
Home » DIABETES (Page 3)
Category Archives: DIABETES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Novartis Molecule for functionally liver selective glucokinase activators for the treatment of type 2 diabetes


(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
(3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide)
cas 866772-52-3
NVP-LBX192
LBX-192

54 Discovery and Evaluation of NVP-LBX192, a Liver Targeted Glucokinase Activator
https://acs.confex.com/acs/nerm09/webprogram/Paper75087.html
| Molecular Formula: | C26H33N5O4S2 |
|---|---|
| Molecular Weight: | 543.70132 g/mol |
Sulfonamide-Thiazolpyridine Derivatives, Glucokinase Activators, Treatment Of Type 2 Diabetes
2009 52 (19) 6142 – 6152
Investigation of functionally liver selective glucokinase activators for the treatment of type 2 diabetes
Journal of Medicinal Chemistry
Bebernitz GR, Beaulieu V, Dale BA, Deacon R, Duttaroy A, Gao JP, Grondine MS, Gupta RC, Kakmak M, Kavana M, Kirman LC, Liang JS, Maniara WM, Munshi S, Nadkarni SS, Schuster HF, Stams T, Denny IS, Taslimi PM, Vash B, Caplan SL
2010 240th (August 22) Medi-198
Glucokinase activators with improved physicochemicalproperties and off target effects
American Chemical Society National Meeting and Exposition
Kirman LC, Schuster HF, Grondine MS et al
2010 240th (August 22) Medi-197
Investigation of functionally liver selective glucokinase activators
American Chemical Society National Meeting and Exposition
Schuster HF, Kirman LC, Bebernitz GC et al
PATENT
http://www.google.com/patents/US7750020
EXAMPLE 1 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

A. Phenylacetic Acid Ethyl Ester
A solution of phenylacetic acid (50 g, 0.36 mol) in ethanol (150 mL) is treated with catalytic amount of sulfuric acid (4 mL). The reaction mixture is refluxed for 4 h. The reaction is then concentrated in vacuo. The residue is dissolved in diethyl ether (300 mL) and washed with saturated aqueous sodium bicarbonate solution (2×50 mL) and water (1×100 mL). The organic layer dried over sodium sulfate filtered and concentrated in vacuo to give phenylacetic acid ethyl ester as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.2 (t, J=7.2, 3H), 3.6 (s, 2H), 4.1 (q, J=7.2, 2H), 7.3 (m, 5H); MS 165 [M+1]+.
B. (4-Chlorosulfonyl-phenyl)-acetic acid ethyl ester
To a cooled chlorosulfonic acid (83.83 g, 48 mL, 0.71 mol) under nitrogen is added the title A compound, phenylacetic acid ethyl ester (59 g, 0.35 mol) over a period of 1 h. Reaction temperature is brought to RT (28° C.), then heated to 70° C., maintaining it at this temperature for 1 h while stirring. Reaction is cooled to RT and poured over saturated aqueous sodium chloride solution (200 mL) followed by extraction with DCM (2×200 mL). The organic layer is washed with water (5×100 mL), followed by saturated aqueous sodium chloride solution (1×150 mL). The organic layer dried over sodium sulfate, filtered and concentrated in vacuo to give crude (4-chlorosulfonyl-phenyl)acetic acid ethyl ester. Further column chromatography over silica gel (60-120 mesh), using 100% hexane afforded pure (4-chlorosulfonyl-phenyl)-acetic acid ethyl ester as a colorless oil.
C. [4-(4-Methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester
A solution of N-methylpiperazine (9.23 g, 10.21 ml, 0.092 mol), DIEA (13 g, 17.4 mL, 0.10 mol) and DCM 80 mL is cooled to 0° C., and to this is added a solution of the title B compound, (4-chlorosulfonyl-phenyl)-acetic acid ethyl ester (22 g, 0.083 mol) in 50 mL of DCM within 30 min. Reaction mixture stirred at 0° C. for 2 h, and the reaction mixture is washed with water (100 mL), followed by 0.1 N aqueous hydrochloric acid solution (1×200 mL). The organic layer dried over sodium sulfate, filtered and concentrated under vacuo to give crude [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester. Column chromatography over silicagel (60-120 mesh), using ethyl acetate afforded pure [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester as white crystalline solid: 1H NMR (400 MHz, CDCl3) δ 1.3 (t, J=7.4, 3H), 2.3 (s, 3H), 2.5 (m, 4H), 3.0 (br s, 4H), 3.7 (s, 2H), 4.2 (q, J=7.4, 2H), 7.4 (d, J=8.3, 2H), 7.7 (d, J=7.3, 2H); MS 327 [M+1]+.
D. 3-Cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester
A solution of the title C compound, [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester (15 g, 0.046 mol) in a mixture of THF (60 mL) and DMTP (10 mL) is cooled to −78° C. under nitrogen. The resulting solution is stirred at −78° C. for 45 min and to this is added LDA (25.6 mL, 6.40 g, 0.059 mol, 25% solution in THF/Hexane). A solution of iodomethylcyclopentane (11.60 g, 0.055 mol) in a mixture of DMTP (12 mL) and THF (20 mL) is added over a period of 15 min at −78° C. and reaction mixture stirred at −78° C. for 3 h further, followed by stirring at 25° C. for 12 h. The reaction mixture is then quenched by the dropwise addition of saturated aqueous ammonium chloride solution (50 mL) and is concentrated in vacuo. The residue is diluted with water (50 mL) and extracted with ethyl acetate (3×100 mL). The organic solution is washed with a saturated aqueous sodium chloride (2×150 mL), dried over sodium sulfate, filtered and concentrated in vacuo. Column chromatography over silica gel (60-120 mesh), using 50% ethyl acetate in hexane as an eluent to afford 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.9-2.1 (m, 11H), 1.2 (t, J=7.1, 3H), 2.3 (s, 3H), 2.5 (br s, 4H), 3.0 (br s, 4H), 3.6 (m, 1H), 4.1 (q, J=7.1, 2H), 7.5 (d, J=8.3, 2H), 7.7 (d, J=8.3, 2H); MS 409 [M+1]+.
E. 3-Cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid
A solution of the title D compound, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester (14 g, 0.034 mol) in methanol:water (30 mL:10 mL) and sodium hydroxide (4.11 g, 0.10 mol) is stirred at 60° C. for 8 h in an oil bath. The methanol is then removed in vacuo at 45-50° C. The residue is diluted with water (25 mL) and extracted with ether (1×40 mL). The aqueous layer is acidified to pH 5 with 3 N aqueous hydrochloric acid solution. The precipitated solid is collected by vacuum filtration, washed with water (20 mL), followed by isopropyl alcohol (20 mL). Finally, solid cake is washed with 100 mL of hexane and dried under vacuum at 40° C. for 6 h to give 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid as a white solid: 1H NMR (400 MHz, CDCl3) δ 1.1-2.0 (m, 11H), 2.4 (s, 3H), 2.7 (br s, 4H), 3.1 (br s, 4H), 3.6 (m, 1H), 7.5 (d, J=8.3, 2H), 7.6 (d, J=8.3, 2H); MS 381 [M+l]+.
F. 5-Methoxy-thiazolo[5,4-b]pyridin-2-ylamine
A solution of 6-methoxy-pyridin-3-ylamine (5.0 g, 0.0403 mol) in 10 mL of acetic acid is added slowly to a solution of potassium thiocyanate (20 g, 0.205 mol) in 100 mL of acetic acid at 0° C. followed by a solution of bromine (2.5 mL, 0.0488 mol) in 5 mL of acetic acid. The reaction is stirred for 2 h at 0° C. and then allowed to warm to RT. The resulting solid is collected by filtration and washed with acetic acid, then partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The insoluble material is removed by filtration and the organic layer is evaporated and dried to afford 5-methoxy-thiazolo[5,4-b]pyridin-2-ylamine as a tan solid.
G. 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
A solution of the title E compound, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (5 g, 0.013 mol) in DCM (250 mL) is cooled to 0° C. and then charged HOBt hydrate (2.66 g, 0.019 mol), followed by EDCI hydrochloride (6 g, 0.031 mol). The reaction mixture is stirred at 0° C. for 5 h. After that the solution of the title F compound, 5-methoxy-thiazolo[5,4-b]pyridin-2-ylamine (2.36 g, 0.013 mol) and D1EA (8 mL, 0.046 mol) in a mixture of DCM (60 mL) and DMF (20 mL) is added dropwise over 30 min. Reaction temperature is maintained at 0° C. for 3 h, then at RT (28° C.) for 3 days. Reaction is diluted with (60 mL) of water and the organic layer is separated and washed with saturated sodium bicarbonate solution (2×50 mL) followed by water washing (2×50 mL) and saturated sodium chloride aqueous solution (1×150 mL). Finally the organic layer is dried over sodium sulfate, filtered, and evaporated under vacuo. The crude product is purified using column chromatography over silica gel (60-120 mesh), using 40% ethyl acetate in hexane as an eluent to afford 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.9-2.1 (m, 11H), 2.2 (s, 3H), 2.5 (br s, 4H), 3.1 (br s, 4H), 3.7 (m, 1H), 4.0 (s, 3H), 6.8 (d, J=8.8, 1H), 7.5 (d, J=8.3, 2H), 7.7 (d, J=8.3, 2H), 7.8 (d, J=8.8, 1H), 8.6 (s, 1H); MS 617 [M+1]+.
H. 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide dihydrochloride
The title G compound, 3-cyclopentyl-2-(4-methyl piperazinyl sulfonyl)phenyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)propionamide (2.8 g, 0.0051 mol) is added to a cooled solution of 10% hydrochloric acid in isopropanol (3.75 mL). The reaction mixture is stirred at 0° C. for 1 h and then at RT for 2 h. The solid is separated, triturated with 10 mL of isopropanol and collected by vacuum filtration and washed with 50 mL of hexane. The solid is dried at 70° C. for 48 h to afford 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide dihydrochloride as an off white solid.
EXAMPLE 2 (R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1,3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4° C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
- Column: Chiralcel OD-R (250×20 mm) Diacel make, Japan;
- Solvent A: water:methanol:acetonitrile (10:80:10 v/v/v);
- Solvent B: water:methanol:acetonitrile (05:90:05 v/v/v);
- Using gradient elution: gradient program (time, min/% B): 0/0, 20/0, 50/100, 55/0, 70/0;
- Flow rate: 6.0 mL/min; and
- Detection: by UV at 305 nm.
EXAMPLE 3 (S)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is prepared analogously to Example 2.
J MED CHEM 2009, 52, 6142-52
Investigation of Functionally Liver Selective Glucokinase Activators for the Treatment of Type 2 Diabetes
http://pubs.acs.org/doi/abs/10.1021/jm900839k

Type 2 diabetes is a polygenic disease which afflicts nearly 200 million people worldwide and is expected to increase to near epidemic levels over the next 10−15 years. Glucokinase (GK) activators are currently under investigation by a number of pharmaceutical companies with only a few reaching early clinical evaluation. A GK activator has the promise of potentially affecting both the β-cells of the pancreas, by improving glucose sensitive insulin secretion, as well as the liver, by reducing uncontrolled glucose output and restoring post-prandial glucose uptake and storage as glycogen. Herein, we report our efforts on a sulfonamide chemotype with the aim to generate liver selective GK activators which culminated in the discovery of 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide (17c). This compound activated the GK enzyme (αKa = 39 nM) in vitro at low nanomolar concentrations and significantly reduced glucose levels during an oral glucose tolerance test in normal mice.


PATENT
EP-1735322-B1
Example 2(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(-)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(-)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4°C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
- Column: Chiralcel OD-R (250 x 20 mm) Diacel make, Japan;
- Solvent A: water:methanol:acetonitrile (10:80:10 v/v/v);
- Solvent B: water:methanol:acetonitrile (05:90:05 v/v/v);
- Using gradient elution: gradient program (time, min / %B): 0/0, 20/0, 50/100, 55/0, 70/0;
- Flow rate: 6.0 mL/min; and
- Detection: by UV at 305 nm.
REFERENCES
US 7750020
WO-2005095418-A1
US-20080103167-A1
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015218151 | 2015-08-06 | NOVEL PHENYLACETAMIDE COMPOUND AND PHARMACEUTICAL CONTAINING SAME |
| US7750020 | 2010-07-06 | Sulfonamide-Thiazolpyridine Derivatives As Glucokinase Activators Useful The Treatment Of Type 2 Diabetes |
///NOVARTIS, DIABETES, Sulfonamide-Thiazolpyridine Derivatives, Glucokinase Activators, Treatment Of Type 2 Diabetes, 866772-52-3, Novartis Molecule, functionally liver selective glucokinase activators, treatment of type 2 diabetes , NVP-LBX192, LBX-192
c1(sc2nc(ccc2n1)OC)NC(C(c3ccc(cc3)S(=O)(=O)N4CCN(CC4)C)CC5CCCC5)=O
Biocon’s Insulin Glargine gets approval in Japan

![]()
Avik Das | TNN | Mar 28, 2016, 02.52 PM IST
BENGALURU: Biopharmaceutical company Biocon said it got approval from Japan’s health ministry to sell its biosimilar Insulin Glargine in the country.
The product, which is a ready-to-use, prefilled disposable pen with 3 ml of 100IU Insulin Glargine, is expected to be launched in Japan in the first quarter of 2017 with its commercial partner FUJIFILM Pharma Co. Ltd, Biocon said on Monday.
The move will help Biocon capture a significant share of the Japanese Glargine market, which is about $144 million and second largest market outside of North America & Europe.
“The Insulin Glargine approval in the highly regulated market like Japan, marks a huge credibility milestone for Biocon. We see this as a significant achievement in our journey of making global impact in diabetes management through our affordable biosimilar insulins,” chairperson and managing director Kiran Mazumdar-Shaw said.

Kiran Mazumdar–Shaw

Biosimilars are biologic products, made inside living cells and has no clinical differences in terms of safety and effectiveness from the main product. They are however not considered duplicates, like generics, by regulators as it is impossible to manufacture exact copies of biotech drugs.



| Public company | |
| Traded as | BSE: 532523 NSE: BIOCON |
| Industry | Biotechnology |
| Founded | 1978 |
| Founder | Kiran Mazumdar-Shaw |
| Headquarters | Bangalore, Karnataka, India |
|
Key people
|
Kiran Mazumdar-Shaw, (Chairman & MD) |
| Products | Pharmaceuticals Enzymes |
| Revenue | ₹22.41 billion (US$330 million) (2014–15)[1] |
|
Number of employees
|
5,585 (Mar 2011)[1] |
| Subsidiaries | Syngene Clinigene |
| Website | www.biocon.com |
//////Biocon, Insulin Glargine, approval, Japan
RO-28-1675 for Type 2 Diabetes

RO-28-1675
- (2R)-3-Cyclopentyl-2-[4-(methanesulfonyl)phenyl]-N-(thiazol-2-yl)propionamide
- Ro 028-1675
- Ro 0281675
- Ro 28-1675
3-Cyclopentyl-2(R)-[4-(methylsulfonyl)phenyl]-N-(2-thiazolyl)propionamide
| MW | 378.51 | .-70.4 °
Conc 0.027 g/100mL; chloroform, 589 nm; 23 °C
|
|
|---|---|---|---|
| Formula | C18H22N2O3S2 | ||
| CAS No | 300353-13-3 |
Glucokinase Activators
Ro 28-1675 (Ro 0281675) is a potent allosteric GK activator with a SC1.5 value of 0.24± 0.0019 uM.
Roche (Innovator)
PHASE 1 Type 2 DIABETES,
IC50 value: 0.24± 0.0019 uM (SC1.5) [1]
Target: Glucokinase activator
The R stereoisomer Ro 28-1675 activated GK with a SC1.5 of 0.24 uM, while the S isomer did not activated GK up to 10 uM. Oral administration of Ro 28-1675 (50 mg/Kg) to male C57B1/6J mice caused a statistically significant reduction in fasting glucose levels and improvement in glucose tolerance relative to the vehicle treated animals [1].
Comparison of rat PK parameters indicated that Ro 28-1675 displayed lower clearance and higher oral bioavailability compared to 9a.
Following a single oral dose, Ro 28-1675 reduced fasting and postprandial glucose levels following an OGTT, was well tolerated, and displayed no adverse effects related to drug administration other than hypoglycemia at the maximum dose (400 mg).
 .
.
RO-28-1675 as glucokinase activator.
Joseph Grimsby et al., of Roche have recently discovered activators of glucokinase that increase kcat and decrease the S0.5 for glucose, and these may offer a treatment for type II diabetes. Glucokinase (GK) plays a key role in whole-body glucose homeostasis by catalyzing the phosphorylation of glucose in cells that express this enzyme, such as pancreatic β cells and hepatocytes.
By screening of a library of 120,000 structurally diverse synthetic compounds, they found one small molecule that increased the enzymatic activity of GK. Chemical optimization of this initial molecule led to the synthesis of RO-28-0450 as a lead GK activator which is a class of antidiabetic agents that act as nonessential, mixed-type GK activators (GKAs) that increase the glucose affinity and maximum velocity (Vmax) of GK. RO-28-0450 is a racemic compound.
Activation of GK was exquisitely sensitive to the chirality of the molecule: The R enantiomer, RO-28-1675, was found to be a potent GKA, whereas the S enantiomer, RO-28-1674, was inactive. RO-28-1675 also reversed the inhibitory action of the human glucokinase regulatory protein (GKRP). The activators binding in a glucokinase regulatory site originally was discovered in patients with persistent hyperinsulinemic hypoglycemi.
The result of RO-28-1675 as a potent small molecule GKA may shed light to the chemical biologists to devise strategy for developing activators. Thus for a success to this end we must focus on highly regulated enzymes, or cooperative enzymes such as glucokinase, where nature has provided binding sites that are designed to modulate catalysis.
.SYNTHESIS


Paper

Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl-propionamide (2.10 g, 74%) as a white foam.
[α] 23 589 = –70.4° (c=0.027, chloroform).
EI-HRMS m/e calcd for C18H22N2O3S2 (M+ ) 378.1072, found 378.1081.
1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 10.48 (br. s., 1 H), 7.88 (d, J=8.6 Hz, 2 H), 7.53 (d, J=8.6 Hz, 2 H), 7.50 (d, J=3.5 Hz, 1 H), 7.06 (d, J=3.5 Hz, 1 H), 3.76 (t, J=7.7 Hz, 1 H), 3.03 (s, 3 H), 2.28 (dt, J=13.6, 7.7 Hz, 1 H), 1.88 – 1.98 (m, 1 H), 1.42 – 1.84 (m, 7 H), 1.07 – 1.19 (m, 2 H).
Anal. Calcd for C18H22N2O3S2: C, 56.94; H, 5.59; N, 7.28. Found: C, 57.12; H, 5.86; N, 7.40.
PATENT
WO 2000058293
http://www.google.com/patents/WO2000058293A2?cl=en
Example 3 (A) 3-CyclopentyI-2-(4-methanesulfonyl-phenyI)-N-thiazol-2-yI-propionamide

A solution of dπsopropylamine (3.3 mL, 23.5 mmol) in dry tetrahydrofuran (50 mL) and 1.3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdιnone (10 mL) was cooled to -78°C under nitrogen and then treated with a 10M solution of n-butyllithium m hexanes (2.35 mL, 23 5 mmol) The yellow reaction mixture was stiπed at -78°C for 30 mm and then treated dropwise with a solution of 4-methylsulfonylphenylacetιc acid (2.40 g, 11.2 mmol) in a small amount of dry tetrahydrofuran. After approximately one-half of the 4- methylsulfonylphenylacetic acid m dry tetrahydrofuran was added, a precipitate formed Upon further addition of the remaining 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture became thick in nature After complete addition of the 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture was very thick and became difficult to stir An additional amount of dry tetrahydrofuran (20 mL) was added to the thick reaction mixture, and the reaction mixture was stirred at –
78 C for 45 mm, at which time, a solution of lodomethylcyclopentane (2.35 g, 11.2 mmol) in a small amount of dry tetrahydrofuran was added dropwise The reaction mixture was allowed to warm to 25°C where it was stiπed for 15 h. The reaction mixture was quenched with water (100 mL), and the resulting yellow reaction mixture was concentrated in vacuo to remove tetrahydrofuran. The aqueous residue was acidified to pH = 2 using concentrated hydrochloπc acid The aqueous layer was extracted with ethyl acetate The organic phase was dπed over magnesium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 230-400 mesh, 1/3 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (1.80 g, 52%) as a white solid: mp 152-154°C; EI-HRMS m/e calcd for C15H20O4S (Nf) 296.1082, found 296.1080
A solution of 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (4.91 g, 16.56 mmol) and tnphenylphosphine (6.52 g, 24.85 mmol) m methylene chloπde (41 mL) was cooled to 0°C and then treated with N-bromosuccinimide (5.01 g, 28.16 mmol) m small portions The reaction mixture color changed from light yellow to a darker yellow then to brown After the complete addition of N-bromosuccinimide, the reaction mixture was allowed to warm to 25°C over 30 min. The brown reaction mixture was then treated with 2-aminothiazole (4.98 g, 49.69 mmol). The resulting reaction mixture was stiπed at 25°C for 19 h. The reaction mixture was then concentrated in vacuo to remove methylene chloride. The remaining black residue was diluted with a 10% aqueous hydrochloric acid solution (400 mL) and then extracted with ethyl acetate (3 x 200 mL). The combined organic layers were washed with a saturated aqueous sodium chloride solution (1 x 200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate then 1/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)-N-thiazol-2- yl-propionamide (4.49 g, 72%) as a white solid: mp 216-217°C; EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1071.
Example 13
(2R)-3-Cyclopentyl-2-(4-methanesuIfonylphenyl)-N-thiazol-2-yl-propionamide

A solution of ^-( ethanesulfonyl)phenyl acetic acid (43 63 g, 0.204 mol) in methanol (509 mL) was treated slowly with concentrated sulfunc acid (2 mL) The resulting reaction mixture was heated under reflux for 19 h The reaction mixture was allowed to cool to 25°C and then concentrated in vacuo to remove methanol The residue was diluted with ethyl acetate (800 mL) The organic phase was washed with a saturated aqueous sodium bicarbonate solution (1 x 200 mL), washed with a saturated aqueous sodium chlonde solution (1 x 200 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 1/1 hexanes/ethyl acetate) afforded 4-(methanesulfonyl)phenyl acetic acid methyl ester (45.42 g, 98%) as a yellow oil which solidified to a cream colored solid upon sitting over time at 25°C mp 78-80°C, EI-HRMS m/e calcd for Cι0H12O4S (M+) 228 0456, found 228 0451.
A mechanical stiπer was used for this reaction A solution of dnsopropylamme (29.2 mL, 0.21 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro- 2(lH)-pyπmιdιnone (62 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (83 4 mL, 0.21 mol) The yellow-orange reaction mixture was stiπed at -78°C for 35 min and then slowly treated with a solution of 4- (methanesulfonyl)phenyl acetic acid methyl ester (45.35 g, 0.20 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdmone (62 mL) The reaction mixture turned dark in color. The reaction mixture was then stiπed at -78°C for 50 mm, at which time, a solution of lodomethylcyclopentane (50.08 g, 0.24 mol) in a small amount of dry tetrahydrofuran was added slowly. The reaction mixture was then stiπed at -78°C for 50 mm, and then allowed to warm to 25°C, where it was stirred for 36 h. The reaction mixture was quenched with water (100 mL), and the resulting reaction mixture was concentrated in vacuo to remove tetrahydrofuran The remaining residue was diluted with ethyl acetate (1.5 L). The organic phase was washed with a saturated aqueous sodium chloπde solution (1 x 500 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid methyl ester (41.79 g, 68%) as a yellow viscous oil EI-HRMS m/e calcd for Cι6H22O4S (M+) 310.1239. found 310.1230.
A solution of 3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid methyl ester (50 96 g, 0.16 mol) in methanol (410 mL) was treated with a IN aqueous sodium hydroxide solution (345 mL, 0.35 mol). The reaction mixture was stirred at 25°C for 24 h. The reaction mixture was concentrated in vacuo to remove methanol. The resulting aqueous residue was acidified to pH = 2 with concentrated hydrochlonc acid and then extracted with ethyl acetate (5 x 200 mL) The combined organic layers were dned over sodium sulfate, filtered, and concentrated in vacuo to afford pure 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (43 61 g, 90%) as a white solid which was used without further puπfication. mp 152-154°C, EI-HRMS m e calcd for C15H20O4S (M+) 296.1082, found 296.1080.
Two separate reactions were setup in parallel: (1) A solution of (R)-(+)-4-benzyl-2- oxazohdmone (3.67 g, 20.73 mmol) m dry tetrahydrofuran (35 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (7.9 mL, 19.86 mmol). The resulting reaction mixture was stiπed at -78°C for 30 mm and then allowed to warm to 25°C, where it was stirred for 1.5 h (2) A solution of racemic 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (5.12 g, 17.27 mmol) in dry tetrahydrofuran (35 mL) was cooled to 0°C and then treated with tnethylamme (2.8 mL, 19.86 mmol). The reaction mixture was stiπed at 0°C for 10 nun and then treated dropwise with tπmethylacetyl chlonde (2.6 mL, 20.73 mmol). The resulting reaction mixture was stiπed at 0°C for 2 h and then cooled to -78°C for the addition of the freshly prepared chiral oxazolidmone. The reaction mixture containing the oxazolidmone was then added to the cooled (-78°C) mixed anhydπde solution The resulting reaction mixture was stiπed as -78°C for 1 h and allowed to gradually warm to 25°C. The reaction mixture was then stiπed at 25°C for 3 d. The resulting reaction mixture was quenched with water (100 mL) and then concentrated in vacuo to remove tetrahydrofuran. The resulting aqueous residue was diluted with ethyl acetate (600 mL). The organic layer was washed with a saturated aqueous sodium chloπde solution (1 x 300 mL), dπed over sodium sulfate, filtered, and concentrated in vacuo Thin layer chromatography using 13/7 hexanes/ethyl acetate as the developing solvent indicated the presence of two products The higher moving product had a Rf =0.32 and the lower moving product had a Rf = 0.19. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 9/1 then 13/7 hexanes/ethyl acetate) afforded two products: (1) The higher Rf product (4R, 2’S)-4-benzyl-3-[3- cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazohdm-2-one (2.12 g, 54%) as a white foam- mp 62-64°C; [c.]23 589 = +6.3° (c=0.24, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M+) 455.1766, found 455.1757. (2) The lower Rf product (4R, 2R)-4- benzyl-3-[3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.88 g, 99%) as a white foam: mp 59-61°C; [α]23 589 = -98.3° (c=0.35, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M +) 455.1766, found 455.1753. The combined mass recovery from the two products was 6.00 g, providing a 76% conversion yield for the reaction
An aqueous solution of lithium hydroperoxide was freshly prepared from mixing a solution of anhydrous lithium hydroxide powder (707.3 mg, 16.86 mmol) m 5.27 mL of water with a 30% aqueous hydrogen peroxide solution (3.44 mL, 33.71 mmol). This freshly prepared aqueous lithium hydroperoxide solution was cooled to 0°C and then slowly added to a cooled (0°C) solution of (4R, 2’R)-4-benzyl-3-[3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.84 g, 8.43 mmol) in tetrahydrofuran (33 mL) and water (11 mL). The reaction mixture was stiπed 0°C for 1.5 h The reaction mixture was then quenched with a 1.5N aqueous sodium sulfite solution (25 mL) The reaction mixture was further diluted with water (300 mL) The resulting aqueous layer was continuously extracted with diethyl ether until thm layer chromatography indicated the absence of the recovered chiral oxazolidmone in the aqueous layer The aqueous layer was then acidified to pH = 2 with a 10% aqueous hydrochlonc acid solution and extracted with ethyl acetate (300 mL) The organic extract was dned over sodium sulfate, filtered, and concentrated in vacuo to afford (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white solid (2.23 g, 89%) which was used without further puπfication Flash chromatography (Merck Silica gel 60, 70-230 mesh, 30/1 methylene chlonde/methanol then 10/1 methylene chlonde/methanol) was used to obtain a punfied sample for analytical data and afforded pure (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white foam- mp 62-64°C (foam to gel), [α]23 589 = -50.0° (c=0.02, chloroform), EI-HRMS m/e calcd for C15H20O4S (M+) 296 1082, found 296 1080
A solution of tnphenylphosphme (3.35 g, 12.79 mmol) m methylene chloπde (19 mL) was cooled to 0°C and then slowly treated with N-bromosuccmimide (2.28 g, 12.79 mmol) in small portions. The reaction mixture was stiπed at 0°C for 30 mm, and dunng this time penod, the color of the reaction mixture changed from light yellow to a darker yellow then to a purple color. The cooled purple reaction mixture was then treated with the (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid (2.23 g, 7.52 mmol) The resulting reaction mixture was then allowed to warm to 25°C over 45 mm, at which time, the reaction mixture was then treated with 2-amιnothιazole (1.88 g, 18.81 mmol) The resulting reaction mixture was stiπed at 25°C for 12 h. The reaction mixture was then concentrated in vacuo to remove methylene chloπde The remaining black residue was diluted with ethyl acetate (300 mL) and then washed well with a 10% aqueous hydrochlonc acid solution (2 x 100 mL), a 5% aqueous sodium bicarbonate solution (3 x 100 mL), and a saturated aqueous sodium chloride solution (1 x 200 mL). The organic layer was then dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl- propionamide (2.10 g, 74%) as a white foam: mp 78-80°C (foam to gel); [α]23 589 = -70.4° (c=0.027, chloroform); EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1081.
REFERENCES
Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
http://www.nature.com/nrd/journal/v8/n5/fig_tab/nrd2850_T2.html
NMR…..http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-NMR-HY-10595-13569-2014.pdf
http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-Lcms_Ms-HY-10595-13569-2014.pdf
///////////RO-28-1675, Ro 0281675
O=C(Nc1nccs1)[C@H](CC2CCCC2)c3ccc(cc3)S(C)(=O)=O

Chemical structures of Roche’s glucokinase activators (GKAs) RO-28-1675 and piragliatin, as well as the related GKA 1.
MELOGLIPTIN

Melogliptin
Phase III
A DP-IV inhibitor potentially for treatment of type II diabetes.
![]()
EMD-675992; GRC-8200
CAS No. 868771-57-7
4(S)-Fluoro-1-[2-[(1R,3S)-3-(1H-1,2,4-triazol-1-ylmethyl)cyclopentylamino]acetyl]pyrrolidine-2(S)-carbonitrile


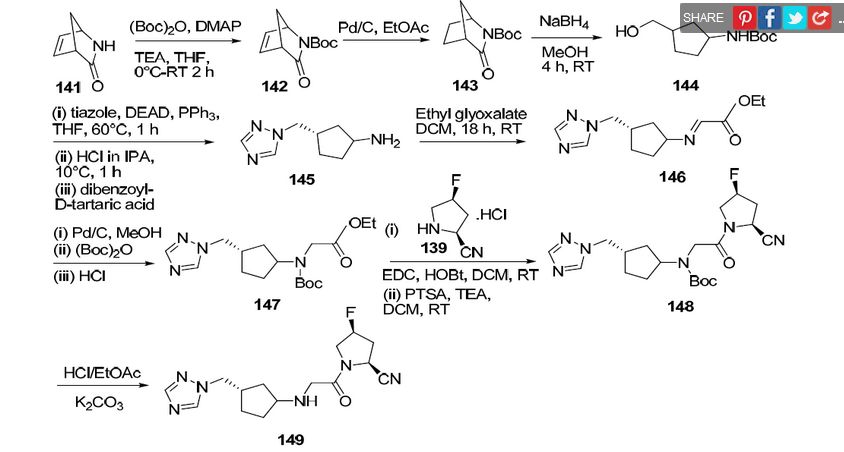
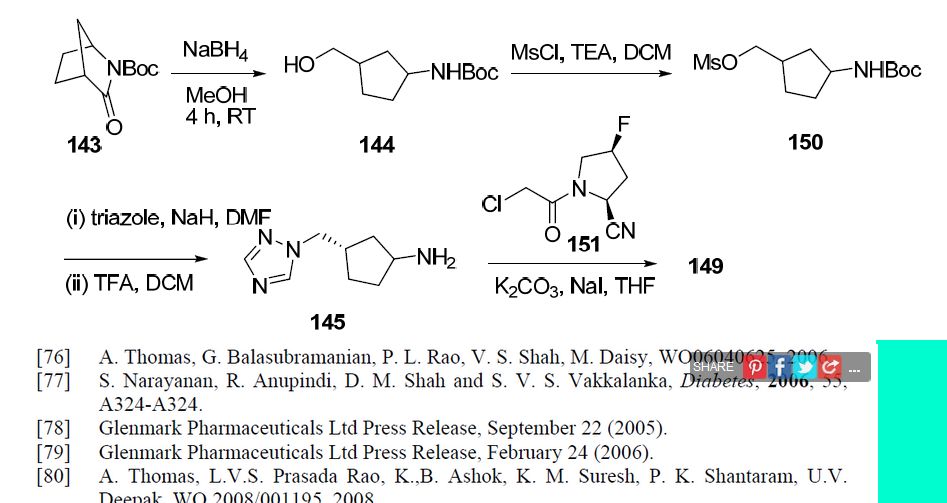
SEE..http://apisynthesisint.blogspot.in/2015/12/melogliptin.html
See more at: http://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html

DISCLAIMER…….The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
/////////
Zydus gets USFDA nod for clinical trials of Saroglitazar

November 19, 2015
New Delhi: Zydus Cadila has received US health regulator’s nod to initiate phase II clinical trials of Saroglitazar, its new drug for treating high fat levels in body due to diabetes, obesity, and sedentary habits.
“United States Food and Drug Administration (USFDA) has endorsed company’s plan to initiate a phase II clinical trial of Saroglitazar in patients with severe hypertriglyceridemia,” Zydus Cadila said in a statement.
http://www.medicaldialogues.in/zydus-gets-usfda-nod-for-clinical-trials-of-sarolitazar/

//////////////
Zydus Cadila’s new 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds in pipeline for diabetes type 2
List of compounds as DPP-IV inhibitors


Watch out on this post as I get to correct structure………..
![]()

2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds

One Example of 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds
CAS 1601479-87-1
(2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[5-(methylsulfonyl)-3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]tetrahydro-2H-pyran-3-amine
MW 399.45, C18 H23 F2 N3 O3 S
INTRODUCTION
Dipeptidyl peptidase IV , CD26; DPP-IV; DP-IV inhibitors acting as glucose lowering agents reported to be useful for the treatment of type 2 diabetes. compound inhibited human DPP-IV enzyme activity (IC50 < 10 nM) in fluorescence based assays.
It lowered glucose levels (with -49.10% glucose change) when administered to C57BL/6J mice at 0.3 mg/kg p.o. in oral glucose tolerance test (OGTT).
Compound displayed the following pharmacokinetic parameters in Wistar rats at 2 mg/kg p.o.: Cmax = 459.04 ng/ml, t1/2 = 59.48 h and AUC = 4751.59 h·ng/ml.
Dipeptidyl peptidase 4 (DPP-IV) inhibitor that inhibited human DPP-IV enzyme activity with an IC50 of < 10 nM in a fluorescence based assay.
Watch out on this post as I get to correct structure………..![]()

PATENT
http://www.google.com/patents/WO2014061031A1?cl=en
Compound 8: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz): 7.32-7.28 (m, IH), 7.26-7.23 (m, 2H), 4.77 (d, IH, J= 10Hz), 4.32(dd, IH, J,= 2.0Hz, J2= 10.8Hz), 4.19 (s, 4H), 3.89-3.83 (m, 4H), 3.70- 3.65 (m, IH), 3.61 (t, IH, J= 11.6Hz), 3.53-3.46 (m, IH), 3.04 (s, 3H), 2.65-2.62 (dd, IH, Ji= 1.2Hz, J2= 12Hz), 1.84 (q, IH, J = 12 Hz); ESI-MS: (+ve mode) 400.0 (M+H)+ (100 %); HPLC: 99.4 %.
Compound 4: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(hexahydropyrrolo[3, 4-c Jpyrrol- 2(lH)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz):

.23-7.20 (m, 2H), 4.64 (d, IH, J= 10.4 Hz), 4.38-4.35 (dd, IH, J,= 2.4Hz, J2= 10.4Hz), 3.69 (t, IH, J= 11Hz), 3.57-3.53 (m, 4H), 3.34-3.30 (m, 8H), 2.68-2.65 (m, IH), 2.04 (q, IH, J = 1 1.6 Hz); ESI-MS: (+ve mode) 323.9 (M+H)+ (100 %), 345.9 (M+Na)+ (20%); HPLC: 98.6 %

PATENT
IN 2012MU03030
“NOVEL DPP-IV INHIBITORS”
3030/MUM/2012
Abstract:
The present invention relates to novel compounds of the general formula (I) their tautomeric forms, their enantiomers, their diastereoisomers, their pharmaceutically accepted salts, or pro-drugs thereof, which are useful for the treatment or prevention of diabetes mellitus (DM), obesity and other metabolic disorders. The invention also relates to process for the manufacture of said compounds, and pharmaceutical compositions containing them and their use.

Pankaj R. Patel (right), Chairman and Managing Director,
////////////2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds, DPP-IV inhibitors
ZYD 1/ZYDPLA 1 From Zydus Cadila, a New NCE in Gliptin class of Antidiabetic agents.

GENERAL STRUCTURE

3-[4-(5-methyl-1,3,4-oxadiazol-2-yl)phenoxy]-5-[[(3R)-1-methyl-2-oxo-3-pyrrolidinyl]oxy]-N-2-thiazolyl- Benzamide
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide……S CONF…..WO2011013141A2
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide…..R CONF…..WO2011013141A2
CAS 1263402-84-1 R CONF
CAS 1263402-76-1 S CONF
ZYD 1/ZYDPLA 1……….Probable Representative structure only, I will modify it as per available info
Watch out on this post as I get to correct structure………..![]()

ZYDPLA1 is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre, the NCE research wing of Zydus. ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents. It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1.
By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
In October 2013, Zydus received IND approval from the US FDA to initiate a phase I trial in type II diabetes
Clinical trials..Type 2 Diabetes Mellitus
NCT01972893; ZYD1/1001;
CTRI/2011/04/001684;
ZYD1
ZYD1/1001
ZYD1 is a novel GLP-1 receptor agonist. The ZYD1 exhibits increased stability to proteolytic cleavage, especially against dipeptidyl peptidase-4 (DPP-IV).ZYD1 is a potent antidiabetic agent without gastrointestinal side-effects. A first in human (FIH) Phase I study intends to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of ZYD1 in normal healthy adult volunteers……..https://clinicaltrials.gov/show/NCT01972893
A randomized, double blind, placebo controlled Phase I clinical study to evaluate the safety, tolerability and pharmacokinetics of ZYD1, a selective GLP-1 agonist, following the subcutaneous administrations in healthy volunteers …………http://www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=2263&EncHid=&modid=&compid=%27,%272263det%27
Some clippings I found

ONE MORE……………

Zydus announces data presentations on ZYDPLA1 “A once-weekly small molecule DPP-IV inhibitor for treating diabetes”, at the ENDO conference in Chicago, Illinois, USA. Ahmedabad, India June 9, 2014 The Zydus group will be presenting data on its molecule ZYDPLA1 a novel compound in the Gliptin class of anti-diabetic agents during the joint meeting of the International Society of Endocrinology and the Endocrine Society: ICE/ENDO 2014 to be held from June 21-24, 2014 in Chicago, Illinois.
ZYDPLA1, currently in Phase I clinical evaluation in USA, is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre. ZYDPLA1 works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP- 1 levels, ZYDPLA1 glucose-dependently increases insulin secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
The Chairman & Managing Director of Zydus, Mr. Pankaj R. Patel said, “Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.”
The abstract of Poster Number: LB-PP02-4 can also be viewed on the ENDO web program at https://endo.confex.com/endo/2014endo/webprogram/authora.html. The Poster Preview is scheduled on Sunday, June 22, 2014 at McCormick Place West.
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPP IV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
About Zydus
Headquartered in Ahmedabad, India, Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 16,000 people worldwide including over 1100 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global researchbased pharmaceutical company by 2020. The group has a strong research pipeline of NCEs, biologics and vaccines which are in various stages of clinical trials including late stage.
About Zydus Research Centre
The Zydus Research Centre has over 20 discovery programmes in the areas of cardio-metabolic disorders, pain, inflammation and oncology. Zydus has in-house capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. The Zydus Research group had identified and developed Lipaglyn™ (Saroglitazar) which has now become India’s first NCE to reach the market. Lipaglyn™ is a breakthrough therapy in the treatment of diabetic dyslipidemia and Hypertriglyceridemia. The company recently announced the commencement of Phase III trials of LipaglynTM (Saroglitazar) in patients suffering from Lipodystrophy.
PATENT
http://www.google.com/patents/WO2011013141A2?cl=en
Rajendra Kharul, Mukul R. Jain, Pankaj R. Patel
Substituted benzamide derivatives as glucokinase (gk) activators

Scheme 2:

Scheme 3:

Scheme 4A:


Scheme 4B.
] Scheme 5 A:

Scheme 5B:

Scheme 6:

Example 1
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
4-(Dimethylamino)pyridine (DMAP) (0.149 g), N-(3-Dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDCI.HC1) (0.524 g) were added to a solution of 3-
( 1 -Methoxypropan-2-yloxy)-5-(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl) phenoxy) benzoic acid (0.5 g) (Intermediate 1) in dry DCM under nitrogen at 0-5 0C. 2-Aminothiazole (0.134 g) was added and the mixture was stirred for 16 h at room temperature. It was diluted with commercially available DCM. Organic phase was washed with dil HCl, saturated solution of NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude residue. The residue was chromatographed using silica gel as stationary phase and MeOH: CHCl3 gradient as mobile phase up to yield the product (0.3 g) as a white solid.
1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.92-2.01 (m, 1 H), 2.59 (s, 3 H), 2.60-2.65 (m,
I H), 2.79 (s, 3 H), 3.31-3.34 (m, 1 H), 3.36-3.44 (m ,1 H), 5.15 (t, J = 7.6 Hz, 1 H),
7.08 (s, 1 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.27-7.29 (m, 1 H), 7.40 (s, 1 H), 7.54 (s, 1 H),
7.62 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H), 12.60 (bs, 1 H); ESI-MS mix (relative intensities): 492.03 (M+H)+ (100 %), 514.02 (M+Na)+(15 %); UPLC Purity: 93.59 %, Rettime: 3.59 min.
Intermediate 1: 3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxo pyrrolidin -3-yloxy)benzoic acid
A solution of Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl- 2-oxopyrrolidin-3-yloxy)benzoate (7 g) (Intermediate 2) in a mixture of THF and methanol (1 :1 ratio) was treated with a solution of sodium hydroxide (2 g) in water and the reaction mixture was stirred for 1 h at room temperature. The resulting solution was concentrated under vacuum to remove THF and methanol, diluted with water, and washed with EtOAc. The aqueous phase was cooled and acidified with 0.1 N HCl and extracted with DCM, combined organic extracts washed with brine, dried over Na2SO4 and concentrated in vacuo to give the product (3.5 g) as white solid.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.20-2.27 (m, 1 H), 2.59-2.67 (m, 1 H), 2.77 (s, 3 H), 2.95 (s, 3 H), 3.38-3.44 (m, 1 H), 3.49-3.54 (m, 1 H), 4.96 (t, J = 7.2 Hz, 1 H), 6.93-6.95 (m, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.32-7.34 (m, 1 H), 7.52 (d, J= 8.8 Hz, 2 H), 9.96-9.98 (m, 2 H); ESI-MS (relative intensities): 431.9 (M+ Na)+ (70%).
Intermediate 2: Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2- oxo- pyrrolidin-3-yloxy)benzoate
To a stirred mixture of Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy) benzoate (15 g) (Intermediate 3), N,N-dimethylglycine hydrochloride (2.3 g), copper (II) iodide (1 g) in dry 1,4-dioxane was added 2-(4-iodophenyl)-5 -methyl- 1,3,4- oxadiazole (15.4 g) (Intermediate 4) under nitrogen. The reaction mixture was refluxed for 24 h. The reaction mixture was cooled, quenched with water and extracted with DCM. Combined organic washings were washed with water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude product. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether (9:1) as mobile phase to give the product (7 g) as thick liquid. 1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.91-1.98 (m, 1 H), 2.49-2.54 (m, 1 H), 2.56 (s, 3 H), 2.77 (s, 3 H), 3.34-3.41 (m, 2 H), 3.81 (s, 3 H), 5.12 (t, J= 7.6 Hz, 1 H), 7.13- 7.15 (m, 2 H), 7.22 (d, J = 8.8 Hz, 2 H), 7.42 (s, 1 H), 7.97 (d, J = 8.8 Hz, 2 H); ESI- MS (relative intensities): 423.9 (M+H)+ (100%), 446.2 (M+ Na)+ (30%).
Intermediate 3: Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy)benzoate
To a stirred solution of Methyl 3, 5-dihydroxybenzoate (20 g) [CAS No. 2150- 44-9] in dry DMF was added potassium carbonate (48 g) and the suspension stirred at ambient temperature under nitrogen. To this 3-Bromo-l-methyl-pyrrolidin-2-one (4Og) (Intermediate 5) [J. Med. Chem., 1987, 30, 1995-98] was added in three equal portions in 4 h intervals at room temperature and stirred overnight at ambient temperature. It was then quenched with water. The aqueous suspension was extracted with DCM. The combined extracts were washed with water, brine, dried over Na2SO4, and filtered, concentrated under reduced pressure to get the thick liquid residue. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether as a mobile phase to yield the product as white solid (15 g).1H NMR (CDCl3, 400 MHz) δ ppm: 2.08-2.10 (m, 1 H), 2.60-2.67 (m, 1 H), 3.04 (s, 3 H), 3.40-
3.43 (m, 1 H), 3.48-3.51 (m, 1 H), 3.87 (s, 3 H), 4.91 (t, J = 7.2 Hz, 1 H), 6.59- 6.61 (m, 1 H), 7.07-7.09 (m, 1 H), 7.09-7.13 (m, 1 H), 8.02 (s, 1 H); ESI-MS (relative intensities): 287.9 (M+ Na)+ (30%).
Example 68…. S CONFIGURATION
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of S-(-)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5- [(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (3.5 g) (Intermediate 13) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, 4- (dimethylamino)pyridine (2.24 g) followed by N-(3-Dimethy lam inopropy I)-N5– ethylcarbodiimide hydrochloride (EDCI. HCl) (3.3 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.94 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (200 mL), washed with dil HCl (20 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (3.5 g). The crude brown solid was purified by solvent trituration.
1H ΝMR (CDCl3, 400 MHz) δ ppm: 2.13-2.22 (m, 1 H), 2.62 (s, 3 H), 2.56-2.64 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.92 (t, J= 7.2 Hz, 1 H), 7.01 (s, 1 H), 7.04 (t, J= 2 Hz, 1 H), 7.21 (d, J = 8.8 Hz, 2 H), 7.26 (s, 1 H), 7.36 (s, 1 H), 7.44 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 513.8 (M+Νa)+ (10 %); UPLC Purity: 98.13 %, Ret. time: 3.577 min. Chiral Purity by HPLC: 97.31 %, Ret. time: 22.93 min. % ee: 94.62 %
Intermediate 13: S-(-)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2- oxo-pyrro- lidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 1.5 g) was added to a stirring mixture of (.S)-(-)-Methyl 3- [4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy] benzoate (5.3g) (Intermediate 14) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (3.5 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.17-2.22 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.99 (t, J= 7.2 Hz, 1 H), 6.89 (t, J = 2.4 Hz, 1 H), 7.07 (d, J = 8.8 Hz, 2 H), 7.28 (s, 1 H), 7.53 (s, 1 H), 7.95 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410 (M+H)+ (100 %); UPLC Purity: 97.85 %, Ret. time: 3.136 min. Chiral Purity by HPLC: 99.59 %, Ret. Time: 57.46 min. % ee: 99.18 %
Intermediate 14: (S) -(-) -Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate
Sodium hydride suspension (0.71 g, 50 %) was added to a stirring solution of (£)-(-)- methyl 3 -(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (5.5 g) (Intermediate 15) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.91 mL) was added and stirred till the reaction completion. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vaccuo to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product (5.3 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.14-2.21 (m, 1 H), 2.58-2.63 (m, 1 H), 2.64 (s, 3 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m , 1 H), 3.89 (s, 3 H), 4.99 (t, J = 7.2 Hz, 1 H), 6.99 (t, J = 2 Hz, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.35 (s, 1 H), 7.53 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 424.1 (M+H)+ (100 %); UPLC Purity: 96.1 1 %, Ret. time: 3.68 min. Chiral Purity by HPLC: 92.05 %, Ret. Time: 39.33 min.
Intermediate 15: (S) -(-) -Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxo pyrrolidin-3-yl)oxy) benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (7 g) (Intermediate 7) and (/?)-(+)-3-hydroxy-2-pyrrolidinone (Intermediate 16) (2.4g) in dry THF (200 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (1 1.3 g) was added. Diisopropyl azodicarboxylate (DIAD) (6.2 mL) in dry THF (10 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (6 g). 1H NMR (CDCl3, 400 MHz) δ ppm: 2.26-2.33 (m, 1 H), 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.47 (m, 1 H), 3.51-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.07 (bs, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (100 %); UPLC Purity: 98.35 %, Ret. time: 3.47 min. Chiral Purity by HPLC: 95.31 %, Ret. Time: 47.97 min. ee: 90.62 %.
Intermediate 16: (R)-(+)-3-Hydroxy-2-pyrrolidinone
To a stirring mixture of 4-Nitrobenzoic acid (21.5 g) and (5)-(-)-3-hydroxy-2- pyrrolidinone (11.8 g) (Intermediate 17) in dry THF (360 mL) taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (61.2 g) was added. To this reaction mixture, diisopropyl diazodicarboxylate (DIAD) (34 mL) was added drop wise in three portions at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was concentrated under vacuum to obtain residue. Methanol (360 mL) was added to the residue followed by potassium carbonate (10 g) at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was diluted with CHCl3 and filtered through celite. Celite bed was successively washed with 1 % MeOH:CHCl3. The filtrates were combined and concentrated to dryness to remove solvents. The residues were partitioned between EtOAc: dil. HCl (200 mL, 9:1) and stirred for 15 min. Layers were separated, aq. layer was washed with EtOAc thrice until all organic impurities were washed out. The aq. Layer was concentrated to dryness to remove the water and solid residues were obtained. The residues obtained were washed with 1-2 % MeOH: CHCl3 (3 x 100 mL), dried over sodium sulfate, filtered trough cotton, concentrated to get brown thick liquid product.
1U NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.46-2.54 (m, 1 H), 3.28-3.35 (m, IH), 3.38-3.48 (m, 1 H), 4.50 (t, J = 8.4 Hz, 1 H), 4.55 (bs, 1 H), 7.02 (bs, 1 H); [α]D25: + 68, c = l, CHCl3
Intermediate 17: (S)-(-)-3-hydroxy-2-pyrrolidinone
Cone. H2SO4 (14.8 g, 8 mL) was added drop wise over 5 min to the stirring solution of (5)-(-)-4-Amino-2-hydroxybutyric acid (15 g) [CAS No. 40371-51-5] in MeOH (95 rnL) under dry conditions using anhydrous CaCl2 guard tube. After refluxing for 4 h, the reaction mixture was allowed to cool to room temperature and diluted with water (15 mL). Potassium carbonate (24 g) was added in portions to the reaction mixture and stirred overnight (20 h). Reaction mixture was diluted with CHCl3, filtered through celite. Celite bed was thoroughly washed with 1 % MeOHiCHCl3. The filtrates were combined and evaporated to dryness to obtain thick liquid residue. The residue was subjected to aging using 1-2 % MeOHiCHCl3 and then filtered. Organic layers were combined, dried over anhydrous sodium sulphate, filtered and concentrated to obtain the white solid. (1 1.8 g)
1H NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.48-2.55 (m, 1 H), 3.30-3.35
(m, IH), 3.36-3.50 (m, 1 H), 4.34 (t, J = 8.4 Hz, 1 H), 6.51 (bs, 1 H); [α]D25: + 98, c =
1, CHCl3
Following examples (Example 70-76) were prepared by using similar procedure as that of example lwith suitable modifications as are well within the scope of a skilled person
Example 77 R CONFIGURATION
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide
CORRECTED AS (R)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of (/?j-(+)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-
[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (0.2 g) (Intermediate 18) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, N.ΛP-dimethylamino pyridine (0.060 g) followed by EDCI. HCl (0.23 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.054 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (20 mL), washed with dil HCl (5 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (0.080 g). The crude brown solid was purified by solvent trituration.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.15-2.20 (m, 1 H), 2.55-2.60 (m, 1 H), 2.62 (s, 3 H), 2.93 (s, 3 H), 3.38-3.43 (m, 1 H), 3.47-3.53 (m, 1 H), 4.91 (t, J= 6.8 Hz, 1 H), 6.99 (d, J= 8.8 Hz, 2 H), 7.10-7.14 (m, 2 H), 7.23-7.26 (m, 1 H), 7.36 (s, 1 H), 7.43 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H), 10.75 (bs, 1 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 514.0 (M+Na)+ (20 %); UPLC Purity: 95.25 %, Ret.time: 3.578 min. Chiral Purity by HPLC: 95.93 %, Ret.time: 14.17min. % ee: 91.86 %
Intermediate 18: (R)-(+)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl- 2-oxo- pyrrolidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 0.35 g) was added To a stirring mixture of (/?)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate (1.1 g) (Intermediate 19) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (0.76 g).
1H NMR (DMSO-J6, 400 MHz) δ ppm: 1.92-1.99 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 3.31 (s, 3 H), 3.32-3.40 (m, 2 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.08 (s, 1 H), 7.14 (s, 1 H), 7.23 (d, J= 8.8 Hz, 2 H), 7.40 (s, 1 H), 7.99 (d, J= 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (65 %), 410.1 (M+H)+ (100 %); UPLC Purity: 96.95 %, Ret. time: 3.12 min. Chiral Purity by HPLC: 89.04 %, Ret. Time: 48.15 min. Intermediate 19: (R)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate:
Sodium hydride suspension (0.16 g, 50 %) was added to a stirring solution of (R)- (+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (1.5 g) (Intermediate 20) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.20 mL) was added and stirred till the reaction completed. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vacuum to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product
(1.2 g).
1U NMR (DMSO-J6, 400 MHz) δ ppm: 1.95-1.98 (m, 1 H), 2.51-2.55 (m, 1 H), 2.56 (s, 3 H), 2.88 (s, 3 H), 3.29-3.34 (m, 1 H), 3.37-3.40 (m ,1 H), 3.81 (s, 3 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.13-7.17 (m, 2 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.41 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 423.9 (M+H)+ (100 %); UPLC Purity: 90.38 %, Ret. time: 3.68 min.
Intermediate 20: (R)-(+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxopyrrolidin -3-yl)oxy)benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (2.5 g) (Intermediate 7) and (5)-(-)-3-hydroxy-2-pyrrolidinone (Intermediate 17) (0.8 g) in dry THF (70 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (3.77 g) was added. Diisopropyl azodicarboxylate (DIAD) (2.1 mL) in dry THF (2 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (2 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.23-2.30 (m, 1 H); 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.46 (m, 1 H), 3.50-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (45 %); UPLC Purity: 96.40 %, Ret. time: 3.48 min. Chiral Purity by HPLC: 90.92 %, Ret. Time: 48.36 min.


http://zyduscadila.com/wp-content/uploads/2015/09/ZYDPLA1-a-Novel-LongActing-DPP-4-Inhibitor.pdf
http://zyduscadila.com/wp-content/uploads/2015/05/PressNote23-10-13.pdf
http://zyduscadila.com/wp-content/uploads/2015/07/annual_report_14-15.pdf
http://pharmaxchange.info/press/2012/08/glucokinase-activators-gkas-in-diabetes-management/
LB-PP02-4 ZYDPLA1, a novel long-acting DPP-4 inhibitor
Jt Int Congr Endocrinol Annu Meet Endocr Soc (ICE/ENDO) (June 21-24, Chicago) 2014, Abst LBSU-1075
LB-PP02-4 ZYDPLA1, a Novel Long-Acting DPP-4 Inhibitor
Session: LBSU 1074-1087-Diabetes & Obesity
Translational
Disclosure: MRJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. AAJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. RB: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. HP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. SK: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. KP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VKR: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PRP: Chairman, Cadila Healthcare Limited, Ahmedabad, India. RD: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India.

////////Dipeptidyl Peptidase IV, CD26, DPP-IV, DP-IV, Inhibitors
GKM 001 in pipeline for Diabetes by Advinus




GKM 001……Several probables
Watch out on this post as I get to correct structure………..![]()
Advinus Therapeutics Private L,
![]()
A glucokinase activator for treatment of type II diabetes
In October 2012, Takeda and Advinus have entered into an agreement to initiate a three-year discovery collaboration program focused on novel targets for inflammation, CNS, and metabolic diseases.
| Company | Advinus Therapeutics Ltd. |
| Description | Activator of glucokinase (GCK; GK) |
| Molecular Target | Glucokinase (GCK) (GK) |
| Mechanism of Action | Glucokinase activator |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase I/II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |

Advinus chief executive officer/MD Dr. Rashmi Barbhaiya.

PATENT
https://www.google.co.in/patents/WO2009047798A2?cl=en
Example Cl : (-)-{5-ChIoro-2-[2-(4-cyclopropanesulfonylphenyI)-2-(2,4- difluorophenoxy)acetylamino]thiazol-4-yl}-acetic acid, ethyl ester


Step I: Preparation of (-)-(4-Cyclopropanesulfonylphenyl)-(2,4- difluorophenoxy)acetic acid (Cl-I):
To a solution of (4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (obtained in example Al -step III) in ethyl acetate was added (S)-(-)-l-phenylethylamine drop wise at -15 °C. After completion of addition the reaction was stirred for 4-6 hours. Solid was filtered and washed with ethyl acetate. The solid was then taken in IN HCl and extracted with ethyl acetate, ethyl acetate layer was washed with brine, dried over anhydrous sodium sulfate. Solvent was removed under reduced pressure to obtain (-)-(4- cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid. Enantiomeric enrichment was done by repeating the diasteriomeric crystallization. [α]23 589 = – 107.1 ° (c = 2%Chloroform) Enantiomeric purity > 99. % (chiral HPLC)
Step II: (-)-{5-Chloro-2-[2-(4-cyclopropanesulfonylphenyl)-2-(2,4- difluorophenoxy)acetyIamino]thiazol-4-yl}-acetic acid ethyl ester : To a solution of (-)-4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (Cl-I) in DCM, was added DMF and cooled to 0 °C, followed by the addition of oxalyl chloride under stirring. Stirring was continued for 1 hour at the same temperature. The resulting mixture was further cooled to -35 °C, and to that, a solution of excess (2- amino-5-chlorothiazol-4-yl)acetic acid ethyl ester in DCM was added drop wise. After completion of reaction, the reaction mixture was poured into IN aqueous HCl under stirring, organic layer was washed with IN HCl, followed by 5% brine, dried over anhydrous sodium sulfate, solvent was removed under reduced pressure to get the crude compound which was purified by preparative TLC to get the title compound. [α]23 589 = – ve (c = 2%Chloroform)
1H NMR(400 MHz, CDCl3): δ 1.06-1.08 (m, 2H), 1.30 (t, J=7.2 Hz, 3H), 1.33-1.38 (m, 2H), 2.42-2.50 (m, IH), 3.73 (d, J=2 Hz, 2H), 4.22 (q, J=7.2 Hz ,2H), 5.75 (s, IH), 6.76- 6.77 (m, IH), 6.83-6.86 (m, IH), 6.90-6.98 (m, IH), 7.73 (d, J=8.4 Hz, 2H), 7.96 (d, J=8.4 Hz, 2H), 9.96 (bs, IH). MS (EI) m/z: 571.1 and 573.1 (M+ 1; for 35Cl and 37Cl respectively).
Examples C2 and C3 were prepared in analogues manner of example (Cl) from the appropriate chiral intermediate:

Example Dl : (+)-{5-Chloro-2-[2-(4-cyclopropanesulfonylphenyl)-2-(2,4- difluorophenoxy)acetylamino]thiazol-4-yl}acetic acid, ethyl ester

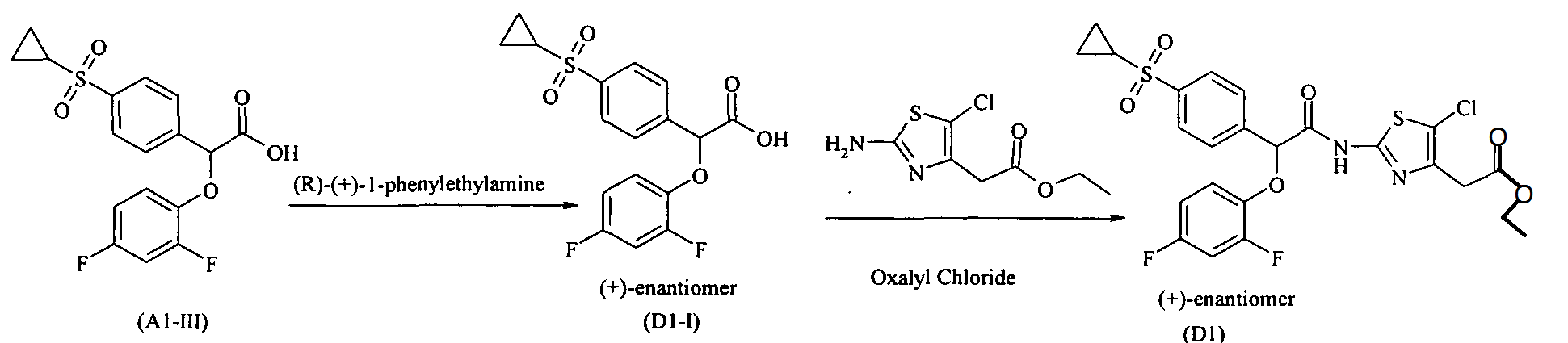
Preparation of (+)-(4-Cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (Dl-I):
To a solution of (4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (obtained in example Al -step III) in ethyl acetate, was added (R) (+)-l- phenylethylamine drop wise at -15 °C. After completion of addition the reaction was stirred for 4-6 hours. Solid was filtered and washed with ethyl acetate. The solid was then taken in IN HCl and extracted with ethyl acetate, ethyl acetate layer was washed with brine, dried over anhydrous sodium sulfate. Solvent was removed under reduced pressure to obtain (+)-(4-Cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid. Enantiomeric enrichment was done by repeating the diasteriomeric crystallization. [α]23 589 = +93.07° (c = 2%Chloroform) Enantiomeric purity > 99. % (by chiral HPLC)
(+)-(4-CyclopropanesuIfonylphenyI)-(2,4-difluorophenoxy)acetic acid ethyl ester (Dl)
The example Dl was prepared using (+)-4-cyclopropanesulfonylphenyl)-(2,4- difluorophenoxy)acetic acid (Dl-I), and following the same reaction condition for amide coupling as described in example Cl, [ot]23 589 = + ve (c = 2%Chloroform)
PATENT
https://www.google.co.in/patents/WO2008104994A2?cl=en
Synthesis Type-P
Example Pl : {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)- propionylamino]-thiazol-4-yI}-acetic acid
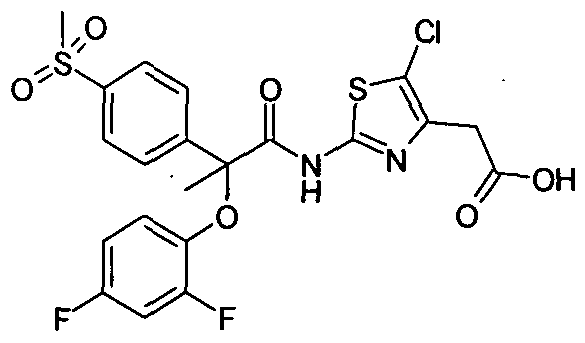
To a solution of {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl- phenyl)-propionylamino]-thiazol-4-yl}-acetic acid methyl ester (0.03 g, 0.05 mmol) in THF: Ethanol: water ( ImI + 0.3ml + 0.3 ml) was added lithium hydroxide (0.0046 g, 0.11 mmol). The resulting mixture was stirred for 5 hours at room temperature followed by removal of solvent under reduced pressure. The residue was suspended in water (15 ml), extracted with ethyl acetate to remove impurities. The aqueous layer was acidified with IN HCl (0.5 ml) and extracted with ethyl acetate (2×10 ml), This ethyl acetate layer was washed with water (15 ml), brine (20 ml), dried over anhydrous sodium sulfate and solvent was removed under reduced pressure to give solid product {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4- methanesulfonyl-phenyl)-propionylamino]-thiazol-4-yl} -acetic acid (9 mg). 1H NMR (400 MHz, CDCl3): δ 1.85 (s, 3H) , 3.07 (s, 3H) , 3.72 ( s, 2H), 6.64-6.69 ( m, 2H ) , 6.89-6.91 (m, IH ), 7.84 ( d, J – 8.4 Hz, 2H), 8.00 ( d, J = 8.8 Hz, 2H). MS (EI) mlz: 530.70 (M + 1), mp: 109-111 0C.
Preparation of {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)- propionylamino)-thiazol-4-yl}-acetic acid methyl ester used in Example Pl:

To a mixture of 2-(2, 4-Difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)-propionic acid (0.110 g, 0.22 mmol), (2-Amino-5-chloro-thiazol-4-yl)-acetic acid methyl ester (0.071 g, 0.32 mmol), HOBt (0.052g, 0.38 mmol), and EDCI (0.074 g, 0.38 mmol) in methylene dichloride (10 ml) was added N-methylmorpholine (0.039 g, 0.38 mmol). The resulting mixture was stirred at room temperature for overnight followed by dilution with 10 ml methylene dichloride. The reaction mixture was poured onto water (20 ml), and organic layer separated, washed with water (2x 20 ml), brine (20 ml), dried over sodium sulfate and solvent evaporated to get residue which was purified by preparative TLC using 50% ethyl acetate in hexane as mobile. To give desired compound (0.30 g). 1H NMR (400 MHz, CDCl3): δ 1.45 (t, J = 7.2 Hz, 3H), 1.93 (s, 3H), 3.14 (s, 3H), 3.77 (d, J = 2.8 Hz, IH), 4.26 (q, J = 7.2 Hz, IH), 6.69-6.77(m, 2H), 6.96-7.02 (m, IH), 7.89 (d, J = 8.4 Hz, 2H), 8.07 (d, J= 8.4Hz, IH).; MS (EI) m/z: 559 .00 (M + 1).
PATENT
http://www.google.com/patents/WO2012020357A1?cl=en
4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]- thiazol-5-yloxy}-benzoic acid, cas 1359151-08-8

Step I: (4-Cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester:
A1C13 (7.98 g, 48.42 mmole) was suspended in DCM (50 mL) and cooled to 0 C under argon atmosphere. To this suspension was added chlorooxo ethylacetate (4.5 mL, 39.98 mmol) at 0 °C and stirred for 45 min. followed by addition of a solution of cyclopropylsulfanyl-benzene (5 g, 33.28 mmol) in DCM (10 mL) and stirred at 25 °C for 2 hr. Reaction mixture was slowly poured over crushed ice, organic layer was separated and aqueous layer was extracted with DCM (3 X 50 mL), combined organic layer was washed with brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain (4- cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester (3.1 g) as an oily product.
*H NMR (400 MHz, CDC13): δ 0.72-0.73 (m, 2H), 1.15-1.17 (m, 2H), 1.40 (t, J = 6.6 Hz, 3H), 2.18-2.21 (m, 1H), 4.41 (q, J = 6.8 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H); MS (EI) m/z: 250.9 (M+l).
Step II: (4-Cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester:
(4-Cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester (3.1 g, 12.53 mmole) in DCM (50 mL) was cooled to 0-5 °C followed by addition of mCPBA (9.8 g , 31.33 mmol) in portion wise at 0 °C. After stirring at 25 °C for 4 hr, the reaction mixture was filtered; filtrate was washed with saturated aq. Na2S203 and satd. aq. sodium bicarbonate solution followed by brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give (4-cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester (3 g).
*H NMR (400 MHz, CDC13): δ 1.05-1.10 (m, 2H), 1.36-1.39 (m, 2H), 1.40 (t, J = 6.8 Hz, 3H), 2.45-2.50 (m, 1H), 4.42 (q, J = 7.2 Hz, 2H), 8.01 (d, J = 8.4 Hz, 2H), 8.20 (d, J = 8.4 Hz, 2H); MS (EI) m/z: 297.1 (M+NH4).
Step III: p-Toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester:
A mixture of (4-cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester (0.5 g, 1.77 mmole) and p-toluene sulfonyl hydrazide (0.48 g , 2.3 mmol) in toluene (15 mL) was refluxed for 16 hr using a Dean-Stark apparatus. Reaction mixture was concentrated to give the crude product which was purified by column chromatography over silica gel using 20-25% ethyl acetate in hexane as eluent to provide p-toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester (0.5 g).
MS (EI) m/z 451.0 (M+l).
Step IV: (4-Cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester:
To a solution of p-toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester (0.5 g, 1.23 mmol) in dry DCM (6 mL), was added triethylamine (0.17 mL, 1.35 mmol) and stirred at 25 °C for 1 hr. Reaction mixture was concentrated to provide (4- cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester (0.5 g) which was used in next reaction without any purification.
MS (EI) m/z: 295.1 (M+l).
Step V: Cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester:
(4-Cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester (1 g, 3.37 mmol) was dissolved in DCM (16 mL) under argon atmosphere. To this solution, cyclopentanol (0.77 mL, 8.44 mmol) was added followed by rhodium(II)acetate dimer (0.062 g, 0.14 mmol). Mixture was stirred at 25 C for 12 hr. Reaction mixture was diluted with DCM (25 mL), organic layer was washed with water followed by brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a crude product which was purified by column chromatography using 25-35% ethyl acetate in hexane as eluent to provide cyclopentyloxy-(4- cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester (0.35 g).
*H NMR (400 MHz, CDC13): δ 1.02-1.05 (m, 2H), 1.24 (t, J = 6.8 Hz, 3H), 1.35-1.37 (m, 2H), 1.53-1.82 (m, 8H), 2.42-2.50 (m, 1H), 4.02-4.04 (m, 1H), 4.15-4.22 (m, 2H), 5.00 (s, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H); MS (EI) m/z: 370.0 (M+18).
Step VI: Cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid:
To cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester (0.35 g, 0.99 mmol) was added a solution of lithium hydroxide (0.208 g, 4.97 mmol) in water (4 mL) followed by THF (2 mL) and methanol (1 drop) and stirred for 12 hours at 25 0 C. Organic solvents were evaporated from the reaction mixture and aqueous layer was acidified IN HCl, extracted with ethyl acetate (3 X 10 mL), organic layer was washed with brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide cyclopentyloxy-(4- cyclopropanesulfonyl-phenyl)-acetic acid (0.210 g).
*H NMR (400 MHz, CDC13): δ 1.02-1.07 (m, 2H), 1.34-1.38 (m, 2H), 1.55-1.62 (m, 2H), 1.69- 1.82 (m, 6H), 2.43-2.47 (m, 1H), 4.08-4.10 (m, 1H), 5.02 (s, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H); MS (EI) m/z: 342.0 (M+18)
Example Al: 4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]-

To a mixture of cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid (Preparation 1) (0.1 g, 0.30 mmol), 4-(2-Amino-thiazol-5-yloxy)-benzoic acid methyl ester (0.085 g, 0.33 mmol), HOBt (0.045 g, 0.33 mmol), and EDCI (0.063 g, 0.33 mmol) in DCM (5 mL), was added N-methyl morpholine (0.033 g, 0.30 mmol). The resulting mixture was stirred at room temperature overnight followed by dilution with methylene chloride (20 mL). The reaction mixture was poured into water; organic layer was washed with water, brine, dried over sodium sulfate, and the organic solvent evaporated to get a residue which was purified by preparative TLC to provide the title compound (0.145 g).
*H NMR (400 MHz, CDC13): δ 1.03-1.05 (m, 2H), 1.34-1.38 (m, 2H), 1.58- 1.65 (m, 2H), 1.76- 1.81 (m, 6H), 2.42-2.45 (m, 1H), 3.89 (s, 3H), 4.05-4.15 (m, 1H), 5.08 (s, 1H), 7.07 (d, J = 8.8 Hz, 2H), 7.15 (s, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.8 Hz, 2H), 9.72 (s, 1H); MS (EI) m/z: 556.9 (M + 1).
Example Bl: 4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]- thiazol-5-yloxy}-benzoic acid:
4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]-thiazol-5-yloxy}- benzoic acid methyl ester (0.145 g, 0.26 mmol, obtained in example Al) was taken in H20: THF (1 :2, 6 mL) to it was added MeOH (1 drop) followed by LiOH (0.054 g, 1.30 mmol) and stirred for 12 hr. After completion of the reaction, organic solvent was removed under reduced pressure. The aqueous layer was washed with diisopropyl ether then acidified with 1 N HC1 to pH 4. The solid formed was filtered, washed with water, diisopropyl ether & dried under vacuum to get the title_compound (0.12 g).
IH NMR- (400 MHz DMSO-ifc):- δ 1.01-1.05 (m, 2H), 1.09-1.13 (m, 2H), 1.22-1.49 (m, 2H), 1.59-1.73 (m, 6H), 2.82-2.86 (m, IH), 3.99-4.01 (m, IH), 5.31 (s, IH), 7.16 (d, J = 8.4 Hz, 2H), 7.37 (s, IH), 7.74 (d, J = 8.4 Hz, 2H), 7.91 (m, 4H), 12.55 (br. s, IH), 12.90 (br.s, IH); MS (EI) m/z: 542.9 (M+l)
CLIPPINGS
Advinus’ GK-activator Achieves Early POC for Diabetes
November 29 2011
Partnership Dialog Actively Underway
Advinus Therapeutics, a research-based pharmaceutical company founded by globally experienced industry executives and promoted by the TATA Group, announced that it has successfully completed a 14-day POC study in 60 Type II diabetic patients on its lead molecule, GKM-001, a glucokinase activator. The results of the trial show effective glucose lowering across all doses tested without any incidence of hypoglycemia or any other clinically relevant adverse events.
The clinical trials on GKM-001 validate the company’s pre-clinical hypothesis that a liver selective Glucokinase activator would not cause hypoglycemia (very low blood sugar), while showing robust efficacy.
“GKM-001 is differentiated from most other GK molecules that are in development, or have been discontinued, due to its novel liver selective mechanism of action. GKM-001 has a prolonged pharmacological effect and a half-life that should support a once a day dosing as both mono and combination therapy.” said Dr. Rashmi Barbhaiya, MD & CEO, Advinus Therapeutics. He added that Advinus is actively exploring partnership options to expedite further development and global marketing of GKM-001.
GKM-001 belongs to a novel class of molecules for treatment of type II diabetes. It is an activator of Glucokinase (GK), a glucose-sensing enzyme found mainly in the liver and pancreas. Being liver selective, GKM-001 mostly activates GK in the liver and not in pancreas, which is its key differentiation from most competitor molecules that activate GK in pancreas as well. The resulting increase in insulin secretion creates a potential for hypoglycemia-a risk GKM-001 is designed to avoid. Advinus has the composition of matter patent on GKM-001 for all major markets globally. Both the Single Ascending Dose data, in healthy and type II diabetics, and the Multiple Ascending Dose Study in Type II diabetics has shown that the molecule shows effective glucose lowering in a dose dependent manner and has excellent safety and tolerability profile over a 40-fold dose range. The pharmacokinetic properties of the molecule support once a day dosing. GKM-001 has the potential to be “First-in-Class” drug to address this large, growing and yet poorly addressed market.
Advinus also has identified a clinical candidate as a back-up to GKM-001, which is structurally different. In its portfolio, the company has a growing pipeline for COPD, sickle cell disease, inflammatory bowel disease, type 2 diabetes, acute and chronic pain and rheumatoid arthritis in various stages of late discovery and pre-clinical development.
About the Diabetes Market:
The present 300 million diabetics population is estimated to jump to 450 million by 2030 worldwide. A large proportion of these patients are poorly controlled despite multiple therapies. Total sales of diabetic prescription products were $32 billion in 2010.
Advinus Therapeutics team discovers novel molecule for treatment of diabetes
- The first glucokinase modulator discovered and developed in India
- A new concept for the management of diabetes for patients, globally
- 100 per cent ‘made in India’ molecule for the treatment of diabetes
- IND approved by DGCI, Phase I clinical trial shows excellent safety and tolerance profiles with efficacy
Bangalore: Advinus Therapeutics (Advinus), the research-based pharmaceutical company founded by leading global pharmaceutical executives and promoted by the Tata group, today, announced the discovery of a novel molecule for the treatment of type II diabetes — GKM-001.The molecule is an activator of glucokinase; an enzyme that regulates glucose balance and insulin secretion in the body.
GKM-001 is a completely indigenously developed molecule and the initial clinical trials have shown excellent results for both safety and efficacy.
“Considering past failures of other companies on this target, our discovery programme primarily focused on identifying a molecule that would be efficacious without causing hypoglycaemia; a side effect associated with most compounds developed for this target.
“Recently completed Phase I data indicate that Advinus’ GKM–001 is a liver selective molecule that has overcome the biggest clinical challenge of hypoglycaemia. GKM-001 is differentiated from most other GK molecules in development due to this novel mechanism of action,” said Dr Rashmi Barbhaiya, MD and CEO, Advinus Therapeutics.
He further added, “We are very proud that GKM-001 is 100 per cent Indian. Advinus’s discovery team in Pune discovered the molecule and entire preclinical development was carried out at our centre in Bangalore. The Investigational New Drug (IND) application was filed with the DGCI for approval to initiate clinical trials in India within 34 months of initiation of the discovery programme. Subsequent to the approval of the IND, we have completed the Phase I Single Ascending Dose study in India within two months.”
GKM-001 is a novel molecule for the treatment of type II diabetes. It is the first glucokinase modulator discovered and developed in India and has potential to be both first or best in class. The success in discovering GKM-001 is attributed to the science-driven efforts in Advinus laboratories and ‘breaking the conventional mold’ for selection of a drug candidate. Advinus has ‘composition of matter’ patent on the molecule for all major markets globally. Glucokinase as a class of target is considered to be novel as currently there is no product in the market or in late clinical trials. The strategy for early clinical development revolved around assessing safety (particularly hypoglycaemia) and early assessment of therapeutic activity (glucose lowering and other biomarkers) in type II diabetics. The Phase I data, in both healthy and type II diabetics, shows excellent safety and tolerability over a 40-fold dose range and desirable pharmacokinetic properties consistent with ‘once a day’ dosing. The next wave of clinical studies planned continues on this strategy of early testing in type II diabetics.
Right behind the lead candidate GKM-001, Advinus has a rich pipeline of back up compounds on the same target. These include several structurally different compounds with diverse potency, unique pharmacology and tissue selectivity. Having discovered the molecule with early indication of wide safety margins, desired efficacy and pharmacokinetic profiles, the company now seeks to out-licence GKM-001 and its discovery portfolio.
Patent
wo 2008104994
| WO2008104994A2 * | 25 Feb 2008 | 4 Sep 2008 | Advinus Therapeutics Private L | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2008104994A2 * | Feb 25, 2008 | Sep 4, 2008 | Advinus Therapeutics Private L | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2009047798A2 * | Oct 7, 2008 | Apr 16, 2009 | Advinus Therapeutics Private L | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
///////GKM 001, pipeline, Diabetes, Advinus, type II diabetes, glucokinase modulator, Rashmi Barbhaiya
Some pics

Annual day party at Advinus !!!with Rashmi Barbhaiya
Dr. Rashmi Barbhaiya, MD & CEO, Advinus Therapeutics Pvt.
 .
.
with Kaushal Joshi, Vishal Pathade, Ramanareddy Jinugu, Mohammed Kakajiwala, Vishal Baxi and Dilip Reddy.
///////
MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan

MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan
KENILWORTH, N.J.–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) has approved MARIZEV® (omarigliptin) 25 mg and 12.5 mg tablets, an oral, once-weekly DPP-4 inhibitor indicated for the treatment of adults with type 2 diabetes. Japan is the first country to have approved omarigliptin……….http://www.mercknewsroom.com/news-release/prescription-medicine-news/marizev-omarigliptin-mercks-once-weekly-dpp-4-inhibitor-type
syn…….https://newdrugapprovals.org/2014/04/18/omarigliptin-mk-3102-in-phase-3-for-type-2-diabetes/

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HEREJoin me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
/////////////MARIZEV, (Omarigliptin), Merck’s, Once-Weekly, DPP-4 Inhibitor, Type 2 Diabetes, Approved, Japan
BEXAGLIFLOZIN


Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
FDA APPROVED
| 1/20/2023 |
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise
Drug Trials Snapshot
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)
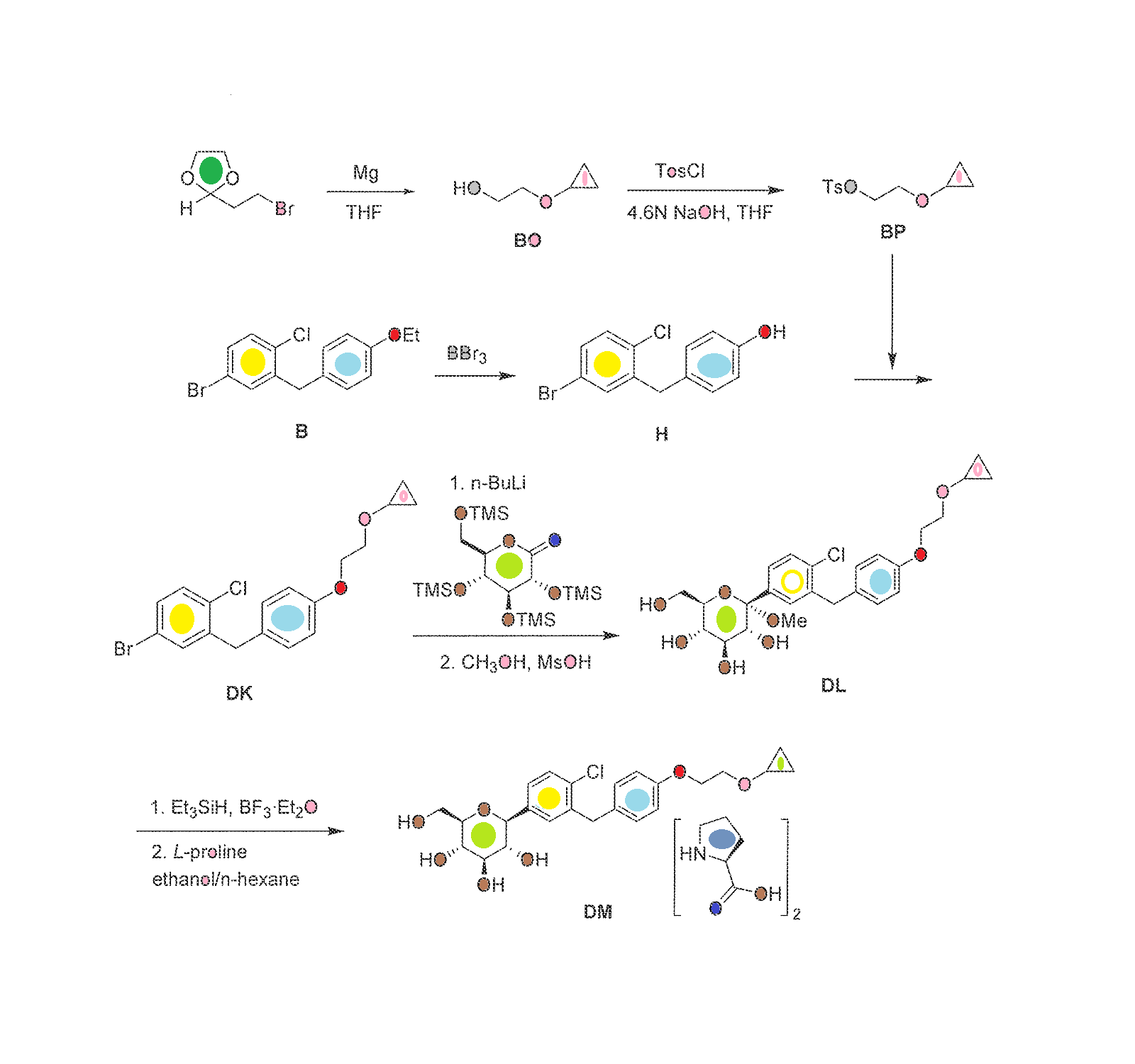

Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.

Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694

Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.

Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.

(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Example 1A
Preparation of 2-cyclopropoxyethanol (1)

To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
////////BEXAGLIFLOZIN, APPROVALS 2023, FDA 2023
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079J.Med.Chem.2025,68,2147−2182
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof
type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting
SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe
reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates,
whichmaybeacommonchallengetootherSGLT2inhibitors.31
Anotherpatent disclosedbyPiramal Enterprises suggesteda
similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34
Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2
whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith
activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal
functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane,
whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was
reported in 92% yield. Details on stereoselectivity of this
approachwerenotdisclosed.
Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol
3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether
3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.

(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447−
453.
(29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium
glucose co-transporter-2 inhibitors for the treatment of diabetes
mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79.
(30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive
review of small-molecule drugs for the treatment of type 2 diabetes
mellitus: Synthetic approaches and clinical applications. Eur. J. Med.
Chem. 2024, 267, No. 116185.
(31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F.
Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A.
Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev.
2018, 22, 467−488.
(32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.;
Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′
position as potent and selective renal sodium-dependent glucose co
transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes.
Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470.
(33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the
preparation of benzyl-benzene C-glycosides via coupling reaction as
potential SGLT2 inhibitors. US 20130267694, 2013.
(34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal,
D. Aprocess for the preparation of SGLT2 inhibitor and intermediates
thereof. WO 2018207113, 2018.
(35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu,
B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside
derivatives as antidiabetic agents. WO 2009026537, 2009.
.
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
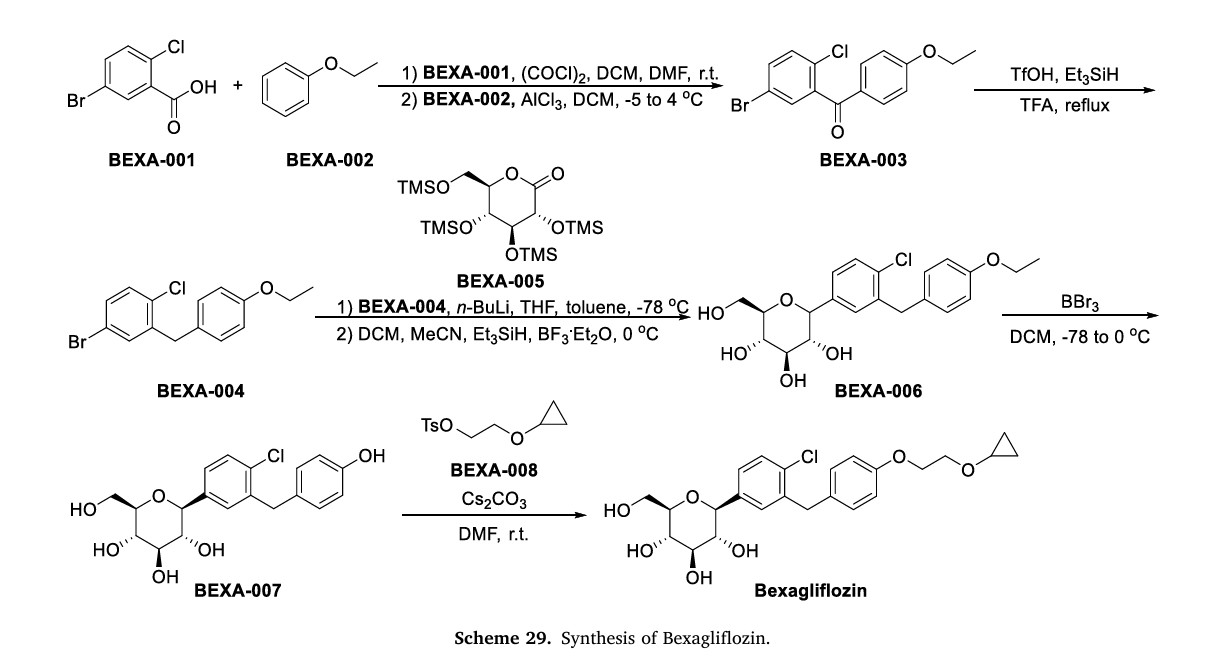
Bexagliflozin (Brenzavvy)
On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin
can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which
lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108].
The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring.
BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol.
Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453.
[105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng,
B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and
prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293.
[106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M.
P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin.
Pharmacother. 22 (2021) 2095–2103.
[107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am.
Soc. Nephrol. 18 (2023) 279–289.
[108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr.
Drug Saf. 13 (2018) 84–91.
[109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd,
J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives
as Antidiabetic Agents, 2009. WO2009026537A1.
.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
