
Home » FDA 2024 (Page 2)
Category Archives: FDA 2024
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ensartinib



Ensartinib
X396, X 396
- 1370651-20-9
- C26H27Cl2FN6O3,
561.4 g/mol - SMA5ZS5B22
6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-N-[4-[(3R,5S)-3,5-dimethylpiperazine-1-carbonyl]phenyl]pyridazine-3-carboxamide
- 3-Pyridazinecarboxamide, 6-amino-5-((1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-N-(4-(((3R,5S)-3,5-dimethyl-1-piperazinyl)carbonyl)phenyl)
- 6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-N-[4-[(3R,5S)-3,5-dimethylpiperazine-1-carbonyl]phenyl]pyridazine-3-carboxamide
- 6-Amino-5-((1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)- N-(4-((3R,5S)-3,5-dimethylpiperazine- 1-carbonyl)phenyl)pyridazine-3-carboxamide
FDA 12/18/2024, Ensacove, To treat non-small cell lung cancer
Ensartinib, sold under the brand name Ensacove, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1] Ensartinib is an Anaplastic lymphoma kinase (ALK) inhibitor used as the salt ensartinib hydrochloride.[1] It is taken by mouth.[1]
The most common adverse reactions include rash, musculoskeletal pain, constipation, cough, pruritis, nausea, edema, pyrexia, and fatigue.[2]
Ensartinib was approved for medical use in the United States in December 2024.[1][2][3][4]
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US90227390&_cid=P11-M9JBTT-36001-1
Synthesis of 6-[bis(tert-butoxycarbonyl)amino]-5-[(1R)-1-(2,6-dichloro-3-fluoro-phenyl)ethoxy]pyridazine-3-carboxylic acid (B)


Synthesis of 6-[bis(tert-butoxycarbonyl)amino]-5-[(1S)-1-(2,6-dichloro-3-fluoro-phenyl)ethoxy]pyridazine-3-carboxylic acid (C)

| Step 1: To a solution of A5 (41.8 g, 200 mmol) in 1,2-dichloroethane (800 mL) was added Boc-L-Pro (26.9 g, 125 mmol) followed by EDCI (31.1 g, 163 mmol) and DMAP (4.12 g, 33.8 mmol) at 0° C. The resulting mixture was stirred at r.t. overnight and then water (350 mL) was added and separated, the water phase was extracted with DCM(150 mL×3), dried over MgSO 4, concentrated and purified by column chromatography to (PE:EA=30:1) to give C1 (13.72 g, yield: 65.6%). |
| Step 2: The procedure from C1 to C was similar to that of B1 to B (9.46 g, yield: 26.4% from C1). |

Medical uses
Ensartinib is indicated for the treatment of adults with anaplastic lymphoma kinase (ALK)-positive locally advanced or metastatic non-small cell lung cancer who have not previously received an ALK-inhibitor.[1][2]
History
Efficacy was evaluated in eXALT3 (NCT02767804), an open-label, randomized, active-controlled, multicenter trial in 290 participants with locally advanced or metastatic ALK-positive non-small cell lung cancer who had not previously received an ALK-targeted therapy.[2] Participants were randomized 1:1 to receive ensartinib or crizotinib.[2]
Society and culture
Legal status
Ensartinib was approved for medical use in the United States in December 2024.[2][3][5]
Name
Ensartinib is the international nonproprietary name.[6]
Ensartinib is sold under the brand name Ensacove.[1][2][3]
References
- ^ Jump up to:a b c d e f g “Ensacove (ensartinib) capsules, for oral use” (PDF). Xcovery Holdings, Inc. U.S. Food and Drug Administration. December 2024.
- ^ Jump up to:a b c d e f g “FDA approves ensartinib for ALK-positive locally advanced or metastatic non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 18 December 2024. Retrieved 20 December 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “FDA Approval of Ensartinib for ALK-Positive Locally Advanced or Metastatic Non-Small Cell Lung Cancer (NSCLC)” (Press release). Xcovery Holdings. 19 December 2024. Retrieved 20 December 2024 – via Business Wire.
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 77”. WHO Drug Information. 31 (1). hdl:10665/330984.
External links
- “Ensartinib (Code C102754)”. NCI Thesaurus.
- Clinical trial number NCT02767804 for “eXalt3: Study Comparing X-396 (Ensartinib) to Crizotinib in ALK Positive Non-Small Cell Lung Cancer (NSCLC) Patients” at ClinicalTrials.gov
Horn L, Infante JR, Reckamp KL, Blumenschein GR, Leal TA, Waqar SN, Gitlitz BJ, Sanborn RE, Whisenant JG, Du L, Neal JW, Gockerman JP, Dukart G, Harrow K, Liang C, Gibbons JJ, Holzhausen A, Lovly CM, Wakelee HA: Ensartinib (X-396) in ALK-Positive Non-Small Cell Lung Cancer: Results from a First-in-Human Phase I/II, Multicenter Study. Clin Cancer Res. 2018 Jun 15;24(12):2771-2779. doi: 10.1158/1078-0432.CCR-17-2398. Epub 2018 Mar 21. [Article]- FDA Approved Drug Products: ENSACOVETM (ensartinib) capsules, for oral use (Dec 2024) [Link]
- NCI Formulary: Ensartinib (X-396) [Link]
| Clinical data | |
|---|---|
| Trade names | Ensacove |
| Other names | X-396 |
| License data | US DailyMed: Ensartinib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1370651-20-9 |
| PubChem CID | 56960363 |
| DrugBank | DB14860 |
| ChemSpider | 58828042 |
| UNII | SMA5ZS5B22 |
| KEGG | D11346 |
| ChEMBL | ChEMBL4113131 |
| ECHA InfoCard | 100.306.918 |
| Chemical and physical data | |
| Formula | C26H27Cl2FN6O3 |
| Molar mass | 561.44 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
\/////////Ensartinib, FDA 2024, APPROVALS 2024, Ensacove, X396, X 396, GLXC-15836, BCP26265, EX-A2941, NSC793150, s8230
Olezarsen


Olezarsen
Olezarsen is an ASO directed inhibitor of Apolipoprotein C-III (apoC-III) mRNA, conjugated to a ligand containing three N-acetyl galactosamine (GalNAc) residues to enable delivery of the ASO to hepatocytes.
TRYNGOLZA contains olezarsen sodium as the active ingredient. Olezarsen sodium is a white to yellow solid and it is freely soluble in water and in phosphate buffer. The molecular formula of olezarsen sodium is C 296H 419N 71O 154P 20S 19Na 20and the molecular weight is 9124.48 daltons. The chemical name of olezarsen sodium is DNA, d(P-thio) ([2′- O-(2-methoxyethyl)] rA-[2′- O-(2-methoxyethyl)] rG-[2′- O-(2-methoxyethyl)] m5rC-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-m5C-T-T-G-T-m5C-m5C-A-G-m5C-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] rA-[2′- O-(2-methoxyethyl)]m5rU), 5′-[26-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[6-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]hexyl]amino]-3-oxopropoxy]methyl]-8,12,19-trioxo-16-oxa-7,13,20-triazahexacos-1-yl hydrogen phosphate], sodium salt (1:20).


Olezarsen
FDA APPROVED 12/19/2024, Tryngolza, To treat familial chylomicronemia syndrome
Drug Trials Snapshot
- AKCEA-APOCIII-LRX
- ALL-P-AMBO-5′-O-(((6-(5-((TRIS(3-(6-(2-ACETAMIDO-2-DEOXY-.BETA.-D-GALACTOPYRANOSYLOXY)HEXYLAMINO)-3-OXOPROPOXYMETHYL))METHYL)AMINO-5-OXOPENTANAMIDO)HEXYL))PHOSPHO)-2′-O-(2-METHOXYETHYL)-P-THIOADENYLYL-(3′-O->5′-O)-2′-O-(2-METHOXYETHYL)-P-THIOGUANYLYL-(3
- DNA, D(P-THIO)((2′-O-(2-METHOXYETHYL))RA-(2′-O-(2-METHOXYETHYL))RG-(2′-O-(2-METHOXYETHYL))M5RC-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETHYL))M5RU-M5C-T-T-G-T-M5C-M5C-A-G-M5C-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETH
- IONIS-APOCIII-LRX
- ISIS-APOCIII-LRX
- ISIS-678354
Olezarsen, sold under the brand name Tryngolza, is a medication used in the treatment of familial chylomicronemia syndrome.[1][2] It is given by injection under the skin.[1]
Olezarsen was approved for medical use in the United States in December 2024.[1][3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9127276 | No | 2015-09-08 | 2034-05-01 |  |
| US9181549 | No | 2015-11-10 | 2034-05-01 | |
| US9593333 | No | 2014-02-14 | 2034-02-14 | |
| US9157082 | No | 2012-04-27 | 2032-04-27 | |
| US9163239 | No | 2014-05-01 | 2034-05-01 | |
Medical uses
Olezarsen is indicated as an adjunct to diet to reduce triglycerides in adults with familial chylomicronemia syndrome.[1]
Pharmacology
Olezarsen is an apolipoprotein C-III-directed antisense oligonucleotide.[1] By binding to apolipoprotein C-III mRNA, it causes its degradation, which in turn increases clearance of plasma triglycerides and very low-density lipoprotein (VLDL).[5]
Adverse effects
In a 66-patient trial, olezarsen was demonstrated to cause following side effects:[5][6]
- injection site reactions
- hypersensitivity reactions (due to immunogenic potential of the medication)
- arthralgia
- thrombocytopenia
- hyperglycemia
- elevation of liver enzymes
History
The US Food and Drug Administration (FDA) granted the application of olezarsen orphan drug designation in February 2024.[7] In August 2024, European Medicines Agency also granted olezarsen this designation.[8]
Society and culture
Legal status
Olezarsen was approved for medical use in the United States in December 2024.[3][9]
Names
Olezarsen is the international nonproprietary name.[10]
Olezarsen is sold under the brand name Tryngolza.[1]
References
^ Jump up to:a b c d e f g “Tryngolza- olezarsen sodium injection, solution”. DailyMed. 19 December 2024. Retrieved 25 January 2025.
- ^ Spagnuolo, Catherine M; Hegele, Robert A (2023). “Recent advances in treating hypertriglyceridemia in patients at high risk of cardiovascular disease with apolipoprotein C-III inhibitors”. Expert Opinion on Pharmacotherapy. 24 (9): 1013–1020. doi:10.1080/14656566.2023.2206015. PMID 37114828.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Jump up to:a b Stroes, Erik S.G.; Alexander, Veronica J.; Karwatowska-Prokopczuk, Ewa; Hegele, Robert A.; Arca, Marcello; Ballantyne, Christie M.; et al. (16 May 2024). “Olezarsen, Acute Pancreatitis, and Familial Chylomicronemia Syndrome”. New England Journal of Medicine. 390 (19): 1781–1792. doi:10.1056/NEJMoa2400201. ISSN 0028-4793.
- ^ Ionis Pharmaceuticals, Inc. (11 December 2024). A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study of AKCEA-APOCIII-LRx Administered Subcutaneously to Patients With Familial Chylomicronemia Syndrome (FCS) (Report). clinicaltrials.gov.
- ^ “Olezarsen Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Retrieved 20 December 2024.
- ^ “EU/3/24/2973 – orphan designation for treatment of familial chylomicronaemia syndrome | European Medicines Agency (EMA)”. http://www.ema.europa.eu. 21 August 2024. Retrieved 22 February 2025.
- ^ “Tryngolza (olezarsen) approved in U.S. as first-ever treatment for adults living with familial chylomicronemia syndrome as an adjunct to diet” (Press release). Ionis Pharmaceuticals. 19 December 2024. Retrieved 20 December 2024 – via PR Newswire.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 87”. WHO Drug Information. 36 (1). hdl:10665/352794.
Further reading
Karwatowska-Prokopczuk, Ewa; Tardif, Jean-Claude; Gaudet, Daniel; Ballantyne, Christie M.; Shapiro, Michael D.; Moriarty, Patrick M.; et al. (2022). “Effect of olezarsen targeting APOC-III on lipoprotein size and particle number measured by NMR in patients with hypertriglyceridemia”. Journal of Clinical Lipidology. 16 (5): 617–625. doi:10.1016/j.jacl.2022.06.005. PMID 35902351.
- Tardif, Jean-Claude; Karwatowska-Prokopczuk, Ewa; Amour, Eric St; Ballantyne, Christie M; Shapiro, Michael D; Moriarty, Patrick M; et al. (6 April 2022). “Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk”. European Heart Journal. 43 (14): 1401–1412. doi:10.1093/eurheartj/ehab820. PMC 8986458. PMID 35025993.
External links
“Olezarsen (Code C180652)”. NCI Thesaurus.
- Clinical trial number NCT04568434 for “A Study of Olezarsen (Formerly Known as AKCEA-APOCIII-LRx) Administered to Patients With Familial Chylomicronemia Syndrome (FCS) (BALANCE)” at ClinicalTrials.gov
- Tardif JC, Karwatowska-Prokopczuk E, Amour ES, Ballantyne CM, Shapiro MD, Moriarty PM, Baum SJ, Hurh E, Bartlett VJ, Kingsbury J, Figueroa AL, Alexander VJ, Tami J, Witztum JL, Geary RS, O’Dea LSL, Tsimikas S, Gaudet D: Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur Heart J. 2022 Apr 6;43(14):1401-1412. doi: 10.1093/eurheartj/ehab820. [Article]
- Karwatowska-Prokopczuk E, Tardif JC, Gaudet D, Ballantyne CM, Shapiro MD, Moriarty PM, Baum SJ, Amour ES, Alexander VJ, Xia S, Otvos JD, Witztum JL, Tsimikas S: Effect of olezarsen targeting APOC-III on lipoprotein size and particle number measured by NMR in patients with hypertriglyceridemia. J Clin Lipidol. 2022 Sep-Oct;16(5):617-625. doi: 10.1016/j.jacl.2022.06.005. Epub 2022 Jun 23. [Article]
- Hooper AJ, Bell DA, Burnett JR: Olezarsen, a liver-directed APOC3 ASO therapy for hypertriglyceridemia. Expert Opin Pharmacother. 2024 Oct;25(14):1861-1866. doi: 10.1080/14656566.2024.2408369. Epub 2024 Sep 26. [Article]
- Bergmark BA, Marston NA, Prohaska TA, Alexander VJ, Zimerman A, Moura FA, Murphy SA, Goodrich EL, Zhang S, Gaudet D, Karwatowska-Prokopczuk E, Tsimikas S, Giugliano RP, Sabatine MS: Olezarsen for Hypertriglyceridemia in Patients at High Cardiovascular Risk. N Engl J Med. 2024 May 16;390(19):1770-1780. doi: 10.1056/NEJMoa2402309. Epub 2024 Apr 7. [Article]
- FDA News: FDA approves drug to reduce triglycerides in adult patients with familial chylomicronemia syndrome [Link]
- FDA Approved Drug Products: TRYNGOLZA (olezarsen) injection, for subcutaneous use [Link]
| Clinical data | |
|---|---|
| Trade names | Tryngolza |
| Other names | IONIS-APOCIII-LRX |
| License data | US DailyMed: Olezarsen |
| Routes of administration | Subcutaneous |
| Drug class | Antisense oligonucleotide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2097587-83-02298451-31-5 |
| DrugBank | DB18728 |
| UNII | S3RS2SA30LNSY2BY6PSB |
| KEGG | D13023 |
////Olezarsen, FDA 2024, APPROVALS 2025, Tryngolza, ISIS-678354, ISIS 678354, familial chylomicronemia syndrome
BENZGALANTAMINE


BENZGALANTAMINE
CAS 224169-27-1
Benzgalantamine gluconate, 1542321-58-3
- 6H-Benzofuro[3a,3,2-ef][2]benzazepin-6-ol, 4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-, benzoate (ester), (4aS,6R,8aS)- (9CI)
- Alpha 1062
- GLN 1062
- Memogain
6h-benzofuro(3a,3,2-ef)(2)benzazepin-6-ol, 4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-, benzoate (ester), (4as,6r,8as)-
| Formula | C24H25NO4 |
|---|---|
| Molar mass | 391.467 g·mol−1 |


External IDs GLN-1062 gluconate
UNIILN7PMJ4P57
CAS Number1542321-58-3
WeightAverage: 587.622
Monoisotopic: 587.236661015
Chemical FormulaC30H37NO11
Benzgalantamine, sold under the brand name Zunveyl, is a medication used for the treatment of mild to moderate dementia of the Alzheimer’s type.[1] It is a cholinesterase inhibitor.[1] Benzgalantamine is a prodrug of galantamine.[1]
The most common side effects include nausea, vomiting, diarrhea, dizziness, headache, and decreased appetite.[1]
Benzgalantamine was approved for medical use in the United States in July 2024.[1][2][3]
compounds that, in addition to enhancing the sensitivity to acetylcholine and choline, and to their agonists, of neuronal cholinergic receptors, and/or acting as cholinesterase inhibitors and/or neuroprotective agents, have enhanced blood-brain barrier permeability in comparison to their parent compounds. The compounds are derived (either formally by their chemical structure or directly by chemical synthesis) from natural compounds belonging to the class of amaryllidaceae alkaloids e.g., Galantamine, Narwedine and Lycoramine, or from metabolites of said compounds. The compounds of the present invention can either interact as such with their target molecules, or they can act as “pro-drugs”, in the sense that after reaching their target regions in the body, they are converted by hydrolysis or enzymatic attack to the original parent compound and react as such with their target molecules, or both. The compounds of this disclosure may be used as medicaments for the treatment of human brain diseases associated with a cholinergic deficit, including the neurodegenerative diseases Alzheimer’s and Parkinson’s disease and the neurological/psychiatric diseases vascular dementia, schizophrenia and epilepsy. Galantamine derivatives disclosed herein have higher efficacy and lower levels of adverse side effects in comparison to galantamine, in treatment of human brain diseases.
Benzgalantamine is a prodrug of galantamine. Gastrointestinal adverse effects are the most frequently reported side effects in patients undergoing treatment with cholinesterase inhibitors, including galantamine, and are often a reason for treatment discontinuation.2 As a prodrug, benzagalantamine remains inert as it passes through the stomach, thereby avoiding many of the gastrointestinal effects associated with peripheral cholinesterase inhibition.4
Benzgalantamine was approved by the FDA in July 2024 for the treatment of mild-to-moderate dementia in Alzheimer’s patients.3,4
SCHEME


US20090253654
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42863485&_cid=P12-M8ZQT3-74791-1

| O-Benzoyl-galantamine(=(4aS,6R,8aS)-4a,5,9,10,11,12-Hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol, benzoate (ester)); yield: 78% |
| O-3,4-Dichlorobenzoyl-galantamine(=(4aS,6R,8aS)-4a,5,9,10,11,12-Hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol, 3,4-dichlorobenzoate (ester)); off-white solid; mp. 69-70° C. |
WO2009127218
US20220220121
https://patentscope.wipo.int/search/en/detail.jsf?docId=US368470159&_cid=P12-M8ZR8V-88578-1
Experiment 1
The gluconate salt of Alpha-1062 was created according to the following previously established general scheme:

AND
US20090253654
Medical uses
Benzgalantamine is indicated for the treatment of mild to moderate dementia of the Alzheimer’s type in adults.[1][2]
Side effects
The most common side effects include nausea, vomiting, diarrhea, dizziness, headache, and decreased appetite.[1]
Society and culture
Legal status
Benzgalantamine was approved for medical use in the United States in July 2024.[1][2]
Names
Benzgalantamine is the international nonproprietary name.[4]
References
- ^ Jump up to:a b c d e f g h i “Zunveyl- benzgalantamine tablet, delayed release”. DailyMed. 8 August 2024. Retrieved 15 August 2024.
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2024/218549Orig1s000ltr.pdf
- ^ “Alpha Cognition’s Oral Therapy Zunveyl Receives FDA Approval to Treat Alzheimer’s Disease” (Press release). Alpha Cognition. 29 July 2024. Archived from the original on 4 August 2024. Retrieved 4 August 2024 – via Business Wire.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
- Baakman AC, ‘t Hart E, Kay DG, Stevens J, Klaassen ES, Maelicke A, Groeneveld GJ: First in human study with a prodrug of galantamine: Improved benefit-risk ratio? Alzheimers Dement (N Y). 2016 Jan 20;2(1):13-22. doi: 10.1016/j.trci.2015.12.003. eCollection 2016 Jan. [Article]
- Bakker C, van der Aart J, Hart EP, Klaassen ES, Bergmann KR, van Esdonk MJ, Kay DG, Groeneveld GJ: Safety, pharmacokinetics, and pharmacodynamics of Gln-1062, a prodrug of galantamine. Alzheimers Dement (N Y). 2020 Oct 13;6(1):e12093. doi: 10.1002/trc2.12093. eCollection 2020. [Article]
- FDA Approved Drug Products: Zunveyl (benzgalantamine) delayed-release tablets for oral use [Link]
- Fierce Pharma: Alpha Cognition’s delayed-release Alzheimer’s drug Zunveyl passes muster with FDA [Link]
- Alpha Cognition: Corporate Presentation Oct 2024 [Link]
External links
- “Benzgalantamine (Code C188656)”. NCI Thesaurus.
| Clinical data | |
|---|---|
| Trade names | Zunveyl |
| Other names | ALPHA-1062 |
| AHFS/Drugs.com | Zunveyl |
| License data | US DailyMed: Benzgalantamine |
| Routes of administration | By mouth |
| Drug class | Cholinesterase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 224169-27-11542321-58-3 |
| DrugBank | DB19353 |
| UNII | XOI2Q0ZF7GLN7PMJ4P57 |
| KEGG | D12930D12931 |
| ChEMBL | ChEMBL5095056 |
| Chemical and physical data | |
| Formula | C24H25NO4 |
| Molar mass | 391.467 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
//////////BENZGALANTAMINE, Alpha 1062, GLN 1062, Memogain, FDA 2024, APPROVALS 2024, Zunveyl
Deuruxolitinib


Deuruxolitinib
C17H18N6, 314.422
Fda approved Leqselvi, 7/25/2024, To treat severe alopecia areata
C-21543, CTP 543, CTP-543, CTP543
(3r)-3-(2,2,3,3,4,4,5,5-d8)cyclopentyl-3-(4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-1h-pyrazol-1-yl)propanenitrile
1h-pyrazole-1-propanenitrile, .beta.-(cyclopentyl-2,2,3,3,4,4,5,5-d8)-4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-, (.beta.r)-D8-ruxolitinib
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Deuruxolitinib phosphate | 8VJ43S4LCM | 2147706-60-1 | JFMWPOCYMYGEDM-NTVOUFPTSA-N |
unii
0CA0VSF91Y
Deuruxolitinib, sold under the brand name Leqselvi, is a medication used for the treatment of alopecia areata.[1] It is a Janus kinase inhibitor selective for JAK1 and JAK2.[2] Although the relative effectiveness of deuruxolitinib and another Janus kinase inhibitor—baricitinib—for alopecia areata may vary depending on the population studied, both drugs are more effective than alternative treatments.[3]
Deuruxolitinib was approved for medical use in the United States in July 2024.[1][4]
Medical uses
Deuruxolitinib is indicated for the treatment of adults with severe alopecia areata.[1]
Side effects
The FDA prescribing label for deuruxolitinib contains a boxed warning for serious infections; malignancies; cardiovascular death, myocardial infarction, and stroke; and thrombosis.[5]
Society and culture
Names
Deuruxolitinib is the international nonproprietary name[6] and the United States Adopted Name.[7]
SYN
20240108633METHOD FOR PREVENTING OR TREATING DISEASE OR CONDITION ASSOCIATED WITH ANTITUMOR AGENT
20240058345TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
2023553253重水素化JAK阻害剤による脱毛障害の治療のためのレジメン
20230390292REGIMENS FOR THE TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
20230322787PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS
1020230093504중수소화된 JAK 억제제를 이용한 탈모 장애의 치료를 위한 요법
WO/2023/018954TREATMENT OF JAK-INHIBITION-RESPONSIVE DISORDERS WITH PRODRUGS OF JAK INHIBITORS
2022171838TREATMENT OF ALOPECIA CAUSED BY DEUTERATED JAK INHIBITOR
2022171838TREATMENT OF ALOPECIA CAUSED BY DEUTERATED JAK INHIBITOR
20220226327Combination therapy comprising JAK pathway inhibitor and rock inhibitor
20220213105PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS
20220202834JAK inhibitor with a vitamin D analog for treatment of skin diseases
20210387991Deuterated JAK inhibitor and uses thereof SUN
WO/2020/163653PROCESS FOR PREPARING ENANTIOMERICALLY ENRICHED JAK INHIBITORS CONCERT
20200222408TREATMENT OF HAIR LOSS DISORDERS WITH DEUTERATED JAK INHIBITORS
2019516684Treatment of Hair Loss Disorders with Deuterated JAK Inhibitors
PATENT
US20210387991
USE OF COMPD NOT SYNTHESIS
https://patentscope.wipo.int/search/en/detail.jsf?docId=US344953814&_cid=P12-M0XGHQ-19840-2
Example 1
Synthesis of Compound 10

| HRMS: Agilent 6530 Q-TOF LC/MS system with electrospray ionization in positive mode. The measured time-of-flight mass-to-charge ratio (m/z) is 333.22839 (theoretical value=333.22735). |
| Clinical data | |
|---|---|
| Trade names | Leqselvi |
| Other names | CTP-543 |
| License data | US DailyMed: Deuruxolitinib |
| Routes of administration | By mouth |
| Drug class | Janus kinase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1513883-39-0as phosphate: 2147706-60-1 |
| PubChem CID | 72704611as phosphate: 154572727 |
| DrugBank | DB18847 |
| ChemSpider | 115010950 |
| UNII | 0CA0VSF91Yas phosphate: 8VJ43S4LCM |
| KEGG | D11866as phosphate: D11867 |
| ChEMBL | ChEMBL4594381 |
| Chemical and physical data | |
| Formula | C17H18N6 |
| Molar mass | 306.373 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
References
King B, Mesinkovska N, Mirmirani P, Bruce S, Kempers S, Guttman-Yassky E, Roberts JL, McMichael A, Colavincenzo M, Hamilton C, Braman V, Cassella JV: Phase 2 randomized, dose-ranging trial of CTP-543, a selective Janus Kinase inhibitor, in moderate-to-severe alopecia areata. J Am Acad Dermatol. 2022 Aug;87(2):306-313. doi: 10.1016/j.jaad.2022.03.045. Epub 2022 Mar 29. [Article]Yan T, Wang T, Tang M, Liu N: Comparative efficacy and safety of JAK inhibitors in the treatment of moderate-to-severe alopecia areata: a systematic review and network meta-analysis. Front Pharmacol. 2024 Apr 10;15:1372810. doi: 10.3389/fphar.2024.1372810. eCollection 2024. [Article]Barati Sedeh F, Michaelsdottir TE, Henning MAS, Jemec GBE, Ibler KS: Comparative Efficacy and Safety of Janus Kinase Inhibitors Used in Alopecia Areata: A Systematic Review and Meta-analysis. Acta Derm Venereol. 2023 Jan 25;103:adv00855. doi: 10.2340/actadv.v103.4536. [Article]Sardana K, Bathula S, Khurana A: Which is the Ideal JAK Inhibitor for Alopecia Areata – Baricitinib, Tofacitinib, Ritlecitinib or Ifidancitinib – Revisiting the Immunomechanisms of the JAK Pathway. Indian Dermatol Online J. 2023 Jun 28;14(4):465-474. doi: 10.4103/idoj.idoj_452_22. eCollection 2023 Jul-Aug. [Article]FDA Approved Drug Products: LEQSELVI (deuruxolitinib) tablets, for oral use [Link]AJMC: FDA Approves Deuruxolitinib for Alopecia Areata [Link]
^ Jump up to:a b c d “Archived copy” (PDF). Archived (PDF) from the original on 29 July 2024. Retrieved 26 July 2024.
- ^ King, Brett; Mesinkovska, Natasha; Mirmirani, Paradi; Bruce, Suzanne; Kempers, Steve; Guttman-Yassky, Emma; et al. (August 2022). “Phase 2 randomized, dose-ranging trial of CTP-543, a selective Janus Kinase inhibitor, in moderate-to-severe alopecia areata”. Journal of the American Academy of Dermatology. 87 (2): 306–313. doi:10.1016/j.jaad.2022.03.045. ISSN 1097-6787. PMID 35364216. S2CID 247866262.
- ^ SEDEH, Farnam Barati; MICHAELSDÓTTIR, Thorunn Elísabet; HENNING, Mattias Arvid Simon; JEMEC, Gregor Borut Ernst; IBLER, Kristina Sophie (25 January 2023). “Comparative Efficacy and Safety of Janus Kinase Inhibitors Used in Alopecia Areata: A Systematic Review and Meta-analysis”. Acta Dermato-Venereologica. 103: 4536. doi:10.2340/actadv.v103.4536. ISSN 0001-5555. PMC 10391778. PMID 36695751.
- ^ “U.S. FDA Approves Leqselvi (deuruxolitinib), an Oral JAK Inhibitor for the Treatment of Severe Alopecia Areata” (Press release). Sun Pharmaceutical. 25 July 2024. Archived from the original on 26 July 2024. Retrieved 26 July 2024 – via PR Newswire.
- ^ http://www.leqselvi.com/&a=Prescribing Information
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
- ^ “Deuruxolitinib”. American Medical Association. Retrieved 27 July 2024.
Further reading
Passeron T, King B, Seneschal J, Steinhoff M, Jabbari A, Ohyama M, et al. (2023). “Inhibition of T-cell activity in alopecia areata: recent developments and new directions”. Frontiers in Immunology. 14: 1243556. doi:10.3389/fimmu.2023.1243556. PMC 10657858. PMID 38022501.
External links
- “Deuruxolitinib (Code C175770)”. NCI Thesaurus.
- “Deuruxolitinib Phosphate (Code C175771)”. NCI Thesaurus.
- Clinical trial number NCT04518995 for “Study to Evaluate the Efficacy and Safety of CTP-543 in Adults With Moderate to Severe Alopecia Areata (THRIVE-AA1) (THRIVE-AA1)” at ClinicalTrials.gov
- Clinical trial number NCT04797650 for “Study to Evaluate the Efficacy and Safety of CTP-543 in Adults With Moderate to Severe Alopecia Areata (THRIVE-AA2) (THRIVE-AA2)” at ClinicalTrials.gov
////Deuruxolitinib, alopecia areata, Leqselvi , approvals 2024, fda 2024, C-21543, CTP 543, CTP-543, CTP543, UNII-0CA0VSF91Y, WHO 11622
Vorasidenib


Vorasidenib
6-(6-chloropyridin-2-yl)-N2,N4-bis[(2R)-1,1,1-trifluoropropan-2-yl]-1,3,5-triazine-2,4-diamine
CAS 1644545-52-7, C14H13ClF6N6, 414.74
FDA APPROVED, 8/6/2024, Voranigo, To treat Grade 2 astrocytoma or oligodendroglioma
UNII 789Q85GA8P
- AG 881
- AG-881
- AG881
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Vorasidenib citrate | X478M962XG | 2316810-02-1 | YOUTVRFNJAAFNS-DLVAHKFUSA-N |
| Vorasidenib citrate anhydrous | W4XG3EQK7B | 2316810-00-9 | OCEHQNOYRLHJCI-WPRTUUMNSA-N |
Vorasidenib, sold under the brand name Voranigo, is an anti-cancer medication used for the treatment of certain forms of glioma.[1][2] Vorasidenib acts to inhibit the enzymes isocitrate dehydrogenase-1 (IDH1) and isocitrate dehydrogenase-2 (IDH2).[1][2]
The most common adverse reactions include fatigue, headache, increased risk of COVID-19 infection, musculoskeletal pain, diarrhea, nausea, and seizures.[2]
Vorasidenib was approved for medical use in the United States in August 2024.[2][3] It is the first approval by the US Food and Drug Administration (FDA) of a systemic therapy for people with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation.[2]
Medical uses
Vorasidenib is indicated for the treatment of people aged twelve years of age and older with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation, following surgery including biopsy, sub-total resection, or gross total resection.[2]
Side effects
The most common adverse reactions include fatigue, headache, increased risk of COVID-19 infection, musculoskeletal pain, diarrhea, nausea, and seizures.[2] The most common grade 3 or 4 laboratory abnormalities include increased alanine aminotransferase, increased aspartate aminotransferase, GGT increased, and decreased neutrophils.[2]
History
Efficacy was evaluated in 331 participants with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation following surgery enrolled in INDIGO (NCT04164901), a randomized, multicenter, double-blind, placebo-controlled trial.[2] Participants were randomized 1:1 to receive vorasidenib 40 mg orally once daily or placebo orally once daily until disease progression or unacceptable toxicity.[2] Isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation status was prospectively determined by the Life Technologies Corporation Oncomine Dx Target Test.[2] Participants randomized to placebo were allowed to cross over to vorasidenib after documented radiographic disease progression.[2] Participants who received prior anti-cancer treatment, including chemotherapy or radiation therapy, were excluded.[2]
Society and culture
Legal status
Vorasidenib was approved for medical use in the United States in August 2024.[2]
The FDA granted the application for vorasidenib priority review, fast track, breakthrough therapy, and orphan drug designations.[2]
SYN
WO/2024/161041NOVEL COMPOUNDS THAT CAN BE USED AS THERAPEUTIC AGENTS
20240254118PRMT5 INHIBITORS AND USES THEREOF
118359585共晶体、其药物组合物以及涉及其的治疗方法
WO/2024/148437USE OF PCLX-001 OR PCLX-002 AS A RADIOSENSITIZER
20240238424HETEROBIFUNCTIONAL COMPOUNDS AND METHODS OF TREATING DISEASE
1020240097895CD73 화합물
WO/2024/137852PRMT5 INHIBITORS AND USES THEREOF
2024057088THERAPEUTICALLY ACTIVE COMPOUNDS AND THEIR METHODS OF USE
20240116928CD73 COMPOUNDS
117586228Preparation method of triazine medicine
20240041892THERAPEUTICALLY ACTIVE COMPOUNDS AND THEIR METHODS OF USE
117529323Therapeutically active compounds and methods of use thereof
WO/2024/006929CD73 COMPOUNDS
PATENT
https://patents.google.com/patent/US10028961B2/en
Step 3: Preparation of 6-(6-chloropyridin-2-yl)-N2,N4-bis((R)-1,1,1-trifluoro propan-2-yl)-1,3,5-triazine-2,4-diamine
A mixture of 2,4-dichloro-6-(6-chloro-pyridin-2-yl)-1,3,5-triazine (0.27 g, 1.04 mol), (R)-1,1,1-trifluoropropan-2-amine hydrochloride (0.39 g, 2.6 mol), and potassium carbonate (0.43 g, 3.1 mol) in dry 1,4-dioxane (2.5 mL) was stirred under the atmosphere of N2 at 50° C. for 36 hr then at 100° C. for another 36 hr until the reaction was complete. The resulting mixture was filtered through Celite and the cake was washed with EtOAc. The filtrate was concentrated and the residue was purified by standard methods to give the desired product.

1H NMR (400 MHz, CDCl3) δ 8.32 (m, 1H), 7.80 (m, 1H), 7.48 (d, J=7.9 Hz, 1H), 5.61 (m, 1.5H), 5.25 (m, 0.5H), 5.09 (m, 0.5H), 4.88 (m, 1.5H), 1.54-1.26 (m, 6H). LC-MS: m/z 415 (M+H)+.
The procedure set forth in Example 10 was used to produce the following compounds using the appropriate starting materials.Compound 6-(6-Chloropyridin-2-yl)-N2,N4-bis((S)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.29-8.16 (m, 1H), 7.72 (d, J=7.6 Hz, 1H), 7.41 (d, J=7.9 Hz, 1H), 5.70-5.13 (m, 2H), 5.09-4.71 (m, 2H), 1.34 (m, 6H). LC-MS: m/z 415 (M+H)+.Compound 6-(6-Chloropyridin-2-yl)-N2—((R)-1,1,1-trifluoropropan-2-yl)-N4—((S)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.41-8.23 (m, 1H), 7.83 (s, 1H), 7.51 (d, J=6.2 Hz, 1H), 5.68-5.20 (m, 2H), 5.18-4.81 (m, 2H), 1.48-1.39 (m, 6H). LC-MS: m/z 415 (M+H)+.Compound 6-(6-Chloropyridin-2-yl)-N2,N4-bis(1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine

1H NMR (400 MHz, CDCl3) δ 8.29-8.16 (m, 1H), 7.72 (d, J=7.6 Hz, 1H), 7.41 (d, J=7.9 Hz, 1H), 5.70-5.13 (m, 2H), 5.09-4.71 (m, 2H), 1.34 (m, 6H). LC-MS: m/z 415 (M+H)+.

| Clinical data | |
|---|---|
| Trade names | Voranigo |
| License data | US DailyMed: Vorasidenib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1644545-52-7 |
| PubChem CID | 117817422 |
| IUPHAR/BPS | 10663 |
| DrugBank | DB17097 |
| ChemSpider | 64835242 |
| UNII | 789Q85GA8P |
| KEGG | D11834 |
| ChEMBL | ChEMBL4279047 |
| Chemical and physical data | |
| Formula | C14H13ClF6N6 |
| Molar mass | 414.74 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
^ Jump up to:a b c “Voranigo- vorasidenib citrate tablet, film coated”. DailyMed. 9 August 2024. Retrieved 15 August 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves vorasidenib for Grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation”. U.S. Food and Drug Administration (FDA). 6 August 2024. Archived from the original on 7 August 2024. Retrieved 7 August 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Servier’s Voranigo (vorasidenib) Tablets Receives FDA Approval as First Targeted Therapy for Grade 2 IDH-mutant Glioma” (Press release). Servier Pharmaceuticals. 6 August 2024. Archived from the original on 7 August 2024. Retrieved 7 August 2024 – via PR Newswire.
Further reading
- Mellinghoff IK, Lu M, Wen PY, Taylor JW, Maher EA, Arrillaga-Romany I, et al. (March 2023). “Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial”. Nature Medicine. 29 (3): 615–622. doi:10.1038/s41591-022-02141-2. PMC 10313524. PMID 36823302.
- Mellinghoff IK, Penas-Prado M, Peters KB, Burris HA, Maher EA, Janku F, et al. (August 2021). “Vorasidenib, a Dual Inhibitor of Mutant IDH1/2, in Recurrent or Progressive Glioma; Results of a First-in-Human Phase I Trial”. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research. 27 (16): 4491–4499. doi:10.1158/1078-0432.CCR-21-0611. PMC 8364866. PMID 34078652.
- Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J, et al. (August 2023). “Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma”. The New England Journal of Medicine. 389 (7): 589–601. doi:10.1056/NEJMoa2304194. PMID 37272516.
- Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. (April 2018). “Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers”. ACS Medicinal Chemistry Letters. 9 (4): 300–305. doi:10.1021/acsmedchemlett.7b00421. PMC 5900343. PMID 29670690.
External links
Clinical trial number NCT04164901 for “Study of Vorasidenib (AG-881) in Participants With Residual or Recurrent Grade 2 Glioma With an IDH1 or IDH2 Mutation (INDIGO)” at ClinicalTrials.gov
- Clinical trial number NCT02481154 for “Study of Orally Administered AG-881 in Patients With Advanced Solid Tumors, Including Gliomas, With an IDH1 and/or IDH2 Mutation” at ClinicalTrials.gov
- Clinical trial number NCT03343197 for “Study of AG-120 and AG-881 in Subjects With Low Grade Glioma” at ClinicalTrials.gov
////////Vorasidenib, Voranigo, FDA 2024, APPROVALS 2024, AG 881, AG-881, AG881
Palopegteriparatide

Palopegteriparatide
Yorvipath , FDA 2024, 8/9/2024, To treat hypoparathyroidism
- G2N64C3385
- 2222514-07-8
- Palopegteriparatide
- UNII-G2N64C3385
- ACP-014
- Mpeg 40000-teriparatide
- Palopegteriparatide [INN]
- Transcon parathyroid hormone (1-34)
- Transcon pth (1-34)
- Palopegteriparatide [USAN]
- TransCon PTH
- WHO 11060


Palopegteriparatide, sold under the brand name Yorvipath, is a hormone replacement therapy used for the treatment of hypoparathyroidism.[1][2] It is a transiently pegylated parathyroid hormone.[4] It is a parathyroid hormone analog.[1]
Palopegteriparatide was approved for medical use in the European Union in November 2023,[2] and in the United States in August 2024.[1][5]
Medical uses
Palopegteriparatide is indicated for the treatment of adults with hypoparathyroidism.[1][2]
Adverse effects
The US Food and Drug Administration (FDA) prescription label for palopegteriparatide includes warnings for a potential risk of risk of unintended changes in serum calcium levels related to number of daily injections and total delivered dose, serious hypocalcemia and hypercalcemia (blood calcium levels that are too high), osteosarcoma (a rare bone cancer) based on findings in rats, orthostatic hypotension (dizziness when standing), and a risk of a drug interaction with digoxin (a medicine for certain heart conditions).[5]
History
The effectiveness of palopegteriparatide was evaluated in a 26-week, randomized, double-blind, placebo-controlled trial that enrolled 82 adults with hypoparathyroidism.[5] Prior to randomization, all participants underwent an approximate four-week screening period in which calcium and active vitamin D supplements were adjusted to achieve an albumin-corrected serum calcium concentration between 7.8 and 10.6 mg/dL, a magnesium concentration ≥1.3 mg/dL and below the upper limit of the reference range, and a 25(OH) vitamin D concentration between 20 to 80 ng/mL.[5] During the double-blind period, participants were randomized to either palopegteriparatide (N = 61) or placebo (N= 21), at a starting dose of 18 mcg/day, co-administered with conventional therapy (calcium and active vitamin D).[5] Study drug and conventional therapy were subsequently adjusted according to the albumin-corrected serum calcium levels.[5] At the end of the trial, 69% of the participants in the palopegteriparatide group compared to 5% of the participants in the placebo group were able to maintain their calcium level in the normal range, without needing active vitamin D and high doses of calcium (calcium dose ≤ 600 mg/day).[5]
The FDA granted the application for palopegteriparatide orphan drug and priority review designations.[5]
Society and culture
Legal status
In September 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Yorvipath, intended for the treatment of chronic hypoparathyroidism in adults.[4][6] The applicant for this medicinal product is Ascendis Pharma Bone Diseases A/S.[4] Palopegteriparatide was approved for medical use in the European Union in November 2023.[2]
Palopegteriparatide was granted an orphan drug designation by the US Food and Drug Administration (FDA) in 2018,[7] and by the EMA in 2020.[8]
Brand names
Palopegteriparatide is the international nonproprietary name.[9][10]
Palopegteriparatide is sold under the brand name Yorvipath.[2]
References
- ^ Jump up to:a b c d e “Yorvipath injection, solution”. DailyMed. 14 August 2024. Retrieved 5 September 2024.
- ^ Jump up to:a b c d e f “Yorvipath EPAR”. European Medicines Agency. 19 October 2020. Archived from the original on 10 December 2023. Retrieved 11 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Yorvipath Product information”. Union Register of medicinal products. 20 November 2023. Archived from the original on 26 November 2023. Retrieved 11 December 2023.
- ^ Jump up to:a b c “Yorvipath: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h “FDA approves new drug for hypoparathyroidism, a rare disorder”. U.S. Food and Drug Administration (FDA) (Press release). 9 August 2024. Archived from the original on 13 August 2024. Retrieved 13 August 2024. This article incorporates text from this source, which is in the public domain.
- ^ “Ascendis Pharma Receives Positive CHMP Opinion for TransCon PTH (palopegteriparatide) for Adults with Chronic Hypoparathyroidism”. Ascendis Pharma (Press release). 14 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ “TransCon Parathyroid Hormone (mPEG conjugated parathyroid hormone 1-34) Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ “EU/3/20/2350”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 89”. WHO Drug Information. 37 (1). hdl:10665/366661.
External links
- Palopegteriparatide Global Substance Registration System
- Palopegteriparatide NCI Thesaurus
- Clinical trial number NCT04701203 for “A Trial Investigating the Safety, Tolerability and Efficacy of TransCon PTH Administered Daily in Adults With Hypoparathyroidism (PaTHway)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Yorvipath |
| Other names | ACP-014, TransCon PTH |
| License data | US DailyMed: Palopegteriparatide |
| Routes of administration | Subcutaneous |
| Drug class | Hormonal agent |
| ATC code | H05AA05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| CAS Number | 2222514-07-8 |
| UNII | G2N64C3385 |
| KEGG | D12395 |
//////Palopegteriparatide, APPRoVALS 2024, FDA 2024, Yorvipath, hypoparathyroidism, UNII-G2N64C3385, ACP-014, TransCon PTH, WHO 11060
Aneratrigine


Aneratrigine
2097163-74-9
5-chloro-2-fluoro-4-[4-fluoro-2-[methyl-[2-(methylamino)ethyl]amino]anilino]-N-(1,3-thiazol-4-yl)benzenesulfonamide
5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
| Benzenesulfonamide, 5-chloro-2-fluoro-4-[[4-fluoro-2-[methyl[2-(methylamino)ethyl]amino]phenyl]amino]-N-4-thiazolyl- |
C19H20ClF2N5O2S2 488.0 g/mol
UNII 6A5ZY5LT78
WHO
SYN

Assignee: Daewoong Pharmaceutical Co., Ltd.
World Intellectual Property Organization, WO2017082688
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017082688&_cid=P11-M0UEPF-95506-1
Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 1) Preparation of tert-butyl (1-(2-amino-5-fluorophenyl)pyridin-3-yl)(methyl)carbamate
2,4-Difluoro-1-nitrobenzene (2.0 g, 12.6 ng/mol) and tert-butyl methyl (pyridin-3-yl)carbamate (2.5 g, 1.0 eq.) were dissolved in DMF (20 mL), and K2C03 ( 2.6 g , 1.5 eq .) was added. The internal temperature was maintained at 60–70 ° C and the mixture was stirred for 2 hours. The completion of the reaction was confirmed by TLC when the reaction solution turned deep yellow. After cooling to room temperature, ethyl acetate (EA)/H20 was added, stirred, and the layers were separated. MgS04 was added to the separated organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was dissolved in EtOH (10 mL) and distilled water (10 mL), and then Na 2 S 2 0 4 (13.0 g, 6 eq.) was added. After stirring for 2 hours while maintaining the internal temperature at 60 to 70 ° C, the completion of the reaction was confirmed by TLC when the yellow color of the reaction solution lightened and became almost colorless. After cooling to room temperature, distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. The filtrate was concentrated under reduced pressure, and the obtained residue was separated by column chromatography (n-Hexane/EA = 3/1) to obtain the title compound (2.0 g, 51. ).
1H NMR (MeOD): 6.73(m, 1H), 6.57(t, 1H), 3.23(m, 1H), 3.10(m, 2H), 2.94(m, 1H), 2.91(s, 3H), 2.25( m, 1H), 1.99(m, 1H)
Step 2) Preparation of tert-butyl thiazol-4-ylcarbamate
Thiazole-4-carboxylic acid (5.0 g, 38.8 vol) was dissolved in t-Bu0H (100 mL), and then TEA (8.1 mL, 1.5 eq.) and DPPA (7.1 mL, 1.5 eq.) were added. The internal temperature was maintained at 90–100 ° C, and the mixture was stirred for 3 days. The completion of the reaction was confirmed by TLC. The product was concentrated under reduced pressure, distilled water (50 mL) was added, and the solution was washed with EA (100 mL).
It was extracted twice. MgSQ 4 was added to the organic layer, stirred, dried, and filtered.
After concentrating the filtrate under reduced pressure, the residue was added to a small amount of EA, slurried, and the resulting solid was filtered to obtain the white title compound (4.0 g, 51.5%).
1H NMR (MeOD): 8.73(s, 1H), 7.24(s, 1H), 1.52(s, 9H)
Step 3) Preparation of tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate
Step 2) The tert-Butyl thiazol-4-ylcarbamate (4.0 g, 20.0 ng ol) prepared in the reaction vessel was placed in a reaction vessel and the interior was replaced with nitrogen gas. After dissolving in THF (32 mL), it was cooled to _78 ° C using dry ice— acetone. After cooling, LiHMDS (22.4 mL, 1.5 eq.) was slowly added and the reaction mass was stirred for 30 minutes. 4-Bromo-5-chloro-2-fluorobenzenesulfonyl chloride (6.0 g, 1.0 eq.) was dissolved in THF (10 mL) and slowly added to the reaction mixture. The reaction mass was stirred overnight and the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was crystallized from THF/n-hexane to obtain the title compound (4.4 g, 59.0%).
1H NMR (MeOD): 9.00(s, 1H), 8.22(d, 1H), 7.90(d, 1H), 7.78(s, 1H), 1.35(s, 9H)
Step 4) Preparation of tert-butyl (l-(2-((4-(N-(tert-butyloxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)-2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate
Tert-butyl (1-(2-amino-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate (0.5 g, 1.1 ng ol) prepared in Step 1) and tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate (0.9 g, 1.2 eq.) prepared in Step 3) were dissolved in 1,4-dioxane (10 mL). Pd(OAc) 2 (0.03 g, 0.1 eq), rac-BINAP (0.19 g, 0.2 eq.), Cs 2 C0 3 (1.5 g, 3.0 eq.) were added to the reaction solution. After reacting at 120 ° C for 30 minutes using a microwave initiator, the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL).
MgS0 4 was added to the organic layer, stirred, filtered and dried. The filtrate was concentrated under reduced pressure, and the residue was separated by column chromatography (EA/n-Hexane = 1/1). This was repeated twice to obtain the title compound (2.0 g, 88.2%).
1H NMR (MeOD): 8.95(s, 1H), 7.94(d, 1H), 7.65(s, 1H), 7.14(t, 1H), 6.70(d, 1H), 6.64(t, 1H), 6.07( d, 1H)ᅳ 3.40(m, 1H), 3.28(m, 2H), 3.16(m, 1H), 2.64(s, 3H), 2.06(m, 1H), 1.89(m, 1H), 1.41(s , 9H), 1.36(s, 9H)
Step 5) Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 4) was prepared by adding 1.25 M HCl in MeOH (15 mL) to tert-butyl (1-(2-((4-(Ν-(tert-butoxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)—2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl) (methyl)carbamate (2.0 g, 2.9 µl). The mixture was heated to 40–50 ° C and stirred overnight, and the completion of the reaction was confirmed by TLC. The product was concentrated, and methylene chloride (15 mL) was added to the residue, which was stirred for 1 hour, and the resulting solid was filtered to obtain the title compound (0.9 g, 58.8%).
1H 證 (MeOD): 8.73(s, 1H), 7.75(d, 1H), 7.12(t, 1H), 7.00(s, 1H), 6.69(d, 1H), 6.67(t, 1H), 6.05( d, 1H), 3.73(m, 1H) , 3.54(m, 1H), 3.45(m, 1H), 3.38(m, 1H), 3.26(m, 1H), 2.63(s, 3H) , 2.31(m , 1H), 1.96(m, 1H)
PATENTS
0002705578SODIUM CHANNEL BLOCKER
20180346459Substituted benzenesulfonamides as sodium channel blockers
2018533606ナトリウムチャネル遮断剤
3375782SODIUM CHANNEL BLOCKER
108349963SODIUM CHANNEL BLOCKER
1020170056461SODIUM CHANNEL BLOCKER

////////////Aneratrigine, DAEWOONG
Seladelpar


Seladelpar
cas 851528-79-5
C21H23F3O5S, 444.47
fda approved 8/14/2024, To treat primary biliary cholangitis (PBC), Livdelzi
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Seladelpar lysine | N1429130KR | 928821-40-3 | WTKSWPYGZDCUNQ-JZXFCXSPSA-N |
- (+)-MBX-8025
- MBX 8025
- MBX-8025
- MBX8025
- RWJ-800025
- ((4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENYL)OXY)ACETIC ACID
- (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)SULFANYL)-2-METHYLPHENOXY)ACETIC ACID PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR (PPAR) AGONIST,ANTIHYPERLIPIDAEMIC
- (R)-2-(4-((2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)-THIO)-2-METHYLPHENOXY)ACETIC ACID
- ACETIC ACID, (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)-
- ACETIC ACID, (4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)- ((4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENYL)OXY)ACETIC ACID
- ACETIC ACID, 2-(4-(((2R)-2-ETHOXY-3-(4-(TRIFLUOROMETHYL)PHENOXY)PROPYL)THIO)-2-METHYLPHENOXY)-
- Seladelpar
Seladelpar, sold under the brand name Livdelzi, is a medication used for the treatment of primary biliary cholangitis.[1] It is used as the lysine dihydrate salt.[1] It is a PPARδ receptor agonist.[1][2][3] The compound was licensed from Janssen Pharmaceutica NV.[4]
Seladelpar was approved for medical use in the United States in August 2024.[1][5]
Seladelpar is a peroxisome proliferator-activated receptor (PPAR)-delta (δ) agonist. Seladelpar is a single enantiomer of the R-configuration.5 On August 14, 2024, seladelpar was granted accelerated approval by the FDA for the treatment of primary biliary cholangitis,6 which is a condition associated with aberrant bile acid metabolism. Seladelpar works to block bile acid synthesis.1
Medical uses
Seladelpar is indicated for the treatment of primary biliary cholangitis in combination with ursodeoxycholic acid in adults who have an inadequate response to ursodeoxycholic acid, or as monotherapy in people unable to tolerate ursodeoxycholic acid.[1]
Clinically, Seladelpar reduces pruritus and IL-31 in patients with primary biliary cholangitis.[6]
- compound 3r [PMID: 17524639]
- Bioorg Med Chem Lett. 2007 Jul 15;17(14):3855-9. doi: 10.1016/j.bmcl.2007.05.007. Epub 2007 May 10.
- 10.1016/j.bmcl.2007.05.007
Drug Discovery, Johnson and Johnson Pharmaceutical Research and Development, LLC, 8 Clarke Drive, Cranbury, NJ 08512, USA
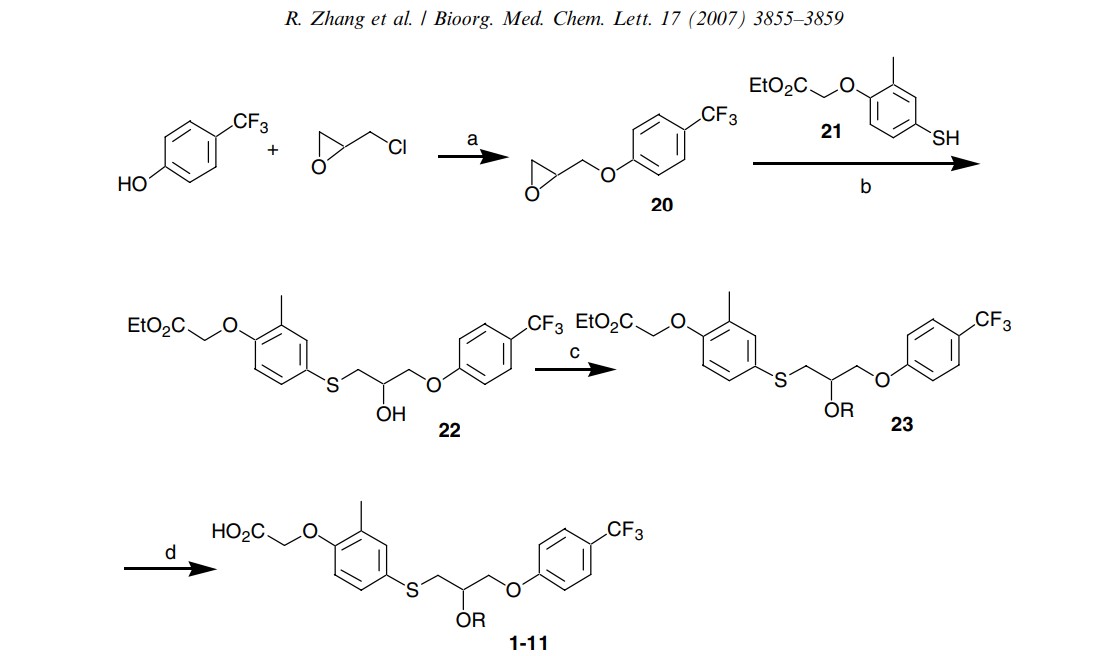
Scheme 1. Reagents and condition: (a) Cs2CO3, dioxane, 100 C 80%; (b) TBAF (cat), THF, 85%; (c) NaH, RI, THF or DMF for esters of 2–5, 8–9, 10–80%; iPr2NEt, RBr or MOMCl, THF for esters of 6–7, 58–79%; ADDP, Ph3P, phenol, CH2Cl2 for esters of 10–11, 68–73%; (d) LiOH, H2O, THF, 90–95%.
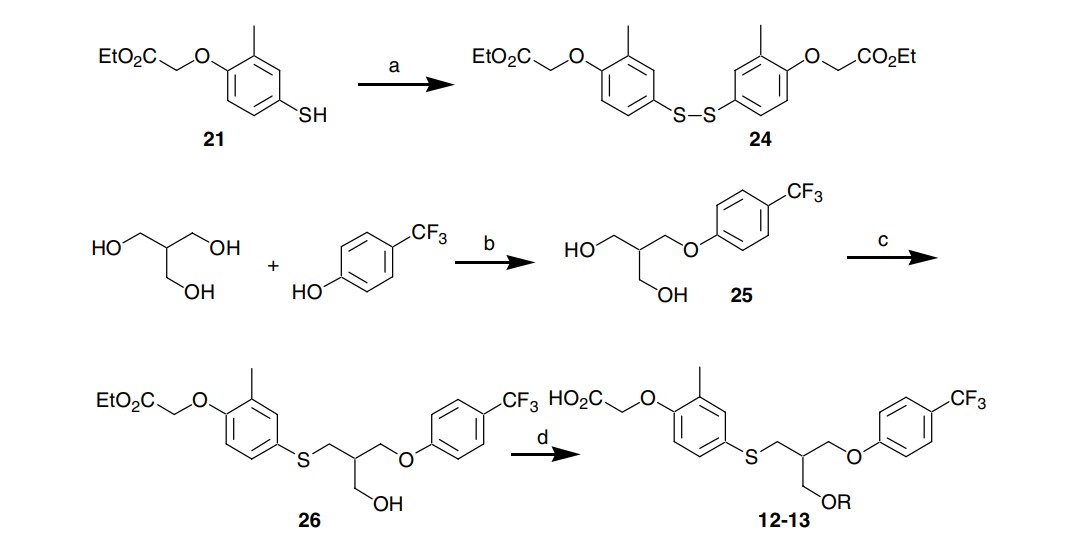
Scheme 2. Reagents: (a) Ba(MnO4)2, CH2Cl2, 89%; (b) DIAD, Ph3P, DMF, THF, 17%; (c) n-Bu3P, 24, Py, 55%; (d) i—NaHMDS, EtOTf, THF for the ethyl ester of 12, 47%; DIAD, Ph3P, para-trifluoromethylphenol for the ethyl ester of 13, 79%; ii—LiOH, H2O, THF, 84–88%.
References
- ^ Jump up to:a b c d e f “Livdelzi- seladelpar lysine capsule”. DailyMed. 14 August 2024. Retrieved 5 September 2024.
- ^ Billin AN (October 2008). “PPAR-beta/delta agonists for Type 2 diabetes and dyslipidemia: an adopted orphan still looking for a home”. Expert Opinion on Investigational Drugs. 17 (10): 1465–1471. doi:10.1517/13543784.17.10.1465. PMID 18808307. S2CID 86564263.
- ^ Bays HE, Schwartz S, Littlejohn T, Kerzner B, Krauss RM, Karpf DB, et al. (September 2011). “MBX-8025, a novel peroxisome proliferator receptor-delta agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin”. The Journal of Clinical Endocrinology and Metabolism. 96 (9): 2889–2897. doi:10.1210/jc.2011-1061. PMID 21752880.
- ^ “Targeting Mixed Dyslipidemia and Metabolic Syndrome”. Metabolex, Inc. 2005. Archived from the original on 17 October 2006.
- ^ “Gilead’s Livdelzi (Seladelpar) Granted Accelerated Approval for Primary Biliary Cholangitis by U.S. FDA” (Press release). Gilead. 14 August 2024. Retrieved 15 August 2024 – via Business Wire.
- ^ Kremer AE, Mayo MJ, Hirschfield GM, Levy C, Bowlus CL, Jones DE, et al. (July 2024). “Seladelpar treatment reduces IL-31 and pruritus in patients with primary biliary cholangitis”. Hepatology. 80 (1): 27–37. doi:10.1097/HEP.0000000000000728. PMC 11191048.
| Clinical data | |
|---|---|
| Trade names | Livdelzi |
| Other names | MBX-8025; RWJ-800025 |
| License data | US DailyMed: Seladelpar |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 851528-79-5 |
| PubChem CID | 11236126 |
| DrugBank | DB12390 |
| ChemSpider | 9411171 |
| UNII | 7C00L34NB9 |
| KEGG | D11256 |
| ChEMBL | ChEMBL230158 |
| CompTox Dashboard (EPA) | DTXSID001045332 |
| Chemical and physical data | |
| Formula | C21H23F3O5S |
| Molar mass | 444.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
///////////////Livdelzi, Seladelpar, (+)-MBX-8025, MBX 8025, MBX-8025, MBX8025, RWJ-800025, FDA 2024, APPROVALS 2024
lazertinib

lazertinib
CAS 1903008-80-9
554.655, C30H34N8O3
FDA APPROVED, 8/19/2024, Lazcluze, To treat non-small cell lung cancer
Drug Trials Snapshot
2-PROPENAMIDE, N-(5-((4-(4-((DIMETHYLAMINO)METHYL)-3-PHENYL-1H-PYRAZOL-1-YL)-2-PYRIMIDINYL)AMINO)-4-METHOXY-2-(4-MORPHOLINYL)PHENYL)-
- N-(5-((4-(4-((DIMETHYLAMINO)METHYL)-3-PHENYL-1H-PYRAZOL-1-YL)PYRIMIDIN-2-YL)AMINO)-4-METHOXY-2-MORPHOLINOPHENYL)ACRYLAMIDE
- C-18112003-G
- GNS 1480
- GNS-1480
- GNS1480
- JNJ-73841937-AAA
- YH 25448
- YH-25448
- YH25448
FDA APPROVED
| 8/19/2024 |
To treat non-small cell lung cancer, Lazcluze
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Lazertinib mesylate monohydrate | WUT449BEG5 | 2411549-88-5 | ZJPNGZUERUYZEG-UHFFFAOYSA-N |
Lazertinib is an oral, third-generation, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI).2,3 Lazertinib was first approved in South Korea on January 18, 2021, for the treatment of EGFR T790M mutation-positive non-small cell lung cancer (NSCLC) with EGFR mutations.1 It was approved by the FDA on August 19, 2024.5 Lazertinib is used alone or in combination with other chemotherapeutic agents.4
Lazertinib, sold under the brand name Lazcluze and Leclaza, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1][2][3] It is a kinase inhibitor of epidermal growth factor receptor.[1]
The most common adverse reactions include rash, nail toxicity, infusion-related reactions (amivantamab), musculoskeletal pain, edema, stomatitis, venous thromboembolism, paresthesia, fatigue, diarrhea, constipation, COVID-19 infection, hemorrhage, dry skin, decreased appetite, pruritus, nausea, and ocular toxicity.[2]
Lazertinib was approved for medical use in South Korea in January 2021,[4][5] and in the United States in August 2024.[2][6]
Medical uses
Lazertinib is indicated in combination with amivantamab for the first-line treatment of adults with locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 19 deletions or exon 21 L858R substitution mutations.[2
History
Efficacy was evaluated in MARIPOSA (NCT04487080), a randomized, active-controlled, multicenter trial of 1074 participants with exon 19 deletion or exon 21 L858R substitution mutation-positive locally advanced or metastatic non-small cell lung cancer and no prior systemic therapy for advanced disease.[2] Participants were randomized (2:2:1) to receive lazertinib in combination with amivantamab, osimertinib monotherapy, or lazertinib monotherapy (an unapproved regimen for non-small cell lung cancer) until disease progression or unacceptable toxicity.[2]
Society and culture
Legal status
Lazertinib was approved for medical use in the United States in August 2024.[2]Names
Lazertinib is the international nonproprietary name.[7]
/////////////////////
References
- ^ Jump up to:a b c “Lazcluze- lazertinib tablet, film coated”. DailyMed. 20 August 2024. Retrieved 5 September 2024.
- ^ Jump up to:a b c d e f g “FDA approves lazertinib with amivantamab-vmjw for non-small lung cancer”. U.S. Food and Drug Administration (FDA). 19 August 2024. Retrieved 21 August 2024. This article incorporates text from this source, which is in the public domain.
- ^ Dhillon S (June 2021). “Lazertinib: First Approval”. Drugs. 81 (9): 1107–1113. doi:10.1007/s40265-021-01533-x. PMC 8217052. PMID 34028784.
- ^ “Yuhan wins approval as MFDS clear T790M EGFR TKI drug ‘Lazertinib'”. 바이오스펙테이터. Retrieved 23 August 2024.
- ^ Dhillon S (2021). “Lazertinib: First Approval”. Drugs. 81 (9): 1107–1113. doi:10.1007/s40265-021-01533-x. ISSN 0012-6667. PMC 8217052. PMID 34028784.
- ^ “Rybrevant (amivantamab-vmjw) plus Lazcluze (lazertinib) approved in the U.S. as a first-line chemotherapy-free treatment for patients with EGFR-mutated advanced lung cancer”. Johnson & Johnson (Press release). 20 August 2024. Retrieved 21 August 2024.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 79”. WHO Drug Information. 32 (1). hdl:10665/330941.
External links
- Clinical trial number NCT04487080 for “A Study of Amivantamab and Lazertinib Combination Therapy Versus Osimertinib in Locally Advanced or Metastatic Non-Small Cell Lung Cancer (MARIPOSA)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lazcluze, Leclaza |
| License data | US DailyMed: Lazertinib |
| Routes of administration | By mouth |
| Drug class | EGFR inhibitor |
| ATC code | L01EB09 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1903008-80-9 |
| PubChem CID | 121269225 |
| IUPHAR/BPS | 10136 |
| DrugBank | DB16216 |
| ChemSpider | 64835231 |
| UNII | 4A2Y23XK11 |
| KEGG | D11980D12245 |
| ChEMBL | ChEMBL4558324 |
| Chemical and physical data | |
| Formula | C30H34N8O3 |
| Molar mass | 554.655 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////lazertinib, C-18112003-G, GNS 1480, GNS-1480, GNS1480, JNJ-73841937-AAA, YH 25448, YH-25448, YH25448, Lazcluze, FDA 2024, APPROVALS 2024
COC1=C(NC2=NC=CC(=N2)N2C=C(CN(C)C)C(=N2)C2=CC=CC=C2)C=C(NC(=O)C=C)C(=C1)N1CCOCC1
ACT-132577, Aprocitentan

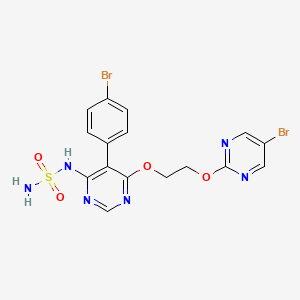
ACT-132577, Aprocitentan,
1103522-45-7
546.19, C16H14Br2N6O4S
3/19/2024 FDA APPROVED, To treat hypertension, Tryvio
N-[5-(4-bromophenyl)-6-{2-[(5-bromopyrimidin-2-yl)oxy]ethoxy}pyrimidin-4-yl]aminosulfonamide
Aprocitentan, sold under the brand name Tryvio, is a medication used to treat hypertension (high blood pressure).[1] It is developed by Idorsia.[2] It is taken by mouth.[1]
Aprocitentan is a dual endothelin-1 antagonist that targets both endothelin A and endothelin B receptors.[3][4]
Aprocitentan was approved for medical use in the United States in March 2024.[1][2][5] It is the first endothelin receptor antagonist to be approved by the US Food and Drug Administration (FDA) to treat systemic hypertension.[2]
Medical uses
Aprocitentan is indicated for the treatment of hypertension in combination with other antihypertensive drugs, to lower blood pressure in adults who are not adequately controlled on other medications.[1]
Adverse effects
Aprocitentan may cause hepatotoxicity (liver damage), edema (fluid retention), anemia (reduced hemoglobin), and decreased sperm count.[1]
Contraindications
Data from animal reproductive toxicity studies with other endothelin-receptor agonists indicate that use is contraindicated in pregnant women.[1]
Mechanism of action
Aprocitentan is an endothelin receptor agonist that inhibits the protein endothelin-1 from binding to endothelin A and endothelin B receptors.[1][4] Endothelin-1 mediates various adverse effects via its receptors, such as inflammation, cell proliferation, fibrosis, and vasoconstriction.[1]
Society and culture
Economics
Aprocitentan is developed by Idorsia, which sold it to Janssen and purchased the rights back in 2023, for US$343 million.[6]
Legal status
Aprocitentan was approved for medical use in the United States in March 2024.[1]
In April 2024, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Jeraygo, intended for the treatment of resistant hypertension in adults.[7] The applicant for this medicinal product is Idorsia Pharmaceuticals Deutschland GmbH.[7]
SYN
US Patent
Trade Name
Application Number
Applicant
IDORSIA PHARMACEUTICALS LTD
IDORSIA PHARMACEUTICALS LTD
IDORSIA PHARMACEUTICALS LTD
IDORSIA PHARMACEUTICALS LTD
IDORSIA PHARMACEUTICALS LTD
PATENT
WO 02/053557
PATENT
Martin Bolli, Christoph Boss, Alexander Treiber. ” 4-pyrimidinesulfamide derivative “, US Patent US8324232B2.
EXAMPLE
Preparation A: Benzylsulfamide Potassium Salt
A.i. Benzylsulfamide
| 1H NMR (D 6-DMSO): δ 4.05 (d, J=6.4 Hz, 2H); 6.60 (s, 2H); 7.04 (s, J=6.4 Hz, 1H); 7.20-7.36 (m, 5H). |
A.ii. Benzylsulfamide Potassium Salt
Preparation B: 5-(4-bromo-phenyl)-4,6-dichloro-pyrimidine
B.i. 4-bromophenylacetic acid methyl ester
B.ii. 2-(4-bromophenyl)-malonic acid dimethyl ester
B.iii. 5-(4-bromophenyl)-pyrimidine-4,6-diol
B.iv. 5-(4-bromo-phenyl)-4,6-dichloro-pyrimidine
Example 1
{5-(4-bromo-phenyl)-6-[2-(5-bromo-pyrimidin-2-yloxy)-ethoxy]-pyrimidin-4-yl}-sulfamide
1.i. Benzyl-sulfamic acid [6-chloro-5-(4-bromophenyl)-pyrimidin-4-yl]-amide
| 1H NMR (CDCl 3): δ 4.23 (d, J=5.9 Hz, 2H); 5.94 (t br., J=6 Hz, 1H); 7.05 (d, J=8.2 Hz, 2H); 7.20-7.35 (m, 5H); 7.68 (d, J=8.2 Hz, 2H); 8.61 (s, 1H). |
1.ii. Benzyl-sulfamic acid [5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl]-amide
1.iii Benzyl-sulfamic acid [5-(4-bromophenyl)-6-{2-(5-bromo-pyrimidin-2-yloxy)-ethoxy}-pyrimidin-4-yl]-amide
1.iv. {5-(4-bromo-phenyl)-6-[2-(5-bromo-pyrimidin-2-yloxy)-ethoxy]-pyrimidin-4-yl}-sulfamide
| 1H NMR (CDCl 3): δ 4.60-4.65 (m, 2H), 4.71-4.74 (m, 2H), 5.50 (s br, 2H), 7.10 (s br, 1H), 7.13-7.17 (m, 2H), 7.55-7.59 (m, 2H), 8.49 (s, 2H), 8.50 (s, 1H). |
| Clinical data | |
|---|---|
| Trade names | Tryvio |
| Other names | ACT-132577 |
| AHFS/Drugs.com | Tryvio |
| Routes of administration | By mouth |
| Drug class | Antihypertensive |
| ATC code | C02KN01 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1103522-45-7 |
| PubChem CID | 25099191 |
| IUPHAR/BPS | 10070 |
| DrugBank | DB15059 |
| ChemSpider | 25027753 |
| UNII | MZI81HV01P |
| KEGG | D11441 |
| ChEBI | CHEBI:76609 |
| ChEMBL | ChEMBL2165326 |
| Chemical and physical data | |
| Formula | C16H14Br2N6O4S |
| Molar mass | 546.19 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESC1=CC(=CC=C1C2=C(N=CN=C2OCCOC3=NC=C(C=N3)Br)NS(=O)(=O)N)Br | |
References
- ^ Jump up to:a b c d e f g h i j “Tryvio- aprocitentan tablet, film coated”. DailyMed. 29 March 2024. Archived from the original on 25 April 2024. Retrieved 25 April 2024.
- ^ Jump up to:a b c “US FDA approves Idorsia’s once-daily Tryvio (aprocitentan) – the first and only endothelin receptor antagonist for the treatment of high blood pressure not adequately controlled in combination with other antihypertensives” (Press release). Idorsia. 20 March 2024. Archived from the original on 28 April 2024. Retrieved 28 April 2024 – via PR Newswire.
- ^ Ojha, Utkarsh; Ruddaraju, Sanjay; Sabapathy, Navukkarasu; Ravindran, Varun; Worapongsatitaya, Pitchaya; Haq, Jeesanul; et al. (2022). “Current and Emerging Classes of Pharmacological Agents for the Management of Hypertension”. American Journal of Cardiovascular Drugs. 22 (3): 271–285. doi:10.1007/s40256-021-00510-9. PMC 8651502. PMID 34878631.
- ^ Jump up to:a b Xu, Jingjing; Jiang, Xiaohua; Xu, Suowen (November 2023). “Aprocitentan, a dual endothelin-1 (ET-1) antagonist for treating resistant hypertension: Mechanism of action and therapeutic potential”. Drug Discovery Today. 28 (11): 103788. doi:10.1016/j.drudis.2023.103788. PMID 37742911.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 29 April 2024. Archived from the original on 30 April 2024. Retrieved 30 April 2024.
- ^ Deswal, Phalguni (6 September 2023). “Idorsia reacquires aprocitentan rights from Janssen for $343m”. Pharmaceutical Technology. Archived from the original on 8 November 2023. Retrieved 8 November 2023.
- ^ Jump up to:a b “Jeraygo EPAR”. European Medicines Agency. 25 April 2024. Archived from the original on 30 April 2024. Retrieved 27 April 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Further reading
- Mahfooz K, Najeed S, Tun HN, Khamosh M, Grewal D, Hussain A, et al. (July 2023). “New Dual Endothelin Receptor Antagonist Aprocitentan in Hypertension: A Systematic Review and Meta-Analysis”. Current Problems in Cardiology. 48 (7): 101686. doi:10.1016/j.cpcardiol.2023.101686. PMID 36893968.
/////ACT-132577, Aprocitentan, Tryvio, FDA 2024, APPROVALS 2024, N-Despropyl-macitentan, WHO 10552

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}