
Home » PHASE 1 (Page 2)
Category Archives: PHASE 1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MEVOCICLIB, SY 1365
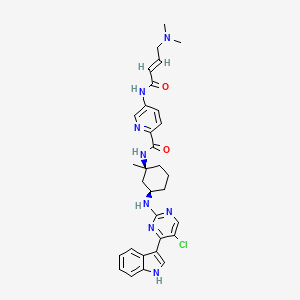
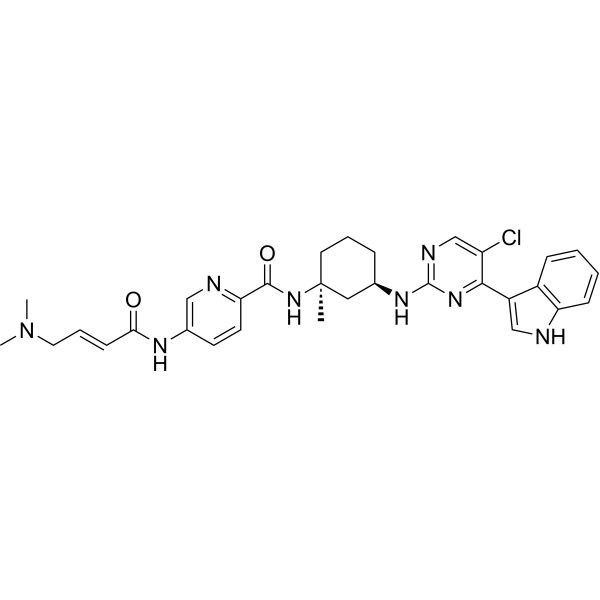
MEVOCICLIB,
CAS 1816989-16-8
SY 1365
N-[(1S,3R)-3-[[5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl]amino]-1-methylcyclohexyl]-5-[[(E)-4-(dimethylamino)but-2-enoyl]amino]pyridine-2-carboxamide
N-((lS,3R)-3-(5-chloro-4-(lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcvclohexyl)-5-((E)-4-(dimethylamino)but-2-enamido)picolinamide
HS Tariff Code: 2934.99.9001
Syros
| Molecular Weight | 587.12 |
|---|---|
| Formula | C₃₁H₃₅ClN₈O₂ |
- OriginatorSyros Pharmaceuticals
- ClassAmides; Amines; Antineoplastics; Chlorinated hydrocarbons; Cyclohexanes; Indoles; Pyridines; Pyrimidines; Small molecules
- Mechanism of ActionCyclin-dependent kinase-activating kinase inhibitors
- DiscontinuedAcute myeloid leukaemia; Breast cancer; Haematological malignancies; Ovarian cancer; Solid tumours
- 23 Oct 2019Discontinued – Preclinical for Haematological malignancies and Acute myeloid leukaemia in the USA (Parenteral); Phase-I for Solid tumours, Ovarian cancer and Breast cancer in the USA (IV) because data obtained did not support an optimal profile for patients and indicated higher or frequent dosing
- 07 Dec 2018Pharmacodynamics data from preclinical trials in Breast cancer presented at the 41st Annual San Antonio Breast Cancer Symposium (SABCS-2018)
- 15 Nov 2018Adverse events, efficacy and pharmacokinetics data from a phase I trial in Solid tumours presented at the 30th EORTC-NCI-AACR Molecular Targets and Cancer Therapeutics Symposium (EORTC-NCI-AACR-2018)
| Clinical Trial | NCT NumberSponsorConditionStart DatePhaseNCT03134638Syros PharmaceuticalsAdvanced Solid Tumors|Ovarian Cancer|Breast CancerMay 12, 2017Phase 1 |
|---|
Mevociclib (SY-1365) is a potent and first-in-class selective CDK7 inhibitor, with a Ki of 17.4 nM. Mevociclib exhibits anti-proliferative and apoptotic effects in solid tumor cell lines. Mevociclib possesses anti-tumor activity in hematological and multiple aggressive solid tumors.
Mevociclib, also known as SY-1365, is a CDK7 inhibitor. In vitro, SY-1365 inhibited cell growth of many different cancer types at nanomolar concentrations. SY-1365 treatment decreased MCL1 protein levels, and cancer cells with low BCL-XL expression were found to be more sensitive to SY-1365. Transcriptional changes in acute myeloid leukemia (AML) cell lines were distinct from those following treatment with other transcriptional inhibitors. SY-1365 demonstrated substantial anti-tumor effects in multiple AML xenograft models as a single agent; SY-1365-induced growth inhibition was enhanced in combination with the BCL2 inhibitor venetoclax.
Syn
WO2015154038
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015154038
Example 16. Synthesis of N-((lS,3R)-3-(5-chloro-4-(lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcvclohexyl)-5-((E)-4-(dimethylamino)but-2-enamido)picolinamide (Compound 267).
[251] (+/-) Benzyl tert-butyl ((lS,3R)-l-methylcvclohexane-l,3-diyl)dicarbamate
(+/-)
[252] A solution of (+/-)-(lS,3R) -3-((ieit-¾itoxycarbonyl)amino)–l -raethylcyclohexanecarboxylic acid prepared as in WO2010/148197 (4.00 g, 15.5 mmol) in toluene (Ϊ 55 mL) was treated with Et3N (2.4 mL, 17.1 mmol) and DPPA (3.68 mL, Ϊ7.1 mmol) and heated at reflux for lh. Benzyl alcohol (8.0 mL, 77.7 mmol) and Et3N (4.4 mL , 31 .4 mmol) were added to the reaction mixture and the solution was heated at 100 °C for 72h. The mixture was cooled to room temperature and then diluted with EtOAc (300 mL) and H20 (300 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 x 200 mL). The combined organics layers were washed with brine (100 mL), filtered and evaporated to dryness. The residue was purified by Si02 chromatography (EtOAc in hexanes, 0 to 50% gradient) and afforded the title compound (3.40 g, 9.38 mmol, 60%) as a white solid.
[253] Benzyl tert-butyl ((lS,3R)-l-methylcvclohexane-l,3-diyl)dicarbamate and benzyl tert- -l-methylcvclohexane-l,3-diyl)dicarbamate
(+/-)
[254] Both enantiomers of (+/-)-Benzyl tert-butyl ((lS,3R)-l-methylcyclohexane-l,3-diyl)dicarbamate (3.40 g, 9.38 mmol) were separated using preparative chiral HPLC (Chiralpak IA, 5 urn, 20×250 mm; hex/MeOH/DCM = 90/5/5) to yield both compounds benzyl tert-butyl ((lS,3R)-l-methylcyclohexane-l,3-diyl)dicarbamate (1.20 g, 3.31 mmol) and benzyl iert-butyl ((lR,3S)-l-methylcyclohexane-l,3-diyl)dicarbamate (1.15 g, 3.17 mmol) as white solids.
255 Benzyl ((lS,3R)-3-amino-l-methylcvclohexyl)carbamate hydrochloride
[256] A solution of benzyl tert-butyl ((lS,3R)-l-methylcyclohexane-l,3-diyl)dicarbamate (700 mg, 1.93 mmol) in DCM (19 mL) was treated with a 4M solution of HCI in dioxane (9.66 mL, 38.6 mmol) and stirred 16h at rt. The mixture was evaporated to dryness and afforded the title compound (577 mg, 1.93 mmol, 100%) as a white solid which was used in the next step without further purification.
[257] (lS,3R)-Benzyl-3-(5-chloro-4-(l-(phenylsulfonyl)-lH ndol-3-yl)pyrimidin-2-ylamino)-1-methylcyclohexylcarbamate
[258] A solution of 3-(2,5-dichloropyrimidin-4-yl)-l-(phenylsulfonyl)-lH-indole (1.02 g, 2.53 mmol), benzyl (( iS,3 )-3- amino- 1 -methylcyclohexyljcarbaniaie hydrochloride (577 mg, 1.93 mmol) and DIPEA (1.15 mL, 6.60 mmol) in NMP (11 mL) was heated at 135 °C (microwave) for 60 min. The cooled mixture was diluted with EtOAc (250 mL), washed with H20 (100 mL), brine (100 mL), dried over MgS04, filtered and evaporated to dryness. The residue was purified by Si02 chromatography (EtOAc in DCM, 0 to 50% gradient) and afforded the title compound (747 mg, 1.19 mmol, 54%) as a yellow foam.
[259] (lS,3R)-N-(5-chloro-4-(l-(phenylsulfonyl)-lH ndol-3-yl)pyrimidin-2-yl)-3-methylcvclohexane-l,3-diamine
[260] A cooled (-78°C) solution of (lS,3R)-benzyl-3-(5-chloro-4-(l-(phenylsulfonyl)-lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcyclohexylcarbamate (747 mg, 1.19 mmol) in DCM (39 mL) was treated with a 1M solution of BBr3 in DCM (2.83 mL, 2.83 mmol) and was slowly warmed up to rt. MeOH (10 mL) was added to the mixture was the resulting solution was stirred lh at rt. The resulting mixture was evaporated to dryness. The residue was purified by reverse phase chromatography (C18, H20/ACN +0.1% HC02H, 0 to 60% gradient) and afforded the title compound (485 mg, 0.978 mmol, 83%) as a yellow solid.
[261] 5-amino-N-( ( lS,3R)-3-( 5-chloro-4-(l-(phenylsulfonyl)-lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcvclohexyl)picolinamide
[262] A solution of (lR,3S)-N-(5-chloro-4-(l-(phenylsulfonyl)-lH-indol-3-yl)pyrimidin-2-yl)-3-methylcyclohexane-l,3-diamine (75.0 mg, 0.150 mmol) and 5-aminopicolinic acid (25.0 mg, 0.180 mmol) in DMF (5.0 mL) was treated with HBTU (86.0 mg, 0.230 mmol) and DIPEA (79 μί, 0.45 mmol). The resulting mixture was stirred 5h at rt and diluted with MeTHF (50 mL) and saturated NaHC03 (50 mL). The layers were separated and the aqueous layer was extracted with MeTHF (2 x 50 mL). The combined organic layers were dried over MgS04, filtered and evaporated to dryness. The residue was purified by Si02 chromatography (EtOAc in DCM, 0 to 50% gradient) and afforded the title compound (74.0 mg, 0.120 mmol, 79%) as a light yellow oil.
[263] 5-amino-N-((lS,3R)-3-(5-chloro-4-(lH ndol-3-yl)pyrimidin-2-ylamino)-l-methylcyclohexyDpicolinamide
[264] A solution of 5-amino-N-((lS,3R)-3-(5-chloro-4-(l-(phenylsulfonyl)-lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcyclohexyl)picolinamide (74.0 mg, 0.120 mmol) in 1,4-dioxane (4.0 mL) was treated with a 2M solution of NaOH in H20 (960 μί, 4.78 mmol) and heated at 60°C for lh. The cooled mixture was diluted with MeTHF (30 mL) and H20 (30 mL). The layers were separated and the aqueous layer was extracted with MeTHF (3 x 30 mL). The combined organic layers were dried over MgS04, filtered and evaporated to dryness affording the title compound (57.0 mg, 0.120 mmol, 100%) as a light yellow oil which was used in the next step without further purification.
[265] N-((lS,3R)-3-(5-chloro-4-(lH ndol-3-yl)pyrimidin-2-ylamino)-l-methylcvd^
( ( E)-4-(dimethylamino)but-2-enamido )picolinamide ( Compound 267)
[266] A cooled (-78°C) solution of 5-amino-N-((lS,3R)-3-(5-chloro-4-(lH-indol-3-yl)pyrimidin-2-ylamino)-l-methylcyclohexyl)picolinamide (57.0 mg, 0.120 mmol) and DIPEA (104 0.598 mmol) in THF/NMP (4.0 mL/1.0 mL) was treated with a 54.2 mg/mL solution of (E)-4-bromobut-2-enoyl chloride in DCM (104 μί, 0.598 mmol). The resulting mixture was stirred 4h at -78°C before addition of a 2M solution of dimethylamine in THF (359 μί, 0.717 mmol). The resulting mixture was warmed up to rt and stirred 45min at this temperature before being evaporated to dryness. The residue was purified by reverse phase chromatography (C18, H20/ACN +0.1% HC02H, 0 to 50% gradient) and afforded the title compound (15.0 mg, 0.026 mmol, 22%) as a white solid after lyophilization. LCMS: Calculated: 587.12; Found (M+H+): 587.39. 1H NMR (500 MHz, DMSO) δ 11.84 (s, 1H), 10.54 (s, 1H), 8.82 (d, J = 2.3 Hz, 1H), 8.64 (s, 1H), 8.47 (s, 1H), 8.25 (dd, J = 8.6, 2.4 Hz, 2H), 7.98 (d, J = 8.9 Hz, 2H), 7.50 (d, J = 7.7 Hz, 1H), 7.25 – 7.07 (m, 3H), 6.81 (dt, J = 15.5, 5.8 Hz, 1H), 6.29 (d, J = 15.4 Hz, 1H), 4.23 – 4.08 (m, 1H), 3.08 (dd, J = 5.7, 1.1 Hz, 2H), 2.46 – 2.37 (m, 1H), 2.18 (s, 6H), 2.04 – 1.95 (m, 2H), 1.87 – 1.70 (m, 3H), 1.63 – 1.46 (m, 4H), 1.39 – 1.26 (m, 1H).
Ref
- [1]. Hu S, et al. Discovery and characterization of SY-1365, a selective, covalent inhibitor of CDK7. Cancer Res. 2019 May 7.[2]. Shanhu Hu, et al. SY-1365, a potent and selective CDK7 inhibitor, exhibits promising anti-tumor activity in multiple preclinical models of aggressive solid tumors.
///////////////
CN(C)C\C=C\C(=O)Nc1ccc(nc1)C(=O)N[C@]1(C)C[C@@H](CCC1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21


NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
SY 5609
[ Fig. 0001] [ Fig. 0002]
[ Fig. 0003]
[ Fig. 0004]
SY 5609
CAS 2519828-12-5
Cancer, solid tumor
PHASE 1
A highly selective and potent oral inhibitor of cyclin-dependent kinase 7 (CDK7) for potential treatment of advanced solid tumors that harbor the Rb pa thway alterations (Syros Pharmaceuticals, Inc., Cambridge, Massachusetts, USA)
SY-5609 is an oral non-covalent CDK7 inhibitor in early clinical development at Syros Pharmaceuticals for the treatment of patients with advanced breast, colorectal, lung or ovarian cancer, or with solid tumors of any histology that harbor Rb pathway alterations.
- OriginatorSyros Pharmaceuticals
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase-activating kinase inhibitors
- Phase IBreast cancer; Solid tumours
- 05 Aug 2021Roche plans the phase I/Ib INTRINSIC trial in Colorectal cancer (Combination therapy, Metastatic disease) in USA, Canada, Italy, South Korea, Spain and United Kingdom (NCT04929223)
- 05 Aug 2021Roche and Syros Pharmaceuticals enters into a clinical trial collaboration to evaluate atezolizumab in combination with SY 5609 in a clinical trial
- 05 Aug 2021Syros Pharmaceuticals plans a phase I trial in Cancer in second half of 2021
- NCT04247126
- https://clinicaltrials.gov/ct2/show/NCT04247126
At #ESMO21, we will be presenting new preclinical and clinical data on SY-5609, our highly selective and potent oral CDK7 inhibitor. #oncology #biotech Learn more: https://lnkd.in/gqYmWYhb
A Promising Approach for Difficult-to-Treat Cancers
SY-5609 is a highly selective and potent oral inhibitor of the cyclin-dependent kinase 7 (CDK7) in a Phase 1 dose-escalation trial in patients with advanced breast, colorectal, lung, ovarian or pancreatic cancer, or with solid tumors of any histology that harbor Rb pathway alterations.
SY-5609 represents a new approach to treating cancer that we believe has potential in a range of difficult-to-treat cancers. It has shown robust anti-tumor activity, including complete regressions, in preclinical models of breast, colorectal, lung and ovarian cancers at doses below the maximum tolerated dose. In preclinical studies of breast, lung and ovarian cancers, deeper and more sustained responses were associated with the presence of Rb pathway alterations. SY-5609 has also shown substantial anti-tumor activity in combination with fulvestrant in treatment-resistant models of estrogen receptor-positive breast cancer, including those resistant to both fulvestrant and a CDK4/6 inhibitor. Early dose-escalation data demonstrated proof-of-mechanism at tolerable doses.
Syros to Present New Data from Phase 1 Clinical Trial of SY-5609 in Oral Presentation at ESMO Congress 2021SEPTEMBER 13, 2021
Management to Host Conference Call on Monday, September 20, 2021 at 4:00 p.m. ET
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Syros Pharmaceuticals (NASDAQ:SYRS), a leader in the development of medicines that control the expression of genes, today announced that it will present new data from the dose-escalation portion of the Phase 1 clinical trial of SY-5609, its highly selective and potent oral cyclin-dependent kinase 7 (CDK7) inhibitor, at the ESMO Congress 2021, taking place virtually September 16-21, 2021. The oral presentation will include safety, tolerability, and initial clinical activity data for SY-5609 in patients with breast, colorectal, lung, ovarian and pancreatic cancers, as well as in patients with solid tumors of any histology harboring Rb pathway alterations.
In separate poster presentations, Syros will present new preclinical data evaluating the antitumor and pharmacodynamic activity of intermittent dosing regimens for SY-5609 in ovarian cancer models, as well as new preclinical data evaluating antitumor activity of SY-5609 as a single agent and in combination with chemotherapy in KRAS-mutant models.
The abstracts for the two poster presentations are now available online on the ESMO conference website at: https://www.esmo.org/meetings/esmo-congress-2021/abstracts, and the presentations will become available for on-demand viewing starting September 16 at 08:30 CEST (September 16 at 2:30 a.m. ET). The abstract for the oral presentation on the Phase 1 dose-escalation data will remain embargoed until September 17 at 00:05 CEST (September 16 at 6:05 p.m. ET).
Details of the oral presentation are as follows:
Presentation Title: Tolerability and Preliminary Clinical Activity of SY-5609, a Highly Potent and Selective Oral CDK7 Inhibitor, in Patients with Advanced Solid Tumors
Session Date & Time: Monday, September 20, 17:30-18:30 CEST (11:30-12:30 p.m. ET)
Presentation Time: 17:55-18:00 CEST (11:55-12:00 p.m. ET)
Session Title: Mini Oral Session: Developmental Therapeutics
Presenter: Manish Sharma, M.D., START Midwest
Abstract Number: 518MO
Details of the poster presentations are as follows:
Presentation Title: Preclinical Evaluation of Intermittent Dosing Regimens on Antitumor and PD Activity of SY-5609, a Potent and Selective Oral CDK7 Inhibitor, in Ovarian Cancer Xenografts
Abstract Number: 14P
Presentation Title: SY-5609, a Highly Potent and Selective Oral CDK7 inhibitor, Exhibits Robust Antitumor Activity in Preclinical Models of KRAS Mutant Cancers as a Single Agent and in Combination with Chemotherapy
Abstract Number: 13P
Conference Call Information
Syros will host a conference call on Monday, September 20, 2021 at 4:00 p.m. ET to discuss the new clinical and preclinical data for SY-5609, which will be presented at the ESMO Congress 2021.
To access the live conference call, please dial 866-595-4538 (domestic) or 636-812-6496 (international) and refer to conference ID 4648345. A webcast of the call will also be available on the Investors & Media section of the Syros website at www.syros.com. An archived replay of the webcast will be available for approximately 30 days following the conference call.
About Syros Pharmaceuticals
Syros is redefining the power of small molecules to control the expression of genes. Based on its unique ability to elucidate regulatory regions of the genome, Syros aims to develop medicines that provide a profound benefit for patients with diseases that have eluded other genomics-based approaches. Syros is advancing a robust clinical-stage pipeline, including: tamibarotene, a first-in-class oral selective RARα agonist in RARA-positive patients with higher-risk myelodysplastic syndrome and acute myeloid leukemia; SY-2101, a novel oral form of arsenic trioxide in patients with acute promyelocytic leukemia; and SY-5609, a highly selective and potent oral CDK7 inhibitor in patients with select solid tumors. Syros also has multiple preclinical and discovery programs in oncology and monogenic diseases.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210913005090/en/
PATENT
CN(C)C\C=C\C(=O)Nc1ccc(cc1)C(=O)Nc1cccc(c1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21
THZ1; 1604810-83-4; THZ-1; HY-80013
CLIP
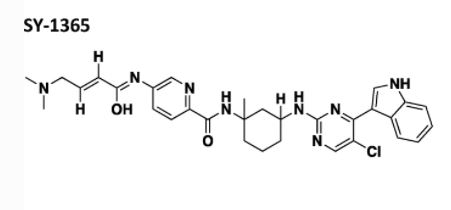
SY 1365 MEVOCICLIB, CAS 1816989-16-8
CN(C)C\C=C\C(=O)Nc1ccc(nc1)C(=O)N[C@]1(C)C[C@@H](CCC1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21
PATENT
PATENT
3-fluoro-4-(methylamino)-N-[(1S,3R)-1-methyl-3-[[4-(7-methyl-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]cyclohexyl]benzamide (Compound 130)
3-chloro-4-[[4-(dimethylamino)-3-hydroxy-butanoyl]amino]-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 129)
4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)-1-methylcyclohexyl)benzamide (Compound 128)
4-amino-3-fluoro-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 127)
4-amino-N-((1S,3R)-3-((5-chloro-4-(2-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 126)
4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 124)
Example 25 Synthesis of N1-(4-(((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)carbamoyl)phenyl)oxalamide (Compound 113)
Example 24 Synthesis of N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)-4-(4-(dimethylamino)butanamido)benzamide (Compound 105)
PATENT
4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)tricyclo[3.3.1.13,7]decanyl)benzamide (Compound 100).
+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-5 hydroxycyclohexyl)benzamide (Compound 101)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 102)
(1S,3R)-N-(4-aminophenyl)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexanecarboxamide (Compound 106)
4-amino-N-((1S,3R)-3-(5-cyclopropyl-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 103)
4-amino-N-((1S,3R)-3-(5-chloro-4-(pyridin-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 108)
4-amino-N-((1S,3R)-3-(5-cyano-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 107)
(+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-5-fluorocyclohexyl)benzamide (Compound 110)
4-amino-N-(5-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)bicyclo[3.1.1]heptan-1-yl)benzamide (Compound 104)
4-amino-N4(1R,5S)-5-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-3,3-difluorocyclohexyl)benzamide (Compound 115)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzenesulfonamide (Compound 109).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-2-fluorobenzamide (Compound 112)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-3-fluorobenzamide (Compound 111).
(+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-1-methylcyclohexyl)benzamide (Compound 116).
N-((1S,3R)-3-(4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-4-aminobenzamide (Compound 114).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-2-morpholinobenzamide(Compound 117).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyridin-2-ylamino)cyclohexyl)benzamide (Compound 118).
3-amino-N-(trans-4-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 119).
(1S,3R)-N1-(R)-1-(4-aminophenyl)-2,2,2-trifluoroethyl)-N3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)cyclohexane-1,3-diamine (Compound 120).
(1S,3R)-N1-(4-aminobenzyl)-N3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)-N1-methylcyclohexane-1,3-diamine.HCl (Compound 122).
4-amino-N-((1S,3R)-3-(5-chloro-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 123).
Synthesis of 5-amino-N-((1S,3R)-3-(5-chloro-4-(1-methyl-1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)picolinamide (Compound 125)
Synthesis of N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)-4-(4-(dimethylamino)butanamido)benzamide (Compound 105)
Synthesis of N1-(4-(((1S,3R)-3-)(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)carbamoyl)phenyl)oxalamide (Compound 113)
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 124)
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(2-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 126)
Synthesis of 4-amino-3-fluoro-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 127).
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl) pyrimidin-2-yl)amino)-1-methylcyclohexyl)benzamide (Compound 128)
Synthesis of 3-chloro-4-[[4-(dimethylamino)-3 hydroxy-butanoyl]amino]-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 129).
Synthesis of 3-fluoro-4-(methylamino)-N-[(1S,3R)-1-methyl-3-[[4-(7-methyl-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]cyclohexyl]benzamide (Compound 130)
//////////////SY 5609, 2519828-12-5, Cancer, solid tumor, PHASE 1, SYROS

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
MAX 40279
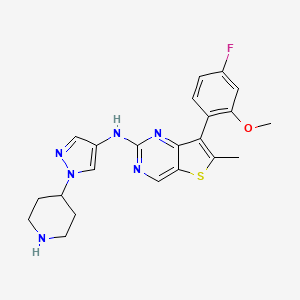
MAX 40279, EX-A4057
Max 4; MAX-40279; MAX-40279-001; MAX-40279-01
UNII-DL772G3NN7
2070931-57-4
C22H23FN6OS, 438.5
7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-ylpyrazol-4-yl)thieno[3,2-d]pyrimidin-2-amine
Thieno[3,2-d]pyrimidin-2-amine, 7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-[1-(4-piperidinyl)-1H-pyrazol-4-yl]-

7-(4-FLUORO-2-METHOXYPHENYL)-6-METHYL-N-(1-(PIPERIDIN-4-YL)-1H-PYRAZOL-4-YL) THIENO (3,2-D)PYRIMIDIN-2-AMINE SEMI-FUMARATE CAS 2388506-43-0
- 7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-[1-(4-piperidinyl)-1H-pyrazol-4-yl]thieno[3,2-d]pyrimidin-2-amine
- Originator Maxinovel Pharmaceuticals
- ClassAntineoplastics
- Mechanism of ActionFibroblast growth factor receptor antagonists; Fms-like tyrosine kinase 3 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia
- Phase IAcute myeloid leukaemia; Solid tumours
Most Recent Events
- 28 Nov 2019Phase-I clinical trials in Solid tumours (Late-stage disease, Metastatic disease) in China (PO) (NCT04183764)
- 16 Apr 2019Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in China (PO) (NCT04187495)
- 23 Jan 2019Guangzhou Maxinovel Pharmaceuticals plans a phase I trial in China (ChiCTR1900020971)
- MaxiNovel Pharmaceuticals, Inc. Announces FDA Orphan Drug Designation for MAX-40279 for the Treatment of Acute Myeloid Leukemia (AML)
March 29, 2018 11:24 AM Eastern Daylight Timehttps://www.businesswire.com/news/home/20180329005826/en/MaxiNovel-Pharmaceuticals-Inc.-Announces-FDA-Orphan-Drug-Designation-for-MAX-40279-for-the-Treatment-of-Acute-Myeloid-Leukemia-AML
GUANGZHOU, China–(BUSINESS WIRE)–MaxiNovel Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (“FDA”) has granted MaxiNovel Orphan Drug Designation for MAX-40279 in the treatment of Acute Myeloid Leukemia (AML).
AML is the most common acute leukemia which accounts for approximately 25% of all adult leukemias worldwide. Approximately one-third of AML patients have a FLT3 gene mutation. Such mutation can result in faster disease progression, higher relapse rates and lower rates of survival than other forms of AML. Inhibition of FLT3 mutation is of high importance in combating AML.
In the preclinical testing, MAX-40279 demonstrated potent inhibition of both FLT3 and FGFR with excellent drug concentration in the bone marrow. It is designed to overcome the observed drug resistance of the current FLT3 inhibitors due to the bone marrow FGF/FGFR pathway activation.
“We are very pleased to receive the ODD,” commented MaxiNovel’s Vice President Dr. Elizabeth Ashraf. “Our objective is to bring the best in class medicine to the patients worldwide.”
The FDA Office of Orphan Products Development grants orphan drug designation to novel drugs and biologics that are intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the United States. The designation allows manufacturers to qualify for various incentives including federal grants, tax credits for qualified clinical trials, a waiver of PDUFA filing fees and 7 years of market exclusivity upon regulatory approval.
About MaxiNovel Pharmaceuticals, Inc:
Maxinovel Pharmaceuticals, Inc. is a biotech company focusing on the discovery and development of Immuno-oncology therapy and targeted therapy. It will use its orally active Immuno-oncology product platform to bring effective combo product of multi-components in a single oral pill to the patients worldwide. For more info: www.maxinovel.com
The JAK-STAT (Janus kinase-signal transducer and activator of transcription) signal pathway is a signal transduction pathway stimulated by cytokines discovered in recent years, and it participates in many important biology such as cell proliferation, differentiation, apoptosis and immune regulation. Process (Aaronson, D Set al. Science 2002, 296, 1653-1655; O’Shea, J Jet al. Nat. Rev. Drug Discovery 2004, 3, 555-564). Compared with other signal pathways, the transmission process of this signal pathway is relatively simple. It mainly consists of three components, namely tyrosine kinase-related receptor, tyrosine kinase JAK and transcription factor STAT. JAK (Janus Kinase), a type of molecule in the cell, is rapidly recruited and activated on the receptor after receiving the signal from the upstream receptor molecule. The activated JAK catalyzes the receptor tyrosine phosphorylation, and the phosphorylation of tyrosine on the receptor molecule Amino acid is the recognition and binding site of a kind of signal molecule STAT SH2. Tyrosine phosphorylation occurs after STAT binds to the receptor. Tyrosine phosphorylated STAT forms a dimer and enters the nucleus. As an active transcription factor, dimeric STAT molecules directly affect the expression of related genes, thereby changing the proliferation or differentiation status of target cells.
The JAK-STAT pathway is widely present in various tissues and cells in the body, and has an important role in the differentiation, proliferation, and anti-infection of lymphocytes, and participates in the interaction of various inflammatory factors and signal transduction (Kiesseleva T. et al. . J. Gene, 2002, 285, 1-24). The abnormal activation of this pathway is closely related to a variety of diseases. Finding and screening JAK inhibitors can help in-depth study of the regulatory mechanism of JAK-STAT, thereby providing new drugs and methods for the prevention and treatment of related diseases
The occurrence, growth, invasion and metastasis of tumors are related to the JAK-STAT signal transduction pathway. In normal signal transduction, the activation of STATs is rapid and transient. The continuous activation of STATs is closely related to the process of malignant transformation of cells (Buettner R. et al. Clin. Cancer Res. 2002, 8(4), 945-954). STAT3 is the focus of multiple oncogenic tyrosine kinase signal channels such as EGFR, IL-6/JAK, Src, etc. It is activated in a variety of tumor cells and tissues, such as breast cancer, ovarian cancer, and head and neck squamous cells. Like cell carcinoma, prostate cancer, malignant melanoma, multiple myeloma, lymphoma, brain tumor, non-small cell lung cancer and various leukemias, etc. (Niu G. et al. Oncogene 2002, 21(13), 2000-2008 ). JAK-STAT pathway inhibitors belong to PTK inhibitors, and this enzyme is a member of the oncogene protein and proto-oncoprotein family, and plays an important role in the normal and abnormal cell proliferation. The occurrence and growth of tumors are inseparable from PTK. Therefore, JAK-STAT pathway inhibitors inhibit tumor growth by antagonizing PTK, and have obvious anti-tumor effects (Mora LBet al.J.Cancer Res.2002,62(22) , 6659-6666).
In addition, the latest research shows that: organ transplant rejection, psoriasis, tissue and organ fibrosis, bronchial asthma, ischemic cardiomyopathy, heart failure, myocardial infarction, blood system diseases, and immune system diseases are all related to JAK-STAT signaling. The pathway is closely related. This signaling pathway is not only important for maintaining the normal physiological functions of cells, but also has an important regulatory role for the occurrence and development of diseases.
The Fibroblast Growth Factor Receptor family belongs to a new type of receptor kinase family, which includes four receptor subtypes (FGFR-1,2,3) encoded by four closely related genes. And 4) and some heterogeneous molecules, which form a ternary complex with fibroblast growth factor (FGF) and heparan sulfate, and then trigger a series of signal transduction pathways to participate in the regulation of physiological processes in the organism. FGFR has a wide range of physiological and pathological effects in the body: (1) Embryo development. Studies have shown that during embryonic development, FGFR signal transduction is essential for most organ development and the formation of embryonic patterns. (2) Cell division, migration and differentiation. FGFR stimulates cell proliferation and participates in the regulation of cell transformation in the pathological process. There are many parallel pathways to achieve FGFR-mediated cell division signal transduction, which has been confirmed by many studies (JKWang et al., Oncogene 1997, 14, 1767 -1778.). (3) Bone diseases. The growth and differentiation of bones are also regulated by the FGF family, and mutations in FGFR can cause bone deformities (R. Shang et al., Cell 1994, 78, 335-342.). (4) The occurrence of tumors. FGFR can promote the migration, proliferation and differentiation of endothelial cells, and plays an important role in the regulation of angiogenesis and angiogenesis. Uncontrolled angiogenesis can lead to the occurrence of tumors and the growth of metastases (J.Folkman.Nat.Med.1995) ,1,27-31.).
FMS-like tyrosine kinase 3 (FMS-like tyrosine kinase 3, FLT3) belongs to the type III receptor tyrosine kinase (receptor tyrosine kinase III, RTK III) family member, it is composed of extracellular domain, intracellular domain and The transmembrane region is composed of 3 parts, which are first expressed in human hematopoietic stem cells. FLT3 interacts with its ligand FL to stimulate or act on stem cells, which is of great significance to the growth and differentiation of stem cells. FLT3 kinase has wild-type FLT3-WT and its main activation mutant FLT3-ITD and FLT3-D835Y. FLT3 is mainly expressed in the precursors of normal myeloid cells, but its abnormal expression is also found in a large part of acute myeloid leukemia (AML) cells.
In recent years, many large-scale studies have confirmed that activating mutations of FLT3 play a very important pathological role in the occurrence and progression of acute myeloid leukemia. FLT3 has become an important target for the treatment of acute myeloid leukemia.
rc family kinase (SFK) is a family of non-receptor tyrosine kinases, including c-Src, LYN, FYN, LCK, HCK, FGR, BLK, YES and YRK, among which LYN kinase has LYNα and LYNβ Both subtypes, LYN kinase and its two subtypes can cause similar intracellular tyrosine phosphorylation. According to the amino acid sequence, SFK can be divided into two sub-families: one family is c-Src, FYN, YES and FGR, which are widely expressed in different tissues; the other family is LCK, BLK, LYN and HCK, which are closely related to hematopoietic cells. SFK is connected to multiple signal transduction pathways in the body, and can be activated by growth factors, cytokines and immune cell receptors, G protein-coupled receptors, integrins and other cell adhesion molecules, and then activate the corresponding signal transduction pathways , Causing a variety of physiological effects of cells. The activity of SFK mainly includes the regulation of cell morphology, cell movement, cell proliferation and survival. The abnormal activation and expression of these kinases leads to the occurrence and development of a wide range of diseases, such as a large number of solid tumors, various hematological malignancies and some neuronal pathologies. Therefore, looking for SFK inhibitors is a promising research topic in the field of medicinal chemistry.

NEW DRUG APPROVALS
ONE TIME
$10.00
Patent
CN106366093A
PATENT
WO 2017012559
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017012559Example 31
N-[7-(4-Fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidin-4-yl)- 1H-pyrazole-4-amine (Compound 31)
Synthesis of compound 31-e
2,4-Dichloro-6-methylthiophene [3,2-d] pyrimidine (10g, 45.6mmol) was dissolved in tetrahydrofuran (100mL) and ethanol (100mL), and the reaction solution was cooled to 0°C and divided Sodium borohydride (12.5 g, 198 mmol) was added in batches. The reaction solution was raised to room temperature and continued to stir for 16 hours, diluted with water (500 mL), and then adjusted to pH=7 with 1N aqueous hydrochloric acid. The aqueous phase was extracted with ethyl acetate (150 mL×3). The organic phase was washed sequentially with water (100mL×3) and saturated brine (100mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-e (7.5g, yield: 88%). The product does not require further purification. LC-MS(ESI): m/z=187[M+H] + .[0492]Synthesis of compound 31-d[0493]Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0°C, active manganese dioxide (35 g, 400 mmol) was added, the reaction solution was raised to room temperature and stirring was continued for 16 hours. The reaction solution was filtered through Celite, and the filter cake was washed with chloroform (100 mL×3). The combined filtrates were concentrated under reduced pressure to obtain white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS(ESI): m/z=185[M+H]+.[0494]Synthesis of compound 31-c[0495]Compound 31-d (3.1g, 16.8mmol) was dissolved in trifluoroacetic acid (30mL) at 0℃, N-iodosuccinimide (5.7g, 25.3mmol) was added in batches, and the reaction solution was raised to Keep stirring at room temperature for 1 hour. Water (50 mL) was added to the reaction solution to quench the reaction, and it was extracted with dichloromethane (50 mL×3). The organic phase was washed successively with water (50mL×3) and saturated brine (50mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain a white solid 31-c (4.9g, yield: 94%). The product does not require further purification. LC-MS(ESI): m/z=311[M+H] + .[0496]Synthesis of compound 31-b[0497]Compound 31-c (615mg, 1.98mmol), 2-methoxy-4-fluorophenylboronic acid (405mg, 2.38mmol) and sodium carbonate (630mg, 5.94mmol) were suspended in dioxane (5mL) water (5mL) ), add [1,1′-bis(diphenylphosphorus)ferrocene]dichloropalladium dichloromethane complex (163mg, 0.2mmol). Replace with nitrogen 3 times, and heat to 80°C to react for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was partitioned with dichloromethane (50mL) and water (50mL). The organic phase was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and purified by silica gel column chromatography (petroleum Ether: dichloromethane=1:1) to obtain a white solid 31-b (240 mg, yield: 39%). LC-MS(ESI): m/z=309[M+H] + .[0498]Synthesis of compound 31-a[0499]Compound 31-b (240mg, 0.78mmol) and compound 32-c (208mg, 0.78mmol) were dissolved in N,N-dimethylformamide (3mL), potassium carbonate (323mg, 2.34mmol) was added, 2- Dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol) and tris(dibenzylideneacetone) dipalladium (134 mg, 0.24 mmol). Under the protection of nitrogen, heat to 110°C to react for 16 hours. After cooling to room temperature, the reaction solution was partitioned with dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin layer chromatography preparation plate (petroleum Ether: ethyl acetate = 1:1) to obtain a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z=539[M+H] + .[0500]Synthesis of compound 31[0501]31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), trifluoroacetic acid (3 mL) was added, and the mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure. The residue was layered with ethyl acetate (50mL) and 1N aqueous hydrochloric acid (50mL). The aqueous phase was adjusted to pH=10 with saturated aqueous potassium carbonate solution. 3) Washing and vacuum drying the solid to obtain a light yellow solid 31 (22 mg, yield: 14%). LC-MS(ESI): m/z=439[M+H] + .[0502]1 H-NMR (400MHz, MeOD) δ: 8.78 (d, J = 5Hz, 1H), 7.87 (s, 1H), 7.48 (s, 1H), 7.35 (m, 1H), 7.05 (dd, J = 11Hz) ,J = 2Hz, 1H), 6.91 (m, 1H), 4.10 (m, 1H), 3.79 (s, 3H), 3.22 (m, 2H), 2.77 (m, 2H), 2.47 (s, 3H), 2.03(m,2H),1.73(m,2H)ppm
PATENT
WO 2019228171
Example 1 Preparation of fumarate of fused ring pyrimidine compound as shown in formula 2
Weigh the compound N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidine-4- Base)-1H-pyrazol-4-amine (synthesized according to Example 31 of patent CN106366093A) 100mg (0.228mmol, 1eq) into the vial, add 10mL 88% acetone-water solution, add the vial at about 50°C and stir until dissolved clear. 1.1 mL of fumaric acid with a concentration of 0.25 mol/L in ethanol (0.275 mmol, 1.2 eq) was slowly added dropwise to the free base solution of fused ring pyrimidine compounds, and stirred at 50 ℃ for 1 hour, and then the solution was The rate of 5°C/h was slowly reduced to room temperature, and the solid was collected and dried under vacuum at 40°C overnight.
1 H-NMR (400MHz, DMSO-d 6 ) δ: 9.45 (s, 1H), 8.94 (s, 1H), 7.75 (s, 1H), 7.78-7.33 (m, 2H), 7.15 (d, J = 6.4Hz, 1H), 6.99 (dd, J = 7.6 Hz, J = 7.2 Hz, 1H), 6.42 (s, 1H), 4.10 (m, 1H), 3.73 (s, 3H), 3.17 (d, J = 12.4 Hz, 2H), 2.77 (dd, J = 12.4 Hz, J = 11.6 Hz, 2H), 2.40 (s, 3H), 1.94 (d, J = 11.6 Hz, 2H), 1.73 (m, 2H) ppm.
PATENT
7-(4-Fluoro-2-methoxyphenyl)-6-methyl-N-(1-piperidin-4-yl)-1hydro-pyrazol-4-yl)thieno[3,2 -D]pyrimidine-2-amino is a strong JAK, FGFR, FLT3 kinase inhibitor, and has a good application prospect in the treatment of tumors, immune system diseases, allergic diseases and cardiovascular diseases. This compound is described in patent CN106366093A and has the following chemical structure:
CN106366093A discloses the preparation method of the compound:
In the above synthetic route, NaBH 4 is sodium borohydride, MnO 2 is manganese dioxide, NIS is N-iodosuccinimide, TFA is trifluoroacetic acid, and Pd(dppf)Cl 2 is [1,1′- Bis(diphenylphosphino)ferrocene]palladium dichloride, DIAD is diisopropyl azodicarboxylate, PPh 3 is triphenylphosphine, Pd/C is palladium on carbon, Pd 2 (dba) 3 is Tris(dibenzylideneacetone)dipalladium, RuPhos is 2-bicyclohexylphosphine-2′,6′-diisopropoxybiphenyl.
However, the above method has the problems of a large number of reaction steps, low yield, and requires column chromatography for separation and purification, and is not suitable for industrial scale-up production. Therefore, it is necessary to improve its preparation method.
The present invention provides a method for preparing a compound represented by formula B, which comprises the following steps: under a protective gas atmosphere, in a solvent, in the presence of a catalyst and a base, a compound represented by formula C is combined with a compound represented by formula K The compound can be subjected to the coupling reaction shown below; the catalyst includes a palladium compound and a phosphine ligand;
The preparation method of the compound represented by formula B may further include the following steps: in an organic solvent, in the presence of a base, the compound represented by formula E and the compound represented by formula D are subjected to the substitution reaction shown below, To obtain the compound represented by formula C;
The present invention provides a method for preparing a compound represented by formula C, which comprises the following steps: in an organic solvent, in the presence of a base, a compound represented by formula E and a compound represented by formula D are subjected to the following steps: Substitution reaction is enough;
Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 500L reactor, add 10% palladium on carbon (4.6Kg), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (24.2Kg, 109.5mol), and tetrahydrofuran (150Kg) in sequence And N,N-diisopropylethylamine (17.0Kg, 131.5mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.5 MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 120 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (58Kg) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (60Kg) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 360Kg of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was separated out, centrifuged, and the filter cake was vacuum dried to obtain the product 2-chloro-6-methylthieno[3,2-D]pyrimidine 18.94Kg, yield: 93.2%. LC-MS(ESI): m/z=185.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.30 (s, 1H), 7.34 (s, 1H), 2.73 (s, 3H).
Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
To a 100mL reaction flask, add 10% palladium on carbon (0.17g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), tetrahydrofuran (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 20 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 2.4 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 82%. The LC-MS and 1 H NMR are the same as in Example 1.
Example 3: 7-Bromo 2-chloro-6-methylthieno[3,2-D]pyrimidine (Compound E)
Add trifluoroacetic acid (150Kg) and 2-chloro-6-methylthieno[3,2-D]pyrimidine (18.90Kg, 102.4mol) into a 500L enamel reactor. Add N-bromosuccinimide (18.33Kg, 103.0mol) under temperature control at 15±5℃. After the addition, the temperature is controlled at 25±5℃ to react for 2 hours. Sampling to monitor the reaction, there is still a small amount of raw materials remaining. Additional N-bromosuccinimide (1.0 Kg, 5.6 mol) was added, stirring was continued for 1 hour, sampling and monitoring showed that the reaction was complete. Control the temperature at 10±5°C, and add 274Kg of water dropwise. After the addition, stir at 10±5°C for 2 hours. After centrifugation, the solid was vacuum-dried to obtain the product, 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine, 24.68Kg, yield: 91.4%. LC-MS(ESI): m/z=265.0[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.33 (s, 1H), 2.64 (s, 3H).
Example 4: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Add pyridine (176Kg) and N-BOC-4-hydroxypiperidine (36.00Kg, 178.9mol) to a 500L enamel reactor. Add p-toluenesulfonyl chloride (50.5Kg, 264.9mol) in batches under temperature control at 10±10°C. After the addition, the temperature is controlled at 25±5°C to react for 18 hours. The reaction solution was transferred to a 1000L reactor, the temperature was controlled at 15±5°C, and 710Kg of water was added dropwise. After the addition, stir at 15±5°C for 2 hours. After filtration, the solid was washed with water and dried in vacuum to obtain the product 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester, 59.3Kg, yield: 93.3%. LC-MS(ESI): m/z=378.0[M+Na] + .
Example 5: 4-(4-Nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound F)
Add N,N-dimethylformamide (252Kg), 4-(p-methylbenzenesulfonyl)-piperidine-1-carbonate tert-butyl ester (59.3Kg, 166.8mol), 4-nitro to the reaction kettle Pyrazole (21.5Kg, 190.1mol), and anhydrous potassium carbonate (34.3Kg, 248.2mol). The temperature was controlled at 80±5°C and the reaction was stirred for 18 hours. Cool down to 15±5°C, add 900Kg of water dropwise, control the dropping rate, and keep the temperature at 15±5°C. After the addition, stir at 5±5°C for 2 hours. After filtering, the solid was washed twice with water and dried in vacuum to obtain the product 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 39.92Kg, yield: 80.8%. LC-MS (ESI): m/z=319.1 [M+Na] + .
1 H NMR (400MHz, d 6 -DMSO): δ8.96(s,1H), 8.27(s,1H), 4.44-4.51(m,1H), 4.06-4.08(m,2H), 2.75-2.91( m, 2H), 2.04-2.07 (m, 2H), 1.80-1.89 (m, 2H), 1.41 (s, 9H).
Example 6: 4-(4-Amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound D)
Add 10% palladium-carbon (2.00Kg), 4-(4-nitro-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (39.94Kg, 134.09mol) to the reaction kettle, nothing Water ethanol (314Kg) and ammonia (20.0Kg, 134.09mol). Fill the kettle with hydrogen, and control the hydrogen pressure at 0.2MPa. Turn on the stirring and keep the temperature at 45±5°C to react for 4 hours. Filter, collect the filtrate, and concentrate the filtrate under reduced pressure. Add ethyl acetate (40Kg) and n-heptane (142Kg) to the concentrate, stir at 25±5°C for 1 hour, and then lower the temperature to 5±5°C and stir for 2 hours. After filtration, the solid was vacuum dried to obtain the product 4-(4-amino-1hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 31.85Kg, yield: 88.6%. LC-MS(ESI): m/z=267.2[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ7.06 (s, 1H), 6.91 (s, 1H), 4.08-4.15 (m, 1H), 3.98-4.01 (m, 2H), 3.81 (brs, 2H), 2.83-2.87 (m, 2H), 1.88-1.91 (m, 2H), 1.63-1.72 (m, 2H), 1.41 (s, 9H).
Example 7: 4-(4-(7-Bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl)piperidine-1 -Tert-butyl carbonate (compound C)
Add n-butanol (117Kg), N,N-diisopropylethylamine (15.00Kg, 116.06mol), 4-(4-amino-1hydro-pyrazol-1-yl)piperidine to the reaction kettle 1-tert-butyl carbonate (32.02Kg, 120.22mol) and 7-bromo-2-chloro-6-methylthieno[3,2-D]pyrimidine (24.68Kg, 93.65mol). Turn on the stirring and keep the temperature at 100±5°C to react for 42 hours. Concentrate under reduced pressure. Methanol was added to the concentrate to be beaten. The solid was filtered and dried under vacuum to obtain the product 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1hydro-pyrazol-1-yl ) Piperidine-1-tert-butyl carbonate 37.26Kg, yield: 80.6%. LC-MS(ESI): m/z=493.1[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.73 (s, 1H), 8.97 (s, 1H), 8.18 (s, 1H), 7.68 (s, 1H), 4.30-4.36 (m, 1H) ,4.01-4.04(m,2H),2.87-2.93(m,2H),2.53(s,3H),2.00-2.03(m,2H),1.70-1.80(m,2H),1.41(s,9H) .
Example 8: 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino)-1 Hydro-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (Compound B)
Add purified water (113Kg), dioxane (390Kg), 4-(4-(7-bromo-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) into the reactor -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate (37.26Kg, 93.65mol), 2-methoxy-4-fluorophenylboronic acid pinacol ester (23.05Kg, 120.22mol) , Anhydrous potassium carbonate (20.95Kg, 151.8mol), palladium acetate (0.18Kg, 0.80mol) and 2-dicyclohexylphosphine-2,4,6-triisopropylbiphenyl (0.90Kg, 1.89mol). Under the protection of nitrogen, the temperature is controlled at 70±5℃ to react for 4 hours. Cool down to 40±5°C, add ammonia water (68Kg), and stir for 8 hours. Cool down to 20±5°C and dilute with water (1110Kg). Dichloromethane extraction twice (244Kg, 170Kg). Combine the organic phases, wash sequentially with water and then with saturated brine. Add 3-mercaptopropyl ethyl sulfide-based silica (4.0Kg, used to remove heavy metal palladium) into the organic phase, and stir at 40±5°C for 20 hours. After filtration, the filtrate was concentrated under reduced pressure. The remainder was slurried sequentially with methyl tert-butyl ether and ethanol. Filter and dry in vacuo to obtain 4-(4-((7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-D]pyrimidin-2-yl)amino) -1H-pyrazol-1-yl)piperidine-1-tert-butyl carbonate 34.6Kg, yield: 68.6%. LC-MS(ESI): m/z=539.3[M+H] + .
1 H NMR (400MHz, d 6 -DMSO): δ9.46 (s, 1H), 8.94 (s, 1H), 7.76 (s, 1H), 7.38 (s, 1H), 7.33 to 7.35 (m, 1H) ,7.08-7.11(m,1H),6.91-6.95(m,1H),4.03-4.12(m,3H),3.73(s,3H),2.85-2.89(m,2H),2.39(s,3H) ,1.90-1.93(m,2H),1.55-1.60(m,2H),1.41(s,9H).
Comparative Example 1: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
Into a 100mL reaction flask, add 10% palladium on carbon (0.1g), 2,4-dichloro-6-methylthieno[3,2-D]pyrimidine (2g, 9.2mmol), methanol (40mL) and N,N-Diisopropylethylamine (1.412 g, 10.9 mmol). Fill the bottle with hydrogen and control the hydrogen pressure at 0.5MPa. Turn on the stirring and keep the temperature at 25±5°C to react for 21 hours. Filter, collect the filtrate, concentrate the filtrate under reduced pressure, add ethanol (2.1 g) to the concentrate, and concentrate again to bring out residual tetrahydrofuran. Add ethanol (2.2g) and stir at 70±5°C until all solids are dissolved. Cool down, control the temperature at 25±5°C, add 13.3g of purified water dropwise to the kettle, control the dropping rate, and keep the temperature at 25±5°C. The solid product was precipitated, centrifuged, and the filter cake was vacuum dried to obtain 1.6 g of 2-chloro-6-methylthieno[3,2-D]pyrimidine as a product, with a yield of 54%. Methoxy substituted impurities in 20% yield.
Comparative Example 2: 2-Chloro-6-methylthieno[3,2-D]pyrimidine (Compound I)
After replacing the solvent tetrahydrofuran in Example 2 with ethyl acetate, the solubility of 2-chloro-6-methylthieno[3,2-D]pyrimidine in ethyl acetate was poor, and only a small amount of product was formed, which was not calculated Specific yield.
Comparative example 3: 4-(p-toluenesulfonyl)-piperidine-1-tert-butyl carbonate (Compound G)
Triethylamine (25mL), N-BOC-4-hydroxypiperidine (5g) were added to a 100mL reaction flask. P-toluenesulfonyl chloride (7.1g) was added in batches while controlling the temperature at 10±10°C. After the addition, the temperature is controlled at 25±5℃ to react for 25 hours. Monitoring by LC-MS showed a large amount of unreacted raw materials and the reaction liquid was black and red.
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019228171-A1 | Salt of fused ring pyrimidine compound, crystal form thereof and preparation method therefor and use thereof | 2018-05-31 | |
| AU-2016295594-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| AU-2016295594-B2 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2020-04-16 |
| EP-3354653-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| EP-3354653-B1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2019-09-04 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| JP-2018520202-A | Fused ring pyrimidine compounds, intermediates, production methods, compositions and applications thereof | 2015-07-21 | |
| KR-20180028521-A | Condensed ring pyrimidine-based compounds, intermediates, methods for their preparation, compositions and applications | 2015-07-21 | |
| US-10494378-B2 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | 2019-12-03 |
| US-2018208604-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 | |
| WO-2017012559-A1 | Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereof | 2015-07-21 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT03412292 | MAX-40279 in Subjects With Acute Myelogenous Leukemia (AML) | Phase 1 | Recruiting | 2021-05-21 |
///////////////Orphan Drug, Acute myeloid leukaemia, MAX 40279, EX-A4057, Max 4, MAX-40279, MAX-40279-001, MAX-40279-01, PHASE 1, Maxinovel Pharmaceuticals
CC1=C(C2=NC(=NC=C2S1)NC3=CN(N=C3)C4CCNCC4)C5=C(C=C(C=C5)F)OC
TRK 700
![1-[4-(Dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=71738795&t=l)
TRK-700
CAS 1463432-16-7C14 H24 N4 O264.371-Propanone, 1-[4-(dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-
1-[4-(dimethylamino)piperidin-1-yl]-3-(1-methylimidazol-2-yl)propan-1-one
- 1-[4-(Dimethylamino)-1-piperidinyl]-3-(1-methyl-1H-imidazol-2-yl)-1-propanone
- OriginatorToray Industries
- ClassAnalgesics
- Mechanism of ActionUndefined mechanism
- Phase IIPostherpetic neuralgia
- PreclinicalPeripheral nervous system diseases
- 12 Sep 2018Pharmacodynamics data from a preclinical trial in Peripheral neuropathy presented at the 17th World Congress on Pain (WCP-2018)
- 01 Jul 2017Toray Industries completes a phase II trial for Postherpetic neuralgia (In adults, In the elderly) in Japan (PO) (NCT02701374)
- 21 May 2017Toray Industries completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (PO) (NCT03043248)
developed by Toray for treating neuropathic pain and investigating for fibromyalgia. In August 2021, this drug was reported to be in phase 1 clinical development.
PATENT
WO 2016136944
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016136944

(Reference Example 22) Synthesis of (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate:
[Chemical 56]
1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, Methyl (triphenylphosphoranylidene) acetate (33.4 g, 99.9 mmol) was added to a solution of 90.8 mmol) in dichloromethane (240 mL) at room temperature, and the mixture was stirred for 16 hours and then concentrated under reduced pressure. The residue was washed with a mixed solvent of hexane / dichloromethane = 19/1, and the washing liquid was concentrated. The residue was purified by silica gel column chromatography (hexane / ethyl acetate) to give (E) -methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate as a white solid (11.9 g, 71. 6 mmol, 79%).
1 H-NMR (400 MHz, CDCl 3 ) δ: 3.76 (3H, s), 3.81 (3H, s), 6.82 (1H, d, J = 15.6 Hz), 6.98 (1H, brs), 7.16 (1H, brs), 7.53 (1H, d, J = 15.6Hz).
ESI-MS: m / z = 167 (M + H) + .
(Reference Example 27) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 61]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propane. -1-one (0.179 g, 0.68 mmol, 63%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Comparative Example 1) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one hydrochloride:
[Chemical 66]
1- (4- (Dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (1.50 g, 5.67 mmol) diethyl ether (60) A dioxane solution of hydrogen chloride (4.0 M, 3.69 mL, 14.8 mmol) was added to the (0.0 mL) solution at 0 ° C. The reaction mixture was stirred at the same temperature for 1 hour and then at room temperature for 30 minutes. The precipitated white solid was collected by filtration, washed with diethyl ether (100 mL), dried at room temperature for 36 hours, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-). Imidazole-2-yl) propan-1-one hydrochloride (1.41 g, 4.18 mmol, 74%) (hereinafter, the compound of Comparative Example 1) was obtained as a white solid.
1 1 H-NMR (400 MHz, D 2 O) δ: 1.53-1.80 (2H, m), 2.12-2.23 (2H, m), 2.68-2.80 (1H, m), 2.88 (6H, s), 3.01- 3.08 (2H, m), 3.15-3.26 (3H, m), 3.47-3.58 (1H, m), 3.84 (3H, s), 4.08-4.16 (1H, m), 4.50-4.59 (1H, m), 7.29-7.33 (2H, m).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) as propan-1-one : m / z = 265 (M + H) + .
(Comparative Example 2) Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one sulfate monohydrate:
[Chemical 67]
1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one (6.72 g, 25.4 mmol) Concentrated sulfuric acid (2.49 g, 25.4 mmol), water (1.83 g, 102 mmol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl) in a DMSO (100 mL) solution. Seed crystals (50 mg, 0.13 mmol) of -1H-imidazol-2-yl) propan-1-one sulfate monohydrate were added at 80 ° C. The reaction was stirred at the same temperature for 2.5 hours, at 50 ° C. for 2.5 hours and at room temperature for 15 hours. The precipitated white solid was collected by filtration, washed successively with DMSO (20 mL) and methyl ethyl ketone (40 mL), dried at room temperature, and then 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl). -1H-imidazol-2-yl) propan-1-one sulfate monohydrate (8.42 g, 22.1 mmol, 87%) (hereinafter, the compound of Comparative Example 2) was obtained as white crystals.
1 1 H-NMR (400 MHz, DMSO-d 6)) δ: 1.36 (1H, m), 1.58 (1H, m), 1.95 (2H, br), 2.44-2.57 (1H, m), 2.65 (6H, s), 2.74-2.88 (4H, m), 3.00 (1H, t, J = 12.0 Hz), 3.22 (1H, m), 3.61 (3H, s), 4.02 (1H, d, J = 14.0 Hz), 4.47 (1H, d, J = 12.8 Hz), 6.87 (1H, d, J = 1.2 Hz), 7.11 (1H, d, J = 1.2 Hz).
ESI-MS; 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-) As 1H-imidazol-2-yl) propan-1-one: m / z = 265 (M + H) + .
NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT
WO-2021153744
PATENT
WO-2021153743
Novel crystalline polymorphic form of 1-(4-(dimethylamino) piperidin-1-yl)-3-(1-methyl-1H-imidazol-2-yl)propan-1-one, useful as an analgesic in treating neuropathic pain and/or fibromyalgia.Pain is an experience with unpleasant sensations and emotions that occurs when or may cause tissue damage. Pain is mainly classified into nociceptive pain, neuropathic pain or psychogenic pain according to its cause. In addition, fibromyalgia is known as pain of unknown cause.
Neuropathic pain is pathological pain caused by dysfunction of the peripheral or central nervous system itself, and is caused by direct damage or compression of nervous tissue even though nociceptors are not stimulated. It refers to the pain that occurs. As a therapeutic agent for neuropathic pain, an anticonvulsant, an antidepressant, anxiolytic, or an antiepileptic drug such as gabapentin or pregabalin is used.
Fibromyalgia is a disease in which systemic pain is the main symptom and neuropsychiatric symptoms and autonomic nervous system symptoms are secondary symptoms. Pregabalin approved in the United States and Japan, duloxetine and milnacipran approved in the United States are mainly used as therapeutic agents for fibromyalgia, and non-approved agents for fibromyalgia are not approved. It has also been used for steroidal anti-inflammatory agents, opioid compounds, antidepressants, anticonvulsants and antiepileptic drugs. However, the therapeutic effects of non-steroidal anti-inflammatory drugs and opioid compounds are generally considered to be low (Non-Patent Document 1).
On the other hand, Patent Document 1 discloses that certain substituted piperidins have cardiotonic activity, and Patent Document 2 discloses that an imidazole derivative exhibits an FXa inhibitory effect. Patent Document 3 suggests that the substituted piperidins may have a medicinal effect on overweight or obesity, and Patent Documents 4 to 6 and Non-Patent Document 2 indicate that the imidazole derivative has an analgesic effect. It is disclosed.
In addition, the quality of pharmaceutical products needs to be maintained over a long period of time such as distribution and storage, and the compound as an active ingredient is required to have high chemical and physical stability. Therefore, as the active ingredient of a pharmaceutical product, a crystal that can be expected to have higher stability than an amorphous substance is generally adopted. Further, if crystals are obtained, a purification effect due to recrystallization during production can be expected. Further, it is preferable to have low hygroscopicity from the viewpoint of maintaining stability and handling during manufacturing, storage, formulation and analysis of the drug substance. In addition, since a drug needs to be dissolved in the digestive tract in order to exhibit its medicinal effect, it is preferable that the drug has excellent solubility, which is a physical property contrary to stability.
In order to obtain crystals of a compound that is an active ingredient of a pharmaceutical product, it is necessary to study various conditions for precipitating crystals from the solution. It is common to carry out crystallization under the condition of being dissolved in.
Patent documents
Patent Document 1: French Patent Invention No. 2567885
Patent Document 2: Japanese Patent Application Laid-Open No. 2006-0083664
Patent Document 3: International Publication No. 2003/031432
Patent Document 4: International Publication No. 2013/147160
Patent Document 5: International Publication No. 2015/046403
Patent Document 6: International Publication No. 2016/136944
Non-patent literature
Non-Patent Document 1: Okifuji et al., Pain and Therapy, 2013, Volume 2, p. 87-104
Non-Patent Document 2: Takahashi et al., Toxicological Pathology, 2019, Vol. 47. p. 494-503
Compound (I) was synthesized by the method described in the following reference example. For the compounds used in the synthesis of the reference example compounds for which the synthesis method is not described, commercially available compounds were used.
(Reference Example 4) Synthesis of amorphous compound (I):
[Chemical formula 2] 2 of
crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanol (5.00 g, 27.4 mmol) Aqueous sodium hydroxide solution (1.0N, 30.2 mL, 30.2 mmol) was added to a solution of -propanol (55 mL) at 0 ° C., and the mixture was stirred at room temperature for 12 hours. 2-Propanol (220 mL) was added to the reaction solution at room temperature, and crude 4- (dimethylamino) piperidine (3.17 g, 24.7 mmol) and DMT-MM (8.35 g, 30.2 mmol) were added at room temperature to react. The liquid was stirred at the same temperature for 3 hours. A 10% aqueous sodium chloride solution and a 1.0N aqueous sodium hydroxide solution were added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give compound (I) (6.98 g) as an amorphous substance.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 (5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz) ), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
(Reference Example 5) Synthesis of crude 4- (dimethylamino) piperidine:
[Chemical
formula 3] 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (20.1 g, 77.0 mmol) in methanol (154.0 mL) Palladium-carbon (10% wet, 2.01 g) was added thereto, and the mixture was stirred at room temperature for 19 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give a crude product of 4- (dimethylamino) piperidine (9.86 g).
(Reference Example 6) Synthesis of crude ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate:
[Chemical
formula 4] Sodium hydride (55%, 4.36 g, 100 mmol) aqueous solution and tetrahydrofuran (150 mL) To the mixture was added triethylphosphonoacetate (19.1 mL, 95.0 mmol) at 0 ° C. After stirring the reaction solution for 20 minutes, a solution of 1-methyl-1H-imidazol-2-carbaldehyde (10.0 g, 91.0 mmol) in tetrahydrofuran (150 mL) was added at 0 ° C., and then ethanol (30 mL) was added in the same manner. The mixture was added at temperature and stirred at room temperature for 2 hours. A 10% aqueous sodium chloride solution was added to the reaction mixture, and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, chloroform / methanol). After adding methanol (310 mL) to the residue, palladium-carbon (10% wet, 1.40 g) was added, and the mixture was stirred at room temperature for 3 hours under a hydrogen atmosphere. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to obtain a crude product (14.2 g) of ethyl 3- (1-methyl-1H-imidazol-2-yl) propanoate.
(Reference Example 7) Synthesis of 1-benzyloxycarbonyl-4- (dimethylamino) piperidine:
[Chemical
formula 5] dichloromethane (55.7 mL) of 1-benzyloxycarbonyl-4-oxopiperidine (13.0 g, 55.7 mmol) ) Solution of dimethylamine in tetrahydrofuran (2.0 M, 34.8 mL, 69.7 mmol), acetic acid (0.32 mL, 5.6 mmol) and sodium triacetoxyborohydride (4.8 g, 22.6 mmol). Added at ° C. After stirring the reaction solution at the same temperature for 30 minutes, sodium triacetoxyborohydride (4.8 g, 22.6 mmol) was added at 0 ° C. The reaction mixture was stirred at the same temperature for 30 minutes, sodium triacetoxyborohydride (8.1 g, 38.2 mmol) was added at 0 ° C., and the mixture was stirred at room temperature for 12 hours. The reaction solution was cooled to 0 ° C. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) and then again by flash chromatography (silica gel, chloroform / methanol) to obtain 1-benzyloxycarbonyl-4- (dimethylamino) piperidine (dimethylamino) piperidine. 13.6 g, 51.8 mmol, 93%) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.34-1.46 (2H, m), 1.78-1.86 (2H, m), 2.28 (6H, s), 2.29-2.34 (1H, m), 2.75-2.85 (2H, m), 4.14-4.28 ( 2H, m), 5.12 (2H, s), 7.29-7.36 (5H, m).
ESI-MS: m / z = 263 (M + H) + .
(Reference Example 8) Synthesis of 1-benzyloxycarbonyl-4-oxopiperidine:
[Chemical
formula 6] Hydrochloride (130 mL) and water (130 mL) of 4-piperidinone hydrochloride monohydrate (10.0 g, 65.1 mmol) Sodium carbonate (13.8 g, 130.2 mmol) and benzyl chloroformate (8.79 mL, 61.8 mmol) were added to the mixed solution with and at 0 ° C., and the mixture was stirred at room temperature for 3 hours. The reaction mixture was extracted with ethyl acetate. The organic layer was washed with 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane / ethyl acetate) to give 1-benzyloxycarbonyl-4-oxopiperidine (13.1 g, 56.2 mmol, 86%) as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3 ) δ: 2.42-2.50 (4H, m), 3.78-3.82 (4H, m), 5.18 (2H, s), 7.32-7.38 (5H, m).
(Example 1) Production of A-type crystal of
compound (I): Amorphous compound (6.98 g) of compound (I) prepared in Reference Example 4 is purified and concentrated with chloroform / methanol by silica gel column chromatography. After that, the wall surface of the flask was rubbed with a spartel and mechanical stimulation was applied to obtain A-type crystals of compound (I) as a powder. For the obtained crystals, measurement of powder X-ray diffraction using a powder X-ray diffractometer (Rigaku Co., Ltd .; 2200 / RINT ultima + PC) and TG-DTA using a TG-DTA device (Rigaku Co., Ltd .; TG8120) Was done. The results of these measurements are shown in FIGS. 1 and 2.
Diffraction angle 2θ: 5.9, 16.5, 17.7, 20.8, 26.7 °
Endothermic peak: 55 ° C
PATENT
WO2013147160
Example 1 Synthesis of 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan-1-one:
[Chemical 27]
(E) )-Methyl 3- (1-methyl-1H-imidazol-2-yl) acrylate (0.180 g, 1.08 mmol) in ethanol (4.0 mL) solution of palladium-carbon (10% wet, 15 mg) at room temperature In a hydrogen atmosphere, the mixture was stirred for 4 hours. The reaction mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure. Methanol (1.0 mL) was added to the obtained residue at room temperature to dissolve it, and the mixture was cooled to 0 ° C. An aqueous sodium hydroxide solution (1.0 N, 1.19 mL, 1.19 mmol) was added to the reaction solution at 0 ° C., the mixture was stirred at room temperature for 2 hours, and then concentrated under reduced pressure. Chloroform (10.0 mL) was added to the obtained residue at room temperature to dissolve it. Add diisopropylethylamine (0.568 mL, 3.25 mmol), HBTU (0.616 g, 1.63 mmol) and 4- (dimethylamino) piperidine (0.125 g, 0.975 mmol) to the reaction solution at room temperature, and add the reaction solution. The mixture was stirred at the same temperature for 16 hours. A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was washed with a 10% aqueous sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (NH silica gel, chloroform / methanol) and 1- (4- (dimethylamino) piperidine-1-yl) -3- (1-methyl-1H-imidazol-2-yl) propan- 1-one (0.179 g, 0.68 mmol, 63%) (hereinafter, the compound of Example 1) was obtained as a colorless oil.
1 1 H-NMR (400 MHz, CDCl 3) δ: 1.29-1.43 (2H, m), 1.80-1.88 (2H, m), 2.27 (6H, s), 2.29-2.38 (1H, m), 2.54-2.63 (1H, m), 2.88-3.04 ( 5H, m), 3.62 (3H, s), 3.98-4.05 (1H, m), 4.57-4.65 (1H, m), 6.79 (1H, d, J = 1.2 Hz), 6.91 (1H, d, J = 1.2 Hz).
ESI-MS: m / z = 265 (M + H) + .
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2016136944-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| JP-WO2013147160-A1 | Cyclic amine derivatives and their pharmaceutical use | 2012-03-29 | |
| TW-201350119-A | Cyclic amine derivatives and their medical uses | 2012-03-29 | |
| WO-2013147160-A1 | Cyclic amine derivative and use thereof for medical purposes | 2012-03-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| RU-2667062-C1 | Dynamic cyclic amine and pharmaceutical application thereof | 2015-02-27 | 2018-09-14 |
| TW-201639826-A | Cyclic amine derivatives and their medical uses | 2015-02-27 | |
| TW-I682927-B | Cyclic amine derivatives and their medical uses | 2015-02-27 | 2020-01-21 |
| US-10173999-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-01-08 |
| US-2018065950-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3263565-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| EP-3263565-B1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-06-26 |
| ES-2744785-T3 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2020-02-26 |
| JP-6569671-B2 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 | 2019-09-04 |
| JP-WO2016136944-A1 | Cyclic amine derivatives and their pharmaceutical use | 2015-02-27 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019189781-A1 | Agent for inhibiting rise in intraneuronal calcium concentration | 2018-03-30 | |
| AU-2016224420-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| AU-2016224420-B2 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | 2019-08-22 |
| CA-2977614-A1 | Cyclic amine derivative and pharmaceutical use thereof | 2015-02-27 | |
| CN-107250128-B | Cyclic amine derivatives and its medical usage | 2015-02-27 | 2019-07-26 |
//////////TRK-700, phase 1, neuropathic pain, fibromyalgia, toray
O=C(CCc1nccn1C)N1CCC(CC1)N(C)C
PRN 473, SAR 444727

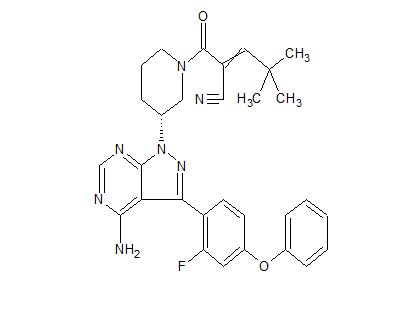
![2-[(3R)-3-[4-Amino-3-(2-fluoro-4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidine-1-carbonyl]-4,4-dimethylpent-2-enenitrile.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=123342594&t=l)
SAR-444727
1414354-91-8C30 H30 F N7 O2 Molecular Weight539.601-Piperidinepropanenitrile, 3-[4-amino-3-(2-fluoro-4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]-α-(2,2-dimethylpropylidene)-β-oxo-, (3R)-
(3R)-3-[4-Amino-3-(2-fluoro-4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]-α-(2,2-dimethylpropylidene)-β-oxo-1-piperidinepropanenitrile
2-(3-(4-amino~3-(2-fiuoro~4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidine-1-carbonyl)-4,4-dimethylpent-2-enenitrile
- OriginatorPrincipia Biopharma
- ClassSmall molecules
- Mechanism of ActionAgammaglobulinaemia tyrosine kinase inhibitors
- Phase IAutoimmune disorders
- DiscontinuedArthritis
- 28 Sep 2020Principia Biopharma has been acquired by Sanofi
- 22 Jun 2020Principia Biopharma plans a pharmacokinetic phase I trial (In volunteers) for Hypersensitivity (for Immunoglobulin E-mediated allergies) in Australia (Topical) (ACTRN12620000693921)
- 10 Mar 2020Phase-I clinical trials in Autoimmune disorders (In volunteers) in Australia (Topical)
- US 8957080
- US 8673925
- WO 2014022569
- WO 2013191965
- WO 2012158764
Useful for treating pemphigus vulgaris, immune thrombocytopenia, inflammatory bowel disease, Sjogren’s syndrome, multiple sclerosis, chronic lymphocytic leukemia and ankylosing spondylitis. Principia Biopharma is developing a topical formulation PRN-473 (presumed to be SAR-444727), a reversible covalent bruton’s (BTK) tyrosine kinase inhibitor, developed based on Principia’s reversible, tailored covalency platform, for treating immune-mediated diseases [phase I, July 2021]. Principia Biopharma was also investigating BTK inhibitors , developed based on Principia’s reversible, tailored covalency platform, for treating hematologic malignancies [no development reported since July 2019]. At the time of publication, Zhu was also affiliated with Nurix Therapeutics , while By and Phiasivongsa were based at Rain Therapeutics and Kronos Bio , respectively.
PATENT
WO-2021142131
Novel crystalline polymorphic forms (I to V) of PRN-473 and their preparation method.
CRYSTALLINE FORMS OF 2- [3- [4- AMINO-3-(2- FLUORO-4-PHENOXY- PHENYL)-1H-PYRAZOLO[3,4-D]PYRIMIDIN-1-YL]PIPERIDINE-1-CARBONYL]- 4,4-DIMETHYLPENT-2-ENENITRILE
This application claims the benefit of priority to U.S. Provisional Application No. 62/958,389, filed January 8, 2020, the contents of which are incorporated by reference herein in their entirety.
Disclosed herein are crystalline forms of 2-(3-(4-amino~3-(2-fiuoro~4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidine-1-carbonyl)-4,4-dimethylpent-2-enenitrile (Compound (I)), methods of using the same, and processes for making Compound (I), including its various crystalline forms. The crystalline forms of Compound (I) are inhibitors of Bruton’s tyrosine kinase (BTK). The enzyme BTK is a member of the Tec family non-receptor tyrosine kinases.
BTK is expressed in most hematopoietic cells, including B cells, mast cells, and macrophages. BTK plays a role in the development and activation of B cells and has been implicated in multiple signaling pathways across a wide range of immune-mediated diseases. BTK activity has been implicated in the pathogenesis of several disorders and conditions, such as B cel1-related hematological cancers (e.g,, non-Hodgkin lymphoma and B cell chronic lymphocytic leukemia) and autoimmune diseases (e.g, rheumatoid arthritis,
Sjogren’s syndrome, pemphigus, IBD, lupus, and asthma).
Compound (I) and various solid forms thereof may inhibit BTK and be useful in the treatment of disorders and conditions mediated by BTK activity. Compound (I) is disclosed as, e.g., Compound 125A in Table 1 of WO 2012/158764 and has the following structure:
Example 1: Preparation of Crystalline Form (I) of Compound (I)
Methyl isobutyl ketone (MIBK; 6 mL) was added to amorphous (R)-2-(3-(4-amino-3- (2-fluoro-4-phenoxyphenyJ)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidine-1-carbonyl)-4,4- dimethylpent-2-enenitrile (1,0 g) and stirred to fonn a solution. After approximately five minutes of agitation, a precipitate began to form. Additional MIBK (10 mL) was charged, and the slurry was stirred. After approximately ten days, the solid was filtered and rinsed with MIBK (10 mL). The solid was dried under vacuum with heating to afford approximately 0.5 g of crystalline Form (I) of Compound (I) as a white solid.
PATENT
WO2012158764 , claiming BTK tyrosine kinase inhibitors, useful for treating cancer.
https://patents.google.com/patent/WO2012158764A1/en
WO 2012/158764 125A

PATENT
US20210205313
PATENT
US20210205312 ,
for concurrently published filings, claiming a gel composition comprising PRN-473 and use of another BTK tyrosine kinase inhibitor ie PRN1008 , respectively.
PATENT
WO2016100914 , claiming use of a BTK inhibitor ie PRN-473, alone or in combination with corticosteroid therapy, for treating pemphigus vulgaris.
PATENT
WO 2014022569
https://patents.google.com/patent/WO2014022569A1/en
//////// PRN-473, PRN 473, SAR 444727, PHASE 1
CC(C)(C)C=C(C#N)C(=O)N1CCC[C@H](C1)n1nc(c2c(N)ncnc21)c1ccc(Oc2ccccc2)cc1F
NEW DRUG APPROVALS
ONE TIME
$10.00
PF-07321332, Nirmatrelvir
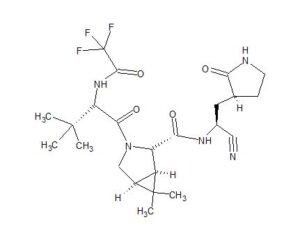
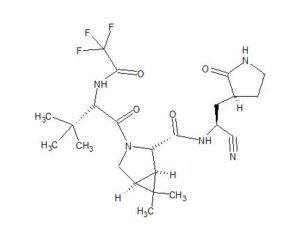

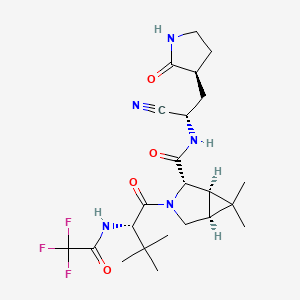
PF-07321332
Nirmatrelvir
UNII-7R9A5P7H32
7R9A5P7H32
PF07321332
CAS 2628280-40-8
C23H32F3N5O4, 499.5
(1R,2S,5S)-N-[(1S)-1-cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]-3-[(2S)-3,3-dimethyl-2-[(2,2,2-trifluoroacetyl)amino]butanoyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide
https://clinicaltrials.gov/ct2/show/NCT04756531

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN
https://pubmed.ncbi.nlm.nih.gov/34726479/
https://www.science.org/doi/10.1126/science.abl4784
Science. 2021 Dec 24;374(6575):1586-1593. doi: 10.1126/science.abl4784. Epub 2021 Nov 2.
An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19
file:///C:/Users/Inspiron/Downloads/science.abl4784_sm.pdf
The worldwide outbreak of COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has become a global pandemic. Alongside vaccines, antiviral therapeutics are an important part of the healthcare response to countering the ongoing threat presented by COVID-19. Here, we report the discovery and characterization of PF-07321332, an orally bioavailable SARS-CoV-2 main protease inhibitor with in vitro pan-human coronavirus antiviral activity and excellent off-target selectivity and in vivo safety profiles. PF-07321332 has demonstrated oral activity in a mouse-adapted SARS-CoV-2 model and has achieved oral plasma concentrations exceeding the in vitro antiviral cell potency in a phase 1 clinical trial in healthy human participants.
Synthesis of PF-07321332 (Compound 6): Anhydrous, MTBE solvate form
(1R,2S,5S)-N-{(1S)-1-Cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl}-6,6-dimethyl-3-[3-methyl-N- (trifluoroacetyl)-L-valyl]-3-azabicyclo[3.1.0]hexane-2-carboxamide (1 eq tert-butyl methyl ether solvate) (6, MTBE solvate). This experiment was carried out in 2 parallel batches. Methyl N- (triethylammoniosulfonyl)carbamate, inner salt (Burgess reagent; 69.3 g, 276 mmol) was added to a solution of T18 (61 g, 111 mmol) in dichloromethane (550 ml). After the reaction mixture had been stirred at 25 °C for 1 h. The reaction mixture was quenched by a mixture of saturated aqueous sodium bicarbonate solution (200 ml) and saturated aqueous sodium chloride solution (100 ml). The separated organic phase was concentrated. The resulting residue was dissolved in 50% ethyl acetate/ tert-butyl methyl ether (600 ml), washed by a mixture of saturated aqueous sodium bicarbonate solution (200 ml) and saturated aqueous sodium chloride solution (100 ml) twice, saturated aqueous sodium chloride solution (200 ml), a mixture of HCl (1 M; 200 ml) and saturated aqueous sodium chloride solution (100 ml) twice. The organic layer was then dried over magnesium sulfate, filtered, and concentrated. The residue was treated with a mixture of ethyl acetate and tert-butyl methyl ether (1:10, 400 ml) and heated to 50 °C; after stirring for 1 hour at 50 °C, it was cooled to 25 °C and stirred overnight. The solid was collected via filtration, dissolved in dichloromethane (100 ml) and filtered through silica gel (200 g); the silica gel was then washed with ethyl acetate (1 Liter), 10% methanol in ethyl acetate (2 Liters). The combined eluates were concentrated. The 2 batches were combined, taken up in a mixture of ethyl acetate and tert-butyl methyl ether (5:95, 550 ml). This mixture was heated to 50 °C for 1 h, cooled to 25 °C, and stirred overnight. Filtration afforded 6, MTBE solvate, as a white solid. Yield: 104 g, 75 %. 1H NMR (600 MHz, DMSO-d6) δ 9.43 (d, J = 8.4 Hz, 1H), 9.03 (d, J = 8.6 Hz, 1H), 7.68 (s, 1H), 4.97 (ddd, J = 10.9, 8.6, 5.1 Hz, 1H), 4.41 (d, J = 8.4 Hz, 1H), 4.15 (s, 1H), 3.91 (dd, J = 10.4, 5.5 Hz, 1H), 3.69 (d, J = 10.4 Hz, 1H), 3.17 – 3.11 (m, 1H), 3.07 (s, 3H, MTBE), 3.04 (td, J = 9.4, 7.1 Hz, 1H), 2.40 (tdd, J = 10.4, 8.4, 4.4 Hz, 1H), 2.14 (ddd, J = 13.4, 10.9, 4.4 Hz,
1H), 2.11 – 2.03 (m, 1H), 1.76 – 1.65 (m, 2H), 1.57 (dd, J = 7.6, 5.5 Hz, 1H), 1.32 (d, J = 7.6 Hz, 1H), 1.10 (s, 9H, MTBE), 1.03 (s, 3H), 0.98 (s, 9H), 0.85 (s, 3H). Anal. Calcd for C23H32F3N5O4 .C5H12O: C, 57.23; H, 7.55; N, 11.92. Found: C, 57.08; H, 7.55; N, 11.85. mp = 118.8 oC
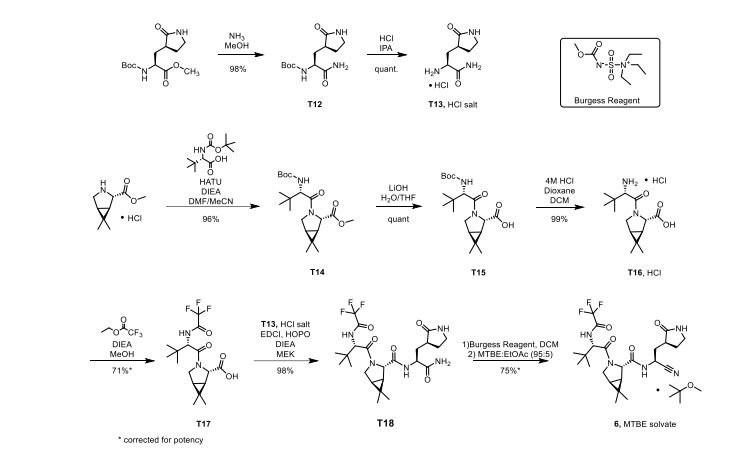
cry
Compound 6 (anhydrous MTBE solvate, 200 g, 332.8 mmol, 83.11 mass%) was charged into a reactor with overhead half-moon stirring at 350 rpm. Heptane (1000 ml) was charged, followed by isopropyl acetate (1000 ml) and the stirring was continued at 20 oC overnight. Additional heptane (1000 ml) was charged over 120 minutes. The reaction vessel was then cooled to 10 oC over 30 min and stirred at that temp for 3 days. The solid was filtered, washing with a mixture of isopropyl acetate (80 ml) and heptane (320 ml). It was then dried under vacuum at 50 °C to provide 6, anhydrous ‘Form 1’, as a white crystalline solid. Yield: 160.93 g, 322 mmol, 97%. 1H NMR (600 MHz, DMSO-d6) δ 9.43 (d, J = 8.4 Hz, 1H), 9.03 (d, J = 8.6 Hz, 1H), 7.68 (s, 1H), 4.97 (ddd, J = 10.9, 8.6, 5.1 Hz, 1H), 4.41 (d, J = 8.4 Hz, 1H), 4.15 (s, 1H), 3.91 (dd, J = 10.4, 5.5 Hz, 1H), 3.69 (d, J = 10.4 Hz, 1H), 3.17 – 3.11 (m, 1H), 3.04 (td, J = 9.4, 7.1 Hz, 1H), 2.40 (tdd, J = 10.4, 8.4, 4.4 Hz, 1H), 2.14 (ddd, J = 13.4, 10.9, 4.4 Hz, 1H), 2.11 – 2.03 (m, 1H), 1.76 – 1.65 (m, 2H), 1.57 (dd, J = 7.6, 5.5 Hz, 1H), 1.32 (d, J = 7.6 Hz, 1H), 1.03 (s, 3H), 0.98 (s, 9H), 0.85 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 177.50, 170.72, 167.45, 156.95 (q, J = 37.0 Hz), 119.65, 115.84 (q, J = 286.9 Hz), 60.08, 58.19, 47.63, 37.77, 36.72, 34.60, 34.15, 30.28, 27.34, 26.86, 26.26, 25.72, 18.86, 12.34. 19F NMR (376 MHz, DMSO-d6) δ -72.94. HRMS (ESI-TOF) m/z calcd. for C23H33F3N5O4 [M + H]+ 500.2474, found 500.2472. Anal. Calcd for C23H32F3N5O4: C, 55.30; H, 6.46; N, 14.02. Found: C, 55.30; H, 6.49; N, 13.96. mp = 192.9 oC







SYN
Bioorganic & medicinal chemistry letters (2021), 50, 128333.
https://www.sciencedirect.com/science/article/pii/S0960894X21005606
https://pubmed.ncbi.nlm.nih.gov/34418570/
pecific anti-coronaviral drugs complementing available vaccines are urgently needed to fight the COVID-19 pandemic. Given its high conservation across the betacoronavirus genus and dissimilarity to human proteases, the SARS-CoV-2 main protease (Mpro) is an attractive drug target. SARS-CoV-2 Mpro inhibitors have been developed at unprecedented speed, most of them being substrate-derived peptidomimetics with cysteine-modifying warheads. In this study, Mpro has proven resistant towards the identification of high-affinity short substrate-derived peptides and peptidomimetics without warheads. 20 cyclic and linear substrate analogues bearing natural and unnatural residues, which were predicted by computational modelling to bind with high affinity and designed to establish structure-activity relationships, displayed no inhibitory activity at concentrations as high as 100 μM. Only a long linear peptide covering residues P6 to P5‘ displayed moderate inhibition (Ki = 57 µM). Our detailed findings will inform current and future drug discovery campaigns targeting Mpro.
SYN
https://pubmed.ncbi.nlm.nih.gov/34498651/
Chemical communications (Cambridge, England) (2021), 57(72), 9096-9099We present a detailed computational analysis of the binding mode and reactivity of the novel oral inhibitor PF-07321332 developed against the SARS-CoV-2 3CL protease. Alchemical free energy calculations suggest that positions P3 and P4 could be susceptible to improvement in order to get a larger binding strength. QM/MM simulations unveil the reaction mechanism for covalent inhibition, showing that the nitrile warhead facilitates the recruitment of a water molecule for the proton transfer step.
PATENT
WO 2021234668
SYNOral inhibitors of the SARS-CoV-2 main protease for the treatment of COVID-19Owen, D., 261st Am Chem Soc (ACS) Natl Meet · 2021-04-05 Abst 243Synthesis of intermediate : Aminolysis of methyl N-Boc-3-[2-oxopyrrolidin-3(S)-yl]-L-alaninate in the presence of NH3 and subsequent N-deprotection using HCl leads to 2(S)-amino-3-[2-oxopyrrolidin-3(S)-yl]propenamide hydrochloride

Condensation of Boc-L-tert-leucine with methyl (1R,2S,5S)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxylate using HATU gives the corresponding amide, which upon hydrolysis of methyl ester moiety in the presence of LiOH and subsequent N-deprotection by means of HCl affords intermediate,. N-Acylation of amine with ethyl trifluoroacetate yields diamide derivative, which upon condensation with 2(S)-amino-3-[2-oxopyrrolidin-3(S)-yl]propenamide hydrochloride using EDC and HOPO generates compound N-1 STEP. Burgess dehydration of amide derivative furnishes PF-7321332 .
In about November to December 2019 a novel coronavirus was identified as the cause of pneumonia cases in Wuhan (China). It spread, resulting in an epidemic throughout China, and thereafter in other countries throughout the world. In February 2020, the World Health Organization designated the disease COVTD-19, which stands for coronavirus disease 2019. The virus is also known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1)
COVID-19 is a betacoronavirus in the same subgenus as the severe acute respiratory syndrome (SARS) virus (as well as several bat coronaviruses), but in a different clade. The structure of the receptor- binding gene region is very similar to that of the SARS coronavirus, and the virus has been shown to use the same receptor, the angiotensin converting enzyme 2 (ACE2), for cell entry (2).
In the situation of rapidly increasing cases, inappropriate management of mild cases could increase the burden of healthcare system and medical costs. Viral clearance is a major standard in the assessment of recovery and discharge from medical care, but early results illustrated that the persistence of viral RNA is heterogeneous despite the rapid remission of symptoms and can last over three weeks even in very mild cases. In addition, long hospitalization stays may increase the risk for hospital-associated mental health problems and unexpected hospital-acquired infections. (9)
At the beginning, the outbreak identified an initial association with a seafood market that sold live animals in Wuhan, China. However, as the outbreak progressed, person-to-person spread became the main mode of transmission.
Person to person transmission is thought to occur mainly via respiratory droplets, resembling the spread of influenza. With droplet transmission, the virus is released in respiratory secretions when an infected person breathes, coughs, sneezes, or talks, and can infect another person if such secretions make direct contact with the mucous membranes. Infection can also occur if a person touches an infected surface and then touches his or her eyes, nose, or mouth. Droplets typically do not travel more than six feet (about two meters) and do not linger in the air. There is still controversy about this topic.
Whether SARS-CoV-2 can be transmitted through the airborne route (through particles smaller than droplets that remain in the air over time and distance) under natural conditions has been controversial.
Reflecting the current uncertainty regarding transmission mechanisms, recommendations on airborne precautions in the health care setting vary by location; airborne precautions are universally recommended when aerosol-generating procedures are performed.
It appears that SARS-CoV-2 can be transmitted prior to the development of symptoms and throughout the course of illness. However, most data informing this issue is from studies evaluating viral RNA detection from respiratory and other specimens, and detection of viral RNA does not necessarily indicate the presence of infectious virus.
A study suggested infectiousness started 2.3 days prior to symptom onset, peaked 0.7 days before symptom onset, and declined within seven days; however, most patients were isolated following symptom onset, which would reduce the risk of transmission later in illness regardless of infectiousness. These findings raise the possibility that patients might be more infectious in the earlier stage of infection, but additional data is needed to confirm this hypothesis (3).
How long a person remains infectious is also uncertain. The duration of viral shedding is variable; there appears to be a wide range, which may depend on severity of the illness. In one study of 21 patients with mild illness (no hypoxia), 90 percent had repeated negative viral RNA tests on nasopharyngeal swabs by 10 days after the onset of symptoms; tests were positive for longer in patients with more severe illness (4). In contrast, in another study of 56 patients with mild to moderate illness (none required intensive care), the median duration of viral RNA shedding from nasal or oropharyngeal specimens was 24 days, and the longest was 42 days (5). However, as mentioned above, detectable viral RNA does not always correlate with isolation of infectious virus, and there may be a threshold of viral RNA level below which infectivity is unlikely. In the study of nine patients with mild COVID-19 described above, infectious virus was not detected from respiratory specimens when the viral RNA level was <106 copies/mL (6).
Risk of transmission from an individual with SARS-CoV-2 infection varies by the type and duration of exposure, use of preventive measures, and likely individual factors (e.g., the amount of virus in respiratory secretions).
Antibodies against the virus are induced in those who have become infected. Preliminary evidence suggests that some of these antibodies are protective, but this remains to be definitively established. It is unknown whether all infected patients develop a protective immune response and how long any protective effect will last.
Diagnosis of COVID-19 is made by detection of SARS-CoV-2 RNA by reverse transcription polymerase chain reaction (RT-PCR). Various RT-PCR assays are used around the world; different assays amplify and detect different regions of the SARSCoV-2 genome. Common gene targets include nucleocapsid (N), envelope (E), spike (S), and RNA-dependent RNA polymerase (RdRp), as well as regions in the first open reading frame (7).
Serologic tests detect antibodies to SARS-CoV-2 in the blood, and those that have been adequately validated can help identify patients who have had COVID-19. However, sensitivity and specificity are still not well defined. Detectable antibodies generally take several days to weeks to develop, for example, up to 12 days for IgM and 14 days for IgG(Si-
………………………………..
PF-07321332 (or nirmatrelvir) is an antiviral drug developed by Pfizer which acts as an orally active 3CLprotease inhibitor. The combination of PF-07321332 with ritonavir has been in phase III trials for the treatment of COVID-19 since September 2021[2][3][4] and is expected to be sold under the brand name Paxlovid.[5] After promising results preventing hospitalization and death if given within the first 3 days of symptoms, Pfizer submitted an application to the U.S. Food and Drug Administration (FDA) for emergency authorization for PF-07321332 in combination with ritonavir in November 2021.[6]
PF-07321332 is an azabicyclohexane that is (1R,5S)-3-azabicyclo[3.1.0]hexane substituted by {(1S)-1-cyano-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl}aminoacyl, 3-methyl-N-(trifluoroacetyl)-L-valinamide, methyl and methyl groups at positions 2S, 3, 6 and 6, respectively. It is an inhibitor of SARS-CoV-2 main protease which is currently under clinical development for the treatment of COVID-19. It has a role as an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor and an anticoronaviral agent. It is a nitrile, a member of pyrrolidin-2-ones, a secondary carboxamide, a pyrrolidinecarboxamide, a tertiary carboxamide, an organofluorine compound and an azabicyclohexane.
Development
Pharmaceutical
Coronaviral proteases cleave multiple sites in the viral polyprotein, usually after glutamine residues. Early work on related human rhinoviruses showed that the flexible glutamine side chain could be replaced by a rigid pyrrolidone.[7][8] These drugs had been further developed prior to the SARS CoV2 pandemic for other diseases including SARS.[9] The utility of targeting the 3CL protease in a real world setting was first demonstrated in 2018 when GC376 (a prodrug of GC373) was used to treat the previously 100% lethal cat coronavirus disease, feline infectious peritonitis, caused by Feline coronavirus.[10]
The Pfizer drug is an analog of GC373, where the aldehyde covalent cysteine acceptor has been replaced by a nitrile.[11][12]
PF-07321332 was developed by modification of an earlier clinical candidate lufotrelvir,[13][14] which is also a covalent inhibitor but its warhead is a phosphate prodrug of a hydroxyketone. However, lufotrelvir needs to be administered intravenously limiting its use to a hospital setting. Stepwise modification of the tripeptide protein mimetic led to PF-0732133, which is suitable for oral administration.[1] Key changes include a reduction in the number of hydrogen bond donors, and the number of rotatable bonds by introducing the rigid bicyclic non-canonical amino acid, which mimics the leucine residue found in earlier inhibitors. This residue had previously been used in the synthesis of boceprevir.[15]
Clinical
In April 2021, Pfizer began phase I trials.[16] In September 2021, Pfizer began a phase II/III trial.[17] In November 2021, Pfizer announced 89% reduction in hospitalizations of high risk patients studied when given within three days after symptom onset.[5]
On December 14, Pfizer announced that Paxlovid, when given within three days of symptom onset, reduced risk of hospitalization or death by 89% compared to placebo in 2,246 high risk patients studied.[18]
Chemistry and pharmacology
Full details of the synthesis of PF-07321332 were first published by scientists from Pfizer.[1]
In the penultimate step, a synthetic homochiral amino acid is coupled with a homochiral amino amide using the water-soluble carbodiimide EDCI as coupling agent. The resulting intermediate is then treated with Burgess reagent, which dehydrates the amide group to the nitrile of the product.
PF-07321332 is a covalent inhibitor, binding directly to the catalytic cysteine (Cys145) residue of the cysteine protease enzyme.[19]
In the drug combination, ritonavir serves to slow down metabolism of PF-07321332 by cytochrome enzymes to maintain higher circulating concentrations of the main drug.[20]
Public health system reactions to development
Despite not being approved yet in any country, the UK placed an order for 250,000 courses after Pfizer´s press release in October 2021,[21][22] and Australia pre-ordered 500,000 courses of the drug.[23]
As of November 2021, the US government was expected to sign a contract to buy around 10 million courses of the combination treatment.[24][25]
Legal status
In November 2021, Pfizer signed a license agreement with the United Nations–backed Medicines Patent Pool to allow PF-07321332 to be manufactured and sold in 95 countries.[26] Pfizer stated that the agreement will allow local medicine manufacturers to produce the pill “with the goal of facilitating greater access to the global population”. However, the deal excludes several countries with major COVID-19 outbreaks including Brazil, China, Russia, Argentina, and Thailand.[27][28]
On 16 November 2021, Pfizer submitted an application to the U.S. Food and Drug Administration (FDA) for emergency authorization for PF-07321332 in combination with ritonavir.[29][30][31]
Misleading comparison with ivermectin
Conspiracy theorists on the internet have claimed that Paxlovid is merely a “repackaged” version of the antiparasitic drug ivermectin, which has been erroneously promoted as a COVID-19 “miracle cure”. Their claims, sometimes using the nickname “Pfizermectin”,[32] are based on a narrative that Pfizer is suppressing the true benefits of ivermectin and rely on superficial correspondences between the drugs and a misunderstanding of their respective pharmacokinetics.[33] Paxlovid is not structurally related or similar to ivermectin, and while both are 3C-like protease inhibitors, Paxlovid is much more potent with an IC50 around 10,000 times lower, allowing for effective oral dosing within the therapeutic margin.[34]
References
- ^ Jump up to:a b c Owen DR, Allerton CM, Anderson AS, Aschenbrenner L, Avery M, Berritt S, et al. (November 2021). “An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19″. Science: eabl4784. doi:10.1126/science.abl4784. PMID 34726479. S2CID 240422219.
- ^ Vandyck K, Deval J (August 2021). “Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection”. Current Opinion in Virology. 49: 36–40. doi:10.1016/j.coviro.2021.04.006. PMC 8075814. PMID 34029993.
- ^ Şimşek-Yavuz S, Komsuoğlu Çelikyurt FI (August 2021). “Antiviral treatment of COVID-19: An update”. Turkish Journal of Medical Sciences. doi:10.3906/sag-2106-250. PMID 34391321. S2CID 237054672.
- ^ Ahmad B, Batool M, Ain QU, Kim MS, Choi S (August 2021). “Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations”. International Journal of Molecular Sciences. 22 (17): 9124. doi:10.3390/ijms22179124. PMC 8430524. PMID 34502033.
- ^ Jump up to:a b “Pfizer’s Novel COVID-19 Oral Antiviral Treatment Candidate Reduced Risk Of Hospitalization Or Death By 89% In Interim Analysis Of Phase 2/3 EPIC-HR Study”. Pfizer Inc. 5 November 2021.
- ^ Mahase E (November 2021). “Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports”. BMJ. 375: n2713. doi:10.1136/bmj.n2713. PMID 34750163. S2CID 243834203.
- ^ Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R (June 2003). “Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs”. Science. 300 (5626): 1763–1767. Bibcode:2003Sci…300.1763A. doi:10.1126/science.1085658. PMID 12746549. S2CID 13031405.
- ^ Dragovich PS, Prins TJ, Zhou R, Webber SE, Marakovits JT, Fuhrman SA, et al. (April 1999). “Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements”. Journal of Medicinal Chemistry. 42 (7): 1213–1224. doi:10.1021/jm9805384. PMID 10197965.
- ^ Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH (July 2016). “An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy”. Journal of Medicinal Chemistry. 59 (14): 6595–6628. doi:10.1021/acs.jmedchem.5b01461. PMC 7075650. PMID 26878082.
- ^ Pedersen NC, Kim Y, Liu H, Galasiti Kankanamalage AC, Eckstrand C, Groutas WC, et al. (April 2018). “Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis”. Journal of Feline Medicine and Surgery. 20 (4): 378–392. doi:10.1177/1098612X17729626. PMC 5871025. PMID 28901812.
- ^ Halford B (7 April 2021). “Pfizer unveils its oral SARS-CoV-2 inhibitor”. Chemical & Engineering News. 99 (13): 7. doi:10.47287/cen-09913-scicon3. S2CID 234887434.
- ^ Vuong W, Khan MB, Fischer C, Arutyunova E, Lamer T, Shields J, et al. (August 2020). “Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication”. Nature Communications. 11 (1): 4282. doi:10.1038/s41467-020-18096-2. PMC 7453019. PMID 32855413.
- ^ Clinical trial number NCT04535167 for “First-In-Human Study To Evaluate Safety, Tolerability, And Pharmacokinetics Following Single Ascending And Multiple Ascending Doses of PF-07304814 In Hospitalized Participants With COVID-19 ” at ClinicalTrials.gov
- ^ Boras B, Jones RM, Anson BJ, Arenson D, Aschenbrenner L, Bakowski MA, et al. (February 2021). “Discovery of a Novel Inhibitor of Coronavirus 3CL Protease for the Potential Treatment of COVID-19”. bioRxiv: 2020.09.12.293498. doi:10.1101/2020.09.12.293498. PMC 7491518. PMID 32935104.
- ^ Njoroge FG, Chen KX, Shih NY, Piwinski JJ (January 2008). “Challenges in modern drug discovery: a case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection”. Accounts of Chemical Research. 41 (1): 50–59. doi:10.1021/ar700109k. PMID 18193821. S2CID 2629035.
- ^ Nuki P (26 April 2021). “Pfizer is testing a pill that, if successful, could become first-ever home cure for COVID-19”. National Post. Archived from the original on 27 April 2021.
- ^ “Pfizer begins dosing in Phase II/III trial of antiviral drug for Covid-19”. Clinical Trials Arena. 2 September 2021.
- ^ Press release (14 December 2021). “Pfizer Announces Additional Phase 2/3 Study Results Confirming Robust Efficacy of Novel COVID-19 Oral Antiviral Treatment Candidate in Reducing Risk of Hospitalization or Death”.
- ^ Pavan M, Bolcato G, Bassani D, Sturlese M, Moro S (December 2021). “Supervised Molecular Dynamics (SuMD) Insights into the mechanism of action of SARS-CoV-2 main protease inhibitor PF-07321332”. J Enzyme Inhib Med Chem. 36 (1): 1646–1650. doi:10.1080/14756366.2021.1954919. PMC 8300928. PMID 34289752.
- ^ Woodley M (19 October 2021). “What is Australia’s potential new COVID treatment?”. The Royal Australian College of General Practitioners (RACGP). Retrieved 6 November 2021.
- ^ “Pfizer Covid pill ‘can cut hospitalisations and deaths by nearly 90%'”. The Guardian. 5 November 2021. Retrieved 17 November 2021.
- ^ Mahase E (October 2021). “Covid-19: UK stockpiles two unapproved antiviral drugs for treatment at home”. BMJ. 375: n2602. doi:10.1136/bmj.n2602. PMID 34697079. S2CID 239770104.
- ^ “What are the two new COVID-19 treatments Australia has gained access to?”. ABC News (Australia). 17 October 2021. Retrieved 5 November 2021.
- ^ “U.S. to Buy Enough of Pfizer’s Covid Antiviral Pills for 10 Million People”. The New York Times. 17 November 2021. Retrieved 17 November 2021.
- ^ Pager T, McGinley L, Johnson CY, Taylor A, Parker C. “Biden administration to buy Pfizer antiviral pills for 10 million people, hoping to transform pandemic”. The Washington Post. Retrieved 16 November 2021.
- ^ “Pfizer and The Medicines Patent Pool (MPP) Sign Licensing Agreement for COVID-19 Oral Antiviral Treatment Candidate to Expand Access in Low- and Middle-Income Countries” (Press release). Pfizer. 16 November 2021. Retrieved 17 November 2021 – via Business Wire.
- ^ “Covid-19: Pfizer to allow developing nations to make its treatment pill”. BBC News. 16 November 2021. Archived from the original on 16 November 2021. Retrieved 17 November 2021.
- ^ “Pfizer Will Allow Its Covid Pill to Be Made and Sold Cheaply in Poor Countries”. The New York Times. 16 November 2021. Retrieved 17 November 2021.
- ^ “Pfizer Seeks Emergency Use Authorization for Novel COVID-19 Oral Antiviral Candidate”. Business Wire (Press release). 16 November 2021. Retrieved 17 November 2021.
- ^ Kimball S (16 November 2021). “Pfizer submits FDA application for emergency approval of Covid treatment pill”. CNBC. Retrieved 17 November 2021.
- ^ Robbins R (5 November 2021). “Pfizer Says Its Antiviral Pill Is Highly Effective in Treating Covid”. The New York Times. ISSN 0362-4331. Archived from the original on 8 November 2021. Retrieved 9 November 2021.
- ^ Bloom J (2 December 2021). “How Does Pfizer’s Pavloxid Compare With Ivermectin?”. American Council on Science and Health. Retrieved 12 December 2021.
- ^ Gorski D (15 November 2021). “Pfizer’s new COVID-19 protease inhibitor drug is not just ‘repackaged ivermectin'”. Science-Based Medicine.
- ^ von Csefalvay C (27 November 2021). “Why Paxlovid is not Pfizermectin”. Bits and Bugs. Chris von Csefalvay. Retrieved 28 November 2021.
External links
- “PF-07321332”. Drug Information Portal. U.S. National Library of Medicine.
- “Early Data Suggest Pfizer Pill May Prevent Severe COVID-19”. National Institutes of Health. 16 November 2021.
Pfizer to make COVID-19 pill available in low- and middle-income nations
If authorized by global health authorities, the drug promises to reduce deaths and hospitalizations linked to
the novel coronavirus.
By Brian Buntz | November 16, 2021FacebookTwitterLinkedInShare
In late October, Merck (NYSE:MRK) and its partner Ridgeback Biotherapeutics agreed to make the COVID-19 antiviral molnupiravir available in the developing world.
Now, Pfizer (NYSE:PFE) is taking a similar approach for its investigational antiviral cocktail Paxlovid, which contains PF-07321332 and ritonavir.
Pfizer, like Merck, struck an agreement with the Medicines Patent Pool (MPP) related to Paxlovid.
MPP’s mission is to expand low- and middle-income countries’ access to vital medicines. The United Nations supports the organization.
Pfizer announced earlier this month that Paxlovid was 89% effective in reducing the risk of hospitalization or death in an interim analysis of the Phase 2/3 EPIC-HR trial.
The collaboration with MPP will enable generic drug makers internationally with sub-licenses to produce Paxlovid for use in 95 countries, which comprise more than half of the world’s population.
“This license is so important because, if authorized or approved, this oral drug is particularly well-suited for low- and middle-income countries and could play a critical role in saving lives, contributing to global efforts to fight the current pandemic,” said Charles Gore, executive director of MPP, in a press release. “PF-07321332 is to be taken together with ritonavir, an HIV medicine we know well, as we have had a license on it for many years, and we will be working with generic companies to ensure there is enough supply for both COVID-19 and HIV.”
At present, MPP has signed agreements with ten patient holders for 13 HIV antiretrovirals and several other drugs.
Filed Under: clinical trials, Drug Discovery, Infectious Disease
Tagged With: Medicines Patent Pool, Merck, PF-07321332, Pfizer, Ridgeback Biotherapeutics, ritonavir
PFIZER INITIATES PHASE 1 STUDY OF NOVEL ORAL ANTIVIRAL THERAPEUTIC AGENT AGAINST SARS-COV-2
Tuesday, March 23, 2021 – 11:00am
- In-vitro studies conducted to date show that the clinical candidate PF-07321332 is a potent protease inhibitor with potent anti-viral activity against SARS-CoV-2
- This is the first orally administered coronavirus-specific investigational protease inhibitor to be evaluated in clinical studies, and follows Pfizer’s intravenously administered investigational protease inhibitor, which is currently being evaluated in a Phase 1b multi-dose study in hospitalized clinical trial participants with COVID-19
NEW YORK–(BUSINESS WIRE)– Pfizer Inc. (NYSE: PFE) announced today that it is progressing to multiple ascending doses after completing the dosing of single ascending doses in a Phase 1 study in healthy adults to evaluate the safety and tolerability of an investigational, novel oral antiviral therapeutic for SARS-CoV-2, the virus that causes COVID-19. This Phase 1 trial is being conducted in the United States. The oral antiviral clinical candidate PF-07321332, a SARS-CoV2-3CL protease inhibitor, has demonstrated potent in vitro anti-viral activity against SARS-CoV-2, as well as activity against other coronaviruses, suggesting potential for use in the treatment of COVID-19 as well as potential use to address future coronavirus threats.
“Tackling the COVID-19 pandemic requires both prevention via vaccine and targeted treatment for those who contract the virus. Given the way that SARS-CoV-2 is mutating and the continued global impact of COVID-19, it appears likely that it will be critical to have access to therapeutic options both now and beyond the pandemic,” said Mikael Dolsten, MD, PhD., Chief Scientific Officer and President, Worldwide Research, Development and Medical of Pfizer. “We have designed PF-07321332 as a potential oral therapy that could be prescribed at the first sign of infection, without requiring that patients are hospitalized or in critical care. At the same time, Pfizer’s intravenous antiviral candidate is a potential novel treatment option for hospitalized patients. Together, the two have the potential to create an end to end treatment paradigm that complements vaccination in cases where disease still occurs.”
Protease inhibitors bind to a viral enzyme (called a protease), preventing the virus from replicating in the cell. Protease inhibitors have been effective at treating other viral pathogens such as HIV and hepatitis C virus, both alone and in combination with other antivirals. Currently marketed therapeutics that target viral proteases are not generally associated with toxicity and as such, this class of molecules may potentially provide well-tolerated treatments against COVID-19.
The Phase 1 trial is a randomized, double-blind, sponsor-open, placebo-controlled, single- and multiple-dose escalation study in healthy adults evaluating the safety, tolerability and pharmacokinetics of PF-07321332.
Initiation of this study is supported by preclinical studies that demonstrated the antiviral activity of this potential first-in-class SARS-CoV-2 therapeutic designed specifically to inhibit replication of the SARS-CoV2 virus. The structure of PF-07321332, together with the pre-clinical data, will be shared in a COVID-19 session of the Spring American Chemical Society meeting on April 6.
Pfizer is also investigating an intravenously administered investigational protease inhibitor, PF-07304814, which is currently in a Phase 1b multi-dose trial in hospitalized clinical trial participants with COVID-19.
About Pfizer: Breakthroughs That Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.Pfizer.com. In addition, to learn more, please visit us on www.Pfizer.com and follow us on Twitter at @Pfizer and @Pfizer News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
.CLIP
https://cen.acs.org/content/cen/articles/99/i13/Pfizer-unveils-oral-SARS-CoV.html

Drugmaker Pfizer revealed its oral COVID-19 antiviral clinical candidate PF-07321332 on Tuesday at the American Chemical Society Spring 2021 meeting. The compound, which is currently in Phase 1 clinical trials, is the first orally administered compound in the clinic that targets the main protease (also called the 3CL protease) of SARS-CoV-2, the virus that causes COVID-19. By inhibiting the main protease, PF-07321332 prevents the virus from cleaving long protein chains into the parts it needs to reproduce itself. Dafydd Owen, director of medicinal chemistry at Pfizer, presented the compound in a symposium of the Division of Medicinal Chemistry.
Last year, Pfizer reported PF-07304814, a different small molecule inhibitor of SARS-CoV-2’s main protease. The work to develop that compound began during the 2002-2003 outbreak of SARS-CoV, severe acute respiratory syndrome. But that molecule can only be given intravenously, which limits its use to hospital settings.
Because PF-07321332 can be taken orally, as a pill or capsule, it could be given outside of hospitals if it proves to be safe and effective. People who have been exposed to SARS-CoV-2 could take it as a preventative measure, for example.
“For the foreseeable future, we will expect to see continued outbreaks from COVID-19. And therefore, as with all viral pandemics, it’s important we have a full toolbox on how to address it,” Charlotte Allerton, Pfizer’s head of medicine design, told C&EN.
PF-07321332 was developed from scratch during the current pandemic. It’s a reversible covalent inhibitor that reacts with one of the main protease’s cysteine residues. Owen also discussed the chemistry involved in scaling up the compound. The first 7 mg of the compound were synthesized in late July 2020. Encouraged by the early biological data, the Pfizer team aimed to scale up the synthesis. By late October, they’d made 100 g of the compound. Just two weeks later, the chemists had scaled up the synthesis to more than 1 kg. Owen said 210 researchers had worked on the project. Ana Martinez, who studies COVID-19 treatments at the Spanish National Research Council CSIC and also presented during the symposium, told C&EN that having a COVID-19 antiviral is of critical importance. She eagerly anticipates the safety and efficacy data from the trials of PF-07321332. “Hopefully we will have a new drug to fight against COVID-19,” Martinez said. And because the molecule targets the main protease, she said that it might be useful for fighting other coronaviruses and preventing future pandemics.Chemical & Engineering News
| Clinical data | |
|---|---|
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2628280-40-8 |
| PubChem CID | 155903259 |
| UNII | 7R9A5P7H32 |
| KEGG | D12244 |
| ChEBI | CHEBI:170007 |
| Chemical and physical data | |
| Formula | C23H32F3N5O4 |
| Molar mass | 499.535 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 192.9[1] °C (379.2 °F) |
| showSMILES | |
| showInChI |
Xray crystal structure (PDB:7SI9 and 7VH8) of the SARS-CoV-2 protease inhibitor PF-07321332 bound to the viral 3CLpro (Mpro) protease enzyme. Ribbon diagram of the protein with the drug shown as sticks. The catalytic residues (His41, Cys145) are shown as yellow sticks.
./////////////////PF-07321332, PF 07321332, COVID 19, CORONA VIRUS, SARS-CoV-2 inhibitor, PHASE 1, nirmatrelvir, PAXLOVID, CORONA VIRUS, COVID 19
C1N(C([C@@H]2C1[C@]2(C)C)C(=O)N[C@@H](CC3C(NCC3)=O)C#N)C(C([C@@](C)(C)C)NC(=O)C(F)(F)F)=O
C1N(C(C2C1C2(C)C)C(=O)N[C@@H](CC3C(NCC3)=O)C#N)C(C([C@@](C)(C)C)NC(=O)C(F)(F)F)=O
NEW DRUG APPROVALS
ONE TIME
$10.00
ABBV 744
ABBV 744
N-Ethyl-4-[2-(4-fluoro-2,6-dimethylphenoxy)-5-(1-hydroxy-1-methylethyl)phenyl]-6,7-dihydro-6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide
1H-Pyrrolo[2,3-c]pyridine-2-carboxamide, N-ethyl-4-[2-(4-fluoro-2,6-dimethylphenoxy)-5-(1-hydroxy-1-methylethyl)phenyl]-6,7-dihydro-6-methyl-7-oxo-
| Molecular Weight |
491.55 |
|---|---|
| Formula |
C₂₈H₃₀FN₃O₄ |
| CAS No. |
2138861-99-9 |
ABBV-744 is a highly BDII-selective BET bromodomain inhibitor, used in the research of inflammatory diseases, cancer, and AIDS.
Acute Myeloid Leukemia (AML)
Phase I, AbbVie is evaluating oral agent ABBV-744 in early clinical trials for the treatment of metastatic castration resistant prostate cancer (CRPC) and for the treatment of relapsed or refractory acute myeloid leukemia (AML).


PATENT
WO 2017177955
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017177955&tab=FULLTEXT
Bromodomains refer to conserved protein structural folds which bind to N-acetylated lysine residues that are found in some proteins. The BET family of bromodomain containing proteins comprises four members (BRD2, BRD3, BRD4 and BRDt) . Each member of the BET family employs two bromodomains to recognize N-acetylated lysine residues typically, but not exclusively those found on transcription factors (Shi, J., et al. Cancer Cell 25 (2) : 210-225 (2014) ) or on the amino-terminal tails of histone proteins. Numbering from the N-terminal end of each BET protein the tandem bromodomains are typically labelled Binding Domain I (BDI) and Binding Domain II (BDII) . These interactions modulate gene expression by recruiting transcription factors to specific genome locations within chromatin. For example, histone-bound BRD4 recruits the transcription factor P-TEFb to promoters, resulting in the expression of a subset of genes involved in cell cycle progression (Yang et al., Mol. Cell. Biol. 28: 967-976 (2008) ) . BRD2 and BRD3 also function as transcriptional regulators of growth promoting genes (LeRoy et al., Mol. Cell 30: 51-60 (2008) ) . BET family members were recently established as being important for the maintenance of several cancer types (Zuber et al., Nature 478: 524-528 (2011) ; Mertz et al; Proc. Nat’l. Acad. Sci. 108: 16669-16674 (2011) ; Delmore et al., Cell 146: 1-14, (2011) ; Dawson et al., Nature 478: 529-533 (2011) ) . BET family members have also been implicated in mediating acute inflammatory responses through the canonical NF-KB pathway (Huang et al., Mol. Cell. Biol. 29: 1375-1387 (2009) ) resulting in the upregulation of genes associated with the production of cytokines (Nicodeme et al., Nature 468: 1119-1123, (2010) ) . Suppression of cytokine induction by BET bromodomain inhibitors has been shown to be an effective approach to treat inflammation-mediated kidney disease in an animal model (Zhang, et al., J. Biol. Chem. 287: 28840-28851 (2012) ) . BRD2 function has been linked to pre-disposition for dyslipidemia or improper regulation of adipogenesis, elevated inflammatory profiles and increased susceptibility to autoimmune diseases (Denis, Discovery Medicine 10: 489-499 (2010) ) . The human immunodeficiency virus utilizes BRD4 to initiate transcription of viral RNA from stably integrated viral DNA (Jang et al., Mol. Cell, 19: 523-534 (2005) ) . BET bromodomain inhibitors have also been shown to reactivate HIV transcription in models of latent T cell infection and latent monocyte infection (Banerjee, et al, J. Leukocyte Biol. doi: 10.1189/jlb. 0312165) . BRDt has an important role in spermatogenesis that is blocked by BET bromodomain inhibitors (Matzuk, et al., Cell 150: 673-684 (2012) ) . Thus, compounds that inhibit the binding of BET family bromodomains to their cognate acetylated lysine proteins are being pursued for the treatment of cancer, inflammatory diseases, kidney diseases, diseases involving metabolism or fat accumulation, and some viral infections, as well as for providing a method for male contraception. Accordingly, there is an ongoing medical need to develop new drugs to treat these indications.
FIDANZE, Steven D., et al. BROMODOMAIN INHIBITORS. WO 2017177955 A1.
////////////ABBV 744, Acute Myeloid Leukemia, AML, Phase 1 , AbbVie
CC(O)(C)C1=CC(C(C2=C3NC(C(NCC)=O)=C2)=CN(C)C3=O)=C(OC4=C(C)C=C(F)C=C4C)C=C1
LNS-8801 (SRR G-1)
LNS-8801 (SRR G-1)
CAS 925419-53-0
Chemokine receptor-like 2 agonist
1-((3aS,4R,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-1-one
- 1-[(3aS,4R,9bR)-4-(6-Bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]ethanone
- G 1
- G 1 (estrogen receptor agonist)
Linnaeus Therapeutics Inc
NOTE 1-((3aR,4S,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-1-one (RSS G-1 or LNS8812)
Novel crystalline forms of an enantiomerically purified LNS-8801 (SRR G-1), or a derivative useful for treating cancer, hearing disorder, depression and myocardial infarction.
Linnaeus Therapeutics is developing LNS-8801, a G-protein coupled estrogen receptor (GPER) agonist, an oral capsule formulation for the treatment of cancer including melanoma, pancreatic ductal adenocarcinoma, non-small cell lung cancer, solid tumor, hematological cancer, advanced cancer and colon carcinoma.
In October 2019, a phase I trial was initiated in patients with locally advanced or metastatic cancer.
PATENT
WO-2020023391
Novel crystalline and amorphous forms of 1-((3aS,4R,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-1-one (here referred to as SRR-G-1 or LNS-8801) and 1-((3aS,4R,9bR)-4-(6-bromobenzo[d][1,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-1-one (RSS G-1 or LNS8812) and its derivative (having chiral purity of >90%; substantially free of its opposite enantiomer; designated as Form A-C) and their co-crystal (eg benzenesulfonic acid) and compositions and combinations comprising them are claimed. Also claimed is their use for treating cancers (disorder expresses GPER), depression, myocardial infarction, osteoporosis, rheumatoid arthritis, insomnia, inflammatory bowel disease, Crohn’s disease, thymic atrophy and hearing disorder and further type-2 diabetes. Further claimed are methods for reducing the likelihood of pregnancy after intercourse, increasing skin pigmentation and skin protection and preventing cancer, reoccurrence of cancer and inhibiting the progression of cancer
Embodiments of the present invention relate to an enantiomerically purified agonist of the G-protein coupled estrogen receptor (GPER), pharmaceutical compositions comprising an enantiomerically purified SRR G-l, or a derivative thereof, and methods of treating disease states and conditions in subjects in need thereof, and methods of treating disease states and conditions mediated through GPER receptors.
[0003] Estrogens mediate multiple complex physiological responses throughout the body. The responses are in turn mediated through the binding of estrogen to receptors. The classical receptors bind steroids, such as estrogen, and are soluble cytoplasmic/nuclear proteins that function as transcription factors. These receptors are known as estrogen receptor alpha and beta (two closely related proteins) that mediate transcriptional activity. GPER is a 7-transmembrane G protein-coupled receptor that also binds to estrogen with high affinity (Kd~6 nM) and mediates rapid cellular responses including cyclic adenosine monophosphate signaling, calcium mobilization and phosphatidylinositol 3,4,5 trisphosphate production.
[0004] Diseases whose development, progression, and or response to therapy, may be influenced by endogenous, and/or pharmacologic activation of GPER signaling include cancer (including the prevention of cancer, prevention of the reoccurrence of cancer, and the inhibition of the progression of cancer; and particularly melanoma, pancreatic, lymphomas, uveal melanoma, non-small cell lung cancer, breast, reproductive and other hormone-dependent cancers, leukemia, colon cancer, prostate, bladder cancer), reproductive (genito-urological) including endometritis, prostatitis, polycystic ovarian syndrome, bladder control, hormone-related disorders, hearing disorders, cardiovascular conditions including hot flashes and profuse sweating, hypertension, stroke, obesity, diabetes, osteoporosis, hematologic diseases, vascular diseases or conditions such as venous thrombosis, atherosclerosis, among numerous others and disorders of the central and peripheral nervous system, including depression, insomnia, anxiety, neuropathy, multiple sclerosis, neurodegenerative disorders such as Parkinson’s disease and Alzheimer’s disease, as well as inflammatory bowel disease, Crohn’s disease, coeliac (celiac) disease and related disorders of the intestine.
Embodiments of the present invention encompass compounds comprising enantiomerically purified G-l and methods of use in the treatment of diseases. G-l is a racemic mixture of the enantiomers l-((3aS,4R,9bR)-4-(6-bromobenzo[if][l,3]dioxol-5-yl)-3a.4.5.9b-tetrahydro-3H-cyclopenta|c |quinolin-8-yl)ethan- 1 -one (henceforth referred to as “SRR G-l” or “LNS8801”) and l-((3aR,4S,9bS)-4-(6-bromobenzo[if][l,3]dioxol-5-yl)-3a.4.5.9b-tetrahydro-3H-cyclopenta|c |quinolin-8-yl)ethan- 1 -one (henceforth referred to as “RSS G-l” or“LNS8812”).
Enantiomerically purified G-l has been purified in favor of its l-((3aS,4R,9bR)-4-(6-bromobenzo[ri][l,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-l-one enantiomer over the corresponding l-((3aR,4S,9bS)-4-(6-bromobenzo[ri][l,3]dioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-l-one enantiomer. Unless specifically described, SRR Gl, or a derivative thereof includes, any physical form, including an amorphous form or any crystalline solid forms such as A, B, C or combinations thereof.
Experimental Section
Scheme 1
[0166] A synthesis of G-l is described in Org. Biomol Client., 2010,8, 2252-2259, which is hereby incorporated by reference, and depicted in Scheme 1. A catalytic amount of Sc(OTf)3 (0.492 g, 1.0 mmol) in anhydrous acetonitrile (2.0 cm3) was added to the mixture of 6-bromopiperonal (2.30 g, 10.0 mmol). / aminoacetophenone (1.30 g, 10.0 mmol) and cyclopentadiene (3.30 g, 50.0 mmol) in acetonitrile (25 cm3). The reaction mixture was stirred at ambient temperature (~23 °C) for 2.0 h. The volatiles were removed in vacuo. The residue was purified by preparative silica gel column chromatography using ethyl acetate-hexanes (10 : 90) to provide G-l (4.03 g, 98%, dr. = 94 : 6) as a white solid. The minor diastereomer was substantially removed by recrystallization to yield a racemic mixure of SRR G-l and RSS G-l.
Example 1: Isolation of the SRR G-l and RSS G-l Enantiomers
[0167] Starting with a highly purified sample of G-l, (±)l-(4-(6-bromobenzo[ri][l,3]dioxol-5-yl)-(3aS*,4R*,9bR*)-tetrahydro-3H-cyclopenta[c]quinolin-8-yl)ethan-l-one, (99.4% purity) purchased from Tocris Bioscience, the material was dissolved in 90: l0:0. l(v/v/v) methyl tert-butyl ether / ethanol / diethyl amine and subjected to preparative chromatography using a column packed with Chrialpak 1A resin. Elution was conducted with 90: l0:0. l(v/v/v) methyl tert-butyl ether / ethanol / diethyl amine and the fractions corresponding to each enantiomer were collected and concentered to a solid. The early eluting enantiomer was determined to be the SRR G-l enantiomer by single crystal x-ray structural analysis.
Example 2: SRR G-l Polymorph Screen
[0168] Starting with SRR G-l prepared according to Example 1, a polymorph screening study was conducted analyzing the solids isolated from slurry of the solid, or from fast and slow evaporation and cooling of solutions (Table 1). Two crystal forms were identified, an anhydrous form designated Form A and mono dichloromethane solvate designated Form B. On exposure to elevated temperature the Form B crystal form desolvates to form the Form C crystal form. Amorphous material was generated from purified SRR G-l by two different methods; quick evaporating a diethyl ether solution of SRR G-l or rotary evaporating from a solution of a dichloromethane solution of SRR G-l.
Table 1
PAPER
Anti-Cancer Agents in Medicinal Chemistry (2019), 19(6), 760-771.

/////////RSS G-1, LNS8812, phase I, Locally advanced, metastatic cancer, LNS-8801, SRR G-1, Linnaeus Therapeutics
CC(=O)c1cc4c(cc1)N[C@@H](c2cc3OCOc3cc2Br)[C@H]5CC=C[C@@H]45
CC-90010
CC-90010
C21 H21 N O4 S, 383.46
CAS 1706738-98-8
1(2H)-Isoquinolinone, 4-[2-(cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]-2-methyl-
- 4-[2-(Cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]-2-methyl-1(2H)-isoquinolinone
- 4-[2-(Cyclopropylmethoxy)-5-(methanesulfonyl)phenyl]-2-methylisoquinolin-1(2H)-one
- 4-[2-(Cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one
Quanticel Pharmaceuticals Inc, Michael John BennettJuan Manuel BetancortAmogh BoloorStephen W. KaldorJeffrey Alan StaffordJames Marvin Veal
![]()
Celgene (now a wholly owned subsidiary of Bristol-Myers Squibb ) , following its acquisition of Quanticel , is developing CC-90010, an oral inhibitor of BET (bromodomain and extraterminal) proteins, for the potential treatment of solid tumors and non-Hodgkin’s lymphoma. In August 2019, a phase I trial for diffuse astrocytoma, grade III anaplastic astrocytoma and recurrent glioblastoma was planned
PATENT
WO2018075796 claiming solid composition comprising a bromodomain inhibitor, preferably 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one in crystalline form A.
PATENT
WO2015058160 (compound 89, page 103).

Example 89: 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
Step 1 : 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one
[00344] A suspension of 4-bromo-2-methylisoquinolin-l-one (100 mg, 0.42 mmol), bis(pinacolato)diboron (214 mg, 0.84 mmol), Pd(dppf)Cl2 (31 mg, 0.04 mmol) and potassium acetate (104 mg, 1.05 mmol) in dioxane (2 mL) under nitrogen was warmed up to 90 °C for 135 minutes. It was then cooled down to room temperature and diluted with ethyl acetate (8 mL). The mixture was washed with aqueous saturated solution of NaHC03 (8 mL) and brine (8 mL). The organic phase was separated, dried over Na2S04, filtered and concentrated under reduced pressure. The residue was purifed by normal phase column chromatography (10-90% EtOAc/Hexanes) to give the title compound (44 mg, 37%). 1H NMR (CDC13, 400 MHz) δ 8.43 (d, J = 7.9 Hz, 1 H), 8.40 (dd, J = 8.2 Hz, 0.9 Hz, 1 H), 7.68 (s, 1 H), 7.65 (ddd, J = 8.2, 8.2, 1.1 Hz, 1 H), 7.46 (t, J = 7.5 Hz, 1 H), 3.63 (s, 3H), 1.38 (s, 12H). LCMS (M+H)+ 286. Step 2: 4-[2-(cyclopropylmethox -5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00345] The title compound was prepared in a manner similar to Example 18, step 3, substituting 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene for 4-bromo-2-methylisoquinolin-l(2H)-one and 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one for N-benzyl-2-methoxy-5-(tetramethyl-l,3,2-dioxaborolan-2-yl)benzamide. 1H NMR (DMSO-d6, 400 MHz) δ 0.09 (m, 2 H), 0.29 (m, 1H), 0.35 (m, 1H),
0.94 (m, 1H), 3.22 (s, 3H), 3.57 (s, 3H), 3.95 (m, 2H), 7.16 (d, J = 7.9 Hz, 1H), 7.37 (d, J =
8.8 Hz, 1H), 7.53 (m, 2H), 7.65 (t, J = 7.6 Hz, 1H), 7.81 (d, J = 2.4 Hz, 1H), 7.97 (dd, J = 8.8,
2.4 Hz, 1H), 8.30 (d, J = 8.1 Hz, 1H). LCMS (M+H)+ 384.
[00346] Alternatively, 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one can be prepared as described below.
Step 1 : 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one
[00347] A mixture of 4-bromo-2-methylisoquinolin-l-one (8.0 g, 33.6 mmol),
bis(pinacolato)diboron (17.1 g, 67.2 mmol), KOAc (6.6 g, 67.2 mmol), Pd2(dba)3 (3.1 g, 3.36 mmol) and X-Phos (1.6 g, 3.36 mmol) in anhydrous dioxane (200 mL) was stirred at 60 °C for 12 h. The reaction mixture was concentrated and the residue was purified by column chromatography on silica gel (PE : EA = 15 : 1) to give the title compound (6.0 g, 62 %) as a solid.
Step 2: 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00348] The title compound from Step 1 (5.0 g, 17.5 mmol), 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene (6.4 g, 21 mmol), K3PO4 (9.3 g, 43.9 mmol) and Pd(dppf)Cl2 (1.4 g, 1.75 mmol) in a dioxane/water (100 mL / 10 mL) mixture were stirred at 60 °C for 12 hrs. The reaction mixture was concentrated under reduced pressure and the residue was purified by column chromatography on silica gel (EA : DCM = 1 : 4).
Appropriate fractions were combined and concentrated under reduce pressure. The resultant solid was recrystallized from DCM / MTBE (1 : 1, 50 mL) to give the title compound (4.0 g, 60 %) as a white solid. 1H NMR: (CDC13, 400 MHz) δ 8.51 (dd, Ji = 8.0 Hz, J2 = 0.8 Hz, 1 H), 7.98 (dd, Ji = 8.4 Hz, J2 = 2.4 Hz, 1 H), 7.86 (d, J = 2.4 Hz, 1 H), 7.53 (m, 2 H), 7.16 (d, J = 7.6 Hz, 1 H), 7.10 (m, 2 H), 3.88 (m, 2 H), 3.66 (s, 3 H), 3.09 (s, 3 H), 1.02-0.98 (m, 1 H), 0.44-0.38 (m, 2 H), 0.11-0.09 (m, 2 H). LCMS: 384.1 (M+H)+
Patent
WO-2020023438
A process for preparing bromodomain inhibitor, particularly 4-[2(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one (having HPLC purity of 99%; compound 1; (hereafter referred to as C-90010)) and its hydrates, solvates, prodrugs and salts comprising the reaction of a substituted 4-(methylsulfonyl)phenol compound with a quinoline derivative, followed by purification is claimed. Also claimed are novel intermediates of CC-90010 and their processes for preparation. Further claimed are novel crystalline form of CC-90010. CC-90010 is known and disclosed to be a bromodomain containing protein inhibitor, useful for treating cancer.
Scheme 10: Synthesis of Compound 1
[0090] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compound 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223g, l.OSmol). Agitation
was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum purged three times with nitrogen. The batch was heated to about 65 to about 75 °C (or any temperature in between and including these two values) and contents stirred for at least about 16 hours until reaction was complete by HPLC analysis. The batch was cooled to about 60 to about 70 °C (or any temperature in between and including these two values), agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at about 60 to about 70°C (or any temperature in between and including these two values). Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at about 60 to about 70°C (or any temperature in between and including these two values). After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at about 60 to about 70°C (or any temperature in between and including these two values). The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to about 60 to about 65°C (or any temperature in between and including these two values) and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at about 60 to about 65°C (or any temperature in between and including these two values). The slurry was cooled to about 15 to about 25°C (or any temperature in between and including these two values) over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at about 40°C with nitrogen purge. Yield: 139g of 1.
[0091] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox et al., Chem. Soc. Rev., 43: 412 (2014). Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used.
Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used.
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cb (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 fCC-900101 bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCh(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 (CC-90010! bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
33 -a
and Form 5. Supporting data (XRPD, DSC, photomicroscopy) for all three forms is provided in the examples below.
[0093] The stmcture of Compound 1 (CC-90010) is shown below:
Example 1: Synthesis of Compound 1
[0217] Synthesis of compound 1 was accomplished according to Scheme 1 below. Referring to Scheme 1, synthesis commenced with bromination of starting material 4-(methylsulfonyl)phenol 4, to produce compound 5. Compound 5 was O-alkylated with (bromomethyl)cyclopropane to produce compound 6. Boronate Compound 2 was then formed by borylation of Compound 6 with Pd catalyst and bis(pinacolato)diboron to produce transient Compound 7, which was subsequenctly treated with diethanolamine (DBA) to afford cross-coupling partner Compound 2. Cross-coupling partner Compound 3 was formed in one pot starting from commercially available Compound 8. Compound 8 was N-methylated and brominated to afford Compound 3. Compounds 2 and 3 were cross-coupled (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)) to afford the target compound 1.
Scheme 1: Synthesis of compound 1
1.1: Bromination of 4
[0218] The bromination of Compound 4 to produce Compound 5 itself is simple, however stopping at the mono-brominated Compound 5 was challenging. The bis-brominated Compound 5-a (see Scheme 2 below) is a particularly pernicious impurity as it couples downstream to form a di ffi cult-to-purge impurity.
Scheme 2: Bromination of Compound 4
[0219] The key to high purity with reasonable yield was to exploit the solubility differences of the starting material Compound 4 (46 mg/ml at 20 °C) and the product Compound 5 (8 mg/ml) in CH2CI2. These solubility differences are summarized in Table 3 below.
[0220] This solubility difference is exploited by performing the reaction at a high
concentration to drive Compound 5 out of solution once formed, thereby minimizing its ability to react further with the brominating reagent to form Compound 5-a diBr. The reaction is seeded with Compound 5 to initiate its crystallization.
[0221] In Fig. 22 (Conversion of Compound 4 to Compound 5: Effect of Sulfuric Acid) it can be seen that in the absence of acid the initial reaction to Compound 5 is rapid, however the conversion plateaus at about 30% Compound 5. The main side product was found to be the impurity Compound 5-a diBr (see Fig. 23: Conversion of Compound 5 and Compound 5-a diBr: No H2SO4). Addition of increasing amounts of sulfuric acid leads to a higher conversion to desired Compound 5.
[0222] Fig. 24 (Compound 4 to Compound 5 Reaction Profile: Portion-wise Addition of NBS, Seeding) depicts further reaction control. The portion-wise addition ofNBS after addition of catalytic sulfuric acid minimizes the temperature rise, and the addition of Compound 5 after an initial NBS charge promotes the reactive crystallization of Compound 5. After about 6 to 7 hours of reaction it can be seen that the major product is Compound 5, with only a small (<5%) of the di-brominated impurity formed. In contrast, in a reaction where Compound 4 and all of the NBS were charged followed by the addition of 4 volumes of methylene chloride, a rapid exotherm resulted and undesired Compound 5-a diBr was found to be the major product.
[0223] Thus, the reaction was run under a high concentration in CH2CI2 with a portion-wise solid addition of NBS (to control both availability of the electrophile and the exotherm). An end of reaction slurry sample typically showed not more than 5% of the starting material Compound 4 remaining. After filtration the crude cake was washed with cold CH2CI2 and the OkCk-washed filter cake contained not more than 0.5% by weight dibrominated Compound 5-a. It also contained a large amount of HPLC-silent succinimide.
[0224] The following procedure was carried out: Compound 4 (25g, 145mmol) followed by CH2CI2 (lOOmL) were added to a reaction vessel and agitated. The batch was adjusted to 17 °C to 23 °C. Sulfuric acid was charged (2.7mL, Slmmol) to the batch maintaining 17 °C to 23°C. The batch was stirred at 17 °C to 23 °C for 10 minutes to 20 minutes. The first portion of A-bromosuccimide (NBS) was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The second portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The batch was seeded with
Compound 5 (0.02wt) and stirred for ca. 30 min at 17 °C to 23 °C to induce crystallization.
[0225] The third portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23 °C and stirred for at least 30 min. NBS (6.5g, 36.5 mmol) was charged to the batch at 17 °C to 23 °C and stirred for at least 30 min. Additional CH2CI2 was charged (50mL) to the batch while maintaining 17 °C to 23 °C to aid in agitation and transfer for filtration. The batch was stirred at 17 °C to 23 °C until complete by HPLC analysis (~20 – 40 h). The product was collected by suction filtration. The filter cake was slurry washed with CH2CI2 (3 x 50mL) at 17 °C to 23 °C (target 20 °C). The filter cake was slurry washed with purified water (3.0vol) at 65 °C to 75 °C for 2 to 3 hours. Then, the filter cake was slurry washed with purified water (3 x 1.0 vol, 3 x 1.0 wt) at 17 °C to 23°C. The wet cake was dried under vacuum with nitrogen bleed at 60 °C. Yield: 27g 5 (74% molar) >97% by weight. ¾ NMR (500 MHz, de-DMSO) 8.01 (1H, d, 4J = 2.1 Hz, RO-Ar meta- H ), 7.76 (1H, dd, J = 8.6 and 4J = 2.1 Hz, RO-Ar meta-H ), 7.14 (1H, d, J = 8.6 Hz, RO-Ar ortho- H), 3.38 (1H, br s, OH), 3.20 (3H, s,
CHJ); MS (ES-) calc. 249/251; found 249/251. Melting point (MP): (DSC) 188 °C.
[0226] The above procedure allowed for the following modifications. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as acetonitrile, tetrahydrofuran, or 2- methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from about 2X vol to about 20 X vol (with respect to Compound 4). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 1.9 equiv. Bromination reagent addition: The brominating reagent can be added all at once, portion wise in about 2 to about 20 portions, or continuously. The addition times can vary from about 0 to about 72 hours. Temperature: Reaction temperatures from about 0 °C to about 40 °C could be used. Acids: Different acids can be envisioned, including benzenesulfonic acid, para-toluenesulfonic acid, triflic acid, hydrobromic acid, and trifluoroacetic acid. Isolation: Instead of directly filtering the product and washing with methylene chloride and water, at the end of reaction an organic solvent capable of dissolving Compound 5 could be charged, followed by an aqueous workup to remove succinimide, and addition of an antisolvent or solvent exchange to an appropriate solvent to crystallize Compound 4. Drying: A temperature range of about 10 to about 60 °C could be used for drying.
[0227] An alternative process to Compound 5 has also been developed. This process is advantageous in that it does not use a chlorinated solvent, and provides additional controls over the formation of the Compound 5-a dibromo impurity. See Oberhauser, T. J Org. Chem 1997, 62, 4504-4506. The process is as follows. Compound 4 (10 g, 58 mmol) and acetonitrile (100 ml) were charged to the reactor and agitated. The batch was cooled to -20 °C. Triflic acid (CF3SO3H or TfOH, 5.5 mL, 62 mmol) was charged while maintaining a batch temperature of -10 to -25 °C. N-bromosuccinimide was charged (NBS, 11.4 g, 64 mmol), stirred at -10 to -25 °C for 30 minutes, then warmed to 20 °C over 3 to 4 hours. Agitation was continued at 15 °C to 25 °C until reaction completion. If the reaction conversion plateaued before completion, the reaction was cooled to -5 to -15 °C, and additional NBS was added, the amount based off of unreacted starting material, followed by warming to 15 °C to 25 °C and reacting until complete.
[0228] After reaction completion, the batch was warmed to 40 °C to 50 °C and concentrated under reduced pressure to 40 mL. The batch was cooled to -5 °C to -15 °C and the resulting product solids were filtered off. The solids were slurry washed three times, each with 20 mL water, for at least 15 minutes. The final cake was dried at 50 °C to 60 °C under reduced pressure to furnish 10 g of 5 containing less than 0.1% MeCN, 0.07% water, and 0.1% triflic acid (TfOH) by weight.
[0229] Alternatives to the above procedure employing MeCN and TfOH are as follows. Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination Reagent Stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 2 equiv. Drying: A temperature range of about 10 °C to about 60 °C could be used for drying.
[0230] The impurity 5-a is was prepared and characterized as follows. 10 g of Compound 4 and sulfuric acid (35 mol%) were dissolved in MeOH (10 vol). The mixture was set to stir at 20 °C to 25 °C for 5-10 min and 2.0 equivalents of NBS were charged in one portion. The resulting yellow mixture was stirred for three days at 20-25 °C. The batch was concentrated under reduced pressure and the resulting solid was slurried in water at 95-100 °C for 3 hours. After a second overnight slurry in CH2CI2 at room temperature, the batch was filtered and dried to give a white solid 5-a (15.0 g, 78%). ¾ NMR (500 MHz, de-DMSO), 8.05 (2H, s, ArH), 3.40 (1H, br s, HO-Ar), 3.28 (3H, s, CH3); MS (ES‘) calc. 327/329/331; found
327/329/331; MP (DSC): 226 °C (onset 221 °C, 102 J/g); lit. 224-226 °C.
1.2: O-alkylation of 5 to produce 6
[0231] Compound 6 was prepared according to Scheme 7 below.
Scheme 7: O-alkylation of 5 to produce 6
[0232] Compound 5 (100 g, 398 mmol) and methyl ethyl ketone (MEK, 700 mL) were charged to the reaction vessel and agitated. Potassium carbonate (K2CO3, 325 mesh 82.56 g, 597 mmol) was then charged to the stirred reaction vessel at 15 °C to 25 °C.
Bromomethylcyclopropane (64.4 mL, 664 mmol) was charged to the reaction vessel over at least 1 hour, maintaining the temperature between 15 °C to 25 °C. MEK (200 mL) was added into the reactor and the reactor heated to 65 to 75 °C. The contents of the reaction vessel were stirred at 65 to 75°C for approximately 10 hours until reaction was complete by HPLC analysis. Water (3.0 vol, 3.0wt) was charged to the vessel maintaining the temperature at 65 to 75 °C. The batch was stirred at 65 to 75 °C. The phases were allowed to separate at 65°C to 75 °C and the lower aqueous phase was removed. Water (300 mL) was charged to the vessel maintaining the temperature at 65 °C to 75 °C. The batch was agitated for at least 10 minutes at 65 to 75 °C. The phases were allowed to separate at 65 °C to 75 °C and the lower aqueous phase was removed. The water wash was repeated once. The temperature was adjusted to 40 to 50°C. The mixture was concentrated to car. 500 mL under reduced pressure. The mixture was distilled under reduced pressure at up to 50 °C with MEK until the water content was <1.0% w/w. n-heptane (500mL) was charged to the vessel maintaining the temperature at 40 to 50 °C. The mixture was continuously distilled under vacuum with n-heptane (300mL), maintaining a 1L volume in the reaction vessel. Compound 6 seeds (0.0 lwt) were added at 40 to 50 °C. The mixture was continuously distilled under reduced pressure at up to 50 °C with n-heptane (300mL) while maintaining 1L volume in the reactor. The batch was cooled to 15 to 25 °C and aged for 2 hours. The product was collected by suction filtration. The filter cake was washed with a solution of 10% MEK in n-heptane (5vol) at 15 to 25°C. The filter cake was dried under reduced pressure at up to 40 °C under vacuum with nitrogen flow to afford 95g of 6. 1H NMR (500 MHz, de-DMSO) 8.07 (1H, d, 4J = 2.2 Hz, ArH), 7.86 (1H, d, J = 8.7 Hz, meta-ArH), 7.29 (1H, d, J = 8.8 Hz, ortho-AiK),
4.04 (2H, d, J = 6.9 Hz, OCH2CH), 3.21 (3H, s, CH3), 1.31-1.24 (1H, m, OCH), 0.62- 0.58 (2H, m, 2 x CHCHaHb), 0.40-0.37 (2H, m, 2 x CHC¾Hb); MS (ES+) calc. 305/307; found 305/307; MP: (DSC) 93 °C.
[0233] The following modifications of the above reaction, synthesis of 6 from 5, may be employed as well. Solvent: Different solvents could be used, for example acetone, methyl isobutyl ketone, ethyl acetate, isopropyl acetate, acetonitrile, or 2-methyl tetrahydrofuran. Reaction volume: Reaction volumes of 3 to 30 volumes with respect to 3 could be used. Base: Different inorganic bases, such as cesium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Also, organic bases, such as trimethylamine or diisopropyldiimide could be used. Base particle size: Different particle sizes of potassium carbonate from 325 mesh could be used. Reaction temperature: A lower temperature, such
as 50 °C could be used. A higher temperature, such as about 100 °C could be used. Any temperature above the boiling point of the solvent could be run in a pressure vessel.
Isolation: Different solvent ratios of MEK to n-heptane could be used. Different amounts of residual water can be left. Different amounts of seeds, from 0 to 50% could be used.
Seeding could take place later in the process and/or at a lower temperature. An un-seeded crystallization can be employed. A different isolation temperature, from 0 °C to 50 °C could be used. A different wash could be used, for example a different ratio of MEK to n-heptane. A different antisolvent from n-heptane could be used, such as hexane, pentane, or methyl tert-butyl ether. Alternatively, the batch could be solvent exchanged into a solvent where Compound 3 has a solubility of less than 100 mg/ml and isolated from this system. Drying: A temperature range of 10 to 60 °C could be used for drying.
[0234] Compound 10, shown below may also be formed as a result of O-alkylation of unreacted 4 present in product 5, or alternatively from or via a palladium mediated proteodesbromination or proteodesborylation in subsequent chemistry discussed in Example 1.3 below.
[0235] Preparation of methylsulfonylphenyl(cyclopropylmethyl) ether 10: Compound 4 (0.86 g, 5.0 mmol) and K2CO3 (1.04 g, 7.5 mmol) were slurried in acetone (17 mL, 20 vols). Cyclopropylmethyl bromide (0.73 mL, 7.5 mmol) was added in several small portions over ~1 minute and the reaction mixture heated to 50 °C for 48 hours, then cooled to 25 °C. Water (5.0 mL) was added with stirring and the acetone was evaporated on a rotary evaporator from which a fine white solid formed which was filtered off and returned to a vessel as a damp paste. A 1 : 1 mixture of MeOH/ water (8 mL) was added and heated to 40 °C with stirring. After 1 hour, the white solid was filtered off. Some residual solid was washed out with fresh water that was also rinsed through the cake, which was then isolated and left to air dry over the two days to give a dense white solid 10 (1.00 g, 88%). ¾ NMR (500 MHz, CDCb) 7.85
(2H, d, J = 8.8 Hz, RO-Ar ortho-H), 7.00 (2H, d, J = 8.8 Hz, RO-Ar meta- H), 3.87 (2H, d, J = 7.0 Hz, OCH2CH), 3.02 (3H, s, CHs), 1.34-1.23 (1H, m, OCH2CH), 0.72-0.60 (2H, m, 2 x CHCHflHb), 0.42-0.31 (2H, m, 2 x CHCH^.
1.3: Synthesis and Isolation Coupling Partner Boronic Ester 2
[0236] The final bond forming step to Compound 1 is a Suzuki-Miyaura coupling between Compounds 2 and 3, as shown in Scheme 3 below (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)). Early studies demonstrated that the boronic ester of the isoquinolinone Compound 3-a had poor physical attributes and solid phase stability (Kaila, N. et al., J. Med Chem., 57: 1299-1322 (2014)). The pinacolatoboronate of the O-alkyl phenol, Compound 7, had acceptable solid phase stability and could be isolated via crystallization.
Scheme 3: Suzuki-Miyaura coupling between 2 and 3
[0237] Process robustness studies for the isolation of Compound 7, however, indicated that Compound 7 has poor solution stability, decomposing primarily to the proteodeborylated compound 10, as shown in Scheme 4 below. This was particularly problematic as the isolation process involved a solvent exchange from 2-MeTHF (2-methyl tetrahydrofuran) to iPrOAc (isopropyl acetate), which is not a fast unit operation on scale.
Scheme 4: Modification of 7
[0238] A search for a more stable boronic ester was undertaken. Early attempts targeted making N-methyliminodiacetic acid (MID A) boronate Compound 2-a (E. Gilis and M. Burke,“Multi step Synthesis of Complex Boronic Acids from Simple MIDA Boronates,” J Am. Chem. Soc., 750(43): 14084-14085 (2008)), however, all attempts resulted in product decomposition. Applicant then turned to a relatively obscure boronate formed by the addition of diethanolamine to Compound 7 (Bonin et al., Tetrahedron Lett., 52: 1132-1135 (2011)). Addition of diethanolamine to a solution of Compound 7 led to rapid ester formation and concomitant crystallization of Compound 2.
[0239] The discovery of boronic ester Compound 2 allowed for a simple, fast, high-yielding, high-purity process comprising the following procedure. Tetrahydrofuran (THF, 1500mL) was charged to a flask containing Compound 6 (100g, 328 mmol), bis(pinacolato)diboron (90.7g, 357 mmol) and cesium acetate (CsOAc, 158g, 822 mmol). The system was vacuum purged three times with nitrogen. Pd(PPh3)2Cl2 (13.8g, 20 mmol) was charged to the reaction and the system was vacuum purged three times with nitrogen. The reaction was then heated to 55 to 65°C.
[0240] The batch was stirred for approximately 8 hours until reaction was complete by HPLC analysis. The batch was cooled to 15 to 25 °C (target 20 °C ) and charged with silica gel (20g) and Ecosorb C-941 (20g). After lh, the mixture was filtered to remove solid. The residual solids were washed twice, each with THF (300mL). The filtrate and washes were combined. In a separate vessel, diethanolamine (34.5mL, 360 mmol) was dissolved in THF (250 mL). The diethanolamine solution in THF (25mL) was then charged to the batch. After 10 minutes, the batch was seeded with 2 (1 g) and aged for 1 to 2 hours. The remaining of the diethanolamine solution in THF was charged to the batch over at least 2 hours and the slurry was stirred for at least 2 hours. The product 2 was collected by suction filtration. The wet cake was washed thrice with THF (200mL). The material was dried under vacuum at 40 °C with nitrogen purge yielding 94.6g of 2.
[0241] The reaction to synthesize Compound 2 from Compound 6 described above may be modified as follows. Solvent: Different solvents from THF could be used, such as 1,4 dioxane or 2-methyltetrahydrofuran. Reaction volume: The reaction volume can be varied from 4 to 50 volumes with respect to compound 2. Catalyst and base: Different palladium catalyst and bases can be used for the borylation. Examples can be found in Chow et al., RSC Adv., 3 : 12518-12539 (2013). Borylation reaction temperature: Reaction temperatures from room temperature (20 °C) to solvent reflux can be used. Carbon/ Silica treatment:
The treatment can be performed without silica gel. The process can be performed without a carbon treatment. Different carbon sources from Ecosorb C-941 can be used. Different amounts of silica, from 0.01X to IX weight equivalents, can be used. Different amounts of Ecosorb C-941, from 0.01X to IX weight equivalents, can be used. Crystallization: A different addition rate of diethanolamine can be used. Different amounts of diethanolamine, from 1.0 to 3.0 molar equivalents can be used. A different cake wash with more or less THF can be used. Different amount of seeds from 0.0001X wt to 50X wt can be used.
Alternatively, the process can be unseeded. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
[0242] The subsequent Suzuki-Miyaura coupling between Compounds 2 and 3 also proceeded well, providing over 20 kg of crude compound 1 with an average molar yield of 80% and LCAP of 99.7%.
1.4: Synthesis of Coupling Partner 3
[0243] Cross-coupling partner 3 was prepared by two different processes corresponding to Schemes 8 and 9 shown below.
Scheme 8: Process A for preparation of 3
[0244] According to Process A, Compound 9 (100g, 628 mmol) was dissolved in acetonitrile (450 mL) at room temperature. In a separate vessel, N-bromosuccinimide (NBS, 112g, 628 mmol) was suspended in acetonitrile (1 L). Compound 9 in acetonitrile was charged to the NBS slurry over at least 45 minutes. The contents of the reaction vessel were warmed to 45 °C to 55 °C and the batch stirred until the reaction was complete by HPLC analysis. The batch was cooled to 35 °C to 45 °C and ensured dissolution. Norit SX plus carbon (lOg) was charged to the mixture and the reaction mixture adjusted to 55 °C to 60 °C. The mixture was stirred at 55 °C to 60 °C for about lh and the mixture filtered at 55 °C to 60 °C to remove solids. The solids were washed with acetonitrile (500mL) at 55 °C to 60 °C. The volume of the combined filtrate was reduced to 900 mL by distilling off acetonitrile under reduced pressure. The batch with Compound 3 (lg) and stirred at 35 °C to 45 °C for at least 60 minutes. The contents of the reaction vessel were cooled to 15 °C to 25 °C over at least 1 hour. Water (2000 mL) was charged to the reaction vessel over at least 90 minutes and the slurry aged for at least 60 minutes. The product was collected by suction filtration. The cake was washed with a premixed 5% solution of acetonitrile in water (300mL). The wet cake was dried under vacuum at 40 °C with nitrogen purge. Yield: 120g of 3.
[0245] The above procedure, Process A for this synthesis of 3, may be practiced with alternative reagents and conditions as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 9). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent Stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
Scheme 9: Process B for preparation of 3
[0246] According to Process B, Compound 3 can be formed starting from 8 via non-isolated compound 9 as follows. Compound 8 (80 g, 55 mmol), cesium carbonate (CS2CO3, 215 g, 66 mmol), and acetonitrile (800 mL) were charged to the reactor. The temperature was adjusted from 15 to 25 °C and iodomethane charged to the reactor (Mel, 86 g, 0.61 mol) while maintaining a batch temperature below 25 °C. The batch was heated to 40 °C and agitated for 10 hours to form Compound 9. The batch was cooled to 25 °C, filtered into a fresh reactor to remove solids, and the solids washed twice with acetonitrile. The combined organic layers were concentrated via atmospheric distillation to about 320 mL.
[0247] In a separate reactor N-bromosuccinimide (NBS, 98.1 g, 0.55 mol) was charged to acetonitrile (800 mL) and agitated. The batch containing Compound 9 was transferred to the NBS solution while maintaining a batch temperature of 15 to 25 °C. The batch was heated to 45 to 55 °C and agitated for at least 4 hours to allow for reaction completion to Compound 3. Upon reaction completion, Norit SX Plus activated carbon (8 g) was charged, and agitated at 45 to 55 °C for one hour. The batch was filtered into a fresh vessel, the Norit SX plus cake was washed with 400 ml of 45 to 55 °C acetonitrile. The acetonitrile layers were combined, cooled to 35 to 45 °C, and distilled under reduced pressure to 720 mL. The batch was adjusted to a temperature of 40 °C, charged with Compound 3 seeds (0.8 g), agitated for one hour, cooled to 15 to 25 °C over at least on hour, then charged with water (1600 mL) over at least two hours. The mixture was agitated for an additional one to two hours, filtered, the cake washed with a premixed 5% solution of acetonitrile in water (240 mL). The wet cake was dried under vacuum at 40°C with nitrogen purge. Yield: 52 g of 3.
[0248] Process B to synthesize Compound 3, described above, may be modified as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 8).
Alkylating reagent: Alternative methylating reagents to methyl iodide can be used such as dimethylsulfate. Alkylating reagent stoichiometry: 1 to 10 molar equivalents of methyl iodide may be used. Base: Different inorganic bases, such as potassium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Brominating agents:
Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. Seeding levels from 0.0001% to 50% can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying.
1.5: Cross-coupling of 2 and 3 to Produce Target Compound 1
[0249] 1 is synthesized by Suzuki cross-coupling of 3 and 2 according to Scheme 10 and as described below.
Scheme 10: Synthesis of 1
[0250] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compovmd 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223 g, l.OSmol). Agitation was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum
purged three times with nitrogen. The batch was heated to 65 to 75°C and contents stirred for at least 16 hours until reaction was complete by HPLC analysis. The batch was cooled to 60 to 70°C, agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at 60 to 70°C. Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at 60 to 70°C. After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at 60 to 70°C. The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to 60 to 65°C and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at 60 to 65°C. The slurry was cooled to 15 to 25°C over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at 40°C with nitrogen purge. Yield: 139g of 1.
[0251] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox, Alister, J.J., Lloyd-Jones, Guy C. Chem. Soc. Rev., 2014, 43, 412. Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1);
cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5); Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compound 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compoxmd 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
[0253] Formic acid is one ICH Class 3 solvent in which Compound 1 is highly soluble, having a solubility greater than 250 mg/ml at 20 °C. The solubility curve of Compound 1 in formic acid-Water is quite steep (see Figure 7), which enables a volumetrically efficient process.
[0254] Initial attempts to recrystallize crude Compound 1 involved dissolving in formic acid, polish filtering, and charging polish filtered water to about 20% supersaturation, followed by seeding with the thermodynamically most stable form (Form 1), followed by slow addition of water to the final solvent ratio, filtration, washing, and drying. Applicant observed that during the initial water charge, if the batch self-seeded it formed a thick slurry. X-ray diffraction (XRD), differential scanning calorimetry (DSC), and photomicroscopy demonstrated that a metastable form was produced. Once seeded with Form 1, the batch converted to the desired form (Form 1) prior to the addition of the remaining water. This process worked well during multiple lab runs, consistently delivering the desired form and purity with about 85% yield.
[0255] Unfortunately, upon scale-up, the batch did not convert to Form 1 after seeding. Additional water was charged and the batch began to convert to the desired form (mix of Form 1 and the metastable form by X-ray powder diffraction (XRPD)). When additional water was charged, the XRPD indicated only the metastable form. After a few hours with no change, Applicant continued the water charge to the final solvent ratio, during which time the batch eventually converted to Form 1. This process is summarized in Figure 8.
[0256] It was subsequently found by closer analysis of the plant and laboratory retains that a new metastable form was formed during scale up, with a similar, but different XRPD pattern. This form (metastable B) could be reproduced in the laboratory, but only when the batch has a high formic acid:water ratio and is seeded with Form 1. Without Form 1 seeds, metastable A is the kinetic form. Both metastable forms converted to Form 1 with additional water and/or upon drying, leading Applicant to believe that the metastable forms are formic acid solvates. These findings are summarized in Figure 9.
[0257] While there is little risk in not being able to control the final form, there is a risk of forming a difficult-to-stir slurry which can lead to processing issues. The crystallization procedure was therefore modified to keep a constant formic acid-water ratio. This was performed by charging 2.4X wt. formic acid and 1.75X wt. water (final solvent composition)
to the crystallizer with 0.03X wt. Form 1 seeds, and performing a simultaneous addition of Compound 1 in 6. IX wt. formic acid and 4.4X wt. water. The batch filtered easily and was washed with formic acid/water, then water, and dried under reduced pressure to yield 8.9 kg of Compound 1 (92% yield) with 99.85% LCAP and N.D. formic acid.
Example 2: Exemplary high throughput experimentation reaction
[0258] The following procedure is an exemplary high throughput experimentation reaction.
[0259] An overview of the reaction is shown below in Scheme 5:
Scheme 5: Reaction conditions tested for cross-coupling reaction of 2 and 3
[0260] Pd catalysts were dosed into the 24-well reactor vial as solutions (100 pL of 0.01 M solution in tetrahydrofuran (THF) or dichloroethane (DCE) depending upon the solubility of the ligand). Plates of these ligands are typically dosed in advance of the reaction, the solvent is removed by evacuation in an evaporative centrifuge and plates are stored in the glovebox. The catalysts screened in the coupling are the following: XPhos, SPhos, CataCXium A, APhos, P(Cy)3, PEPPSI-IPent. For the first five ligands, these were initially screened as the Buchwald Pd G2/G3 precatalysts.
[0261] To the plates was then added a stock solution of Compound 3 (10 pmol) and Compound 2 (12 pmol) dissolved in the following solvents: dimethylformamide (DMF),
tetrahydrofuran (THF), butanol (/r-BuOH), and toluene. The base was then added as a stock solution (30 mmol) in 20 mL of water.
[0262] A heatmap summarizing catalyst performance is shown in Figs. 10A and 10B. High performance liquid chromatography (HPLC) yields for this screening span from <5% up to -85%. Larger circles indicate higher yield. Lighter circles indicate higher cleanliness.
[0263] A similarly designed screening of base and solvent also indicate that a range of alcoholic solvents (methanol, ethanol, propanol, 2-butanol, 2-propanol, and /-amyl alcohol) are also all viable in this coupling chemistry. Bases such as potassium phosphate, potassium carbonate, potassium acetate, and potassium hydroxide were all successful in achieving the coupling. Fig. 10B shows a heatmap with HPLC yields ranging from -50 – 95%. Larger, darker circles indicate higher yield.
[0264] This chemistry from microvial screening has been scaled to a laboratory process. To a 3 -necked jacketed 250 mL flask equipped with overhead stirring, nitrogen inlet, and thermocouple was added Compound 3 (1.0 eq, 4.00 grams), Compound 2 (1.2 eq, 1.71 x wt), potassium carbonate (3.0 eq, 1.74 x wt). The reactor was inerted three times and then degassed 2-propanol (24 x vol.) followed by degassed water (6 x vol) was then added.
Stirring was then initiated at 300 rpms. The reactor was then stirred and blanketed with nitrogen for 1 hour. The catalyst was then added (0.01 eq, 0.028 x wt) and stirring continued (300 rpms) and the reactor was heated into the Tj = 65 °C.
[0265] After 2 hours, with full conversion confirmed analytically, trioctylphosphine (0.1 eq, 0.16 x wt) dosed, and reaction mixture allowed to cool slowly to room temperature hours.
The reaction mixture was then filtered, washed with 2-propanol (4 x vol), 2-propanol: water (4: 1, 4 x vol), and then with water (4 x vol). Note: If 2 is dimer present in cake, an additional ethyl acetate (EtOAc) wash (4 x vol) can be added for purging. The cake was then transferred to a vacuum oven to dry overnight at 40 °C, -40 cm Hg, under nitrogen flow. After transfer to a bottle, 6.03 grams of 1 were isolated, 98.6% assay, 91% overall yield.
Scheme 6: Alternative reagents and solvents for cross-coupling
[0266] Based on the previously delineated results, it was expected that a variety of monodentate (PPI13 [triphenylphosphine], PBu3 [tributylphosphine], etc) and bidentate phosphines (dppf [1,1 ‘-bis(diphenylphosphino)ferrocene], BINAP [2,2 -bis(diphenylphosphino)- 1 , 1 -binaphthyl], Xantphos [4,5-bis(diphenylphosphino)-9,9-dimethylxanthene], dppe [l,2-bis(diphenylphosphino)ethane], etc) ligated to any number of Pd sources (Pd halides, Pd(H) precatalyts, Pd(0) sources) could reasonably be employed to arrive at the Compound 1 crude material. A range of organic solvents ranging from non-polar (heptane, benzene), protic (alcohols), polar aprotic (dimethylsulfoxide, dimethylformamide, dimethylacetamide, acetonitrile) as well as a variety of esters and ketones (acetone, 2-butanone, ethylacetate) should also serve as effective solvents for this reactivity. Finally, inorganic bases of varying strength (phosphates, carbonates, acetates, etc) along with organic variants such as triethylamine, l,8-diazabicyclo(5.4.0)undec-7-ene, and others in a wide pKa range are viable as stoichiometric basic additives.
Example 3: Exemplary Compound 5 process
[0267] The purpose of this example was to describe an exemplary process for making Compound 5.
[0268] Charge 4 (lOg, 58mmol) and acetonitrile (lOOmL) to a reaction vessel and start the stirrer. Adjust the batch to -18 °C to -22 °C (target -20 °C). Charge triflic acid (5.5mL, 62mmol) to the batch maintaining -10 °C to -25 °C (target -20 °C). Stir the batch at -10 °C to -25 °C (target -20 °C) for 10 to 20 minutes. Charge NBS (11.38g, 64mmol) to the batch at -10 °C to -25 °C (target -20 °C) and stir for ca. 30 min at -10 °C to -25 °C (target -20 °C). Warm the batch to 20 °C over 3-4 hours (reaction will occur when internal temp is between 5 °C and 15 °C). Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and sample for reaction completion.
[0269] If Compound 4 relative to Compound 5 is more than 5%:
[0270] Cool the bath to -5 °C to -15 °C (target -10 °C) (cooling below 0 °C to ensure selectivity). Charge NBS to the batch according to the follow formula: Mass of NBS = (% Compound 4 x lOg). Warm the batch to 20 °C over 1-2 hours. Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and check reaction for completion. Proceed to next line.
[0271] If Compound 4 relative to Compound 5 is less than 5%:
[0272] Warm the batch to 40 °C to 50 °C (target 48 °C). Concentrate the batch under reduced pressure to a final volume of ~40mL. Cool the batch to -15 °C to -5 °C (target -10 °C) and stir for ca. lh. Filter the batch by suction filtration. Slurry wash the filter cake with purified water (3 x 20mL) at 15 °C to 25 °C (target 20 °C) for 10 to 15 minutes each wash. Remove a sample of the filter cake for analysis by ¾ NMR. Continue washing cake until the residual succimide is below 1.0%mol% relative to 5. Dry the filter cake at up to 60°C under vacuum and nitrogen purge. Analyse the 5 by HPLC analysis (97%w/w to 99%w/w). Expected yield: 60-85% theory (90-110% w/w).
Example 4: Purification of Compound 1 (CC-90010) by crystallization from formic acid and water.
[0273] This example describes a method for the purification of Compound 1 by
crystallization from formic acid and water. Also detailed are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1.
[0274] Figure 11 shows XH NMR of Compound 1 (CC-90010). Solvent: d6DMSO; and Figure 12 shows microscopy of Compound 1 (CC-90010) Form I. Figure 13 shows XRPD of Compound 1 (CC-90010) Form I, with peak information detailed in Table 6:
PATENT
US 20190008852
WO 2018081475
US 20180042914
WO 2016172618
WO 2015058160
/////////CC-90010, solid tumors , non-Hodgkin’s lymphoma, PHASE 1, CANCER, QUANTICEL
CS(=O)(=O)c4cc(C1=CN(C)C(=O)c2ccccc12)c(OCC3CC3)cc4
IIIM-290

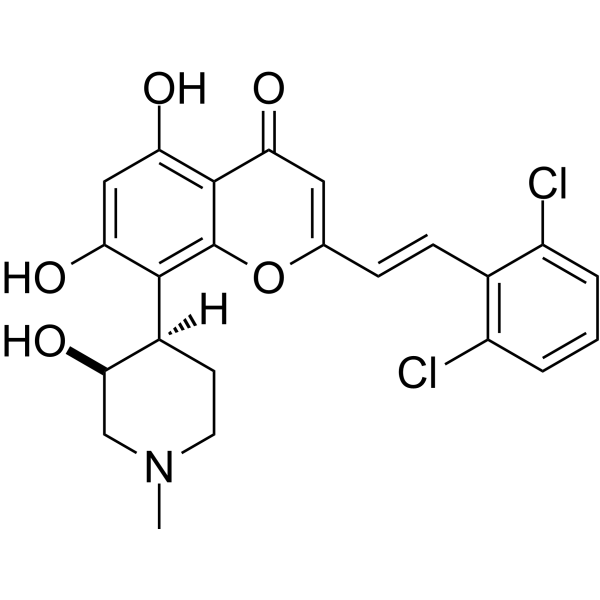
IIIM-290
4H-1-Benzopyran-4-one, 2-[2-(2,6-dichlorophenyl)ethenyl]-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidinyl]-
| Molecular Weight |
462.32 |
|---|---|
| Formula |
C₂₃H₂₁Cl₂NO₅ |
| CAS No. |
2213468-64-3 |
CSIR-IIIM Jammu has filed an IND Application of “IIIM-290” to Drug Controller General of India for conducting Phase I/Phase II clinical trial of its capsule formulation in patients with locally advanced or metastatic pancreatic cancer. This IND candidate has emerged from the eight years of medicinal chemistry/ preclinical efforts of IIIM Jammu in the area of small molecule kinase inhibitors. IIIM-290 (NCE) is an orally bioavailable CDK inhibitor, obtained via semisynthetic modification of a natural product rohitukine. Institute has already secured a patent on this small molecule as well as on its oral capsule formulation.

IIIM-290 is a potent and oral CDK inhibitor with IC50s of 90 and 94 nM for CDK2/A and CDK9/T1.


PAPER
https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.7b01765
Discovery and Preclinical Development of IIIM-290, an Orally Active Potent Cyclin-Dependent Kinase Inhibitor
- Sandip B. Bharate
- Vikas Kumar
- Shreyans K. Jain
- Mubashir J. Mintoo
- Santosh K. Guru
- Vijay K. Nuthakki
- Mohit Sharma
- Sonali S. Bharate
- Sumit G. Gandhi
- Dilip M. Mondhe
- Shashi Bhushan
- Ram A. Vishwa
Abstract

Rohitukine (1), a chromone alkaloid isolated from Indian medicinal plant Dysoxylum binectariferum, has inspired the discovery of flavopiridol and riviciclib, both of which are bioavailable only via intravenous route. With the objective to address the oral bioavailability issue of this scaffold, four series of rohitukine derivatives were prepared and screened for Cdk inhibition and cellular antiproliferative activity. The 2,6-dichloro-styryl derivative IIIM-290 (11d) showed strong inhibition of Cdk-9/T1 (IC50 1.9 nM) kinase and Molt-4/MIAPaCa-2 cell growth (GI50 < 1.0 μM) and was found to be highly selective for cancer cells over normal fibroblast cells. It inhibited the cell growth of MIAPaCa-2 cells via caspase-dependent apoptosis. It achieved 71% oral bioavailability with in vivo efficacy in pancreatic, colon, and leukemia xenografts at 50 mg/kg, po. It did not have CYP/efflux-pump liability, was not mutagenic/genotoxic or cardiotoxic, and was metabolically stable. The preclinical data presented herein indicates the potential of 11d for advancement in clinical studies.


Patent
IN201811026240



Patent
InventorRam A. VishwakarmaSandip B. BharateShashi BhushanDilip M. MondheShreyans K. JainSamdarshi MeenaSantosh K. GuruAnup S. PathaniaSuresh KumarAkanksha BehlMubashir J. MintooSonali S. BharatePrashant Joshi Current Assignee Council of Scientific and Industrial Research (CSIR)
https://patents.google.com/patent/US9932327B2/en
The disruption of any internal and external regulation of cellular growth leads to tumorogenesis by uncontrolled proliferation. This loss of control occurs at multiple levels in most of the cancer cases. Cyclin-dependent kinases (CDKs) have been recognized as key regulators of cell cycle progression. Alteration and deregulation of CDK activity have pathogenic link to the cancer. Number of cancers are associated with hyper-activation of CDKs as a result of mutation of the CDK genes or CDK inhibitor genes. Therefore, CDK inhibitors or modulators are of great interest to explore as novel therapeutic agents against cancer (Senderowicz, A. M. Leukemia 2001, 15, 1). Several classes of chemical inhibitors of CDK activity have been described (Zhang, J. et. al. Nat Rev Cancer. 2009, 9, 28) and some of them have reached to clinical pipeline for cancer.
Because CDK inhibitors are ATP competitive ligands; hence earlier they were typically described as purine class of compounds for example dimethylaminopurine, a first substance to be known as a CDK inhibitor (Neant, I. et al. Exp. Cell Res. 1988, 176, 68), olomoucine (Vesely, J. et al. Eur. J. Biochem. 1994, 224, 771) and roscovitine (Meijer, L. et al. Eur. J. Biochem. 1997, 243, 527). The IC50values of these purine class of compounds for CDK1/cyclin B are 120, 7 and 0.2-0.8 μM respectively (Gray, N. et al. Curr. Med. Chem. 1999, 6, 859). Some of the more potent members of this series have been prepared by the Schultz group using combinatorial approaches (Gray, N. S. et al. Science 1998, 281, 533). Number of synthetic flavoalkaloids having potent CDK inhibitory activity has been reviewed recently (Jain, S. K. et al. Mini–Rev. Med. Chem. 2012, 12, 632).
Specific CDKs operate in distinct phases of the cell cycle. CDK complexes with their respective type cyclin partners such as, complex of CDK2 and cyclin A is responsible for the cell’s progression from G1 phase to S phase (Sherr, C. J. Science 1996, 274, 1672). DNA synthesis (S phase) begins with the CDK mediated phosphorylation of Rb (retinoblastoma) protein. Phosphorylated Rb is released from its complex with E2F. The released E2F then promotes the transcription of numerous genes required for the cell to progress through S phase, including thymidylate synthase and dihydrofolate reductase which are required for cell progression (Hatakeyama, M. et. al, Cell Cycle Res. 1995, 1, 9; Zhang, H. S. et. al. Cell 1999, 97, 53). Majority of human cancers have abnormalities in some component of the Rb pathway because of hyper-activation of CDKs resulting from the over-expression of positive cofactors (cyclins/CDKs) or a decrease in negative factors (endogenous CDK inhibitors) or Rb gene mutations (Sausville, E. A. et. al, Pharmacol. Ther. 1999, 82, 285).
The CDK-9 is a member of the Cdc2-like family of kinases. Its cyclin partners are members of the family of cyclin T (T1, T2a and T2b) and cyclin K. The CDK-9/cyclin T complexes appear to be involved in regulating several physiological processes. CDK9/cyclin T1 belongs to the P-TEFb complex, and is responsible for the phosphorylation of carboxyl terminal domain of the RNA Polymerase II, thus promoting general elongation. CDK-9 has also been described as the kinase of the TAK complex, which is homologous to the P-TEFb complex and is involved in HIV replication. CDK9 also appears to be involved in the differentiation program of several cell types, such as muscle cells, monocytes and neurons, suggesting that it may have a function in controlling specific differentiative pathways. In addition, CDK-9 seems to have an anti-apoptotic function in monocytes, that may be related to its control over differentiation of monocytes. This suggests the involvement of CDK-9 in several physiological processes in the cell, the deregulation of which may be related to the genesis of transforming events that may in turn lead to the onset of cancer. In addition, since the complex CDK-9/cyclin T1 is able to bind to the HIV-1 product Tat, the study of the functions of CDK-9/cyclin T may be of interest in understanding the basal mechanisms that regulate HIV replication (Falco, G. D. and Giordano A. Cancer Biol. Therapy 2002, 1, 337).
Rohitukine belongs to a class of chromone alkaloids and it was isolated by chemists at Hoechst India Ltd. in the early 1990’s from Dysoxylum binectariferum Hook. which is phylogenetically related to the Ayurvedic plant, D. malabaricum Bedd., used for rheumatoid arthritis. Rohitukine was isolated as the constituent responsible for anti-inflammatory and immunomodulatory activity (Naik, R. G. et. al. Tetrahedron 1988, 44, 2081; U.S. Pat. No. 4,900,727, 1990). Medicinal chemistry efforts around this nature-derived flavone alkaloid led to discovery of two promising clinical candidates for treatment of cancer viz. flavopiridol of Sanofi-Aventis and P-276-00 of Piramal life sciences. Recently FDA has granted the orphan drug status to flavopiridol for treatment of chronic lymphocytic leukemia (CLL).
The molecular formula of rohitukine is C16H19NO5 and the structure has a molecular weight of 305.32 g/mol. The chemical structure of rohitukine (1) is shown below. The present invention reports new semi-synthetic analogs of rohitukine as promising inhibitors of cyclin-dependent kinases such as CDK-2 and CDK-9.

Synthesis of styryl analog 2-(2,6-dichlorostyryl)-5,7-dihydroxy-8-(3-hydroxy-1-methylpiperidin-4-yl)-4H-chromen-4-one (33)
This compound was synthesized using the procedure as described in example 4. Yellow solid; 1H NMR (DMSO-d6, 400 MHz): δ 7.68 (m, 2H), 7.61 (d, J=16 Hz, 1H), 7.49 (t, J=8 Hz, 1H), 7.14 (d, J=16 Hz, 1H), 6.41 (s, 1H), 5.85 (s, 1H), 4.53 (brs, 1H), 3.10-2.50 (m, 6H of piperidine), 2.65 (s, 3H), 1.62 (m, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 179.68. 171.27, 159.20, 158.02, 154.03, 133.12, 131.49, 129.75, 128.35 (2C), 128.20, 127.90, 108.81, 106.79, 100.88, 100.52, 66.35, 59.82, 54.45, 43.15, 35.79, 22.01, 20.33, ESI-MS: m/z 462.01 [M+H]+; IR (CHCl3): νmax 3400, 2921, 1652, 1577, 1550, 1417, 1380, 1191, 1085 cm−1.
///////////IIIM-290, nda, india, phase 1, dcgi, CSIR, ROHITUKINE
|
OC1=C2C(OC(/C=C/C3=C(Cl)C=CC=C3Cl)=CC2=O)=C([C@]4([H])[C@H](O)CN(C)CC4)C(O)=C1 |

{kind=link}
{kind=link}
{kind=link}