
Home » GENERIC DRUG (Page 5)
Category Archives: GENERIC DRUG
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
HEPARIN SODIUM



HEPARIN SODIUM
LAUNCHED 1937
9041-08-1 NA SALT
9005-49-6 (heparin)
Thromboliquine, Calciparine, Certoparin, Dalteparin, Fraxiparin, Heparinate, Multiparin, Novoheparin, Parnaparin
Unfractionated heparin (UH) is a heterogenous preparation of anionic, sulfated glycosaminoglycan polymers with weights ranging from 3000 to 30,000 Da. It is a naturally occurring anticoagulant released from mast cells. It binds reversibly to antithrombin III (ATIII) and greatly accelerates the rate at which ATIII inactivates coagulation enzymes thrombin (factor IIa) and factor Xa. UH is different from low molecular weight heparin (LMWH) in the following ways: the average molecular weight of LMWH is about 4.5 kDa whereas it is 15 kDa for UH; UH requires continuous infusions; activated partial prothrombin time (aPTT) monitoring is required when using UH; and UH has a higher risk of bleeding and higher risk of osteoporosis in long term use. Unfractionated heparin is more specific than LMWH for thrombin. Furthermore, the effects of UH can typically be reversed by using protamine sulfate.
Unfractionated heparin is indicated for prophylaxis and treatment of venous thrombosis and its extension, prevention of post-operative deep venous thrombosis and pulmonary embolism and prevention of clotting in arterial and cardiac surgery. In cardiology, it is used to prevent embolisms in patients with atrial fibrillation and as an adjunct antithrombin therapy in patients with unstable angina and/or non-Q wave myocardial infarctions (i.e. non-ST elevated acute coronary artery syndrome) who are on platelet glycoprotein (IIb/IIIa) receptor inhibitors. Additionally, it is used to prevent clotting during dialysis and surgical procedures, maintain the patency of intravenous injection devices and prevent in vitro coagulation of blood transfusions and in blood samples drawn for laboratory values.
Indication: For anticoagulant therapy in prophylaxis and treatment of venous thrombosis and its extension, for prevention of post-operative deep venous thrombosis and pulmonary embolism and for the prevention of clotting in arterial and cardiac surgery.
Mechanism of action: The mechanism of action of heparin is antithrombin-dependent. It acts mainly by accelerating the rate of the neutralization of certain activated coagulation factors by antithrombin, but other mechanisms may also be involved. The antithrombotic effect of heparin is well correlated to the inhibition of factor Xa. Heparin interacts with antithrombin III, prothrombin and factor X.
Heparin (from Ancient Greek ηπαρ (hepar), liver), also known as unfractionated heparin, a highly sulfated glycosaminoglycan, is widely used as an injectable anticoagulant, and has the highest negative charge density of any known biological molecule.[3] It can also be used to form an inner anticoagulant surface on various experimental and medical devices such as test tubes and renal dialysis machines.
Although it is used principally in medicine for anticoagulation, its true physiological role in the body remains unclear, because blood anticoagulation is achieved mostly by heparan sulfate proteoglycans derived from endothelial cells.[4] Heparin is usually stored within the secretory granules of mast cells and released only into the vasculature at sites of tissue injury. It has been proposed that, rather than anticoagulation, the main purpose of heparin is defense at such sites against invading bacteria and other foreign materials.[5] In addition, it is conserved across a number of widely different species, including some invertebrates that do not have a similar blood coagulation system.
 HEPARIN
HEPARIN
Heparin structure

Native heparin is a polymer with a molecular weight ranging from 3 to 30 kDa, although the average molecular weight of most commercial heparin preparations is in the range of 12 to 15 kDa.[6] Heparin is a member of the glycosaminoglycan family of carbohydrates (which includes the closely related molecule heparan sulfate) and consists of a variably sulfated repeating disaccharide unit.[7] The main disaccharide units that occur in heparin are shown below. The most common disaccharide unit is composed of a 2-O-sulfated iduronic acid and 6-O-sulfated, N-sulfated glucosamine, IdoA(2S)-GlcNS(6S). For example, this makes up 85% of heparins from beef lung and about 75% of those from porcine intestinal mucosa.[8] Not shown below are the rare disaccharides containing a 3-O-sulfated glucosamine (GlcNS(3S,6S)) or a free amine group (GlcNH3+). Under physiological conditions, the ester and amide sulfate groups are deprotonated and attract positively charged counterions to form a heparin salt. Heparin is usually administered in this form as an anticoagulant.
One unit of heparin (the “Howell unit”) is an amount approximately equivalent to 0.002 mg of pure heparin, which is the quantity required to keep 1 ml of cat’s blood fluid for 24 hours at 0°C.[9]
-
 GlcA-GlcNAc
GlcA-GlcNAc -
 GlcA-GlcNS
GlcA-GlcNS -
 IdoA-GlcNS
IdoA-GlcNS -
 IdoA(2S)-GlcNS
IdoA(2S)-GlcNS -
 IdoA-GlcNS(6S)
IdoA-GlcNS(6S) -
 IdoA(2S)-GlcNS(6S)
IdoA(2S)-GlcNS(6S)



-GlcNS.png)
.png)
-GlcNS(6S).png)
Abbreviations
- GlcA = β-D–glucuronic acid
- IdoA = α-L–iduronic acid
- IdoA(2S) = 2-O-sulfo-α-L-iduronic acid
- GlcNAc = 2-deoxy-2-acetamido-α-D-glucopyranosyl
- GlcNS = 2-deoxy-2-sulfamido-α-D-glucopyranosyl
- GlcNS(6S) = 2-deoxy-2-sulfamido-α-D-glucopyranosyl-6-O-sulfate
Three-dimensional structure
The three-dimensional structure of heparin is complicated because iduronic acid may be present in either of two low-energy conformations when internally positioned within an oligosaccharide. The conformational equilibrium is influenced by sulfation state of adjacent glucosamine sugars.[10] Nevertheless, the solution structure of a heparin dodecasaccharide composed solely of six GlcNS(6S)-IdoA(2S) repeat units has been determined using a combination of NMR spectroscopy and molecular modeling techniques.[11] Two models were constructed, one in which all IdoA(2S) were in the 2S0 conformation (A and B below), and one in which they are in the 1C4 conformation (C and D below). However, no evidence suggests that changes between these conformations occur in a concerted fashion. These models correspond to the protein data bank code 1HPN.

In the image above:
- A = 1HPN (all IdoA(2S) residues in 2S0 conformation) Jmol viewer
- B = van der Waals radius space filling model of A
- C = 1HPN (all IdoA(2S) residues in 1C4 conformation) Jmol viewer
- D = van der Waals radius space filling model of C
In these models, heparin adopts a helical conformation, the rotation of which places clusters of sulfate groups at regular intervals of about 17 angstroms (1.7 nm) on either side of the helical axis.
Medical use

A sample of Heparin Sodium for injection
Heparin is a naturally occurring anticoagulant produced by basophils and mast cells.[12] Heparin acts as an anticoagulant, preventing the formation of clots and extension of existing clots within the blood. While heparin does not break down clots that have already formed (unlike tissue plasminogen activator), it allows the body’s natural clot lysis mechanisms to work normally to break down clots that have formed. Heparin is generally used for anticoagulation for the following conditions:
- Acute coronary syndrome, e.g., NSTEMI
- Atrial fibrillation
- Deep-vein thrombosis and pulmonary embolism
- Cardiopulmonary bypass for heart surgery
- ECMO circuit for extracorporeal life support
- Hemofiltration
- Indwelling central or peripheral venous catheters
Mechanism of action
Heparin and its low-molecular-weight derivatives (e.g., enoxaparin, dalteparin, tinzaparin) are effective at preventing deep vein thromboses and pulmonary emboli in patients at risk,[13][14] but no evidence indicates any one is more effective than the other in preventing mortality.[15] Heparin binds to the enzyme inhibitor antithrombin III (AT), causing a conformational change that results in its activation through an increase in the flexibility of its reactive site loop.[16] The activated AT then inactivates thrombin and other proteases involved in blood clotting, most notably factor Xa. The rate of inactivation of these proteases by AT can increase by up to 1000-fold due to the binding of heparin.[17]
AT binds to a specific pentasaccharide sulfation sequence contained within the heparin polymer:
GlcNAc/NS(6S)-GlcA-GlcNS(3S,6S)-IdoA(2S)-GlcNS(6S)
The conformational change in AT on heparin-binding mediates its inhibition of factor Xa. For thrombin inhibition, however, thrombin must also bind to the heparin polymer at a site proximal to the pentasaccharide. The highly negative charge density of heparin contributes to its very strong electrostatic interaction with thrombin.[3] The formation of a ternary complex between AT, thrombin, and heparin results in the inactivation of thrombin. For this reason, heparin’s activity against thrombin is size-dependent, with the ternary complex requiring at least 18 saccharide units for efficient formation.[18] In contrast, antifactor Xa activity requires only the pentasaccharide binding site.

Chemical structure of fondaparinux
This size difference has led to the development of low-molecular-weight heparins (LMWHs) and, more recently, to fondaparinux as pharmaceutical anticoagulants. LMWHs and fondaparinux target antifactor Xa activity rather than antithrombin activity, with the aim of facilitating a more subtle regulation of coagulation and an improved therapeutic index. The chemical structure of fondaparinux is shown above. It is a synthetic pentasaccharide, whose chemical structure is almost identical to the AT binding pentasaccharide sequence that can be found within polymeric heparin and heparan sulfate.
With LMWH and fondaparinux, the risk of osteoporosis and heparin-induced thrombocytopenia (HIT) is reduced. Monitoring of the activated partial thromboplastin time is also not required and does not reflect the anticoagulant effect, as APTT is insensitive to alterations in factor Xa.
Danaparoid, a mixture of heparan sulfate, dermatan sulfate, and chondroitin sulfate can be used as an anticoagulant in patients having developed HIT. Because danaparoid does not contain heparin or heparin fragments, cross-reactivity of danaparoid with heparin-induced antibodies is reported as less than 10%.[19]
The effects of heparin are measured in the lab by the partial thromboplastin time (aPTT), one of the measures of the time it takes the blood plasma to clot. Partial thromboplastin time should not be confused with prothrombin time, or PT, which measures blood clotting time through a different pathway of the coagulation cascade.
Administration
Heparin is given parenterally because it is not absorbed from the gut, due to its high negative charge and large size. It can be injected intravenously or subcutaneously (under the skin); intramuscular injections (into muscle) are avoided because of the potential for forming hematomas. Because of its short biologic half-life of about one hour, heparin must be given frequently or as a continuous infusion. Unfractionated heparin has a half-life of about one to two hours after infusion, [20] whereas LMWH has a half-life of four to five hours.[21] The use of LMWH has allowed once-daily dosing, thus not requiring a continuous infusion of the drug. If long-term anticoagulation is required, heparin is often used only to commence anticoagulation therapy until an oral anticoagulant e.g. warfarin takes effect.
Details of administration are available in clinical practice guidelines by the American College of Chest Physicians:[22]
Production
Pharmaceutical-grade heparin is derived from mucosal tissues of slaughtered meat animals such as porcine (pig) intestines or bovine (cattle) lungs.[23] Advances to produce heparin synthetically have been made in 2003 and 2008.[24]
Protamine sulfate (1 mg per 100 units of heparin that had been given over the past four hours) has been given to counteract the anticoagulant effect of heparin.[26]
Heparin is one of the oldest drugs currently in widespread clinical use. Its discovery in 1916 predates the establishment of the Food and Drug Administration of the United States, although it did not enter clinical trials until 1935.[27] It was originally isolated from canine liver cells, hence its name (hepar or “ήπαρ” is Greek for “liver”). Heparin’s discovery can be attributed to the research activities of Jay McLean and William Henry Howell.
In 1916, McLean, a second-year medical student at Johns Hopkins University, was working under the guidance of Howell investigating procoagulant preparations, when he isolated a fat-soluble phosphatide anticoagulant in canine liver tissue. In 1918, Howell coined the term ‘heparin’ for this type of fat-soluble anticoagulant. In the early 1920s, Howell isolated a water-solublepolysaccharide anticoagulant, which was also termed ‘heparin’, although it was distinct from the phosphatide preparations previously isolated. McLean’s work as a surgeon probably changed the focus of the Howell group to look for anticoagulants, which eventually led to the polysaccharide discovery.
In the 1930s, several researchers were investigating heparin. Erik Jorpes at Karolinska Institutet published his research on the structure of heparin in 1935,[28] which made it possible for the Swedish company Vitrum AB to launch the first heparin product for intravenous use in 1936. Between 1933 and 1936, Connaught Medical Research Laboratories, then a part of the University of Toronto, perfected a technique for producing safe, nontoxic heparin that could be administered to patients in a salt solution. The first human trials of heparin began in May 1935, and, by 1937, it was clear that Connaught’s heparin was a safe, easily available, and effective blood anticoagulant. Prior to 1933, heparin was available, but in small amounts, and was extremely expensive, toxic, and, as a consequence, of no medical value.[29]
A posthumous attempt to nominate McLean for a Nobel Prize failed
Heparin Sodium Injection, USP is a sterile, nonpyrogenic solution of heparin sodium (derived from porcine intestinal mucosa) in water for injection. Each container contains 10000, 12500, 20000 or 25,000 USP Heparin Units; 40 or 80 mg sodium chloride added to render isotonic (see HOW SUPPLIEDsection for various sizes and strength). May contain sodium hydroxide and/or hydrochloric acid for pH adjustment. pH 6.0 (5.0 to 7.5).
The solution contains no bacteriostat, antimicrobial agent or added buffer and is intended for use only as a single-dose injection. When smaller doses are required, the unused portion should be discarded.
Heparin sodium in the ADD-Vantage™ system is intended for intravenous administration only after dilution.
Heparin Sodium, USP is a heterogenous group of straight-chain anionic mucopolysaccharides, called glycosamino-glycans having anticoagulantproperties. Although others may be present, the main sugars occurring in heparin are: (1) α- L-iduronic acid 2-sulfate, (2) 2-deoxy-2-sulfamino-α-D-glucose-6-sulfate, (3) β-D-glucuronic acid, (4) 2-acetamido-2-deoxy-α-D-glucose, and (5) α-L-iduronic acid. These sugars are present in decreasing amounts, usually in the order (2) > (1) > (4) > (3) > (5), and are joined by glycosidic linkages, forming polymers of varying sizes. Heparin is strongly acidic because of its content of covalently linked sulfate and carboxylic acid groups. In heparin sodium, the acidic protons of the sulfate units are partially replaced by sodium ions. The potency is determined by a biological assay using a USP reference standard based on units of heparin activity per milligram.
Structure of Heparin Sodium (representative subunits):

………………..

http://www.medgadget.com/2008/08/on_the_road_to_a_fully_synthetic_heparin.html
…………

The chemoenzymatic synthesis of heparin from E. coli’s carbohydrate coat
Now, Linhardt’s team – who were also the first to identify the contaminant in the tainted batches as oversulfated chondroitin sulfate – have come up with a potentially safer way to produce heparin. The researchers grew flasks of the gut bacteria E. coli, then converted its naturally produced carbohydrate coat to heparin in just a few steps using enzymes and chemical treatment.
Linhardt says the key to the procedure was starting with the carbohydrate capsule that E coli produces to hide itself from the human immune system. The capsule is made from heparosan – a polysachharide that is already quite similar to heparin.
The team first chemically removed acetyl groups from the heparosan with sodium hydroxide and added a sulfate group using sulfur trioxide trimethylamine. Then, using four enzymes found in all mammals that produce heparin, they introduced further modifications, including the addition of three more sulfates at different positions on the molecule to get to heparin.
The checked the structure of the compound using NMR and showed that the synthetic compound could stop blood clotting as well as heparin derived from animals. To date, however, the team have only made a total of around 100mg of pure heparin – barely enough for a single dose. That is still a million times more than produced by a 2003 total synthesis of heparin, from researchers at the Massachusetts Institute of Technology, US.
- Heparin Sodium injection
- heparin. In: Lexi-Drugs Online [database on the Internet]. Hudson (OH): Lexi-Comp, Inc.; 2007 [cited 2/10/12]. Available from: http://online.lexi.com. subscription required to view.
- Cox, M.; Nelson D. (2004). Lehninger, Principles of Biochemistry (4). Freeman. p. 1100. ISBN 0-7167-4339-6.
- Marcum JA, McKenney JB. et al. (1986). “Anticoagulantly active heparin-like molecules from mast cell-deficient mice”.Am. J. Physiol. 250 (5 Pt 2): H879–888. PMID 3706560.
- Nader, HB et al.; Chavante, S.F.; Dos-Santos, E.A.; Oliveira, F.W.; De-Paiva, J.F.; Jerônimo, S.M.B.; Medeiros, G.F.; De-Abreu, L.R.D. et al. (1999). “Heparan sulfates and heparins: similar compounds performing the same functions in vertebrates and invertebrates?”. Braz. J. Med. Biol. Res. 32 (5): 529–538. doi:10.1590/S0100-879X1999000500005. PMID 10412563.
- Francis CW, Kaplan KL (2006). “Chapter 21. Principles of Antithrombotic Therapy”. In Lichtman MA, Beutler E, Kipps TJ, et al. Williams Hematology (7th ed.). ISBN 978-0-07-143591-8.
- Bentolila, A. et al.. “Synthesis and heparin-like biological activity of amino acid-based polymers” (Subscription required). Wiley InterScience. Retrieved 2008-03-10.
- Gatti, G., Casu, B. et al. (1979). “Studies on the Conformation of Heparin by lH and 13C NMR Spectroscopy” (PDF). Macromolecules 12 (5): 1001–1007. Bibcode:1979MaMol..12.1001G.doi:10.1021/ma60071a044.
- “Online Medical Dictionary”. Centre for Cancer Education. 2000. Retrieved 2008-07-11.
- Ferro D, Provasoli A, et al. (1990). “Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences”. Carbohydr. Res. 195 (2): 157–167.doi:10.1016/0008-6215(90)84164-P. PMID 2331699.
- Mulloy B, Forster MJ, Jones C, Davies DB. (1 January 1993). “N.m.r. and molecular-modelling studies of the solution conformation of heparin”. Biochem. J. 293 (Pt 3): 849–858. PMC 1134446. PMID 8352752.
- Guyton, A. C.; Hall, J. E. (2006). Textbook of Medical Physiology (11). Elsevier Saunders. p. 464. ISBN 0-7216-0240-1.
- Agnelli G, Piovella F, Buoncristiani P et al. (1998). “Enoxaparin plus compression stockings compared with compression stockings alone in the prevention of venous thromboembolism after elective neurosurgery”. N Engl J Med 339 (2): 80–5. doi:10.1056/NEJM199807093390204.PMID 9654538.
- Bergqvist D, Agnelli G, Cohen AT et al. (2002). “Duration of prophylaxis against venous thromboembolism with enoxaparin after surgery for cancer”. N Engl J Med 346(13): 975–980. doi:10.1056/NEJMoa012385.PMID 11919306.
- Handoll HHG, Farrar MJ, McBirnie J, Tytherleigh-Strong G, Milne AA, Gillespie WJ (2002). “Heparin, low molecular weight heparin and physical methods for preventing deep vein thrombosis and pulmonary embolism following surgery for hip fractures”. In Handoll, Helen HG. Cochrane Database Syst Rev 4 (4): CD000305.doi:10.1002/14651858.CD000305. PMID 12519540.
- Chuang YJ, Swanson R. et al. (2001). “Heparin enhances the specificity of antithrombin for thrombin and factor Xa independent of the reactive center loop sequence. Evidence for an exosite determinant of factor Xa specificity in heparin-activated antithrombin”. J. Biol. Chem. 276 (18): 14961–14971. doi:10.1074/jbc.M011550200. PMID 11278930.
- Bjork I, Lindahl U. (1982). “Mechanism of the anticoagulant action of heparin”. Mol. Cell. Biochem. 48 (3): 161–182.doi:10.1007/BF00421226. PMID 6757715.
- Petitou M, Herault JP, Bernat A, Driguez PA et al. (1999). “Synthesis of Thrombin inhibiting Heparin mimetics without side effects”. Nature 398 (6726): 417–422.Bibcode:1999Natur.398..417P. doi:10.1038/18877.PMID 10201371.
- Shalansky, Karen. DANAPAROID (Orgaran) for Heparin-Induced Thrombocytopenia. Vancouver Hospital & Health Sciences Centre, February 1998 Drug & Therapeutics Newsletter. Retrieved on 8 January 2007.
- Eikelboom JW, Hankey GJ (2002). “Low molecular weight heparins and heparinoids”. The Medical Journal of Australia 177 (7): 379–383. PMID 12358583.
- Weitz JI (2004). “New anticoagulants for treatment of venous thromboembolism”. Circulation 110 (9 Suppl 1): I19–26. doi:10.1161/01.CIR.0000140901.04538.ae.PMID 15339877.
- Hirsh J, Raschke R (2004). “Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy”. Chest 126 (3 Suppl): 188S–203S.doi:10.1378/chest.126.3_suppl.188S. PMID 15383472.
- Linhardt RJ, Gunay NS. (1999). “Production and Chemical Processing of Low Molecular Weight Heparins”. Sem. Thromb. Hem. 3: 5–16. PMID 10549711.
- Bhattacharya, Ananyo (August 2008). “Flask synthesis promises untainted heparin”. Chemistry World. Royal Society of Chemistry. Retrieved 6 February 2011.
- Kusmer, Ken (20 September 2006). “3rd Ind. preemie infant dies of overdose”. Fox News (Associated Press). Retrieved 2007-01-08.
- Internal medicine, Jay H. Stein, page 635
- Linhardt RJ. (1991). “Heparin: An important drug enters its seventh decade”. Chem. Indust. 2: 45–50.
- Jorpes E (August 1935). “The chemistry of heparin”. The Biochemical Journal 29 (8): 1817–30. PMC 1266692.PMID 16745848.
- Rutty, CJ. “Miracle Blood Lubricant: Connaught and the Story of Heparin, 1928–1937”. Health Heritage Research Services. Retrieved 2007-05-21.
|
9-1-2004
|
Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy.
|
Chest
|
|
|
4-1-1999
|
Synthesis of thrombin-inhibiting heparin mimetics without side effects.
|
Nature
|
|
|
1-1-1999
|
Production and chemical processing of low molecular weight heparins.
|
Seminars in thrombosis and hemostasis
|
|
|
8-1-1993
|
N.m.r. and molecular-modelling studies of the solution conformation of heparin.
|
The Biochemical journal
|
|
|
1-15-1990
|
Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences.
|
Carbohydrate research
|
Heparin, a highly sulfated glycosaminoglycan (GAG), is used extensively as an anticoagulant. It consists of repeating disaccharide units, containing iduronic acid (or glucuronic acid) and glucosamine, exhibiting variable degrees of sulfation. Heparin, and its analogues, are used during surgery and dialysis, and are often used to coat indwelling catheters and other devices where the vascular system is exposed. Administered parenterally, often continuously due to its short half-life, over 0.5 billion doses are required per year. Currently obtained from mucosal tissue of meat animals, mainly porcine intestine, and to a lesser extent bovine lung, its early stage production is poorly controlled, due to the source of the material (Figure 1). This problem came into sharp focus in 2008 when the presence of contaminating over-sulfated chondroitin sulfate in heparin, sourced from pigs, resulted in almost 100 deaths in the USA. This, coupled with the fact that only two doses are obtained per animal means that the demand for alternative and more controlled sources of heparin is high.




Figure.3. (A) The structure of heparosan disaccharide unit. (B) the structures of the major and minor variable repeating disaccharides comprising heparin where X = SO3- or H and Y = SO3- or COCH3.1

Figure.4. Time course of dry cell weight (g/L) and heparosan concentration in the fermentation supernatant (g/L) during the fermentation in a 20 L fermentor1
3. Z.Wang, M.Ly, F.Zhang, W. Zhong, A.Suen, A.M.Hickey, J.S.Dordick, R.J.Linhardt,”E. coli K5 fermentation and the preparation of heparosan, a bioengineered heparin precursor“, Biotechnol. Bioeng. 107, 964-973 (2010).
- M.Ly, Z.Wang, T.N.Laremore, F.Zhang, W.Zhong, D.Pu, D.V.Zagorevski, J.S.Dordick, R.J.Linhardt, “Analysis of E. coli K5 capsular polysaccharide heparosan.” Analytical and Bioanalytical Chemistry 399, 737-745 (2011).
FONDAPARINUX

FONDAPARINUX
Fondaparinux is a drug belonging to the group of the antithrombotic agents and are used to prevent deep vein thrombosis in patients undergoing orthopedic surgery. It is also used for the treatment of severe venous thrombosis and pulmonary
114870-03-0 ………..10x SODIUM SALT
| CAS number | 114870-03-0 FREE FORM |
|---|
| MF | C31H43N3Na10O49S8 10X SODIUM |
|---|---|
| MW | 1726.77 g/mol 10X SODIUM |
GSK-576428 Org-31540 SR-90107SR-90107A
launched 2002
Arixtra, Quixidar, Fondaparinux sodium, Fondaparin sodium, Arixtra (TN), Fondaparinux, Org-31540, AC1LCS4W, SR-90107A
Fondaparinux (Arixtra) is a synthetic pentasaccharide anticoagulant. Apart from the O-methyl group at the reducing end of the molecule, the identity and sequence of the five monomeric sugar units contained in fondaparinux is identical to a sequence of five monomeric sugar units that can be isolated after either chemical or enzymatic cleavage of the polymeric glycosaminoglycan heparin and heparan sulfate (HS). This monomeric sequence in heparin and HS is thought to form the high affinity binding site for the natural anti-coagulant factor, antithrombin III (ATIII).
Binding of heparin/HS to ATIII has been shown to increase the anti-coagulant activity of antithrombin III 1000-fold. Fondaparinux potentiates the neutralizing action ofATIII on activated Factor X 300-fold. Fondaparinux may be used: to prevent venous thromboembolism in patients who have undergone orthopedic surgery of the lower limbs (e.g. hip fracture, hip replacement and knee surgery); to prevent VTE in patients undergoing abdominal surgery who are are at high risk of thromboembolic complications; in the treatment of deep vein thrombosis (DVT) and pumonary embolism (PE); in the management of unstable angina (UA) and non-ST segment elevation myocardial infarction (NSTEMI); and in the management of ST segment elevation myocardial infarction (STEMI).

FONDAPARINUX
Fondaparinux (trade name Arixtra) is an anticoagulant medication chemically related to low molecular weight heparins. It is marketed byGlaxoSmithKline. A generic version developed by Alchemia is marketed within the US by Dr. Reddy’s Laboratories.
Fondaparinux is a synthetic pentasaccharide Factor Xa inhibitor. Apart from the O-methyl group at the reducing end of the molecule, the identity and sequence of the five monomeric sugar units contained in fondaparinux is identical to a sequence of five monomeric sugar units that can be isolated after either chemical or enzymatic cleavage of the polymeric glycosaminoglycans heparin and heparan sulfate (HS). Within heparin and heparan sulfate this monomeric sequence is thought to form the high affinity binding site for the anti-coagulant factor antithrombin III (ATIII). Binding of heparin/HS to ATIII has been shown to increase the anti-coagulant activity of antithrombin III 1000 fold. In contrast to heparin, fondaparinux does not inhibit thrombin.
Fondaparinux is given subcutaneously daily. Clinically, it is used for the prevention of deep vein thrombosis in patients who have had orthopedic surgery as well as for the treatment of deep vein thrombosis and pulmonary embolism.
One potential advantage of fondaparinux over LMWH or unfractionated heparin is that the risk for heparin-induced thrombocytopenia (HIT) is substantially lower. Furthermore, there have been case reports of fondaparinux being used to anticoagulate patients with established HIT as it has no affinity to PF-4. However, its renal excretion precludes its use in patients with renal dysfunction.
Unlike direct factor Xa inhibitors, it mediates its effects indirectly through antithrombin III, but unlike heparin, it is selective for factor Xa.[1]
Fondaparinux is similar to enoxaparin in reducing the risk of ischemic events at nine days, but it substantially reduces major bleeding and improves long term mortality and morbidity.[2]
It has been investigated for use in conjunction with streptokinase.[3]
Fondaparinux sodium, a selective coagulation factor Xa inhibitor, was first launched in the U.S. in 2002 by GlaxoSmithKline in a subcutaneous injection formulation for the prophylaxis of deep venous thrombosis (DVT) which may lead to pulmonary embolism in patients at risk for thromboembolic complications who are undergoing hip replacement, knee replacement, hip fracture surgery or abdominal surgery. The product is available in Japan for the treatment of acute deep venous thrombosis and acute pulmonary thromboembolism. In 2004, GlaxoSmithKline launched fondaparinux as an injection to be used in conjunction with warfarin sodium for the subcutaneous treatment of acute pulmonary embolism and DVT.
In 2007, GlaxoSmithKline received approval in the E.U. for the treatment of acute coronary syndrome (ACS), specifically unstable angina or non-ST segment elevation myocardial infarction (UA/NSTEMI) and ST-segment elevation myocardial infarction (STEMI), while in the U.S. an approvable letter was received for this indication. Currently, the drug is in clinical development at GlaxoSmithKline for the treatment of venous limb superficial thrombosis.

Fondaparinux Molecule
GlaxoSmithKline had filed a regulatory application in the E.U. seeking approval of fondaparinux sodium for the prevention of venous thromboembolic events (VTE), however; in 2008, the application was withdrawn for commercial reasons. Commercial launch in Japan for the product for the prevention of venous thromboembolism in high risk patients undergoing surgery in the abdomen took place in 2008.
In 2010, the EMA approved a regulatory application filed by GlaxoSmithKline seeking approval of a once-daily formulation of fondaparinux sodium for the treatment of adults with acute symptomatic spontaneous superficial-vein thrombosis (SVT) of the lower limbs without concomitant DVT. Product launch took place in the U.K. for this indication the same year.
The antithrombotic activity of fondaparinux is the result of antithrombin III (ATIII)-mediated selective inhibition of Factor Xa. By selectively binding to ATIII, the drug potentiates (about 300 times) the innate neutralization of Factor Xa by ATIII. Neutralization of Factor Xa, in turn, interrupts the blood coagulation cascade and thus inhibits thrombin formation and thrombus development. Fondaparinux does not inactivate thrombin (activated Factor II) and has no known effect on platelet function. At the recommended dose, no effects have been demonstrated on fibrinolytic activity or bleeding time.
Originally developed by Organon and Sanofi (formerly known as sanofi-aventis), fondaparinux sodium is currently available in approximately 30 countries. In 2004, Organon transferred its rights to the drug to Sanofi in exchange for revenues based on future sales from jointly developed antithrombotic products and in early 2005, GlaxoSmithKline also acquired the antithrombotic.
At the beginning of 2005, GlaxoSmithKline signed a two-year agreement with Adolor (acquired by Cubist in 2011) for the copromotion of fondaparinux sodium in the U.S. In Sepetember 2013, Aspen Pharmacare acquired Arixtra global rights (excluding China, India and Pakistan) from GlaxoSmithKline for the treatment of thrombosis with GlaxoSmithKline commercializing the product in Indonesia under licence from Aspen.
Chemical structure
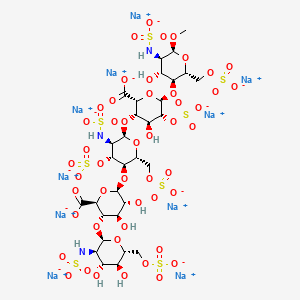
Abbreviations
- GlcNS6S = 2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside
- GlcA = β-D-glucopyranuronoside
- GlcNS3,6S = 2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl
- IdoA2S = 2-O-sulfo-α-L-idopyranuronoside
- GlcNS6SOMe = methyl-O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside
The sequence of monosaccharides is D-GlcNS6S-α-(1,4)-D-GlcA-β-(1,4)-D-GlcNS3,6S-α-(1,4)-L-IdoA2S-α-(1,4)-D-GlcNS6S-OMe, as shown in the following structure:
ARIXTRA (fondaparinux sodium) Injection is a sterile solution containing fondaparinux sodium. It is a synthetic and specific inhibitor of activatedFactor X (Xa). Fondaparinux sodium is methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-β-D-glucopyranuronosyl-( 1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-Osulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt.
The molecular formula of fondaparinux sodium is C31H43N3Na10O49S8 and its molecular weight is 1728. The structural formula is provided below:
ARIXTRA is supplied as a sterile, preservative-free injectable solution for subcutaneous use.
Each single-dose, prefilled syringe of ARIXTRA, affixed with an automatic needle protection system, contains 2.5 mg of fondaparinux sodium in 0.5 mL, 5.0 mg of fondaparinux sodium in 0.4 mL, 7.5 mg of fondaparinux sodium in 0.6 mL, or 10.0 mg of fondaparinux sodium in 0.8 mL of an isotonic solutionof sodium chloride and water for injection. The final drug product is a clear and colorless to slightly yellow liquid with a pH between 5.0 and 8.0.
……………….
INTRODUCTION
In U.S. Patent No. 7,468,358, Fondaparinux sodium is described as the “only anticoagulant thought to be completely free of risk from HIT-2 induction.” The biochemical and pharmacologic rationale for the development of a heparin pentasaccharide in Thromb. Res., 86(1), 1-36, 1997 by Walenga et al. cited the recently approved synthetic pentasaccharide Factor Xa inhibitor Fondaparinux sodium. Fondaparinux has also been described in Walenga et al., Expert Opin. Investig. Drugs, Vol. 11, 397-407, 2002 and Bauer, Best Practice & Research Clinical Hematology, Vol. 17, No. 1, 89-104, 2004.
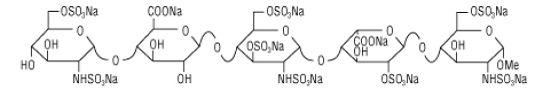
Fondaparinux sodium is a linear octasulfated pentasaccharide (oligosaccharide with five monosaccharide units ) molecule having five sulfate esters on oxygen (O-sulfated moieties) and three sulfates on a nitrogen (N- sulfated moieties). In addition, Fondaparinux contains five hydroxyl groups in the molecule that are not sulfated and two sodium carboxylates. Out of five saccharides, there are three glucosamine derivatives and one glucuronic and one L-iduronic acid. The five saccharides are connected to each other in alternate α and β glycosylated linkages (see Figure 1).
Figure 1 Fondaparinux Sodium

Monosaccharide E Monosaccharide D Monosaccharide C Monosaccharide B Monosaccharide A derived from derived from derived from derived from derived from
Monomer E Monomer D Monomer C Monomer B1 Monomer A2
Fondaparinux Sodium
Fondaparinux sodium is a chemically synthesized methoxy derivative of the natural pentasaccharide sequence, which is the active site of heparin that mediates the interaction with antithrombin (Casu et al., J. Biochem., 197, 59, 1981). It has a challenging pattern of O- and N- sulfates, specific glycosidic stereochemistry, and repeating units of glucosamines and uronic acids (Petitou et al, Progress in the Chemistry of Organic Natural Product, 60, 144-209, 1992).
The monosaccharide units comprising the Fondaparinux molecule are labeled as per the convention in Figure 1, with the glucosamine unit on the right referred to as monosaccharide A and the next, an uronic acid unit to its left as B and subsequent units, C, D and E respectively. The chemical synthesis of Fondaparinux starts with monosaccharides of defined structures that are themselves referred to as Monomers A2, Bl, C, D and E, for differentiation and convenience, and they become the corresponding monosaccharides in fondaparinux sodium.
Due to this complex mixture of free and sulfated hydroxyl groups, and the presence of N- sulfated moieties, the design of a synthetic route to Fondaparinux requires a careful strategy of protection and de-protection of reactive functional groups during synthesis of the molecule. Previously described syntheses of Fondaparinux all adopted a similar strategy to complete the synthesis of this molecule. This strategy can be envisioned as having four stages.
The strategy in the first stage requires selective de-protection of five out of ten hydroxyl groups. During the second stage these five hydroxyls are selectively sulfonated. The third stage of the process involves the de -protection of the remaining five hydroxyl groups. The fourth stage of the process is the selective sulfonation of the 3 amino groups, in the presence of five hydroxyl groups that are not sulfated in the final molecule. This strategy can be envisioned from the following fully protected pentasaccharide, also referred to as the late-stage intermediate.

In this strategy, all of the hydroxyl groups that are to be sulfated are protected with an acyl protective group, for example, as acetates (R = CH3) or benzoates (R = aryl) (Stages 1 and 2) All of the hydroxyl groups that are to remain as such are protected with benzyl group as benzyl ethers (Stage 3). The amino group, which is subsequently sulfonated, is masked as an azide (N3) moiety (Stage 4). R1 and R2 are typically sodium in the active pharmaceutical compound (e.g., Fondaparinux sodium).
This strategy allows the final product to be prepared by following the synthetic operations as outlined below: a) Treatment of the late- stage intermediate with base to hydrolyze (deprotect) the acyl ester groups to reveal the five hydroxyl groups. The two R1 and R2 ester groups are hydrolyzed in this step as well.

b) Sulfonation of the newly revealed hydroxyl groups.

c) Hydrogenation of the O-sulfated pentasaccharide to de-benzylate the five benzyl- protected hydroxyls, and at the same time, unmask the three azides to the corresponding amino groups.

d) On the last step of the operation, the amino groups are sulfated selectively at a high pH, in the presence of the five free hydroxyls to give Fondaparinux (Figure 1). While the above strategy has been shown to be viable, it is not without major drawbacks. One drawback lies in the procedure leading to the fully protected pentasaccharide (late stage intermediate), especially during the coupling of the D-glucuronic acid to the next adjacent glucose ring (the D-monomer to C-monomer in the EDCBA nomenclature shown in Figure 1). Sugar oligomers or oligosaccharides, such as Fondaparinux, are assembled using coupling reactions, also known as glycosylation reactions, to “link” sugar monomers together. The difficulty of this linking step arises because of the required stereochemical relationship between the D-sugar and the C-sugar, as shown below:

The stereochemical arrangement illustrated above in Figure 2 is described as having a β- configuration at the anomeric carbon of the D-sugar (denoted by the arrow). The linkage between the D and C units in Fondaparinux has this specific stereochemistry. There are, however, competing β- and α-glycosylation reactions.
The difficulties of the glycosylation reaction in the synthesis of Fondaparinux is well known. In 1991 Sanofi reported a preparation of a disaccharide intermediate in 51% yield having a 12/1 ratio of β/α stereochemistry at the anomeric position (Duchaussoy et al., Bioorg. & Med. Chem. Lett., 1(2), 99-102, 1991).
In another publication (Sinay et al, Carbohydrate Research, 132, C5-C9, 1984) yields on the order of 50% with coupling times on the order of 6- days are reported. U.S. Patent No. 4,818,816 {see e.g., column 31, lines 50-56) discloses a 50% yield for the β-glycosylation.
Alchemia’s U.S. Patent No. 7,541,445 is even less specific as to the details of the synthesis of this late-stage Fondaparinux synthetic intermediate. The ‘445 Patent discloses several strategies for the assembly of the pentasaccharide (1+4, 3+2 or 2+3) using a 2-acylated D-sugar (specifically 2-allyloxycarbonyl) for the glycosylation coupling reactions. However, Alchemia’s strategy involves late-stage pentasaccharides that all incorporate a 2-benzylated D- sugar.
The transformation of acyl to benzyl is performed either under acidic or basic conditions. Furthermore, these transformations, using benzyl bromide or benzyl trichloroacetimidate, typically result in extensive decomposition and the procedure suffers from poor yields. Thus, such transformations (at a disaccharide, trisaccharide, and pentasaccharide level) are typically not acceptable for industrial scale production.
Examples of fully protected pentasaccharides are described in Duchaussoy et al, Bioorg. Med. Chem. Lett., 1 (2), 99-102, 1991; Petitou et al, Carbohydr. Res., 167, 67-75, 1987; Sinay et al, Carbohydr. Res., 132, C5-C9, 1984; Petitou et al., Carbohydr. Res., 1147, 221-236, 1986; Lei et al., Bioorg. Med. Chem., 6, 1337-1346, 1998; Ichikawa et al., Tet. Lett., 27(5), 611-614, 1986; Kovensky et al, Bioorg. Med. Chem., 1999, 7, 1567-1580, 1999.
These fully protected pentasaccharides may be converted to the O- and N-sulfated pentasaccharides using the four steps (described earlier) of: a) saponification with LiOHZH2CVNaOH, b) O-sulfation by an Et3N- SO3 complex; c) de-benzylation and azide reduction via H2/Pd hydrogenation; and d) N-sulfation with a pyridine-SO3 complex.
Even though many diverse analogs of the fully protected pentasaccharide have been prepared, none use any protective group at the 2-position of the D unit other than a benzyl group. Furthermore, none of the fully protected pentasaccharide analogs offer a practical, scaleable and economical method for re-introduction of the benzyl moiety at the 2-position of the D unit after removal of any participating group that promotes β-glycosylation.
Furthermore, the coupling of benzyl protected sugars proves to be a sluggish, low yielding and problematic process, typically resulting in substantial decomposition of the pentasaccharide (prepared over 50 synthetic steps), thus making it unsuitable for a large [kilogram] scale production process.

Ref. 1. Sinay et al, Carbohydr. Res., 132, C5-C9, 1984.
Ref. 2. Petitou et al., Carbohydr. Res., 147, 221-236. 1986
It has been a general strategy for carbohydrate chemists to use base-labile ester-protecting group at 2-position of the D unit to build an efficient and stereoselective β-glycosidic linkage. To construct the β-linkage carbohydrate chemists have previously acetate and benzoate ester groups, as described, for example, in the review by Poletti et al., Eur. J. Chem., 2999-3024, 2003.
The ester group at the 2-position of D needs to be differentiated from the acetate and benzoates at other positions in the pentasaccharide. These ester groups are hydrolyzed and sulfated later in the process and, unlike these ester groups, the 2-hydroxyl group of the D unit needs to remain as the hydroxyl group in the final product, Fondaparinux sodium.
Some of the current ester choices for the synthetic chemists in the field include methyl chloro acetyl and chloro methyl acetate [MCA or CMA] . The mild procedures for the selective removal of theses groups in the presence of acetates and benzoates makes them ideal candidates. However, MCA/CMA groups have been shown to produce unwanted and serious side products during the glycosylation and therefore have not been favored in the synthesis of Fondaparinux sodium and its analogs. For by-product formation observed in acetate derivatives see Seeberger et al., J. Org. Chem., 2004, 69, 4081-93.
Similar by-product formation is also observed using chloroacetate derivatives. See Orgueira et al., Eur. J. Chem., 9(1), 140-169, 2003.
The levulinyl group can be rapidly and almost quantitatively removed by treatment with hydrazine hydrate as the deprotection reagent as illustrated in the example below. Under the same reaction conditions primary and secondary acetate and benzoate esters are hardly affected by hydrazine hydrate. See, e.g., Seeberger et al, J. Org. Chem., 69, 4081-4093, 2004.

The syntheses of Fondaparinux sodium described herein takes advantage of the levulinyl group in efficient construction of the trisaccharide EDC with improved β- selectivity for the coupling under milder conditions and increased yields.

Substitution of the benzyl protecting group with a THP moiety provides its enhanced ability to be incorporated quantitatively in position-2 of the unit D of the pentasaccharide. Also, the THP group behaves in a similar manner to a benzyl ether in terms of function and stability. In the processes described herein, the THP group is incorporated at the 2-position of the D unit at this late stage of the synthesis (i.e., after the D and C units have been coupled through a 1,2-trans glycosidic (β-) linkage). The THP protective group typically does not promote an efficient β- glycosylation and therefore is preferably incorporated in the molecule after the construction of the β-linkage.
Fondaparinux and sodium salt thereof can be prepared from pure compound of Formula II by following the teachings from Bioorganic and Medicinal Chemistry Letters, 1(2), p. 95-98 (1991). A second aspect of the present invention provides a process for the preparation of 4-0- -D-glucopyranosyl-l,6-anhydro- -D-glucopyranose, represented by STR BELOW

……………………………..
SYNTHESIS
EP2464668A2 AND US8288515
The scheme below exemplifies some of the processes of the present invention disclosed herein.

The tetrahydropyranyl (THP) protective group and the benzyl ether protective group are suitable hydroxyl protective groups and can survive the last four synthetic steps (described above) in the synthesis of Fondaparinux sodium, even under harsh reaction conditions. Certain other protecting groups do not survive the last four synthetic steps in high yield.
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:


Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; ACN is acetonitrile; MS is molecular sieves; DMF is dimethyl formamide; PMB is p-methoxybenzyl; Bn is benzyl; DCM is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; CSA is camphor sulfonic acid; TEA is triethylamine; MeOH is methanol; DMAP is dimethylaminopyridine; RT is room temperature; CAN is ceric ammonium nitrate; Ac2O is acetic anhydride; HBr is hydrogen bromide; TEMPO is tetramethylpiperidine-N-oxide; TBACl is tetrabutyl ammonium chloride; EtOAc is ethyl acetate; HOBT is hydroxybenzotriazole; DCC is dicyclohexylcarbodiimide; Lev is levunlinyl; TBDPS is tertiary-butyl diphenylsilyl; TCA is trichloroacetonitrile; O-TCA is O-trichloroacetimidate; Lev2O is levulinic anhydride; DIPEA is diisopropylethylamine; Bz is benzoyl; TBAF is tetrabutylammonium fluoride; DBU is diazabicycloundecane; BF3.Et2O is boron trifluoride etherate; TMSI is trimethylsilyl iodide; TBAI is tetrabutylammonium iodide; TES-Tf is triethylsilyl trifluoromethanesulfonate (triethylsilyl triflate); DHP is dihydropyran; PTS is p-toluenesulfonic acid.
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:


The ester moieties in EDCBA Pentamer were hydrolyzed with sodium and lithium hydroxide in the presence of hydrogen peroxide in dioxane mixing at room temperature for 16 hours to give the pentasaccharide intermediate API1. The five hydroxyl moieties in API1 were sulfated using a pyridine-sulfur trioxide complex in dimethylformamide, mixing at 60° C. for 2 hours and then purified using column chromatography (CG-161), to give the pentasulfated pentasaccharide API2. The intermediate API2 was then hydrogenated to reduce the three azides on sugars E, C and A to amines and the reductive deprotection of the five benzyl ethers to their corresponding hydroxyl groups to form the intermediate API3. This transformation occurs by reacting API2 with 10% palladium/carbon catalyst with hydrogen gas for 72 hours. The three amines on API3 were then sulfated using the pyridine-sulfur trioxide complex in sodium hydroxide and ammonium acetate, allowing the reaction to proceed for 12 hours. The acidic work-up procedure of the reaction removes the THP group to provide crude fondaparinux which is purified and is subsequently converted to its salt form. The crude mixture was purified using an ion-exchange chromatographic column (HiQ resin) followed by desalting using a size exclusion resin or gel filtration (Biorad Sephadex G25) to give the final API, fondaparinux sodium
Experimental Procedures Preparation of IntD1 Bromination of Glucose Pentaacetate
To a 500 ml flask was added 50 g of glucose pentaacetate (C6H22O11) and 80 ml of methylene chloride. The mixture was stirred at ice-water bath for 20 min HBr in HOAc (33%, 50 ml) was added to the reaction mixture. After stirring for 2.5 hr another 5 ml of HBr was added to the mixture. After another 30 min, the mixture was added 600 ml of methylene chloride. The organic mixture was washed with cold water (200 ml×2), Saturated NaHCO3(200 ml×2), water (200 ml) and brine (200 ml×2). The organic layer was dried over Na2SO4 and the mixture was evaporated at RT to give white solid as final product, bromide derivative, IntD1 (˜95% yield). C14H19BrO9, TLC Rf=0.49, SiO2, 40% ethyl acetate/60% hexanes; Exact Mass 410.02.
Preparation of IntD2 by Reductive Cyclization
To a stirring mixture of bromide IntD1 (105 g), tetrabutylammonium iodide (60 g, 162 mmol) and activated 3 Å molecular sieves in anhydrous acetonitrile (2 L), solid NaBH4 (30 g, 793 mmol) was added. The reaction was heated at 40° C. overnight. The mixture was then diluted with dichloromethane (2 L) and filtered through Celite®. After evaporation, the residue was dissolved in 500 ml ethyl acetate. The white solid (Bu4NI or Bu4NBr) was filtered. The ethyl acetate solution was evaporated and purified by chromatography on silica gel using ethyl acetate and hexane as eluent to give the acetal-triacetate IntD2 (˜60-70% yield). TLC Rf=0.36, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD3 by De-Acetylation
To a 1000 ml flask was added triacetate IntD2 (55 g) and 500 ml of methanol. After stirring 30 min, the reagent NaOMe (2.7 g, 0.3 eq) was added and the reaction was stirred overnight. Additional NaOMe (0.9 g) was added to the reaction mixture and heated to 50° C. for 3 hr. The mixture was neutralized with Dowex 50Wx8 cation resin, filtered and evaporated. The residue was purified by silica gel column to give 24 g of trihydroxy-acetal IntD3. TLC Rf=0.36 in SiO2, 10% methanol/90% ethyl acetate.
Preparation of IntD4 by Benzylidene Formation
To a 1000 ml flask was added trihydroxy compound IntD3 (76 g) and benzaldehyde dimethyl acetate (73 g, 1.3 eq). The mixture was stirred for 10 min, after which D(+)-camphorsulfonic acid (8.5 g, CSA) was added. The mixture was heated at 50° C. for two hours. The reaction mixture was then transferred to separatory funnel containing ethyl acetate (1.8 L) and sodium bicarbonate solution (600 ml). After separation, the organic layer was washed with a second sodium bicarbonate solution (300 ml) and brine (800 ml). The two sodium carbonate solutions were combined and extracted with ethyl acetate (600 ml×2). The organic mixture was evaporated and purified by silica gel column to give the benzylidene product IntD4 (77 g, 71% yield). TLC Rf=0.47, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD5 by Benzylation
To a 500 ml flask was added benzylidene acetal compound IntD4 (21 g,) in 70 ml THF. To another flask (1000 ml) was added NaH (2 eq). The solution of IntD4 was then transferred to the NaH solution at 0° C. The reaction mixture was stirred for 30 min, then benzyl bromide (16.1 ml, 1.9 eq) in 30 ml THF was added. After stirring for 30 min, DMF (90 ml) was added to the reaction mixture. Excess NaH was neutralized by careful addition of acetic acid (8 ml). The mixture was evaporated and purified by silica gel column to give the benzyl derivative IntD5. (23 g) TLC Rf=0.69, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD6 by Deprotection of Benzylidene
To a 500 ml flask was added the benzylidene-acetal compound IntD5 (20 g) and 250 ml of dichloromethane, the reaction mixture was cooled to 0° C. using an ice-water-salt bath. Aqueous TFA (80%, 34 ml) was added to the mixture and stirred over night. The mixture was evaporated and purified by silica gel column to give the dihydroxy derivative IntD6. (8 g, 52%). TLC Rf=0.79, SiO2 in 10% methanol/90% ethyl acetate.
Preparation of IntD7 by Oxidation of 6-Hydroxyl
To a 5 L flask was added dihydroxy compound IntD6 (60 g), TEMPO (1.08 g), sodium bromide (4.2 g), tetrabutylammonium chloride (5.35 g), saturated NaHCO3 (794 ml) and EtOAc (1338 ml). The mixture was stirred over an ice-water bath for 30 min To another flask was added a solution of NaOCl (677 ml), saturated NaHCO3 (485 ml) and brine (794 ml). The second mixture was added slowly to the first mixture (over about two hrs). The resulting mixture was then stirred overnight. The mixture was separated, and the inorganic layer was extracted with EtOAc (800 ml×2). The combined organic layers were washed with brine (800 ml). Evaporation of the organic layer gave 64 g crude carboxylic acid product IntD7 which was used in the next step use without purification. TLC Rf=0.04, SiO2 in 10% methanol/90% ethyl acetate.
Preparation of Monomer D by Benzylation of the Carboxylic Acid
To a solution of carboxylic acid derivative IntD7 (64 g) in 600 ml of dichloromethane, was added benzyl alcohol (30 g) and N-hydroxybenzotriazole (80 g, HOBt). After stirring for 10 min triethylamine (60.2 g) was added slowly. After stirring another 10 min, dicyclohexylcarbodiimide, (60.8 g, DCC) was added slowly and the mixture was stirred overnight. The reaction mixture was filtered and the solvent was removed under reduced pressure followed by chromatography on silica gel to provide 60.8 g (75%, over two steps) of product, Monomer D. TLC Rf=0.51, SiO2 in 40% ethyl acetate/60% hexanes.
Synthesis of the BA Dimer
Step 1. Preparation of BMod1, Levulination of Monomer B1
A 100 L reactor was charged with 7.207 Kg of Monomer B1 (21.3 moles, 1 equiv), 20 L of dry tetrahydrofuran (THF) and agitated to dissolve. When clear, it was purged with nitrogen and 260 g of dimethylamino pyridine (DMAP, 2.13 moles, 0.1 equiv) and 11.05 L of diisopropylethylamine (DIPEA, 8.275 kg, 63.9 moles, 3 equiv) was charged into the reactor. The reactor was chilled to 10-15° C. and 13.7 kg levulinic anhydride (63.9 mol, 3 equiv) was transferred into the reactor. When the addition was complete, the reaction was warmed to ambient temperature and stirred overnight or 12-16 hours. Completeness of the reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. When the reaction was complete, 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. The mixture was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L of water, 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight. The yield of the isolated syrup of BMod1 was 100%.
Synthesis of the BA Dimer
Step 2. Preparation of BMod2, TFA Hydrolysis of BMod1
A 100 L reactor was charged with 9296 Kg of 4-Lev Monomer B1 (BMod1) (21.3 mol, 1 equiv). The reactor chiller was turned to <5° C. and stirring was begun, after which 17.6 L of 90% TFA solution (TFA, 213 mole, 10 equiv) was transferred into the reactor. When the addition was complete, the reaction was monitored by TLC and HPLC. The reaction took approximately 2-3 hours to reach completion. When the reaction was complete, the reactor was chilled and 26.72 L of triethylamine (TEA, 19.4 Kg, 191.7 mole, 0.9 equiv) was transferred into the reactor. An additional 20 L of water and 20 L ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer was extracted (EtOAc layer) with 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 50:50, 80:20 (EtOAc/heptane), 100% EtOAc, 5:95, 10:90 (MeOH/EtOAc). The pure fractions were pooled and evaporated to a syrup. The yield of the isolated syrup, BMod2 was 90%.
Synthesis of the BA Dimer
Step 3. Preparation of BMod3, Silylation of BMod2
A 100 L reactor was charged with 6.755 Kg 4-Lev-1,2-DiOH Monomer B1 (BMod2) (17.04 mol, 1 equiv), 2328 g of imidazole (34.2 mol, 2 equiv) and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., then 5.22 L tert-butyldiphenylchloro-silane (TBDPS-Cl, 5.607 Kg, 20.4 mol, 1.2 equiv) was transferred into the reactor. When addition was complete, the chiller was turned off and the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (40% ethyl acetate/hexane) and HPLC. The reaction took approximately 3 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. Dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The yield of BMod3 was about 80%.
Synthesis of the BA Dimer
Step 4. Preparation of BMod4, Benzoylation
A 100 L reactor was charged with 8.113 Kg of 4-Lev-1-Si-2-OH Monomer B1 (BMod3) (12.78 mol, 1 equiv), 9 L of pyridine and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., after which 1.78 L of benzoyl chloride (2155 g, 15.34 mol, 1.2 equiv) was transferred into the reactor. When addition was complete, the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (40% ethyl acetate/heptane) and HPLC. The reaction took approximately 3 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The DCM solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). Isolated syrup BMod4 was obtained in 91% yield.
Synthesis of the BA Dimer
Step 5. Preparation of BMod5, Desilylation
A 100 L reactor was charged with 8.601 Kg of 4-Lev-1-Si-2-Bz Monomer B1 (BMod4) (11.64 mol, 1 equiv) in 30 L terahydrofuran. The reactor was purged with nitrogen and chilled to 0° C., after which 5.49 Kg of tetrabutylammonium fluoride (TBAF, 17.4 mol, 1.5 equiv) and 996 mL (1045 g, 17.4 mol, 1.5 equiv) of glacial acetic acid were transferred into the reactor. When the addition was complete, the reaction was stirred at ambient temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. The reaction took approximately 6 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20 (EtOAc/heptane) and 200 L 100% EtOAc. Pure fractions were pooled and evaporated to a syrup. The intermediate BMod5 was isolated as a syrup in 91% yield.
Synthesis of the BA Dimer
Step 6: Preparation of BMod6, TCA Formation
A 100 L reactor was charged with 5.238 Kg of 4-Lev-1-OH-2-Bz Monomer B1 (BMod5) (10.44 mol, 1 equiv) in 30 L of DCM. The reactor was purged with nitrogen and chilled to 10-15° C., after which 780 mL of diazabicyclo undecene (DBU, 795 g, 5.22 mol, 0.5 equiv) and 10.47 L of trichloroacetonitrile (TCA, 15.08 Kg, 104.4 mol, 10 equiv) were transferred into the reactor. Stirring was continued and the reaction was kept under a nitrogen atmosphere. After reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by HPLC and TLC (40:60 ethyl acetate/heptane). The reaction took approximately 2 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of dichloromethane were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was separated with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/Heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of BMod6 was 73%.
Synthesis of the BA Dimer
Step 7. Preparation of AMod1, Acetylation of Monomer A2
A 100 L reactor was charged with 6.772 Kg of Monomer A2 (17.04 mole, 1 eq.), 32.2 L (34.8 Kg, 340.8 moles, 20 eq.) of acetic anhydride and 32 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C. When the temperature reached −20° C., 3.24 L (3.63 Kg, 25.68 mol, 1.5 equiv) of boron trifluoride etherate (BF3.Et2O) was transferred into the reactor. After complete addition of boron trifluoride etherate, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 3-5 hours for completion. When the reaction was complete, extraction was performed with 3×15 L of 10% sodium bicarbonate and 20 L of water. The organic phase (DCM) was evaporated to a syrup (bath temp. 40° C.) and allowed to dry overnight. The syrup was purified in a 200 L silica column using 140 L each of the following gradient profiles: 5:95, 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of AMod1 was 83%.
Synthesis of the BA Dimer
Step 8. Preparation of AMod3,1-Methylation of AMod1
A 100 L reactor was charged with 5891 g of acetyl Monomer A2 (AMod1) (13.98 mole, 1 eq.) in 32 L of dichloromethane. The reactor was purged with nitrogen and was chilled to 0° C., after which 2598 mL of trimethylsilyl iodide (TMSI, 3636 g, 18 mol, 1.3 equiv) was transferred into the reactor. When addition was complete, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 2-4 hours to reach completion. When the reaction was complete, the mixture was diluted with 20 L of toluene. The solution was evaporated to a syrup and was co-evaporated with 3×6 L of toluene. The reactor was charged with 36 L of dichloromethane (DCM), 3.2 Kg of dry 4 Å Molecular Sieves, 15505 g (42 mol, 3 equiv) of tetrabutyl ammonium iodide (TBAI) and 9 L of dry methanol. This was stirred until the TBAI was completely dissolved, after which 3630 mL of diisopropyl-ethylamine (DIPEA, 2712 g, 21 moles, 1.5 equiv) was transferred into the reactor in one portion. The completion of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 16 hours for completion. When the reaction was complete, the molecular sieves were removed by filtration. Added were 20 L EtOAc and extracted with 4×20 L of 25% sodium thiosulfate and 20 L 10% NaCl solutions. The organic layer was separated and dried with 8-12 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 5:95, 10:90, 20:80, 30:70 and 40:60 (EtOAc/heptane). The pure fractions were pooled and evaporated to give intermediate AMod3 as a syrup. The isolated yield was 75%.
Synthesis of the BA Dimer
Step 9. Preparation of AMod4, DeAcetylation of AMod3
A 100 L reactor was charged with 4128 g of 1-Methyl 4,6-Diacetyl Monomer A2 (AMod3) (10.5 mol, 1 equiv) and 18 L of dry methanol and dissolved, after which 113.4 g (2.1 mol, 0.2 equiv) of sodium methoxide was transferred into the reactor. The reaction was stirred at room temperature and monitored by TLC (40% ethyl acetate/hexane) and HPLC. The reaction took approximately 2-4 hours for completion. When the reaction was complete, Dowex 50Wx8 cation resin was added in small portions until the pH reached 6-8. The Dowex 50Wx8 resin was filtered and the solution was evaporated to a syrup (bath temp. 40° C.). The syrup was diluted with 10 L of ethyl acetate and extracted with 20 L brine and 20 L water. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight at the same temperature. The isolated yield of the syrup AMod4 was about 88%.
Synthesis of the BA Dimer
Step 10. Preparation of AMod5,6-Benzoylation
A 100 L reactor was charged with 2858 g of Methyl 4,6-diOH Monomer A2 (AMod4) (9.24 mol, 1 equiv) and co-evaporated with 3×10 L of pyridine. When evaporation was complete, 15 L of dichloromethane, 6 L of pyridine were transferred into the reactor and dissolved. The reactor was purged with nitrogen and chilled to −40° C. The reactor was charged with 1044 mL (1299 g, 9.24 mol, 1 equiv) of benzoyl chloride. When the addition was complete, the reaction was allowed to warm to −10° C. over a period of 2 hours. The reaction was monitored by TLC (60% ethyl acetate/hexane). When the reaction was completed, the solution was evaporated to a syrup (bath temp. 40° C.). This was co-evaporated with 3×15 L of toluene. The syrup was diluted with 40 L ethyl acetate. Extraction was carried out with 20 L of water and 20 L of brine solution. The Ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 5:95, 10:90, 20:80, 25:70 and 30:60 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup. The isolated yield of the intermediate AMod5 was 84%.
Synthesis of the BA Dimer
Step 11. Preparation of BA1, Coupling of Amod5 with BMod6
A 100 L reactor was charged with 3054 g of methyl 4-Hydroxy-Monomer A2 (AMod5) from Step 10 (7.38 mol, 1 equiv) and 4764 g of 4-Lev-1-TCA-Monomer B1 (BMod6) from Step 6 (7.38 mol, 1 equiv). The combined monomers were dissolved in 20 L of toluene and co-evaporated at 40° C. Co evaporation was repeated with an additional 2×20 L of toluene, after which 30 L of dichloromethane (DCM) was transferred into the reactor and dissolved. The reactor was purged with nitrogen and was chilled to below −20° C. When the temperature was between −20° C. and −40° C., 1572 g (1404 mL, 11.12 moles, 1.5 equiv) of boron trifluoride etherate (BF3.Et2O) were transferred into the reactor. After complete addition of boron trifluoride etherate, the reaction was allowed to warm to 0° C. and stirring was continued. The completeness of the reaction was monitored by HPLC and TLC (40:70 ethyl acetate/heptane). The reaction required 3-4 hours to reach completion. When the reaction was complete, 926 mL (672 g, 6.64 mol, 0.9 equiv) of triethylamine (TEA) was transferred into the mixture and stirred for an additional 30 minutes, after which 20 L of water and 10 L of dichloromethane were transferred into the reactor. The solution was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was separated with 2×20 L water and 20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and used in the next step. The isolated yield of the disaccharide BA1 was about 72%.
Synthesis of the BA Dimer
Step 12, Removal of Levulinate Methyl [(methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl]-2-deoxy-α-D-glucopyranoside
A 100 L reactor was charged with 4.104 Kg of 4-Lev BA Dimer (BA1) (4.56 mol, 1 equiv) in 20 L of THF. The reactor was purged with nitrogen and chilled to −20 to −25° C., after which 896 mL of hydrazine hydrate (923 g, 18.24 mol, 4 equiv) was transferred into the reactor. Stirring was continued and the reaction was monitored by TLC (40% ethyl acetate/heptane) and HPLC. The reaction took approximately 2-3 hour for the completion, after which 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (ETOAc layer) was extracted with 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). The pure fractions were pooled and evaporated to dryness. The isolated yield of the BA Dimer was 82%. Formula: C42H43N3O13; Mol. Wt. 797.80.
Synthesis of the EDC Trimer
Step 1. Preparation of EMod1, Acetylation
A 100 L reactor was charged with 16533 g of Monomer E (45 mole, 1 eq.), 21.25 L acetic anhydride (225 mole, 5 eq.) and 60 L of dichloromethane. The reactor was purged with nitrogen and was chilled to −10° C. When the temperature was at −10° C., 1.14 L (1277 g) of boron trifluoride etherate (BF3.Et2O, 9.0 moles, 0.2 eq) were transferred into the reactor. After the complete addition of boron trifluoride etherate, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by TLC (30:70 ethyl acetate/heptane) and HPLC. The reaction took approximately 3-6 hours to reach completion. When the reaction was completed, the mixture was extracted with 3×50 L of 10% sodium bicarbonate and SOL of water. The organic phase (DCM) was evaporated to a syrup (bath temp. 40° C.) and allowed to dry overnight. The isolated yield of EMod1 was 97%.
Synthesis of the EDC Trimer
Step 2. Preparation of EMod2, De-Acetylation of Azidoglucose
A 100 L reactor was charged with 21016 g of 1,6-Diacetyl Monomer E (EMod1) (45 mole, 1 eq.), 5434 g of hydrazine acetate (NH2NH2.HOAc, 24.75 mole, 0.55 eq.) and 50 L of DMF (dimethyl formamide). The solution was stirred at room temperature and the reaction was monitored by TLC (30% ethyl acetate/hexane) and HPLC. The reaction took approximately 2-4 hours for completion. When the reaction was completed, 50 L of dichloromethane and 40 L of water were transferred into the reactor. This was stirred for 30 minutes and the layers were separated. This was extracted with an additional 40 L of water and the organic phase was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight at the same temperature. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of intermediate EMod2 was 100%.
Synthesis of the EDC Trimer
Step 3. Preparation of EMod3, Formation of 1-TCA
A 100 L reactor was charged with 12752 g of 1-Hydroxy Monomer E (EMod2) (30 mole, 1 eq.) in 40 L of dichloromethane. The reactor was purged with nitrogen and stirring was started, after which 2.25 L of DBU (15 moles, 0.5 eq.) and 15.13 L of trichloroacetonitrile (150.9 moles, 5.03 eq) were transferred into the reactor. Stirring was continued and the reaction was kept under nitrogen. After the reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (30:70 ethyl acetate/Heptane) and HPLC. The reaction took approximately 2-3 hours to reach completion. When the reaction was complete, 40 L of water and 20 L of DCM were charged into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 40 L water and the DCM solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90 (DCM/EtOAc/heptane), 20:5:75 (DCM/EtOAc/heptane) and 20:10:70 DCM/EtOAc/heptane). Pure fractions were pooled and evaporated to give Intermediate EMod3 as a syrup. Isolated yield was 53%.
Synthesis of the EDC Trimer
Step 4. Preparation of ED Dimer, Coupling of E-TCA with Monomer D
A 100 L reactor was charged with 10471 g of 6-Acetyl-1-TCA Monomer E (EMod3) (18.3 mole, 1 eq., FW: 571.8) and 6594 g of Monomer D (16.47 mole, 0.9 eq, FW: 400.4). The combined monomers were dissolved in 20 L toluene and co-evaporated at 40° C. This was repeated with co-evaporation with an additional 2×20 L of toluene, after which 60 L of dichloromethane (DCM) were transferred into the reactor and dissolved. The reactor was purged with nitrogen and was chilled to −40° C. When the temperature was between −30° C. and −40° C., 2423 g (2071 mL, 9.17 moles, 0.5 eq) of TES Triflate were transferred into the reactor. After complete addition of TES Triflate the reaction was allowed to warm and stirring was continued. The completeness of the reaction was monitored by HPLC and TLC (35:65 ethyl acetate/Heptane). The reaction required 2-3 hours to reach completion. When the reaction was completed, 2040 mL of triethylamine (TEA, 1481 g, 0.8 eq.) were transferred into the reactor and stirred for an additional 30 minutes. The organic layer (DCM layer) was extracted with 2×20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 15:85, 20:80, 25:75, 30:70 and 35:65 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The ED Dimer was obtained in 81% isolated yield.
Synthesis of the EDC Trimer
Step 5. Preparation of ED1 TFA, Hydrolysis of ED Dimer
A 100 L reactor was charged with 7.5 Kg of ED Dimer (9.26 mol, 1 equiv). The reactor was chilled to <5° C. and 30.66 L of 90% TFA solution (TFA, 370.4 mol, 40 equiv) were transferred into the reactor. When the addition was completed the reaction was allowed to warm to room temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexanes) and HPLC. The reaction took approximately 3-4 hours to reach completion. When the reaction was completed, was chilled and 51.6 L of triethylamine (TEA, 37.5 Kg, 370.4 mole) were transferred into the reactor, after which 20 L of water & 20 L ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. Ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 20:80, 30:70, 40:60, 50:50, 60:40 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup. Isolated yield of ED1 was about 70%.
Synthesis of the EDC Trimer
Step 6. Preparation of ED2, Silylation of ED1
A 100 L reactor was charged with 11000 g of 1,2-diOH ED Dimer (ED1) (14.03 mol, 1 equiv), 1910.5 g of imidazole (28.06 mol, 2 equiv) and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., after which 3.53 L butyldiphenylchloro-silane (TBDPS-Cl, 4.628 Kg, 16.835 mol, 1.2 equiv) was charged into the reactor. When the addition was complete, the chiller was turned off and the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (50% ethyl acetate/hexane) and HPLC. The reaction required 4-6 hours to reach completion. When the reaction was completed, 20 L of water and 10 L of dichloromethane were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. Dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). Intermediate ED2 was obtained in 75% isolated yield.
Synthesis of the EDC Trimer
Step 7. Preparation of ED3, D-Levulination
A 100 L reactor was charged with 19800 g of 1-Silyl ED Dimer (ED2) (19.37 moles, 1 equiv) and 40 L of dry tetrahydrofuran (THF) and agitated to dissolve. The reactor was purged with nitrogen and 237 g of dimethylaminopyridine (DMAP, 1.937 moles, 0.1 equiv) and 10.05 L of diisopropylethylamine (DIPEA, 63.9 moles, 3 equiv) were transferred into the reactor. The reactor was chilled to 10-15° C. and kept under a nitrogen atmosphere, after which 12.46 Kg of levulinic anhydride (58.11 moles, 3 eq) was charged into the reactor. When the addition was complete, the reaction was warmed to ambient temperature and stirred overnight or 12-16 hours. The completeness of the reaction was monitored by TLC (40:60 ethyl acetate/hexane) and by HPLC. 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L of water, 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The ED3 yield was 95%.
Synthesis of the EDC Trimer
Step 8. Preparation of ED4, Desilylation of ED3
A 100 L reactor was charged with 19720 g of 1-Silyl-2-Lev ED Dimer (ED3) (17.6 mol, 1 equiv) in 40 L of THF. The reactor was chilled to 0° C., after which 6903 g of tetrabutylammonium fluoride trihydrate (TBAF, 26.4 mol, 1.5 equiv) and 1511 mL (26.4 mol, 1.5 equiv) of glacial acetic acid were transferred into the reactor. When the addition was complete, the reaction was stirred and allowed to warm to ambient temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. The reaction required 3 hours to reach completion. When the reaction was completed, 20 L of water and 10 L of dichloromethane were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified using a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20 (EtOAc/heptane) and 200 L 100% EtOAc. The pure fractions were pooled and evaporated to a syrup and used in the next step. The isolated yield of ED4 was about 92%.
Synthesis of the EDC Trimer
Step 9. Preparation of ED5, TCA Formation
A 100 L reactor was charged with 14420 g of 1-OH-2-Lev ED Dimer (ED4) (16.35 mol, 1 equiv) in 30 L of dichloromethane. The reactor was purged with nitrogen and stirring was begun, after which 1222 mL of diazabicycloundecene (DBU, 8.175 mol, 0.5 equiv) and 23.61 Kg of trichloroacetonitrile (TCA, 163.5 mol, 10 equiv) were transferred into the reactor. Stirring was continued and the reaction was kept under nitrogen. After reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by HPLC and TLC (40:60 ethyl acetate/heptane). The reaction took approximately 2 hours for reaction completion. When the reaction was completed, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified using a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup and used in the next step. The isolated yield of intermediate ED5 was about 70%.
Synthesis of the EDC Trimer
Step 10.
Preparation of EDC Trimer, Coupling of ED5 with Monomer C 6-O-acetyl-2-azido-3,4-di-O-benzyl-2-deoxy-α-D-glucopyranosyl-(1→4)-benzyl (3-O-benzyl-2-O-levulinoyl)-β-D-glucopyranosyluronate-(1→4)-(3-O-acetyl-1,6-anhydro-2-azido)-2-deoxy-β-D-glucopyranose
A 100 L reactor was charged with 12780 g of 2-Lev 1-TCA ED Dimer (ED5) (7.38 mole, 1 eq., FW) and 4764 g of Monomer C (7.38 mole, 1 eq). The combined monomers were dissolved in 20 L toluene and co-evaporated at 40° C. Repeated was co-evaporation with an additional 2×20 L of toluene, after which 60 L of dichloromethane (DCM) was transferred into the reactor and dissolved. The reactor was purged with nitrogen and chilled to −20° C. When the temperature was between −20 and −10° C., 2962 g (11.2 moles, 0.9 eq) of TES Triflate were transferred into the reactor. After complete addition of TES Triflate the reaction was allowed to warm to 5° C. and stirring was continued. Completeness of the reaction was monitored by HPLC and TLC (35:65 ethyl acetate/Heptane). The reaction required 2-3 hours to reach completion. When the reaction was completed, 1133 g of triethylamine (TEA, 0.9 eq.) were transferred into the reactor and stirred for an additional 30 minutes. The organic layer (DCM layer) was extracted with 2×20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. Dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 15:85, 20:80, 25:75, 30:70 and 35:65 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of EDC Trimer was 48%. Formula: C55H60N6O18; Mol. Wt. 1093.09. The 1H NMR spectrum (d6-acetone) of the EDC trimer is shown in FIG. 3.

Preparation of EDC1
Step 1:
Anhydro Ring Opening & Acetylation 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-1,3,6-tri-O-acetyl-β-D-glucopyranose
7.0 Kg (6.44 mol) of EDC Trimer was dissolved in 18 L anhydrous Dichloromethane. 6.57 Kg (64.4 mol, 10 eq) of Acetic anhydride was added. The solution was cooled to −45 to −35° C. and 1.82 Kg (12.9 mol, 2 eq) of Boron Trifluoride etherate was added slowly. Upon completion of addition, the mixture was warmed to 0-10° C. and kept at this temperature for 3 hours until reaction was complete by TLC and HPLC. The reaction was cooled to −20° C. and cautiously quenched and extracted with saturated solution of sodium bicarbonate (3×20 L) while maintaining the mixture temperature below 5° C. The organic layer was extracted with brine (1×20 L), dried over anhydrous sodium sulfate, and concentrated under vacuum to a syrup. The resulting syrup of EDC1 (6.74 Kg) was used for step 2 without further purification. The 1H NMR spectrum (d6-acetone) of the EDC-1 trimer is shown in FIG. 4.

Preparation of EDC2
Step 2:
Deacetylation 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-β-D-glucopyranose
The crude EDC1 product obtained from step 1 was dissolved in 27 L of Tetrahydrofuran and chilled to 15-20° C., after which 6 Kg (55.8 mol) of benzylamine was added slowly while maintaining the reaction temperature below 25° C. The reaction mixture was stirred for 5-6 hours at 10-15° C. Upon completion, the mixture was diluted with ethyl acetate and extracted and quenched with 10% citric acid solution (2×20 L) while maintaining the temperature below 25° C. The organic layer was extracted with 10% NaCl/1% sodium bicarbonate (1×20 L). The extraction was repeated with water (1×10 L), dried over anhydrous sodium sulfate and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel yielded 4.21 Kg (57% yield over 2 steps) of EDC2[ also referred to as 1-Hydroxy-6-Acetyl EDC Trimer]. The 1H NMR spectrum (d6-acetone) of the EDC-2 trimer is shown in FIG. 5.

Preparation of EDC3
Step 3:
Formation of TCA Derivative 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-1-O-trichloroacetimidoyl-β-D-glucopyranose
4.54 Kg (3.94 mol) of EDC2 was dissolved in 20 L of Dichloromethane. 11.4 Kg (78.8 mol, 20 eq) of Trichloroacetonitrile was added. The solution was cooled to −15 to −20° C. and 300 g (1.97 mol, 0.5 eq) of Diazabicycloundecene was added. The reaction was allowed to warm to 0-10° C. and stirred for 2 hours or until reaction was complete. Upon completion, water (20 L) was added and the reaction was extracted with an additional 10 L of DCM. The organic layer was extracted with brine (1×20 L), dried over anhydrous sodium sulfate, and concentrated under vacuum to a syrup. Column chromatographic separation using silica gel and 20-60% ethyl acetate/heptane gradient yielded 3.67 Kg (72% yield) of 1-TCA derivative, EDC3. The 1H NMR spectrum (d6-acetone) of the EDC-3 trimer is shown in FIG. 6.

Preparation of EDCBA1
Step 4:
Coupling of EDC3 with BA Dimer Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl)-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
3.67 Kg (2.83 mol) of EDC3 and 3.16 Kg (3.96 mol, 1.4 eq) of BA Dimer was dissolved in 7-10 L of Toluene and evaporated to dryness. The resulting syrup was coevaporated with Toluene (2×15 L) to remove water. The dried syrup was dissolved in 20 L of anhydrous Dichloromethane, transferred to the reaction flask, and cooled to −15 to −20° C. 898 g (3.4 mol, 1.2 eq) of triethylsilyl triflate was added while maintaining the temperature below −5° C. When the addition was complete, the reaction was immediately warmed to −5 to 0° C. and stirred for 3 hours. The reaction was quenched by slowly adding 344 g (3.4 mol, 1.2 eq) of Triethylamine. Water (15 L) was added and the reaction was extracted with an additional 10 L of DCM. The organic layer was extracted with a 25% 4:1 Sodium Chloride/Sodium Bicarbonate solution (2×20 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. The resulting syrup of the pentasaccharide, EDCBA1 was used for step 5 without further purification. The 1H NMR spectrum (d6-acetone) of the EDCBA-1 pentamer is shown in FIG. 7.

Preparation of EDCBA2
Step 5:
Hydrolysis of Levulinyl moiety Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl)-(1→4)—O-[benzyl 3-O-benzyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl)-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
The crude EDCBA1 from step 4 was dissolved in 15 L of Tetrahydrofuran and chilled to −20 to −25° C. A solution containing 679 g (13.6 mol) of Hydrazine monohydrate and 171 g (1.94 mol) of Hydrazine Acetate in 7 L of Methanol was added slowly while maintaining the temperature below −20° C. When the addition was complete, the reaction mixture was allowed to warm to 0-10° C. and stirred for several hours until the reaction is complete, after which 20 L of Ethyl acetate was added and the reaction was extracted with 10% citric acid (2×12 L). The organic layer was washed with water (1×12 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel and 10-45% ethyl acetate/heptane gradient yielded 2.47 Kg (47.5% yield over 2 steps) of EDCBA2. The 1H NMR spectrum (d6-acetone) of the EDCBA-2 pentamer is shown in FIG. 8.

Preparation of EDCBA Pentamer
Step 6:
THP Formation Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
2.47 Kg (1.35 mol) of EDCBA2 was dissolved in 23 L Dichloroethane and chilled to 10-15° C., after which 1.13 Kg (13.5 mol, 10 eq) of Dihydropyran and 31.3 g (0.135 mol, 0.1 eq) of Camphorsulfonic acid were added. The reaction was allowed warm to 20-25° C. and stirred for 4-6 hours until reaction was complete. Water (15 L) was added and the reaction was extracted with an additional 10 L of DCE. The organic layer was extracted with a 25% 4:1 Sodium Chloride/Sodium Bicarbonate solution (2×20 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel and 10-35% ethyl acetate/heptane gradient yielded 2.28 Kg (88.5% yield) of fully protected EDCBA Pentamer. The 1H NMR spectrum (d6-acetone) of the EDCBA pentamer is shown in FIG. 9.

Preparation of API1
Step 1:
Saponification Methyl O-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-azido-2-deoxy-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-α-L-Idopyranosyluronosyl-(1→4)-2-azido-3-O-benzyl-2-deoxy-α-D-glucopyranoside disodium salt
To a solution of 2.28 Kg (1.19 mol) of EDCBA Pentamer in 27 L of Dioxane and 41 L of Tetrahydrofuran was added 45.5 L of 0.7 M (31.88 mol, 27 eq) Lithium hydroxide solution followed by 5.33 L of 30% Hydrogen peroxide. The reaction mixture was stirred for 10-20 hours to remove the acetyl groups. Then, 10 L of 4 N (40 mol, 34 eq) sodium hydroxide solution was added. The reaction was allowed to stir for an additional 24-48 hours to hydrolyze the benzyl and methyl esters completely. The reaction was then extracted with ethyl acetate. The organic layer was extracted with brine solution and dried with anhydrous sodium sulfate. Evaporation of the solvent under vacuum gave a syrup of API1 [also referred to as EDCBA(OH)5] which was used for the next step without further purification.
Preparation of API2
Step 2:
O-Sulfonation Methyl O-2-azido-2-deoxy-3,4-di-O-benzyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-azido-2-deoxy-3,6-di-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-azido-2-deoxy-6-O-sulfo-α-D-glucopyranoside, heptasodium salt
The crude product of API1 [aka EDCBA(OH)5] obtained in step 1 was dissolved in 10 L Dimethylformamide. To this was added a previously prepared solution containing 10.5 Kg (66 moles) of sulfur trioxide-pyridine complex in 10 L of Pyridine and 25 L of Dimethylformamide. The reaction mixture was heated to 50° C. over a period of 45 min. After stiffing at 1.5 hours at 50° C., the reaction was cooled to 20° C. and was quenched into 60 L of 8% sodium bicarbonate solution that was kept at 10° C. The pH of the quench mixture was maintained at pH 7-9 by addition of sodium bicarbonate solution. When all the reaction mixture has been transferred, the quench mixture was stirred for an additional 2 hours and pH was maintained at pH 7 or greater. When the pH of quench has stabilized, it was diluted with water and the resulting mixture was purified using a preparative HPLC column packed with Amberchrom CG161-M and eluted with 90%-10% Sodium Bicarbonate (5%) solution/Methanol over 180 min. The pure fractions were concentrated under vacuum and was then desalted using a size exclusion resin or gel filtration (Biorad) G25 to give 1581 g (65.5% yield over 2 steps) of API2 [also referred to as EDCBA(OSO3)5]. The 1H NMR spectrum (d6-acetone) of API-2 pentamer is shown in FIG. 10.

Preparation of API3
Step 3:
Hydrogenation Methyl O-2-amino-2-deoxy-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-amino-2-deoxy-3,6-di-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-amino-2-deoxy-6-O-sulfo-α-D-glucopyranoside, heptasodium salt
A solution of 1581 g (0.78 mol) of O-Sulfated pentasaccharide API2 in 38 L of Methanol and 32 L of water was treated with 30 wt % of Palladium in Activated carbon under 100 psi of Hydrogen pressure at 60-65° C. for 60 hours or until completion of reaction. The mixture was then filtered through 1.0μ and 0.2μ filter cartridges and the solvent evaporated under vacuum to give 942 g (80% yield) of API3 [also referred to as EDCBA(OSO3)5(NH2)3]. The 1H NMR spectrum (d6-acetone) of API-3 pentamer is shown in FIG. 11.

Preparation of Fondaparinux Sodium
Step 4:
N-Sulfation & Removal of THP Methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)—O-β-D-glucopyranuronosyl-(1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt
To a solution of 942 g (0.63 mol) of API3 in 46 L of water was slowly added 3.25 Kg (20.4 mol, 32 eq) of Sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2 N sodium hydroxide solution. The reaction was allowed to stir for 4-6 hours at pH 9.0-9.5. When reaction was complete, the pH was adjusted to pH 7.0 using 50 mM solution of Ammonium acetate at pH 3.5. The resulting N-sulfated EDCBA(OSO3)5(NHSO3)3 mixture was purified using Ion-Exchange Chromatographic Column (Varian Preparative 15 cm HiQ Column) followed by desalting using a size exclusion resin or gel filtration (Biorad G25). The resulting mixture was then treated with activated charcoal and the purification by ion-exchange and desalting were repeated to give 516 g (47.6% yield) of the purified Fondaparinux Sodium form.
Analysis of the Fondaparinux sodium identified the presence of P1, P2, P3, and P4 in the fondaparinux. P1, P2, P3, and P4 were identified by standard analytical methods.
INTERMEDIATES
The monomers used in the processes described herein may be prepared as described in the art, or can be prepared using the methods described herein.

The synthesis of Monomer A-2 (CAS Registry Number 134221-42-4) has been described in the following references: Arndt et al., Organic Letters, 5(22), 4179-4182, 2003; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; and Sakairi et al., Journal of the Chemical Society, Chemical Communications, (5), 289-90, 1991, and the references cited therein, which are hereby incorporated by reference in their entireties.

Monomer C(CAS Registry Number 87326-68-9) can be synthesized using the methods described in the following references: Ganguli et al., Tetrahedron: Asymmetry, 16(2), 411-424, 2005; Izumi et al., Journal of Organic Chemistry, 62(4), 992-998, 1997; Van Boeckel et al., Recueil: Journal of the Royal Netherlands Chemical Society, 102(9), 415-16, 1983; Wessel et al.,Helvetica Chimica Acta, 72(6), 1268-77, 1989; Petitou et al., U.S. Pat. No. 4,818,816 and references cited therein, which are hereby incorporated by reference in their entireties.

Monomer E (CAS Registry Number 55682-48-9) can be synthesized using the methods described in the following literature references: Hawley et al., European Journal of Organic Chemistry, (12), 1925-1936, 2002; Dondoni et al., Journal of Organic Chemistry, 67(13), 4475-4486, 2002; Van der Klein et al., Tetrahedron, 48(22), 4649-58, 1992; Hori et al., Journal of Organic Chemistry, 54(6), 1346-53, 1989; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; Tailler et al.,Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio–Organic Chemistry, (23), 3163-4, (1972-1999) (1992); Paulsen et al., Chemische Berichte, 111(6), 2334-47, 1978; Dasgupta et al., Synthesis, (8), 626-8, 1988; Paulsen et al., Angewandte Chemie, 87(15), 547-8, 1975; and references cited therein, which are hereby incorporated by reference in their entireties.

Monomer B-1 (CAS Registry Number 444118-44-9) can be synthesized using the methods described in the following literature references: Lohman et al., Journal of Organic Chemistry, 68(19), 7559-7561, 2003; Orgueira et al., Chemistry—A European Journal, 9(1), 140-169, 2003; Manabe et al., Journal of the American Chemical Society, 128(33), 10666-10667, 2006; Orgueira et al., Angewandte Chemie, International Edition, 41(12), 2128-2131, 2002; and references cited therein, which are hereby incorporated by reference in their entireties.
Synthesis of Monomer D
Monomer D was prepared in 8 synthetic steps from glucose pentaacetate using the following procedure:

Pentaacetate SM-B was brominated at the anomeric carbon using HBr in acetic acid to give bromide derivative IntD1. This step was carried out using the reactants SM-B, 33% hydrogen bromide, acetic acid and dichloromethane, stirring in an ice water bath for about 3 hours and evaporating at room temperature. IntD1 was reductively cyclized with sodium borohydride and tetrabutylammonium iodide in acetonitrile using 3 Å molecular sieves as dehydrating agent and stirring at 40° C. for 16 hours to give the acetal derivative, IntD2. The three acetyl groups in IntD2 were hydrolyzed by heating with sodium methoxide in methanol at 50° C. for 3 hours and the reaction mixture was neutralized using Dowex 50WX8-100 resin (Aldrich) in the acid form to give the trihydroxy acetal derivative IntD3.
The C4 and C6 hydroxyls of IntD3 were protected by mixing with benzaldehyde dimethyl acetate and camphor sulphonic acid at 50° C. for 2 hours to give the benzylidene-acetal derivative IntD4. The free hydroxyl at the C3 position of IntD4 was deprotonated with sodium hydride in THF as solvent at 0° C. and alkylated with benzyl bromide in THF, and allowing the reaction mixture to warm to room temperature with stirring to give the benzyl ether IntD5. The benzylidene moiety of IntD5 was deprotected by adding trifluoroacetic acid in dichloromethane at 0° C. and allowing it to warm to room temperature for 16 hours to give IntD6 with a primary hydroxyl group. IntD6 was then oxidized with TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxide) in the presence of tetrabutylammonium chloride, sodium bromide, ethyl acetate, sodium chlorate and sodium bicarbonate, with stirring at room temperature for 16 hours to form the carboxylic acid derivative IntD7. The acid IntD7 was esterified with benzyl alcohol and dicyclohexylcarbodiimide (other reactants being hydroxybenzotriazole and triethylamine) with stirring at room temperature for 16 hours to give Monomer D.
Synthesis of the BA Dimer
The BA Dimer was prepared in 12 synthetic steps from Monomer B1 and Monomer A2 using the following procedure:


The C4-hydroxyl of Monomer B-1 was levulinated using levulinic anhydride and diisopropylethylamine (DIPEA) with mixing at room temperature for 16 hours to give the levulinate ester BMod1, which was followed by hydrolysis of the acetonide with 90% trifluoroacetic acid and mixing at room temperature for 4 hours to give the diol BMod2. The C1 hydroxyl of the diol BMod2 was silylated with tert-butyldiphenylsilylchloride by mixing at room temperature for 3 hours to give silyl derivative BMod3. The C2-hydroxyl was then benzoylated with benzoyl chloride in pyridine, and mixed at room temperature for 3 hours to give compound BMod4. The silyl group on BMod4 was then deprotected with tert-butyl ammonium fluoride and mixing at room temperature for 3 hours to give the C1-hydroyl BMod5. The C1-hydroxyl is then allowed to react with trichloroacetonitrile in the presence of diazobicycloundecane (DBU) and mixing at room temperature for 2 hours to give the trichloroacetamidate (TCA) derivative BMod6, which suitable for coupling, for example with Monomer A-2.
Monomer A-2 was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1.
Monomer A2 was prepared for the coupling reaction by opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1. This transformation occurs using boron trifluoride etherate, acetic anhydride and dichloromethane, between −20° C. and room temperature for 3 hours. The C1-Acetate of AMod1 was then hydrolyzed and methylated in two steps to give the diacetate AMod3. That is, first AMod1 was reacted with trimethylsilyl iodide and mixed at room temperature for 2 hours, then reacted with and tetrabutyl ammonium iodide. This mixture was reacted with diisoproylethylamine and methanol and stirred for 16 hours at room temperature, thus forming AMod3. The C4 and C6 acetates of AMod3 are hydrolyzed with sodium methoxide to give the diol Amod4. The AMod3 mixture was also subjected to mixing at room temperature for 3 hours with Dowex 50 Wx4x8-100 resin in the acid form for neutralization. This formed Amod4. The C6-hydroxyl of AMod4 is then benzoylated by treating with benzoyl chloride in pyridine at −40° C. and then allowing it to warm up to −10° C. over 2 hours to give AMod5.
Coupling of monomer AMod5 with the free C4-hydroxyl group of BMod6 was performed in the presence of BF3.Et2O and dichloromethane with mixing between −20° C. and room temperature for 3 hours to provide disaccharide BA1. The C4-levulinyl moiety of the disaccharide was then hydrolyzed with hydrazine to give the BA Dimer, which is suitable for subsequent coupling reactions.
Synthesis of EDC Trimer
The EDC Trimer was prepared in 10 synthetic steps from Monomer E, Monomer D and Monomer C using the following procedure:


Monomer E was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give diacetate EMod1. This occurs by the addition of Monomer E with boron trifluoride etherate, acetic anhydride and dichloromethane at −10° C., and allowing the reaction to warm to room temperature with stirring for 3 hours. The C1-Acetate of EMod1 is then hydrolyzed to give the alcohol, EMod2. This occurs by reacting Emod1 with hydrazine acetate and dimethylformamide and mixing at room temperature for 3 hours. The C1-hydroxyl of Emod2 is then reacted with trichloroacetonitrile to give the trichloro acetamidate (TCA) derivative EMod3 suitable for coupling, which reaction also employs diazabicycloundecane and dichloromethane and mixing at room temperature for 2 hours.
Monomer D, having a free C4-hydroxyl group, was coupled with monomer EMod3 in the presence of triethylsilyl triflate with mixing at −40° C. for 2 hours to give the disaccharide ED Dimer. The acetal on ring sugar D of the ED Dimer is hydrolyzed to give the C1,C2-diol ED1. This occurs by reacting the ED Dimer with 90% trifluoro acetic acid and mixing at room temperature for 4 hours. The C1-hydroxyl moiety of ED1 was then silylated with tert-butyldiphenylsilyl chloride to give the silyl derivative ED2. The C2-hydroxyl of ED2 was then allowed to react with levulinic anhydride in the presence of dimethylaminopyridine (DMAP) and diethylisopropylamine for approximately 16 hours to give the levulinate ester ED3. The TBDPS moiety is then deprotected by removal with tert-butylammonium fluoride in acetic acid with mixing at room temperature for 3 hours to give ED4 having a C1-hydroxyl. The C1-hydroxyl moiety of ED4 was then allowed to react with trichloroacetonitrile to give the TCA derivative ED5, which is suitable for coupling.
The C1-hydroxyl moiety of ED4 is then allowed to react with trichloroacetonitrile to give the TCA derivative ED5 suitable for coupling using diazabicycloundecane and dichloromethane, and mixing at room temperature for 2 hours. Monomer C, havinga free C4-hydroxyl group, was then coupled with the disaccharide ED5 in the presence of triethylsilyl triflate and mixed at −20° C. for 2 hours to give the trisaccharide EDC Trimer.
Synthesis of the EDCBA Pentamer
The EDCBA Pentamer was prepared using the following procedure:

The preparation of EDCBA Pentamer is accomplished in two parts as follows. In part 1, the EDC Trimer, a diacetate intermediate, is prepared for the coupling reaction with Dimer BA by initially opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the tetraacetate derivative EDC1. This occurs by reacting the EDC Trimer with boron trifluoride etherate, acetic anhydride and dichlormethane and stirring between −10° C. and room temperature for 3 hours. The C1-Acetate of EDC1 is then hydrolyzed to give the alcohol, EDC2, by reacting EDC1 with benzylamine [BnNH2] and tetrahydrofuran and mixing at −10° C. for 3 hours. The C1-hydroxyl of EDC2 is then reacted with trichloroacetonitrile and diazabicycloundecane, with mixing at room temperature for 2 hours, to give the trichloro acetamidate (TCA) derivative EDC3 suitable for coupling.


In Part 2 of the EDCBA Pentameter synthesis, the Dimer BA, having a free C4-hydroxyl group, is coupled with trisaccharide EDC3 in the presence of triethylsilyltriflate at −30° C. mixing for 2 hours to give the pentasaccharide EDCBA1. The levulinyl ester on C2 of sugar D in EDCBA1 is hydrolyzed with a mixture of deprotecting agents, hydrazine hydrate and hydrazine acetate and stiffing at room temperature for 3 hours to give the C2-hydroxyl containing intermediate EDCBA2. The C2-hydroxyl moiety on sugar D of EDCBA2 is then alkylated with dihydropyran (DHP) in the presence of camphor sulfonic acid (CSA) and tetrahydrofuran with mixing at room temperature for 3 hours to give the tetrahydropyranyl ether (THP) derivative, EDCBA Pentamer.
…………………………
A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodium, Org Process Res Dev 2013, 17: 869, http://pubs.acs.org/doi/full/10.1021/op300367c

An improved method for the simultaneous removal of O-benzyl and N-carboxybenzyl groups as well as reducing azide groups to amines in protected heparin-like pentasaccharides, a key process in fondaparinux sodium synthesis, is reported. Under catalytic transfer hydrogenation conditions, using readily available and inexpensive ammonium formate, the hydrogenolysis is done in less than an hour in good yield and purity. This procedure represents a major advantage over the previously published procedures, the latter of which involve several hours/days of hydrogenation reaction under catalytic reduction using gaseous hydrogen.

Synthesis of Compound 1 (FONDAPARINUX)
………………
SYNTHESIS
In the synthesis of Fondaparinux sodium, the monomers XII, XVIII, XXVII, XXXVIII, XXXXI and dimers XIX, XX, XL described herein may be made either by processes described in the art or, by a process as described herein. The XII and XVIII monomers may then linked to form a disaccharide XX, XXXIX and XXVII monomers may then linked to form a disaccharide XL, XLIII and XX dimers may then linked to form a tetrasaccharide, XLVII tetramer and XLV monomer may be linked to form a pentasaccharide (XLVIII) pentamer. The XLVIII pentamer is an intermediate that may be converted through a series of reactions to fondaparinux sodium. This strategy described herein provides an efficient method for multi-kilogram preparation of fondaparinux in high yields and highly stereoselective purity.
Fondaparinux sodium (LIII) was prepared in 3 synthetic steps from O – S pentasaccharide (L) using the following procedure:

Fondaparinux Sodium (LIII)
Preparation of Fondaparinux sodium (LIII)—
N- sulfonation of Deprotected Pentasaccharide (LI) methyl 0-2-deoxy-3,6-di-0- sulfo-2-(sulfoamino)-a-D-glucopyranosyl-(l— >4)-0-2-0-sulfo-a-L- idopyranurosyl-( 1— >4)-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D-glucopyranoside,decasodium salt
A solution of deprotected pentasaccharide (LI) (145 gm) in water (2.54 V) was adjusted to a pH of 9.5 – 10.5 with 1 N NaOH solution. S03-pyridine complex (156 gm) was added into 3 lots every 15 min, the pH being maintained at 9.5-10.5 by automatic addition of 1 N NaOH. The mixture was stirred for 2 hrs at RT, during this aqueous NaOH (IN solution) was added to maintain pH at 9.5 – 10.5. After neutralization to pH 7 – 7.5 by addition of HC1 solution, the mixture was evaporated using vacuum. The residue was dissolved in water (1.6 L) at RT, to this solution was added acetone (1.6 L) at RT. The reaction mass was cooled to 5°C – 1 0 °C and stirred for 1 hr. The solid was filtered and washed with cold acetone: water (1 :1). The clear filtrate was distilled off completely under vacuum below 55°C. The residue was dissolved in water (1.6 L) at RT, and to this solution was added acetone(1.6 L) at RT. The mixture was cooled to 5 to 10°C and stirred for 1 hr. The solid was filtered and washed with cold acetone/water (1 :1). The clear filtrate was distilled off completely under vacuum below 55°C. The residue was dissolved in water (0.7 L) and charcoal (40 gm) was added at RT. The mixture was stirred for 30 min at RT then filtered. To the filtrate was added charcoal (40 gm) at RT. The mixture was stirred for 30 min at RT then filtered. To the filtrate was added charcoal (40 gm) at RT. The mixture was stirred for 30 min at RT then filtered. The pH of the clear filtrate was adjusted to 8.0 – 8.5 with IN NaOH solution and distilled off completely under vacuum below 55 °C. The residue was dissolved in 0.5 M NaCl solution and layered onto a column of Dowex® 1×2 -400 resins using a gradient of NaCl solution (0.5 to 10M). The product fractions were combined and distilled off under vacuum below 55 °C up to 1 – 2 L volume. The solid was filtered off and the clear filtrate was distilled off under vacuum below 55 °C up to slurry stage and subjected to azeotropic distillation with methanol two times. The solid residue was stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The wet solid was again stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The wet solid was again stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The above solid was dissolved in water and the pH adjusted to 4 – 4.5 with IN HC1 solution and charcoalized three times with 26 gm of charcoal at RT for 15-30 minutes and filtered off. To the clear filtrate was added 0.39 kg of NaCl, then methanol was added (35 volume) at RT and the mixture was stirred for 15-30 minutes. The solution was decanted and the sticky mass was stirred with methanol (0.65 L) at RT for 15-30 minutes. The solid was filtered off and dissolved in water, and the pH adjusted to 8 – 8.5 with IN NaOH solution. The solution was filtered through 0.45 micron paper & distilled off completely under vacuum below 55°C. The solution was subjected to azeotropic distillation with methanol to give highly pure fondaparinux sodium (97.17 gm) (HPLC purity 99.7%).
SOR Results
Three batches of product made in accordance with the present processes provided the following stereoisomeric optical rotation results:
Specification: Between +50.0° and +60.0°.
Batch- 1 = +55.1°
Batch-2 = +55.7° Batch-3 = +55.4°.
INTERMEDIATES
Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; MS is molecular sieve; DMF is dimethyl formamide; Bn is benzyl; MDC is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; MeOH is methanol; RT is room temperature; Ac2O is acetic anhydride; HBr is hydrogen bromide; EtOAc is ethyl acetate; Cbz is benzyloxycarbonyl; CADS is chloro acetyl disaccharide; HDS is hydroxy disaccharide; NMP is N-methylpyrrolidone.
Methyl 3-O-benzyl-4-O-monochloro acetyl-β-L-idopyranuronate

Route of Synthesis for α-Methyl-6-o-acetyl-3-o-benzyl-2-(benzyloxy carbonyl)amino-2-deoxy-α-D-glucopyranoside

Methyl 6-O-acetyl-3-O-benzyl-2-(benzyloxy carbonyl)amino-2-deoxy-4-O-(methyl-2-O acetyl-3-O-benzyl-α-L-idopyranosyluronate)-glucopyranoside

Route of Synthesis for 1,6-Anhydro-2-azido-3-O-acetyl-2-deoxy-beta-D-glucopyranose

Route of synthesis for Methyl 2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranuronate

Route of synthesis for 3-O-Acetyl-1,6-anhydro-2-azido-4-O-2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranosyl methyluronate-beta-D-glucopyranose
(or)
3-O-Acetyl-1,6-anhydro-2-azido-2-deoxy-4-O-(methyl 2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranosyluronate)-beta-D-glucopyranose

Route of Synthesis for 1,6-Anhydro-2-azido-3,4-di-O-benzyl-2-deoxy-beta-D-glucopyranose

Synthesis of Disaccharide XLIII
Disaccharide XLIII was prepared in 2 synthetic steps from CADS sugar (XL) using the following procedure:

CADS sugar XL was acetylated at the anomeric carbon using AC2O and TFA to give acetyl derivative XLII. This step was carried out using the reactants CADS, AC2O and TFA, stirring in an ice water bath for about 5-24 hours, preferably 20 hours, and evaporating to residue under vacuum. Residue was recrystallized in ether. Acetyl CADS (XLII) was brominated at the anomeric carbon using titanium tetra bromide in MDC andethylacetate and stirring at 20° C.-50° C. for 6-16 hours, preferably 6 hours, to give the bromo derivative, (XLIII) after work-up and recrystallization from solvent/alcohol.
Synthesis of the Monosaccharide (XLV)
The monosaccharide (XLV) was prepared in 2 synthetic steps from monomer (XLI) using the following procedure:

Mono sugar (XLI) was acetylated at the anomeric carbon using AC2O and TFA to give acetyl derivative (XLIV). This step was carried out using the reactants Mono sugar (XLI), AC2O and TFA, stirring in an ice water bath for about 5-24 hours, preferably 24 hours, and evaporating to residue under vacuum. Residue was recrystallized in ether. Acetyl Mono sugar (XLIV) was brominated at the anomeric carbon using titanium tetra bromide in MDC and ethyl acetate and stirring at 20° C.-50° C. for 6-20 hours, preferably 16 hours, to give the bromo derivative, (XLV) after work-up and recrystallization from ether.
Synthesis of the Hydroxy Tetrasaccharide (XLVII)
The hydroxy tetrasaccharide (XLVII) was prepared in 2 synthetic steps from disaccharide (XLIII) and HDS (XX) using the following procedure:

Disaccharide (XLIII), was coupled with disaccharide (XX) in the presence of silver carbonate, silver per chlorate and 4 A° MS in MDC and stirred at ambient temperature for 5-12 hrs, preferably 4-6 hours, in the dark followed by work-up and purification in water/methanol to give the tetrasaccharide (XLVI). The d echloroacetylation of tetrasaccharide (XLVI) was carried out in THF, ethanol and pyridine in the presence of thiourea at reflux for 6 to 20 hrs, preferably 12 hours, to give the hydroxy tetrasaccharide (XLVIII).
Synthesis of the Pentasaccharide (XLVIII)
The pentasaccharide (XLVIII) was prepared in 2 synthetic steps from monosaccharide (XLV) and tetrasaccharide (XLVII) using the following procedure:

Monosaccharide (XLV), was coupled with tetrasaccharide (XLVII) in the presence of 2,4,6-collidine, silver triflate and 4 A° MS in MDC and stirred at −10° C. to −20° C. for 1 hr in the dark followed by work-up and purification by column chromatography to give the pentasaccharide (XLVIII).
Synthesis of OS Pentasaccharide (L)
The OS pentasaccharide (L) was prepared in 2 synthetic steps from pentasaccharide (XLVIII) using the following procedure:

Pentasaccharide (XLVIII) was deacetylated in the presence of NaOH in mixture of solvents of MDC, methanol and water at 0° C. to 35° C., for 1-2 hrs followed by work-up and distillation to obtain deacetylated pentasaccharide (XLIX) which was subjected to O-sulfonation in DMF in the presence of SO3-trimethylamine (TMA) at 50° C. to 100° C., preferably 50° C.-55° C., for 6-24 hrs, preferably 12 hours, followed by salt removal through Sephadex® resin and column chromatography purification, then pH adjustment by dilute NaOH to give OS pentasaccharide (L).
…………………………
INTERMEDIATE
highly pure 4-Ο-β-ϋ- glucopyranosyl- 1 ,6-anhydro- -D-glucopyranose

Example 1 : Preparation and purification of 4-0- -D-grucopyranosyl-L6-anhvdro- -D- glucopyranose
A solution of pentachlorophenyl 2,3,6,2′,3′,4′,6′-hepta-(9-acetyl- -D-ceilobioside represented by Formula I;

(400 g) in isopropyl alcohol (4 L) at ambient temperature was cooled to 2°C to 5°C and pulverized potassium hydroxide (355 g) was added to it. This reaction mixture was stirred and the temperature was allowed to rise to ambient temperature. At ambient temperature, the mixture was stirred until the reaction was complete (about 2 hours). The mixture was then heated to 50°C to 55°C and stirred for 30 minutes. The solid obtained was filtered and washed with isopropyl alcohol (400 mL). The solid was stirred with isopropyl alcohol (2.8 L) at 50°C for 30 minutes followed by filtering and washing with isopropyl alcohol (400 mL). The resultant solid was suspended into methanol (800 mL to 1600 mL) followed by cooling to 2°C to 5°C. The pH of the suspension was adjusted to 2 to 3 using 15% methanolic hydrochloride. The solid so obtained was filtered and washed with methanol (400 mL). Solvent was recovered from the filtrate to dryness under vacuum to obtain the pure compound of Formula II as foamy solid.
Yield: 142 g
Example 2: Preparation and purification of 4-Q- -D-grucopyranosyl-l,6-anhvdro- -D- glucopyranose
A solution of pentachlorophenyl 2,3,6,2 ,3 ^ ^‘-hepta-O-acetyl- -D-cellobioside of Formula I (100 g) in methanol (300 mL) at ambient temperature was cooled to 2°C to 5°C and pulverized potassium hydroxide (88.6 g) was added to it. This reaction mixture was stirred and the temperature was allowed to rise to ambient temperature. At ambient temperature, the mixture was stirred until the reaction was complete (about 2 hours). The mixture was cooled to 2°C to 5°C and 15% methanolic hydrogen chloride was added to it until the pH of the mixture reached 2 to 3. At this pH, the reaction mixture was filtered and the residual solid was washed with methanol (100 mL). The solvent was recovered from the filtrate under vacuum. The solid material so obtained was stirred with dichloromethane (500 mL) followed by removal of solvent through decantation/filtration. The resultant solid was stirred with isopropyl alcohol (500 mL), filtered and dried to obtain the pure compound of Formula II.
Yield: 29 g
………………………
SYNTHESIS
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:
Conversion of FPP (also referred to a Fully Protected Pentamer) to FondaparinuxSodium:

Reagents: 1. NaOH, H202, LiOH, Dioxane, RT, 24-48 h; 2. Py.S03, DMF, 60°C, 2h, CG-161 purification; 3. 10% Pd/C, H2, 72h; 4. (a) Py.S03, NaOH, NH4OAc, 12h, (b) HiQ NH4OAc/ NaCl ion-exchange, Sephadex Desalt and (c) HiQ NaCl ion-exchange, Sephadex Desalt. The ester moieties in EDCBA Pentamer-CB were hydrolyzed with sodium and lithium hydroxide in the presence of hydrogen peroxide in dioxane mixing at room temperature for 24- 48 hours to give the pentasaccharide intermediate API1-CB. The five hydroxyl moieties in API1-CB were sulfated using a pyridine-sulfur trioxide complex in dimethylformamide, mixing at 60°C for 2 hours and then purified using column chromatography (CG-161), to give the pentasulfated pentasaccharide API2-CB. The intermediate API2-CB was then hydrogenated to reduce the three azides on sugars E, C and A to amines and the reductive deprotection of the six benzyl ethers to their corresponding hydroxyl groups to form the intermediate API3-CB. This transformation occurs by reacting API2-CB with 10% palladium/carbon catalyst with hydrogen gas for 72 hours. The three amines on API3-CB were then sulfated using the pyridine-sulfur trioxide complex in sodium hydroxide and ammonium acetate, allowing the reaction to proceed for 12 hours . The crude fondaparinux is purified and is subsequently converted to its salt form. The crude mixture was purified using an ion-exchange chromatographic column (HiQ resin) followed by desalting using a size exclusion resin or gel filtration (Biorad Sephadex G25) to give the final product, fondaparinux sodium.
Preparation of Fondaparinux Sodium – Step 4: N-Sulfation of API-3-CB:
Methyl 0-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D-glucopyranosyl-(l→4)-0^-D- glucopyranuronosyl-(l→4)-0-2-deoxy-3,6-di-0-sulfo-2-(sulfoamino)-a-D-glucopyranosyl- (l→4)-0-2-0-sulfo-a-L-idopyranuronosyl-(l→4)-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D- glucopyranoside, decasodium salt
To a solution of 25.4 gram (16.80 mmol, leq) of API-3-CB in 847 mL of water was slowly added 66.85 gram (446.88 mmol, 25eq) of sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2N sodium hydroxide solution. The reaction was allowed to stir for 4 hours at pH 9.0 – 9.5. When reaction was completed, the pH was adjusted 7.0 by using 70 mL of 50 mmol Ammonium acetate solution pH -3.5. The resulting N-Sulfated Cellobiose mixture was purified using Ion-Exchange
Chromatographic Column followed by desalting using size exclusion resin to gave gram ( %) of the purified Fondaparinux Sodium form.
To a solution of 942 g (0.63 mol) of API3 in 46 L of water was slowly added 3.25 Kg (20.4 mol, 32 eq) of Sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2 N sodium hydroxide solution. The reaction was allowed to stir for 4-6 hours at pH 9.0-9.5. When reaction was complete, the pH was adjusted to pH 7.0 using 50 mM solution of Ammonium acetate at pH 3.5. The resulting N- sulfated EDCBA(OS03)5(NHS03)3 mixture was purified using Ion-Exchange Chromatographic Column (Varian Preparative 15 cm HiQ Column) followed by desalting using a size exclusion resin or gel filtration (Biorad G25). The resulting mixture was then treated with activated charcoal and the purification by ion-exchange and desalting were repeated to give 516 g (47.6% yield) of the purified Fondaparinux sodium form.
INT
SCHEME 1 – Synthesis of Monomer A-2 & AMod5 fBuildinq Block Al

Reagents: 1. NaOMe, MeOH, RT, 2hr, 50wx resin; 2. (Bu3Sn)20 (0.8equiv), ACN, MS, reflux, 3h; 3.l2 (1.5 equiv), 5°C to RT, 2h; 4. NaH (2 equiv), DMF, p-MeOC6H4CH2Br (PMB-Br, 2.5 equiv), -20°C to RT, 2h; 5. NaN3, DMF, 120°C, 12h; 6. NaH, DMF, BnBr, 0°C to RT, 3h.; 7. BF3.Et20, Ac20, DCM, -20°C to RT, 3h; 8. (a) TMS-I, TBAI, RT, 2h; (b) DIPEA, MeOH, 16h, RT; 9. NaOMe, Dowex 50WX8-100 resin H+ form, RT, 3h; 10. Pyridine, Bz-CI, -40°C to -10°C, 2h;
Scheme 2 – Synthesis of Monomer B-1 and BMod6 fBuildinq Block B1

Reagents: 1. NaH, BnBr, THF, DMF, 0° to 65°C, 3h; 2. 66% Acetic Acid/H20, 40 °C, 16h; 3. Nal04, (Bu)4NBr, DCM, H20, Dark, 3h; 4. (PhS)3CH, n-BuLi, THF, -78 °C, 3h; 5. CuCI2/CuO, MeOH, H20, 3h; 6. 90% TFA/H20, DCM, RT, 2h; 7. DMF, CSA 2-methoxypropene, 0° to RT, 16hrs; MeOH, TEA. 8. Lev20, DIPEA, RT, 16h; 9. 90% TFA, RT, 4h; 10. Imidazole, TBDPSi-CI, RT, 3h; 11. Pyridine, BzCI, RT, 3h; 12. TBAF, RT, 3h; 13. TCA, DBU, RT, 2h; Also see, e.g., Bonnaffe et al., Tetrahedron Lett., 41, 307-311, 2000; Bonnaffe et al., Carbohydr. Res., 2003, 338, 681-686, 2003; and Seeberger et al., J. Org. Chem., 2003, 68, 7559- 7561, 2003.
……………………..
Carbohydrate Research, 2012 , vol. 361, p. 155 – 161
1H NMR (D2O) δ: 5.68 (d, J = 3.8 Hz, 1H, H-1D), 5.56 (d, J = 3.4 Hz, 1H, H-1F), 5.24 (d, J = 3.8 Hz, 1H, H-1G), 5.07 (d, J = 3.5 Hz, 1H, H-1H), 4.68 (d, J = 7.9 Hz, 1H, H-5G), 4.54 (dd, J = 11.4, 2.2 Hz, 1H, H-1E), 4.48-4.34 (m, 6H, H-6F, 6H, 6’H, 6D, 3E, 2G), 4.33-4.30 (m, 1H, H-6’F), 4.25-4.17 (m, 4H, H-4G, 3G, 6’D, 5F), 4.06-3.98 (m, 2H, H-4F, 5H), 3.94 (dd, J = 9.7, 2.2 Hz, 1H, H-5D), 3.92-3.86 (m, 2H,H-3E, H-4E), 3.85-3.80 (m, 2H, H-5E, 4H), 3.73-3.60 (m, 3H, H-3H, 3D, 4D), 3.53-3.44 (m, 2H, H-2F, H-2E), 3.47 (s, 3H, OMe), 3.34 (dd, J = 10.2, 3.7 Hz, 1H, H-2H), 3.31(dd, J = 10.2, 3.7 Hz, 1H, H-2D)

FONDAPARINUX
……………………………………..
Synthesis of intermediates
Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; ACN is acetonitrile; MS is molecular sieves; DMF is dimethyl formamide; PMB is p-methoxybenzyl; Bn is benzyl; DCM is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; CSA is camphor sulfonic acid; TEA is triethylamine; MeOH is methanol; DMAP is dimethylaminopyridine; RT is room temperature; CAN is ceric ammonium nitrate; Ac2O is acetic anhydride; HBr is hydrogen bromide; TEMPO is tetramethylpiperidine-N-oxide; TBACl is tetrabutyl ammonium chloride; EtOAc is ethyl acetate; HOBT is hydroxybenzotriazole; DCC is dicyclohexylcarbodiimide; Lev is levunlinyl; TBDPS is tertiary-butyl diphenylsilyl; TCA is trichloroacetonitrile; O-TCA is O-trichloroacetimidate; Lev2O is levulinic anhydride; DIPEA is diisopropylethylamine; Bz is benzoyl; TBAF is tetrabutylammonium fluoride; DBU is diazabicycloundecane; BF3.Et2O is boron trifluoride etherate; TMSI is trimethylsilyl iodide; TBAI is tetrabutylammonium iodide; TES-Tf is triethylsilyl trifluoromethanesulfonate (triethylsilyl triflate); DHP is dihydropyran; PTS is p-toluenesulfonic acid.
The monomers used in the processes described herein may be prepared as described in the art, or can be prepared using the methods described herein.
The synthesis of Monomer A-2 (CAS Registry Number 134221-42-4) has been described in the following references: Arndt et al., Organic Letters, 5(22), 4179-4182, 2003; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; and Sakairi et al., Journal of the Chemical Society, Chemical Communications, (5), 289-90, 1991, and the references cited therein, which are hereby incorporated by reference in their entireties.
Monomer C(CAS Registry Number 87326-68-9) can be synthesized using the methods described in the following references: Ganguli et al., Tetrahedron: Asymmetry, 16(2), 411-424, 2005; Izumi et al., Journal of Organic Chemistry, 62(4), 992-998, 1997; Van Boeckel et al., Recueil: Journal of the Royal Netherlands Chemical Society, 102(9), 415-16, 1983; Wessel et al.,Helvetica Chimica Acta, 72(6), 1268-77, 1989; Petitou et al., U.S. Pat. No. 4,818,816 and references cited therein, which are hereby incorporated by reference in their entireties.
Monomer E (CAS Registry Number 55682-48-9) can be synthesized using the methods described in the following literature references: Hawley et al., European Journal of Organic Chemistry, (12), 1925-1936, 2002; Dondoni et al., Journal of Organic Chemistry, 67(13), 4475-4486, 2002; Van der Klein et al., Tetrahedron, 48(22), 4649-58, 1992; Hori et al., Journal of Organic Chemistry, 54(6), 1346-53, 1989; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; Tailler et al.,Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio–Organic Chemistry, (23), 3163-4, (1972-1999) (1992); Paulsen et al., Chemische Berichte, 111(6), 2334-47, 1978; Dasgupta et al., Synthesis, (8), 626-8, 1988; Paulsen et al., Angewandte Chemie, 87(15), 547-8, 1975; and references cited therein, which are hereby incorporated by reference in their entireties.
Monomer B-1 (CAS Registry Number 444118-44-9) can be synthesized using the methods described in the following literature references: Lohman et al., Journal of Organic Chemistry, 68(19), 7559-7561, 2003; Orgueira et al., Chemistry—A European Journal, 9(1), 140-169, 2003; Manabe et al., Journal of the American Chemical Society, 128(33), 10666-10667, 2006; Orgueira et al., Angewandte Chemie, International Edition, 41(12), 2128-2131, 2002; and references cited therein, which are hereby incorporated by reference in their entireties.
Synthesis of Monomer D
Monomer D was prepared in 8 synthetic steps from glucose pentaacetate using the following procedure:
Pentaacetate SM-B was brominated at the anomeric carbon using HBr in acetic acid to give bromide derivative IntD1. This step was carried out using the reactants SM-B, 33% hydrogen bromide, acetic acid and dichloromethane, stirring in an ice water bath for about 3 hours and evaporating at room temperature. IntD1 was reductively cyclized with sodium borohydride and tetrabutylammonium iodide in acetonitrile using 3 Å molecular sieves as dehydrating agent and stirring at 40° C. for 16 hours to give the acetal derivative, IntD2. The three acetyl groups in IntD2 were hydrolyzed by heating with sodium methoxide in methanol at 50° C. for 3 hours and the reaction mixture was neutralized using Dowex 50WX8-100 resin (Aldrich) in the acid form to give the trihydroxy acetal derivative IntD3.
The C4 and C6 hydroxyls of IntD3 were protected by mixing with benzaldehyde dimethyl acetate and camphor sulphonic acid at 50° C. for 2 hours to give the benzylidene-acetal derivative IntD4. The free hydroxyl at the C3 position of IntD4 was deprotonated with sodium hydride in THF as solvent at 0° C. and alkylated with benzyl bromide in THF, and allowing the reaction mixture to warm to room temperature with stirring to give the benzyl ether IntD5. The benzylidene moiety of IntD5 was deprotected by adding trifluoroacetic acid in dichloromethane at 0° C. and allowing it to warm to room temperature for 16 hours to give IntD6 with a primary hydroxyl group. IntD6 was then oxidized with TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxide) in the presence of tetrabutylammonium chloride, sodium bromide, ethyl acetate, sodium chlorate and sodium bicarbonate, with stirring at room temperature for 16 hours to form the carboxylic acid derivative IntD7. The acid IntD7 was esterified with benzyl alcohol and dicyclohexylcarbodiimide (other reactants being hydroxybenzotriazole and triethylamine) with stirring at room temperature for 16 hours to give Monomer D.
Synthesis of the BA Dimer
The BA Dimer was prepared in 12 synthetic steps from Monomer B1 and Monomer A2 using the following procedure:
The C4-hydroxyl of Monomer B-1 was levulinated using levulinic anhydride and diisopropylethylamine (DIPEA) with mixing at room temperature for 16 hours to give the levulinate ester BMod1, which was followed by hydrolysis of the acetonide with 90% trifluoroacetic acid and mixing at room temperature for 4 hours to give the diol BMod2. The C1 hydroxyl of the diol BMod2 was silylated with tert-butyldiphenylsilylchloride by mixing at room temperature for 3 hours to give silyl derivative BMod3. The C2-hydroxyl was then benzoylated with benzoyl chloride in pyridine, and mixed at room temperature for 3 hours to give compound BMod4. The silyl group on BMod4 was then deprotected with tert-butyl ammonium fluoride and mixing at room temperature for 3 hours to give the C1-hydroyl BMod5. The C1-hydroxyl is then allowed to react with trichloroacetonitrile in the presence of diazobicycloundecane (DBU) and mixing at room temperature for 2 hours to give the trichloroacetamidate (TCA) derivative BMod6, which suitable for coupling, for example with Monomer A-2.
Monomer A-2 was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1.
Monomer A2 was prepared for the coupling reaction by opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1. This transformation occurs using boron trifluoride etherate, acetic anhydride and dichloromethane, between −20° C. and room temperature for 3 hours. The C1-Acetate of AMod1 was then hydrolyzed and methylated in two steps to give the diacetate AMod3. That is, first AMod1 was reacted with trimethylsilyl iodide and mixed at room temperature for 2 hours, then reacted with and tetrabutyl ammonium iodide. This mixture was reacted with diisoproylethylamine and methanol and stirred for 16 hours at room temperature, thus forming AMod3. The C4 and C6 acetates of AMod3 are hydrolyzed with sodium methoxide to give the diol Amod4. The AMod3 mixture was also subjected to mixing at room temperature for 3 hours with Dowex 50 Wx4x8-100 resin in the acid form for neutralization. This formed Amod4. The C6-hydroxyl of AMod4 is then benzoylated by treating with benzoyl chloride in pyridine at −40° C. and then allowing it to warm up to −10° C. over 2 hours to give AMod5.
Coupling of monomer AMod5 with the free C4-hydroxyl group of BMod6 was performed in the presence of BF3.Et2O and dichloromethane with mixing between −20° C. and room temperature for 3 hours to provide disaccharide BA1. The C4-levulinyl moiety of the disaccharide was then hydrolyzed with hydrazine to give the BA Dimer, which is suitable for subsequent coupling reactions.
Synthesis of EDC Trimer
The EDC Trimer was prepared in 10 synthetic steps from Monomer E, Monomer D and Monomer C using the following procedure:
Monomer E was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give diacetate EMod1. This occurs by the addition of Monomer E with boron trifluoride etherate, acetic anhydride and dichloromethane at −10° C., and allowing the reaction to warm to room temperature with stirring for 3 hours. The C1-Acetate of EMod1 is then hydrolyzed to give the alcohol, EMod2. This occurs by reacting Emod1 with hydrazine acetate and dimethylformamide and mixing at room temperature for 3 hours. The C1-hydroxyl of Emod2 is then reacted with trichloroacetonitrile to give the trichloro acetamidate (TCA) derivative EMod3 suitable for coupling, which reaction also employs diazabicycloundecane and dichloromethane and mixing at room temperature for 2 hours.
Monomer D, having a free C4-hydroxyl group, was coupled with monomer EMod3 in the presence of triethylsilyl triflate with mixing at −40° C. for 2 hours to give the disaccharide ED Dimer. The acetal on ring sugar D of the ED Dimer is hydrolyzed to give the C1,C2-diol ED1. This occurs by reacting the ED Dimer with 90% trifluoro acetic acid and mixing at room temperature for 4 hours. The C1-hydroxyl moiety of ED1 was then silylated with tert-butyldiphenylsilyl chloride to give the silyl derivative ED2. The C2-hydroxyl of ED2 was then allowed to react with levulinic anhydride in the presence of dimethylaminopyridine (DMAP) and diethylisopropylamine for approximately 16 hours to give the levulinate ester ED3. The TBDPS moiety is then deprotected by removal with tert-butylammonium fluoride in acetic acid with mixing at room temperature for 3 hours to give ED4 having a C1-hydroxyl. The C1-hydroxyl moiety of ED4 was then allowed to react with trichloroacetonitrile to give the TCA derivative ED5, which is suitable for coupling.
The C1-hydroxyl moiety of ED4 is then allowed to react with trichloroacetonitrile to give the TCA derivative ED5 suitable for coupling using diazabicycloundecane and dichloromethane, and mixing at room temperature for 2 hours. Monomer C, having a free C4-hydroxyl group, was then coupled with the disaccharide ED5 in the presence of triethylsilyl triflate and mixed at −20° C. for 2 hours to give the trisaccharide EDC Trimer.
Synthesis of the EDCBA Pentamer
The EDCBA Pentamer was prepared using the following procedure:
The preparation of EDCBA Pentamer is accomplished in two parts as follows. In part 1, the EDC Trimer, a diacetate intermediate, is prepared for the coupling reaction with Dimer BA by initially opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the tetraacetate derivative EDC1. This occurs by reacting the EDC Trimer with boron trifluoride etherate, acetic anhydride and dichlormethane and stirring between −10° C. and room temperature for 3 hours. The C1-Acetate of EDC1 is then hydrolyzed to give the alcohol, EDC2, by reacting EDC1 with benzylamine [BnNH2] and tetrahydrofuran and mixing at −10° C. for 3 hours. The C1-hydroxyl of EDC2 is then reacted with trichloroacetonitrile and diazabicycloundecane, with mixing at room temperature for 2 hours, to give the trichloro acetamidate (TCA) derivative EDC3 suitable for coupling.
In Part 2 of the EDCBA Pentameter synthesis, the Dimer BA, having a free C4-hydroxyl group, is coupled with trisaccharide EDC3 in the presence of triethylsilyltriflate at −30° C. mixing for 2 hours to give the pentasaccharide EDCBA1. The levulinyl ester on C2 of sugar D in EDCBA1 is hydrolyzed with a mixture of deprotecting agents, hydrazine hydrate and hydrazine acetate and stiffing at room temperature for 3 hours to give the C2-hydroxyl containing intermediate EDCBA2. The C2-hydroxyl moiety on sugar D of EDCBA2 is then alkylated with dihydropyran (DHP) in the presence of camphor sulfonic acid (CSA) and tetrahydrofuran with mixing at room temperature for 3 hours to give the tetrahydropyranyl ether (THP) derivative, EDCBA Pentamer.
………………………………
Intermediates listed on the internet
Fondaparinux sodium Intermediates
Fondaparinux sodium N-4

……………………………….
Fondaparinux sodium N-3
114903-05-8

a-D-Glucopyranoside, Methyl O-2-azido-2-deoxy-3,4-bis-O-(phenylMethyl)-a-D-glucopyranosyl-(14) -O-2,3-bis-O-(phenylMethyl)-b-D-glucopyranuronosyl-(14)-O-2-azido- 2-deoxy-a-D-glucopyranosyl-(14)-O-3-O-(phenylMethyl)-a-L-idopyranu ronosyl-(14)-2-deoxy-2
FSC

114903-05-8

|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||
|
|||||||
| Description | |||||||
|
|||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
References
- “Medscape.com”. Retrieved 2009-01-23.
- “NEJM — Comparison of Fondaparinux and Enoxaparin in Acute Coronary Syndromes”. Retrieved 2009-01-23.
- Peters RJ, Joyner C, Bassand JP, et al. (February 2008). “The role of fondaparinux as an adjunct to thrombolytic therapy in acute myocardial infarction: a subgroup analysis of the OASIS-6 trial”.Eur. Heart J. 29 (3): 324–31. doi:10.1093/eurheartj/ehm616. PMID 18245119.
- WO 2013003001
- Synthesis of heparin fragments: A methyl alpha-pentaoside with high affinity for antithrombin III
Carbohydr Res 1987, 167: 67 - A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodiumOrg Process Res Dev 2013, 17: 869, http://pubs.acs.org/doi/full/10.1021/op300367c
- WO 2012047174
- US 2012116066
- WO 2013011460 RANBAXY
- WO 2013115817
- The unique antithrombin III binding domain of heparin: A lead to new synthetic antithrombotics
Angew Chem Int Ed Engl 1993, 32(12): 1671 - Bioorganic and Medicinal Chemistry Letters, 1(2), p. 95-98 (1991).
- Carbohydrate Research, 101, p. 148-151 (1982),
- Chemistry – A European Journal, 2012 , vol. 18, 34 pg. 10643 – 10652
- Carbohydrate Research, 2012 , vol. 361, p. 155 – 161
- Analytical Chemistry, 2006 , vol. 78, 6 pg. 1774 – 1779
PATENTS
| US4818816 * | Oct 26, 1987 | Apr 4, 1989 | Choay, S.A. | Process for the organic synthesis of oligosaccharides and derivatives thereof |
| US6376663 * | Nov 29, 1996 | Apr 23, 2002 | Macquarie Research Ltd. | Desalting and purification of oligosaccharides and their derivatives |
| US7541445 * | Sep 6, 2002 | Jun 2, 2009 | Alchemia Limited | Synthetic heparin pentasaccharides |
| US20040048785 * | Jun 18, 2003 | Mar 11, 2004 | Societe L’oreal S.A. | C-glycoside compounds for stimulating the synthesis of glycosaminoglycans |
| US20040149200 * | Jun 11, 2002 | Aug 5, 2004 | Tsuyoshi Shimose | Crystals of an oligosaccharides and process for preparation thereof |
| US20110105418 * | Jul 30, 2010 | May 5, 2011 | Reliable Biopharmaceutical Corporation | Process for preparing fondaparinux sodium and intermediates useful in the synthesis thereof |
| WO2011014793A2 * | Jul 30, 2010 | Feb 3, 2011 | Reliable Biopharmaceutical Corporation | Process for preparing fondaparinux sodium and intermediates useful in the synthesis thereof | |
| AU2008200616A1 | Title not available | ||||
| JPS63218691A * | Title not available | ||||
| US4818816 | Oct 26, 1987 | Apr 4, 1989 | Choay, S.A. | Process for the organic synthesis of oligosaccharides and derivatives thereof | |
| US7468358 | Oct 27, 2004 | Dec 23, 2008 | Paringenix, Inc. | Method and medicament for sulfated polysaccharide treatment of heparin-induced thrombocytopenia (HIT) syndrome | |
| US84771910 | Title not available | ||||
| USPP23055709 | Title not available |
FONDAPARINUX



The three specialties available in the United States – dalteparin (Fragmin, Pfizer), enoxaparin (Lovenox, Sanofi-Aventis) and tinzaparin (Innohep, Bristol-Myers Squibb) – the first two are found in Brazil, enoxaparin under the names Lovenox, Cutenox and Dripanina.

Zanamivir, Relenza…For the prevention and treatment of influenza A and B.

Zanamivir
139110-80-8
APPROVED 26-7-96……. GSK NDA 021036
A guanido-neuraminic acid that is used to inhibit neuraminidase.
Zanamivir INN /zəˈnæmɨvɪər/ is a neuraminidase inhibitor used in the treatment and prophylaxis of influenza caused by influenza A virus andinfluenza B virus. Zanamivir was the first neuraminidase inhibitor commercially developed. It is currently marketed by GlaxoSmithKline under the trade name Relenza as a powder for oral inhalation.
The drug is approved for use for the prevention and treatment of influenza in those over the age of 7 in the United States, Canada, European Union, and many other countries. It is not recommended for people with respiratory problems and ailments.
| United States | 6294572 | APPROVED 1994-12-15 | EXPIRY 2014-12-15 |
| United States | 5360817 | 1993-07-26 | 2013-07-26 |
| Canada | 2291994 | 2003-10-14 | 2011-04-24 |
| Canada | 2081356 | 2000-02-22 | 2011-04-24 |
| Patent No | PatentExpiry | use code |
|---|---|---|
| 5360817 | Jul 26, 2013 | |
| 5648379 | Jul 15, 2014 | U-274 |
| 5648379 | Jul 15, 2014 | U-721 |
| 5648379 | Jul 15, 2014 | U-722 |
| 6294572 | Dec 15, 2014 |
Zanamivir was discovered in 1989 by scientists led by Peter Malcolm Colman and Joseph Varghese at the CSIRO, in collaboration with theVictorian College of Pharmacy, Monash University, and scientists at Glaxo, UK. Zanamivir was the first of the neuraminidase inhibitors. The discovery was initially funded by the Australian biotechnology company Biota and was part of Biota’s ongoing program to develop antiviral agents throughrational drug design. Its strategy relied on the availability of the structure of influenza neuraminidase, by X-ray crystallography. It was also known, as far back as 1974, that 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA), a sialic acid analogue, is an inhibitor of neuraminidase. Sialic acid (N-acetyl neuraminic acid, NANA), the substrate of neuraminidase, is itself a mild inhibitor of the enzyme, but the dehydrated derivative DANA, a transition-state analogue, is a better inhibitor.
Computational chemistry techniques were used to probe the active site of the enzyme, in an attempt to design derivatives of DANA that would bind tightly to the amino acid residues of the catalytic site, and so would be potent and specific inhibitors of the enzyme. The GRID software by Molecular Discovery was used to determine energetically favourable interactions between various functional groups and residues in the catalytic site canyon. This investigation showed that there is a negatively charged zone in the neuraminidase active site that aligns with the C4hydroxyl group of DANA. This hydroxyl is, therefore, replaced with a positively charged amino group; the 4-amino DANA was shown to be 100 times better as an inhibitor than DANA, owing to the formation of a salt bridge with a conserved glutamic acid (119) in the active site. It was also noticed that Glu 119 is at the bottom of a conserved pocket in the active site, just big enough to accommodate a more basic functional positively charged group, such as a guanidino group, which was also larger than the amino group. Zanamivir, a transition-state analogue inhibitor of neuraminidase, was the result.
As Biota was a small company, it did not have the resources to bring zanamivir to market by itself. In 1990, zanamivir patent rights were licensed to Glaxo, now GlaxoSmithKline (GSK). In 1999, the product was approved for marketing in the US and subsequently has been registered by GSK in a total of 70 countries (GlaxoSmithKline News release, 2006). Zanamivir is delivered via Glaxo’s proprietary Diskhaler inhalation device. The license agreement entitled Biota to receive a 7% royalty on Glaxo’s sales of zanamivir.
|
Chemical name: |
5- Acetamido- 2, 6- anhydro- 3, 4, 5- trideoxy- 4- guanidino- D- glycero- D- galacto- non- 2- enonic acid |
| Synonyms: | Zanamivir, GG167, 4-guanidino-Neu5Ac2en and 2,3- Didehydro- 2, 4- dideoxy- 4- guanidino- N- acetyl- D- neuraminic acid(2R,3R,4S)-4-guanidino-3-(prop-1-en-2-ylamino)-2-((1R,2R)-1,2,3-trihydroxypropyl)-3,4-dihydro-2H-pyran-6-carboxylic acid |
| Empirical formula: |
C12H20N4O7 |
| Structural formula: | |
| Molecular weight: | 332.31g |
| Beilstein number: | 7083099 |
| Normal State: | Powder |
| Colour: | White to ‘off white’ |
| Melting point: | 325oC |
| Optical rotary power: | Type [�]Conc: 0.9g/100mlSolvent: H2OOptical rotary power: 41 degWavelength: 589nmTemp: 20oC |
| CAS number: | 139110-80-8 |
| Solubility: | 18mg/mL in water at 20oC |
Zanamivir is used for the treatment of infections caused by influenza A virus and influenza B virus. There is low to moderate evidence that it decreases the risk of one’s getting influenza by 1% to 12% in those exposed. In otherwise-healthy individuals, benefits overall appear to be small.It is unclear whether it affects the risk of one’s need to be hospitalized or the risk of death. An independent analysis of its effects by the Cochrane collaboration was awaiting release of trial data as of 2012. The evidence for a benefit in preventing influenza is weak in children with concerns of publication bias in the literature. As of 2009 no influenza has shown any signs of resistance. Since then genes expressing resistance to were found in patients infected with Influenza A H7N9 and who were treated with corticosteroids.
 ZANAMIVIR
ZANAMIVIR
Mass

| 1H NMR |
| Hydrogen | Chemical shift /ppm |
| (1H, d, 3-H) | 5.53 |
| (2H, 2dd, 4- and 6-H) | 4.50 – 4.38 |
| (1H, dd, 5-H) | 4.21 |
| (2H, dd+ddd, 9-Ha and 8-H) | 4.00-3.88 |
| (2H, 2dd, 9-Hb and 7-H) | 3.70-3.62 |
| (3H, s, Ac) | 2.05 |
|
|
| 13C NMR |
| Carbon | Shift /ppm |
| (C=O, Ac) | 177.3 |
| (C-1) | 172.1 |
| (guanidino) | 159.9 |
| (C-2) | 152.1 |
| (C-3) | 106.8 |
| (C-6) | 78.3 |
| (C-8) | 72.6 |
| (C-7) | 71.0 |
| (C-9) | 65.9 |
| (C-4) | 54.0 |
| (C-5) | 50.6 |
| (Me) | 24.8 |
ref 12
IR spectra:
| The following peaks are present in the IR spectra of Relenza: 3332cm-1, 1676cm-1, 1600cm-1, 1560cm-1, 1394cm-1, 1322cm-1 and 1281cm-1. |
UV spectra
| The maximum peak is 235nm giving E = 199 dm-3 mol-1cm-1 |
ref 13for above

Although zanamivir was the first neuraminidase inhibitor to the market, it had only a few months lead over the second entrant, oseltamivir (Tamiflu), with an oral tablet formulation.
According to the CDC, Tamiflu, zanamivir’s main competitor, is not as effective at treating the influenza viruses as zanamivir, especially in H1N1 seasonal flu. In fact, tests showed 99.6% of the tested strains of seasonal H1N1 flu and 0.5% of 2009 pandemic flu were resistant to Tamiflu, while no flu samples, seasonal or pandemic, showed any resistance to zanamivir.
When first marketed in the US in 1999/2000, zanamivir captured only 25% of the influenza antiviral market, despite a huge promotional campaign. By the end of that season, Tamiflu was outselling zanamivir 3:1. During that season, zanamivir experienced worldwide safety warnings involving the risk of bronchospasm and death. Glaxo then reduced the marketing of zanamivir, and Tamiflu’s dominance increased. More than US$20 million worth of zanamivir sold by Glaxo in the first US season was returned to the company in the next two seasons because zanamivir’s sales to patients were far less than expected.
Biota commenced legal proceedings in 2004 alleging Glaxo’s reduced marketing of zanamivir to be a breach of contract. Biota claimed approximately A$700m from Glaxo. After Biota spent four years trying to progress its case, and incurring A$50m in legal costs, the company abandoned the claim in July 2008, recovering only A$20 million, including legal costs following settlement at mediation. Biota had refused an earlier tactical offer from Glaxo of A$75 million plus legal costs.
In August 2006, Germany announced it would buy 1.7 million doses of zanamivir, as part of its preparation strategy against bird flu. “Germany’s purchase shows that countries are starting to take a balanced view of influenza preparedness,” says Simon Tucker, head of research at Melbourne-based Biota, where zanamivir was originally developed.
In April 2009, many cases of swine flu (H1N1-type virus) were reported in US and Mexico. Zanamivir is one of only two drugs prescribed to treat it. A study published in June 2009 emphasized the urgent need for augmentation of oseltamivir (Tamiflu) stockpiles, with additional antiviral drugs including zanamivir, based on an evaluation of the performance of these drugs in the scenario that the 2009 H1N1 swine flu neuraminidase (NA) were to acquire the Tamiflu-resistance (His274Tyr) mutation, which is currently widespread in 99.6% of all tested seasonal H1N1 strains.n January 2011, GSK announced that it would commence phase III trials for intravenous zanamivir in a study that will span 20 countries in the Northern and Southern Hemispheres.
Recently, the reported oseltamivir-resistance H5N1 virus neuraminidase still retaining susceptibility to zanamivir indicates that the structure of zanamivir has some advantages over oseltamivir in binding to the active pocket of H5N1 neuraminidase.
As a proven anti-influenza drug target, neuraminidase continues to be attractive for the development of new inhibitors. The crystal structure of H5N1 avian influenza neuraminidase (PDB code: 2HTY) provides the three-dimensional structural information and opportunity for finding new inhibitors in this regard, because the existing inhibitors, such as oseltamivir and zanamivir, were developed based on different structures of neuraminidase, such as subtypes N9 and N2, and type B genus of influenza virus.
ZANAMIVIR
Chemistry

- Scheigetz, J.; Zamboni, R.; Bernstein, M. A.;Roy, B. (December 1995). “A syntheses of 4-a-guanidino-2-deoxy-2,3-didehydro n-acetylneuraminic acid”. Organic Letters 27 (6): 637–644.doi:10.1021/ol901511x. Retrieved 2010-11-14.
Zanamivir synthetic process in the world
Together with oseltamivir, zanamivir is the only medicine which can prevent influenza on humans caused by H5N1 and H1N1 virus. Vietnam prepared oseltamivir (Tamiflu) medicine. But there was no zanamivir – the first influenza medicine belonging N1 kind, discovered and commercialized before oseltamivir. The scientific name of zanamivir is acid 5-acetamido-4-guanidino-6-(1,2,3-trihydroxy-propyl)-5,6-dihydro-4H-pyran-2-carboxylic. The discovery of zanamivir opens research possibilities for new medicines which have the same effect on enzyme neuraminidase inhibitor to prevent and treat influenza.
Acid sialic is an input to synthetize zanamivir. The name acid sialic (Neu5Ac2en) is used to indicate derivation at O- and N- positions of acid neuraminic, just for acid N-axetylneuraminic. Acid sialic of carbohydrate groups is on animal cells and microorganism, especially in glycoprotein and gangliosid. The commercial acid sialic is extracted from whey of the cheese and milk process as well as egg yolk, and costs about 5,000 USD per kilo.
In 1994, zanamivir was first synthesized and made public by Von Itzstein and other scientists from the Department of Pharmaceutical Chemistry under Monash University (Australia). Then, Chandler and co-workers of Glaxo company (GSK, Britain) acquired results, improved reaction steps and made them public in 1995. Accordingly, this method produced 8.3% of general output. The synthetic process is described in Figure 1.

Figure 1: Zanamivir synthetic process according to Chandler
Up to now, the research of Chandler has been the only publication about zanamivir synthetic method, the output of which is greater than milligrams, and it reproduces details about reaction conditions and physiochemical properties of the requisite substances.
Recently, a research group of Yao (China) proposed a new approach to synthetize into intermediate compound 5. Researchers started from another material – D-glucono-δ-lactone, which is cheaper than acid sialic. However, the synthetic process is longer and much complicated, including 24 steps, with lower productivity (0.2%).
Researching on synthesizing Zanamivir from Acid sialic by Institute of Chemistry
Synthetizing methyl N-acetylneuraminate (2) and O-pentaacetoxy (3) from acid sialic
Scientists from the Institute of Chemistry used acid sialic (axit N-acetylneuraminic) 98% from China as the input for the zanamivir synthetic process. They decided to use the method of Warner, using ion exchange resin Dowex-H, with the role of catalyst. Reaction was performed in the room in 10 hours. The output was metyl (2) este product of acid N-acetylneuraminic with a productivity of 99%.
Then, to synthetize O-pentaacetoxy (3), scientists applied axetyl effective chemistry method recently published, using BF3.OEt2catalysis at 00C. Productivity in this case exceeded 95%.

Figure 2: The diagram of O-pentaacetoxy 3 derivative making
The use of catalysts which were ion exchange resin Dowex-H (for este chemical reaction) and BF3.OEt2 (for axetyl chemical reaction) had more advantages than the method by scientists from Glaxo.
Synthesizing intermediate compound – oxazoline (4) key from O-pentaacetoxy (3)

Figure 3: Diagram to synthesize oxazoline (4) from O-pentaacetoxy (3) according to a and b methods
Firstly, scientists conducted a survey on oxazoline (4) synthetic process according to Chandler’s process. O-pentaacetoxy (3) compound was separated from two types of OAc and formed oxazoline round thanks to the effect of strong acid Lewis, which was TMSOTf at 520C in 2.5 hour. The productivity of this reaction achieved 40%. The pilot instead of TMSOTf by BF3.OEt2 catalysis in dichloromethane at room temperature at night, the productivity of the reaction to form oxazoline round from penta-acetoxy (5) was similar to the method using TMSOTf (42%). To increase productivity, scientists made a survey on one-pot method, directly from metyl este (2) to oxazoline (4), without passing O-pentaacetoxy (3), gave the highest productivity (73,3%) and was the most economic effectiveness.
Synthesizing zanamivir from oxazoline (4) intermediate compound
The next, scientists successfully conducted reactions from oxazoline (4) intermediate compound to Zanamivir (9) final product (Figure 1). Zanamivir product had IR and NMR data which were compatible with their structure.
Therefore, scientists from the Institute of Chemistry under Vietnam Academy of Science and Technology built a stable process, including seven major steps, synthesizing from acid sialic with the general productivity of 6.6% (the productivity made public in the world was 8.3%). Especially, in the first period, from acid sialic to oxazolin (4) was optimized and gave a general productivity of 74%, higher than the productivity made public by (61.7%). However, the productivity gained in the later period is still low. Now, synthesizing zanamivir influenza medicine still continues to be researched.
……………………
Beau and coworkers assembled the core dihydropyran framework of zanamivir congeners via a combination of PBM reaction and Iron(III)-promoted deprotection-cyclization sequence. A stereochemically-defined α-hydroxyaldehyde 2, diallylamine and a dimethylketal-protected boronic acid 1 is coupled to form the acyclic, stereochemically-defined amino-alcohol 3, which then undergoes an Iron(III)-promoted cyclization to form a bicyclic dihydropyran 4. Selective opening of the oxazoline portion of the dihydropyran intermediate 4 with water or timethylsilyl azide then furnish downstream products that have structures resembling the Zanamivir family members.

| Reaction scheme part 1: |
| The commercially available N-acetyl-neuraminic acid 1 is the starting reagent for the most direct approach to the synthesis of 4-guanidino-Neu5Ac2en (Relenza). In reaction scheme 1 the steps for the conversion of N-acetyl-neuraminic acid 1 to its 4-amino analogue is shown. Step 1 is the addition of methanolic HCl (MeOH and HCl gas), which produces the methyl ester of 1, followed by acetic anhydride in pyridine with 4-(dimethylamino)pyridine catalysis, which produces the penta-acetoxy compound, 2. In step 2, 2 is converted into the oxazoline 3 at high yield using trimethylsilyl trifluoromethanesulfonate (TMSOTf) in ethyl acetate at 52oC. In step 3, the azido compound, 4, is produced by the reaction of 3 with trimethylsilyl azide in tert-butyl alcohol at 80oC. In step 4 catalytic sodium methoxide in methanol was used to remove the acetate protecting groups from 4 to give triol 5. The 4-amino analogue, 6 was made in step 5, by hydrolysis using triethylamine in water, hydrogenolysis with a Lindlar catalyst and finally the addition of Dowex 2 * 8 resin. The triethylamine salt of the 6 was made during hydrogenolysis and the purpose of the Dowex 2 * 8 resin was to desalt this intermediate. The chemical names of the compounds are: |
| 1: N-acetyl-neuraminic acid |
| 2: 5- Acetamido- 3,5- dideoxy- D- glycero- �- D- galacto- 2- nonulo- pyranosonic acid methyl ester |
| 3: Methyl (3aR, 4R, 7aR)- 2- Methyl- 4- [(1’S, 2’R)- 1′, 2′, 3′ – triacet- oxypropyl]- 3a, 7a- dihydro- 4H- pyrano [3, 4-d] oxazole- 6- carboxlate. |
| 4: 5- Acetamido- 7, 8, 9- tri- O- acetyl- 2, 6- anhydro- 4- azido- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid methyl ester. |
| 5: 5- Acetamido- 2, 6- anhydro- 4- azido- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid methyl ester. |
| 6: 5- Acetamido- 4- amino- 2, 6- anhydro- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid. |

Part one of reaction scheme
| Synthesis of reactant necessary for part 2 of reaction: |
| Aminoiminomethane-sulfonic acid (AIMSA), 7, which is necessary for the conversion of compound 6 into Relenza, 9, is synthesised in Reaction scheme 2. The oxidizing solution necessary for the reaction is prepared by the addition of peracetic acid to 30% hydrogen peroxide and then conc. sulfuric acid. This is followed by acetic anhydride and, once the reaction has completed, methanol. Thiourea is dissolved in methanol and added slowly to the oxidizing solution.to produce compound 7. Note that any crystals that form are removed and that the reaction needs to be carried out under cooled conditions. See the reference source for more experimental details. |

Synthesis of AIMSA
| Reaction scheme part 2: |
| Reaction scheme 3 shows the conversion of compound 6 into Relenza For route A, 3 mol equivalent of AIMSA, 7, and 3 mol equivalent of potassium carbonate are added in a portionwise manner to compound 6 over an eight hour period. A yield of about 48% of the crystalline product 8 should be obtained for this method. An alternative route is to treat compound 6 with 1.1 mol equivalent of cyanogen bromide in the presence of sodium acetate in methanol. Route B step 1 gives compound 9, which can be converted into the final product 8 by treating it with ammonium hydroxide and ammonium formate at 85oC. A 36% yield of the purified product can be obtained after purification with ion-exchange chromatography and crystallisation. The chemical names of the compounds in this scheme are: |
| 8. 5- Acetamido- 2, 6- anhydro- 3, 4, 5- trideoxy- 4- guanidino- D- glycero- D- galacto- non- 2- enonic acid. (Relenza) |
| 9. 5- Acetylamino- 2, 6- anhydro- 4- cyanoamino- 3, 4, 5- trideoxy- D- glycero- D- galacto- non- 2- enonic acid |

Part 2 of reaction scheme
ref are 13 and 14
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
SYNTHESIS FROM PATENT EP2276479A2
ZANAMIVIR AND BOC PROTECTED ZANAMIVIR
The synthesis of zanamivir is shown in Scheme 1. The starting material used for zanamivir synthesis is sialic acid 1, which was converted to the methyl ester 2, in presence of Dowex H+ as described in detail in reference 104. The hydroxyl groups of 2 are protected with acetyl groups to give compound 3, which was then converted to the oxazoline derivative 4 in the presence of trimethyltrifluoromethanesulfonate as described in detail in reference 105. Azide 5 was synthesized from 4 in presence of azidotrimethylsilane as described in detail in reference 105. The azide is reduced to the corresponding amine 6 by using Lindlar’s catalyst, and the amine is in turn converted to the guanidine derivative 7 as described in detail in reference 106. The final step involves the deprotection of the methyl ester and acetyl groups in the presence of methanolic sodium hydroxide to give Boc-protected zanamivir 8 as described in detail in reference 106. 8, 1H NMR (CD3OD) δ (ppm) 5.6 (d, J = 2.0 Hz, IH), 5.01 (dd, J = 9.6, 2.1 Hz, IH), 4.25 (dd, J = 10.8, 1.1 Hz, IH), 4.18 (dd, J = 10.6, 9.6 Hz, IH), 3.89 (ddd, J = 9.4, 6.2, 2.7 Hz, IH), 3.84 (dd, J = 11.3, 2.8 Hz, IH), 3.67 (dd, J = 11.3, 5.8 Hz, IH), 3.57(d, J = 9.3 Hz, IH), 1.9 (s, 3H), 1.55 (s, 9H), 1.50 (s, 9H); ESI-MS: 533 (M+H)+.
Scheme 1



a) Dowex H Methanol b) Aceticanhydride DMAP pyridine c) trimethylsilyl tπfluorαmethane sulfonate ethylacetate d) azidotrimethylsilane butanol e) Lindlar’s catalyst ethanol f) N N’-bis-tert-butoxycarbonyMH-pyrazole-i carboxamidine tetrahydrofuran g) sodium hydroxide methanol
104. Martin, R., K.L. Witte, and C-H. Wong, The synthesis and enzymatic incorporation of sialic acid derivatives for use as tools to study the structure, activity, and inhibition of glycoproteins and other glycoconjugates. Bioorganic & Medicinal Chemistry, 1998. 6(8): p. 1283-1292.
105. Malcolm Chandler, M.J.B., Richard Conroy, Brian Lamount, Bina Patel, Vipulkumar K. Patel, Ian P. Steeples, Richard Storer, Naill G. Weir, Michael
Wrightm Christopher Williamson, Synthesis of the potent influenza neuraminidase inhibitor 4-guanidino Neu5Ac2en. X-Ray molecular structure of S-acetamido^-amino^^-anhydro-S^^-trideoxy-D-erythro-L-gluco- nononic acid. J. Chem. Soc, Perkin Trans. 1, 1995: p. 1173 – 1180.
106. Masuda, T., et al., Synthesis and anti-influenza evaluation of polyvalent sialidase inhibitors bearing 4-guanidino-Neu5Ac2en derivatives. Chem Pharm Bull (Tokyo), 2003. 51(12): p. 1386-98
,,,,,,,,,,,,,,,,,,,,,,,,,,,,,
The active component of RELENZA is zanamivir. The chemical name of zanamivir is 5- (acetylamino)-4-[(aminoiminomethyl)-amino]-2,6-anhydro-3,4,5-trideoxy-D-glycero-D-galacto non-2-enonic acid. It has a molecular formula of C12H20N4O7 and a molecular weight of 332.3. It has the following structural formula:
 |
Zanamivir is a white to off-white powder for oral inhalation with a solubility of approximately 18 mg/mL in water at 20°C.
RELENZA is for administration to the respiratory tract by oral inhalation only. Each RELENZA ROTADISK contains 4 regularly spaced double-foil blisters with each blister containing a powder mixture of 5 mg of zanamivir and 20 mg of lactose (which contains milk proteins). The contents of each blister are inhaled using a specially designed breath-activated plastic device for inhaling powder called the DISKHALER. After a RELENZA ROTADISK is loaded into the DISKHALER, a blister that contains medication is pierced and the zanamivir is dispersed into the air stream created when the patient inhales through the mouthpiece. The amount of drug delivered to the respiratory tract will depend on patient factors such as inspiratory flow. Under standardized in vitro testing, RELENZA ROTADISK delivers 4 mg of zanamivir from the DISKHALER device when tested at a pressure drop of 3 kPa (corresponding to a flow rate of about 62 to 65 L/min) for 3 seconds.
CLIP
On Zanamivir
Total Synthesis of Anti-Influenza Agents Zanamivir and Zanaphosphor via Asymmetric Aza-Henry Reaction

The potent anti-influenza agents, zanamivir and its phosphonate congener, are synthesized by using a nitro group as the latent amino group at C4 for asymmetric aza-Henry reaction with a chiral sulfinylimine, which is derived from inexpensive d-glucono-δ-lactone to establish the essential nitrogen-containing substituent at C5. This method provides an efficient way to construct the densely substituted dihydropyran core of zanamivir and zanaphosphor without using the hazardous azide reagent.
Zanamivir as the TFA salt (40 mg, 90 %). C14H21F3N4O9; colorless solid, mp 260262 oC;
1H NMR (400 MHz, D2O) δ 5.67 (1 H, d, J = 2.1 Hz), 4.48 (1 H, dd, J = 9.3, 2.1 Hz), 4.41 (1H, d, J = 10.6 Hz), 4.26 (1 H, dd, J = 10.6, 9.3 Hz), 3.98–3.90 (2 H, m), 3.71–3.66 (2 H, m),2.06 (3 H, s);
13C NMR (100 MHz, D2O) δ 174.5, 166.4, 162.9 (CO2 of TFA, q, J = 35.4 Hz ),157.0, 146.2, 116.3 (CF3 of TFA, q, J = 290.2 Hz ), 107.2, 75.8, 69.8, 67.9, 62.9, 50.8, 47.6,21.9;
ESI–HRMS calcd for C12H20N4O7Na: 355.1230, found: m/z 355.1288 [M + Na]+.
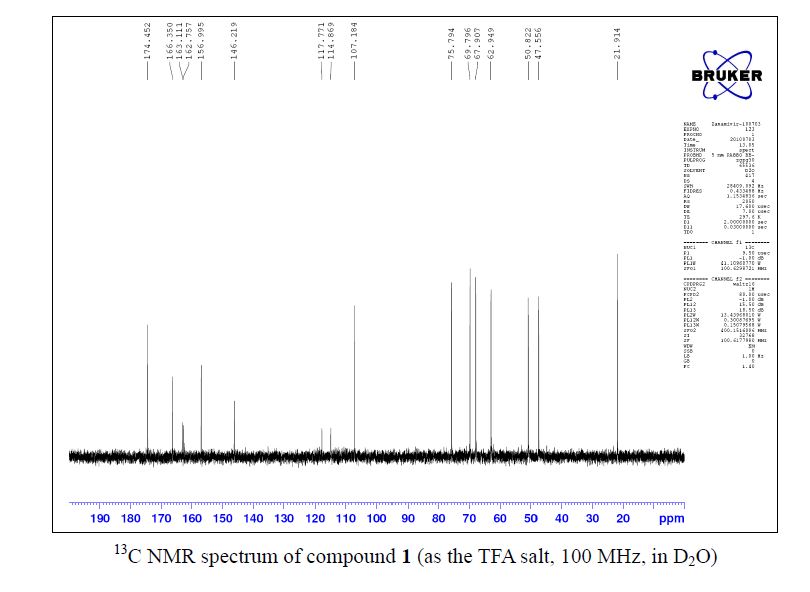
Introduction
Relenza (Zanamivir for oral inhalation) is the first in a new generation of influenza virus-specific drugs known as neuraminidase inhibitors, which work by interferring with the life cycles of influenza viruses A and B. It prevents the virus spreading infection to other cells by blocking the neuraminidase enzyme present on the surface of the virus. Relenza is available as a powder that is administered by inhalation of 2 blisters from the rotadisk inside the diskhaler (Fig. 1) twice daily for five days. This means that 20mg of Relenza is delivered to the principal site of viral replication each day.The main method for preventing influenza since the 1960s is by vaccination and although this and anti-viral drugs such as amantadine and its analogue rimantadine have long been available (since 1976 and 1993 respectively), they are only of limited use because of the constant mutation of the virus. This chameleon-like nature also means that the virus can become unrecognizable to the human immune system and thus repeatedly infect millions of people year after year.

Fig 1: The diskhaler used to administer Relenza. Each blister in the Rotadisk contains 5mg of the drug
Why there is a need for a more effective influenza treatment: At present influenza is basically an uncontrolled disease and an effective method is needed for both the prevention and treatment of it. In the 20th century there were some major pandemics such as the 1918-1919 Spanish ‘flu which killed 20 million people world wide, the 1957 Asian ‘flu, the 1968 Hong Kong ‘flu and the 1977 Russian ‘flu12 These viruses also affect different animals, especially domesticated chickens and turkeys and in Hong Kong in 1997 a virulent bird flu virus, started infecting and killing people for the first time ever. Of the 18 people affected 6 died, although there was no evidence that the virus was able to spread between people. Given the antigenic properties of the influenza virus, in the future the virus may be passed from person to person, and because human immune systems are not prepared for avian viruses the effects on the population could be grave. It would not be possible to prepare vaccines in time and anti-viral drugs are not always adequate.
Advantages of Relenza over previous treatments:
Relenza has a number of advantages over the existing treatments for influenza. It does not cause significant side effects and the development of zanamivir-resistant viruses is not expected to occur readily in patients. This is because selection of drug-resistant mutants characterized by changes in neuraminidase requires prolonged passage in tissue culture and may be a biological cripple. If started within two days of the onset of influenza symptoms and if a fever is present, the duration of illness is decreased by an average of 1.5 days. It appears to decrease the severity of flu symptoms for the remainder of the illness, as well as decreasing the number of complications from the flu. It is also possible that Relenza could be used as a method of ‘flu prevention although it has not yet been approved for this use.
Comparison of the symptoms of the ‘flu with that of a common cold:
| People infected by an influenza virus suffer a lot more than those with a cold. As you can see from the table below, some of the symptoms are similar, but with a cold they are less severe.Influenza also becomes more serious when it leads to secondary bacterial pneumonia or primary influenza viral pneumonia or when it exacerbates underlying medical conditions such as pulmonary or cardiac disease. In children, the symptoms are similar to those observed in adults, however children often have higher fevers and younger ones may develop gastrointestinal manifestations. It should be noted that Relenza is not effective on people with colds or other viral illnesses. |
| Influenza | Cold |
| Sore throat | Mild sore throat |
| High fever and chills | Low-grade fever |
| Non-productive cough | Cough |
| Severe muscle aches | Congestion |
| Headache | |
| Intense fatigue. |
The effect of Relenza on patients with respiratory diseases:Relenza is not generally recommended for the treatment of patients with respiratory dieseases such as asthma or chronic obstructive pulmonary disease (COPD) and has carried an approval since its approval in July 1999. Some patients with underlying airway diseases have experienced serious adverse events following treatment, with some fatal outcomes although causality has been difficult to establish. It has been recommended that patients with asthma have a fast-acting bronchodilator inhaler available and use it about 15 minutes before taking Relenza
Successfulness of Relenza:The sialidase inhibitory activities (determined by methods described in reference 7) of Relenza compared to the more recent neuraminidase inhibitor Oseltamivir are shown in the table below9.IC50 is the concentration that reduces enzyme activity by 50%.
| Compound | Influenza A IC50 (�M) | Influenza B IC50 (�M) |
| Relenza | 0.005 | 0.004 |
| Oseltamivir | 0.002 | 0.032 |
The results demonstrate that both compounds are good inhibitors of influenza A and B, with Oseltamivir being more selective towards Influenza A and Relenza showing a better overall performance. In phase I and II tests reported by the Lancet5, no important adverse effects were found in healthy patients or those reported to have mild to moderate asthma following an inhaled administration of 40mg/day of Relenza. There was a significant improvement of the symptoms of people taking Relenza compared to those taking the placebo.
1940s: Discovery that the influenza virus’s enzyme was destroying receptors on red blood cellsF.This was discovered by George Hirst, who noticed that when red blood cells were mixed with fluids from influenza infected chicken embryos in cold conditions the cells were very heavily agglutinated by the virus. These red cells dispersed when warmed up and could not be re-agglutinated in the cold with fresh virus. This led him to the conclusion that the influenza virus’s enzyme was destroying receptors on red blood cells.
The finding of sialidase (also known as neuraminidase):Alfred Gottschalk heard of Hirst’s experiment and interpretation of results, and this led him to believe that there was a “split product”. He discovered sialic or neuraminic acid (Fig 2), a type of sugar, and the enzyme on the virus was called neuraminidase (or sialidase). At this time it was thought that it was the neuraminidase which was responsible for the observations made by Hirst, but it was later shown by Robin Valentine, W. Graeme Laver, Norbert Bischofberger and Robert G. Webster that the hemagglutinin (receptor-binding) and neuraminidase (receptor-destroying) activities of the virus resided in two quite different spikes on the surface of the virus.

Fig 2: Sialic Acid
Discovery of how new pandemic strains of ‘flu A occured.
Ed Kilbourne, W. Graeme Laver, Norbert Bischofberger and Robert G. Webster realised that hybrid viruses could be formed by infecting cells simultaneously with two different Type A flu viruses. This was because the RNA pieces coding the various virus proteins reassorted, some of the viruses contained the hemagglutinin from one parent and the neuraminidase from the other. This “mating” of two parent viruses to give a hybrid virus explained how new pandemic strains of ‘flu A occurred, and led to a very good way of producing influenza viruses with any desired combination of hemagglutinin and neuraminidase spikes. This helped towards finding a way of producing pure neuraminidase which was later essential for crystal growth and drug design experiments.
The crystallization of neuraminidase:
Laver, Bischofberger and Webster isolated one type of influenza virus by sucking off the allantoic fluid surrounding the embryo of infected chicken eggs and purifying this. The virus particles were incubated with an enzyme capable of digesting proteins. This enzyme was selected to split the “heads” of the neuraminidase spikes off the virus particle without destroying them and to leave behind or destroy the hemagglutinin spike. The neuraminidase “heads” obtained were concentrated using high-speed centrifugation. The tiny pellet of neuraminidase heads examined had a crystalline appearance, and X-ray diffraction analysis of larger crystals showed that they were made of protein.
Neu5Ac2en (DANA) was shown to inhibit influenza neuraminidase:
Different variants of ‘flu neuraminidase were known to exist, each containing an amino acid sequence that varies between types of neuraminidase apart from one small sequence.It was seen that the conserved amino acids came together when the neuraminidase polypeptide folded up to form the active enzyme. This formed a well conserved cavity which was the active catalytic site of the neuraminidase enzyme. It became apparent that a plug-drug could be made to exactly fit into the active site and inhibit the neuraminidase activity from other influenza viruses. A synthetic analog of sialic acid called Neu5Ac2en (DANA) (Fig 3) was shown to inhibit the influenza virus neuraminidase, but not sufficiently enough to be used treatment for the ‘flu in humans.
Fig 3: Neu5Ac2en (DANA)

Fig 3: Neu5Ac2en (DANA)
The plug drug.Mark von Itzstein and colleagues discovered that replacing the OH at the 4 position of sialic acid with a positively charged amino group made a better inhibitor than sialic acid or its analogue, DANA. Replacing the OH at the 4 position of sialic acid with a guanidino group led to a potent inhibitor of ‘flu neuraminidase. This compound was given the names GG167 and Zanamivir and is now more commonly known as Relenza. Peter Colman soaked the substrate for sialic acid in neuraminidase crystals and used X-ray crystallography to determine the three-dimensional structure of the crystals. The strong binding of Relenza by ‘flu neuraminidase which was seen is due to the positively charged guanidino group being anchored by the negatively charged glutamic acids. More details about this are provided in the immunology section.
Immunology

|
Fig 4: The influenza viruses as seen under the electron microscope. Neuraminidase and haemagglutin spikes are visible. |
||
Structure of the flu virus:Influenza (Fig 4) is an RNA virus which may exist as any shape from round balls to long, spaghetti-like filaments. The genome of this virus is associated with five different viral proteins and is surrounded by a lipid membrane, which means that influenza belongs to the “enveloped” group of viruses. Eight separate pieces of ribonucleic acid (RNA) make up the influenza virus genome and each piece of RNA specifies the amino acid sequence of one and sometimes two of the virus’s proteins. The segmented nature of the RNA allows differenet flu viruses to easily “mate” with each other to form hybrid progeny viruses with bits of RNA from each parent virus.Two glycoprotein molecules, known as hemagglutinin (HA) and neuraminidase (NA) (Fig 5) are stuck onto the lipid envelope of the virus and both play a crucial role in the infection of the epithelial cells of the upper respiratory tract. HA is a rod-shaped triangular molecule.and NA exists as a mushroom shaped spike with a box-like head on top of a long stalk, containing a hydrophobic region by which it is embedded in the viral membrane.. |
||
|
Fig 5: The Neuraminidase enzyme |
||
| The enzyme Neuraminidase, also known as sialidase, is a tetramer with C-4 symmetry and an approximate molecular weight of 250 000. It contains a symmetrical folding pattern of six four-stranded antiparallel �-sheets arranged like propeller blades. Nine types of neuraminidase have been identified for influenza A and only one subtype for influenza B, and only 30% of the overall amino acid sequence is conserved between all known types of neuraminidase8 – these are the amino acids which line and surround the walls of the binding pocket. If they mutate, the enzyme is inactivated, so the virus could not mutate to escape from a drug which interfered with this site. So neuraminidase offers an attractive site for therapeutic intervention in influenza infections. | ||
|
|
||
How the influenza virus works:The influenza virus (like all viruses) can only replicate after invading selected living cells and growing inside them. It makes thousands of new virus particles from the cellular machinery and then goes on to infect other cells.. Hemagglutinin allows the virus to infect the epithelial cells of the upper respiratory tract by attaching it to cells through receptors on the cell containing sialic acid, it fuses the cell membrane with the membrane of the virus, allowing the RNA of the virus to get inside the cell and thus instruct the cell to make thousands of new virus particles. After this viral replication, the progeny virions must be released from the cell to repeat the cell cycle of infection.Neuraminidase removes the sialic acid receptors from the host cell and other newly made virus particles by cleavage of -glycosidic bonds. This enables the virus to escape from the cell in which it grew and spread in the body to infect other cells. The action of NA may also facilitate viral mobility through the mucus of the respiratory tract. 
|
| Fig. 6: The life cycle of the influenza virus. Click once on this image to see a larger version | ||
|
The life cycle of the influenza virusG begins with the individual virus entering the cell lining of the respiratory tract (letter a in Fig. 6), and the cell being induced to take up the virus because hemagglutinin on the virus binds to the sialic acid (b and c in Fig 6). The virus then dispatches its genetic material (made up of RNA) and its internal proteins to the nucleus of the cell (e and f). Messenger RNA is produced when some of the internal proteins duplicate the RNA (f). This messenger RNA is used by the cell as a template for making viral proteins (g and h) and genes which become new viral particles and leave the cell covered in sialic acid. This sialic acid needs to be removed so that the hemagglutinin molecules on one particle don’t attach to the sialic acid on other ones, thus causing the new viruses to clump together and stick to the cell. The sialic acid is removed from the surface of the new viral particle by neuraminidase (j) and the new viral particles are able to travel and invade other cells (k). |
||
| How Relenza works:
Relenza adopts a position within the active site of the enzyme and copies the geometry of the sialoside hydrolysis transition state9. It can achieve very good binding through appropriate presentation of its four pendent substituents and contains a hydrogen bonding glycerol sidechain. The guanidino group in Relenza is believed to form salt bridges with Glu 119 in the neuraminidase active site and add a strong charge interaction with Glu 2278. Two hydroxyl groups of the 6-glycerol side chain are hydrogen bonded to Glu276 and the 4-hydroxyl is oriented towards Glu119. The NH group of the 5-N acetyl side chain interacts with a bound water molecule on the floor of the active site. The carbonyl oxygen of the same side chain is hydrogen bonded to Arg152 and the methyl group enters a hydrophobic pocket lined by Ile222 and Trp178. The glycosidic oxygen projects into bulk solvent.
|

|
Fig 7. Relenza bound to neuraminidase |
||
|
The binding involved in Fig 7 is shown more clearly in Fig 8 below. Neuraminidase can no longer remove the sialic acid receptors from the host cell and newly made virus particles because of this binding. Therefore the virsuse ‘clump’ together or to the host cell and cannot go on to effect new cells. |
||
 |
||
|
Fig 8: Depiction of interaction of Relenza (GG 167) in the neuraminidase binding site6 |
||
References
| 1): K. J. Lui and A. P. Kendal, Am. J. Public Health, 1987, 77, 712 |
| 2): Scheiget, Zambonis, Bernstein and Roy, Org. Prep. Proced. Int., 1995, 27, 637- 644 |
| 3): Glaxo Wellcome Inc. Relenza� (zanamivir for inhalation) [package insert]. Research Triangle Park, NC: Glaxo Wellcome, Inc., 1999 |
| 4): N Seppa, Scientific American, July 10th 1999, Volume 156 |
| 5): L. Gubareva, Lancet, March 4th 2000, 355: 827-35 |
| 6): J. Medicinal Chemistry. 1999, 42, 2332-2343 |
| 7):P Smith, S Sollis, P Howes, P Cherry, I Starkey, K Cobley, H Weston, J Scicinski, A Merritt, A Whittington, P Wyatt, N Taylor, D Green, R Bethall, S Madar, R Fenton, P Morley, T Pateman, A Beresford. A. J. Med. Chem, 41, 1998, 787-797 |
| 8): C Kim, W Lew, M Williams, H Liu, L Zhang, S Swaminathan, N Bischofberger, M Chen, D Mendel, C Tai, G Laver, R Stevens, J Am Chem Soc, 1997, 119, 681-690 |
| 9): P Smith, J Robinson, D Evans, S Sollis, P Howes, N Trivedi and R Bethell, Bioorganic and Medicinal Chemistry Letters 9, 1999, 601-604 |
| 10): A. J. Hay, A. J. Wolstenholme, J. J. Skehel and M. H. Smith. EMBO J,. 1985, 4, 3021: L. J. Holsinger and R. A. Lamb, Cell, 1992, 69, 517 |
| 11): J. C. Stoof, J. Booij, B. Drukarch and E. C. Wolters, Eur. J. Pharmacol., 1992, 213, 439 |
| 12): W. Graeme Laver, Norbert Bischofberger, and Robert G. Webster, Perspectives in Biology and Medicine 43.2 (2000) 173-192. This can be seen by visitinghttp://www.press.jhu.edu/journals/perspectives_in_biology_and_medicine/v043/43.2laver.html nmr |
| 13): M. Chandler, M. J. Bamford, R. Conroy, B. Lamont, B. Patel, V. K. Patel, I. P. Steeples, R. Storer, N. G. Weir, M. Wright, C. Williamson, J. Chem. Soc. Perkin Trans. 1, 1995, 1173- 1180 nmr synth |
| 14): A. E. Miller, J. J. Bischoff, Synthesis, 1986, 777- 779 |
| 15): G. D. Allena, S. T. Brookesa, A. Barrow, b, J. A. Dunnc and C. M. Grossec, Journal of Chromatography B: 1999, 732, 383-393 |
Zanamivir
139110-80-8
APPROVED 26-7-96……. GSK NDA 021036
A guanido-neuraminic acid that is used to inhibit neuraminidase.
READ AT
AVANAFIL …..A PDE5 inhibitor.

AVANAFIL
A phosphodiesterase (PDE5) inhibitor, used to treat erectile dysfunction.

Avanafil is a new phosphodiesterase-5 inhibitor that is faster acting and more selective than other drugs belonging to the same class. Chemically, it is a derivative of pyrimidine and is only available as the S-enantiomer. FDA approved on April 27, 2012.
CAS RN: 330784-47-9
4-{[(3-chloro-4-methoxyphenyl)methyl]amino}-2-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide
| (S)-2-(2-Hydroxymethyl-1-pyrrolidinyl)-4-(3-chloro-4-methoxybenzylamino)-5-[(2-pyrimidinylmethyl)carbamoyl]pyrimidine |
| 4-[[(3-Chloro-4-methoxyphenyl)methyl]amino]-2-[(2S)-2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide |
| TA 1790 |
Molecular Formular: C23H26ClN7O3
Molecular Mass: 483.95064
- Stendra
- TA 1790
- TA-1790
- UNII-DR5S136IVO
- NDA 202276
INNOVATOR — VIVUS
APPROVED FDA 27/4/2-12
| Patent No | Patent Expiry | patent use code |
|---|---|---|
| 6656935 | Sep 13, 2020 | U-155 |
| 7501409 | May 5, 2023 |
U 155… TREATMENT OF ERECTILE DYSFUNCTION
| Exclusivity Code | Exclusivity_Date |
|---|---|
| NCE | Apr 27, 2017 |
Stendra (avanafil) was given the green light by the US Food and Drug Administration 27/4/2012, but there has been no launch yet as Vivus has been seeking a partner. The latest data should be attractive to potential suitors and could help Stendra take on other phosphodiesterase type 5 (PDE5) inhibitors, notably Pfizer’s Viagra (sildenafil) but also Eli Lilly’s Cialis (tadalafil) and Bayer’s Levitra (vardenafil).
read all at
http://www.pharmatimes.com/Article/13-06-20/Vivus_ED_drug_gets_to_work_in_less_than_15_mins.aspx
STENDRA (avanafil) is a selective inhibitor of cGMP-specific PDE5.
。

Avanafil is designated chemically as (S)-4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2pyrimidinylmethyl)-5-pyrimidinecarboxamide and has the following structural formula:
 |
Avanafil occurs as white crystalline powder, molecular formula C23H26ClN7O3 and molecular weight of 483.95 and is slightly soluble in ethanol, practically insoluble in water, soluble in 0.1 mol/L hydrochloric acid. STENDRA, for oral administration, is supplied as oval, pale yellow tablets containing 50 mg, 100 mg, or 200 mg avanafil debossed with dosage strengths. In addition to the active ingredient, avanafil, each tablet contains the following inactive ingredients: mannitol, fumaric acid, hydroxypropylcellulose, low substituted hydroxypropylcellulose, calcium carbonate, magnesium stearate, and ferric oxide yellow.
AVANAFIL

Avanafil is a PDE5 inhibitor approved for erectile dysfunction by FDA on April 27, 2012 [1] and by EMA on June 21, 2013.[2] Avanafil is known by the trademark names Stendra and Spedra and was developed by Vivus Inc. In July 2013 Vivus announced partnership with Menarini Group, which will commercialise and promote Spedra in over 40 European countries plus Australia and New Zealand.[3] Avanafil acts by inhibiting a specificphosphodiesterase type 5 enzyme which is found in various body tissues, but primarily in the corpus cavernosum penis, as well as the retina. Other similar drugs are sildenafil, tadalafil and vardenafil. The advantage of avanafil is that it has very fast onset of action compared with other PDE5 inhibitors. It is absorbed quickly, reaching a maximum concentration in about 30–45 minutes.[4] About two-thirds of the participants were able to engage in sexual activity within 15 minutes.[4]
Avanafil is a highly selective PDE5 inhibitor that is a competitive antagonist of cyclic guanosine monophosphate. Specifically, avanafil has a high ratio of inhibiting PDE5 as compared with other PDE subtypes allowing for the drug to be used for ED while minimizing adverse effects. Absorption occurs quickly following oral administration with a median Tmax of 30 to 45 minutes and a terminal elimination half-life of 5 hours. Additionally, it is predominantly metabolized by cytochrome P450 3A4. As such, avanafil should not be co-administered with strong cytochrome P450 3A4 inhibitors. Dosage adjustments are not warranted based on renal function, hepatic function, age or gender. Five clinical trials suggest that avanafil 100 and 200 mg doses are effective in improving the Sexual Encounter Profile and the Erectile Function Domain scores among men as part of the International Index of Erectile Function. A network meta-analysis comparing the PDE5 inhibitors revealed avanafil was less effective on Global Assessment Questionnaire question 1 while safety data indicated no major differences among the different PDE5 inhibitors. The most common adverse effects reported from the clinical trials associated with avanafil were headache, flushing, nasal congestion, nasopharyngitis, sinusitis, and dyspepsia.
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
…………………………………
DESCRIPTION IN A PATENT
EXAMPLE 92-145
The corresponding starting compounds are treated in a similar manner to give the compounds as listed in the following Table 7.
| TABLE 7 |
 |
 |
 |
Amorphous MS(m/z): 484(MH+) |
ENTRY 98 IS AVANAFIL
…………………………………………………….
The invention discloses a preparation method of Avanafil (Avanafil, I), which comprises the following steps: carrying out a substitution reaction on 6-amino-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (XII) and 3-chloro-4-methoxy benzyl chloride (XIII) so as to obtain 6-(3-chloro-4-methoxy benzyl amino)-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (IXV); carrying out condensation on the compound (IXV) and S-hydroxymethyl pyrrolidine (II) so as to generate 4-[(3-chloro-4-methoxy benzyl) amino]-2-[2-(hydroxymethyl)-1-pyrrole alkyl] pyrimidine-5-carboxylic acid ethyl ester (XI); and carrying out hydrolysis on the compound (XI) and then carrying out an acylation reaction on the compound (XI) and the compound (XI) so as to obtain Avanafil (I). The preparation method is simple in process, economic and environmental-friendly, suitable for the requirements of industrialization amplification.
……………………………………………………
The invention discloses a method for preparing avanafil (Avanafil, I). The method comprises the steps of taking cytosine as an initial material; and orderly carrying out replacement, halogen addition and condensation reaction on a side chain 3-chlorine-4-methoxy benzyl halide (III), N-(2-methylpyrimidine) formamide (IV) and S-hydroxymethyl pyrrolidine (II), so as to obtain a target product avanafil (I). The preparation method is available in material, concise in technology, economic and environment-friendly, and suitable for the demands of industrial amplification.
…………………………………………………….
SYNTHESIS
Avanafil can be synthesized from a benzylamine derivative and a pyrimidine derivative REF 5:Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709

- ………………………………………………………
- SYNTHESIS
- A cutting that phenanthrene by a methylthio urea ( a ) and ethoxy methylene malonate ( 2 ) cyclization of 3 , chloride, phosphorus oxychloride get 4 , 4 with benzyl amine 5 occurred SNAr the reaction product after oxidation with mCPBA 6 . In pyrimidine, if the 2 – and 4 – positions are active simultaneously the same leaving group in the case, SNAr reaction occurs preferentially at 4 – position, but does not guarantee the 2 – side reaction does not occur. Here is an activity of the poor leaving group sulfide spans 2 – bit, and a good leaving group active chlorine occupy four – position, thus ensuring a high regioselectivity of the reaction. 4 – position after completion of the reaction, then the 2 – position of the group activation, where sulfide sulfoxide better than the leaving group. Amino alcohols 7 and 6 recurrence SNAr reaction 8 , 8 after alkaline hydrolysis and acid alpha amidation get that phenanthrene.
AVANAFIL
- …………………………….

-
- FDA approves Stendra for erectile dysfunction” (Press release). Food and Drug Administration (FDA). April 27, 2012.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002581/human_med_001661.jsp&mid=WC0b01ac058001d124
- http://ir.vivus.com/releasedetail.cfm?releaseid=775706
- Kyle, Jeffery; Brown, Dana (2013). “Avanafil for Erectile Dysfunction”. Annals of Pharmacotherapy (Sage Publishing). doi:10.1177/1060028013501989. Retrieved 28 September 2013.
- Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709
-
- Peterson CA. Hemodynamic effect of avanafil and glyceryl trinitrate coadministration. , Drugs Context , Volume 2013 , 2013 Feb 26
- Gur S. The Effect of Intracavernosal Avanafil, a Newer Phosphodiesterase-5 Inhibitor, on Neonatal Type 2 Diabetic Rats With Erectile Dysfunction. , Urology , 2013 Dec 9
- Hill JK. Avanafil for erectile dysfunction. , Ann Pharmacother , Volume 47 , Issue 10 , 2013 Oct
- Sanford M. Avanafil: a review of its use in patients with erectile dysfunction. , Drugs Aging , Volume 30 , Issue 10 , 2013 Oct
- Hellstrom WJ. PDE5 inhibitors: considerations for preference and long-term adherence. , Int J Clin Pract , Volume 67 , Issue 8 , 2013 Aug
- Aversa A. An update on pharmacological treatment of erectile dysfunction with phosphodiesterase type 5 inhibitors. , Expert Opin Pharmacother , Volume 14 , Issue 10 , 2013 Jul
- Oelke M. Phosphodiesterase inhibitors in clinical urology. , Expert Rev Clin Pharmacol , Volume 6 , Issue 3 , 2013 May
- Kukreja RC. Sildenafil and cardioprotection. , Curr Pharm Des , Volume 19 , Issue 39 , 2013
- Day WW. An open-label, long-term evaluation of the safety, efficacy and tolerability of avanafil in male patients with mild to severe erectile dysfunction. , Int J Clin Pract, Volume 67 , Issue 4 , 2013 Apr
- Tang J. Comparative effectiveness and safety of oral phosphodiesterase type 5 inhibitors for erectile dysfunction: a systematic review and network meta-analysis. , Eur Urol , Volume 63 , Issue 5 , 2013 May
United States APPROVED 6656935 2012-04-27 EXPIRY 2020-09-13 United States 7501409 2012-04-27 2023-05-05 - Faster-Working Erectile Dysfunction Drug?. CBS News. November 24, 2009.
- Vivus says men taking avanafil were more likely to be ready for sex within 15 minutes. The Gaea Times. January 11, 2010.
- “Avanafil is the New Player in The Erectile Dysfunction Field”. June 28, 2011.
-
- • Hatzimouratidis, K., et al.: Drugs, 68, 231 (2008)
-
4-20-2011Tablets quickly disintegrated in oral cavity7-16-2010Combination treatment for diabetes mellitus8-28-2009Roflumilast for the Treatment of Pulmonary Hypertension1-32-2008Cyclic compounds
US5242391 Oct 30, 1991 Sep 7, 1993 ALZA Corporation Urethral insert for treatment of erectile dysfunction US5474535 Jul 19, 1993 Dec 12, 1995 Vivus, Inc. Dosage and inserter for treatment of erectile dysfunction US5773020 Oct 28, 1997 Jun 30, 1998 Vivus, Inc. Treatment of erectile dysfunction US6656935 Aug 10, 2001 Dec 2, 2003 Tanabe Seiyaku Co., Ltd. Aromatic nitrogen-containing 6-membered cyclic compounds Update nov 2015
NEW PATENT WO 2015177807

WO 2015177807
Suryakant Shivaji Pol; Nitin Sharadchandra Pradhan; Shashikant Balu Padwal; Vihar Raghunath Telange; Nitn Shankar Bondre
Wanbury ltd

The present invention relates to a novel compound of Formula (II), and its use in preparation of Avanafil, [Formula should be inserted here] wherein R is -OH, -CI or -OR1 and R1 is C1 to C3 alkyl group
It having been developed and launched by VIVUS and JW Pharmaceutical, under license from Mitsubishi Tanabe Pharma, and Auxilium Pharmaceuticals, for treating ED.
A process for preparation of Avanafil was first disclosed in US 6,797,709 (depicted in Scheme I), wherein 4-chloro-5-ethoxycarbonyl-2-methylthio-pyrimidine is coupled with 3-chloro-4-methoxybenzylamine in presence of triethylamine to provide compound of Formula (A), which on oxidization provides a sulfonyl compound of Formula (B). Said compound of Formula (B) is reacted with L-prolinol and exert compound of Formula (C). The resulting compound of Formula (C) undergoes column chromatographic purification and crystallization, while further subjected to hydrolysis to obtain compound of Formula (D). The compound of Formula (D) is coupled with 2-aminomethylpyrimidine to obtain Avanafil of Formula (I). The final product obtained is purified by column chromatography. The need to purify the intermediate compound of Formula (C) and final product, by column chromatography makes this process cumbersome, time consuming and unviable for large scale production thereby contributing to main disadvantages of the process.
Scheme I

Formula (A)m-CPBA/chloroform

Formula (C) Formula (B)
NaOH/DMSO

Formula (D) Formula (I)CN 103254179, discloses a process for preparation of Avanafi, wherein 3-chloro-4-methoxybenzylhalide is coupled with cytosine to result compound of Formula (E), later on condensation with L-prolinol yields 4-[(3-chloro-4-methoxy benzyl)amino-2-(2-hydroxymethyl)-l -pyrrolinyl]pyrimidine of Formula (F). The compound of Formula (F) is then condensed with N-(2-pyrimidylmethyl)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme II
Scheme II

Formula (F) Formula (I)
CN 103254180 describes an alternate process for preparation of Avanafil of Formula (I), wherein a substitution reaction on 6-amino-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester and 3-chloro-4-methoxybenzylchloride provides 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester of Formula (G) which on condensation with L-prolinoI generates 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid ethyl ester of Formula (H). The compound of Formula (H) is then hydrolysed and coupled with N-(2-pyrimidylmethyI)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme III
Scheme III

Formula (H) Formula (Γ)
In all the prior art discussed above, chiral compound L-prolinol is coupled in molecule in earlier steps of synthesis. This approach seems to be less feasible for large scale production; the insertion of L-prolinol in early stage may need to exert number of purifications for intermediates. Further the main shortcoming in such process is that the chirality of molecule is disturbed by inserting L-prolinol in early stages because there are number of operations in line in process to obtain the target compound.
CN 103483323, discloses a synthetic method for preparation of avanafil, wherein amidation of pyrimidine-5-carbonyl chlorides with 2-(aminomethyl)pyrimidine at temperature ranging from -10 to 5°C resulted an amide (intermediates A); which underwent condensation with 3-chloro-4-methoxybenzylamine at the temperature ranging from 0 -3°C to give 4-[(3-chloro-4-methoxybenzyl)amino]-5-
pyrimidinecarboxamides (intermediates B), which further on condensation with L-prolinol gave avanafil. The disadvantage of this process is the need to maintain the reaction temperature in range of – 10 to 5°C which adds up to cost of process and makes the process complicated. The process is depicted in Scheme IV.
Scheme IV

Intermediate (A)

wherein, R’ & R2 are independently, hydrogen, halogen, alkoxy, alkoxyalkyl, cyno group, amino group
Hence, to overcome shortcomings of prior art the inventors of present invention have skillfully designed a process with novel intermediate which concomitantly result Avanafil compound of Formula (I), substantially free from impurities. Further this invention encompass L-proline in last stage of molecule in order to avoid the number of purifications of intermediate which relent the economic significances by taking into account yield of each stage.
Object of the invention
1. The main object of the invention is to provide a novel compound of Formula
(ID-
2. Another object of present invention is to provide a process for preparation of a novel compound of Formula (II).
3. Yet another object of present invention is to provide a process for preparation of Avanafil of Formula (I), in high yield and purity using a novel compound of Formula (II).
4. Yet another object of the present invention to provide simple, economic and industrially scalable process for the preparation of Avanafil o Formula (I).
Summary of the invention
According to an aspect of present invention, there is provided a novel compound of Formula (II).

Formula (II)
wherein R is -OH, -CI or -OR and R is Q to C3 alkyl group
The invention will be specifically described below with reference to Examples but it should not be construed that the scope of the invention is limited thereto. Since the starting compound was produced by a modified method from that described in prior art, it will be described as Referential Example 1 to 3. Here synthesis routes of Referential Example 1 to 3 and Example 1 to 10 are illustrated below in Scheme (V).
Scheme (V)

Formula (I) Referential Examples
Referential Example 1 – Preparation of ethyl 4-[(3-chloro-4-methoxybenzyl)amino]-2-(methyl sulfanyl)pyrimidine-5-carboxylate
To 600ml of methylene dichloride was added l OOg of ethyl 4-chloro-2-(methylsulfanyl) pyrimidine-5-carboxylate and 91.2g of 3-chloro-4-methoxybenzylamine. The reaction mixture was stirred and 500ml of water, 48g of sodium carbonate and Ig of tetra-butylammonium bromide were added to it. The reaction mixture was then maintained overnight at 25-30°C. After completion of reaction, methylene dichloride layer was separated, washed with water and evaporated to obtain 145g of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate having 95% of HPLC purity.
Above reaction can also be carried out using ammonia or triethylamine in same reaction conditions and parameters, in place of sodium carbonate.
Referential Example 2 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid
To 600ml of methanol was added l OOg of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate (Referential Example 1) and an aqueous solution of sodium hydroxide (15g of NaOH in 140ml of water). The reaction mixture was heated to reflux temperature. After completion of reaction, the pH of mixture was adjusted to 1 -2 using concentrated hydrochloric acid followed by stirring the mixture for 1 hour at 10-15°C. The solid product obtained was filtered, washed sequentially with water and methanol, and dried overnight at 70-75°C to get 87g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid.
Referential Example 3 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III)
To a mixture of 400ml of toluene and 0.5ml of dimethyl formamide was added 50g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid (Referential Example 2) and 70g of thionyl chloride, and the reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain yellow solid mass. The solid mass thus obtained, was cooled to 15-20°C followed by addition of 1 75ml of methylene dichloride, 36. l g of 2-amino methyl pyrimidine mesylate and 35.55g of triaethylamine. The reaction mixture was stirred overnight at 25-30°C. After completion of reaction, methylene dichloride was distilled out to get residue. The residue was washed sequentially with 2.5% sodium carbonate solution and water. The residue was then treated with methanol to obtain 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) having HPLC purity of more than 95% (yield: 80%)
Referential Example 4 – Preparation of 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[(2S)-2-(hydroxymethyl)-l -pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide (Avanafil)
Step i)
To 200ml of dichloromethane was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyI)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting dichloromethane layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the dichloromethane layer obtained in step i), was added 2.57g of triethylamine followed by slow addition of 125ml solution of L-prolinol in dichloromethane (2.46g of L-prolinol in 125ml of dichlromethane). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water followed by evaporation of dichloromethane to obtain an oily mass. The oily mass thus obtained was treated with methanol to yield 8g of Avanafil.
Examples
Example 1 : Preparation of Compound of Formula (II) (wherein R is -OH)
Step i)
To 200ml of methylene dichloride was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting methylene dichloride layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the methylene dichloride layer comprising compound of Formula (IV) obtained in step i), was added 5g of triethylamine followed by slow addition of 125ml solution of L-proline in methylene dichloride (2.8g of L-proline in 125ml of methylene dichloride). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water and 5% sodium carbonate solution, followed by evaporation of methylene dichloride to obtain an oily mass. The oily mass obtained was stripped with 50ml acetone to yield 9g of compound of Formula (II) having HPLC purity 98%.
Example 2: Preparation of Compound of Formula (II) (wherein R is -OC2H5)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filtrate containing product. The ethanol in filtrate is completely distilled out to isolate 10.45g of esterified compound of Formula (II).
Example 3 : Preparation of Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of toluene and 0.5ml of dimethylformamide was added 50g of compound of Formula (II) obtained in example 1 , and 70g of thionyl chloride. The reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain 50.5g of oily carboxylic acid chloride compound of Formula (II).
Example 4: Preparation of Avanafil of Formula (I)
In an inert atmosphere, a solution of 30g of compound of Formula (II) obtained in example 1 or 2, in 150 ml of tetrahydrofuran was dropwise added to 180ml of suspension of 1.0M lithium aluminium hydride solution in tetrahydrofuran, The reaction mixture was refluxed for 5 hours. After completion of reaction, the mixture was cooled in ice-bath and saturated aqueous solution of sodium sulfate was added to decompose excess of lithium aluminium hydride. The mixture was then diluted with 200ml of methylene dichloride and thus formed organic layer was separated. The organic layer was washed with water (3 χ 100 ml), dried over MgS04 and concentrated to collect crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 22.8g of Avanafil of Formula (I) having HPLC purity of 99.20%.
Example 5 : Preparation of Avanafil of Formula (I)
To a mixture of 1.3g sodium borohydride, 1 ml methanesulfonic acid and 50ml ethanol was added l Og of compound of Formula (II) obtained in example 1 or 2, and the mixture was stirred at 25-30°C for 5 hours. After completion of reaction, 100ml water was added and the mixture was extracted with 1 00ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with ethyl acetate at 45- 50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 6 to Example 8
The procedure is carried out as in example 5 except for instead of methanesulfonic acid other reducing agents are used in combination with sodium borohydride. The results are given in Table I
Table I

Example 9: Preparation of Avanafil of Formula (I)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filterate containing product. To the fi Iterate was added 1.2g of sodium borohydride and 2.6g of lithium bromide, and the mixture was stirred for 5 hours. After complete conversion of ester to final product, l OOml water was added and the mixture was extracted with 100ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with 25ml ethyl acetate at 45-50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7.5g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 10: Preparation of Avanafil of Formula (I) from Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of tetrahydrofuran and 50g of carboxylic acid chloride compound of Formula (II) obtained in example 3, was added 12g sodium borohydride at 0-5°C. After completion of reaction, water was added to reaction mixture to decompose excess of sodium borohydride present. The reaction mixture was then concentrated and a solution of 30g of potassium hydroxide in 200 ml of water was added. The mixture was heated to 60-70°C and maintained for 15-18 hours. The mixture was then cooled to 25-30°C and 500 ml of methylene dichloride was added. The organic layer thus formed, was separated and evaporated to yield crude Avanafil
of Formula (I) which was then subjected to purification using methanol as solvent to obtain 40g of Avanafil of Formula (I) having HPLC purity of 99.01%.

Mr. K. Chandran Wholetime Director & Vice Chairman EXTRAS
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof. In one aspect, the PDE5 inhibitor is tadalafil.
“Tadalafil” or “TAD” is described in U.S. Pat. Nos. 5,859,006 and 6,821,975. It refers to the chemical compound, (6R-trans)-6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione and has the following chemical formula:

Tadalafil is currently marketed in pill form for treating erectile dysfunction (ED) under the trade name Cialis® and under the trade name Adcirca® for the treatment of PAH.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
“Lodenafil” refers to the chemical compound, bis-(2-{4-[4-ethoxy-3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-benzenesulfonyl]piperazin-1-yl}-ethyl)carbonate and has the following chemical formula:

More information about lodenafil is available at Toque H A et al., (2008) European Journal of Pharmacology, 591(1-3):189-95. Lodenafil is manufactured by Cristália Produtos Químicose Farmacêuticos in Brazil and sold there under the brand-name Helleva®. It has undergone Phase III clinical trials, but is not yet approved for use in the United States by the U.S. FDA.
“Mirodenafil” refers to the chemical compound, 5-Ethyl-3,5-dihydro-2-[5-([4-(2-hydroxyethyl)-1-piperazinyl]sulfonyl)-2-propoxyphenyl]-7-propyl-4H-pyrrolo[3,2-d]pyrimidin-4-one and has the following chemical formula:

More information about mirodenafil can be found at Paick J S et al., (2008) The Journal of Sexual Medicine, 5 (11): 2672-80. Mirodenafil is not currently approved for use in the United States but clinical trials are being conducted.
“Sildenafil citrate,” marketed under the name Viagra®, is described in U.S. Pat. No. 5,250,534. It refers to 1-[4-ethoxy-3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)phenylsulfonyl]-4-methylpiperazine and has the following chemical formula:

Sildenafil citrate, sold as Viagra®, Revatio® and under various other trade names, is indicated to treat erectile dysfunction and PAH.
“Vardenafil” refers to the chemical compound, 4-[2-Ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-9-methyl-7-propyl-3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one and has the following chemical formula:

Vardenafil is described in U.S. Pat. Nos. 6,362,178 and 7,696,206. Vardenafil is marketed under the trade name Levitra® for treating erectile dysfunction.
“Udenafil” refers to the chemical compound, 3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide and has the following chemical formula:

More information about udenafil can be found at Kouvelas D. et al., (2009) Curr Pharm Des, 15(30):3464-75. Udenafil is marketed under the trade name Zydena® but not approved for use in the United States.



THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family







VORINOSTAT

Vorinostat, MK0683
CAS 149647-78-9
Zolinza, SAHA, suberoylanilide hydroxamic acid, Suberanilohydroxamic acid, N-hydroxy-N’-phenyloctanediamide
US patent 5369108, PDT PATENT
For the treatment of cutaneous manifestations in patients with cutaneous T-cell lymphoma who have progressive, persistent or recurrent disease on or following two systemic therapies. Inhibits histone deacetylase I & 3.
- CCRIS 8456
- HSDB 7930
- M344
- N-Hydroxy-N’-phenyloctanediamide
- SAHA
- SAHA cpd
- Suberanilohydroxamic acid
- suberoylanilide hydroxamic acid
- UNII-58IFB293JI
- MK0683
| Average: 264.3202 Monoisotopic: 264.147392516 |
|
| Chemical Formula | C14H20N2O3 |
|---|
| N-hydroxy-N‘-phenyl-octanediamide | |
|---|---|
| Trade names | Zolinza, 100 MG, CAPSULE, ORAL |
| ZOLINZA (VORINOSTAT) [Merck Sharp & Dohme Corp.] | |
| MedlinePlus | a607050 |
| Licence data | US FDA:link |
| LAUNCHED 2006 MERCKhttp://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021991s002lbl.pdf | |
| Legal status | ℞-only (US) |
| Routes | Oral |
| Pharmacokinetic data | |
| Protein binding | 71% |
| Metabolism | Hepatic glucuronidation andoxidation CYP system not involved |
| Half-life | 2 hours |
| Excretion | Renal (negligible) |
| Identifiers | |
| CAS number | 149647-78-9 |
| ATC code | L01XX38 |
| Chemical data | |
| Formula | C14H20N2O3 |
| Mol. mass | 264.32 g/mol |
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=Vorinostat
Vorinostat (rINN) also known as suberanilohydroxamic acid (suberoyl+anilide+hydroxamic acid abbreviated as SAHA) is a member of a larger class of compounds that inhibit histone deacetylases (HDAC). Histone deacetylase inhibitors (HDI) have a broad spectrum of epigenetic activities.
Vorinostat is marketed under the name Zolinza for the treatment of cutaneous T cell lymphoma (CTCL) when the disease persists, gets worse, or comes back during or after treatment with other medicines.[1] The compound was developed by Columbia University chemist, Ronald Breslow.
Vorinostat was the first histone deacetylase inhibitor[2] approved by the U.S. Food and Drug Administration (FDA) for the treatment of CTCL on October 6, 2006. It is manufactured by Patheon, Inc., in Mississauga, Ontario, Canada, for Merck & Co., Inc., White House Station, New Jersey.[3]
ZOLINZA contains vorinostat, which is described chemically as N-hydroxy-N’-phenyloctanediamide. The empirical formula is C14H20N2O3. The molecular weight is 264.32 and the structural formula is:
 |
Vorinostat is a white to light orange powder. It is very slightly soluble in water, slightly soluble in ethanol, isopropanol and acetone, freely soluble in dimethyl sulfoxide and insoluble in methylene chloride. It has no chiral centers and is non-hygroscopic. The differential scanning calorimetry ranged from 161.7 (endotherm) to 163.9°C. The pH of saturated water solutions of vorinostat drug substance was 6.6. The pKa of vorinostat was determined to be 9.2.
Each 100 mg ZOLINZA capsule for oral administration contains 100 mg vorinostat and the following inactive ingredients: microcrystalline cellulose, sodium croscarmellose and magnesium stearate. The capsule shell excipients are titanium dioxide, gelatin and sodium lauryl sulfate.
Vorinostat has been shown to bind to the active site of histone deacetylases and act as a chelator for Zinc ions also found in the active site of histone deacetylases [4] Vorinostat’s inhibition of histone deacetylases results in the accumulation of acetylated histones and acetylated proteins, including transcription factors crucial for the expression of genes needed to induce cell differentiation. [4]
SAHA inhibits class I and class II HDACs at nanomolar concentrations and arrests cell growth in a wide variety of transformed cells in culture at 2.5-5.0 µM. This compound efficiently suppressed MES-SA cell growth at a low dosage (3 µM) already after 24 hours treatment. Decrease of cell survival was even more pronounced after prolonged treatment and reached 9% and 2% after 48 and 72 hours of treatment, respectively. Colony forming capability of MES-SA cells treated with 3 µM vorinostat for 24 and 48 hours was significantly diminished and blocked after 72 hours.

Vorinostat has also been used to treat Sézary syndrome, another type of lymphoma closely related to CTCL.[5]
A recent study suggested that vorinostat also possesses some activity against recurrent glioblastoma multiforme, resulting in a median overall survival of 5.7 months (compared to 4 – 4.4 months in earlier studies).[6] Further brain tumor trials are planned in which vorinostat will be combined with other drugs.
Including vorinostat in treatment of advanced non-small-cell lung cancer (NSCLC) showed improved response rates and increased median progression free survival and overall survival (although the survival improvements were not significant at the P=0.05 level).[7]
It has given encouraging results in a phase II trial for myelodysplastic syndromes in combination with Idarubicin and Cytarabine.[8]
Vorinostat is an interesting target for scientists interested in eradicating HIV from infected persons.[9] Vorinostat was recently shown to have both in vitro and in vivo effects against latently HIV infected T-cells.[10][11]
Vorinostat, represented by structural formula (I) and chemically named as N-hydroxy-N’- phenyl-octanediamide or suberoylanilide hydroxamic acid (SAElA), is a member of a larger class of compounds that inhibit histone deacetylases (HDAC). Histone deacetylase inhibitors (HDI) have a broad spectrum of epigenetic activities and vorinostat is marketed, under the brand name Zolinza®, for the treatment of a type of skin cancer called cutaneous T-cell lymphoma (CTCL). Vorinostat is approved to be used when the disease persists, gets worse, or comes back during or after treatment with other medicines. Vorinostat has also been used to treat Sέzary’s disease and, in addition, possesses some activity against recurrent glioblastoma multiforme.

Vorinostat was first described in US patent 5369108, wherein four different synthetic routes for the preparation of vorinostat are disclosed (Schemes 1 to 4).
The single step process illustrated in Scheme 1 involves coupling of the diacid chloride of suberic acid with aniline and hydiOxylamine hydrochloride. However, the yield of this reaction is only 15-30%.

Scheme 1
The multistep process illustrated in Scheme 2 begins with the monomethyl ester of suberic acid, which undergoes conversion to the corresponding acid chloride. Further coupling with aniline gives the methyl ester of suberanilic acid. Hydrolysis of the ester and further coupling with benzyl protected hydroxylamine gives benzyl protected vorinostat which on deprotection gives vorinostat.
HO. (CH2J6 OMe . ,OOMM e
O O



Scheme 2
In addition to the disadvantage of being a five-step process with overall yields reported as 35-65%, this process suffers from further disadvantages such as the use of the expensive monomethyl ester of suberic acid.

Scheme 3
The two step process illustrated in Scheme 3 involves coupling of the diacid chloride of suberic acid with aniline and O-benzyl hydroxylamine and then deprotection. However, the overall yield of this reaction is only 20-35%.

Scheme 4
The process illustrated in Scheme 4 is similar to that illustrated in Scheme 3, with the exception that O-trimethylsilyl hydroxylamine was used instead of O-benzyl hydroxylamine. The overall yield of this reaction is reported as 20-33%.
Another process for the preparation of vorinostat has been reported in J. Med. Chem.,
1995, vol. 38(8), pages 1411-1413. The reported process, illustrated in Scheme 5, begins with the conversion of suberic acid to suberanilic acid by a high temperature melt reaction.
Suberanilic acid is further converted to the corresponding methyl ester using Dowex resin and the methyl ester of suberanilic acid thus formed is converted to vorinostat by treatment with hydroxylamine hydrochloride. However, this process employs high temperatures (1900C) in the preparation of vorinostat which adds to the inefficiency and high processing costs on commercial scale. The high temperatures also increase the likelihood of impurities being formed during manufacture and safety concerns. The overall yield reported was a poor 35%.

MeOH, Dowex, 22 hours


Scheme 5
Another process for the preparation of vorinostat has been reported in OPPI Briefs, 2001, vol. 33(4), pages 391-394. The reported process, illustrated in Scheme 6, involves conversion of suberic acid to suberic anhydride, which on treatment with aniline gives suberanilic acid. Coupling of this suberanilic acid with ethyl chloroformate gives a mixed anhydride which upon treatment with hydroxylamine gives vorinostat in an overall yield of 58%. In the first step, there is competition between the formation of suberic anhydride and the linear anhydride and consequently isolation of pure suberic anhydride from the reaction mixture is very difficult. This process step is also hindered by the formation of process impurities and competitive reactions. In the second step, there is formation of dianilide by reaction of two moles of aniline with the linear anhydride. In the third step, suberanilic acid is an inconvenient by-product as the suberanilic acid is converted to a mixed anhydride with ethyl chloroformate, which is highly unstable and is converted back into suberanilic acid. Consequently, it is very difficult to obtain pure vorinostat from the reaction mixture. Although the reported yield was claimed to be 58%, when repeated a yield of only 38% was obtained.

Scheme 6
A further process for the preparation of vorinostat has been reported in J. Med. Chem., 2005, vol. 48(15), pages 5047-5051. The reported process, illustrated in Scheme 7, involves conversion of monomethyl suberate to monomethyl suberanilic acid, followed by coupling with hydroxylamine hydrochloride to afford vorinostat in an overall yield of 79%. However, the process uses the expensive monomethyl ester of suberic acid as starting material.
HOBt, DCC, DMF, RT, 4 hours



CLIP
Vorinostat (ZolinzaTM) Vorinostat, a histone deacetylase (HDAC) inhibitor from Merck, was approved for the treatment of cutaneous T-cell lymphoma (CTCL), a type of non-Hodgkin’s lymphoma.
Vorinostat was shown to inhibit HDAC1, HDAC2, HDAC3 and HDAC6 at nanomolar concentrations. HDAC inhibitors are potent differentiating agents toward a variety of neoplasms, including leukemia and breast and prostate cancers [58].
Commercially available monomethyl ester 125 wasVorinostat (ZolinzaTM) Vorinostat, a histone deacetylase (HDAC) inhibitor from Merck, was approved for the treatment of cutaneous T-cell lymphoma (CTCL), a type of non-Hodgkin’s lymphoma.
Vorinostat was shown to inhibit HDAC1, HDAC2, HDAC3 and HDAC6 at nanomolar concentrations. HDAC inhibitors are potent differentiating agents toward a variety of neoplasms, including leukemia and breast and prostate cancers [58].
Commercially available monomethyl ester 125 was reacted with aniline in the presence of DCC and HOBt in DMF to give amide 127 in 89%yield [59] (Scheme 16).
Methyl ester amide 127 was then reacted with hydroxylamine HCl salt and potassium hydroxide in methanol to give vorinostat(XVI) in 90% yield.
[58] Breslow, R.; Marks, P.A.; Rifkind, R. A.; Jursic, B. WO9307148,2003.
[59] Gediya, L. K.; Chopra, P.; Purushottamachar, P.; Maheshwari, N.;Njar, V. C. O. J. Med. Chem., 2005, 48, 5047.
PATENT
VORINOSTAT
http://www.google.com/patents/EP2349985A2
A preferred embodiment of the first aspect of the present invention is illustrated in Scheme

suberic acid subefanilic acid NH2OHHCl, CDI

suberoylanilide hydroxamic acid (T)
Scheme 8
Optionally, an activating agent can be used in step (a) and/ or step (b) to afford products with high yields and purity. Preferably, the activating agent is selected from cyanuric chloride, cyanuric fluoride, catecholborane, or a mixture thereof. The activating agent is preferably used in combination with the coupling agent. A preferred embodiment of the process according to the first aspect of the present invention comprises the following steps:
(i) taking a mixture of THF, CDI and DCC;
(ii) adding suberic acid; (iii) adding aniline in THF to the solution from step (ii);
(iv) stirring at 25-30°C;
(v) filtering off the solid dicyclohexyl urea formed in the reaction;
(vi) concentrating the filtrate in vacuo;
(vii) adding a solution of KOH in water; (vϋi) filtering off the solid by-product;
(ix) heating the filtrate;
(x) adding aq. HCl;
(xi) isolating suberanilic acid;
(xii) mixing the suberanilic acid and CDI in DMF; (xiii) adding hydroxylamine hydrochloride as solid to the mixture from step (xii);
(xiv) isolating vorinostat from the mixture obtained in step (xiii);
(xv) adding acetonitrile and aq. ammonia to the vorinostat from step (xiv);
(xvi) heating the mixture;
(xvii) cooling the mixture to 20-27°C; and (xvϋi) isolating pure vorinostat from the mixture obtained in step (xvii).
Preferably, by utilising the same organic solvent in steps (a) and (b), pure vorinostat can be obtained without isolation of any synthetic intermediate^).
A preferred embodiment of the second aspect of the present invention is illustrated in Scheme 9.

suberic acid N-hydtoxy-7-carboxy-heptanamide

Example 1
Stage 1 : Conversion of suberic acid to suberanilic acid
A mixture of CDI (0.5eq) and DCC (0.8eq) in THF (15 vol) was stirred for 1 hour at 25- 3O0C. Suberic acid (leq) and aniline (leq) in THF (1 vol) was added and the mixture stirred for a further 16-20 hours. The solid by-product was removed by filtration and the filtrate was concentrated in vacuo at 5O0C. The solid residue obtained was treated with a solution of KOH (2eq) in water (10 vol) and stirred for 30 minutes at 25-300C and any solid byproduct formed was removed by filtration. The filtrate obtained was heated at 6O0C for 3-4 hours and cooled to 200C before addition of an aqueous solution of HCl (17.5%, 3 vol). The mixture was stirred for 30 minutes and the solid filtered, washed with water (2×5 vol) and dried under vacuum at 60-650C. Molar Yield = 60-65% Purity by HPLC = 99.5%
Stage 2: Conversion of suberanilic acid to crude vorinostat The suberanilic acid (leq) obtained in stage 1 was dissolved in DMF (5 vol) and CDI (2eq) was added at 25-3O0C and maintained for 30 minutes under stirring. Hydroxylamine hydrochloride (4eq) was added and stirring continued for 30 minutes. Water (25 vol) was then added and the mixture stirred for 2 hours. The precipitated solid was filtered, washed with water (2×5 vol) and dried under vacuum at 500C. Molar Yield = 70-75% Purity by HPLC = 99% Stage 3: Purification of crude vorinostat
Aqueous ammonia (2.5 vol) was added to the crude vorinostat (leq) in acetonitrile (15 vol) at 25-30°C. The mixture was then maintained at 55-60°C for 1 hour before being cooled to 20-25°C and being stirred for a further hour. The resulting solid was filtered, washed with acetonitrile (2×0.5 vol) and dried under vacuum at 45-5O0C for 5 hours. Molar Yield = 55-60% Purity by HPLC > 99.8%
Example 2
Stage 1 : Conversion of suberic acid to crude vorinostat
A mixture of CDI (0.5eq) and DCC (0.8eq) in THF (15 vol) was stirred for 1 hour at 25- 30°C. Suberic acid (leq) and hydroxylamine (leq) in THF (1 vol) was added and the mixture stirred for a further 1 hour. Then CDI (0.5eq), DCC (0.8eq) and aniline (leq) were added to the mixture and the mixture was stirred for a further 16-20 hours. The solid byproduct was removed by filtration and the filtrate was concentrated in vacuo at 50°C to obtain crude vorinostat. Molar Yield = 55-60% Purity by HPLC > 95.8%
Stage 2: Purification of crude vorinostat
Aqueous ammonia (2.5 vol) was added to the crude vorinostat (leq) in acetonitrile (15 vol) at 25-3O0C. The mixture was then maintained at 55-600C for 1 hour before being cooled to 20-250C and being stirred for a further hour. The resulting solid was filtered, washed with acetonitrile (2×0.5 vol) and dried under vacuum at 45-500C for 5 hours. Molar Yield = 35-40% Purity by HPLC > 99.8%
PATENT
SYNTHESIS
Scheme V. – –

Vorinostat
Suberic acid (l.Oeq) was dissolved in tetrahydrofuran (15vol) and the clear solution was chilled to 0-5°C. Methyl chloro formate (l.leq) and triethylamine (1.1 eq) were added to the solution at the same temperature and the mixture was stirred for 15 minutes. The triethylamine.HCl salt formed was filtered off, then aniline (leq) was added to the reaction mixture at 0-50C and stirring was continued for 15 minutes. Methyl chloroformate (l.leq) and triethylamine (l.leq) were added to the clear solution and stirring was continued for a further 15 minutes at 0-5°C. This chilled reaction mixture was added to a freshly prepared hydroxylamine solution in methanol (*see below) chilled to 0-5°C and stirred for 15 minutes at 0-5°C. The solvent was removed under vacuum at 40°C and the residue obtained was taken in methylene dichloride and the organic solution was washed with water and dried over anhydrous sodium sulfate. Methylene dichloride was removed under vacuum at 40°C and acetonitrile was added to the residue. This mixture was stirred for 15 minutes before the solid was filtered under vacuum and dried under vacuum at 60°C to afford the product as a white solid. Molar yield = 35-41%; HPLC purity = 99.90%.
VORINOSTAT
1H-NMR (DMSO-d6): 1.27 (m, 4H, 2 x -CH2-), 1.53 (m, 4H, 2 x -CH2-), 1.94 (t, J = 7.3 Hz, 2H, -CH2-), 2.29 (t, J = 7.4 Hz, 2H, -CH2-), 7.03 (t, J = 7.35 Hz, IH, aromatic para position), 7.27 (t, J = 7.90 Hz, 2H, aromatic meta position), 7.58 (t, J = 7.65 Hz, 2H, aromatic ortho position), 8.66 (s, IH, -OH, D2O exchangeable), 9.85 (s, IH, amide -NH-, D2O exchangeable), 10.33 (s, IH, -NH-OH, D2O exchangeable).
13C-NMR (DMSO-d6): 25.04 (2C, 2 x -CH2-), 28.43 (2C, 2 x -CH2-), 32.24 (1C, -CH2-), 36.34 (1C, -CH2-), 119.01 (2C, Ar-C), 122.96 (1C, Ar-C), 128.68 (2C, Ar-C), 139.24 (1C, Ar- C, =CNH-), 169.23 (1C, -CO-), 171.50 (1C, -CO-).
*Preparation of hydroxylamine solution:
Potassium hydroxide (l.leq) was added to methanol (8vol) and the solution was chilled to 0-5°C. Similarly hydroxylamine hydrochloride (l.leq) was added to methanol (8vol) and chilled to 0-5°C. The chilled amine solution was added to the chilled alkali solution and stirred for 15 minutes at 0-50C. The white potassium chloride salt was filtered off and the filtrate was used as such.
SPECTRAL DATA AND SYNTHESIS
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
for structures see above link
Suberoylanilide hydroxamic acid (26, SAHA, vorinostat).
Suberic acid monomethyl ester (23) (15.09 g, 80.2 mmol) and DMF (0.10 mL) in anhydrous
DCM (300 mL) was added SOCl2 (34.6 mL, 0.481 mol), and the reaction mixture was refluxed for 3
h. The mixture was then concentrated. Toluene (300 mL) was added to the residue and evaporated
to afford crude acid chloride 24. Crude 24 was dissolved in DCM (240 mL), and followed by
addition of aniline (7.3 mL, 80.2 mmol) and Et3N (16.9 mL, 0.120 mol). The reaction mixture was
stirred for 90 min at room temp. The course of reaction was monitored by TLC (30% EtOAc in
hexanes) and LC–MS. DCM was removed, and ethyl acetate (500 mL) was added to dissolve the
residue. The organic layer was washed with aqueous NaHCO3 (500 mL × 2), 1 N HCl (400 mL × 2),
water, dried (Na2SO4), and evaporated to dryness under reduced pressure. The residue was purified
by vacuum liquid chromatography (silica, 20% EtOAc in hexanes) to afford compound 25as white crystalline solids (20.15 g, 96 %). NaOMe in MeOH solution (5.4 M, 106 mL, 0.573 mol) was added to a solution of compound 25 (10.05 g, 38.2 mmol) and NH2OH·HCl (26.54 g, 0.382 mol) in
dry MeOH (375 mL). The reaction mixture was stirred for 40 min at room temp. The reaction was
quenched by adding of 1 N HCl to pH 7–8. MeOH was removed under reduced pressure and water
(1 L) was added to the residue. The precipitated solid was filtered and washed with water (300 mL)
and EtOAc (150 mL) to afford crude 26 which was further purified by recrystallization. MeOH (200
mL) was added to crude 26 (5 g) and warmed to dissolve all solids. The MeOH solution was filtered,
and deionized water (400 mL) was added to the filtrate, the resulting solution was placed at 4 oC
overnight. Crystals obtained were filtered and washed with deionized water (100 mL) to afford pure
26 (vorinostat, SAHA) as off-white crystals. Overall yield: 80–85% from compound 23. Compound
26,
LC–MS m/z 265.1 ([M + H]+).
1H NMR (DMSO-d6) 10.35 (1H, s), 9.86 (1H, s), 8.68 (1H, s),
7.58 (2H, d, J = 7.6 Hz), 7.28 (2H, t, J = 7.5 Hz), 7.02 (1H, t, J = 7.4 Hz), 2.29 (2H, t, J = 7.4 Hz),
1.94 (2H, t, J = 7.4 Hz), 1.57 (2H, m), 1.49 (2H, m), 1.33 – 1.20 (2H, m); 13C NMR (DMSO-d6)
171.2, 169.1, 139.3, 128.6, 122.9, 119.0, 36.3, 32.2, 28.4, 28.3, 25.0. Anal. (C10H20N2O3) C, H, N.
CLIP

Suberic acid monomethyl ester (23) (15.09 g, 80.2 mmol) and DMF (0.10 mL) in anhydrous DCM (300 mL) was added SOCl2 (34.6 mL, 0.481 mol), and the reaction mixture was refluxed for 3 h. The mixture was then concentrated. Toluene (300 mL) was added to the residue and evaporated to afford crude acid chloride 24. Crude 24 was dissolved in DCM (240 mL), and followed by addition of aniline (7.3 mL, 80.2 mmol) and Et3N (16.9 mL, 0.120 mol). The reaction mixture was stirred for 90 min at room temp. The course of reaction was monitored by TLC (30% EtOAc in hexanes) and LC–MS. DCM was removed, and ethyl acetate (500 mL) was added to dissolve the residue. The organic layer was washed with aqueous NaHCO3 (500 mL × 2), 1 N HCl (400 mL ×2), water, dried (Na2SO4), and evaporated to dryness under reduced pressure. The residue was purified by vacuum liquid chromatography (silica, 20% EtOAc in hexanes) to afford compound 25 as white crystalline solids (20.15 g, 96 %). NaOMe in MeOH solution (5.4 M, 106 mL, 0.573 mol) was added to a solution of compound 25 (10.05 g, 38.2 mmol) and NH2OH·HCl (26.54 g, 0.382 mol) in dry MeOH (375 mL). The reaction mixture was stirred for 40 min at room temp. The reaction was quenched by adding of 1 N HCl to pH 7–8. MeOH was removed under reduced pressure and water (1 L) was added to the residue. The precipitated solid was filtered and washed with water (300 mL) and EtOAc (150 mL) to afford crude 26 which was further purified by recrystallization. MeOH (200 mL) was added to crude 26 (5 g) and warmed to dissolve all solids. The MeOH solution was filtered, S37 and deionized water (400 mL) was added to the filtrate, the resulting solution was placed at 4 oC overnight. Crystals obtained were filtered and washed with deionized water (100 mL) to afford pure 26 (vorinostat, SAHA) as off-white crystals. Overall yield: 80–85% from compound 23.
. Compound 26,
LC–MS m/z 265.1 ([M + H] + ).
1H NMR (DMSO-d6) 10.35 (1H, s), 9.86 (1H, s), 8.68 (1H, s), 7.58 (2H, d, J = 7.6 Hz), 7.28 (2H, t, J = 7.5 Hz), 7.02 (1H, t, J = 7.4 Hz), 2.29 (2H, t, J = 7.4 Hz), 1.94 (2H, t, J = 7.4 Hz), 1.57 (2H, m), 1.49 (2H, m), 1.33 – 1.20 (2H, m);
13C NMR (DMSO-d6) 171.2, 169.1, 139.3, 128.6, 122.9, 119.0, 36.3, 32.2, 28.4, 28.3, 25.0.
Anal. (C10H20N2O3) C, H, N.


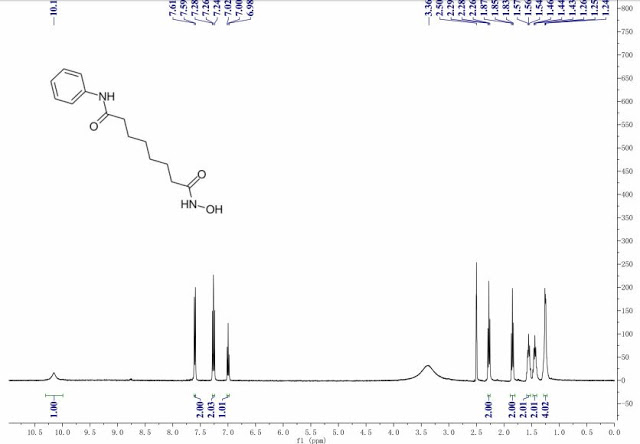
References
- “ZOLINZA, Merck’s Investigational Medicine for Advanced Cutaneous T-Cell Lymphoma (CTCL), To Receive Priority Review from U.S. Food and Drug Administration” (Press release). Merck & Co. June 7, 2006. Retrieved 2006-10-06.
- HDAC Inhibitors Base (vorinostat)
- “FDA Approves New Drug for Skin Cancer, Zolinza” (Press release). Food and Drug Administration. October 6, 2006. Retrieved 2006-10-06.
- Richon, Victoria. “Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor”. British Journal of Cancer. Retrieved 3 May 2012.
- Cuneo A, Castoldi. “Mycosis fungoides/Sezary’s syndrome”. Retrieved 2008-02-15.
- “Vorinostat shows anti-cancer activity in recurrent gliomas” (Press release). Mayo Clinic. June 3, 2007. Retrieved 2007-06-03.
- http://www.rtmagazine.com/reuters_article.asp?id=20091209clin013.html Dec 2009. URL dead Jan 2012
- “Zolinza, Idarubicin, Cytarabine Combination Yields High Response Rates In MDS Patients (ASH 2011)”.
- “Study of the Effect of Vorinostat on HIV RNA Expression in the Resting CD4+ T Cells of HIV+ Pts on Stable ART”. ClinicalTrials.gov. 2011-03-21.
- Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM (2009). “Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid.”. AIDS Res Hum Retroviruses 25 (2): 207–12. doi:10.1089/aid.2008.0191. PMC 2853863. PMID 19239360.
- Contreras X, Schweneker M, Chen CS, McCune JM, Deeks SG, Martin J et al. (2009). “Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells.”. J Biol Chem 284 (11): 6782–9.doi:10.1074/jbc.M807898200. PMC 2652322. PMID 19136668.
- Vorinostat bound to proteins in the PDB
- J. Med. Chem.,1995, vol. 38(8), pages 1411-1413.
- A new simple and high-yield synthesis of suberoylanilide hydroxamic acid and its inhibitory effect alone or in combination with retinoids on proliferation of human prostate cancer cells
J Med Chem 2005, 48(15): 5047 - A new facile and expeditious synthesis of N-hydroxy-N’-phenyloctanediamide, a potent inducer of terminal cytodifferentiation
Org Prep Proced Int 2001, 33(4): 391 - US patent 5369108, PDT PATENT
- WO2007/22408………
- WO 1993007148
- CN 102344392
| United States | 7456219 | APPROVAL 2006-11-14 | EXPIRY 2026-11-14 |
| United States | 6087367 | 1994-10-04 | 2011-10-04 |
| Canada | 2120619 | 2006-11-21 | 2012-10-05 |
| Patent | Patent Expiry | pat use code |
|---|---|---|
| 7399787 | Feb 9, 2025 | U-892 |
| 7456219 | Mar 11, 2027 | |
| 7652069 | Mar 4, 2023 | |
| 7732490 | Mar 4, 2023 | U-892 |
| 7851509 | Feb 21, 2024 | U-892 |
| 8067472 | Mar 4, 2023 | U-892 |
| 8093295 | May 16, 2026 | |
| 8101663 | Mar 4, 2023 | U-892 |
| RE38506 | Nov 29, 2013 |
U 892 =TREATMENT OF CUTANEOUS MANIFESTATIONS IN PATIENTS WTIH CUTANEOUS T-CELL LYMPHOMA (CTCL)
| Exclusivity Code | Exclusivity_Date |
|---|---|
| ODE | Oct 6, 2013 |
| WO2009098515A1 * | Feb 6, 2009 | Aug 13, 2009 | Generics Uk Ltd | Novel process for the preparation of vorinostat |
Marks, P.A., Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotech 25(1) 84-90 (2007). DOI: 10.1038/nbt1272
Takashi Kumagai, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. International Journal of Cancer. 2007 Aug 1;121(3):656-65. DOI: 10.1002/ijc.22558
Hrzenjak A, et al. Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol Cancer. 2010 Mar 4;9:49. DOI: 10.1186/1476-4598-9-49
………………………………………………………………………………………
|

| US7148257 | Aug 26, 2003 | Dec 12, 2006 | Merck Hdac Research, Llc | Methods of treating mesothelioma with suberoylanilide hydroxamic acid |
| US7375137 | Mar 28, 2006 | May 20, 2008 | Merck Hdac Research, Llc | Methods of treating cancer with HDAC inhibitors |
| US7399787 | Jul 9, 2003 | Jul 15, 2008 | Merck Hdac Research, Llc | Methods of treating cancer with HDAC inhibitors |
| US7456219 | Jun 19, 2003 | Nov 25, 2008 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7652069 | Oct 30, 2007 | Jan 26, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7732490 | Sep 11, 2007 | Jun 8, 2010 | Merck Hdac Research, Llc | Methods of treating cancer |
| US7847122 | Mar 18, 2008 | Dec 7, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7851509 | Mar 18, 2008 | Dec 14, 2010 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US7879865 | Nov 18, 2005 | Feb 1, 2011 | Sloan-Kettering Institute For Cancer Research | Treatment of cancer of the brain using histone deacetylase inhibitors |
| US7998957 | Feb 6, 2008 | Aug 16, 2011 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicylcoheptenes, their preparation and use |
| US8058268 | Jul 29, 2009 | Nov 15, 2011 | Lixte Biotechnology, Inc. | Neuroprotective agents for the prevention and treatment of neurodegenerative diseases |
| US8067472 | Apr 23, 2010 | Nov 29, 2011 | Merck Hdac Research, Llc | Methods of treating Hodgkin’s and non-Hodgkin’s lymphoma |
| US8088951 | Nov 30, 2007 | Jan 3, 2012 | Massachusetts Institute Of Technology | Epigenetic mechanisms re-establish access to long-term memory after neuronal loss |
| US8093295 | May 16, 2006 | Jan 10, 2012 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing the same |
| US8101663 | Dec 7, 2009 | Jan 24, 2012 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| US8143445 | Oct 1, 2008 | Mar 27, 2012 | Lixte Biotechnology, Inc. | HDAC inhibitors |
| US8227473 | Jul 17, 2009 | Jul 24, 2012 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8288440 * | Jan 13, 2010 | Oct 16, 2012 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US8329719 | Aug 1, 2011 | Dec 11, 2012 | Lixte Biotechnology, Inc. | Neuroprotective agents for the prevention and treatment of neurodegenerative diseases |
| US8426444 | Jun 30, 2011 | Apr 23, 2013 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8450372 * | Jan 13, 2010 | May 28, 2013 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US8455688 | Mar 21, 2012 | Jun 4, 2013 | Lixte Biotechnology, Inc. | HDAC inhibitors |
| US8541458 | Jun 11, 2012 | Sep 24, 2013 | Lixte Biotechnology, Inc. | Oxabicycloheptanes and oxabicycloheptenes, their preparation and use |
| US8563615 | Nov 1, 2010 | Oct 22, 2013 | Massachusetts Institute Of Technology | Use of CI-994 and dinaline for the treatment of memory/cognition and anxiety disorders |
| US20100112046 * | Jan 13, 2010 | May 6, 2010 | Jeannie Chow Wong | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20100113829 * | Jan 13, 2010 | May 6, 2010 | Cote Aaron S | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20100119596 * | Jan 13, 2010 | May 13, 2010 | Jeannie Chow Wong | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| US20110263712 * | Oct 14, 2009 | Oct 27, 2011 | Generics (Uk) Limited | Process for the preparation of vorinostat |
| US20110313044 * | Jun 16, 2011 | Dec 22, 2011 | Urquima S.A. | Polymorphs of Suberoylanilide Hydroxamic Acid |
| EP2079304A1 * | Sep 24, 2007 | Jul 22, 2009 | Merck & Co., Inc. | Amine base salts of saha and polymorphs thereof |
| EP2229941A1 * | May 16, 2006 | Sep 22, 2010 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| EP2292221A2 * | May 16, 2006 | Mar 9, 2011 | Merck Sharp & Dohme Corp. | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2006127319A2 * | May 16, 2006 | Nov 30, 2006 | Merck & Co Inc | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2006127321A2 * | May 16, 2006 | Nov 30, 2006 | Merck & Co Inc | Formulations of suberoylanilide hydroxamic acid and methods for producing same |
| WO2008039421A2 * | Sep 24, 2007 | Apr 3, 2008 | Arlene E Mckeown | Pharmaceutical compositions of hdac inhibitors and chelatable metal compounds, and metal-hdac inhibitor chelate complexes |
| WO2008042146A1 * | Sep 24, 2007 | Apr 10, 2008 | Arlene E Mckeown | Amine base salts of saha and polymorphs thereof |
| WO2008097654A1 * | Feb 8, 2008 | Aug 14, 2008 | Nancie M Archin | Methods of using saha for treating hiv infection |
| WO2009020565A1 * | Aug 1, 2008 | Feb 12, 2009 | Lixte Biotechnology Inc | Use of phosphatases to treat neuroblastomas and medulloblastomas |
| WO2010061220A2 * | Nov 25, 2009 | Jun 3, 2010 | Generics [Uk] Limited | Novel processes and pure polymorphs |
EXTRAS
MS-275 (Entinostat); CI-994 (Tacedinaline); BML-210; M344; MGCD0103 (Mocetinostat); PXD101 (Belinostat); LBH-589 (Panobinostat); Tubastatin A; Scriptaid; NSC 3852; NCH 51; HNHA; BML-281; CBHA; Salermide; Pimelic Diphenylamide; ITF2357 (Givinostat); PCI-24781; APHA Compound 8; Droxinostat; SB939.
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
//////////////149647-78-9, MK0683, VORINOSTAT, Zolinza
ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1
Rapamycin (Sirolimus) For the prophylaxis of organ rejection in patients receiving renal transplants.

Rapamycin (Sirolimus)
(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25, 26,27,32,33,34,34a-Hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone
Wyeth Pharmaceuticals (Originator)
M.Wt:914.18
Formula:C51H79NO13
53123-88-9 cas no
Antifungal and immunosuppressant. Specific inhibitor of mTOR (mammalian target of Rapamycin). Complexes with FKBP-12 and binds mTOR inhibiting its activity. Inhibits interleukin-2-induced phosphorylation and activation of p70 S6 kinase. Induces autophagy in yeast and mammalian cell lines.
Rapamycin is a triene macrolide antibiotic, which demonstrates anti-fungal, anti-inflammatory, anti-tumor and immunosuppressive properties. Rapamycin has been shown to block T-cell activation and proliferation, as well as, the activation of p70 S6 kinase and exhibits strong binding to FK-506 binding proteins. Rapamycin also inhibits the activity of the protein, mTOR, (mammalian target of rapamycin) which functions in a signaling pathway to promote tumor growth. Rapamycin binds to a receptor protein (FKBP12) and the rapamycin/FKB12 complex then binds to mTOR and prevents interaction of mTOR with target proteins in this signaling pathway. Rapamycin name is derived from the native word for Easter Island, Rapi Nui.
- (-)-Rapamycin
- Antibiotic AY 22989
- AY 22989
- AY-22989
- CCRIS 9024
- HSDB 7284
- NSC 226080
- Rapammune
- Rapamune
- Rapamycin
- SILA 9268A
- Sirolimus
- UNII-W36ZG6FT64
- WY-090217
- A 8167
A macrolide compound obtained from Streptomyces hygroscopicus that acts by selectively blocking the transcriptional activation of cytokines thereby inhibiting cytokine production. It is bioactive only when bound to IMMUNOPHILINS. Sirolimus is a potent immunosuppressant and possesses both antifungal and antineoplastic properties.
Sirolimus (INN/USAN), also known as rapamycin, is an immunosuppressant drug used to prevent rejection in organ transplantation; it is especially useful in kidney transplants. It prevents activation of T cells and B cells by inhibiting their response to interleukin-2 (IL-2). Sirolimus is also used as a coronary stent coating. Sirolimus works, in part, by eliminating old and abnormal white blood cells.[citation needed] Sirolimus is effective in mice with autoimmunity and in children with a rare condition called autoimmune lymphoproliferative syndrome (ALPS).

A macrolide, sirolimus was discovered by Brazilian researchers as a product of the bacterium Streptomyces hygroscopicus in a soil sample fromEaster Island[1] — an island also known as Rapa Nui.[2] It was approved by the FDA in September 1999 and is marketed under the trade nameRapamune by Pfizer (formerly by Wyeth).
Sirolimus was originally developed as an antifungal agent. However, this use was abandoned when it was discovered to have potent immunosuppressive and antiproliferative properties. It has since been shown to prolong the life of mice and might also be useful in the treatment of certain cancers.
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response tointerleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12(FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits themammalian target of rapamycin (mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.
 rapamycin
rapamycin
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response to interleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12 (FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits the mammalian target of rapamycin(mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.

Rapamycin and its preparation are described in US Patent No. 3,929,992, issued December 30, 1975. Alternatively, rapamycin may be purchased commercially [Rapamune®, Wyeth].
Rapamycin (Sirolimus) is a 31-member natural macrocyclic lactone [C51H79N1O13; MWt=914.2] produced by Streptomyces hygroscopicus and found in the 1970s (U.S. Pat. No. 3,929,992; 3,993,749). Rapamycin (structure shown below) was approved by the Food and Drug Administration (FDA) for the prophylaxis of renal transplant rejection in 1999.

Rapamycin resembles tacrolimus (binds to the same intracellular binding protein or immunophilin known as FKBP-12) but differs in its mechanism of action. Whereas tacrolimus and cyclosporine inhibit T-cell activation by blocking lymphokine (e.g., IL2) gene transcription, sirolimus inhibits T-cell activation and T lymphocyte proliferation by binding to mammalian target of rapamycin (mTOR). Rapamycin can act in synergy with cyclosporine or tacrolimus in suppressing the immune system.
Rapamycin is also useful in preventing or treating systemic lupus erythematosus [U.S. Pat. No. 5,078,999], pulmonary inflammation [U.S. Pat. No. 5,080,899], insulin dependent diabetes mellitus [U.S. Pat. No. 5,321,009], skin disorders, such as psoriasis [U.S. Pat. No. 5,286,730], bowel disorders [U.S. Pat. No. 5,286,731], smooth muscle cell proliferation and intimal thickening following vascular injury [U.S. Pat. Nos. 5,288,711 and 5,516,781], adult T-cell leukemia/lymphoma [European Patent Application 525,960 A1], ocular inflammation [U.S. Pat. No. 5,387,589], malignant carcinomas [U.S. Pat. No. 5,206,018], cardiac inflammatory disease [U.S. Pat. No. 5,496,832], anemia [U.S. Pat. No. 5,561,138] and increase neurite outgrowth [Parker, E. M. et al, Neuropharmacology 39, 1913-1919, 2000].
Although rapamycin can be used to treat various disease conditions, the utility of the compound as a pharmaceutical drug has been limited by its very low and variable bioavailability and its high immunosuppressive potency and potential high toxicity. Also, rapamycin is only very slightly soluble in water. To overcome these problems, prodrugs and analogues of the compound have been synthesized. Water soluble prodrugs prepared by derivatizing rapamycin positions 31 and 42 (formerly positions 28 and 40) of the rapamycin structure to form glycinate, propionate, and pyrrolidino butyrate prodrugs have been described (U.S. Pat. No. 4,650,803). Some of the analogues of rapamycin described in the art include monoacyl and diacyl analogues (U.S. Pat. No. 4,316,885), acetal analogues (U.S. Pat. No. 5,151,413), silyl ethers (U.S. Pat. No. 5,120,842), hydroxyesters (U.S. Pat. No. 5,362,718), as well as alkyl, aryl, alkenyl, and alkynyl analogues (U.S. Pat. Nos. 5,665,772; 5,258,389; 6,384,046; WO 97/35575).
………………………………………..
Synthesis
ref are independent of body…see below for this clip
Several total synthese of rapamycin have been reported3,4as well as many fragments and part-syntheses. Rapamycin is a complicated molecule comprising a 31-membered ring including a pipecolinyl group and pyranose ring, a conjugated triene system and a tri-carbonyl region. It also has 15 chiral centres, meaning the number of possible stereoisomers is enormous. The synthesis of rapamycin therefore presents a huge challenge to synthetic chemists.
In the following synthesis, published in three separate papers5,6,7two fragments of C10-C21 and C22-C42 are prepared separately, before being combined to give the total synthesis of rapamycin. Only the main outline of the synthesis will be shown as it is too long and complicated to show in great detail. For the full experimental details of the synthesis see the literature (ref. nos. given above).

In the retro-synthesis shown the molecule is disconnected at the ester group next to carbon 1 and the C21-C22 double bond of the triene to give the synthetic precursors 2 and 3. Further disconnections of 3 will be shown later. First the C10-C21 fragment is synthesised.
Synthesis of C10-C21 fragment
The synthesis uses (R)-methyl 3-hydroxy-2-methylpropionate (8) as a starting material.

The starting material 8 is converted to an alcohol by a four-step process; protection of the alcohol as aTHP ether followed by reduction, ether formation and deprotection steps. Substitution of the hydroxyl group in the product for a bromine leads to the formation of the bromide 9. Reaction of 9 with methyl acetoacetate gave ester 10.

Catalytic reduction of 10 using the conditions of Noyori produced ester 11, which was then converted to its Weinreb amide 12. Overall, compound 12 was produced in 54% yield from an inexpensive starting material. Vinyl bromide 13 was metalated with t-BuLi and the resulting vinyllithium was combined with 12 and the PMB-protecting group was removed to give 14. The remaining carbonyl group in 14 was selectively reduced to a hyrdoxy group. In order to differentiate the 1,3-diol a lactol was formed, where one hydroxy group ended up in the ring. To acheive this an oxidation was performed using RuCl2(PPh3)3 resulting in formation of a lactol. The two remaining alcohol groups could then be methylated using MeI forming 15.

The lactol ring opening was achieved using TiCl4 and thiol HS(CH2)2SH to form a dithiolane. The freed alcohol was then protected as its TBS ether and the same protecting group selectively removed from the primary alcohol to form 16. To avoid removing the dithiolane group at a later stage in the synthesis the thio-acetal was converted to the dimethyl acetal 17 using PhI(OCOCF3)2 and methanol.

The next stage in the synthesis was to extend 17 for the building of the triene region. The terminal alcohol was oxidised to its aldehyde using BaMnO4 , then a Wittig reaction was carried out using Ph3P=CHCO2Et and CH2Cl2 to form the second double bond. Reduction of the ester group to an alcohol was carried out using DIBAL-H, then treatment with PPh3 and exposure to the air gave rapamycin fragment 2.
Synthesis of C22-C42 fragment
Here the retro-synthesis of 3 is shown, giving the three synthetic precursors 5, 6 and 7

It was thought 4 could be obtained by alkylative coupling of a vinyllithium species generated from 7 to the Weinreb amide 6. The nucleophilic opening of epoxide 5 by the lithiated sulfone from phenyl sulfone 4 would then produce the desired fragment.
The ester 18 was used as a starting material to make fragment 6.

A Wittig reaction followed by reduction and protection steps produced 19. This was hydrogenated using a rhodium catalyst to give syn-dimethyl product 20. The minor anti diastereomer was successfully separated off. 20 was oxidised then underwent an aldol condensation to give adduct 21.

Transamination of 21 and protection of the alcohol with PMB resulted in amide 6, corresponding to the C22-C28 segment of rapamycin.
The vinyl bromide 7 was prepared using ester 22 as a starting material.

Reduction of 22 followed by dibromoolefination resulted in product 23. Acetylene 24 was prepared using n-BuLi, THF and MeI, then sulfenylation with Ph2S2 and bromination gave fragment 7.

Iodination and alkylation of starting material 25 with the lithiated allylic sulfide shown followed by a number of further steps resulted in its conversion to fragment 5.

Fragments 7 was first converted to its vinyllithium using t-BuLi then combined with 6 forming an enone in 78% yield. Stereoselective reduction of the carbonyl group using Zn(BH4)2 gave an alcohol which was protected with DEIPS giving 28. The phenyl sulfide was oxidised to a sulfone using m-CPBA in excess pyridine.

Lithiation and addition of the epoxide 5 resulted in the hydroxy sulfone in a 4:1 ratio of two diastereomers which were separated by HPLC. Metalation using n-BuLi followed by oxidation formed the total C22-C42 fragment.
Total synthesis of rapamycin through the combination of C10-C21 and C22-C42 fragments.
Fragment 3 (C22-C42) was treated with (S)-Boc-pipecolinal, followed by a Swern oxidation resulted in the aldehyde 29.

Condensation with the lithium salt of phosphine oxide 2 (C10-C21) produced the triene shown below.

The triene was hydrolysed with pyridinium p-toluenesulfonic acid and an aldol reaction was performed. Treatment with triethylsilyl triflate produced an amino acid which was subjected to Mukaiyama macrocyclization conditions to form the 31-membered ring. Finally, deprotection steps were performed to give synthetic rapamyin (1). This was judged to be identical to natural rapamycin by comparison of physical properties, 1H-NMR, 13C-NMR, IR and UV spectral data.
3. K. C. Nicolaou, T. K. Chakraborty, A. D. Piscopio, N. Minowa, P. Bertinato; J. Am. Chem. Soc.; 115; 1993; 4419
4. C. M. Hayward, D. Yohannes, S. J. Danishefsky; J. Am. Chem. Soc.; 115; 1993; 9345
5. S. D. Meyer, T. Miwa, M. Nakatsuka, S. L. Schreiber; J. Org. Chem.; 57; 1992; 5058-5060
6. D. Romo, D. D. Johnson, L. Plamondon, T. Miwa, S. L. Schreiber; J. Org. Chem.; 57; 1992; 5060-5063
7. S. D. Meyer, D. Romo, D. D. Johnson, S. L. Schreiber; J. Am. Chem. Soc.; 115; 1993; 7906-7907
………………………………………….
Synthesis
PREPARATION
CUT PASTE FROM TEXT
In one embodiment of this invention rapamycin is prepared in the followingmanner: 4
A suitable fermenter is charged with production meis reached in the fermentation mixture after 2-8 days,
usually after about 5 days, as determined by the cup plate method and Candida albicans as the test organism. The mycelium is harvested by filtration with diatomaceous earth. Rapamycin is then extracted from the mycelium with a water-miscible solvent, for example a lower alkanol, preferably methanol or ethanol. The latter extract is then concentrated, preferably under reduced pressure, and the resulting aqueous phase is extracted with a water-immiscible solvent. A preferred water-immiscible solvent for this purpose is methylene dichloride although chloroform, carbon tetrachloride, benzene, n-butanol and the like may also be used. The latter extract is concentrated, preferably under reduced pressure, to afford the crude product as an oil.
The product may be purified further by a variety of methods. Among the preferred methods of purification is to dissolve the crude product in a substantially nonpolar, first solvent, for example petroleum ether or hexane, and to treat the resulting solution with a suit able absorbent, for example charcoal or silica gel, so that the antibiotic becomes absorbed on the absorbant. The absorbant is then separated and washed or eluted with a second solvent more polar than the first solvent, for example ethyl acetate, methylene dichloride, or a mixture of methylene dichloride and ether (preferred). Thereafter, concentration of the wash solution or eluate affords substantially pure rapamycin. Further purification is obtained by partial precipitation with a nonpolar solvent, for example, petroleum ether, hexane, pentane and the like, from a solution of the rapamycin in a more polar solvent, for example, ether, ethyl acetate, benzene and the like. Still-further purification is obtained by column chromatography, preferably employing silica gel, and by crystallization of the rapamycin from ether.
In another preferred embodiment of this invention a first stage inoculum of S treptomyces hygroscopicus NRRL 5491 is prepared in small batches in a medium containing soybean flour, glucose, ammonium sulfate, and calcium carbonate incubated at about 25C at pH 7.l-7.3 for 24 hrs. with agitation, preferably on a gyrotary shaker. The growth thus obtained is used to inoculate a number of somewhat larger batches of the same medium as described above which are incubated at about 25C and pH 7.1-7.3 for 18 hrs. with agitation, preferably on a reciprocating’shaker, to obtain a sec- “ond stagc inoculum which is used to inoculate the production stage fermenters.
6 5.86′.2.-The fermenters are inoculated with the second stage inoculum described above and incubated at about 25C with’ agitationand aeration while controlling and ‘mai’ntaining the mixture at approximately pH 6.0 by
addition offa base, for example, sodium hydroxide, potassium hydroxide or preferably ammonium hydroxide, as required from time to time. Addition of a source -of assimilable carbon, preferably glucose, is started when theconcentrationof the latter in the broth has dropped to about 0.5% wt/vol, normally about 48 hrs after. the start of fermentation, and is maintained until the end ofthe particular run. In this manner a fermentation broth containing about 60 ug/ml of rapamycin as determined by the assay method described above is obtained in 45 days, when fermentation is stopped.
‘ Filtration of the’mycelium, mixing the latter with a watef-miscible ‘lower’ alkanol, preferably methanol, followed by extraction with a halogenated aliphatic hydrocarbon, preferably trichloroethane, and evaporation of the solvents yields a first oily residue. This first oily residue is dissolved in a lower aliphatic ketone, preferably acetone, filtered from insoluble impurities, the filtrate evaporated to yield a second oily residue which is extractedjwith a water-miscible lower alkanol,
preferably methanol, and the latter extract is evaporated to yield crude rapamycin as a third oily residue. This third oily residue is dissolved in a mixture of a lower aliphatic ketone and a lower aliphatic hydrocarbon, preferably acetone-hexane, an absorbent such as charcoal or preferably silica gel is added to adsorb the rapamycin, the latter is eluted from the adsorbate with a similar but more polar solvent mixture, for example a mixture as above but containing a higher proportion of the aliphatic ketone, the eluates are evaporated and the residue is crystallized from diethyl ether, to yield pure crystalline rapamycin. In this manner a total of 45-5 8% of the rapamycin initially present in the fermentation mixture is recovered as pure crystalline rapamycin.
CHARACTERIZATION solvent systems; for example, ether-hexane 40:60 (Rf 0.42), ‘isopropyl alcoholvbenzene 15:85 (Rf= 0.5) and ethanol-benzene 20:80 (Rf f 0.43);
d. rapamycin obtained from four successive fermentation batchesgave the following values on repeated The production stage fermenters are equipped with 7 devices for controlling and maintaining pH at a predetermined level and for continuous metered addition of elemental analyses:
AVER- e. rapamycin exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum ethanol):
f. the infrared absorption spectrum of rapamycin in chloroform is reproduced in FIG. 1 and shows characteristic absorption bands at 3560, 3430, 1730, 1705 and 1630-1610 cm;
Further infrared absorption bands are characterized by the following data given in reciprocal centimeters with (s) denoting a strong, (m) denoting a medium, and ![]() denoting a weak intensity band. This classification is arbitrarily selected in such a manner that a band is denoted as strong (s) if its peak absorption is more than two-thirds of the background in the same region; medium (m) if its peak is between one-third and twothirds of the background in the same region; and weak
denoting a weak intensity band. This classification is arbitrarily selected in such a manner that a band is denoted as strong (s) if its peak absorption is more than two-thirds of the background in the same region; medium (m) if its peak is between one-third and twothirds of the background in the same region; and weak ![]() if its peak is less than one-third of the background in the same region.
if its peak is less than one-third of the background in the same region.
2990 cm (m) 1158 cm” (m) 2955 cm (s) 1129 cm (s) 2919 cm (s) 1080 cm (s) 2858 cm (s) 1060 cm (s) 2815 cm (m) 1040 cm (m) 1440 cm (s) 1020 crn’ (m) 1365 cm (m) 978 cm” (s) 1316 cm (in) 905 cm (m) 1272 cm (m) 888 cm” ![]() 1178 cm (s) 866 cm-
1178 cm (s) 866 cm- ![]() g. the nuclear magnetic resonance spectrum of rapamycinin deuterochloroform is reproduced in FIG. 2; SEE PATENT
g. the nuclear magnetic resonance spectrum of rapamycinin deuterochloroform is reproduced in FIG. 2; SEE PATENT
CLAIMS
l. Rapamycin, an antibiotic which a. is a colourless, crystalline compound with a melting point of 183 to l8SC, after recrystallization from ether;
b. is soluble in ether, chloroform, acetone, methanol and dimethylformamide, very sparingly soluble in hexane and petroleum ether and substantially insoluble in water;
c. shows a uniform spot on thin layer plates of silica gel”,
d. has a characteristic elemental analysis of about C,
e. exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum (95% ff has ‘a characteristic infrared absorption spectrum shown in accompanying FIG. 1; SEE PATENT
……………………………………………..
Rapamycin synthetic studies. 1. Construction of the C(27)-C(42) subunit. Tetrahedron Lett 1994, 35, 28, 4907

A partial synthesis of rapamycin has been reported: The condensation of sulfone (I) with epoxide (II) by means of butyllithium followed by desulfonation with Na/Hg gives the partially protected diol (III), which is treated with methanesulfonyl chloride and NaH to afford the epoxide (IV). Ring opening of epoxide (IV) with LiI and BF3.Et2O followed by protection of the resulting alcohol with PMBOC(NH)CCl3 yields the primary iodo compound (V). The condensation of (V) with the fully protected dihydroxyaldehyde (VI) (see later) by means of butyllithium in THF/HMPT gives the fully protected trihydroxyketone (VII), which is hydrolyzed with camphorsulfonic acid (CSA) to the corresponding gemdiol and reprotected with pivaloyl chloride (the primary alcohol) and tert-butyldimethylsilyl trifluoromethanesulfonate (the secondary alcohol), yielding a new fully protected trihydroxyketone (VIII). Elimination of the pivaloyl group with DIBAL and the dithiane group with MeI/CaCO3 affords the hydroxyketone (IX), which is finally oxidized with oxalyl chloride to the ketoaldehyde (X), the C(27)-C(42) fragment [the C(12)-C(15) fragment with the C(12)-substituent based on the IUPAC nomenclature recommendations]. The fully protected dihydroxyaldehyde (VI) is obtained as follows: The reaction of methyl 3-hydroxy-2(R)-methylpropionate (XI) with BPSCl followed by reduction with LiBH4 to the corresponding alcohol and oxidation with oxalyl chloride gives the aldehyde (XII), which is protected with propane-1,3-dithiol and BF3.Et2O to afford the dithiane compound (XIII). Elimination of the silyl group with TBAF followed by esterification with tosyl chloride, reaction with NaI and, finally, with sodium phenylsulfinate gives the sulfone (XIV), which is condensed with the partially protected dihydroxyaldehyde (XV), oxidized with oxalyl chloride and desulfonated with Al/Hg to afford the dithianyl ketone (XVI). The reaction of (XVI) with lithium hexamethyldisilylazane gives the corresponding enolate, which is treated with dimethyllithium cuprate to yield the fully protected unsaturated dihydroxyaldehyde (VI).
……………………………………………
……………………………
The Ley Synthesis of RapamycinRapamycin (3) is used clinically as an immunosuppressive agent. The synthesis of 3 (Angew. Chem. Int. Ed. 2007, 46, 591. DOI: 10.1002/anie.200604053) by Steven V. Ley of the University of Cambridge was based on the assembly and subsequent coupling of the iododiene 1 and the stannyl alkene 2.
The lactone of 1 was prepared by Fe-mediated cyclocarbonylation of the alkenyl epoxide 5, following the protocol developed in the Ley group.
The cyclohexane of 2 was constructed by SnCl4-mediated cyclization of the allyl stannane 9, again employing a procedure developed in the Ley group. Hydroboration delivered the aldehyde 11, which was crotylated with 12, following the H. C. Brown method. The alcohol so produced (not illustrated) was used to direct the diastereoselectivity of epoxidation, then removed, to give 13. Coupling with 14 then led to 2.
Combination of 1 with 2 led to 15, which was condensed with catechol to give the macrocycle 16. Exposure of 16 to base effected Dieckmann cyclization, to deliver the ring-contracted macrolactone 17, which was carried on to (-)-rapamycin (3).
|




……………………………….
Total Synthesis of Rapamycin
Angewandte Chemie International Edition
Volume 46, Issue 4, pages 591–597, January 15, 2007

PREVIEW THIS ARTICLE WITH READCUBE
![]()
……………………..

Ley, Maddess, Tackett, Watanabe, Brennan, Spilling, Scott and Osborn. ACIEE, 2006, EarlyView. DOI:10.1002/anie.200604053.
It’s been in the works for quite a while, but Steve Ley’s synthesis of Rapamycin has just been published. This complex beast has a multitude of biological activities, including an interesting immunosuppressive profile, resulting in clinical usage following organ transplantation. So, unsurprisingly, it’s been the target of many projects, with complete total syntheses published by Smith, Danishefsky, Schreiber and KCN.
So what makes this one different? Well, it does have one of the most interesting macrocyclisations I’ve seen since Jamison’s paper, and a very nice demonstration of the BDA-aldol methodology. The overall strategy is also impressive, so on with the retro:

First stop is the BDA-aldol; this type of chemistry is interesting, because the protecting group for the diol is also the stereo-directing group. The stereochemistry for this comes from a glycolic acid, and has been usedin this manner by the group before. The result is as impressive as ever, with a high yield, and presumably a very high d.r. (no mention of actual numbers).

The rest of the fragment synthesis was completed in a succinct and competent manner, but using relatively well known chemistry. However, I was especially impressed with the macrocyclisation I mentioned:

Tethering the free ends of the linear precursor with a simple etherification/esterification onto catechol gave then a macrocycle holding the desired reaction centres together. Treatment of this with base then induces a Dieckmann-condensation type cyclisation to deliver the desired macrocycle. Of course, at this stage, only a few more steps were required to complete the molecule, and end an era of the Wiffen Lab.
………………………………
Drugs Fut 1999, 24(1): 22
DOI: 10.1358/dof.1999.024.01.474036

In CDCl3 rapamycin exists as a mixture of conformers in a 3:1 ratio, which complicates the NMR spectrum. In the table below the chemical shifts of the carbons and hydrogens of the major isomer only are given.
| Carbon No. | Carbon Type | Major carbon | Major proton | Carbon No. | Carbon Type | Major carbon | Major proton |
|
1
|
C=O | 169.2 |
–
|
28
|
CH-OH | 77.3 | 4.17 |
|
2
|
CH | 51.3 | 5.29 |
29
|
C=C | 136.1 |
–
|
|
3
|
CH2 | 27.0 | 2.34, 1.76 |
30
|
CH=C | 126.8 | 5.42 |
|
4
|
CH2 | 20.6 | 1.78, 1.47 |
31
|
CH | 46.6 | 3.33 |
|
5
|
CH2 | 25.3 | 1.75, 1.48 |
32
|
C=O | 208.2 |
–
|
|
6
|
CH2 | 44.2 | 3.59, 3.44 |
33
|
CH2 | 40.7 | 2.74, 2.60 |
|
8
|
C=O | 166.8 |
–
|
34
|
CH-OCO | 75.7 | 5.17 |
|
9
|
C=O | 192.5 |
–
|
35
|
CH | 33.1 | 1.98 |
|
10
|
O-C-OH | 98.5 |
–
|
36
|
CH2 | 38.4 | 1.22, 1.12 |
|
11
|
CH | 33.7 | 1.98 |
37
|
CH | 33.2 | 1.39 |
|
12
|
CH2 | 27.3 | 1.60, 1.60 |
38
|
CH2 | 34.2 | 2.10, 0.68 |
|
13
|
CH2 | 31.3 | 1.62, 1.33 |
39
|
CH-OCH3 | 84.4 | 2.93 |
|
14
|
67.2 | 3.86 |
40
|
CH-OH | 73.9 | 3.37 | |
|
15
|
CH2 | 38.8 | 1.85, 1.52 |
41
|
CH2 | 31.3 | 1.99, 1.33 |
|
16
|
CH-OCH3 | 84.4 | 3.67 |
42
|
CH2 | 31.7 | 1.70, 1.00 |
|
17
|
C=C | 135.5 |
–
|
43
|
11-CH3 | 16.2 | 0.95 |
|
18
|
CH=C | 129.6 | 5.97 |
44
|
17-CH3 | 10.2 | 1.65 |
|
19
|
CH=C | 126.4 | 6.39 |
45
|
23-CH3 | 21.5 | 1.05 |
|
20
|
CH=C | 133.6 | 6.32 |
46
|
25-CH3 | 13.8 | 1.00 |
|
21
|
CH=C | 130.1 | 6.15 |
47
|
29-CH3 | 13.0 | 1.74 |
|
22
|
CH=C | 140.2 | 5.54 |
48
|
31-CH3 | 16.0 | 1.11 |
|
23
|
CH | 35.2 | 2.32 |
49
|
35-CH3 | 15.9 | 0.92 |
|
24
|
CH2 | 40.2 | 1.50, 1.20 |
50
|
16-OCH3 | 55.8 | 3.13 |
|
25
|
CH | 41.4 | 2.74 |
51
|
27-OCH3 | 59.5 | 3.34 |
|
26
|
C=O | 215.6 |
–
|
52
|
39-OCH3 | 56.5 | 3.41 |
|
27
|
CH-OCH3 | 84.9 | 3.71 |
REFERENCES
- Vézina C, Kudelski A, Sehgal SN (October 1975). “Rapamycin (AY-22,989), a new antifungal antibiotic”. J. Antibiot. 28 (10): 721–6. doi:10.7164/antibiotics.28.721. PMID 1102508.
- Pritchard DI (2005). “Sourcing a chemical succession for cyclosporin from parasites and human pathogens”. Drug Discovery Today 10 (10): 688–691. doi:10.1016/S1359-6446(05)03395-7. PMID 15896681.
Wu X, Wang L, Han Y, Regan N, Li PK, Villalona MA, Hu X, Briesewitz R, Pei D.
ACS Comb Sci. 2011 Sep 12;13(5):486-95. doi: 10.1021/co200057n. Epub 2011 Jul 28.
Gibbons JJ, Abraham RT, Yu K.
Semin Oncol. 2009 Dec;36 Suppl 3:S3-S17. doi: 10.1053/j.seminoncol.2009.10.011. Review.
Ayral-Kaloustian S, Gu J, Lucas J, Cinque M, Gaydos C, Zask A, Chaudhary I, Wang J, Di L, Young M, Ruppen M, Mansour TS, Gibbons JJ, Yu K.
J Med Chem. 2010 Jan 14;53(1):452-9. doi: 10.1021/jm901427g.
6. Fluorescent probes to characterise FK506-binding proteins.
Kozany C, März A, Kress C, Hausch F.
Chembiochem. 2009 May 25;10(8):1402-10. doi: 10.1002/cbic.200800806.
7. Recent advances in the chemistry, biosynthesis and pharmacology of rapamycin analogs.
Graziani EI.
Nat Prod Rep. 2009 May;26(5):602-9. doi: 10.1039/b804602f. Epub 2009 Mar 5. Review.
8 Total synthesis of rapamycin.
Ley SV, Tackett MN, Maddess ML, Anderson JC, Brennan PE, Cappi MW, Heer JP, Helgen C, Kori M, Kouklovsky C, Marsden SP, Norman J, Osborn DP, Palomero MA, Pavey JB, Pinel C, Robinson LA, Schnaubelt J, Scott JS, Spilling CD, Watanabe H, Wesson KE, Willis MC.
Chemistry. 2009;15(12):2874-914. doi: 10.1002/chem.200801656.
Evans AC, Longbottom DA, Matsuoka M, Davies JE, Turner R, Franckevicius V, Ley SV.
Org Biomol Chem. 2009 Feb 21;7(4):747-60. doi: 10.1039/b813494d. Epub 2009 Jan 6.
Maddess ML, Tackett MN, Ley SV.
Prog Drug Res. 2008;66:13, 15-186. Review.
Zhang J, Rodila R, Watson P, Ji Q, El-Shourbagy TA.
Biomed Chromatogr. 2007 Oct;21(10):1036-44.
Sormani R, Yao L, Menand B, Ennar N, Lecampion C, Meyer C, Robaglia C.
BMC Plant Biol. 2007 Jun 1;7:26.
13 Total synthesis of rapamycin.
Maddess ML, Tackett MN, Watanabe H, Brennan PE, Spilling CD, Scott JS, Osborn DP, Ley SV.
Angew Chem Int Ed Engl. 2007;46(4):591-7. No abstract available.
14 Drug evaluation: AP-23573–an mTOR inhibitor for the treatment of cancer.
Elit L.
IDrugs. 2006 Sep;9(9):636-44.
15 lipase-catalyzed regioselective esterification of rapamycin: synthesis of temsirolimus (CCI-779).
Gu J, Ruppen ME, Cai P.
Org Lett. 2005 Sep 1;7(18):3945-8.
Elit L.
Curr Opin Investig Drugs. 2002 Aug;3(8):1249-53. Review.
Dumont FJ.
Curr Opin Investig Drugs. 2001 Sep;2(9):1220-34. Review.
18 Kuo et al (1992) Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358 70. PMID:1614535.
19 Huang et al (2003) Rapamycins: mechanism of action and cellular resistance. Cancer Biol.Ther. 2 221. PMID:12878853.
20 Kobayashi et al (2007) Rapamycin, a specific inhibitor of the mammalian target of rapamycin, suppresses lymphangiogenesis and lymphatic metastasis. Cancer Sci. 98 726. PMID: 17425689.
21 Fleming et al (2011) Chemical modulators of autophagy as biological probes and potential therapeutics. 7 9. PMID:21164513.
22 J Am Chem Soc1993,115,(10):4419
23 Tetrahedron Lett1994,35,(28):4911
24 Chemistry (Weinheim)1995,1,(5):318
24
 SIROLIMUS
SIROLIMUS
FEMALE FERTILITY
PATENTS
| Canada | 2293793 | APPROVED2006-07-11 | EXP 2018-06-11 |
| Canada | 2103571 | 2003-04-29 | 2012-02-21 |
| United States | 5989591 | 1998-09-11 | 2018-09-11 |
| United States | 5212155 | 1993-05-18 | 2010-05-18 |
| WO1998054308A2 * | May 28, 1998 | Dec 3, 1998 | Biotica Tech Ltd | Polyketides and their synthesis and use |
| EP0589703A1 * | Sep 23, 1993 | Mar 30, 1994 | American Home Products Corporation | Proline derivative of rapamycin, production and application thereof |
| US20010039338 * | Jun 7, 2001 | Nov 8, 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO2007067560A2 * | Dec 6, 2006 | Jun 14, 2007 | Clifford William Coughlin | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| WO2012131019A1 | Mar 30, 2012 | Oct 4, 2012 | Sandoz Ag | Regioselective acylation of rapamycin at the c-42 position |
| US7622578 | Dec 6, 2006 | Nov 24, 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US3929992 | Apr 12, 1974 | Dec 30, 1975 | Ayerst Mckenna & Harrison | Rapamycin and process of preparation |
| US5646160 | May 26, 1995 | Jul 8, 1997 | American Home Products Corporation | Method of treating hyperproliferative vascular disease with rapamycin and mycophenolic acid |
| US5665772 | Sep 24, 1993 | Sep 9, 1997 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5728710 | Jul 16, 1993 | Mar 17, 1998 | Smithkline Beecham Corporation | Rapamycin derivatives |
| US5957975 | Dec 15, 1997 | Sep 28, 1999 | The Centre National De La Recherche Scientifique | Stent having a programmed pattern of in vivo degradation |
| US5985890 | Jun 5, 1996 | Nov 16, 1999 | Novartis Ag | Rapamycin derivatives |
| US6001998 | Oct 13, 1995 | Dec 14, 1999 | Pfizer Inc | Macrocyclic lactone compounds and their production process |
| US6015815 | Sep 24, 1998 | Jan 18, 2000 | Abbott Laboratories | Tetrazole-containing rapamycin analogs with shortened half-lives |
| US6187568 | Aug 20, 1999 | Feb 13, 2001 | Pfizer Inc | Macrocyclic lactone compounds and their production process |
| US6273913 | Apr 16, 1998 | Aug 14, 2001 | Cordis Corporation | Modified stent useful for delivery of drugs along stent strut |
| US6585764 | Jun 4, 2001 | Jul 1, 2003 | Cordis Corporation | Stent with therapeutically active dosage of rapamycin coated thereon |
| US6641611 | Nov 26, 2001 | Nov 4, 2003 | Swaminathan Jayaraman | Therapeutic coating for an intravascular implant |
| US6805703 | Sep 18, 2001 | Oct 19, 2004 | Scimed Life Systems, Inc. | Protective membrane for reconfiguring a workpiece |
| US7025734 | Sep 28, 2001 | Apr 11, 2006 | Advanced Cardiovascular Systmes, Inc. | Guidewire with chemical sensing capabilities |
| US7056942 | Jan 16, 2004 | Jun 6, 2006 | Teva Pharmaceutical Industries Ltd. | Carvedilol |
| US7820812 * | Jul 23, 2007 | Oct 26, 2010 | Abbott Laboratories | Methods of manufacturing crystalline forms of rapamycin analogs |
| US20010027340 | Jun 4, 2001 | Oct 4, 2001 | Carol Wright | Stent with therapeutically active dosage of rapamycin coated thereon |
| US20010029351 | May 7, 2001 | Oct 11, 2001 | Robert Falotico | Drug combinations and delivery devices for the prevention and treatment of vascular disease |
| US20020005206 | May 7, 2001 | Jan 17, 2002 | Robert Falotico | Antiproliferative drug and delivery device |
| US20020007213 | May 7, 2001 | Jan 17, 2002 | Robert Falotico | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| US20020082680 | Sep 7, 2001 | Jun 27, 2002 | Shanley John F. | Expandable medical device for delivery of beneficial agent |
| US20020123505 | Sep 10, 2001 | Sep 5, 2002 | Mollison Karl W. | Medical devices containing rapamycin analogs |
| US20030129215 | Sep 6, 2002 | Jul 10, 2003 | T-Ram, Inc. | Medical devices containing rapamycin analogs |
| US20040072857 | Jul 2, 2003 | Apr 15, 2004 | Jacob Waugh | Polymerized and modified rapamycins and their use in coating medical prostheses |
| US20050033417 | Jul 1, 2004 | Feb 10, 2005 | John Borges | Coating for controlled release of a therapeutic agent |
| US20050101624 | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20050152842 | Dec 22, 2004 | Jul 14, 2005 | Chun Li | Poly (L-glutamic acid) paramagnetic material complex and use as a biodegradable MRI contrast agent |
| US20050175660 | Oct 29, 2004 | Aug 11, 2005 | Mollison Karl W. | Medical devices containing rapamycin analogs |
| US20050208095 | Nov 22, 2004 | Sep 22, 2005 | Angiotech International Ag | Polymer compositions and methods for their use |
| US20050209244 | Feb 27, 2003 | Sep 22, 2005 | Prescott Margaret F | N{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]-2-methylphenyl}-4-(3-pyridyl)-2-pyrimidine-amine coated stents |
| US20050239178 | Apr 25, 2005 | Oct 27, 2005 | Wyeth | Labeling of rapamycin using rapamycin-specific methylases |
| US20060094744 | Sep 28, 2005 | May 4, 2006 | Maryanoff Cynthia A | Pharmaceutical dosage forms of stable amorphous rapamycin like compounds |
| US20060229711 | Apr 4, 2006 | Oct 12, 2006 | Elixir Medical Corporation | Degradable implantable medical devices |
| US20070015697 | Nov 1, 2005 | Jan 18, 2007 | Peyman Gholam A | Enhanced ocular neuroprotection and neurostimulation |
| US20070059336 | Feb 27, 2006 | Mar 15, 2007 | Allergan, Inc. | Anti-angiogenic sustained release intraocular implants and related methods |
| US20070207186 | Mar 3, 2007 | Sep 6, 2007 | Scanlon John J | Tear and abrasion resistant expanded material and reinforcement |
| US20080086198 | May 24, 2007 | Apr 10, 2008 | Gary Owens | Nanoporous stents with enhanced cellular adhesion and reduced neointimal formation |
| EP1236478A1 | Feb 27, 2002 | Sep 4, 2002 | Medtronic Ave, Inc. | Peroxisome proliferator-activated receptor gamma ligand eluting medical device |
| EP1588727A1 | Apr 20, 2005 | Oct 26, 2005 | Cordis Corporation | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO1993016189A1 | Feb 11, 1993 | Aug 19, 1993 | Pfizer | Novel macrocyclic lactones and a productive strain thereof |
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO1996041807A1 | Jun 5, 1996 | Dec 27, 1996 | Sylvain Cottens | Rapamycin derivatives |
| WO1998007415A2 | Aug 18, 1997 | Feb 26, 1998 | Ciba Geigy Ag | Methods for prevention of cellular proliferation and restenosis |
| WO2001087263A2 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery systems for treatment of vascular disease |
| WO2001087342A2 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087372A1 | Apr 25, 2001 | Nov 22, 2001 | Cordis Corp | Drug combinations useful for prevention of restenosis |
| WO2001087373A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087374A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery systems for treatment of vascular disease |
| WO2001087375A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087376A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO2002056790A2 | Dec 18, 2001 | Jul 25, 2002 | Avantec Vascular Corp | Delivery of therapeutic capable agents |
| WO2002065947A2 | Feb 18, 2002 | Aug 29, 2002 | Jomed Gmbh | Implants with fk506 for prophylaxis and treatment of restonoses |
| WO2003064383A2 | Feb 3, 2003 | Aug 7, 2003 | Ariad Gene Therapeutics Inc | Phosphorus-containing compounds & uses thereof |
| WO2006116716A2 | Apr 27, 2006 | Nov 2, 2006 | William A Dunn | Materials and methods for enhanced degradation of mutant proteins associated with human disease |

![]()
A plaque, written in Brazilian Portuguese, commemorating the discovery of sirolimus on Easter Island, near Rano Kau
mTOR inhibitor
temsirolimus (CCI-779), everolimus (RAD001), deforolimus (AP23573), AP21967, biolimus, AP23102, zotarolimus (ABT 578), sirolimus (Rapamune), and tacrolimus (Prograf).\
SIROLIMUS
1H NMR

13 C NMR

HPLC

Iloprost (ciloprost)
– See more at: http://worlddrugtracker.blogspot.in/2014/01/iloprost-ciloprost-used-to-treat.html
TELMISARTAN ..Actavis’ Generic Version of Micardis Receives FDA Approval
DUBLIN, Jan. 8, 2014 /PRNewswire/ — Actavis plc today announced that it has received approval from the U.S. Food and Drug Administration (FDA) on its Abbreviated New Drug Application (ANDA) for Telmisartan Immediate-Release Tablets, 20 mg, 40 mg and 80 mg, a generic equivalent to Boehringer Ingelheim’s Micardis. Actavis intends to launch the product immediately.
Perampanel

Perampanel
5′-(2-cyanophenyl)-1′-phenyl-2,3′-bipyridinyl-6′(1’H)-one
cas no 380917-97-5
FDA-approved drug to treat epilepsy. Trade name Fycompa, Eisai (Eisai) research and development.
FYCOMPA tablets contain perampanel, a non-competitive AMPA receptorantagonist. Perampanel is described chemically as 2-(2-oxo-1-phenyl-5-pyridin-2-yl-1,2-dihydropyridin-3-yl) benzonitrile hydrate (4:3).
The molecular formula is C23H15N3O •3/4H2O and the molecular weight is 362.90 (3/4 hydrate). The chemical structure of perampanel is:
 |
Perampanel is a white to yellowish white powder. It is freely soluble in N-methylpyrrolidone, sparingly soluble in acetonitrile and acetone, slightly soluble in methanol, ethanol and ethyl acetate, very slightly soluble in 1-octanol and diethyl ether and practically insoluble in heptane and water.
Perampanel (INN/USAN, trade name Fycompa) is an antiepileptic drug developed by Eisai Co. that acts as a selective noncompetitive antagonist of AMPA receptors, the major subtype of ionotropic glutamate receptors.[1][2]
Perampanel was found to be effective in the treatment of refractory partial-onset seizures in three pivotal (Phase 3) clinical trials[3][4] and has been approved for marketing under the brand name Fycompa by the European Medicines Agency.[5] The minimum effective dose is 4 mg once daily; doses of 8 mg and 12 mg daily provide a greater therapeutic benefit with a corresponding increase in adverse events. Dizziness and somnolence/sedation/fatigue are the most frequent dose-related adverse events. The drug is currently approved, for the control of partial-onset seizures, in those of both sexes who suffer from epilepsy and who are 12 years of age and older, by the Food and Drug Administration, and is considered to be a scheduled drug (an agent with the potential for addiction). Perampanel has been studied in other clinical indications includingParkinson’s disease.[6][7]
It has high potency (IC50 in vitro in functional studies of about 100-250 nM) and a prolonged terminal half-life in humans of approximately 105 hours. The drug is 95% bound to plasma protein. Its primary route of metabolism is by CYP3A4. It does not induce or inhibit P450 enzymes. About 70% of the dose is excreted in the feces and 30% in the urine; less than 2% of the dose is excreted unchanged into the urine.
In clinical trials, perampanel was generally well tolerated although the incidence of adverse events increased in a dose-dependent fashion. There was no increase in serious adverse events compared with placebo. According to the Food and Drug Administration, most common adverse reactions reported by patients receiving Fycompa in clinical trials include dizziness, drowsiness, fatigue, irritability, falls, upper respiratory tract infection,weight increase, vertigo, loss of muscle coordination (ataxia), gait disturbance, balance disorder, anxiety, blurred vision, stuttering (dysarthria), weakness (asthenia), aggression, and excessive sleep (hypersomnia).[8]
Fycompa’s label has a boxed warning to alert prescribers and patients about the risk of serious neuropsychiatric events. Some of these events were reported as serious and life-threatening. Violent thoughts or threatening behavior (including homicidal ideation) was also observed in a few patients. Patients and caregivers should alert a health care professional immediately if changes in mood or behavior that are not typical for the patient are observed. Health care professionals should closely monitor patients during the titration period when higher doses are used.[9]
- Rogawski, M. A. (2011). “Revisiting AMPA Receptors as an Antiepileptic Drug Target”. Epilepsy Currents 11 (2): 56–63. doi:10.5698/1535-7511-11.2.56. PMC 3117497. PMID 21686307. edit
- Rogawski MA, Hanada T. Preclinical pharmacology of perampanel, a selective non-competitive AMPA receptor antagonist. Acta Neurol Scand 2013;127 (Suppl. 197): 19–24.Rogawski, M. A.; Kaukinen, T.; Collin, P.; Krekelä, I.; Patrikainen, H.; Tillonen, J.; Nyrke, T.; Laurila, K.; Haimila, K.; Partanen, J.; Valve, R.; Mäki, M.; Luostarinen, L. (2013). “Preclinical pharmacology of perampanel, a selective non-competitive AMPA receptor antagonist”. Acta Neurologica Scandinavica 127 (1): 19–25. doi:10.1111/ane.12100. PMID 22494246. edit
- Krauss, G. L.; Serratosa, J. M.; Villanueva, V.; Endziniene, M.; Hong, Z.; French, J.; Yang, H.; Squillacote, D.; Edwards, H. B.; Zhu, J.; Laurenza, A. (2012). “Randomized phase III study 306: Adjunctive perampanel for refractory partial-onset seizures”. Neurology 78 (18): 1408–1415.doi:10.1212/WNL.0b013e318254473a. PMID 22517103. edit
- French, J. A.; Krauss, G. L.; Biton, V.; Squillacote, D.; Yang, H.; Laurenza, A.; Kumar, D.; Rogawski, M. A.; Campanille, V.; Floridia, J.; Ilari, R.; Consalvo, D. E.; Thomson, A.; Sfaello, I.; Pociecha, J.; Nieto, F.; Firstenfeld, A.; Zuin, D.; Mesri, J.; Silva, W.; Nofal, P.; Cristalli, D.; Clement, J. F.; Hwang, P.; McLachlan, R.; Pillay, N.; Lasso, J.; Peralta, B. L.; Hernandez, M. L.; Tenhamm, E. (2012). “Adjunctive perampanel for refractory partial-onset seizures: Randomized phase III study 304”. Neurology 79 (6): 589–596. doi:10.1212/WNL.0b013e3182635735. PMC 3413761. PMID 22843280. edit
- “European Medicines Agency Report on Perampanel”.
- Gottwald MD, Aminoff MJ (July 2008). “New frontiers in the pharmacological management of Parkinson’s disease”. Drugs Today 44 (7): 531–45.doi:10.1358/dot.2008.44.7.1217105. PMID 18806903.
- http://www.webmd.com/epilepsy/news/20121024/epilepsy-drug-fycompa-approved
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm325038.htm
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm325038.htm
Perampanel structure is formed by the coupling of an aromatic ring . Pyridone centrally located, surrounded by connecting two benzene rings and a pyridine ring. The synthesis of 2,5 – dibromopyridine (1) Start with sodium methoxide to produce 2-substituted, and an organic tin compound occurs Stille Coupling 3 4 4 HBr generated after acid hydrolysis and coupling of benzyl bromide with NBS to give 5,5. After 6 coupling of boronic ester and get Perampanel.
Perampanel is a pharmaceutically active agent, currently in clinical phase 3. It can be used to treat Parkinson’s disease, epilepsy and multiple sclerosis.
Perampanel, having the following chemical formula

is also known as E 2007, ER 155055-90 and 3-(2-cyanophenyl)-1-phenyl-5-(2-pyridil)-1,2-dihydropyridin-2-one
Various methods of synthesis of such molecules are known, such as those reported in EP1300396, EP 1465626, EP 1772450, EP 1764361 and EP 1970370.
Many of the methods of synthesis of such active substances reported by the prior art use the key intermediate 5-(2-pyridil)-1,2-dihydropyridin-2-one also known as 2,3′-bipyridin-6′(1′H)-one having the following chemical formula:

Other methods use the synthetic precursor of this intermediate known as 2-methoxy-5-(pyridin-2-yl)pyridine or 6′-methoxy-2,3′-bipyridine having the formula:

2,3′-bipyridin-6′(1′H)-one. it is in fact prepared by simple acid-catalysed demethylation of the 6′-methoxy-2,3′-bipyridine as is reported in the prior art.
Various ways of synthesising 2-methoxy-5-(pyridin-2-yl)pyridine are known. The process summarised in Diagram (I) below is described in WO 2001096308:

Such process highlights clear disadvantages such as the need to operate in cryogenic conditions (T=−78° C.) using special equipment and the need to isolate boronic acid via work-up. In addition the use of 2-Bromopyridine is required, which exacerbates the production of waste compared to 2-chloropyridine.
Another process described in WO 2004009553 is summarised in Diagram (II):

Disadvantages of this process include the use of high molecular weight benzene-sulfonyl pyridine entailing a scarce atom-economy of the process and the need to operate at low temperature T (−78° C.) using special equipment.
Lastly, a completely different process is described in WO20087093392 for the preparation of 2,3′-bipyridin-6′(1′H)-one (Diagram (III)) which however does not include the preparation of the intermediate precursor 2-methoxy-5-(pyridin-2-yl)pyridine:

Perampanel and other 1 ,2-dihydropyridine compounds which possess antagonistic action against AMPA receptor and/or inhibitory action against kainate receptor are described in WO 01/96308. Example 7 in WO 01/96308 discloses a process for producing perampanel by reacting 3-(2-cyanophenyl)-5-(2-pyridyl)-2(lH)-pyridone with phenyl boronic acid, copper acetate and triethylamine in methylene chloride, followed by addition of concentrated aqueous ammonia, water and ethyl acetate. After work-up (phase separation, washing the organic phase and drying over magnesium sulfate), the solvent was concentrated in vacuo and the residue was purified by a silica gel column chromatography (ethyl acetate:hexane=l :2) to give the title product as pale yellow powder. There is no disclosure regarding the polymorphic nature of the product.
A new crystalline or amorphous form of a compound may possess physical properties that differ from, and are advantageous over, those of other crystalline or amorphous forms. These include, packing properties such as molar volume, density and hygroscopicity; thermodynamic properties such as melting temperature, vapor pressure and solubility; kinetic properties such as dissolution rate and stability under various storage conditions; surface properties such as surface area, wettability, interfacial tension and shape; mechanical properties such as hardness, tensile strength, compactibility, handling, flow and blend; and filtration properties. Variations in any one of these properties may affect the chemical and pharmaceutical processing of a compound as well as its bioavailability and may often render the new form advantageous for pharmaceutical and medical use.
EP 1764361 (US 2010/324297) discloses three anhydrous crystalline forms ofperampanel, designated Form I, Form III and Form V and a hydrate form ofperampanel. Anhydrous Form I is prepared in accordance with Example Dl by dissolving perampanel in ethyl acetate (EtOAc) under reflux, cooling the solution, seeding with anhydrous perampanel crystals, continued cooling and collecting the precipitated crystals. Anhydrous Form V is prepared in accordance with Example CI, by dissolving perampanel in acetone, heating to reflux and concentrating the solution to solidification, dissolving the solids in acetone-water, refluxing then cooling and collecting the precipitate. The hydrate form is prepared in accordance with Example Bl by dissolving perampanel in acetone-water, heating, cooling the solution, seeding with perampanel hydrate crystals, continued cooling and collecting the precipitated crystals. US 2009/0088574 discloses a crystalline form of perampanel designated Form IV, which is prepared by slurring perampanel in an acetone/water mixture.
US 7,803,818 discloses an amorphous form of perampanel which is prepared by spray drying perampanel from an acetone solution.
US 7,718,807 discloses acid addition salts of perampanel or a hydrate thereof, wherein the acid is selected from the group consisting of benzenesulfonic acid, p- toluenesulfonic acid, hydrochloric acid, hydrobromic acid, sulfuric acid, methanesulfonic acid, fumaric acid, tartaric acid, succinic acid and benzoic acid.
…………………………………………………………………
Perampanel aromatic ring structure is made of highly coupled. Pyridone centrally located, surrounded by connecting two benzene ring and a pyridine ring. The synthesis of 2,5 – dibromo pyridine ( 1) Start (Synthesis, 2012, 57), sodium methoxide instead of generating 2 , and organotin compounds 3 Stille Coupling occurs to generate 4 . 4 in HBr phenylboronic acid after hydrolysis and coupling to get 5 , 5 after bromination with NBS and borate 6 coupled to get Perampanel.
…………………………….
nmr
A Practical, Laboratory-Scale Synthesis of Perampanel
……………………
updated info
-
Perampanel is a pharmaceutical active substance, currently in clinical phase 3, used to treat Parkinson’s disease, epilepsy and multiple sclerosis.
-
[0003]Perampanel, having the following chemical formula
is also known as E 2007, ER 155055-90 and 3-(2-cyanophenyl)-1-phenyl-5-(2-pyridil)-1,2-dihydropyridin-2-one
-
[0004]Various methods of synthesis of such molecule are known, such as those reported in the patent publications EP1300396 , EP1465626 ,EP1772450 , EP1764361 and EP 1970370 .
-
[0005]Many of the methods of synthesis of such active substance reported by the prior art use the key intermediate 5-(2-pyridil)-1,2-dihydropyridin-2-one also known as 2,3′-bipyridin-6′(1’H)-one having the following chemical formula:
or use the synthetic precursor thereof named 2-methoxy-5-(pyridin-2-yl)pyridine or 6′-methoxy-2,3′-bipyridine having the formula:

2,3′-bipyridin-6′(1’H)-one is in fact prepared by simple acid-catalysed demethylation of the 6′-methoxy-2,3′-bipyridine as thoroughly reported in the prior art.
-
[0006]Various ways of synthesising 2-methoxy-5-(pyridin-2-yl)pyridine are known. The process summarised in the diagram (I) below is described in the publication WO 2001096308 :
Diagram (I)
-
[0007]
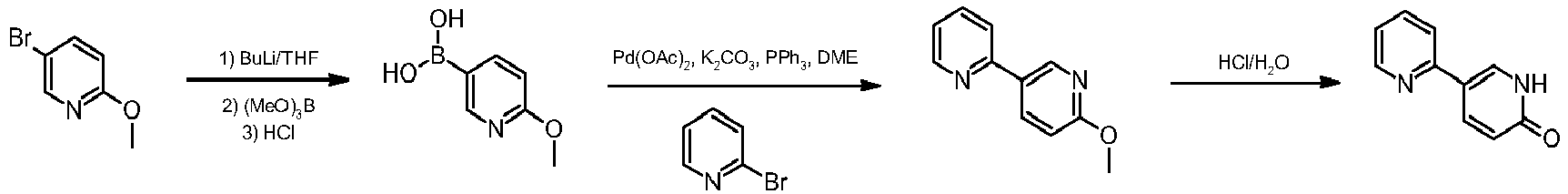
-
[0008]Such process highlights clear disadvantages such as the need to operate in cryogenic conditions (T=-78°C) using special equipment and the need to isolate the boronic acid via work-up; in addition the use of 2-Bromopyridine is envisaged, which is less convenient as regards the production of waste compared to 2-chloropyridine.
-
[0009]Another process described in WO 2004009553 is summarised in the diagram (II) :
Diagram (II)
-
[0010]
-
[0011]It presents clear disadvantages such as the use of high molecular weight benzenesulfonyl pyridine entailing a scarce atom-economy of the process and the need to operate at low temperature T (-78°C) using special equipment.
-
[0012]Lastly, a completely different process is described in WO20087093392for the preparation of 2,3′-bipyridin-6′(1’H)-one which however does not include the preparation of the intermediate precursor named 2-methoxy-5-(pyridin-2-yl)pyridine, process shown in the diagram (III) :
diagram (III)
-
[0013]

LOSARTAN

E-3340
L-158086
MK-0954
MK-954
Ex-89 (free acid)
COZAAR (losartan potassium, cas 124750-99-8) is an angiotensin II receptor (type AT1)antagonist. Losartan potassium, a nonpeptide molecule, is chemically described as 2-butyl-4-chloro-1-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]imidazole-5-methanol monopotassium salt. Its empirical formula is C22H22ClKN6O, and its structural formula is:
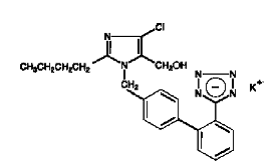 |
Losartan potassium is a white to off-white free-flowing crystalline powder with a molecular weight of 461.01. It is freely soluble in water, soluble in alcohols, and slightly soluble in common organic solvents, such as acetonitrile and methyl ethyl ketone. Oxidation of the 5-hydroxymethyl group on the imidazole ring results in the active metabolite of losartan.
COZAAR is available as tablets for oral administration containing either 25 mg, 50 mg or 100 mg of losartan potassium and the following inactive ingredients: microcrystalline cellulose, lactose hydrous, pregelatinized starch, magnesium stearate, hydroxypropyl cellulose, hypromellose, and titanium dioxide.
COZAAR 25 mg, 50 mg and 100 mg tablets contain potassium in the following amounts: 2.12 mg (0.054 mEq), 4.24 mg (0.108 mEq) and 8.48 mg (0.216 mEq), respectively. COZAAR 25 mg, COZAAR 50 mg, and COZAAR 100 mg may also contain carnauba wax.
|
Losartan (rINN) /loʊˈsɑrtən/ is an angiotensin II receptor antagonist drug used mainly to treat high blood pressure (hypertension). Losartan was the first angiotensin II antagonist to be marketed. Losartan potassium is marketed by Merck & Co. Inc. under the trade nameCozaar. Losartan is available in generic form.
As with all angiotensin II type 1 receptor (AT1) antagonists, losartan is indicated for the treatment of hypertension. It may also delay progression of diabetic nephropathy, and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes, hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).
Although clinical evidence shows calcium channel blockers and thiazide-type diuretics are preferred first-line treatments for most patients (from both efficacy and cost points of view), an angiotensin II receptor antagonist such as losartan is recommended as first-line treatment in patients under the age of 55 who cannot tolerate an ACE inhibitor.The LIFE study demonstrated losartan was significantly superior to atenolol in the primary prevention of adverse cardiovascular events (myocardial infarction or stroke), with a significant reduction in cardiovascular morbidity and mortality for a comparable reduction in blood pressure. A study hints that losartan has a beneficial effect on mitochondria by reversing age related dysfunction in maintaining normal blood pressure and cellular energy usage. The maximal effects on blood pressure usually occur within 3–6 weeks upon starting losartan.
Losartan is also available as hydrochlorothiazide/losartan, a combination drug with a low dose thiazide diuretic to achieve an additive antihypertensive effect.

-
Activation of AT1 receptors in the outer membrane of vascular smooth muscle cells of the heart and arteries causes those tissues to constrict. Blocking of vasoconstriction mediated by AT1 receptors has been found to be beneficial to patients with hypertension.
-
[0003]AT1 receptors are activated by an octa-peptide, angiotensin II. Angiotensin II helps to maintain constant blood pressure despite fluctuations in a person’s state of hydration, sodium intake and other physiological variables. Angiotensin II also performs the regulatory tasks of inhibiting excretion of sodium by the kidneys, inhibiting norephedrin reuptake and stimulating aldosterone biosynthesis.
-
[0004]Inhibiting angiotensin II binding to AT1 receptors with an AT1 receptor antagonist disrupts the vasoconstriction mediated by AT1 receptors that contributes to hypertension.
-
[0005]In the early 1970s, it was discovered that certain oligopeptides competitively inhibited angiotensin receptors (at that time the existence of two receptor subtypes, AT1 and AT2, was unknown). This discovery spurred interest in development of therapeutic oligopeptides with increased potency, but interest in peptide analogs waned due in part to their poor oral bioavailability.
-
[0006]In 1982, Furukawa. Kishimoto and Nishikawa of Taketa Chemical Indus. discovered a class of non-peptide-containing imidazoles that also inhibited the vasoconstriction effect of angiotensin II. See U.S. Patents Nos. 4,340,598 and 4,355,040. Later, U.S. Patent No. 5,138,069 was obtained by Carini, Denucia and Pancras of E.I. DuPont de Nemours on another class of imidazoles, which encompasses the compound losartan. In 1995, losartan (CA Index: 2-butyl-4-chloro-1-[[2′-(1H-tetrazol-5-yl) [1,1′-biphenyl] -4-yl]methyl]-1H-imidazole-5-methanol) (formula I):

became the first nonpeptide AT1 antagonist approved by the U.S. Food and Drug Administration for clinical use. Losartan can be administered orally as its monopotassium salt. Losartan potassium is available by prescription in tablet form as a sole active ingredient (Cozaar®: Merck) and as a co-active ingredient with hydrochlorothiazide (Hyzaar®: Merck).
-
[0007]Losartan has been prepared by a variety of synthetic pathways. In several of these synthetic pathways, the penultimate product is 2-butyl-4-chloro-1-[[2′-(2-triphenylmethyl-2H-tetrazol-5-yl) [1,1′-biphenyl] -4-yl]methyl]-1H-imidazole-5-methanol (“trityl losartan”). Trityl losartan is an intermediate in processes described in U.S. Patents Nos. 5,138,069; 5,962,500 and 5,206,374.
-
[0008]In a process described in Example 316 of U.S. Patent No. 5,138,069, the tetrazole ring of losartan is formed by reacting 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-butyl-4-chloro-5-hydroxymethylimidazole with trimethyltin azide. The reaction gives a trimethylstannyl substituted tetrazole compound directly. The trimethylstannyl group is cleaved from the product by reacting with trityl chloride. This reaction results in attachment of the trityl group to the tetrazole ring. In the last step, the trityl group is cleaved with acid to give losartan (Scheme 1).

-
[0009]In the last step, trityl losartan was suspended in methanol and cooled to ~10°C. 3.4 N Hydrochloric acid was added to the slurry. After a period of time, the pH of the reaction mixture was raised to 13 with 10 N NaOH. Methanol was then distilled off while makeup water was added. After distillation, additional water and toluene were added. The toluene phase was separated and the aqueous phase was extracted once more with toluene. Ethyl acetate and acetic acid were then added to the aqueous phase. Losartan was recovered from the aqueous phase as a solid and further purified by slurrying in ethyl acetate. Losartan was obtained in 88.5% yield and 98.8% purity as determined by HPLC. This process is also described in U.S. Patents Nos. 5,128,355 and 5,155,188.
-
[0010]U.S. Patent No. 5,962,500, Examples 3-5, describe a process for preparing losartan in which the tetrazole ring of losartan is present in the starting material, 5-phenyltetrazole. The ‘500 patent process, depicted in Scheme 2, is convergent and uses a Suzuki coupling reaction (Miyaura, N.; Suzuki, A. Chem. Rev., 1995, 95, 2457) in the convergent step. On one branch of the synthesis, 5-phenyltetrazole is converted into the boronic acid coupling partner for the Suzuki reaction by ortho metalation with n-butyl lithium, followed by reaction with trisopropylborate. The tetrazole ring is protected from reacting with the strong allcyl lithium base with a trityl group. The trityl group is conventionally attached by reacting the tetrazole with trityl chloride in the presence of a non-nucleophilic base. On the other branch of the convergent synthesis, 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde is alkylated with 4-bromobenzylbromide, followed by reduction of the aldehyde with sodium borohydride to yield the other Suzuki coupling partner.

-
[0011]The direct product of Suzuki coupling is trityl losartan. In the next and last step, the tetrazole ring of trityl losartan is deprotected with 4N H2SO4 in THF. In that step, the acidic solution was aged overnight at 20 to 25°C. The solution was then extracted with isopropyl acetate and residual organic solvent was removed from the aqueous phase under vacuum. The solution was then carried forward to from the potassium salt without intermediate isolation of losartan. This process is also described in U.S.Patents Nos, 5,206,374, Example 21, and 5,310,928, Example 21.
-
[0012]Larsen, R.D et al. [J. Org. Chem. (1994), 59, 6391-6394] discloses a similar convergent synthesis of lasartan, whereby the trityl lasartan, generated by Suzuki coupling, is deprotected using 0.7 M H2SO4 in a 50 : 50 mixture of acetonitrile /water.
-
[0013]U.S. Patent No. 5,206,374 Examples 1 and 4-8 describe another process for making Iosartan that also involves a Suzuki coupling reaction. However, unlike the ‘500 patent process, the ‘374 patent process is not convergent. The ‘374 patent process is depicted in Scheme 3.

-
[0014]In the ‘374 patent process, as in the `500 patent process, the tetrazole ring of 5-phenyltetrazole is protected with a trityl group before orthometallation of the phenyl moiety with n-butyl lithium in preparation for making the boronic acid Suzuki coupling partner. In the Suzuki coupling step, the boronic acid is reacted with 4-bromotoluene. The methyl group attached to one of the phenyl rings of the Suzuki product is then halogenated with N-bromosuccinamide and the benzylic bromine atom of that product is displaced with 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde. Reduction of the aldehyde group with sodium borohydride yields trityl losartan. The tetrazole group of trityl losartan was deprotected with 12% aqueous HCl in THF. After 12 hours, the pH of the reaction mixture was raised to 12.5 with 30% NaOH. The THF was then distilled off while make-up water was added to the mixture. After distillation, the mixture was cooled and the triphenyl methanol byproduct of deprotection, which had precipitated, was removed by filtration. The filtrate and rinsate, with which it was combined, were extracted with toluene. Then, ethyl acetate was added and 36% HCI was added until the pH of the reaction mixture was lowered to 3.8. The mixture was cooled, causing losartan to precipitate from the solution. Losartan was obtained in 83% theoretical yield starting from trityl losartan.




EP 253310 discloses a process, wherein 2-n-butyl-4-chloro-1H-imidazolyl-5-methanol (III) is coupled with 5-(4′-bromomethyl-1,1′-biphenyl-2-yl)-2-triphenylmethyl-2H-tetrazole (IV) in N,N-dimethylformamide as solvent in presence of sodium methoxide as the base to furnish trityl losartan. The other bases that have been claimed are sodium hydride, alkali metal carbonates such as sodium carbonate and potassium carbonate and amine bases such as triethyl amine and pyridine.

The coupling reaction results in a mixture of trityl losartan and its regio isomer (V). These are separated by column chromatography.
U.S. Pat. Nos. 5,130,439 and 5,310,928 disclose a method for coupling (IV) and (VI) in N,N-dimethylacetamide solvent in the presence of anhydrous potassium carbonate as base. The imidazole aldehyde (VI) gives predominantly the desired regio isomer (VII). The intermediate VII is then reduced with sodium borohydride to furnish the trityl losartan. The product is isolated by extraction into toluene from aqueous N,N-dimethylacetamide, concentration of the toluene solution and crystallization using ethyl acetate or ethanol as solvent. The synthesis steps are depicted as follows.

In a process published in J. Med. Chem. (1991), 34, 2525-2547, Losartan is prepared by coupling (III) and (IV) in N,N-dimethylformamide in the presence of sodium methoxide. The desired compound is isolated after vacuum distillation of solvent followed by extractive work-up. The resultant product mixture is purified by chronmatography.
The U.S. Pat. Nos. 5,138,069, 5,128,355 and 5,155,118 describe a process for the preparation of losartan, wherein the tetrazole ring of losartan is formed by reacting 1-((2′-cyanobiphenyl-4-yl)methyl)-2-butyl-4-chloro-5-hydroxymethylimidazole with trimethyltin azide. The reaction results in trimethylstannyl substituted tetrazole compound, which is then reacted with trityl chloride and sodium hydroxide.

The trityl losartan thus formed is treated with 3.4N hydrochloric acid in methanol at about 10° C. to give losartan.
The U.S. Pat. Nos. 5,138,069, 5,128,355 and 5,155,118 also disclose another process for making trityl losartan, where in the coupling between IV and VI is carried out in a biphasic solvent system comprising of chlorinated solvent and water. The reaction is carried out at room temperature in presence of sodium hydroxide as the base and aliquat 336 as the phase transfer catalyst. The resulting intermediate VII is then reduced in situ with sodium borohydride to furnish trityl losartan.

U.S. Pat. No. 5,206,374, 5,310,928 and 5,962,500 disclose another process for preparing losartan in which 5-phenyltetrazole (X) is converted into the boronic acid coupling partner (XII) for the Suzuki reaction by tritylation of phenyltetrazole with trityl chloride in presence of a non-nucleophilic base, ortho metalation with n-butyl lithium, followed by reaction with triisopropylborate. 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde (VI) is alkylated with 4-bromobenzylbromide, followed by reduction of the aldehyde with sodium borohydride to yield the other Suzuki coupling partner (XIII). The product of Suzuki coupling is trityl losartan. This process is published in J. Org. Chem. (1994), 59, 6391-6394.


European patents EP 470,794 and EP 470,795 describe a method for the manufacture of biphenyl carbonitriles (XVI). These patents also describe a method of preparation of trityl losartan by coupling of intermediates (III) and (IV) employing the procedure described in EP 253,310.

Losartan potassium exhibits polymorphism. Several polymorphic forms have been prepared and characterized. The following paragraphs briefly describe various polymorphs.
U.S. Pat. No. 5,608,075 discloses the polymorphic forms of losartan, wherein the trityl losartan is deprotected with H2SO4 in 50:50 acetonitrile:water and the free acid is treated with KOH solution. The aqueous solution containing losartan potassium is added slowly to a refluxing azeotropic mixture of cyclohexane/iso propanol and the ternary azeotrope cyclohexane/iso propanol/water is distilled till the water content of the pot is less than 0.05%. The white crystalline solid thus obtained is polymorphic form-I, which is characterized by DSC, XRD and IR. Polymorphic form-II is prepared by heating form-I in a DSC cell. This process is also described in U.S. Pat. No. 5,859,258.
U.S. Pat. No. 6,710,183 discloses the synthesis of losartan potassium starting from trityl losartan, wherein trityl losartan is reacted in an alcohol of formula R—OH (where R is C1 to C4 straight chain alkyl group) with 0.1 to 1 equivalent KOH. Losartan potassium thus formed is isolated after crystallizing out by changing the solvent to an aprotic or weakly protic solvent. The alcohol used is preferably methanol and the protic dipolar solvent used for the crystallization of the final product is preferably acetonitrile or straight or branched chain or cyclic aliphatic hydrocarbons.
EP 1294712 (WO 02/094816) discloses the process to manufacture losartan potassium form-I, wherein trityl losartan or losartan is suspended in a solvent and KOH is added to obtain a clear solution, which is then concentrated under reduced pressure to remove most of the solvent. An anti solvent is added to crystallize losartan potassium. The solvents to prepare losartan potassium include methanol, ethanol, and butanol but preferably the salt formation is carried out in methanol. Anti solvent is selected from common solvents such as ethyl acetate, acetonitrile, toluene and acetone, but the preferred anti solvent is acetone.
US application 2004/0006237 (WO 03/048135) relates to novel amorphous and novel crystalline forms III, IV, V of losartan potassium and the processes for their preparation. The patent also discloses novel processes for preparing losartan potassium forms I and II. The preparation of amorphous losartan includes the step of dissolving losartan potassium in a solvent to form a solution and distilling the solvent form the solution to dryness. Losartan form III (hydrated) is obtained by exposing losartan potassium amorphous or form I to an atmosphere having high relative humidity. Losartan potassium form IV is obtained by treating a saturated solution of losartan potassium in ethanol with methylene chloride. Losartan form V is obtained by treating a saturated solution of losartan potassium in ethanol with hexane. Losartan potassium form II is obtained by adding a saturated solution of losartan potassium in ethanol to xylene to form a mixture and evaporating ethanol from the mixture. Losartan form I is obtained by treating a saturated solution of losartan potassium in ethanol or iso propanol, with less soluble solvent like ethyl acetate, toluene, acetone, methyl ethyl ketone, methylene chloride, acetonitrile, dimethyl carbonate or hexane.
US application 2004/0034077 (WO 03/093262) discloses a process for preparing losartan and losartan potassium, wherein trityl losartan is treated with an acid in a diluent comprising a ketone. Especially preferred liquid ketones are acetone, methyl ethyl ketone and methyl isobutyl ketone, and acetone being the most preferred. Acids, which have been found suitable, include hydrochloric acid, sulphuric acid, acetic acid, trifluoroacetic acid, hydrobromic acid and formic acid. After the trityl losartan has been substantially converted to losartan, reaction mixture is basified. Preferred bases are alkali metal hydroxides and alkoxides. After addition of the base, the liquid ketone is evaporated under vacuum. After separation of triaryl methyl alcohol the residue is acidified to yield losartan. Free losartan is suspended in an alcohol and treated with a solution of potassium ions. Finally losartan potassium is precipitated from the alcohol. The alcohol is selected from the group consisting of isopropyl alcohol, butyl alcohol and isobutyl alcohol. The potassium ion solution is prepared by dissolving potassium iso propoxide, potassium butoxide and potassium iso butoxide or potassium hydroxide in the diluent.
US application 2004/0097568 discloses a process for preparing form III of losartan potassium, wherein trityl losartan is treated with aqueous solution of potassium hydroxide in methanol to obtain losartan potassium. The solvent is evaporated under vacuum and traces of water are removed as an azeotrope with toluene. Methanol and carbon are added to the resulting mixture. The carbon is filtered and the methanol is distilled. The resulting mixture is cooled to 20-25° C. to obtain crystalline form III losartan potassium.
US 5,138,069 and
WO 93/10106. The advantages provided by pharmaceutical products in the crystalline form in terms of easiness of processes for the preparation of related medicaments are well known. Crystalline compounds are in fact known to be more suited to the formulation of galenic forms, thanks both to their flowability in the form of powders or granulates, and to the surface properties of the crystals which promote adhesion, for example during the preparation of tablets. Furthermore, the solubility of crystalline compounds in aqueous solutions, in particular in the gastric juices, can also be significantly different than that of the corresponding amorphous compounds. There is therefore the need to discriminate between the crystalline and the amorphous forms of biologically active compounds, so as to fulfil the various pharmaceutical requirements.
A number of crystalline and amorphous forms of losartan potassium are known from
WO 95/17396 and
WO 03/048135. According to
WO 95/17396, crystalline losartan potassium is prepared by salification of acid losartan with an alkali hydroxide. The losartan potassium aqueous solution is then added to a isopropanol-cyclohexane azeotropic mixture under reflux. Water is then removed by azeotropic distillation of the resulting water-isopropanol-cyclohexane ternary mixture, which boils at 64°C. When the solution is anhydrous, the head temperature raises to 69°C and losartan potassium crystallizes.
US 5,859,258 discloses another crystallization process which comprises dissolution of losartan potassium in isopropanol-water, distillation of the binary azeotrope to an approx. 2.6% water content, precipitation by addition of a losartan potassium suspension in cyclohexane, subsequent distillation of the ternary azeotrope to a water content ranging from 0.02 to 0.11 %, and finally drying crystalline losartan potassium under vacuum at a temperature of approx. 45-50°C.

 , Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author email
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author emailThe imidazole ring of losartan, an antihypertensive and angiotensin II blocker is formed in a condensation reaction between valeroamidine 160 and dihydroxyacetone [50]. It was found that direct chlorination of the imidazole 162also forms the dichlorination product 164 (as shown in Scheme 33) with formaldehyde as a by-product which proved difficult to suppress and made purification of the reaction mixture problematic. Hence, a sequence involving silyl protection, chlorination and deprotection was established which gave the desired product in 90% overall yield (Scheme 33).
![[1860-5397-7-57-i33]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i33.png)
Alternatively, glycine can be reacted with methyl pentanimidate 169 to form the corresponding amidine 171 in high yield. Cyclisation, followed by a Vilsmeier-type reaction then furnishes the key chloroimidazolyl building block 172in good yield (Scheme 34) [51].
![[1860-5397-7-57-i34]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i34.png)
- 50———Shi, Y.-J.; Frey, L. F.; Tschaen, D. M.; Verhoeven, T. R. Synth. Commun. 1993, 23, 2623–2630.doi:10.1080/00397919308012598
- 51—-Griffiths, G. J.; Hauck, M. B.; Imwinkelried, R.; Kohr, J.; Roten, C. A.; Stucky, G. C.; Gosteli, J. J. Org. Chem. 1999,64, 8084–8089. doi:10.1021/jo9824910
- 52–Zhong, Y.-L.; Lee, J.; Reamer, R. A.; Askin, D. Org. Lett. 2004, 6, 929–931. doi:10.1021/ol036423y
NMR: (1H, DMSO, 300 mHz): δ 0.80 (3H, t, J=10. CH3), 1.25 (2H, sext, J=10. CH3CH2), 1.45 (2H, quin, J=10. CH3CH2CH2), 2.45-2.55 (2H, m, CH3CH2CH2CH2), 4.25 (2H, d, J= 3, CH2OH), 5.15-5.25 (3H, m, CH2Ar and OH), 6.88 (d, 2H, J=12, ArH), 7.08 (d, 2H, J=12, ArH), 7.23-7.36 (3H, m, ArH), 7.50-7.55 (1H, ArH).
SEACOND SET
http://www.google.co.in/patents/US7915425
IR v max (KBR): 3201.01, 1580.73, 1460.18, 764.81, 540.09
1H NMR (MeOD) δ, 0.87 (t, 3H), 1.33 (sext, 2H), 1.53 (quint, 2H), 2.56 (t, 2H), 4.43 (s, 2H), 5.24 (s, 2H), 6.89-7.53 (m, 8H).
13C NMR (MeOD) δ, 14.07, 23.24, 27.40, 30.92, 126.71, 126.86, 127.35, 128.21, 130, 130.8, 131, 131.19, 131.81, 136.09, 142.21, 149.97, 162.72
MS (m/z)=423.3 (M+1).
……………………………..
Melting point: 179-180.2
IR, v max (KBR): 3376.27, 1579.77, 1468.86, 762.88, 556.4
1H NMR (CDCl3) δ, 0.87 (t, 3H), 1.31 (sext, 2H), 1.54 (quint, 2H), 2.57 (t, 2H), 4.45 (s, 2H), 5.30 (s, 2H), 7.01-7.68 (m, 8H).
13C NMR (CDCl3) δ, 14.07, 23.24, 27.40, 30.92, 126.71, 126.86, 127.35, 128.21, 130, 130.8, 131, 131.19, 131.81, 136.09, 142.21, 149.97, 162.72
MS (m/z)=423.5 (M+1).
……………………………..
ADDITIONAL WRITEUP FOR READERS, NUMBERINGS ARE ALL NEW
Losartan and its potassium salt, having the formulae (1) & (2) respectively are angiotensin-II receptor (Type AT1) antagonists.

In adults Losartan is currently indicated for the treatment of hypertension (in hypertensive patients with left ventricular hypertrophy, it is also indicated to reduce the risk of stroke).
Losartan Potassium having the formula 2 and its principle active metabolite block the vasoconstrictor and aldosterone. Secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor found in many tissues (e.g., vasicular smooth muscle, adrenal gland) otherwise called as angiotensin receptor blockers (ARBs).
The present invention relates to a short, simple and practical process for the preparation of Losartan 1 which belongs to a novel class of tetrazole-imidazole compounds.
There are many processes recorded in literature. The latest prior art information for the preparation of Losartan is the disclosure made in the patent application of Novartis in their PCT WO 2005/014602 dated 17 Feb. 2005.
The process described in the application comprises the reaction of 4′-(Bromomethyl)-2-cyanobiphenyl (BromoOTBN) of the formula 3 with 2-n-butyl-4-chloro-5-formyl imidazole (BCFI) of the formula of 4 in the presence of Potassium carbonate and acetonitrile to give ‘cyano aldehyde’ of the formula 5. The Cyano aldehyde of the formula 5 is reduced with sodium borohydride to get ‘cyano alcohol’ of the formula 6. The Cyano alcohol is reacted with diethyl aluminium azide in the presence of triethyl aluminium to give Losartan of the formula 1.
The reaction scheme of the process is shown in the Scheme 1

Even though the process is simple, handling of triethyl aluminium used needs special attention like very anhydrous conditions, reactions are to be performed under nitrogen or argon and transferring of triethyl aluminium from the containers needs anhydrous systems. The neat liquid and dense solutions of triethyl aluminium are known to ignite very easily at room temperature in presence of air (Pyrophoric). So handling of both triethyl aluminium and diethyl aluminium needs special attention like anhydrous conditions, nitrogen atmosphere etc.,
In EP 0578125A1 of Takeda Chemical Industries dated 12 Jan. 1994, yet another method for the preparation of Losartan has been disclosed in which Trioctadecyl or Trioctyl tin azide has been used as a tetrazole-forming agent. This method also uses the Cyano alcohol of the formula (6). The process comprises reacting the cyano alcohol of the formula (6) with tri-n-octyl tin azide in presence of toluene to give tri-n-octyl tetrazole derivative, which was treated with nitrous acid to give Losartan of the formula (1) in 94.7% yield. The process is shown in the reaction scheme 2

Even though the yields are better (94.7%) in this process again handling of tri-n-octyl tin azide is involved.
Dupont/Merck in their patents and papers always described that trityl Losartan of the formula 7 is detritylated to get Losartan 1 For example they described in J. Med. Chem., 1991, 34, 2525-2547, the preparation of Losartan of the formula 1, from trityl Losartan of the formula 7 using mineral acids such as Hydrochloric acid and sulfuric acid in 93% yield. The reaction scheme of the process is shown in the scheme 3

In this paper ‘Aldehyde Tetrazole’ of the formula 8 is isolated from trityl tetrazole aldehyde of the formula 21 and were further used for preparing derivatives of aldehyde such as benzene sulfonyl hydrazones of the formula 9 but not for Losartan. This process is shown in the scheme 4

In J. Org. Chem 1994, 59, 6391-6394 again by Merck team reported Trityl Losartan and Losartan synthesis by coupling of boronic acid derivative 11 with 3-(4-bromobenzyl) derivative of BCBMI of the formula 10. The formed trityl Losartan of the formula 7 is converted to Losartan of the formula 1 with acid. The whole process is described in Scheme 5

The Compound of the formula 10 is prepared from the reaction of BCFI of the formula 4 with p-bromo benzyl bromide of the formula 12 in potassium carbonate and Dimethyl formamide followed by reduction with sodium borohydride (NaBH4). The details are given in the Scheme 6

The Compound of the formula 11 is prepared from 5-phenyl tetrazole of the formula 14 by reacting with trityl chloride to get N-trityl-5-phenyl tetrazole of the formula 13, which on reaction with butyl lithium and triisopropyl borate followed by hydrolysis to give compound of the formula 11. This process is shown in the Scheme 7

In one of the first patent filed by Dupont/Merck (date of filing 9 Jul. 1987, priority 11 January 1986 EP0253310) reported a procedure for the preparation of Losartan. Bromo OTBN of the formula 3 is reacted with BCHMI of the formula 15 in the presence of a base to give cyano alcohol of the formula 6, and its regioisomer of the formula 14. Separation of the isomer needs column chromatography. The cyano alcohol 6 is reacted with sodium/ammonium azide in DMF for 13 days to get Losartan 1 in 21% yield. The process is shown in the Scheme 8

The drawbacks of the above process are
- 1). Separation of the regioisomer using column chromatography which is industrially not feasible for the preparation of large scale (ton) material/product
- 2). The tetrazole formation takes 13 days with 21% yield, which is unproductive.
- 3). Dupont/Merck uses BCHMI 15 as the starting material for preparing cyano alcohol of the formula 6. BCHMI 15 is an expensive intermediate compared to BCFI 4, and also the formation of unwanted regio isomer 14 is higher. The process is schematically described in scheme 8. Even though the process looks simple it has two problems.
First: Cyano alcohol is produced as a mixture of regioisomers and needs column chromatography for purification.
Second: Tetrazole formation. This takes 13 days with 21% yield, which limits commercialization of the process.
In U.S. Pat. No. 4,820,843 and U.S. Pat. No. 4,879,186, Dupont prepares Losartan by reaction of BCFI of the formula 4 and N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16 in the presence of base, followed by reduction with sodium borohydride to give Trityl Losartan of the formula 7, which is treated with mineral acid to give Losartan 1.
The process is shown in scheme 9

In U.S. Pat. No. 4,874,867 of Dupont/Merck, a process for the preparation of N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16 is described by the reaction of OTBN of the formula 20 with trimethyl tin azide to give the compound 17, which is treated with Hydrochloric acid to give tetrazole derivative of OTBN of the formula 18. The tetrazole derivative of OTBN of the formula 18 is protected with trityl chloride to give compound of the formula 19, followed by bromination with N-bromosuccinimide to give N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16.
The process is shown in the scheme 10.

In all the above papers and patents by Dupont/Merck, the process yields in many steps are good 75-95% and in some steps are less to moderate 21-49%. The drawbacks, or the problems in all these processes is, the number of unit operations.
For example:
- 1). In J. Med. Chem 1991, 34, 2525-2547 the number of steps are six (6) to prepare Losartan of the formula 1 from the readily available intermediates.
- 2). In J. Org. Chem 1994, 59, 6391-6394 the number of steps are nine (9) to prepare Losartan of the formula 1 from the readily available intermediates.
- 3). In EP 0253310 patent the number of operations are two (2) but the problem is time & yields i.e., 13 days and poor yield (21%), also the uneconomical column chromatographic separation of regioisomer.
- 4). In U.S. Pat. Nos. 4,820,843 and 4,879,186 the number of steps are six (6).
- 5). In U.S. Pat. No. 4,874,867 the number of steps are seven (7).
………………………..
INTERMEDIATES
(1-(2′-Cyano biphenyl-4-methyl)-2-butyl-4-chloro-5-formyl imidazole) of the formula 5.

Melting point: 107-108° C.
HPLC Purity: >98%
IR. v max (KBR): 2218 (—CN), 1662.40 (—CHO)
1H NMR (CDCl3) δ, 0.91 (t, 3H), 1.38 (sext, 2H), 1.73 (quint, 2H), 2.67 (t, 2H), 5.61 (s, 2H), 7.16-7.77 (m, 8H), 9.77 (s, 1H).
13C NMR (CDCl3) δ, 13.51, 22.18, 26.33, 29.04, 47.74, 110.05, 118.36, 124.11, 126.59, 127.65, 129.16, 129.81, 132.76, 133.61, 136.01, 137.69, 142.96, 144.33, 154.46, 177.73