
Home » CHINA 2022
Category Archives: CHINA 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
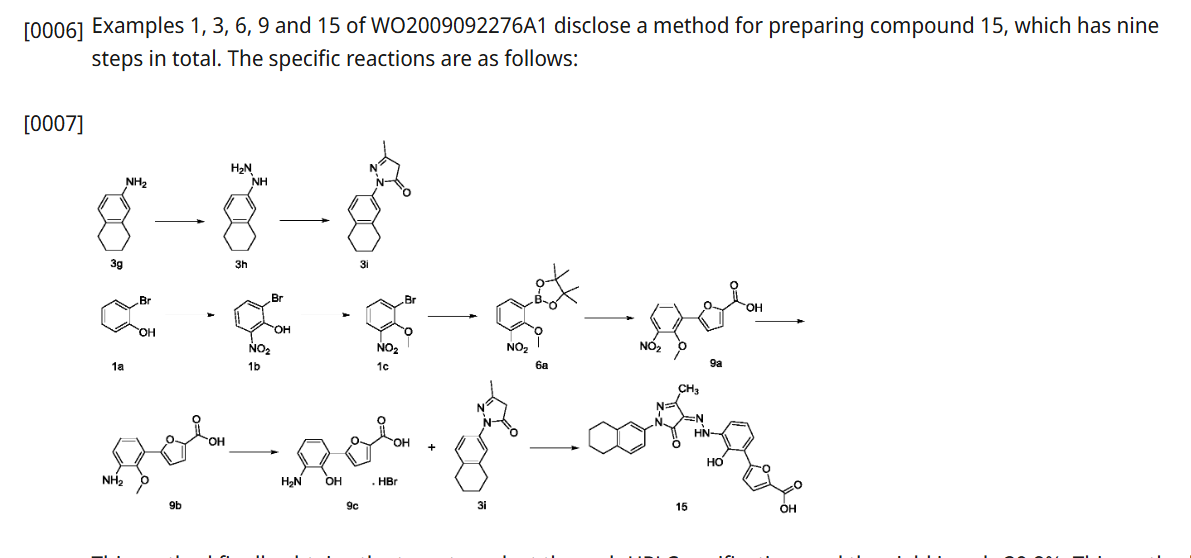
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pamiparib
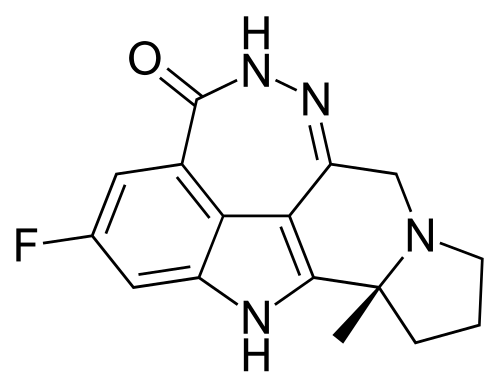
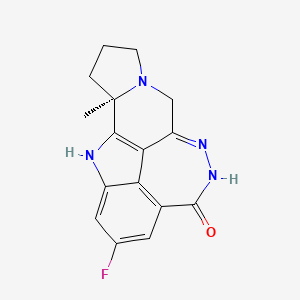
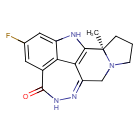
Pamiparib
BGB-290 APPROVED CHINA 2022, BEIGENE
(2R)-14-fluoro-2-methyl-6,9,10,19-tetrazapentacyclo[14.2.1.02,6.08,18.012,17]nonadeca-1(18),8,12(17),13,15-pentaen-11-one
- 1446261-44-4
- 8375F9S90C
- 5,6,7a,11-Tetraazacyclohepta(def)cyclopenta(a)fluoren-4(7H)-one, 2-fluoro-5,8,9,10,10a,11-hexahydro-10a-methyl-, (10aR)-
- 5,6,7a,11-Tetraazacyclohepta[def]cyclopenta[a]fluoren-4(7H)-one, 2-fluoro-5,8,9,10,10a,11-hexahydro-10a-methyl-, (10aR)-
- 298.31 g/mol, C16H15FN4O
Pamiparib, sold under the brand name Partruvix, is a pharmaceutical drug used for the treatment of various types of cancer. Pamiparib is a member of the PARP inhibitor drug class.[1]
In China, it is approved for the treatment of germline BRCA mutation-associated recurrent advanced ovarian, fallopian tube, and primary peritoneal cancers previously treated with two or more lines of chemotherapy.[2]
It is currently under investigation for the treatment of other forms of cancer.[3][1]
Pamiparib is under investigation in clinical trial NCT03933761 (Pamiparib in Fusion Positive, Reversion Negative High Grade Serous Ovarian Cancer or Carcinosarcoma With BRCA1/2 Gene Mutations If Progression on Substrate Poly ADP Ribose Polymerase Inhibitbor (PARPI) or Chemotherapy).
Pamiparib is an orally bioavailable inhibitor of the nuclear enzyme poly(ADP-ribose) polymerase (PARP), with potential antineoplastic activity. Upon administration, pamiparib selectively binds to PARP and prevents PARP-mediated repair of single-strand DNA breaks via the base-excision repair (BER) pathway. This enhances the accumulation of DNA strand breaks, promotes genomic instability, and eventually leads to apoptosis. PARP is activated by single-strand DNA breaks and, subsequently, catalyzes post-translational ADP-ribosylation of nuclear proteins which then transduce signals to recruit other proteins to repair damaged DNA. Pamiparib may both potentiate the cytotoxicity of DNA-damaging agents and reverse tumor cell chemo- and radioresistance.
REF
https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-0040-1719372
REF
J. Med. Chem. 2020, 63, 15541−15563.
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.0c01346
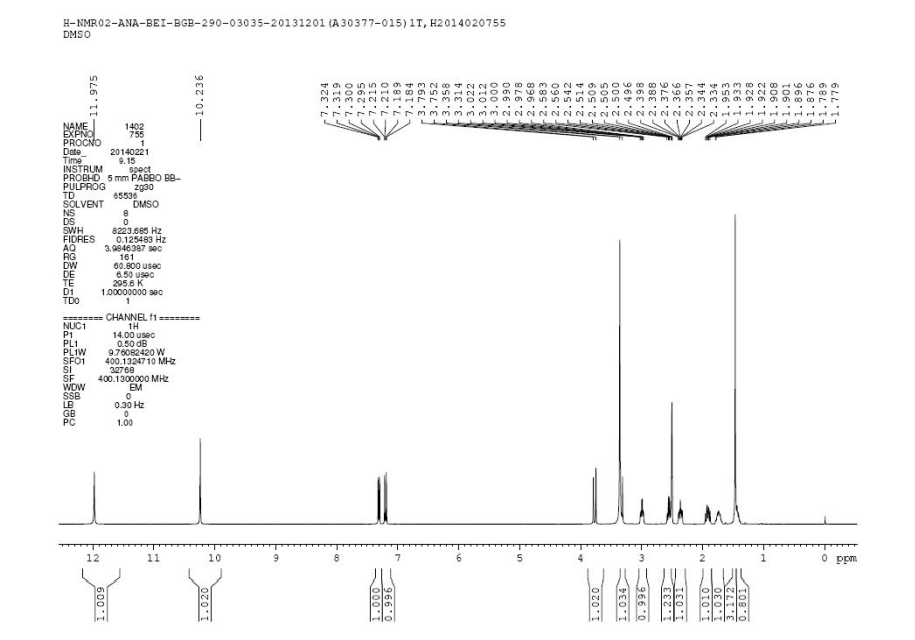
1HNMR 400, DMSO D6
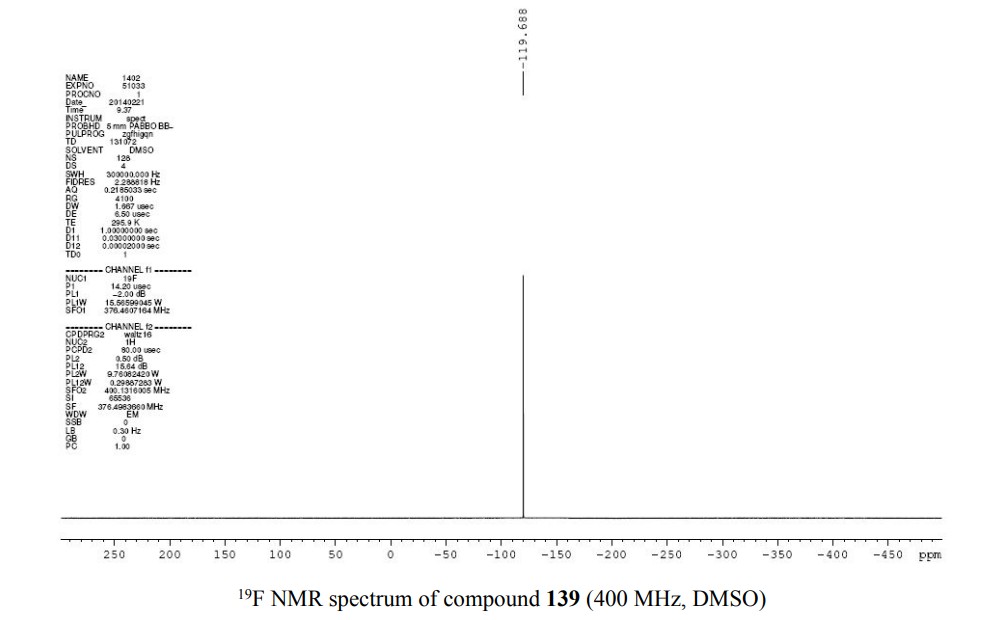
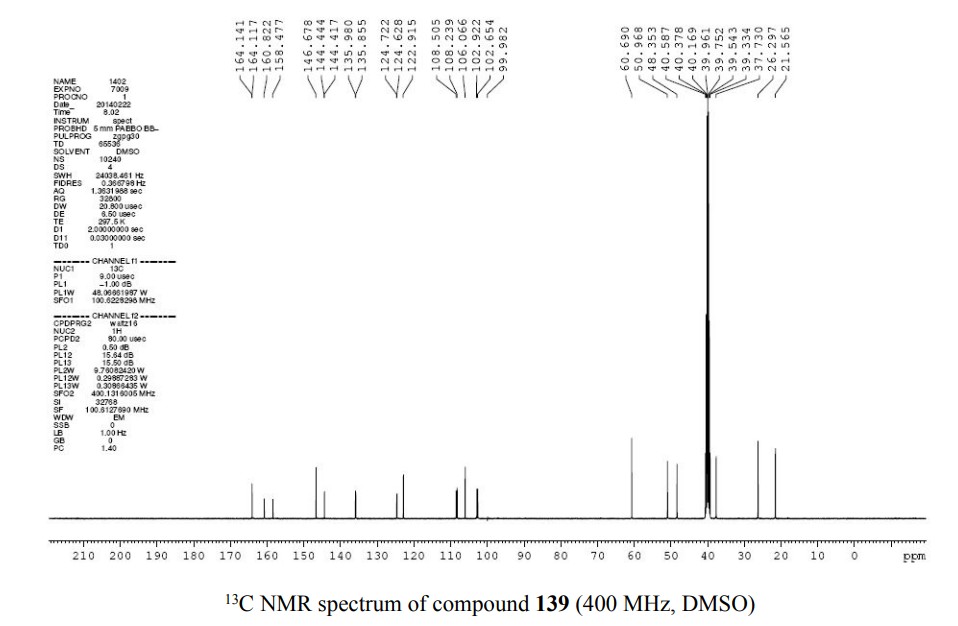
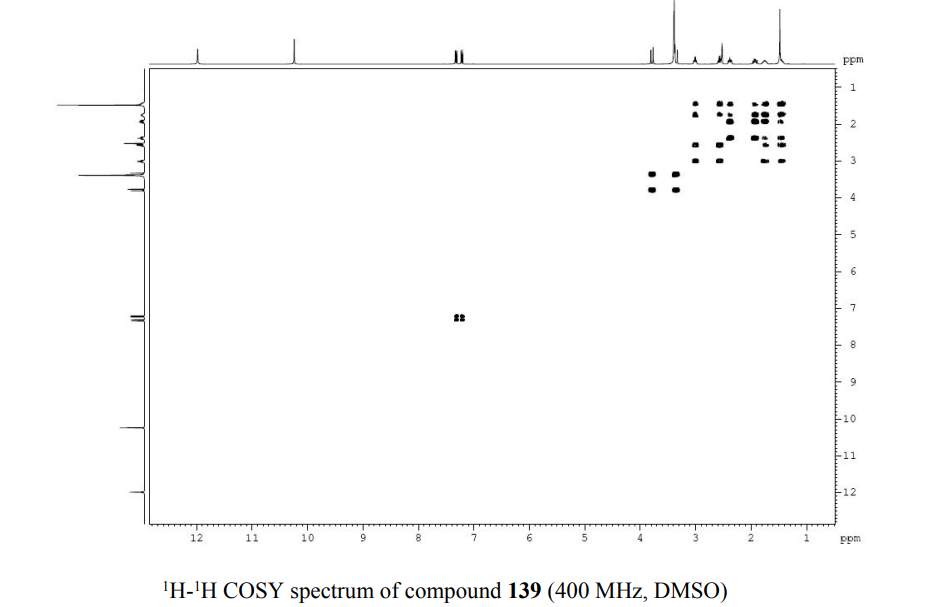
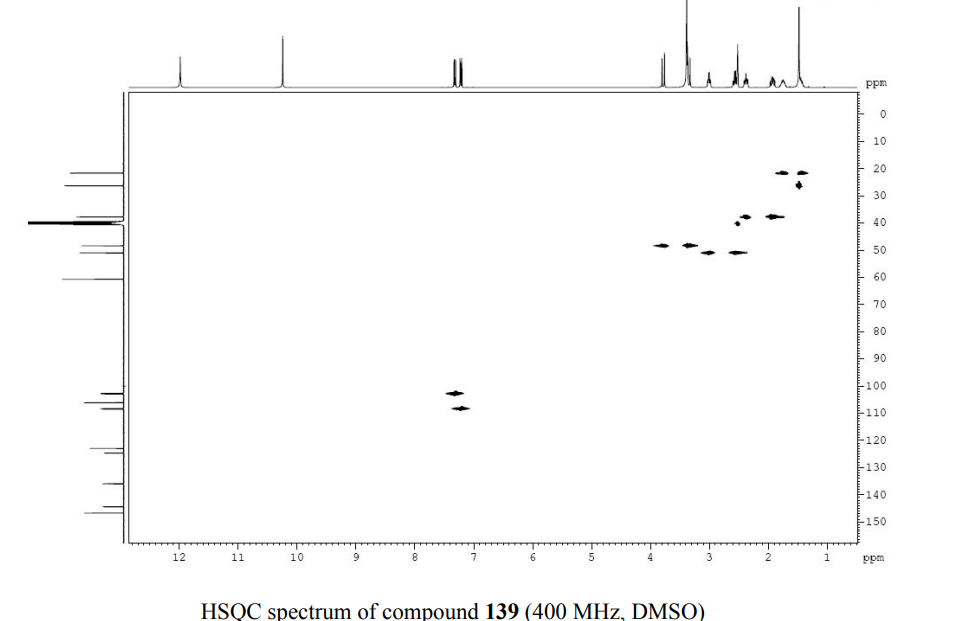
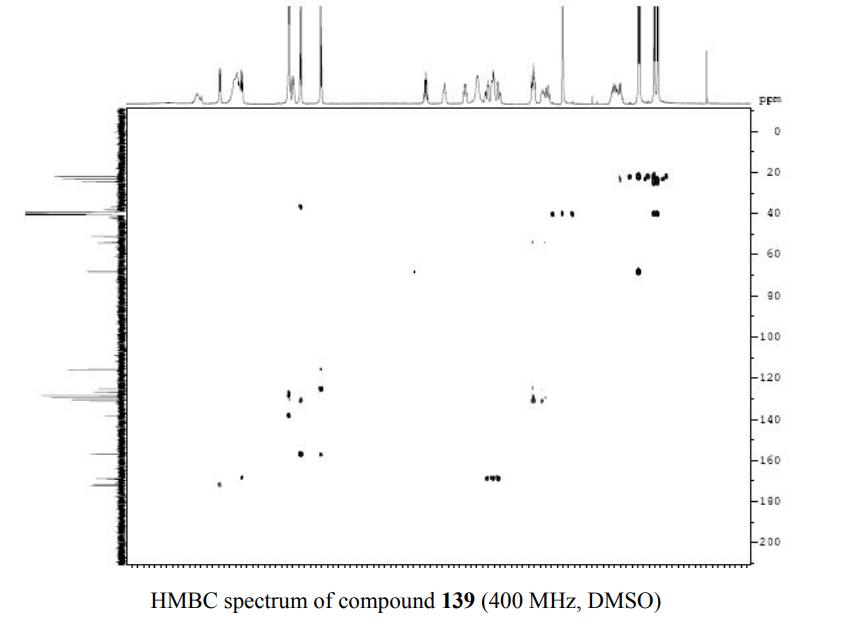
PATENT
WO 2018157794
https://patents.google.com/patent/WO2018157794A1/en
(R) -2-fluoro-10a-methyl-7, 8, 9, 10, 10a, 11-hexahydro-5, 6, 7a, 11-tetraazacyclohepta [def] cyclopenta – [a] fluoren-4 (5H) -one (hereafter Compound 1) , has been disclosed as a highly selective and potent Parp1/2 inhibitor, See WO 2013/097225 A1, which is incorporated herein by reference.

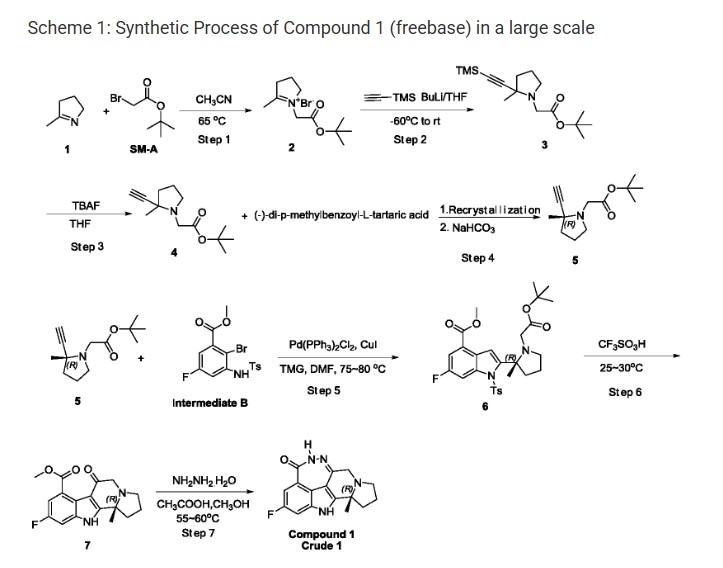
Step 1: Synthesis of Compound-2

t-Butyl bromoacetate (51.7 Kg) was dissolved in anhydrous acetonitrile (72 Kg) . The temperature was raised to 65-75 ℃, then methyl pyrroline (22 Kg) was added. The reaction mixture was condensed after the reaction was completed, the residual acetonitrile was removed by adding THF and then condensing. After GC showed a complete removal of acetonitrile, more THF was added and stirred. The resulting solid was filtered and collected. 44.1 Kg of off white solid Compound-2 was obtained. 1H NMR (400 MHz, DMSO-d6) δ 4.91 (s, 2H) , 4.15 (m, 2H) , 3.29 (m, 2H) , 2.46 (s, 3H) , ) , 2.14 (m, 2H) , 1.46 (s, 9H) ppm.
Step 2: Synthesis of Compound-3

To a cool (-60 ℃) solution of trimethylsilyl acetyne (12.4 Kg) in THF was added a solution of n-butyl lithium in hexane (43.4 Kg) . After complete addition of n-butyl lithium solution, the resulting mixture was stirred for additional 1-2 h and then the entire solution was transferred into a suspension of Compound-2 (31 Kg) in THF cooled at -60 ℃. After transfer completion, the resulting mixture was warmed to room temperature and stirred for 1 h. The reaction was quenched with water, extracted with petroleum. The organic phase was washed with brine, dried over sodium sulfate, condensed to give 25.1 Kg of Compound-3. 1H NMR (400 MHz, DMSO-d6) δ 3.34 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.78 (d, J = 16.0 Hz, 1H) , 2.27 (m, 1H) , 1.93 (m, 1H) , 1.68 (m, 3H) , 1.41 (s, 9H) , 1.24 (s, 3H) , 0.13 (s, 9 H) ppm.
Step 3: Synthesis of Compound-4

To a cool (0-5 ℃) solution of 70.1 Kg of Compound-3 in THF was added tetrabutylammonium fluoride (13.3 Kg) in THF. After de-silylation was completed, the reaction was quenched with water, extracted with petroleum (290 Kg) and the organic phase was condensed and passed through a pad of silica gel. The filtrate was condensed to give 48 Kg of Compound-4. 1H NMR (400 MHz, DMSO-d6) δ 3.36 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.82 (d, J = 16.0 Hz, 1H) , 2.28 (m, 1H) , 1.97 (m, 1H) , 1.70 (m, 3H) , 1.41 (s, 9H) , 1.26 (s, 3H) ppm.
Step 4: Syntheses of Compound-5

A solution of Compound-4 (48 Kg) in THF was warmed to 50-60 ℃. To the above solution was added a solution of (-) -di-p-methylbenzoyl-L-tartaric acid (69.6 Kg) in THF. The resulting mixture was stirred at 50-60 ℃ 1-2 h and then gradually cooled to 0-10 ℃. The resulting salt solid was filtered and re-suspended in methyl tert-butyl ether and heated at 50-60 ℃ for 1 h. The mixture was gradually cooled to 0-5 ℃. The resulting solid was filtered to give 13.1 Kg of off-white solid. The solid was treated with aqueous sodium hydroxide, extracted with petroleum, condensed to give 13.1 Kg of Compound-5 (ee≥96%) . 1H NMR (400 MHz, DMSO-d6) δ 3.36 (d, J = 16.0 Hz, 1H) , 3.15 (m, 1H) , 2.82 (d, J = 16.0 Hz, 1H) , 2.29 (m, 1H) , 1.97 (m, 1H) , 1.70 (m, 3H) , 1.41 (s, 9H) , 1.26 (s, 3H) ppm.
Step 5: Syntheses of Compound-6

Intermediate B (14 Kg) , bis (triphenyl) palladium dichloride (0.7 Kg) , CuI (0.42 Kg) and tetramethyl guanidine (11.5 Kg) were dissolved in DMF (48.1 Kg) . The resulting solution was stirred and de-gassed and then heated under nitrogen. A solution of Compound-5 (9.24 Kg) in DMF (16 Kg) was added dropwise. After coupling, the organic phase was condensed, the resiue was stirred with water (145 Kg) and methyl t-butyl ether (104 Kg) , the entire mixture passed trough a pad of celite, separated. The organic phase was washed with a solution of thiourea (14 Kg) in water (165 kg) and brine (100 Kg) , condensed. The residue was dissolved in a mixture of n-heptane (120 Kg) and ethyl acetate (28 Kg) . The solution was mixed with charcoal (1.4 kg) , heated at 40-50 ℃ for 1-2 h, fltered though a pad of silica gel. The filtrate was condensed to give Compound-6 solid (14.89 Kg) and the liquid filtrate (13 Kg heptane solution, contains 1.24 Kg of Compound-6) . 1H NMR (400 MHz, DMSO-d6) δ 7.85 (d, J = 9.6 Hz, 1H) , 7.55 (m, 3H) , 7.32 (m, 2H) , 3.87 (s, 3H) , 3.37 (d, J = 16.0 Hz, 1H) , 3.22 (m , 1H) , 2.94 (d, J = 16.0, Hz, 1H) , 2.60 (m, 1H) , 2.48 (m, 1H) , 2.29 (s, 3h) , 2.26 (m, 1 H) , 1.82 (m, 2H) , 1.49 (s, 3H) , 1.43 (s, 9H) ppm.
Step 6: Syntheses of Compound-7

The above heptane solution of Compound-6 was added into a cold trifluoromethane sulfonic acid (66.1 Kg) while maintaining the internal temperature below 25 ℃. Then solid Compound-6 (14.87 Kg) was added batchwise. After complete addition of Compound-6, the reaction mixture was warmed to 25-30℃ and stiired until the reaction was completed. The entire mixture was poured into a solution of sodium acetate (123.5 Kg) in water (240 Kg) . pH of the solution was then adjusted to 7-8 by adding solid potassium carbonate (46.1 Kg) . The mixture was extracted wuth dichloromethane (509 Kg) , condensed. The residue was mixed with n-heptane (41 Kg) , condensed again to give the precipitate which was filtered and washed by n-heptane (8 Kg) and dried. 8.78 Kg of Compound-7 was obtained. 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H) , 7.35 (dd, J = 9.2, 1.6 Hz, 1H) , 7.08 (dd, J = 9.2, 1.6 Hz, 1H) , 3.79 (s, 3H) , 3.68 (d, J = 17.2 Hz, 1H) , 3.21 (d, J = 17.2 Hz, 1H) , 3.06 (m, 1H) , 2.68 (m, 1H) , 1.96 (m, 1H) , 1.74 (m, 1H) , 1.49 (s, 3H) ppm.
Step 7: Syntheses of Compound 1 –Crude 1

Compound-7 (8.76 Kg) was dissolved in methanol (69 Kg) and internally cooled below 25 ℃. Acetic acid (9.3 Kg) and hydrazine hydrate (7.4 Kg, 85%) were added while maintaining internal temperature below 25 ℃. After de-gassed and re-filled with nitrogen (repeated three times) , the reaction mixture was stirred at 55-60 ℃ for 4 h. After a complete reaction, the mixture was mixed with water (29 Kg) . The organic phase was condensed and potassium carbonate (12.5 Kg) in water (40 Kg) was added. The resulting solid was filtered, washed with water (18.3 Kg) . The solid was slurred with water (110 Kg) , centrifuged, dried and slurred with ethanol (9.4 Kg) , centrifuged, filtered, washed with ethanol, dried in vacuum to give Compound 1-Crude 1 (7.91 Kg) . 1H-NMR (600 MHz, DMSO-d 6) δ 12.0 (s, 1H) , 10.2 (s, 1H) , 7.31 (dd, 1H, J=9.6, 2.0 Hz) , 7.19 (dd, 1H, J=9.6, 2.0 Hz) , 3.77 (d, 1H, J=16.4 Hz) , 3.34 (d, 1H, J=16.4 Hz) , 2.97-3.02 (m, 1H) , 2.54-2.58 (m, 1H) , 2.35-2.40 (m, 1H) , 1.90-1.94 (m, 1H) , 1.73-1.75 (m, 1H) , 1.47 (s, 3H) , 1.43-1.45 (m, 1H) ppm. MS (ESI) m/e [M+1] + 299.
Step 8: Synthesis of Compound 1-Crude 2

Under nitrogen protection, Compound 1 (Crude 1) (7.88 Kg) was stirred with isopropanol (422 Kg) and heated at 70-80 ℃ for 1-2 h until the solid disappeared completely. A solution of (+) -di-p-methylbenzoyl-D-tartaric acid (10.25 Kg) in isopropanol (84.4 Kg) was added. The mixture was stirred for 14-16 h, filtered and washed with isopropanol (16 Kg) , dried. The resulting salt was added into a stirred solution of potassium carbonate (6.15 Kg) in water (118 Kg) . The precipitate was centrifuged, filtered, washed with water (18 Kg) . The solid was slurred with water (110 Kg) , centrifuged, dried. The solid was dissolved in THF (75 Kg) , active carbon (0.8 Kg) was added. The mixture was degassed and re-protected by nitrogen, stirred and heated at 40-45 ℃ for 1-2 h, cooled, filtered through celite, condensed to give the solid which was further slurred with ethanol (6.5 Kg) , filtered to give 5.6 Kg of Compound
1 crude
2. 1H NMR (400 MHz, DMSO-d6) δ 12.0 (s, 1H) , 10.2 (s, 1H) , 7.31 (dd, 1H, J=9.6, 2.0 Hz) , 7.19 (dd, 1H, J=9.6, 2.0 Hz) , 3.77 (d, 1H, J=16.4 Hz) , 3.34 (d, 1H, J=16.4 Hz) , 2.97-3.02 (m, 1H) , 2.54-2.58 (m, 1H) , 2.35-2.40 (m, 1H) , 1.90-1.94 (m, 1H) , 1.73-1.75 (m, 1H) , 1.47 (s, 3H) , 1.43-1.45 (m, 1H) ppm. MS (ESI) m/e [M+1] + 299.
PATENT
WO 2017032289
https://patents.google.com/patent/WO2017032289A1/en
Scheme 1: Synthetic Process of Compound A in a large scale

PATENT
WO 2013097225
https://patents.google.com/patent/WO2013097225A1/en
Example 36: Synthesis of Compound 69 Compound 69: (RV2-fluoro-10a-methyl-7,8,9 JO .10a.l l-hexahydro-5,6,7a,l 1- tetraazacvcloheptardeflcyclopentara1fluoren-4(5H)-one

Step 1 : Methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate

To a solution of methyl 3-amino-2-bromo-5-fluorobenzoate (25. Og, 100 mmol) and K2CO3 (42.0g, 302 mmol) in DCM (250mL) were added 2,2,2-trifluoroacetic anhydride (249.0g, 1.197mol) at 5 -10°C under nitrogen atmosphere. The mixture was stirred for overnight at 25°C. The reaction mixture was diluted with DCM, washed with H20 (200mLx2) and saturared
NaHCC”3 aq (200mLx2), dried over anhydrousNa2S04, and concentrated to give 34.0 g (98%) of methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate as white solid. 1H NMR (CDCI3– dl) δ 8.87 (s, 1H), 8.36 (d, 1H,J=6.4 Hz), 7.43 (d, 1H,J=5.2 Hz), 3.98 (s, 3H).
Step 2: (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6- (2,2,2trifluoroacetamido)phenyl)ethvnyl)-2-methylpyrrolidine-l-carboxylate

A mixture of methyl 2-bromo-5-fluoro-3-(2,2,2-trifluoroacetamido)benzoate (27.52g, 80 mmol), (PPh3)2PdCl2 (2.8 g, 4 mmol), (R)-benzyl 2-ethynyl-2-methylpyrrolidine-l-carboxylate (19.44 g, 80 mmol),copper(I) iodide (764 mg, 4 mmol) and tetramethylguanidine (27.6 g, 240 mmol) in DMF (200 mL) was heated at 80 °C with nitrogen protection system for 16 hours. The cooled reaction mixture was diluted with EA (3×200 mL) and water (800 mL). The organic layer was separated, washed with water (2×200 mL), dried (Na2S04), and concentrated. The remaining residue was chromatographed on silica gel, eluted with gradient 0-30% EtOAc in hexane to give the product (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6-
(2,2,2trifluoroacetamido)phenyl)ethynyl)-2-methylpyrrolidine-l-carboxylate (21 g, 53%) as white solid. 1H NMR (DMSO-dl) δ 11.01 (s, 1H), 7.64-7.77 (m, 1H), 7.36 (m, 5H),7.19-7.31 (m, 1H), 5.04-5.12 (m, 2H), 3.85(s, 3H ), 3.44-3.47 (m, 2H), 2.0-2.29 (m, 2H), 1.90-1.97 (m, 2H), and 1.69 (s, 3H).MS (ESI) m/e [M+l]+ 507.0.
Step 3: (R)-methyl 6-fluoro-2-(2 -methyl- l-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl)-lH-indole-4- carboxylate

To a solution of (R)-benzyl 2-((4-fluoro-2-(methoxycarbonyl)-6- (2,2,2trifiuoroacetamido)phenyl)ethynyl)-2-methylpyrrolidine- 1 -carboxylate(5.0g, 1 Ommol) in toluene was added zinc(II) bromide(l 1.25g, 50 mmol) at room temperture. The reaction mixture was heated at 80 °C with nitrogen protection system for 15 hours. The solvent was removed under reduced pressure, and the residue was treated with DCM (500 mL) and water (800 mL). The organic layer was separated, washed with water (2×200 mL), dried (Na2S04), and
concentrated. The remaining residue was chromatographed on silica gel ,eluted with gradient 0- 50% EtOAc in hexane to give the product(R)-methyl 6-fluoro-2-(2 -methyl- 1 -(2,2,2- trifluoroacetyl)pyrrolidin-2-yl)-lH-indole-4-carboxylate (1.9 g, 51%) as yellow solid. 1H NMR (CDCls-dl) δ 9.97 (s, 1H), 7.62 (d,lH, J=10.2 Hz), 7.27 (d,lH, J=9.6 Hz), 7.05 (d,lH, J=1.2 Hz), 3.98 (s, 3H), 3.86-3.88 (m,2H),2.91-2.96 (m,lH), 2.25-2.28 (m,lH), 2.12-2.16 (m, 2H), and 1.99 (s, 3H). MS (ESI) m/e [M+l]+ 507.0.
Step 4: (R)-methyl 6-fluoro-2-(2-methylpyrrolidin-2-yl)-lH-indole-4-carboxylate

To a solution of (R)-methyl 6-fluoro-2-(2 -methyl- l-(2,2,2-trifluoroacetyl)pyrrolidin-2-yl)- lH-indole-4-carboxylate (1.0 g, 1.9 mmol) in MeOH was added NaBH4 (706 mg, 11.4 mmol) at room temperature. The reaction mixture was refluxed for 4 hours with nitrogen protection system. The solvent was removed under reduced pressure. The residue was dissolved in DCM (200 mL), which was washed with water (200 mL)and brine (200 mL), dried over Na2S04, and concentrated to give the desire product as yellow oil. (R)-methyl 6-fluoro-2-(2-methylpyrrolidin- 2-yl)-lH-indole-4-carboxylate (727 mg, 98%). 1H NMR (CD3OD-dl) δ 7.50(dd,lH, J=10.2, 2.4 Hz), 7.32 (d,lH, J=9.0, 2.4 Hz), 6.93 (s, 1H),3.97 (s, 3H), 3.03-3.12 (m, 2H), 2.27-2.32 (m, 1H),1.88-1.98 (m, 3H), and 1.60 (s, 3H). MS (ESI) m/e [M+l]+ 276.0.
Step 5: (R)-Methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate

To a stirred mixture of (R)-methyl 6-fluoro-2-(2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate (1.0, 1.27 mol), CH3CN (50 ml) and methylbromoacetate (0.58 g, 3.82mmol) was added DIPEA(0.82 g, 6.35 mmol). The reaction mixture was stirred at room temperature for about 20 hours. The reaction mixture was then diluted with CH2CI2 (15 ml) and washed with water three times. The organic layer was dried with MgS04 and concentrated to give 0.85 g of (R)-methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2-yl)-lH-indole-4- carboxylate. 1H NMR (CD3OD-d4) δ 7.47 (dd, 1H, J=2.4, 12.0 Hz), 7.27 (dd, 1H, J=2.4, 9.0 Hz), 6.89 (s,lH), 3.95 (s, 3H), 3.66-3.68 (m, 1H), 3.64 (s, 3H), 3.16-3.17 (m, 2H), 2.72-2.75 (m, 1H), 1.88-2.02 (m, 4H), and 1.44 (s, 3H).MS (ESI) m/e [M+l]+ 349.0.
Step 6: (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3, 5,6, 11,1 lb-hexahydro-lH-indolizinor8,7- blindole-7-carboxylate

In a 25 -mL flask, (R)-methyl 6-fluoro-2-(l-(2-methoxy-2-oxoethyl)-2-methylpyrrolidin-2- yl)-lH-indole-4-carboxylate (100 mg) was treated with anhydrous MeS03H (6 mL). The flask was fitted with a reflux condenser and heated at 60 °C for 1 h. Then, the reaction mixture was cooled in an ice-bath and diluted with distilled water (6.0 mL). The pH of the solution was increased to pH~10 by the addition of saturated aq. NaHC03. The reaction mixture was then extracted with EtOAc (3×5 mL). Theorganic extracts were combined and washed with brine (lx5mL), dried over Na2S04, filtered, and concentrated. The residue was purified by Pre-TLC to give (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3, 5,6,11,1 lb-hexahydro-lH-indolizino[8,7- b]indole-7-carboxylate(30 mg). 1H NMR (CDCl3-d) δ 7.14-7.224 (m, 2H), 4.03 (s, 3H), 3.81- 3.84 (m, 1H), 3.57-3.59 (m, 1H), 3.22-3.24 (m, 1H), 2.92-2.94 (m, 1H), 2.39-2.40 (m,lH), 2.16- 2.17 (m,lH),1.93-1.94 (m, 1H), 1.63 (s, 3H), and 1.56-1.57 (m, 1H).MS (ESI) m e [M+l]+ 317.0. Step 7: (RV2-fluoro-10a-methyl-7,8,9 JO JOa.l l-hexahvdro-5.6.7a.l 1- tetraazacyclohepta[def|cyclopenta[alfluoren-4(5H)-one

A solution of compound (R)-methyl 9-fluoro-l lb-methyl-6-oxo-2,3,5,6,l 1,1 lb-hexahydro- lH-indolizino[8,7-b]indole-7-carboxylate (90 mg), acetic acid (0.54 g), and hydrazine hydrate (0.28g) in methanol (30 mL) was heated at reflux. After 5 h, the reaction was cooled and water (5 mL) was added.The mixture was extracted with EtOAc (3×5 mL). The combined organic layers were washed with brine (10 mL) and driedover MgSC^. The mixture was filtered, and the filtrate was evaporated to dryness, and the residue was purified by Pre-TLC using CH2CI2 as eluent to give 80 mg of (R)-2-fluoro-10a-methyl-7,8,9,10,10a,l l-hexahydro-5,6,7a,l l- tetraazacyclohepta[defJcyclopenta[a]fluoren-4(5H)-one. 1H NMR (DMSO-d6) δ 11.9 (s, 1H), 10.2 (s, 1H), 7.30 (d, 1H, J=9.6 Hz), 7.20 (d, 1H, J=10.2 Hz), 3.76 (d, 1H, J=16.4 Hz), 3.34 (d, 1H, J=16.4 Hz), 2.99-3.02 (m, 1H), 2.54-2.58 (m, 1H), 2.35-2.40 (m, 1H), 1.90-1.94 (m, 1H), 1.73-1.75 (m, 1H), 1.48 (s, 3H), and 1.43-1.45(m, 1H). MS (ESI) m/e [M+l]+ 299.
SYN
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
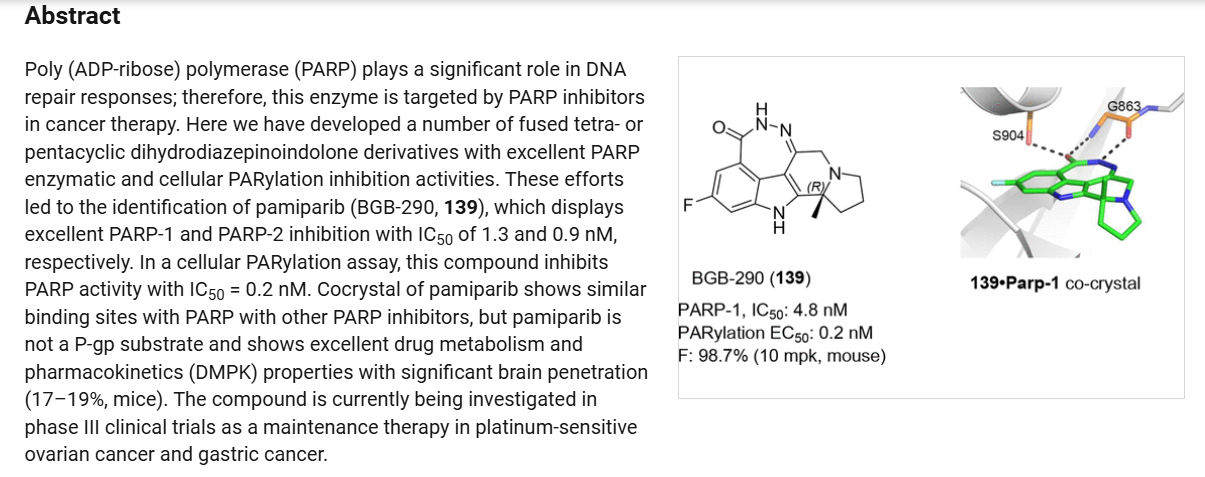
Pamiparib (Partruvix). Pamiparib (27) is an orally active, potent, and highly selective PARP1 and PARP2
inhibitor being developed by BeiGene Limited.190 The drug was approved in China in 2022 for the treatment of germline BRCA-mutated recurrent advanced ovarian, fallopian tube, or primary peritoneal cancer.190BRCA1 and BRCA2 are critical tumor suppressors that help DNA double-strand break (DSB)
repair by functional homologous recombination (HR). 191,192 192 It was claimed that pamiparib showed good brain penetration ability for the treatment of cancer patients with brain metastasis. A small-scale synthesis of pamiparib (27) was first disclosed by BeiGene Limited in 2013.193 Later, they reported a
modified route for industrial scale preparation of the API which is described below. 194,195
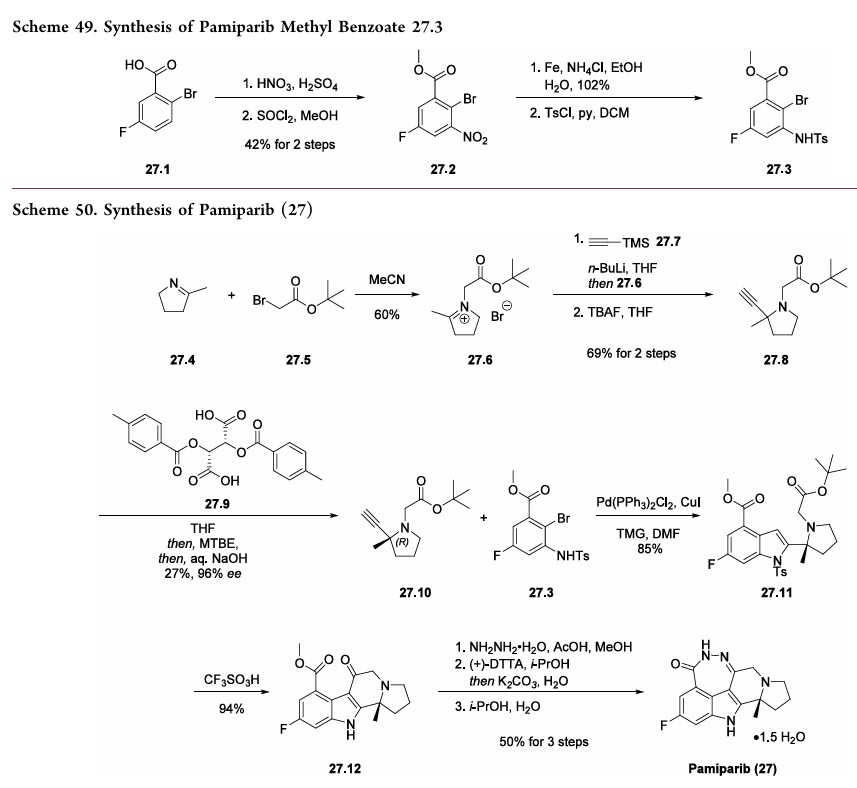
The synthesis commenced with 2-bromo-5-fluorobenzoic acid (27.1) which was subjected to nitration followed by esterification to deliver methyl benzoate 27.2 in 42% overall yield (Scheme 49). The nitro
derivative 27.2 was reduced to an aniline and subsequently protected as a tosylate to obtain the key aryl bromide fragment 27.3. It should be noted that a yield for the tosylation step was not provided by the inventors.
Preparation of the other key fragment 27.10 and endgame of the pamiparib synthesis are described in Scheme 50. First,pyrroline 27.4 was treated with t-butyl bromoacetate 27.5 to generate iminium bromide salt 27.6. An acetylide derived from trimethylsilyl acetylene 27.7 was then added to the iminium to
install the tertiary center. A TBAF-mediated removal of the silyl moiety delivered racemic alkyne 27.8 in 69% yield over two steps. The enantiomers were separated via a classical salt resolution with (−)-di-p-methylbenzolyl-L-tartaric acid (27.9). The desired (R)-enantiomer 27.10 was obtained in 96%
enantiomeric excess (ee) after isolation as the free-base amine. The authors explored several routes to access 27.10; however,the salt resolution approach was selected due to its scalability and reproducibility.
192With the alkyne subunit 27.10 and bromide subunit 27.3 in hand, the next objective was combining them in a convergent manner. This was achieved via an efficient Larock heteroannulation reaction, affording indole 27.11 in 85% yield. Treatment of diester 27.11 with triflic acid triggered removal of both t-butyl ester and N-tosyl protecting groups, as well as cyclization to generate tetracycle 27.12 in 94% yield. The ketoester 27.12 was subjected to hydrazine hydrate in the presence of acetic acid to deliver the
crude cyclized material which was purified via salt formation with (+)-DTTA. Finally, treatment of the amine precursor with water in hot isopropanol delivered pamiparib (27) as a sesquihydrate crystalline solid in 50% over 3 steps.
(190) Markham, A. Pamiparib: First approval. Drugs 2021, 81,1343−1348.
(191) Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.;Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent andselective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431−440.
(192) Wang, H.; Ren, B.; Liu, Y.; Jiang, B.; Guo, Y.; Wei, M.; Luo,L.; Kuang, X.; Qiu, M.; Lv, L.; et al. Discovery of pamiparib (BGB290), a potent and selective poly (ADP-ribose) polymerase (PARP)inhibitor in clinical development. J. Med. Chem. 2020, 63, 15541−15563.
(193) Zhou, C.; Ren, B.; Wang, H. Fused tetracyclic and pentacyclic
dihydrodiazepinocarbazolones as PARP inhibitors and their prepara
tion. WO 2013097225 A1, 2013.
(194) Wang, H.; Zhou, C.; Ren, B.; Kuang, X. Process for preparing
(R)-2-fluoro-10a-methyl-7,8,9,10,10a,11-hexahydro-5,6,7a,11
tetraazacyclohepta[def]cyclopenta[a]fluoren-4(5H)-one as PARP in
hibitor, crystalline forms, and uses thereof. WO 2017032289 A1,
2017.
(195) Wang, H.; Kuang, X.; Zhou, C. Crystalline forms of salts of
fused tetra or penta-cyclic dihydrodiazepinocarazolones, and uses
thereof. WO 2018157794 A1, 2018.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Xiong Y, Guo Y, Liu Y, Wang H, Gong W, Liu Y, et al. (September 2020). “Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor”. Neoplasia. 22 (9): 431–440. doi:10.1016/j.neo.2020.06.009. PMC 7350150. PMID 32652442.
- Markham A (July 2021). “Pamiparib: First Approval”. Drugs. 81 (11): 1343–1348. doi:10.1007/s40265-021-01552-8. PMID 34287805.
- Friedlander M, Mileshkin L, Lombard J, Frentzas S, Gao B, Wilson M, et al. (September 2023). “Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-expansion stage of a multicentre, open-label, phase I trial”. British Journal of Cancer. 129 (5): 797–810. doi:10.1038/s41416-023-02349-0. PMC 10449784. PMID 37474720.
| Clinical data | |
|---|---|
| Trade names | Partruvix |
| Other names | BGB-290 |
| ATC code | L01XK06 (WHO) |
| Legal status | |
| Legal status | US: Investigational New DrugRx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1446261-44-4 |
| PubChem CID | 135565554 |
| DrugBank | DB14769 |
| ChemSpider | 58805610 |
| UNII | 8375F9S90C |
| KEGG | D11426 |
| ChEMBL | ChEMBL4112930 |
| Chemical and physical data | |
| Formula | C16H15FN4O |
| Molar mass | 298.321 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Pamiparib, APPROVALS 2022, CHINA 2022, BeiGene, BGB 290
Dorzagliatin
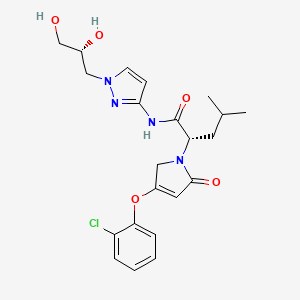
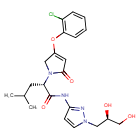
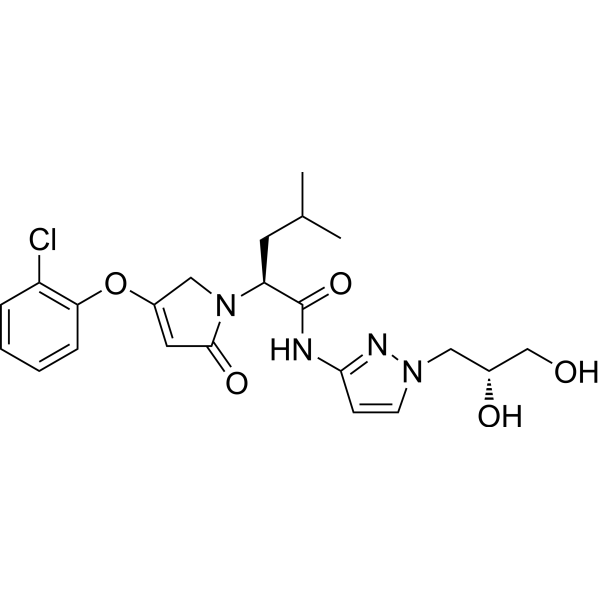
Dorzagliatin
- CAS 1191995-00-2
- HMS5552
- Sinogliatin
- HMS-5552
- MW 462.9 g/mol MF C22H27ClN4O5
- (2S)-2-[3-(2-chlorophenoxy)-5-oxo-2H-pyrrol-1-yl]-N-[1-[(2R)-2,3-dihydroxypropyl]pyrazol-3-yl]-4-methylpentanamide
- RO5305552
- RO-5305552
- X59W6980E8
- (2S)-2-[3-(2-chlorophenoxy)-5-oxo-2H-pyrrol-1-yl]-N-[1-[(2R)-2,3-dihydroxypropyl]pyrazol-3-yl]-4-methyl-pentanamide
- 1H-PYRROLE-1-ACETAMIDE, 4-(2-CHLOROPHENOXY)-N-(1-((2R)-2,3-DIHYDROXYPROPYL)-1H-PYRAZOL-3-YL)-2,5-DIHYDRO-.ALPHA.-(2-METHYLPROPYL)-2-OXO-, (.ALPHA.S)-
Dorzagliatin(18)was developed by Hua Medicine as a treatment for diabetic kidney disease(DKD), type1diabetes mellitus(T1DM), and type2 diabetes mellitus (T2DM). CHINA 2022
Dorzagliatin is a glucokinase activator that is being developed to treat diabetes.[1] Unlike other diabetes drugs, it is intended to increase insulin sensitivity.[2]
Dorzagliatin is under investigation in clinical trial NCT03173391 (Long-term Efficacy and Safety of HMS5552 in T2DM).
PATENT
https://patents.google.com/patent/CN112062754A/en
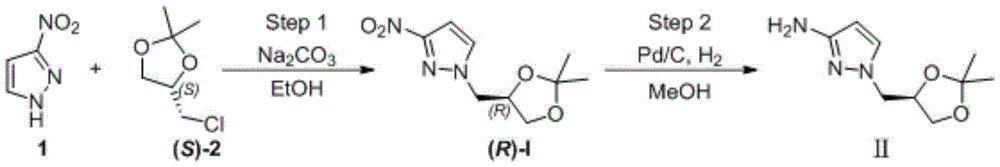
(R) -1- ((2, 2-dimethyl-1, 3-dioxolane-4-yl) methyl) -1H-pyrazole-3-ammonia (II) is a very important medical intermediate for synthesizing Dorzagliatin. Dorzagliatin is a novel medicine for treating type 2 diabetes mellitus, and (R) -1- ((2, 2-dimethyl-1, 3-dioxolane-4-yl) methyl) -1H-pyrazole-3-ammonia (II) is an essential intermediate in the synthetic process of the medicine, and along with the steady promotion of new Dorzagliatin medicines to the market, the demand of the chiral intermediate in the market is required to be rapidly increased.
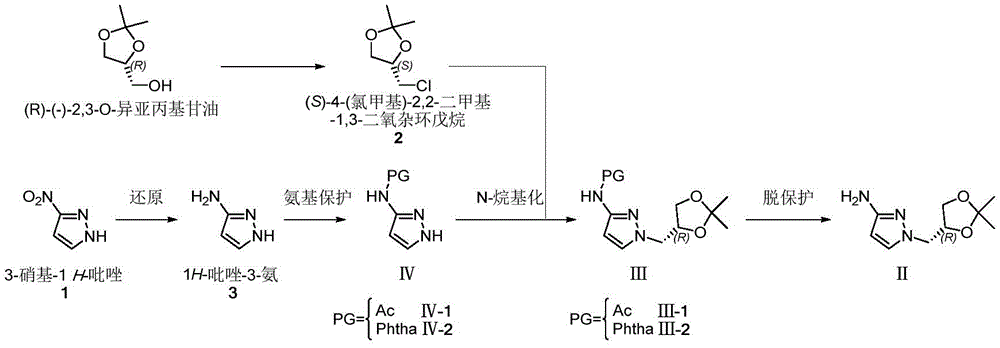
The main production method of the key chiral intermediate is shown as follows: reducing nitro in 3-nitro-1H-pyrazole substrate into amino, protecting free amino, carrying out N-alkylation reaction with (R) – (-) -2, 3-O-isopropylidene glycerol-OH derivative active intermediate, and deprotecting to obtain the final product. The synthetic route needs to be subjected to an N-protection process, so that route steps are added, and the cost is increased. The synthesis of N-protected substrate iv is reported: in the patent US2013203802, 1H-pyrazole-3-ammonia is protected by acetic anhydride, and in WO2017040757, N-acetyl-1H-pyrazole-3-ammonia is obtained by an N- (1-benzyl-1H-pyrazole-3-yl) acetamide debenzylation method; the protection of the N-benzoyl group of 1H-pyrazol-3-amine is reported in the patent US 6118008; in addition, WO2009106209, US2012095064, mention the phthalimide protection strategy of 1H-pyrazole-3-ammonia with phthalic anhydride.
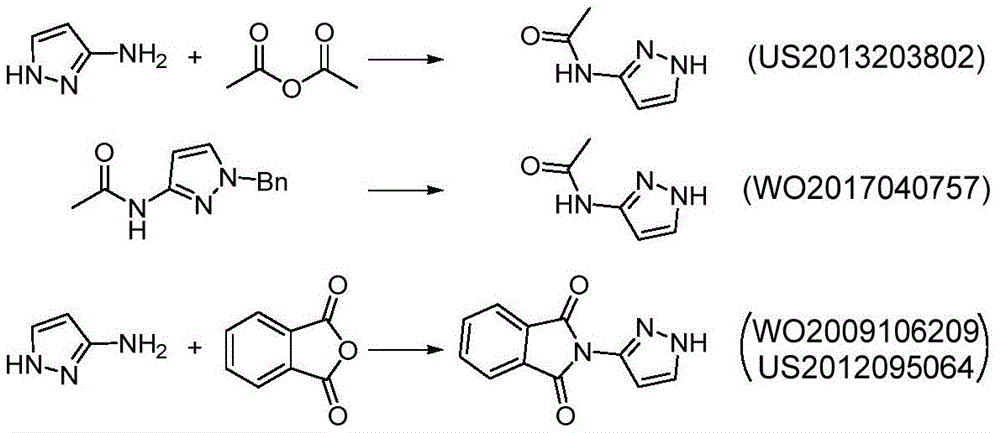
Example 1
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine
The first step is as follows: intermediate (R) -I preparation:
under the protection of nitrogen, 3-nitro-1H-pyrazole (1) (100.00g,0.884mol), ethanol (1.0L) and sodium carbonate (133.90g, 1.26mol) are sequentially added into a 3L reaction bottle, and the system is stirred for 0.5H at room temperature; (S) – (-) -4-chloromethyl-2, 2-dimethyl-1, 3-dioxolane ((S) -2) (126.84g, 0.842mol) was dissolved and diluted with 634ml of ethanol and then added dropwise to the reaction flask. After the dropwise addition, the temperature is raised to 50 ℃ and the reaction is stirred for 5 hours. Ethanol was distilled off under reduced pressure, and the residue was diluted with (1.0L) of water and then extracted twice with dichloromethane (500ml × 2); the organic phase was washed with water and then with saturated sodium chloride brine. Concentrating under reduced pressure to remove dichloromethane to obtain crude oily substance; the crude product was purified by silica gel column chromatography (eluent: n-hexane/ethyl acetate mixed system) to give 166.5g of a pale yellow oily product, with a yield of 87% and an ee value of 98% or more.
The second step is that: reducing nitro to obtain target product
A2L autoclave was charged with (R) -I substrate (150g, 0.66mol), methanol (750mL), Pd/C (0.75g, 0.5% W/W), and the mixture was subjected to nitrogen substitution three times, then hydrogen substitution three times, under a hydrogen-charging pressure of 2.0MPa, at a temperature of 50 ℃ for reaction for 8 hours. Filtering, filtering to remove Pd/C catalyst, concentrating the filtrate to remove methanol to obtain 123.70g of light yellow oily matter, wherein the yield is 95%, and the ee value is more than or equal to 98%.
Example 2
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine by Raney-Ni reduction system
The first step is the same as in example 1.
The second step is that: reduction of nitro groups by Rany-Ni
The intermediate (R) -I (150g, 0.66mol) obtained in the first step was charged into a 2L reactor, and ethanol (1.2L) was added thereto and stirred, followed by adding Rany-Ni (75g) and stirring at room temperature for reaction for 15 hours. Filtering, filtering to remove the solid catalyst, and concentrating the filtrate to dryness to obtain 106.77g of light yellow oily substance with yield of 82% and ee value of more than or equal to 97%.
Example 3
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine by hydrazine hydrate system
The first step is the same as in example 1.
The second step is that: A2L reaction flask was charged with intermediate (R) -I (150g, 0.66mol), ferric trichloride (528mg, 3.3mmol), and ethanol (1.2L), stirred, charged with hydrazine hydrate (39.5g, 0.79mol), and heated to reflux for 6 h. Ethanol was removed by concentration under reduced pressure, the residue was diluted with 750ml of water and extracted twice with ethyl acetate (250 ml. times.2). The organic phase was washed with water and then with saturated brine. The ethyl acetate is removed by concentration to obtain 110.7g of crude light yellow oily substance, the yield is 85 percent, and the ee value is more than or equal to 97 percent.
SYN
https://doi.org/10.1021/acs.jmedchem.3c02374J.Med.Chem.2024,67,4376−4418
Dorzagliatin(HuaTangNing).
Dorzagliatin(18)was developed by Hua Medicine as a treatment for diabetic kidney disease(DKD), type1diabetes mellitus(T1DM), and type2 diabetes mellitus (T2DM).133 This first-in-class, small
molecule,oral,glucokinaseactivator(GKA)wasfirst approved in ChinainSeptember2022foradultpatientswithT2DMasa monotherapy and in combination with metformin (an antidiabetic medication).134 Expression of glucokinase is reduced for individuals with T2DM, thus GKAs such as dorzagliatin serve as a novel class of antidiabetic treatment options.135,136 Theinitialpatent thatdisclosesthesynthesisofdorzagliatin (18)began fromreadily availablematerials 3-aminopyrazole
(18.1) and 2-chlorophenol (18.5). The synthetic strategy reliedonaconvergentamidecouplingofamine18.4(Scheme32) and carboxylic acid 18.9 (Scheme 33).137 A later disclosure provided an updated route toward amine 18.4 (Scheme 32), detailing the synthetic improvements with respect to yield and purity.138 This later disclosure also detailed the synthesis of dorzagliatinonmultikilogramscale fromtheamidationofacid18.9withamine18.4,yieldingover
10kgoftheactivepharmaceutical ingredient.Acetylationof3 aminopyrazole (18.1) with acetic anhydride provided the protectedpyrazole18.2(Scheme32). Subsequent alkylation with alkyl chloride 18.3 followed by base-mediated deprotectionyieldedamine18.4. The synthesis of acid 18.9 began with base-mediated
alkenylationof2-chlorophenol (18.5)withethyl 2-butynoate toprovideester18.6(Scheme33). Subsequentbromination withNBSandAIBNyieldsallylbromide18.7.Next,subjection
ofL-leucinemethylesterhydrochloride(18.8)tobaseresulted ina freeamine thatunderwent allylationwithbromide18.7. Acid 18.9was subsequently generated froma cyclization
condensation sequence and saponification reaction with NaOH. Final amidebondformationwas facilitatedbyEDCI andHOBt toprovideamide18.10, anddorzagliatin(18)was generatedonthemultikilogramscale followingacid-mediated acetonidedeprotectiontoreveal the1,2-diol.
(133) Syed, Y. Y. Dorzagliatin: First approval. Drugs 2022, 82,
1745−1750.
(134) Xu, H.; Sheng, L.; Chen, W.; Yuan, F.; Yang, M.; Li, H.; Li, X.;
Choi, J.; Zhao, G.; Hu, T.; et al. Safety, tolerability, pharmacokinetics,
and pharmacodynamics of novel glucokinase activator HMS5552:
results from a first-in-human single ascending dose study. Drug Des.
Devel. Ther. 2016, 10, 1619−26.
(135) Ren, Y.; Li, L.; Wan, L.; Huang, Y.; Cao, S. Glucokinase as an
emerging anti-diabetes target and recent progress in the development
of its agonists. J. Enzyme Inhib. Med. Chem. 2022, 37, 606−615.
(136) Toulis, K. A.; Nirantharakumar, K.; Pourzitaki, C.; Barnett, A.
H.; Tahrani, A. A. Glucokinase activators for type 2 diabetes:
Challenges and future developments. Drugs 2020, 80, 467−475.
(137) Berthel, S. J.; Brinkman, J. A.; Hayden, S.; Haynes, N.-E.;
Kester, R. F.; McDermott, L. A.; Qian, Y.; Sarabu, R.; Scott, N. R.;
Tilley, J. W. Pyrrolidinone as glucokinase activators and their
preparation, pharmaceutical compositions and use in the treatment
of metabolic disorders. WO 2009127546, 2009.
(138) Chen, J.; Ren, Y.; She, J.; Wang, L. Process for the preparation
of 1-([1,3]dioxolan-4-ylmethyl)-1h-pyrazol-3-ylamine. U.S. Patent US
20150315176, 2015.
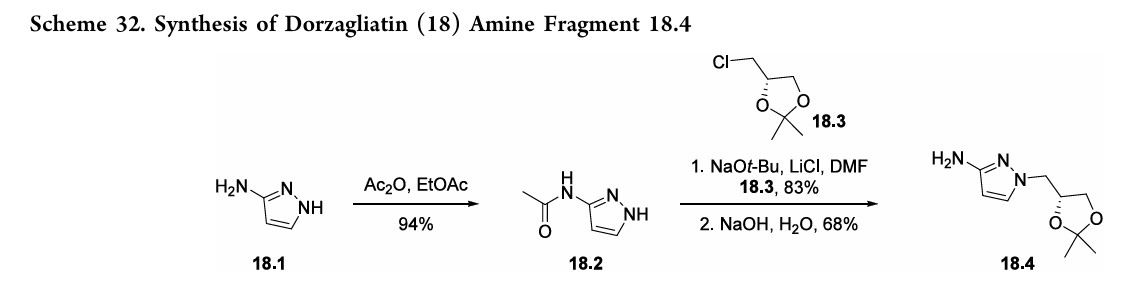
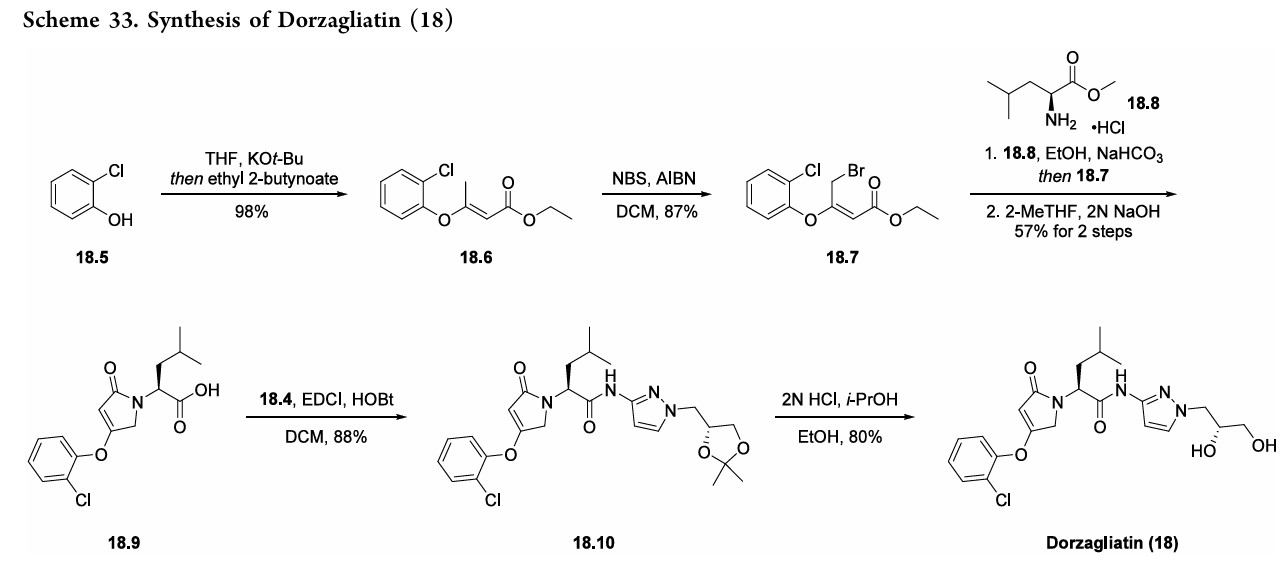
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Chow, Elaine; Wang, Ke; Lim, Cadmon K.P.; Tsoi, Sandra T.F.; Fan, Baoqi; Poon, Emily; Luk, Andrea O.Y.; Ma, Ronald C.W.; Ferrannini, Ele; Mari, Andrea; Chen, Li; Chan, Juliana C.N. (1 February 2023). “Dorzagliatin, a Dual-Acting Glucokinase Activator, Increases Insulin Secretion and Glucose Sensitivity in Glucokinase Maturity-Onset Diabetes of the Young and Recent-Onset Type 2 Diabetes”. Diabetes. 72 (2): 299–308. doi:10.2337/db22-0708. PMC 9871194.
- Zhu, Dalong; Li, Xiaoying; Ma, Jianhua; Zeng, Jiao’e; Gan, Shenglian; Dong, Xiaolin; Yang, Jing; Lin, Xiaohong; Cai, Hanqing; Song, Weihong; Li, Xuefeng; Zhang, Keqin; Zhang, Qiu; Lu, Yibing; Bu, Ruifang; Shao, Huige; Wang, Guixia; Yuan, Guoyue; Ran, Xingwu; Liao, Lin; Zhao, Wenjuan; Li, Ping; Sun, Li; Shi, Lixin; Jiang, Zhaoshun; Xue, Yaoming; Jiang, Hongwei; Li, Quanmin; Li, Zongbao; Fu, Maoxiong; Liang, Zerong; Guo, Lian; Liu, Ming; Xu, Chun; Li, Wenhui; Yu, Xuefeng; Qin, Guijun; Yang, Zhou; Su, Benli; Zeng, Longyi; Geng, Houfa; Shi, Yongquan; Zhao, Yu; Zhang, Yi; Yang, Wenying; Chen, Li (May 2022). “Dorzagliatin in drug-naïve patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial”. Nature Medicine. 28 (5): 965–973.
- [1]. Zhu XX, et al. Dorzagliatin (HMS5552), a novel dual-acting glucokinase activator, improves glycaemic control and pancreatic β-cell function in patients with type 2 diabetes: A 28-day treatment study using biomarker-guided patient selection. Diabetes Obes Metab. 2018 Sep;20(9):2113-2120. [Content Brief][2]. Wang P, et al. Effects of a Novel Glucokinase Activator, HMS5552, on Glucose Metabolism in a Rat Model of Type 2 Diabetes Mellitus. J Diabetes Res. 2017;2017:5812607. [Content Brief]
//////////Dorzagliatin, APPROVALS 22, CHINA 22, DIABETES, Hua Medicine, 1191995-00-2, HMS 5552, Sinogliatin, HMS-5552, RO 5305552, RO-5305552, X59W6980E8
Hetrombopag Olamine
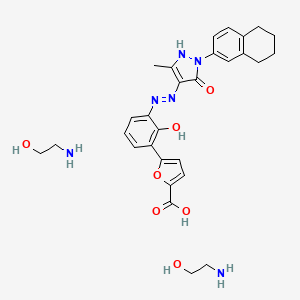
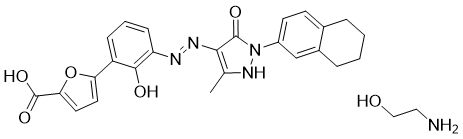
Hetrombopag Olamine, RAFUTROMBOPAG OLAMINE
- Hetrombopag diolamine
- SHR8735 olamine
- Hetrombopag ethanolamine
- SHR-8735 olamine
580.6 g/mol, C29H36N6O7, V45T2I862X
2-aminoethanol;5-[2-hydroxy-3-[[5-methyl-3-oxo-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4-yl]diazenyl]phenyl]furan-2-carboxylic acid
CAS 1257792-42-9
1257792-41-8 (free acid) 1257792-41-8 (ethanolamine) 1257792-42-9 (olamine)
Jiangsu Hengrui Pharmaceutical, was approved in China in June 2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemia who have not responded well to other treatments
Hetrombopag Olamine is the orally active ethanolamine salt of hetrombopag, a small-molecule, nonpeptide thrombopoietin receptor (TPO-R or CD110) agonist, with megakaryopoiesis-stimulating activity. Upon oral administration, hetrombopag targets, binds to and stimulates the transmembrane domain of the platelet TPO-R, a member of the hematopoietin receptor superfamily. Activation of TPO-R leads to the proliferation and differentiation of cells in the megakaryocytic lineage and an increase in platelet production. This may prevent or treat chemotherapy-induced thrombocytopenia.
- OriginatorJiangsu Hengrui Medicine Co.
- DeveloperAtridia; Jiangsu Hengrui Medicine Co.
- ClassAntianaemics; Antihaemorrhagics; Aza compounds; Carboxylic acids; Furans; Pyrazolones; Small molecules; Tetrahydronaphthalenes
- Mechanism of ActionThrombopoietin receptor agonists
- Orphan Drug StatusYes – Thrombocytopenia
- MarketedAplastic anaemia; Idiopathic thrombocytopenic purpura
- Phase IIIThrombocytopenia
- No development reportedUnspecified
- 07 Dec 2024Efficacy and adverse events data from a phase-III trial in Aplastic anaemia presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)
- 31 Jul 2024Phase-III clinical trials in Thrombocytopenia in China (PO) (NCT06507436)
- 25 Jul 2024Jiangsu Hengrui Medicine plans a phase III trial in Thrombocytopenia (PO) in July 2024 (NCT06507436)
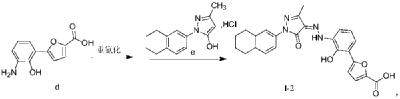
SYN
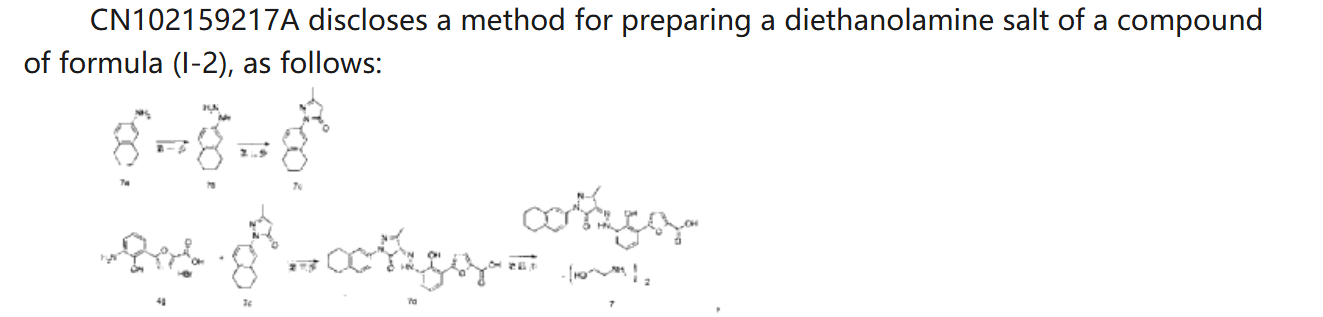
CN 113929668
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN349207982&_cid=P21-MDCUSL-44897-1
| Example 1. Synthesis of 5-(2-carbonyl-2,3-dihydrobenzoxazol-7-yl)furan-2-carboxylic acid |
| |
| Add purified water to the batching barrel, add 4.0kg of compound a under stirring, then add 10L of hydrochloric acid, stir, pump the material into a 50L reactor, add 10L of purified water to the batching barrel and pump it into the reactor. Turn on stirring, start cooling, the temperature drops to -5~2°C, start adding sodium nitrite aqueous solution (6.4L purified water, 1840g sodium nitrite), keep the temperature in the reactor no higher than 5°C during the process; after adding, continue stirring for 10~20min; add 800g of urea, continue stirring for 10~20min, the obtained diazonium salt solution is ready for use, and the temperature in the whole process is kept no higher than 5°C. |
| 44kg of acetone was pumped into a 200L reactor, and 15.0kg of compound b and 463.5g of copper chloride dihydrate were added in sequence under stirring. The temperature was raised to 30-35°C, and the obtained diazonium salt solution was added. The temperature was maintained at 30-40°C during the period. After the addition was completed, the temperature was maintained at 30-40°C and the reaction was continued with stirring for 1-1.5h. 120.0L of purified water was added, the temperature was raised to 40-45°C, and stirring was continued for a period of time. Filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. 80L of purified water was added to the reactor, stirring was started, and the filter cake was added. Sodium hydroxide aqueous solution was added to the reactor to adjust the pH, the pH value was maintained at 8-10 for a period of time, and the filtrate was pumped into the reactor, and the filter was pressed into the material barrel through the filter press. Then 10L of purified water was pumped into the reactor and filtered into the material barrel. The material in the material barrel was pumped into the reactor, and then ethyl acetate was pumped in, stirred, and allowed to stand for 30-40 minutes. The aqueous phase was separated and collected, and the aqueous phase was pumped into the reactor, and the pH was adjusted to 3-4 with hydrochloric acid solution, and the filter cake was washed with purified water until the filtrate was neutral, and then the filter cake was collected. The filter cake was dried to obtain compound c. The yield of this step was 3.59 kg, and the yield was 55%. |
| Example 2: Synthesis of 5-(3-amino-2-hydroxyphenyl)furan-2-carboxylic acid |
| |
| Purified water was pumped into the 50L reactor, stirring was started, 3.53kg of sodium hydroxide was added, and compound c obtained in the previous step was added. Under nitrogen protection, the reaction mixture was heated to reflux in the reactor for reaction. After the reaction, the reaction solution was cooled, the temperature was lowered to 0-10°C, and hydrochloric acid solution was added to adjust the pH value to 5-6. The filter cake was filtered, and the filtrate was washed with purified water until neutral, and then filtered again to collect the filter cake. The filter cake was dried to obtain compound d. The yield in this step was 2.78kg, with a yield of 90%. |
| Example 3. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| Purified water was added to the batching barrel, and compound d was added in sequence under stirring, and then 6.3L hydrochloric acid was added, and the materials were pumped into a 200L reactor. Purified water was added to the batching barrel again, and then pumped into the reactor. Stirring was started, and the temperature was lowered to -5 to 2°C. Sodium nitrite aqueous solution (sodium nitrite to compound d molar ratio is 1:1) was added, and the internal temperature was kept at no more than 5°C during the process. After the addition was completed, stirring was continued; urea was added, and stirring was continued to obtain a diazonium salt solution for use, and the internal temperature was kept at no more than 5°C during the whole process. |
| Add 36L purified water and 4000g sodium hydroxide to the batching barrel, stir to dissolve, and set aside. Take 26kg of the above sodium hydroxide aqueous solution, add compound e (the molar ratio of compound e to compound d is 0.9:1), stir, and add the resulting solution to the diazonium salt solution, keeping the temperature not exceeding 8°C. Add the above-prepared sodium hydroxide aqueous solution dropwise, adjust the pH to 8-10, and keep the temperature at 5-10°C for 3-4h. Add hydrochloric acid solution dropwise to adjust the pH to 2-3, keep the temperature not exceeding 25°C, filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. Pump 48.0kg of tetrahydrofuran aqueous solution (22.5kg tetrahydrofuran, 25.5L purified water) into the reactor, add the above-obtained filter cake, beat, filter, wash the filter cake with tetrahydrofuran aqueous solution, wash the filter cake with purified water, filter again, and collect the filter cake. Dry the filter cake. |
| Ethyl acetate was pumped into the reactor, and the above-obtained materials were added to the reactor for slurrying, and the filter cake was washed with ethyl acetate, and the filter cake was washed until no obvious droplets flowed out of the mirror, and the filter cake was collected and dried to obtain the compound of formula (I-2). The yield in this step was 5.34 kg, and the yield was 97.5%. |
| Example 4. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the equivalent of compound e was adjusted from 0.9 in Example 3 to the current 0.95, other conditions remained unchanged). |
| Comparative Example 1: Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazole-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the step of adding urea was changed to starch potassium iodide test paper to indicate the reaction endpoint, and other conditions remained unchanged). |
| Test Example 1: Effect of urea on the preparation process of the compound of formula (I-2) |
| HPLC conditions: |
| Chromatographic column: Welch Ultimate |
| Flow rate: 1.0ml/min |
| Injection volume: 10 μl |
| Detector: UV detector |
| Detection wavelength: 251nm |
| Mobile phase: 0.1% trifluoroacetic acid aqueous solution was used as mobile phase A, acetonitrile was used as mobile phase B, and elution was performed at a ratio of 50%/50% of mobile phase A/mobile phase B. |
PATENT
EP 2441457
PATENT
WO 2010142137
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010142137&_cid=P21-MDCUXF-51461-1
PATENT
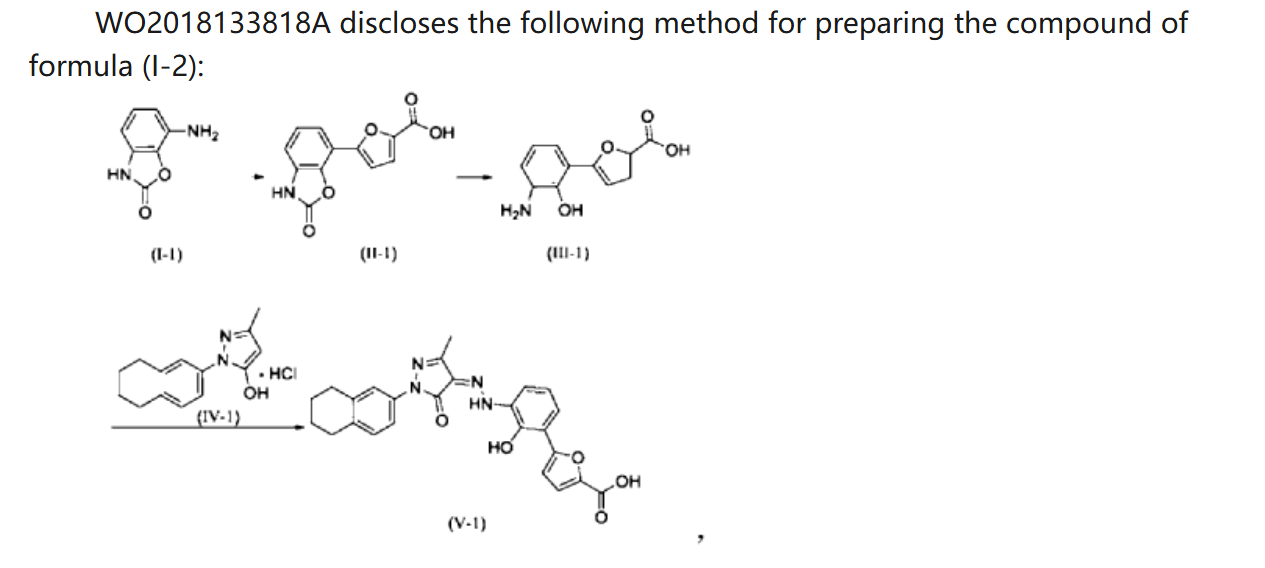
WO 2018133818
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018133818&_cid=P21-MDCUYN-53075-1
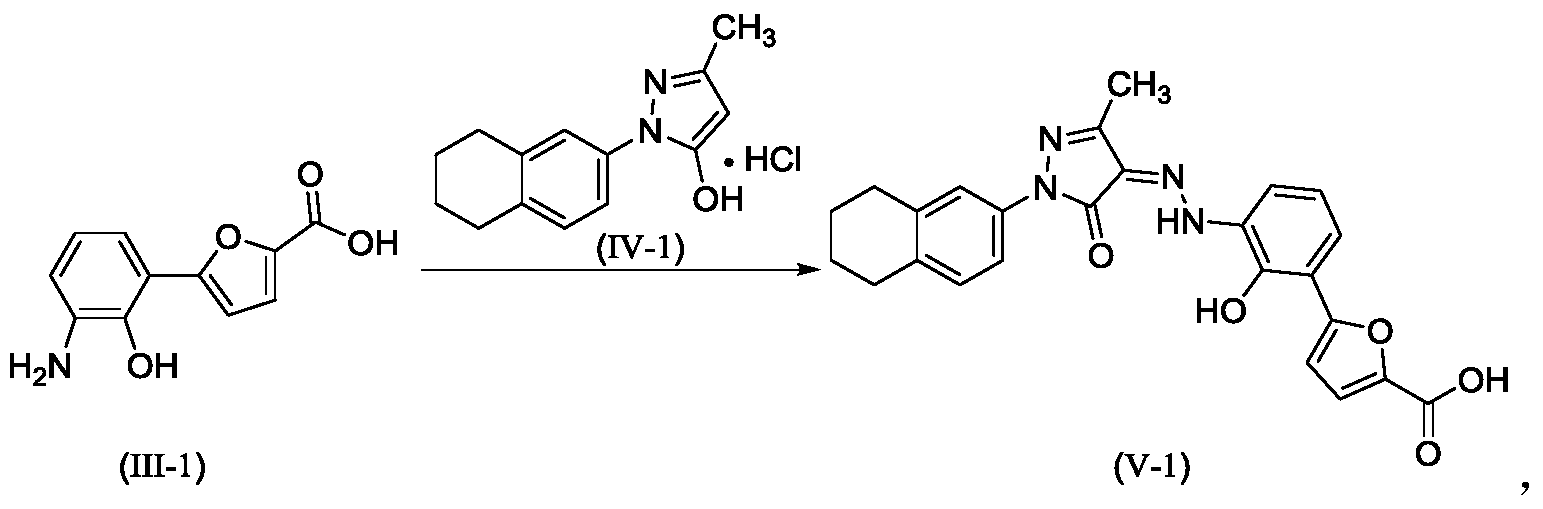
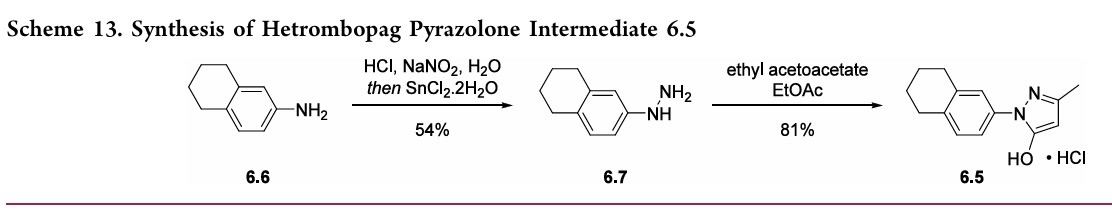
Example 1. Preparation of 3-methyl-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-5-ol hydrochloride
[0107]
[0108](5,6,7,8-tetrahydronaphthalene-2-yl)hydrazine hydrochloride (1.3 kg, prepared according to the method in patent application WO2009092276A1) and ethyl acetoacetate (1.17 L) were added to ethyl acetate (5.2 L). The mixture was heated under reflux for 2 hours. The reaction solution was cooled to room temperature, then cooled to 0-5°C, stirred for 1 hour, filtered, and the solid was washed with a small amount of ethyl acetate to obtain a white solid product (1.4 kg, yield 81%).
[0109]
[0110]Example 2. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid (V-1)
[0111]
[0112]Step 1: Synthesis of intermediate (II-1)
[0113]Purified water (14.80 kg), 7-aminobenzo[d]oxazol-2(3H)-one (2.00 kg, prepared according to the method in patent application WO2005016898A2), and hydrochloric acid (5.33 kg) were added to the reaction kettle, the temperature was raised to 40-45°C, stirred for 10 min, cooled to -3-5°C, and sodium nitrite aqueous solution (sodium nitrite 940 g, water 3.20 kg) was added dropwise, the internal temperature was kept at no more than 5°C, the end point was controlled by starch potassium iodide test paper, and stirring was continued for 15 min;
[0114]Add acetone (28L) to the reactor, then add furoic acid (4.57kg) and cupric chloride dihydrate (232g), stir at 35-40℃ until dissolved, add diazonium salt solution dropwise, keep the internal temperature at 35-40℃, and continue stirring for 1.5h. Add purified water (60L), heat to 35-40℃ and stir for 30min. Filter, wash the filter cake with 45-50℃ purified water. Add the filter cake to purified water (40kg), adjust the pH to 8-9 with 15% sodium hydroxide aqueous solution, filter, adjust the pH of the filtrate to 3-4 with 6mol/L hydrochloric acid, filter, wash the filter cake with purified water, and dry to obtain a solid (1.63kg, yield 50%).
[0115]Step 2: Synthesis of intermediate (III-1)
[0116]The product from the previous step (1.4 kg) and 15% aqueous sodium hydroxide solution (9.7 kg) were heated to reflux under argon protection and reacted for 28 hours. The reaction solution was poured into ice water (5-6 kg), and hydrochloric acid (6N, 3 L) was slowly added to adjust the pH value to 5-6. The temperature was maintained below 20°C. During this period, ethyl acetate was added to eliminate bubbles. The mixture was filtered, washed with purified water, and dried to obtain a solid (1.18 kg, yield 94%).
[0117]Step 3: Synthesis of intermediate (V-1)
[0118]Add the product of the previous step (1.10kg), purified water (27.5kg), and hydrochloric acid (2.92kg) to the reactor in sequence, stir and dissolve, cool to -4 to -1°C, add sodium nitrite aqueous solution (346g sodium nitrite, 5.5kg water), and continue to react for 15min after the addition is completed. Cool to -8 to -5°C. Dissolve sodium hydroxide (1.48kg) in purified water (13.2kg) to obtain a 10% sodium hydroxide aqueous solution. Add 5-methyl-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-2H-pyrazole-3-ol hydrochloride (1.26kg) to the above sodium hydroxide aqueous solution (10kg) to dissolve, and add the resulting solution to the diazonium salt solution at once, keeping the temperature not higher than 10°C. Add the remaining 10% sodium hydroxide aqueous solution, adjust the pH to 8 to 9, naturally heat to 8 to 12°C for reaction, and react for 4h. Add 6N hydrochloric acid, adjust pH=2-3, keep the temperature not more than 20°C, filter, and wash the filter cake with water until pH=6-7. Add the filter cake to 50% tetrahydrofuran aqueous solution (19kg), slurry at room temperature for 2h, filter, wash with 50% tetrahydrofuran aqueous solution, wash with water, and dry. Add ethyl acetate (20kg) to the solid, slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, add the solid to ethyl acetate (20kg), slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, and dry to obtain a solid (2.18kg, yield 95%, purity 99.5%).
[0120]Example 3. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid ethanolamine salt (1:2)
[0121]
[0122]Preparation of crude product
[0123]The compound of formula (V-1) (1.8 kg) was suspended in a tetrahydrofuran/ethanol (14.5 kg, V/V = 2:1) mixed solvent at room temperature, stirred for 0.5 h, cooled to 10-15 ° C, and a tetrahydrofuran ethanol solution of ethanolamine (479.6 g) (tetrahydrofuran 91 g and ethanol 41 g) was added dropwise. The mixture was naturally heated to room temperature and reacted for 20 h. Filtered, washed with a tetrahydrofuran/ethanol (V/V = 2:1) mixed solvent, washed with ethyl acetate, filtered, and dried to obtain a dark red solid (1.73 kg, yield 76%, purity 99.7%).
[0124]
1H-NMR(500MHz,D 2O+NaOH)δ7.725-7.741(d,1H),7.298-7.316(d,3H),7.183-7.198(d,1H),7.131-7.149(m,2H),6.612-6.643(t,1H),3.574-3.596(t,4H),2.759-2.778(br,4H),2.698-2.721(t,4H),2.428(s,3H),1.772(br,4H).
SYN
J.Med.Chem.2024,67,4376−4418
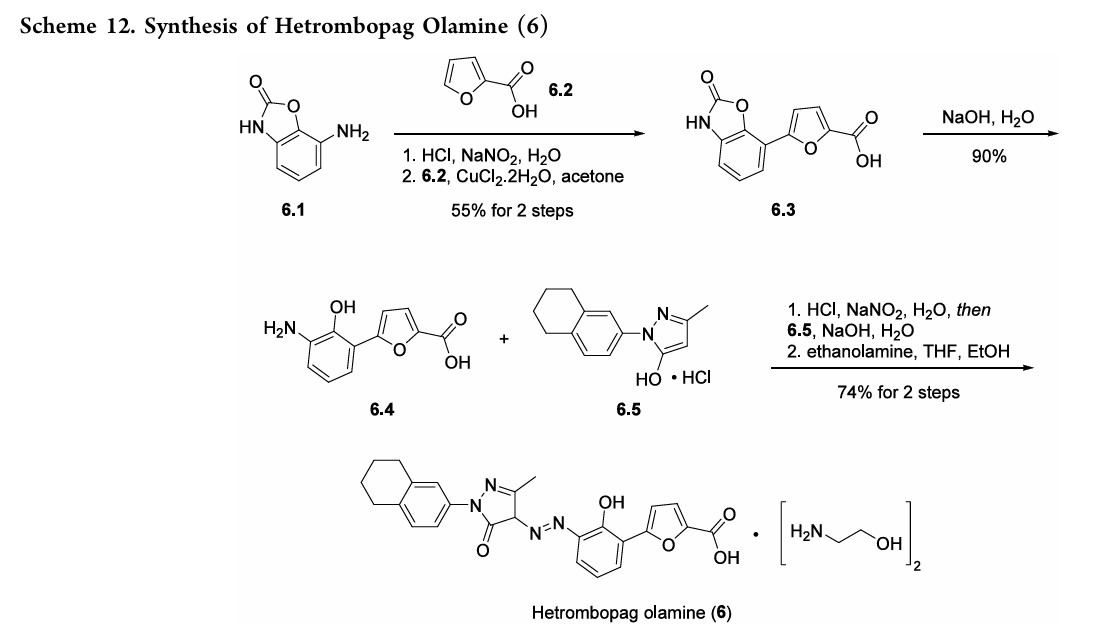
HetrombopagOlamine (Hengqu).
Hetrombopag olamine (6), an oral nonpeptide thrombopoietin receptor
(TpoR)agonistdevelopedby JiangsuHengruiPharmaceutical, was approved in China in June2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemiawhohave not responded well to other treatments.46Hetrombopag, like other TpoR agonists, increases platelet production by binding to the transmembranedomainofTpoRinprogenitorcells, inducing
megakaryocytes.Theeffectisadditivewiththeactionofnative thrombopoietin, whichbinds to the extracellular domainof TpoR.Hetrombopag is structurallyrelatedtoeltrombopag, a previously approvedTpoR, withmodifications to enhance potencyandminimizetoxicity.46−48InaPhaseIIIclinicaltrial, ITPpatients demonstratedadurableplatelet response, with reducedbleedingriskanduseof rescuetherapycomparedto
placebo.49 Akilo-scale, chromatography-freesynthesisofhetrombopag has been reported by researchers at Jiangsu Hengrui Pharmaceutical in the Chinese-language patent literature (Scheme 12).50,51 Commercially available aniline 6.1 was coupledwith furoic acid (6.2) using aMeerwein arylation reaction togive intermediate6.3.This process first involves diazotizationof the anilineusing sodiumnitrite andhydrochloricacid.Ureawasusedtoquenchtheresidualnitrousacid, animprovement thatultimatelygavetheproductwithhigher purity and lower levels of specific impurities; the crude
diazoniumsalt solutionwas carried forwarddirectlywithout furthermanipulation.Furoicacid(6.2)inacetonewastreated withcopper(II)chloridedihydratefollowedbyadditionofthe
diazonium salt solution to affect the arylation. The crude productwaspurifiedbyacid−baseextractionandisolatedby filtrationtoprovide6.3 in55%yield.Basichydrolysisof the
cycliccarbamateunveiledthefreeanilineandphenolmoieties in arene 6.4. Nucleophilic attack of the enolate anion of pyrazolone 6.5 (see Scheme 13) on the diazoniumsalt of aniline6.4 formed the central hydrazonemoiety ina JappKlingemann-like reaction. The crude product was triturated withethylacetatetorapidlyprovidehetrombopagfreebasein
97.5%yield.TreatmentwithethanolamineinTHFandEtOH thengeneratedhetrombopagolamine (6) in76%yieldand 99.7%purity.51 Pyrazolone intermediate6.5was synthesized in two steps
(Scheme 13).52,53 5,6,7,8-Tetrahydronaphthalen-2-yl amine (6.6)was converted to the diazoniumion and reduced in situ to the corresponding hydrazine 6.7 using stannous chloridedihydrate.Condensationof thehydrazinewithethyl acetoacetate in ethyl acetate and in situ cyclization gave pyrazolone6.5.While the synthesis fromaniline6.1 to the activepharmaceutical ingredient(API)6wasreportedonthe
kilo-scale, thesynthesisofpyrazolone6.5wasreportedonlyon gram-scale
(46) Syed, Y. Y. Hetrombopag: First approval. Drugs 2021, 81, 1581−1585.
(47) Xie, C.; Zhao, H.; Bao, X.; Fu, H.; Lou, L. Pharmacological characterization of hetrombopag, a novel orally active human thrombopoietin receptor agonist. J. Cell. Mol. Med. 2018, 22, 5367−5377.
(48) Zheng, L.; Liang, M.-z.; Zeng, X.-l.; Li, C.-z.; Zhang, Y.-f.; Chen, X.-y.; Zhu, X.; Xiang, A.-b. Safety, pharmacokinetics and pharmacodynamics of hetrombopag olamine, a novel TPO-R agonist, in healthy individuals. Basic Clin. Pharmacol. Toxicol. 2017, 121, 414−422.
(49) Mei, H.; Liu, X.; Li, Y.; Zhou, H.; Feng, Y.; Gao, G.; Cheng, P.; Huang, R.; Yang, L.; Hu, J.; Hou, M.; Yao, Y.; Liu, L.; Wang, Y.; Wu, D.; Zhang, L.; Zheng, C.; Shen, X.; Hu, Q.; Liu, J.; Jin, J.; Luo, J.; Zeng, Y.; Gao, S.; Zhang, X.; Zhou, X.; Shi, Q.; Xia, R.; Xie, X.; Jiang, Z.; Gao, L.; Bai, Y.; Li, Y.; Xiong, J.; Li, R.; Zou, J.; Niu, T.; Yang, R.;
Hu, Y. A multicenter, randomized phase III trial of hetrombopag: a novel thrombopoietin receptor agonist for the treatment of immune thrombocytopenia. J. Hematol. Oncol. 2021, 14, 37.
(50) Shi, A.; Diao, A.; Du, Y. Preparation of bicyclic substituted pyrazolone azo derivatives. China Patent CN 113929668, 2022.
(51) Diao, A.; Gao, X.; Bian, L. Method for preparing bicyclo substituted pyrazolone azo derivatives and intermediates. WO 2018133818, 2018.
(52) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Preparation of pyrazole derivatives as thrombopoietin receptor agonists. WO 2010142137, 2010.
(53) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Salts of bicyclo substituted pyrazolon azo derivatives, preparation method and use
thereof. European Patent EP 2441457, 2014.

.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Hetrombopag Olamine, CHINA 2021, APPROVALS 2021, Hetrombopag diolamine, SHR 8735 olamine, Hetrombopag ethanolamine, SHR-8735 olamine, V45T2I862X, RAFUTROMBOPAG OLAMINE
AZVUDINE
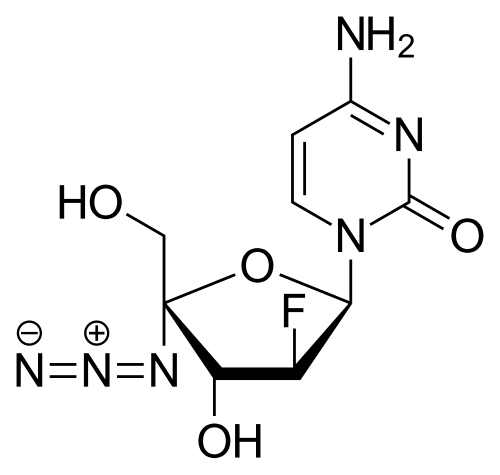
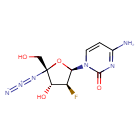
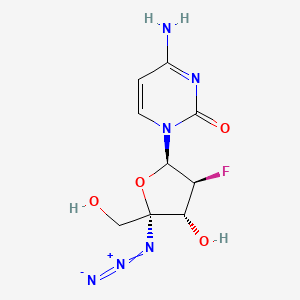
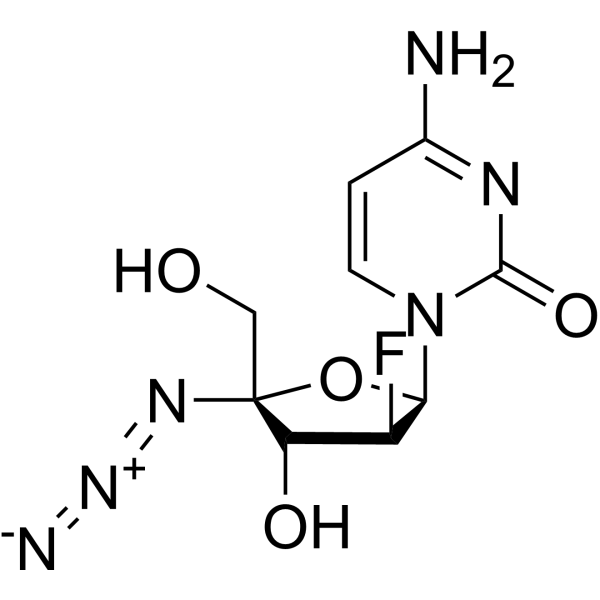
AZVUDINE
CAS
WeightAverage: 286.223
Monoisotopic: 286.082581021
Chemical FormulaC9H11FN6O4
- FNC
- HY-19314
- RO 0622
- RO-0622
- SB17040
IJ2XP0ID0K- DTXSID901027757
4-amino-1-[(2R,3S,4R,5R)-5-azido-3-fluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2-dihydropyrimidin-2-one
- 2′-Deoxy-2′-beta-fluoro-4′-azidocytidine
- 2(1H)-Pyrimidinone, 4-amino-1-(4-c-azido-2-deoxy-2-fluoro-beta-D-arabinofuranosyl)-
- 4-amino-1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-5-((imino-l,5-azanylidene)amino)tetrahydrofuran-2-yl)pyrimidin-2-one
- 4-amino-1-((2R,3S,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)-5-((imino-l,5-azanylidene)amino)tetrahydrofuran-2-yl)pyrimidin-2-one
- 4-Amino-1-(4-c-azido-2-deoxy-2-fluoro-beta-D-arabinofuranosyl)-2(1H)-pyrimidinone
- 4′-C-azido-2′-deoxy-2′-fluoro-beta-D-arabinocytidine
- 4-amino-1-[(2R,3S,4R,5R)-5-azido-3-fluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one
- AZVUDINE [WHO-DD]
- 4-amino-1-((2R,3S,4R,5R)-5-azido-3-fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidin-2(1H)-one
- 4-AMINO-1-(4-C-AZIDO-2-DEOXY-2-FLUORO-.BETA.-D-ARABINOFURANOSYL)-2(1H)-PYRIMIDINONE
- 2(1H)-PYRIMIDINONE, 4-AMINO-1-(4-C-AZIDO-2-DEOXY-2-FLUORO-.BETA.-D-ARABINOFURANOSYL)-
- 4′-C-Azido-2′-deoxy-2′-fluoro-b-D-arabinocytidine
Azvudine is an antiviral drug which acts as a reverse transcriptase inhibitor.[3] It was discovered for the treatment of hepatitis C[4] and has since been investigated for use against other viral diseases such as AIDS and COVID-19,[2][5] for which it was granted conditional approval in China.[6][7]
Azvudine was first discovered in 2007.[8] It costs 350 Chinese yuan per 7 days for COVID, as of November 2022.[9]
Azvudine is under investigation in clinical trial NCT04668235 (Study on Safety and Clinical Efficacy of AZVUDINE in COVID-19 Patients (Sars-cov-2 Infected)).
Azvudine (RO-0622) is a potent nucleoside reverse transcriptase inhibitor (NRTI), with antiviral activity on HIV, HBV and HCV. Azvudine exerts highly potent inhibition on HIV-1 (EC50s ranging from 0.03 to 6.92 nM) and HIV-2 (EC50s ranging from 0.018 to 0.025 nM). Azvudine inhibits NRTI-resistant viral strains. Azvudine is a click chemistry reagent, it contains an Azide group and can undergo copper-catalyzed azide-alkyne cycloaddition reaction (CuAAc) with molecules containing Alkyne groups. It can also undergo strain-promoted alkyne-azide cycloaddition (SPAAC) reactions with molecules containing DBCO or BCN groups.
SYN
https://patents.google.com/patent/CN111892636A/en
According to the inventor’s research and understanding, at present, the synthesis of azvudine mainly includes the following methods according to different raw materials:
1. It is prepared by using a ribonucleotide as a raw material. The method requires a total of 15 steps of reaction to obtain the target product. The inventor’s research found that DAST is used as a fluorination reagent in the fluorination process of this method, which has large steric hindrance and is difficult to fluoride, and the route process is complicated and the route is long. Low cost, high cost, not suitable for industrial production.
2. It is prepared by using 1,3,5-O-tribenzoyl-2-deoxy-2-fluoro-D-arabinofuranoside as raw material. The inventors have found that the preparation raw materials are not easy to obtain, and the route involves uridine to In the conversion reaction of cytidine, the reaction route is further extended, thus limiting the further application of this method.
3. Using ribonucleotides as raw materials to synthesize, the inventors found that this method requires 12 steps to synthesize the target product, and DAST is also used as a fluorination reagent in the fluorination process, which has large steric hindrance and low fluorination efficiency.
4. It is prepared by using uracil nucleotide as raw material. The inventors have found that the raw material cost of this method is relatively high, it involves the conversion process of uridine to cytidine, and the reaction yield is not high.
To sum up, the inventors have found that the currently known processes for preparing azvudine have the following disadvantages: synthesizing uracil nucleotides with ribonucleotides as raw materials, and then converting uracil nucleotides into target products, synthesizing uracil nucleotides. The process routes all exceed 12 steps, and the fluorination reaction uses DAST (diethylaminosulfur trifluoride) reagent, which increases the difficulty of the reaction due to steric hindrance, reduces the yield, and brings difficulties to industrial production.
Example 1

Accurately weigh 4.86 g of cytidine (compound 1), dissolve it in 20 mL of ethanol, then add 4.52 g of benzoic anhydride, raise the temperature to 60° C., and stir overnight. After the reaction was completed, the solvent was removed under reduced pressure, and 100 mL of deionized water was added to wash and filter to obtain compound 2 (96.5% yield). The structural characterization is shown in Figure 1 .
Example 2

Weigh 3.5 g of compound 2, dissolve in 25 mL of pyridine, ice-water bath at 0°C, add 3.0 mL of TIPDS under nitrogen protection, react for 1 h, decompose the reaction mixture with water, remove pyridine under reduced pressure, extract with chloroform, and wash with saturated aqueous sodium bicarbonate solution , and dried over anhydrous sodium sulfate to obtain compound 3 (83.6% yield). The structural characterization is shown in Figure 2.
Example 3

Weigh 5.9 g of compound 3, dissolve it in 100 mL of tetrahydrofuran, add 2.82 g of trifluoromethanesulfonic anhydride (Tf 2 O), and react at room temperature for 2 h under nitrogen protection. Then, the reaction temperature was lowered to -20°C, 2.46 g of tetrabutylammonium fluoride (Bu 4 NF) was added, and the reaction was continued for 10 h. After the reaction was completed, tetrahydrofuran was removed under reduced pressure, extracted with chloroform, washed with saturated aqueous sodium bicarbonate solution, and dried over anhydrous sodium sulfate to obtain compound 4 (86.8% yield). The structural characterization is shown in Figure 3.
Example 4

5.9 g of compound 3 was weighed, dissolved in 100 mL of dichloromethane and 10 mL of anhydrous pyridine, cooled to -50°C under nitrogen protection, added with 1.61 g of DAST, and reacted for 12 h. After the reaction was completed, the solvent was removed under reduced pressure, extracted with chloroform, washed with saturated aqueous sodium bicarbonate solution, and dried over anhydrous sodium sulfate to obtain compound 4 (76.0% yield).
Example 5

Weigh 3.5g of compound 4, add 100mL of tetrahydrofuran, add 0.5g of imidazole, 0.5g of triphenylphosphine, slowly add 3.75g of tetrahydrofuran solution containing 10wt% iodine, stir at room temperature for 5h, the reaction is complete, remove the solvent under reduced pressure, Compound 5 was prepared (84% yield) and the structural characterization is shown in Figure 4 .
Example 6

Weigh 4.59g of compound 5, dissolve it in 100mL of methanol, add 0.5g of DBU, control the temperature to 60°C, react for 12h, cool to room temperature, add saturated aqueous sodium chloride solution, adjust the acidic pH=3 with 1M hydrochloric acid, extract with ethyl acetate, It was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain compound 6 (77.4% yield). The structural characterization is shown in Figure 5 .
Example 7

Weigh 3.3 g of compound 6, add 50 mL of DMF solution dissolved with 0.6 g of sodium azide, add 50 mL of DMF solution dissolved with 0.6 g of ICl, control the temperature to 0 ° C, and react for 12 h. After the reaction is completed, add sodium bisulfite until The color of iodine disappears completely. The solvent was removed under reduced pressure to obtain compound 7 (77% yield). The structural characterization is shown in FIG. 6 .
Example 8

Weigh 2.5 g of compound 7, dissolve it in 50 mL of DMF, add 0.65 g of benzoic acid, add 0.5 g of silver acetate, and stir at room temperature for 12 h. After the reaction was completed, the mixture was filtered, and the solvent was removed under reduced pressure to obtain compound 8 (71.2% yield). The structural characterization is shown in FIG. 7 .
Example 9

Weigh 4.82 g of compound 8, add 100 mL of methanol, 10 mL of deionized water, 3 mL of triethylamine, stir at room temperature for 5 h, and remove the solvent under reduced pressure to obtain compound 9 (88.7% yield). The structural characterization is shown in Figure 8.
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00166
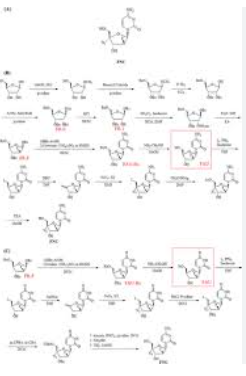
Azvudine was approved for the treatment of adult HIV-1 infection in China in 2021, and it was approved for conditional marketing for the treatment of SARS-CoV-2 in China in 2022. In this work, we describe a fully continuous flow synthesis of 2′-deoxy-2′-fluoroarabinoside, a key intermediate for azvudine. The process was accomplished via six chemical transformations, including chlorination, hydrolysis, fluorination, bromination, condensation, and deprotection in six sequential continuous flow devices. Under the optimized process conditions, the total yield was 32.3% with a total residence time of 156 min. Compared with batch conditions, the total yield was doubled, the total reaction time was shortened 16 times, and the E factor was reduced 1.63 times.
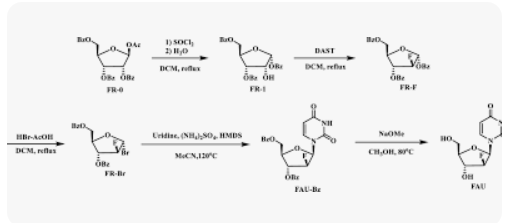
1,3,5-Tri-O-benzoyl-D-ribofuranose (FR-1)
To a stirred solution of O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose (FR-0,
1.008 g, 2.0 mmol) in anhydrous dichloromethane (DCM, 10 mL) was added
dropwise thionyl chloride (0.713 g,6 mmol) at 0 oC, and the resulting mixture
allowed to stir at room temperature for 14 h. The solution was evaporated,
dissolved with toluene (5 mL× 3), and then evaporated. To the residue was
added DCM (10 mL) and water (5 mL), and and continued stirring at room
temperature for 2 h. The solution was then washed with saturated aqueous
NaHCO3 (15 mL) and dried over Na2SO4, filtered and evaporated. DCM (2 mL)
and n-Hexane (20 mL) was added to the residue and the mixture was stirred at
room. A white solid was collected by filtration to give FR-1 (yield: 47.8%).
2-Deoxy-2-fluoro-1,3,5-tri-O-benzoyl-D-ribofuranose (FR-F)
To a stirred solution of FR-1 (0.924 g, 2.0 mmol) in anhydrous DCM (10 mL) was
added dropwise DAST (0.976 g, 6.0 mmol) for 30 min, and the resulting mixture
allowed to stir at 40 °C (16 h). The reaction was cooled and quenched with
saturated aqueous NaHCO3 (15 mL). The solution was extracted with DCM and
water, and then washed with saturated aqueous NaHCO3 (20 mL). This was then
dried over Na2SO4, filtered and evaporated. The crude product was purified by
silica gel column chromatography (ethyl acetate: petroleum ether =1:10) to
obtain a white solid (yield: 41.2%).
α-D-Arabinofuranosyl bromide, 2-deoxy-2-fluoro-, 3,5-dibenzoate (FR-Br)
To a stirred solution of FR-F (0.928 g, 2.0 mmol) in anhydrous DCM (10 mL) was
added dropwise 33.3% HBr-HOAc (1.47 g, 6.0 mmol) at 0 oC, and the resulting
mixture were allowed to stir at room temperature for 7 h. The reaction was
quenched with saturated aqueous NaHCO3 (10 mL), dried over Na2SO4, filtered
and evaporated to yield the product (FR-Br) as a brown oil (yield: 99.8%).
1-(2-deoxy-2-fluoro-3,5-di-O-benzoyl-β-D-arabino-furanosyl)uracil (FAU-Bz)
A mixture of uracil (0.336 g, 3 mmol) and (NH4)2SO4 (10 mg, 0.075 mmol) in
hexamethyldisilazane (HMDS) (6 mL) was refluxed under nitrogen for 5 h. To
the silylated uracil solution was added a solution of FR-Br in dry acetonitrile (10
mL) and the mixture was refluxed under nitrogen for 5 h. The solution was
evaporated, extracted with DCM (20 mL) and washed with saturated aqueous
NaHCO3 (15 mL). This was then dried over Na2SO4, filtered and evaporated.
Ethyl acetate (10 mL) and petroleum ether (50 mL) was added to the residue
and the mixture was stirred at room temperature. A light yellow solid was
collected by filtration to give FAU-Bz (yield: 83.2%).
2′-deoxy-2′-fluoro- arabinoside (FAU)
To a solution of FUA-Bz (0.908 g, 2.0 mmol) in anhydrous methanol (MeOH, 10
mL) was added NH3-MeOH (5 mL), stirred at room temperature for 15 h and
evaporated to dryness under reduced pressure. White solid
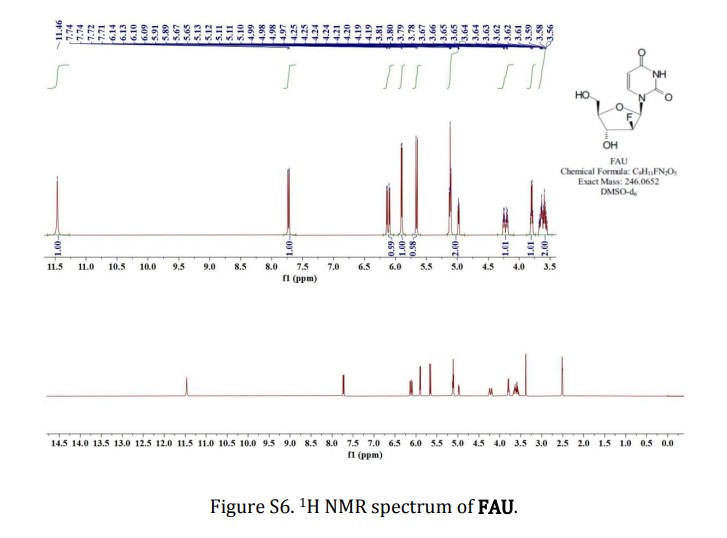
SYN
: J. Med. Chem. 2024, 67, 4376−4418
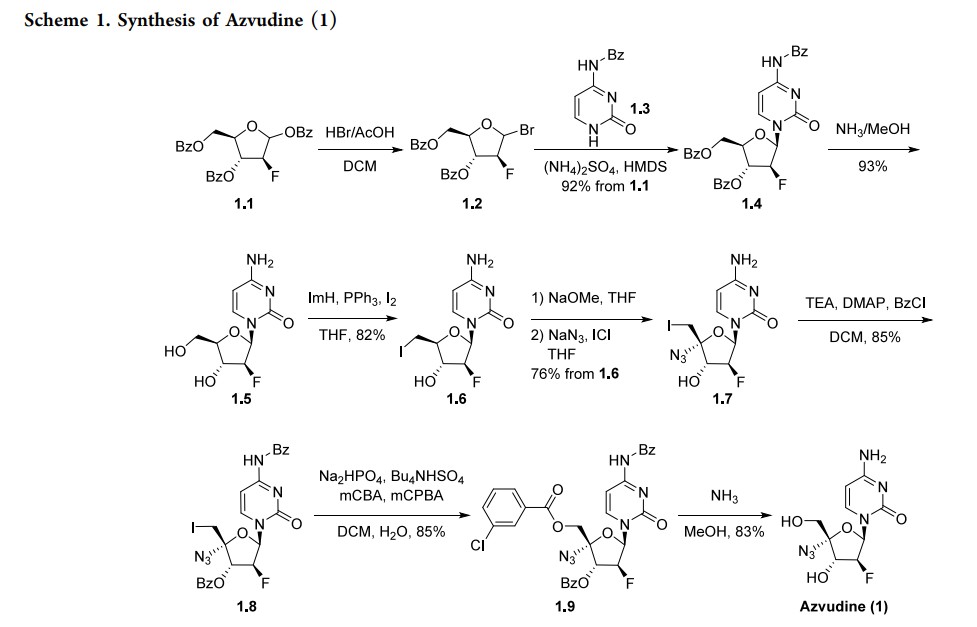
Azvudine (1). Azvudine (1) is an antiviral manufactured by China-based Genuine Biotech. It was
approved in China in 2021 as a first-in-class treatment for human immunodeficiency virus (HIV). It has a dual function, acting as a reverse transcriptase inhibitor and targeting the viral infectivity factor/apolipoprotein B mRNA-editing enzyme, catalytic subunit 3G (Vif/A3G) protein−protein interaction.6 Azvudine has activity against both wild-type and drug-resistant strains of HIV due to the presence of a 3′-hydroxy group and substitution in the 4′-position of the ribose core.7 Due to its known antiviral activity, azvudine was repurposed as a treatment for COVID-19 and approved for this indication inChina in 2022. It acts as an RNA-dependent RNA polymerase (RdRp) inhibitor, the same mechanism as the previously approved molnupiravir and remdesivir. In addition to its antiviral activity, concentration of the drug in the thymus has suggested immune-targeting activity; this dual function is unique among RdRp inhibitors.8 Several syntheses of azvudine have been reported in the scientific and patent literature. Scheme 1 highlights a 100 g scale synthesis from a patent filed by Shandong University.9 Other syntheses are similar, containing the furanose functional group manipulations in 1.5−1.8, though these routes differ in choice of nucleobase and protecting group strategy, and were reported on a smaller scale.10−12 The synthesis began from benzoyl-protected fluoro-furanose 1.1. Bromination with HBr in acetic acid followed by displacement of the bromide 1.2 with protected cytosine 1.3 yielded intermediate 1.4. Deprotection of the benzoyl groups with ammonia in MeOH formed diol 1.5, and a Mitsunobu reaction converted the
primary alcohol to alkyl iodide 1.6. Elimination of the iodide with sodium methoxide followed by addition of sodium azide and iodine monochloride across the resulting alkene produced substitution in the 4′-position of the ribose core.7 Due to its known antiviral activity, azvudine was repurposed as a treatment for COVID-19 and approved for this indication in China in 2022. It acts as an RNA-dependent RNA polymerase (RdRp) inhibitor, the same mechanism as the previously approved molnupiravir and remdesivir. In addition to its antiviral activity, concentration of the drug in the thymus has suggested immune-targeting activity; this dual function is unique among RdRp inhibitors.8 Several syntheses of azvudine have been reported in the scientific and patent literature. Scheme 1 highlights a 100 g scale synthesis from a patent filed by Shandong University.9 Other syntheses are similar, containing the furanose functional group manipulations in 1.5−1.8, though these routes differ in choice of nucleobase and protecting group strategy, and were reported on a smaller scale.10−12 The synthesis began from
benzoyl-protected fluoro-furanose 1.1. Bromination with HBr in acetic acid followed by displacement of the bromide 1.2 with protected cytosine 1.3 yielded intermediate 1.4. Deprotection of the benzoyl groups with ammonia in MeOH formed diol 1.5, and a Mitsunobu reaction converted the primary alcohol to alkyl iodide 1.6. Elimination of the iodide with sodium methoxide followed by addition of sodium azide
and iodine monochloride across the resulting alkene producedazide 1.7. Both the alcohol and amine were reprotected with benzoyl chloride, and the iodide was displaced with metachlorobenzoic acid in an oxidative nucleophilic substitution reaction to yield penultimate intermediate 1.9. All protecting
groups were then removed with ammonia in MeOH to yield
azvudine (1).
(6) Sun, L.; Peng, Y.; Yu, W.; Zhang, Y.; Liang, L.; Song, C.; Hou, J.;
Qiao, Y.; Wang, Q.; Chen, J.; et al. Mechanistic insight into
antiretroviral potency of 2’-deoxy-2’-beta-fluoro-4’-azidocytidine
(FNC) with a long-lasting effect on HIV-1 prevention. J. Med.
Chem. 2020, 63, 8554−8566.
(7) Chang, J. 4’-Modified nucleosides for antiviral drug discovery:
achievements and perspectives. Acc. Chem. Res. 2022, 55, 565−578.
(8) Zhang, J.-L.; Li, Y.-H.; Wang, L.-L.; Liu, H.-Q.; Lu, S.-Y.; Liu, Y.;
Li, K.; Liu, B.; Li, S.-Y.; Shao, F.-M.; Wang, K.; Sheng, N.; Li, R.; Cui,
J.-J.; Sun, P.-C.; Ma, C.-X.; Zhu, B.; Wang, Z.; Wan, Y.-H.; Yu, S.-S.;
Che, Y.; Wang, C.-Y.; Wang, C.; Zhang, Q.; Zhao, L.-M.; Peng, X.-Z.;
Cheng, Z.; Chang, J.-B.; Jiang, J.-D. Azvudine is a thymus-homing
anti-SARS-CoV-2 drug effective in treating COVID-19 patients. Signal
Transduct. Target Ther. 2021, 6, 414.
(9) Chen, X.; Wang, Z.; Yu, H.; Liu, X. Preparation method of
azvudine and its intermediates. China Patent CN 115960147, 2023.
(10) Smith, D. B.; Kalayanov, G.; Sund, C.; Winqvist, A.; Maltseva,
T.; Leveque, V. J.-P.; Rajyaguru, S.; Pogam, S. L.; Najera, I.;
Benkestock, K.; Zhou, X.-X.; Kaiser, A. C.; Maag, H.; Cammack, N.;
Martin, J. A.; Swallow, S.; Johansson, N. G.; Klumpp, K.; Smith, M.
The design, synthesis, and antiviral activity of monofluoro and
difluoro analogues of 4’-azidocytidine against hepatitis C virus
replication: The discovery of 4’-azido-2’-deoxy-2’-fluorocytidine and4’-azido-2’-dideoxy-2’,2’-difluorocytidine. J. Med. Chem. 2009, 52,
2971−2978.
(11) Wang, Q.; Hu, W.; Wang, S.; Pan, Z.; Tao, L.; Guo, X.; Qian,
K.; Chen, C. H.; Lee, K. H.; Chang, J. Synthesis of new 2’-deoxy-2’-
fluoro-4’-azido nucleoside analogues as potent anti-HIV agents. Eur. J.
Med. Chem. 2011, 46, 4178−83.
(12) Deng, W.; Jiang, S.; Yu, T.; Zhai, D. Synthesis method of
azvudine. China Patent CN 111892636, 2020.
Medical uses
In July 2021, azvudine became conditionally approved in China for the following indication: “to treat high-viral-load cases of HIV-1, in combination with a nucleoside reverse-transcriptase inhibitor and a non-nucleoside reverse-transcriptase inhibitor”. The approval text describes it as a dual reverse transcriptase and Vif inhibitor.[10]
In July 2022, azvudine received emergency conditional approval for COVID-19 in adults.[11] It is believed to work by inhibiting the RNA-dependent RNA polymerase (RdRp) enzyme in the SARS-CoV-2 virus.[12][13]
Adverse effects
According to the manufacturer, phase II trials of azvudine in combination with doravirine and tenofovir disoproxil fumarate in HIV patients found an adverse effect profile similar to, but milder, than lamivudine combined with the two drugs. Very common (> 10%) side effects include dizziness, elevated liver enzymes, vomiting, and elevated alkaline phosphatase. Common (> 1%) side effects include nausea, elevated blood lipids, fever, insomnia, tiredness, and diarrhea. Detailed numbers are provided by Genuine in the slides and the medication package insert.[14][15] A boxed warning is present at the beginning of the Chinese package insert, describing a risk of “decrease in absolute neutrophil count, increase in total bilirubin, increase in glutathione aminotransferase, and increase in blood glucose”.[15]
The small (n=10) open-label pilot study for azvudine used alone in COVID reported no adverse events.[16]
Non-human models
Azvudine is found to be mutagenic in in vitro in the Ames test, CHL test, and in vitro in the mice micronucleus test.[17]
Azvudine is toxic to the reproductive system of rats and rabbit. The minimum reproductive NOAEL found for males is 5.0 mg/kg/d and for females 0.5 mg/kg/d. It is excreted in rat breast milk; the NOAEL for rat pups is 1.5 mg/kg/d.[17]
Azvudine is mainly toxic to the immune system, bone marrow, and digestive system of model animals. The chronic NOAELs are 0.5 mg/kg/d (rat, 3 months), 0.3 mg/kg/d (rat, 26 weeks), and 0.1 mg/kg/d (beagle dog, 1 month and 39 weeks).[17] For comparison, the chronic human dose for HIV treatment is 0.05 mg/kg/d, using the reference 3 mg dose and an average Chinese body mass of 59.5 kg (2014).
History
Azvudine was first found in literature in a patent filed by Chang Jun-biao of Zhengzhou University.[8] It received its current name in 2009, when researchers at Roche independently discovered it as a Hep C RNA polymerase inhibitor in vitro.[4] In the following years, Chinese scientists tested it in vitro for a number of targets, most importantly HBV (human and duck) and HTLV-1, two viruses with a reverse transcriptase.[18][19][20]
It was first proposed as an HIV treatment in 2011, when in vitro tests by the Chang group provided positive results.[21] In 2014, its oral pharmokinetics in rats was elucidated.[1] A phase II study (NCT04109183) was finished in March 2019 by Genuine Biotech. In August 2020, the Chang group found that the substance inhibits vif in vitro.[22] In the same month, China’s drug regulator (NMPA) decided to fast-track the approval process, labelling it a first-in-class medication.[14] In July 2021, NMPA granted conditional approval for HIV-1.[7] It was included in the 2021 HIV treatment recommendnation by the Chinese Medical Association and Chinese CDC, published October that year.[14] Curiously, no full results of the trial have been made available for this study in any journal detailing the experiment design as of December 2022.[23] Parts of the results are shown on the drug monograph as well as a 2022 slides deck produced by Genuine for the NHSA available on the latter’s website.[14]
Azvudine was found to inhibit some coronaviruses in vitro around 2020, leading to an interest in its use in COVID. An open-label pilot study on mild and moderate cases was performed in 2020, with mildly positive results.[16] A phase III trial was performed in 2022 in China. In July 2022, China’s drug regulator granted conditional approval for it to be used to treat COVID-19, following a local phase III trial.[6] Initially, no detailed description of the said trial was published in any journals, but state media quoted some numbers from the developer: “40% clinical improvement in 7 days by FNC group, compared to 11% in control”.[7] It is unclear how such “improvement” is defined.
Four phase III clinical trials investigated azvudine’s efficacy and safety in adults with mild-to-moderate COVID-19. The findings indicate that azvudine may reduce the time to eliminate detectable levels of virus (viral load) and improve symptoms faster than standard treatment. In trials, it was reported to be safe with few side effects. However, some studies produced inconsistent results in terms of symptom improvement and severe illness prevention. Additionally, the studies tended to use a smaller number of participants than other major COVID-19 drug trials.[12][13]
Society and culture
Genuine owns two different tradenames for this medication: 双新艾克 (literally “dual new AIDS inhibitor”) for HIV use[14] and 捷倍安 (literally “fast extra safe”) for COVID use.[7] No generics are available.
Geniune holds one patent related to the drug: the original 2007 patent on the entire class of 2′-fluorine-4′-substituted nucleotides, purchased from Zhengzhou University.[8] Two other Chinese patents on synthesizing the drug are found on Google Patents, but the owners do not appear to be connected to Geniune.[24] Roche held one 2002 patent, CNA028118480A (CN1516590A), over the broader class of 4′-substituted nucleotides. The patent was voided in 2019 after Riboscience, its new holder, stopped paying fees.[25]
References
- Peng Y, Cheng T, Dong L, Zhang Y, Chen X, Jiang J, et al. (September 2014). “Quantification of 2′-deoxy-2′-β-fluoro-4′-azidocytidine in rat and dog plasma using liquid chromatography-quadrupole time-of-flight and liquid chromatography-triple quadrupole mass spectrometry: Application to bioavailability and pharmacokinetic studies”. Journal of Pharmaceutical and Biomedical Analysis. 98: 379–386. doi:10.1016/j.jpba.2014.06.019. PMID 24999865.
- Liu Y, Liu B, Zhang Y, Peng Y, Huang C, Wang N, et al. (July 2017). “Intestinal absorption mechanisms of 2′-deoxy-2′-β-fluoro-4′-azidocytidine, a cytidine analog for AIDS treatment, and its interaction with P-glycoprotein, multidrug resistance-associated protein 2 and breast cancer resistance protein”. European Journal of Pharmaceutical Sciences. 105: 150–158. doi:10.1016/j.ejps.2017.05.009. PMID 28487144. S2CID 4252337.
- Wang RR, Yang QH, Luo RH, Peng YM, Dai SX, Zhang XJ, et al. (2014). “Azvudine, a novel nucleoside reverse transcriptase inhibitor showed good drug combination features and better inhibition on drug-resistant strains than lamivudine in vitro”. PLOS ONE. 9 (8): e105617. Bibcode:2014PLoSO…9j5617W. doi:10.1371/journal.pone.0105617. PMC 4140803. PMID 25144636.
- Smith DB, Kalayanov G, Sund C, Winqvist A, Maltseva T, Leveque VJ, et al. (May 2009). “The design, synthesis, and antiviral activity of monofluoro and difluoro analogues of 4′-azidocytidine against hepatitis C virus replication: the discovery of 4′-azido-2′-deoxy-2′-fluorocytidine and 4′-azido-2′-dideoxy-2′,2′-difluorocytidine”. Journal of Medicinal Chemistry. 52 (9): 2971–2978. doi:10.1021/jm801595c. PMID 19341305.
- Harrison C (April 2020). “Coronavirus puts drug repurposing on the fast track”. Nature Biotechnology. 38 (4): 379–381. doi:10.1038/d41587-020-00003-1. PMID 32205870.
- Ye Y (July 2022). “China approves first homegrown COVID antiviral”. Nature. doi:10.1038/d41586-022-02050-x. PMID 35883009. S2CID 251104078.
- “首个国产抗新冠口服药附条件获批上市” [First domestic oral anti-Covid drug conditionally approved]. 新华网. 证券日报. 2022-07-26. Archived from the original on 2022-08-09. Retrieved 2022-07-26.
- Chang J, Bao X, Wang Q, Guo X, Wang W, Qi X. Preparation of 2′-fluoro-4′-substituted nucleoside analogs as antiviral agents. 20070807. Chinese Patent Application No: CN 2007-10137548. Chinese Patent No: CN 101177442A, 20080514.
- “新冠口服药阿兹夫定片线上开售, 每瓶售价350元” [Oral COVID drug Azvudine tablet available online at 350 yuan/bottle]. Xinmin Evening News. 19 November 2022. Retrieved 28 December 2022 – via Beijing Daily (repost).
- “国家药监局附条件批准阿兹夫定片上市” [NMPA conditionally approvals azvudine tablets]. http://www.nmpa.gov.cn (in Chinese). 2021-07-21.
- “国家药监局应急附条件批准河南真实生物科技有限公司阿兹夫定片增加新冠肺炎治疗适应症注册申请” [NMPA grants emergency conditional approval for additional indication registration of azvudine tablets (Hebei Genuine Biotech Co., Ltd.)]. http://www.nmpa.gov.cn. 2022-07-25.
- Zhu KW (2023). “Efficacy and safety evaluation of Azvudine in the prospective treatment of COVID-19 based on four phase III clinical trials”. Frontiers in Pharmacology. 14: 1228548. doi:10.3389/fphar.2023.1228548. PMC 10484631. PMID 37693894.
- Wang Y, Xie H, Wang L, Fan J, Zhang Y, Pan S, Zhou W, Chen Q, Liu X, Wu A, Zhang H, Wang J, Tian X (February 2024). “Effectiveness of azvudine in reducing mortality of COVID-19 patients: a systematic review and meta-analysis”. Virology Journal. 21 (1): 46. doi:10.1186/s12985-024-02316-y. PMC 10893615. PMID 38395970.
- Genuine Biotech (July 11, 2022). “阿兹夫定片(双新艾克)” [Azvudine Tablets (Shuāngxīnàikè)]. NHSA.gov.cn. Archived from the original on 2022-09-06.
- “阿兹夫定片说明书” [Azvudine Tablets, Monograph] (PDF). WUXU DATA. Retrieved 2023-01-03.
- Ren Z, Luo H, Yu Z, Song J, Liang L, Wang L, et al. (October 2020). “A Randomized, Open-Label, Controlled Clinical Trial of Azvudine Tablets in the Treatment of Mild and Common COVID-19, a Pilot Study”. Advanced Science. 7 (19): e2001435. doi:10.1002/advs.202001435. PMC 7404576. PMID 35403380.
- Drug Review Center (China) (June 30, 2022). “阿兹夫定片 (CXHS2000016-17) 申请上市技术审评报告” [Azvudine tabs (CHXS2000016-17) request for marketing technical review report] (PDF). WUXU DATA. Retrieved 2023-01-03.
- Wang Q, Liu X, Wang Q, Zhang Y, Jiang J, Guo X, et al. (April 2011). “FNC, a novel nucleoside analogue inhibits cell proliferation and tumor growth in a variety of human cancer cells”. Biochemical Pharmacology. 81 (7): 848–855. doi:10.1016/j.bcp.2011.01.001. PMID 21219886.
- Zheng L, Wang Q, Yang X, Guo X, Chen L, Tao L, et al. (2012). “Antiviral activity of FNC, 2′-deoxy-2′-β-fluoro-4′-azidocytidine, against human and duck HBV replication”. Antiviral Therapy. 17 (4): 679–687. doi:10.3851/IMP2094. PMID 22452880. S2CID 25576607.
- Zhou Y, Zhang Y, Yang X, Zhao J, Zheng L, Sun C, et al. (2012). “Novel nucleoside analogue FNC is effective against both wild-type and lamivudine-resistant HBV clinical isolates”. Antiviral Therapy. 17 (8): 1593–1599. doi:10.3851/IMP2292. PMID 22910281. S2CID 29382902.
- Wang Q, Hu W, Wang S, Pan Z, Tao L, Guo X, et al. (September 2011). “Synthesis of new 2′-deoxy-2′-fluoro-4′-azido nucleoside analogues as potent anti-HIV agents”. European Journal of Medicinal Chemistry. 46 (9): 4178–4183. doi:10.1016/j.ejmech.2011.06.020. PMC 3164908. PMID 21745701.
- Sun L, Peng Y, Yu W, Zhang Y, Liang L, Song C, et al. (August 2020). “Mechanistic Insight into Antiretroviral Potency of 2′-Deoxy-2′-β-fluoro-4′-azidocytidine (FNC) with a Long-Lasting Effect on HIV-1 Prevention”. Journal of Medicinal Chemistry. 63 (15): 8554–8566. doi:10.1021/acs.jmedchem.0c00940. PMID 32678592. S2CID 220631451.
- Li G, Wang Y, De Clercq E (April 2022). “Approved HIV reverse transcriptase inhibitors in the past decade”. Acta Pharmaceutica Sinica. B. 12 (4): 1567–1590. doi:10.1016/j.apsb.2021.11.009. PMC 9279714. PMID 35847492.
- Google Patents Search, “阿兹夫定” (with quotes), CN114149475A, CN111892636A.
- Guokr.com (10 August 2022). “真实生物的真实面目”. Huxiu.com. Retrieved 30 December 2022.
Further reading
- Zhu KW (2023). “Efficacy and safety evaluation of Azvudine in the prospective treatment of COVID-19 based on four phase III clinical trials”. Frontiers in Pharmacology. 14: 1228548. doi:10.3389/fphar.2023.1228548. PMC 10484631. PMID 37693894.
| Clinical data | |
|---|---|
| Trade names | 捷倍安, 双新艾克 |
| Other names | 2′-Deoxy-2′-β-fluoro-4′-azidocytidine (FNC), RO-0622 |
| Legal status | |
| Legal status | US: Investigational drugCN: Conditional use Rx |
| Pharmacokinetic data | |
| Bioavailability | 83% (rat, dog)[1] |
| Metabolism | liver (CYP3A)[2] |
| Elimination half-life | 4 hours (dog)[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1011529-10-4 |
| PubChem CID | 24769759 |
| DrugBank | DB16407 |
| ChemSpider | 24717759 |
| UNII | IJ2XP0ID0K |
| ChEMBL | ChEMBL519846 |
| Chemical and physical data | |
| Formula | C9H11FN6O4 |
| Molar mass | 286.223 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////AZVUDINE, Genuine Biotech, APPROVALS 2022, CHINA 2022, FNC, HY 19314, RO 0622, RO-0622, SB17040, IJ2XP0ID0K, DTXSID901027757
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Henagliflozin


Henagliflozin, SHR-3824 ,
CAS 1623804-44-3
C22-H24-Cl-F-O7, 454.8756
PHASE 2 for the treatment of type 2 diabetes
China 20222, approvals 2022
HengRui (Originator)
| Jiangsu Hengrui Medicine Co Ltd |
UNII-21P2M98388; 21P2M98388; Henagliflozin; SHR3824; SHR-3824;

- HENAGLIFLOZIN PROLINE
- 4IO819SW6M
- 570.0 g/mol
- C27H33ClFNO9
- (1R,2S,3S,4R,5R)-5-[4-chloro-3-[(4-ethoxy-3-fluorophenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol;(2R)-pyrrolidine-2-carboxylic acid
In April 2016, Jiangsu Hengrui Medicine is developing henagliflozin (phase 2 clinical trial), a sodium-glucose cotransporter-2 (SGLT-2) inhibitor, for treating type 2 diabetes.
SGLT1 and SGLT2 inhibitors, useful for treating eg diabetes.
Henagliflozin proline is in phase II clinical trials by Jiangsu Hengrui (江苏恒瑞) for the treatment of type 2 diabetes.
1,6-dehydrated-1-C{4-chloro-3-[(3-fluoro-4-ethoxyphenyl)methyl]phenyl}-5-C-(hydroxymethyl)-β-L-idopyranose L-proline
(1 ^ 2345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) 6,8 – alcohol dioxide
(1R,2S,3S,4R,5R)-5-[4-chloro-3-[(4-ethoxy-3-fluorophenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
Henagliflozin is a pharmaceutical drug for the treatment of type 2 diabetes.[1] In China, it is approved for adult patients with type 2 diabetes to improve the glycemic control.[2][3]
Henagliflozin, like other drugs of the gliflozin class, inhibits the transporter protein sodium/glucose cotransporter 2 (SGLT2) which leads to a reduction in blood glucose levels.[4]
Shanghai Hengrui Pharmaceutical Co., Ltd., 上海恒瑞医药有限公司, Jiangsu Hengrui Medicine Co., Ltd., 江苏恒瑞医药股份有限公司, Less «
- 01 May 2015 Jiangsu HengRui Medicine Co. initiates enrolment in a phase I drug interaction trial in volunteers in China (NCT02500485)
- 12 Feb 2015 Jiangsu HengRui Medicine plans a phase I trial for Type-2 diabetes mellitus in China (NCT02366377)
- 01 Feb 2015 Jiangsu HengRui Medicine initiates enrolment in a phase I trial for Type-2 diabetes mellitus in China (NCT02366351)
Henagliflozin is a novel sodium-glucose transporter 2 inhibitor and presents a complementary therapy to metformin for patients with T2DM due to its insulin-independent mechanism of action. This study evaluated the potential pharmacokinetic drug-drug interaction between henagliflozin and metformin in healthy Chinese male subjects. 2. In open-label, single-center, single-arm, two-period, three-treatment self-control study, 12 subjects received 25 mg henagliflozin, 1000 mg metformin or the combination. Lack of PK interaction was defined as the ratio of geometric means and 90% confidence interval (CI) for combination: monotherapy being within the range of 0.80-1.25. 3. Co-administration of henagliflozin with metformin had no effect on henagliflozin area under the plasma concentration-time curve (AUC0-24) (GRM: 1.08; CI: 1.05, 1.10) and peak plasma concentration (Cmax) (GRM: 0.99; CI: 0.92, 1.07). Reciprocally, co-administration of metformin with henagliflozin had no clinically significant on metformin AUC0-24 (GRM: 1.09, CI: 1.02, 1.16) although there was an 11% increase in metformin Cmax (GRM 1.12; CI 1.02, 1.23). All monotherapies and combination therapy were well tolerated. 4. Henagliflozin can be co-administered with metformin without dose adjustment of either drug.
PATENT
PATENT
WO2012019496
https://www.google.com/patents/WO2012019496A1?cl=en
Example 4
(1 ^ 2345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) 6,8 – alcohol dioxide


first step
1-ethoxy-2-fluoro – benzene
A mixture of 2-fluoro-phenol 4a (6.7 g, 60 mmol) was dissolved in 66 mL of acetone, was added iodoethane (6.3 mL,
78 mmol) and potassium carbonate (12.4 g, 90 mmol), at reflux in an oil bath for 5 hours. The reaction solution was concentrated under reduced pressure, was added 100 mL of ethyl acetate and 60 mL of water, separated, the aqueous phase was extracted with ethyl acetate (30 mLx2), the organic phases combined, dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, to give the title product 1-ethoxy-2-fluoro – benzene 4b (6.9 g, red oil). yield: 82.1%.
MS m / z (ESI): 280.2 [2M + 1]
The second step
(5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanone A mixture of 5-bromo-2-chloro – benzoyl chloride 2a (12.4 g, 48.8 mmol) was dissolved a 100 mL of dichloromethane was added 1-ethoxy-2-fluoro – benzene 4b (6.84 g, 48.8 mmol), cooled to 0 ° C, was added portionwise aluminum (5.86 g, 44 mmol) chloride, 16 h. Was added dropwise under ice-cooling to the reaction mixture 20 mL of 2 M HCl solution, separated, the aqueous phase was extracted with 30 mL of dichloromethane, and the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title The product (5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanone 4c (12.7 g, yellow solid), yield: 72.6%.
MS m / z (ESI): 358.9 [M + l] Step
(5 – bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanol (5-Bromo-2-chloro – phenyl) – (4-ethoxy -3 – fluoro – phenyl) -methanone 4c (12.7 g, 35.5 mmol) was dissolved in methanol and a 100 mL of tetrahydrofuran (ν: ν = 1: 1) mixed solvent, under an ice bath was added portionwise sodium borohydride (2.68 g, 70 mmol), and reacted at room temperature for 30 minutes. Add 15 mL of acetone, the reaction solution was concentrated under reduced pressure, 150 mL of ethyl acetate was added to dissolve the residue, washed with saturated sodium chloride solution (50 mLx2). The combined organic phase was dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure The filtrate, to give the title product (5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanol 4d (12.7 g, orange oil), was used directly without isolation next reaction.
the fourth step
4 – [(5-bromo-2-chloro-phenyl) – methyl] Small-ethoxy-2-fluoro – benzene (5-bromo-2-chloro – phenyl) – (4-ethoxy -3 – fluoro – phenyl) methanol 4d (12.7 g, 35.3 mmol) was dissolved in a 100 mL of dichloromethane was added triethylsilane (16.9 mL, 106 mmol), was added dropwise boron trifluoride etherate (8.95 mL, 70.6 mmol ), for 3 hours. Was added 50 mL of saturated sodium bicarbonate solution, separated, the aqueous phase was extracted with ethyl acetate (100 mLx2), the organic phases combined, dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography to elute B surfactant system resulting residue was purified to give the title product 4 – [(5-bromo-2-chloro – phenyl) methyl] -1-ethoxy-2-fluoro – benzene 4e (10 g, as a pale yellow oil ) yield: 82.4%.
1H NMR (400 MHz, CDC1 3 ): δ 7.33-7.27 (m, 3H), 6.95-6.90 (m, 3H), 4.14 (q, 2H), 4.01 (s, 2H), 1.49 (t, 3H)
the fifth step
(2 3R, 4S, 5 ^ 6R) -2- [4- chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) – 2-methoxy – tetrahydro-pyran-3,4,5-triol
4 – [(5-bromo-2-chloro – phenyl) methyl] -1-ethoxy-2-fluoro – benzene 4e (7.36 g, 21.4 mmol) was dissolved in 30 mL of tetrahydrofuran, cooled to -78 ° C, was added dropwise a solution of n-butyllithium in hexane (10.27 mL, 25.7 mmol), at -78 ° C to react 1 hour, a solution of 20 mL (3R, 4S, 5R, 6R) -3,4,5 – tris (trimethylsilyloxy) -6- (trimethylsilyloxy) tetrahydropyran-2-one 2f (llg, 23.6 mmol) in tetrahydrofuran at -78 ° C under reaction 2 h, 2.8 mL of methanesulfonic acid and 71 mL of methanol, the reaction at room temperature for 16 hours. Was added 100 mL of saturated sodium carbonate solution, the reaction solution was concentrated under reduced pressure, to the residue was added 50 mL of saturated sodium chloride solution, extracted with ethyl acetate (100 mLx3), organic phases were combined, dried over anhydrous magnesium sulfate, filtered, The filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product (2 3R, 4S, 5 6R) -2- [4- chloro-3 – [(4-ethoxyphenyl 3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) -2-methoxy – tetrahydro-pyran-3,4,5-triol 4f (5.7 g, white solid ) yield: 58.3%.
1H NMR (400 MHz, CD 3 OD): δ 7.56 (s, 1H), 7.48 (dd, 1H), 7.37 (dd, 1H), 6.95-6.87 (m, 3H), 4.08-4.07 (m, 4H) , 3.91 (m, 1H), 3.93-3.73 (m, 2H), 3.56-3.53 (m, 1H), 3.45-3.43 (m, 1H), 3.30 (s, 2H), 3.08 (s, 3H), 1.35 (t, 3H)
The sixth step
(2 3R, 4S, 5 6R) -6- [(tert-butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro – phenyl) methyl] phenyl] -2-methoxy – tetrahydro-pyran-3,4,5-triol the (2 3R, 4S, 5 6R) -2- [4- chloro-3- [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) -2-methoxy – 4f tetrahydropyran-3,4,5-triol (5.7 g, 12.5 mmol) was dissolved in 50 mL of pyridine, followed by adding tert-butyldimethylsilyl chloride (2.26 g, 15 mmol) and 4-dimethylaminopyridine (305 mg, 2.5 mmol), for 16 hours. The reaction solution was concentrated under reduced pressure, was added 200 mL of ethyl acetate, washed with a saturated copper sulfate solution (50 mLx3). The combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (2 3R, 4S, 5 6R) -6- [(tert-butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2-methoxy – tetrahydro-pyran-3,4,5-triol 4g (7.14 g, colorless oil), without isolation directly used for the next reaction.
Seventh Step
[[(2R, 3R, 4S, 5R, 6 ^ -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl yl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethyl-silane (2 3R, 4S, 5 6R) -6- [(tert butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2-methoxy yl – tetrahydro-pyran-3,4,5-triol 4g (7.14 g, 12.5 mmol) was dissolved in 100 mL N, N- dimethylformamide was added 60% sodium hydride under ice-cooling (2.5 g , 62.5 mmol), and reacted at room temperature for 40 minutes completed the opening force, was added benzyl bromide (7.5 mL, 62.5 mmol), reaction of 16 hours. 20 mL of methanol, the reaction solution was concentrated under reduced pressure, was added 200 mL of ethyl acetate and 50 mL of water to dissolve the residue, separated, the aqueous phase was extracted with ethyl acetate (50 mL), the organic phase was washed with water (50 mL), washed with saturated sodium chloride solution (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate , filtered, and the filtrate was concentrated under reduced pressure to give the title product [[(2R, 3R, 4S, 5R, 6 ^ -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4- ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethylsilane 4h (10.5 g , yellow oil) yield: 99.8%.
Step Eight
[(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol
The [[(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl yl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethylsilane 4h (10.52 g, 12.5 mmol) was dissolved in 50 mL of methanol dropwise add acetyl chloride CO.13 mL, 1.9 mmol), for 1 hour. The reaction solution was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems B resultant residue was purified to give the title product [(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy–6 – [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol 4i (7.6 g , yellow oil yield: 83.6%.
Step Nine
(2 ^ 3456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 6-methoxy – tetrahydropyran-2-carbaldehyde
Oxalyl chloride (1.17 mL, 13.6 mmol) was dissolved in 20 mL of dichloromethane, cooled to -78 ° C, were added dropwise 20 mL of dimethyl sulfoxide (1.56 mL, 21.9 mmol) in methylene chloride and 50 mL [(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol 4i (7.6 g, 10.45 mmol) in methylene chloride, and reacted at -78 ° C for 30 min, triethylamine (7.25 mL, 52.3 mmol), 2 hours at room temperature was added 50 mL 1 M HCl solution, separated, the organic phase was washed with saturated sodium chloride solution (50 mL x 2), the aqueous phase was extracted with dichloromethane (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (2 ^ 3456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4 – ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-carbaldehyde 4j (7.58 g, colorless oil), was used directly without isolation next reaction.
The tenth step
(2S, 3 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl ] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-carbaldehyde
The (23456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 6-methoxy – tetrahydropyran-2-carbaldehyde 4j (7.6 g, 10.45 mmol) was dissolved in 80 mL 1,4- dioxane, followed by adding 15.8 mL 37% aqueous formaldehyde and sodium hydroxide solution (31.35 mL, 31.35 mmol), reacted at 70 ° C for 16 h. Add 50 mL of saturated sodium chloride solution, extracted with ethyl acetate (50 mLx4), the organic phase was washed with saturated sodium bicarbonate solution (50 mL), washed with saturated sodium chloride solution (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (23,456 benzyloxy-3,4,5-tris – 6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran – 2- formaldehyde 4k (7.9g, as a colorless oil), without isolation directly used for the next reaction.
Step Eleven
[(3 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-yl] methanol
The (23456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-carbaldehyde 4k (7.9 g, 10.45 mmol) was dissolved in 50 mL of tetrahydrofuran and methanol (v: v = 2: 3) mixed solvent , was added sodium borohydride (794 mg, 20.9 mmol), for 30 minutes. Add a small amount of acetone, the reaction solution was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product, 5R, 6 -3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2- (hydroxymethyl ) -6-methoxy – tetrahydropyran-2-yl] methanol 4m (l.ll g, colorless oil). yield: 14.1%.
Step Twelve
[(12345 ^ -2,3,4-tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol
The [(3S, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] benzene yl] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-yl] methanol 4m (l.ll g, 1.46 mmol) was dissolved in 20 mL of dichloromethane, cooled to -10 ° C, was added trifluoroacetic acid (0.23 mL, 3 mmol), and reacted at room temperature for 2 hours. 20 mL of saturated sodium bicarbonate solution, separated, the aqueous phase was extracted with dichloromethane (20 mL> <2), and the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems B resultant residue was purified to give the title product [(1 2 3 4R, 5 -2,3,4- tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol 4nC830 mg, colorless oil). yield: 78.3%.
MS m / z (ESI): 742.3 [M + 18]
Thirteenth Step
(12345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) -6,8 dioxa-bicyclo [3.2.1] octane-2,3,4-triol
The [(1 2 3 4R, 5S) -2,3,4- tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] benzene yl] -6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol 4n (830 mg, 1.14 mmol) was dissolved in 20 mL of tetrahydrofuran and methanol (v: v = l: l) the a mixed solvent of o-dichlorobenzene was added (1.3 mL, 1 1.4 mmol) and Pd / C (500 mg, 10%), purged with hydrogen three times, the reaction for 3 hours. The reaction solution was filtered, rinsed with a small amount of ethyl acetate, the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product (1S, 2 3S, 4R, 5 -5- [ 4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) -6,8-dioxa-bicyclo [3.2.1] octane-2,3,4-triol 4 (420 mg, white solid), yield: 81.0% MS m / z (ESI):. 472.2 [m + 18]
1H NMR (400 MHz, CD 3 OD): δ 7.47 (s, 1H), 7.42-7.35 (m, 2H), 6.95-6.87 (m, 3H), 4.16-4.14 (m, 1H), 4.06-4.02 ( m, 4H), 3.85-3.70 (m, 2H), 3.67-3.54 (m, 4H), 1.37 (t, 3H)
References
- Weng J, Zeng L, Zhang Y, Qu S, Wang X, Li P, et al. (August 2021). “Henagliflozin as add-on therapy to metformin in patients with type 2 diabetes inadequately controlled with metformin: A multicentre, randomized, double-blind, placebo-controlled, phase 3 trial”. Diabetes, Obesity & Metabolism. 23 (8): 1754–1764. doi:10.1111/dom.14389. PMID 33769656.
- Wang G (17 February 2022). “Monthly Report: New Drug Approvals in China, January 2022”. BaiPharm.
Henagliflozin Proline Tablets
- “Henagliflozin – Jiangsu HengRui Medicine”. AdisInsight. Springer Nature Switzerland AG.
- He X, Liu G, Chen X, Wang Y, Liu R, Wang C, et al. (July 2023). “Pharmacokinetic and Pharmacodynamic Interactions Between Henagliflozin, a Novel Selective SGLT-2 Inhibitor, and Warfarin in Healthy Chinese Subjects”. Clinical Therapeutics. 45 (7): 655–661. doi:10.1016/j.clinthera.2023.06.002. PMID 37451912.
 |
|
| Clinical data | |
|---|---|
| Trade names | Rui Qin; 瑞沁 |
| Other names | SHR3824; SHR-3824 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| Chemical and physical data | |
| Formula | C22H24ClFO7 |
| Molar mass | |
////////Henagliflozin, SHR-3824 , PHASE 2, type 2 diabetes, UNII-21P2M98388, 21P2M98388, SHR 3824, SHR3824, approvals 2022, china 2022, Henagliflozin proline
CCOc1ccc(cc1F)Cc2cc(ccc2Cl)[C@]34[C@@H]([C@H]([C@@H]([C@](O3)(CO4)CO)O)O)O
SYN
Synthesis 2024, 56, 906–943
Henagliflozin (12) (also known as SHR3824), developed by Lexicon Pharmaceuticals (Princeton, NJ, USA), is a potent and selective SGLT inhibitor administered orally. In 2013, the first synthetic route for the preparation of henagliflozin (12) was described and claimed by two pharmaceutical companies: Shanghai Hengrui Pharmaceutical Co., Ltd., and Jiangsu Hengrui Medicine Co., Ltd. Several other C-aryl-glucoside-type derivatives were prepared and registered in the United States under patent application number US8609622B2.67 Among these derivatives, the synthesis of henagliflozin (12) was carried out using a thirteen-step process, resulting in an overall yield of 3% (Schemes 40 and 41). The process consisted of the formation of the key intermediate 215 starting from commercially available 2-fluorophenol (211). In the first step, phenolic compound 211 was converted into 212 in 82% yield using ethyl bromide and po
tassium carbonate in acetone. The Friedel–Crafts reaction of acid chloride 26c′ using AlCl3 in DCM afforded intermediate 213 in 72% yield, which was further reduced to 214 using NaBH4 in a mixture of THF/MeOH. Without further isolation, the reduction of 214 was carried out using Et3SiH and BF3·Et2O in DCM to give 215 (Scheme 40). The intermediate 215 was taken forward for lithium halogen exchange using n-BuLi followed by addition of the lithiated compound to O-silyl-protected compound 22 at
low temperature to afford a lactol intermediate. The obtained lactol intermediate was protected using
MsOH/MeOH to give the desired product 216 in 58% yield. Under the above conditions, deprotection of the O-silylgroups of the C-glucoside 22 was also observed. Further, under basic conditions, the secondary hydroxy group of 216 was silyl protected using tert-butyldimethylsilyl chloride (TBSCl) and DMAP to afford compound 217, which was treated with NaH and BnBr to give benzylated compound
218 in excellent yield. In methanol solution, deprotection of the silyl protecting group of compound 218 using acetylchloride afforded 219. Swern oxidation of the hydroxy compound 219 in the presence of oxalyl chloride and DMSO gave intermediate 220, which was used for the next step without isolation. The crude compound 220 was treated with NaOH and 37% formaldehyde solution to afford 221.
Dihydroxy intermediate 222 was then obtained in low yield via reduction of the aldehyde group of compound 221 with sodium borohydride in THF/MeOH mixture. Next, treatment of 222 with trifluoroacetic acid gave compound 223. Debenzylation of compound 223 was carried out by Pd/C
catalytic hydrogenation to afford the final product henaglifozin (12) (Scheme 41).
The highlight of the synthesis is the design of the route with minimal isolation stages and intermediates possessing unstable functional groups were subjected to subsequent transformations in situ. The drawbacks of the above synthetic process are the use of a protection and deprotection
strategy that led to low throughput and the final compound being obtained in low yield. Reduction of the aldehyde in 221 mediated by sodium borohydride resulted in a poor yield of product 222, and this procedure is not recommend ed for scale-up due to safety concerns. Additionally, the use
of palladium in the last step of the synthesis involves the risk of this toxic metal leaching into the final product. To address the issue with the discovery route, Yongjun and co-workers reported an alternative approach to obtain compound 12 (Scheme 42).68 The authors published the synthesis of henagliflozin proline (12a) starting from TMS protected D-glucolactone 22 and aglycone intermediate The diol 226 was obtained after carrying out a disproportionation reaction on the aldehyde using paraformaldehyde under strong alkaline conditions. Intramolecular etherification of diol 226 using 30% HCl gave henagliflozin
(12) in 95% yield, which was further treated with L-proline to give henagliflozin proline monohydrate 12a. The authors reported several advantages such as easy steps, cost-effective procedures, simple product purification and an overall method that was amenable for commercialization. This Addition of the aglycone intermediate 215 was carried out with 22 followed by mesylation of the OH group to provide 216 in 65% yield. Further, all the secondary hydroxy groups of intermediate 216 were selectively protected us ing TMSCl, imidazole and PPTS to give 224 in 95% yield. The free primary hydroxy group of 224 was oxidized using pyridine sulfur trioxide in triethylamine and DMSO to afford process involves 10 steps and gave an overall yield of 22% of henagliflozin proline (12a) (Schemes 40 and 42)
REF 67, 68
(67) Yang, F.; Tang, P. C.; Dong, Q.; Tu, W.; Fan, J.; Guan, D.; Shen, G.;Wang, Y.; Yuan, J.; Zhang, L. US8609622B2, 2013.
(68) Chun, K.; Peng, Z.; Qichao, L.; Bo, Z.; Zhen, W.; Guorong, Z.;Yongjun, T. CN 112375087A, 2020.
.

{kind=link}
{kind=link}