
Home » FDA 2023
Category Archives: FDA 2023
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Velagliflozin
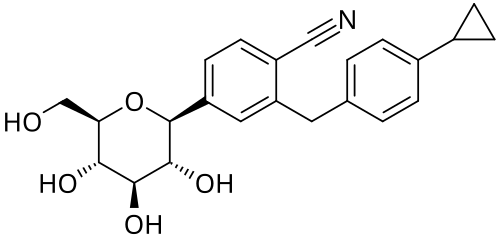
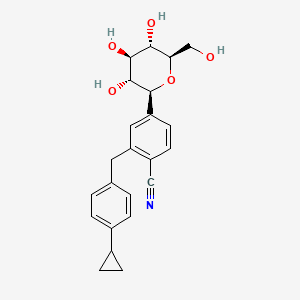
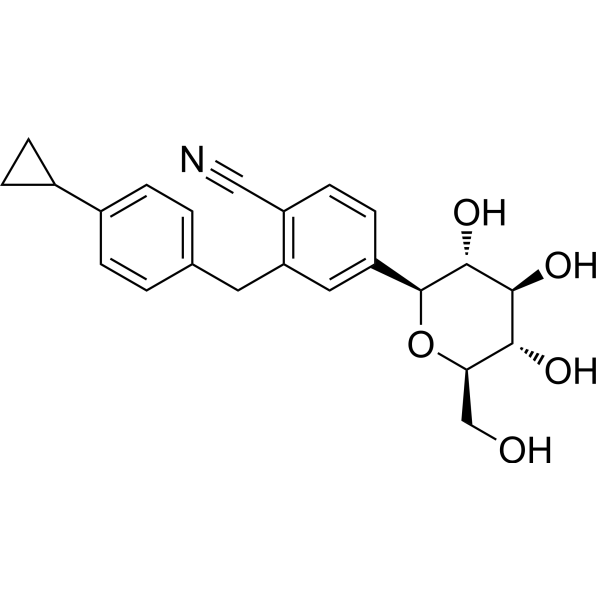
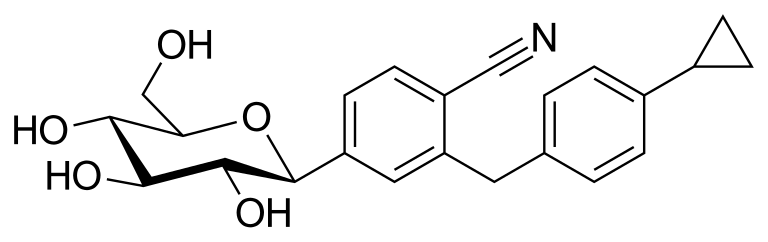
Velagliflozin
VETERINARY DRUG
- Cas 946525-65-1
- FV2YU8SL0P
- 2-((4-cyclopropylphenyl)methyl)-4-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl)benzonitrile
- 2-((4-Cyclopropylphenyl)methyl)-4-beta-D-glucopyranosylbenzonitrile
- 395.4 g/mol, C23H25NO5
2-[(4-cyclopropylphenyl)methyl]-4-[(2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]benzonitrile
- 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYLBENZONITRILE
- BENZONITRILE, 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYL-
Velagliflozin L-proline H2O
Velagliflozin, sold under the brand name Senvelgo, is an antidiabetic medication used for the treatment of cats.[2][4][5] Velagliflozin is a sodium-glucose cotransporter 2 (SGLT2) inhibitor.[6] It is taken by mouth.[2]
Velagliflozin is the active ingredient of the first oral liquid medication approved by the Food and Drug Administration for the treatment of diabetes in cats. This compound belongs to the known class of sodium-glucose cotransporter 2 inhibitors approved to treat diabetes in human.
- Application: NADA 141-568Drug: Senvelgo®Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Patent(s): 7776830 (Exp: 05/01/2027); 8557782 (Exp: 05/01/2027); 9145434 (Exp: 09/07/2033); 10617666 (Exp: 06/06/2035); 11896574 (Exp: 12/17/2034); 10220017 (Exp: 09/29/2036); 10709683 (Exp: 08/24/2036); 11225500 (Exp: 12/17/2038)
- [Indication for Use] To improve glycemic control in otherwise healthy cats with diabetes mellitus not previously treated with insulin.Application: NADA 141-568Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Freedom of Information: FOIA Summary 14320Approval Date: August 10, 2023
APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO
Velagliflozin (brand name Senvelgo) is a veterinary medication approved for treating diabetes in cats, not humans.
Approved countries and years for velagliflozin:
- United States (US): Approved by the FDA in August 2023.
- European Union (EU): Received marketing authorization in November 2023.
- Switzerland: Approved in 2023.
- Great Britain: Approved in 2023.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US310904480&_cid=P11-METCZG-99171-1
SYN
US7776830
https://patentscope.wipo.int/search/en/detail.jsf?docId=US41880220&_cid=P11-METD0X-00376-1
| The following compound is obtained analogously to Example XXIV: |
(1) 1-Cyano-2-(4-cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzene
EXAMPLE 17
2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile
| The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner. |
SYN
WO2007128749
https://patents.google.com/patent/WO2007128749A1/en
The following compound is obtained analogously to Example XXIV:
(1 ) 1 -Cvano-2-(4-cvclopropyl-benzyl)-4-(3-D-glucopyranos-1 -vD-benzene

Mass spectrum (ESI“): m/z = 413 [M+H] + Advantageously, the reduction of the anomeric carbon center of the appropriate intermediate obtained during the synthesis of this compound is conducted with the oxygen functionalities on the pyranose ring protected. Preferred protective groups are benzyl, p-methoxybenzyl, trimethylsilyl, triethylsilyl, terfbutyldimethylsilyl, triisopropylsilyl and allyl.
Example XXV

1-Cyano-2-(4-cyclopropyl-benzyl)-4-(tetra-O-acetyl-β-D-glucopyranos-1-yl)-benzene To a flask charged with a stir bar, 4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1-yl)-2-(4- trifluoromethylsulfonyloxy-benzyl)-benzonitrile (4.4 g), degassed toluene (12 ml.) and degassed water (8 ml.) and kept under argon atmosphere is added cyclopropylboronic acid (0.20 g), potassium phosphate (5.0 g), tricyclohexylphosphine (0.19 g) and at last palladium(ll)acetate (76 mg). The mixture is stirred at 1 10 °C for 6 h meanwhile cyclopropylboronic acid is added after each hour (5x 0.20 g). After cooling to room temperature, the mixture is diluted with aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is removed under reduced pressure. The residue is chromatographed on silica gel (cyclohexane/ethyl acetate 20:1 -> 1 :1 ). Yield: 3.2 g (87% of theory ) Mass spectrum (ESI+): m/z = 581 [M+NH4] +
Example XXVI

4-(1 -Hvdroxy-cvclopropyD-phenylboronic acid A 3.0 M solution of ethylmagnesium bromide in diethylether (7.6 ml.) is added to a stirred solution of titanium(IV) isopropoxide (2.2 ml.) in diethylether (70 ml.) chilled to -78 °C. The resultant solution is stirred at -78 °C for 1.5 h, before 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa borolan-2-yl)-benzoic acid methyl ester (2.0 g) is added. The reaction mixture is warmed to ambient temperature and stirred for an additional 12 h. Then, 1 M aqueous hydrochloric acid is added and the resulting mixture is extracted with ethyl acetate. The combined organic extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is dissolved in acetone (60 ml.) and 0.1 M aqueous NH4OAc solution (50 ml.) followed by NaIO4 (2.3 g) is added. The resulting reaction mixture is stirred at room temperature for 18 h. After removal of the acetone, the residue is extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is purified by chromatography on silicagel (cyclohexane/ethyl acetate). Yield: 0.45 g (33% of theory) Mass spectrum (ESI“): m/z = 223 [M+HCOO]“ Preparation of the end compounds:
Example 17: 2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile

Mass spectrum (ESI+): m/z = 413 [M+NH4]+
The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner.
Yield: 83% of theory
Alternatively this compound is obtained as described in Example XXIV(I ).
The compound of example 17 is also obtained by employing the following procedure:
A solution of 2-(4-cyclopropyl-benzyl)-4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1 -yl)- benzonitrile (0.80 g) in methanol (5 ml.) and THF (5 ml.) is treated with aqueous potassium hydroxide solution (4 mol/l, 5 ml_). The reaction solution is stirred at ambient temperature for 1 h and then neutralized with 1 M hydrochloric acid. The organic solvents are evaporated and the residue is diluted with brine and extracted with ethyl acetate. The organic extracts are dried (sodium sulphate) and the solvent is removed. The residue is chromatographed on silica gel (dichloromethane/methanol 1 :0 -> 9:1 ). Yield: 0.54 g (96% of theory)
SYN
Synthesis 2024, 56, 906–943
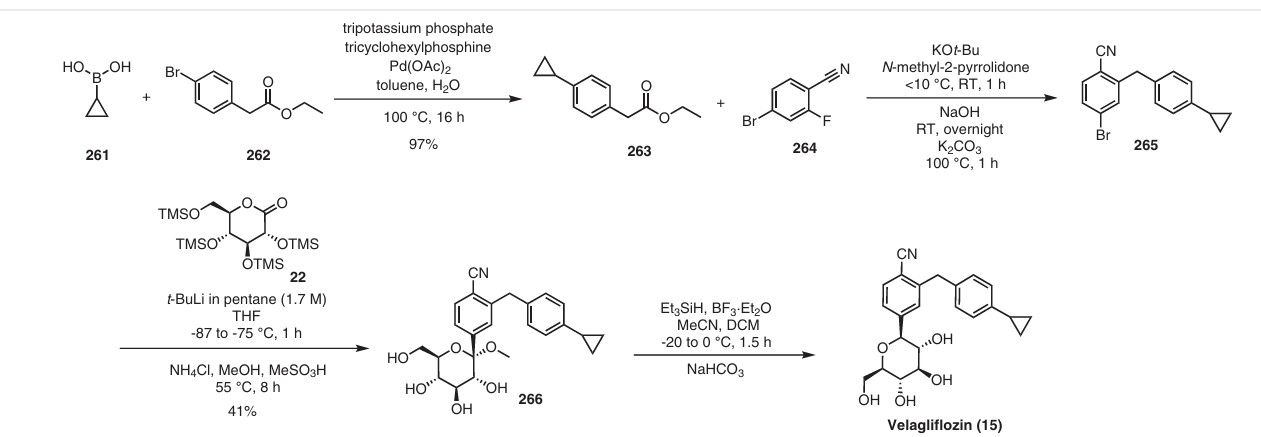
In 2007, Boehringer-Ingelheim Vetmedica GmbH pioneered the development of velagliflozin (15), subsequently submitting a patent application in the United States with the identification number US7776830B2.72a More recently, through clinical investigations, this compound has demonstrated its efficacy as an SGLT2 inhibitor, proving adept at curtailing glucose reabsorption, encouraging glucosuria,
and leading to reductions in both blood glucose and insulin levels.
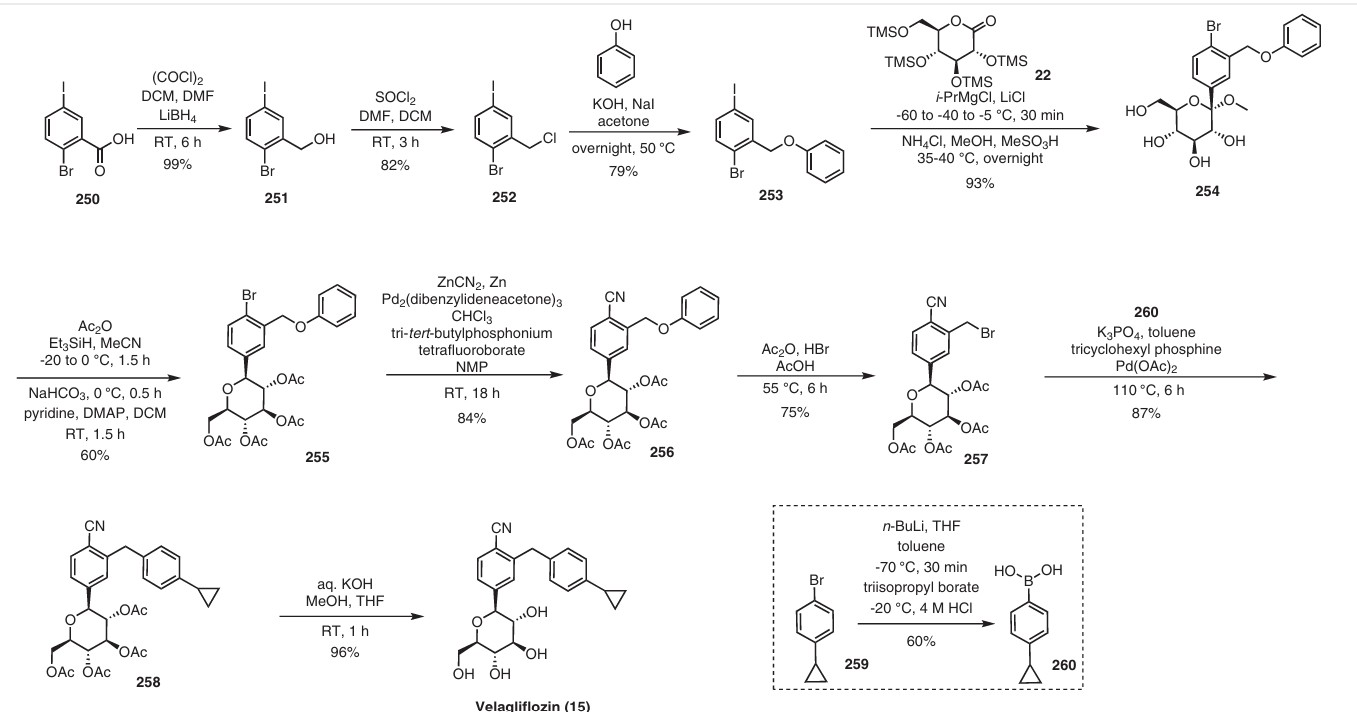
The initial synthesis of velagliflozin (15) was also disclosed in the above patent,72a and in patent
WO2007128749A1.72b The synthesis, depicted in Scheme46, comprises of nine-steps starting with the readily available raw material 2-bromo-5-iodobenzoic acid (250), which undergoes reduction using LiBH4 to form the corresponding alcohol 251. Subsequently, chlorination is carried out using thionyl chloride, resulting in the formation of chloride 252. O-Alkylation of phenol with compound 252 is
then conducted in a basic medium, yielding intermediate 253.The C-glycosylation of 253 with 2,3,4,6-tetrakis-O(trimethylsilyl)-D-glucopyranone 22 in the presence of turbo Grignard reagent (isopropylmagnesium chloride and LiCl) and methanesulfonic acid in methanol gives compound
254 with an impressive 93% yield. The hydroxy group of in termediate 254 is protected using acetic anhydride, and themethoxy group is subsequently removed via Lewis acid (BF3·Et2O, Et3SiH) treatment, providing compound 255 in a yield of 60%. A metal-catalyzed cyano group installation is then performed on intermediate 255, leading to the formation of compound 256 in 84% yield. The subsequent steps involve benzylic bromination followed by coupling with cyclopropylphenyl boronic acid 260, resulting in the formation of intermediate 258. Finally, deacetylation of intermediate 258 using aqueous KOH produces the desired product
The overall yield obtained for velagliflozin (15) is calculated to be 11.3%, with this synthetic route providing a systematic and efficient approach. The highlight of the route is high-yielding chemical transformations. However, the drawback is the use of two palladium-mediated couplings
that increase the possibility of leaching of the toxic metal in scale-up batches. Additionally, the synthetic route requires a large number of chemical transformations and not best suited for commercial production.
The same authors reported an alternative method (Scheme 47) for the synthesis of velagliflozin (15) in the product patent.72 The aglycone intermediate 265 is accessed in two steps starting from ethyl 2-(4-bromophenyl)acetate (262). O-Glycosylation takes place with the aglycone
4-bromo-2-(4-cyclopropylbenzyl)benzonitrile (265) using 2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone 22 in the presence of tert-butyllithium in pentane (1.7 M), resulting in the formation of compound 266. Reduction of compound 266 using boron trifluoride–diethyl etherate yields
the final API velagliflozin (15). This truncated synthetic route is well suited for scale-up due to the significantly low er number of transformations compared to the previous route. Unfortunately, the specific yields were not clearly in dicated for this process. This method presents an alternative approach to the synthesis of velagliflozin (15), providing a potential pathway for its preparation in 5 steps with
an overall yield of 40%.
(72) (a) Eckhardt, M.; Himmelsbach, F.; Eickelmann, P.; Sauer, A.;
Thomas, L. US7776830B2, 2010. (b) Eckhardt, M.; Himmelsbach,
F.; Eickelmann, P.; Sauer, A.; Thomas, L. WO2007128749A1,
2007.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Medical uses
Velagliflozin is indicated to improve glycemic control in otherwise healthy cats with diabetes not previously treated with insulin.[2][4][6]
References
- “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-10-18]”. Health Canada. 18 October 2024. Retrieved 25 October 2024.
- “Senvelgo- velagliflozin solution”. DailyMed. 8 November 2023. Retrieved 13 December 2023.
- “Senvelgo Product information”. Union Register of veterinary medicinal products. 22 November 2023. Retrieved 29 August 2024.
- “NADA 141-568 Senvelgo (velagliflozin oral solution) Cats”.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Cook AK, Behrend E (January 2025). “SGLT2 inhibitor use in the management of feline diabetes mellitus”. Journal of Veterinary Pharmacology and Therapeutics. 48 Suppl 1 (Suppl 1): 19–30. doi:10.1111/jvp.13466. PMC 11736986. PMID 38954371.
- “Dear Veterinarian Letter regarding important safety conditions associated with the use of Senvelgo (velagliflozin oral solution) for improving glycemic control in certain cats with diabetes mellitus”. U.S. Food and Drug Administration. 4 December 2023. Retrieved 13 December 2023. This article incorporates text from this source, which is in the public domain.
| Clinical data | |
|---|---|
| Trade names | Senvelgo |
| License data | US DailyMed: Velagliflozin |
| Routes of administration | By mouth |
| ATCvet code | QA10BK90 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 946525-65-1 |
| PubChem CID | 24862817 |
| ChemSpider | 58827717 |
| UNII | FV2YU8SL0PEQE2P2T77I |
| Chemical and physical data | |
| Formula | C23H25NO5 |
| Molar mass | 395.455 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- SGLT2 inhibitors: a novel therapy for cognitive impairment via multifaceted effects on the nervous systemPublication Name: Translational NeurodegenerationPublication Date: 2024-08-09PMCID: PMC11312905PMID: 39123214DOI: 10.1186/s40035-024-00431-y
- Demographic, morphologic, hormonal and metabolic factors associated with the rate of improvement from equine hyperinsulinaemia-associated laminitisPublication Name: BMC Veterinary ResearchPublication Date: 2022-01-18PMCID: PMC8764787PMID: 35042535DOI: 10.1186/s12917-022-03149-z
- The efficacy and safety of velagliflozin over 16 weeks as a treatment for insulin dysregulation in poniesPublication Name: BMC Veterinary ResearchPublication Date: 2019-02-26PMCID: PMC6390376PMID: 30808423DOI: 10.1186/s12917-019-1811-2
- The sodium-glucose co-transporter 2 inhibitor velagliflozin reduces hyperinsulinemia and prevents laminitis in insulin-dysregulated poniesPublication Name: PLOS ONEPublication Date: 2018-09-13PMCID: PMC6136744PMID: 30212530DOI: 10.1371/journal.pone.0203655
- Effects of the sodium‐glucose cotransporter 2 (<scp>SGLT</scp>2) inhibitor velagliflozin, a new drug with therapeutic potential to treat diabetes in catsPublication Name: Journal of Veterinary Pharmacology and TherapeuticsPublication Date: 2017-11-15PMID: 29139146DOI: 10.1111/jvp.12467
/////////Velagliflozin, APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO, DIABETES, SENVELGO,
Ropotrectinib
Ropotrectinib
- CAS 1802220-02-5
- TPX-0005
- Augtyro
- 08O3FQ4UNP
WeightAverage: 355.373
Monoisotopic: 355.144453003
Chemical FormulaC18H18FN5O2
- repotrectinibum
- (3R,11S)-6-fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentazatetracyclo[13.5.2.04,9.018,22]docosa-1(21),4(9),5,7,15(22),16,19-heptaen-14-one
- 1,15-Etheno-1H-pyrazolo(4,3-F)(1,4,8,10)benzoxatriazacyclotridecin-4(5H)-one, 11-fluoro-6,7,13,14-tetrahydro-7,13-dimethyl-, (7S,13R)-
- 1,15-Etheno-1H-pyrazolo(4,3-f)(1,4,8,10)benzoxatriazacyclotridecin-4(5H)-one, 11-fluoro-2,6,7,13-tetrahydro-7,13-dimethyl-, (14Z)-
- (1Z)-6-Fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentaazatetracyclo(13.5.2.04,9.018,22)docosa-1,4,6,8,15,19,21-heptaen-14-one
- (3R,11S)-6-fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentaazatetracyclo[13.5.2.0,.0,]docosa-1(21),4(9),5,7,15(22),16,19-heptaen-14-one
- (7S,13R)-11-fluoro-7,13-dimethyl-6,7,13,14- tetrahydro-1,15-ethenopyrazolo[4,3- f][1,4,8,10]benzoxatriazacyclotridecin-4(5H)- one
- (3R,6S,)-45-FLUORO-3,6-DIMETHYL-5-OXA-2,8-DIAZA-1(5,3)-PYRAZOLO(1,5-A)PYRIMIDINA-4(1,2)-BENZENANONAPHAN-9-ONE
- (7S,13R)-11-Fluoro-7,13-Dimethyl-6,7,13,14-Tetrahydro-1,15-Ethenopyrazolo[4,3-F][1,4,8,10]Benzoxatriazacyclotridecin-4(5H)-One
- 1,15-ETHENO-1H-PYRAZOLO(4,3-F)(1,4,8,10)BENZOXATRIAZACYCLOTRIDECIN-4(5H)-ONE, 11-FLUORO-6,7,13,14-TETRAHYDRO-7,13-DIMETHYL-, (7S,13R)-
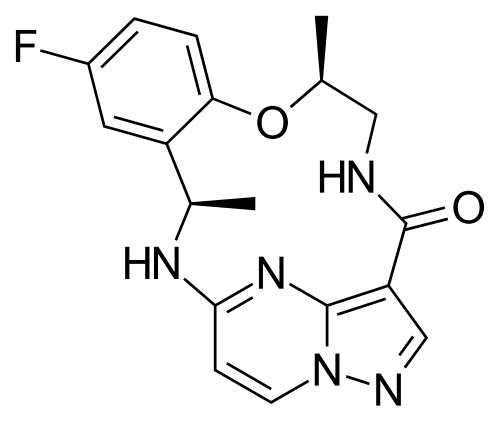
Repotrectinib, sold under the brand name Augtyro, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[2][5] It is taken by mouth.[2] Repotrectinib is an inhibitor of proto-oncogene tyrosine-protein kinase ROS1 (ROS1) and of the tropomyosin receptor tyrosine kinases (TRKs) TRKA, TRKB, and TRKC.[2]
The most common adverse reactions include dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, ataxia, fatigue, cognitive disorders, and muscular weakness.[5]
Repotrectinib was approved for medical use in the United States in November 2023,[5][6] and in the European Union in January 2025.[3][4] CHINA 2024
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/slct.202405153
Synthesis of Repotrectinib
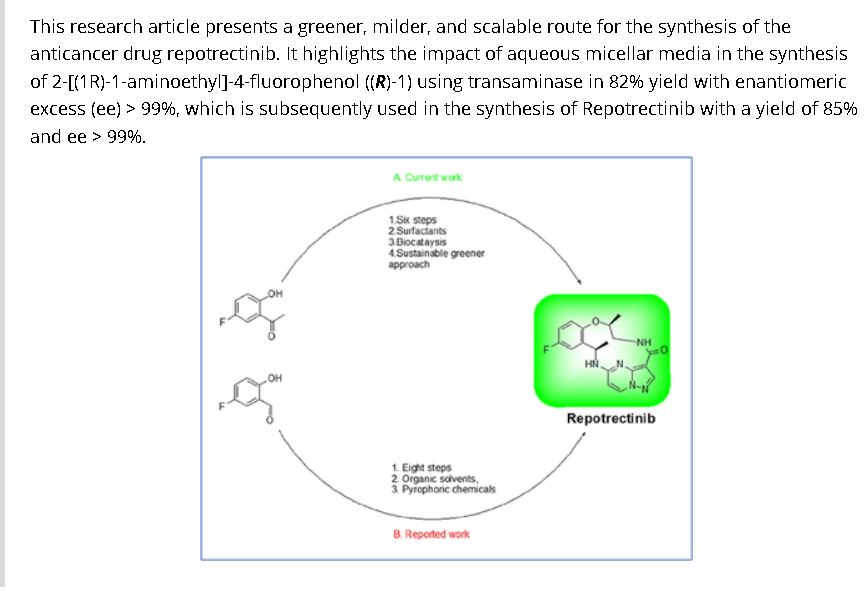
To a stirred solution of 5-{[(1R)-1-(2-{[(2S)-1-aminopropan-2-yl]oxy}-5-fluorophenyl)ethyl]amino}pyrazolo[1,5-a]pyrimidine-3-carboxylic acid 15 (0.25 g, 0.000611 mol, 1.0 eq.) in DMF (4.0 mL, 16V) was slowly added to solution of DIPEA (0.6 mL, 0.00488 mol, 8.0 eq.) in DCM (1.8 mL, 7V) at 0-5 °C. Then FDPP (0.25 g, 0.000672 mol, 1.1 eq.) was added at 0-5 °C. The reaction mixture was allowed to stirr for 1-2h at 25-30 °C. The reaction was monitored by TLC for disappearance of starting material. Then the resulting reaction mixture was diluted with ethyl acetate (50 mL), washed with water (20 mL) and brine solution (20 mL). The separated organic layer was dried over sodium sulphate and concentrated under reduced pressure at 45 °C. The obtained crude product was purified by silica gel (60-120 mesh) column chromatography to get repotrectinib asawhite solid (0.18 g, 85%).

HRMS
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.3c00152
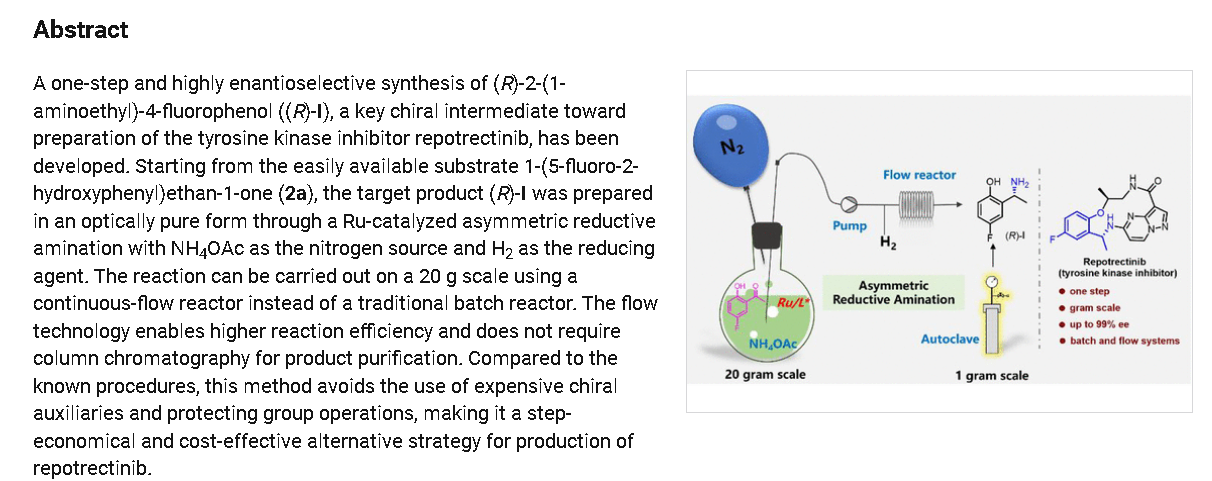
REF
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00061
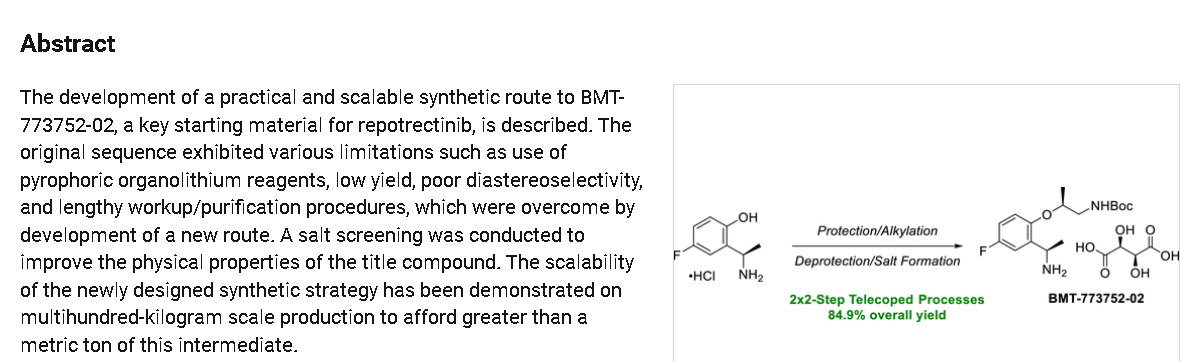
REF
US20180194777
https://patentscope.wipo.int/search/en/detail.jsf?docId=US222923082&_cid=P11-ME283N-03701-1
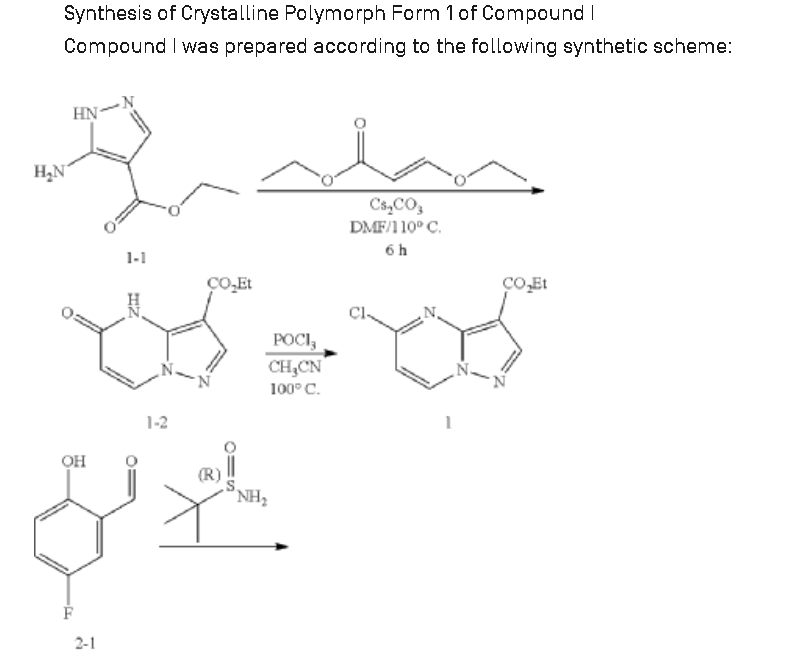
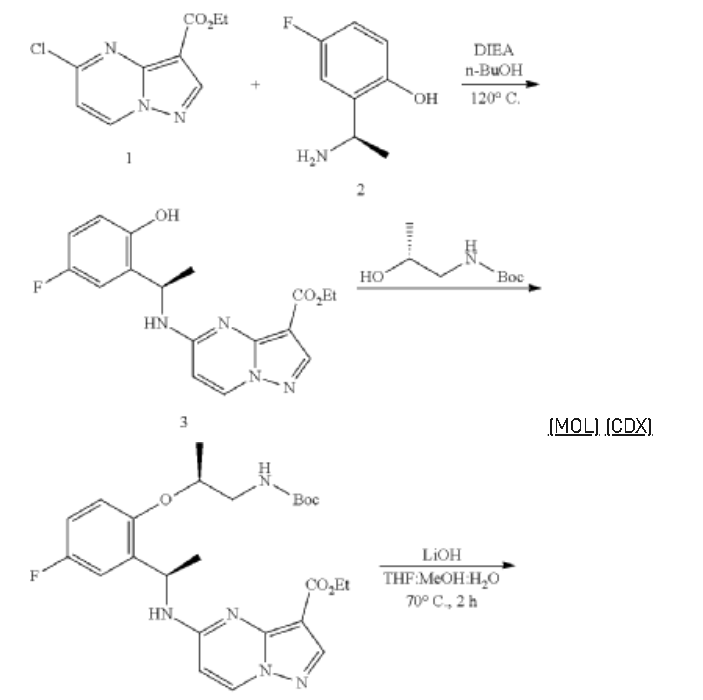
Example 1: Preparation of 5-chloropyrazolo[1,5-a]pyrimidine-3-carboxylate (1)
Step 1: Preparation of ethyl 5-oxo-4H-pyrazolo[1,5-a]pyrimidine-3-carboxylate (1-2)
Step 2: Preparation of 5-chloropyrazolo[1,5-a]pyrimidine-3-carboxylate (1)
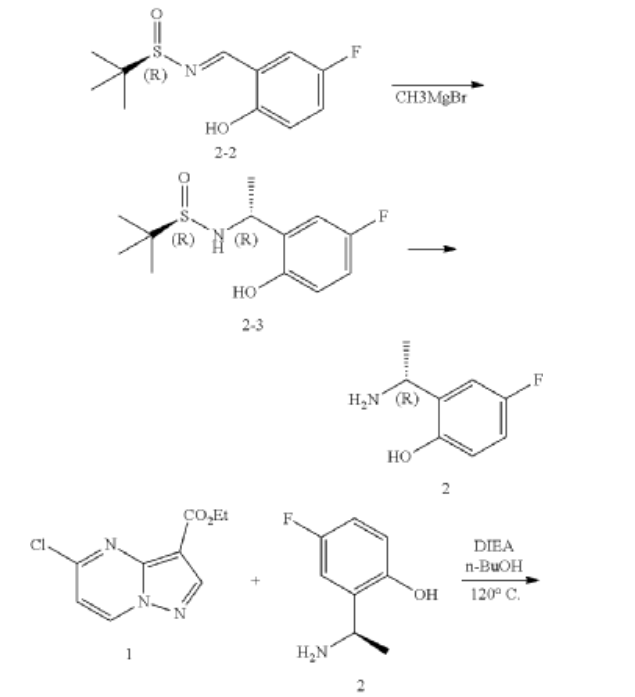
PATENT
https://patents.google.com/patent/US10246466B2/en
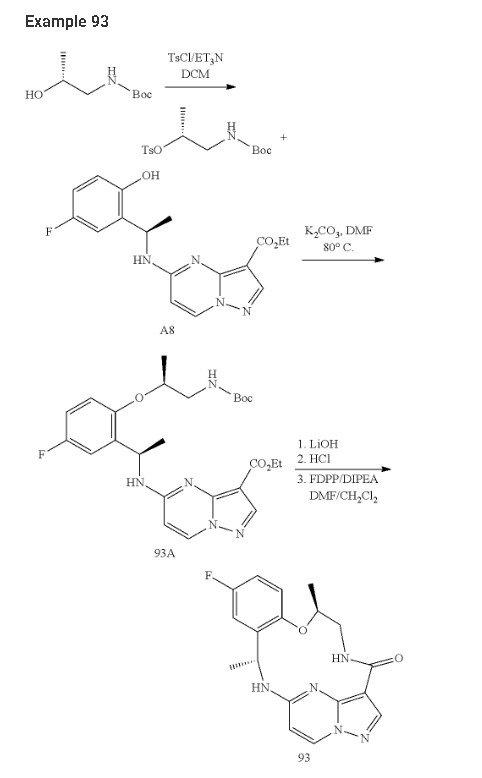
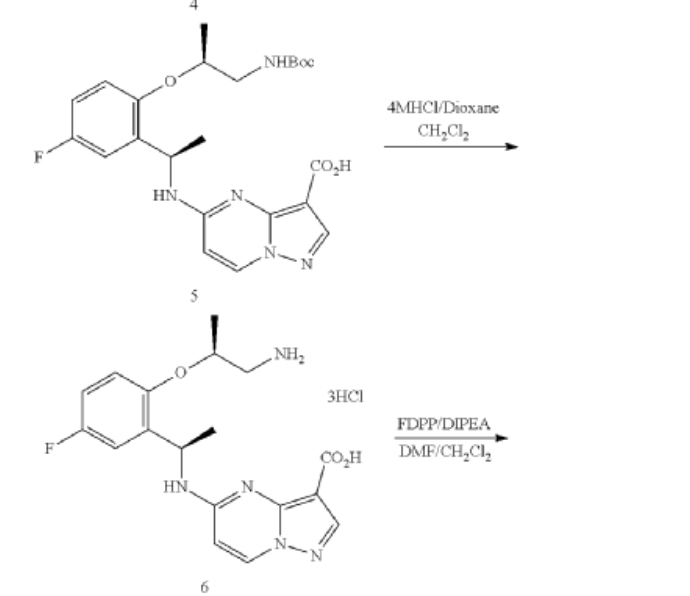
Step 1. To a solution of tert-butyl (R)-(2-hydroxypropyl)carbamate (1.00 g, 5.71 mmol) and tosyl chloride (1.14 g, 6.00 mmol) in DCM (29 mL) was added triethylamine (1.44 g, 14.28 mmol and the mixture was stirred at room temp for 48 hour. The reaction solution was concentrated under reduced pressure and the residue was purified with flash chromatography (ISCO system, silica (40 g), 0-20% ethyl acetate in hexane) to provide (R)-1-((tert-butoxycarbonyl)amino)propan-2-yl 4-methylbenzenesulfonate (1.12 g, 3.40 mmol, 59.54% yield).
Step 2. To a solution of A8 (100.00 mg, 0.290 mmol) and (R)-1-((tert-butoxycarbonyl)amino)propan-2-yl 4-methylbenzenesulfonate (143.50 mg, 0.436 mmol) in DMF (1.45 mL) was added K2CO3 (200.7 mg, 1.45 mmol) and heated at 80° C. with stirring for 16 hour. The reaction was cooled to ambient temperature and diluted with DCM (3 mL), filtered through a syringe filter, and concentrated under reduced pressure. Flash chromatography (ISCO system, silica (12 g), 0-60% ethyl acetate in hexane) provided 93A (32.90 mg, 0.0656 mmol, 22.59% yield).
Step 3. To a solution of 93A (32.90 mg, 0.0656 mmol) in MeOH (3 mL) and THF (2 mL) was added LiOH aqueous solution (2M, 2 mL) at ambient temperature. The reaction solution was heated at 70° C. for 2 hours The reaction flask was cooled to ambient temperature, diluted with water and methanol, and then quenched with HCl aqueous solution (2 M, 2 mL) to pH<5. The mixture was extracted with DCM (3×5 mL), dried with Na2SO4, concentrated under reduced and dried on high vacuum overnight. To a solution of the acid product in DCM (4 mL) was added 4 M HCl in 1,4-dioxane (2.0 mL). The mixture was stirred at room temperature for 3 hours, and then concentrated under reduced pressure and dried on high vacuum. To a solution of the de-Boc product and FDPP (27.62 mg, 0.0719 mmol) in DMF (1.6 mL) was added Hunig’s base (42.23 mg, 0.327 mmol) at room temperature. The mixture was stirred for 2.5 hours, and then quenched the reaction with 2 M Na2CO3 solution (2 mL). The mixture was stirred for 15 min then extracted with DCM (4×10 mL). The combined extracts were dried with Na2SO4 and concentrated under reduced pressure. The residue was purified with flash chromatography (ISCO system, silica (12 g), 0-10% methanol in dichloromethane) to provide 93 (10.1 mg, 0.0284 mmol, 43.49% yield for three steps).
PATENT
SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
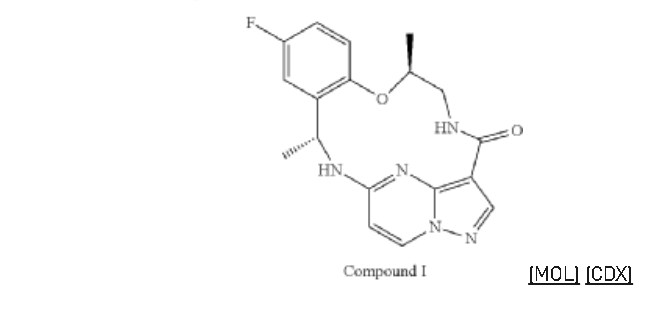
Repotrectinib (Augtyro) Repotrectinib, developed by Turning Point Therapeutics, Inc., was granted FDA approval on November 15, 2023. It is indicated to treat locally advanced or metastatic ROS proto-oncogene 1, receptor tyrosine kinase (ROS1)-positive non-small cell lung cancer (NSCLC). Repotrectinib is a highly effective inhibitor of ROS1 (ICtyrosine receptor kinase (TRK) (IC5050= 0.07 nM) and
=0.83/0.05/0.1 nM for TRKA/B/C) [87]. After undergoing currently approved targeted therapies, patients with tumors containing ROS1 and neurotrophic tyrosine kinase receptor (NTRK) gene fusions frequently acquire resistance mutations [88,89]. These mutations restrict the ability of drugs to bind to their
targets, ultimately resulting in the advancement of tumors. Repotrectinib, a novel tyrosine kinase inhibitor (TKI), is the pioneering drug developed to specifically target ROS1 or NTRK-positive metastatic
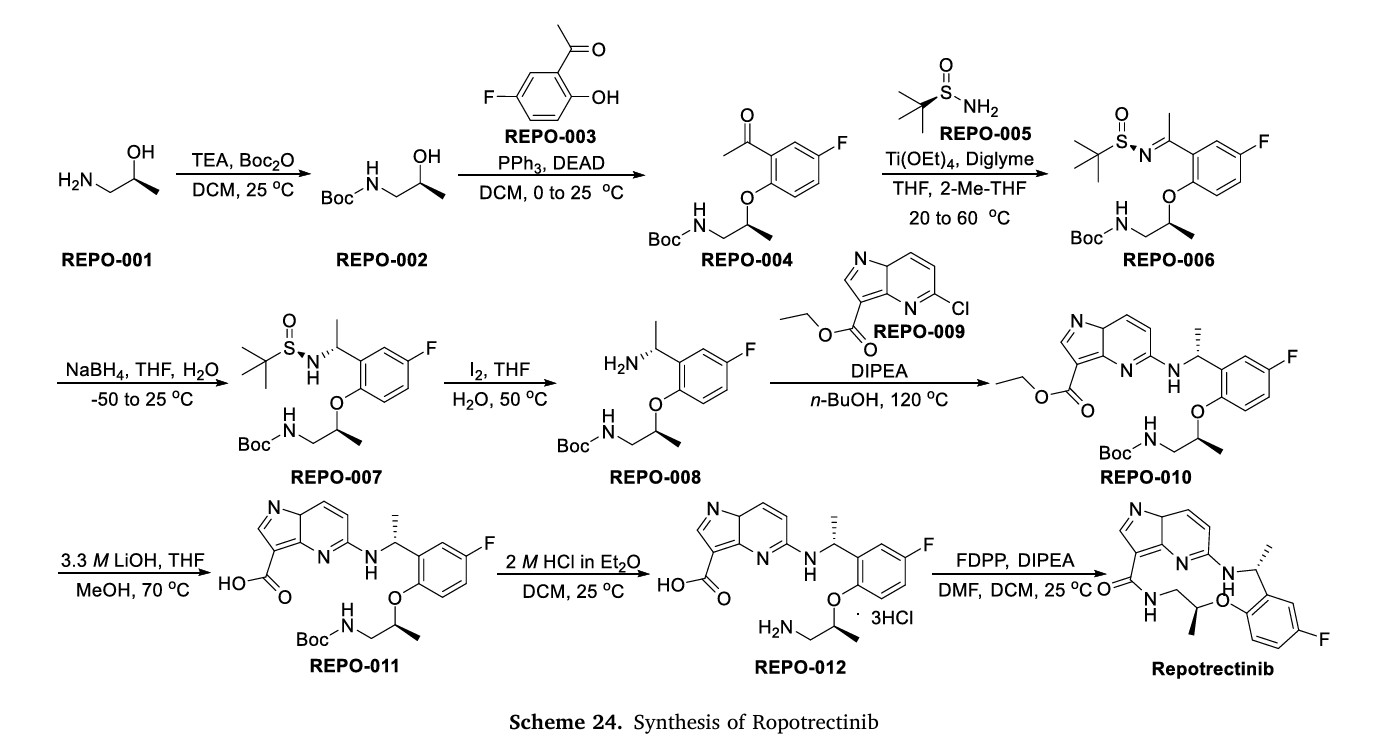
NSCLC and effectively combat the primary factors contributing to disease advancement [90].Preparation of Repotrectinib is described as Scheme 24 [91].Protecting the amino group of REPO-001 with Boc group in the presence of Kgave REPO-002, followed by intermolecular dehydration with
1-(5-fluoro-2-hydroxyphenyl)ethan-1-one (REPO-003) to give the ester REPO-004. REPO-004 was reacted with chiral auxiliary REPO-005 to give REPO-006, which was reduced by NaBH4
to obtain REPO-007. Then REPO-008 was obtained by removing the chiral auxiliary under iodine conditions. Substitution of REPO-008 with REPO-009 gave REPO-010, which was further hydrolyzed under alkaline conditions to obtain REPO-011. Salt formation of REPO-011 with hydrochloric acid
yielded REPO-012, which underwent intramolecular condensation to obtain the product Repotrectinib.
[87] D. Zhai, W. Deng, Z. Huang, E. Rogers, J.J. Cui, The novel, rationally-designed,
ALK/SRC inhibitor TPX-0005 overcomes multiple acquired resistance
mechanisms to current ALK inhibitors, Cancer Res. 76 (2016) 2132.
[88] C. Keddy, P. Shinde, K. Jones, S. Kaech, R. Somwar, U. Shinde, M.A. Davare,
Resistance profile and structural modeling of next-generation ROS1 tyrosine
kinase inhibitors, Mol. Cancer Therapeut. 21 (2022) 336–346.
[89] E. Cocco, M. Scaltriti, A. Drilon, NTRK fusion-positive cancers and TRK inhibitor
therapy, Nat. Rev. Clin. Oncol. 15 (2018) 731–747.
[90] A. Drilon, S.I. Ou, B.C. Cho, D.W. Kim, J. Lee, J.J. Lin, V.W. Zhu, M.J. Ahn, D.
R. Camidge, J. Nguyen, D. Zhai, W. Deng, Z. Huang, E. Rogers, J. Liu, J. Whitten,
J.K. Lim, S. Stopatschinskaja, D.M. Hyman, R.C. Doebele, J.J. Cui, A.T. Shaw,
Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that
potently inhibits ROS1/TRK/ALK solvent-front mutations, Cancer Discov. 8
(2018) 1227–1236.
[91] J.J. Cui, E.W. Rogers, Gialir Macrocyclic Polymorph, 2018. US20180194777A1.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
Repotrectinib, developed by Bristol-Myers Squibb and marketed under the brand name Augtyro, is an oral tyrosine kinase inhibitor (TKI) targeting ROS1 and TRK oncogenic drivers. In 2024, NMPA condition
ally approved Repotrectinib for adult patients with ROS1-positive locally advanced or metastatic NSCLC [15]. Repotrectinib exerts its antitumor activity by inhibiting ROS1 and TRK kinases, thereby disrupting the downstream signaling pathways that facilitate tumor cell proliferation and survival [16]. This argeted mechanism is particularly effective against tumors that harbor ROS1 or NTRK gene fusions. The clinical efficacy of Repotrectinib has been through validated the Phase 1/2 TRIDENT-1 trial (NCT03093116) [17]. In the study cohort, treat ment-naïve patients harboring ROS1-positive NSCLC exhibited an overall response rate (ORR) of 79 %, characterized by a median duration of response (DOR) reaching 34.1 months. Conversely, among those who had previously received ROS1 TKI therapy, the ORR was documented at 38 %, accompanied by a median DOR of 14.8 months. With respect to safety profiles, the adverse event spectrum commonly encompassed dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and nausea
[18,19]. These side effects are generally manageable, but patients should be monitored for potential severe adverse events.
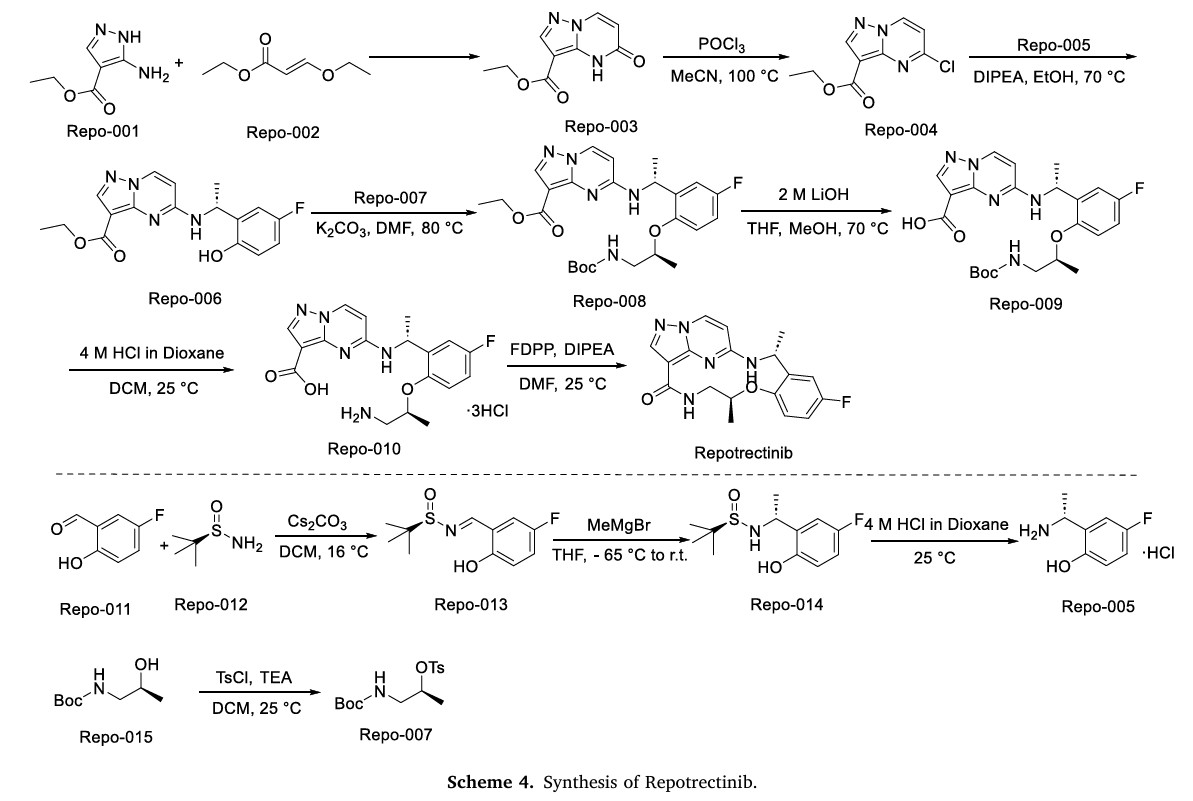
The synthetic route of Repotrectinib, shown in Scheme 4, begins with condensation reaction between Repo-001 and Repo-002 to afford Repo-003, which is chlorinated to yield Repo-004 [20]. This intermediate undergoes nucleophilic substitution with Repo-005 to form Repo-006,
followed by second nucleophilic substitution with Repo-007 to produce Repo-008. Ester hydrolysis of Repo-008 affords Repo-009, which undergoes acid-mediated deprotection to generate Repo-010. Final
intramolecular amidation of Repo-010 delivers Repotrectinib. In parallel, Repo-011 and Repo-012 undergo condensation to form imine Repo-013, which undergoes Grignard addition to afford Repo-014.
Acidification of Repo-014 then yields Repo-005. Concurrently, Repo-015 undergoes nucleophilic substitution to generate Repo-007.
[15] S. Dhillon, Repotrectinib: first approval, Drugs 84 (2024) 239–246.
[16] T. Rais, A. Shakeel, L. Naseem, N. Nasser, M. Aamir, Repotrectinib: a promising
new therapy for advanced nonsmall cell lung cancer, Ann Med Surg (Lond) 86
(2024) 7265–7269.
[17] A. Drilon, S.I. Ou, B.C. Cho, D.W. Kim, J. Lee, J.J. Lin, V.W. Zhu, M.J. Ahn, D.
R. Camidge, J. Nguyen, D. Zhai, W. Deng, Z. Huang, E. Rogers, J. Liu, J. Whitten, J.
K. Lim, S. Stopatschinskaja, D.M. Hyman, R.C. Doebele, J.J. Cui, A.T. Shaw,
Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that
potently inhibits ROS1/TRK/ALK solvent-front mutations, Cancer Discov. 8 (2018)
1227–1236.
[18] Repotrectinib, Drugs and Lactation Database (Lactmed®), National Institute of
Child Health and Human Development, Bethesda (MD), 2006.
[19] H. Zhong, J. Lu, M. Wang, B. Han, Real-world studies of crizotinib in patients with
ROS1-positive non-small-cell lung cancer: experience from China, J Comp Eff Res
14 (2024) e240043.
[20] J.J. Cui, E.W. Rogers, Preparation of
Fluorodimethyltetrahydroethenopyrazolobenzoxatriazacyclotridecinone
Derivatives for Use as Antitumor Agents, 2017. US20180194777A1.
Repotrectinib is indicated for the treatment of adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer.[2][5]
In June 2024, the US Food and Drug Administration (FDA) expanded the indication to include the treatment of people twelve years of age and older with solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion, are locally advanced or metastatic or where surgical resection is likely to result in severe morbidity, and that have progressed following treatment or have no satisfactory alternative therapy.[7][8]
References
- “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 23 May 2025.
- “Augtyro- repotrectinib capsule”. DailyMed. 15 November 2023. Archived from the original on 12 December 2023. Retrieved 12 December 2023.
- “Augtyro EPAR”. European Medicines Agency (EMA). 14 November 2024. Retrieved 16 November 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Augtyro PI”. Union Register of medicinal products. 14 January 2025. Retrieved 16 January 2025.
- “FDA approves repotrectinib for ROS1-positive non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 15 November 2023. Archived from the original on 16 November 2023. Retrieved 17 November 2023. This article incorporates text from this source, which is in the public domain.
- “U.S. Food and Drug Administration Approves Augtyro (repotrectinib), a Next-Generation Tyrosine Kinase Inhibitor (TKI), for the Treatment of Locally Advanced or Metastatic ROS1-Positive Non-Small Cell Lung Cancer (NSCLC)” (Press release). Bristol Myers Squibb. 16 November 2023. Archived from the original on 16 November 2023. Retrieved 17 November 2023 – via Business Wire.
- “FDA grants accelerated approval to repotrectinib for adult and pediatric participants with neurotrophic tyrosine receptor kinase gene fusion-positive solid tumors”. U.S. Food and Drug Administration. 13 June 2024. Archived from the original on 13 June 2024. Retrieved 13 June 2024. This article incorporates text from this source, which is in the public domain.
- “Cancer Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 6 December 2024.
- Turning Point Therapeutics, Inc. (5 February 2024). A Phase 1/2, Open-Label, Multi-Center, First-in-Human Study of the Safety, Tolerability, Pharmacokinetics, and Anti-Tumor Activity of TPX-0005 in Patients With Advanced Solid Tumors Harboring ALK, ROS1, or NTRK1-3 Rearrangements (TRIDENT-1) (Report). clinicaltrials.gov. Archived from the original on 18 June 2024. Retrieved 18 June 2024.
- “Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 11-14 November 2024”. European Medicines Agency (EMA). 15 November 2024. Retrieved 16 November 2024.
Further reading
- Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. (October 2018). “Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations”. Cancer Discovery. 8 (10): 1227–1236. doi:10.1158/2159-8290.CD-18-0484. PMID 30093503.
External links
- “Repotrectinib (Code C133821)”. NCI Thesaurus. 25 September 2023. Retrieved 17 November 2023.
| Clinical data | |
|---|---|
| Trade names | Augtyro |
| Other names | TPX-0005 |
| AHFS/Drugs.com | Augtyro |
| License data | US DailyMed: Repotrectinib |
| Routes of administration | By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code | L01EX28 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3][4] |
| Identifiers | |
| CAS Number | 1802220-02-5 |
| PubChem CID | 135565923 |
| DrugBank | DB16826 |
| ChemSpider | 64853849 |
| UNII | 08O3FQ4UNP |
| KEGG | D11454 |
| ChEBI | CHEBI:229220 |
| ChEMBL | ChEMBL4298138 |
| PDB ligand | 7GI (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H18FN5O2 |
| Molar mass | 355.373 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- (3R,6S,)-45-FLUORO-3,6-DIMETHYL-5-OXA-2,8-DIAZA-1(5,3)-PYRAZOLO(1,5-A)PYRIMIDINA-4(1,2)-BENZENANONAPHAN-9-ONE
- (7S,13R)-11-Fluoro-7,13-Dimethyl-6,7,13,14-Tetrahydro-1,15-Ethenopyrazolo[4,3-F][1,4,8,10]Benzoxatriazacyclotridecin-4(5H)-One
- 1,15-ETHENO-1H-PYRAZOLO(4,3-F)(1,4,8,10)BENZOXATRIAZACYCLOTRIDECIN-4(5H)-ONE, 11-FLUORO-6,7,13,14-TETRAHYDRO-7,13-DIMETHYL-, (7S,13R)-
////////Ropotrectinib, FDA 2023, APPROVALS 2023, Turning Point , EU 2025, APPROVALS 2025, EMA 2025, Augtyro, TPX 0005, CHINA 2024, APPROVALS 2024
Vamorolone
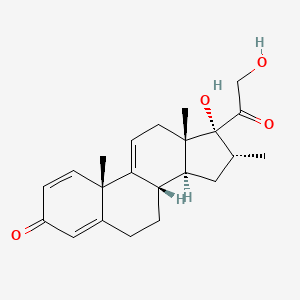

Vamorolone
CAS 13209-41-1
| Molecular Weight | 356.46 |
|---|---|
| Formula | C22H28O4 |
- Agamree
- 17,21-Dihydroxy-16alpha-methylpregna-1,4,9(11)-triene-3,20-dione
- VBP-15 free alcohol
- (8S,10S,13S,14S,16R,17R)-17-hydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-7,8,12,14,15,16-hexahydro-6H-cyclopenta[a]phenanthren-3-one
- (16alpha)-17,21-dihydroxy-16-methylpregna-1,4,9(11)-triene-3,20-dione
- (8S,10S,13S,14S,16R,17R)-17-hydroxy-17-(2-hydroxyacetyl)-10,13,16-trimethyl-7,8,12,14,15,16-hexahydro-6H-cyclopenta(a)phenanthren-3-one
- DTXCID601356317
- (1R,2R,3aS,3bS,9aS,11aS)-1-hydroxy-1-(2-hydroxyacetyl)-2,9a,11a-trimethyl-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,11H,11aH-cyclopenta(a)phenanthren-7-one
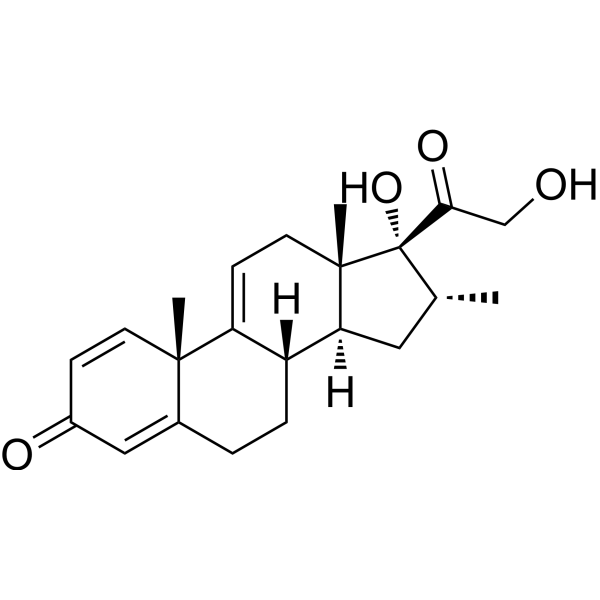
Vamorolone (VBP15) is a first-in-class, orally active dissociative steroidal anti-inflammatory agent and membrane-stabilizer. Vamorolone improves muscular dystrophy without side effects. Vamorolone shows potent NF-κB inhibition and substantially reduces hormonal effects.
Vamorolone, sold under the brand name Agamree, is a synthetic corticosteroid, which is used for the treatment of Duchenne muscular dystrophy.[4][5][6][7][8] It is taken by mouth.[1] It is a dual atypical glucocorticoid and antimineralocorticoid.[9]
The most common adverse reactions include cushingoid features, psychiatric disorders, vomiting, increased weight, and vitamin D deficiency.[10]
Vamorolone was approved for medical use in the United States in October 2023,[11][10] and in the European Union in December 2023.[2][3]
Vamorolone is a novel and fully synthetic glucocorticoid developed by Santhera Pharmaceuticals. It is used to manage inflammation and immune dysregulation in patients with Duchenne muscular dystrophy (DMD), a neuromuscular disorder characterized by the insidious regression of neuromuscular function and the most common form of muscular dystrophy in the United States. Corticosteroid therapy is the current standard of care for DMD despite relatively high rates of adverse effects. Vamorolone is positioned as having a more tolerable adverse effect profile than other corticosteroids owing to its unique receptor binding profile, thus providing an additional treatment option in patients for whom corticosteroid adverse effects are intolerable or otherwise unacceptable. Vamorolone was approved by the FDA in October 2023 for the management of DMD in patients ≥2 years of age. In December 2023, it was approved in the EU for the treatment of patients ≥4 years of age.
PATENT
https://patents.google.com/patent/WO2023016817A1/en
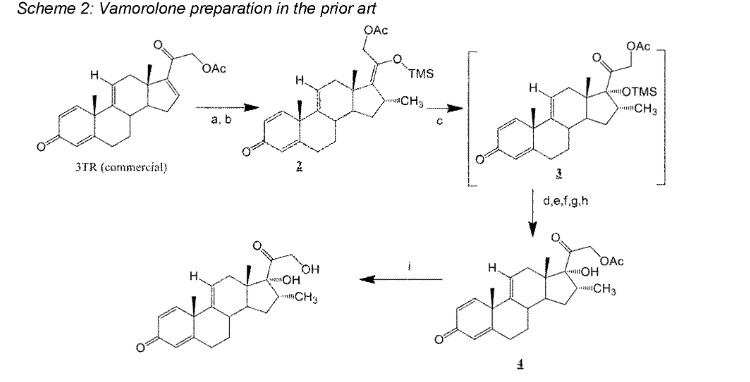
Vamorolone is currently produced from the commercially available 3TR (Tetraene acetate) – see Scheme 2. In step a, TMS imidazole, MeMgCI and THF are added to 3TR, with subsequent addition of CuAc2, H2O, DMPU, MeMgCI and THF in step b. Under treatment with peracetic acid in Toluene from compound 2 the intermediate 3 is formed in step c. After treatment with NaHSCO3 and TFA (step d), EtOAc and heptane (step e) and acetonitrile trituration (step f) HBr in CH2CI2 is added (step g) and MeOH (step h) is used for crystallization to form Acetyl- Vamorolone 4. Acetyl-Vamorolone is deacetylated with K2CO3 in MeOH, followed by HCI to obtain Vamorolone (step i). The synthesis is disclosed in Bioorganic & Medicinal Chemistry, Volume 21 , Issue 8, 15 April 2013, Pages 2241-2249.
Example 2: Synthesis of the present invention
Scheme C: Route of Synthesis of Vamorolone from 8-DM

Vamorolone was synthesized in three synthetic steps from commercially available 8-DM.
The synthetic route started with the acetylation of 8-DM using acetic anhydride and catalytic DMAP in THF, followed by crystallization of 8-DM Acetate after aqueous quench. Then, a deoxygenation reaction converted 8-DM Acetate directly into Vamorolone Acetate. This deoxygenation proceeded via initial formation of an iodohydrin with excess aq. HI, followed by simultaneous I2 and H2O-elimination to give Vamorolone Acetate. During the reaction, partial de-acetylation occurred (20-25%) and therefore re-acetylation with acetic anhydride was necessary. After completed re-acetylation, Vamorolone Acetate was directly crystallized by addition of H2O. Finally, the acetate group is cleaved under basic conditions to give crude Vamorolone, which was recrystallized from iPrOH to obtain the pure product.
2.1 Acetylation

A 10 L glass dj ( double jacketed reactor )-reactor was charged with 8-DM (490 g, 1.32 mol, 1.0 eq.) and DMAP (16.1 g, 0.132 mmol, 0.10 eq.). THF (1.25 L, 2.5 vol.) was added at IT = 20-25 °C. Then, AC2O (201 g, 187 mL, 1.97 mol, 1.5 eq.) was added dropwise over 20-40 min, keeping IT below 30 °C during the addition. After complete addition, the reaction mixture was stirred at IT = 20-25 °C for 30 min. IPC control by LC/MS indicated >99% conversion of 8-DM to 8-DM Acetate.
The reaction mixture was quenched by dropwise addition of H2O (4.9 L, 10 vol.) over 30-45 min, keeping IT below 25 °C. The resulting aqueous suspension was aged at IT = 20-25 °C for 1 h. The product was filtered off, washed with H2O (3 x 0.5 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide 8-DM Acetate (539 g, 1.30 mol, 99% yield, >99% a/a, 98% w/w) as a white solid (cryst 1#1).
Analytical Data:
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of 8-DM relative to formation of 8- DM Acetate.
Detected mass: [M+1]= 373.19 for 8-DM and [M+1] = 415,19 8-DM Acetate. 2.2 Deoxygenation with HI

A 10 L glass dj-reactor was charged with 8-DM Acetate (500 g, 1.21 mol, 1.0 eq.). Toluene (2.5 L, 5 vol.) was added. The suspension was cooled to IT = 0-5 °C and then a solution of 57% aqueous HI (1 .08 kg, 637 mL, 4.83 mol, 4.0 eq.) in AcOH (1.25 L, 2.5 vol.) was added via peristaltic pump over 45-60 min, keeping IT below 5 °C during the addition. The resulting dark purple to brown solution was stirred at IT = 3-5 °C for 24 h. IPC control by LC/MS indicated >98% conversion of 8-DM Acetate/intermediate iodohydrin to Vamorolone Acetate/Vamorolone.
The reaction mixture was quenched by dropwise addition of 25% aq. Na2SO3 solution (2.0 L, 4 vol.) over 10-20 min, keeping IT below 15 °C. After complete addition, EtOAc (1.0 L, 2 vol.) was added and the biphasic mixture was warmed to IT = 15-20 °C. Stirring was stopped and the phases were separated (Organic Phase 1 and aqueous Phase 1 ; goal pH of the aqueous Phase 1 : 2; aqueous Phase 1 disposed). 25% aq. Na2SO3 solution (1.25 L, 2.5 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 2; goal pH aqueous Phase 2: 4-5; aqueous Phase 2 disposed). 25% aq. Na2SO3 solution (1.25 L, 2.5 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 3; goal pH aqueous Phase 3: 5-6; aqueous Phase 3 disposed). H2O (0.5 L, 1.0 vol.) was added to Organic Phase 1 and the biphasic mixture was stirred at IT = 15-20 °C for 5 min, stirring was stopped and phases separated (Organic Phase 1 and aqueous Phase 4; goal pH aqueous Phase 4: 5-6; aqueous Phase 4 disposed).
A slight vacuum was applied to the double-jacketed reactor (100-150 mbar), containing Organic Phase 1 , and toluene was distilled off at 70 °C jacket temperature (ET) from the reaction mixture with continuous addition of MeCN, and the distillation continued until target residual toluene value has been reached (goal: less than 5% toluene according to 1 H-NMR of reaction mixture. Final volume in reactor after distillation: ca. 3.5 L (7.5 vol.).
Once toluene was removed, the vacuum was broken with N2 and resulting fine suspension cooled to IT = 20-25 °C. At this point, the amount of Vamorolone was assessed by IPC (typical ratio: Vamorolone Acetate to Vamorolone: 75:25; x = 25% a/a). DMAP (3.7 g, 0.0302 mol, 0.025 eq.) was added, followed by slow addition of AC2O (61.6 g, 57 mL, 0.603 mol, 0.5 eq.) over 5-10 min at IT = 20-25 °C. After complete addition of AC2O, the reaction mixture was stirred for 30 min at IT = 20-25 °C. IPC control by LC/MS indicated ≤ 2% a/a Vamorolone (ratio: Vamorolone Acetate to Vamorolone: 98.5:1.5).
The reaction mixture was quenched by slow addition of H2O (4.9 L, 10 vol.) over 15-30 min, keeping IT below 25 °C. The resulting aqueous suspension was cooled to IT = 0-5 °C and aged at this temperature for 2 h. The product was filtered off, washed with H2O/MeCN 4:1 (2 x 0.5 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone Acetate (301 g, 0.76 mol, 63% yield, 98% a/a, 98% w/w) as an off-white solid (cryst 1#1).
Over the course of the reaction, partial de-acetylation of Vamorolone Acetate to Vamorolone was observed (between 20-25% a/a). Therefore, after aq. workup and solvent switch to MeCN, the ratio of Vamorolone Acetate to Vamorolone was assessed by LC/MS (in % a/a), and the following amounts of DMAP and Ac2O were added: x = amount of Vamorolone in % a/a (e.g. x = 20% a/a)
DMAP eq. = (0.1 -x)/100 (e.g. 0.02 eq.)
Ac2O eq. = (2.0-x)/100 (e.g. 0.40 eq.)
Analytical Data
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of the sum of (8-DM Acetate + intermediate iodohydrin) relative to the sum of (Vamorolone Acetate + Vamorolone). Detected mass: [M+1] = 415,19 for 8-DM Acetate, [M+1]= 357,28 for Vamorolone, 399,20 for
Vamorolone Acetate and 543,12 for intermediate lodohydrin
2.3 De-Acetylation

A 10 L glass dj-reactor was charged with Vamorolone Acetate (280 g, 0.703 mol, 1.0 eq.). MeOH (1.54 L, 5.5 vol.) was added. The suspension was cooled to IT = 0-5 °C and then a solution of K2CO3 (107 g, 0.773 mol, 1.1 eq.) in H2O (0.7 L, 2.5 vol.) was added dropwise via peristaltic pump over 20-40 min, keeping IT below 10 °C during the addition. After complete addition, the reaction mixture was warmed IT = 20-25 °C and stirred for 5 h. IPC control by LC/MS indicated 99.3% conversion of Vamorolone Acetate to Vamorolone.
The reaction mixture was cooled to IT = 15-17 °C and quenched by dropwise addition of 1 M aq. HCI (950 mL, 0.95 mol, 1.35 eq.) over 20-40 min, keeping IT below 20 °C during the addition (goal pH: 5-6). The resulting aqueous suspension was aged at IT = 15-20 °C for 12 h. The product was filtered off, washed with H2O/MeOH 2:1 (3 x 0.3 L), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone (241.5 g, 0.68 mol, 96% yield, >99% a/a, 98% w/w) as a slightly yellow solid (crude 1#1).
Analytical Data
LC/MS column: Zorbax RRHD SB-Aq, 2.1x50mm, 1.8pm
Program: G_005%B_TFA_0,800ml_2,00min
Eluent A: Water/TFA 100:0.04, Eluent B: Acetonitrile
IPC preparation for LC/MS
10 microliter in 1 mL H2O:MeCN 1 :1
Conversion was determined with respect to consumption of Vamorolone Acetate relative to formation of Vamorolone. 2.4 Recrystallization

A 10 L glass dj-reactor was charged with Vamorolone (230 g, 0.645 mol, 1.0 eq.). iPrOH (5 L, 22 vol.) was added. The suspension was heated to reflux (jacket temperature ET = 97 °C) and stirred until complete dissolution of Vamorolone occurred (10-15 min on this scale).
After complete dissolution, the clear yellow solution was slowly cooled to IT = 0-5 °C over the course of 12 h and then aged at IT = 0-5 °C for 1 h. The recrystallized product was filtered off, washed with cold iPrOH (2 x 250 mL), and dried on a rotary evaporator (900-10 mbar, 65 °C bath temperature) to provide Vamorolone (201 g, 87% recovery, >99% a/a, 99% w/w) as an off white glimmery solid (cryst 1#1).
Iso-propanol (iPrOH) was found to the best solvent for recrystallization with excellent purity upgrading properties (by rejection of impurities), although a high dilution is necessary to completely dissolve the crude Vamorolone at reflux temperature. Higher concentrations for the recrystallization satisfactory results are obtainable using mixtures of isopropanol and water. Maximum solubility of Vamorolone was determined to be at reflux of a 80:20 (isopropanol : water) mixture.
PATENT
https://patents.google.com/patent/US20200281942A1/en
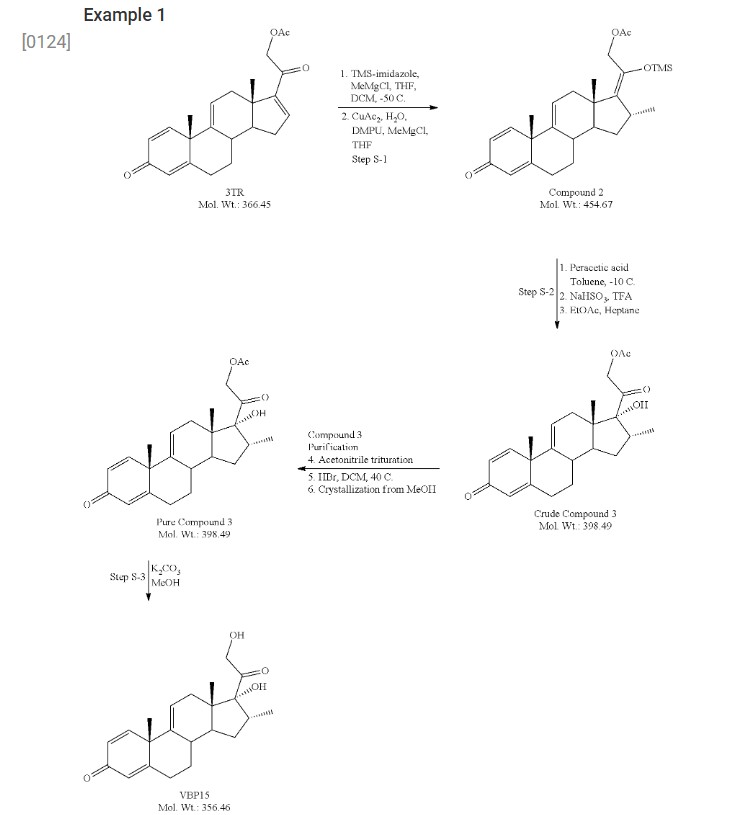
- [0124]
- [0125]3-TR (100 g, 273 mmol), dichloromethane (DCM, 500 mL) and tetrahydrofuran (THF, 400 mL) were charged to a reaction flask under nitrogen. To this was charged trimethylsilyl imidazole (TMS-imidazole, 65.3 g, 466 mmol, 1.7 eq). The resulting mixture was stirred at room temperature for 3 hours.
- [0126]In a separate flask, copper acetate monohydrate (5.4 g, 27 mmol), tetrahydrofuran (400 ml) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU, 53.3 g, 416 mmol) were combined and stirred at room temperature for approximately 3 hours. The blue mixture was subsequently cooled to −50° C., and to this was added methyl magnesium chloride solution (27 ml, 3.0 M in THF, 82 mmol) dropwise. After 30 minutes, the mixture had formed a deep blue, sticky “ball.”
- [0127]The 3-TR/TMS-imidazole mixture was cooled to −50° C. and to this was charged the copper acetate/DMPU solution above via canula. The residual sticky mass from the copper acetate/DMPU mixture was dissolved using DCM (50 mL) and also transferred.
- [0128]Methyl magnesium chloride (123.2 mL, 3.0 M solution in THF, 368 mmol) was added dropwise over 45 minutes to the combined reaction mixtures, which were then allowed to stir for 2 hours at −50° C. Subsequent HPLC analysis showed complete consumption of starting material. The mixture was allowed to warm to room temperature overnight, with stirring.
- [0129]Toluene (800 mL) was added to the mixture, followed by 5% acetic acid solution (600 mL). The aqueous layer was removed and discarded. The acetic acid wash was repeated. The organic layer was washed with brine (400 mL), 5% sodium bicarbonate solution (400 mL×2), followed by a brine wash (400 mL). The organic solution was dried over sodium sulfate, then concentrated to dryness under reduced pressure. The product was recovered as a viscous, light golden oil. Mass recovery was 146 grams (119% of theoretical).
- [0130]Compound 2 (92 g, 202 mmol) and toluene (1000 mL, 10.9 vol) were charged to a reaction flask under nitrogen and the solution was cooled to −10° C. A 32 wt % solution of peracetic acid in acetic acid (60 mL, 283 mmol, 1.4 eq) was added dropwise over about 30 min maintaining the temperature at −10° C. The reaction was held for approximately 20 h (HPLC showed 75% Cmpd 3, Cmpd 2 1.5%, 6% diastereomer; 5% epoxide). Starting at −10° C., a 20% aqueous solution of sodium bisulfite (920 mL, 10 vol) was added carefully via addition funnel, keeping the temperature below 10° C. Trifluoroacetic acid (16 mL, 202 mmol, 1 eq) was added and the mixture was held for 3 h at 0-5° C. to complete desilylation (endpoint by HPLC). The lower aqueous layer was drained, and the organic layer was washed with a saturated solution of sodium bicarbonate (3×250 mL), followed by water (1×250 mL), and brine (1×150 mL). The organic layer was then dried over Na2SO4, filtered and concentrated to a pasty solid (89 g). The residue was taken up in 1.5 vol of EtOAc and transferred to neat heptane (19 vol) to precipitate crude Cmpd 3 as an off-white solid (50 g, 62.5% yield; HPLC 79% Cmpd 3, 5.6% epoxide, 1.7% diastereomer). The crude Cmpd 3 (48.5 g) was triturated in hot acetonitrile (2 vol) at 60° C. for 4 h, and then gradually cooled to ambient temperature overnight. The mixture was filtered using the recycled filtrate to rinse and wash the wet cake. After drying, the recovery was 64.3% (31.2 g; HPLC 93.5% Cmpd 3, 3.3% epoxide). To remove the epoxide impurity, the 31 Cmpd 3 was dissolved in DCM (250 mL, 8 vol) and a solution of 48% HBr in water was added (7.5 mL). The mixture was heated at 40° C. for 1 h (HPLC<0.3% epoxide). The mixture was cooled and transferred to a separatory funnel. The lower aqueous layer (brown) was removed and the upper organic layer was washed with water (200 mL), saturated NaHCO3 (150 mL), and brine (100 mL). The organic layer was dried over Na2SO4, filtered, and concentrated to a tan foam (32 g, ˜100% recovery). Methanol (64 mL, 2 vol) was added to the 32 g foam forming a slurry. To this was added a 1:1 solution of MeOH:water (60 mL, 2 vol) dropwise. The slurry cooled to slightly below ambient temperature and filtered using recycled filtrate to rinse and wash the wet cake. The solids were dried to constant weight, affording 26.1 g Cmpd 3 (81% recovery; HPLC 97.8%). The overall yield for Step 2 was 32.5%.
- [0131]Compound 3 (26 g, 65 mmol) and MeOH (156 mL, 6 vol) were mixed in a reaction flask and cooled to 0-5° C. A solution of K2CO3 (9.9 g, 72 mmol, 1.1 eq) in water (65 mL) was added dropwise, and the mixture was allowed to gradually warm to ambient temperature overnight. Analysis by HPLC showed 2.5% SM and another 5 mol % K2CO3 was added and the mixture stirred for another day (HPLC endpoint 1.1% Cmpd 3). The mixture was neutralized to pH 7 with 1.5 M HCl (53 mL) and ˜25% of the MeOH (30 g) was removed under vacuum to maximize recovery. After stirring for 2 days, the product was isolated by filtration using the recycled filtrate to aid transferring the wet cake to the funnel. The wet cake was dried under vacuum, affording 19.3 g VBP15 (83% yield) as an off-white powder. Analysis of the solids by HPLC showed 98.8% purity with 0.6% Cmpd 3 as the only major impurity.
- [0132]Power X-Ray Diffraction (pXRD)
- [0133]The solid samples were examined using X-ray diffractometer (Bruker D8 advance). The system is equipped with highly-parallel x-ray beams (Gobel Mirror) and LynxEye detector. The samples were scanned from 3 to 40°2θ, at a step size 0.02°2θ and a time per step of 19.70 seconds. The tube voltage and current were 45 kV and 40 mA, respectively. The sample was transferred from sample container onto zero background XRD-holder and gently ground.
Syn
EuropeanJournalofMedicinalChemistry265(2024)116124
Vamorolone (Agamree)
On October 26, 2023, Vamorolone, developed jointly by Santhera Pharmaceuticals and ReveraGen BioPharma, has received FDA approval to treat DMD in patients aged 2 years and older [1]. DMD is a prevalent neuromuscular disorder in childhood, ranking among the most common.
This condition is caused by mutations in the gene responsible for producing the dystrophin protein, which plays a crucial role in maintaining muscle integrity. Moreover, DMD is an X-linked genetic disorder [69]. Vamorolone is a novel steroidal anti-inflammatory and membrane-stabilizing agent that can be taken orally. The distinction between it and traditional corticosteroid drugs lies in its capacity to
specifically activate particular signaling pathways of corticosteroids. In individuals diagnosed with DMD, the primary mechanism through which corticosteroid drugs exhibit their effectiveness is by exerting
anti-inflammatory effects. However, the secondary activities of corticosteroids can lead to adverse effects that impact the overall well-being of patients. Vamorolone has the ability to decrease the occurrence of
adverse effects while still preserving the therapeutic effectiveness of corticosteroids in individuals with DMD [70].
Preparation of Vamorolone is depicted in Scheme 19, which began with commercially available steroid 3 TR VAMO-001 [71]. Copper catalyzed addition of VAMO-001 with trimethylsilyl chloride (TMSCl)
gave silyl enol ether VAMO-002. VAMO-002 was oxidized by peracetic acid in acetic acid to yield intermediate VAMO-003, which was deprotected and hydrolyzed to obtain Vamorolone.
[69] D. Duan, N. Goemans, S. Takeda, E. Mercuri, A. Aartsma-Rus, Duchenne muscular
dystrophy, Nat. Rev. Dis. Prim. 7 (2021) 13.
[70] M. Guglieri, P.R. Clemens, S.J. Perlman, E.C. Smith, I. Horrocks, R.S. Finkel, J.
K. Mah, N. Deconinck, N. Goemans, J. Haberlova, V. Straub, L.J. Mengle-Gaw, B.
D. Schwartz, A.D. Harper, P.B. Shieh, L. De Waele, D. Castro, M.L. Yang, M.
M. Ryan, C.M. McDonald, M. Tulinius, R. Webster, H.J. McMillan, N.L. Kuntz, V.
K. Rao, G. Baranello, S. Spinty, A.M. Childs, A.M. Sbrocchi, K.A. Selby,
M. Monduy, Y. Nevo, J.J. Vilchez-Padilla, A. Nascimento-Osorio, E.H. Niks, I.J.
M. de Groot, M. Katsalouli, M.K. James, J. van den Anker, J.M. Damsker,
A. Ahmet, L.M. Ward, M. Jaros, P. Shale, U.J. Dang, E.P. Hoffman, Efficacy and
safety of vamorolone vs placebo and prednisone among boys with duchenne
muscular dystrophy: a randomized clinical trial, JAMA Neurol. 79 (2022)
1005–1014.
[71] E.K.M. Reeves, E.P. Hoffman, K. Nagaraju, J.M. Damsker, J.M. McCall, VBP15:
preclinical characterization of a novel anti-inflammatory delta 9,11 steroid,
Bioorg. Med. Chem. 21 (2013) 2241–2249.

Syn
J. Med. Chem. 2025, 68, 2147−2182
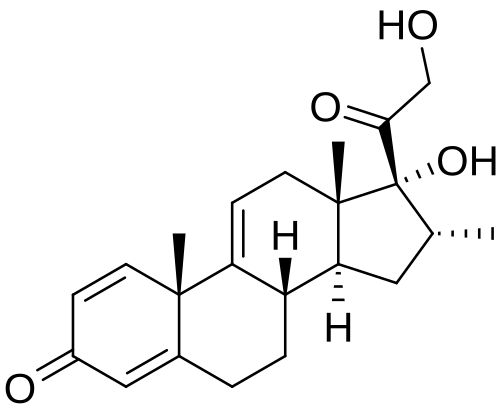
Vamorolone (Agamree). Developed by Santhera and ReveraGen BioPharma, the corticosteroid vamorolone (9) was approved for the treatment of Duchenne muscular dystrophy in October 2023.
70 Traditional corticosteroid treatment has been hampered by safety concerns including decreased bone mineral density and increased muscle atrophy. 71−73 Vamorolone is structurally distinct from other corticosteroids such as prednisone (Figure 3). 74 Removalofthe11βcarbonylmaintains binding to the glucocorticoid receptor but results in mineralocorticoid receptor antagonism; prednisone is a
mineralocorticoid receptor agonist. 75,76 This also results in decreased glucocorticoid receptor-drive transactivation, ultimately improving the safety profile of vamorolone as compared to other corticosteroid therapies. 74 The synthesis of vamorolone (9) as disclosed by ReveraGen BioPharma is summarized in Scheme 13. 77 Readily available steroid 9.1 was subjected to copper-catalyzed Michael addition.
Thein situ generated enolate was trapped using TMS-imidazole 9.2, providing the silyl enol ether 9.3. Treatment of crude 9.3 with peracetic acid 9.4 resulted in oxidized intermediate 9.5. Quenching of the peracetic acid and silyl deprotection afforded the protected steroid 9.6 in 54% yield from 9.1. Finally, K2CO3 mediated acetate deprotection of 9.6, neutralization and methanol/water crystallization provided vamorolone (9) in 79% yield over three steps.
(70) Keam, S. J. Vamorolone: first approval. Drugs 2024, 84, 111−
117.
(71) Hoffman, E. P.; Nader, G. A. Balancing muscle hypertrophy and
atrophy. Nat. Med. 2004, 10, 584−585.
(72) Hoffman, E. P.; Reeves, E.; Damsker, J.; Nagaraju, K.; McCall, J.
M.; Connor, E. M.; Bushby, K. Novel approaches to corticosteroid
treatment in Duchennemusculardystrophy.Phys.Med.Rehabil. Clin. N.
Am. 2012, 23, 821−828.
(73) Singh, A.; Schaeffer, E. K.; Reilly, C. W. Vertebral fractures in
Duchenne muscular dystrophy patients managed with Deflazacort. J.
Pediatr. Orthop. 2018, 38, 320−324.
(74) Liu, X.; Wang, Y.; Gutierrez, J. S.; Damsker, J. M.; Nagaraju, K.;
Hoffman, E. P.; Ortlund, E. A. Disruption of a key ligand-H-bond
network drives dissociative properties in vamorolone for Duchenne
muscular dystrophy treatment. Proc. Natl. Acad. Sci. U. S. A. 2020, 117,
24285−24293.
(75) Heier, C. R.; Yu, Q.; Fiorillo, A. A.; Tully, C. B.; Tucker, A.;
Mazala, D. A.; Uaesoontrachoon, K.; Srinivassane, S.; Damsker, J. M.;
Hoffman, E.P.; et al. Vamorolone targets dual nuclear receptors to treat
inflammation and dystrophic cardiomyopathy. Life Sci. Alliance 2019, 2,
No. e201800186.
(76)Boger, D.L.Thedifferenceasingleatomcanmake:synthesisand
design at the chemistry−biology interface. J. Org. Chem. 2017, 82,
11961−11980.
(77) Reeves, E. K. M.; Hoffman, E. P.; Nagaraju, K.; Damsker, J. M.;
McCall, J. M. VBP15: Preclinical characterization of a novel anti
inflammatory delta 9,11 steroid. Bioorg. Med. Chem. 2013, 21, 2241−
2249.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
PATENTS
Drugs 2024, 84, 111−117.
Nat. Med. 2004, 10, 584−585.
Clin. N.Am. 2012, 23, 821−828.
Pediatr. Orthop. 2018, 38, 320−324.
Proc. Natl. Acad. Sci. U. S. A. 2020, 117,24285−24293.
Life Sci. Alliance 2019, 2,No. e201800186.
J. Org. Chem. 2017, 82,11961−11980.
Bioorg. Med. Chem. 2013, 21, 2241−2249.
Neurol. 79 (2022)1005–1014
Bioorg. Med. Chem. 21 (2013) 2241–2249
Nat. Rev. Dis. Prim. 7 (2021) 13
Chemistry
Vamorolone is a synthetic corticosteroid and is also known by the chemical name 17α,21-dihydroxy-16α-methylpregna-1,4,9(11)-triene-3,20-dione or as 16α-methyl-9,11-dehydroprednisolone. It is a derivative of cortisol (hydrocortisone) and prednisolone (1,2-dehydrocortisol).
Anti-inflammatory drugs of the corticosteroid class show a carbonyl (=O) or hydroxyl (-OH) group on the C11 carbon of the steroid backbone. In contrast, vamorolone contains a Δ9,11 double bond between the C9 and C11 carbons. This change in structure has been shown to remove a molecular contact site with the glucocorticoid receptor, and leads to dissociative properties.[12]
History
In phase I clinical trials of adult volunteers, vamorolone was shown to be safe and well tolerated, with blood biomarker data suggesting possible loss of safety concerns of the corticosteroid class.[13]
In phase IIa dose-ranging clinical trial of 48 children with Duchenne muscular dystrophy (2 weeks on drug, 2 weeks off drug), vamorolone was shown to be safe and well tolerated, and showed blood biomarker data consistent with a myofiber membrane stabilization and anti-inflammatory effects, and possible loss of safety concerns.[14] These children continued on to a 24-week open-label extension study at the same doses, and this showed dose-dependent improvement of motor outcomes, with 2.0 and 6.0 mg/kg/day suggesting benefit.[15] These same children continued on a long-term extension study with dose escalations, and this suggested continued clinical improvement through 18-months treatment.[16]
Population pharmacokinetics (PK) of vamorolone was shown to fit to a 1-compartment model with zero-order absorption, with both adult men and young boys showing dose-linearity of PK parameters for the doses examined, and no accumulation of the drug during daily dosing. Apparent clearance averaged 2.0 L/h/kg in men and 1.7 L/h/kg in boys. Overall, vamorolone exhibited well-behaved linear PK, with similar profiles in healthy men and boys with DMD, moderate variability in PK parameters, and absorption and disposition profiles similar to those of classical glucocorticoids.[17] Exposure/response analyses have suggested that the motor outcome of time to stand from supine velocity showed the highest sensitivity to vamorolone, with the lowest AUC value providing 50% of maximum effect (E50 = 186 ng·h/mL), followed by time to climb 4 stairs (E50 = 478 ng·h/mL), time to run/walk 10 m (E50 = 1220 ng·h/mL), and 6-minute walk test (E50 = 1770 ng·h/mL). Week 2 changes of proinflammatory PD biomarkers showed exposure-dependent decreases. The E50 was 260 ng·h/mL for insulin-like growth factor-binding protein 2, 1200 ng·h/mL for matrix metalloproteinase 12, 1260 ng·h/mL for lymphotoxin α1/β2, 1340 ng·h/mL for CD23, 1420 ng·h/mL for interleukin-22-binding protein, and 1600 ng·h/mL for macrophage-derived chemokine/C-C motif chemokine 22.[18]
A trial titled “Efficacy and Safety of Vamorolone Over 48 Weeks in Boys With Duchenne Muscular Dystrophy” published in March 2024 found vamorolone (Agamree) at a dose of 6 mg/kg/d showed maintenance of improvement for all motor outcomes to week 48. There was also significant improvement in linear growth after crossover in the prednisone to vamorolone 6 mg/kg/d group, and quick reversal of prednisone-induced decline in bone turnover biomarkers in each crossover group.[19]
The US Food and Drug Administration (FDA) approved vamorolone based on evidence from a single clinical trial of 121 boys with DMD who were 4 to <7 years of age. The trial (Study 1) was conducted at 33 sites in 11 countries in Australia, Belgium, Canada, the Czech Republic, Spain, the United Kingdom, Greece, Israel, Netherlands, Sweden, and the United States.[10] In addition to Study 1, safety was also evaluated in a separate, open-label study of children with DMD aged 2 to <4 years (N=16) and children with DMD aged 7 to <18 years (N=16).[10]
Society and culture
Legal status
Santhera Pharmaceuticals signed an agreement with Catalyst Pharmaceuticals for the North American commercialization of vamorolone in July 2023.[20]
In October 2023, the FDA approved vamorolone (Agamree; Catalyst Pharmaceuticals) for the treatment of Duchenne muscular dystrophy.[11][21][22]
In October 2023, the Committee for Medicinal Products for Human Use adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Agamree, intended for the treatment of Duchenne muscular dystrophy.[2] The applicant for this medicinal product is Santhera Pharmaceuticals (Deutschland) GmbH.[2] Vamorolone was approved for medical use in the European Union in December 2023.[2][3]
Brand names
Vamorolone is the international nonproprietary name.[23]
Vamorolone is sold under the brand name Agamree.[1][2][3] Agamree (vamorolone) is a dissociative steroid that selectively binds to the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Vamorolone also inhibits mineralocorticoid receptor activation by aldosterone.[24]
References
- “Agamree- vamorolone kit”. DailyMed. 26 October 2023. Retrieved 20 November 2023.
- “Agamree EPAR”. European Medicines Agency. 12 October 2023. Retrieved 27 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Agamree Product information”. Union Register of medicinal products. 15 December 2023. Retrieved 26 December 2023.
- “Vamorolone – ReveraGen Biopharma”. AdisInsight. Springer Nature Switzerland AG. Archived from the original on 7 October 2017. Retrieved 2 July 2017.
- Reeves EK, Hoffman EP, Nagaraju K, Damsker JM, McCall JM (April 2013). “VBP15: preclinical characterization of a novel anti-inflammatory delta 9,11 steroid”. Bioorganic & Medicinal Chemistry. 21 (8): 2241–2249. doi:10.1016/j.bmc.2013.02.009. PMC 4088988. PMID 23498916.
- Heier CR, Damsker JM, Yu Q, Dillingham BC, Huynh T, Van der Meulen JH, et al. (October 2013). “VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects”. EMBO Molecular Medicine. 5 (10): 1569–1585. doi:10.1002/emmm.201302621. PMC 3799580. PMID 24014378.
- Dadgar S, Wang Z, Johnston H, Kesari A, Nagaraju K, Chen YW, et al. (October 2014). “Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy”. The Journal of Cell Biology. 207 (1): 139–158. doi:10.1083/jcb.201402079. PMC 4195829. PMID 25313409.
- Damsker JM, Conklin LS, Sadri S, Dillingham BC, Panchapakesan K, Heier CR, et al. (September 2016). “VBP15, a novel dissociative steroid compound, reduces NFκB-induced expression of inflammatory cytokines in vitro and symptoms of murine trinitrobenzene sulfonic acid-induced colitis”. Inflammation Research. 65 (9): 737–743. doi:10.1007/s00011-016-0956-8. PMID 27261270. S2CID 18698831.
- Heier CR, Yu Q, Fiorillo AA, Tully CB, Tucker A, Mazala DA, et al. (February 2019). “Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy”. Life Sci Alliance. 2 (1): e201800186. doi:10.26508/lsa.201800186. PMC 6371196. PMID 30745312.
- “Drug Trials Snapshots: Agamree”. U.S. Food and Drug Administration (FDA). 16 February 2024. Archived from the original on 18 February 2024. Retrieved 14 March 2024. This article incorporates text from this source, which is in the public domain.
- “Drug Approval Package: Agamree”. U.S. Food and Drug Administration (FDA). 7 November 2023. Archived from the original on 13 November 2023. Retrieved 13 November 2023.
- Liu X, Wang Y, Gutierrez JS, Damsker JM, Nagaraju K, Hoffman EP, et al. (September 2020). “Disruption of a key ligand-H-bond network drives dissociative properties in vamorolone for Duchenne muscular dystrophy treatment”. Proceedings of the National Academy of Sciences of the United States of America. 117 (39): 24285–24293. Bibcode:2020PNAS..11724285L. doi:10.1073/pnas.2006890117. PMC 7533876. PMID 32917814.
- Hoffman EP, Riddle V, Siegler MA, Dickerson D, Backonja M, Kramer WG, et al. (June 2018). “Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes”. Steroids. 134: 43–52. doi:10.1016/j.steroids.2018.02.010. PMC 6136660. PMID 29524454.
- Conklin LS, Damsker JM, Hoffman EP, Jusko WJ, Mavroudis PD, Schwartz BD, et al. (October 2018). “Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug”. Pharmacological Research. 136: 140–150. doi:10.1016/j.phrs.2018.09.007. PMC 6218284. PMID 30219580.
- Hoffman EP, Schwartz BD, Mengle-Gaw LJ, Smith EC, Castro D, Mah JK, et al. (September 2019). “Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function”. Neurology. 93 (13): e1312 – e1323. doi:10.1212/WNL.0000000000008168. PMC 7011869. PMID 31451516.
- Smith EC, Conklin LS, Hoffman EP, Clemens PR, Mah JK, Finkel RS, et al. (September 2020). “Efficacy and safety of vamorolone in Duchenne muscular dystrophy: An 18-month interim analysis of a non-randomized open-label extension study”. PLOS Medicine. 17 (9): e1003222. doi:10.1371/journal.pmed.1003222. PMC 7505441. PMID 32956407.
- Mavroudis PD, van den Anker J, Conklin LS, Damsker JM, Hoffman EP, Nagaraju K, et al. (July 2019). “Population Pharmacokinetics of Vamorolone (VBP15) in Healthy Men and Boys With Duchenne Muscular Dystrophy”. Journal of Clinical Pharmacology. 59 (7): 979–988. doi:10.1002/jcph.1388. PMC 6548694. PMID 30742306.
- Li X, Conklin LS, van den Anker J, Hoffman EP, Clemens PR, Jusko WJ (October 2020). “Exposure-Response Analysis of Vamorolone (VBP15) in Boys With Duchenne Muscular Dystrophy”. Journal of Clinical Pharmacology. 60 (10): 1385–1396. doi:10.1002/jcph.1632. PMC 7494537. PMID 32434278.
- “Efficacy and Safety of Vamorolone Over 48 Weeks in Boys With Duchenne Muscular Dystrophy: A Randomized Controlled Trial”. PMID 38335499.
{{cite web}}: Missing or empty|url=(help) - Deswal P. “Santhera and Catalyst to market DMD drug vamorolone in North America”. Pharmaceutical Technology.
- “FDA Approves Vamorolone for Treatment of Duchenne Muscular Dystrophy in Patients Aged 2 Years and Older”. Pharmacy Times. 26 October 2023. Archived from the original on 27 October 2023. Retrieved 27 October 2023.
- “Santhera Receives U.S. FDA Approval of Agamree (vamorolone) for the Treatment of Duchenne Muscular Dystrophy” (Press release). Santhera Pharmaceuticals Holding AG. 27 October 2023. Archived from the original on 31 October 2023. Retrieved 13 November 2023 – via GlobeNewswire.
- World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 77”. WHO Drug Information. 31 (1). hdl:10665/330984.
- “Agamree for the Treatment of Duchenne Muscular Dystrophy, US”. Clinicaltrials Arena. Retrieved 11 February 2025.
External links
Clinical trial number NCT03439670 for “A Study to Assess the Efficacy and Safety of Vamorolone in Boys With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
- [1]. Heier CR, et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med. 2013 Oct;5(10):1569-85. [Content Brief][2]. Dillingham BC, et al. VBP15, a novel anti-inflammatory, is effective at reducing the severity of murine experimental autoimmune encephalomyelitis. Cell Mol Neurobiol. 2015 Apr;35(3):377-387. [Content Brief][3]. Heier CR, et al. Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci Alliance. 2019 Feb 11;2(1). pii: e201800186. [Content Brief]
| Clinical data | |
|---|---|
| Trade names | Agamree |
| Other names | VBP; VBP-15; 17α,21-Dihydroxy-16α-methylpregna-1,4,9(11)-triene-3,20-dione |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624005 |
| License data | US DailyMed: Vamorolone |
| Routes of administration | By mouth |
| ATC code | H02AB18 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 13209-41-1 |
| PubChem CID | 3035000 |
| DrugBank | DB15114 |
| ChemSpider | 2299335 |
| UNII | 8XP29XMB43 |
| KEGG | D11000 |
| ChEBI | CHEBI:228304 |
| ChEMBL | ChEMBL2348780 |
| CompTox Dashboard (EPA) | DTXSID60927527 |
| ECHA InfoCard | 100.032.874 |
| Chemical and physical data | |
| Formula | C22H28O4 |
| Molar mass | 356.462 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////////Vamorolone, VBP 15, APPROVALS 2023, EMA 2023, FDA 2023, 8XP29XMB43, AGAMREE, EU 2023
Zilucoplan
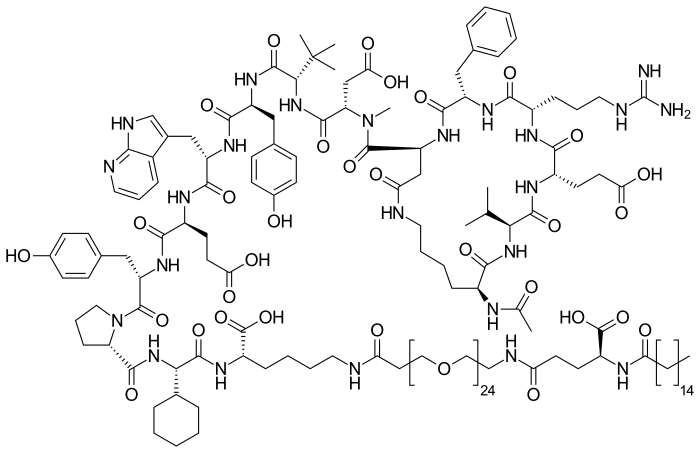
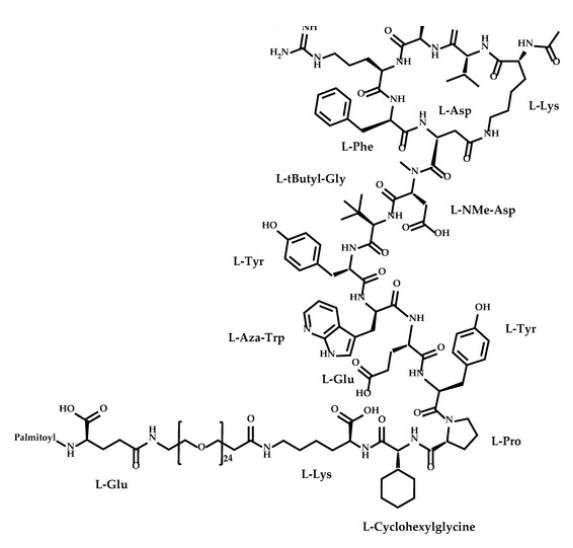
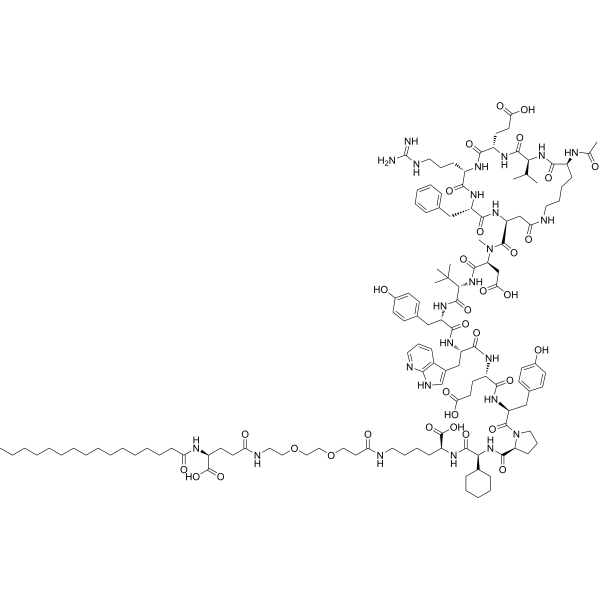
Zilucoplan
CAS 1841136-73-9
YG391PK0CC, RA101495, WHO 10602
3562 g/mol, C172H278N24O55
Zilucoplan lso designated as RA101495, is the active principle of Zilbrysq®, commercialized by UCB Pharma S.A. It is a 3.5 kDa synthetic macrocyclic peptide composed of 15 amino acid residues, including four unnatural amino acids [27]. The amino acid residues composition is: L-Lys, L-Val, L-Glu, L-Arg, L-Phe, L-Asp, L-L-NMe-Asp, L-tButyl-Gly, L-Tyr, L-7-aza-Trp, L-Glu, L-Tyr, L-Pro, L-Cyclohexyl-Gly, and L-Lys.
N2-ACETYL-L-LYSYL-L-VALYL-L-.ALPHA.-GLUTAMYL-L-ARGINYL-L-PHENYLALANYL-L-.ALPHA.-ASPARTYL-N-METHYL-L-.ALPHA.-ASPARTYL-3-METHYL-L-VALYL-L-TYROSYL-3-(1H-PYRROLO(2,3-B)PYRIDIN-3-YL)-L-ALANYL-L-.ALPHA.-GLUTAMYL-L-TYROSYL-L-PROLYL-(2S)-2-CYCLOHEXYLGLYCYL-N6-(3
POLY(OXY-1,2-ETHANEDIYL), ALPHA-(2-(((4S)-4-CARBOXY-1-OXO-4-(1-OXOHEXADECYL)BUTYL)AMINO)ETHYL)-OMEGA-HYDROXY-, 15-ETHER WITH N-ACETYL-L-LYSYL-L-VALYL-L-ALPHA-GLUTAMYL-L-ARGINYL-L-PHENYLALANYL-L-ALPHA-ASPARTYL-N-METHYL-L-ALPHA-ASPARTYL-3-METHYL-
(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,5S,8S,11S,14S,22S)-22-acetamido-11-benzyl-8-(3-carbamimidamidopropyl)-5-(2-carboxyethyl)-3,6,9,12,16,23-hexaoxo-2-propan-2-yl-1,4,7,10,13,17-hexazacyclotricosane-14-carbonyl]-methylamino]-3-carboxypropanoyl]amino]-3,3-dimethylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(1H-pyrrolo[2,3-b]pyridin-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]pyrrolidine-2-carbonyl]amino]-2-cyclohexylacetyl]amino]-6-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[(4S)-4-carboxy-4-(hexadecanoylamino)butanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]hexanoic acid
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Zilucoplan Sodium | Not Available | Not Available | FUSMWKLQHKXKHI-WHKBRXDJSA-J |
FDA 10/17/2023, Zilbrysq, To treat generalized myasthenia gravis in adults who are anti-acetylcholine receptor (AChR) antibody positive
Drug Trials Snapshot
Zilucoplan, sold under the brand name Zilbrysq, is a medication used for the treatment of generalized myasthenia gravis.[6][9][10] It is a complement inhibitor that is injected subcutaneously (under the skin).[6]
Zilucoplan is a cyclic peptide that binds to the protein complement component 5 (C5) and inhibits its cleavage into C5a and C5b.[11]
Zilucoplan was approved for medical use in the United States in October 2023,[6][12] in the European Union in December 2023,[7] and in Australia in July 2024.[1]
Zilucoplan is a 15 amino-acid, synthetic macrocyclic peptide with formula C172H278N24O55. Its sodium salt is used for the treatment of generalised myasthenia gravis (a disease that leads to muscle weakness and tiredness) in adults whose immune system produces antibodies against acetylcholine receptors. It has a role as a complement component 5 inhibitor and an immunosuppressive agent. It is a macrocycle, a homodetic cyclic peptide and a polyether. It is a conjugate acid of a zilucoplan(4-).
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US11752190 | No | 2023-09-12 | 2035-06-12 |  |
| US11014965 | No | 2021-05-25 | 2035-06-12 | |
| US10435438 | No | 2019-10-08 | 2035-06-12 | |
| US10208089 | No | 2019-02-19 | 2035-06-12 | |
| US10106579 | No | 2018-10-23 | 2035-06-12 | |
| US10835574 | No | 2020-11-17 | 2035-06-12 | |
| US11535650 | No | 2022-12-27 | 2035-06-12 | |
| US10562934 | No | 2020-02-18 | 2035-06-12 | |
| US11965040 | No | 2024-04-23 | 2035-06-12 | |
PAPER
https://www.mdpi.com/2813-2998/3/2/18
References
- ^ Jump up to:a b c “Zilbrysq (zilucoplan)”. Therapeutic Goods Administration (TGA). 24 September 2024. Retrieved 12 October 2024.
- ^ “Therapeutic Goods (Poisons Standard—June 2024) Instrument 2024”. Federal Register of Legislation. 30 May 2024. Retrieved 10 June 2024.
- ^ “Zilbrysq (UCB Australia Pty Ltd T/A UCB Pharma Division of UCB Australia)”. Therapeutic Goods Administration (TGA). 13 September 2024. Retrieved 15 September 2024.
- ^ “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-08-13]”. Health Canada. 13 August 2024. Retrieved 15 August 2024.
- ^ “Regulatory Decision Summary for Zilbrysq”. Drug and Health Products Portal. 11 July 2024. Retrieved 27 December 2024.
- ^ Jump up to:a b c d e “Zilbrysq- zilucoplan injection, solution”. DailyMed. 19 July 2024. Retrieved 15 September 2024.
- ^ Jump up to:a b c d “Zilbrysq EPAR”. European Medicines Agency. 1 December 2023. Retrieved 11 December 2023.
- ^ “Zilbrysq Product information”. Union Register of medicinal products. 4 December 2023. Archived from the original on 11 December 2023. Retrieved 11 December 2023.
- ^ Howard JF, Kaminski HJ, Nowak RJ, Wolfe GI, Benatar MG, Ricardo A, et al. (April 2018). “RA101495, a subcutaneously administered peptide inhibitor of complement component 5 (C5) for the treatment of generalized myasthenia gravis (gMG): Phase 1 results and phase 2 design (S31. 006)”. Neurology. 90 (15 Supplement). doi:10.1212/WNL.90.15_supplement.S31.006. S2CID 56969245. Archived from the original on 22 February 2022. Retrieved 24 June 2021.
- ^ Howard JF, Vissing J, Gilhus NE, Leite MI, Utsugisawa K, Duda PW, et al. (May 2021). “Zilucoplan: An Investigational Complement C5 Inhibitor for the Treatment of Acetylcholine Receptor Autoantibody-Positive Generalized Myasthenia Gravis”. Expert Opinion on Investigational Drugs. 30 (5): 483–493. doi:10.1080/13543784.2021.1897567. hdl:11250/2770699. PMID 33792453. S2CID 232482753.
- ^ Ricardo A, Arata M, DeMarco S, Dhamnaskar K, Hammer R, Fridkis-Hareli M, et al. (2015). “Preclinical Evaluation of RA101495, a Potent Cyclic Peptide Inhibitor of C5 for the Treatment of Paroxysmal Nocturnal Hemoglobinuria”. Blood. 126 (23): 939. doi:10.1182/blood.V126.23.939.939.
- ^ “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 22 December 2023. Archived from the original on 8 January 2023. Retrieved 27 December 2023.
- ^ Jump up to:a b “Zilbrysq: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 26 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Zilucoplan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Archived from the original on 17 October 2023. Retrieved 19 October 2023.
- ^ “EU/3/22/2650: Orphan designation for the treatment of myasthenia gravis”. European Medicines Agency. 15 September 2023. Archived from the original on 29 January 2023. Retrieved 24 September 2023.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3). hdl:10665/330907.
External links
- Clinical trial number NCT04115293 for “Safety, Tolerability, and Efficacy of Zilucoplan in Subjects With Generalized Myasthenia Gravis (RAISE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zilbrysq |
| Other names | RA101495 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624002 |
| License data | US DailyMed: Zilucoplan |
| Pregnancy category | AU: D[1] |
| Routes of administration | Subcutaneous |
| Drug class | Complement inhibitor |
| ATC code | L04AJ06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[2][3][1]CA: ℞-only[4][5]US: ℞-only[6]EU: Rx-only[7][8] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1841136-73-9 |
| PubChem CID | 133083018 |
| DrugBank | DB15636 |
| ChemSpider | 71115966 |
| UNII | YG391PK0CC |
| KEGG | D12357 |
| ChEBI | CHEBI:229659 |
| Chemical and physical data | |
| Formula | C172H278N24O55 |
| Molar mass | 3562.229 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////zilucoplan, Zilbrysq, FDA 2023, APPROVALS 2023, EU 2023, EMA 2023, RA101495, RA 101495, WHO 10602
Gepirone
Gepirone
CAS 83928-76-1
BMY 13805, MJ 13805, ORG 13011, Gepirona,
JW5Y7B8Z18
FDA 9/22/2023, Gepirone is indicated for the treatment of major depressive disorder (MDD) in adults
Exxua |
Average: 359.474
Monoisotopic: 359.232125194
Chemical Formula
C19H29N5O2
4,4-dimethyl-1-{4-[4-(pyrimidin-2-yl)piperazin-1-yl]butyl}piperidine-2,6-dione
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Gepirone Hydrochloride | 80C9L8EP6V | 83928-66-9 | DGOCVISYYYQFEP-UHFFFAOYSA-N |
Gepirone, sold under the brand name Exxua, is a medication used for the treatment of major depressive disorder.[1] It is taken orally.[1]
Side effects of gepirone include dizziness, nausea, insomnia, abdominal pain, and dyspepsia (indigestion).[1] Gepirone acts as a partial agonist of the serotonin 5-HT1A receptor.[1][2] An active metabolite of gepirone, 1-(2-pyrimidinyl)piperazine, is an α2-adrenergic receptor antagonist.[1][3] Gepirone is a member of the azapirone group of compounds.[2]
Gepirone was synthesized by Bristol-Myers Squibb in 1986 and was developed and marketed by Fabre-Kramer Pharmaceuticals.[4] It was approved for the treatment of major depressive disorder in the United States in September 2023.[4] This came after the drug had been rejected by the Food and Drug Administration (FDA) three times over two decades due to insufficient evidence of effectiveness.[5]
History
Gepirone was developed by Bristol-Myers Squibb in 1986,[5] but was out-licensed to Fabre-Kramer in 1993. The FDA rejected approval for gepirone in 2002 and 2004.[5] It was submitted for the preregistration (NDA) phase again in May 2007 after adding additional information from clinical trials as the FDA required in 2009. However, in 2012 it once again failed to convince the FDA of its qualities for treating anxiety and depression.[5] In December 2015, the FDA once again gave gepirone a negative review for depression due to concerns of efficacy.[12] However, in March 2016, the FDA reversed its decision and gave gepirone ER a positive review.[13] Gepirone ER was finally approved for the treatment of major depressive disorder in the United States in September 2023.[5]
SYN
Synthesis

Ormaza, V. A.; 1986, ES 8606333.
SYN
https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/gepirone
Gepirone (Exxua)
Gepirone, a selective and affinitive 5-hydroxytryptamine 1A (5-HT1A) agonist, received FDA approval on September 22, 2023, to treat major depressive disorder in adults [37]. Gepirone, an azapirone compound, is a pharmacological derivative of buspirone that exhibits specific activity on both pre- and post-synaptic 5-HT1A receptors. Despite the promising results observed in previous clinical trials for Gepirone, the need for frequent administration is a requirement due to its formulation as an immediate-release tablet and its short half-lives. Gepirone did not become a potential candidate for a new antidepressant until an extended-release formulation of it was developed [38–40].
An efficient approach of Gepirone has been disclosed in Scheme 11 [41]. Substitution of 2-(piperazin-1-yl)pyrimidine (GEPI-001) with 1,4-dibromobutane (GEPI-002), followed by ring opening and addition, generated the final product Gepirone.
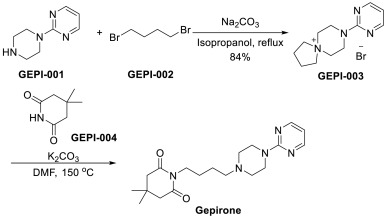
PATENT
https://patents.google.com/patent/WO2020148621A1/en
Gepirone (4,4-dimethyl-l-[4-[4-(2-pyrimidinyl)-l-piperazinyl]butyl]-2,6- piperidindione) is an antidepressant and anxiolytic medicament belonging to the azapirone group, currently at the pre-registration stage in the USA. Like other azapirones, gepirone is a selective partial agonist of the 5-HT1A receptor.
The prior art includes some synthesis strategies for the preparation of gepirone (I); they are mainly multi-step reactions which present various drawbacks such as economic inefficiency, low yield and low industrial applicability.
The synthesis of gepirone (I) is described in J. Med. Chem. 1988, 31, 1967-1971; WO 2012/016569; EP 0680961; Dier Junyi Daxue Xuebao, 26(2), 223-224; 2005; Patentschrift (CH), 682564, 15 October 1993; Heterocycles, 36(7), 1463-9, 1993; and Bioorganic & Medicinal Chemistry Letters, 14(7), 1709-1712, 2004.
The scientific article published in J. Med. Chem. 1988, 31, 1967-1971 describes the synthesis of gepirone (I) from N-bromobutyl phthalimide (2) and l-(pyrimidin-2- yl)piperazine (3) in the presence of potassium carbonate to give the intermediate 2-(4-(4- (pyrimidin-2-yl)piperazin-l-yl)butyl)isoindoline-l,3-dione (4), from which the phthalimide protecting group is removed with hydrazine hydrate. The compound 4-(4-(pyrimidin-2-yl)piperazin- 1 -yl)butan- 1 -amine (5) thus synthesised is used in the reaction with 4,4-dimethyldihydro-2H-pyran-2,6(3H)-dione (6) to obtain gepirone (I) (Scheme 1).
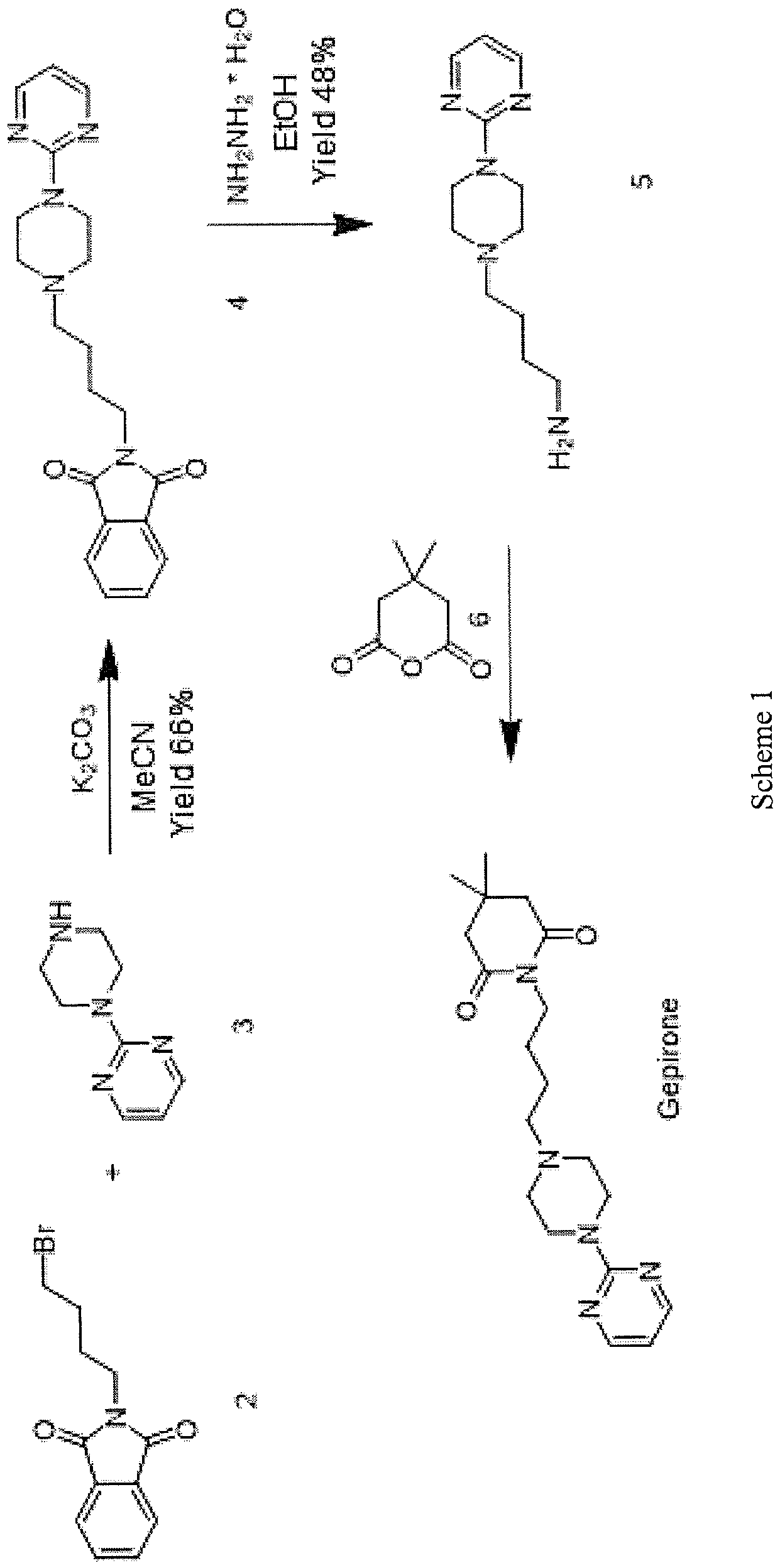
SCHEME 2
In WO 2012/016569, gepirone (I) is synthesised in four synthesis steps from 1- (pyrimidin-2-yl)pipetazine (3) and 4-((tert-butoxycarbonyl)amino)butanoic acid (7) with the use of condensing agents, such as HATU, and strong reducing agents such as lithium aluminium hydride. The use of condensing agents makes the process practically unusable on an industrial scale because of their high economic impact and the formation of countless by-products which are difficult to remove during the work-up step (Scheme 2).
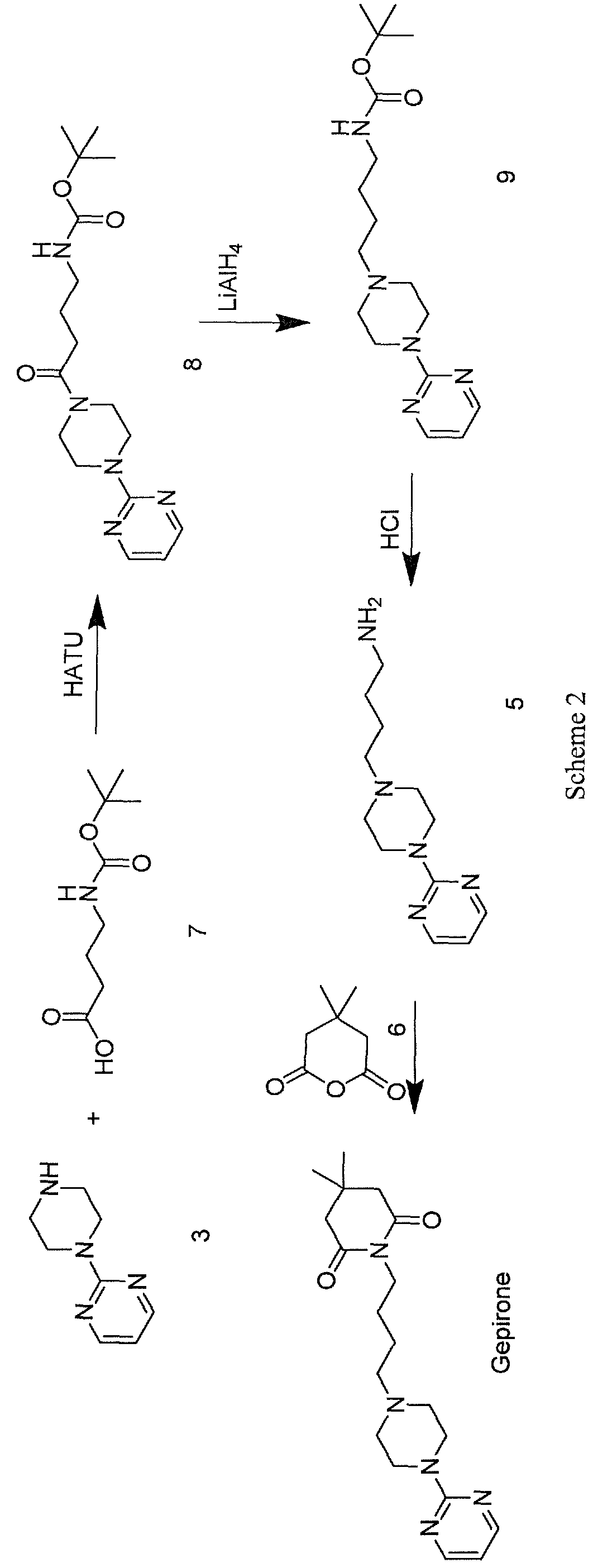
A further approach for the synthesis of gepirone is described in the literature (Ί). This strategy, disclosed in EP 0680961, initially involves synthesising (i) a spiranic intermediate (8-(pyrimidin-2-yl)- 5 ,8-diazaspiro [4,5] decan- 5 -ium bromide) (11) from 1- (pyrimidin-2-yl)piperazine (3) and (ii) 1,4-dibromobutane (10), then opening the spiranic compound (11) with the use of potassium 4,4-dimethyl-2,6-dioxopiperidin-l-ide (13), a secondary amine characterised by a high level of nucleophilicity (Scheme 3)
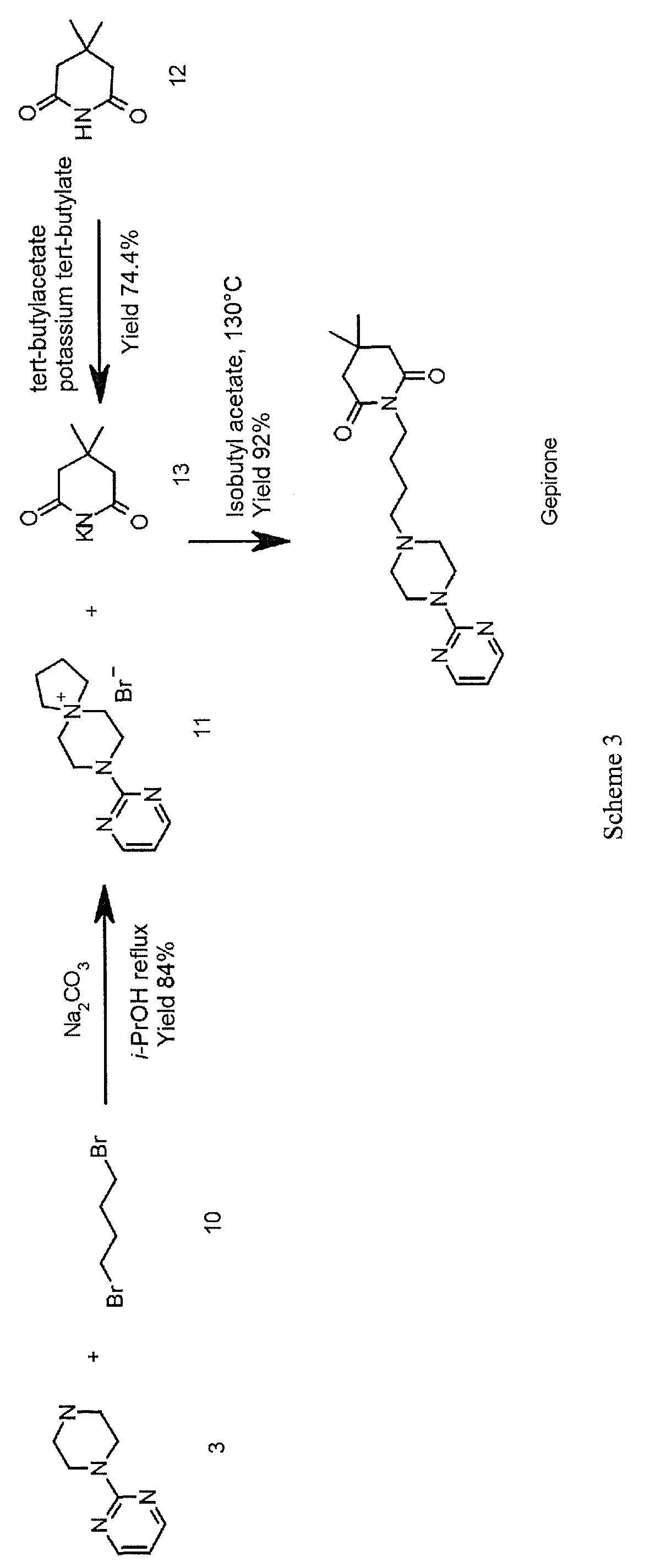
A novel approach to the synthesis of gepirone (I) has now been found, which involves opening spiranic derivative (11) to give 4-(4-(pyrimidin-2-yl)piperazin-l- yl)butan- 1 -amine (5), using suitable nitrogen nucleophile precursors of a primary amino group having the following characteristics: moderate nucleophilicity, so as to prevent reaction by-products, and easy generation of a primary amino group by means of a mild work-up (Scheme 4).
synthesis of gepirone (I) from
8-(pyrimidin-2-yl)-5,8-diazaspiro[4,5]decan-5-ium bromide (11), which is commercially available or easily obtainable by well-known procedures, such as those described in US 4351939.
Spiranic derivative (11) initially undergoes selective opening by suitable nitrogen nucleophile precursors of a primary amino group, such as di-tert-butyl iminodicarboxylate (14) and 2,2,2-trifluoroacetamide (15), in the presence of an organic and/or inorganic base. The opening of spiranic ring (11), followed by a simple, mild acid- base work-up, produces, in a single high-yield synthesis step, the intermediate 4-(4-(pyrimidin-2-yl)piperazin- 1 -yl)butan- 1 -amine (5), which is converted to gepirone (I) by reaction with 4,4-dimethyldihydro-2H-pyran-2,6(3H)-dione (6) (Scheme 5).
Example 1 – 4-(4-(pyrimidin-2-yl)piperazin- 1 -yl)butan- 1 -amine (5)
10.0 g of 8-(pyrimidin-2-yl)-5,8-diazaspiro[4,5]decan-5-ium bromide (11) (0.0334 moles), obtained according to US 4423049, is suspended in xylene (150 mL). 21.78 g of caesium carbonate (0.0668 moles) is then added. The resulting mixture is heated to 130°C and left under stirring for 60 minutes. 12.7 g of di-tert-butyl iminodicarboxylate (0.0584 moles) is then added and left under stirring until the reaction is complete. The mixture is cooled to about 80°C and filtered under vacuum, and the solid filtrate is washed with xylene (100 mL). 50 mL of 37% HC1 is added to the organic phase, and the resulting mixture is left under stirring for 10 min. The phases are then separated, and the organic phase is washed with a mixture of 50 mL of water and 5 mL of 37% HC1. 130 mL of dichloromethane is added to the aqueous acid phase and basified with 30% NaOH until pH = 13 is reached. The resulting mixture is left under stirring for 10 min., and the phases are separated. The aqueous phase is re-extracted with 200 mL of dichloromethane, and the combined organic phases are washed with 300 mL of water and 50 mL of brine, dried on sodium sulphate, filtered, and finally concentrated under vacuum to give 7.8 g of 4-(4-(pyrimidin-2-yl)piperazin-l-yl)butan-l-amine (5) (orange oil; yield 99%).
1H NMR (400 MHz, chloroform-d) d 8,17 (d, J = 4,7 Hz, 2H), 6,34 (t, J = 4,7 Hz, 1H), 3,76 – 3,63 (m, 4H), 2,60 (t, J = 6,9 Hz, 2H), 2,46 – 2,32 (m, 4H), 2,32 – 2,21 (m, 2H), 1,53 – 1,24 (m, 6H).
13C NMR (101 MHz, chloroform-d) d 161,55, 157,55, 109,65, 58,48, 53,02, 43,57, 42,01, 31,63, 24,18.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am “EXXUA (gepirone) extended-release tablets, for oral use” (PDF). Mission Pharmacal Company. U.S. Food and Drug Administration. 2023. Archived from the original (PDF) on 28 September 2023. Retrieved 28 September 2023.
- ^ Jump up to:a b c Kishi T, Meltzer HY, Matsuda Y, Iwata N (August 2014). “Azapirone 5-HT1A receptor partial agonist treatment for major depressive disorder: systematic review and meta-analysis” (PDF). Psychological Medicine. 44 (11): 2255–2269. doi:10.1017/S0033291713002857. PMID 24262766. S2CID 20830020. Archived from the original (PDF) on 18 February 2019.
- ^ Jump up to:a b Halbreich U, Montgomery SA (1 November 2008). Pharmacotherapy for Mood, Anxiety, and Cognitive Disorders. American Psychiatric Pub. pp. 375–. ISBN 978-1-58562-821-6.
- ^ Jump up to:a b c d e “Gepirone – Fabre-Kramer Pharmaceuticals”. AdisInsight. Springer Nature Switzerland AG. Archived from the original on 11 April 2023. Retrieved 28 September 2023.
- ^ Jump up to:a b c d e Becker Z (28 September 2023). “Decades long regulatory odyssey ends with FDA nod for Fabre-Kramer’s depression med Exxua”. Fierce Pharma.
- ^ Firth S (30 November 2015). “Controversial Antidepressant Comes Up for FDA OK — Again”. MedPage Today.
- ^ Kirsch I (2014). “Antidepressants and the Placebo Effect”. Zeitschrift für Psychologie. 222 (3): 128–134. doi:10.1027/2151-2604/a000176. PMC 4172306. PMID 25279271.
- ^ “FDA Rules Favorably On Efficacy Of Travivo (Gepirone ER) For Treatment Of Major Depressive Disorder”. Fabre-Kramer Pharmaceuticals, Inc. Cision PR Newswire. 17 March 2016.
- ^ “Gepirone”. Drugs and Lactation Database. National Institute of Child Health and Human Development. 2006. PMID 37856644. Retrieved 11 December 2023.
- ^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing Textbook of Psychopharmacology. American Psychiatric Pub. pp. 494–. ISBN 978-1-58562-309-9.
- ^ Kaur Gill A, Bansal Y, Bhandari R, Kaur S, Kaur J, Singh R, et al. (July 2019). “Gepirone hydrochloride: a novel antidepressant with 5-HT1A agonistic properties”. Drugs of Today. 55 (7): 423–437. doi:10.1358/dot.2019.55.7.2958474. PMID 31347611. S2CID 198911377.
- ^ “Gepirone ER”. Adis Insight. Archived from the original on 6 August 2016. Retrieved 13 January 2016.
- ^ “FDA Rules Favorably On Efficacy Of Travivo (Gepirone ER) For Treatment Of Major Depressive Disorder” (Press release). 17 March 2016. Archived from the original on 24 September 2017. Retrieved 23 January 2018.
- ^ Jump up to:a b Fabre LF, Brown CS, Smith LC, Derogatis LR (May 2011). “Gepirone-ER treatment of hypoactive sexual desire disorder (HSDD) associated with depression in women”. The Journal of Sexual Medicine. 8 (5): 1411–1419. doi:10.1111/j.1743-6109.2011.02216.x. PMID 21324094.
- ^ Jump up to:a b Fabre LF, Clayton AH, Smith LC, Goldstein I, Derogatis LR (March 2012). “The effect of gepirone-ER in the treatment of sexual dysfunction in depressed men”. The Journal of Sexual Medicine. 9 (3): 821–829. doi:10.1111/j.1743-6109.2011.02624.x. PMID 22240272.
- Robinson DS, Sitsen JM, Gibertini M: A review of the efficacy and tolerability of immediate-release and extended-release formulations of gepirone. Clin Ther. 2003 Jun;25(6):1618-33. doi: 10.1016/s0149-2918(03)80159-5. [Article]
- Jenkins SW, Robinson DS, Fabre LF Jr, Andary JJ, Messina ME, Reich LA: Gepirone in the treatment of major depression. J Clin Psychopharmacol. 1990 Jun;10(3 Suppl):77S-85S. doi: 10.1097/00004714-199006001-00014. [Article]
- Yocca FD: Neurochemistry and neurophysiology of buspirone and gepirone: interactions at presynaptic and postsynaptic 5-HT1A receptors. J Clin Psychopharmacol. 1990 Jun;10(3 Suppl):6S-12S. [Article]
- FDA Approved Drug Products: EXXUA (gepirone) extended-release tablets, for oral use [Link]
- Gepirone US Patent Application Publication [Link]
- Fabre-Kramer Pharmaceuticals Announces FDA Approval of EXXUA™, the First and Only Oral Selective 5HT1a Receptor Agonist for the Treatment of Major Depressive Disorder in Adults [Link]
| Clinical data | |
|---|---|
| Trade names | Exxua |
| Other names | BMY-13805; MJ-13805; ORG-13011 |
| Routes of administration | By mouth[1] |
| ATC code | N06AX19 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Pharmacokinetic data | |
| Bioavailability | 14–17%[1] |
| Protein binding | 72%[1] |
| Metabolism | CYP3A4[1] |
| Metabolites | 3′-OH-gepirone; 1-(2-Pyrimidinyl)piperazine[1] |
| Elimination half-life | IRTooltip Instant release: 2–3 hours ERTooltip Modified-release dosage: 5 hours[1] |
| Excretion | Urine: 81%[1] Feces: 13%[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 83928-76-1 83928-66-9 |
| PubChem CID | 55191 |
| DrugBank | DB12184DBSALT002148 |
| ChemSpider | 49836 49835 |
| UNII | JW5Y7B8Z1880C9L8EP6V |
| KEGG | D04314 |
| ChEBI | CHEBI:135990 |
| ChEMBL | ChEMBL284092 ChEMBL1204187 |
| CompTox Dashboard (EPA) | DTXSID90232813 |
| Chemical and physical data | |
| Formula | C19H29N5O2 |
| Molar mass | 359.474 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////Gepirone, FDA 2023, APPROVALS 2023, Exxua, BMY 13805, MJ 13805, ORG 13011, BMY-13805, MJ-13805, ORG-13011, Gepirona, JW5Y7B8Z18
Lotilaner



Lotilaner
- CAS 1369852-71-0
- Credelio
- XDEMVY
- lotilanerum
- 596.8 g/mol, C20H14Cl3F6N3O3S
- TP 03
- TP-03
- TP03
3-methyl-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]-5-[(5S)-5-(3,4,5-trichlorophenyl)-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]thiophene-2-carboxamide
FDA Xdemvy, 7/25/2023, To treat Demodex blepharitis
Drug Trials Snapshot
Lotilaner, sold under the brand name Xdemvy, is an ectoparasiticide (anti-parasitic) medication used for the treatment of blepharitis (inflammation of the eyelid) caused by infestation by Demodex (tiny mites).[1][7] It is used as an eye drop.[1]
It was approved for medical use in the United States in July 2023.[1][7][8][9] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[10]
SYN
https://patents.google.com/patent/WO2022016490A1/en
Scheme 1

Scheme 1 depicts coupling the compound of formula (5) with an appropriate amine to give lotilaner. An appropriate amine refers to either 2-amino-2′, 2′, 2′-trifluoroethyl-acetamide or the sequential reaction of glycine optionally carboxyl protected, followed by coupling with 2, 2, 2-trifluorethylamine. Such coupling reactions of carboxylic acids or activated carboxylic acid derivatives such as acid halides with amines to form amides are well known in the art. The use of carboxyl protected glycine, deprotection, and an amide coupling with 2, 2, 2-trifluorethylamine is likewise readily accomplished. See WO 2010/070068 and WO 2014/090918.
Example 1
(5S) -3- (5-Bromo-4-methyl-2-thienyl) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H- isoxazole

Combined (Z/E) 1- (5-bromo-4-methyl-2-thienyl) -4, 4, 4-trifluoro-3- (3, 4, 5-trichlorophenyl) but-2-en-1-one (50.0 g, 104.5 mmol) and (R) – [ (2S) -1- [ [3, 5-bis (tert-butyl) phenyl] methyl] -5-vinyl-quinuclidin-1-ium-2-yl] – (6-methoxy-4-quinolyl) methanol bromide (0.11 eq. ) in dichloromethane (100 mL) and ethyl t-butyl ether (400 mL) . The reaction mixture was stirred at 30℃ for 30 minutes and then cooled to the range of-20℃ then slowly added a solution of hydroxylamine in water (50%, 40 mL, 313 mmol, 3.0 eq. ) and sodium hydroxide (34.5 mL, 345 mmol, 10 M, 3.3 eq. ) maintaining an internal temperature in the range of-15℃ to-20℃. After stirring 18 hours at-15℃ to-20℃ aqueous hydrochloric acid (1N, 500 mL) was added and the reaction mixture was stirred at 15℃ to 20℃ then the stirring was stopped and after 30 minutes the phases were separated. The organic layer was extracted with aqueous hydrochloric acid (1N, 75 mL) , the layers separated and the organic layer again extracted with aqueous hydrochloric acid (1N, 100 mL) . The organic layer was separated and extracted with saturated aqueous sodium bicarbonate (75 mL) and the layers were separated and again the organic layer was extracted with saturated aqueous sodium bicarbonate (100 mL) . The layers were separated and the organic layer was dried over sodium sulfate (10 g) . The organic layer was filtered, the cake washed with ethyl t-butyl ether (50 mL) and then montmorillonite clay (50 g) was added and the mixture was stirred at 10℃ to 20℃. After 2 hours the reaction mixture was filtered, the cake rinsed with ethyl t-butyl ether (50 mL) and the filtrate was concentrated to about 100 mL, twice added THF and concentrated again to about 100 mL, and then added THF (150 mL) to obtain the title compound as a solution in THF. The solution was evaluated by chiral HPLC which indicated 96.5%S-isomer and 3.5%R-isomer.
Example 2
3-Methyl-5- [ (5S) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H-isoxazol-3- yl] thiophene-2-carboxylic acid

A 22%solution of (5S) -3- (5-bromo-4-methyl-2-thienyl) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H-isoxazole (185.0 g, 374.8 mmol) in THF was cooled to 0℃ to 5℃. A solution of ethyl magnesium chloride in THF (2 M, 300 mL, 1.6 eq) was added dropwise maintaining an internal temperature below 10℃. The reaction mixture was stirred at 15℃ to 20℃ for 2 to 4 hours. Then carbon dioxide gas (58 g, 3.5 eq) was introduced subsurface at 0℃ to 5℃ after passing through concentrated sulfuric acid (50 mL) . The reaction mixture was stirred at 0℃ to 5℃ for 2 hours and an 8%aqueous sodium chloride solution (601 g) was added dropwise at below 10℃, followed by addition of 37%aqueous HCl solution (92.5 g) at below 0℃. The reaction mixture was stirred at 10℃ to 15℃ for 30 minutes then the stirring was stopped and after 30 minutes the phases were separated. The organic layer was concentrated to about 370 mL under vacuum, followed by three iterations of THF (1850 mL) addition and concentration under vacuum to about 370 mL to 555 mL. After confirming the reaction mixture was dry, three cycles of acetonitrile (925 mL) addition followed by vacuum concentration to about 555 mL to 740 mL were performed. The reaction mixture was heated to 75℃ and gradually cooled to 50℃ over one hour. Product seeds (1.85 g) were added at 50℃ and the reaction mixture was stirred at 50℃ for 30 minutes. The batch was gradually cooled to-10℃ over three hours and kept at-10℃ for two hours. The batch was filtered and the cake was washed with cold acetonitrile (93 to 185 mL) . 110 g of the title compound was obtained after drying the wet cake at 50℃ under vacuum for 12 hours. The product was evaluated by chiral HPLC which indicated>99.9%S-isomer.
Above-referenced product seeds were prepared as follows. A solution of (5S) -3- (5-bromo-4-methyl-2-thienyl) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H-isoxazole (48.93 g, 99.1 mmol) in 300mL of THF was cooled to 0℃ to 5℃. A solution of ethyl magnesium chloride in THF (2 M, 80 mL) was added dropwise maintaining an internal temperature below 10℃. The reaction mixture was stirred at 15℃ to 20℃ for 2 to 4 hours. Then carbon dioxide gas (25 g, 3.5 eq) was introduced subsurface at 0℃ to 5℃ after passing through concentrated sulfuric acid (50 mL) . The reaction mixture was stirred at 0℃ to 5℃ for 6 hours and an 5%aqueous sodium chloride solution (157 g) was added dropwise at below 10℃, followed by addition of 37%aqueous HCl solution (25 g) at below 0℃. The reaction mixture was stirred at 10℃ to 15℃ for 30 minutes then the stirring was stopped and after 30 minutes the phases were separated. The organic layer was concentrated to remove the solvent. 50ml of heptane was added into the mixture then removed the solvent. The crude product was dissolved in 50mL of EA and 100mL of heptane at 40℃. Additional 1000mL of heptane was charged dropwise into the mixture slowly. Then the mixture was stirred at 40℃ for 15h. The mixture was filtered and the wet cake was obtained. The wet cake was slurried by acetone at 20℃. The mixture was filter and the wet cake was dried at 50℃ under vacuum for 3h to afford 9.7g of product. The product was evaluated by chiral HPLC which indicated>99.9%S-isomer.
Example 3
3-Methyl-N- [2-oxo-2- (2, 2, 2-trifluoroethylamino) ethyl] -5- [ (5S) -5- (3, 4, 5-trichlorophenyl) – 5- (trifluoromethyl) -4H-isoxazol-3-yl] thiophene-2-carboxamide

A solution of 3-methyl-5- [ (5S) -5- (3, 4, 5-trichlorophenyl) -5- (trifluoromethyl) -4H-isoxazol-3-yl] thiophene-2-carboxylic acid (101.5 g, 221.3 mmol) in DCM (1000 mL) was heated to 40℃. Thionyl chloride (50 g, 1.9 eq) was added dropwise and the reaction mixture was stirred at reflux for 2 to 4 hours. The reaction mixture was concentrated to 100 to 200 mL and DCM (500 mL) was added. Two more cycles of concentration followed by DCM addition were performed. In a separate vessel, a suspension of 2-amino-trifluoroethyl-acetamide HCl (50.26 g, 1.2 eq) in DCM (500 mL) was cooled to 0℃ to 5℃, triethylamine (70.15 g, 3.1 eq) was added, and the reaction mixture was stirred at 0℃ to 5℃ for 30 minutes. The acid chloride solution in DCM was then transferred to the reaction mixture containing 2-amino-trifluoroethyl-acetamide maintaining the internal temperature below 5℃. The reaction mixture was stirred at 0℃ to 5℃ for 2 to 4 hours. 1 N HCl (500 mL) was added dropwise and the reaction mixture was stirred at 15 to 25℃ for 30 minutes. The stirring was stopped and after 30 minutes the phases were separated. The organic layer was extracted with saturated sodium bicarbonate solution (1N, 1000 mL) , the layers separated and the organic layer extracted with water (1000 mL) . The layers were separated and the organic layer was concentrated under vacuum to 200 to 300 mL. Twice ethyl acetate (500 mL) was added and the batch was concentrated to 200 mL. The reaction mixture was heated to 55℃ and n-heptane (700 mL) was added dropwise at 55℃. Product seeds (1.0 g) was added and the reaction mixture was stirred at 55℃ for one hour. n-Heptane (1000 mL) was added dropwise and the mixture was stirred at 55℃ for three hours. The batch was gradually cooled to 35℃ over three hours, then to 20℃ over three hours. The batch was filtered and the cake was washed with n-heptane (200 mL) . 113 g of the title compound was obtained after drying at 50℃ under vacuum for 12 hours.
Above-referenced product seeds are prepared as follows. The crude product was dissolved in 7.9 wt-parts of cumene to obtain a solution at<150℃. Then 2.3 wt-parts of heptanes was added to the hot solution until slight haze was observed. The heating was turned off and the mixture was cooled to ambient temperature and stirred overnight. The desired polymorph G was obtained as powder after filtration and drying under vacuum, which was used as seeds to induce crystallization of polymorph G in future batches.
PATENT
https://patents.google.com/patent/WO2017176948A1/en
PATENT
CN112457267
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN320823634&_cid=P10-MBBMZO-15915-1
REFSatish Kumar RangarajuSatish Kumar Rangaraju •
The first FDA-approved treatment for Demodex Blepharitis, a common eyelid disease caused by microscopic mites living in the eyelashes’ hair follicles. hashtag#Lotilaner
For more information blog:
https://lnkd.in/gFTeKEtv

Research
Tarsus Pharmaceuticals has conducted phase II studies of lotilaner as a remedy to prevent tick bites in humans.[16][11]
References
- ^ Jump up to:a b c d e “Xdemvy- lotilaner ophthalmic solution solution/ drops”. DailyMed. 26 July 2023. Retrieved 23 August 2023.
- ^ Jump up to:a b c “Credelio- lotilaner tablet, chewable”. DailyMed. Archived from the original on 5 August 2021. Retrieved 4 August 2021.
- ^ Jump up to:a b c “Credelio- lotilaner tablet, chewable”. DailyMed. Archived from the original on 5 August 2021. Retrieved 4 August 2021.
- ^ Jump up to:a b “Credelio EPAR”. European Medicines Agency (EMA). 17 September 2018. Archived from the original on 27 January 2022. Retrieved 4 August 2021.
- ^ Jump up to:a b c “Lotimax EPAR”. European Medicines Agency. 13 March 2024. Retrieved 20 March 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Lotimax Product information”. Union Register of veterinary medicinal products. 29 April 2024. Retrieved 29 August 2024.
- ^ Jump up to:a b “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 25 July 2023. Archived from the original on 21 January 2023. Retrieved 5 August 2023.
- ^ “Xdemvy: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Archived from the original on 27 July 2023. Retrieved 5 August 2023.
- ^ “FDA Approves Xdemvy (lotilaner ophthalmic solution) 0.25% for the treatment of Demodex blepharitis” (Press release). Tarsus Pharmaceuticals. 25 July 2023. Retrieved 5 August 2023 – via GlobeNewswire.
- ^ New Drug Therapy Approvals 2023. U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original (PDF) on 10 January 2024. Retrieved 9 January 2024.
- ^ Jump up to:a b c “A Pill That Kills Ticks Is a Promising New Weapon Against Lyme Disease”. Wired. 15 March 2024. Retrieved 17 March 2024.
- ^ Jump up to:a b “Lotimax PI”. Union Register of medicinal products. 29 April 2024. Retrieved 17 June 2024.
- ^ Kuntz EA, Kammanadiminti S (November 2017). “Safety evaluation of lotilaner in dogs after oral administration as flavoured chewable tablets (Credelio)”. Parasites & Vectors. 10 (1): 538. doi:10.1186/s13071-017-2468-y. PMC 5664904. PMID 29089043.
- ^ “Freedom Of Information Summary, Supplemental New Animal Drug Application, NADA 141-494, Credelio, Lotilaner, Chewable Tablets, Dogs”. 3 September 2019. Archived from the original on 18 May 2022. Retrieved 1 December 2019.
- ^ Jump up to:a b “Credelio Plus EPAR”. European Medicines Agency. 19 February 2021. Archived from the original on 7 December 2022. Retrieved 4 August 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Tarsus Announces Positive Topline Results from Carpo, a Phase 2a Proof-of-Concept “Tick-Kill” Trial Evaluating TP-05 (lotilaner) for the Prevention of Lyme Disease (press release)”. Tarsus Pharmaceuticals. 22 February 2024. Archived from the original on 17 March 2024. Retrieved 17 March 2024.
| Clinical data | |
|---|---|
| Trade names | Credelio, Xdemvy, others |
| Other names | TP-03 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Lotilaner |
| Routes of administration | By mouth, eye drops |
| Drug class | Antiparasitic |
| ATC code | S01AX25 (WHO) QP53BE04 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1][2][3]EU: Rx-only[4][5][6] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1369852-71-0 |
| PubChem CID | 76959255 |
| DrugBank | DB17992 |
| ChemSpider | 32699771 |
| UNII | HEH4938D7K |
| KEGG | D11212 |
| ChEBI | CHEBI:229657 |
| ChEMBL | ChEMBL3707310 |
| CompTox Dashboard (EPA) | DTXSID701027551 |
| Chemical and physical data | |
| Formula | C20H14Cl3F6N3O3S |
| Molar mass | 596.75 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Toutain CE, Seewald W, Jung M: Pharmacokinetics of lotilaner following a single oral or intravenous administration in cats. Parasit Vectors. 2018 Jul 13;11(1):412. doi: 10.1186/s13071-018-2966-6. [Article]
- Rufener L, Danelli V, Bertrand D, Sager H: The novel isoxazoline ectoparasiticide lotilaner (Credelio): a non-competitive antagonist specific to invertebrates gamma-aminobutyric acid-gated chloride channels (GABACls). Parasit Vectors. 2017 Nov 1;10(1):530. doi: 10.1186/s13071-017-2470-4. [Article]
- Lamassiaude N, Toubate B, Neveu C, Charnet P, Dupuy C, Debierre-Grockiego F, Dimier-Poisson I, Charvet CL: The molecular targets of ivermectin and lotilaner in the human louse Pediculus humanus humanus: New prospects for the treatment of pediculosis. PLoS Pathog. 2021 Feb 18;17(2):e1008863. doi: 10.1371/journal.ppat.1008863. eCollection 2021 Feb. [Article]
- FDA Approved Drug Products: XDEMVY (lotilaner) 0.25%, Ophthalmic Solution (July 2023) [Link]
- Business Wire: FDA Approves XDEMVY™ (lotilaner ophthalmic solution) 0.25% for the treatment of Demodex blepharitis [Link]
- EMA Medicines Overview: Credelio [Link]
///////////Lotilaner, Xdemvy, FDA 2023, APPROVALS 2023, Credelio, XDEMVY, lotilanerum
Flotufolastat F 18



Flotufolastat F 18
- Flotufolastat F-18 Gallium
- POSLUMA
- CAS 2305081-64-3
- 18FrhPSMA-7.3
- 18F-rhPSMA-7.3
- 1537.3 g/mol
- C63H96FGaN12O25Si
gallium;2-[7-[(1S)-1-carboxy-4-[[(2R)-1-[[(1R)-1-carboxy-5-[[4-[[(4R)-4-carboxy-4-[[(4S)-4-carboxy-4-[[(1S)-1,3-dicarboxypropyl]carbamoylamino]butanoyl]amino]butyl]amino]-4-oxobutanoyl]amino]pentyl]amino]-3-[[4-[ditert-butyl(fluoranyl)silyl]benzoyl]amino]-1-oxopropan-2-yl]amino]-4-oxobutyl]-4,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate

WeightAverage: 1470.63
Monoisotopic: 1469.662295938
Chemical FormulaC63H99FN12O25Si
2639294-14-5 CAS
FDA 2023, Posluma, 5/25/2023, To use with positron emission tomography imaging in certain patients with prostate cancer
Drug Trials Snapshot
Flotufolastat (18F), sold under the brand name Posluma, is a radioactive diagnostic agent for use with positron emission tomography (PET) imaging for prostate cancer.[1] The active ingredient is flotufolastat (18F).[1]
Flotufolastat (18F) was approved for medical use in the United States in May 2023.[1][2]
SYNTHESIS
Bejot, R., et al. (2022). Methods of preparation of 18F labelled silyl-fluoride compounds (WO 2023047138 A1). World Intellectual Property Organization. https://patents.google.com/patent/WO2023047138A1/en?oq=WO2023047138A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023047138&_cid=P12-MB54HA-36492-1
EXAMPLES
rhPMSA-7, rhPMSA-10 & 2C013
Synthesis protocols for the 19F compounds 19F-rhPSMA-7.1 , 19F-rhPSMA-7.2, 19F-rhPSMA-7.3, 19F-rhPSMA-7.4, 19F-rhPSMA-10.1 , 19F-rhPSMA-10.2 and 19F-2C013 (shown below) are provided in WO2019/020831 , W02020/157177, WO2020/157184 and EP21157154.2.
rhPMSA-7.1
rhPMSA-7.2
rhPMSA-7.4
2C013
18F-Fluorination of rhPSMA-7.3
Aqueous 18F’ was passed through a quaternary methyl ammonium carbonate anion exchange cartridge (Sep-Pak Accell Plus QMA Carbonate), which was preconditioned with 5 mL of water. 18F’ was eluted with a 15 mg/mL cryptand 222 and 2.0 mg/mL potassium carbonate solution in acetonitrile/water (9/1 v/v). The resulting [18F]fluoride, cryptand and potassium carbonate solution was then azeotropically dried by heating at approx. 100 °C. Before radiolabelling, a 160 mM solution of acetic acid in DMSO was used to dissolve 0.27 pmol of rhPSMA-7.3. The resulting rhPSMA-7.3 solution was added to azeotropically-dried [18F]fluoride and the reaction mixture was incubated for 5 minutes at room temperature. For purification, a solid-phase extraction cartridge containing a hydrophobic resin (Sep-Pak Plus Short tC18 cartridge), preconditioned with 5 mL EtOH, followed by 10 mL of H2O was used. The labelling mixture was diluted with 5 mL citrate buffer (pH 5) and passed through the cartridge followed by 24 mL of citrate buffer. The 18F-rhPSMA-7.3 was eluted with 3 mL of a 1 :1 mixture (v/v) of EtOH in water.
Previously the process made use of oxalic acid and the impact of oxalic acid content, with on-cartridge drying of alkaline [18F]fluoride/K222, on radionuclide incorporation with rhPSMA-7.3 and similar silicon-fluorine acceptors (Kostikov, A. P. et al. Bioconjugate Chem. 2012, 23, 106-114) was evaluated. Maximum 18F-radiolabelling was reached when using approx.
30 pmol oxalic acid for 90 pmol of potassium hydroxide (acid-base molar ratio -0.6:1) (Wurzer, A. et al. EJNMMI radiopharm. chem. 6, 4 (2021)).
Although used in a limited quantity, oxalic acid may be toxic. Hence further development was conducted to replace oxalic acid with acetic acid, a common excipient for parenteral administration. Therefore, oxalic acid (dicarboxylic acid, 30 pmol) was replaced with 2 molar equivalents of acetic acid (monocarboxylic acid 60 pmol) and was shown to yield 18F-rhPSMA- 7.3 successfully using the Scintomics GRP synthesis module.
Implementation of the process with azeotropic drying of [18F]fluoride requires inverse addition, i.e., addition of acidified precursor solution to alkaline [18F]fluoride/K222 instead of addition of alkaline [18F]fluoride/K222 to the acidic precursor solution. As shown in Figure 1 , a higher amount of acid was required to prevent isomerisation of 18F-rhPSMA-7.3 or 19F-rhPSMA-7.3 in the presence of carbonate to related Compound A shown below. A decrease of radiolabelling conversion was also observed with increasing acid content. The optimised acetic acid amount for each process is provided in Table 1.
Compound A
Table 1 : Nominal acetic acid amounts for 18F-radiolabelling
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 : Impact of acetic acid content on isomerisation (formation of Related Compound A) and yield.
Medical uses
Flotufolastat (18F) is indicated for positron emission tomography of prostate-specific membrane antigen positive lesions in men with prostate cancer.[1][3]
References
- ^ Jump up to:a b c d e “Posluma- flotufolastat f-18 injection”. DailyMed. 2 June 2023. Retrieved 25 June 2023.
- ^ “U.S. FDA Approves Blue Earth Diagnostics’ Posluma (Flotufolastat F 18) Injection, First Radiohybrid PSMA-targeted PET Imaging Agent for Prostate Cancer” (Press release). Blue Earth Therapeutics. 30 May 2023. Retrieved 25 June 2023 – via Business Wire.
- ^ Heo YA (September 2023). “Flotufolastat F 18: Diagnostic First Approval”. Molecular Diagnosis & Therapy. 27 (5): 631–636. doi:10.1007/s40291-023-00665-y. PMID 37439946. S2CID 259843992.
External links
- Clinical trial number NCT04186819 for “Imaging Study to Investigate the Safety and Diagnostic Performance of rhPSMA 7.3 (18F) in Newly Diagnosed Prostate Cancer (LIGHTHOUSE)” at ClinicalTrials.gov
- Clinical trial number NCT04186845 for “Imaging Study to Investigate Safety and Diagnostic Performance of rhPSMA 7.3 (18F) PET Ligand in Suspected Prostate Cancer Recurrence (SPOTLIGHT)” at ClinicalTrials.gov
| Flotufolastat F-18 gallium | |
| Clinical data | |
|---|---|
| Trade names | Posluma |
| Other names | 18F-rhPSMA-7.3, flotufolastat F18 (USAN US) |
| License data | US DailyMed: Flotufolastat f-18 |
| Routes of administration | Intravenous |
| ATC code | V09IX18 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 2639294-14-5 |
| PubChem CID | 166177191 |
| DrugBank | DB17851 |
| UNII | 811W19E3OL |
| KEGG | D12606 |
| Chemical and physical data | |
| Formula | C63H9918FN12O25Si |
| Molar mass | 1537.3 g·mol−1 |
| 3D model (JSmol) | Interactive imageInteractive image |
| showSMILES | |
| showInChI | |
/////////Flotufolastat F 18, Posluma, FDA 2023, APPROVALS 2023, Flotufolastat F-18 Gallium, 18FrhPSMA-7.3, 18F-rhPSMA-7.3
Durlobactam
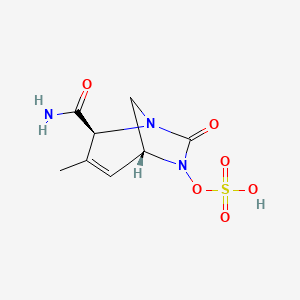
Durlobactam
CAS 1467829-71-5
WeightAverage: 277.25
Monoisotopic: 277.03685626
Chemical FormulaC8H11N3O6S
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Durlobactam sodium | F78MDZ9CW9 | 1467157-21-6 | WHHNOICWPZIYKI-IBTYICNHSA-M |
FDA 5/23/2023, Xacduro, To treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex
Press Release
Drug Trials Snapshots
(2S,5R)-2-CARBAMOYL-3-METHYL-7-OXO-1,6-DIAZABICYCLO(3.2.1)OCT-3-EN-6-YL SULFATE
SULFURIC ACID, MONO((2S,5R)-2-(AMINOCARBONYL)-3-METHYL-7-OXO-1,6-DIAZABICYCLO(3.2.1)OCT-3-EN-6-YL) ESTER
ETX 2514, ETX-2514, ETX2514, WHO 10824
Durlobactam is a non-beta-lactam, beta-lactamase inhibitor used to treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia.
Durlobactam is a beta-lactamase inhibitor used in combination with sulbactam to treat susceptible strains of bacteria in the genus Acinetobacter[1] It is an analog of avibactam.
The combination therapy sulbactam/durlobactam was approved for medical use in the United States in May 2023.[1]
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9309245 | No | 2016-04-12 | 2033-04-02 | |
| US9623014 | No | 2017-04-18 | 2033-04-02 | |
| US9968593 | No | 2018-05-15 | 2035-11-17 | |
| US10376499 | No | 2019-08-13 | 2035-11-17 | |
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Durlobactam (1) is a copackaged antibiotic combination being developed by Entasis Therapeutics for the treatment of infections caused by Acinetobacter baumannii-calcoaceticus. 13,14
Entasis Therapeutics obtained the worldwide development rights for durlobactam (1) from AstraZeneca in 2015.13 The drug combination was approved by the USFDA in 2023 for use in patients 18 years of age and older as an intravenous infusion.13 Acinetobacter baumannii is a critical bacterial pathogen that has
become highly resistant to various β-lactam antibiotics for Gram-negative infections, including penicillin.15,16 The inventors targeted β-lactam resistance via coadministration of a β-lactamase inhibitor to restore the activity of β-lactam antibiotics. Sulbactam is a β-lactam antibiotic that inhibits penicillin binding proteins (PBP 1 and 3) essential for cell wall synthesis. Durlobactam is a β-lactamase inhibitor that protects sulbactam from degradation by Ambler class A, C, and D serine β-lactamases produced byAcinetobacter baumannii-calcoaceticus. Durlobactam binds covalently with these β-lactamases by
carbamoylating the active site serines, thus safeguarding sulbactam from enzymatic degradation.17,18 The covalent bond between durlobactam and the active site serine isreversible due to the ability of sulfated amine of durlobactam to recyclize back into urea. This allows durlobactam to exchange from one
enzyme molecule to another via a mechanism known as acylation exchange
(13) Keam, S. J. Sulbactam/Durlobactam: first approval. Drugs 2023, 83, 1245−1252.
(14) El-Ghali, A.; Kunz Coyne, A. J.;Caniff, K.; Bleick,C.; Rybak, M. J.Sulbactam-durlobactam: a novel β-lactam-β-lactamase inhibitor combination targeting carbapenem-resistant Acinetobacter baumannii
infections. Pharmacotherapy 2023, 43, 502−513.
(15) O’Donnell, J.; Tanudra, A.; Chen, A.; Miller, A. A.; McLeod, S.M.; Tommasi, R. In vitro pharmacokinetics/pharmacodynamics of the β-lactamase inhibitor, durlobactam, in combination with sulbactam against Acinetobacter baumannii-calcoaceticus complex. Antimicrob.Agents Chemother. 2024, 68, e00312-23.
(16) Arya, R.; Goldner, B. S.; Shorr, A. F. Novel agents in development for multidrug-resistant Gram-negative infections: potential new options facing multiple challenges. Curr. Opin. Infect. Dis. 2022, 35, 589−594.
(17) Shapiro, A. B.; Moussa, S. H.; McLeod, S. M.; Durand-Réville, T.; Miller, A. A. Durlobactam, a new diazabicyclooctane β-lactamase inhibitor for the treatment of Acinetobacter infections in combination
with Sulbactam. Front. Microbiol. 2021, 12, No. 709974.
(18) Iyer, R.; Moussa, S. H.; Durand-Reville, T. F.; Tommasi, R.; Miller, A. Acinetobacter baumannii OmpA is a selective antibiotic permeant porin. ACS Infect. Dis. 2018, 4, 373−381.
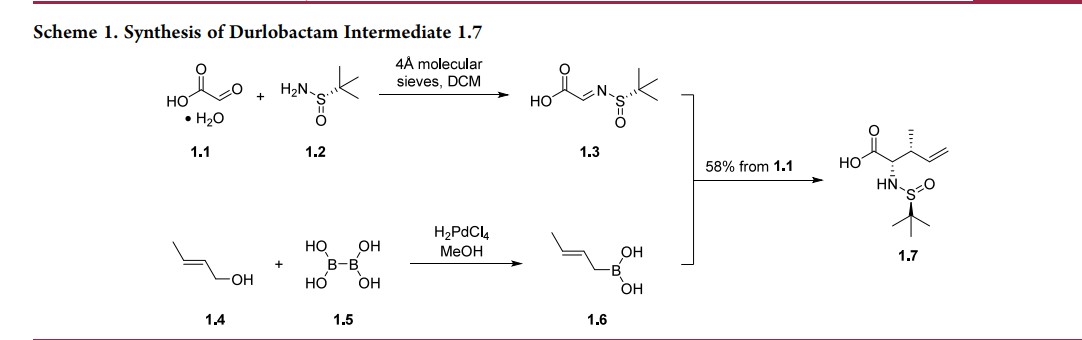
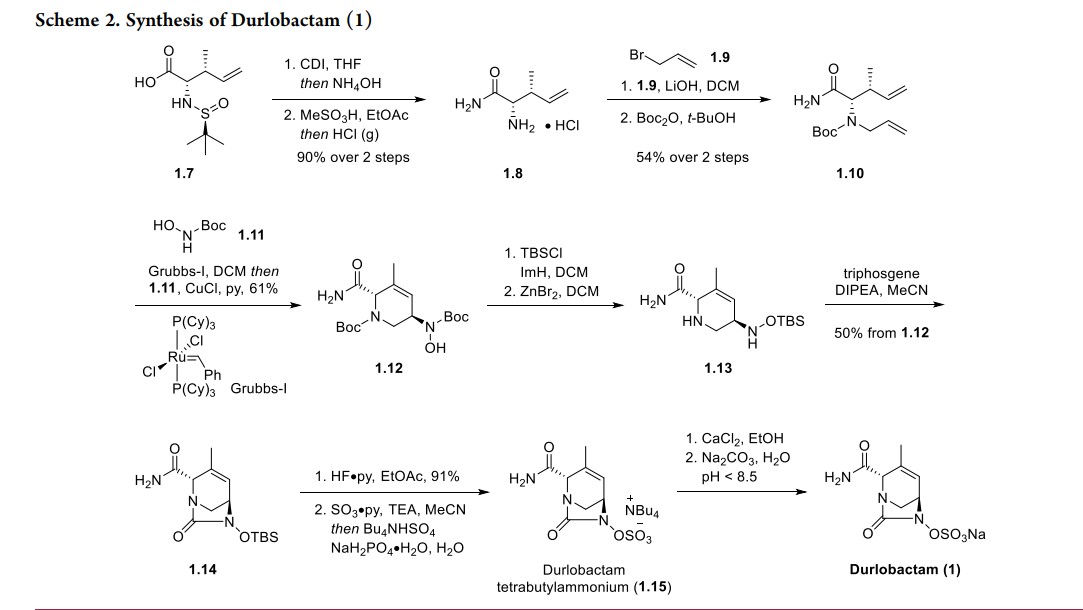
The route below was chosen as it was demonstrated on a multikilogram scale (Scheme 1), although some reagents (e.g.,triphosgene) are not typical for large-scale manufacturing.20,22 The synthesis commenced with the condensation of glyoxylic acid monohydrate (1.1) with (S)-tert-butylsulfinamide (1.2) to generate a solution of 2-(tert-butylsulfinylimino)acetic acid 1.3. In parallel, commercially available trans-crotyl alcohol (1.4) was treated with diboronic acid (1.5) in the presence of a palladium catalyst to produce a solution of crotylboronic acid 1.6. These two solutions were mixed to afford chiral α-amino acid 1.7 in
58% overall yield. Diastereo- and enantioselectivity were not reported for the transformation.
Conversion of 1.7 into durlobactam sodium (1) is described in Scheme 2. First, the carboxylic acid 1.7 was converted to an amide and the sulfinamide was removed to afford amino amide 1.8 as an HCl salt. The primary amine in 1.8 was subsequently alkylated with allyl bromide (1.9) and the resulting allyl amine
was protected with Boc anhydride to provide olefin metathesis precursor 1.10. Bisolefin 1.10 was then subjected to Grubbs first-generation catalyst (Grubbs-I) to generate a tetrahydropyridine precursor, which participated in a one-pot nitroso-ene reaction with N-Boc hydroxylamine (1.11) to produce allyl
hydroxylamine 1.12 in 61% overall yield. This key transformation efficiently installed the amine stereocenter required for formation of the bridged urea. Next, the hydroxyl moiety in 1.12 was protected as the TBS ether and the two Boc groups were removed with ZnBr2 to unveil bis-amine 1.13. The intramolecular urea formation was accomplished by the treatment with triphosgene to generate diazabicyclooctene 1.14 in 50% yield over 3 steps. The TBS ether was then removed, and the hydroxyl urea intermediate was treated with sulfur trioxide-pyridine complex and tetrabutylammonium
hydrogen sulfate to afford durlobactam tetrabutylammonium salt 1.15. Finally, tetrabutylammonium durlobactam 1.15 was converted to a calcium salt and subsequently to the targeted sodium salt providing 1. The authors mentioned that the salt formations were required to improve the purity of the final API
(>99%), however, the yields of these steps were not reported.22
(19) McGuire, H.; Bist, S.; Bifulco, N.; Zhao, L.; Wu, Y.; Huynh, H.; Xiong, H.; Comita-Prevoir, J.; Dussault, D.; Geng, B.; et al. Preparation of oxodiazabicyclooctenyl hydrogen sulfate derivatives for use as betalactamase inhibitors. WO 2013150296, 2013.
(20) Basarab, G. S.; Moss, B.; Comita-Prevoir, J.; Durand-Reville, T. F.; Gauthier, L.; O’Donnell, J.; Romero, J.; Tommasi, R.; Verheijen, J.C.; Wu, F.; et al. Preparation of substituted 2-(1,6-diazabicyclo[3.2.1]-
oct-3-en-6-yloxy)acetates as beta-lactamase inhibitors. WO2018053215, 2018.
(21) Durand-Reville, T. F.;Comita-Prevoir, J.; Zhang, J.; Wu, X.; MayDracka, T. L.; Romero, J. A. C.; Wu, F.; Chen, A.; Shapiro, A. B.; Carter, N. M.; et al. Discovery of an orally available diazabicyclooctane
inhibitor (ETX0282) of class A, C, and D serine β-lactamases. J. Med.Chem. 2020, 63, 12511−12525.
(22) Durand-Reville, T. F.; Wu, F.; Liao, X.; Wang, X.; Zhang, S.Preparation of Durlobactam crystalline forms. WO 2023206580, 2023
syn
https://www.mdpi.com/1424-8247/15/3/384
Synthesis of Durlobactam
Chemically, durlobactam is [(2S,5R)-2-carbamoyl-3-methyl-7-oxo-1,6-diazabicyclo [3.2.1] oct-3-en-6-yl] hydrogen sulfate which can be prepared from the key intermediate hydroxyurea 6-hydroxy-3-methyl-7-oxo-1,6-diaza-bicyclo [3.2.1] oct-3-ene-2-carboxylic acid amide I, which is the structural isomer of III prepared to synthetize ETX-1317 [101]. Then, according to Scheme 15, compound 1 obtained in the synthesis of III (Scheme 14) was reacted with penta-1,3-diene in place of isoprene, and, by an aza-Diels−Alder reaction, compound 2 was obtained.
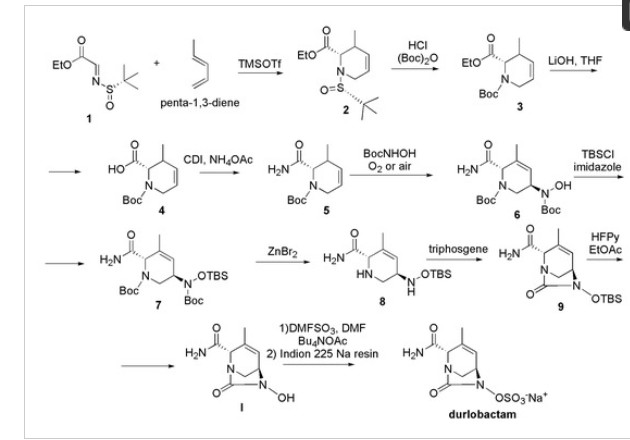
Scheme 15. Synthesis of durlobactam (C8H11N3O6S, MW = 277.36, IUPAC name, [(2S,5R)-2-carbamoyl-3-methyl-7-oxo-1,6-diazabicyclo [3.2.1] oct-3-en-6-yl] hydrogen sulphate.
Compound 2 underwent deprotection of the tert-butyl sulfinyl group to afford 3, subsequently Boc protected, to give compound 4. The saponification of the ester followed by amide coupling using ammonium acetate afforded compound 5. The reaction of alkene 5 with N-Boc-hydroxylamine in the presence of oxygen or air gave the desired compound 6 in a single step. Compound 6 was then protected with TBS group, using TBSCl to afford 7, which was Boc deprotected using zinc bromide obtaining compound 8. Cyclization of the diamine 8 with tri-phosgene provided the corresponding cyclic urea 9, which was TBS deprotected with HFPy to give the key intermediate I. This compound was then immediately sulfated with the DMF:SO3 complex to obtain the sulfate, which was isolated as its tetrabutylammonium salt 10 by reacting with tetrabutylammonium acetate. The tetrabutylammonium salt was converted to durlobactam in the form of sodium salt by passing 10 through a column filled with Indion 225 sodium resin.
REF
https://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract350784.shtml
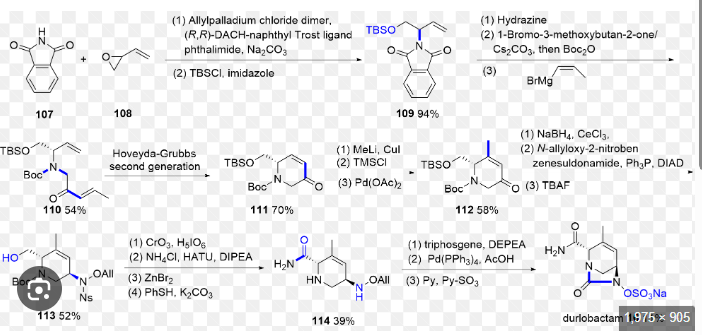
References
- ^ Jump up to:a b “FDA Approves New Treatment for Pneumonia Caused by Certain Difficult-to-Treat Bacteria”. U.S. Food and Drug Administration (Press release). 24 May 2023. Retrieved 24 May 2023. This article incorporates text from this source, which is in the public domain.
Further reading
- Shapiro AB, Moussa SH, McLeod SM, Durand-Réville T, Miller AA (2021). “Durlobactam, a New Diazabicyclooctane β-Lactamase Inhibitor for the Treatment of Acinetobacter Infections in Combination With Sulbactam”. Frontiers in Microbiology. 12: 709974. doi:10.3389/fmicb.2021.709974. PMC 8328114. PMID 34349751.
- Papp-Wallace KM, McLeod SM, Miller AA (May 2023). “Durlobactam, a Broad-Spectrum Serine β-lactamase Inhibitor, Restores Sulbactam Activity Against Acinetobacter Species”. Clinical Infectious Diseases. 76 (Supplement_2): S194 – S201. doi:10.1093/cid/ciad095. PMC 10150275. PMID 37125470.
| Clinical data | |
|---|---|
| Other names | ETX2514 |
| Routes of administration | Intravenous |
| Drug class | Antibacterial, beta-lactamase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only co-packaged with sulbactam |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1467829-71-5 |
| PubChem CID | 89851852 |
| DrugBank | DB16704DBSALT003190 |
| ChemSpider | 5761778471060725 |
| UNII | PSA33KO9WAF78MDZ9CW9 |
| KEGG | D11591D11592 |
| ChEMBL | ChEMBL4298137ChEMBL4297378 |
| Chemical and physical data | |
| Formula | C8H11N3O6S |
| Molar mass | 277.25 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Durlobactam, Xacduro, FDA 2023, APPROVED 2023, ETX 2514, ETX-2514, ETX2514, WHO 10824
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
Durlobactam, developed by Entasis Therapeutics, is a novel β-lactamase inhibitor designed to combat multidrug-resistant (MDR) Acinetobacter baumannii infections [83]. It is co-formulated with sulbactam, a
β-lactam antibiotic, and marketed under the brand name XACDURO. In 2024, the NMPA approved XACDURO for the treatment of hospital-acquired bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP) caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex in adults [84]. Durlobactam inhibits a broad spectrum of β-lactamases, including class A, C, and D enzymes, which are commonly produced by A. baumannii. By protecting sulbactam from enzymatic degradation, it restores sulbactam’s antibacterial activity against these resistant pathogens. The
clinical efficacy of sulbactam-durlobactam was demonstrated in the PhaseIII ATTACK trial (NCT03894046), a randomized, active-controlled study comparing sulbactam-durlobactam to colistin in patients with infections caused by carbapenem-resistant A. baumannii [85]. In this trial, the primary efficacy endpoint was achieved. It demonstrated non-inferiority in terms of 28-day all-cause mortality. The mortality rate in the sulbactam – durlobactam group was 19.0 %, while that in the colistin group reached 32.3 %. Moreover, the incidence of nephrotoxicity was remarkably lower in the sulbactam-durlobactam
group. From the perspective of toxicity, sulbactam-durlobactam was typically well-tolerated by the subjects. The most common adverse reactions included liver function test abnormalities, diarrhea, and hypokalemia. Notably, the incidence of nephrotoxicity was lower compared to colistin, highlighting a more favorable safety profile. The approval of XACDURO provides a targeted therapeutic option for managing severe infections caused by MDR A. baumannii, addressing a critical need in the
treatment of these challenging pathogens [86–88].
The synthetic route of Durlobactam, shown in Scheme 20, commences with a Grignard substitution between Durl-001 and Durl-002, affording Durl-003 [89]. This intermediate undergoes Diels-Alder
cyclization to form Durl-004, followed by reduction to Durl-005. Mitsunobu reaction of Durl-005 generates Durl-006, which is subjected to sequential deprotections yielding Durl-007 and subsequently Durl-008. Amidation of Durl-008 produces Durl-009, followed by TBAF-mediated deprotection to afford Durl-010. Oxidation of Durl-010Ngives carboxylic acid Durl-011, which undergoes amidation to form
Durl-012. Palladium-catalyzed coupling of Durl-012 produces Durl-013, with final ion exchange affording Durlobactam.
83-89
[83] A.B. Shapiro, S.H. Moussa, S.M. McLeod, T. Durand-R´ eville, A.A. Miller,
Durlobactam, a new diazabicyclooctane β-Lactamase inhibitor for the treatment of
acinetobacter infections in combination with sulbactam, Front. Microbiol. 12
(2021) 709974.
[84] G. Granata, F. Taglietti, F. Schiavone, N. Petrosillo, Durlobactam in the treatment
of multidrug-resistant Acinetobacter baumannii infections: a systematic review, J. Clin. Med. 11 (2022) 3258.
[85] K.M. Papp-Wallace, S.M. McLeod, A.A. Miller, Durlobactam, a broad-spectrum
serine β-lactamase inhibitor, restores sulbactam activity against acinetobacter
species, Clin. Infect. Dis. 76 (2023) S194–s201.
[86] Sulbactam and Durlobactam, Drugs and Lactation Database (Lactmed®), National
Institute of Child Health and Human Development, Bethesda (MD), 2006.
[87] S.J. Keam, Sulbactam/durlobactam: first approval, Drugs 83 (2023) 1245–1252.
[88] Y. Fu, T.E. Asempa, J.L. Kuti, Unraveling sulbactam-durlobactam: insights into its
role in combating infections caused by Acinetobacter baumannii, Expert Rev. Anti
Infect. Ther. 23 (2024) 1–12.
[89] H. McGuire, S. Bist, N. Bifulco, L. Zhao, Y. Wu, H. Huynh, H. Xiong, J. Comita-
Prevoir, D. Dussault, B. Geng, B. Chen, T. Durand-Reville, S. Guler, Preparation of
Oxodiazabicyclooctenyl Hydrogen Sulfate Derivatives for Use as beta-lactamase
Inhibitors, 2013 US9623014B2.
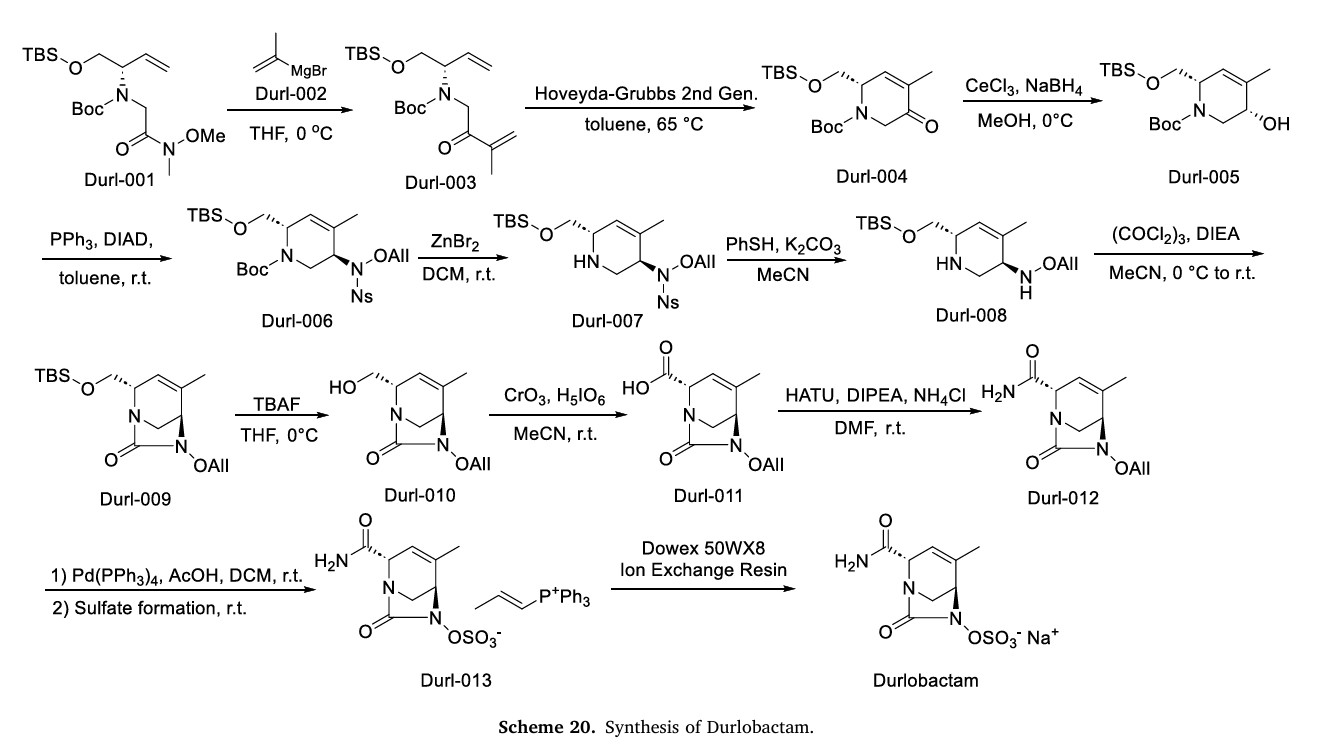
Sulbactam



Sulbactam
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Sulbactam benzathine | 49MU89FVBV | 83031-43-0 | YSEPFTSCLHUBNH-HFKSPEPWSA-N |
| Sulbactam sodium | DKQ4T82YE6 | 69388-84-7 | NKZMPZCWBSWAOX-IBTYICNHSA-M |
WeightAverage: 233.242
Monoisotopic: 233.035793157
Chemical FormulaC8H11NO5S
(2S,5R)-3,3-dimethyl-4,4,7-trioxo-4λ6-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
Betamaze, Penicillanic Acid Sulfone, Sulbactamum, CP 45899, CP-45899, CP45899
FDA 2023, Xacduro, 5/23/2023, To treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex
Press Release
Drug Trials Snapshots
Sulbactam is a β-lactamase inhibitor. This drug is given in combination with β-lactam antibiotics to inhibit β-lactamase, an enzyme produced by bacteria that destroys the antibiotics.[1]
It was patented in 1977 and approved for medical use in 1986.[2]
Sulbactam is a beta (β)-lactamase inhibitor and a derivative of the basic penicillin nucleus. When given in combination with β-lactam antibiotics, sulbactam produces a synergistic effect as it blocks the enzyme responsible for drug resistance by hydrolyzing β-lactams.
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9309245 | No | 2016-04-12 | 2033-04-02 | |
| US9623014 | No | 2017-04-18 | 2033-04-02 | |
| US9968593 | No | 2018-05-15 | 2035-11-17 | |
| US10376499 | No | 2019-08-13 | 2035-11-17 | |
doi:10.1016/S0040-4039(00)89275-8

SYN
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
On May 23, 2023, the FDA granted approval to Xacduro for the treatment of Baumannii-sensitive strains causing hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia in
patients aged 18 years or older [4]. Xacduro consists of Sulbactam and Durlobactam. Sulbactam, a medication with a similar structure to Penicillin, has the ability to eliminate Acinetobacter baumannii. On the other hand, Durlobactam shields Sulbactam from being broken down by enzymes that may be produced by Acinetobacter baumannii [5].
The production process of Sulbactam started with 6-aminopenicilanic acid (6-APA) (SULB-001) as the starting material (Scheme 1) [6]. It underwent bromination reaction with sodium nitrite and bromine
in the presence of sulfuric acid. Then, SULB-002 was oxidized by potassium permanganate to obtain sulfone SULB-003. Finally, the sulfonewas catalytically hydrogenated and dehalogenated in the presence of Raney nickel to get Sulbactam
[5] A. El-Ghali, A.J. Kunz Coyne, K. Caniff, C. Bleick, M.J. Rybak, Sulbactamdurlobactam: a novel β-lactam-β-lactamase inhibitor combination targeting
carbapenem-resistant Acinetobacter baumannii infections, Pharmacotherapy 43
(2023) 502–513.
[6] Z.M. Song, W. Liu, J. Yang, Y. Sun, Improvement on the synthetic process of
sulbactam, Chin. J
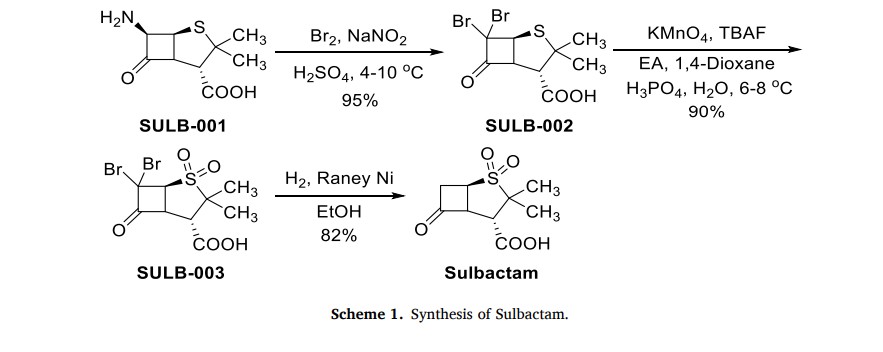
PATENT
https://patents.google.com/patent/CN101967155A/en
Embodiment 1
In the four-hole boiling flask of 2000ML, add 600ML methylene dichloride and 180ML2.5N sulfuric acid, stirring is cooled to below 0 ℃, add 28ML bromine and 25g Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 40g 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 100ML dichloromethane extraction 3 times, merge organic phase, with 100ML saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
To go up 6, the 6-dibromo penicillanic acid changes in the 2000ML beaker mutually. and add 250ML distilled water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 80ML deionized water, merge water, water changes in the 2000ML four-hole boiling flask, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (44g KMn04+10.8ML H3P04+700MLH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 500ML ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add the color fade of 27.5% hydrogen peroxide (about 45g) to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 250ML ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 100ML saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam;
To go up 6,6-dibromo sulbactam acid organic phase changes in the 2000ML four-hole boiling flask, adds 350ML water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 25ML methyl alcohol, add the 26g zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 25ML ethyl acetate and 25ML water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 150ML ethyl acetate extraction 4 times, washs to redness with the 50ML-100ML5% potassium permanganate solution earlier at organic layer and does not take off, again with 150ML saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 2g activated carbon decolorizing, the 15g anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 32g, the product yield is 74%, the product colour pure white was placed 30 days the color no change under the room temperature.
Embodiment 2
In the reactor of 2000L, add 600L methylene dichloride and 180L2.5N sulfuric acid, stirring is cooled to below 0 ℃, add 28L bromine and 25Kg Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 40Kg 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 100L dichloromethane extraction 3 times, merge organic phase, with 100L saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
Will on obtain 6, the 6-dibromo penicillanic acid changes in the 2000L reactor mutually. add the 250L tap water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 80L deionized water, merge water, water changes in the 2000L reactor, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (44Kg KMn04+10.8L H3P04+700LH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 500L ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add 28% hydrogen peroxide (about 44Kg) color fade to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 250L ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 100L saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam;
To go up organic phase and contain 6, the acid of 6-dibromo sulbactam changes in the 2000L reactor, adds 350L water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 25L methyl alcohol, add the 26Kg zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 25L ethyl acetate and 25L water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 150L ethyl acetate extraction 4 times, washs to redness with the 30-50L10% potassium permanganate solution earlier at organic layer and does not take off, again with 150L saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 2Kg activated carbon decolorizing, the 15Kg anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 31.5Kg, the product yield is 72.8%, the product colour pure white was placed 30 days the color no change under the room temperature.
Embodiment 3
In the four-hole boiling flask of 1000ML, add 300ML methylene dichloride and 90ML2.5N Hydrogen bromide, stirring is cooled to below 0 ℃, add 14ML bromine and 12.5g Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 20g 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 50ML dichloromethane extraction 3 times, merge organic phase, with 50ML saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
To go up 6, the 6-dibromo penicillanic acid changes in the 1000ML beaker mutually. and add 125ML distilled water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 40ML deionized water, merge water, water changes in the 1000ML four-hole boiling flask, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (22g KMn04+5.4ML H3P04+300MLH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 250ML ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add 25% hydrogen peroxide (about 29g) color fade to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 125ML ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 50ML saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam.
To go up organic phase and contain 6, the acid of 6-dibromo sulbactam changes in the 1000ML four-hole boiling flask, adds 175ML water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 12.5ML methyl alcohol, add the 13g zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 12.5ML ethyl acetate and 12.5ML water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 75ML ethyl acetate extraction 4 times, washs to redness with the 15ML-35ML7% potassium permanganate solution earlier at organic layer and does not take off, again with 75ML saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 1g activated carbon decolorizing, the 7.5g anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 15.9g, the product yield is 73.5%, the product colour pure white was placed 30 days the color no change under the room temperature.
PATENT
https://patents.google.com/patent/US4420426A/en
Medical uses
The combination ampicillin/sulbactam (Unasyn) is available in the United States.[3]
The combination cefoperazone/sulbactam (Sulperazon) is available in many countries but not in the United States.[4]
The co-packaged combination sulbactam/durlobactam was approved for medical use in the United States in May 2023.[5]
Mechanism
Sulbactam is primarily used as a suicide inhibitor of β-lactamase, shielding more potent beta-lactams such as ampicillin.[6] Sulbactam itself contains a beta-lactam ring, and has weak antibacterial activity by inhibiting penicillin binding proteins (PBP) 1 and 3, but not 2.[7]
References
- ^ Totir MA, Helfand MS, Carey MP, Sheri A, Buynak JD, Bonomo RA, Carey PR (August 2007). “Sulbactam forms only minimal amounts of irreversible acrylate-enzyme with SHV-1 beta-lactamase”. Biochemistry. 46 (31): 8980–8987. doi:10.1021/bi7006146. PMC 2596720. PMID 17630699.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 492. ISBN 9783527607495.
- ^ “Unasyn- ampicillin sodium and sulbactam sodium injection, powder, for solution”. DailyMed. U.S. National Library of Medicine. 29 March 2023. Retrieved 25 May 2023.
- ^ “Sulperazon”. drugs.com.
- ^ “FDA Approves New Treatment for Pneumonia Caused by Certain Difficult-to-Treat Bacteria”. U.S. Food and Drug Administration (Press release). 24 May 2023. Retrieved 24 May 2023.
- ^ Crass RL, Pai MP (February 2019). “Pharmacokinetics and Pharmacodynamics of β-Lactamase Inhibitors”. Pharmacotherapy. 39 (2): 182–195. doi:10.1002/phar.2210. PMID 30589457. S2CID 58567725.
- ^ Penwell WF, Shapiro AB, Giacobbe RA, Gu RF, Gao N, Thresher J, et al. (March 2015). “Molecular mechanisms of sulbactam antibacterial activity and resistance determinants in Acinetobacter baumannii”. Antimicrobial Agents and Chemotherapy. 59 (3): 1680–1689. doi:10.1128/AAC.04808-14. PMC 4325763. PMID 25561334.
Further reading
Singh GS (January 2004). “Beta-lactams in the new millennium. Part-II: cephems, oxacephems, penams and sulbactam”. Mini Reviews in Medicinal Chemistry. 4 (1): 93–109. doi:10.2174/1389557043487547. PMID 14754446.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a693021 |
| Routes of administration | Intravenous, intramuscular |
| ATC code | J01CG01 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 29% |
| Elimination half-life | 0.65–1.20 hrs |
| Excretion | Mainly kidneys (41–66% within 8 hrs) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 68373-14-8 |
| PubChem CID | 130313 |
| ChemSpider | 115306 |
| UNII | S4TF6I2330 |
| KEGG | D08533 |
| ChEBI | CHEBI:9321 |
| ChEMBL | ChEMBL403 |
| CompTox Dashboard (EPA) | DTXSID1023605 |
| ECHA InfoCard | 100.063.506 |
| Chemical and physical data | |
| Formula | C8H11NO5S |
| Molar mass | 233.24 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 148 to 151 °C (298 to 304 °F) |
| showSMILES | |
| showInChI | |
//////////Sulbactam, Xacduro, FDA 2023, APPROVALS 2023, Betamaze, Penicillanic Acid Sulfone, Sulbactamum, CP 45899, CP-45899, CP45899
Perfluorhexyloctane



WeightAverage: 432.269
Monoisotopic: 432.112266666Chemical FormulaC14H17F13
Perfluorhexyloctane
- 133331-77-8
- MIEBO
- Tetradecane, 1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluoro-
- 7VYX4ELWQM
- NOV03, NOV 03
- 1-(perfluorohexyl)octane
- F6H8
- NOV03
- Perfluorohexyloctane
1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluorotetradecane
FDA APPROVED 8/16/2023, Sohonos, To reduce the volume of new heterotopic ossification in adults and pediatric patients (aged 8 years and older for females and 10 years and older for males) with fibrodysplasia ossificans progressiva
Drug Trials Snapshot
Perfluorohexyloctane is a fluoroalkane that is tetradecane in which all of the hydrogen atoms at positions 1, 2, 3, 4, 5, and 6 have been replaced by fluorine atoms. It is an ophthalmic solution used to treat the signs and symptoms of dry eye disease. It has a role as an ophthalmology drug and a nonionic surfactant. It is a fluorohydrocarbon and a fluoroalkane. It derives from a hydride of a tetradecane.
Perfluorohexyloctane (branded as Evotears, Miebo,[a] and Novatears, among others) is a medication used for the treatment of dry eye disease.[4] It is a semifluorinated alkane.[4]
Perfluorohexyloctane has been available in multiple markets since 2015 under the brand names Evotears and Novatears,[5] and was additionally approved for medical use in the United States in May 2023 under the brand name Miebo.[4][6] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[7]
PATENT
Show 102550100 entries
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US11357738 | No | 2022-06-14 | 2036-09-29 | |
| US10058615 | No | 2018-08-28 | 2033-09-12 | |
| US10369117 | No | 2019-08-06 | 2033-09-12 | |
| US10449164 | No | 2019-10-22 | 2033-09-12 | |
| US10507132 | No | 2019-12-17 | 2037-06-21 | |
| US10576154 | No | 2020-03-03 | 2033-09-12 | |
SYN
https://www.scientificupdate.com/process-chemistry-articles/not-a-dry-eye-in-the-house



Medical uses
Perfluorohexyloctane is indicated for the treatment of the signs and symptoms of dry eye disease.[4][8][9]
Availability
Perfluorohexyloctane is sold as an over-the-counter medication under the brand names Evotears and Novatears in multiple countries,[10] costing around NZ$34.00, A$30, and €30 for a one-month supply.
In the US, perfluorohexyloctane is sold under the brand name Miebo; a prescription is required.
Notes and references
- ^ “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-10-18]”. Health Canada. 18 October 2024. Retrieved 25 October 2024.
- ^ “Miebo product information”. Health Canada. 4 September 2024. Retrieved 27 December 2024.
- ^ “Regulatory Decision Summary for Miebo”. Drug and Health Products Portal. 4 September 2024. Retrieved 27 December 2024.
- ^ Jump up to:a b c d e “Miebo- perfluorohexyloctane solution”. DailyMed. 18 May 2023. Retrieved 8 June 2023.
- ^ “URSAPHARM GmbH and Novaliq GmbH Announce European Partnership Agreement” (Press release). Retrieved 15 February 2024.
- ^ “Bausch + Lomb and Novaliq Announce FDA Approval of Miebo (Perfluorohexyloctane Ophthalmic Solution) for the Treatment of the Signs and Symptoms of Dry Eye Disease” (Press release). Bausch + Lomb Corporation. 18 May 2023. Retrieved 8 June 2023 – via Business Wire.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Ballesteros-Sánchez A, De-Hita-Cantalejo C, Sánchez-González MC, Jansone-Langine Z, de Sotomayor MA, Culig J, et al. (October 2023). “Perfluorohexyloctane in dry eye disease: A systematic review of its efficacy and safety as a novel therapeutic agent”. The Ocular Surface. 30: 254–262. doi:10.1016/j.jtos.2023.10.001. hdl:11441/151762. PMID 37813152. S2CID 263802332.
- ^ Sheppard JD, Evans DG, Protzko EE (November 2023). “A review of the first anti-evaporative prescription treatment for dry eye disease: perfluorohexyloctane ophthalmic solution”. The American Journal of Managed Care. 29 (14 Suppl): S251 – S259. doi:10.37765/ajmc.2023.89464. PMID 37930231. S2CID 265032840.
- ^ “In Australia, NovaTears Eye Drops Are Available on the Pharmaceutical Benefits Scheme (PBS) from Now On” (Press release). Retrieved 15 February 2024.
Further reading
- Azhar A, Taimuri MA, Oduoye MO, Sumbal A, Sheikh A, Iqbal A, et al. (September 2024). “MEIBO (perfluorohexyloctane): a novel approach to treating dry eye disease”. Annals of Medicine and Surgery (2012). 86 (9): 5292–5298. doi:10.1097/MS9.0000000000002322. PMC 11374244. PMID 39239035.
| Clinical data | |
|---|---|
| Trade names | Evotears Miebo (/ˈmaɪboʊ/ MY-bow) Novatears |
| Other names | NOV03; 1-(perfluorohexyl)octane |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623054 |
| License data | US DailyMed: Perfluorohexyloctane |
| Routes of administration | Eye drops |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1][2][3]US: ℞-only[4] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 133331-77-8 |
| PubChem CID | 10477896 |
| DrugBank | DB17823 |
| ChemSpider | 8653305 |
| UNII | 7VYX4ELWQM |
| KEGG | D12604 |
| ChEBI | CHEBI:229658 |
| CompTox Dashboard (EPA) | DTXSID20440585 |
| Chemical and physical data | |
| Formula | C14H17F13 |
| Molar mass | 432.269 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////Perfluorhexyloctane, Sohonos, APPROVALS 2023, FDA 2023, NOV03, NOV 03, MIEBO, 1-perfluorohexyl)octane, F6H8, NOV03, Perfluorohexyloctane

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}