colorless solid. The solid was purified by preparative SFC chromatography (Lux-Cellulose-2 (3x25cm), 8% methanol in CO2, 140ml/min @220nm and 35 °C; Sample: 3.6g in 50cc methanol, conc.=70mg/ml, Stack injection:
0.5cc/9.2min). Fractions containing product were concentrated, dried overnight under vacuum. Obtained Example 1 (2.74 g, 79%) as a colorless solid (Crystal Form -1): HPLC: RT=9.601 min (H20/CH3CN with TFA, Sunfire CI 8 3.5um, 4.6x150mm, 4.6x150mm, gradient = 15 min, wavelength = 220 and 254 nm). MS (ES): m/z= 557 [M+H]+; XH NMR (400 MHz, DMSO-d6) δ ppm 9.54 (1 H, d, J=7.28 Hz), 7.71-7.80 (1 H, m), 7.68 (2 H, d, J=8.78 Hz), 7.50-7.62 (3 H, m), 7.45 (2 H, t, J=7.28 Hz), 7.29-7.40 (2 H, m), 7.15 (1 H, br. s.), 5.30 (1 H, d, J=7.28 Hz), 3.39 (3 H, s), 2.74-2.86 (1 H, m), 2.02-2.32 (3 H, m), 1.45-1.79 (4 H, m); [a]D = -107.0° (5.73 mg/mL, DMSO).
[00231] Crystal Form A-2 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of acetone/acetonitrile/water solution (2:2: 1). A mixture of colorless needles and thin blades crystals were obtained after one day of slow evaporation of the solution at room temperature. The thin blade crystals were separated to provide crystal Form A-2.
[00232] Crystal Form EA-3 was prepared by adding approximately 1 mg of Example 1 to approximately 0.7 mL of ethyl acetate/heptane solution (1 : 1). Colorless blade crystals were obtained after three days of slow evaporation of the solution at room temperature.
[00233] Crystal Form THF-2 was obtained by adding approximately 5 mg of Example 1 to approximately 0.7 mL of THF/water solution (4: 1). Colorless blade-like crystals were obtained after one day of solvent evaporation at room temperature.
Alternate Procedure to Make Example 1 : Preparation 1M: 3,3,3-Trifluoropropyl trifluoromethanesulfonate

[00234] To a cold (-25 °C), stirred solution of 2,6-lutidine (18.38 mL, 158 mmol) in CH2CI2 (120 mL) was added Tf20 (24.88 mL, 147 mmol) over 3 min, and stirred for 5 min. To the reaction mixture was added 3,3,3-trifluoropropan-l-ol (12 g, 105 mmol) over an interval of 3 min. After 2 hr, the reaction mixture was warmed to room temperature and stirred for 1 hr. The reaction mixture was concentrated to half volume, then purified by loading directly on silica gel column (330g ISCO) and eluted with CH2C12. Obtained Preparation 1M (13.74 g, 53%) as a colorless oil. XH NMR (400 MHz, CDCI3) δ ppm 4.71 (2 H, t, J=6.15 Hz), 2.49-2.86 (2 H, m).
Preparation IN: (45)-4-Benzyl- -(5,5,5-trifluoropentanoyl)-l,3-oxazolidin-2-one

[00235] Preparation IN was prepared from 5,5,5-trifluoropentanoic acid (3.35 g, 21.46 mmol) and (45)-4-benzyl-l,3-oxazolidin-2-one (3.80 g, 21.46 mmol) by the general methods shown for Preparation IB. Preparation IN (5.67 g, 84%) was obtained as a colorless viscous oil: XH NMR (400 MHz, CDC13) δ ppm 7.32-7.39 (2 H, m), 7.30 (1 H, d, J=7.05 Hz), 7.18-7.25 (2 H, m), 4.64-4.74 (1 H, m), 4.17-4.27 (2 H, m), 3.31 (1 H, dd, J=13.35, 3.27 Hz), 3.00-3.1 1 (2 H, m), 2.79 (1 H, dd, J=13.35, 9.57 Hz), 2.16-2.28 (2 H, m), 1.93-2.04 (2 H, m).
Preparation 10: tert-Butyl (3R)-3-(((45)-4-benzyl-2-oxo-l,3-oxazolidin-3-yl)carbonyl)- 6,6,6-trifluorohexanoate

[00236] To a cold (-78 °C), stirred solution of Preparation IN (3.03 g, 9.61 mmol) in THF (20 mL) was added NaHMDS (1.0M in THF) (10.6 mL, 10.60 mmol) under nitrogen atmosphere. After 2 hours, tert-butyl 2-bromoacetate (5.62 g, 28.8 mmol) was added neat via syringe at -78 °C and stirring was maintained at the same temperature. After 6 hours, the reaction mixture was warmed to room temperature. The reaction mixture was partitioned between saturated NH4C1 and EtOAc. The organic phase was separated, and the aqueous was extracted with EtOAc (3x). The combined organics were washed with brine, dried (Na2S04), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (Teledyne ISCO CombiFlash Rf, 5% to 100% solvent A/B=hexanes/EtO Ac, REDISEP® Si02 120g). Concentration of appropriate fractions provided Preparation 10 (2.79 g, 67.6%) as a colorless viscous oil: XH NMR (400 MHz, CDC13) δ ppm 7.34 (2 H, d, J=7.30 Hz), 7.24-7.32 (3 H, m), 4.62- 4.75 (1 H, m, J=10.17, 6.89, 3.43, 3.43 Hz), 4.15-4.25 (3 H, m), 3.35 (1 H, dd, J=13.60, 3.27 Hz), 2.84 (1 H, dd, J=16.62, 9.57 Hz), 2.75 (1 H, dd, J=13.35, 10.07 Hz), 2.47 (1 H, dd, J=16.62, 4.78 Hz), 2.1 1-2.23 (2 H, m), 1.90-2.02 (1 H, m), 1.72-1.84 (1 H, m), 1.44 (9 H, s). -2-(2-tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid

[00237] Preparation IP was prepared from Preparation 10 (2.79 g, 6.50 mmol) by the general methods shown for Preparation IE. Preparation IP (1.45 g, 83%) was obtained as a colorless oil: XH NMR (400 MHz, CDC13) δ ppm 2.83-2.95 (1 H, m), 2.62-2.74 (1 H, m), 2.45 (1 H, dd, J=16.62, 5.79 Hz), 2.15-2.27 (2 H, m), 1.88-2.00 (1 H, m), 1.75-1.88 (1 H, m), 1.45 (9 H, s). Preparation IE: (2R,35′)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00238] To a cold (-78 °C), stirred solution of Preparation IP (5.44 g, 20.13 mmol) in THF (60 mL) was slowly added LDA (24.60 mL, 44.3 mmol) over 7 min. After stirring for 2 hr, Preparation 1M (6.44 g, 26.2 mmol) was added to the reaction mixture over 3 min. After 45 min, the reaction mixture was warmed to -25 °C bath (ice/MeOH/dry ice) for 1 hr, and then warmed to 0 °C. After 45 min, Preparation 1M (lg) was added and the reaction mixture was stirred for 20 min. The reaction was quenched with water and IN NaOH and was extracted with (¾(¾. The organic layer was again extracted with IN NaOH (2x) and the aqueous layers were combined. The aqueous layer was cooled in ice/water bath and then acidified with concentrated HCl to pH 2. Next, the aqueous layer was extracted with EtOAc. The combined organics were washed with brine, dried over anhydrous sodium sulphate, and concentrated under reduced pressure. The residue was dried under high vacuum to provide a 1 :5 (IE: IF) mixture (as determined by XH NMR) of Preparation IE and Preparation IF (5.925 g, 80%) as a pale yellow solid. XH NMR (500 MHz, CDC13) 8 ppm 2.81 (1 H, ddd, J=10.17, 6.32, 3.85 Hz), 2.63-2.76 (1 H, m), 2.02- 2.33 (4 H, m), 1.86-1.99 (2 H, m), 1.68-1.85 (2 H, m), 1.47 (9 H, s).
Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid, and
Preparation IF: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

(1 E) (1 F)
[00239] A mixture of Preparation IE and Preparation IF (64 mg, 1.758 mmol) was taken in THF (6 mL) to give a colorless solution which was cooled to -78 °C. Then, LDA (2.149 mL, 3.87 mmol) (1.8M in heptane/THF/ethylbenzene) was slowly added to the reaction mixture over 10 min. After stirring for 15 min the reaction mixture was placed in a room temperature water bath. After 15 min the reaction mixture was placed back in -78 °C bath and then diethylaluminum chloride (3.87 mL, 3.87 mmol) (1M in hexane) was added slowly over 5 min. The reaction mixture was stirred at -78 °C. After 15 min the reaction mixture was placed in a room temperature water bath for 10 min and then cooled back to -78 °C bath. After 15 min the reaction was quenched with MeOH (8 mL, 198 mmol), removed from the -78 °C bath and concentrated. To the reaction mixture was added ice and HC1 (16 mL, 16.00 mmol), followed by extraction with EtOAc (2x). The organic layer was washed with potassium fluoride (920 mg, 15.84 mmol) (in 25 mL FLO) and HC1 (4.5 mL, 4.50 mmol). The organics were dried over anhydrous magnesium sulphate and concentrated under reduced pressure to provide a 9: 1 (IE: IF) enriched mixture of Preparation IE and Preparation IF (540 mg, 1.583 mmol, 90% yield) as light yellow/orange solid. ¾ NMR (400 MHz, CDC13) δ ppm 2.64-2.76 (2 H, m), 2.04-2.35 (4 H, m), 1.88-2.00 (2 H, m), 1.71-1.83 (2 H, m), 1.48 (9 H, s). It was converted to Example 1 by the sequence of reactions as outlined above.
Alternate procedure to make Preparation IE:
Preparation 1Q: (2R,35)- -Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate

(1Q) [00240] A clean and dry 5 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler at room temperature was charged with Ν,Ν-dimethyl formamide (2.07 L), a 1.2: 1 mixture of Preparation IE and Preparation IF (207 g, 0.5651 moles), potassium carbonate (1 17.1 g, 0.8476 moles) followed by benzyl bromide (116 g, 0.6781 moles) over 15-20 min. The reaction mixture was stirred for 2-3 hr. After completion of the reaction, the reaction mixture was concentrated to dryness at 50-55 °C under vacuum. Ethyl acetate (3.1 L, 30 Vol.) was charged into the concentrated reaction mass and then washed with water (2.07 L), brine (0.6 L) then dried over anhydrous sodium sulfate (207 g), filtered and concentrated to dryness at 40-45 °C under vacuum. The residue was dissolved in dichloromethane (1.035 L, 5 vol.) and then absorbed onto silica gel (60-120) (607 g, 3.0 w/w), then was purified with column chromatography using petroleum ether and ethyl acetate as solvents. After pooling several batches, Preparation 1Q (235 g) was obtained. HPLC purity: 99.77%, Preparation IE: (2R,35)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00241] A clean and dry 2 L autoclave was charged with methanol (540 mL) and was purged with nitrogen for 5-10 minutes. To the autoclave was added 10% palladium on carbon (12 g, 20%), purged with nitrogen once again for 5-10 min then was charged with Preparation 1Q (60g, 0.1315 moles), the autoclave was flushed with methanol (60mL) and stirred for 4-6 hr at 20-25 °C under 5Kg hydrogen pressure. After completion of the reaction, the reaction mass was filtered through CELITE®, washed with methanol (180 mL), dried with anhydrous sodium sulfate (60 g), filtered and concentrated to dryness at 45-50 °C under vacuum. Obtained Preparation IE (45.8 g, 95%) as a colorless solid: HPLC purity: 98.9%.
Alternate procedure to make Preparation IE: Preparation IE: (2R,35)-3-(te^Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3- trifluoropropyl)hexanoic acid

[00242] Preparation IE was prepared in a procedure identical as above from a mixture of Preparations IE and IF (200g, 0.5460 moles) using LDA (1.8 M solution in THF, ethyl benzene and heptane) (698mL, 2.3equiv.) and diethyl aluminum chloride (1.0 M solution in hexane) (1256mL, 2.3equiv) in THF (2.0L). After workup as explained above, the resulting residue was treated as follows: The crude material was added to a 2L four neck round bottom flask, followed by the addition of MTBE (1.0L) charged below 30 °C. The resulting mixture was stirred for 5-10 minutes to obtain a clear solution.
Hexanes (600mL) was charged to the reaction mixture at a temperature below 30 °C. The reaction mixture was stirred for 10 min. Next, tert-butylamine (43.8g, l. leq) was charged slowly over a period of 15 minutes below 30 °C. This addition was observed to be exothermic. The reaction mixture was stirred for 2 hrs below 30 °C and filtered. The solid material was washed with 5:3 MTBE: hexane (200mL), the filtrate was
concentrated and transferred to an amber color bottle. The filtered solid was dissolved in dichloromethane (2.0L), washed with IN HC1 (2.0), the organic layer was washed with brine (1.0L x 2), then was concentrated under reduced pressure below 45 °C. This material was found to be 91.12% pure. The material was repurified by the above t- butylamine crystallization purification procedure. Obtained Preparation IE (78 g, 39%): HPLC purity: 99.54%.
Alternate procedure to make Example 1 :
Preparation II: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3- dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate

[00243] A clean and dry 2 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with N,N- dimethylformamide (457 mL), Preparation IE (45.7g, 0.1248moles) and Preparation lG’CSA (62.08g, 0.1248moles) under nitrogen atmosphere at 20-25 °C. The reaction mixture was stirred for 15-20 minutes to make clear solution at 20-25 °C. To the reaction mixture was added TBTU (48.16g, 0.1498 moles) at 20-25 °C followed by triethylamine (50.51g, 0.4992 moles) over 15-20 minutes at 20-25 °C. The reaction mixture was stirred for 60-120 minutes at 20-25 °C under nitrogen atmosphere. After completion of the reaction, the reaction was quenched into water (1.37L, 30 Vol.) at 20-25 °C under stirring. The reaction mixture was stirred for 30 minutes at 20-25 °C. The reaction mixture was filtered and washed with water (228 mL). The resulting solid material was dissolved in ethyl acetate (457 mL), washed with water (2×137 mL), brine (137 mL), and then dried with anhydrous sodium sulfate (45.7g). Activated charcoal (9.14 g, 20%) was charged into the reaction mixture and stirred for 30 minutes. The mixture was filtered through CELITE® bed and 1 micron filter cloth, washed charcoal bed with ethyl acetate (137 mL), concentrated to 1.0 Vol. stage and then petroleum ether (457 mL, 10 Vol.) was charged and stirred for 30 minutes at 20-25 °C. The solid was collected by filtration, washed with petroleum ether (137 mL) and then dried under vacuum at 40-45 °C for 8 hr until loss on drying was less than 3.0%. Obtained Preparation II (65.2 g, 85%): HPLC purity: 98.26%.
Preparation IK: (25,,3R)-6,6,6-Trifluoro-3-(((35)-l-methyl-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoic acid

[00244] A clean and dry 3 L four neck round bottom flask equipped with mechanical stirring, thermometer socket and nitrogen bubbler was charged with dichloromethane (980 mL) under nitrogen atmosphere followed by Preparation II (140 g, 0.2282 moles) at 20-25 °C. The reaction mixture was cooled to 0-5 °C and trifluoroacetic acid (980 mL) was charged slowly for 30-40 minutes. The resulting mixture was stirred for 2 hr at 0-5 °C under nitrogen atmosphere. The reaction temperature was raised to 20 to 25 °C, and the reaction mixture was stirred for 1-2 hr at 20 to 25 °C. After completion of the reaction, the reaction mixture was concentrated to dryness at 50 to 55 °C under vacuum. Toluene (3×700 mL,) was charged into the concentrated reaction mass, and then distilled off at 50 to 55 °C under vacuum. After complete concentration from toluene, ethyl acetate (280 mL) was charged into the reaction mass at 20 to 25 °C, stirred for 60 minutes, then the solid was collected by filtration, washed with ethyl acetate (140 mL), dried under vacuum at 50 to 55 °C for 12 hr until loss on drying was less than 2.0%. Obtained Preparation IK (106 g, 84%): HPLC purity: 98.43%.
Example 1 :
[00245] A reaction vessel was charged with Preparation IK (30 g, 53.81 mmol), HOBt (8.7g, 64.38 mmol), and THF (150 mL) at room temperature. To the homogeneous solution was added EDCI (12.4g, 64.68 mmol), stirred for 15 min, then cooled to 8 °C. To the reaction mixture was added ammonia (2M in IP A) (81 mL, 162 mmol) over 5 min so as to maintain a temperature below 10 °C. The resulting heavy slurry was stirred for 10 min, warmed to room temperature over 30 min, then stirred for 4 hr. At the completion of the reaction, water (230 mL) was slowly added over 15 min to maintain a temperature below 20 °C, and then stirred for 2 hr. The solid was collected by filtration, washed with water (3X60 mL), then dried under vacuum 48 hr at 55 °C. The above crude product was charged into a 1 L 3 -necked round flask. IP A (200 mL) was added, then heated to 80 °C resulting in a homogeneous solution. Water (170 mL) was slowly added (15 min) to maintain an internal temperature >75 °C. The resulting slurry was stirred and cooled to room temperature for 2 hr. The solid was collected by filtration, washed with water (2 X 50 mL), then dried under vacuum (55 °C for 24 h, and 30 °C for 48 h).
Obtained Example 1 (23.4 g, 78% yield): HPLC purity: 99.43%.
Example 2 NOT SAME
WITHOUT METHYL GROUP
(2R,35)-N-((35)-2-Oxo-5-phenyl-2,3-dihydro-lH-l,4-benzodiazepin-3-yl)-2,3-bis(3,3,3- trifluoropropyl)succinamide

Preparation 2A: (35)-3-Amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one, and Preparation 2B: -3-Amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one

(2A) (2B)
[00246] Racemic 3-amino-5-phenyl-l,3-dihydro-2H-l,4-benzodiazepin-2-one (J. Med. Chem., 49:231 1-2319 (2006), compound# 5) was prepared according to the literature procedure. The enantiomers were separated on Berger SFC MGIII Column: Lux 25X3 cm, 5cm; Mobile phase: 30% MeOH+ 0.1% DEA in C02; Flow rate: 150 mL/min;
Temperature: 40 °C; Detector wavelength: 250 nM. Obtained the S-enantiomer
Preparation 2A as a white solid: XH NMR (400 MHz, DMSO-d6) δ ppm 10.67 (1 H, br. s.), 7.58 (1 H, td, J=7.65, 1.76 Hz), 7.37-7.53 (5 H, m), 7.23-7.30 (2 H, m), 7.14-7.22 (1 H, m), 4.23 (1 H, s), 2.60 (2 H, br. s.); HPLC: RT=3.0625 min (30% MeOH + 0.1% DEA in C02 on OD-H Column, 3 mL/min, 35 °C, 96 bar, 230 nm, ΙΟμΙ inj); [a]D = -208.3° (5.05 mg/niL, MeOH). Also obtained the R-enantiomer Preparation 2B as an off white solid: HPLC: RT=3.970 min; [a]D = 182.1° (2.01 mg/mL, MeOH).
Preparation 2C: tert-Butyl (25,,3R)-6,6,6-trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro- 1 H- 1 ,4-benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate, and
Preparation 2D: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro- 1 H- -benzodiazepin-3 -yl)carbamoyl)-2-(3 ,3 ,3 -trifluoropropyl)hexanoate

(2C) (2D)
[00247] Preparation 2C was prepared from Preparation 2A (564 mg, 2.244 mmol) and a mixture of Preparation IE and Preparation IF (822 mg, 2.244 mmol) according to the general procedure shown for Preparation II. Obtained Preparation 2C and Preparation 2D (1.31 g, 97%): HPLC: RT=3.443 min (CHROMOLITH® ODS 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.% TFA, 4 mL/min, monitoring at 220 nm); MS (ES): m/z= 600.3 [M+H]+.
Preparation 2E: (25′,3R)-6,6,6-Trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro-lH-l,4- benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 2F: (2R,3R)-6,6,6-Trifluoro-3-(((35)-2-oxo-5-phenyl-2,3-dihydro-lH-l,4- benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid

(2E) (2F) [00248] A mixture of Preparation 2E and Preparation 2F was prepared from a mixture of Preparation 2C and Preparation 2D (1.3 lg, 2.185 mmol) by the general methods shown for Preparation IK. Obtained a mixture of Preparation 2E and Preparation 2F (1.18 g, 99%): HPLC: RT=2.885 min (CHROMOLITH® ODS 4.6 x 50 mm (4 min grad) eluting with 10-90% aqueous MeOH over 4 minutes containing 0.% TFA, 4 mL/min, monitoring at 220 nm). MS (ES): m/z= 544.2 [M+H]+.
Example 2:
[00249] Example 2 was prepared from a mixture of Preparation 2E and Preparation 2F (354 mg, 0.651 mmol) by the general methods shown for Example 1. After separation of the diastereoisomers, Example 2 was obtained (188 mg, 52%) as a white solid: HPLC: RT=9.063 min (H20/CH3CN with TFA, Sunfire C18 3.5um, 4.6x150mm, 4.6x150mm, gradient = 15 min, wavelength = 220 and 254 nm); MS (ES): m/z= 543 [M+H]+; XH NMR (400 MHz, DMSO-d6) δ ppm 10.87 (1 H, br. s.), 9.50-9.55 (1 H, m), 7.62-7.69 (2 H, m), 7.40-7.57 (5 H, m), 7.29-7.36 (2 H, m), 7.22-7.28 (1 H, m), 7.16 (1 H, br. s.), 5.25 (1 H, d), 3.30-3.32 (1 H, m), 2.75-2.86 (1 H, m), 2.44-2.48 (1 H, m), 2.06-2.34 (3 H, m), 1.51- 1.77 (4 H, m); [a]D = -114.4° (8.04 mg/mL, DMSO).
[00250] Crystal Form M2- 1 was prepared by adding approximately 1 mg of Example 2 to approximately 0.7 mL of MeOH/fluorobenzene solution (3 : 1). Colorless plate-like crystals were obtained after 2 days of solvent evaporation at room temperature.
PATENT







Example 1
(2R,3S)—N-((3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)-2,3-bis(3,3,3-trifluoropropyl)succinamide
Preparation 1A: tert-Butyl 5,5,5-trifluoropentanoate
Preparation 1B: (4S)-4-(Propan-2-yl)-3-(5,5,5-trifluoropentanoyl)-1,3-oxazolidin-2-one
Preparation 1C: (2S,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate, and
Preparation 1D: (2R,3R)-tert-Butyl 6,6,6-trifluoro-3-((S)-4-isopropyl-2-oxooxazolidine-3-carbonyl)-2-(3,3,3-trifluoropropyl)hexanoate
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
Preparation 1G: (3S)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, and
Preparation 1H: (3R)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one
Alternate Procedure to Make Preparation 1G
Preparation 1G•CSA salt: (3S)-3-Amino-1-methyl-5-phenyl-1,3-dihydro-2H-1,4-benzodiazepin-2-one, (1 S)-(+)-10-camphorsulfonic acid salt
Preparation 1I: tert-Butyl (2S,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate, and
Preparation 1J: tert-Butyl (2R,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate
Preparation 1K: (2S,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1L: (2R,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid
Example 1
Alternate Procedure to Make Example 1
Preparation 1M: 3,3,3-Trifluoropropyl trifluoromethanesulfonate
Preparation 1N: (4S)-4-Benzyl-3-(5,5,5-trifluoropentanoyl)-1,3-oxazolidin-2-one
Preparation 1O: tert-Butyl (3R)-3-(((4S)-4-benzyl-2-oxo-1,3-oxazolidin-3-yl)carbonyl)-6,6,6-trifluorohexanoate
Preparation 1P: (2R)-2-(2-tert-Butoxy-2-oxoethyl)-5,5,5-trifluoropentanoic acid
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid, and
Preparation 1F: (2R,3R)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
Alternate Procedure to Make Preparation 1E
Preparation 1Q: (2R,3S)-1-Benzyl 4-tert-butyl 2,3-bis(3,3,3-trifluoropropyl)succinate
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
Alternate Procedure to Make Preparation 1E
Preparation 1E: (2R,3S)-3-(tert-Butoxycarbonyl)-6,6,6-trifluoro-2-(3,3,3-trifluoropropyl)hexanoic acid
Alternate Procedure to Make Example 1
Preparation 1I: tert-Butyl (2S,3R)-6,6,6-trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoate
Preparation 1K: (2S,3R)-6,6,6-Trifluoro-3-(((3S)-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl)carbamoyl)-2-(3,3,3-trifluoropropyl)hexanoic acid
Example 1
PATENTS
Clip
For some disease targets, an indirect approach may be best. Or so Ashvinikumar V. Gavai and his colleagues atBristol-Myers Squibbfound in their quest toward a potential cancer drug. Gavai unveiled BMS-906024, which is an experimental—and slightly roundabout—treatment for a number of cancers, including breast, lung, and colon cancers, and leukemia.
Cancers have a tendency to relapse or to become resistant to treatments that once worked. Research at BMS and elsewhere had suggested that a family of proteins called Notch is implicated in that resistance and in cancer progression more generally. Gavai, director of oncology chemistry at BMS in Princeton, N.J., and his team set out to block Notch family signaling.
Notch family members lack enzymatic activity, so blocking them directly is difficult. Instead, BMS developed inhibitors of an enzyme that is essential for activating Notch signaling—γ-secretase.

Company: Bristol-Myers Squibb
Target: pan-Notch
Disease: breast, lung, colon cancer; leukemia
Interfering with Notch, even in this indirect way, can have detrimental effects on the gastrointestinal tract. Only two of the four Notch family members are linked to that side effect, Gavai says. But he and his team think their drug will be most effective if it acts on all four family members roughly equally—a so-called pan-Notch inhibitor. By selecting a molecule that’s well tolerated in animals and carefully scheduling doses of the drug in humans, it could be possible to minimize side effects, he says.
The BMS team relied on Notch signaling assays in leukemia and breast cancer cell lines to find leads. They soon learned that for their molecules to work, three chiral centers had to be in the S,R,Sconfiguration. After that, they strove to make the molecules last in the bloodstream. They removed an isobutyl group and tweaked some other parts of their candidate’s succinamide side chain. It was tough to retain both a long half-life and activity against Notch, Gavai told C&EN. “You’d optimize one and lose the other.”
His team threaded the needle with BMS-906024. Their studies with mice suggest that a dose of 4–6 mg once a week could be effective in people. That’s lower than doses being tested for other Notch-targeted agents, according to the website clinicaltrials.gov. The mouse studies also back the idea that Notch is involved in cancer drug resistance and suggest that Notch could be a target for taking on cancer stem cells, which are notoriously resistant to chemotherapy.
BMS-906024 is in Phase I clinical trials, both alone and in combination with other agents. Patients with colon, lung, breast, and other cancers are receiving intravenous doses of the compound to determine its safety and optimum dose ranges.

(From left, front row) Gavai, Weifeng Shan, (second row) Aaron Balog, Patrice Gill, Gregory Vite, (third row) Francis Lee, Claude Quesnelle, (rear row) Wen-Ching Han, Richard Westhouse.
Credit: Catherine Stroud Photography
http://cen.acs.org/articles/91/i16/BMS-906024-Notch-Signaling-Inhibitor.html

clip

BMS-906024
Company: Bristol-Myers Squibb
Meant to treat: cancers including breast, lung, colon, and leukemia
Mode of action: pan-Notch inhibitor
Medicinal chemistry tidbit: The BMS team used an oxidative enolate heterocoupling en route to the candidate– a procedure from Phil Baran’s lab at Scripps Research Institute. JACS 130, 11546
Status in the pipeline: Phase I
Relevant documents: WO 2012/129353
PAPER

An enantioselective synthesis of (S)-7-amino-5H,7H-dibenzo[b,d]azepin-6-one (S–1) is described. The key step in the sequence involved crystallization-induced dynamic resolution (CIDR) of compound 7 using Boc-d-phenylalanine as a chiral resolving agent and 3,5-dichlorosalicylaldehyde as a racemization catalyst to afford S–1 in 81% overall yield with 98.5% enantiomeric excess.
† Department of Discovery Synthesis, Biocon Bristol-Myers Squibb Research Centre, Biocon Park, Bommasandra IV Phase, Jigani Link Road, Bengaluru 560099, India
‡Bristol-Myers Squibb Company, P.O Box 4000, Princeton, New Jersey 08543-4000, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00207, http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.6b00207
*E-mail: anuradha.gupta@syngeneintl.com.
References
- Jump up^ C. Drahl, Liveblogging First-Time Disclosures of Drug Structures from #ACSNOLA, 2013, http://cenblog.org/the-haystack/2013/04/liveblogging-first-time-disclosures-of-drug-structures-from-acsnola/
- Jump up^ http://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012129353
- Jump up^ http://clinicaltrials.gov/show/NCT01653470
- Jump up^ http://clinicaltrials.gov/show/NCT01292655
1. Quesnelle, Claude; Kim, Soong-Hoon; Lee, Francis; Gavai, Ashvinikumar. Bis(fluoroalkyl)-1,4-benzodiazepinone compounds as Notch receptor inhibitors and their preparation and use in the treatment of cancer. PCT Int. Appl. (2012), WO 2012129353 A1 20120927.
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016060232 | 2016-03-03 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US2016022723 | 2016-01-28 | COMBINATION THERAPY FOR THE TREATMENT OF PROLIFERATIVE DISEASES |
| US2016008316 | 2016-01-14 | USE OF DIANHYDROGALACTITOL AND ANALOGS OR DERIVATIVES THEREOF IN COMBINATION WITH PLATINUM-CONTAINING ANTINEOPLASTIC AGENTS TO TREAT NON-SMALL-CELL CARCINOMA OF THE LUNG AND BRAIN METASTASES |
| US2016009785 | 2016-01-14 | NOVEL FUSION MOLECULES AND USES THEREOF |
| US2015284342 | 2015-10-08 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US2015232491 | 2015-08-20 | PRODRUGS OF 1, 4-BENZODIAZEPINONE COMPOUNDS |
| US8968741 | 2015-03-03 | Anti-CD22 antibodies and immunoconjugates and methods of use |
| US2014357605 | 2014-12-04 | BIS(FLUOROALKYL)-1, 4-BENZODIAZEPINONE COMPOUNDS |
| US8822454 | 2014-09-02 | Bisfluoroalkyl-1, 4-benzodiazepinone compounds |
| US8629136 | 2014-01-14 | Bisfluoroalkyl-1, 4-benzodiazepinone compounds |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2R,3S)-N-[(3S)-1-Methyl-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepin-3-yl]-2,3-bis(3,3,3-trifluoropropyl)succinamide
|
|
| Identifiers | |
| PubChem | CID 66550890 |
| ChemSpider | 28536138 |
| Chemical data | |
| Formula | C26H26F6N4O3 |
| Molar mass | 556.500 g/mol |
///////////////3,5-dichlorosalicylaldehyde, Alzheimer’s disease, Boc-D-phenylalanine, CIDR;dibenzoazepenone, DKR; Notch inhibitors, Notch inhibitor, SAR, T-acute lymphoblastic leukemia, triple-negative breast cancer, γ-secretase inhibitor, PHASE 1, BMS, Bristol-Myers Squibb, 1401066-79-2, Ashvinikumar Gavai
CN1c2ccccc2C(=N[C@@H](C1=O)NC(=O)[C@H](CCC(F)(F)F)[C@H](CCC(F)(F)F)C(=O)N)c3ccccc3

Patent US8377886 – Use of gamma secretase inhibitors and notch …
Figure US08377886-20130219-C00003. gamma secretase inhibitor

RO4929097 | γ-secretase inhibitor – Cellagen Technology
RO4929097 | γ-secretase inhibitor

YO-01027 (Dibenzazepine) | gamma-secretase inhibitor – Cellagen …
YO-01027 (Dibenzazepine) | gamma-secretase inhibitor

Semagacestat (LY450139) | Gamma-secretase inhibitor | Read Reviews …
Semagacestat (LY450139) Chemical Structure
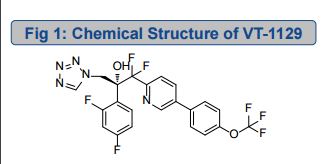
VT 1129, QUILSECONAZOLE
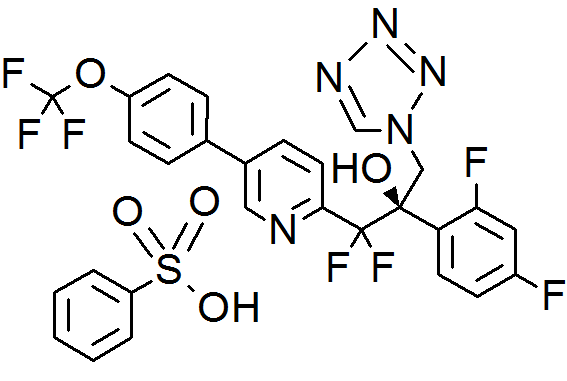
VT 1129 BENZENE SULFONATE
CAS 1809323-18-9

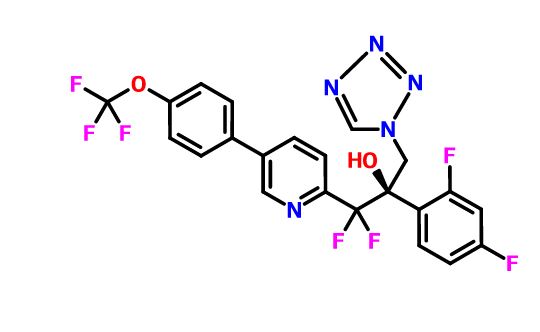
VT 1129
QUILSECONAZOLE
1340593-70-5 CAS
MF C22 H14 F7 N5 O2, MW 513.37
2-Pyridineethanol, α-(2,4-difluorophenyl)-β,β-difluoro-α-(1H-tetrazol-1-ylmethyl)-5-[4-(trifluoromethoxy)phenyl]-, (αR)-
R ISOMER
ROTATION +
- Originator Viamet Pharmaceuticals
- Class Antifungals; Small molecules
- Mechanism of Action 14-alpha demethylase inhibitors
- Orphan Drug Status Yes – Cryptococcosis
- On Fast track Cryptococcosis
- Phase I Cryptococcosis
-
Most Recent Events
- 01 Jun 2016 VT 1129 receives Fast Track designation for Cryptococcosis [PO] (In volunteers) in USA
- 30 May 2016 Viamet Pharmaceuticals plans a phase II trial for Cryptococcal meningitis in USA (Viamet Pharmaceuticals pipeline; May 2016)
- 27 May 2016 Phase-I clinical trials in Cryptococcosis (In volunteers) in USA (PO) before May 2016 (Viamet Pharmaceuticals pipeline; May 2016)

| William J. Hoekstra, Stephen William Rafferty,Robert J. Schotzinger | |
| Applicant | Viamet Pharmaceuticals, Inc. |

Viamet, in collaboration with Therapeutics for Rare and Neglected diseases, is investigating VT-1129, a small-molecule lanosterol demethylase inhibitor, developed using the company’s Metallophile technology, for treating fungal infections, including Cryptococcus neoformans meningitis.
VT-1129 is a novel oral agent that we are developing for the treatment of cryptococcal meningitis, a life-threatening fungal infection of the brain and the spinal cord that occurs most frequently in patients with HIV infection, transplant recipients and oncology patients. Without treatment, the disease is almost always fatal.
 VT-1129 has shown high potency and selectivity in in vitro studies and is an orally administered inhibitor of fungal CYP51, ametalloenzyme important in fungal cell wall synthesis. In preclinical studies, VT-1129 has demonstrated substantial potency against Cryptococcus species, the fungal pathogens that cause cryptoccocal meningitis, and has also been shown to accumulate to high concentrations within the central nervous system, the primary site of infection.
VT-1129 has shown high potency and selectivity in in vitro studies and is an orally administered inhibitor of fungal CYP51, ametalloenzyme important in fungal cell wall synthesis. In preclinical studies, VT-1129 has demonstrated substantial potency against Cryptococcus species, the fungal pathogens that cause cryptoccocal meningitis, and has also been shown to accumulate to high concentrations within the central nervous system, the primary site of infection.
In in vitro studies, VT-1129 was significantly more potent against Cryptococcus isolates than fluconazole, which is commonly used for maintenance therapy of cryptococcal meningitis in the United States and as a primary therapy in the developing world. Oral VT-1129 has also been studied in a preclinical model of cryptococcal meningitis, where it was compared to fluconazole. At the conclusion of the study, there was no detectable evidence of Cryptococcus in the brain tissue of the high dose VT-1129 treated groups, in contrast to those groups treated with fluconazole. To our knowledge, this ability to reduce the Cryptococcus pathogen in the central nervous system to undetectable levels in this preclinical model is unique to VT-1129.
Opportunity
An estimated 3,400 hospitalizations related to cryptococcal meningitis occur annually in the United States and the FDA has granted orphan drug designation to VT-1129 for the treatment of this life-threatening disease. In addition, the FDA has granted Qualified Infectious Disease Product designation to VT-1129 for the treatment of Cryptococcus infections, which further underscores the unmet medical need. In developing regions such as Africa, cryptococcal meningitis is a major public health problem, with approximately one million cases and mortality rates estimated to be as high as 55-70%.
Current Status
VT-1129 has received orphan drug and Fast Track designations for the treatment of cryptococcal meningitis and has been designated a Qualified Infectious Disease Product (QIDP) by the U.S. Fod and Drug Administration. We are currently conducting a Phase 1 single-ascending dose study of VT-1129 in healthy volunteers.
- Activity of VT-1129 against Cryptococcus neoformans Clinical Isolates with High Fluconazole MICs
- VT1129 Binds Potently and Selectively to Recombinant Cryptococcal CYP51 Consistent with Its In Vitro Anti-Cryptococcal Activity (ICAAC 2015)
- Investigational CYP51 Inhibitors VT-1161 and VT-1129 Show Strong In Vitro Activity Against Candida glabrata Isolates Clinically Resistant to Azole and Echinocandin Compounds (ICAAC 2015)
- The Novel Fungal Cyp51 Inhibitor VT-1129 Demonstrates Potent In Vivo Activity In Mice Against Cryptococcal Meningitis with a Loading/Maintenance Dose Strategy (ECCMID 2015)
- Susceptibility testing of VT-1129, a novel fungal CYP51 inhibitor, against Cryptococcus neoformans and Cryptococcus gattii (ICCC 2014)
- The Novel Fungal Cyp51 Inhibitor VT-1129 Demonstrates Potent In vivo Activity Against Cryptococcal Meningitis with an Improved Formulation (ICCC 2014)
- The Fungal Cyp51 Inhibitors VT-1161 and VT-1129 Maintain in vitro Activity Against Candida albicans Isolates with Reduced Antifungal Susceptibility (2011 ICAAC)
- In Vitro Activity of Two Metalloenzyme Inhibitors Compared to Caspofungin and Fluconazole Against a Panel of 74 Candida spp. (2010 ICAAC)
- Novel Metalloenzyme Inhibitors, VT-1161 and VT-1129, Exhibit Efficacy and Survival Benefit in a Murine Systemic Candidiasis Model (2010 ICAAC)
Conclusions
• VT-1129 has robust activity against Cryptococcus isolates with elevated fluconazole MICs and may be a viable option in persons infected with such strains.
• A Phase 1 study of VT-1129 in healthy volunteers is scheduled to begin by the end of 2015. Phase 2 trials in persons with cryptococcal meningitis are targeted to begin by the end of 2016.

Living organisms have developed tightly regulated processes that specifically import metals, transport them to intracellular storage sites and ultimately transport them to sites of use. One of the most important functions of metals such as zinc and iron in biological systems is to enable the activity of metalloenzymes. Metalloenzymes are enzymes that incorporate metal ions into the enzyme active site and utilize the metal as a part of the catalytic process. More than one-third of all characterized enzymes are metalloenzymes.
The function of metalloenzymes is highly dependent on the presence of the metal ion in the active site of the enzyme. It is well recognized that agents which bind to and inactivate the active site metal ion dramatically decrease the activity of the enzyme. Nature employs this same strategy to decrease the activity of certain metalloenzymes during periods in which the enzymatic activity is undesirable. For example, the protein TIMP (tissue inhibitor of metalloproteases) binds to the zinc ion in the active site of various matrix metalloprotease enzymes and thereby arrests the enzymatic activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain a l-(l,2,4-triazole) group that binds to the heme iron present in the active site of the target enzyme lanosterol demethylase and thereby inactivates the enzyme.
In the design of clinically safe and effective metalloenzyme inhibitors, use of the most appropriate metal-binding group for the particular target and clinical indication is critical. If a weakly binding metal-binding group is utilized, potency may be suboptimal. On the other
hand, if a very tightly binding metal-binding group is utilized, selectivity for the target enzyme versus related metalloenzymes may be suboptimal. The lack of optimal selectivity can be a cause for clinical toxicity due to unintended inhibition of these off-target metalloenzymes. One example of such clinical toxicity is the unintended inhibition of human drug metabolizing enzymes such as CYP2C9, CYP2C19 and CYP3A4 by the currently- available azole antifungal agents such as fluconazole and voriconazole. It is believed that this off-target inhibition is caused primarily by the indiscriminate binding of the currently utilized l-(l,2,4-triazole) to iron in the active site of CYP2C9, CYP2C19 and CYP3A4. Another example of this is the joint pain that has been observed in many clinical trials of matrix metalloproteinase inhibitors. This toxicity is considered to be related to inhibition of off-target metalloenzymes due to indiscriminate binding of the hydroxamic acid group to zinc in the off-target active sites.
Therefore, the search for metal-binding groups that can achieve a better balance of potency and selectivity remains an important goal and would be significant in the realization of therapeutic agents and methods to address currently unmet needs in treating and preventing diseases, disorders and symptoms thereof. Similarly, methods of synthesizing such therapeutic agents on the laboratory and, ultimately, commercial scale is needed. Addition of metal-based nucleophiles (Zn, Zr, Ce, Ti, Mg, Mn, Li) to azole-methyl substituted ketones have been effected in the synthesis of voriconazole (M. Butters, Org. Process Res. Dev.2001, 5, 28-36). The nucleophile in these examples was an ethyl-pyrimidine substrate. Similarly, optically active azole-methyl epoxide has been prepared as precursor electrophile toward the synthesis of ravuconazole (A. Tsuruoka, Chem. Pharm. Bull.1998, 46, 623-630). Despite this, the development of methodology with improved efficiency and selectivity is desirable.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011133875
Scheme 1

EXAMPLE 7

2-(2, 4-Difluorophenyl)-l, l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4- (trifluoromethoxy) phenyl) pyridin-2-yl) propan-2-ol (7)
To a stirred solution of bromo epoxide C (0.5 g, 1.38 mmol) in THF (30 mL) and water (14 mL) were added 4-(trifluoromethoxy) phenylboronic acid (0.22 g, 1.1 mmol), Na2C03 (0.32 g, 3.1 mmol) and Pd(dppf)2Cl2 (0.28 g, 0.34 mmol) at RT under inert atmosphere. After purged with argon for a period of 30 min, the reaction mixture was heated to 75°C and stirring was continued for 4 h. Progress of the reaction was monitored by TLC. The reaction mixture was cooled to RT and filtered through a pad of celite. The filtrate was concentrated under reduced pressure; obtained residue was dissolved in ethyl acetate (30 mL). The organic layer was washed with water, brine and dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude compound was purified by column chromatography to afford the coupled product (0.45 g, 1.0 mmol, 73%) as solid. 1H NMR (200 MHz, CDC13): δ 8.87 (s, 1 H), 7.90 (dd, / = 8.2, 2.2 Hz, 1 H), 7.66-7.54 (m, 3 H), 7.49-7.34 (m, 3 H), 6.90-6.70 (m, 2 H), 3.49 (d, / = 5.0 Hz, 1 H), 3.02-2.95 (m, 1 H). Mass: m/z 444 [M++l].
To a stirred solution of the coupled product (0.45 g, 1.0 mmol) in DMF (10 mL) was added K2C03 (70 mg, 0.5 mmol) followed by IH-tetrazole (70 mg, 1.0 mmol) at RT under inert atmosphere. The reaction mixture was stirred for 4 h at 80 °C. The volatiles were removed under reduced pressure and obtained residue was dissolved in water (15 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with water, brine and dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude compound was purified by column chromatography to afford 7 (0.19 g, 0.37 mmol, 36 %) as white solid. 1H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.97 (dd, / = 8.0, 2.0 Hz, 1 H), 7.68 (d, / = 8.5 Hz, 1 H), 7.60-7.56 (m, 3 H), 7.43-7.36 (m, 3 H), 6.80-6.76 (m, 1 H), 6.70-6.67 (m, 1 H), 5.57 (d, / = 14.5 Hz, 1 H), 5.17 (d, / = 14.5 Hz, 1 H). HPLC: 98.3%. Mass: m/z 513.9 [M++l].
Chiral preparative HPLC of enantiomers:
The enantiomers of 7 (17.8 g, 34.6 mmol) were separated by normal-phase preparative high performance liquid chromatography (Chiralpak AD-H, 250 x 21.2 mm, 5μ; using (A) n-hexane – (B) IPA (A:B : 70:30) as a mobile phase; Flow rate: 15 mL/min) to obtain 7(+) (6.0 g) and 7(-) (5.8 g).
Analytical data for 7 (+):
HPLC: 99.8%.
Chiral HPLC: Rt = 9.88 min (Chiralpak AD-H, 250 x 4.6mm, 5μ; mobile phase (A) n-Hexane (B) IPA (7/3): A: B (70:30); flow Rate: 1.00 mL/min)
Optical rotation [a]D25: + 19° (C = 0.1 % in MeOH).
Patent
WO2015143137,
Examples
The present invention will now be demonstrated using specific examples that are not to be construed as limiting.
General Experimental Procedures
Definitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.
Synthesis of 1 or la

A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of 1 or la may be accomplished using the example syntheses that are shown below (Schemes 1-9). The preparation of precursor ketone 8 is performed starting with reaction of dibromo-pyridine 2-Br with ethyl 2-bromo-difluoroacetate to produce ester 3-Br. This ester is reacted with tetrazole reagent 4 via Claisen reaction to furnish 5-Br. Decarboxylation of 5-Br via a two-step process produces compound 6-Br. Suzukin coupling of 6-Br with boronate 7 furnishes 8.
Scheme 1. Synthesis of ketone 8

Ketone 8 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared using according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).
Scheme 2. Synthesis of ketone 8


= halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.
Compounds 6 or 8 may be reacted with a series of metallated derivatives of 2,4-difluoro-bromobenzene and chiral catalysts/reagents (e.g. BINOL) to effect enantiofacial-selective addition to the carbonyl group of 6 or 8 (Scheme 3). These additions can be performed on 6 or 8 to furnish 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof), respectively.
Scheme 3. Synthesis of 1 or la

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.
Alternatively, ketone 8 can be synthesized from aldehyde 10 (Scheme 4). Aldehyde 10 is coupled with 7 to produce 11. Compound 11 is then converted to 12 via treatment with diethylaminosulfurtrifluoride (DAST).
Scheme 4. Alternate synthesis of ketone 8


Scheme 5 outlines the synthesis of precursor ketone 15-Br. The ketone is prepared by conversion of 2-Br to 3-Br as described above. Next, ester 3-Br is converted to 15-Br by treatment via lithiation of 2,4-difluoro-bromobenzene.
Scheme 5. Synthesis of ketone 15-Br

Ketone 15 may be prepared in an analogous fashion as described for 15-Br in Scheme 5 starting from corresponding substituted 2-bromo-pyridines, which can be prepared using according to synthetic transformations known in the art and contained in the references cited herein (Scheme 6).
Scheme 6. Synthesis of ketone 15

F = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.
Ketone 15 may be used to prepare 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) by the following three-step process (Scheme 7). In the presence of a chiral catalyst/reagent (e.g. proline derivatives), base-treated nitromethane is added to 15 or 16 to furnish 17 (or 17a, the enantiomer of 17, or mixtures thereof) or 18 (or 18a, the enantiomer of 18, or mixtures thereof), respectively. Reduction of 17 (or 17a, the enantiomer of 17, or mixtures thereof) or 18 (or 18a, the enantiomer of 18, or mixtures thereof) (e.g. lithium aluminum hydride) produces 19 (or 19a, the enantiomer of 19, or mixtures thereof) or 20 (or 20a, the enantiomer of 20, or mixtures thereof). Annulation of 19 (or 19a, the enantiomer of 19, or mixtures thereof) or 20 (or 20a, the enantiomer of 20, or mixtures thereof) by treatment with sodium azide/triethylorthoformate furnishes tetrazoles 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof). Suzuki coupling of 9 (or 9a, the enantiomer of 9, or mixtures thereof) with 4-trifluoromethoxyphenyl-boronic acid produces 1 (or la, the enantiomer of 1, or mixtures thereof).
Scheme 7. Asymmetric Henry reaction

R-ι = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted a 0(S02)-aryl, or -0(S02)-substituted aryl.
Ketone 21 may be employed to prepare optically-active epoxides via Horner-Emmons reaction of a difluoromethyl substrate to produce 22 or 22a. Ketones related to 21 have been prepared (M. Butters, Org. Process Res. Dev. 2001, 5, 28-36). Nucleophilic addition of metalated 5-(4-trifluoromethoxy)phenyl-2-pyridine (M = metal) to epoxide 22 or 22a may furnish compound
1 or la.
Scheme 8. Enantioselective epoxidation strategy

Ketone 15 or 16 may be used to prepare 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) by an alternative three-step process to Scheme 7 (Scheme 9). In the presence of a chiral catalyst/reagent, trimethylsilyl-cyanide is added to 15 or 16 to furnish 23 (or 23a, the enantiomer of 23, or mixtures thereof) or 24 (or 24a, the enantiomer of 24, or mixtures thereof), respectively (S.M. Dankwardt, Tetrahedron Lett. 1998, 39, 4971-4974). Reduction of 23 (or 23a, the enantiomer of 23, or mixtures thereof) or 24 (or 24a, the enantiomer of 24, or mixtures thereof) (e.g. lithium aluminum hydride) produces 19 (or 19a, the enantiomer of 19, or mixtures thereof) or 20 (or 20a, the enantiomer of 20, or mixtures thereof). Annulation of 19 (or 19a, the enantiomer of 19, or mixtures thereof) or 20 (or 20a, the enantiomer of 20, or mixtures thereof) by treatment with sodium azide/triethylorthoformate furnishes tetrazoles 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof). Suzuki coupling of 9 (or 9a, the enantiomer of 9, or mixtures thereof) with 4-trifluoromethoxyphenyl-boronic acid produces 1 (or la, the enantiomer of 1, or mixtures thereof).
Scheme 9. Asymmetric cyanohydrin strategy

R’ = H or trimethylsilyl
Suzuki

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.

1
2-(2, 4-Difluorophenyl)-l, l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(trifluoromethoxy) phenyl) pyridin-2-yl) propan-2-ol (1 or la)
White powder: *H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.97 (dd, J = 8.0, 2.0 Hz, 1 H), 7.68 (d, / = 8.5 Hz, 1 H), 7.60-7.56 (m, 3 H), 7.43-7.36 (m, 3 H), 6.80-6.76 (m, 1 H), 6.70-6.67 (m, 1 H), 5.57 (d, J = 14.5 Hz, 1 H), 5.17 (d, J = 14.5 Hz, 1 H). HPLC: 98.3%. Mass: m/z 513.9 [M++l]. HPLC: 99.8%. Optical rotation [a]D25: + 19° (C = 0.1 % in MeOH).
INTERMEDIATE 3-Br Ri = Br)
To a clean and dry 100 L jacketed reactor was added copper powder (1375 g, 2.05 equiv, 10 micron, sphereoidal, SAFC Cat # 326453) and DMSO (17.5 L, 7 vol). Next, ethyl bromodifluoroacetate (2.25 kg, 1.05 equiv, Apollo lot # 102956) was added and the resulting slurry stirred at 20-25 °C for 1-2 hours. Then 2,5-dibromopyridine (2-Br, 2.5 kg, 1.0 equiv, Alfa Aesar lot # F14P38) was added to the batch and the mixture was immediately heated (using the glycol jacket) to 35 °C. After 70 hours at 35 °C, the mixture was sampled for CG/MS analysis. A sample of the reaction slurry was diluted with 1/1 CH3CN/water, filtered (0.45 micron), and the filtrate analyzed directly. Ideally, the reaction is deemed complete if <5% (AUC) of 2,5-dibromopyridine remains. In this particular batch, 10% (AUC) of 2,5-dibromopyridine remained. However due to the already lengthy reaction time, we felt that prolonging the batch would not help the conversion any further. The reaction was then deemed complete and diluted with EtOAc (35 L). The reaction mixture was stirred at 20-35 °C for 1 hour and then the solids (copper salts) were removed by filtration through a pad of Celite. The residual solids inside the reactor were rinsed forward using EtOAc (2 x 10 L) and then this was filtered through the Celite. The filter cake was washed with additional EtOAc (3 x 10 L) and the EtOAc filtrates were combined. A buffer solution was prepared by dissolving NH4CI (10 kg) in DI water (100 L), followed by the addition of aqueous 28% NH4OH (2.0 L) to reach pH = 9. Then the combined EtOAc filtrates were added slowly to a pre-cooled (0 to 15 °C) solution of NH4C1 and NH4OH (35 L, pH = 9) buffer while maintaining T<30 °C. The mixture was then stirred for 15-30 minutes and the phases were allowed to separate. The aqueous layer (blue in color) was removed and the organic layer was washed with the buffer solution until no blue color was discernable in the aqueous layer. This experiment required 3 x 17.5 L washes. The organic layer was then washed with a 1/1 mixture of Brine (12.5 L) and the pH = 9 NH4C1 buffer solution (12.5 L), dried over MgS04, filtered, and concentrated to dryness. This provided crude compound 3-Br [2.29 kg, 77% yield, 88% (AUC) by GC/MS] as a yellow oil. The major impurity present in crude 3-Br was unreacted 2,5-dibromopyridine [10% (AUC) by GC/MS]. ‘ll NMR (CDC13) was consistent with previous lots of crude compound 3-Br. Crude compound 3-Br was then combined with similar purity lots and purified by column chromatography (5/95 EtO Ac/heptane on S1O2 gel).
INTERMEDIATE 15-Br (R, = Br)
To a clean and dry 72 L round bottom flask was added l-bromo-2,4-difluorobenzene (1586 g,
1.15 equiv, Oakwood lot # H4460) and MTBE (20 L, 12.6 vol). This solution was cooled to -70 to -75 °C and treated with n-BuLi (3286 mL, 1.15 equiv, 2.5 M in hexanes, SAFC lot # 32799MJ), added as rapidly as possible while maintaining -75 to -55 °C. This addition typically required 35-45 minutes to complete. (NOTE: If the n-BuLi is added slowly, an white slurry will form and this typically gives poor results). After stirring at -70 to -65 °C for 45 minutes, a solution of compound 3-Br (2000 g, 1.0 equiv, AMRI lot # 15CL049A) in MTBE (3 vol) was added rapidly (20-30 min) by addition funnel to the aryl lithium solution while maintaining -75 to -55 °C. After stirring for 30-60 minutes at -75 to -55 °C, the reaction was analyzed by GC/MS and showed only trace (0.5% AUC) l-bromo-2,4-difluorobenzene present. The reaction was slowly quenched with aqueous 2 M HC1 (3.6 L) and allowed to warm to room temperature. The mixture was adjusted to pH = 6.5 to 8.5 using NaHCC>3 (4 L), and the organic layer was separated. The MTBE layer was washed with brine (5% NaCl in water, 4 L), dried over MgS04, filtered, and concentrated. In order to convert the intermediate hemi-acetal to 4-Br, the crude mixture was heated inside the 20 L rotovap flask at 60-65 °C for 3 hours (under vacuum), at this point all the hemi-acetal was converted to the desired ketone 4 by !Η NMR (CDC13). This provided crude compound 4-Br [2.36 kg, 75% (AUC) by HPLC] as a brown oil that solidified upon standing. This material can then be used “as-is” in the next step without further purification.
PATENT FOR VT1161 SIMILAR TO VT 1129
Synthesis of 1 or la

EXAMPLE 1
Preparation of Compound 1 X-Hydrate
Compound 1 and its preparation are described in the art, including in US Patent 8,236,962 (incorporated by reference herein). Compound 1 can then be partitioned between ethanol and water to afford Compound 1 X-hydrate.
EXAMPLE 2
Compound 1 Anhydrous Form Recrystallization
Compound 1 X-hydrate (29.1 g, 28.0 g contained 1) was suspended in 2-propanol (150 ml) and heated to 56 °C. The solution was filtered through a 0.45 μιη Nylon membrane with 2-propanol rinses. The combined filtrate was concentrated to 96.5 g of a light amber solution. The solution was transferred to a 1-L flask equipped with overhead stirring, thermocouple and addition funnel, using 2-propanol (30 ml total) to complete the transfer. The combined solution contained about 116 ml 2-propanol.
The solution was heated to 50 °C and n-heptane (234 ml) was added over 22 minutes. The resulting hazy mixture was seeded with 1 anhydrous form. After about 1 hour a good
suspension had formed. Additional n-heptane (230 ml) was added over 48 minutes. Some granular material separated but most of the suspension was a finely divided pale beige solid. After about ½ hour at 50 °C the suspension was cooled at 10 °C/h to room temperature and stirred overnight. The product was collected at 22 °C on a vacuum filter and washed with 1:4 (v/v) 2-PrOH/ n-heptane (2 x 50 ml). After drying on the filter for 1-2 hours the weight of product was 25.5 g. The material was homogenized in a stainless steel blender to pulverize and blend the more granular solid component. The resulting pale beige powder (25.37 g) was dried in a vacuum oven at 50 °C. The dry weight was 25.34 g. The residual 2-propanol and n- heptane were estimated at <0.05 wt% each by 1H NMR analysis. The yield was 90.5% after correcting the X-hydrate for solvent and water content. Residual Pd was 21 ppm. The water content was 209 ppm by KF titration. The melting point was 100.7 °C by DSC analysis.
Table 1: Data for the isolated and dried Compound 1 – X-hydrate and anhydrous forms

M.P. by DSC; Pd by ICP; Purity by the API HPLC method; Chiral purity by HPLC; water content by KF titration; residual solvent estimated from :H NMR.
Table 2: Characterisation Data for Compounds 1 (X-hydrate) and 1 (anhydrous)

Needle like crystals Needle like crystals and agglomerates
PLM
particle size >100μιη particle size range from 5μπι-100μιη
0.59%w/w water uptake at 90%RH. 0.14%w/w water uptake at 90%RH.
GVS
No sample hysteresis No sample hysteresis
XRPD
No form change after GVS experiment No form change after GVS experiment post GVS
KF 2.4%w/w H20 Not obtained
<0.001mg/ml <0.001mg/ml
Solubility
pH of saturated solution = 8.6 pH of saturated solution = 8.7
Spectral Pattern 1 Spectral Pattern 2
Charcteristic bands/ cm“1: Charcteristic bands/ cm 1:
FT-IR 3499, 3378, 3213, 3172 3162
1612, 1598, 1588, 1522, 1502 1610, 1518, 1501 931, 903, 875, 855, 828, 816 927, 858, 841, 829, 812

The structure solution of Compound 1 anhydrous form was obtained by direct methods, full-matrix least-squares refinement on F 2 with weighting w‘1 = <52{F02) + (0.0474P)2 + (0.3258P), where P = (F02+2F 2)/3, anisotropic displacement parameters, empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. Final wR2
= {∑[w(F02-Fc2)2]/∑[w(F02)2]m} = 0.0877 for all data, conventional Ri = 0.0343 on F values of 8390 reflections with F0 > 4a( F0), S = 1.051 for all data and 675 parameters. Final Δ/a (max) 0.001, A/a(mean), 0.000. Final difference map between +0.311 and -0.344 e A“3.
Below shows a view of two molecules of Compound 1 in the asymmetric unit of the anhydrous form showing the numbering scheme employed. Anisotropic atomic displacement ellipsoids for the non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are displayed with an arbitrarily small radius. The absolute configuration of the molecules has been determined to be R.

EXAMPLE 3
Compound 1 Ethanol Solvate Recrystallization
Compound 1 X-hydrate (50 mg) was suspended in -40 volumes of 15% H20/EtOH. The suspension was then placed in an incubation chamber for maturation. The maturation protocol involved treating the suspension to a two-temperature cycle of 50 °C/ ambient temperature at 8 hours per cycle for 3 days with constant agitation. After maturation, the suspension was cooled in a fridge at 4°C for up to 2 days to encourage the formation of crystals. Then, the solvent was removed at RT and the sample was vacuum dried at 30°C -35°C for up to 1 day. Suitable crystals formed on cooling were harvested and characterized.
Table 4: Single Crystal Structure of 1 Ethanol solvate
Molecular formula C25H22F7N5O3

The structure solution of Compound 1 ethanol solvate was obtained by direct methods, full-matrix least-squares refinement on F 2 with weighting w‘1 = σ2^2) + (0.0450P)2 + (0.5000P), where P = (F02+2F 2)/3, anisotropic displacement parameters, empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. Final wR2 = {∑[w(F02-F 2)2]/∑[w(F02)2]m} = 0.0777 for all data, conventional Ri = 0.0272 on F values of 4591 reflections with F0 > 4σ( F0), S = 1.006 for all data and 370 parameters. Final Δ/σ (max) 0.000, A/a(mean), 0.000. Final difference map between +0.217 and -0.199 e A“3.
Below shows a view of the asymmetric unit of the ethanol solvate from the crystal structure showing the numbering scheme employed. Anisotropic atomic displacement ellipsoids for the non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are displayed with an arbitrarily small radius. The asymmetric unit shows stoichiometry of 1 : 1 for solvent of crystallisation to Compound 1.

EXAMPLE 4
Compound 1 1.5 Hydrate Recrystallization
Compound 1 X-hydrate (50 mg) was suspended in -40 volumes of 15% Η20/ΙΡΑ. The suspension was then placed in an incubation chamber for maturation. The maturation protocol involved treating the suspension to a two-temperature cycle of 50 °C/ ambient temperature at 8 hours per cycle for 3 days with constant agitation. After maturation, the suspension was cooled in a fridge at 4°C for up to 2 days to encourage the formation of crystals. Then, the solvent was removed at RT and the sample was vacuum dried at 30°C -35°C for up to 1 day. Suitable crystals formed on cooling were harvested and characterized.
Table 5: Single Crystal Structure of 1 1.5 Hydrate


The structure solution of Compound 1 1.5 hydrate was obtained by direct methods, full-matrix least-squares refinement on F 2 with weighting w‘1 = ^(F 2) + (0.1269P)2 + (0.0000P), where P = (F02+2F 2)/3, anisotropic displacement parameters, empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. Final wR2 = {∑[w(F 2-F 2)2]/∑[w(F 2)2] m} = 0.1574 for all data, conventional Ri = 0.0668 on F values of 2106 reflections with F0 > 4σ( F0), S = 1.106 for all data and 361 parameters. Final Δ/σ (max) 0.000, A/a(mean), 0.000. Final difference map between +0.439 and -0.598 e A“3.
Below shows a view of the asymmetric unit of the 1.5 hydrate from the crystal structure showing the numbering scheme employed. Anisotropic atomic displacement ellipsoids for the non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are displayed with an arbitrarily small radius. The asymmetric unit shows stoichiometry of 1.5: 1 for water to Compound 1.

EXAMPLE 5
Human Pharmacokinetic Comparison of Compound 1 X-Hydrate and Compound 1 Anhydrous Form
Table 6 compares human multiple-dose pharmacokinetic (PK) parameters between dosing with Compound 1 X-hydrate and Compound 1 Anhydrous form. Compound 1 X-hydrate was dosed at 600 mg twice daily (bid) for three days followed by dosing at 300 mg once daily (qd) for 10 days. Compound 1 Anhydrous form was dosed at 300 mg qd for 14 days. Despite the higher initial dosing amount and frequency (i.e., 600 mg bid) of Compound 1 X-hydrate, Compound 1 Anhydrous form surprisingly displayed higher maximal concentration (Cmax) and higher area-under-the-curve (AUC) than Compound 1 X-hydrate.
Table 6. Comparison of Multiple Dose PK between Compound 1 X-Hydrate and Compound 1
Anhydrous Polymorph

Further characterization of the various polymorph forms of compound 1 are detailed in the accompanying figures.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015143154
Examples
General Experimental Procedures
Definitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.
Synthesis of 1 or la

la
A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of lor la may be accomplished using the example syntheses that are shown below (Schemes 1-4). The preparation of precursor ketone 3-Br is performed starting with reaction of 2,5-dibromo-pyridine with ethyl 2-bromo-difluoroacetate to produce ester 2-Br. This ester is reacted with morpholine to furnish morpholine amide 2b-Br, followed by arylation to provide ketone 3-Br.
Scheme 1. Synthesis of ketone 3-Br

Ketone 3 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared using according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).
Scheme 2. Synthesis of ketone 3

R1 = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, 0(S02)-aryl, or -0(S02)-substituted aryl.
Alternatively, compound 1 (or la, the enantiomer of 1, or mixtures thereof) can be prepared according to Scheme 3 utilizing amino-alcohols ±4b or ±1-6. Epoxides 4 and 5 can be prepared by reacting ketones 3 and 1-4 with trimethylsulfoxonium iodide (TMSI) in the presence of a base (e.g., potassium i-butoxide) in a suitable solvent or a mixture of solvents (e.g., DMSO or THF). Also, as indicated in Scheme 3, any of pyridine compounds, 3, 4, ±4b, 4b, or 6, can be converted to the corresponding 4-CF3O-PI1 analogs (e.g., 1-4, 5, ±1-6, 1-6*, or 1 or the corresponding enantiomers, or mixtures thereof) by cross-coupling with (4-trifluoromethoxyphenyl)boronic acid (or the corresponding alkyl boronates or pinnacol boronates or the like), in a suitable solvent system (e.g., an organic-aqueous solvent mixture), in the presence of a transition metal catalyst (e.g., (dppf)PdCl2; dppf = 1,1′-(diphenylphosphino)ferrocene), and in the presence of a base (e.g., KHCO3, K2CO3, CS2CO3, or Na2CC>3, or the like). Epoxides 4 and 5 can then be converted into amino-alcohols ±4b and ±1-6 through ammonia-mediated epoxide opening using ammonia in a suitable solvent (e.g., MeOH, EtOH, or water). Racemic amino-alcohols ±4b and ±1-6 can then be enantio-enriched by exposure to a chiral acid (e.g., tartaric acid, di-benzoyltartaric acid, or di-p-toluoyltartaric acid or the like) in a suitable solvent (e.g., acetonitrile, isopropanol, EtOH, or mixtures thereof, or a mixture of any of these with water or MeOH; preferably acetonitrile or a mixture of acetonitrile and MeOH, such as 90:10, 85: 15, or 80:20 mixture) to afford compounds 4b (or 4c, the enantiomer of 4b, or mixtures thereof) or 1-6* (or 1-7*, the enantiomer of 1-6*, or mixtures thereof). Subsequent treatment with TMS-azide in the presence of trimethylorthoformate and sodium acetate in acetic acid would yield compounds 20 (or 20a, the enantiomer of 20, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) (US 4,426,531).
Scheme 3. Synthesis of 1 or la via TMSI Epoxidation Method


R-ι = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)- substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0- aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.
Compound 1 (or la, the enantiomer of 1, or mixtures thereof) prepared by any of the methods presented herein can be converted to a sulfonic salt of formula IX (or IXa, the enantiomer of
IX, or mixtures thereof), as shown in Scheme 4. This can be accomplished by a) combining compound 1 (or la, the enantiomer of 1, or mixtures thereof), a crystallization solvent or crystallization solvent mixture (e.g., EtOAc, i‘PrOAc, EtOH, MeOH, or acetonitrile, or o
Z-S-OH
combinations thereof), and a sulfonic acid o (e.g., Z = Ph, p-tolyl, Me, or Et), b) diluting the mixture with an appropriate crystallization co-solvent or crystallization co-solvent mixture (e.g., pentane, methyl i-butylether, hexane, heptane, or toluene, or combinations thereof), and c) filtering the mixture to obtain a sulfonic acid salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof).
Scheme 4. Synthesis of a Sulfonic Acid Salt of Compound 1 or la

EXAMPLE 1: Preparation of l-(2,4-difluorophenyl)-2,2-difluoro-2-(5-(4- (trifluoromethoxy)phenyl)pyridin-2-yl)ethanone (1-4).
la. ethyl 2-(5-bromopyridin-2-yl)-2,2-difluoroacetate (2)

2-Br
Typical Procedure for Preparing 2-Br
Copper ( 45μιη, 149g, 0.198moles, 2.5 equiv) was placed into a 3L, 3-neck round bottom flask equipped with a condenser, thermocouple, and an overhead stirrer. DMSO (890 mL, 4.7 vol. based on ethyl 2-bromo-2,2-difluoroacetate) and 14mL of concentrated sulfuric acid was added and the mixture stirred for 30 minutes. The mixture self-heated to about 31°C during the stir time. After cooling the contents to 23°C, 2,5-dibromopyridine 1 (277g, 1.17 moles, 1.5 eq) was added to the reaction mixture. The temperature of the contents decreased to 16°C during a 10 minute stir time. 2-bromo-2,2-difluoroacetate (190 g, 0.936 moles, 1.0 eq) was added in one portion and the mixture stirred for 10 min. The flask contents were warmed to 35°C and the internal temperature was maintained between 35-38° for 18 h. In-process HPLC showed 72% desired 2-Br. The warm reaction mixture was filtered through filter paper and the collected solids washed with 300mL of 35°C DMSO. The solids were then washed with 450mL of n-heptane and 450mL of MTBE. The collected filtrate was cooled to about 10°C and was slowly added 900mL of a cold 20% aqueous NH4C1 solution, maintaining an internal temperature of <16°C during the addition. After stirring for 15 minutes, the layers were settled and separated. The aqueous layer was extracted 2 X 450mL of a 1: 1 MTBE: n-heptane mixture. The combined organic layers were washed 2 X 450mL of aqueous 20% NH4CI and with 200mL of aqueous 20% NaCl. The organic layer was dried with 50g MgS04 and the solvent removed to yield 2-Br as a dark oil. Weight of oil = 183g ( 70% yield by weight) HPLC purity ( by area %) = 85%. *H NMR (400 MHz, d6-DMSO) : 58.86 (m, 1H), 8.35 ( dd, J= 8.4, 2.3Hz, 1H), 7.84 (dd, J= 8.3, 0.6Hz, 1H), 4.34 ( q, J= 7.1Hz, 2H), 1.23 ( t, J= 7.1Hz, 3H). MS m/z 280 ( M+H+), 282 (M+2+H+).
lb. 2-(5-bromopyridin-2-yl)-2,2-difluoro-l-morpholinoethanone (2b-Br)

Table 2 illustrates the effects of the relative proportions of each of the reagents and reactants, and the effect of varying the solvent had on the overall performance of the transformation as measured by the overall yield and purity of the reaction.
Table 2. Process Development for the Preparation of compound 2b-Br

Note: All reactions were conducted at 22- 25°C
Typical Procedure for Converting 2-Br to 2b-Br
Crude ester 2-Br (183g, 0.65moles) was dissolved in 1.5L of n-heptane and transferred to a 5L 3-neck round bottom flask equipped with a condenser, an overhead stirrer and a thermocouple. Morpholine ( 248g, 2.85 moles, 4.4 equiv.) was charged to the flask and the mixture warmed to 60°C and stirred for 16 hours. In-process HPLC showed <1 % of ester 2-Br. The reaction mixture was cooled to 22-25 °C and 1.5L of MTBE was added with continued cooling of the mixture to 4°C and slowly added 700mL of a 30%, by weight, aqueous citric acid solution. The temperature of the reaction mixture was kept < 15°C during the addition. The reaction was stirred at about 14°C for one hour and then the layers were separated. The organic layer was washed with 400mL of 30%, by weight, aqueous citric acid solution and then with 400mL of aqueous 9% NaHC03. The solvent was slowly removed until 565g of the reaction mixture
remained. This mixture was stirred with overhead stirring for about 16 hours. The slurry was filtered and the solids washed with 250mL of n-heptane. Weight of 2b-Br = 133g. HPLC purity (by area %) 98%.
This is a 44% overall yield from 2,5-dibromopyridine.
*H NMR (400 MHz, d6-DMSO): 58.86 (d, J= 2.3Hz, 1H), 8.34 (dd, J= 8.5, 2.3Hz, 1H), 7.81 (dd, J = 8.5, 0.5Hz, 1H), 3.63-3.54 ( m, 4H), 3.44-3.39 (m, 2H), 3.34-3.30 ( m, 2H). MS m/z 321 (M+H+), 323 (M+2+H+).
lc. 2-(5-bromopyridin-2-yl)-l-(2,4-difluorophenyl)-2,2-difluoroethanone (3-Br)
Process Development

Table 3 illustrates the effects of the relative proportions of each of the reagents and reactants, and the effect of varying the temperature had on the overall performance of the transformation as measured by the overall yield and purity of the reaction.
Table 3. Process Development for the Preparation of bromo-pyridine 3-Br

Typical Procedure for Converting 2b-Br to 3-Br
Grignard formation:
Magnesium turnings (13.63 g, 0.56 moles) were charged to a 3-neck round bottom flask equipped with a condenser, thermocouple, addition funnel, and a stir bar. 540 mL of anhydrous tetrahydrofuran was added followed by l-Bromo-2,4-difluorobenzene (16.3 mL, 0.144 moles). The contents were stirred at 22-25°C and allowed to self -heat to 44°C. 1- Bromo-2,4-difluorobenzene ( 47mL, 0.416 moles) was added to the reaction mixture at a rate that maintained the internal temperature between 40-44°C during the addition. Once the addition was complete, the mixture was stirred for 2 hours and allowed to cool to about 25° during the stir time.
This mixture was held at 22-25°C and used within 3-4 hours after the addition of l-bromo-2,4-difluorobenzene was completed.
Coupling Reaction
Compound 2b-Br (120 g, 0.0374 moles) was charged to a 3-neck round bottom flask equipped with a condenser, thermocouple, and an overhead stirrer. 600 mL of anhydrous
tetrahydrofuran was added. The flask contents were stirred at 22°C until a clear solution was obtained. The solution was cooled to 0-5°C. The previously prepared solution of the Grignard reagent was then added slowly while maintaining the reaction temperature at 0-2°C. Reaction progress was monitored by HPLC. In-process check after 45 minutes showed <1% amide 2b-Br remaining. 2 N aqueous HC1 (600 mL, 3 vol) was added slowly maintaining the temperature below 18°C during the addition. The reaction was stirred for 30 minutes and the layers were separated. The aqueous layer was extracted with 240mL MTBE. The combined organic layers were washed with 240mL of aqueous 9% NaHCC>3 and 240mL of aqueous 20% NaCl. The organic layer was dried over 28g of MgS04 and removed the solvent to yield 3-Br (137g) as an amber oil.
HPLC purity ( by area %) = -90%; *H NMR (400 MHz, d6-DMSO) : 58.80 (d, J= 2.2Hz, 1H), 8.41 ( dd, J= 8.3, 2.3Hz, 1H), 8.00 (m, 2H), 7.45 ( m, 1H), 7.30 ( m, 1H). MS m/z 348 (M+H+), 350 (M+2+H+).
Id. l-(2,4-difluorophenyl)-2,2-difluoro-2-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)ethanone (1-4)

Typical Procedure for Converting 3-Br to 1-4
Into a 250 mL reactor were charged THF (45 mL), water (9.8 mL), bromo-pyridine 3-Br (6.0 g, 17.2 mmoles), 4-(trifluoromethoxy)phenylboronic acid (3.57 g, 17.3 mmoles), and Na2CC>3 (4.55 g, 42.9 mmoles). The stirred mixture was purged with nitrogen for 15 min. The catalyst (Pd(dppf)Cl2 as a CH2C12 adduct, 0.72 g, 0.88 mmoles) was added, and the reaction mixture was heated to 65 °C and held for 2.5 h. The heat was shut off and the reaction mixture was allowed to cool to 20-25 °C and stir overnight. HPLC analysis showed -90% ketone 1-4/hydrate and no unreacted bromo-pyridine 3-Br. MTBE (45 mL) and DI H20 (20 mL) were added, and the quenched reaction was stirred for 45 min. The mixture was passed through a plug of Celite (3 g) to remove solids and was rinsed with MTBE (25 mL). The filtrate was transferred to a separatory funnel, and the aqueous layer drained. The organic layer was washed with 20% brine (25 mL). and split into two portions. Both were concentrated by rotovap to give oils (7.05 g and 1.84 g, 8.89 g total, >100% yield, HPLC purity -90%). The larger aliquot was used to generate hetone 1-4 as is. The smaller aliquot was dissolved in DCM (3.7 g, 2 parts) and placed on a pad of Si02 (5.5 g, 3 parts). The flask was rinsed with DCM (1.8 g), and the rinse added to the pad. The pad was eluted with DCM (90 mL), and the collected filtrate concentrated to give an oil (1.52 g). To this was added heptanes (6 g, 4 parts) and the mixture stirred. The oil crystallized, resulting in a slurry. The slurry was stirred at 20-25 °C overnight. The solid was isolated by vacuum filtration, and the cake washed with heptanes (-1.5 mL). The cake was dried in the vacuum oven (40-45 °C) with a N2 sweep. 0.92 g of ketone 1-4 was obtained, 60.1% yield (corrected for aliquot size), HPLC purity = 99.9%.
TMSI Epoxidation Method
3d. 2-((2-(2,4-difluorophenyl)oxiran-2-yl)difluoromethyl)-5-(4-(trifluoromethoxy)phenyl)pyridine (5)

Typical Procedure for Converting 1-4 to 5
i-BuOK (2.22 g, 19.9 mmoles), TMSI (4.41 g, 20.0 mmoles), and THF (58.5 mL) were charged to a reaction flask, and the cloudy mixture was stirred. DMSO (35.2 mL) was added, and the clearing mixture was stirred at 20-25°C for 30 min before being cooled to 1-2°C.
Ketone 1-4 (crude, 5.85 g, 13.6 mmoles) was dissolved in THF (7.8 mL), and the 1-4 solution was added to the TMSI mixture over 12.75 min, maintaining the temperature between 1.5 and 2.0°C. The reaction was held at 0-2°C. After 1 h a sample was taken for HPLC analysis, which showed 77.6% epoxide 5, and no unreacted ketone 1-4. The reaction was quenched by the slow addition of 1 N HC1 (17.6 mL), keeping the temperature below 5°C. After 5 min 8% NaHCC>3 (11.8 mL) was added slowly below 5°C to afford a pH of 8. The reaction mixture was transferred to a separatory funnel, and the layers were separated. The aqueous layer was extracted with MTBE (78 mL), and the combined organic layers were washed with 20% NaCl (2 x 20 mL). After concentration, 7.36 g of a dark oil was obtained. HPLC of the crude oil shows it contained 75% epoxide 5. The oil was dissolved in DCM (14.7 g, 2 parts) and the solution placed on a pad of Si02 (22 g, 3 parts). The flask was rinsed with DCM (7.4 g, 1 part) and the rinse placed on the pad. The pad was eluted with DCM (350 mL) to give an amber filtrate. The filtrate was concentrated by rotovap, and when space in the flask allowed, heptane (100 mL) was added. The mixture was concentrated until 39.4 g remained in the flask, causing solid to form. The suspension was stirred for 70 min at 20-25°C. Solid was isolated by vacuum filtration, and the cake washed with heptane (10 mL) and pulled dry on the funnel. After drying in a vacuum oven (40-45 °C) with a N2 sweep, 3.33 g solid was obtained, 55.1% yield from bromo-pyridine 3, HPLC purity = 99.8%.
3e. 3-amino-2-(2,4-difluorophenyl)-l,l-difluoro-l-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)propan-2-ol (±1-6)

Process Development
Table 8 illustrates the effects of the relative proportions of each of the reagents and reactants, the effect of varying the solvent, and the effect of varying the temperature had on the overall performance of the transformation as measured by the overall yield and purity of the reaction. Table 8. Process Development for the Preparation of ±1-6

Typical Procedure for Converting 5 to +1-6
Epoxide 5 (2.17 g, 4.89 mmoles) was combined in a glass pressure tube with methanol (48 mL) and aqueous ammonia (19.5 mL). The tube was sealed and placed in an oil bath held at 54°C, with stirring. After 15 h the tube was removed from the bath, cooled, and the reaction sampled for HPLC, which showed 93.6% amino-alcohol ±1-6 and 6.0% di-adducts. To the reaction were added MTBE (48 mL) and 20% NaCl (20 mL). The layers were separated and the aqueous layer extracted with MTBE (20 mL). The combined organic layers were washed with H20 (20 mL) and transferred to a rotovap flask. Heptane (20 mL) was added, and the solution was concentrated until 16.9 g remained in the flask. An H20 layer appeared in the flask, and was pipetted out, leaving 12.8 g. Compound 1-6 seed was added, and the crystallizing mixture was stirred at 20-25 °C overnight. The flask was cooled in an ice bath for 2 h prior to filtration, and the isolated solid was washed with cold heptane (5 mL), and pulled dry on the funnel. After drying in a vacuum oven (40-45°C) for several hours 1.37 g of amino-alcohol ±1-6 was obtained, 60.8% yield, HPLC purity = 98.0%.
3f . 3-amino-2-(2,4-difluorophenyl)- 1 , 1-difluoro- 1 -(5-(4-(trifluoromethoxy)phenyl)pyridin-2- yl)propan-2-ol (1-6* or 1-7*)

Process Development
Table 9 illustrates the initial screen performed surveying various chiral acid/solvent combinations. All entries in Table 9 were generated using 0.1 mmoles of amino-alcohol ±1-6, 1 equivalent of the chiral acid, and 1ml of solvent.
Table 9. Resolution of ±1-6 (Initial Screen)

Since the best results from Table 9 were generated using tartaric acid and di-p-toluoyltartaric acid, Table 10 captures the results from a focused screen using these two chiral acids and various solvent combinations. All entries in Table 10 were performed with 0.2 mmoles of amino-alcohol ±1-6, 87 volumes of solvent, and each entry was exposed to heating at 51 °C for lh, cooled to RT, and stirred at RT for 24h.
Table 10. Resolution of ±1-6 (Focused Screen)

Each of the three entries using di-p-toluoyltartaric acid in Table 10 resulted in higher levels of enantio-enrichment when compared to tartaric acid. As such, efforts to further optimize the enantio-enrichment were focusing on conditions using di-p-toluoyltartaric acid (Table 11).

Ό.6 equivalents used
ee sense was opposite from the other entries in the table (i.e., enantiomer of 1-6*)
Typical Procedure for Converting +1-6 to 1-6* or 1-7*
(This experimental procedure describes resolution of ±1-6, but conditions used for DPPTA resolution of 1-6 or 1-7 are essentially the same.)
Amino-alcohol ±1-6 (7.0 g, 15 mmoles) was dissolved in a mixture of acetonitrile (84 mL) and methanol (21 mL). (D)-DPTTA (5.89 g, 15 mmoles) was added, and the reaction was warmed to 50°C and held for 2.5 h. The heat was then removed and the suspension was allowed to cool and stir at 20-25 °C for 65 h. The suspension was cooled in an ice bath and stirred for an additional 2 h. Solid was isolated by vacuum filtration, and the cake was washed with cold 8:2 ACN/MeOH (35 mL). After drying at 50°C, 5.18 g of 1-6* or l-7*/DPPTA salt was isolated, HPLC purity = 99.0, ee = 74.
The 1-6* or l-7*/DPPTA salt (5.18 g) was combined with 8:2 ACN/MeOH (68 mL) and the suspension was heated to 50°C and held for 20 min. After cooling to 20-25 °C the mixture was stirred for 16 h. Solids were isolated by vacuum filtration, and the cake washed with cold 8:2 ACN/MeOH (30 mL), and pulled dry on the funnel. 2.82 g of 1-6* or l-7*/DPPTA salt was obtained, 44.4% yield (from crude ±1-6), ee = 97.5. The resulting solids were freebased to provide 1-6* or 1-7* with the same achiral and chiral purity as the DPPTA salt.
EXAMPLE 4: Preparation of 2-(2.4-difluorophenyl -l.l-difluoro-3-(lH-tetrazol-l-yl -l-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la).


The procedure used to generate compound 1 or la is as described in US 4,426,531. Table 13 illustrates the efficient and quantitative nature of this procedure as performed on amino- alcohol 1-6* or 1-7* produced from both the TMS-cyanohydrin method and the TMSI- epoxidation method.
Table 13. Formation of Compound 1 or la

EXAMPLE 5: 2-(2.4-difluorophenyl -l.l-difluoro-3-(lH-tetrazol-l-yl -l-(5-(4- (trifluoromethoxy)phenyl)pyridin-2-yl)propan-2-ol benzenesulfonate (1 or la-BSA).

Typical Procedure for Converting 1 or la to 1 or la-BSA
46.6 g of compound 1 or la was dissolved in ethylacetate (360ml). The solution was filtered through a glass microfiber filter and placed in a 2 L reaction flask equipped with an overhead stirrer, condenser, and a J-Kem thermocouple. Pharma-grade benzenesulfonic acid (BSA, 14.39g, leq) was dissolved in ethyl acetate (100ml). The BSA solution was filtered through a glass microfiber filter and added to the stirred 1 or la solution in one portion. The mixture was warmed to 60-65 °C; precipitation of the 1 or la/BSA salt occurred during the warm up period. The slurry was held for 60 minutes at 60-65 °C. The suspension was allowed to slowly cool to 22 °C and was stirred at 20-25 °C for 16 hours. n-Heptane (920ml) was charged in one portion and the suspension was stirred at 22 °C for an additional 90 minutes. The slurry was filtered and the collected solids washed with n-heptane (250ml). The isolated solids were placed in a vacuum oven at 50 °C for 16 hours. 52.26g (86% yield) of 1 or la
benzenesulfonate was obtained.
*H NMR (400 MHz, DMSO-d6 + D20): 89.16 (s, 1H), 8.95 (d, J = 2.1 Hz, 1H), 8.26 (dd, J = 8.2, 2.3 Hz, 1H), 7.96-7.89 (m, 2H), 7.66-7.61 (m, 2H), 7.59 (dd, J = 8.3, 0.4 Hz, 1H), 7.53 (br d, J = 8.0 Hz, 2H), 7.38-7.15 (m, 5H), 6.90 (dt, J = 8.3, 2.5 Hz, 1H), 5.69 (d, J = 14.8 Hz, 1H), 5.15 (d, J = 15.2 Hz, 1H).
Further results are in Table 14.
Table 14. Formation of 1 or la-BSA
![]()
( ) (%ee) Yield Purity (%) ee
97.9 95.9 84% 98.2 97.1
Figures 1-2 contain the analytical data for 1 or la-BSA prepared by the TMSI-epoxidation process.
EXAMPLE 6: 5-bromo-2-((2-(2,4-difluorophenyl)oxiran-2-yl)difluoromethyl)pyridine -Br).

Typical Procedure for Converting 3-Br to 4-Br
KOtBu ( 41.7g, 0.372moles, 1.05 equiv) and trimethylsulfoxonium iodide ( 85.7g,
0.389moles, 1.1 equiv) were charged to a 3L 3-neck round bottom flask equipped with an overhead stirrer, a thermocouple and an addition funnel. 1.2L of anhydrous THF and 740mL of DMSO were added to the flask and stirred at 22-25 °C for 70 minutes. The contents were cooled to 0°C. Crude ketone 3 was dissolved in 250mL of anhydrous THF and slowly added the ketone 3-Br solution to the reaction mixture over 20 minutes while maintaining a reaction temperature at < 3°C during the addition and stirred at 0°C for one hour. In-process HPLC showed <1% ketone 3-Br remaining. 200mL of IN HC1 was slowly added maintaining a reaction temperature of < 6°C during the addition. After stirring for 30 minutes the layers were separated and the aqueous layer was extracted with 375mL of MTBE. The combined organic layers were washed with 375mL of aqueous 9% NaHCC>3 and with 375mL of aqueous 20% NaCl. The solvent was removed to yield 4-Br as a brown waxy solid.
Weight of crude epoxide 4-Br = 124.6g; *H NMR (400 MHz, d6-DMSO) : 58.82 (d, J= 2.3Hz, 1H), 8.21 ( dd, J= 8.3, 2.3Hz, 1H), 7.50 (dd, J= 8.3, 0.5Hz, 1H), 7.41 ( m, 1H), 7.25 ( m, 1H), 7.10 (m,lH), 3.40 ( d, J= 4.5Hz, 1H), 3.14 ( m, 1H). MS m/z 362 (M+H+), 364 (M+2+H+).
EXAMPLE 7: 3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan-2-ol (4b-Br).

Typical Procedure for Converting 4-Br to 4b-Br
Crude epoxide 4-Br ( 54.4g, 0.15moles) was placed into a Schott autoclave bottle equipped with a stir bar. 550mL of MeOH was added to the bottle and stirred for 90 minutes at 22-25 °C. Concentrated NH4OH ( 550mL, 7.98 moles, 53 equiv) was added to the epoxide 4-Br
solution. The bottle was sealed and placed in an oil bath at 55 °C. The mixture was stirred at 55°C for 17 hours. The bottle was removed from the oil bath and cooled to 22-25°C. In-process HPLC showed <1% epoxide 4-Br remaining. The solvent was removed via rotary evaporation until 362g ( 37%) of the reaction mass remained. 500mL of MTBE was added and cooled the mixture to 8°C. 500mL of 6N HCl was slowly added maintaining the reaction temperature between 8 – 12°C during the addition. After stirring for 10 minutes, the layers were separated. The MTBE layer was extracted with 350mL of 6N HCl. The combined aqueous layers were washed with 250mL MTBE and 2 X 250mL heptane. MTBE, 250mL, was added to the aqueous layer and the mixture was cooled to 2°C. 344g of KOH was dissolved in 500mL of water. The KOH solution was slowly added to the reaction mixture over one hour while maintaining the temperature at <19°C. After stirring for 15 minutes, the layers were separated. The aqueous layer was extracted with 250mL MTBE. The combined organic layers were washed with 250mL of aqueous 20% NaCl and the solvent was removed to yield ±4b-Br as a dark oil. Weight of crude amino alcohol ±4b-Br = 46.0g. HPLC purity ( by area %) = 92%; *H NMR (400 MHz, d6-DMSO) : 58.67 (d, J= 2.2Hz, 1H), 8.15 ( dd, J= 8.6, 2.4Hz, 1H), 7.46 (m, 1H), 7.40 ( dd, J= 8.5, 0.7Hz, 1H), 7.10 ( m, 1H), 7.00 (m,lH), 3.37 (dd, J= 13.7, 2.1Hz, 1H), 3.23 ( dd, J= 13.7, 2.7, 1H). MS m/z 379 (M+H+), 381 (M+2+H+).
EXAMPLE 8: 3-amino-l-(5-bromopyridin-2-yl -2-(2.4-difluorophenyl -l.l-difluoropropan-2-ol (4b-Br or 4c-Br).

Typical Procedure for Converting 4-Br to 4b-Br or 4c-Br
Crude amino alcohol ±4b-Br ( 42.4, O. llmoles) was dissolved in 425mL of 8:2 IPA: CH3CN. The solution was charged to a 1L 3-neck round bottom flask equipped with a condenser, overhead stirrer and a thermocouple. Charged di-p-toluoyl-L-tartaric acid ( 21.6g, 0.056moles, 0.5 equiv) to the flask and warmed the contents to 52°C. The reaction mixture was stirred at 52°C for 5 hours, cooled to 22-25°C and stirred for 12 hours. The slurry was cooled to 5-10°C and stirred for 90 minutes. The mixture was filtered and collected solids washed with 80mL of cold CH3CN. The solids were dried in a vacuum oven 45-50°C. Weight of amino alcohol/ DPTTA salt = 17.4g
Chemical purity by HPLC ( area %) = 98.5%; Chiral HPLC= 98.0% ee.
13.60g of the amino alcohol/ DPTTA salt was placed into a 250mL flask with a stir bar and to this was added lOOmL of MTBE and lOOmL of 10% aqueous K2CO3solution. The reaction was stirred until complete dissolution was observed. The layers were separated and the aqueous layer was extracted with 50mL of MTBE. The combined MTBE layers were washed with 50mL of 20% aqueous NaCl and the solvent removed to yield 8.84 (98%) of 4b-Br or 4c-Br as a light yellow oil.
EXAMPLE 9: 3-amino-2-(2,4-difluorophenyl)-l J-difluoro-l-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)propan-2-ol (1-6* or 1-7*).

Typical Procedure for Converting 4b-Br or 4c-Br to 1-6* or 1-7*
Amino alcohol 4b-Br or 4c-Br (8.84g, 0.023moles, 1 equiv) was dissolved in 73mL of n-propanol. The solution was transferred to a 250mL 3-neck round bottom flask equipped with a condenser, thermocouple, stir bar and septum. 17mL of water was added and stirred at 22-25°C for 5 minutes. To the reaction was added K2CO3 ( 9.67g, 0.07moles, 3 equiv), 4-(trifluoromethoxy)phenylboronic acid ( 5.76g, 0.028moles, 1.2 equiv.) and Pd(dppf)Cl2 as a CH2Cl2 adduct ( 0.38g, 0.47mmoles, 0.02 equiv) to the flask. After the mixture was purged with nitrogen for 10 minutes, the reaction was then warmed to 85-87°C and stirred at 85-87°C for 16 hours. HPLC analysis showed < 1% of the amino alcohol 4b-Br or 4c-Br remaining. The mixture was cooled to 22-25 °C, then 115mL of MTBE and 115mL of water were added and stirred for 30 minutes. The layers were separated and the organic layer was washed with 2 X 60mL of 20% aqueous NaCl. The solvent was removed to yield 12.96g ( 121% yield) of 1-6* or 1-7* as a crude dark oil. It should be noted that the oil contains residual solvent, Pd and boronic acid impurity.
‘ll NMR (400 MHz, d6-DMSO) : 58.90 (d, J= 2.2Hz, 1H), 8.22 ( dd, J= 8.3, 2.3Hz, 1H), 7.91 (m, 2H), 7.54 ( m, 4H), 7.14 ( m, 1H), 7.02 (m,lH), 3.41 (m, 1H), 3.27 ( dd, J= 14.0, 2.7, 1H). MS m/z 461 (M+H+)
CLIP
Med. Chem. Commun., 2016,7, 1285-1306
DOI: 10.1039/C6MD00222F
Fungal infections directly affect millions of people each year. In addition to the invasive fungal infections of humans, the plants and animals that comprise our primary food source are also susceptible to diseases caused by these eukaryotic microbes. The need for antifungals, not only for our medical needs, but also for use in agriculture and livestock causes a high demand for novel antimycotics. Herein, we provide an overview of the most commonly used antifungals in medicine and agriculture. We also present a summary of the recent progress (from 2010–2016) in the discovery/development of new agents against fungal strains of medical/agricultural relevance, as well as information related to their biological activity, their mode(s) of action, and their mechanism(s) of resistance.

CLIP
Volume 24, Issue 15, 1 August 2014, Pages 3455–3458
Design and optimization of highly-selective fungal CYP51 inhibitors
- Viamet Pharmaceuticals Inc., Durham, NC 27703, USA


| Compound | R | C. albicans MICa | T. rubrum MICa | CYP3A4 IC50b | Selectivity indexc |
|---|---|---|---|---|---|
| 7a | Cl | ⩽0.001 | 0.004 | 36 | 9000 |
| 7b | CF3 | ⩽0.001 | 0.002 | 54 | 27,000 |
| 7c
VT 1129 |
OCF3 | ⩽0.001 | ⩽0.001 | 79 | >79,000 |
| 7d
VT 1161 |
OCH2CF3 | ⩽0.001 | ⩽0.001 | 65 | >65,000 |
| Itraconazole | — | 0.016 | 0.062 | 0.07 | 1.1 |
- aMinimum concentration that achieved 50% inhibition of fungal growth; MIC units in μg/mL.5
- bInhibition of CYP3A4 measured in microsomes obtained from pooled human hepatocytes, IC50 units in μM.8
- cIn vitro selectivity calculated as CYP3A4 IC50/T. rubrum MIC.
- (R)-(+)-Enantiomers (7a–7d) were isolated from racemates using chiral chromatography.
- 16 Hoekstra, W.J.; Schotzinger, R.J.; Rafferty, S.W. U.S. Patent 8,236,962 issued Aug. 7, 2012.
update………….
QUILSECONAZOLE, VT 1129, New Patent, WO, 2017049080, Viamet

<p>Formula (I)</p> <p>Crizotinib is a potent small-molecule inhibitor of c-Met/HGFR (hepatocyte growth factor receptor) kinase and ALK (anaplastic lymphoma kinase) activity. Enantiomerically pure compound of formula I was first disclosed in US Patent No. 7,858,643. Additionally, the racemate of compound of formula I was disclosed in U.S. patent application 2006/0128724, both of these references discloses similar methods for the synthesis of Compound of Formula I.</p> <p>Conventionally, the compounds of formula I are prepared by reacting Bis(pinacolato)diboron with protected 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-pyridin-2-ylamine in the presence of Pd catalyst. The obtained product after deprotection is reacted with N- protected 4-(4-bromo-pyrazol-l-yl)-piperidine in the presence of Pd Catalyst. The obtained product is filtered through celite pad and purified by Column Chromatography. The final product of formula I was obtained by deprotection of the purified compound by using HCl/dioxane. US Patent No. 7,858,643 provides enantiomerically pure aminoheteroaryl compounds, particularly aminopyridines and aminopyrazines, having protein tyrosine kinase activity. More particularly, US 7,858,643 describes process for the preparation of 3-[(lR)-l-(2,6- dichloro-3-fluorophenyl)ethoxy]-5-(l-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine. The Scheme is summarized below in Scheme- 1 :</p>


<p>Scheme-1</p> <p>wherein, “Boc” means tert-butoxycarbonyl; and a) (Boc)<sub>2</sub>, DMF, Dimethylaminopyridine b) Pd(dppf)Cl<sub>2</sub>, KOAc, Dichloromethane; c) HC1, Dioxane, Dichloromethane; d) Pd(PPh<sub>3</sub>)<sub>2</sub>Cl<sub>2</sub>, Na<sub>2</sub>C0<sub>3</sub>, DME/H<sub>2</sub>0; e) 4M HCl/Dioxane, Dichloromethane</p> <p>A similar process has been disclosed in the U.S. patent application 2006/0128724 for the preparation of Crizotinib. J. Jean Cui et. al. in J. Med. Chem. 2011, 54, 6342-6363, also provides a similar process for the preparation of Crizotinib and its derivatives.</p> <p>However, above mentioned synthetic process requires stringent operational conditions such as filtration at several steps through celite pad. Also column chromatography is required at various steps which is not only tedious but also results in significant yield loss. Another disadvantage of above process involves extensive use of palladium catalysts, hence metal scavengers are required to remove palladium content from the desired product at various steps which makes this process inefficient for commercial scale.</p> <p>Yet another disadvantage of above process is the cost of Bis(pinacolato)diboron. This reagent is used in excess in the reaction mixture resulting in considerable cost, especially during large-scale syntheses.</p> <p>US Patent No. 7,825,137 also discloses a process for the preparation of Crizotinib where Boc protected 4-(4-iodo-pyrazol-l-yl)-piperidine is first reacted with Bis(pinacolato)diboron in the presence of Pd catalyst. The reaction mixture is filtered through a bed of celite and the obtained filtrate is concentrated and purified by silica gel chromatography to give to form tert-butyl-4-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]piperidine-l- carboxylate. To this compound, 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]- pyridin-2-ylamine is added in the presence of a Pd catalyst. The reaction mixture is stirred for 16h at 87°C. The reaction mixture is filtered through celite pad and the concentrated filtrate is purified on silica gel column to obtain (4-{6-amino-5-[(R)-l-(2,6-dichloro-3-fluoro- phenyl)-ethoxy]-pyri- din-3-yl}-pyrazol-l-yl)-piperidine-l-carboxylic acid tert-butyl ester of 95% purity. To the solution of resulting compound in dichloromethane 4N HCl/Dioxane is added and thereby getting the reaction suspension is filtered in Buchner funnel lined with filter paper. The obtained solid is dissolved in HPLC water and pH is adjusted to 10 with the addition of Na<sub>2</sub>C0<sub>3</sub> Compound is extracted using dichloroform and is purified on a silica gel column by eluting with CH<sub>2</sub>Cl<sub>2</sub> MeOH/NEt<sub>3</sub> system to obtain Crizotinib. The scheme is summarized below in scheme 2:</p>

<p>Formula (i) Formula (ii)</p>

<p>Formula (iii) Formula (ii) ula (iv)</p>

<p>Formula (v) Formula (I)</p> <p>Scheme-2</p> <p><span style=”color:#ff0000;”>Preparation of Crizotinib:</span></p> <p>To a stirred solution of Tert-butyl 4-(4-{ 6-amino-5-[(li?)-l-(2,6-dichloro-3- fluorophenyl)ethoxy]pyridin-3 -yl } – lH-pyrazol- 1 -yl)piperidine- 1 -carboxylate (material obtained in Example 3) (l.Og, 0.00181 moles) in dichloromethane (-13 ml) at 0°C was added 4.0 M dioxane HQ (6.7 ml, 0.0272 moles). Reaction mixture was stirred at room temperature for 4h. After the completion of reaction monitored by TLC, solid was filtered and washed with dichloromethane (10 ml). The obtained solid was dissolved in water (20 ml); aqueous layer was extracted with dichloromethane (10×2). The pH of aqueous layer was adjusted to 9-10 with Na<sub>2</sub>C03 and compound was extracted with dichloromethane (10 x 3), combined organic layers were washed with water (20 ml), evaporated under vacuum to get solid product. The solid was stirred with ether (10 ml), filtered off, washed well with ether, dried under vacuum to get <span style=”color:#ff0000;”>Crizotinib.</span></p> <p>Yield: 0.45g (55 %)</p> <p>HPLC Purity: 99.35 %</p> <p><span style=”color:#ff0000;”>1HNMR (400 MHz, CDC1<sub>3</sub>) δ: 7.76 (d, J = 1.6 Hz, 1H), 7.56 (s, 1H), 7.49 (s, 1H), 7.30 (dd, J = 9.2 Hz), 7.0 (m, 1H), 6.86 (d, J = 1.6 Hz, 1H), 6.09 ( q, J= 6.8 Hz, 1H), 4.75 (brs, 1H), 4.19 (m, 1H), 3.25 (m, 2H), 2.76 (m, 2H), 2.16 (m, 2H), 1.92 (m, 2H), 1.85 (d, J= 6.8 Hz, 3H), 1.67 (brs, 1H)</span></p> <p>…………………………</p> <p><a href=”http://www.sciencedirect.com/science/article/pii/S0040403914000872″>http://www.sciencedirect.com/science/article/pii/S0040403914000872</a></p>
Abstract
A novel approach for the synthesis of Crizotinib (1) is described. In addition, new efficient procedures have been developed for the preparation of (S)-1-(2,6-dichloro-3-fluorophenyl)ethanol (2) and tert-butyl 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)piperidine-1-carboxylate (4), the key intermediates required for the synthesis of Crizotinib.
<hr id=”absgraphicalab0051″ class=”artHeader” />
Graphical abstract
textboxdefaultfig
“>

- …………………
- http://www.sciencedirect.com/science/article/pii/S0040403911021745
-
Abstract
4-(4-Iodo-1H-pyrazol-1-yl)piperidine is a key intermediate in the synthesis of Crizotinib. We report a robust three-step synthesis that has successfully delivered multi-kilogram quantities of the key intermediate. The process includes nucleophilic aromatic substitution of 4-chloropyridine with pyrazole, followed by hydrogenation of the pyridine moiety and subsequent iodination of the pyrazole which all required optimization to ensure successful scale-up.
<hr id=”absgraphical1″ class=”artHeader” />
Graphical abstract
textboxdefaultfig
“>
</div> </div> </dt> </dl> </div> </div> </div> <p>……………………</p>
Org. Process Res. Dev., 2011, 15 (5), pp 1018–1026DOI: 10.1021/op200131n
<p class=”articleBody_abstractText”>A robust six-step process for the synthesis of crizotinib, a novel c-Met/ALK inhibitor currently in phase III clinical trials, has been developed and used to deliver over 100 kg of API. The process includes a Mitsunobu reaction, a chemoselective reduction of an arylnitro group, and a Suzuki coupling, all of which required optimization to ensure successful scale-up. Conducting the Mitsunobu reaction in toluene and then crystallizing the product from ethanol efficiently purged the reaction byproduct. A chemoselective arylnitro reduction and subsequent bromination reaction afforded the key intermediate <b>6</b>. A highly selective Suzuki reaction between <b>6</b> and pinacol boronate <b>8</b>, followed by Boc deprotection, completed the synthesis of crizotinib <b>1</b>.</p> </div> <p><span id=”d43162769e1806″ class=”title2″>3-[(1<i>R</i>)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1<i>H</i>-pyrazol-4-yl]pyridin-2-amine <b>1</b></span></p> <p><span style=”color:#ff0000;”> <i>crizotinib</i><b>1</b> (20.7 kg, 80%) as a white solid. </span></p> <p><span style=”color:#ff0000;”>Mp 192 °C;</span></p> <p><span style=”color:#ff0000;”><sup>1</sup>H NMR (400 MHz, CDCl<sub>3</sub>) δ: 7.78 (d, <i>J</i> = 1.8 Hz, 1H), 7.58 (s, 1H), 7.52 (s, 1H), 7.31 (dd, <i>J</i> = 9.0, 4.9 Hz, 1H), 7.06 (m, 1H), 6.89 (d, <i>J</i> = 1.7 Hz, 1H), 6.09 (q, 1H), 4.79 (br s, 2H), 4.21 (m, 1H), 3.26 (m, 2H), 2.78 (m, 2H), 2.17 (m, 2H), 1.90 (m, 2H), 1.87 (d, <i>J</i> = 6.7 Hz, 3H), 1.63 (br s, 1H).</span></p> <p><span style=”color:#ff0000;”> <sup>13</sup>C NMR (100.6 MHz, CDCl<sub>3</sub>) δ: 157.5 (d, <i>J</i> = 250.7 Hz), 148.9, 139.8, 137.0, 135.7, 135.6, 129.9, 129.0 (d, <i>J</i> = 3.7 Hz), 122.4, 122.1 (d, <i>J</i> = 19.0 Hz), 119.9, 119.3, 116.7 (d, <i>J</i> = 23.3 Hz), 115.0, 72.4, 59.9, 45.7, 34.0, 18.9.</span></p> <p><span style=”color:#ff0000;”> LC-MS: found <i>m</i>/<i>z</i> 450.0, 451.0, 452.0, 453.0, 454.0, 455.0. </span></p> <p><span style=”color:#ff0000;”>Anal. Calcd for C<sub>21</sub>H<sub>22</sub>Cl<sub>2</sub>FN<sub>5</sub>O: C, 56.01; H, 4.92; N, 15.55. Found: C, 56.08; H, 4.94; N, 15.80.</span></p>
Cui, J. J.; Botrous, I.; Shen, H.; Tran-Dube, M. B.; Nambu, M. D.; Kung, P.-P.; Funk, L. A.; Jia, L.; Meng, J. J.; Pairish, M. A.; McTigue, M.; Grodsky, N.; Ryan, K.; Alton, G.; Yamazaki, S.; Zou, H.; Christensen, J. G.; Mroczkowski, B.Abstracts of Papers; 235th ACS National Meeting, New Orleans, LA, United States, April 6–10, 2008.
</div>
Cui, J. J.; Funk, L. A.; Jia, L.; Kung, P.-P.; Meng, J. J.; Nambu, M. D.; Pairish, M. A.; Shen, H.; Tran-Dube, M. B. U.S. Pat. Appl. U. S. 2006/0046991 A1, 2006.
 Cosy predict above1H NMR PREDICT
Cosy predict above1H NMR PREDICT![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-1h.png) 13C NMR PREDICT
13C NMR PREDICT![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-13c.png)
WO2006021881A2 * 15 Aug 2005 2 Mar 2006 Pfizer Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors WO2006021884A2 * 15 Aug 2005 2 Mar 2006 Pfizer Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors WO2013181251A1 * 29 May 2013 5 Dec 2013 Ratiopharm Gmbh Crizotinib hydrochloride salt in crystalline EP2620140A1 * 26 Jan 2012 31 Jul 2013 ratiopharm GmbH Crizotinib containing compositions textboxdefaultfig
“>
-
textboxdefaultfig
“>
WO2010048131A1 * Oct 20, 2009 Apr 29, 2010 Vertex Pharmaceuticals Incorporated C-met protein kinase inhibitors WO2011042389A2 * Oct 4, 2010 Apr 14, 2011 Bayer Cropscience Ag Phenylpyri(mi)dinylazoles US7825137 Nov 23, 2006 Nov 2, 2010 Pfizer Inc. Method of treating abnormal cell growth US7858643 Aug 26, 2005 Dec 28, 2010 Agouron Pharmaceuticals, Inc. Crizotinib, a c-Met protein kinase inhibitor anticancer agent; 3-[(R)-1-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-5-(1-piperidin-4-yl-1H-pyrazol-4-yl)-pyridin-2-ylamine is crizotinib US20060128724 Aug 26, 2005 Jun 15, 2006 Agouron Pharmaceuticals, Inc. Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors 1 J. JEAN CUI J. MED. CHEM. vol. 54, 2011, pages 6342 – 6363 2 ORG. PROCESS RES. DEV. vol. 15, 2011, pages 1018 – 1026 3 * PIETER D. DE KONING ET AL: “Fit-for-Purpose Development of the Enabling Route to Crizotinib (PF-02341066)“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 15, no. 5, 16 September 2011 (2011-09-16), pages 1018-1026, XP055078841, ISSN: 1083-6160, DOI: 10.1021/op200131n 
VT 1129 BENZENE SULFONATE
CAS 1809323-18-9

VT 1129
1340593-70-5 CASMF C22 H14 F7 N5 O2, MW 513.372-Pyridineethanol, α-(2,4-difluorophenyl)-β,β-difluoro-α-(1H-tetrazol-1-ylmethyl)-5-[4-(trifluoromethoxy)phenyl]-, (αR)-R ISOMERROTATION +QUILSECONAZOLE, VT-1129
Viamet, in collaboration with Therapeutics for Rare and Neglected diseases, is investigating quilseconazole benzenesulfonate (VT-1129), a small-molecule lanosterol demethylase (CYP51) inhibitor, developed using the company’s Metallophile technology, for treating fungal infections, including Cryptococcus neoformans meningitis.
WO-2017049080



////////VT 1129, VIAMET, WO 2016149486, Viamet Pharmaceuticals, Antifungals, Small molecules, 14-alpha demethylase inhibitors, Orphan Drug Status, Cryptococcosis, On Fast track, PHASE 1, VT-1129, QUILSECONAZOLE
O[C@@](Cn1cnnn1)(c2ccc(F)cc2F)C(F)(F)c3ccc(cn3)c4ccc(OC(F)(F)F)cc4



Nacubactam, A diazabicyclooctane beta-lactamase inhibitor, for treating bacterial infection


Nacubactam
RG-6080, FPI-1459, OP-0595, WK ?, WK-?, WK?
CAS 1452458-86-4, MF C9 H16 N4 O7 S, MW 324.31
Sulfuric acid, mono[(1R,2S,5R)-2-[[(2-aminoethoxy)amino]carbonyl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester,
(2S,5R)-N-(2-amino ethoxy)-6-(sulfooxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
Beta lactamase inhibitor
Roche, under license from Meiji Seika Pharma and Fedora Pharmaceuticals is developing nacubactam hydrate
Meiji Seika Pharma Co., Ltd., Meiji Seikaファルマ株式会社
A diazabicyclooctane beta-lactamase inhibitor, for treating bacterial infection. In July 2016, nacubactam was reported to be in phase 1 clinical development
PATENTS , IN2015MU287, WO2016116878, WO 2016120752, INDICATE INTEREST FROM WOCKHARDT


Sulfuric acid, mono[(1R,2S,5R)-2-[[(2-aminoethoxy)amino]carbonyl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
A β-lactamase inhibitor potentially for the treatment of bacterial infections.
![]()
RG-6080; FPI-1459; OP-0595
CAS No. 1452458-86-4
| Molecular Formula | C9 H16 N4 O7 S |
| Formula Weight | 324.31 |
- Originator Fedora Pharmaceuticals
- Developer Meiji Seika Pharma
- Class Antibacterials; Azabicyclo compounds
- Mechanism of Action Beta lactamase inhibitors
- Phase I Bacterial infections
Most Recent Events
- 13 Jan 2015 OP 0595 licensed to Roche worldwide, except Japan ,
- 30 Nov 2014 Meiji Seika Pharma completes a phase I trial in Healthy volunteers in Australia (NCT02134834)
- 01 May 2014 Phase-I clinical trials in Bacterial infections (in volunteers) in Australia (IV)

In September 2014, preclinical data were presented at the 54th ICAAC Meeting in Washington, DC. Nacubactam hydratedemonstrated Ki values of 0.24, 3 and 0.79 microM against AmpC P99 derived from Enterobacter cloacae, KPC-3, and CTX-M-15 enzymes, respectively; the Ki values were lower than that of cefepime
Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate.
The persistent exposure of bacterial strains to a multitude of beta- lactam antibacterial agents has led to overproduction and mutation of beta-lactamases. These new extended spectrum beta-lactamases (ESBL) are capable of hydrolyzing penicillins, cephalosporins, monobactams and even carbapenems. Such a wide spread resistance to many of the existing beta-lactam antibacterial agents, either used alone or in combination with other agents, is posing challenges in treating serious bacterial infections.
Due to various reasons, the oral therapeutic options for treating bacterial infections (including those caused by ESBL strains) are limited. For example, a combination of amoxicillin and clavulanic acid is effective against Class A ESBLs producing bacteria. However, the usefulness of this combination is compromised against bacteria producing multiple or mixed beta-lactamase enzymes (such as, for example, bacteria producing Class A and Class C ESBLs concurrently), and Klebsiella pneumoniae carbapenemases (KPCs). Therefore, oral antibacterial agents or combinations with activity against a range of bacterial strains (including those producing multiple ESBLs and KPCs) are urgently desired.
Cephalosporin antibacterial agents are known for treatment for various bacterial infections. Surprisingly, it has been found that pharmaceutical compositions comprising a cephalosporin antibacterial agent and certain nitrogen containing bicyclic compound (disclosed in PCT/IB2013/053092, PCT/JP2013/064971 and PCT/IB 2012/002675) exhibit unexpectedly synergistic antibacterial activity, even against highly resistant bacterial strains.
SYNTHESIS
WO 2015046207,
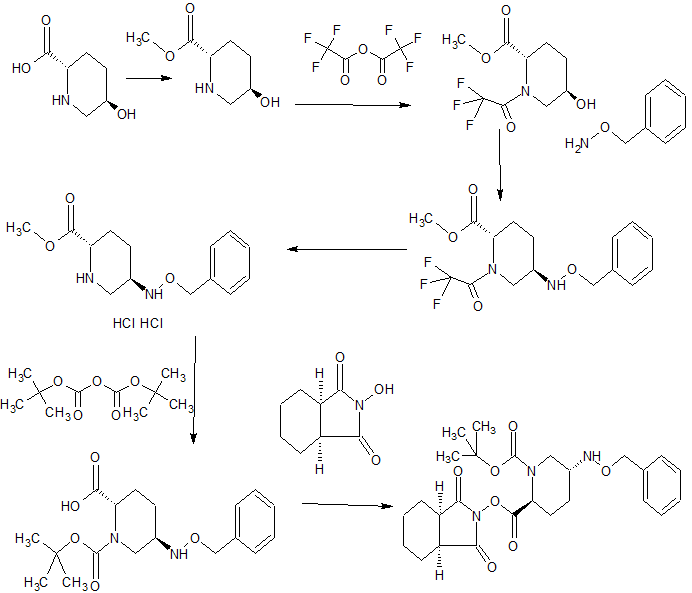
CONTD…………………..
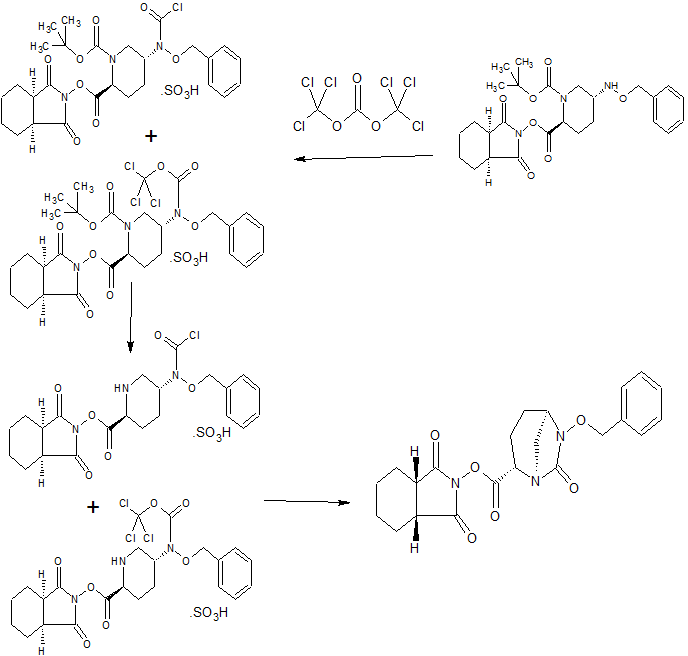
CONTD………………………………..
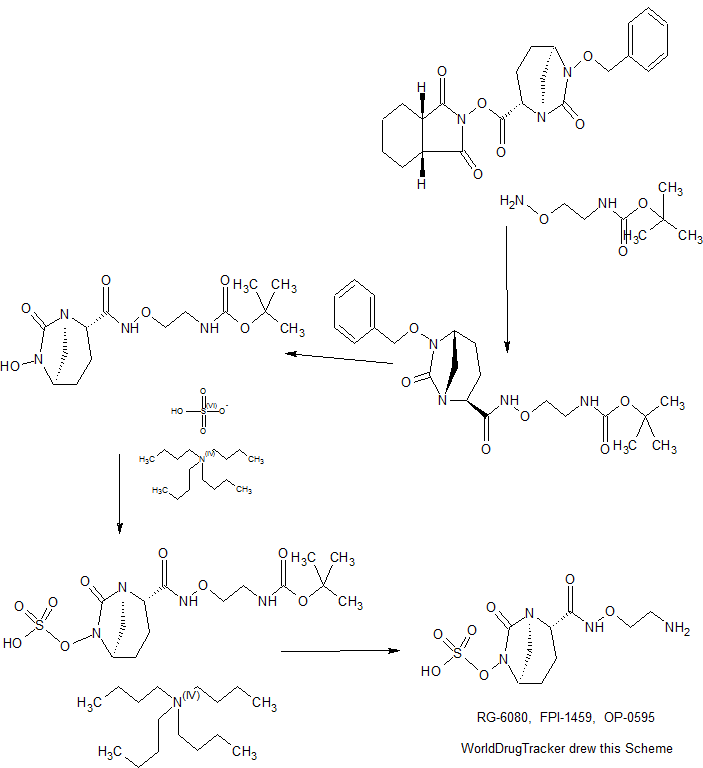
Patent
The novel heterocyclic compound in Japanese Patent 4515704 (Patent Document 1), preparation and shown for their pharmaceutical use, sodium trans-7-oxo-6- (sulfooxy) as a representative compound 1,6-diazabicyclo [3 .2.1] discloses an octane-2-carboxamide (NXL104). Preparation in regard to certain piperidine derivatives which are intermediates Patent 2010-138206 (Patent Document 2) and JP-T 2010-539147 (Patent Document 3) are shown at further WO2011 / 042560 (Patent Document 4) NXL104 to disclose a method for producing the crystals.
In Patent 5038509 (Patent Document 5) (2S, 5R) -7- oxo -N- (piperidin-4-yl) -6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane – 2- carboxamide (MK7655) is shown, discloses the preparation of certain piperidine derivatives with MK7655 at Patent 2011-207900 (Patent Document 6) and WO2010 / 126820 (Patent Document 7).
The present inventors also disclose the novel diazabicyclooctane derivative represented by the following formula (VII) in Japanese Patent Application 2012-122603 (Patent Document 8).
Patent Document 1: Japanese Patent No. 4515704 Pat
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
[Chemical formula 1] (In the formula, R 3 are the same as those described below)
Reference Example
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
step 1 tert-butyl {2 – [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate (IV-1)(2S, 5R)-6-(benzyloxy) -7-oxo-1,6-diazabicyclo [3.2.1] octane-2-carboxylic acid (4 .30g, dehydrated ethyl acetate (47mL) solution of 15.56mmol) was cooled to -30 ℃, isobutyl chloroformate (2.17g, washing included dehydration ethyl acetate 1mL), triethylamine (1.61g, washing included dehydration ethyl acetate 1 mL), successively added dropwise, and the mixture was stirred 1 hour at -30 ° C.. To the reaction solution tert- butyl 2-dehydration of ethyl acetate (amino-oxy) ethyl carbamate (3.21g) (4mL) was added (washing included dehydration ethyl acetate 1mL), raising the temperature over a period of 1.5 hours to 0 ℃, It was further stirred overnight. The mixture of 8% aqueous citric acid (56 mL), saturated aqueous sodium bicarbonate solution (40 mL), sequentially washed with saturated brine (40 mL), dried over anhydrous magnesium sulfate, filtered, concentrated to 5 mL, up to 6mL further with ethanol (10 mL) It was replaced concentrated. Ethanol to the resulting solution (3mL), hexane the (8mL) in addition to ice-cooling, and the mixture was stirred inoculated for 15 minutes. The mixture was stirred overnight dropwise over 2 hours hexane (75 mL) to. Collected by filtration the precipitated crystals, washing with hexane to give the title compound 5.49g and dried in vacuo (net 4.98 g, 74% yield). HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 50/50, 1.0 mL / min, UV 210 nm, Retweeted 4.4 min; 1 H NMR (400 MHz, CDCl 3 ) [delta] 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m, 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br.d., J = 11.6 Hz , 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br. . s, 1H), 7.34-7.48 (yd, 5H), 9.37 (Br.S., 1H); MS yd / z 435 [M + H] + .
Step 2
tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate
(V-1) tert-butyl {2 – [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
Step 3
Tetrabutylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide – pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
Step 4 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
tetra butylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
PATENT
Example
64 tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]
tert- butyl {2 – [({[(2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate (example 63q, net 156.42g, 360mmol) in methanol solution (2.4L) of 10% palladium carbon catalyst (50% water, 15.64g) was added, under an atmosphere of hydrogen, stirred for 1.5 hours did. The catalyst was filtered through celite, filtrate was concentrated under reduced pressure until 450mL, concentrated to 450mL by adding acetonitrile (1.5 L), the mixture was stirred ice-cooled for 30 minutes, collected by filtration the precipitated crystals, washing with acetonitrile, and vacuum dried to obtain 118.26g of the title compound (net 117.90g, 95% yield). Equipment data of the crystals were the same as those of the step 2 of Reference Example 3.
Example
65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)
tert- butyl {2 – [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (example 64,537.61g, 1.561mol) in acetonitrile (7.8L) solution of 2,6-lutidine (512.08g), sulfur trioxide – pyridine complex (810.3g) was added, at room temperature in the mixture was stirred overnight. Remove insolubles and the mixture was filtered, the filtrate concentrated to 2.5 L, diluted with ethyl acetate (15.1L). The mixture was extracted with 20% phosphoric acid 2 hydrogencarbonate aqueous solution (7.8L), the resulting aqueous layer into ethyl acetate (15.1L), added tetrabutylammonium hydrogen sulfate (567.87g), was stirred for 20 min. The organic layer was separated layers, dried over anhydrous magnesium sulfate (425 g), after filtration, concentration under reduced pressure, substituted concentrated tetrabutylammonium tert- butyl with dichloromethane (3.1L) {2 – [({[(2S, 5R ) -7-oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 758g (net 586.27g, Osamu rate 84%).
The tetra-butyl ammonium salt 719g (net 437.1g, 0.656mol) in dichloromethane (874mL) solution was cooled to -20 ℃, dropping trifluoroacetic acid (874mL) at 15 minutes, 1 the temperature was raised to 0 ℃ It was stirred time. The reaction was cooled to -20 ° C. was added dropwise diisopropyl ether (3.25L), and the mixture was stirred for 1 hour the temperature was raised to 0 ° C.. The precipitate is filtered, washed with diisopropyl ether to give the title compound 335.36g of crude and vacuum dried (net 222.35g, 99% yield).
The title compound of crude were obtained (212.99g, net 133.33g) and ice-cold 0.2M phosphate buffer solution of pH5.3 mix a little at a time, alternating between the (pH6.5,4.8L). The solution was concentrated under reduced pressure to 3.6L, it was adjusted to pH5.5 at again 0.2M phosphate buffer (pH6.5,910mL). The solution resin purification (Mitsubishi Kasei, SP207, water ~ 10% IPA solution) is subjected to, and concentrated to collect active fractions, after lyophilization, to give the title compound 128.3 g (96% yield). Equipment data of the crystals were the same as those of step 3 of Reference Example 3.
PATENT
US 20140288051
WO 2014091268
WO 2013180197
US 20130225554
PATENT
IN2015MU287
PATENT
Example 59
(2S, 5R) -N- (2- aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (II-059)

Step 1
tert- butyl {2 – [({[(2S, 5R) -6- Benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate

Acid of Example 9 or 16 (6b, 1.34g, 4.87mmol) in methylene chloride (35mL) solution of triethylamine (2.71mL), N- ethyl -N ‘- (3- dimethylaminopropyl) carbodiimide hydrochloride (1.41g), 1- hydroxybenzotriazole monohydrate (1.15g), were added tert- butyl of Reference Example 9, wherein 2- (amino-oxy) ethyl carbamate (1.12g), room temperature It was stirred overnight Te.Water was added to the reaction solution to a residue obtained by concentration under reduced pressure, and extracted with ethyl acetate. The resulting organic layer with 0.1M hydrochloric acid, saturated aqueous sodium bicarbonate solution, washed with saturated aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered and concentrated.The resulting residue was purified by silica gel column and purified by chromatography (hexane / ethyl acetate = 8 / 2-0 / 10) to give the title compound 1.77g (84% yield).
[Α] D 20 -0.08 ° (c 0.29, CHCl 3); 1 H NMR (400 MHz, CDCl 3), δ: 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m , 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br d, J = 11.6 Hz, 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br s, 1H), 7.34-7.48 (m, 5H), 9.37 (br s, 1H); MS m / z 435 [M + H] +; enantiomeric excess of 99.9% or higher ee (CHIRALPAK AD-H, 4.6x150mm, hexane / ethanol = 2/1, UV210nm, flow rate 1mL / min, retention time 4.95min (2R, 5S), 6.70min (2S, 5R).
Step 2
tert- butyl {2 – [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate

Compound of the above Step 1 (3.91g, 9.01mmol) in methanol (80mL), 10% palladium on carbon catalyst (50% water, 803mg) was added, under hydrogen atmosphere and stirred for 45 minutes. The reaction mixture was filtered through Celite, then concentrated under reduced pressure, to give 3.11g of the title compound (quantitative).
1 H NMR (400 MHz, CD 3 OD), δ: 1.44 (s, 9H), 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 ( br dd, J = 15.0, 7.0 Hz, 1H), 3.03 (d, J = 12.0 Hz, 1H), 3.12 (br d, J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS m / z 345 [M + H] +.
Step 3
(2S, 5R) -N- (2- aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide The above step 2 compound (3. 09g, in methylene chloride (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide – was added pyridine complex (3.58g), and stirred at room temperature overnight. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, and washed the aqueous layer with chloroform, and tetrabutylammonium hydrogen sulfate (3.47g) and chloroform (30mL) was added to the aqueous layer and stirred for 10 minutes. After extracting the aqueous layer with chloroform, drying the resulting organic layer over anhydrous sodium sulfate, filtered, concentrated under reduced pressure tetrabutylammonium tert- butyl {2 – [({[(2S, 5R) -7- oxo – 6- (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 5.46g (91% yield).
1 H NMR (400 MHz, CDCl 3), δ: 1.01 (t, J = 7.4 Hz, 12H), 1.37-1.54 (m, 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30-2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85- 3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s, 1H), 9.44 (br s, 1H); MS m / z 425 [M-Bu 4 N + 2H] +.
The tetrabutyl ammonium salt (5.20g, 7.82mmol) in methylene chloride (25mL) solution of under ice-cooling trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed resulting residue with diethyl ether, at aqueous sodium bicarbonate was adjusted to pH7, it performs an octadecyl silica gel column chromatography (water), after freeze-drying, 1.44g of the title compound The obtained (57% yield).
[Α] D 24 -63.5 ° (c 0.83, H 2 O); 1 H NMR (400 MHz, D 2 O), δ: 1.66-1.76 (m, 1H), 1.76-1.88 (m, 1H), 1.91 -2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz, 2H), 3.18 (br d, J = 12.0 Hz , 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4, 3.2 Hz, 1H); MS m / z 325 [ M + H] +.
PATENT

ANTIBACTERIAL COMPOSITIONS OF A BETA-LACTAMASE INHIBITOR WITH A CEPHALOSPORINAbstract:
Pharmaceutical compositions comprising: (a) at least one cephalosporin antibacterial agent and (b) a compound of Formula (I) or a stereoisomer or a pharmaceutically acceptable derivative thereof are disclosed. Formula (I)
PATENT
WO 2016120752, WOCKHARDT, NEW PATENT, Nacubactam
![]()
Formula (I), chemically known as (25, 5i?)-N-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide has antibacterial properties and is disclosed in PCT International Patent Application No. PCT/IB2013/053092, PCT/JP2013/064971 and PCT/IB2012/002675. The present invention discloses a process for preparation of a compound of Formula (I).
Formula (I)

(VII) (VIII) (IX)
Scheme 2
Example 1
Synthesis of fert-butyl-r2-(aminooxy) ethyllcarbamate (III)
Preparation of fert-butyl-2-hydroxy ethylcarbamate (VIII):
Formula (VIII)

To a stirred solution of ethanolamine (50.0 g, 0.8186 mol) in dichloromethane (1000 ml), was added triethylamine (124 g, 1.228 mol) at 0°C. After 10 minutes, di-teri-butyl dicarbonate (VII, 214.15 g, 0.9823 mol) was added drop wise at 0°C under continuous stirring. Then reaction mass was allowed to warm to 25°C and stirred further for 3 hours. After completion of reaction, the resulting reaction mixture was poured into water (250 ml) and the organic layer was separated and dried over anhydrous sodium sulfate. The dried organic layer was concentrated under reduced pressure to obtain 130 g of the titled product as colorless oil in 98% yield.
Analysis:
Mass: 162 (M+l); for Molecular Weight of 161.2 and Molecular Formula of C7H15NO3.
1H NMR (400MHz, CDC13): δ 4.92(br s,lH), 3.72-3.68(q,2H), 3.30-3.26(q,2H), 2.33(br s,lH), 1.44(s,9H).
Preparation of A7-Boc-2-(2-aminoethoxy)isoindoline-l,3-dione (IX):

To a stirred solution of teri;butyl-2-hydroxy-ethylcarbamate (VIII, 50 g, 0.3106 mol) in tetrahydrofuran (500 ml), was added triphenylphosphine (89.5 g, 0.3416 mol) at 25°C. After stirring for 10 minutes, a solution of N-hydroxyphthalimide (50.66 g, 0.3106 mol) in dichloromethane (250 ml) was added to the reaction mass at 25 °C over a period of 10 minutes. After stirring for further 10 minutes, diisopropyl azodicarboxylate (69.1 g, 0.3416 mol) was added to the reaction mass in small portions (exothermic reaction was observed up to 34°C). The resulting reaction mass was stirred further at 25°C. After 16 hours, the reaction mass was concentrated under reduced pressure to obtain colorless oily material. The oily residue was diluted with diisopropyl ether (200 ml) and stirred for 30 minutes. The separated solid was filtered under suction. The filtrate was evaporated under reduced pressure and the residue subjected to di-isopropyl ether treatment (200 ml). This procedure was repeated once again. The filtrate was concentrated to obtain a solid product. The obtained solid was washed with diisopropyl ether (50 ml) and dried under reduced pressure. This solid contains small amount of triphenylphosphine oxide, along with the product. This was used as such for the next reaction without further purification.
Analysis:
Mass: 307.2 (M+l); for Molecular Weight of 306.3 and Molecular Formula of Ci5Hi8N205; 1H NMR of purified material (400MHz, CDC13): 7.85-7.25 (m,4H), 5.62(br s,lH), 4.26-4.23(t,2H), 3.46-3.42(q,2H), 1.46(s,9H).
Step 3: Preparation of fert-butyl-[ -(aminooxy) ethyl]carbamate (III):
Formula (III)

To a stirred solution of N-Boc-2-(2-aminoethoxy)isoindoline-l ,3-dione (IX, 97 g, 0.3167 mol) in dichloromethane (970 ml) was added hydrazine hydrate (31.7 g, 0.6334 mol) , at 0°C, drop wise, over a period of 45 minutes and the stirring continued further. After 2 hours, the reaction mass was filtered under suction. Filtrate was washed with water (485 ml), and the organic layer was diluted with an aq. solution of 10% potassium hydrogen sulfate (485 ml) and stirred for 15 minutes. The aqueous layer was separated, neutralized with solid sodium hydrogen carbonate and extracted with dichloromethane (2 x 485 ml). The organic layer was separated, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain colorless oil, this was used as such for further reaction immediately (28g, overall yield of step II and step III was 60%)
Analysis:
Mass: 177.2 (M+l) for Molecular Weight of 176.2 and Molecular Formula of C7H16N2O3.
Example 2
Synthesis of (25,5R)-jV-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicvclor3.2.11octane-2- carboxamide (I)
Step 1: Preparation of (25,5R)-iV-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (IV):

To a clear solution of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II, 42.67 g, 0.143 mol; prepared according to the procedure disclosed in Indian Patent Application No. 699/MUM/2013) in water (426 ml) was added EDC.HC1 (67.1 g, 0.349 mol) at 15°C
under stirring. After 10 minutes, a solution of teri-butyl-[2-(aminooxy) ethyl]carbamate (III, 28.0g, 0.159 mol; prepared as per the literature procedure depicted in Scheme 2) in dimethylformamide (56 ml) was added drop wise at 10°C under continuous stirring. The temperature of the reaction mass was allowed to warm to 25°C and then HOBt (21.5g, 0.159 mol) was added in small portions over a period of 15 minutes and the resulting mixture was further stirred at room temperature for 16 hours. The reaction was continuously monitored using thin layer chromatography using mixture of acetone and hexane (35 :65) as solvent system. After completion of reaction, the resulting mixture was filtered and the residue was washed with water (130 ml). The obtained white residue was suspended in water (130 ml) and the mixture stirred at 50°C for 3 hours. The resulting suspension was filtered, the residue dried under reduced pressure to obtain 51 g of (2S,5R)-N-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (IV) as off white solid in 73% yield.
Analysis:
Mass: 433.4 (M-l ); for Molecular Weight of 434.5 and Molecular Formula of C21H30N4O6;
1H-NMR (400MHz, CDC13): δ 9.32 (br s, 1H), 7.41 -7.26(m,5H), 5.41(br s, 1H), 5.06-4.88(dd, 2H), 3.98-3.96(d,lH), 3.91-3.90(m,2H), 3.39(m, 1H), 3.31-3.26(m, 2H), 3.04-3.01(d,lH), 2.77-2.74(d, 1H), 2.33-2.28(m, 1H), 2.03-1.93(m, 2H), 1.67-1.64(m, 1H), 1.44(s, 9H);
Purity as determined by HPLC: 99.4%.
Step 2: Preparation of (2S,5R)-iV-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (V):

A solution of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1] octane-2-carboxamide (IV, 38 g, 0.0875 mol) in a mixture of dimethylformamide and dichloromethane (2: 8, 76 ml: 304 ml), containing 10% Pd/C (7.6 g, 50% wet) was hydrogenated at 50 psi hydrogen atmosphere at 25°C for 3 hours. The resulting mixture was filtered through a celite pad. The residue was washed with dichloromethane (75 ml). The solvent from the combined filtrate was evaporated
under reduced pressure to obtain 30 g (25,5i?)-N-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide (V) as an oil, which was used as such for the next reaction without further purification.
Analysis:
Mass: 343.3 (M-l ) for Molecular Weight of 344.3 and Molecular Formula of C14H24N4O6.
Step 3: Preparation of (25,5R)-iV-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide,tetrabutyl ammonium salt (VI):

To a stirred solution of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(hydroxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide (V, 30.0 g, 0.0875 mol) in dimethylformamide (150 ml) was added sulphur trioxide dimethylformamide complex (16.06 g, 0.105 mol) in one portion, at 10°C. The reaction mass was stirred at the same temperature for 30 minutes and then allowed to warm to room temperature. After 2 hours, a solution of tetrabutylammonium acetate (31.6 g, 0.105 mol) in water (95 ml) was slowly added to the reaction mixture and stirred for another 2 hours. The solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2 x 60 ml) to obtain thick mass. This mass was partitioned between 1 : 1 mixture of dichloromethane (300 ml) and water (300 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (150 ml). The combined organic extracts were washed with water (3 x 150 ml) and dried over anhydrous sodium sulphate. The solvent was evaporated under reduced pressure and the resulting oily mass was triturated with ether (3 x 60 ml). Each time the ether layer was decanted and the residue was finally concentrated under reduced pressure to obtain the sticky mass. The so obtained material was purified by column chromatography over silica gel using mixture of methanol and dichloromethane as elution solvent. The solvent from the combined fractions was evaporated to obtain 47.5 g of (25,5i?)-N-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide,tetrabutyl ammonium salt as white foam in 70% yield.
Analysis:
Mass: 423.4 (M-l) as free sulphonic acid; for Molecular Weight of 665.9 and Molecular Formula of C30H59N5O9 S;
1H- NMR (400MHz, CDC13): δ 9.52(br s, 1H), 5.53(br s, 1H), 4.33(s, 1H), 3.95-3.92(m,3H), 3.37-3.27(m, 1 1H), 2.87-2.84(d, 1H), 2.35-2.30(m, 1H), 2.17(m, 1H), 1.96-1.88(m, 2H), 1.74-1.60(m,8 H), 1.47-1.40(m, 17H), 1.02-0.98(m, 12H).
Step 4: Preparation of (2S R)-iV-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (I):
Formula (I)

To a stirred solution of (2S,5i?)-N-(2-Boc-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide, tetrabutyl ammonium salt (VI, 17 g, 0.0225 mol) in dichloromethane (85 ml) was added trifluoroacetic acid (85 ml) drop wise at -10°C over a period of 45 minutes. The resulting mass was further stirred at same temperature for 1 hour. The resulting reaction mixture was poured into cyclohexane (850 ml), stirred well for 30 minutes and the separated oily layer was collected. This procedure was repeated one more time and finally the separated oily layer was added to tert-butyl methyl ether (170 ml) under vigorous stirring at 25°C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with tert-butyl methyl ether (2 x 170 ml). The solid thus obtained was stirred with fresh dichloromethane (170 ml) for 30 minutes and filtered. The residual solid was dried at 45°C under reduced pressure to yield 7.3g of the titled compound in crude form. The obtained solid was further dissolved in water, (7.3 ml) and to this solution was added basic resin (Amberlyst A-26 -OH ion exchange resin, 4.4 g) under stirring. After 0.5 hour, the resin was filtered and to the filtrate isopropanol (51 ml) was added slowly at 25°C. The solution was further stirred for 12 hours. The separated solid was filtered and washed with additional isopropanol (7.5 ml) and dried under reduced pressure to obtain 4.3 g of (2S ,5R)-N-(2-aminoethoxy)-6-(sulfooxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1 ]octane-2-carboxamide as off-white solid in 52 % yield.
Analysis:
Mass: 323.1 (M-l); for Molecular Weight of 324.31 and Molecular Formula of C9H16N4O7S; 1H-NMR (400MHz, D20): δ 4.07-4.06(d, 1H), 4.05-4.03(t, 2H), 3.96-3.94(d, 1H), 3.20(br s, 1H), 3.16-3.13(t, 2H), 3.02-2.99(d, 1H), 2.04-1.68(m, 4H);
Purity as determined by HPLC: 94.88%.

REF
| WO2015110969A3 * | Jan 21, 2015 | Nov 26, 2015 | Wockhardt Limited | Nitrogen containing compounds and their use as antibacterial agents |
| WO2015150941A1 * | Mar 12, 2015 | Oct 8, 2015 | Wockhardt Limited | A process for preparation of sodium (2s, 5r)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate |
| WO2016088863A1 * | Dec 4, 2015 | Jun 9, 2016 | Meiji Seikaファルマ株式会社 | Method for producing crystals of diazabicyclooctane derivative and stable lyophilized preparation |
| EP2931723A4 * | Dec 11, 2012 | Jun 1, 2016 | Fedora Pharmaceuticals Inc | New bicyclic compounds and their use as antibacterial agents and -lactamase inhibitors |
| US8933232 | Mar 29, 2013 | Jan 13, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| US8933233 | Mar 29, 2013 | Jan 13, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US8940897 | Mar 29, 2013 | Jan 27, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US8962843 | Mar 29, 2013 | Feb 24, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| US8962844 | Mar 29, 2013 | Feb 24, 2015 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US9120795 | Mar 14, 2014 | Sep 1, 2015 | Cubist Pharmaceuticals, Inc. | Crystalline form of a β-lactamase inhibitor |
| US9120796 | Oct 2, 2014 | Sep 1, 2015 | Cubist Pharmaceuticals, Inc. | B-lactamase inhibitor picoline salt |
| US9309245 | Apr 2, 2013 | Apr 12, 2016 | Entasis Therapeutics Limited | Beta-lactamase inhibitor compounds |
| US9393239 | Apr 15, 2014 | Jul 19, 2016 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and betalactamase inhibitors |
/////////////IN2015MU287, WO-2016120752, nacubactam, WOCKHARDT, NEW PATENT, WK ?, WK-?, WK?, CAS 1452458-86-4, C9 H16 N4 O7 S, 324.31, Beta lactamase inhibitor, Roche, Meiji Seika Pharma, Fedora Pharmaceuticals, nacubactam hydrate , PHASE 1, A diazabicyclooctane beta-lactamase inhibitor, bacterial infection, July 2016, phase 1 clinical development, RG-6080, 1452458-86-4, FPI-1459, OP-0595, Phase I , β-lactamase inhibitor, bacterial infections, Fedora parmaceuticals, Meiji Seika Pharma
NCCONC(=O)[C@@H]2CC[C@@H]1C[N@]2C(=O)N1OS(=O)(=O)O
RG 6080, Nacubactam
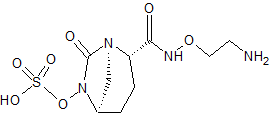

RG-6080
Sulfuric acid, mono[(1R,2S,5R)-2-[[(2-aminoethoxy)amino]carbonyl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
Phase I
A β-lactamase inhibitor potentially for the treatment of bacterial infections.
![]()
RG-6080; FPI-1459; OP-0595
CAS No. 1452458-86-4
| Molecular Formula | C9 H16 N4 O7 S |
| Formula Weight | 324.31 |
- Originator Fedora Pharmaceuticals
- Developer Meiji Seika Pharma
- Class Antibacterials; Azabicyclo compounds
- Mechanism of Action Beta lactamase inhibitors
- Phase IBacterial infections
Most Recent Events
- 13 Jan 2015 OP 0595 licensed to Roche worldwide, except Japan ,
- 30 Nov 2014 Meiji Seika Pharma completes a phase I trial in Healthy volunteers in Australia (NCT02134834)
- 01 May 2014 Phase-I clinical trials in Bacterial infections (in volunteers) in Australia (IV)
![]()
SYNTHESIS
WO 2015046207,
CONTD…………………..
CONTD………………………………..
Patent
The novel heterocyclic compound in Japanese Patent 4515704 (Patent Document 1), preparation and shown for their pharmaceutical use, sodium trans-7-oxo-6- (sulfooxy) as a representative compound 1,6-diazabicyclo [3 .2.1] discloses an octane-2-carboxamide (NXL104). Preparation in regard to certain piperidine derivatives which are intermediates Patent 2010-138206 (Patent Document 2) and JP-T 2010-539147 (Patent Document 3) are shown at further WO2011 / 042560 (Patent Document 4) NXL104 to disclose a method for producing the crystals.
In Patent 5038509 (Patent Document 5) (2S, 5R) -7- oxo -N- (piperidin-4-yl) -6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane – 2- carboxamide (MK7655) is shown, discloses the preparation of certain piperidine derivatives with MK7655 at Patent 2011-207900 (Patent Document 6) and WO2010 / 126820 (Patent Document 7).
The present inventors also disclose the novel diazabicyclooctane derivative represented by the following formula (VII) in Japanese Patent Application 2012-122603 (Patent Document 8).
Patent Document 1: Japanese Patent No. 4515704 Pat
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
[Chemical formula 1] (In the formula, R 3 are the same as those described below)

Reference Example
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
step 1 tert-butyl {2 – [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate (IV-1)(2S, 5R)-6-(benzyloxy) -7-oxo-1,6-diazabicyclo [3.2.1] octane-2-carboxylic acid (4 .30g, dehydrated ethyl acetate (47mL) solution of 15.56mmol) was cooled to -30 ℃, isobutyl chloroformate (2.17g, washing included dehydration ethyl acetate 1mL), triethylamine (1.61g, washing included dehydration ethyl acetate 1 mL), successively added dropwise, and the mixture was stirred 1 hour at -30 ° C.. To the reaction solution tert- butyl 2-dehydration of ethyl acetate (amino-oxy) ethyl carbamate (3.21g) (4mL) was added (washing included dehydration ethyl acetate 1mL), raising the temperature over a period of 1.5 hours to 0 ℃, It was further stirred overnight. The mixture of 8% aqueous citric acid (56 mL), saturated aqueous sodium bicarbonate solution (40 mL), sequentially washed with saturated brine (40 mL), dried over anhydrous magnesium sulfate, filtered, concentrated to 5 mL, up to 6mL further with ethanol (10 mL) It was replaced concentrated. Ethanol to the resulting solution (3mL), hexane the (8mL) in addition to ice-cooling, and the mixture was stirred inoculated for 15 minutes. The mixture was stirred overnight dropwise over 2 hours hexane (75 mL) to. Collected by filtration the precipitated crystals, washing with hexane to give the title compound 5.49g and dried in vacuo (net 4.98 g, 74% yield). HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 50/50, 1.0 mL / min, UV 210 nm, Retweeted 4.4 min; 1 H NMR (400 MHz, CDCl 3 ) [delta] 1.44 (s, 9H), 1.56-1.70 (m, 1H), 1.90-2.09 (m, 2H), 2.25-2.38 (m, 1H), 2.76 (d, J = 11.6 Hz, 1H), 3.03 (br.d., J = 11.6 Hz , 1H), 3.24-3.47 (m, 3H), 3.84-4.01 (m, 3H), 4.90 (d, J = 11.6 Hz, 1H), 5.05 (d, J = 11.6 Hz, 1H), 5.44 (br. . s, 1H), 7.34-7.48 (yd, 5H), 9.37 (Br.S., 1H); MS yd / z 435 [M + H] + .

Step 2
tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate
(V-1) tert-butyl {2 – [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
(V-1) tert-butyl {2 – [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
Step 3
Tetrabutylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide – pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
Tetrabutylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide – pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
Step 4 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
tetra butylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
tetra butylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
PATENT
Example
64 tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]

64 tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]
tert- butyl {2 – [({[(2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate (example 63q, net 156.42g, 360mmol) in methanol solution (2.4L) of 10% palladium carbon catalyst (50% water, 15.64g) was added, under an atmosphere of hydrogen, stirred for 1.5 hours did. The catalyst was filtered through celite, filtrate was concentrated under reduced pressure until 450mL, concentrated to 450mL by adding acetonitrile (1.5 L), the mixture was stirred ice-cooled for 30 minutes, collected by filtration the precipitated crystals, washing with acetonitrile, and vacuum dried to obtain 118.26g of the title compound (net 117.90g, 95% yield). Equipment data of the crystals were the same as those of the step 2 of Reference Example 3.
Example
65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)
[of 125]

65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)
[of 125]
tert- butyl {2 – [({[(2S, 5R) -1,6- -6- hydroxy-7-oxo-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (example 64,537.61g, 1.561mol) in acetonitrile (7.8L) solution of 2,6-lutidine (512.08g), sulfur trioxide – pyridine complex (810.3g) was added, at room temperature in the mixture was stirred overnight. Remove insolubles and the mixture was filtered, the filtrate concentrated to 2.5 L, diluted with ethyl acetate (15.1L). The mixture was extracted with 20% phosphoric acid 2 hydrogencarbonate aqueous solution (7.8L), the resulting aqueous layer into ethyl acetate (15.1L), added tetrabutylammonium hydrogen sulfate (567.87g), was stirred for 20 min. The organic layer was separated layers, dried over anhydrous magnesium sulfate (425 g), after filtration, concentration under reduced pressure, substituted concentrated tetrabutylammonium tert- butyl with dichloromethane (3.1L) {2 – [({[(2S, 5R ) -7-oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate was obtained 758g (net 586.27g, Osamu rate 84%).
The tetra-butyl ammonium salt 719g (net 437.1g, 0.656mol) in dichloromethane (874mL) solution was cooled to -20 ℃, dropping trifluoroacetic acid (874mL) at 15 minutes, 1 the temperature was raised to 0 ℃ It was stirred time. The reaction was cooled to -20 ° C. was added dropwise diisopropyl ether (3.25L), and the mixture was stirred for 1 hour the temperature was raised to 0 ° C.. The precipitate is filtered, washed with diisopropyl ether to give the title compound 335.36g of crude and vacuum dried (net 222.35g, 99% yield).
The title compound of crude were obtained (212.99g, net 133.33g) and ice-cold 0.2M phosphate buffer solution of pH5.3 mix a little at a time, alternating between the (pH6.5,4.8L). The solution was concentrated under reduced pressure to 3.6L, it was adjusted to pH5.5 at again 0.2M phosphate buffer (pH6.5,910mL). The solution resin purification (Mitsubishi Kasei, SP207, water ~ 10% IPA solution) is subjected to, and concentrated to collect active fractions, after lyophilization, to give the title compound 128.3 g (96% yield). Equipment data of the crystals were the same as those of step 3 of Reference Example 3.
PATENT
US 20140288051
WO 2014091268
WO 2013180197
US 20130225554
///////////RG-6080, 1452458-86-4, FPI-1459, OP-0595, Phase I , β-lactamase inhibitor, bacterial infections, Fedora parmaceuticals, Meiji Seika Pharma
TAK-243, AOB 87172, MLN-7243

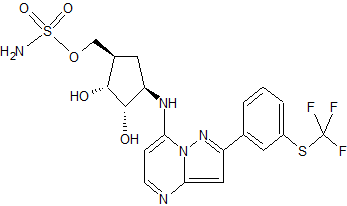
TAK-243, AOB 87172, MLN-7243
CAS 1450833-55-2
Chemical Formula: C19H20F3N5O5S2
Molecular Weight: 519.5142
Sulfamic acid, [(1R,2R,3S,4R)-2,3-dihydroxy-4-[[2-[3-[(trifluoromethyl)thio]phenyl]pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]methyl ester
((lR,2R,3S,4R)-2,3-dihydroxy-4-(2-(3-(trifluoromethylthio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamate
methyl ((1S,2R,3S,4R)-2,3-dihydroxy-4-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclopentyl)sulfamate
Phase I
Millennium Pharmaceuticals, Inc. INNOVATOR
Roushan AFROZE, Indu T. Bharathan,Jeffrey P. CIAVARRI, Paul E. Fleming,Jeffrey L. Gaulin, Mario Girard, Steven P. Langston, Francois R. SOUCY, Tzu-Tshin WONG, Yingchun Ye,
A UAE inhibitor potentially for the treatment of solid tumors.
TAK-243, also known as MLN7243 and AOB87172, is a small molecule inhibitor of ubiquitin-activating enzyme (UAE), with potential antineoplastic activity. UAE inhibitor MLN7243 binds to and inhibits UAE, which prevents both protein ubiquitination and subsequent protein degradation by the proteasome. This results in an excess of proteins in the cells and may lead to endoplasmic reticulum (ER) stress-mediated apoptosis. This inhibits tumor cell proliferation and survival. UAE, also called ubiquitin E1 enzyme (UBA1; E1), is more active in cancer cells than in normal, healthy cells.
![]()
Research Code TAK-243; MLN-7243, TAK-243; TAK 243; TAK243; MLN7243; MLN-7243; MLN 7243; AOB87172; AOB-87172; AOB 87172.
CAS No. 1450833-55-2(MLN 7243)
- Originator Millennium
- Developer Takeda Oncology
- Class Antineoplastics
- Mechanism of Action Ubiquitin-protein ligase inhibitors
- Phase I Solid tumours

Most Recent Events
- 01 Feb 2014 Phase-I clinical trials in Solid tumours (late-stage disease, second-line therapy or greater) in USA (IV)
- 18 Dec 2013 Preclinical trials in Solid tumours in USA (IV)
- 18 Dec 2013 Millennium plans a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA (NCT02045095)
Cancer is the second most common cause of death in the U.S. and accounts for one of every eight deaths globally (American Cancer Society, Cancer Facts and Figures, 2014). The American Cancer Society expects that in 2014 at least 1,665,540 new cancer cases will be diagnosed in the US and 585,720 Americans are expected to die of cancer, almost 1 ,600 people per day. Currently available paradigms for treating solid tumors may include systemic treatment such as chemotherapy, hormonal therapy, use of targeted agents and biological agents, either as single agents or in combination. These treatments can be delivered in combination with localized treatments such as surgery or radiotherapy. These anti-cancer paradigms can be use in the curative setting as adjuvant or neo-adjuvant treatments or in the metastatic setting as palliative case for prolonged survival and to help manage symptoms and side-effects. In hematological cancers, stem cell transplants may also be an option in certain cancers as well as chemotherapy and/or radiation. Although medical advances have improved cancer survival rates, there remains a continuing need for new and more effective treatments.
Ubiquitin is a small 76-amino acid protein that is the founding member of a family of posttranslational modifiers known as the ubiquitin-like proteins (Ubls). Ubls play key roles in controlling many biological processes including cell division, cell signaling and the immune response. There are 8 known human Ubl activating enzymes (known as Els) (Schulman, B.A., and J.W. Harper, 2009, Ubiquitin-like protein activation by El enzymes: the apex for downstream signalling pathways, Nat Rev Mol Cell Biol 10:319-331). Ubiquitin and other Ubls are activated by a specific El enzyme which catalyzes the formation of an acyl-adenylate intermediate with the C-terminal glycine of the Ubl. The activated Ubl molecule is then transferred to the catalytic cysteine residue within the El enzyme through formation of a thioester bond intermediate. The El -Ubl intermediate and an E2 enzyme interact, resulting in a thioester exchange wherein the Ubl is transferred from the El to active site cysteine on the E2. The Ubl is then conjugated, i.e. transferred, to the target protein, either directly or in conjunction with an E3 ligase enzyme, through isopeptide bond formation with the amino group of a lysine side chain in the target protein. Eukaryotic cells possess ~35 ubiquitin E2 enzymes and >500 ubiquitin E3 enzymes. The E3 enzymes are the specificity factors of the ubiquitin pathway which mediate the selective targeting of specific cellular substrate proteins (Deshaies, R.J., and C.A. Joazeiro, 2009, RING domain E3 ubiquitin ligases, Annu Rev Biochem 78:399-434; Lipkowitz, S., and A.M. Weissman, 2011, RTNGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis, Nat Rev Cancer 11 :629-643; Rotin, D., and S. Kumar, 2009, Physiological functions of the HECT family of ubiquitin ligases, Nat Rev Mol Cell Biol 10:398-409).
Two El enzymes have been identified for ubiquitin, UAE (ubiquitin-activating enzyme) and UBA6 (Jin, J., et al., 2007, Dual El activation systems for ubiquitin differentially regulate E2 enzyme charging, Nature 447: 1135-1138). UAE is the El responsible for the majority (>99%) of ubiquitin flux within the cell. UAE is capable of charging each of the approximately -35 E2 enzymes with the exception of Usel, which is the only E2 known to exclusively work with UBA6 (Jin et al., 2007). Inhibition of UAE is sufficient to dramatically impair the great majority of ubiquitin-dependent cellular processes (Ciechanover, A., et al., 1984, Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85, Cell 37:57-66; Finley, D., A. et al., 1984, Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85, Cell 37:43-55).
The cellular signals generated by ubiquitin are diverse. Ubiquitin can be attached to substrates as a single entity or as polyubiquitin polymers generated through isopeptide linkages between the C-terminus of one ubiquitin and one of the many lysines on a second ubiquitin. These varied modifications are translated into a variety of cellular signals. For example, conjugation of a lysine 48 -linked polyubiquitin chain to a substrate protein is predominantly associated with targeting the protein for removal by the 26S proteasome. A single ubiquitin modification, or monoubiquination, typically affects protein localization and/or function. For example, monoubiquitination modulates the following: 1) the function of Histones 2a and 2b (Chandrasekharan, M.B., et al., 2010, Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation, Epigenetics 5:460-468), 2) controls the nucleo-cytoplasmic shuttling of PTEN (Trotman, L,C, et al., 2007, 3) ubiquitination regulates PTEN nuclear import and tumor suppression, Cell 128: 141-156), 4) drives localization of the FANCD2 protein to sites of DNA damage (Gregory, R.C., et al., 2003, Regulation of the Fanconi anemia pathway by monoubiquitination, Semin Cancer Biol 13:77-82) and 5) promotes the internalization and endosomal/lysosomal turnover of some cell surface receptors, like EGFR (Mosesson, Y., and Y. Yarden, 2006, Monoubiquitylation: a recurrent theme in membrane proteintransport. Isr Med Assoc J 8:233-237). Other forms of polyubiquitination chains occur on lysine positions 11, 29 and 63, impacting various cellular roles including cell cycle, DNA repair and autophagy (Behrends, C, and J.W. Harper, 2011, Constructing and decoding unconventional ubiquitin chains, Nat Struct Mol Biol 18:520-528; Bennett, E.J., and J.W. Harper, 2008, DNA damage: ubiquitin marks the spot, Nat Struct Mol Biol 15:20-22; Komander, D., 2009, The emerging complexity of protein ubiquitination, Biochem Soc Trans 37:937-953).
UAE-initiated ubiquitin conjugation plays an important role in protein homeostasis, cell surface receptor trafficking, transcription factor turnover and cell cycle progression. Many of these processes are important for cancer cell survival and it is believed that tumor cells may have increased sensitivity to UAE inhibition as a result of their rapid growth rate, increased metabolic demands and oncogene fueled protein stress. Preclinical studies with PYZD-4409, a UAE inhibitor, demonstrated this compound induced cell death in both leukemia and myeloma cell lines and induced anti-tumor activity in a mouse acute myeloid leukemia (AML model). (Xu, W.G., et al., 2010, The ubiquitin-activating enzyme El as a therapeutic target for the treatment of leukemia and multipie myeloma, Blood, 115:2251-59). Thus, UAE represents a protein homeostasis target opportunity for the treatment of cancer.
Abstracts: AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; November 5-9, 2015; Boston, MA
Abstract
Clinical results of VELCADE® (bortezomib) For Injection have prompted evaluation of other enzymes within the ubiquitin proteasome system (UPS) as druggable targets for human cancer. We have identified a first in class investigational drug, TAK-243 (MLN7243), which targets the ubiquitin activating enzyme, UAE (UBA1), an essential cellular enzyme responsible for activating > 99% of all cellular ubiquitin. Ubiquitin is involved in multiple cellular processes including ubiquitin-dependent protein turnover, cell cycle progression, regulation of apoptosis, protein localization and response to DNA damage. Experiments combining targeted siRNA knockdown with TAK-243 identified DNA damage repair genes necessary for UAE inhibitor-induced cell death. A more focused approach revealed TAK-243 treatment blocked essential monoubiquitination events within the Translesion synthesis (TLS), Fanconi Anemia (FA) and Homologous recombination (HR) pathways. Inhibition of UAE prevented mono-ubiquitin signaling of key mediators within these pathways, including PCNA and FANCD2, by blocking formation of their specific E2-ubiquitin thioesters. In vitro cell-based assays combining TAK-243 with ultraviolet (UV) and radiation, both known to induce DNA damage, yielded inhibition of cell growth and enhanced DNA damage as observed through colony formation assays and Comet assay detection, respectively. Xenograft tumor bearing mice were treated with carboplatin or docetaxel, combined with TAK-243, to evaluate combination benefits in vivo. Synergistic and additive anti-tumor combination benefits were observed in animals treated with TAK-243 + carboplatin and TAK-243 + docetaxel. These important mechanistic in vitro and in vivo studies indicate the dependency of ubiquitination signaling in DNA damage repair and provide a mechanistic rationale for combining radiation, carboplatin or docetaxel with TAK-243 in the clinical setting. Currently, TAK-243 is being evaluated in a solid tumor phase I clinical trial evaluating safety, tolerability, pharmacokinetics, pharmacodynamics and anti-tumor activity (ClinicalTrials.gov identifier: NCT02045095).
Citation Format: Michael A. Milhollen, Judi Shi, Tary Traore, Jessica Huck, Darshan Sappal, Jennifer Duffy, Eric Lightcap, Yuko Ishii, Jeff Ciavarri, Paul Fleming, Neil Bence, Marc L. Hyer. The small molecule UAE inhibitor TAK-243 (MLN7243) prevents DNA damage repair and reduces cell viability/tumor growth when combined with radiation, carboplatin and docetaxel. [abstract]. In: Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2015 Nov 5-9; Boston, MA. Philadelphia (PA): AACR; Mol Cancer Ther 2015;14(12 Suppl 2):Abstract nr A164.
PATENT
WO 2013123169
https://www.google.com/patents/WO2013123169A1?cl=en
Scheme 1 : General route for 2-substituted ((1R,2R,3S,4R)-2,3-dihydroxy-4- (pyrazolo[1,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates

A genera! route for the synthesis of compounds represented by structure iv wherein Z is an optionally substituted fused or non-fused aryl or heteroaryl ring is outlined above in Scheme 1. Compound i (obtained by coupling an appropriately protected cyclopentylamine or salt thereof with 2-bromo-7-chloropyrazolo[1 ,5-a]pyrimidine in the presence of a suitable base as described below in the procedure of Examples 1a and 1b) is transformed to a compound of formula iii by coupling with a metal substituted compound Z-M via a palladium catalyzed reaction. A compound of formula iii can also be obtained by first transforming i to a metal substituted compound of formula ii using suitable boron or tin containing reagents, and then coupling with a halogen substituted compound Z-X via a palladium catalyzed reaction. Compounds of formula iv are then obtained by reaction with an appropriate sulfamating reagent (for example chlorosulfonamide or see Armitage, I. et. al. U.S. Patent Application US2009/0036678, and Armitage, I. et. al. Org. Lett., 2012, 14 (10), 2626-2629) followed by appropriate deprotection conditions.
Scheme 2: General route for 5-halogen substituted, 2 -substituted ((1R,2R,3S,4R)- 2,3-dihydroxy-4-(pyrazolo[1,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates


A general route for the synthesis of compounds represented by structure ix wherein Z is an optionally substituted fused or non-fused aryl or heteroaryl ring and X is a halogen is outlined above in Scheme 2. Cyclization of amino-pyrazole v with a suitable diester and an appropriate base at an elevated temperature is followed by reaction with an appropriate halogenating reagent such as POCI3 at an elevated temperature to give compounds of formula vii. Compounds of formula viii are then obtained by reaction with an appropriately protected cyc!opentylamine or a salt thereof in the presence of a suitable base. Sulfamation and deprotection following Method 1 as described previously provides compounds of formula ix.
Scheme 3: General route for 5-alkyl substituted, 2-substituted ((1R,2R,3S,4R)-2,3- dihydroxy-4-(pyrazolo[1 ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates

SIMILAR COMPD
Example 17. Synthesis of (s.e.)-{(1 ,2R,3S,4R)-4-[(3,6-dichloro-2-{3- [(trifluoromethyl)sulfanyl]phenyl}pyrazolo[1,5-a]pyrimidin-7-yl)amino]-2,3- dihydroxycyclopentyl}methyl sulfamate (1-124) and (s.e.)-{(1 ,2R,3S,4R)-4-[(6-chloro-2-{3- [(trifluoromethyl)sulfanyl]phenyl}pyrazolo[1,5^]pyrimidin-7-y[)arnino]-2,3- dihydroxycyclopentyl}methyl sulfamate 0-125).

SIMILAR NOT SAME
Step 1. To a vial containing s.e {(1 ,2 ,3S,4 )-2,3-dihydroxy-4-t(2-{3- [(t rif I u orometh y l)sulf a nyl] phen l}p^
sulfamate (0.82 g, 0.0015 mol) and cooled to 0 °C is added N-chlorosuccinimide (126 mg, 0.000943 mol) as a solution in 12 mL of N,N-dimethy)formamide. The reaction mixture is stirred overnight with warming to rt. Saturated sodium bicarbonate solution is added and the reaction mixture is extracted with ethyl acetate, washed with brine, dried over sodium sulfate and concentrated in vacuo. The crude material is first purified by column chromatography (eluent: methanol/methylene chloride) and then purified by HPLC to afford both the dichloro (LCMS: (FA) +1 588) and mono chloro (LCMS: (FA) M+1 554) titlecompounds.
PATENT
UAE inhibitors are disclosed in patent application publications WO2013/123169 and US 2014/0088096. In one embodiment, the UAE inhibitor is a compound having the following structure (Compound 1):

(Compound 1);
or a pharmaceutically acceptable salt thereof. The Compound 1 is named ((lR,2R,3S,4R)-2,3-dihydroxy-4-(2-(3-(trifluoromethylthio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamate.
process for making Compound 1 :

Compound 1;
or pharmaceutically acceptable salt thereof, comprising the steps of:
a) contacting Compound 9 or a salt, solvate or hydrate thereof with 2,2-dimethyl-l,3-dioxane-4,6-dione (Meldrum’s acid):

Compound 9
under coupling conditions to provide compound 8 or a salt, solvate or hydrate thereof:

Compound 8
b) subjecting compound 8 or a salt, solvate or hydrate thereof to cyclization conditions to provide compound 7 or a salt, solvate or hydrate thereof

Compound 7
c) contacting Compound 7 or a salt, solvate or hydrate thereof with benzotriazole under chlorination displacement conditions to provide Compound 5 or a salt, complex, solvate or hydratei thereof

; Compound 5
d) contacting Compound 5 or a salt, complex, solvate or hydrate thereof with Compound 6 or a solvate or hydrate thereof:

; Compound 6
under displacement reaction conditions to provide Compound 3 or a salt, solvate or hydrate thereof
solvate or hydrate thereof with Compound

Cl ; Compound 4
under sulfamoylating reaction conditions to provide Compound 2 or a salt, solvate or hydrate thereof

; Compound 2
f) contacting Compound 2 or a salt, solvate or hydrate thereof with an acid under sulfamoylation conditions to provide Compound 1 or a pharmaceutically acceptable salt thereof
 COMPD1
COMPD1
Example 1: Synthesis of S-iB-Ktrifluoromethyltsulfanyllphenyll-lH-pyrazol-S-amine
Step A: 3-((trifluoromethyl)thio)benzoate
[0148] To dimethylcarbonate (68 mL) was added 3-((trifluoromethyl)thio)benzoic acid (100 g, Beta Pharma Scientific) and a catalytic amount of sulfuric acid (2.4 mL). The mixture was then heated to 90°C for 5h. The reaction was then cooled to room temperature and quenched with sodium bicarbonate (1.0 L). To the aqueous layer was with ethyl acetate (1.0 L). The phases were separated and this process was repeated with ethyl acetate (1.0 L). The organic layers were combined and concentrated with a rotovap to give a light orange oil. The methyl 3-((trifluoromethyl)thio)benzoate (105g, 99%) was taken on crude to the next reaction. Ή NMR (300 MHz, CHLOROFORM-^ δ ppm 3.99 (s, 3 H) 7.49 – 7.58 (m, 1 H) 7.85 (d, J=l.62 Hz, 1 H) 8.17 (dt, J=7.69, 1.43 Hz, 1 H) 8.32 – 8.44 (m, 1 H).
[0149] Step B: 3-oxo-3-(3-((trifluoromcthvnthio)phcnyl>proDaneiiitrilc
[0150] Methyl 3-((trifluoromethyl)thio)benzoate (100.0 g) in tetrahydrofuran (1.0 L) was added acetonitrile (44.2 mL, 847 rnmol) and 1M (in THF) potassium tert-butoxide (95.01 g). The reaction was complete in 10 min by HPLC analysis. The reaction was quenched with 1M HC1 (1.0 L) and then extracted with three times with (1.0 L) of ethyl acetate. The organic layers with 3-oxo-3-(3-((trifluoromethyl)thio)phenyl)propanenitrile were then concentrated to dryness. This material (lOO.Og, 96.3%) was taken on crude with further purification. Ή NMR (300 MHz, CHLOROFORM-rf) δ ppm 4.12 (s, 2 H) 7.51 – 7.75 (m, 1 H) 7.89 – 8.01 (m, 1 H) 8.01 – 8.10 (m, 1 H) 8.20 (s, 1 H)
[0151] Step C: 3-}3-htrifliioromethv sulfan llphenyl}-lH-pyrazol-5-amine
[0152] To 3-oxo-3-{3-[(trifluoromethyl)sulfanyl]phenyl}propanenitrile (100.0 g,) in ethanol (1000.0 mL) was added hydrazine hydrate (59.52 mL). The reaction was heated to 100°C for lh at which point HPLC analysis showed the reaction was complete. The reaction was concentrated to dryness on a rotovap to give a brown oil. The oil was taken up in ethyl acetate (1.0 L) and extracted with water (1.0 L). The phases were separated and the organic phase was concentrated. Upon concentration 3-{3-[(trifluoromethyl)sulfanyl]phenyl}-lH-pyrazol-5-amine was obtained (80.8 g; Yield = 76.4%) . !H NMR (300 MHz, CHLOROFORM-^ δ ppm 5.95 (s, 1 H) 6.73 (br s, 1 H) 7.13 – 7.34 (m, 2 H) 7.42 – 7.74 (m, 3 H) 7.85 (s, 1 H).
[0153] Example 2: f R.2R.3St4RV2.3-dihvdroxy-4-ff2-r3- ((trifluoromethylHhio)phenvnpyrazolo[l,5-alpyrimidin-7-yl¼mino)cvclopentyl)metliyl sulfamate
[0154] Step 1: f2.2-dimethyl-5-ffl3-(3-((triiluoromethvnthio phenvn-lH-pyrazol-5- amino methyleBC>-1.3-dioxane-4,6-dione)
[0155] To trimethoxy orthoformate (2.0 L), at 20°C and under a blanket of nitrogen, was added 2,2-dimethyl-l,3-dioxane-4,6-dione (361.35 g). The resulting white suspension went clear within minutes and was heated to 85°C over 15 minutes. The reaction was held at 85°C for 120 minutes. While the reaction was heated and stirred another solution of 3-(3-((trifluoromethyl)thio)pheny])-lH-pyrazol-5-amine (500.0 g) was made. To a 4L RBF was added 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (500.0 g) and then trimethoxy orthoformate (1.4 L) added into this solid. This solution was mixed to dissolve the solids and resulted a dark brown solution. The solution of 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (-1.8L in trimethoxy orthoformate) was added to the reactor over 30 minutes while maintaining the reaction temperature at 85°C. The reaction was then stirred for 20 minutes with white solids forming in the solution. After 20 minutes the reaction was sampled and the UPLC showed the complete conversion of 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5 -amine to 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l ,3-dioxane-4,6-dione. The reaction was cooled to 20 °C over 20 minutes and maintained at that temperature for 20 additional minutes. At this point, a thick white slurry had formed and the reaction was filtered using a Nutche Filter over 15 minutes. The reactor was washed with 1L of ethyl acetate and this solution was then mixed with the filter cake and removed by filtration. The cake was dried for -40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16 hours). The reaction was then analyzed by FfPLC and NMR to give 2,2-dimethyl-5-(((3-(3 -((trifluoromethyl)thio)phenyi lH-pyrazol-5-yl)amino)methylene)- 1 ,3-dioxane-4,6-dione (635.3 g, 79%) XH NMR (300 MHz, DMSO-cfe) δ ppm 1.68 (s, 6 H) 7.05 (d, J=2.05 Hz, 1 H) 7.64 -7.77 (m, 2 H) 7.77 – 8.03 (m, 1 H) 8.12 (s, 1 H) 8.72 (d, J=14.36 Hz, 1 H) 1 1.35 (d, J=14.66 Hz, 1 H) 13.47 (s, 1 H).
[0156] Step 2: 2-( 3-f(trifluoromethyl)thio phenyl)pyrazoIo [1,5-al pyrimidin-7-ol
[0157] A solution of 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l,3-dioxane-4,6-dione (615.00 g) in 1,2-dichIorobenzene (6.3 L) was stirred at ambient temperature for 10 minutes. The solution was then heated to 150°C over 75 minutes. The reaction was maintained at this temperature for 16 hours. An sample was taken after 16 hours and the UPLC analysis showed the complete conversion of 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yI)amino)methylene)-l,3-dioxane-4,6-dione to 2-(3- ((trifluoromethyl)tmo)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol. The reaction was cooled to 20°C over 130 minutes. At this point, a thick white slurry had formed and the reaction was filtered using a Nutche Filter over 15 minutes. The reactor was washed with 1.8 L of acetonitrile and this solution was then mixed with the filter cake and then the solvent was removed by filtration. The cake was dried for ~40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16 hours). The reaction was then analyzed by HPLC and NMR to give 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (331.2 g, 72%) Ή NMR (300 MHz, METHANOL-^) δ ppm 6.55 (d, J=7.33 Hz, 1 H) 7.59 (s, 1 H) 8.40 – 8.52 (m, 1 H) 8.53 – 8.64 (m, 1 H) 8.69 (d, J=7.62 Hz, 1 H) 9.01 (dt, J=7.77, 1.39 Hz, 1 H) 9.12 (s, 1 H) 13.34 (s, 1 H).
[0158] Step 3: l-(2-(3-(f trffluoromethvmhiotohenvnpyrazolo n.5-al pyrimidin-7-vn-lH-benzofdiri.2.31triazole: triethylamine: hydrochloride complex (1:1.25:1.25 molesimolestmolest
[0159] To a solution of 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (30.00 g), benzotriazole (287.02 g) in acetonitrile (3000 mL) and triethylamine (403.00 mL) at 0°C, was added phosphoryl chloride (108 mL) slowly under a blanket of nitrogen, maintaining < 10°C. The reaction was then warmed to 80°C over 45 minutes and stirred for 240 minutes. HPLC indicated complete
consumption of starting material. To the reaction mixture was added acetonitrile (3000 mL) while maintaining the temperature at 80°C. The reaction was then cooled to 20°C over 80 minutes. The reaction was then stirred at ambient temperature for 14 hours. At this point, a thick slurry had formed and the reaction was filtered using a Nutche filter over 15 minutes. The reactor was washed twice with 900 mL of acetonitrile and this solution was then mixed with the filter cake and then the solvent was removed by filtration. The cake was dried for -40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16h). The reaction was then analyzed by HPLC and NMR to give l-(2-(3-((trifluorometJiyl)thio)phe
triethylamine: hydrochloride complex (1:1.25:1.25 moles:moles:moles) (438.1 g, 83%). ¾ NMR (300 MHz, DMSO-</6) δ ppm 1.19 (t, J=7.33 Hz, 12 H) 3.07 (qd, J=7.28, 4.84 Hz, 8 H) 7.60 – 7.78 (m, 6 H) 7.80 – 7.87 (m, 1 H) 8.15 (dt, J=7.99, 1.28 Hz, 1 H) 8.24 (s, 1 H) 8.33 (dt, J=8.14, 0.92 Hz, 1 H) 8.85 (d, J=4.69 Hz, 1 H).
[0160] Step 4: ff3aR4R.6R.6aS 2.2-dimethyl-6-ff2-f3~mrifluoromethyl)thio)phenvnpyrazoloil.5-alD\timidin-7-yl¼mino)tctralivdro-3aH-cvcLoDentaldlll,31dioxol-4-vnincthanol
[0161] To the reactor was added l-(2 3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)-lH-benzo[d][l,2,3]triazole: triethylamine: hydrochloride complex (1 :1.25: 1.25 moles :moles:moles) (430.0 g) and ((3aR,4R,6R,6aS)-6-amino-2,2-dimethyltetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol hydrochloride (209.0 g) and then triethylamine (2103 mL) was added. The reaction was then heated to 80°C, under a blanket of nitrogen. After 360 minutes, HPLC analysis indicated that the reaction mixture contained <1% starting material and the reaction was cooled to 20°C over 60 minutes. To the reaction was added ethyl acetate (3.5 L) and water (3.5 L). After stirring for 10 minutes the phases were separated and the aqueous layer was back extracted with ethyl acetate (3.5 L). The organic layers were combined and concentrated to form a dark, brown oil. Acetonitrile (4.5 L) was added and the solution was concentrated to dryness to give an orange solid. The solids was transferred back to the reaction with water (4.3 L), heated to 50°C, and stirred for 20 minutes. White solids formed in this hot solution and were isolated by filtration using a Nutche Filter over 15 minutes. The solids were dried under vacuum for 15 minutes on the filter and then dissolved in acetonitrile (4.0 L) at 0°C. The solution was stirred for 1 minutes. The solution was then filtered through a fritted funnel to remove the hydrolysis solid by product and the solution was concentrated to dryness. The solids were dried in a vacuum oven at full vacuum overnight (40°C, 16 hours). The reaction was then analyzed by HPLC and NMR to give ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5 -a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanoI (349.2 g, 88%). Ή NMR (300 MHz, DMSO-<¾) δ ppm 1.25 (s, 3 H) 1.47 (s, 3 H) 1.76 – 1.90 (m, 1 H) 2.25 (br d, J-3.22 Hz, 1 H) 2.33 – 2.47 (m, 1 H) 3.46 – 3.67 (m, 2 H) 4.08 (br d, J=5.57 Hz, 1 H) 4.48 – 4.64 (m, 2 H) 5.19 (t, J=4.40 Hz, 1 H) 6.28 (d, J=5.28 Hz, 1 H) 7.06 (s, 1 H) 7.58 – 7.71 (m, 1 H) 7.72 – 7.80 (m, 1 H) 8.12 – 8.24 (m, 2 H) 8.31 (d, J=7.62 Hz, 1 H) 8.42 (s, 1 H).
[0162] Step 5: ((3aR.4R.6R.6aS 2.2-dimethyl-6-ff2-f3-fftrifluoroinethYmhio)phenvnpyrazolo[1.5-al Dyrimidin-7-vnan] iiio>tetrahvdro-3aH-cvclonen ta [dl [1,31 dioTOl-4-yl )meth yl tert-bntoxycarbonylsulfamate
[0163] ((3aR,4R,6R,6aS)-2,2-dime l-6-((2-(3-((trifluorome
7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol (6.0 g) was dissolved in 2-methyltetrahedrafuran (60.0 mL) and to this solution was added pyridinium p-toluenesulfonate (5.9 g). This formed a precipitated and to this white slurry was added (4-aza-l-azoniabicyclo[2.2.2]oct-l-ylsulfonyl)(tert-butoxycarbonyl)azanide-l,4-diazabicyclo[2.2.2]octane (1 :1) hydrochloride1 (17.0 g). The mixture was stirred at ambient temperature until the HPLC showed <1% ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol remaining starting material (-300 minutes). The reaction was quenched with water (60 mL) and the phases were separated. To the organic layer was added acetonitrile (60 mL) and the mixture was concentrated using a rotovap at 50°C to ~60 mL. The mixture was allowed to cool to room temperature and stirred overnight. During this time a white slurry formed. White solids were filtered using a medium fritted filter. The solid was dried in a vacuum oven at full vacuum overnight (40 °C). The reaction was then analyzed by HPLC and NMR to give ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyI)tM^
cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (5.03 g, 68%). [H NMR (300 MHz, DMSO- 6) δ ppm 1.26 (s, 3 H) 1.42 (s, 9 H) 1.51 (s, 3 H) 2.33 – 2.48 (m, 2 H) 3.30 (br s, 1 H) 4.06 – 4.21 (m, 1 H) 4.29 (d, J=5.28 Hz, 2 H) 4.52 (dd, J=7.18, 5.13 Hz, 1 H) 4.76 (dd, J=7.18, 4.54 Hz, 1 H) 6.35 (d, J=5.57 Hz, 1 H) 7.08 (s, 1 H) 7.63 – 7.72 (m, 1 H) 7.74 – 7.82 (m, 1 H) 8.01 (d, ^=7. 2 Hz, 1 H) 8.21 (d, J=5.28 Hz, 1 H) 8.31 (dt, J=7.84, 1.36 Hz, 1 H) 8.48 (s, 1 H) 1 1.92 (br s, 1 H)
[0164] Step 6: f R,2R3S.4R)-2J-dihvdroxy-4-((2-(3-fftrifluoromethvDthio^phenvnpyrazolori.5-a]pyrimidin-7-yl)aminokvcl nent\l)methyl sulfamate
[0165] To a solution of ((3aR,4R,6R!6aS)-2,2-dimethyl-6-((2-(3- ((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (2.0 g) in acetonitrile (11 mL) at 0°C was added phosphoric acid (1 1 mL) while maintaining the temperature below 10°C. This mixture was warmed to ambient temperature and stirred for 4 hours. At this time HPLC analysis showed that <1% ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate starting material or reaction intermediates remained. To the reaction was added ethyl acetate (1 1 mL) and water (11 mL) and saturated Na2C03 (10 mL) dropwise. After this addition was complete saturated Na2C03 was added until the pH was between 6-7. The phases were separated and to the organic layer was added acetonitrile (30 mL) and the mixture was concentrated on a rotovap to ~16 mL. The mixture was stirred overnight. During this time a white slurry formed. The white solids were filtered using a medium filtted filter. The solid was dried in a vacuum oven at full vacuum overnight (40°C). The reaction was then analyzed by HPLC and NMR to give ((lR,2R,3S,4R)-2,3-dihydroxy-4-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)cyclopentyl)methyl sulfamate (1.5g ,84%). lH NMR (300 MHz, DMSO-c¾) δ ppm 1.44 – 1.61 (m, 1 H) 2.20 – 2.42 (m, 2 H) 3.78 (q, J-4.50 Hz, 1 H) 3.90 – 4.09 (m, 3 H) 4.09 – 4.22 (m, 1 H) 4.80 (d, ^5.28 Hz, 1 H) 5.03 (d, J=5.28 Hz, 1 H) 6.31 (d, J=5.57 Hz, 1 H) 7.05 (s, 1 H) 7.48 (s, 2 H) 7.62 – 7.72 (m, 1 H) 7.77 (d, J=7.92 Hz, 2 H) 8.17 (d, J=5.28 Hz, 1 H) 8.31 (dt, ^7.70, 1.43 Hz, 1 H) 8.47 (s, 1 H).
[0166] Example 3: fflR.2R.3S.4RV2.3-dihvdrosy-4-ff2-f3- ( ( trifluoroniethyl )thio)ph en vDpyrazolo 11,5-a I pyi Lmidin-7-Yl)amino)cvclopcntyl>m ethyl sulfama te
[0167] Step 1: .2-dimethyl-5-ff -(3-frtrifluoromethvnthio)phenvn-lH-pyrazol-5-yl)ainino)methylene -l,3-dioxane-4,6-dione)
[0168] Under a blanket of nitrogen at 20°C, Meldrum’s acid (18.6 Kg) and isopropanol (33 L) were placed in a 100 L glass-lined reactor. Trimethyl orthoformate (15.5 Kg (16.0L)) and isopropanol (11 L) were added and the mixture was heated to 80 °C for 40 min, whereby a small amount of methanol distilled off (<0.5 L). The mixture was stirred for 2 h at 80 °C. in a separate 160 L glass-lined reactor under nitrogen at 20 °C, 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (prepared in the manner described above) was mixed with isopropanol ( 10.9 kg, 42.0 mmol) and heated up to 80 °C within 60 min. The content of the 100 L reactor was transferred into the reaction mixture in the 160 L reactor at 80 °C, which was completed after 3 min. The reaction mixture was stirred for 30 min at 78 °C, the reaction was then cooled to 60 °C. HPLC analysis showed the reaction was 99.56% complete (product%/(product%+starting material0/.). The reaction mixture was cooled to 20 °C within 100 min, then the mixture was stirred for further 100 min at 20 °C. The suspension was then transferred onto a pressure filter. At 1.2 bar nitrogen, the solids were collected on the filter. The filter cake was washed 4 x with ethyl acetate (18 L each time). The wet cake was dried on the filter for 17 h at 20°C using a slight stream of nitrogen/vacuum (200-100 mbar). The wet product (14.7 kg) was further dried at the rotavap for approx. 24 h at 40-50 °C. 11,75 kg of the crude title compound was obtained (68% yield). NMRspectrum was consistent with that described above in Example 2.
[0169] Step 2: 2-(3-fftrifluoromethvnthio)phenYnpyrazolori.S-a1pyrimidin-7-ol
[0170] Under nitrogen at 20 °C, (2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l ,3-dioxane-4,6-dione) was placed in the reactor. 1 ,2-Dichlorobenzene (117 L) was added. The suspension was heated to 147°C for 90 min to give a solution, then it was stirred at 147°C for 18 h. Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 92.28% completion (product%/(product%+starting material%). The mixture was heated up again to 147°C and stirred for further 5 h at this temperature. HPLC analysis showed the reaction was 96.51% complete (product%/(product%+starting material%). The mixture was then stirred for 48 hours at 20°C, then it was heated again to 147°C und stirred at this temperature for 5 h. Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 98.47% completion (product%/(product%+starting material%). The mixture was heated up again to 146°C and stirred for further 5 h at this temperature.
Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 99.35% complete (product%/(product%+starting material%). The reaction was cooled to 20°C and the suspension was transferred in a pressure filter. The solids were collected on the filter at 1.8-3 bar N2 over a greater than 10 hour period. The filter cake was washed 4 x with acetonitrile (17 L), then it was dried on the filter for 18 h at 20°C/200-100 mbar, using a slight stream of N2. The material was transferred to a 50 L flask and dried on a rotavap at 50-60°C / 24-14 mbar for 2 d. 6.118 kg of the crude title compound was obtained (70% yield). NMR spectrum was consistent with that described above in Example 2.
[0171] Step 3: l-f2-f3- trifluoromethYnthio^phenvnpyrazoIo[1.5-alpyriinidiii-7-vn-lH-benzofdl [1.2.31 triazolc: triethylamine: hydrochloride complex ( 1 :0.21:0.21 moles:moles:moles)
[0172] Under N2 at 20°C, acetonitrile (30 L) was placed in the reactor, 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (6.00 kg) and lH-benzotriazol (5.83 kg) was added. A further portion of acetonitrile (30 L) was added, then the mixture was stirred at 20°C. Stirring proceeded over night. Triethylamine (8.16 L) was added at 20°C over 6 min. The yellow suspension was heated up to 45°C for 40 min. While stirring at 150 rpm, phosphoryl chloride (4.562 kg) was slowly added for 45 min. By controlling the addition, the reagent was dropped directly into the mixture to avoid the formation of lumps. The addition was exothermic, a maximum temperature of 53°C was observed. The brown suspension was heated up to 80°C over 1 h, then the reaction mixture was stirred for 5 h at this temperature. Acetonitrile (30 L) was added over 20 min keeping the internal temperature between 75-80°C. HPLC analysis showed the reaction was 98.31% completion (product%/(product%+starting material%).The mixture (brown suspension) was further stirred at 80°C for 70 min. HPLC analysis showed the reaction was 99.48% completion (product%/(product%+starting material%). Acetonitrile (61 L) was added over 30 min maintaining the temperature between 75-80°C. The pale brown suspension was stirred at 80°C for 90 min, then it was cooled to 20°C over 2.5 h. The mixture was stirred for 12 h at 20°C. The mixture was transferred in a pressure filter. The filter cake was washed twice with acetonitrile ( 18 L). Both wash steps were done at 3.5-4 bar N2. Each of these filtrations took overnight to go to completion. The filter cake was dried on the filter for 7.5 h. The material was transferred in a 50 L flask and dried at the rotavap at Ta 40-50°C / 50-11 mbar for 3 d to get a dry mass of 99.88% . The yield of l -(2-(3-((trifluoromethyl)t]iio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-yl)-lH-benzo[d][l,2,3]triazole: triethylamine: hydrochloride complex (1 :0.21 :0.21 moles:moles:moles) was 7.948 kg (75%). NMR spectrum was consistent with that described above in Example 2.
[0173] Step 4: 3aR4R.6R,6aS)-2,2-dimethYl-6-f(2-f3-ffMfluoromethvnthio phenvnpyrazolori.5-alDyrimidin-7-yl)amino)tetrahvdro-3aH- vclopenta Idl [1.31 dioxol-4-vDmethanol
[0174] Under N2 in a 160 L glasslined reactor, triethylamine (21%) compound with l -(2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5 -a] pyrimidin-7-yI) – 1 H-benzo [d] [ 1 ,2,3 Jtriazole (21 %) hydrochloride (7.86 kg) was dissolved in triethylamine (23.3 L) at 20°C. ((3aR,4R,6R,6aS)-6-amino-2,2-dimethyltetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol hydrochloride (4.49 kg) was added, followed by triethylamine (23 L). The reaction mixture was heated up to 80°C over 1 h, and then the mixture was stirred for 8 h at 80°C. The mixture was then cooled to 20°C. HPLC analysis showed the reaction was 99.97% complete (product%/(product%+starting material%). Water (66 L) was then added over 30 min at 20-25°C (exotherm), whereby a brown suspension was obtained. The mixture was concentrated at 60°C, 150-95 mbar, until 42 L solvent was distilled off. The suspension was heated to 50°C, and the solids were collected on a 90 L pressure filter (1.2 bar N2), which took 40 min. During this process, the material on the filter was not actively heated. The remaining solids in the reactor were rinsed with 15 L of the mother liquor. The wet filter cake was transferred back in the reactor. Water (64 L) was added. The mixture was heated up to 50°C over 30 min. The washed solids were collected on the 90 L pressure filter. Remaining mother liquor in the filter cake was pressed off at 1.2 bar N2 for 50 min (50 L mother liquor was used to rinse the reactor). The filter cake was dried on the pressure filter for 13.5 h, applying a slight stream of N2 / vac at 20°C to afford 10.247 kg of crude ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)tWo)phenyl)pyrazolo[l ,5-a]pyrimidin-7-yl)ammo)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol. The wet filter cake was isolated. The wet filter cake was loaded into the reactor. Acetonitrile (65 L) was added, followed by activated charcoal (6.59 kg). The mixture was heated to 50°C for 30 min and stirred for 2 h at 50°C. Meanwhile a bed of celite (4.25 kg) had been prepared in the 90 L pressure filter, using acetonitrile (20 L) for conditioning. The bed was heated at 50°C. The black suspension was transferred on the filter and pushed through the Celite plug at 2 bar. The filtrate was transferred to a 200 L stirring tank via a heat resistant tube and a 0.45 μιη inline filter. The operation needed 18 min for completion. For washing, acetonitrile (50 L) which had been warmed up in the reactor to 50°C and transferred over the warmed filter cake and pushed through at 2 bar. Again, the filtrate was transferred in the 200 L stirring tank via a heat resistant tube and a 0.45 μιη inline filter. The operation needed 10 min for completion. The reactor was cleaned to remove attached charcoal (abrasive cleaning, using NaCl /acetone). The filtrate in the stirring tank was transferred in the reactor and concentrated at 50°C / 120 mbar until 63 L were distilled off. While well stirring (300 rpm) and 50°C, Water (1 10 L) was slowly added over 2 h. A pale yellow suspension was formed. The concentrate was cooled to 20°C for 3 h, then stirred at this temperature for 13 h. The solids were collected on a 50 L filter, using 1.2 bar N2 to push the filtrate through. The filter cake was washed twice with water (18 L), then dried on the filter for 24 h at 200-100 mbar, using a slight stream of N2. 4.563 kg of the title compound was obtained 55% yield. NMR spectrum was consistent with that described above in Example 2.
[0175] Step 5: (f3aR,4R,6R,6aS)-2^-dimethyl-6-(f2-f3-fftrifluorQmethvnthio phenvnpyrazolo[1.5- |pyrimidm-7-vnamino)teti ahYclro-3aH-cvclopenta|d||1.3ldioxol-4-yl mcthyl tert-butoxycarbonylsutfamatc
[0176] Under N2 at 20°C, ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3- ((trifluoromethyl)thio)phenyl)pyrazolo[ 1 , 5 -a]pyrimidin-7-yl)amino)tetrahydro-3 aH-cyclopenta[d][l,3]dioxol-4-yl)methanol (4.019 kg) was placed in a 160 L glasslined reactor, then 2-methyl-tetrahydrofuran (40 L) was added. The mixture was stirred at 150 rpm for 30 min at 20°C, whereby a clear solution was formed. A KF measurement was taken and showed the water content to be 0.036% H20. The solution was stirred over night at 20 °C. The next morning, PPTS (2.2 kg) was loaded into the reactor. At 20°C, (4-aza-l-azoniabicyclo[2.2.2]oct-l-yIsulfonyl)(tert-butoxyc£u-bonyl)azanide-l,4-diazabicyclo[2.2.2]octane (1:1) hydrochloride (10.2 kg) was added. Stirring of the heterogeneous mixture was started at 130 rpm. The reaction was stirred with 200 rpm for 1 h at 20°C, then with increased speed of 250 rpm for an additional hour. HPLC analysis showed the conversion to be 87.3%. The reaction mass was stirred with 300 rpm for 2 h at 20°C. HPLC analysis showed the conversion to be 95.6%. The reaction mass was stirred with 300 rpm for 2 h at 20°C. HPLC analysis showed the conversion to be 97.7%. NaHC03 3.7% (40 L) was added to the mixture at 20°C and the reaction was stirred at 300 rpm for 10 min. Most of the solids from the reaction mixture went into solution. To dissolve remaining material which was attached at the top of the reactor, the bilayered mixture was stir up shortly by a N2 stream from the bottom. The layers were separated, which was completed after 13 min. The aqueous layer was discharged, the organic layer remained in the reactor. The org. layer was a brown solution, the aqueous layer was colorless and turbid. The pH of aqueous layer was approx. 8 (pH stick). NaHC03 3.7% (40 L) was added to the mixture at 20°C and it was stirred at 300 rpm for 10 min. The layers were separated, which was completed after 27 min. The aqueous layer was discharged, the organic layer remained in the reactor. The organic layer was a brown solution, the aqueous layer was colorless and turbid. The pH of aqueous layer was approx. 8-9 (pH stick) and the pH of organic layer was approx. 8 (pH stick, wet). The product in organic layer was transferred in the feeding tank and stored temporarily (approx. 30 min) at 20°C. The reactor was optically cleaned using a mixture of 2-methyltetrahydrofuran (30 L) and H20 (20 L). The org. layer was placed in the reactor and stored at -20°C for 14.5 h . While stirring at 150 rpm, the org. layer (suspension) was diluted with acetonitrile (16 L) and water (15 L) and warmed up to 5°C. At 5°C, acetic acid (0.172 kg) was added over 5 min. to a pH of 6; resulting in a mixture that was a pale brown solution. ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (2.0 g; prepared in a similar manner to that described above Example 2, Step 5) was added as seed. At 5°C, acetic acid (0.515 kg) was added over 15 min. to pH 4-5; a suspension formed. The feeding tank was rinsed with water (1.6 L). The mixture was stirred at 5°C with 90 rpm for 1.5 h, then it was transferred in a 50 L filter and filtered at 1.2 bar N2, in only 4 min. The filter cake was washed 4 x with cold acetonitrile (8 L, 0-5°C), then it was dried on the filter at 20°C for 8 h at 200 mbar, using a slight stream of N2. The yield of the title compound was 3.594 kg (62%). MR spectrum was consistent with that described above in Example 2.
[0177] Step 6: friR.2R.3S.4R 2.3-dihvdroxY-4-ff2-f3-fftrifluoromethvntliio phenvnDyrazolori.5-alpyrimidin-7-yl)aminokvciopent>T)mcthyl sulfamate Compound 1
[0178] 3.538 kg of ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate was suspended in 13.5 kg of acetonitrile and cooled to 5°C. To this mixture was added 27.3 kg of H3PO4 over 1 hour and 50 minutes. The reaction was warmed to 20°C over 50 minutes and then stirred for 8h at 22°C. HPLC analysis showed the reaction was 99.69% complete. To the first portion (50% of the reaction mixture) was added 8.9 kg of water and 7.95 kg of ethyl acetate. The pH was then adjusted to 6.5 with 48 L of saturated sodium carbonate. 7.7 kg of ethyl acetate was added and the phases were separated. To the second portion (50% of the reaction mixture) was added 8.9 kg of water and 7.95 kg of ethyl acetate. The pH was then adjusted to 6.15 with 48 L of saturated sodium carbonate. 7.7 kg of ethyl acetate was added and the phases were separated. The organic phases were combined in a vessel (rinsed with 1.8 kg of ethyl acetate) and washed with 17.8 kg of water. The phases were separated and 17.8 kg of water and 0.237 kg of NaCl were added and the phases were separated. A repeat of wash with 17.8 kg of water and 0.237 kg of NaCl was added and the phases were separated. The organic layers were then combined and the temperature of the mixture was raised to 40°C and the pressure was reduced to 300-142 mbar. 27 L of liquid was distilled off over 4h. 31.7 kg of acetonitrile were then added to the solution and the temperature of the mixture was raised to 38°C and the pressure was reduced to 320-153 mbar. 26 L of liquid was distilled over 3h. 31.7 kg of acetonitrile were then added to the solution and the temperature of the mixture was raised to 37°C and the pressure was reduced to 320-153 mbar. 34 L of liquid was distilled over 2h. The suspension was stirred for lh at 50°C and then cooled to 20-25°C over 3h. The reaction was stirred overnight and the product was filtered and washed with 8.9 kg of acetonitrile twice. The cake was dried for 2h at 20°C (33 mbar) then at 40-45°C (1 mbar) to afford 2.08 kg (75.8%) of the title compound. 2.066 kg of ((lR,2R,3S,4R)-2,3-dihydroxy-4-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 , 5 -a]pyrimidin-7-yl)amino)cyclopenty l)methy 1 sulfamate was loaded into a reactor with 9.76 kg of acetronitrile and 4.12 kg of water and heated at a temperature of 56 °C for 1 hour and 10 minutes until dissolved. The solution was polished filtered and the filter was
rinsed with 3.16 kg acetonitrile and 1.37 kg of water. To the resulting solution was added with 11.0 kg of water over 45 minutes while maintaining the reaction temperature between 52-55°C. 0.009 kg of (( 1 R,2R,3S,4R)-2,3 -dihydroxy-4-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)cyclopentyl)methyl sulfamate was added as seed (prepared in a similar manner to that described above Example 2, Step 5). A suspension was visible after 10 minutes of stirring. To the solution was added 9.62 kg of water over 3h while maintaining the reaction temperature between 50-55°C. The suspension was then cooled over 3h to 20°C and stirred for 12h at 22-23°C. The suspension was then filtered and washed twice with 13.7 kg of water. The product was dried at 40°C. 1.605 kg of the title compound was obtained in 78% yield. NMR spectrum was consistent with that described above in Example 2.
PATENT
WO2016069392
SYNTHESIS
![]()
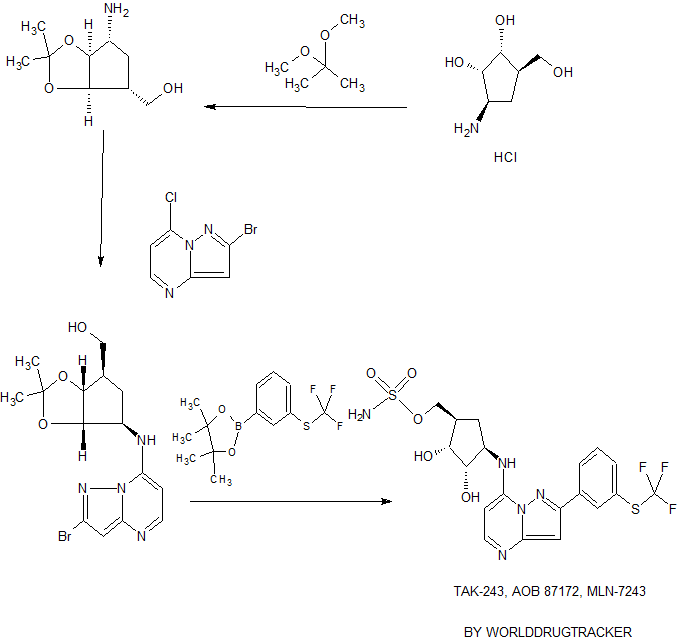
![]()
///////////////1450833-55-2, MLN 7243, TAK-243, TAK 243, TAK243, MLN7243; MLN-7243, MLN 7243, AOB87172, AOB-87172, AOB 87172, Millennium Pharmaceuticals, Inc., PHASE 1, TAKEDA ONCOLOGY
COS(=O)(=O)N[C@H]1C[C@H]([C@@H]([C@@H]1O)O)NC2=CC=NC3=CC(=NN23)C4=CC(=CC=C4)SC(F)(F)F
PF-06282999

PF 6282999
Alternative Names: PF-06282999; PF-6282999, PF-06282999
Cas 1435467-37-0
[2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide]
2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide
MF C13H12ClN3O3S
Molecular Weight: 325.767
Elemental Analysis: C, 47.93; H, 3.71; Cl, 10.88; N, 12.90; O, 14.73; S, 9.84
Irreversible inactivator of myeloperoxidase
Currently in clinical trials for the potential treatment of cardiovascular diseases.
Phase I
- Phase I Acute coronary syndromes
Most Recent Events
- 01 Mar 2015 Pfizer terminates phase I trial in Healthy volunteers in USA (NCT01965600)
- 10 Sep 2014 Pfizer completes enrolment in its phase I trial in Healthy volunteers in USA (NCT01965600)
- 01 Feb 2014 Phase-I clinical trials in volunteers in USA (PO)
A drug potentially for the treatment of acute coronary syndrome (ACS).

![]()
![]()
PF-06282999 is a potent and selective myeloperoxidase Inhibitor which is potential useful for the Treatment of Cardiovascular Diseases. PF-06282999 displayed excellent oral pharmacokinetics in preclinical species and robust irreversible inhibition of plasma MPO activity both in human blood stimulated exogenously and in plasma collected after oral (po) administration to lipopolysaccharide (LPS)-treated cynomolgus monkeys.
PF-06282999 has been advanced into first-in-human pharmacokinetics and safety studies. Myeloperoxidase (MPO) is a heme peroxidase that catalyzes the production of hypochlorous acid. Clinical evidence suggests a causal role for MPO in various autoimmune and inflammatory disorders including vasculitis and cardiovascular and Parkinson’s diseases, implying that MPO inhibitors may represent a therapeutic treatment option
The thiouracil derivative PF-06282999 [2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide] is an irreversible inactivator of myeloperoxidase and is currently in clinical trials for the potential treatment of cardiovascular diseases. Concerns over idiosyncratic toxicity arising from bioactivation of the thiouracil motif to reactive species in the liver have been largely mitigated through the physicochemical (molecular weight, lipophilicity, and topological polar surface area) characteristics of PF-06282999, which generally favor elimination via nonmetabolic routes.

To test this hypothesis, pharmacokinetics and disposition studies were initiated with PF-06282999 using animals and in vitro assays, with the ultimate goal of predicting human pharmacokinetics and elimination mechanisms. Consistent with its physicochemical properties, PF-06282999 was resistant to metabolic turnover from liver microsomes and hepatocytes from animals and humans and was devoid of cytochrome P450 inhibition. In vitro transport studies suggested moderate intestinal permeability and minimal transporter-mediated hepatobiliary disposition. PF-06282999 demonstrated moderate plasma protein binding across all of the species.
Pharmacokinetics in preclinical species characterized by low to moderate plasma clearances, good oral bioavailability at 3- to 5-mg/kg doses, and renal clearance as the projected major clearance mechanism in humans. Human pharmacokinetic predictions using single-species scaling of dog and/or monkey pharmacokinetics were consistent with the parameters observed in the first-in-human study, conducted in healthy volunteers at a dose range of 20-200 mg PF-06282999.
In summary, disposition characteristics of PF-06282999 were relatively similar across preclinical species and humans, with renal excretion of the unchanged parent emerging as the principal clearance mechanism in humans, which was anticipated based on its physicochemical properties and supported by preclinical studies.
PAPER
Journal of Medicinal Chemistry (2015), 58(21), 8513-8528.
Discovery of 2-(6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999): A Highly Selective Mechanism-Based Myeloperoxidase Inhibitor for the Treatment of Cardiovascular Diseases
Worldwide Research and Development, Pfizer, Inc., Groton, Connecticut 06340, United States
J. Med. Chem., 2015, 58 (21), pp 8513–8528
DOI: 10.1021/acs.jmedchem.5b00963

Myeloperoxidase (MPO) is a heme peroxidase that catalyzes the production of hypochlorous acid. Clinical evidence suggests a causal role for MPO in various autoimmune and inflammatory disorders including vasculitis and cardiovascular and Parkinson’s diseases, implying that MPO inhibitors may represent a therapeutic treatment option. Herein, we present the design, synthesis, and preclinical evaluation of N1-substituted-6-arylthiouracils as potent and selective inhibitors of MPO. Inhibition proceeded in a time-dependent manner by a covalent, irreversible mechanism, which was dependent upon MPO catalysis, consistent with mechanism-based inactivation. N1-Substituted-6-arylthiouracils exhibited low partition ratios and high selectivity for MPO over thyroid peroxidase and cytochrome P450 isoforms. N1-Substituted-6-arylthiouracils also demonstrated inhibition of MPO activity in lipopolysaccharide-stimulated human whole blood. Robust inhibition of plasma MPO activity was demonstrated with the lead compound 2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999, 8) upon oral administration to lipopolysaccharide-treated cynomolgus monkeys. On the basis of its pharmacological and pharmacokinetic profile, PF-06282999 has been advanced to first-in-human pharmacokinetic and safety studies.
tan solid (mp = 165.3 °C).
1H NMR (500 MHz, DMSO-d6) δ 12.85 (s, 1 H), 7.57 (dd, J = 9.03, 2.68 Hz, 1 H), 7.33 (s, 1 H), 7.17–7.23 (m, 2 H), 7.10 (s, 1 H), 5.89 (d, J = 1.71 Hz, 1 H), 5.41 (br s, 1 H), 3.89 (br s, 1 H), 3.84 (s, 3 H).
MS (ES+) m/z: 326.0 [M + H]+. HRMS: m/z calcd for C13H13ClN3O3S [M + H]+ 326.0366, found 326.0361.
Anal. Calcd for C13H12ClN3O3S: C, 47.93; H, 3.71; N, 12.90; S, 9.84. Found: C, 47.81; H, 3.70; N, 12.83; S, 9.83. HPLC purity: >95%.
PATENT
WO 2013068875
http://www.google.co.in/patents/WO2013068875A1?cl=en
Beta Keto Ester Route Section
A. Carboxylic Acid Route Section
Preparation 1

Ethyl 3-(5-chloro-2-methoxyphenyl)-3-oxopropanoate
A 3000 mL 3-necked round-bottomed flask flushed with nitrogen was charged with magnesium ethoxide (67.46 g, 589.51 mmoles) and THF (1 100 mL), and the resulting mixture was stirred as ethyl hydrogen malonate (162.26 g, 1 .18 moles; 145.00 mL diluted in 100 ml of THF) was added and the mixture was heated at 45 °C for 4 hours. Meanwhile, a 2000 mL 3-necked round-bottomed flask flushed with nitrogen was charged with 5-chloro-2-methoxybenzoic acid (100 g, 536 mmoles) and THF (600 mL). To this mixture stirring at room temperature was added 1 , 1 ‘-carbonyldiimidazole (95.59 g, 589.5 mmoles) in portions to avoid excess foaming. After stirring for 3 hours at room temperature the second solution was added gradually to the first solution. After addition the reaction mixture was heated to 45 °C. After 20 hours, the reaction mixture was concentrated under reduced pressure before adding ethyl acetate (1 L) followed by 2 N HCI (500 mL). After mixing, the layers were separated and the organic phase was washed sequentially with 2 N HCI (500 mL), saturated sodium bicarbonate (500 mL), and water (500 mL). The organic phase was concentrated under reduced pressure, the residue taken up in ethyl acetate (1000 mL) and concentrated again to afford the title compound (104.94 g).
MS (ES+) 257.2 [M+1 ]+. 1 H NMR showed product as a 7.5:1 keto:enol mixture. For the keto tautomer: 1 H NMR (500 MHz, CDCI3) δ ppm 7.85 (d, J=2.93 Hz, 1 H) 7.45 (dd, J=8.90, 2.81 Hz, 1 H) 6.92 (d, J=8.78 Hz, 1 H) 4.18 (q, J=7.16 Hz, 2 H) 3.95 (s, 2 H) 3.90 (s, 3 H) 1 .24 (t, J=7.07 Hz, 3 H). Preparation 2

(Z)-Ethyl 3-((2-amino-2-oxoethyl)amino)-3-(5-chloro-2-methoxyphenyl)acrylate A 5-L reaction vessel was charged with methanol (3.3 L), sodium methoxide (102.4 g, 1.8 moles), and glycinamide hydrochloride (202 g, 1.8 moles). The mixture was heated at 65 °C for 1 hour before cooling to 50 °C and adding acetic acid (514.25 mmoles, 30.88 g, 29.47 ml.) and ethyl 3-(5-chloro-2-methoxyphenyl)-3-oxopropanoate (300 g, 1.03 mole). After heating to reflux for 16 hours, the reaction mixture was stirred as it was cooled to 10 °C. After 30 min the resulting solid was collected by vacuum filtration, pulling dry to form a cake that was dried in a vacuum oven (20 mm Hg, 65 °C) for 14 hours to afford the title compound (339.4 g).
MS (ES+) 313.2 [M+1]+. 1H NMR (500 MHz, DMSO-d6) δ ppm 8.80 (t, J=5.00 Hz, 1 H) 7.47 (dd, J=8.90, 2.81 Hz, 1 H) 7.27 (br. s., 1 H) 7.22 (d, J=2.68 Hz, 1 H) 7.14 (d, J=8.78 Hz, 1 H) 7.09 (br. s., 1 H) 4.30 (s, 1 H) 4.03 (q, J=7.07 Hz, 2 H) 3.80 (s, 3 H) 3.56 (br. s., 1 H) 3.45 (br. s., 1 H) 1.18 (t, J=7.07 Hz, 3 H).
Example 1

2-( 6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3, 4-dihydropyrimidin
acetamide
A reaction vessel equipped with an efficient stirrer was charged with (Z)-ethyl 3-((2- amino-2-oxoethyl)amino)-3-(5-chloro-2-methoxyphenyl)acrylate (15 g, 50.2 mmol), butyl acetate (150 ml.) and trimethylsilyl isothiocyanate (160.7 mmole, 21 .1 g, 22.7 ml.) and the mixture was heated to reflux. After 15 hours, the mixture was cooled to 30 °C and treated with 1 N aqueous sodium hydroxide (1 12.5 ml_, 1 12.5 mmoles). After 30 min, the organic layer was separated and extracted with another portion of 1 N sodium hydroxide (37.5 ml_, 37.5 mmoles). The combined aqueous phases were extracted twice with dichloromethane (2 x 45 mL), filtered, and treated with 6N HCI until a pH of 2.5 was achieved. After stirring for 1 hour, the resulting solid was isolated by vacuum filtration, resuspended in 100 mL of a 1 :1 methanol-water solution, heated with stirring at 50 °C for 2 hours, and cooled to room temperature before collecting the solid by vacuum filtration, pulling dry and drying in a vacuum oven (20 mm Hg, 50 °C) for 12 hours to afford 8.7 g of the desired product as a tan solid.
MS (ES+) 326.0 [M+1]+. 1H NMR (500 MHz, DMSO-d6) δ ppm 12.85 (s, 1 H) 7.57 (dd, J=9.03, 2.68 Hz, 1 H) 7.33 (s, 1 H) 7.17 – 7.23 (m, 2 H) 7.10 (s, 1 H) 5.89 (d, J=1.71 Hz, 1 H) 5.41 (br. s, 1 H) 3.89 (br. s, 1 H) 3.84 (s, 3 H).
Alternative Preparation of Example 1

2-( 6-( 5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3, 4-dihydropyrimidin- 1 ( 2H)-yl) acetamide A slurry of (Z)-ethyl 3-((2-amino-2-oxoethyl)amino)-3-(5-chloro-2- methoxyphenyl)acrylate (20 g, 63 mmol) in a mixture of butyl acetate (140 mL) and DMF (38 mL) was treated with trimethylsilyl isothiocyanate (16.8 g, 125 mmol) and the mixture was heated at 1 15-120 °C for 5-6 hours. The mixture was cooled to 0-5 °C, butyl acetate (100 mL) was added and the mixture was slurried for 8 hours. The formed solids were filtered, and the filter cake was washed with butyl acetate (2 x 100 mL). The solid was dried in a vacuum oven at 50 °C for 12 hours to a tan solid. The solid was dissolved in a 5:1 mixture of DMF and water at room temperature and additional water was added slowly to crystallize the material. The slurry was cooled to 10 °C and stirred for 8 hours, followed by filtration and washing with water. The filter cake was dried in a vacuum oven at 50 °C for 8 hours. The solid was dissolved in a 1 :1 mixture of methanol and water and the slurry was heated to 50 °C and held at this temperature for 2 hours. After cooling to 10 °C over 30 minutes, the slurry was held at this temperature for 1 hour, filtered and washed with water and dried in a vacuum oven at 50 °C for 8 hours to give the title compound as a white solid. MS (ES+) 326.0 [M+1]+.1H NMR (500 MHz, DMSO-d6) δ ppm 12.85 (s, 1 H) 7.57 (dd, J=9.03, 2.68 Hz, 1 H) 7.33 (s, 1 H) 7.17 – 7.23 (m, 2 H) 7.10 (s, 1 H) 5.89 (d, J=1.71 Hz, 1 H) 5.41 (br. s, 1 H) 3.89 (br. s, 1 H) 3.84 (s, 3 H).
REFERENCES
1: Ruggeri RB, Buckbinder L, Bagley SW, Carpino PA, Conn EL, Dowling MS, Fernando DP, Jiao W, Kung DW, Orr ST, Qi Y, Rocke BN, Smith A, Warmus JS, Zhang Y, Bowles D, Widlicka DW, Eng H, Ryder T, Sharma R, Wolford A, Okerberg C, Walters K, Maurer TS, Zhang Y, Bonin PD, Spath SN, Xing G, Hepworth D, Ahn K, Kalgutkar AS. Discovery of 2-(6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999): A Highly Selective Mechanism-Based Myeloperoxidase Inhibitor for the Treatment of Cardiovascular Diseases. J Med Chem. 2015 Oct 28. [Epubahead of print] PubMed PMID: 26509551.
////////////PF 06282999, 1435467-37-0, PFIZER, PHASE 1, PF-06282999; PF-6282999, PF06282999, ACUTE CORONARY SYNDROME
O=C(N)CN(C(N1)=S)C(C2=CC(Cl)=CC=C2OC)=CC1=O
RO-5126766


RO-5126766
| 946128-88-7 | |
| MW | 471.46 |
|---|---|
| MF | C21H18FN5O5S |
Phase I
3- [[2-[(Methylaminosulfonyl)amino]-3- fluoropyridin-4-yl]methyl]-4-methyl-7-[(pyrimidin-2-yl)oxy]- 2H-1-benzopyran-2-one
3-[[3-fluoro-2-(methylsulfamoylamino)pyridin-4-yl]methyl]-4-methyl-7-pyrimidin-2-yloxychromen-2-one
| Chugai Seiyaku Kabushiki Kaisha |
![]()
Hoffmann-La Roche
Royal Marsden NHS Foundation Trust
Collaborators:
Institute of Cancer Research, United Kingdom
Chugai Pharmaceutical
A MEK1/Raf inhibitor potentially for the treatment of solid tumors and multiple myeloma.
![]()
RO-5126766; RG-7304; CH-5126766; CKI-27; R-7304
CAS No. 946128-88-7

Although melanoma is the most aggressive skin cancer, recent advances in BRAF and/or MEK inhibitors against BRAF-mutated melanoma have improved survival rates. Despite these advances, a treatment strategy targeting NRAS-mutated melanoma has not yet been elucidated. We discovered CH5126766/RO5126766 as a potent and selective dual RAF/MEK inhibitor currently under early clinical trials. We examined the activity of CH5126766/RO5126766 in a panel of malignant tumor cell lines including melanoma with a BRAF or NRAS mutation. Eight cell lines including melanoma were assessed for their sensitivity to the BRAF, MEK, or RAF/MEK inhibitor using in vitro growth assays. CH5126766/RO5126766 induced G1 cell cycle arrest in two melanoma cell lines with the BRAF V600E or NRAS mutation. In these cells, the G1 cell cycle arrest was accompanied by up-regulation of the cyclin-dependent kinase inhibitor p27 and down-regulation of cyclinD1. CH5126766/RO5126766 was more effective at reducing colony formation than a MEK inhibitor in NRAS- or KRAS-mutated cells. In the RAS-mutated cells, CH5126766/RO5126766 suppressed the MEK reactivation caused by a MEK inhibitor. In addition, CH5126766/RO5126766 suppressed the tumor growth in SK-MEL-2 xenograft model
A method for producing a coumarin derivative of general formula (VII) is disclosed in Patent document 1 or 2. Patent document 1 or 2 discloses a method represented by the scheme below [In the scheme, DMF represents N,N-dimethylformamide, TBS represents a tert-butyldimethylsilyl group, dba represents dibenzylideneacetone, and BINAP represents 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl. Also, the numerical values (%) and “quant.” given below some structural formulas indicate the yields of the respective compounds], for example (see the manufacturing example for “compound 1j-2-16-2K” in Patent document 1 or 2).


CITATION LIST Patent Literature
Patent document 1: WO 2007/091736
Patent document 2: WO 2009/014100
PATENT
http://www.google.co.in/patents/EP1982982A1?cl=en
- Compound 1j-2-16-2:
3-{2-(Methylaminosulfonyl)amino-3-fluoropyridin-4-ylmethyl}-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran
Methylamine (158 µL, 317 µmol) and DMAP (38.7 mg, 317 µmol) were added at -78 °C to a solution of sulfuryl chloride (28 µL, 340 µmol) in dichloromethane (2 mL), and the mixture was then stirred at room temperature for 2 hours to yield the corresponding sulfamoyl chloride. 3-(2-Amino-3-fluoropyridin-4-ylmethyl)-7-(pyrimidin-2-yloxy)-4-methyl-2-oxo-2H-1-benzopyran (compound 1h-2-16) (60 mg, 159 µmol), pyridine (65 µL, 795 µmol) and dichloromethane (2 mL) were added to the reaction solution, and the mixture was stirred at room temperature for 4 hours. After addition of water, the organic layer was extracted with dichloromethane. After washing with sodium hydrogen carbonate solution and saturated saline, the organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled away under reduced pressure. The resultant residue was purified by silica gel column chromatography to yield the title compound (32 mg, 43%).
1H NMR (CD3OD, 270 MHz) δ (ppm): 2.54 (3H, s), 2.62 (3H, s), 4.22 (2H, s), 6.84 (1H, dd, J = 5.4 Hz), 7.20-7.30 (3H, m), 7.80-7.95 (2H, m), 8.63 (2H, d, J = 4.9 Hz)
ESI (LC/MS positive mode) m/z: 472 (M + H).
- Compound 1j-2-16-2Na:
3-(2-(N-Methylsulfamoyl)amino-3-fluoropyridin-4-ylmethyl)-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran sodium salt
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-1Na, except that compound 1j-2-16-2 was used instead of compound 1j-1-5-1.
1H NMR (DMSO-d6, 270 MHz) δ (ppm): 2.30 (3H, s), 2.46 (3H, s), 3.89 (2H, s), 5.68 (1H, brs), 6.09-6.23 (1H, m), 7.20 (1H, dd, J = 2.4, 8.7 Hz), 7.34 (1H, t, J = 4.8 Hz), 7.38 (1H, d, J = 2.4 Hz), 7.55 (1H, d, J = 5.3 Hz), 7.90 (1H, d, J = 8.7 Hz), 8.69 (1H, d, J = 4.8 Hz).
ESI (LC/MS positive mode) m/z: 472 (M + 2H – Na).
- Compound 1j-2-16-2K:
3-(2-(N-Methylsulfamoyl)amino-3-fluoropyridin-4-ylmethyl)-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran potassium salt
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-1Na, except that compound 1j-2-16-2 was used instead of compound 1j-1-5-1, and that KOH was used instead of NaOH.
1H NMR (DMSO-d6, 270 MHz) δ (ppm): 2.36 (3H, s), 2.47 (3H, s), 3.93 (2H, s), 6.26-6.40 (1H, m), 7.27 (1H, dd, J = 2.3, 8.6 Hz), 7.34 (1H, t, J = 4.8 Hz), 7.39 (1H, d, J = 2.3 Hz), 7.64 (1H, d, J = 4.8 Hz), 7.91 (1H, d, J = 8.6 Hz), 8.69 (1H, d, J = 4.8 Hz).
ESI (LC/MS positive mode) m/z: 472 (M + 2H – K).
PAPER
ACS Medicinal Chemistry Letters (2014), 5(4), 309-314.
Optimizing the Physicochemical Properties of Raf/MEK Inhibitors by Nitrogen Scanning
† Research Division, Chugai Pharmaceutical Co., Ltd., 200 Kajiwara, Kamakura, Kanagawa 247-8530, Japan
‡ Research Division, Chugai Pharmaceutical Co., Ltd., 1-135 Komakado, Gotemba, Shizuoka 412-8513, Japan
ACS Med. Chem. Lett., 2014, 5 (4), pp 309–314
DOI: 10.1021/ml400379x
Publication Date (Web): January 22, 2014

Substituting a carbon atom with a nitrogen atom (nitrogen substitution) on an aromatic ring in our leads 11a and 13g by applying nitrogen scanning afforded a set of compounds that improved not only the solubility but also the metabolic stability. The impact after nitrogen substitution on interactions between a derivative and its on- and off-target proteins (Raf/MEK, CYPs, and hERG channel) was also detected, most of them contributing to weaker interactions. After identifying the positions that kept inhibitory activity on HCT116 cell growth and Raf/MEK, compound 1(CH5126766/RO5126766) was selected as a clinical compound. A phase I clinical trial is ongoing for solid cancers.
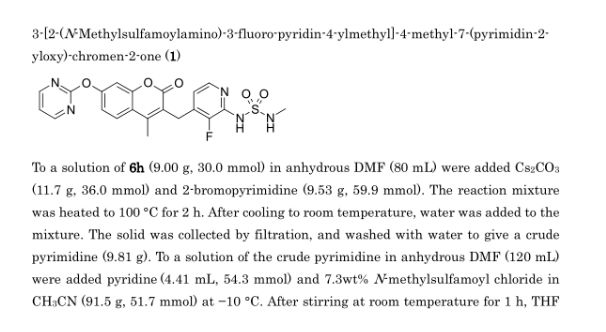

PATENT
https://www.google.com/patents/US20140213786
Step 5 Synthesis of 4-methyl-3-(3-fluoro-2-aminopyridin-4-ylmethyl)-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran
Under a nitrogen atmosphere, potassium carbonate (2.3 g, 17 mmol) was added to a solution of the solid product of step 4 (3.0 g) and 2-bromopyrimidine (1.6 g, 9.8 mmol) in DMF (48 mL), and the mixture was stirred at 115° C. for 2.5 hours. The reaction mixture was cooled to 28° C., water (48 mL) was added dropwise over a period of 5 minutes at that temperature, and after cooling to 0° C., the mixture was stirred for 2 hours. The precipitated crystals were collected by filtration, washed with water (24 mL) and acetonitrile (24 mL) in that order, and dried under reduced pressure to obtain crude crystals (2.3 g). DMF (65 mL) was added to the crude crystals (2.3 g), and after heating to 60° C. and confirming the dissolution, the mixture was cooled to 25° C. Water (65 mL) was added at 25° C., and the mixture was further cooled to 0° C. and stirred for 4 hours. The precipitated crystals were collected by filtration, washed with water (22 mL) and acetonitrile (22 mL) in that order, and dried under reduced pressure to obtain the title compound (2.1 g). The title compound is a compound disclosed in WO 2007/091736.
Yield (overall yield from the 2-acetylamino-5-chloro-3-fluoropyridine used in step 2): 27%
Patent
https://www.google.com/patents/US20100004233
Compound 1h-2-16:
3-(3-Fluoro-2-aminopyridin-4-ylmethyl)-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1h-2-4 (synthesis scheme 2), except that compound 5d-0-16 was used instead of compound 4a-0-4.
1H NMR (DMSO-d6, 270 MHz) δ (ppm): 2.45-2.55 (3H, m), 3.94 (2H, s), 6.12 (2H, brs), 6.28 (1H, dd, J=4.7 Hz), 7.27 (1H, dd, J=8.6 Hz, J=2.1 Hz), 7.34 (1H, dd, J=4.9 Hz), 7.38 (1H, d, J=2.1 Hz), 7.58 (1H, d, J=4.7 Hz), 7.91 (1H, d, J=8.6 Hz), 8.68 (2H, d, J=4.7 Hz).
ESI (LC/MS positive mode) m/z: 479 (M+H).
Compound 1j-2-4-2:
3-{2-Fluoro-3-(methylaminosulfonyl)aminobenzyl}-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-2, except that compound 1h-2-4 was used instead of compound 1h-1-5.
1H NMR (270 MHz, DMSO-d6) δ (ppm): 2.45 (3H, s), 3.99 (2H, s), 6.83-6.92 (1H, m), 6.97-7.06 (1H, m), 7.17 (1H, brs), 7.34-7.40 (4H, m), 7.91 (1H, d, J=8.4 Hz), 8.69 (2H, dd, J=4.8, 1.2 Hz), 9.38 (1H, br.s).
One of the CH3 peaks was overlapped with the DMSO peak.
ESI (LC/MS positive mode) m/z: 471 (M+H).
Compound 1j-2-4-2Na:
3-{2-Fluoro-3-(methylaminosulfonyl)aminobenzyl}-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran sodium salt
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-1Na, except that compound 1j-2-4-2 was used instead of compound 1j-1-5-1.
1H NMR (270 MHz, DMSO-d6) δ (ppm): 2.33 (3H, d, J=3.3 Hz), 2.43 (3H, s), 3.89 (2H, s), 6.10-6.19 (1H, m), 6.58-6.66 (1H, m), 7.17 (1H, ddd, J=8.3, 1.5 Hz, JHF=8.3 Hz), 7.25 (1H, dd, J=8.7, 2.3 Hz), 7.33 (1H, t, J=4.8 Hz), 7.37 (1H, d, J=2.3 Hz), 7.88 (1H, d, J=8.7 Hz), 8.69 (2H, d, J=4.8 Hz)
ESI (LC/MS positive mode) m/z: 471 (M+2H—Na).
Compound 1j-2-4-2K:
3-{2-Fluoro-3-(methylaminosulfonyl)aminobenzyl}-4-methyl-7-(pyrimidin-2-yl-oxy)-2-oxo-2H-1-benzopyran potassium salt
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-1Na, except that compound 1j-2-4-2 was used instead of compound 1j-1-5-1, and that KOH was used instead of NaOH.
1H NMR (270 MHz, DMSO-d6) δ (ppm): 8.69 (d, 2H, J=4.8 Hz), 7.88 (d, 1H, J=8.7 Hz), 7.36 (d, 1H, J=2.3 Hz), 7.33 (t, 1H, J=4.8 Hz), 7.25 (dd, 1H, J=8.7, 2.3 Hz), 7.16 (td, 1H, J=8.5, 1.4 Hz), 6.59 (t, 1H, J=7.8 Hz), 6.10 (t, 1H, J=6.3 Hz), 4.76 (q, 1H, J=5.8 Hz), 3.88 (s, 2H), 2.43 (s, 3H), 2.32 (d, 3H, J=5.6 Hz).
ESI (LC-MS positive mode) m/z: 471 (M+2H—K).
PATENT
Method for producing a coumarin derivative of formula (VII) are described in Patent Documents 1 and 2. Patent Documents 1 and 2, for example, in the following scheme [scheme, DMF is N, represents a N- dimethylformamide, TBS represents a tert- butyldimethylsilyl group, dba represents dibenzylideneacetone, BINAP is 2, I represents a 2′-bis (diphenylphosphino) -1,1′-binaphthyl. Further, numerical values given under the formula (%) or “quant.” Indicates the yield of the compound. Methods have been described that are shown in (see Preparation of “Compound 1j-2-16-2K” in Patent Documents 1 and 2).

WO2007 / 091736 WO2009 / 014100
While coumarin derivatives of the general formula (VII) can be prepared by the methods described in Patent Documents 1 and 2, in the method described in Patent Documents 1 and 2, after the formylation reaction and a reduction reaction, and unintended Reaction To suppress, it is necessary to perform the introduction and removal steps of the protecting group for hydroxy group. Also, during the formylation reaction, from the viewpoint of cryogenic conditions of the reaction control (eg, -95 ℃ ~ -65 ℃) is required. Furthermore, the alkylation reaction (the seventh step in the above scheme), it is preferred that an excess amount of use of ethyl acetoacetate in terms of efficient synthesis, in which case, requires complicated operation of removing residual reagents become.
[Example 1]
Step 1:
Synthesis of 2-acetylamino-5-chloro-3-fluoropyridine:

Under a nitrogen atmosphere, acetamide (94.8g, 1.61mol) in DMF with (200mL) and THF (830mL) was added and heated to 50 ℃. The resulting solution was a THF solution of 40wt% sodium hexamethyldisilazide (629g, 1.37mol) was added dropwise and stirred at the same temperature for 2 hours. 5-chloro-2,3-difluoro pyridine (100.0g, 0.67mol) After adding, THF and (20mL), and the mixture was stirred at the same temperature for 3 hours. After cooling to 0 ℃, it is added to 2.8M HCl (500mL) to the reaction mixture, and the organic layer was separated and the temperature was raised to room temperature.The organic layer was washed with 20wt% sodium chloride solution (500mL), and evaporated under reduced pressure. The residue in THF (500mL) was added, and the residue was dissolved by heating at 70 ℃. After confirming the solid precipitated by cooling to room temperature, n- heptane (1500mL) was added and further cooled to 0 ℃, followed by stirring at the same temperature for 3 hours. The The precipitated crystals were collected by filtration, to give after washing with a mixed solvent of THF (100mL) and n- heptane (500mL), and dried under reduced pressure to give the title compound (91.2g).
Yield: 72%
1 H-NMR (CDCl 3) δ (ppm): 2.36 (3H, s), 7.49 (1H, dd, J = 2.0,9.5Hz), 7.78 (1H, br), 8.17 (1H, d, J = 2.0Hz).
MS (ESI +): 189 [M + 1] +
Step 2:
Synthesis of 2-acetylamino-5-chloro-3-fluoro-4-formyl pyridine:

Under a nitrogen atmosphere, and dissolved at room temperature 2-acetylamino-5-chloro-3-fluoropyridine (70.0g, 0.37mol) and 4-formyl-morpholine (128.2g, 1.11mol) to THF (840mL) It was. The solution was cooled to -20 ℃ and was added dropwise a THF solution of 24wt% of lithium hexamethyldisilazide (595g, 0.85mol), and stirred 5.5 hours at the same temperature. The reaction mixture, citric acid monohydrate (257g) and sodium chloride (70g) in an aqueous solution dissolved in water (420mL), and I was added at stirring at 0 ℃. The organic layer was separated and the resulting organic layer was successively washed with 50wt% phosphoric acid aqueous solution of potassium dihydrogen (350mL) and 20wt% sodium chloride solution (350mL) to (1458g). The portion of the organic layer was taken for analysis (292g), and evaporated remainder (1166g) at reduced pressure. The residue in THF (350mL) was added, and the solvent was distilled off under reduced pressure. Again, the residue in THF (350mL) was added to and evaporated under reduced pressure to give a solid (81.4g) containing the title compound. The product was used in the next step without further purification.
Some of the organic layer which had been collected (292g) to (29g), and evaporated under reduced pressure. The residue was purified by silica gel column chromatography: subjected to [eluent AcOEt / hexane (1 / 4-9 / 1)], I give the title compound (1.05g, 4.85mmol) as a white powdery solid.
Yield: 66%
1 H-NMR (CDCl 3) δ (ppm): 2.40 (3H, s), 7,59 (1H, br), 8.34 (1H, br), 10.42 (1H, s).
MS (ESI +): 217 (M + 1)
Step 3:
2 – [(4-2-acetylamino-3-fluoro-pyridin-yl) methyl] -3-oxobutanoic acid ethyl ester:

Under a nitrogen atmosphere to dissolve the solid product of Step 2 (81.4g) in 2,2,2-trifluoroethanol (448mL), piperidine (4.4g, 51.7mmol), acetic acid (3.1g, 51 .7mmol) and 3-oxobutanoic acid ethyl (37.0g, 0.28mol) was added and stirred for 3 hours after raising the temperature to 50 ℃. After cooling the reaction mixture to room temperature, triethylamine (758mL, 5.5mol) and formic acid (172mL, 4.6mol) of 2-propanol (1248mL) solution and 20% Pd (OH) 2 carbon (21.2g, moisture content 46.2%) were added, followed by stirring for 4 hours the temperature was raised to 50 ℃. The reaction mixture was filtered through Celite, and the residue was washed with 2-propanol (679mL). Combined filtrate and washings (2795g), and evaporated under reduced pressure a part of the (399g) (remaining (2396g) I was saved). Ethyl acetate (24.2mL) was added to the residue obtained by evaporation of the solvent, and evaporated under reduced pressure. Again, the residue ethyl acetate (182mL) was added to the washed successively with an organic layer 20wt% brine (61mL), 10wt% of potassium dihydrogen phosphate solution (61mL) and 20wt% sodium chloride solution (61mL), under a reduced pressure The solvent was evaporated. Furthermore, in addition to the residue of 2,2,2-trifluoroethanol (24mL), and the solvent evaporated under reduced pressure to obtain oil containing the title compound (15.0g). The product was used in the next step without further purification.
1 H-NMR (CDCl 3) δ (ppm): 1.24 (3H, t, J = 7.0Hz), 2.27 (3H, s), 2.37 (3H, s), 3.16- 3.26 (2H, m), 3.86 (1H, t, J = 7.5Hz), 4.15-4.22 (2H, m), 6.98 (1H, t, J = 5.0Hz ), 7.68 (1H, br), 8.05 (1H, d, J = 5.0Hz).
MS (ESI +): 297 (M + 1)
Step 4:
Synthesis of 3- (3-fluoro-2-amino-pyridin-4-ylmethyl) -7-hydroxy-4-methyl-2-oxo -2H-1- benzopyran methanesulphonate:

Under a nitrogen atmosphere, oily product of Step 3 (15.0g) and I were dissolved in 2,2,2-trifluoroethanol (33mL). The solution of resorcinol (5.3g, 47.9mmol) and methane sulfonic acid (11.7mL, 181mmol) was added at 24 ℃, and stirred for 4 hours at 90 ℃. And allowed to stand for 13 hours and cooled to room temperature and ethanol (33mL) and water (11mL), and the mixture was stirred for 4.5 hours at 90 ℃. After adding 2-propanol (105mL) was cooled to 55 ℃, and allowed to stand for 14 hours then cooled to room temperature. The The precipitated crystals were collected by filtration to give 2-propanol was washed twice with (33mL), and dried under reduced pressure to give the title compound (8.2g).
(Total from 2-acetylamino-5-chloro-3-fluoropyridine was used in step 2 Yield) Yield: 49%
MS (ESI +): 301 [M + 1-MsOH] +
Step 5:
4-methyl-3- (3-fluoro-2-amino-pyridin-4-ylmethyl) -7- (pyrimidin-2-yloxy) -2-oxo -2H-1- benzopyran Synthesis:

Under a nitrogen atmosphere, 3- (3-fluoro-2-amino-pyridin-4-ylmethyl) -7-hydroxy-4-methyl-2-oxo -2H-1- benzopyran methanesulphonate (7.6g, 19.2mmol) and 2-bromo-pyrimidine (4.0g, 24.9mmol) was dissolved in DMF (122mL), potassium carbonate (5.8g, 42.2mmol) was added, and the mixture was stirred for 3.5 hours at 115 ℃. After cooling the reaction mixture to 28 ℃, water (122mL) was added dropwise over the same temperature for 0.5 hours, and stirred for 2 minutes. In addition, after cooling to 0 ℃, and the mixture was stirred for 1 hour, and the precipitated crystals were collected by filtration. The obtained crystals were washed successively with water (61mL) and acetonitrile (61mL), to give the title compound was dried under reduced pressure and crystals (6.5g).
The resultant was taken for analysis a portion of the crystals (0.1g), it was suspended remainder (6.4g) in DMF (70mL). The resulting suspension was stirred 60 ℃ and heated for 5 minutes and stirred for 80 minutes by the addition of acetonitrile (185mL) at the same temperature. Then, it was stirred for 0.5 hours and then cooled to 40 ℃, and the mixture was stirred for 0.5 hours and further cooled to 25 ℃. After a further 1.5 hours with stirring and cooled to 0 ℃, the precipitated crystals were collected by filtration. After washing the resulting crystals in acetonitrile (46mL), was obtained by drying under reduced pressure to the title compound (5.5g). Incidentally, the title compound is a compound described in WO2007 / 091736.
Yield: 76%
Step 6:
3- {2- (methyl-aminosulfonyl) amino-3-fluoro-pyridin-4-ylmethyl} -4-methyl-7- (pyridin-2-yloxy) -2-oxo -2H-1- benzopyran Synthesis:

Under a nitrogen atmosphere, 4-methyl-3- (3-fluoro-2-amino-pyridin-4-ylmethyl) -7- (pyrimidin-2-yloxy) -2-oxo -2H-1- benzopyran (1.7g, 4 the .5mmol) it was suspended in DMF (18mL). To this solution pyridine (0.8mL, 9.9mmol) was cooled to In 10 ℃ added, N- methyl-sulfamoyl chloride (1.05g, 8.1mmol) in acetonitrile (18mL) solution of the internal temperature of 15 ℃ it was dropped so as to maintain below. After stirring for 90 minutes at the same temperature, acetonitrile (3.4mL) was added and further water (50mL), was added dropwise the inner temperature so as to maintain the 20 ℃ below. It was cooled to an external temperature of 0 ℃, and the mixture was stirred for an internal temperature of 5 ℃ 2 hours after arrival. The precipitated crystals were collected by filtration, washed with water (8.5mL), and dried to give the title compound (1.9g, 4.0mmol) was obtained.
Yield: 88%
MS (ESI +): 472 [M + 1] +
Step 7:
Synthesis of 3- {2- (methyl-aminosulfonyl) amino-3-fluoro-pyridin-4-ylmethyl} -4-methyl-7- (pyridin-2-yloxy) -2-oxo -2H-1- benzopyran potassium salt:

Under a nitrogen atmosphere, 3- {2- (methyl-aminosulfonyl) amino-3-fluoro-pyridin-4-ylmethyl} -4-methyl-7- (pyridin-2-yloxy) -2-oxo -2H-1- benzopyran ( 1.6g, was suspended 3.4mmol) in THF (10mL), water (3mL) was added. The suspension in 2.0M aqueous potassium hydroxide (1.8mL, 3.6mmol) was added dropwise over 10 min at 25 ℃, after raising the temperature to 60 ℃, and the mixture was stirred for 2 hours at the same temperature. After cooling the reaction mixture to 20 ℃, it was added dropwise over a period of THF (8mL) 30 min. After completion of the dropwise addition, the mixture was cooled to an external temperature of -5 ℃, and the mixture was stirred for an internal temperature of 0 ℃ reached after 160 minutes. The precipitated crystals were collected by filtration, then washed with a mixture of THF (14mL) and water (1.6mL) (pre-cooled to 5 ℃), further washed with THF (8mL), and dried to give the title compound (0 .72g, we got 1.4mmol).
Yield: 42%
MS (ESI +): 472 [M + 2H-K] +
CLIP
RO5126766 (CH5126766) is a first-in-class dual inhibitor of Raf/MEK [1].
The RAS/RAF/MEK/ERK signaling pathway is an important signal transduction system and participates in cell differentiation, movement, division and death. Activated Ras activates RAF kinase, which then phosphorylates and activates MEK (MEK1 and MEK2) [1]. The mutations in BRAF, RAS, and NF1 are associated with many human tumors [2].
RO5126766 (CH5126766) is a first-in-class dual Raf/MEK inhibitor. In cell-free kinase assays, CH5126766 effectively inhibited the phosphorylation of MEK1 protein by RAF and the activation of ERK2 protein by MEK1 with IC50 values of 0.0082-0.056 and 0.16 μM, respectively. In NCI-H460 (KRAS Q61H) human lung large cell carcinoma cell line, RO5126766 induced cell-cycle inhibitor p27Kip1 protein expression and caused G1 arrest. In HCT116 KRAS-mutant colorectal cancer cells, RO5126766 CH5126766 completely inhibited the phosphorylation of MEK and ERK [2].
In Japanese patients with advanced solid tumors, RO5126766 exhibited the maximum tolerable dose (MTD) of 2.25 mg/day once daily [1]. In a HCT116 (G13D KRAS) mouse xenograft model, RO5126766 (1.5 mg/kg) inhibited pERK and ERK signaling and exhibited ED50 value of 0.056 mg/kg [2].
References:
[1]. Honda K, Yamamoto N, Nokihara H, et al. Phase I and pharmacokinetic/pharmacodynamic study of RO5126766, a first-in-class dual Raf/MEK inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol, 2013, 72(3): 577-584.
[2]. Ishii N, Harada N, Joseph EW, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res, 2013, 73(13): 4050-4060.
| WO2007091736A1 | 9 Feb 2007 | 16 Aug 2007 | Chugai Seiyaku Kabushiki Kaisha | Novel coumarin derivative having antitumor activity |
| WO2009014100A1 | 18 Jul 2008 | 29 Jan 2009 | Chugai Seiyaku Kabushiki Kaisha | p27 PROTEIN INDUCER |
| JPH0236145A * | Title not available |
| Reference | ||
|---|---|---|
| 1 | BIOORGANIC MEDICINAL CHEMISTRY, vol. 13, 2005, pages 1393 – 1402 | |
| 2 | JOURNAL OF MEDICINAL CHEMISTRY, vol. 47, 2004, pages 6447 – 6450 | |
| 3 | ORGANIC PREPARATIONS AND PROCEDURES INTERNATIONAL, vol. 36, 2004, pages 347 – 351 | |
| 4 | * | See also references of EP2754654A1 |
| 5 | * | STANCHO STANCHEV, ET AL.: “Synthesis and Inhibiting Activity of Some 4-Hydroxycoumarin Derivatives on HIV-1 Protease. Art 137637“, ISRN PHARMACEUTICS, vol. 63, no. 10, 2011, pages 1 – 9, XP055145297 |
| 6 | * | STANCHO STANCHEV, ET AL.: “Synthesis, computational study and cytotoxic activity of new 4-hydroxycoumarin derivatives“, EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 43, no. 4, 2008, pages 694 – 706, XP022576473 |
| 7 | SYNTHETIC COMMUNICATIONS, vol. 34, 2004, pages 4301 – 4311 | |
| Patent ID | Date | Patent Title |
|---|---|---|
| US7897792 | 2011-03-01 | Coumarin derivative having antitumor activity |
| US2011009398 | 2011-01-13 | p27 Protein Inducer |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016024051 | 2016-01-28 | SALTS AND SOLID FORMS OF ISOQUINOLINONES AND COMPOSITION COMPRISING AND METHODS OF USING THE SAME |
| US2015290207 | 2015-10-15 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
| US2015283142 | 2015-10-08 | TREATMENT OF CANCERS USING PI3 KINASE ISOFORM MODULATORS |
| US2015225410 | 2015-08-13 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
| US2015111874 | 2015-04-23 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
| US2014377258 | 2014-12-25 | Treatment Of Cancers Using PI3 Kinase Isoform Modulators |
| US2014213786 | 2014-07-31 | Method for Producing Coumarin Derivative |
| US2014038920 | 2014-02-06 | TFEB PHOSPHORYLATION INHIBITORS AND USES THEREOF |
| US2011092700 | 2011-04-21 | Novel Coumarin Derivative Having Antitumor Activity |
| US7897792 | 2011-03-01 | Coumarin derivative having antitumor activity |
//////////////RO-512676, RG-7304, CH-5126766, CKI-27, R-730, 946128-88-7, PHASE 1, MEK1/Raf inhibitor, treatment of solid tumors and multiple myeloma, CANCER
CC(C1=C(O2)C=C(OC3=NC=CC=N3)C=C1)=C(C2=O)CC4=C(F)C(NS(NC)(=O)=O)=NC=C4
MK 8876
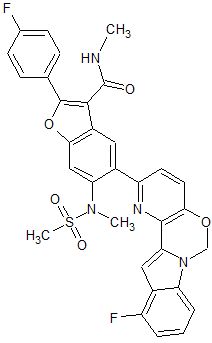

MK 8876
CAS 1426960-33-9
2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-(N-methylmethanesulfonamido)-1-benzofuran-3-carboxamide
| 2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-[methyl(methylsulfonyl)amino]-3-benzofurancarboxamide |
| Molecular Formula | C32H24F2N4O5S | |
| Molecular Weight | 614.62 |
- Originator Merck & Co
- Class Antivirals
- Phase I Hepatitis C
Most Recent Events
- 11 Oct 2013 Phase-I clinical trials in Hepatitis C in Germany (PO)
- 11 Oct 2013 Phase-I clinical trials in Hepatitis C in Moldova (PO)
- 23 Aug 2013 Preclinical trials in Hepatitis C in USA (PO)
DATA
2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-(N-methylmethanesulfonamido)-1-benzofuran-3-carboxamide
MK-8876 off-white solid
1H NMR (500 MHz, DMSO-d6) δ 8.56 (q, J = 4.7 Hz, 1H), 8.06–8.01 (m, 2H), 8.05 (s, 1H), 7.86 (s, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 8.3 Hz, 1H), 7.46–7.40 (m, 2H), 7.29–7.22 (m, 1H), 7.11 (s, 1H), 6.94 (dd, J = 10.6, 7.9 Hz, 1H), 6.27 (s, 2H), 3.31 (s, 3H), 2.96 (s, 3H), 2.85 (d, J = 4.7 Hz, 3H);
13C NMR (125.7 MHz, DMSO-d6) δ 162.86, 162.82 (d, JC–F = 248.5 Hz), 155.74 (d, JC–F = 246.1 Hz), 153.80, 152.43, 152.28, 147.20, 137.08, 137.00 (d, JC–F = 10.8 Hz), 136.36, 136.20, 132.37, 129.50 (d, JC–F = 8.6 Hz), 127.17, 125.45 (d, JC–F = 3.1 Hz), 125.08, 125.02, 123.70 (d, JC–F = 7.7 Hz), 122.28, 117.23 (d, JC–F = 22.4 Hz), 116.01 (d, JC–F = 21.9 Hz), 113.65, 111.76, 106.90 (d,JC–F = 3.5 Hz), 105.32 (d, JC–F = 18.5 Hz), 94.16, 73.57, 39.39, 37.24, 26.16;
HR-ESI-MS m/zcalcd for C32H25N4O5SF2+ [M + H]+ 615.1514, found 615.1500.
. HPLC Method and Retention Time Data
| HPLC Method | |
|---|---|
| column | Ascentis Express C18 2.7 μm (fused core), 100 mm × 4.6 mm |
| detection | UV at 210 nm |
| column temperature | 40 °C |
| flow rate | 1.8 mL/min |
| injection volume | 5.0 μL |
| gradient | 90% A to 5% A over 11 min, hold at 5% A for 2 min, 5% A back to 90% A over the next 0.1 min, and then hold at 90% A for 2.9 min |
| run time | 16 min |
| data collection | acquisition for the first 13 min |
| mobile phases | solvent A: water with 0.1% H3PO4 |
| solvent B: acetonitrile | |
| Retention Time Data | |
|---|---|
| identity | tR (min) |
| boronic acid 27 | 4.24 |
| desbromoarene 28 | 5.33 |
| MK-8876 (1) | 7.89 |
| chloropyridine starting material 2 | 8.03 |
| BHT | 10.22 |
SYNTHESIS



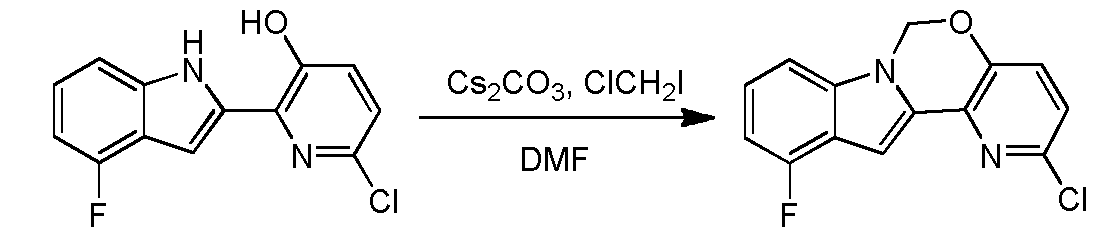
CONTD……………
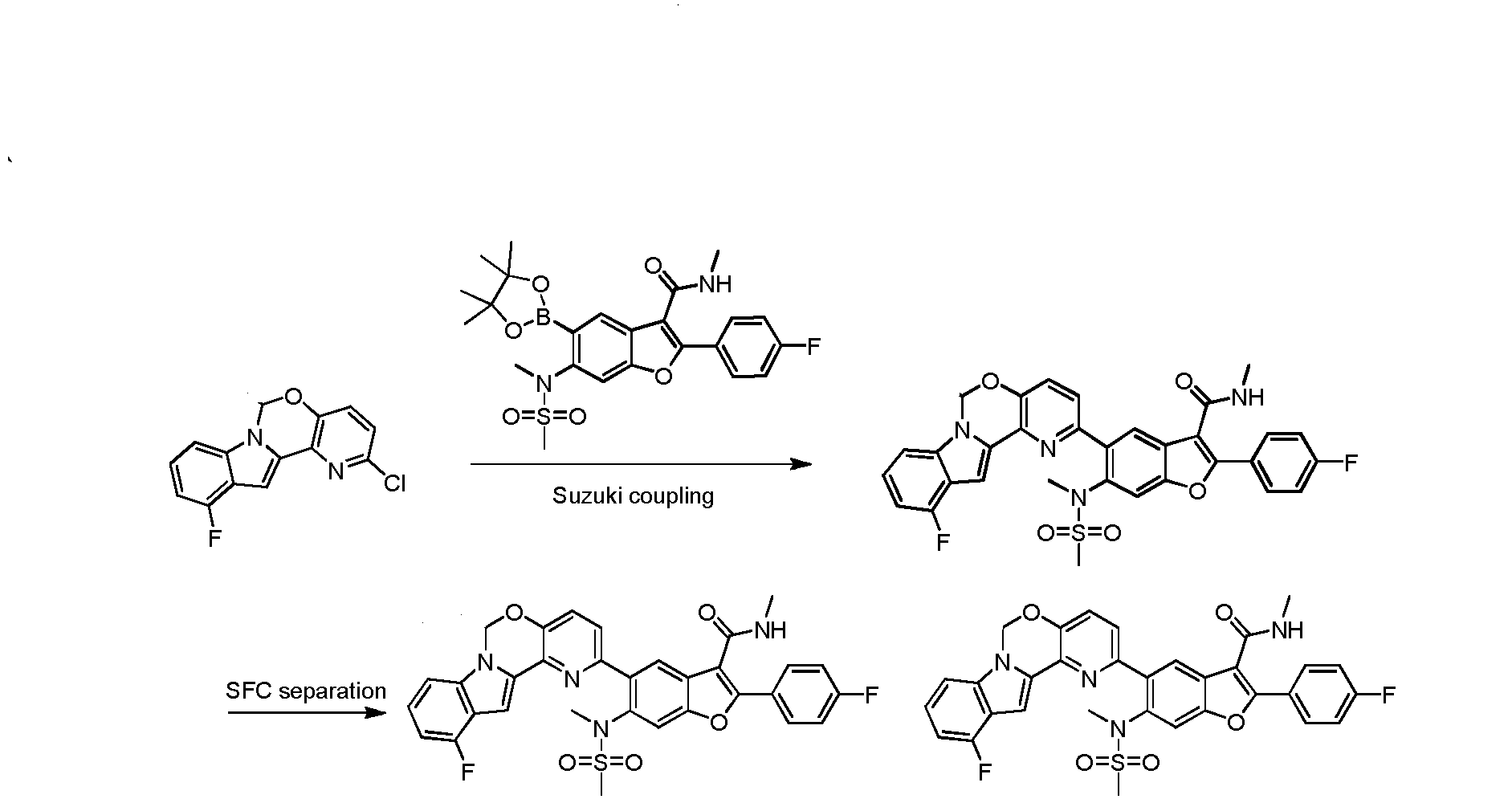
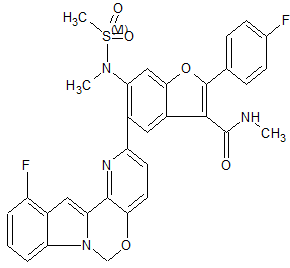
MK 8876










MK 8876
Patent
Scheme 1

Scheme 2

Scheme 3

Q
Scheme 4

EXAMPLES
Example 1
Preparation of Compound 1
 THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876
THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876Step 1 – Synthesis of 2,6-dichloropyridin-3-ol

Η202 (1.60 g, 47.12 mmol) was added slowly to the solution of compound 2,6- dichloropyridin-3-ylboronic acid (3 g, 15.71 mmol) in CH2CI2 (30 mL) at 0 °C. After stirred at room temperature for about 15 hours, the mixture was quenched with sat. Na2S203 aqueous (50 mL) and adjusted to pH < 7 with IN HC1. The mixture was extracted with EtOAc (40 mL x 3). The organic layer was washed with brine (100 mL), dried over Na2S04, filtered and the solvent was evaporated to provide2,6-dichloropyridin-3-ol (2.34 g, yield: 91.4%). 1H-NMR (CDC13, 400 MHz) δ 7.30 (d, / = 8.4 Hz, 1H), 7.19 (d, / = 8.4 Hz, 1H), 5.70 (br, 1H).
– Synthesis of 2,6-dichloro- -methoxypyridine
To a solution of 2,6-dichloropyridin-3-ol (16.3 g, 0.1 mol) and K2C03 (41.4 g, 0.3 mol) in DMF (200 mL) were added Mel (21.3 g, 0.15 mol). The mixture was allowed to stir at 80 °C for 2 hours. The mixture was then diluted with water (200 mL) and extracted with EtOAc (200 mL x 3). The organic layer was washed with brine (200 mL x 3), dried over Na2S04, filtered and the solvent was evaporated to provide 2,6-dichloro-3-methoxypyridine (17.0 g, yield: 96.0%). 1H-NMR (CDC13, 400 MHz) δ 7.12-7.18 (m, 2H), 3.86 (s, 3H). Step 3 – Synthesis of2-(6-chloro-3-methoxypyridin-2-yl)-lH-indole

To a degassed solution of compound 2,6-dichloro-3-methoxypyridine (8.9 g, 0.05 mol), (l-(tert-butoxycarbonyl)-lH-indol-2-yl)boronic acid (13 g, 0.05 mol) and K3PO4 (31.8 g, 3.0 mol) in DMF (100 mL) was added Pd(dppf)Cl2 (3.65 g, 0.005 mol) under N2. The mixture was heated at 60 °C for about 15 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc and filtered. The filtrate was washed with H20, brine, dried over Na2S04. After being concentrated in vacuo, the resulting residue was purified using prep-HPLC to provide the desired product of 2-(6-chloro-3-methoxypyridin-2-yl)-lH-indole (9.0 g, yield:
69.8%). 1H-NMR (CDC13, 400 MHz) δ 9.52 (s, 1H), 7.65 (d, / = 7.6 Hz, 1H), 7.38-7.43 (m, 2H), 7.07-7.26 (m, 4H), 4.03 (s, 3H).
Step 4 – Synthesis of6-chlor -2-(lH-indol-2-yl)pyridin-3-ol

BBr3 (0.4 mL, 0.39 mmol) was added to the solution of 2-(6-chloro-3- methoxypyridin-2-yl)-lH-indole (50 mg, 0.194 mmol) in CH2C12 (0.5 mL) at -78 °C under N2. The mixture was allowed to stir at room temperature for 3 hours. The mixture was then quenched with CH3OH (10 mL) at -78 °C. After being concentrated in vacuo, the resulting residue was purified using prep-TLC (PE : EtOAc = 2.5 : 1) to afford the desired product of 6- chloro-2-(lH-indol-2-yl)pyridin-3-ol (40 mg, yield: 85.1%). 1H-NMR (CDC13, 400 MHz) δ 10.09 (s, 1H), 9.72 (s, 1H), 7.50 (d, / = 7.9 Hz, 1H), 7.17-7.32 (m, 3H), 7.08-7.14 (m, 1H), 6.87-6.96 (m, 2H).
Step 5 – Synthesis of 2-chlo -6H-pyrido[2′ ,3′ : 5 ,6] [ 1 ,3]oxazino[3 ,4-a]indole

To a solution of chloroiodomethane (3.51 g, 20.0 mmol) and K2CO3 (1.38 g, 10.0 mmol) in DMF (50 mL) was allowed to stir at 100 °C, 6-chloro-2-(lH-indol-2-yl)pyridin-3-ol (480 mg, 2.0 mmol) in DMF (50 mL) was added dropwise. After addition, the mixture was allowed to stir for another 0.5 hours. The mixture was then diluted with water (100 mL) and extracted with EtOAc (100 mL x 3). The organic layer was washed with brine (100 mL x 3), dried over Na2S04 and concentrated. The residue was purified using prep-TLC (PE : EtOAc = 3 1) to afford the desired product of 2-chloro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (260 mg, yield: 50.7%). 1H-NMR (CDC13, 400 MHz) δ 7.63 (d, / = 8.0 Hz, 1H), 7.22-7.27 (m, 3H), 7.19 (d, / = 2.4 Hz, 1H), 7.08-7.12 (m, 2H), 5.86 (s, 2H).
Step 6 – Synthesis of2-(4-fluowphenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(6H- pyridol 2 ‘,3’:5,6][ l, mpound 1 )
 THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876
THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876To a degassed solution of 2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzofuran-3- carboxamide (502 mg, 1.0 mmol), 2-chloro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (256 mg, 1.0 mmol) and K3PO4 (636 mg, 3.0 mmol) in dioxane : H20 (1.5 mL : 0.4 mL) was added Pd2(dba)3 (91 mg, 0.1 mmol) and X-phos (91 mg, 0.2 mmol) under N2. The mixture was heated to 110 °C for 3 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc and filtered. The filtrate was washed with H20, brine, dried over Na2S04. After being concentrated in vacuo, the resulting residue was purified using prep-HPLC to provide the desired product of Compound 1 (275 mg, yield: 46.1%). 1H-NMR (CDC13, 400 MHz) δ 7.88-7.94 (m, 3H), 7.61-7.63 (m, 2H), 7.40 (s, 2H), 7.09-7.28 (m, 6H), 5.94 (s, 2H), 5.86 (d, / = 4.4 Hz, 1H), 3.29 (s, 3H), 2.92 (d, / = 5.2 Hz, 3H), 2.65 (s, 3H). MS (M+H)+: 596.
Compounds 2-15, depicted in the table below, were prepared using the method described above.
COMPD 2 IS MK 8876

PATENT
Example 81
Preparation of Compound 2
Synthesis of ethyl 3- 4-fluorophenyl)-3-oxopropanoate
Diethyl carbonate (130 g, 1.1 mol) was dissolved in a suspension ofNaH (60% in oil, 50.2 g, 1.3 mol) in anhydrous tetrahydrofuran (1.5 L), and then l-(4-fluorophenyl)ethanone (150 g, 1.09 mol) was added dropwise at 70 °C. The resulting mixture was stirred at 70 °C for 3 hours. After the reaction mixture was cooled to room temperature and poured into HCl (1 N). The mixture was extracted with EtOAc, the organic phase was dried with anhydrous NaS04 and concentrated in vacuo. The resulting residue was purified using column chromatography (eluted with petroleum ether / EtOAc = 50 / 1) to provide ethyl 3-(4-fluorophenyl)-3-oxopropanoate (217 g, yield: 95%). 1H-NMR (CDC13, 400 MHz) δ 7.92-7.97 (m, 2H), 7.07-7.13 (m, 2H), 4.14-4.20 (m, 2H), 3.93 (s, 2H), 1.22 (d, J= 7.2 Hz, 3H). MS (M+H)+: 211. Step 2 – Synthesis of ethyl 5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate
A solution of ethyl 3-(4-fluorophenyl)-3-oxopropanoate (130 g, 0.6 mol), 4- bromophenol (311 g, 1.8 mol) and FeCl3-6H20 (19.5 g, 0.09 mol) in DCE (700 mL) was heated to reflux, and then 2-(tert-butylperoxy)-2-methylpropane (193 g, 1.32 mol) was added dropwise under nitrogen. After 6 hours of refluxing, the mixture was cooled to RT, quenched with saturated NaHS03 and extracted with dichloromethane. The organic phases were washed with water, brine and dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether / dichloromethane = 15 / 1) to provide the crude product, which was crystallized from cold MeOH to provde ethyl 5-bromo-2- (4-fluorophenyl)benzofuran-3-carboxylate (37 g, yield: 14.3%) as solid. 1H- MR (CDC13, 400 MHz) δ 8.12 (s, 1H), 7.97-8.01 (m, 2H), 7.37 (d, J= 4.0 Hz, 1H), 7.32 (d, J= 8.0 Hz, 1H), 7.11 (t, J= 8.0 Hz, 2H), 4.32-4.38 (m, 2H), 1.36 (t, J= 8.0 Hz, 3H). MS (M+H)+: 363 / 365.
Step 3 – Synthesis of eth l 5-bromo-2-(4-fluorophen -6-nitrobenzofuran-3-carboxylate
To a solution of ethyl 5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate (50 g,
137.6 mmol) in CHC13 (500 mL), fuming HN03 (50 mL) was added dropwise at -15 °C and the mixture was stirred for 0.5 hour. The reaction mixture was poured into ice water and extracted with CH2C12. The organic layer was washed with a.q. sat. NaHC03 and brine, after removed the most of solvent, the resulting residue was crystallized with petroleum ether / dichloromethane = 20 / 1 to provide product of ethyl 5-bromo-2-(4-fluorophenyl)-6-nitrobenzofuran-3-carboxylate (35 g, yield: 66%). 1H- MR (CDC13, 400 MHz) δ 8.36 (s, 1H), 8.02-8.04 (m, 3H), 7.13-7.18 (m, 2H), 4.36-4.41 (m, 2H), 1.37 (t, J= 4.0 Hz, 3H). MS (M+H)+: 408 / 410.
Step 4 – Synthesis of ethyl 6-amino-5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate
A mixture of ethyl 5-bromo-2-(4-fluorophenyl)-6-nitrobenzofuran-3-carboxylate (52 g, 127 mmol), iron filings (21.3 g, 382.2 mmol) and H4C1 (41 g, 764.4 mmol) in MeOH / THF / H20 (2 / 2 / 1, 500 mL) was stirred at reflux for 3 hour. After filtered and concentrated, the resulting residue was purified using column chromatography (petroleum ether / EtOAc / dichloromethane = 20 : 1 : 20) to provide ethyl 6-amino-5-bromo-2-(4-fluorophenyl) benzofuran-3-carboxylate (40 g, yield: 82%). 1H- MR (CDC13, 400 MHz) δ 8.01 (s, 1H), 7.94-7.98 (m, 2H), 7.08 (t, J= 8.0 Hz, 2H), 6.83 (s, 1H), 4.32-4.36 (m, 2H), 4.18 (s, 2H), 1.35 (t, J= 8.0 Hz, 3H). MS (M+H)+: 378 / 380.
Step 5 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid eth l ester
MsCI (31.7 g, 277.5 mmol) was added to a solution of ethyl 6-amino-5-bromo-2- (4-fluorophenyl)benzofuran-3-carboxylate (35 g, 92.5 mmol) and pyridine (60 mL) in
dichloromethane (300 mL) at 0 °C. After stirred overnight at room temperature, the mixture was diluted with water and extracted with dichloromethane. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated in vacuo, the resulting residue was purified using crystallized with EtOAc to provde the pure product of ethyl 5-bromo-2-(4-fluorophenyl)-6- (methylsulfonamido)benzofuran-3-carboxylate (35 g, yield: 82%). 1H- MR (CDC13, 400 MHz) δ 8.27 (s, 1H), 8.01-8.05 (m, 2H), 7.87 (s, 1H), 7.15-7.19 (m, 2H), 6.87 (s, 1H), 4.38-4.43 (m, 2H), 3.00 (s, 3H), 1.40 (t, J= 40 Hz, 3H). MS (M+H)+: 456 / 458.
Step 6 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid
To a solution of ethyl 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido) benzofuran-3-carboxylate (53 g, 0.23 mol) in dioxane / H20 (5 / 1, 600 mL) was added
LiOH-H20 (25 g, 1.17 mol), and the mixture was stirred at 100 °C for 3 hours. After
concentrated, the resulting residue was dissolved in H20, 1 N HCl was added until pH reached 3, and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04 and filtered. The solvent was removed to provide the product of 5-bromo-2-(4- fluorophenyl)-6-(methylsulfonamido)benzofuran-3-carboxylic acid (48 g, yield: 96%).1H- MR (DMSO- e, 400 MHz) δ 13.49 (s, 1H), 9.67 (s, 1H), 8.30 (s, 1H), 8.12-8.17 (m, 2H), 7.87 (s, 1H), 7.45-7.50 (m, 2H), 3.16 (s, 3H). MS (M+H)+: 428 / 430. Step 7 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid methylamide

A solution of 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido) benzofuran-3- carboxylic acid (33 g, 77 mmol), HOBT (15.6 g, 115.5 mmol) and EDCI (22.2 g, 115.5 mmol) in DMF (250 mL) was stirred at room temperature. After 2 hours, Et3N (50 mL) and CH3 H2 (HC1 salt, 17.7 g, 231 mmol) was added to the mixture, and the mixture was stirred overnight. After the solvent was removed, H20 was added and the mixture was extracted with ethyl acetate. The combined organic layer was washed with H20, brine and concentrated in vacuo. The resulting residue was washed with EtOAc to provide the product of 5-bromo-2-(4-fluorophenyl)-N- methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (32 g, yield: 94%). 1H- MR (DMSO- ck, 400 MHz) δ 9.55 (br s, 1H), 8.46-8.48 (m, 1H), 8.12-8.17 (m, 2H), 7.96 (s, 1H), 7.87 (s, 1H), 7.45-7.50 (m, 2H), 3.16 (s, 3H), 2.93 (d, J= 8.4 Hz, 3H). MS (M+H)+: 441 / 443.
Step 8 – Synthesis of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido benzofuran-3-carboxamide

CH3I (31.6 g, 223 mmol) was added to a mixture of 5-bromo-2-(4-fluorophenyl)- N-methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (32 g, 74 mmol), K2C03 (25.6 g, 186 mmol) and KI (246 mg, 1.5 mmol) in DMF (150 mL) under N2 protection. The mixture was stirred at 80-90 °C overnight. After concentrated in vacuo, the resulting residue was washed with water (200 mL) and EtOAc (200 mL) to provide the product of 5-bromo-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (31.5 g, 94%). 1H- MR (CDCI3, 400 MHz) δ 8.16 (s, 1H), 7.88-7.92 (m, 2H), 7.70 (s, 1H), 7.18-7.23 (m, 2H), 5.78 (br s, 1H), 3.34 (s, 3H), 3.09 (s, 3H), 3.00 (d, J= 4.8 Hz, 3H). MS (M+H)+: 455 / 457. Step 9 – Synthesis of 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(4, 4, 5, 5- tetramethyl-1 -dioxaborolan-2-yl)benzofuran-3-carboxamide
a degassed solution of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (1.0 g, 2.2 mmol) and pinacol diborane (2.79 g, 11.0 mmol) in 1,4-Dioxane (25 mL) was added KOAc (647 mg, 6.6 mmol) under N2 and stirred for 4 hours at room temperature. Then Pd(dppf)Cl2 (60 mg) was added, and the mixture was stirred for another 30 minutes. Then the mixture was put into a pre-heated oil-bath at 130 °C and stirred for another 1 hour under N2. The reaction mixture was cooled to room
temperatureand concentrated and extracted with EtOAc. The organic layers were washed with brine, dried over Na2S04. After concentrated, the crude product of the boronic ester was purified using column chromatography (petroleum ether / EtOAc = 5 / 1 to 2 / 1) to obtain 2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)benzofuran-3-carboxamide as white solid (700 mg, yield: 64%). 1H- MR (CDCI3, 400 ΜΗζ) δ 8.17 (s, 1H), 7.87-7.91 (m, 2H), 7.52 (s, 1H), 7.11 (t, 7= 7.6 Hz, 2H), 5.81 (d, 7= 2.8 Hz, 1H), 3.30 (s, 3H), 2.97 (d, 7= 5.2 Hz, 3H), 2.90 (s, 3H), 1.31 (s, 12H). MS (M+H)+: 503.
Step 10 – Synthesis of tert-butyl 4-fluoro-lH-indole-l -car boxy late
To a solution of 4-fluoro-lH-indole (5 g, 0.11 mol) and DMAP (150 mg, 3%Wt) in THF (50 mL) was added (Boc)20 (8.5 g, 0.04 mol) dropwise. The mixture was stirred at room temperature for 2 hours. The organic solvent was removed in vacuo, and the resulting residue was purified using column chromatography (pure petroleum ether) to provide tert-butyl 4-fluoro- lH-indole-l-carboxylate (8.3 g, yield: 96%). 1H- MR (CDC13, 400 MHz) δ 7.92 (d, J= 8.4 Hz, 1H), 7.55 (d, J= 3.6 Hz, 1H), 7.23 (m, 1H), 6.90 (m, 1H), 6.66 (d, J= 3.6 Hz, 1H), 1.67 (s, 9H). MS (M+H)+: 236.
Step 11 – Synthesis of (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid

To a solution of diisopropylamine (7.5 mL, 0.11 mol) in THF (35 mL) at 0 °C was added «-BuLi (21 mL, 0.055 mol) dropwise. The mixture was stirred at 0 °C for 40 minutes. Then the mixture was cooled to -78 °C. Tert-butyl 4-fluoro-lH-indole-l-carboxylate (5 g, 0.02 mol) in THF (13 mL) was added dropwise slowly. After addition, the mixture was stirred at -78 °C for 2 hours. Then triisopropyl borate (3.29 g, 0.03 mol) was added. The mixture was stirred at -78 °C for another 40 minutes. The reaction was monitored using TLC. When the reaction was completed, the mixture was adjusted to pH = 6 with 1 N HC1. After extracted with EtOAc (25 mL x 3), the combined organic layers were washed with brine (50 mL), dried over Na2S04, filtered and concentrated in vacuo. The obtained solid was recrystallized with EtOAc and petroleum ether to provide (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid (4.5 g, yield: 76.7%, which might be unstable at high temp, work up, store in fridge). 1H- MR (CDC13, 400 MHz) δ 7.77 (d, J= 8.4 Hz, 1H), 7.57 (s, 1H), 7.44 (s, 2H), 7.24 (m, 1H), 6.90 (m, 1H), 1.66 (s, 9H). MS (M+H)+: 280.
Step 12 – Synthesis of 6-chloro-2-iodopyridin-3-ol
6-chloropyridin-3-ol (5.0 g, 38.6 mmol) was dissolved in water (50 mL) and placed under an N2 atmosphere. Na2C03 (8.2 g, 77.4 mmol) was added followed by iodine (9.8 g, 38.8 mmol). The reaction mixture was stirred at room temperature for 2 hours. The mixture was poured into 1M Na2S203 and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04 and concentrated to provide the product of 6-chloro-2- iodopyridin-3-ol (7.0 g, yield: 70.9%). 1H- MR (CDC13, 400 MHz) δ 7.17 (d, J= 8.4 Hz, 1H), 7.06 (d, J= 8.4 Hz, 1H). MS (M+H)+: 256 / 258.
Step 13 – Synthesis of 6-chloro-2-(4-fluoro-lH-indol-2-yl)pyridin-3-ol
A mixture of (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid (5 g, 18.0 mmol), 6-chloro-2-iodopyridin-3-ol (3.82 g, 15.0 mol) and NaHC03 (3.78 g, 45.0 mol) in 1, 4-dioxane (76 mL) and water (7 mL) was stirred at room temperature for 15 minutes. Then Pd(PPh3)2Cl2 (527 mg, 0.75 mmol) was added under nitrogen atmosphere, and the mixture was heated at 100 °C under N2 for 16 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc (50 mL), filtered and concentrated in vacuo. The resulting residue was diluted with H20 (60 mL) and EtOAc (30 mL), and the layer was separated, the aqueous layer was extracted with EtOAc (3*30 mL). The combined organic layers were washed with brine (50 mL), dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether / EtOAc = 20 / 1 ~ 3 / 1) to provide 6-chloro-2- (4-fluoro-lH-indol-2-yl)pyridin-3-ol (3 g, yield: 76.5%). 1H- MR (MeOD, 400 MHz) δ 7.36 (s, 1H), 7.23-7.27 (m, 2H), 7.03-7.11 (m, 2H), 6.63-6.68 (m, 1H). MS (M+H)+: 263 / 265.
Ste 14 – Synthesis of 2-chloro-ll-fluoro-6H-pyrido[2′,3′:5, 6][l,3]oxazino[3,4-a]indole
A solution of 6-chloro-2-(4-fluoro-lH-indol-2-yl)pyridin-3-ol (2 g, 7.6 mmol) and Cs2C03 (7.46 g, 22.89 mmol) in DMF (100 mL) was stirred at 100 °C (internal temperature) for 15 min, and then chloroiodomethane (2.85 g, 15.3 mmol) in DMF (2 mL) was added dropwise. After the reaction was completed, the mixture was filtered and concentrated in vacuo. The resulting residue was diluted with water (50 mL) and extracted with ethyl acetate (30 mL x 3). The organic layer was washed with brine, dried over Na2S04 and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether:EA=10: l) to provde 2-chloro-l l-fluoro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (1.8 g, yield: 86.1%). 1H- MR (DMSO-i¾, 400 MHz) δ 7.64 (d, J= 8.8 Hz, 1H), 7.39-7.46 (m, 2H), 7.21-7.25 (m, 1H), 7.06 (s, 1H), 6.88-6.92 (m, 1H), 6.18 (s, 2H). MS (M+H)+: 275 / 277. Step 15 – Synthesis of5-(ll-fluoro-6H-pyrido[2 3′:5, 6][l,3]oxazino[3,4-a]indol-2-yl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxam
To a degassed solution of 2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzofuran-3- carboxamide (100 mg, 0.199 mmol), 2-chloro-l l-fluoro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4- a]indole (56 mg, 0.199 mmol) and Κ3Ρ04·3Η20 (159 mg, 0.597 mmol) in dioxane / H20 (0.8 mL / 0.2 mL) was added Pd2(dba)3 (9 mg, 0.01 mmol) and X-Phos (9 mg, 0.02 mmol) under N2. The mixture was heated at 80 °C for 1 hour. The mixture was then diluted with water (30 mL) and extracted with EtOAc (15 mL x 3). The organic layer was washed with brine (20 mL), dried over Na2S04 and concentrated in vacuo. The resulting residue was purified using prep-TLC (petroleum ether / EtOAc = 1 : 1.5) to provde the pure product of 5-(l l-fluoro-6H- pyrido [2′, 3 ‘ : 5 , 6] [ 1 , 3 ]oxazino [3 ,4-a]indol-2-yl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (60 mg, 48.8%). 1H- MR (CDC13, 400 MHz) δ: 7.99 (s, 1H), 7.93-7.96 (m, 2H), 7.65 (s, 1H), 7.45-7.50 (m, 2H), 7.17-7.21 (m, 4H), 7.10 (d, J= 8.0 Hz, 1H), 6.81-6.85 (m, 1H), 5.98 (s, 3H), 3.35 (s, 3H), 2.98 (d, J= 4.8 Hz, 3H), 2.72 (s, 3H). MS (M+H)+: 615.
Paper

We describe the route development and multikilogram-scale synthesis of an HCV NS5B site D inhibitor, MK-8876. The key topics covered are (1) process improvement of the two main fragments; (2) optimization of the initially troublesome penultimate step, a key bis(boronic acid) (BBA)-based borylation; (3) process development of the final Suzuki–Miyaura coupling; and (4) control of the drug substance form. These efforts culminated in a 28 kg delivery of the desired active pharmaceutical ingredient.
Process Development of the HCV NS5B Site D Inhibitor MK-8876
† Department of Process Research and Development, Merck Research Laboratories, Rahway, New Jersey 07065, United States
‡ Department of Process Chemistry, Merck Sharp & Dohme Ltd., Hertford Road, Hoddesdon, Hertfordshire EN11 9BU, United Kingdom
§ Werthenstein BioPharma GmbH (MSD Switzerland), Industrie Nord 1, CH-6105 Schachen, Switzerland
∥ WuXi AppTec Co., Ltd., No. 1 Building, #288 FuTe ZhongLu, WaiGaoQiao Free Trade Zone, Shanghai 200131, China
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00405
*E-mail: qinghao.chen@merck.com
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00405
PAPER

Using the Teasdale method, purge factor estimates for six impurities identified as mutagenic alerts in the synthesis of MK-8876 are compared to actual measured amounts of these impurities determined via appropriate analytical methods. The results from this comparison illustrate the conservative nature of purge factor estimates, meaning that overprediction of mutagenic impurity purging is unlikely when using this method. Industry and regulatory acceptance of the purge factor estimation method may help minimize analytical burden in pharmaceutical development projects.
Evaluation and Control of Mutagenic Impurities in a Development Compound: Purge Factor Estimates vs Measured Amounts
† Merck and Co., Rahway, New Jersey 07065, United States
‡ Advanced Polymer Technology, The Dow Chemical Company, 400 Arcola Road, Collegeville, Pennsylvania 19426, United States
Org. Process Res. Dev., 2015, 19 (11), pp 1531–1535
DOI: 10.1021/acs.oprd.5b00263
*E-mail: mark_mclaughlin@merck.com.
This article is part of the Genotoxic Impurities 2015 special issue.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00263?journalCode=oprdfk
| WO2004041201A2 * | Oct 31, 2003 | May 21, 2004 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| WO2011106992A1 * | Mar 2, 2011 | Sep 9, 2011 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| WO2004041201A2 * | Oct 31, 2003 | May 21, 2004 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| WO2010030592A1 * | Sep 8, 2009 | Mar 18, 2010 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2011106992A1 * | Mar 2, 2011 | Sep 9, 2011 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014123794A1 * | Feb 3, 2014 | Aug 14, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| WO2014123795A2 * | Feb 3, 2014 | Aug 14, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| WO2014123795A3 * | Feb 3, 2014 | Oct 30, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| US9242998 | Feb 3, 2014 | Jan 26, 2016 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis C |
//////MK-8876, 1426960-33-9, Merck & Co, Antivirals, Phase I, Hepatitis C
Fc7cccc6c7cc2n6COc1ccc(nc12)c3cc4c(cc3N(C)S(C)(=O)=O)oc(c4C(=O)NC)c5ccc(F)cc5
Tenatoprazole, テナトプラゾール
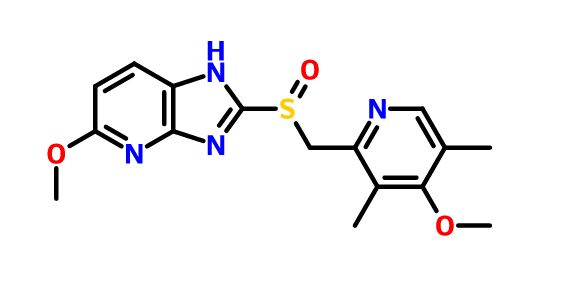
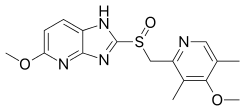
Tenatoprazole
テナトプラゾール
泰妥拉唑
| Tenatoprazole; 113712-98-4; Ulsacare; Protop; TU 199; TU-199; | |
| Molecular Formula: | C16H18N4O3S |
|---|---|
| Molecular Weight: | 346.40412 g/mol |
5-methoxy-2-[(4-methoxy-3,5-dimethylpyridin-2-yl)methylsulfinyl]-1H-imidazo[4,5-b]pyridine
2-[2-(3,5-Dimethyl)pyridylmethylsulfinyl]-5-methoxyimidazo[4,5-b]pyridine
Phase I
PHASE 1 FOR ………..A proton pump inhibitor potentially for the treatment of gastroesophageal reflux disease.
![]()
Research Code TU-199
CAS No. 113712-98-4
Mitsubishi Tanabe Pharma and was licensed to Negma Laboratories

Tenatoprazole is a proton pump inhibitor drug candidate that was undergoing clinical testing as a potential treatment for refluxoesophagitis and peptic ulcer as far back as 2003.[1] The compound was invented by Mitsubishi Tanabe Pharma and was licensed to Negma Laboratories (part of Wockhardt as of 2007[2]).[3]:22
Mitsubishi reported that tenatoprazole was still in Phase I clinical trials in 2007[4]:27 and again in 2012.[3]:17
Tenatoprazole has an imidazopyridine ring in place of the benzimidazole moiety found in other proton pump inhibitors, and has a half-life about seven times longer than other PPIs.[5]

Tenatoprazole is a novel imidazopyridine derivative and has an imidazopyridine ring in place of the benzimidazole moiety found in other proton pump inhibitors. It is activated more slowly than other proton pump inhibitor, but its inhibition is resistant to reversal.Tenatoprazole has an extended plasma half-life in comparison with those of all other proton pump inhibitors; this makes it more potent in the treatment of nocturnal acid breakthrough than esomeprazole, one of the most popular proton pump inhibitors.
Tenatoprazole belongs to the class of covalent proton pump inhibitors (PPIs), which is converted to the active sulfenamide or sulfenic acid by acid in the secretory canaliculus of the stimulated parietal cell of the stomach.This active species binds to luminally accessible cysteines of the gastric H+,K+-ATPase, resulting in disulfide formation and acid secretion inhibition.Tenatoprazole binds at the catalytic subunit of the gastric acid pump with a stoichiometry of 2.6 nmol mg−1 of the enzyme in vitro. In vivo, maximum binding of tenatoprazole was 2.9 nmol mg−1of the enzyme at 2 h after intravenous (IV) administration.
Tenatoprazole, or (+)-5-methoxy-2-{[(4-methoxy-3,5-dimethyl-2-pyridyl) methyl] sulfinyl} imidazo-[4,5-b] pyridine, is described in Patent No. EP 254,588. It belongs to the group of drugs considered as proton pump inhibitors, which inhibit the secretion of gastric acid and are useful in the treatment of gastric and duodenal ulcers. It can also be used to treat gastro-oesophageal reflux, digestive bleeding and dyspepsia, because of its relatively long elimination half-life, as described in the application for French patent No. FR 02. 13113.
The first known derivative of this series of proton pump inhibitors was omeprazole, described in Patent No. EP 001,529, which is endowed with properties which inhibit the secretion of gastric acid and is widely employed as an anti-ulcerative in human therapeutics.
In addition to omeprazole, other proton pump inhibitors are well known, and particular mention can be made of rabeprazole, pantoprazole and lansoprazole, which all exhibit structural analogy and lansoprazole, which all exhibit structural analogy and belong to the group of pyridinyl methyl sulfinyl benzimidazoles. These compounds are sulfoxides presenting with asymmetry at the level of the sulphur atom, and therefore generally take the form of a racemic mixture of two enantiomers.
Like omeprazole and other sulfoxide with an analogue structure, tenatoprazole has an asymmetric structure and may therefore be present in the form of a racemic mixture or of its enantiomers. Thus it may exist in the form of its two enantiomers with R and S configurations, or (+) or (−), respectively.
Recent studies have shown that, unlike all the other proton pump inhibitors such as, for example, omeprazole or lansoprazole, and unexpectedly, tenatoprazole is endowed with a markedly prolonged duration of action, resulting from a plasma half-life which is about seven times longer. Thus the clinical data collected have shown that tenatoprazole enables a degree of symptom relief and healing of gastric lesions which is superior to that achieved by other drugs belonging to the same therapeutic category of proton pump inhibitors, which thus allows its effective use in the treatment of atypical and oesophageal symptoms of gastro-oesophageal reflux, digestive bleeding and dyspepsia, as indicated above.
Studies performed by the application have made it possible to show that the two enantiomers contribute differently to the properties of tenatoprazole, and that the two enantiomers, (+) and (−) exhibit significantly different pharmacokinetic properties. Thus it is possible to prepare medicinal products with specific activity by isolating the enantiomers, and these enantiomers themselves exhibit a different pharmacokinetic profile from that of the known racemic mixture. It then becomes possible to use each of these enantiomers more effectively in precise indications for the treatment of perfectly identified pathologies.
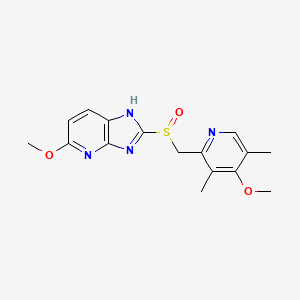
Anti-ulcer drug
tenatoprazole (tenatoprazole) is a new proton pump inhibitor, by the Japanese company Tokyo Tanabe, Japan’s Mitsubishi Corporation and Japan’s Hokuriku pharmaceutical companies jointly developed, has passed Phase II clinical trials. It acts on gastric parietal cells, reducing treatment of gastric ulcer, duodenal ulcer, reflux wall cell H + / K + -ATP activity, inhibition of gastric acid secretion, and H. pylori antibacterial activity, mainly for esophagitis and Zhuo – Ellison syndrome and gastric acid secretion disorders related diseases. Compared with the same types of drugs, Tenatoprazole suppress H + / K + -ATP enzyme activity is stronger, more stable, its efficacy than similar products currently widely used in clinical omeprazole strong 7 times. It has not been in the domestic market, nor ratified the production, with broad market prospects and development potential.
Proton pump inhibitors (proton pump inhibitors) for the treatment of acid-related diseases, the past ten years a wide range of clinical applications, better effect of the drug. It can quickly pass through the stomach wall membrane, gathered in a strongly acidic secretory tubules, and H + / K + -ATP enzyme (proton pump) thiol groups covalently bonded to form a disulfide bond, proton pump inactivation, inhibition of the enzyme H + / K + transport, so as to achieve the effect of acid suppression. Proton pump inhibitors and conventional clinical application of gastric acid suppression drugs H2 receptor antagonists compared with different sites of action and have different characteristics, namely acid-suppressing effect at night is good, rapid onset of acid inhibition strong and long time, easy to take these drugs can quickly and efficiently inhibit gastric acid secretion and clearance of Helicobacter pylori, it is widely used gastric ulcer, duodenal ulcer, reflux esophagitis and Zhuo – Ellison syndrome and other diseases treatment. Currently, proton pump inhibitors has been listed on the main omeprazole, lansoprazole, pantoprazole, rabeprazole and esomeprazole.Physical and
chemical properties ofwhite or white crystalline powder, melting point 174 ~ 175 ℃. Soluble in chloroform, insoluble in alcohol and water.
This product and other proton pump inhibitors as compared to chemically stable. China had 34 omeprazole preparations from Portugal, Brazil, India, China and other 13 countries, the stability of the measurements were made. The results showed that only six products (18%) during the trial showing good physical and chemical stability of. 27 products (79%) less (including Chinese product), the active ingredient a significant chemical decomposition, color and physical properties such as dissolution, are also a corresponding change. The results of a stability test designed to compare the various proton pump inhibitors show investigated eight days at 60 ℃, relative humidity of 75%, after omeprazole decomposition only 3% of the active ingredient, the tenatoprazole 77% of the data, said Alpha pantoprazole stability far superior to omeprazole, is already developed similar products in the most promising products.
Synthesis
Matsuishi, N.; Takeda, H.; Iizumi, K.; Murakami, K.; Hisamitsu, A. US Patent 4,808,596, 1989
Synthesis of Tenatoprazole 1 commences with the coupling of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 in the presence of potassium hydroxide affords 4 with 73% yield in ethanol and chloroform. The oxidation of the penultimate sulfide intermediate4 with m-CPBA in chloroform (100 vol) afforded 1
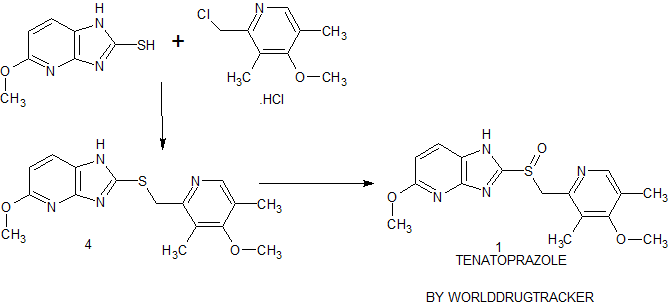
Syn 2
Org. Process Res. Dev., 2009, 13 (4), pp 804–806
DOI: 10.1021/op800173u
synthesis of 1 begins with the solvent-free condensation of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 to deliver the sulfide intermediate4 with 98% yield.
The final step of the synthesis is the oxidation of the sulfide intermediate with m-CPBA to form tenatoprazole 1. The sulfide intermediate 4 on treatment with 0.9 equiv of m-chloroperbenzoic acid (m-CPBA) at −10 to −15 °C afforded the crude tenatoprazole which was isolated as its sodium salt. The sodium salt of tenatoprazole 5 was purified by recrystallsation using dimethyl formamide and ethyl acetate (2:1 ratio) to yield the pure crystalline tenatoprazole sodium 5. Treatment of tenatoprazole sodium 5 with dil. HCl in the presence of acetone and water afforded the pure tenatoprazole 1
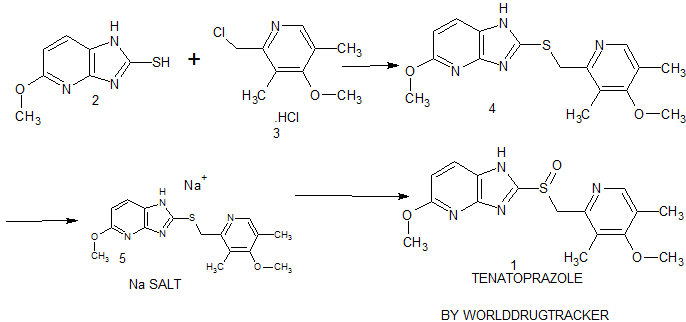
PATENT
CN 1861600
CN 1982311
WO 2009116072
CN 101429192
WO 2010043601
IN 2010CH00462
IN 251400
CN 102304127
WO 2012004802
CN 102703922
IN 2009DE01392
WO 2014111957
IN 2013MU00181
IN 2014CH01419
PAPER
Dai, Liyan; Synthetic Communications 2008, V38(4), P576-582
Advanced Materials Research (Durnten-Zurich, Switzerland) (2011), 233-235(Pt. 1, Fundamental of Chemical Engineering), 160-164.
Organic Process Research & Development (2013), 17(10), 1293-1299
Enantiomeric separation of proton pump inhibitors on new generation chiral columns using LC and supercritical fluid chromatography
Journal of Separation Science (2013), 36, (18), 3004-3010………http://onlinelibrary.wiley.com/doi/10.1002/jssc.201300419/abstract
PATENT
CN 102304127
https://www.google.com/patents/CN102304127A?cl=en
Tenatoprazole is a new type of gastric H + / K + -ATP enzyme inhibitors (proton pump inhibitor PPI), the chemical name 5-methoxy-2- (4-methoxy-3, 5-dimethyl-2-methylsulfinyl) imidazole and W, 5-b] pyridine, useful in the treatment of gastric ulcer, duodenal ulcer, reflux esophagitis and Zhuo – Ai syndrome and gastric acid secretion disorders related diseases. The drug was developed by Japan’s Tokyo Tanabe, Japan’s Mitsubishi Corporation and Japan’s Hokuriku pharmaceutical companies. Compared with other varieties of the same type, which inhibit H + / K + -ATP enzyme activity is stronger, ulcers of various tests are effective, and significantly improve the stability compared with other proton pump inhibitors.
US patent US4808596 “hidazo [4,5_b] pyridine compounds and pharmaceutical compositions containing same)) synthesis process disclosed Tenatoprazole the below formula:

By The route of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride with 2-mercapto-5-methoxy-imidazole, 5-b] pyridine under basic conditions condensation of Intermediate 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine, and then oxidizing the Thai duly omeprazole. This route for the synthesis of pull azole classic line, many pull azoles such as omeprazole can be synthesized by a similar route, this route mild condition, simple operation. But the route condensation, oxidation treatment after use of large amounts of toxic solvent chloroform, is not conducive to industrial scale; lower oxidation yields, the resulting Tenatoprazole containing unreacted starting materials 2- [2_ (3,5 – dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine, further comprising a sulfone by-product, N- oxide, N- oxide sulfone, These by-products may interfere with purification of tenatoprazole.
Japanese Patent invention Wo 丨 J JP05222038 “5_methoxy-2- [[(4_methoxy-3, 5-dimethyl-2-pyridyl) methyl] thio] imidazo [4,5 ~ b] pyridine and intermediates)) male
Synthesis open Tenatoprazole the below formula:

4-chloro-2-chloromethyl-3,5-dimethylpyridine -N- oxide 2_ mercapto _5_ methoxy-imidazo – [4, 5-b] pyridine in alkaline under condensation of Intermediate 5-Methoxy-2- (4-chloro-3,5-dimethyl-2-methylthio Bi) imidazo W, 5-b] pyridine-oxide -N- ( yield 82%), then refluxed in a solution of sodium methoxide in methanol to give 5-methoxy-2- (4-oxo-3,5-dimethyl-2-methyl sulfide) imidazo W , 5-b] pyridine -N- oxide (income ¥ 71%), and then at room temperature in methylene chloride, phosphorus trichloride treated with deoxy (yield 95%), and finally oxidation in Tenatoprazole (income Rate not reported). The novel synthetic route, mild reaction conditions, simple operation, the yield of each step is higher, but the route is too long resulting in a total yield is not high, prolonged and rising production costs.
Reaction route is as follows:

Example 1:
] a) 2- [2- (3,5-dimethyl) -4-methoxy-picolyl thioether _5_ methoxy] imidazo [4,5_b] pyridine:
To a reaction flask was added 2-mercapto-5-methoxy-imidazole, 5-b] pyridine 18. lg, 12g of sodium hydroxide and water 144. 8g, stirred and dissolved at 25 ° C, was added dropwise within Ih 20g of the 2-chloromethyl-dimethyl-4-methoxy _3,5- pyridine hydrochloride and 60g of water were mixed solution dropwise at 25 ° C the reaction 2h, the reaction is completed, filtered, washed with water 144. 8g, 36. 2mL ethanol and washed to obtain a wet powder; wet powder was dried at 50 ° C in vacuo to constant weight to give 2- [2_ (3,5-dimethyl) -4-methoxy-pyridylmethyl sulfide -5 – methoxy] imidazo [4,5-b] pyridine 32. Og;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine 30g, dichloromethane 300g, methanol 15g, and dissolved with stirring; cooled to -10 ° C, was added dropwise the 15g and 485g m-chloroperbenzoic acid in methylene chloride mixed solution, dropwise addition the reaction temperature was controlled at -10 ° C, the dropping time of the pool; the dropwise addition, the temperature control at -10 ° C, the reaction 30min; completion of the reaction, at 10 ° C by the dropwise addition of lithium hydroxide and 135g water 15g mixed solution, drip complete, insulation stirred Ih; filtered cake was washed with acetone 60mL, get wet powder; wet powder was dried at 35 ° C under vacuum to constant weight to give Tenatoprazole lithium salt ^ g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, acetone 63mL, water IOOmL, cooling M0 ° C, dropping lmol within lh / L hydrochloric pH7 0, drops. Albert, stirring 30min; the filter cake washed with water 50mL, washed with acetone and 50mL, wet powder was dried at 35 ° C under vacuum to constant weight to give Tenatoprazole 19. Sg.
Example 2:
a) 2- [2- (3,5-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5_b] pyridine (4) Preparation: To the reaction flask was added 2-mercapto-5-methoxy-imidazo 44,5-b] pyridine 18. lg, 11. 2g of potassium hydroxide and water 217mL, stirred and dissolved at! 35 ° C, was added dropwise within 2h by the 33. 3g of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride and 133. 2mL water mixed solution, dropwise at 35 ° C the reaction 4h, the reaction is completed, filtration, water 217mL, 72. 4mL ethanol and washed to obtain a wet powder; wet powder was dried at 60 ° C in vacuo to constant weight to give 2- [2- (3,5-dimethyl) -4-methoxy-pyridylmethyl sulfide -5-methoxy-yl] imidazo W, 5-b] pyridine 33. Ig;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 400mL, methanol 50mL, stirring to dissolve; cooled to _15 ° C, was added drop by the m-chloroperoxybenzoic acid 16g of mixed solution of dichloromethane and 400mL , the process reactor temperature control was added dropwise at -20 ° C, the dropping time 2. 5h; the dropwise addition, the temperature control _15 ° C, the reaction 35min; completion of the reaction, at 15 ° C by the dropwise addition of 20g of hydrogen Lithium oxide and 200mL water mixed solution, drip completed, insulation mixing 1. 5h; filtration, the filter cake washed with acetone 90mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 6g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, ethanol 75mL, water 150mL, cooled to 10 ° C, dropping 6mol / L hydrochloric pH8 0 within 2h,. drops Albert, stirring 40min; the filter cake washed with water 100mL, washed with acetone IOOmL, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 5g.
Example 3:
a) 2- [2- (3,5-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5_b] pyridine (4) Preparation: To the reaction flask was added 2-mercapto-5-methoxy-imidazo 44,5-b] pyridine 18. lg, 8.4g of lithium hydroxide and water 180ml, stirred and dissolved at 30 ° C, was added dropwise within 1. 5h by the Guang .6g 2-chloro-3,5-dimethyl-4-methoxy-pyridine hydrochloride and 90mL water mixed solution, drop end at 30 ° C reaction 3h, the reaction is complete, filtration, water 217mL , washed with 85mL ethanol to obtain a wet powder; wet powder was dried at 55 ° C in vacuo to constant weight to give 2- [2- (3,5-dimethyl) -4-methoxy-5-pyridylmethyl sulfide oxy] imidazo [4,5-b] pyridine 32. 4g;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 600mL, methanol 60mL, stirring to dissolve; cooled to -20 ° C, was added drop by the m-chloroperoxybenzoic acid 18g of mixed solution of dichloromethane and 600mL , dropwise addition the reaction temperature is controlled at _20 ° C, the dropping time of the pool; the dropwise addition, the temperature control at _20 ° C, the reaction 40min; completion of the reaction, at 20 ° C by the dropwise addition of lithium hydroxide and 300mL 30g water mixed solution, drip complete insulation mixing tank; filter, the filter cake washed with acetone and 120mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 7g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, methanol 75mL, water 120mL, cooled to 5 ° C, dropping dilute hydrochloric acid within 1 5h tune pH7 5,.. drops Albert, stirring 35min; the filter cake washed with water 75mL, 75mL acetone washed, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 6g.
Example 4:
a) 2- [2- (3,5-dimethyl) -4-methoxy-picolyl thioether _5_ methoxy] imidazo [4,5_b] pyridine ⑷ Preparation of: To a solution The reaction flask was added 2-mercapto-5-methoxy imidazole -½, 5-b] pyridine 18. lg, IOg sodium hydroxide and water 150ml, stirred and dissolved at 30 ° C, the 1. 5h dropwise added from 21 . 5g of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride and 90mL water mixed solution, dropwise at 30 ° C the reaction 3h, completion of the reaction, was filtered, washed with water 217mL, The wet powder was washed with ethanol to give 85mL; wet powder was dried at 55 ° C in vacuo to constant weight to give 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide ] imidazo [4,5-b] pyridine 32. 3g;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 500mL, methanol 60mL, stirring to dissolve; cooled to -20 ° C, was added drop by the m-chloroperoxybenzoic acid 18g of mixed solution of dichloromethane and 500mL , the process reactor temperature control was added dropwise at -20 ° C, the dropping time pool; the dropwise addition, the temperature control in -20 ° C, the reaction 40min; completion of the reaction, at 20 ° C by the dropwise addition of lithium hydroxide 30g and 300mL water mixed solution, drip complete insulation mixing tank; filter, the filter cake washed with acetone and 120mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 6g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, isopropanol 75mL, water 120mL, cooled to 5 ° C, dropping 3mol / L hydrochloric within 1 5h. . pH7 5, drops Albert, stirring 35min; the filter cake washed with water 75mL, 75mL acetone washed, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 7g.
PAPER
An Improved Synthesis of Antiulcerative Drug: Tenatoprazole
http://pubs.acs.org/doi/full/10.1021/op800173u
Department of Research and Development, Srini Pharmaceuticals Ltd., Plot No. 10, Type-C, Road No. 8, Film Nagar, Jubilee Hills, Hyderabad-500033, Andhra Pradesh, India, Department of Chemistry, Osmania University, Tarnaka, Hyderabad-500007, Andhra Pradesh, India and Research and Development, Integrated Product Development Organization, Innovation Plaza, Dr. Reddy’s Laboratories Ltd., Bachupally, Qutubullapur, R. R. Dist. 500 072, Andhra Pradesh, India
Org. Process Res. Dev., 2009, 13 (4), pp 804–806
DOI: 10.1021/op800173u
Publication Date (Web): November 12, 2008
Copyright © 2008 American Chemical Society
* To whom correspondence should be addressed. Telephone: +91 9490783736. E-mail: drkvr_ou@yahoo.com;kvgr1951@rediffmail.com., †Srini Pharmaceuticals Ltd.
, ‡Osmania University.
, §Dr. Reddy’s Laboratory Ltd.
, ‡Osmania University.
, §Dr. Reddy’s Laboratory Ltd.

An efficient, cost-effective and multikilogram-scale process for the synthesis of tenatoprazole 1, an antiulcerative drug, is described. The key steps in this synthesis involve the coupling of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 to yield 4 and its subsequent oxidation with m-CPBA to produce sulfoxide 1. The process has been scaled up for the multikilogram-scale of compound 1 with an overall yield of 72%. The new process requires no purification process and affords the target compound 1 with 99.8% purity by HPLC.
2-[2-(3,5-dimethyl)pyridylmethylsulfinyl]-5-methoxyimidazo[4,5-b]pyridine (1, 15.5 kg, 74%). Purity by HPLC 99.8%; 1H NMR (200 MHz, DMSO) δ 2.2 (s, 6H), 3.8 (s, 6H), 4.8 (s, 2H), 6.6 (d, 1H), 7.8 (d, 1H), 8.2 (s, 1H), 13.0 (s, 1H).
PATENT
http://www.google.co.in/patents/US7507746
the (+) enantiomer of tenatoplazole can be obtained by using chloroform, an industrially acceptable solvent, in accordance with the method proposed by Umemura et al. (J. Org. Chem. 1993, 58, 4592) as follows:

Example 1 (−)-5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridineThe conditions for preparative chromatography, shown as an example, are as follows:
Column: 265×110 mm ChiralPak®
Chiral Stationary Phase selector of the Amylose tris type [(S)-a methylbenzylcarbamate]
Flow rate: 570 ml/min
Detection: UV 240 nm
Temperature: Ambient temperature
These conditions are implemented on a liquid preparative chromatography apparatus.
Introduce approximately 2 g of the racemic mixture if tenatoprazole exhibiting purity higher than 99.5%. The (−) enantiomer is identified by measuring the angle of optical rotation, which must be laevogyre. This measurement can be performed directly on the column, the product being dissolved in the solvent (acetonitrile).
Example 2 (+)-5-methoxy-2-{(4-methoxy-3, 5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridine(R)-(+)-binaphthol 85 g (0.311 mol, 0.2 equivalence), ortho titanic acid isopropyl 42 g (0.148 mol, 0.1 equivalence), water 55 g (3.06 mol) and chloroform 7.5 L were stirred for 1 hour at room temperature. To the resultant, 5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]thio}imidazo[4,5-b]pyridine (MPI), 0.5 kg, was added and stirred for 0.5 hours at room temperature. The thus-prepared mixture was cooled to 5° C. and then 70% aqueous solution of tert-butylhydroperoxide, 0.4 L (approx. 3.0 mol, 2.0 equivalence) was added and stirred for 72 hours at the same temperature as above. After the reaction endpoint was confirmed by HPLC, an aqueous solution of sodium hydroxide was added thereto to separate the aqueous layer, thus removing foreign matter. Then, the resultant was concentrated. Ethyl acetate was added to concentrated residues, which were then heated and suspended. The thus-prepared crude crystalline substances were dissolved in water and neutralized to pH 6.8 with a diluted sulfuric acid solution which was chilled with ice. Deposited crystals were filtered, dried and recrystallized by addition of ethanol to obtain (+)-5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridine {(+)-TU-199}
Yield: 77%
Optical purity: 96.6% ee
Chemical purity: 94.5%
Melting point: 135° C.
Optical rotation: +184° (conditions: C=1.0, N,N-dimethylformaldehyde solution)
Ultraviolet absorption spectrum: (10 μg/mL)λmax (nm): 316, 273, 206
When measurements were carried out, for a solubility of (+), (−) forms and a racemic form (±) of tenatoprazole in relation to water, it was found that the (+) form dissolved almost 3 times greater than the racemic body and (−) form dissolved over 2 times greater than the racemic form, exhibiting favorable physical properties in preparing drugs (refer to Table 2 below).
| TABLE 2 | |||
| (+) form | (−) form | (±)racemic form | |
| Solubility (water) μg/mL | 93.0 | 74.4 | 34.6 |
CLIPS

Tenatoprazole is a pyridinylmethylsulfinyl imidazopyridine compound, which is a weak base. This compound has three pKas. One is the pyridine pKa of pyridinylmethyl moiety and the others are the imidazole pKa and the pyridine pKa of the imidazopyridine moiety. The pyridine pKa1 enables tenatoprazole accumulation in the acidic canaliculus of the parietal cell. Protonation of the imidazopyridine ring enhances electron deficiency at the C-2 position, allowing intramolecular rearrangement to the active form. The active form is the sulfenic acid and/or cyclic sulfonamide, and reacts with luminal cysteine thiols of the enzyme to inhibit the enzyme activity
Synthesis route
from 2-mercapto-5-methoxy-imidazo [4,5-b] pyridine (2) and 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride ( 3) by nucleophilic substitution synthesis of 2- (4-methoxy-3,5-dimethyl-2-methylthio) -5-methoxy-imidazo [4,5-b] pyridine (4) the oxidation of 4 1. Figure 1 is a synthesis route of tenatoprazole

References
- DataMonitor. March 2003. Gastrointestinal Disease Update: Digestive Disease Week 2003
- Economic Times. 3 March, 2011. Investors unwilling to forgive Wockhardt, promoter for failings
- Mitsubishi Tanabe Pharma State of New Product Development (as of May 8, 2012)
- Mitsubishi Tanabe Pharma FY2007 Interim Financial Results
- Li H et al. H+/K+-ATPase inhibitors: a patent review. Expert Opin Ther Pat. 2013 Jan;23(1):99-111. PMID 23205582
| US4808596 * | 24 Jul 1987 | 28 Feb 1989 | Tokyo Tanabe Company, Ltd. | Imidazo[4,5-b]pyridine compounds and pharmaceutical compositions containing same |
| US5753265 * | 7 Jun 1995 | 19 May 1998 | Astra Aktiebolag | Multiple unit pharmaceutical preparation |
| US5798120 * | 6 Oct 1994 | 25 Aug 1998 | Tokyo Tanabe Company Limited | Enteric granule-containing tablets |
| EP0124495A2 | 28 Feb 1984 | 7 Nov 1984 | Aktiebolaget Hässle | Omeprazole salts |
| EP0254588A1 | 24 Jul 1987 | 27 Jan 1988 | Tokyo Tanabe Company Limited | Imidazo[4,5-b] pyridine compounds, process for preparing same and pharmaceutical compositions containing same |
| Reference | ||
|---|---|---|
| 1 | * | Andersson et al., Pharmacology & Therapeutics, 2005, vol. 108, pp. 294-307. |
| 2 | * | Anon et al., Drugs in R&D, 2002, vol. 3, pp. 276-277. |
| 3 | Kakinoki et al., Methods and Findings in Experimental and Clinical Pharmacology, 21(3): 179-187 (1999). | |
| 4 | Komatsu et al., J. Org. Chem., 58(17): 4529-4533 (1993). | |
| 5 | Uchiyama et al., Journal of Pharmacy and Pharmacology, 51(4): 457-464 (1999). | |
| 6 | Uchiyama et al., Methods and Findings in Experimental and Clinical Pharmacology, 21(2): 115-122 (1999). | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20120220623 * | 30 Aug 2012 | Mitsubishi Tanabe Pharma Corporation | The enantiomer of tenatoprazole and the use thereof in therapy |
| CN1453278A * | May 10, 2002 | Nov 5, 2003 | 中国人民解放军军事医学科学院放射医学研究所 | Omprazole compound and its prepn and application |
| CN1861600A * | Jun 14, 2006 | Nov 15, 2006 | 浙江大学 | Preparation process of taytrolazole |
| Reference | ||
|---|---|---|
| 1 | * | 《Organic Process Research & Development》 20081112 Somaiah Sripathi et al. An Improved Synthesis of Antiulcerative Drug:Tenatoprazole 第804-806页 1-6 第13卷, |
| 2 | * | 《Synthetic Communication》 20080101 Liyan Dai et al. Improved Synthetic Approach to Tenatoprazole 第576-582页 1-6 第38卷, |
| 3 | * | 《中国药物化学杂志》 20061231 赵冬梅等 抗溃疡药泰妥拉唑的合成 第360-362页 1-6 第16卷, 第6期 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-methoxy-2-[(4-methoxy-3,5-dimethylpyridin-2-yl)methylsulfinyl]-1H-imidazo[4,5-b]pyridine
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Metabolism | Hepatic (CYP2C19-mediated) |
| Biological half-life | 4.8 to 7.7 hours |
| Identifiers | |
| CAS Number | 113712-98-4 |
| ATC code | none |
| PubChem | CID 636411 |
| ChemSpider | 552196 |
| UNII | RE0689TX2K |
| Chemical data | |
| Formula | C16H18N4O3S |
| Molar mass | 346.405 g/mol |
| Chirality | Racemic mixture |
テナトプラゾール
Tenatoprazole

C16H18N4O3S : 346.4
[113712-98-4]
/////////////Tenatoprazole, 113712-98-4, TU-199, proton pump inhibitor, treatment of gastroesophageal reflux disease, Mitsubishi Tanabe Pharma, Negma Laboratories, PHASE 1, テナトプラゾール
CC1=CN=C(C(=C1OC)C)CS(=O)C2=NC3=C(N2)C=CC(=N3)OC
Recilisib Sodium, EX-RAD

Recilisib Sodium
Phase I
| C16H12ClNaO4S | |
| Molecular Weight: | 358.771849 g/mol |
|---|
A protein kinase inhibitor potentially for the treatment of acute radiation syndrome.
sodium;4-[(E)-2-[(4-chlorophenyl)methylsulfonyl]ethenyl]benzoate
![]()
Onc-01210; ON-01210.Na, Ex-RAD; ON 01210.Na; ON-01210; ON-01210-Na; Recilisib
CAS No. 334969-03-8(free)
CAS 922139-31-9(Recilisib sodium)
Benzoic acid, 4-[(1E)-2-[[(4-chlorophenyl)methyl]sulfonyl]ethenyl]-, sodium salt (1:1)
Onconova Therapeutics Inc, Univ Temple INNOVATOR
Stephen C Cosenza, Lawrence Helson,Premkumar E Reddy, Ramana M V Reddy INVENTORS
![]()
![]()

| Latest Stage of Development | Phase I |
| Standard Indication | Poisoning |
| Indication Details | Prevent radiation poisoning; Provide radation protection; Treat and prevent acute radiation syndrome (ARS) |
- Originator Onconova Therapeutics
- Class Radioprotectives; Small molecules; Sulfonamides
- Mechanism of Action Apoptosis inhibitors; Protein kinase inhibitors
- Orphan Drug Status Yes – Acute radiation syndrome
- Phase I Acute radiation syndrome
Most Recent Events
- 22 Apr 2016 Phase I development is ongoing in the US (PO & SC)
- 20 Mar 2014 Recilisib receives Orphan Drug status for Acute radiation syndrome in USA
- 03 Oct 2012 Phase-I clinical trials in Acute radiation syndrome in USA (PO)

Ex-Rad (or Ex-RAD), also known by the code name ON 01210.Na, or recilisib sodium (INN, USAN) is a drug developed by Onconova Therapeutics and the U.S. Department of Defense.[1][2] This newly developed compound is said to be a potent radiation protection agent. Chemically, it is the sodium salt of 4-carboxystyryl-4-chlorobenzylsulfone.[3]
Clinical trials
The results of two Phase I clinical studies in healthy human volunteers indicate that subcutaneously injected Ex-Rad is safe and well tolerated, with “no evidence of systemic side effects”.[4] A study in mice demonstrated the efficacy of Ex-Rad by increasing the survival rate of mice exposed to typically lethal whole-body irradiation. The study tested oral and parenteral administration of Ex-Rad for both pre- and post-exposure radiomitigation.[1]
Research on Ex-Rad has involved collaboration with the Armed Forces Radiobiology Research Institute (AFRRI), the Department of Biochemistry and Molecular & Cellular Biology at Georgetown University, Long Island University‘s Arnold & Marie Schwartz College of Pharmacy, and the Department of Oncological Sciences at the Mt. Sinai School of Medicine.[1]
Mechanism of action
Onconova suggests that Ex-Rad protects cells exposed to radiation against DNA damage, and that the drug’s mechanism of action does not involve scavenging free radicals or arresting the cell cycle. Instead, they claim it employs a “novel mechanism” involving “intracellular signaling, damage sensing, and DNA repair pathways”.[4] Ex-RAD is a chlorobenzylsulfone derivative that works after free radicals have damaged DNA. Onconova CEO Ramesh Kumar believes this is a better approach than trying to scavenge free radicals. “Free radicals are very short-lived, and so the window of opportunity to give a drug is very narrow,” he says. In cell and animal models, Ex-RAD protects hematopoieticand gastrointestinal tissues from radiation injury when given either before or after exposure.[5]
While anti-radiation suits or other protective gear may be effective at reducing radiation exposure, such gear is expensive, unwieldy, and generally not available to public. Moreover, radioprotective gear will not protect normal tissue adjacent to a tumor from stray radiation exposure during radiotherapy. Pharmaceutical radioprotectants offer a cost-efficient, effective and easily available alternative to radioprotective gear. However, previous attempts at radioprotection of normal cells with pharmaceutical compositions have not been entirely successful. For example, cytokines directed at mobilizing the peripheral blood progenitor cells confer a myeloprotective effect when given prior to radiation (Neta et al., Semin. Radiat. Oncol. 6:306-320, 1996), but do not confer systemic protection. Other chemical radioprotectors administered alone or in combination with biologic response modifiers have shown minor protective effects in mice, but application of these compounds to large mammals was less successful, and it was questioned whether chemical radioprotection was of any value (Maisin, J. R., Bacq and Alexander Award Lecture. “Chemical radioprotection: past, present, and future prospects”, Int J. Radiat Biol. 73:443-50, 1998). Pharmaceutical radiation sensitizers, which are known to preferentially enhance the effects of radiation in cancerous tissues, are clearly unsuited for the general systemic protection of normal tissues from exposure to ionizing radiation.
The major biological effects of radiation exposure are the destruction of bone marrow cells, gastrointestinal (GI) damage, lung pneumonitis, and central nervous system (CNS) damage. The long-term effects of radiation exposure include an increase in cancer rates. It has been estimated that the exposure of 100 rems (roentgen equivalent man: a measurement used to quantify the amount of radiation that would produce harmful biological effects) would produce ARS symptoms. Exposure levels above 300 rems would result in the death of approximately 50% of the exposed population.
The α,β-unsaturated aryl sulfones, in particular benzyl styryl sulfones, provide significant and selective systemic protection of normal cells from radiation-induced damage in animals. When used in radiotherapy techniques, these compounds also exhibit independent toxicity to cancer cells. These α,β-unsaturated aryl sulfones, in particular benzyl styryl sulfones, are described in U.S. Pat. Nos. 6,656,973 and 6,667,346, which are particularly incorporated herein by reference in their entirety. Although these compounds are stable in solid state their aqueous formulations for parenteral administration are pH sensitive and pose challenging hurdles to overcome physical stability. The most likely causative factor may be attributed to the reactive styryl sulfone conjugated double bond, which is prone to Michael addition by nucleophiles and eventual fallout of the conjugated addition product.
U.S. Patent No. 6,656,973, describes in vitro pharmacological effects of DMSO solubilization of a benzyl styryl sulfone (e.g. ON 01210.NA) but fails to disclose a composition comprising ON 01210. NA formulation and specifically, a shelf stable formulation which is suitable for administration to humans.
PCT Application WO 2007/016201 describes pharmaceutical solution compositions for parenteral administration for reducing toxic effects of ionizing radiation in a subject, comprising an effective amount of at least one radioprotective α,β-Unsaturated aryl sulfone, and at least one component selected from the group consisting of a) a water soluble polymer in an amount between about 0.5% and about 90% w/v, b) at least one chemically modified cyclodextrin in an amount between about 20% and about 60% w/v, and c) DMA in an amount between 10% and about 50% w/v.
U.S. Patent Application 20090247624, and corresponding PCT Application WO 2008/105808, are directed to aqueous solutions, which comprise between about 20 mg/ml to about 100 mg/ml of at least one α,β-unsaturated aryl sulfone (e.g., the compound ON 01210. Na ((E)-4-Carboxystyryl-4-chlorobenzylsulfone sodium salt, a cosolvent in an amount between about 25% and about 90% w/v (e.g., about 50% PEG 400), wherein the composition is buffered and exists within the range of about pH 7.0 to about pHIO (e.g., 0.2M Tris-EDTA, pH about 8.5). The aforementioned solution formulations have exhibited a sub-optimal shelf life and lack a preferred degree of solubility and/or stability. These formulations evolved progressively as a result of addressing the most challenging aspects in the formulation and drug development field, namely, solubility and stability parameters that defined the long term viability of these formulations. There seems to be a delicate balance between pH, solubility and stability of the active moiety in aqueous milieu, wherein achieving such balance and development of a shelf stable aqueous formulation has presented a formidable challenge. Therefore, a shelf stable effective solution formulation that prevents the breakdown of the therapeutically active entity and keeps the drug in the solution at the desired pH was most desired and significant effort was directed towards this goal.
What is needed therefore, is a shelf stable effective solution formulation of radioprotective α,β-unsaturated aryl sulfones that prevents the breakdown of the therapeutically active entity and keeps the drug in the solution at the desired pH. This invention solves these and other long felt needs by providing improved solution formulation of radioprotective α,β- unsaturated aryl sulfones having improved physical and chemical stability and enhanced shelf life.
SYNTHESIS BY WORLDDRUGTRACKER
PATENT
WO 2011119863
An exemplary species of a radioprotective α,β-unsaturated aryl sulfone is ON 01210.Na. ON 01210.Na is a derivative of chlorobenzylsulfone. This compound is described in U.S. Pat. Nos. 6,656,973 and 6,667,346 as exhibiting valuable prophylactic properties which mitigate the effects of accidental and intentional exposure to life-threatening levels of irradiation. Hence, a systematic development of this compound is described with the objective of developing a shelf stable formulation.
Table 1 describes the general physical properties of ON. 1210. Na. The exemplary compound is a sodium salt of (E)-4-Carboxystyryl-4-chlorobenzylsulfone.
TABLE 1
Physical Properties of ON.1210.Na
Chemical Structure

Chemical Name (E)-4-Carboxystyryl-4-chlorobenzylsulfone,
Sodium Salt
Empirical Formula C16H12ClNa04S
Molecular Weight 358.79
Physical Nature White crystalline flakes
Melting Point 354-356° C.
Solubility Soluble in water at 8-10 mg/ml
The compound ON 01210. Na appears to form at least one polymorph. X-ray diffraction pattern, for example, of precipitated ON 01210. Na is different from that of the originally synthesized compound. Polymorphs of ON 01210.Na are intended to be within the scope of the claims appended hereto.
EXAMPLE 1
Preparation of ON 01210. Na
4-Chlorobenzyl-4-carboxystyryl sulfone (ON 01210) (49 g; 0.145 mol) was taken in a one-liter conical flask and 500 ml of distilled water was added. Sodium hydroxide solution (16 ml: 10 M stock) (0.150 mol.) was added to the conical flask. The contents of the flask were then boiled with stirring till ON 01210 was completely dissolved. The solution was then cooled to room temperature and shining crystals separated were filtered through a fluted filter paper. The crystalline material was dried under vacuum to yield (48 g) (92% yield) of pure ON 1210. Na.
EXAMPLE II
Preparation of ON 01210. Na Formulation A (Without Vitamin E TPGS)
TRIS (968.0 mg), EDTA (233.8 mg), and deionized (DI) water (24 ml) were combined in a beaker equipped with a Teflon coated stirring bar. The mixture was stirred until complete dissolution occurred, and the resulting solution was covered with aluminum foil and allowed to stir gently overnight at room temperature. The following morning, PEG 400 NF (40.0 ml) was added to the TRIS/EDTA aqueous solution with continued stirring. The vessel containing PEG 400 NF was rinsed with DI water (2 x 3.2 ml), and the rinsate added to the formulation mixture. After stirring the mixture to homogeneity (approx. 10 minutes), the pH was measured to be 9.46 using a calibrated electronic pH meter. The pH was adjusted to 8.37 (target pH = 8.40) by the careful addition of 98 pipet drops of 1.0 M HCl (aq) with stirring and allowed to fully equilibrate over a 10-15 minute period. Once the pH steadied at 8.37, ON 01210. Na (4.0 g) was added to the stirring formulation mixture. Complete dissolution required vigorous stirring and brief periodic sonication to break up ON 01210.Na clumps over a two hour period. After complete dissolution of ON 01210. Na, DI water (approx. 5 ml) was added to bring the final volume to approximately 80 milliliters. The pH of the resulting solution was determined to be 8.31, and thus 20 pipet drops of 1.0N NaOH(aq) were added to adjust the final formulation batch (defined as ON 01210.Na Formulation A) pH to 8.41-8.42. Formulation A was 0.22 micron filtered using a 100 ml Gastight Syringe equipped with a Millex®GP filter unit (Millipore Express® PES Membrane; Lot No R8KN13888).
PATENT
PATENT
PATENT
The α,β unsaturated aryl sulfones are characterized by cis-trans isomerism resulting from the presence of one or more double bonds. The compounds are named according to the Cahn-Ingold-Prelog system, the IUPAC 1974 Recommendations, Section E: Stereochemistry, in Nomenclature of Organic Chemistry, John Wiley & Sons, Inc., New York, NY, 4th ed., 1992, p.
127-138. Stearic relations around a double bond are designated as “Z” or “E”.
(E)-α,β unsaturated aryl sulfones may be prepared by Knoevenagel condensation of aromatic aldehydes with benzylsulfonyl acetic acids or arylsulfonyl acetic acids. The procedure is described by Reddy et al, Ada. Chim. Hung. 115:269-71 (1984); Reddy et al, Sulfur Letters 13:83-90 (1991); Reddy et al, Synthesis No. 4, 322-23 (1984); and Reddy et al, Sulfur Letters 7:43-48 (1987), the entire disclosures of which are incorporated herein by reference.
According to the Scheme 1 below, Ra and Rb each represent from zero to five substituents on the depicted aromatic nucleus. For purposes of illustration, and not limitation, the aryl groups are represented as phenyl groups, that is, the synthesis is exemplified by the preparation of styryl benzylsulfones. Accordingly, the benzyl thioacetic acid B is formed by the reaction of sodium thioglycollate and a benzyl chloride A. The benzyl thioacetic acid B is then oxidized with 30% hydrogen peroxide to give a corresponding benzylsulfonyl acetic acid C. Condensation of the benzylsulfonyl acetic acid C with an aromatic aldehyde D via a Knoevenagel reaction in the presence of benzylamine and glacial acetic acid yields the desired (E)-styryl benzylsulfone E.

Scheme 1
The following is a more detailed two-part synthesis procedure for preparing (E)-styryl benzylsulfones according to the above scheme.
General Procedure 1: Synthesis (E)-Styryl Benzylsulfones
Part A. To a solution of (8g, 0.2 mol) sodium hydroxide in methanol (200 ml), thioglycollic acid (0.1 mol) is added slowly and the precipitate formed is dissolved by stirring the contents of the flask. Then an appropriately substituted benzyl chloride (0.1 mol) is added stepwise and the reaction mixture is refluxed for 2-3 hours. The cooled contents are poured onto crushed ice and neutralized with dilute hydrochloric acid (200 ml). The resulting corresponding benzylthioacetic acid (0.1 mol) is subjected to oxidation with 30% hydrogen peroxide (0.12 mol) in glacial acetic acid (125 ml) by refluxing for 1 hour. The contents are cooled and poured onto crushed ice. The separated solid is recrystalized from hot water to give the corresponding pure benzylsulfonylacetic acid.
Part B. A mixture of the benzylsulfonyl acetic acid (10 mmol), an appropriately substituted aromatic aldehyde (10 mmol), and benzylamine (0.2 ml) in glacial acetic acid (12 ml) is refluxed for 2-3 hours. The contents are cooled and treated with cold ether (50 ml). Any product precipitated out is separated by filtration. The filtrate is diluted with more ether and washed successively with a saturated solution of sodium bicarbonate (20 ml), sodium bisulfite (20 ml), dilute hydrochloric acid (20 ml) and finally with water (35 ml). Evaporation of the dried ethereal layer yields styryl benzylsulfones as a solid material.
According to an alternative to Part A, the appropriate benzylsulfonylacetic acids may be generated by substituting a thioglycollate
HSCH2COOR for thioglycollic acid, where R is an alkyl group, typically C1-C6 alkyl. This leads to the formation of the alkylbenzylthioacetate intermediate (F),
![]()
which is then converted to the corresponding benzyl thioacetic acid B by alkaline or acid hydrolysis.
(E)-styryl phenyl sulfones (formula I: n=zero; Qls Q2 = substituted or unsubstituted phenyl) are prepared according to the method of General Procedure 1, replacing the benzylsulfonyl acetic acid in Part B with the appropriate substituted or unsubstituted phenylsulfonyl acetic acid.
(Z)-Styryl benzylsulfones are prepared by the nucleophilic addition of the appropriate thiols to substituted phenylacetylene with subsequent oxidation of the resulting sulfide by hydrogen peroxide to yield the (Z)-styryl benzylsulfone. The procedure is generally described by Reddy et al., Sulfur Letters 13:83-90 (1991), the entire disclosure of which is incorporated herein as a reference.
In the first step of the (Z)-styryl benzylsulfones synthesis, the sodium salt of benzyl mercaptan or the appropriate substituted benzyl mercaptan is allowed to react with phenylacetylene or the appropriate substituted phenylacetylene forming the pure (Z)-isomer of the corresponding styryl benzylsulfide in good yield.
In the second step of the synthesis, the (Z)-styryl benzylsulfide intermediate is oxidized to the corresponding sulfone in the pure (Z)-isomeric form by treatment with hydrogen peroxide.
The following is a more detailed two-part synthesis procedure for preparing (Z)-styryl benzylsulfones:
Procedure 2: Synthesis of (Z)-Styryl Benzylsulfones
Part A. To a refluxing methanolic solution of substituted or unsubstituted sodium benzylthiolate prepared from 460 mg (0.02g atom) of (i) sodium, (ii) substituted or unsubstituted benzyl mercaptan (0.02 mol) and (iii) 80 ml of absolute methanol, is added freshly distilled substituted or unsubstituted phenylacetylene. The mixture is refluxed for 20 hours, cooled and then poured on crushed ice. The crude product is filtered, dried and recrystalized from methanol or aqueous methanol to yield a pure (Z)- styryl benzylsulfide.
Part B. An ice cold solution of the (Z)- styryl benzylsulfide (3.0g) in 30 ml of glacial acetic acid is treated with 7.5 ml of 30% hydrogen peroxide. The reaction mixture is refluxed for 1 hour and then poured on crushed ice. The separated solid is filtered, dried, and recrystalized from 2-propanol to yield the pure (Z)-styryl benzylsulfone. The purity of the compounds is ascertained by thin layer chromatography and geometrical configuration is assigned by analysis of infrared and nuclear magnetic resonance spectral data.
The bis(styryl) sulfones of formula IN are prepared according to Procedure 3:
Procedure 3
Synthesis of (E)(E)- and (E)(Z)-bis(Styryl) Sulfones
To freshly distilled phenyl acetylene (51.07 g, 0.5 mol) is added sodium thioglycollate prepared from thioglycollic acid (46 g, 0.5 mol) and sodium hydroxide (40 g, 1 mol) in methanol (250 ml). The mixture is refluxed for 24 hours and poured onto crushed ice (500 ml) after cooling. The styrylthioacetic acid, formed after neutralization with dilute hydrochloric acid (250 ml), is filtered and dried; yield 88 g (90%); m.p. 84-86°C.
The styrylthioacetic acid is then oxidized to styrylsulfonylacetic acid as follows. A mixture of styrylthioacetic acid (5 g, 25 mmol) in glacial acetic acid (35 ml) and 30% hydrogen peroxide (15 ml) is heated under reflux for 60 minutes and the mixture is poured onto crushed ice (200 ml) after cooling. The compound separated is filtered and recrystalized from hot water to give white crystalline flakes of (Z)-styrylsulfonylacetic acid; yield 2.4 g (41%); m.p. 150-51°C.
A solution of (Z)-styrylsulfonylacetic acid (2.263 g, 10 m mol) in glacial acetic acid (6 ml) is mixed with an aromatic aldehyde (10 mmol) and benzylamine (0.2 ml) and refluxed for 3 hours. The reaction mixture is cooled, treated with dry ether (50 ml), and any product separated is collected by filtration. The filtrate is diluted with more ether and washed successively with a saturated solution of sodium hydrogen carbonate (15 ml), sodium bisulfite (15 ml), dilute hydrochloric acid (20 ml) and finally with water (30 ml). Evaporation of the dried ethereal layer yields (E)(Z)-bis(styryl)sulfones.
(E),(E)-bis(styryl)sulfones are prepared following the same procedure as described above with exception that sulfonyldiacetic acid is used in place of (Z)-styrylsulfonylacetic acid, and twice the amount of aromatic aldehyde (20 mmol) is used.
The styryl sulfones of formula N, which are systematically identified as 2-(phenylsulfonyl)-l-phenyl-3-phenyl-2-propen-l-ones, may be prepared according to either Method A or Method B of Procedure 4:
Procedure 4
Synthesis of 2-(Phenylsulfonyl)-l-phenyl-3-phenyl-2-propen-l-ones
These compounds are synthesized by two methods which employ different reaction conditions, solvents and catalysts.
Method A: Phenacyl aryl sulfones are made by refluxing α-bromoacetophenones (0.05 mol) and sodium arylsulfinates (0.05 mol) in absolute ethanol (200 ml) for 6-8 hours. The product which separates on cooling is filtered and washed several times with water to remove sodium bromide. The product is then recrystalized from ethanol: phenacyl-phenyl sulfone, m.p. 90-91°C; phenacyl-p-fluorophenyl sulfone, m.p. 148-149°C; phenacyl-p-bromophenyl sulfone, m.p. 121-122°C; phenacyl-p-methoxyphenyl sulfone, m.p. 104-105°C; p-nitrophenacyl-phenyl sulfone, m.p. 136-137°C.
A solution of phenacyl aryl sulfone (0.01 mol) in acetic acid (10 ml) is mixed with an araldehyde (0.01 mol) and benzylamine (0.02 ml) and refluxed for 3 hours. The solution is cooled and dry ether (50 ml) is added. The ethereal solution is washed successively with dilute hydrochloric acid, aqueous 10% NaOH, saturated NaHSO3 solution and water. Evaporation of the dried ethereal layer gives a solid product which is purified by recrystallization.
Method B: Dry tetrahydrofuran (200 ml) is taken in a 500 ml conical flask flushed with nitrogen. To this, a solution of titanium (IN) chloride (11 ml, 0.01 mol) in absolute carbon tetrachloride is added dropwise with continuous stirring. The contents of the flask are maintained at -20°C throughout the course of the addition. A mixture of phenacyl aryl sulfone (0.01 mol) and aromatic aldehyde (0.01 mol) is added to the reaction mixture and pyridine (4 ml, 0.04 mol) in tetrahydrofuran (8 ml) is added slowly over a period of 1 hour. The contents are stirred for 10-12 hours, treated with water (50 ml) and then ether (50 ml) is added. The ethereal layer is separated and washed with 15 ml of saturated solutions of 10% sodium hydroxide, sodium bisulfite and brine. The evaporation of the dried ethereal layer yields 2-(phenylsulfonyl)-l-phenyl-3-phenyl-2 propen-l-ones.
PATENT
https://www.google.com/patents/CN104817488A?cl=en
The structure of this medicine formula (I) shown below,

Wherein, R1 is absent or is halogen, C1-3 alkyl, alkoxy and -CF3; R2 is absent or is halogen, C1-3 alkyl, alkoxy and -cf3; structural formula (I) The method for the preparation of compounds as follows:

| WO2007016201A2 | Jul 28, 2006 | Feb 8, 2007 | Onconova Therapeutics, Inc. | FORMULATION OF RADIOPROTECTIVE α, β UNSATURATED ARYL SULFONES |
| WO2008105808A2 | Jul 27, 2007 | Sep 4, 2008 | Onconova Therapeutics, Inc. | FORMULATIONS OF RADIOPROTECTIVE α, β UNSATURATED ARYL SULFONES |
| US6656973 | Nov 27, 2002 | Dec 2, 2003 | Temple University – Of The Commonwealth System Of Higher Education | (E)-4-carboxystyrl-4-chlorobenzyl sulfone and pharmaceutical compositions thereof |
| US6667346 | Feb 28, 2002 | Dec 23, 2003 | Temple University – Of The Commonwealth System Of Higher Education | Method for protecting cells and tissues from ionizing radiation toxicity with α, β unsaturated aryl sulfones |
| US6982282 * | May 17, 2002 | Jan 3, 2006 | Sonus Pharmaceuticals, Inc. | Emulsion vehicle for poorly soluble drugs |
| US20090247624 | Jul 27, 2007 | Oct 1, 2009 | Onconova Therapeutics Inc. | Formulations of radioprotective alpha beta unsaturated aryl sulfones |
References
- “Onconova Therapeutics presents new data demonstrating radioprotection by Ex-RAD at RRS annual meeting” (Press release). EurekAlert. 2010-09-27. Archived from the originalon 2011-03-22. Retrieved 2011-03-22.
- Hipp, Van (2011-03-16). “Ex-Rad, the U.S. Military’s Radiation Wonder Drug”. FoxNews.com (FOX News Network). Archived from the original on 2011-03-26. Retrieved 2011-03-26.
- Ghosh, Sanchita P.; Perkins, Michael W.; Hieber, Kevin; Kulkarni, Shilpa; Kao, Tzu-Cheg; Reddy, E. Premkumar; Reddy, M. V Ramana; Maniar, Manoj; Seed, Thomas; Kumar, K. Sree (2009). “Radiation Protection by a New Chemical Entity, Ex-Rad™: Efficacy and Mechanisms”. Radiation Research 171 (2): 173–9. doi:10.1667/RR1367.1. PMID 19267542.
- “Ex-RAD® for Protection from Radiation Injury”. Onconova Therapeutics. 2009. Archived from the original on 2011-03-22. Retrieved 2011-03-22.
- http://cen.acs.org/articles/90/i26/Drugs-Never-Used.html[full citation needed]
- Kouvaris, J. R.; Kouloulias, V. E.; Vlahos, L. J. (2007). “Amifostine: The First Selective-Target and Broad-Spectrum Radioprotector”. The Oncologist 12 (6): 738–47.doi:10.1634/theoncologist.12-6-738. PMID 17602063.
- http://www.news-medical.net/news/20110323/Cellerant-commences-CLT-008-Phase-III-trial-in-patients-with-leukemia.aspx
- Reliene, Ramune; Pollard, Julianne M.; Sobol, Zhanna; Trouiller, Benedicte; Gatti, Richard A.; Schiestl, Robert H. (2009). “N-acetyl cysteine protects against ionizing radiation-induced DNA damage but not against cell killing in yeast and mammals”. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 665: 37. doi:10.1016/j.mrfmmm.2009.02.016.
- Mansour, Heba H.; Hafez, Hafez F.; Fahmy, Nadia M.; Hanafi, Nemat (2008). “Protective effect of N-acetylcysteine against radiation induced DNA damage and hepatic toxicity in rats”.Biochemical Pharmacology 75 (3): 773–80. doi:10.1016/j.bcp.2007.09.018. PMID 18028880.
- Demirel, C; Kilçiksiz, S; Ay, OI; Gürgül, S; Ay, ME; Erdal, N (2009). “Effect of N-acetylcysteine on radiation-induced genotoxicity and cytotoxicity in rat bone marrow”. Journal of radiation research 50 (1): 43–50. doi:10.1269/jrr.08066. PMID 19218780.
- Demirel, C; Kilciksiz, S; Evirgen-Ayhan, S; Gurgul, S; Erdal, N (2010). “The preventive effect of N-acetylcysteine on radiation-induced dermatitis in a rat model”. Journal of the Balkan Union of Oncology 15 (3): 577–82. PMID 20941831.
- Geiger, Hartmut; Pawar, Snehalata A; Kerschen, Edward J; Nattamai, Kalpana J; Hernandez, Irene; Liang, Hai Po H; Fernández, Jose Á; Cancelas, Jose A; Ryan, Marnie A; Kustikova, Olga; Schambach, Axel; Fu, Qiang; Wang, Junru; Fink, Louis M; Petersen, Karl-Uwe; Zhou, Daohong; Griffin, John H; Baum, Christopher; Weiler, Hartmut; Hauer-Jensen, Martin (2012).“Pharmacological targeting of the thrombomodulin–activated protein C pathway mitigates radiation toxicity”. Nature Medicine 18 (7): 1123–9. doi:10.1038/nm.2813. PMC 3491776.PMID 22729286.
External links
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015265549 | 2015-09-24 | STABLE AQUEOUS FORMULATION OF (E)-4-CARBOXYSTYRYL-4-CHLOROBENZYL SULFONE |
| US2015238448 | 2015-08-27 | FORMULATION OF RADIOPROTECTIVE ALPHA, BETA UNSATURATED ARYL SULFONES |
| US2013012588 | 2013-01-10 | COMPOSITIONS AND METHODS FOR PREVENTION AND TREATEMENT OF WOUNDS |
| US2013012589 | 2013-01-10 | STABLE AQUEOUS FORMULATION OF (E)-4-CARBOXYSTYRYL-4-CHLOROBENZYL SULFONE |
| US2011250184 | 2011-10-13 | METHODS FOR DETERMINING EFFICACY OF A THERAPEUTIC REGIMEN AGAINST DELETERIOUS EFFECTS OF CYTOTOXIC AGENTS IN HUMAN |
| US2011028504 | 2011-02-03 | Formulation of radioprotective alpha beta unsaturated aryl sulfones |
| US2009247624 | 2009-10-01 | FORMULATIONS OF RADIOPROTECTIVE ALPHA BETA UNSATURATED ARYL SULFONES |
 |
|
| Identifiers | |
|---|---|
| 922139-31-9 |
|
| PubChem | 23668369 |
| Properties | |
| C16H12ClNaO4S | |
| Molar mass | 358.77 g·mol−1 |
//////////Onc-01210, ON-01210.Na, 334969-03-8, 922139-31-9, Recilisib Sodium, Phase I , A protein kinase inhibitor, treatment of acute radiation syndrome, Orphan Drug Status, Ex-RAD
C1=CC(=CC=C1CS(=O)(=O)C=CC2=CC=C(C=C2)C(=O)[O-])Cl.[Na+]
