
Home » cancer (Page 15)
Category Archives: cancer
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Onion extract slows colon cancer growth just as effectively as chemo drug

(NaturalNews) Researchers have just discovered that flavonoids extracted from common onions slow the rate of colon cancer growth in mice just as effectively as a chemotherapy drug. And while the mice on chemo saw their LDL cholesterol go up (a possible side effect of the drug), the mice on onion extract actually saw their LDL levels drop.
Onion flavonoids slow colon tumor growth by 67% in vivo
Learn more: http://www.naturalnews.com/044318_onion_extract_colon_cancer_chemotherapy_drug.html##ixzz2wD3udzfF
http://www.naturalnews.com/044318_onion_extract_colon_cancer_chemotherapy_drug.html#

The US FDA has issued full approval for Israeli drugmaker Teva’s Synribo (omacetaxine mepesuccinate)高三尖杉酯碱 for chronic myeloid leukaemia (CML).


Omacetaxine mepesuccinate 高三尖杉酯碱
Alkaloid from Cephalotaxus harringtonia; FDA approved orphan drug status for Ceflatonin in the treatment of chronic myeloid leukemia due to being an inducer of apoptosis in myeloid cells and inhibitor of angiogenesis.
26833-87-4 CAS NO
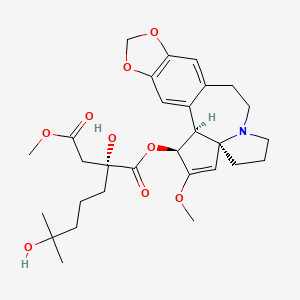
1-((1S,3aR,14bS)-2-Methoxy-1,5,6,8,9,14b-hexahydro-4H-cyclopenta(a)(1,3)dioxolo(4,5-h)pyrrolo(2,1-b)(3)benzazepin-1-yl) 4-methyl (2R)-2-hydroxy-2-(4-hydroxy-4-methylpentyl)butanedioate
1-((11bS,12S,14aR)-13-methoxy-2,3,5,6,11b,12-hexahydro-1H-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-d]cyclopenta[b]pyrrolo[1,2-a]azepin-12-yl) 4-methyl 2-hydroxy-2-(4-hydroxy-4-methylpentyl)succinate
Also known as: NSC-141633,
- BRN 5687925
- Ceflatonin
- CGX-635
- Homoharringtonine
- Myelostat
- NSC 141633
- Omacetaxine mepesuccinate
- Omapro
- Synribo
- UNII-6FG8041S5B
- 高三尖杉酯碱
CGX-635-14 (formulation), CGX-635, HHT, ZJ-C, Myelostat, Ceflatonin
USFDA on 26th October 2012 APPROVED
| Formula | C29H39NO9 |
|---|---|
| Mol. mass | 545.62 g/mol |
The US Food and Drug Administration has now issued full approval for Israeli drugmaker Teva’s Synribo (omacetaxine mepesuccinate) for chronic myeloid leukaemia (CML).
Synribo is indicated for adult patients with chronic phase (CP) or accelerated phase (AP) CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs).
Read more at: http://www.pharmatimes.com/Article/14-02-17/US_green_light_for_Teva_s_CML_drug_Synribo.aspx#ixzz2tdkbGFcw
Homoharringtonine is an angiogenesis-inhibiting and apoptosis-inducing alkaloid which was approved in October 2012 by the FDA for the treatment of adult patients with chronic or accelerated phase chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKI). In November 2012, the product was commercialized as Synribo(R) on the U.S. market by Teva.
The original developer, ChemGenex, selected homoharringtonine for the combination trials due to its complementary mechanism of action that can reduce Bcr-Abl protein expression associated with resistance to imatinib mesylate.
In 2004, the compound received orphan drug designation from the EMEA for the treatment of AML and CML. Orphan drug designation was granted by the FDA for the treatment of CML in 2006 and for the treatment of myelodysplasia in 2009. Fast track designation was assigned to homoharringtonine for CML in 2006. In 2009, the product was licensed to Hospira by ChemGenex Pharmaceuticals for development and marketing in Europe, the Middle East and parts of Africa.
Homoharringtonine, AKA HHT or omacetaxine mepesuccinate, is a cephalotaxine ester and protein synthesis inhibitor with established clinical activity as a single agent in hematological malignancies. Homoharringtonine is synthesized from cephalotaxine, which is an extract from the leaves of the plant, Cephalotaxus species. In October 2005, homoharringtonine received Orphan Drug designation from the EMEA for the treatment of chronic myeloid leukemia (CML). Then in March 2006, homoharringtonine received Orphan Drug status from the FDA for the treatment of CML. In November 2006, homoharringtonine, for the treatment of CML, was granted Fast Track designation by the FDA. Most recently, in October 2012, homoharringtonine was marketed under the brand name Synribo” and FDA approved for patients who are intolerant and/or resistant to two or more tyrosine kinase inhibitors used to treat accelerated or chronic phase CML
Omacetaxine mepesuccinate is administered subcutaneously and acts differently from TKIs. It may have a therapeutic advantage for patients who have failed TKIs. Omacetaxine is currently in global phase 2/3 clinical trials for CML and has been granted Orphan Drug designations by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMEA) as well as Fast Track status by the FDA. In vitro and animal model trails are promising and recent results showed that omacetaxine has potential to treat resistant leukemia mainly CML and ALL.
| PATENT | ||
|---|---|---|
|
3-25-2011
|
CEPHALOTAXUS ESTERS, METHODS OF SYNTHESIS, AND USES THEREOF
|
Tetrahedron Letters,Vo1.23,No.34,pp 3431-3434 … – Brock University
Omacetaxine mepesuccinate (INN, trade name Synribo) is a semi-synthetic analogue of an alkaloid from Cephalotaxus harringtonia that is indicated for treatment of chronic myelogenous leukemia (CML). It was approved by the US FDA in October 2012 for the treatment of adult patients with CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs).[1]
Omacetaxine mepesuccinate is a semisynthetic derivative of the cytotoxic plant alkaloid homoharringtonine isolated from the evergreen tree Cephalotaxus with potential antineoplastic activity. Omacetaxine mepesuccinate binds to the 80S ribosome in eukaryotic cells and inhibits protein synthesis by interfering with chain elongation. This agent also induces differentiation and apoptosis in some cancer cell types. Omacetaxine mepesuccinate (INN, or homoharringtonine, trade name Synribo) is an alkaloid from Cephalotaxus harringtonia that is indicated for treatment of Chronic Myelogenous Leukemia. It was approved by the USFDA on 26th October 2012 for the treatment of adult patients with chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs)
Omacetaxine is indicated for use as a treatment for patients with chronic myeloid leukaemia who are intolerant of tyrosine kinase inhibitors.[2][3]
In June 2009, results of a long-term open label Phase II study were published, which investigated the use of omacetaxine infusions in CML patients. After twelve months of treatment, about one third of patients showed a cytogenetic response.[4] A study in patients who had failed imatinib and who had the drug resistant T315I mutation achieved cytogenetic response in 28% of patients and haematological response in 80% of patients, according to preliminary data.[5]
Phase I studies including a small number of patients have shown benefit in treating myelodysplastic syndrome (MDS, 25 patients)[6] and acute myelogenous leukaemia (AML, 76 patients).[7] Patients with solid tumors did not benefit from omacetaxine.[8]
Omacetaxine is a protein translation inhibitor. It inhibits protein translation by preventing the initial elongation step of protein synthesis. It interacts with the ribosomal A-site and prevents the correct positioning of amino acid side chains of incoming aminoacyl-tRNAs. Omacetaxine acts only on the initial step of protein translation and does not inhibit protein synthesis from mRNAs that have already commenced translation.[9]
Omacetaxine mepesuccinate
SYNRIBO contains the active ingredient omacetaxine mepesuccinate, a cephalotaxine ester. It is a protein synthesis inhibitor. Omacetaxine mepesuccinate is prepared by a semi-synthetic process from cephalotaxine, an extract from the leaves of Cephalotaxus sp. The chemical name of omacetaxine mepesuccinate is cephalotaxine, 4-methyl (2R)-hydroxyl-2-(4-hydroxyl-4-methylpentyl) butanedioate (ester).
Omacetaxine mepesuccinate has the following chemical structure:
 |
The molecular formula is C29H39NO9 with a molecular weight of 545.6 g/mol. SYNRIBO for injection is a sterile, preservative-free, white to off-white, lyophilized powder in a single-use vial. Each vial contains 3.5 mg omacetaxine mepesuccinate and mannitol.
SYNRIBO is intended for subcutaneous administration after reconstitution with 1.0 mL of 0.9% Sodium Chloride Injection, USP. The pH of the reconstituted solution is between 5.5 and 7.0.
…………………………………..
INTRODUCTION
Harringtonines 3 are particular cephalotaxanes formed by attachement of a branched hydroxyacyloxy side-chain at the 3-position of various cephalotaxines moieties. Harringtoriines are natural esters of cephalotaxines exhibiting generally a strong cytotoxic activity. However the lost only one atom of this minimal structure lead to a dramatic lost of activity (see below). Some example of harringtonines are harringtonine
3a, homoharringtonine 3b, drupangtonine 3c, anhydroharringtonine 3d and neoharringtonine 3e.
SCHEME 1 DEFINITION NOMENCLATURE AND NUMBERING OF CEPHALOTAXANES


Examples of harringtonines

Examples of cephalotaxines

Harringtonine 3a (n = 2) Anhydroharringtonine 3d Homoharringtonine 3b (n = 3)

(-)-Cephalotaxine 2a


Drupacine 2b Drupangtonine 3c Neoharringtonine 3e (n = 2)
…………………………………
The term “cephalotaxanes” refers to compounds or salts thereof which have a basic skeleton of formula

where p is equal to 1 or 2 (it being possible for the two units to be identical or different and linked via a single bond or an oxygen atom), which can contain various oxygenated substituents (aliphatic or aromatic ethers, free or esterified alcohols, substituted or free enols and/or phenols, bridged ethers, and more generally any substituent usually encountered in the natural state on compounds of this type).
Harringtonines are alkaloids which are of high interest in anticancer chemotherapy, in particular on certain haematosarcomas which are multi-resistant to the existing therapies. The selectivity of harringtonines, which is based on a novel mechanism of action relating to protein synthesis, is such that this series is favoured with a great future in anticancer therapy.
Several literature compilations give a seemingly exhaustive review of all of the knowledge relating to cephalotaxanes, these compilations being, chronologically: [C. R. Smith, Jr, R. G. Powell and K. L. Mikolajczack, Cancer Treat. Rep., Vol. 60, 1157 (1976); C. R. Smith, Jr, L. Kenneth, K. L. Mikolajczack and R. G. Powell in “Anticancer Agent Based on Natural Product Model”, 391 (1980); Liang Huang and Zhi Xue in “The Alkaloids”, Vol. XXIII (A. Brossi Ed.), 157 (1984); M. Suffness and G. A. Cordell in “The Alkaloids, Chemistry and Pharmacology” (A. Brossi Ed.), Vol. 25, 57-69, 295-298 (1’987); P. J. O’Dwyer, S. A. King, D. F. Hoth, M. Suffness and B. Leyland-Jones, Journal of Clinical Oncology, 1563 (1986); T. Hudlicky, L. D. Kwart and J. W. Reed, in “Alkaloid: Chemical and Biological Perspectives” (S. W. Pelletier Ed.), Vol. 5, 639 (1987); M. A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, 199 (1998)].
Antiparasitic activities, in particular on the haematozoon of malaria, have also been recognized [J. M. Whaun and N. D. Brown, Ann Trop. Med. Par., Vol. 84, 229 (1990)].
Homo-harringtonine (HHT), the most active member of the series, is active at and above daily doses of 2.5 mg/m2 of body area per 24 hours, i.e., as a guide, at doses twenty times lower than that for Taxol. HHT has already undergone fourteen phase I and II clinical trials and it is the only known product capable of a 70% reinduction of full haematological remissions in patients suffering from chronic myeloid leukaemias that have become resistant to alpha-interferon [S. O’Brien, H. Kantarjian, M. Keating, M. Beran, C. Koler, L. E. Robertson, J. Hester, M. Rios, M. Andreeff and M. Talpaz, Blood, 332 (1995); Leukemia Insights, Vol. 3, No. 1 (1998)].
Harringtonines were extracted over 35 years ago from an exclusively Asiatic cephalotaxacea known as Cephalotaxus harringtonia, following the programme of research into novel anticancer agents in the plant kingdom developed by the National Cancer Institute. In fact, the Cephalotaxus alkaloids consist essentially (at least 50%) of cephalotaxine, a biosynthetic precursor of the harringtonines, the latter individually representing only a few percent of the total alkaloids.
Besides their low concentration in the natural state in plant starting material, harringtonines are mixed with many congeners which have very similar chemical structures. Thus, in a high resolution high performance liquid chromatography (HPLC) chromatogram of a semi-purified alkaloid extract, no less than several tens of cephalotaxine esters are counted.
Numerous antileukemia drugs have been investigated but so far, there is no single drug that is effective and safe. As discussed in U.S. 3,497,593, an alkaloid from Tylophora plant is said to have antitumor activity against mouse leukemia (L-1210). U.S. 3,928,584 discloses an organic composition derived from tree sap and is said to have activity against mouse leukemia P-388. Also U.S. 4,431,639 discloses that an extract of Rhisoma Stractylis promotes the production of lymphocytes in the circulating blood, consequently eliminating cancer growth
-
Harringtonine or Homoharringtonine, hereinafter referred to as HH, has been known to be effective against acute chronic granulocytic and monocytic leukemia (Journal of Chinese Internal Medicine 3:162-164, 1978). However, it is highly toxic and causes damage to heart and hematopoietic organs. The results of experiments in animals, such as mice, rabbits and dogs, indicate that most of them die from cardiotoxicity after receiving the drug. Therefore, there is a need to improve the HH drug for safe use against leukemia. This drug is of special importance in that all known antileukemia drugs are effective against lymphatic leukemia and there are no effective drugs for treating nonlymphatic leukemia
All the literature from 1972 to the present date [Mikolajczack et al., Tetrahedron, 1995 (1972); T. Hudlicky, L. D. Kwart and J. W. Reed in “Alkaloid: Chemical and Biological Perspectives” (S. W. Pelletier Ed.), Vol. 5, 639 (1987); M. A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, p. 236 (1998)] mention the impossibility hitherto of esterifying the highly sterically hindered secondary hydroxyl of cephalotaxane 2a with the tertiary carboxyl of the alkanoyl chain of harringtonic acid 3 totally preformed to give a harringtonine 4b, i.e. the conversion 2a+3e(4b as described in the example featured in the scheme below

- ……………………………………………………..
SYNTHESIS
Tetrahedron Lett 1982,23(34),3431, J Org Chem 1983,48(26),5321

The oxidation of 2-methyl-1-cyclopentene-1-carbaldehyde (I) with O3 and Ag2O gives 2,6-dioxoheptanoic acid (II), which is esterified with cephalotaxine (III) by means of (COCl)2, yielding the ester (IV). Reformatsky reaction of (IV) with methyl bromoacetate (V) and Zn affords the adduct (VI), which is treated with an excess of methylmagnesium iodide to provide the target homoharringtonine (as a single diastereomer), along with some starting cephalotaxine that is separated by chromatography.
………………………………
SYNTHESIS
EP 1064285; FR 2776292; WO 9948894, Tetrahedron Lett 1999,402931

The intermediate (racemic)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (VIII) has been obtained by several related methods: 1. The Grignard condensation of 4-methyl-3-pentenyl bromide (I) with diethyl oxalate (II) in HF gives the 2-oxoheptenoate (III), which is condensed with methyl acetate (IV) by means of LiHMDS in THF to yield 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V).
The cyclization of (V) by means of Ts-OH in hot toluene or by means of hot aqueous formic acid affords 2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid ethyl ester (VI), which is hydrolyzed with KOH in boiling water to provide the corresponding dicarboxylic acid (VII). Finally, this compound is regioselectively monoesterified by means of BF3/MeOH in methanol to furnish the intermediate (racemic)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (VIII). 2.
The reaction of 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V) with HCl in hot methanol gives 3-(ethoxycarbonyl)-3,7-dihydroxy-7-methyloctanoic acid methyl ester (IX), which is then cyclized by means of ZnCl2 in hot dichloroethane to yield the previously described intermediate (VIII). 3. The hydrolysis of 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V) with KOH in refluxing methanol/water gives the corresponding diacid (X), which is regioselectively monoesterified by means of BF3/MeOH in methanol to yield 3-carboxy-3-hydroxy-7-methyl-6-octenoic acid methyl ester (XI).
Finally, this compound is cyclized by means of Ts-OH in hot toluene to afford the previously described carboxylic intermediate (VIII). The racemic acid (VIII) is submitted to optical resolution by esterification with quinine (XII) by means of 2,4,6-trichlorobenzoyl chloride and TEA or DCC to give a diastereomeric mixture of esters (XIII) that is separated by preparative HPLC to obtain the desired diastereomer (XIV).
The hydrolysis of (XIV) with KOH in refluxing ethanol/water gives the corresponding chiral dicarboxylic acid (XV), which is regioselectively monoesterified with BF3/MeOH in methanol to yield the chiral (R)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (XVI).
The esterification of (XVI) with cephalotaxine (XVII) by means of 2,4,6-trichlorobenzoyl chloride and TEA in toluene affords the corresponding ester (XVIII), which is treated with HBr in dichloromethane/HOAc, providing the bromoester (XIX). Finally, this compound is treated with NaHCO3, CaCO3 or BaCO3 in acetone/water to give the target hydroxyester.
………………………………………….
EXTRACTION
-
Throughout the specification, the concentration of the solvent is the same as first given unless stated otherwise. Redeuced pressure means about 2,27 kPa (17 mm Hg. abs), l is liter, kg is kilogram. ml is milliliter. Yield in weight %.
Example 1. HH is extracted from the skins, stems, leaves and seeds of Cephalotaxus fortunel Hook and other related species, such as Cephalotaxus sinensis Li, C. hainanensis, and C. wilsoniana, including C.oliveri mast and C.harringtonia. -
1 kg of finely ground Cephalotaxus fortunel Hook is extracted with 8 l of 90% ethanol at room temperature for 24 hrs. The solution is filtered to yield a filtrate A and filtercake. The filtercake is percolated with ethanol and filtered again to yield filtrate B. A and B are combined and distilled under reduced pressure to recover ethanol and an aqueous residue. To this residue, 2% HCl is added to adjust the pH to 2.5. The solids are separated from the solution by filtration to yield a filtrate C. The solids are washed once with 2% HCl and filtered to yield a filtrate D. C and D are combined and the pH adjusted to 9.5 by adding saturated sodium carbonate solution. The alkaline filtrate is extracted with chloroform and the chloroform layer separated from the aqueous layer. This extration process is repeated five times. All the chloroform extracts are combined and distilled at reduced pressure to recover chloroform and alkaloid as a solid residue respectively.
-
The solid alkaloid is then dissolved in 20 ml. of 6% citric acid in water. The solution is divided into three equal portions. These are adjusted to pH 7,8 and 9 by adding saturated sodium carbonate solution.
-
The portions having pH 8 and 9 are combined and extracted with chloroform. The chloroform extracts are distilled under reduced pressure, whereby chloroform is removed and recovered and a solid residue of crude Harringtonine is obtained.
-
The crude Harringtonine is dissolved in pure ethanol i.e. alkaloid : anhydrous ethanol 1:10 , and crystallized. The crystals are refined by recrystalliation in diethyl ether. Overall yield of Harringtonine is about 0.1% including yield from mixed HH from the subsequent process.
Harringtonine has the following chemical structure:
wherein R is

- melting point:
- 135° – 137°C
- crystal:
- colorless
- infrared spectrum:
- 3750, 1660, 1505, 1490, 1050, and 945 cm⁻¹.

-
The portion having a pH of 7 and the mother liquors from the foregoing crystallization of Harringtonine are combined and passed through a liquid chromatographic column of diameter to height ratio 1:50 packed with alumina. The column is finally flushed with chloroform and followed by chloroform-methanol of 9:1 mixture. The resulting alkaloids are mixture of HH. The mixed HH is then separated from each other by countercurrent distribution employing chloroform and pH 5 buffer. The first fraction of the countercurrent distribution is Homoharringtonine and the last fraction of the countercurrent distribution is Harringtonine. Homoharringtonine is purified by crystallization in methyl alcohol.
Homoharringtonine has the following chemical structure:
wherein R is

- yield:
- 0.02%
- melting point:
- 144° – 146°C
- infrared spectrum:
- 3500∼3400, 1750, 1665, 1030 and 940 cm⁻¹.







…………………………………………………………………………..
EXTRACTION
All the literature from 1972 to the present date [Mikolajczack et al.,Tetrahedron, 1995 (1972); T. Hudlicky, L.D. Kwart and J.W. Reed in “Alkaloid: Chemical and Biological Perspectives” (S.W. Pelletier Ed.), Vol. 5, 639 (1987); M.A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, p. 236 (1998)] mention the impossibility hitherto of esterifying the highly sterically hindered secondary hydroxyl of cephalotaxine 2a with the tertiary carboxyl of the alkanoyl chain of harringtonic acid 3e totally preformed to give a harringtonine 4b , i.e. the conversion 2a + 3e ( 4b as described in the example featured in the scheme below

Example 46
Preparation of purified (-) cephalotaxine from total alkaloidic extract of Cephalotaxus sp
-
[0319]

-
Partially racemized cephalotaxine [H. Wenkui; L. Yulin; P. Xinfu, Scientia Sinica,; 23; 7; 835 (1980)]
-
1H NMR of two batches of cephalotaxine (extracted in the same conditions as above) with the optically active NMR shift reagent europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate (1 éq) showed the following results:
- Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine) ; 5.99 (1H, OCH2O (-)-cephalotaxine) and 5.76 (1H, OCH2O (-)-cephalotaxine).
Presence of 11 ± 5 % de (+)-cephalotaxine.
[α]22 = -134,0° (c = 0,214; CHCl3) : calculated rate 25 ± 5 % - Batch B: slightly racemized (1%)
[α]19 = -173,3° (c = 0,208; CHCl3)
- Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine) ; 5.99 (1H, OCH2O (-)-cephalotaxine) and 5.76 (1H, OCH2O (-)-cephalotaxine).

Enantiomeric enrichment of the natural cephalotaxine:
-
Crude chromatographied cephalotaxine (20g) was dissolved at 55°C in dry methanol (100 ml). Crystallization occurs by cooling with rotary evaporator and after filtration the product thus obtained showed 99.9% of HPLC purity.
[α]20 D =-130° (C1, CHD3) corresponding to 10 % of racemization. The crystallized product thus obtained (20g) was dissolved again in hot methanol (100 ml).
Slowly cooling the solution allows translucent prisms composed of pure enantiomeric (-)-cephalotaxine [α]20 D= -185°(C1,CHCl3).
After filtration, the mother liquors was allowed to slowly evaporate at room temperature and crystals in the form of macled needles exclusively composed of racemic cephalotaxine [α]D 20 = 0,5° (C1 ; CHCl3) were obtained.
After filtration, the second mother liquors allowed prisms composed of (-)-cephalotaxine identical to this obtained at the first crystallization.
After filtration, the third mother liquors still allowed macled needles (urchins) composed of (±)-cephalotaxine.
The cycle is repeated three times. The combined prismatic crystals was recrystallized once to give enantiomerically pure (-)-cephalotaxine, while the combined macled needles treated in the same way gives 100% racemic cephalotaxine.
Chemical evaluation of the enantiomeric purity of natural cephalotaxine:
-
A sample of partially racemized natural cephalotaxine was inserted in the process, which sequence is described in the Examples 1,2,3,4,5,6,15,19 and 21, by using a pure (2R)-homoharrintonic acid resulting from Example 19.
The HPLC analysis of the diastereomeric mixture of anhydro-homoharrintonine thus obtained showed a significant enantio-epi-homoharringtonine rate (11% ± 3%) corresponding to the (+)-cephalotaxine content in the racemic mixture of origin (it has been demonstrated that the two antipodes of the homoharringtonic acid react in a stoechiometric way comparable to the pure enantiomeric cephalotaxine).
Example 47Preparation of homoharringtonine, from anhydro-homoharringtonine:

1)° Method A
-
A commercial solution of hydrobromic acid in acetic acid (17.4 ml, 86.6 mmol, HBr 30% w/w) was added to a stirred solution of anhydrohomoharringtonine resulting from Example 21 (50.8 g, 9.63 mmol) in anhydrous dichloromethane (25.6 ml) at -10°C. After stirring at -10°C for 3 hours was added water (240 ml) and the reaction mixture was become viscous. The temperature was allowed to rise to room temperature and after stirring for 2.5 hours was added sodium carbonate 0.76M (406 ml) to pH 8. The resulting aqueous layer was saturated with sodium chloride, then was extracted with dichloromethane (3 × 230 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness to afford a foam. After phase reverse chromatography below-mentioned were obtained 4.03g of homoharringtonine (77%). The product thus obtained showed identical characteristics to this resulting from Example 25.
2°) Method B
-
To a stirred solution of anhydrohomoharringtonine resulting from Example 21 (214 mg, 0.406 mmol) in anhydrous dichloromethane (1.1 ml) was added at -10°C a commercial solution of hydrobromic acid in acetic acid (0.728 ml, 3.6 mmol, HBr 30% w/w). After stirring at -10°C for 3 hours, was added water (13 ml) and then the temperature was raised to 20°C. After stirring at 20°C for 3 hours, was added a sodium carbonate solution (0.76M; 31.5 ml) up to pH 8. The resulting aqueous layer, after saturation with sodium chloride, was extracted with dichloromethane (3 × 20 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by phase reverse chromatography below-mentioned to provide homoharringtonine (166 mg, 75%). The product thus obtained showed identical characteristics to this resulting from Example 25.




……………………
SEMISYNTHESIS
EXAMPLE 27 Preparation of homoharringtonine as a pharmaceutical use from crude semi-synthetic homoharringtonine resulting from example 25 by preparative high-performance liquid chromatography

1°) Method A
Crude homoharringtonine (35 g) is dissolved in buffer (triethylamine (1.55/1000) in deionised water and orthophosphoric acid to adjust pH to 3. The solution was filtered then injected on a preparative high-performance liquid chromatograph equipped with axial compression and high pressure pump (stationary phase: n-octadecylsilane, 15 μm, porosity 100, 1 kg; mobile phase; buffer/tetrahydrofurane 85/15). Elution was performed at a flow rate of 0.2 l/min. Fractions contain was monitored by U.V. detector and TLC. Retained fraction were finally checked by HPLC then combined, alkalinised with 2.5% aqueous ammonia and extracted with dichloromethane (4×400 ml). After concentration under reduced pressure homoharringtonine is obtained as a pale yellow resin which on trituration in a 8/2 water-methanol mixture gave pure homoharringtonine as a white crystalline solid (mp=127° C.), HPLC purity was higher than 99.8%.
2°) Method B
Same procedure of purification as method A was performed but mobile phase buffer/methanol (68/32) was used instead buffer/tetrahydrofurane.
3°) Method C
Same procedure of purification as method A was performed but mobile phase buffer/acetonitrile (85/15) was used instead buffer/tetrahydrofurane.
EXAMPLE 28 Preparation of homoharringtonine as a pharmaceutical use from semi-purified natural cephalotaxine
Crude homoharringtonine, prepared according to Example 25 from a partially racemized natural cephalotaxine and purified by chromatography and crystallisation according to the method A of Example 27, gave an homoharringtonine showing a non natural enantiomeric epi-homoharringtonine content less than 0.05%.
EXAMPLE 46 Preparation of purified (−) cephalotaxine from total alkaloidic extract of cephatotaxus sp

Partially racemized cephalotaxine [H. Wenkui; L. Yulin; P. Xinfu, Scientia Sinica; 23; 7; 835 (1980)]
1H NMR of two batches of cephalotaxine (extracted in the same conditions as above) with the optically active NMR shift reagent europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate (1éq) showed the following results:
Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine); 5.99 (1H, OCH2O (−)-cephalotaxine) and 5.76 (1H, OCH2O (−)-cephalotaxine). Presence of 11±5% de (+)-cephalotaxine. [α]22=−134,0°(c=0,214; CHCl3): calculated rate 25±5%
Batch B: slightly racemized (1%) [α]19=−173,3°(c=0,208; CHCl3)
Enantiomeric Enrichment of the Natural Cephalotaxine:
Crude chromatographied cephalotaxine (20 g) was dissolved at 55° C. in dry methanol (100 ml). Crystallization occurs by cooling with rotary evaporator and after filtration the product thus obtained showed 99.9% of HPLC purity, [α]20 D=−130°(C1, CHD3) corresponding to 10% of racemization. The crystallized product thus obtained (20 g) was dissolyed again in hot methanol (100 ml).
Slowly cooling the solution allows translucent prisms composed of pure enantiomeric (-−)-cephalotaxine [α]20 D=−185°(C1, CHCl3).
After filtration, the mother liquors was allowed to slowly evaporate at room temperature and crystals in the form of macled needles exclusively composed of racemic cephalotaxine [α]D 20=0,5°(C1; CHCl3) were obtained.
After filtration, the second mother liquors allowed prisms composed of (−)-cephalotaxine identical to this obtained at the first crystallization.
After filtration, the third mother liquors still allowed macled needles (urchins) composed of (±)-cephalotaxine.
The cycle is repeated three times. The combined prismatic crystals was recrystallized once to give enantiomerically pure (−)-cephalotaxine, while the combined macled needles treated in the same way gives 100% racemic cephalotaxine.
Chemical Evaluation of the Enantiomeric Purity of Natural Cephalotaxine:
A sample of partially racemized natural cephalotaxine was inserted in the process, which sequence is described in the Examples 1,2,3,4,5,6,15,19 and 21, by using a pure (2R)-homoharrintonic acid resulting from Example 19. The HPLC analysis of the diastereomeric mixture of anhydro-homoharrintonine thus obtained showed a significant enantio-epi-homoharringtonine rate (11%±3%) corresponding to the (+)-cephalotaxine content in the racemic mixture of origin (it has been demonstrated that the two antipodes of the homoharringtonic acid react in a stoechiometric way comparable to the pure enantiomeric cephalotaxine).
EXAMPLE 47
Preparation of homoharringtonine, from anhydro-homoharringtonine

1°) Method A
A commercial solution of hydrobromic acid in acetic acid (17.4 ml, 86.6 mmol, HBr 30% w/w) was added to a stirred solution of anhydrohomoharringtonine resulting from Example 21 (50.8 g, 9.63 mmol) in anhydrous dichloromethane (25.6 ml) at −10° C. After stirring at −10° C. for 3 hours was added water (240 ml) and the reaction mixture was become viscous. The temperature was allowed to rise to room temperature and after stirring for 2.5 hours was added sodium carbonate 0.76M (406 ml) to pH 8. The resulting aqueous layer was saturated with sodium chloride, then was extracted with dichloromethane (3×230 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness to afford a foam. After phase reverse chromatography below-mentioned were obtained 4.03 g of homoharringtonine (77%). The product thus obtained showed identical characteristics to this resulting from Example 25.
2°) Method B
To a stirred solution of anhydrohomoharringtonine resulting from Example 21 (21.4 mg, 0.406 mmol) in anhydrous dichloromethane (1.1 ml) was added at −10° C. a commercial solution of hydrobromic acid in acetic acid (0.728 ml, 3.6 mmol, HBr 30% w/w). After stirring at −10° C. for 3 hours, was added water (13 ml) and then the temperature was raised to 20° C. After stirring at 20° C. for 3 hours, was added a sodium carbonate solution (0.76M; 31.5 ml) up to pH 8. The resulting aqueous layer, after saturation with sodium chloride, was extracted with dichloromethane (3×20 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by phase reverse chromatography below-mentioned to provide homoharringtonine (166 mg, 75%). The product thus obtained showed identical characteristics to this resulting from Example 25.
…………………………………
EXTRACTION
The remarkable clinical efficacy of Homoharringtonine (HHT) resulting in lot of observations of complete remission of leukemia and other solid cancer in human being since 1988. Recently, research articles reported that the HHT efficacy in glaucoma, inhibition of Hepatities B virus replication and using in bone marrow transplantation. For example, the University of Texas M.D. Anderson Cancer Center and National Cancer Institute reported that “Ninety-two percent of patients achieved CHR with HHT.” [Susan O’Brien, at al.; Sequential homoharringtonine and interferon-α in the treatment of early chronic phase chronic myelogenous leukemia; Blood, Vol 93, No 12 (June 15), 1999: pp 4149-4153]. Another article reported that “the median number of days on HHT per month was 2 days with a median follow-up of 26 months; the estimated 2-year survival rate was 90%.” (Susan O’Brien, at al.; Simultaneous homoharringtonine and interferon-α in the treatment of patients with chronic-phase chronic myelogenous leukemia; American Cancer Society; Apr. 1, 2002, Vol 94, No. 7).
On Nov. 8, 1988, U.S. Pat. No. 4,783,454 titled Process for producing harringtonine and homoharringtonine disclosed the technique of isolation of a purified HHT from bark of Cephalotaxus. However, the natural source ofCephalotaxus is very limited. Trees of Cephalotaxus grow slowly. Bark ofCephalotaxus has very low content of HHT. Extracting HHT from bark ofCephalotaxus the yield was about 0.02% only. More important to harvest bark ofCephalotaxus will kill and destroy trees. Supply of HHT is very short now. Therefore, it is necessary to find a new manufacturing method.
DETAILED DESCRIPTION
Great progress has been made in research on Homoharringtonine (HHT) production and on future generation HHT drug since 1988. For example, the University of Texas M.D. Anderson Cancer Center and National Cancer Institute reported that “Ninety-two percent of patients achieved CHR with HHT.” Another article reported that “the median number of days on HHT per month was 2 days with a median follow-up of 26 months; the estimated 2-year survival rate was 90%.”
The good clinical results of HHT in treating cancer brought to the major problem, which is the supply of HHT both short term and long term. It is apparent that a huge amount of bark of Cephalotaxus is needed for collection, extraction and purification of HHT. It is clear that due to the slow growth of the trees ofCephalotaxus, which is a nature source of HHT, and the killing of trees by harvesting bark is not a sustainable resource for HHT production.
Present invention disclosed new methods for producing HHT. The new methods of producing HHT are shown as follows.
1. Tissue Culture (Plant Cell Culture):
Culture manipulation to promote secretion of HHT is a new way for an extracellular product HHT. The biosynthetic methods can yield more HHT through precursor of HHT feeding. The production of HHT increased significantly after the addition of the precursors and special biochemical agents. Content of precursor of HHT abounds in tree and it is very cheap. The present methods include several significant developments in technique of culture plant tissues that are
-
- (a) yields of HHT selected from rapid growth, resistance to infections organisms; and
- (b) HHT can excrete into media.
Traditional method of plant culture is very difficult to overcome the problem of high cost. Therefore, traditional method appears too long to have commercial value. HHT is secondary metabolite of Cephalotaxus. Secondary compound acts in defense against the harmful effects of toxins, carcinogens or mutagens found in the plant. In fact, traditional method is very difficult to increase HHT contenting in plant tissues. The present new method uses a special biochemical agent for increasing content of HHT and more easily to purify HHT from other metabolites.
More important is that the key of the present new technique for producing high content of HHT in plant cell culture is to increase production of HHT by directed fermentation through precursor of HHT feeding. The present new methods are used special metabolite of Cephalotaxus for markedly enhance production of HHT. Therefore, the present invention disclosed a new source for the long term of producing HHT.
2. Using Precursor of HHT:
Recent research’s results have established that direct production of HHT from its precursor and advances in biosynthetic understanding for HHT metabolism. Biosynthesis or semisynthesis of HHT from major nonactivity ingredients is well established through great advances in special biochemistry reactions. Using precursor of HHT for semisynthesis and increase of production in plant cell culture are new developing methods for producing HHT.
3. Using Leaves:
Our new method use leaves of tree of Cephalotaxus not use the bark. So far, the extraction of HHT is used bark. The leaves are harvested from the trees ofCephalotaxus, which grow in mountains of South China. The natural source of leaves is very abundance. The new methods do not use bark. Therefore, it can avoid destroy trees. The natural source of Cephalotaxus tree is very limited and slow growing. In fact, bark of Cephalotaxus has very low yield of HHT. The yield of HHT from Cephalotaxus bark is about 50-100 mg/kg of dried bark. The present new method, therefore, has a great economic and environmental value.
4. Semisynthesis:
HHT has received important chemical studies particularly in regard to structure and anticancer activity relationship and semisynthesis.
A great progress in biochemistry allows semisynthesis to use precursor of HHT from leaves of Cephalotaxus and to produce HHT. The total chemical synthesis of HHT appears too long to have commercial value too. Semisynthesis method can yield a high efficient conversion of precursor to HHT. It is other better biological source for manufacturing HHT. This new method uses closing chemical analogues to convert to HHT. This analogue is produced from leaves or other organ of Cephalotaxus. The present invention disclosed that new methods and techniques of manufacturing HHT could avoid chopping down Cephalotaxus trees which governmental environmentalists are trying to have declared a threatened species.
5. Using Taxol Residual
The anticancer drug Taxol is the most promising new chemotherapeutic agents that developed for cancer treatment in the past twenty years. Taxol has a unique mechanism of action. It has been shown to promote tubulin polymerization and stabilize microtubules against depolymerization. The FDA approved the clinical use of Taxol for several types of cancer. So far, annual sales of Taxol are more than $2 billion in market. Taxol is extracted from bark or leaves of an evergreen tree named Taxus species including Taxus brevifolia (or called Pacific yew). After Taxol has been extracted from bark or leaves, all residual materials of Taxus brecifolia named Taxus residual, which are waste.
Both taxol and HHT can be extracted from yew tree. The content of taxol is less than 0.01% in yew tree. The content of HHT in yew tree is about 0.01% -0.22%. The content of HHT is much higher than content of Taxol. Taxol extracted from bark of yew is difficult and expensive. One reason is that the presences of closely related congeners are similar to Taxol. A major congener is Cephalomannine (CPM), which is a waster of process in manufacturing of Taxol.
The chemical and physical characters are very close between Taxol and Cephalomannine (CPM).
CPM characterized by the same ring structure as Taxol and distinguishes from them only in C-13 ester structure. The present invention disclosed that CPM and related derivative are used to produce HHT.
The following specific examples will provide detailed illustrations of methods of producing relative drugs, according to the present invention and pharmaceutical dosage units containing demonstrates its effectiveness in treatment of cancer cells. These examples are not intended, however, to limit or restrict the scope of the invention in any way, and should not be construed as providing conditions, parameters, reagents, or
EXAMPLE 1
Production of HHT by Culture Cells
So far, HHT is extracted from bark and skins of Cephalotaxus species. However, growth of Cephalotaxus species is very slow and concentration of HHT in plant is extremely low. Furthermore, it is difficult to harvest the plants because of their low propagation rate and the danger of drastic reduced in plant availability. Also, cost of total chemical synthesis of HHT is very expensive and is not available for commerce now. For the reasons given above it is more difficult to obtainCephalotaxus on a large scale for long time. Therefore, Cephalotaxus cell cultures are one of best methods for obtaining HHT. In this present invention, special elicitation is disclosed and it will significantly increase production of HHT.
The methods of cell and tissue culture are disclosed as below.
Parts of bark, stems, leaves, or roots of Cephalotaxus species were surface disinfected by treatment in 70% ethanol for 10 minutes and followed by 0.1 HgCl2for 3 minutes. Plant materials were washed five times for 10 minutes each by sterilized water. Parts of plant were cut into small pieces (0.5-1 mm) and put pieces to Murashige and Skoog’s (MS) medium and supplemented with derivative of new active ingredient of phylum mycota (IPM), precursor of HHT which is a derivative of Cephalotaxus (CEP), tyrosine (TYR) naphthaleneacetic acid (NAA), Kinetin (3 mg/L), and 3% sucrose (w/v). PH of medium was adjusted to 5.7˜5.8. Agar (10 g/L) added to medium. Callus tissues are collected from agar media and suspension cultured cells were harvested by filtration and cultured in MS medium.
The cultures were kept in a culture room at 26° C.±1° C. Friable callus tissues were obtained. The callu was inoculated into 4 L of MS liquid medium containing sucrose, derivative of CEP, PHE, TYR, NAA and Kinetin. Then callus tissues were cultivated 26° C. for 35 days on rotary shaker operated at 120 rpm in the dark. Cells were subcultured into fresh medium of same composition every 2 weeks and maintained at 120 rpm at 26°±1° C. Packed cell volume (PCV), fresh weight (FW), dry weight (DW), concentration of HHT and concentration of sugar were determined every 5th day. The cells were harvested and dried.
In general, callus and suspension cultures of cephalotaxus species grow very slow and no production of free or esterified HHT. However, according to the present invention, addition of IPM to cultures cause a drastic increasing in HHT after 30 days of incubation. For example, in control group (no IPM), HHT in cultured cells is 0.020 mg/g dry weight, but in treatment group (addition of IPM) HHT is about 0.050 mg/g dry weight. Therefore, IPM can increase 250% of content of HHT. It has resulted in plant cell culture systems that producing HHT at concentration higher than those produced by the mother plant. The production of HHT increases significantly after the addition of precursors (CEP). Addition of CEP can increase HHT. Obviously, the present invention provided a new commercial and economic method for producing HHT. The IPM and precursors (CEP) play key role in cultured cells.
EXAMPLE 2
Semi-Synthesis of HHT
HHT shows a significant inhibitory activity against leukemia and other cancer. Concentration of HHT, however, has only 0.01% in natural sources. Cephalotazine (CEP) is major alkaloids present in plant extracts and the concentration ofCephalotaxus has about 1%. Therefore, concentration of CEP is about 100 times higher then HHT in nature plant sources. But CEP is inactive. For the reason given above, semisynthesis of HHT from CEP will increase huge natural sources of HHT.
-
- (1) Extraction of CEP
10 kg of dried stems or leaves or roots of Cephalotaxus species were milled, placed in a percolator, along 80 L of 95% of ethanol, and allowed to stand 24 hours. The ethanol was recovered under reduced pressure (below 40° C.). 20 L of 5% tartaric acid was added to concentrated ethanol solution. The ammonia water was added to the acidic solution and adjusted pH to 9. The solution of pH 9 was filtered and yielded a filtrate. The filtrate was extracted with CHCl3. CHCl3 was recovered under reduced pressure and residue was obtained. The residue was chromatographed packed with alumna and eluted by CHCl3-MeOH (9:1). Eluate was concentrated under reduced pressure. Residue was dried under vacuum. The product is CEP.
-
- (2) Semisynthesized HHT from CEP
Materials and Methods
Melting points were determined on a Fisher-Johns apparatus. Infrared spectra were obtained on a Perkin-Elmer 567 infrared spectrophotometer or on a Beckman 4230 IR spectrophotometer. Peak positions were given in cm−1. The IR spectra of solid samples were measured as potassium bromide dispersions, and the spectra of liquids were determined in chloroform or carbon tetrachloride solutions. NMR spectra were measured on a Varian A-60, Perkin-Elmer R-32, Varian EM-390, or Brüker WH-90 NMR spectrometer. Chemical-shift values were given in parts per million downfield from Me4Si as an internal standard. Mass spectra were run on an AE1 MS-12 Finnigan 3300, or CEC21-110B mass spectrometer.
Preparative thin-layer chromatography was accomplished using 750-μm layers of aluminum oxide HF-254 (type E), aluminum oxide 60 PF-254 (type E), silica gel HF-254 (type 60 PF-254), or silica gel GF-254. Visualization was by short-wave ultraviolet light. Grace silica gel, Grade 923, and Woelm neutral aluminum oxide, activity III, were used for column chromatography. Analytical thin-layer chromatography was run on plastic sheets precoated with aluminum oxide F-254 neutral (type T), 200-μm thick, and on Polygram Sil G/UV254 (silica gel), 250 μm on plastic sheets. Visualization was usually by short-wave ultraviolet light, phosphomolybdic acid, or iodoplatinate.
Preparation of α-Ketoester-Harringtonine
1 g of Benzene-α-acetone Na was put into 10 L of benzene. Mixture was stirred at room temperature then was dissolved in 10 L of pyridine and stirred at 0° C. Oxalic chloride was added from a dropping funnel to solution of pyridine. Stirring was continued while the solution warmed to room temperature and stand overnight. Excess reagent was removed. This solution was dissolved in CH2Cl2and cooled to near 0° C. in an ice water bath. 5 g of CEP, 2.5 L of CH2Cl2 and 2.5 L of pyridine were added to cold CH2Cl2 solution. Manipulations were done in a dry N2 atmosphere and all glassware heat-dried just before use. The suspension was stirred at room temperature and overnight. The mixture was washed with 10% Na2CO3 and saturated aqueous NaCl, then dried with auhydrous magenesium sulfate, and filtered and the solvents were removed in vacuo. Evaporation provided as an amorphous solid α-ketoester-harringtonine (mp 143˜145° C.).
Semi-Synthesis of HHT
10 L of CH3CHBrCOOEt and activated zin dust and THF were added to the α-ketoester-harringtonine (at −78° C.) for 6 hours followed by slow warming to room temperature with stirred. The reaction mixture was diluted with 10 L CHCl3 and 10 L H2O and solid Na2CO3 was added. CHCl3 was evaporated under reduced pressure and residue was obtained.
The residue was purified by chromatography on alumina. The column was flushed with chloroform and followed by chloroform-methanol (9:1). The solvents were recovered under reduced pressure to provide as a solid. Solid was dissolved in pure ethanol and crystallized. The crystals were refined by recrystalization in diethyl ether. The crystals dried under vacuum. The product is HHT, which has the following characters:
[α]D −119° (C=0.96),
MSm/e (%): 689 (M+, 3), 314 (3), 299 (20), 298 (100), 282 (3), 266 (4), 20 (3), 150 (8), 131 (12), 73 (18)
EXAMPLE 3
HHT Extracted from Plant Tissue
Extraction of HHT has several major methods which including extraction by organic solvent, chromatograph and adjust pH.
HHT was extracted from plant tissue culture, plant cells or leaves of Cephalotaxusspecies.
1 kg of ground Cephalotaxus fortunei Hook was extracted with 10 liters of water at room temperature for 24 hrs. To filtered the solution to yield a filtrate. Ten liters of 90% ethanol added to filtrate. The mixture was Centrifugalized to yield a sediment. Percolated the sediment with ethanol and filter again to yield filtrate, combined filtrates, and distilled under reduced pressure to recover ethanol and an aqueous residue. To this residue, added 10% of HCl to adjust the pH to 2.5. To separated the solids from the solution by filtration to yield a filtrate (1). Washed the solids once with 2% HCl and filtered to yield a filtrate (2). Combined (1) and (2) and adjusted the pH to 9.5 by adding saturated sodium carbonate solution. Extracted the alkaline filtrate with chloroform and separated the chloroform layer from the aqueous layer. To repeated this extraction process five times. Combined all the chloroform extracts and distilled at reduced pressure to recover chloroform and alkaloid as a solid residue obtained. The solid alkaloid was then dissolved in 6% citric acid in water. The solution was divided into three equal portions. These were adjusted to pH 7, 8 and 9 by adding saturated sodium carbonate solution. The portions having pH 8 and 9 were combined and extracted with chloroform. The chloroform extracts were distilled under reduced pressure, whereby chloroform was removed and recovered and crude HHT was obtained. The crude HHT was dissolved in pure ethanol and crystallized. The crystals were refined by recrystallization in diethyl ether. The crude HHT obtained.
The portion having a pH of 7 passed through a liquid chromatographic column packed with alumina of diameter to height 1:50. The column was finally flushed with chloroform and followed by chloroform-methanol of 9:1 mixture. The resulting alkaloids were mixture crude of HHT. Combined crude HHT and then separated from each other by countercurrent distribution employing chloroform and pH 5 buffers. The first fraction of the countercurrent distribution was HHT. HHT was purified by crystallization in methyl alcohol. The crystallization was purified by recrystallization in methyl alcohol and dried under vacuum.
…………………….
Example 1 : Preparation of harringtonine drug substance by purification of commercial natural harringtonine
A. Analytical profile of starting product
By combination of HPLC analysis with UV detection (see Figure 6) and mass spectrometry detection (see figure 7 and 8) a total of 6.5% of related compound (identified as b,c: position isomer of harringtonine = 3.4%; d: homoharringtonine = 3%; e: 4′-demethyl harringtonine = 0.01%; f: drupacine derivative: 0.05%) are found in the starting product.
B. Chromatography of natural harringtonine
Natural harringtonine (5 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 80 mm; length: 1000 mm) containing 1000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi). Unwanted fractions are discarded based upon in-line UV spectrophotometric detection. Kept fractions are collected in 16 separate containers which each are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase). During the development phase, a dual in-line UV-MS detection is used. After discarding of the fractions representing more than 0.5 % of the total content of harringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure. Then crude concentrated solution of harringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 1.5 %. C. Crystallization of raw harringtonine
Under a laminar flow hood, the above raw harringtonine (4.1 grams) is dissolved in methanol (5ml), at 30°C. The resulting alcoholic solution was filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized rotary flask. Then, desionized water (50mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of harringtonine is kept under vacuum and rotation is continued during appearance of white crystals of pure harringtonine. The stirring is continued until no more crystal occurs. Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water (10 mL x 2). The white translucent crystals are then dried using high vacuum at 40°C for 24 hours. Overall yield is 76%. All operations were documented prior to start the process and full current Good Manufacturing Practices were applied. This clinical batch corresponds to 400 therapeutic units dosed at 10mg.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 7 and 9 compare HPLC chromatogram before and after purification in using this process. Table II shows the comparison of the corresponding related compound content.

For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC) thermogravimetry, 2D NMR, solid NMR and X-ray powder diffractometry.
Infrared Spectrometry:
Identical IR spectra were obtained by either the KBr pellet and/or mineral oil mull preparation technique. Figure 5 shows typical infrared spectrum (KBr) for unambiguous identification at the solid state of the crystalline harringtonine obtained by this process. A series of sharp absorption bands are noted at 615, 654, 674, 689, 709, 722, 750, 761 805, 850, 928, 989, 1022, 1033, 1062, 1083, 1112, 1162, 1205, 1224, 1262, 1277, 1308, 1340, 1364, 1382, 1438 1486, 1508, 1625, 1656, 1725, 1745, 2883, 2936, 2972, 3079, 3353, 3552 and 3647 cm“1
Differential Scanning Calorimetry (DSC) And Thermogravimetry (TG) Measurement of DSC and TG were obtained on a Mettler Toledo STAR System. Approximately 12 mg of harringtonine drug substance were accurately weighed (12.4471 mg) into a DSC pan. The sample was heated from 25°C to 200°C at a rate of 10°C/min. The DSC data were obtained following a standard method in the art. The DSC curve of crystalline harringtonine drug substance ((Figure 4), exhibits a melting endotherm at 79.5 °C . No subsequent decomposition occurred under the upper tested temperature 200°C. Simultaneous TG measurement, indicated a loss on drying of 1.3 % which did not correspond to a lost of structural molecule of solvent or water.
Example 2: Preparation of homoharringtonine drug substance by purification of raw semi- synthetic (hemi-synthetic) homoharringtonine
A. Analytical profile of starting product
Crude reaction mixture of raw homoharringtonine contains a potential of 250 grams of homoharringtonine DS together with process impurities such as catalyst, unchanged starting product (anhydro-homo-harringtonine), and some related side product. HPLC analysis with UV detection (see left-side chromatogram on Figure 10) indicated a total of 9 % of related impurities. B. Chromatography of semi-synthetic homoharringtonine
Raw semi-synthetic homoharringtonine (550 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 450 mm; length: 1000 mm) containing 48,000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi, flow-rate 540 L/hour). Unwanted fractions are discarded based upon by- passed in-line UV spectrophotometric detector. Kept fractions are collected in 30 separate stainless steel containers (20 or 50 L each) which are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase) and equipped with a diode array detector. After discarding of the fractions representing more than 0.5 % of the total content of homoharringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure in using a mechanically stirred thin film evaporator. Then crude concentrated solution of homoharringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 0.5 % (see rigth-side chromatogram on Figure 10)
C. Crystallization of homoharringtonine DS
In a controlled clean room, under a laminar flow hood, the above raw homoharringtonine DS (210 grams) is dissolved in methanol (240 mL), at 30°C. The resulting alcoholic solution is filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized pilot rotary flask. Then, desionized water (2400mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated pilot rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of homoharringtonine DS is kept under vacuum and rotation is continued during appearance of white crystals of pure homoharringtonine. The stirring is continued until no more crystal occurs. Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water (450 mL x 2). The white cryitals are then dried using high vacuum at 60°C for 48 hours. Overall yield is 88% from potential content of homoharringtonine in raw semi-synthetic homoharringtonine. All operations were documented prior to start the process and full current Good Manufacturing Practices were applied. This clinical batch corresponds to 40,000 therapeutic units dosed at 5mg.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 11 shows HPLC chromatogram before and after crystallization. Total of related impurities of homoharringtonine DS is 0.03%.
For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC), thermogravimetry (TD), 2D NMR, solid NMR and X-ray powder diffractometry.
Infrared Spectrometry:
Identical IR spectra were obtained by either the KBr pellet and/or mineral oil mull preparation technique. Figure 3 shows typical infrared spectrum (KBr) for unambiguous identification at the solid state of the crystalline homoharringtonine obtained by this process. A series of sharp absorption bands are noted at 612, 703, 771 , 804, 826, 855, 879, 932, 1029, 1082, 1119,
1135, 1161 , 1191 , 1229, 1274, 1344, 1367, 1436, 1457, 1488, 1505, 1653, 1743, 2814, 2911 ,
2958, 3420, and 3552 cm“1
Differential Scanning Calorimetry (DSC) And Thermogravimetry (TG)
Measurement of DSC and TG were obtained on a Mettler Toledo STAR System. Approximately 11 mg of homoharringtonine drug substance were accurately weighed (10.6251 mg) into a DSC pan. The sample was heated from 25°C to 250°C at a rate of 5°C/min. The
DSC data were obtained following a standard method in the art. The DSC curve of crystalline homoharringtonine drug substance (Figure 1), exhibits a melting endotherm at 145.6 °C.
Melting range performed by the capillary method (Bucchi Apparatus) gave 143-145°C. Literature indicated 144-146°C [Anonymous, Acta Bot. Sin. 22, 156 (1980) cited by L. Huang and Z. Xue, Cephalotaxus Alkaloids, in “The Alkaloids”, vol. XXIII, pp157, (1988).
Crystallization medium was not published. This is the only literature reference regarding melting point of a crystalline form of HHT] X-Ray Powder Diffraction
X-ray powder diffraction pattern was collected on a INEL microdiffractomer, model
DIFFRACTINEL. Powdered homoharringtonine DS was packed in a glass capillary tube and was analyzed according to a standard method in the art. The X-ray generator was opered at 45 kV and 40 mA, using the copper Kalpha line as the radiation source. The sample was rotated along the chi axis and data was collected between 0 and 120 deg 2-theta. A collection time of 1200 sec was used. As showed on Figure 2, the x-ray powder diffraction for this crystalline form of homoharringtonine shows a typical pattern including major reflection peaks at approximately 7.9, 9.2, 10.9, 14.9 16.0, 17.7, 19.5, 19.7, 21.78, 23.1 , 25.3, 25.4 and 25.7 deg 2-theta.
Example 3: Preparation of homoharringtonine drug substance by purification of a commercial sample of impure homoharringtonine from Chinese source
A. Analytical profile of starting product
Analytical HPLC chromatogram of natural homoharringtonine (China National Pharmaceutical) is displayed on Figure 12 (bottom left).
B. Chromatography of Natural Homoharringtonine
Natural homoharringtonine (25 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 200 mm; length: 1000 mm) containing 12,000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi, flow-rate 120 IJhour). Unwanted fractions are discarded based upon bypassed in-line UV spectrophotometric detector. Kept fractions are collected in 22 separate stainless steel containers which are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase) and equipped with a diode array detector. After discarding of the fractions representing more than 0.5 % of the total content of homoharringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure in using a mechanically stirred thin film evaporator. Then crude concentrated solution of homoharringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 0.5 %.
C. Crystallization of homoharringtonine DS
In a controlled clean room, under a laminar flow hood, the above chromatographied homoharringtonine DS (18 grams) is dissolved in methanol (35 mL), at 30°C. The resulting alcoholic solution is filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized pilot rotary flask. Then, desionized water (300 mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated pilot rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of homoharringtonine DS is kept under vacuum and rotation is continued during appearance of white crystals of pure homoharringtonine. The stirring is continued until no more crystal occurs.
Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water
(50 mL x 2). The white crystals are then dried using high vacuum at 60°C for 48 hours. Overall yield is 84% from potential content of homoharringtonine in raw semi-synthetic homoharringtonine. All operations were documented prior to start the process and full current
Good Manufacturing Practices were applied.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 12 (bottom right) shows HPLC chromatogram after crystallization. Total of related impurities of homoharringtonine DS is 0.05%.
For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC), thermogravimetry (TD), 2D NMR, solid NMR and X-ray powder diffractometry. Infrared Spectra, Differential Scanning Calorimetry (DSC) and X-Ray Powder Diffraction gave patterns strictly superimposable to the one of example 2 obtained from semi-synthetic homoharringtonine (Figure 3, 1 , and 2, respectively).
………………………………….
KOREAN PAPER.. LINK
Title: 한국산 개비자(Cephalotaxus koreans)에서의 Harringtonine과 Homoharringtonine의 확인 및 함량 분석
Author: 박호일 ; 이연 (한국생물공학회)
Source: 한국생물공학회지 = Korean journal of biotechnology and bioengineering; ISSN:1225-7117 @ 1225-7117 @ ; VOL.11; NO.6; PAGE.689-695; (1996)
Pub.Country: Korea
Language: Korean
Abstract: Harringtonine and homoharringtonine known as anti-cancer agents were isolated from Korean native plumyew(Cephalotaxus koreana) using column chromatography(CHCl3:MeOH=19:1, Rf=0.28). The structure of the mixture of two compounds was characterized by 1H-NMR. Comparison of our spectra of harringtonine and homoharringtonine with previously reported ones indicated that the two are identical. The contents of harringtonine and homoharringtonine in the needles, stems, and roots of Korean native plumyew were determined by high performance liquid chromatography(HPLC). The contents of both compounds varied with the site of location and the part of plant. The content of harringtonine was higher in needles and roots than in stems, whereas the content of homoharringtonlne was lower than harringtonine. Homoharringtonine contents in needles at Mt. Palgong, Mt. Dukyu, Mt. Baekyang, Mt. Jiri, and Namhae were higher than in stems and roots. But homoharringtonine contents in needles al Mt. Jokye and Jindo were lower than in stems and roots.
http://img.kisti.re.kr/originalView/originalView.jsp
……………………………………………………………………………….
SYNTHESIS OF HOMOHARRINGTONINE AND SEPARATION OF ITS STEREOMERS
-
[PDF]
Chapter 1 Drug Discovery from Plants – Springer
LC-NMR-MS and LC-SPE-NMR to accelerate their future discovery. Keywords …..Ceflatonine (34), a synthetic version of homoharringtonine produced by.
…………………………………………………………………………….
References
- “Synribo (omacetaxine) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 18 February 2014.
- “SYNRIBO (omacetaxine mepesuccinate) injection, powder, lyophilized, for solution [Cephalon, Inc.]”. DailyMed. Cephalon, Inc. October 2012. Retrieved 18 February 2014.
- Sweetman, S, ed. (14 November 2012). Omacetaxine Mepesuccinate. “Martindale: The Complete Drug Reference”. Medicines Complete(Pharmaceutical Press).
- Li, Y. F.; Deng, Z. K.; Xuan, H. B.; Zhu, J. B.; Ding, B. H.; Liu, X. N.; Chen, B. A. (2009). “Prolonged chronic phase in chronic myelogenous leukemia after homoharringtonine therapy”. Chinese medical journal122 (12): 1413–1417. PMID 19567163. edit
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J. (2009). “Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009”. Cancer 115 (23): 5382–5393.doi:10.1002/cncr.24601. PMID 19739234. edit
- Wu, L.; Li, X.; Su, J.; Chang, C.; He, Q.; Zhang, X.; Xu, L.; Song, L.; Pu, Q. (2009). “Effect of low-dose cytarabine, homoharringtonine and granulocyte colony-stimulating factor priming regimen on patients with advanced myelodysplastic syndrome or acute myeloid leukemia transformed from myelodysplastic syndrome”. Leukemia & Lymphoma50 (9): 1461. doi:10.1080/10428190903096719. edit
- Gu, L. F.; Zhang, W. G.; Wang, F. X.; Cao, X. M.; Chen, Y. X.; He, A. L.; Liu, J.; Ma, X. R. (2010). “Low dose of homoharringtonine and cytarabine combined with granulocyte colony-stimulating factor priming on the outcome of relapsed or refractory acute myeloid leukemia”.Journal of Cancer Research and Clinical Oncology 137 (6): 997–1003.doi:10.1007/s00432-010-0947-z. PMID 21152934. edit
- Kantarjian, H. M.; Talpaz, M.; Santini, V.; Murgo, A.; Cheson, B.; O’Brien, S. M. (2001). “Homoharringtonine”. Cancer 92 (6): 1591–1605.doi:10.1002/1097-0142(20010915)92:6<1591::AID-CNCR1485>3.0.CO;2-U. PMID 11745238. edit
- Wetzler M, Segal D. Omacetaxine as an Anticancer Therapeutic: What is Old is New Again. Current Pharmaceutical Design 2011;17:59-64
- Concise total synthesis of (±)-cephalotaxine via a transannulation strategy: Development of a facile reductive oxy-nazarov cyclization
Org Lett 2011, 13(13): 3538 - The first semi-synthesis of enantiopure homoharringtonine via anhydrohomoharringtonine from a preformed chiral acyl moiety
Tetrahedron Lett 1999, 40: 2931 - Synthesis of homoharringtonine and its derivative by partial esterification of cephalotaxine
Tetrahedron Lett 1982, 23(34): 3431 - Construction of chiral tertiary alcohol stereocenters via the (2,3)-Meisenheimer rearrangement: Enantioselective synthesis of the side-chain acids of homoharringtonine and harringtonine
J Org Chem 2013, 78(2): 339 - Studies in Cephalotaxus alkaloids. Stereospecific total synthesis of homoharringtonine
J Org Chem 1983, 48(26): 5321 - Chemistry – A European Journal, 2008 , vol. 14, 14 pg. 4293 – 4306
| WO2000040269A2 * | Jan 5, 2000 | Jul 13, 2000 | Clarence C Lee | Pharmaceutical compositions for treatment of diseased tissues |
| WO2002032904A1 * | Oct 17, 2000 | Apr 25, 2002 | Oncopharm Corp | New cephalotaxanes, their method of preparation and their use in treatment of cancers, leukemias, parasites including thus resistant to usual chemotherapeutic agents and as reversal agents |
| EP0393575A1 * | Apr 17, 1990 | Oct 24, 1990 | G.D. Searle & Co. | Neoplasia treatment compositions containing antineoplastic agent and side-effect reducing protective agent |
| USH271 * | Dec 18, 1985 | May 5, 1987 | The United States Of America As Represented By The Secretary Of The Army | Treatment of malaria with esters of cephalotaxine |
| US7169774 | Jun 25, 2004 | Jan 30, 2007 | Stragen Pharma S.A. | Cephalotaxane derivatives and their processes of preparation and purification |
| US7842687 | May 25, 2006 | Nov 30, 2010 | Chemgenex Pharmaceuticals, Inc. | Cephalotaxane derivatives and their processes of preparation and purification |
| US8466142 | Mar 3, 2009 | Jun 18, 2013 | Sloan-Kettering Institute For Cancer Research | Cephalotaxus esters, methods of synthesis, and uses thereof |
| Reference | ||
|---|---|---|
| 1 | * | KANTARJIAN H.M. ET AL: “Chronic myelogenous leukemia – Progress at the M. D. Anderson Cancer Center over the past two decades and future directions: First Emil J Freireich Award Lecture.” CLINICAL CANCER RESEARCH, (1997) 3/12 II (2723-2733). , XP001095529 |
| 2 | * | LEVY, VINCENT (1) ET AL: “Subcutaneous homoharringtonine (SQ HHT ): 1. Pharmacokinetic study in dogs and HHT determination in blood in using LC-MS method.” BLOOD, (NOVEMBER 16, 2001) VOL. 98, NO. 11 PART 2, PP. 179B. HTTP://WWW.BLOODJOURNAL.ORG/. PRINT. MEETING INFO.: 43RD ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY, PART 2 ORLANDO, FLORIDA, USA DECEMBER 07-11, 2001 , XP001095449 |
| 3 | * | LEVY, VINCENT (1) ET AL: “Subcutaneous homoharringtonine (SQ HHT ): 2. Tolerance in humans and case report of a refractory patient with AML treated by very small dose of SQ HHT.” BLOOD, (NOVEMBER 16, 2001) VOL. 98, NO. 11 PART 2, PP. 202B. HTTP://WWW.BLOODJOURNAL.ORG/. PRINT. MEETING INFO.: 43RD ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY, PART 2 ORLANDO, FLORIDA, USA DECEMBER 07-11, 2001 , XP001095450 |
| 4 | * | WHAUN J M ET AL: “TREATMENT OF CHLOROQUINE -RESISTANT MALARIA WITH ESTERS OF CEPHALOTAXINE HOMOHARRINGTONINE.” ANN TROP MED PARASITOL(1990) 84(3), 229-237, XP008006193 |
1H NMR

13 CNMR

HPLC

Garden Cress Extract Kills 97% of Breast Cancer Cells in Vitro

Garden Cress Extract Kills 97% of Breast Cancer Cells in Vitro: Garden cress, like broccoli, is a cruciferous-family vegetable but is unique because it contains very high amounts of BITC (benzyl isothiocyanate) which has emerged as a powerful anti-cancer compound. In this study, BITC was seen to kill 97% of ER- breastcancer cells (MDA-MB-231) after 24 hours of treatment. For comparison, the same dose of sulforaphane from broccoli killed only 75% of the cancer cells.

In other research, BITC has been found to slow the rate of breast cancer metastasizing by 86% and when given to mice, resulted in breast tumors 53% smaller than in untreated mice. BITC is now being intensively studied for a variety of cancers and has been shown in lab studies to be active against melanoma, glioma, prostate cancer, lung cancer, ovarian cancer, pancreatic cancer and others. Garden cress is one of the best sources of BITC. Other good sources include cabbage, Indian cress, Japanese radish (in particular Karami daikon) and, quite surprisingly, papaya seeds. As with othercruciferous vegetables, the best way to eat cress is raw in order to maximize the delivery of BITC.
http://www.ncbi.nlm.nih.gov/pubmed/17121941
http://extension.usu.edu/files/publications/publication/HG_Garden_2006-05.pdf
and
http://nopr.niscair.res.in/bitstream/123456789/12732/1/IJNPR%202(3)%20292-297.pdf
BITC

Botanical name: Lepidium sativum L.
Family: Brassicaceae = Cruciferae
Common names. English: cress, common cress, garden cress, land cress, pepper cress; Spanish: mastuerzo, mastuerzo hortense, lepidio, berro de jardín (Spain), berro de sierra, berro hortense (Argentina), escobilla (Costa Rica); Catalan: morritort, morrisà, Portuguese and Galician: masturco, mastruco, agrião-mouro, herba do esforzo; Portuguese: mastruco do Sul, agrião (Brazil); Basque: buminka, beatzecrexu
Synonyms/Common Names/Related Substances:
- Alpha-linolenic acid (ALA), agrião (Portuguese), agrião-mouro (Portuguese, Galician), beatzecrexu (Basque), berro de jardín (Spanish), berro de tierra (Spanish), berro hortense (Spanish), benzyl isothiocyanate (BITC), Brassicaceae (family), bran, buminka (Basque), common cress, cress, docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), escobilla (Spanish), endosperm, fiber, garden cress seed oil (GCO), garden pepper grass, glucosinolates, glutamic acid, herba do esforzo (Portuguese, Galician), hurf (Arabic), indoles, isothiocyanates, kardamon (Greek), land cress, linoleic acid (LA), lectin, lepidio (Spanish), Lepidium sativium, Lepidium sativum, leucine, mastruco (Portuguese, Galician), mastruco do sul (Portuguese), mastuerzo (Spanish), mastuerzo hortense (Spanish), methanol, morrisá (Catalan), morritort (Catalan), nasturtium (Latin), nasum torcere (Latin), omega-3 fatty acid, pepper cress, pepper grass, pepperwort, sulforaphane, tuffa’ (Arabic), turehtezuk (Persian), water cress, whole meal.
- Combination product example: SulforaWhite (a liposomal preparation that contains Lepidium sativum sprout extract, glycerin, lecithin, phenoxyethanol, and water).
Garden cress [commonly known as aliv in Marathi or halim in Hindi] is a green, cool-season perennial plant used as a leafy vegetable, typically used as a garnish. Undisturbed, the plant can grow to a height of two feet with minimal maintenance. When mature, garden cress produces white or light-pink flowers, and small seed pods. It has long leaves at the bottom of the stem and small, bright-green, feather-like leaves arranged on opposite sides of its stalks at the top.
Garden Cress, also called Pepper Wort, is an herb that is botanically known as Lepidium Sativum. It is referred to as ‘Aliv’ in Marathi and ‘Halim’ in Hindi. Belonging to the family Cruciferae, it is grown in all parts of India and is often used in the Indian cuisine. The leaves, roots, as well as seeds of this plant are used in cooking as they are extremely nutritious and also therapeutic in nature. The flowers of this plant are either white or light-pink in color.
This herb is the best source of iron and is hence recommended in the treatment of iron-deficiency anemia. It is also rich in folate, calcium, ascorbic acid, tocopherol, and beta-carotene. Garden Cress seeds are loaded with not just protein, but also linoleic and arachidic fatty acids. Since they contain phytochemicals that mimic estrogen to some extent, intake of these seeds is known to regulate menstruation and stimulate milk production in lactating mothers. That is precisely why women are given foods containing Garden Cress following childbirth.
The blood-purifying as well as antioxidant properties of this amazing plant are well documented. Hence, its regular consumption can greatly help to boost one’s immunity and prevent a gamut of diseases. It acts as a general tonic and can also help to increase the libido naturally. Since the testae of these seeds contain mucilage, they are invaluable in the management of both dysentery and constipation. The whole plant, along with its seeds, is said to be good for the eyes too. Hence, it is advisable to add it raw to salads, sandwiches, and chutneys, or to simply use it as a garnish along with coriander leaves for any food item.
Pregnant women should avoid taking Garden Cress in any form because it has the ability to induce uterine contractions and thereby trigger a spontaneous abortion. Also, since it is goitrogenic in nature, it may not be suitable for patients suffering from hypothyroidism. The oil derived from Garden Cress seeds is edible and can therefore be used as a cooking medium; however, some people may experience symptoms of indigestion due to its use. Such individuals should discontinue using this oil or mix it with some other edible oil, so as to dilute it and reduce its adverse effects.
Cress (Lepidium sativum), sometimes referred to as garden cress to distinguish it from similar plants also referred to as cress, is a rather fast-growing, edible herb. Garden cress is genetically related to watercress and mustard, sharing their peppery, tangy flavor and aroma. In some regions, garden cress is known as mustard and cress, garden pepper cress, pepperwort pepper grass, or poor man’s pepper.[1][2]
This annual plant can reach a height of 60 cm (~24 inches), with many branches on the upper part. The white to pinkish flowers are only 2 mm (1/12 of an inch) across, clustered in branched racemes.[3][4]
Origin of the name
Cultivation of this species, which is native to Southwest Asia (perhaps Persia) and which spread many centuries ago to western Europe, is very old, as is shown by the philological trace of its names in different Indo-European languages. These include the Persian word turehtezuk, the Greek kardamon, the Latin nasturtium and Arabic tuffa’ and hurf. In some languages there is a degree of confusion with watercress. It seems that the meaning of the word nasturtium (nasum torcere, because its smell causes the nose to turn up) must have been applied initially to garden cress, as both Pliny and Isidoro de Sevilla explain. The confusion remains with the terms used by the Hispano-Arabs. The word hurf is applied without distinction to watercress and garden cress (several species certainly of up to three different genera: Nasturtium, Lepidium and Cardaria). Thus the medieval agronomists of Andalusia went as far as differentiating between several hurf, such as hurf abyad, hurf babili, hurf madani….
Garden cress in agriculture
Garden cress is commercially grown in England, France, the Netherlands and Scandinavia.[5]
Cultivation of garden cress is practical on both mass scales and on the individual scale. Garden cress is suitable for hydroponic cultivation and thrives in slightly alkaline water. In many local markets, the demand for hydroponically grown cress can exceed available supply, partially because cress leaves are not suitable for distribution in dried form, so can be only partially preserved. Consumers commonly acquire cress as seeds or (in Europe) from markets as boxes of young live shoots.[5]
Edible shoots are typically harvested in one to two weeks after planting, when they are 5–13 cm (2 – 5 inches) tall.[6]
Properties, uses and cultivation
Xenophon (400 BC) mentions that the Persians used to eat this plant even before bread was known. It was also familiar to the Egyptians and was very much appreciated by the Greeks and Romans, who were very fond of banquets rich in spices and spicy salads. Columela (first century) makes direct reference to the cultivation of garden cress. In Los doce libros de Agricultura, he writes: ” …immediately after the calends of January, garden cress is sown out… when you have transplanted it before the calends of March, you will be able to harvest it like chives, but less often… it must not be cut after the calends of November because it dies from frosts, but can resist for two years if it is hoed and manured carefully… there are also many sites where it lives for up to ten years” (Book XI). The latter statements seem to indicate that he is also speaking of the perennial species L. Iatifolium, as L. sativum is an annual.
Almost all of the Andalusian agronomists of the Middle Ages (Ibn Hayyay, Ibn Wafid, Ibn al-Baytar, Ibn Luyun, Ibn al-Awwam) and many of the doctors, such as Maimonides, mention garden cress. Ibn al-Awwam also includes references from Abu al-Jair, Abu Abdalah as well as from Nabataean agriculture and, among other comments, he says: “Garden cress is sown between February and April (in January in Seville). It has small seeds which are mixed with earth for sowing to prevent the wind carrying them away…. It is harvested in May and is grown between ridges, in combination/conjunction with flax cultivation.”
Many of the authors of the old oriental and Mediterranean cultures emphasized the medicinal properties of cress, especially as an antiscorbutic, depurative and stimulant. Columela notes its vermifugal powers. Ibn al-Awwam refers to certain apparently antihistaminic properties, since it was used against insect bites and also as an insect repellent, in the form of a fumigant. It was perhaps Ibn al-Baytar, an Andalusian botanist (eighth century), who collected most information on its properties, summarizing the opinions of other authors such as El Farcy, who says that it incites coitus and stimulates the appetite; Ibn Massa, according to whom it dissipates colic and gets rid of tapeworms and other intestinal worms; or Ibn Massouih, who mentions that it eliminates viscous humours. Ibn al-Baytar also says that it is administered against leprosy, is useful for renal “cooling” and that, if hair is washed with garden cress water, it is “purified” and any loss is arrested.
In Iran and Morocco, the seeds are used as an aphrodisiac. In former Abyssinia, an edible oil was obtained from the seeds. In Eritrea, it was used as a dyestuff plant. Some Arab scholars have attributed garden cress’s reputation among Muslims to the fact that it was directly recommended by the Prophet.
Garden cress’s main use was always as an aromatic and slightly pungent plant. Not only in antiquity but also in the Middle Ages it enjoyed considerable prestige on royal tables. The young leaves were used for salads. The ancient Spartans ate them with bread. This use still continues and they are also eaten with bread and butter or with bread to which lemon, vinegar or sugar is added. However, it is mainly used nowadays in the seedling stage, the succulent hypocotyls being added to salads and as a garnish and decoration for dishes.
The roots, seeds and leaves have been used as a spicy condiment. Columela explains how oxygala, a type of curd cheese with herbs, was prepared: “Some people, after collecting cultivated or even wild garden cress, dry it in the shade and then, after removing the stem, add its leaves to brine, squeezing them and placing them in milk without any other seasoning, and adding the amount of salt they consider sufficient…. Others mix fresh leaves of cultivated cress with sweetened milk in a pot…”.
L. Iatifolium L. stands out for its horticultural interest; although it grows spontaneously on the edges of rivers and lakes, it is also occasionally grown in the same way as L. sativum. Its young leaves can be used for salads; the ancient Greeks and Romans used to grow it for this purpose. Its leaves and seeds were also used as a spicy condiment. Several sauces are prepared with its leaves, including in particular the bitter sauce of the paschal lamb of the Jews. The seeds of this species were known in England as the poor people’s pepper. The roots have been used on occasion as a substitute for radish.
In the fifteenth century, we know through Alonso de Herrera that garden cress was one of the vegetables most widely eaten in Castile. During the sixteenth century, obstinate attempts were made to introduce it into America. Right up to the beginning of the nineteenth century, its cultivation in Spain continued to be important, since Boutelou and Boutelou (1801) deal specifically with this crop in their Tratado de la huerta, commenting on the existence of several cultivars. At present, the cultivation of cress is very occasional in countries such as Spain and France. Water cress, in competition with garden cress, has eclipsed the cultivation of the latter. However, this is not the case in other central European countries or the United Kingdom, where its use is normal and the system of cultivation has changed substantially.
Botanical description
Cress is an annual, erect herbaceous plant, growing up to 50 cm. The basal leaves have long petioles and are lyrate-pinnatipartite; the caulinar leaves are laciniate-pinnate while the upper leaves are entire. The inflorescences are in dense racemes. The flowers have white or slightly pink petals, measuring 2 mm. The siliquae measure 5 to 6 x 4 mm, are elliptical, elate from the upper half, and glabrous. Cress flowers in the wild state between March and June.
It is an allogamous plant with self-compatible and self-incompatible forms and with various degrees of tolerance to prolonged autogamy. There are diploid forms, 2n = 2x = 16, and tetraploid forms, 2n = 4x =32. A degree of variability is noted in the character of the basal leaves which are cleft or split to a greater or lesser degree, a character which is controlled by a single incompletely dominant gene.
Ecology and phytogeography
Cress is a plant that is well suited to all soils and climates, although it does not tolerate frosts. In temperate conditions, it has a very rapid growth rate. It grows subspontaneously in areas transformed by humans, close to crops or human settlements. It appears in this way on the Iberian peninsula, mainly in the eastern regions.
Wild cress extends from the Sudan to the Himalayas. Most authors consider it to be a native of western Asia, whence it passed very quickly to Europe and the rest of Asia as a secondary crop, probably associated with cultivars of flax. Vavilov considers its main centre to be Ethiopia, where he found the widest variability; the Near East, central Asia and the Mediterranean are considered secondary centres. It is now naturalized in numerous parts of Europe, including the British Isles.
Cress in cookery
Genetic diversity
The genus Lepidium is made up of about 150 species, distributed throughout almost all temperate and subtropical regions of the world. On the Iberian peninsula and the Balearic Islands, at least 20 species or subspecies exist among the autochthonous and allochthonous taxa, some genetically close to L. sativum. Seven of them are exclusively endemic to the peninsula or, at the very most, are common with North Africa. Other close species are L. campestre (L.) R. Br. and L. ruderale L. which also have edible leaves. The leaves of L. campestre are used to prepare excellent sauces for fish.
Common cress (L. sativum L.), with regard to the anatomy of the leaf, stem and root, has been divided into three botanical varieties: vulgare, crispum and latifolium. The latter is the most mesomorphic, crispum the most xeromorphic and vulgare intermediate.
At present, most of the studies on the variability and development of new cultivars are being carried out in liaison with the VIR of St Petersburg, where there is a good collection of material. Of the 350 forms of garden cress studied in the Ukraine, Uzkolistnyti 3 was the best, being highly productive and of good quality. It is being used as the basis of improvement programmes, as it appreciably surpasses the best Soviet varieties in production and quality. Other cultivars well suited to European Russia are Tuikers Grootbladige (broad-leaved) and the lines Mestnyi k 137, k 106 and k 115. Of the types most cultivated in Europe, Early European, Eastern, Dagestan and Entire Leaved stand out, being distinguished by the length and shape of the leaf, earliness and susceptibility to cold. In Western Europe, one broad leaved type is especially appreciated (Broad Leaved French) as are curly types (Curly Leaved), the latter being used extensively to garnish dishes. In Africa, there are red, white and black varieties.
This crop is also arousing interest in Japan, and collecting expeditions to Nepal have been organized. Some specimens collected during an expedition to Iraq in 1986 are now stored in Abu Ghraib and in Gratersleben, Germany. There are also small collections of L. sativum in the PGRC in Addis Ababa (Ethiopia), at the ARARI of Izmir in Turkey and in Bari, Italy. At the Universidad Politécnica de Madrid there are accessions of 20 species of Lepidium, while the BGV of the Córdoba Botanical Garden keeps germplasm of the southern Iberian species of the genus.
| Nutritional value per 100 g (3.5 oz) | |
|---|---|
| Energy | 134 kJ (32 kcal) |
| Carbohydrates | 5.5 g |
| – Sugars | 4.4 g |
| – Dietary fiber | 1.1 g |
| Protein | 2.6 g |
| Vitamin A equiv. | 346 μg (43%) |
| – beta-carotene | 4150 μg (38%) |
| – lutein and zeaxanthin | 12500 μg |
| Thiamine (vit. B1) | 0.08 mg (7%) |
| Riboflavin (vit. B2) | 0.26 mg (22%) |
| Niacin (vit. B3) | 1 mg (7%) |
| Pantothenic acid (B5) | 0.247 mg (5%) |
| Vitamin B6 | 0.247 mg (19%) |
| Folate (vit. B9) | 80 μg (20%) |
| Vitamin C | 69 mg (83%) |
| Vitamin E | 0.7 mg (5%) |
| Vitamin K | 541.9 μg (516%) |
| Calcium | 81 mg (8%) |
| Iron | 1.3 mg (10%) |
| Magnesium | 38 mg (11%) |
| Manganese | 0.553 mg (26%) |
| Phosphorus | 76 mg (11%) |
| Potassium | 606 mg (13%) |
| Link to USDA Database entry Percentages are roughly approximated using US recommendations for adults. Source: USDA Nutrient Database |
|
Garden cress is added to soups, sandwiches and salads for its tangy flavor.[6] It is also eaten as sprouts, and the fresh or dried seed pods can be used as a peppery seasoning (haloon).[5] In England, cut cress shoots are commonly used in sandwiches with boiled eggs, mayonnaise and salt.

Garden cress can grow almost anywhere.
Nutrition profile
Garden cress is an important source of iron, folic acid, calcium, vitamins C, E and A. The seed contains arachidic and linoleic fatty acids. The seeds are high in calories and protein, whereas the leaves are an excellent source of vitamin A, C and folate.
| Energy | 30 Kcal |
| Carbohydrates | 5.5 g |
| Dietary fibre | 1.1 g |
| Protein | 2.6 g |
| Fat | 0.7 g |
| Vitamin A | 346 mcg |
| Folate | 80 mcg |
| Vitamin C | 69 mg |
| Calcium | 81 mg |
| Iron | 1.3 mg |

Both the leaves and stems of cress can be eaten raw in salads or sandwiches, and are sometimes called cress sprouts. When buying cress, look for firm, evenly coloured, rich green leaves. Avoid cress with any signs of slime, wilting, or discoloration. If stored in plastic, it can last up to five days in a refrigerator. To prolong the life of cress, place the stems in a glass container with water and cover them, refrigerating the cress until it is needed.
Cultivation practices
Cress is an easily grown plant with few requirements. It can be broadcast after the winter frosts or throughout the year in temperate climates. However, Boutelou and Boutelou (1801) were already recommending sowing in shallow furrows, which enables surplus plants to be thinned out and facilitates hoeing. Sowing has to be repeated every 15 to 20 days so that there is no shortage of young shoots and new leaves for salads – the leaves of earlier sowings begin to get tough and are no longer usable. The seed sprouts four or six days after sowing, depending on the season, and the leaves are ready for consumption after two or three weeks.
The usual form of cultivation continues to be as described, with 15 to 20 cm between rows and the use of irrigation in the summer, since they are lightly rooted seedlings which can dry up in a few days. Its growth is very rapid and harvesting can begin in the same month as sowing, with yields reaching 6 tonnes per hectare.
Health benefits of garden cress
For women’s health
Emenagogue: Garden cress has mild oestrogenic properties. It helps to regulate the menstrual cycle.
Galactogogue: Kheer made of garden cress seeds increases milk production and secretion in lactating mothers. Because of its high iron and protein content, it is often given post-partum to lactating mothers.
Aphrodisiac: Garden cress helps to improve libido.
For the gastro intestinal tract
Garden cress helps purify blood and stimulate appetite. It is used during constipation as a laxative and a purgative. Paste made of the seeds can be taken internally with honey to treat amoebic dysentery. The mucilage of the germinating seeds allays the irritation of the intestines in dysentery and diarrhoea. Garden cress crushed and drunk with hot water is beneficial to treat colic especially in infants.
For the respiratory tract
Garden cress seeds are good expectorants and when chewed they treat sore throat, cough, asthma and headache. The aerial parts are used in the treatment of asthma and cough.
For anaemia
Garden cress seeds being the richest source of non-haeme iron [iron found in haemoglobin which is an easily absorbed dietary iron.] help to increase the haemoglobin levels. When taken regularly, it helps to alleviate anaemia. It is advisable to have vitamin C half an hour after consumption of these seeds as it enhances iron absorption.

For diabetes
The seed coat of germinating seeds contains mucilage, which has a phytochemical called lepidimoide. Studies show that seeds of the plant lower the glycemic response to a test meal.
note sodium 2-O-rhamnopyranosyl-4-deoxy-threo-hex-4-enopyranosiduronate (designated lepidimoide)
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1075667/
epi-Lepidimoide
Sodium 6-deoxy-2-O-(4-deoxy-β-L-threo-hex-4-enopyranuronosyl)-α-L-glucopyranose
 cas 145039-76-5 and 157676-09-0
cas 145039-76-5 and 157676-09-0
The total synthesis of the unsaturated disaccharide, lepidimoide 4-deoxy--l-threo-hex-4-enopyranuronosyl-(1->2)-l-rhamnopyranose sodium salt, has been carried out from d-glucose and l-rhamnose (Tetrahedron Lett. 1993, 34, 2653), but the process is very long and complicated. A method for more easily producing this compound and in large quantities is necessary for further research. We have succeeded in conveniently synthesizing lepidimoide from okra (Hibiscus esculentus L.) fruit mucilage. At the same time, the isomer (epi-lepidimoide) was obtained as a byproduct. The structure was determined as the 4-deoxy--l-threo-hex-4-enopyranuronosyl-(1->2)-6-deoxy-l-glucopyranose sodium salt by spectral analysis. We found that lepidimoide easily epimerized to epi-lepidimoide in alkaline media. Both lepidimoide and epi-lepidimoide exhibited the same high activity in the cockscomb hypocotyls elongation test….Carbohydrate Research, Volume 339, Number 1, 2 January 2004 , pp. 9-19(11)
-
Dictionary of Natural Products, Supplement 3
books.google.co.in/books?isbn=0412604302John Buckingham – 1996 – ScienceLepidimoide. L-30020. 6-Deoxy-2-0-(4-deoxy-fi-L-lhreo-hex-4-enopyranuronosyl)-L- mannose, 9CI [157676-09-0] HOA—O …
lepidimoide
Sodium 2-O-L-rhamnopyranosyl-4-deoxy-alpha-L-threo-hex-4-eno-pyranosiduronateMolecular Formula: C12H17NaO10 Molecular Weight: 344.247149sodium;(2S,3R,4S)-3,4-dihydroxy-2-[(2S,3R,4R,5R,6S)-2,4,5-trihydroxy-6-methyloxan-3-yl]oxy-3,4-dihydro-2H-pyran-6-carboxylateSodium2-O-L-rhamnopyranosyl-4-deoxy-α-L-threo-hex-4-eno-pyranosiduronate
For cancer
Garden cress seeds contain antioxidants like vitamin A and E which help protect cells from damage by free radicals. Hence, these seeds have a chemoprotective [drugs which protect healthy tissue from the toxic effects of anticancer drugs] nature.
Anti-Cancer:
Being a family of Brassica family it has good anti cancer property. Garden cress seeds contain antioxidants like vitamin A and E which help protect cells from damage by free radicals. Hence, these seeds have a chemo protective nature.

Few years back garden cress seeds/ halim/ aliv was not a common food item or a familiar to be heard. But as years passed it’s popularity and it’s importance have been realized and now people are aware of some of the facts of these seeds. Though these facts are also accompanied by some myths. So I chose to write and clear few myths and doubts of these seeds so that maximum people can make use of it in their lives and improve quality of their diet and nutrition.
Nutritive value of these seeds is very high. It is available in almost all parts of the world. Its high nutritive value and cheaper availability makes it possible for people of all the sections of society to include in the diet and increase nutritive value of their meals. Garden cress seeds are very high in Iron and Folic acid content. These seeds are use as herbal medicine to treat iron deficiency anemia. People consuming 2tsp/day have seen to have good increased levels of hemoglobin over a period of 1-2 months. Garden cress seeds also contains calcium, ascorbic acid, tocopherol, and beta-carotene which helps to improve body’s immunity.Garden Cress seeds are loaded with not just protein, but also linoleic and arachidic fatty acids. Since they contain phytochemicals that resemble estrogen to some extent, intake of these seeds helps to regulate menstruation and stimulate milk production in lactating mothers. That is why women are given foods containing Garden Cress following childbirth.
Traditionally garden cress seeds were considered to be useful only during last few weeks of gestation and post delivery. It is considered to be hot food. But the truth is that these seeds have ability too increase uterine contraction. So in later stages of pregnancy it helps in inducing labour but if in case consumed in early stage of pregnancy (1st trimester) it leads to spontaneous abortion. It is also very carefully prescribed to a hypothyroid patients because it belongs to cruciferous family and is a goitrogen that prevent iodine absorption.

How to eat:
1. Roasted slightly with added salt.
2. Soaked in water then added to milk or juice.
3. Chikki or laddoo can be made. (preparation similar to til laddoo/chikki).
How much to eat:
Start with 1 tsp/day and then an be taken 1 tsp/2c a day.
Cress seeds have many more medicinal properties and researches are still on to find its benefits on health. Garden cress should be eaten in moderation. Excess consumption of these seeds may hv adverse effect on health.
For other things
Garden cress seeds are memory boosters because they contain arachidic and linoleic acids. They help gaining lean body mass because they are a good source of iron and protein. Research has proved that 60 per cent women have hair loss due to low iron levels and poor protein. A teaspoon of garden cress seeds soaked in lime water helps in iron absorption, which in turn strengthens hair. The plant is also used in treating bleeding piles. The leaves are mildly stimulant and diuretic, useful in scorbutic [related to or resembling scurvy] diseases and liver complaints. A paste of the seeds with water is applied to chapped lips, and against sunburn.
Side-effects
It is an abortifacient [substance that induces abortion], if had in excess. It contains goitrogens that prevent iodine absorption in thyroids and hence can lead to hypothyroidism. If large quantities of garden cress are consumed, the mustard oil it contains may cause digestive difficulties in some people who are sensitive to it. Therefore, garden cress should be eaten in moderation.
Other uses
Garden cress, known as chandrashoor, and the seeds, known as halloon[7] in India, are commonly used in the system of Ayurveda to prevent postnatal complications.[citation needed]
Garden cress seeds, since ancient times, have been used in local traditional medicine of India.[8] Garden cress seeds are bitter, thermogenic, depurative, rubefacient, galactogogue, tonic, aphrodisiac, ophthalmic, antiscorbutic, antihistaminic and diuretic. They are useful in the treatment ofasthma, coughs with expectoration, poultices for sprains, leprosy, skin disease, dysentery, diarrhoea, splenomegaly, dyspepsia, lumbago, leucorrhoea, scurvy and seminal weakness. Seeds have been shown to reduce the symptoms of asthma and improve lung function in asthmatics.[9]The seeds have been reported as possessing a hypoglycemic property[10] and the seed mucilage is used as a substitute for gum arabic and tragacanth.
Cress may be given to budgerigars.[11] The seeds are employed as poultice for removing pain, swelling etc.Some use it in the belief that it can cure asthma, bronchitis bleeding piles.[12]
Some use Lepidium sativum seeds for indigestion and constipation.[13]
Prospects for improvement
Most of the genetic improvement work on garden cress is being carried out in the CIS, with little or no work being done at present in the countries of western Europe. Mainly early cultivars with a prolonged production period and better cold tolerance are being developed.
Cress can be grown and used like white mustard. It germinates more slowly at low temperatures, the emergence period being three or four days longer. Shortening this period is an interesting improvement objective.
However, cress’s recovery and its greater presence on markets mainly depends on a modification of cultivation and marketing techniques. In countries such as the United Kingdom, where this vegetable is normally to be found at the markets, cultivation takes place in greenhouses throughout the year. The whole succulent hypocotyls of the very young seedlings are eaten. The seed is placed on the soil surface on soft, level beds. It is finely sprinkled with water and then covered with sackcloth which has been steam-sterilized and moistened. The latter is frequently wetted to maintain moisture and is removed when the seedlings reach 4 to 5 cm in height (after approximately seven days in spring and autumn and ten days in winter). The yellowish leaves turn green after two to three days.
The cress is harvested when the first pair of cotyledon leaves have developed and it is marketed in small bags or trays, sometimes together with seedlings of white mustard.
Garden cress and white pepper are sometimes sown in the plastic trays or bags in which they will be sold, generally in peat with a nutrient solution.
References
- Cassidy, Frederic Gomes and Hall, Joan Houston. Dictionary of American regional English, Harvard University Press, 2002. Page 97. ISBN 0-674-00884-7, ISBN 978-0-674-00884-7
- Staub, Jack E, Buchert, Ellen. 75 Exceptional Herbs for Your Garden Published by Gibbs Smith, 2008. ISBN 1-4236-0251-X, 9781423602514
- Vegetables of Canada. Published by NRC Research Press. ISBN 0-660-19503-8, ISBN 978-0-660-19503-2
- Boswell, John T. and Sowerby, James. English Botany: Or, Coloured Figures of British Plants. Robert Hardwicke, 1863. Page 215.
- Vegetables of Canada. NRC Research Press. ISBN 0-660-19503-8, ISBN 978-0-660-19503-2
- Hirsch, David P.. The Moosewood Restaurant kitchen garden: creative gardening for the adventurous cook. Ten Speed Press, 2005. ISBN 1-58008-666-7, ISBN 978-1-58008-666-0
- http://www.organicindia.com/PR_OH_chandrashoor.php
- The Wealth of Indian Raw Materials ,. New Delhi: Publication and information Directorate. 1979. pp. CSIR Vol 9, Page 71–72.
- NP, Archana; Anita, AM (2006). “A study on clinical efficacy of Lepidium sativum seeds in treatment of bronchial asthma”. Iran J Pharmacol Ther 5: 55–59.
- M, Eddouks; Maghrani M, Zeggwagh NA, Michel JB (2005). “Study of the hypoglycaemic activity of Lepidium sativum L. aqueous extract in normal and diabetic rats”. J Ethnopharmacol 97: 391–395.
- Budgerigars – Diets, PDSA.
- Bhatiya, KN (1996). Modern Approach to Batany. India: Surya publications. p. 516.
- Najeeb-Ur-Rehman, Mehmood MH, Alkharfy KM, Gilani AU, “Prokinetic and laxative activities of Lepidium sativum seed extract with species and tissue selective gut stimulatory actions. J Ethnopharmacol. 2011 Feb 2;







MARIZOMIB, Salinosporamide A
MARIZOMIB
http://www.ama-assn.org/resources/doc/usan/marizomib.pdf
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. 6-Oxa-2-azabicyclo[3.2.0]heptane-3,7-dione, 4-(2-chloroethyl)-1-[(S)-(1S)-2-
cyclohexen-1-ylhydroxymethyl]-5-methyl-, (1R,4R,5S)-
2. (1R,4R,5S)-4-(2-chloroethyl)-1-{(S)-[(1S)-cyclohex-2-en-1-yl]hydroxymethyl}-5-methyl-
6-oxa-2-azabicyclo[3.2.0]heptane-3,7-dione
MOLECULAR FORMULA C15H20ClNO4
MOLECULAR WEIGHT 313.8
MANUFACTURER Nereus Pharmaceuticals, Inc.
NOTE….Nereus Pharmaceuticals was acquired by Triphase Research and Development in 2012.
CODE DESIGNATION NPI-0052
CAS REGISTRY NUMBER 437742-34-2
Scripps Institution of Oceanography (Originator)
mp, 168–170° C. (authentic sample: 168–170° C., 169–171° C. in Angew. Chem. Int. Ed., 2003, 42, 355–357); mixture mp, 168–170C.
[α]23 D −73.2 (c 0.49, MeOH), −72.9 (c 0.55, MeOH, in Angew. Chem. Int. Ed., 2003, 42, 355–357);
FTIR (film) νmax: 3406, 2955, 2920, 2844, 1823, 1701, 1257, 1076, 1012, 785, 691 cm−1;
1H NMR (CDCl3, 500 MHz): δ 10.62 (1H, br), 6.42 (1H, d, J=10.5 Hz), 5.88 (1H, m), 4.25 (1H, d, J=9.0 Hz), 4.14 (1H, m), 4.01 (1H, m), 3.17 (1H, t, J=7.0 Hz), 2.85 (1H, m), 2.48 (1H, m), 2.32 (2H, m), 2.07 (3H, s), 1.91 (2H, m), 1.66 (2H, m), 1.38 (1H, m);
13C NMR (CDCl3, 125 MHz): δ 176.92, 169.43, 129.08, 128.69, 86.32, 80.35, 70.98, 46.18, 43.28, 39.31, 29.01, 26.47, 25.35, 21.73, 20.00;
HRMS (ESI) calcd. for (M−H)− C15H19ClNO4 312.1003, found 312.1003.
Marizomib, a highly potent proteasome inhibitor, is in early clinical development at Triphase Research and Development I Corp for the treatment of relapsed or relapsed/refractory multiple myeloma. Phase I clinical trials have also been carried out for the treatment of solid tumors and lymphoma; however, no recent developments have been reported for these studies.
HDAC inhibitors halt tumor cell differentiation and growth, and when combined with marizomib in preclinical in vitro and in vivo studies, show additive and synergistic antitumor activities.
The compound was discovered from a new marine-obligate gram-positive actinomycete (Salinispora tropica). Preclinical studies suggest that this next-generation compound may be superior to other proteasome inhibitors, with broader target inhibition, faster onset and longer duration of action, higher potency, and oral and intravenous availability. By inhibiting proteasomes, marizomib prevents the breakdown of proteins involved in signal transduction, which blocks growth and induces apoptosis in cancer cells.
In 2013, orphan drug designation was assigned in the U.S. for the treatment of multiple myeloma.
The compound was originally developed by Nereus Pharmaceuticals, which was acquired by Triphase Research and Development in 2012.
marizomib is a naturally-occurring salinosporamide, isolated from the marine actinomycete Salinospora tropica, with potential antineoplastic activity. Marizomib irreversibly binds to and inhibits the 20S catalytic core subunit of the proteasome by covalently modifying its active site threonine residues; inhibition of ubiquitin-proteasome mediated proteolysis results in an accumulation of poly-ubiquitinated proteins, which may result in the disruption of cellular processes, cell cycle arrest, the induction of apoptosis, and the inhibition of tumor growth and angiogenesis. This agent more may more potent and selective than the proteasome inhibitor bortezomib
Marizomib (NPI-0052) is an oral, irreversible ββ-lactone derivative that binds selectively to the active proteasomal sites. In vivo studies with marizomib demonstrate reduced tumor growth without significant toxicity in myeloma xenograft models. A phase I trial in refractory and relapsed MM is under way.
Salinosporamide A is a potent proteasome inhibitor used as an anticancer agent that recently entered phase I human clinical trials for the treatment of multiple myeloma only three years after its discovery.[1][2] This novel marine natural product is produced by the recently described obligate marine bacteria Salinispora tropica and Salinispora arenicola, which are found in ocean sediment. Salinosporamide A belongs to a family of compounds, known collectively as salinosporamides, which possess a densely functionalized γ-lactam-β-lactone bicyclic core.
Salinosporamide A was discovered by William Fenical and Paul Jensen from Scripps Institution of Oceanography in La Jolla, CA. In preliminary screening, a high percentage of the organic extracts of cultured Salinospora strains possessed antibiotic and anticancer activities, which suggests that these bacteria are an excellent resource for drug discovery.Salinospora strain CNB-392 was isolated from a heat-treated marine sediment sample and cytotoxicity-guided fractionation of the crude extract led to the isolation of salinosporamide A. Although salinosporamide A shares an identical bicyclic ring structure with omuralide, it is uniquely functionalized. Salinosporamide A displayed potent in vitro cytotoxicity against HCT-116 human colon carcinoma with an IC50 value of 11 ng mL-1. This compound also displayed potent and highly selective activity in the NCI’s 60-cell-line panel with a mean GI50 value (the concentration required to achieve 50% growth inhibition) of less than 10 nM and a greater than 4 log LC50 differential between resistant and susceptible cell lines. The greatest potency was observed against NCI-H226 non-small cell lung cancer, SF-539 CNS cancer, SK-MEL-28 melanoma, and MDA-MB-435 breast cancer (all with LC50 values less than 10 nM). Salinosporamide A was tested for its effects on proteasome function because of its structural relationship to omuralide. When tested against purified 20S proteasome, salinosporamide A inhibited proteasomal chymotrypsin-like proteolytic activity with an IC50 value of 1.3 nM.[3] This compound is approximately 35 times more potent than omuralide which was tested as a positive control in the same assay. Thus, the unique functionalization of the core bicyclic ring structure of salinosporamide A appears to have resulted in a molecule that is a significantly more potent proteasome inhibitor than omuralide.[1]
Salinosporamide A inhibits proteasome activity by covalently modifying the active site threonine residues of the 20S proteasome.
Biosynthesis

Salinosporamide A and B building blocks

Proposed biosynthesis of the nonproteinogenic amino-acid beta-hydroxycyclohex-2′-enylanine (3) (R = H or S~PCP) via a shunt in the phenylalanine biosynthetic pathway

Biosynthesis
It was originally hypothesized that salinosporamide B was a biosynthetic precursor to salinosporamide A due to their structural similarities.
It was thought that the halogenation of the unactivated methyl group was catalyzed by a non-heme iron halogenase.[4][5]Recent work using 13C-labeled feeding experiments reveal distinct biosynthetic origins of salinosporamide A and B.[4][6]
While they share the biosynthetic precursors acetate and presumed β-hydroxycyclohex-2′-enylalanine (3), they differ in the origin of the four-carbon building block that gives rise to their structural differences involving the halogen atom. A hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) pathway is most likely the biosynthetic mechanism in which acetyl-CoA and butyrate-derived ethylmalonyl-CoA condense to yield the β-ketothioester (4), which then reacts with (3) to generate the linear precursor (5).
The first stereoselective synthesis was reported by Rajender Reddy Leleti and E. J.Corey.[7] Later several routes to the total synthesis of salinosporamide A have been reported.[7][8][9][10]
In vitro studies using purified 20S proteasomes showed that salinosporamide A has lower EC50 for trypsin-like (T-L) activity than does Bortezomib. In vivo animal model studies show marked inhibition of T-L activity in response to salinosporamide A, whereas bortezomib enhances T-L proteasome activity.
Initial results from early-stage clinical trials of salinosporamide A in relapsed/refractory multiple myeloma patients were presented at the 2011 American Society of Hematology annual meeting.[11] Further early-stage trials of the drug in a number of different cancers are ongoing.[12]
- Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W (2003). “Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora”. Angew. Chem. Int. Ed. Engl. 42 (3): 355–7.doi:10.1002/anie.200390115. PMID 12548698.
- Chauhan D, Catley L, Li G et al. (2005). “A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib”. Cancer Cell 8 (5): 407–19.doi:10.1016/j.ccr.2005.10.013. PMID 16286248.
- K. Lloyd, S. Glaser, B. Miller, Nereus Pharmaceuticals Inc.
- Beer LL, Moore BS (2007). “Biosynthetic convergence of salinosporamides A and B in the marine actinomycete Salinispora tropica”. Org. Lett. 9 (5): 845–8.doi:10.1021/ol063102o. PMID 17274624.
- Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT (2006). “Nature’s inventory of halogenation catalysts: oxidative strategies predominate”. Chem. Rev.106 (8): 3364–78. doi:10.1021/cr050313i.PMID 16895332.
- Tsueng G, McArthur KA, Potts BC, Lam KS (2007). “Unique butyric acid incorporation patterns for salinosporamides A and B reveal distinct biosynthetic origins”. Applied Microbiology and Biotechnology 75 (5): 999–1005. doi:10.1007/s00253-007-0899-7.PMID 17340108.
- Reddy LR, Saravanan P, Corey EJ (2004). “A simple stereocontrolled synthesis of salinosporamide A”. J. Am. Chem. Soc. 126 (20): 6230–1. doi:10.1021/ja048613p.PMID 15149210.
- Ling T, Macherla VR, Manam RR, McArthur KA, Potts BC (2007). “Enantioselective Total Synthesis of (-)-Salinosporamide A (NPI-0052)”.Org. Lett. 9 (12): 2289–92. doi:10.1021/ol0706051. PMID 17497868.
- Ma G, Nguyen H, Romo D (2007). “Concise Total Synthesis of (±)-Salinosporamide A, (±)-Cinnabaramide A, and Derivatives via a Bis-Cyclization Process: Implications for a Biosynthetic Pathway?”. Org. Lett. 9 (11): 2143–6. doi:10.1021/ol070616u. PMC 2518687.PMID 17477539.
- Endo A, Danishefsky SJ (2005). “Total synthesis of salinosporamide A”. J. Am. Chem. Soc. 127 (23): 8298–9.doi:10.1021/ja0522783. PMID 15941259.
- “Marizomib May Be Effective In Relapsed/Refractory Multiple Myeloma (ASH 2011)”. The Myeloma Beacon. 2012-01-23. Retrieved 2012-06-10.
- ClinicalTrials.gov: Marizomib
……………………………………………………
IMPORTANT PAPERS
Total synthesis of salinosporamide A
Org Lett 2008, 10(19): 4239
Entry to heterocycles based on indium-catalyzed conia-ene reactions: Asymmetric synthesis of (-)-salinosporamide A
Angew Chem Int Ed 2008, 47(33): 6244
A concise and straightforward total synthesis of (+/-)-salinosporamide A, based on a biosynthesis model
Org Biomol Chem 2008, 6(15): 2782
Formal synthesis of salinosporamide A starting from D-glucose
Synthesis (Stuttgart) 2009, 2009(17): 2983
Stereoselective functionalization of pyrrolidinone moiety towards the synthesis of salinosporamide A
Tetrahedron 2012, 68(32): 6504
………………
Salinosporamide A(1) was recently discovered by Fenical et al. as a bioactive product of a marine microorganism that is widely distributed in ocean sediments. Feeling, R. H.; Buchanan, G. O.; Mincer, T. J.; Kauffman, C. A.; Jensen, P. R.; Fenical, W., Angew. Chem. Int. Ed., 2003, 42, 355–357.

Structurally Salinosporamide A closely resembles the terrestrial microbial product omuralide (2a) that was synthesized by Corey et al. several years ago and demonstrated to be a potent inhibitor of proteasome function. See, (a) Corey, E. J.; Li, W. D., Z. Chem. Pharm. Bull., 1999, 47, 1–10; (b) Corey, E. J., Reichard, G. A.; Kania, R., Tetrahedron Lett., 1993, 34, 6977–6980; (c) Corey, E. J.; Reichard, G. A., J. Am. Chem. Soc., 1992, 114, 10677–10678; (d) Fenteany, G.; Standaert, R. F.; Reichard, G. A.; Corey, E. J.; Schreiber, S. L., Proc. Natl. Acad. Sci. USA, 1994, 91, 3358–3362.
Omuralide is generated by β-lactonization of the N-acetylcysteine thiolester lactacystin (2b) that was first isolated by the Omura group as a result of microbial screening for nerve growth factor-like activity. See, Omura, S., Fujimoto, T., Otoguro, K., Matsuzaki, K., Moriguchi, R., Tanaka, H., Sasaki, Y., Antibiot., 1991, 44, 113–116; Omura, S., Matsuzaki, K., Fujimoto, T., Kosuge, K., Furuya, T., Fujita, S., Nakagawa, A., J. Antibiot., 1991, 44, 117–118.
Salinosporamide A, the first compound Fenical’s group isolated from Salinospora, not only had a never-before-seen chemical structure 1, but is also a highly selective and potent inhibitor of cancer-cell growth. The compound is an even more effective proteasome inhibitor than omuralide and, in addition, it displays surprisingly high in vitro cytotoxic activity against many tumor cell lines (IC50values of 10 nM or less). Fenical et al. first found the microbe, which they’ve dubbed Salinospora, off the coasts of the Bahamas and in the Red Sea. See,Appl. Environ. Microbiol., 68, 5005 (2002).
Fenical et al. have shown that Salinospora species requires a salt environment to live. Salinospora thrives in hostile ocean-bottom conditions: no light, low temperature, and high pressure. The Fenical group has now identified Salinosporain five oceans, and with 10,000 organisms per cm3 of sediment and several distinct strains in each sample; and according to press reports, they’ve been able to isolate 5,000 strains. See, Chemical & Engineering News, 81, 37 (2003).
A great percentage of the cultures Fenical et al. have tested are said to have shown both anticancer and antibiotic activity. Like omuralide 2a, salinosporamide A inhibits the proteasome, an intracellular enzyme complex that destroys proteins the cell no longer needs. Without the proteasome, proteins would build up and clog cellular machinery. Fast-growing cancer cells make especially heavy use of the proteasome, so thwarting its action is a compelling drug strategy. See, Fenical et al., U.S. Patent Publication No. 2003-0157695A1
PATENTS
WO 2005113558
http://www.google.com/patents/US7183417
Part I. Synthesis of the Salinosporamide A(1)
EXAMPLE 1

(4S, 5R) Methyl 4,5-dihydro-2 (4-methoxyphenyl)-5-methyloxazole-4-carboxylate (4)
A mixture of (2S, 3R)-methyl 2-(4-methoxybenzamido)-3-hydroxybutanoate (3) (35.0 g, 131 mmol) and p-TsOH.H2O (2.5 g, 13.1 mmol) in toluene (400 mL) was heated at reflux for 12 h. The reaction mixture was diluted with water (200 mL) and extracted with EtOAc (3×200 mL). The combined organic layers were washed with water, brine and dried over Na2SO4. The solvent was removed in vacuo to give crude oxazoline as yellow oil. Flash column chromatography on silica gel (eluent 15% EtOAc-Hexanes) afforded the pure oxazoline (26.1 g, 80%) as solid.
Rf=0.51 (50% ethyl acetate in hexanes), mp, 86–87° C.; [α]23 D+69.4 (c 2.0, CHCl3); FTIR (film) νmax: 2955, 1750, 1545, 1355, 1187, 1011, 810 cm−1; 1HNMR(CDCl3, 400 MHz): δ 7.87 (2H, d, J=9.2 Hz), 6.84 (2H, d, J=8.8 Hz), 4.90 (1H, m), 4.40 (1H, d, J=7.6 Hz), 3.79 (3H,s), 3.71 (3H, s), 1.49 (3H, d, J=6.0 Hz); 13C NMR (CDCl3, 100 MHz): δ 171.93, 165.54, 162.64, 130.52, 119.80, 113.85, 78.91, 75.16, 55.51, 52.73, 21.14; HRMS (ESI) calcd for C13H16NO4 (M+H)+.250.1079, found 250.1084.
EXAMPLE 2

(4R, 5R)-Methyl 4-{(benzyloxy) methyl)}-4,5-dihydro-2-(4-methoxyphenyl)-5-methyloxazole-4-carboxylate (5)
To a solution of LDA (50 mmol, 1.0 M stock solution in THF) was added HMPA (24 mL, 215 mmol) at −78° C. and then oxazoline 4 (12.45 g, 50 mmol, in 20 mL THF) was added dropwise with stirring at −78° C. for 1 h to allow complete enolate formation. Benzyloxy chloromethyl ether (8.35 mL, 60 mmol) was added at this temperature and after stirring the mixture at −78° C. for 4 h, it was quenched with water (50 mL) and warmed to 23° C. for 30 min. Then the mixture was extracted with ethyl acetate (3×50 mL) and the combined organic phases were dried (MgSO4) and concentrated in vacuo. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:4 then 1:3) to give the benzyl ether 5 (12.7 g, 69%).
Rf=0.59 (50% ethyl acetate in hexanes). [α]23 D−6.3 (c 1.0, CHCl3); FTIR (film) (νmax; 3050, 2975, 1724, 1642, 1607, 1252, 1027, 745, 697 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.96 (2H, d, J=9.2 Hz), 7.26 (5H, m), 6.90 (2H, J=8.8 Hz), 4.80 (1H, m), 4.61 (2H, s), 3.87 (3H, m), 3.81 (3H, s), 3.73 (3H, s), 1.34 (3H, d, J=6.8 Hz); 13C NMR (CDCl3, 100 MHZ): 6171.23, 165.47, 162.63, 138.25, 130.64, 128.52, 127.87, 127.77, 120.15, 113.87, 81.40, 79.92, 73.91, 73.43, 55.58, 52.45, 16.92; HRMS (ESI) calcd for C21H24O5 (M+H)+370.1654, found 370.1644.
EXAMPLE 3

(2R,3R)-Methyl 2-(4-methoxybenzylamino)-2-((benzyloxy)methyl)-3hydroxybutanoate (6)
To a solution of oxazoline 5 (18.45 g, 50 mmol) in AcOH (25 mL) at 23° C. was added in portions NaCNBH3 (9.3 g, 150 mmol). The reaction mixture was then stirred at 40° C. for 12 h to allow complete consumption of the starting material. The reaction mixture was diluted with water (100 mL), neutralized with solid Na2CO3 and the aqueous layer was extracted with ethyl acetate (3×100 mL). The combined organic phases were dried over NaSO4 and concentrated in vacuo. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5) to give the N-PMB amino alcohol 6 (16.78 g, 90%).
Rf=0.50 (50% ethyl acetate in hexanes). [α]23 D−9.1(c 1.0, CHCl3); FTIR (film) νmax; 3354, 2949, 1731, 1511, 1242, 1070, 1030, 820, 736, 697 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.32 (7H, m), 6.87 (2H, d, J=8.8 Hz), 4.55 (2H, m), 4.10 (1H, q, J=6.4 Hz), 3.85 (2H, dd, J=17.2, 10.0 Hz), 3.81 (3H, s,), 3.77 (3H, s), 3. 69 (2H, dd, J=22.8, 11.6 Hz), 3.22 (2H, bs), 1.16 (3H, d, J=6.0 Hz); 13C NMR (CDCl3, 100 MHz): δ 173.34, 159.03, 137.92, 132.51, 129.78, 128.67, 128.07, 127.98, 114.07, 73.80, 70.55, 69.82, 69.65, 55.51, 55.29, 47.68, 18.15; HRMS (ESI) calcd. for C21H28NO5 (M+H)+ 374.1967, found 374.1974.
EXAMPLE 4

(2R,3R)-Methyl-2-(N-(4-methoxybenzyl)acrylamido)-2-(benzyloxy)methyl)-3-hydroxybutanoate (7)
A solution of amino alcohol 6 (26.2 g, 68.5 mmol) in Et2O (200 mL) was treated with Et3N (14.2 mL, 102.8 mmol) and trimethylchlorosilane (10.4 mL, 82.2 mmol) at 23° C. and stirred for 12 h. After completion, the reaction mixture was diluted with ether (200 mL) and then resulting suspension was filtered through celite. The solvent was removed to furnish the crude product (31.2 g, 99%) in quantitative yield as viscous oil. A solution of this crude trimethylsilyl ether (31.1 g) in CH2Cl2 (200 mL) was charged with diisopropylethylamine (14.2 mL, 81.6 mmol) and then cooled to 0° C. Acryloyl chloride (6.64 mL, 82.2 mmol) was added dropwise with vigorous stirring and the reaction temperature was maintained at 0° C. until completion (1 h). The reaction mixture was then diluted with CH2Cl2 (100 mL) and the organic layer was washed with water and brine. The organic layer was separated and dried over Na2SO4. The solvent was removed to afford the crude acrylamide 7 as a viscous oil. The crude product was then dissolved in Et2O (200 mL) and stirred with 6N HCl (40 mL) at 23° C. for 1 h. The reaction mixture was diluted with water (100 mL) and concentrated to provide crude product. The residue was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5 to 1:1) to give pure amide 7 (28.3 g, 96%) as colorless solid, mp 88–89° C.
Rf=0.40 (50% ethyl acetate in hexanes), [α]23 D−31.1 (c 0.45, CHCl3), FTIR (film) νmax; 3435, 2990, 1725, 1649, 1610, 1512, 1415, 1287, 1242, 1175, 1087, 1029, 732, 698 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.25 (5H, m), 7.15 (2H, d, J=6.0 Hz), 6.85 (2H, d, J=7.5 Hz), 6.38 (2H, d, J=6.0 Hz), 5.55 (1H, t, J=6.0 Hz), 4.81 (2H, s), 4.71 (1H, q, J=6.5 Hz), 4.35 (2H, s), 4.00 (1H, d, J=10.0 Hz), 3.80 (1H, d, J=10.0 Hz), 3.76 (3H, s), 3.75 (3H, s), 3.28 (1H, bs), 1.22 (3H, d, J=6.0 Hz); 13C NMR (CDCl3, 125 MHz): δ 171.87, 168.74, 158.81, 137.73, 131.04, 129.68, 128.58, 128.51, 127.94, 127.72, 127.20, 127.14, 114.21, 73.71, 70.42, 69.76, 67.65, 55.45, 52.52, 49.09, 18.88; HRMS (ESI) calcd. for C24H30NO6 (M+H)+428.2073, found 428.2073.
EXAMPLE 5

(R)-Methyl-2-(N-(4-methoxybenzyl)acrylamido)-2-(benzyloxy)methyl)-3-oxybutanoate (8)
To a solution of amide 7 (10.67 g, 25.0 mmol) in CH2Cl2 (100 mL) was added Dess-Martin periodinane reagent (12.75 g, 30.0 mmol, Aldrich Co.) at 23° C. After stirring for 1 h, the reaction mixture was quenched with aq NaHCO3—Na2S2O3 (1:1, 50 mL) and extracted with ethyl acetate (3×50 mL). The organic phase was dried and concentrated in vacuo to afford the crude ketone. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes) to give pure keto amide 8 (10.2 g, 96%).
Rf=0.80 (50% ethyl acetate in hexanes), mp 85 to 86° C.; [α]23 D−12.8 (c 1.45, CHCl3); FTIR (film) νmax: 3030, 2995, 1733, 1717, 1510, 1256, 1178, 1088, 1027, 733, 697 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.30 (2H, d, J=8.0), 7.25 (3H, m), 7.11 (2H, m), 6.88 (2H, d, J=9.0 Hz), 6.38 (2H, m), 5.63 (1H, dd, J=8.5, 3.5 Hz), 4.93 (1H, d, J=18.5 Hz), 4.78 (1H, d, J=18.5, Hz), 4.27 (2H, m), 3.78 (3H, s), 3.76 (3H, s), 2.42 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 198.12, 169.23, 168.62, 158.01, 136.95, 130.64, 130.38, 128.63, 128.13, 127.77, 127.32, 114.33, 77.49, 73.97, 70.66, 55.49, 53.09, 49.03, 28.24; HRMS (ESI) calcd. for C24H28NO6 (M+H)+ 426.1916, found 426.1909.
EXAMPLE 6

(2R,3S)-Methyl-1-(4-methoxybenzyl)-2-((benzyloxy)methyl)-3-hydroxy-3-methyl-4-methylene-5-oxopyrrolidine-2-carboxylate (9+10)
A mixture of keto amide 8 (8.5 g, 20.0 mmol) and quinuclidine (2.22 g, 20.0 mmol) in DME (10 mL) was stirred for 5 h at 23° C. After completion, the reaction mixture was diluted with ethyl acetate (50 mL) washed with 2N HCl, followed by water and dried over Na2SO4. The solvent was removed in vacuo to give the crude adduct (8.03 g, 94.5%, 3:1 ratio of 9 to 10 dr) as a viscous oil. The diastereomeric mixture was separated at the next step, although small amounts of 9 and 10 were purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:10 to 1:2) for analytical purposes.
Major Diastereomer (9).
[α]23 D−37.8 (c 0.51, CHCl3); FTIR (film) vmax: 3450, 3055, 2990, 1733, 1683, 1507, 1107, 1028, 808,734 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.29 (5H, m), 7.15 (2H, d, J=7.5 Hz), 6.74 (2H, d, J=8.5 Hz), 6.13 (1H, s), 5.57 (1H, s), 4.81 (1H, d, J=14.5 Hz), 4.45(1H, d, J=15.0 Hz), 4.20 (1H, d, J=12.0 Hz), 4.10 (1H, d, J=12.0 Hz) 3.75 (3H, s), 3.70 (1H, d, J=10.5 Hz), 3.64 (3H, s), 3.54 (1H, d, J=10.5 Hz), 2.55 (1H, bs, OH), 1.50 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 169.67, 168.42, 158.97, 145.96, 137.57, 130.19, 130.12, 128.53, 127.83, 127.44, 116.79, 113.71, 76.32, 76.00, 73.16, 68.29, 55.45, 52.63, 45.36, 22.64; HRMS (ESI) calcd. for C24H28NO6 (M+H)+ 426.1916, found 426.1915.
Minor Diastereomer (10).
[α]23 D−.50.1 (c 0.40, CHCl3); FTIR (film) νmax: 3450, 3055, 2990, 1733, 1683, 1507, 1107, 1028, 808, 734 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.29 (5H, m), 7.12 (2H, d, J=7.5 Hz), 6.73 (2H, d, J=8.5 Hz), 6.12 (1H, s), 5.57 (1H, s), 4.88 (1H, d, J=15.5 Hz), 4.31 (1H, d, J=15.0 Hz), 4.08 (3H, m), 3.99 (1H, d, J=12.0 Hz) 3.73 (3H, s), 3.62 (3H, s), 3.47 (1H, bs, OH), 3.43 (1H, d, J=10.0 Hz), 1.31 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 169.65, 167.89, 159.13, 147.19, 136.95, 130.29, 129.76, 128.74, 128.19, 127.55, 116.80, 113.82, 76.21, 75.66, 73.27, 68.02, 55.45, 52.52, 45.24, 25.25; HRMS (ESI) calcd. for (M+H)+ C24H28NO6 426.1916, found 426.1915.
EXAMPLE 7
Silylation of 9 and 10 and Purification of 11.
To a solution of lactams 9 and 10 (7.67 g, 18 mmol) in CH2Cl2 (25 ml) was added Et3N (7.54 ml, 54 mmol), and DMAP (2.2 g, 18 mmol) at 0° C., and then bromomethyl-dimethylchlorosilane (5.05 g, 27 mmol) (added dropwise). After stirring the mixture for 30 min at 0° C., it was quenched with aq NaHCO3 and the resulting mixture was extracted with ethyl acetate (3×50 mL). The combined organic layers were washed with water, brine and dried over Na2SO4. The solvent was removed in vacuo to give a mixture of the silated derivatives of 9 and 10 (9.83 g, 95%). The diastereomers were purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5 to 1:4) to give pure diastereomer 11 (7.4 g, 72%) and its diastereomer (2.4 g, 22%).
Silyl Ether (11).
Rf=0.80 (30% ethyl acetate in hexanes). [α]23 D−58.9 (c 0.55, CHCl3); FTIR (film) νmax; 3050, 2995, 1738, 1697, 1512, 1405, 1243, 1108, 1003, 809, 732 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.27 (5H, m), 7.05 (2H, d, J=7.0 Hz), 6.71 (2H, d, J=8.5 Hz), 6.18 (1H, s), 5.53 (1H, s), 4.95 (1H, d, J=15.5 Hz), 4.45 (1H, d, J=15.0 Hz), 4.02 (1H, J=12.0 Hz), 3.86 (1H, d, J=11.5 Hz) 3.72 (3H, s), 3.68 (3H, s), 3.65 (1H, d, J=10.5 Hz), 3.30 (1H, d, J=10.0 Hz), 2.34 (2H, d, J=2.0 Hz), 1.58 (3H, s), 0.19 (3H, s), 0.18 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 168.62, 168.12, 158.93, 145.24, 137.53, 130.32, 130.30, 128.49, 127.76,127.22, 117.26, 113.60, 78.55, 78.03, 72.89, 68.45, 55.43, 52.37, 45.74, 21.87, 17.32, −0.72, −0.80; HRMS (ESI) Calcd. for C27H35BrNO6Si (M+H)+ 576.1417, found 576.1407.
EXAMPLE 8

Conversion of (11) to (12).
To a solution of compound 11 (5.67 g 10 mmol) in benzene (250 mL) at 80° C. under nitrogen was added a mixture of tributyltin hydride (4.03 ml, 15 mmol) and AIBN (164 mg, 1 mmol) in 50 ml benzene by syringe pump over 4 h. After the addition was complete, the reaction mixture was stirred for an additional 4 h at 80° C. and the solvent was removed in vacuo. The residue was dissolved in hexanes (20 mL) and washed with saturated NaHCO3 (3×25 mL), water and dried over Na2SO4. The solvent was removed in vacuo to give crude product. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5) to afford the pure 12 (4.42 g, 89%).
Rf=0.80 (30% ethyl acetate in hexanes). [α]23 D−38.8 (c 0.25, CHCl3); FTIR (film) νmax; 3025, 2985, 1756, 1692, 1513, 1247, 1177, 1059, 667 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.28 (5H, m), 7.09 (2H, d, J=7.0 Hz), 6.73 (2H, d, J=9.0 Hz), 4.96(1H, d, J=15.0 Hz), 4.35 (1H, d, J=15.5 Hz), 3.97 (1H, d, J=12.5 Hz), 3.86 (1H, d, J=12.0 Hz), 3.80 (1H, d, J=10.0 Hz), 3.72 (3H, s), 3.65 (3H, s), 3.27 (1H, d, J=10.5 Hz), 2.67 (1H, t, J=4.0 Hz), 2.41 (1H, m), 1.79 (1H, m), 1.46 (3H, s), 0.77 (1H, m), 0.46 (1H, m), 0.10 (3H, s), 0.19 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 175.48, 169.46, 158.76, 137.59, 131.04, 129.90, 128.58, 127.88, 127.52, 113.59, 113.60, 81.05, 78.88, 73.12, 69.03, 55.45, 51.94, 48.81, 45.50, 22.79, 17.06, 7.76, 0.54; HRMS (ESI) calcd. for (M+H)+ C27H36NO6Si 498.2312, found 498.2309.
EXAMPLE 9

Debenzylation of (12).
A solution of 12 (3.98 g, 8 mmol) in EtOH (50 ml) at 23° C. was treated with 10% Pd—C (˜1 g) under an argon atmosphere. The reaction mixture was evacuated and flushed with H2 gas (four times) and then stirred vigorously under an atmosphere of H2 (1 atm, H2 balloon) at 23° C. After 12 h, the reaction mixture was filtered through Celite and concentrated in vacuo to give the crude debenzylation product (3.08 g, 95%) which was used for the next step. A small amount crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:3) for analytical purposes. Rf=0.41 (50% ethyl acetate in hexanes).
mp, 45–47° C.; [α]23 D−30.9 (c 0.55, CHCl3); FTIR (film) νmax: 3432, 3020, 2926, 1735, 1692, 1512, 1244, 1174, 1094, 1024, 870, 795 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.36 (2H, d, J=8.5 Hz), 6.83 (2H, d, J=8.5 Hz), 5.16 (1H, d, J=15.0 Hz), 4.29 (1H, d, J=15.0 Hz), 3.92 (1H, m), 3.78 (3H, s), 3.68 (3H, s), 3.45 (1H, m), 2.53 (1H, t, J=4.0 Hz), 2.42 (1H, m), 1.82 (1H, m), 1.50 (3H, s), 1.28 (1H, m), 0.75 (1H, m), 0.47 (1H, m), 0.11 (3H, s), 0.02 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 175.82, 169.51, 159.32, 131.00, 129.72, 114.52, 80.79, 80.13, 61.85, 55.48, 51.99, 49.29, 45.06, 23.11, 17.03, 7.44, 0.54; HRMS (ESI) calcd. for C20H30NO6Si (M+H)+ 408.1842, found 408.1846.
EXAMPLE 10
Oxidation to Form Aldehyde (13).
To a solution of the above alcohol from debenzylation of 12 (2.84 g, 7 mmol) in CH2Cl2 (30 mL) was added Dess-Martin reagent (3.57 g, 8.4 mmol) at 23° C. After stirring for 1 h at 23° C., the reaction mixture was quenched with aq NaHCO3—Na2S2O3 (1:1, 50 mL) and extracted with ethyl acetate (3×50 mL). The organic phase was dried and concentrated in vacuo to afford the crude aldehyde. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5) to give pure aldehyde 13 (2.68 g, 95%). Rf=0.56 (50% ethyl acetate in hexanes).
mp, 54–56° C.; [α]23 D−16.5 (c 0.60, CHCl3); FTIR (film) νmax: 3015, 2925, 1702 1297, 1247, 1170, 1096, 987, 794 cm−1; 1H NMR (CDCl3, 500 MHz): δ 9.62 (1H, s), 7.07 (2H, d, J=8.0 Hz), 6.73 (2H, d, J=8.5 Hz), 4.49 (1H, quart, J=8.5 Hz), 3.70 (3H, s), 3.67 (3H, s), 2.36 (2H, m), 1.75 (1H, m), 1.37 (3H, s), 0.73 (1H, m), 0.48 (1H, m), 0.07 (3H, s), 0.004 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 197.26, 174.70, 167.36, 158.07, 130.49, 128.96, 113.81, 83.97, 82.36, 55.34, 52.43, 47.74, 46.32, 23.83, 16.90, 7.52, 0.56, 0.45; HRMS (ESD calcd. for C20H28NO6Si (M+H)+ 406.1686, found 406.1692.
EXAMPLE 11

Conversion of (13) to (14).
To a solution of freshly prepared cyclohexenyl zinc chloride (10 mL, 0.5 M solution in THF, 5 mmol) (see Example 15 below) at −78° C. under nitrogen was added a −78° C. solution of aldehyde 13 (1.01 g, in 3 ml of THF, 2.5 mmol). After stirring for 5 h at −78° C. reaction mixture was quenched with water (10 mL) then extracted with ethyl acetate (3×10 mL). The combined organic layers were dried over Na2SO4 and solvent was removed in vacuo to give crude product (20:1 dr). The diastereomers were purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:10 to 1:2 affords the pure major diastereomer 14 (1.0 g, 83%) and a minor diastereomer (50 mg 5%). For 14: Rf=0.56 (50% ethyl acetate in hexanes).
mp, 79–81° C.; [a]23 D−28.5 (c 1.45, CHCl3); FTIR (film) νmax: 3267, 2927, 2894, 2829, 1742, 1667, 1509, 1248, 1164, 1024, 795 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.34 (2H, d, J=8.5 Hz), 6.81 (2H, d, J=9.0 Hz), 5.84 (1H, m), 5.73 (1H, m), 4.88 (1H, d, J=15.5 Hz), 4.39 (1H, d, J=14.5 Hz), 4.11 (1H, t, J=6.5 Hz), 3.77 (3H, s), 3.58 (3H, s), 3.00 (1H, m), 2.95 (1H, d, J=9.0 Hz), 2.83 (1H, t, J=3.5 Hz), 3.36 (1H, m), 2.27 (1H, m), 1.98 (2H, m), 1.74 (3H, m), 1.62 (3H, s), 1.14 (2H, m), 0.59 (1H, m), 0.39 (11H, m), 0.13 (3H, s), 0.03 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 176.80, 170.03, 158.27, 131.86, 131.34, 128.50, 126.15, 113.40, 83.96, 82.45, 77.17, 55.45, 51.46, 48.34, 48.29, 39.08, 28.34, 25.29, 22.45, 21.09, 17.30, 7.75, 0.39, 0.28; HRMS (ESI) calcd. for C26H38NO6Si (M+H)+ 488.2468, found 488.2477.
EXAMPLE 12

Tamao-Fleming Oxidation of (14) to (15).
To a solution of 14 (0.974 g, 2 mmol) in THF (5 mL) and MeOH (5 mL) at 23° C. was added KHCO3 (0.8 g, 8 mmol) and KF (0.348 g, 6 mmol). Hydrogen peroxide (30% in water, 5 mL) was then introduced to this mixture. The reaction mixture was vigorously stirred at 23° C. and additional hydrogen peroxide (2 ml) was added after 12 h. After 18 h, the reaction mixture was quenched carefully with NaHSO3 solution (15 mL). The mixture was extracted with ethyl acetate (3×25 mL) and the combined organic layers were washed with water and dried over Na2SO4. The solvent was removed in vacuo to give the crude product. The crude product was purified by column chromatography (silica gel, ethyl acetate) to give the pure triol 15 (0.82 g, 92%).
Rf=0.15 (in ethyl acetate). mp, 83–84° C.; [α]23 D: +5.2 (c 0.60, CHCl3); FTIR (film) νmax; 3317, 2920, 2827, 1741, 1654, 1502, 1246, 1170, 1018, 802 cm−1; 1HNMR(CDCl3, 500 MHz): δ 7.77 (2H, d, J=8.0 Hz), 6.28 (2H, d, J=8.0 Hz), 5. 76 (1H, m), 5.63 (1H, d, J=10.0 Hz), 4.74 (1H, d, J=15.5 Hz), 4.54 (1H, d, J=15.0 Hz), 4.12 (1H, d, J=2.5 Hz), 3.80 (1H, m), 3.76 (3H, s), 3.72 (1H, m), 3.68 (3H, s), 3.00 (1H, m), 2.60 (1H, br), 2.20 (1H, m), 1.98 (2H, s), 1.87 (1H, m), 1.80 (1H, m), 1.71 (2H, m), 1.61 (3H, s), 1.14 (2H, m); 13C NMR (CDCl3, 125 MHz): δ 178.99, 170.12, 158.27, 131.30, 130.55, 128.13, 126.39, 113.74, 81.93, 80.75, 76.87, 61.61, 55.45, 51.97, 51.32, 48.07, 39.17, 27.71, 27.13, 25.22, 21.35, 21.22; HRMS (ESI) calcd. for C24H34NO7 (M+H)+ 448.2335, found 448.2334.
EXAMPLE 13

Deprotection of (15) to (16).
To a solution of 15 (0.670 g, 1.5 mmol) in acetonitrile (8 mL) at 0° C. was added a pre-cooled solution of ceric ammonium nitrate (CAN) (2.46 g 4.5 mmol in 2 mL H2O). After stirring for 1 h at 0° C. the reaction mixture was diluted with ethyl acetate (50 mL), washed with saturated NaCl solution (5 mL) and organic layers was dried over Na2SO4. The solvent was removed in vacuo to give the crude product which was purified by column chromatography (silica gel, ethyl acetate) to give the pure 16 (0.4 g, 83%).
Rf=0.10 (5% MeOH in ethyl acetate). mp, 138 to 140° C.; [α]23 D+14.5 (c 1.05, CHCl3); FTIR (film) νmax 3301, 2949, 2911, 2850, 1723, 1673, 1437, 1371, 1239, 1156, 1008, 689 cm−1; 1H NMR (CDCl3, 600 MHz): δ 8.48 (1H, br), 6.08 (1H, m), 5. 75 (1H, d, J=9.6 Hz), 5.29 (1H, br), 4.13 (1H, d, J=6.6 Hz), 3.83 (3H, m), 3.79 (1H, m), 3.72 (1H, m), 2.84 (1H, d, J=10.2 Hz), 2.20 (1H, m), 2.16 (1H, br), 1.98 (3H, m), 1.77 (3H, m), 1.59 (1H, m), 1.54 (3H, s), 1.25 (1H, m). 13C NMR (CDCl3, 125 MHz): δ 180.84, 172.95, 135.27, 123.75, 82.00, 80.11, 75.56, 62.39, 53.14, 51.78, 38.95, 28.79, 26.48, 25.04, 20.66, 19.99; HRMS (ESI) calcd. (M+H)+ for C16H26NO6 328.1760, found 328.1752.
EXAMPLE 14

Conversion of (16) to Salinosporamide A(1).
A solution of triol ester 16 (0.164 g, 0.5 mmol) in 3 N aq LiOH (3 mL) and THF (1 mL) was stirred at 5° C. for 4 days until hydrolysis was complete. The acid reaction mixture was acidified with phosphoric acid (to pH 3.5). The solvent was removed in vacuo and the residue was extracted with EtOAc, separated, and concentrated in vacuo to give the crude trihydroxy carboxylic acid 16a (not shown). The crude acid was suspended in dry CH2Cl2 (2 mL), treated with pyridine (0.5 mL) and stirred vigorously at 23° C. for 5 min. To this solution was added BOPCl (152 mg, 0.6 mmol) at 23° C. under argon, and stirring was continued for 1 h. The solvent was removed under high vacuum and the residue was suspended in dry CH3CN (1 mL) and treated with pyridine (1 mL). To this solution was added PPh3Cl2 (333 mg, 1.0 mmol) at 23° C. under argon with stirring. After 1 h the solvent was removed in vacuo. The crude product was purified by column chromatography (silica gel, ethyl acetate-CH2Cl2, 1:5) to give the pure β-lactone 1 (100 mg, 64%) as a colorless solid.
Rf=0.55 (50% ethyl acetate in hexane). mp, 168–170° C. (authentic sample: 168–170° C., 169–171° C. in Angew. Chem. Int. Ed., 2003, 42, 355–357); mixture mp, 168–170C. [α]23 D −73.2 (c 0.49, MeOH), −72.9 (c 0.55, MeOH, in Angew. Chem. Int. Ed., 2003, 42, 355–357); FTIR (film) νmax: 3406, 2955, 2920, 2844, 1823, 1701, 1257, 1076, 1012, 785, 691 cm−1; 1H NMR (CDCl3, 500 MHz): δ 10.62 (1H, br), 6.42 (1H, d, J=10.5 Hz), 5.88 (1H, m), 4.25 (1H, d, J=9.0 Hz), 4.14 (1H, m), 4.01 (1H, m), 3.17 (1H, t, J=7.0 Hz), 2.85 (1H, m), 2.48 (1H, m), 2.32 (2H, m), 2.07 (3H, s), 1.91 (2H, m), 1.66 (2H, m), 1.38 (1H, m);13C NMR (CDCl3, 125 MHz): δ 176.92, 169.43, 129.08, 128.69, 86.32, 80.35, 70.98, 46.18, 43.28, 39.31, 29.01, 26.47, 25.35, 21.73, 20.00; HRMS (ESI) calcd. for (M−H)− C15H19ClNO4 312.1003, found 312.1003.
| 544814 | Oct 1, 2007 | Jun 9, 2009 | Nereus Pharmaceuticals, Inc. | [3.2.0] Heterocyclic compounds and methods of using the same |
| US7579371 | Jun 15, 2006 | Aug 25, 2009 | Nereus Pharmaceuticals, Inc. | Methods of using [3.2.0] heterocyclic compounds and analogs thereof |
| US7824698 | Feb 4, 2008 | Nov 2, 2010 | Nereus Pharmaceuticals, Inc. | Lyophilized formulations of Salinosporamide A |
| US7842814 | Apr 6, 2007 | Nov 30, 2010 | Nereus Pharmaceuticals, Inc. | Total synthesis of salinosporamide A and analogs thereof |
| US7910616 | May 12, 2009 | Mar 22, 2011 | Nereus Pharmaceuticals, Inc. | Proteasome inhibitors |
| US8003802 | Mar 6, 2009 | Aug 23, 2011 | Nereus Pharmaceuticals, Inc. | Total synthesis of Salinosporamide A and analogs thereof |
| US8067616 | Oct 27, 2010 | Nov 29, 2011 | Nereus Pharmaceuticals, Inc. | Total synthesis of salinosporamide A and analogs thereof |
| US8168803 | Jun 10, 2008 | May 1, 2012 | Nereus Pharmaceuticals, Inc. | Methods of using [3.2.0] heterocyclic compounds and analogs thereof |
| US8217072 | Jun 18, 2004 | Jul 10, 2012 | The Regents Of The University Of California | Salinosporamides and methods for use thereof |
| US8222289 | Dec 15, 2009 | Jul 17, 2012 | The Regents Of The University Of California | Salinosporamides and methods for use thereof |
| US8227503 | Mar 21, 2011 | Jul 24, 2012 | Nereus Pharmaceuticals, Inc. | Proteasome inhibitors |
| US8314251 | Jul 15, 2011 | Nov 20, 2012 | Nereus Pharmaceuticals, Inc. | Total synthesis of salinosporamide A and analogs thereof |
| US8389564 | May 14, 2012 | Mar 5, 2013 | Venkat Rami Reddy Macherla | Proteasome inhibitors |
| US8394816 | Dec 5, 2008 | Mar 12, 2013 | Irene Ghobrial | Methods of using [3.2.0] heterocyclic compounds and analogs thereof in treating Waldenstrom’s Macroglobulinemia |
| Name: | Marizomib | |
| Synonyms: | 6-Oxa-2-azabicyclo[3.2.0]heptane-3,7-dione, 4-(2-chloroethyl)-1-[(S)-(1S)-2-cyclohexen-1-ylhydroxymethyl]-5-methyl-, (1R,4R,5S)-; Other Names: (-)-Salinosporamide A; ML 858; Marizomib; NPI 0052; Salinosporamide A | |
| CAS Registry Number: | 437742-34-2 | |
| Molecular Formula: | C15H20ClNO4 | |
| Molecular Weight: | 313.1 | |
| Molecular Structure: |  |
Ixabepilone for breast cancer

Ixabepilone, 219989-84-1 cas
(1R,5S,6S,7R,10S,14S,16S)-6,10-dihydroxy-1,5,7,
9,9-pentamethyl-14-[(E)-1-(2-methyl-1,3-thiazol-
4-yl)prop-1-en-2-yl]-17-oxa-13-azabicyclo[14.1.0]
heptadecane-8,12-dione
Ixabepilone (INN; also known as azaepothilone B, codenamed BMS-247550) is an epothilone B analog developed byBristol-Myers Squibb as a chemotherapeutic medication for cancer.
It is produced by Sorangium cellulosum.
It acts to stabilize microtubules. It is highly potent agent, capable of damaging cancer cells in very low concentrations, and retains activity in cases where tumor cells are insensitive to paclitaxel.
On October 16, 2007, the U.S. Food and Drug Administration approved ixabepilone for the treatment of aggressive metastaticor locally advanced breast cancer no longer responding to currently available chemotherapies. In November 2008, the EMEAhas refused a marketing authorisation for Ixabepilone.
Ixabepilone is administered through injection, and is marketed under the trade name Ixempra.

patent approval expiry
| United States | 7312237 | 2004-08-21 | 2024-08-21 |
| United States | 6605599 | 1998-05-26 | 2018-05-26 |
| Applicant | Tradename | Generic Name | Dosage | NDA | Approval Date | Type | RLD | US Patent No. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Bristol Myers Squibb
|
IXEMPRA KIT
|
ixabepilone
|
INJECTABLE;IV (INFUSION) | 022065 | Oct 16, 2007 | RX | Yes | RE41911*PED | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Bristol Myers Squibb
|
IXEMPRA KIT
|
ixabepilone
|
INJECTABLE;IV (INFUSION) | 022065 | Oct 16, 2007 | RX | Yes | RE41393*PED | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Bristol Myers Squibb
|
IXEMPRA KIT
|
ixabepilone
|
INJECTABLE;IV (INFUSION) | 022065 | Oct 16, 2007 | RX | Yes | 7,312,237*PED | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Bristol Myers Squibb
|
IXEMPRA KIT
|
ixabepilone
|
INJECTABLE;IV (INFUSION) | 022065 | Oct 16, 2007 | RX | Yes | 7,125,899*PED | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Patent No | Patent Expiry | patent use code |
|---|---|---|
| 6670384 | Jan 23, 2022 | U-959 |
| 6670384 | Jan 23, 2022 | U-960 |
| 6670384*PED | Jul 23, 2022 | |
| 7022330 | Jan 23, 2022 | U-958 |
| 7022330*PED | Jul 23, 2022 | |
| 7125899 | May 26, 2018 | U-957 |
| 7125899*PED | Nov 26, 2018 | |
| 7312237 | Aug 21, 2024 | U-965 |
| 7312237*PED | Feb 21, 2025 | |
| RE41393 | Feb 8, 2022 | U-961 |
| RE41393*PED | Aug 8, 2022 | |
| RE41911 | Sep 28, 2020 | U-961 |
| RE41911*PED | Mar 28, 2021 |
| Exclusivity Code | ExclusivityDate |
|---|---|
| NCE | Oct 16, 2012 |
| PED | Apr 18, 2015 |
| M-61 | Oct 18, 2014 |
| PED | Apr 16, 2013 |
| Exclusivity Code | ExclusivityDate |
|---|---|
| NCE | Oct 16, 2012 |
Ixabepilone, in combination with capecitabine, has demonstrated effectiveness in the treatment of metastatic or locally advanced breast cancer in patients after failure of an anthracycline and a taxane.
It has been investigated for use in treatment of non-Hodgkin’s lymphoma. In pancreatic cancer phase two trial it showed some promising results (used alone). Combination therapy trials are ongoing.
Ixabepilone is an anti cancer agent acting as a microtubule inhibitor, and which in particular are efficient in the treatment of cancer not reacting to other anti cancer agents, such as e.g. paclitaxel. Ixabepilone is marketed under the trade name Ixempra® and are approved for the treatment of aggressive metastatic or locally advanced breast cancer which not responding to the current prevailing chemotherapies.
Ixabepilone known under the CAS no. 219989-84-1 has the following structure:

Ixabepilone
Ixabepilone may be prepared from a starting material named epothilone B having the structural formula:

Epothilone B Ixabepilone as a compound is described in the USRE4191 1. USRE4191 1 furthermore disclose a process for synthesizing Ixabepilone.
The US 6,365,749 describes a process for making ixabepilone by reacting epothilone B with a palladium catalyst in the presence of a nucleophilic donor.
The USRE39356 do also describe a process for making Ixabepilone by reacting epothilone B with an azide donor agent and a reducing agent in the presence of a phase transfer catalyst and a palladium catalyst.

Ixabepilone is the treatment of metastatic and advanced breast cancer drugs.Ixabepilone as anticancer drugs alone or in combination with capecitabine (Capecitabine) in combination. October 16, 2007 approved for marketing by the FDA, trade name Ixempra, by the Bristol-Myers Squibb Company’s development.
Ixabepilone is an anti-mitotic drugs that are inhibitors of tubulin, the mechanism and paclitaxel (Taxol) the same class of drugs. Epothilone (Epothilone) by colistin (myxobacterium) Sorangium cellulosum fermentation of several macrolide metabolites in general. Anticancer activity in vitro experiments, epothilone A and epothilone B showed good activity, even in the paclitaxel-resistant cells also showed good activity. But its activity in vivo experiments in general, this is probably due to the body of the ester hydrolases that macrolide ring opening induced inactivation. In a series of epothilone derivatives activity test, it was found with the lactam bond instead of the original product of ester bonds – ixabepilone anticancer activity can be well retained.
Ixabepilone is epothilone B semi-synthetic derivatives. Epothilone B is a macrocyclic lactone, a hydroxyl moiety is allyl alcohol, the Pd catalyst can be obtained by ring-opening Pd complexes 1 , 1 received azide nucleophile attacking the anion generated with three azide product phosphorus reduction to give methyl amino acids 2 . Here we must point out that the attack was completely azide stereoselectivity, which is determined by two consecutive trans-attack lead, Pd (0)-trans lactone generate offensive allyl Pd complexes, to accept anti-azide anion type attack, to maintain the configuration of the product obtained. Amino acids 2 HoBt and EDCI generated by an amide bond to get ixabepilone.

IXEMPRA (ixabepilone) is a microtubule inhibitor belonging to a class of antineoplastic agents, the epothilones and their analogs. The epothilones are isolated from the myxobacterium Sorangium cellulosum. Ixabepilone is a semisynthetic analog of epothilone B, a 16-membered polyketide macrolide, with a chemically modified lactam substitution for the naturally existing lactone.
The chemical name for ixabepilone is (1S,3S,7S,10R,11S,12S,16R)-7,11dihydroxy-8,8,10,12,16-pentamethyl-3-[(1E)-1-methyl-2-(2-methyl-4-thiazolyl)ethenyl]17-oxa-4-azabicyclo[14.1.0] heptadecane-5,9-dione, and it has a molecular weight of 506.7. Ixabepilone has the following structural formula:
 |
IXEMPRA (ixabepilone) for injection is intended for intravenous infusion only after constitution with the supplied DILUENT and after further dilution with a specified infusion fluid . IXEMPRA (ixabepilone) for injection is supplied as a sterile, non-pyrogenic, single-use vial providing 15 mg or 45 mg ixabepilone as a lyophilized white powder. The DILUENT for IXEMPRA is a sterile, non-pyrogenic solution of 52.8% (w/v) purified polyoxyethylated castor oil and 39.8% (w/v) dehydrated alcohol, USP. The IXEMPRA (ixabepilone) for injection and the DILUENT for IXEMPRA are co-packaged and supplied as IXEMPRA Kit.
….

back to home for more updates


DR ANTHONY MELVIN CRASTO Ph.D
Current Oncology Pipeline Trends

Oncology drug development far outpaces drug development for other therapeutic areas and the magnitude of that difference is significant. Here’s a current review of what is in the pipeline and an analysis of where oncology research is headed.

Stacey Ness, PharmD, RPh, MSCS, AAHIVP, has worked in both national specialty pharmacy and payer organizations and has experience in clinical management, adherence, and persistency programs, as well as chronic disease cost optimization strategies. Dr. Ness is active in the Consortium of Multiple Sclerosis Centers, Academy of Managed Care Pharmacy, National Home Infusion Association, National Association of Specialty Pharmacy, Specialty Pharmacy Certification Board, and Hematology and Oncology Pharmacy Association, and has served on the Minnesota Medicaid Drug Formulary Committee since 2008. She is a multiple sclerosis certified specialist, a credentialed HIV pharmacist, and currently serves as the director of specialty clinical services at Managed Health Care Associates, Inc, a health care services organization based in Florham Park, New Jersey.
VINCRISTINE……..Chemistry, Isolation

VINCRISTINE
(3aR,3a1R,4R,5S,5aR,10bR)-methyl 4-acetoxy-3a-ethyl-9-((5S,7S,9S)-5-ethyl-5-hydroxy-9-(methoxycarbonyl)-2,4,5,6,7,8,9,10-octahydro-1H-3,7-methano[1]azacycloundecino[5,4-b]indol-9-yl)-6-formyl-5-hydroxy-8-methoxy-3a,3a1,4,5,5a,6,11,12-octahydro-1H-indolizino[8,1-cd]carbazole-5-carboxylate
…………………………………………………………..
………………………………………………………………….


Vincristine (brand name, Oncovin), formally known as leurocristine, sometimes abbreviated “VCR”, is a vinca alkaloid from the Catharanthus roseus (Madagascar periwinkle), formerly Vinca rosea and hence its name. It is amitotic inhibitor, and is used in cancer chemotherapy. Vincristine is created by the coupling of indole alkaloids vindoline and catharanthine in the vinca plant.[1]
Mechanism
Tubulin is a structural protein that polymerizes to microtubules. The cell cytoskeleton and mitotic spindle, among other things, are made of microtubules. Vincristine binds to tubulin dimers, inhibiting assembly of microtubule structures. Disruption of the microtubules arrests mitosis in metaphase. Therefore, the vinca alkaloids affect all rapidly dividing cell types including cancer cells, but also those of intestinal epithelium and bone marrow.

Uses
Vincristine is delivered via intravenous infusion for use in various types of chemotherapy regimens. Its main uses are in non-Hodgkin’s lymphoma as part of the chemotherapy regimen CHOP, Hodgkin’s lymphoma as part of MOPP, COPP, BEACOPP, or the less popular Stanford V chemotherapy regimen, in acute lymphoblastic leukemia, and in treatment for nephroblastoma (Wilms tumor, a kidney tumor most common in young children). It is also used to induce remission in ALL with Dexamethasone and L-Asparaginase. Vincristine is occasionally used as an immunosuppressant, for example, in treating thrombotic thrombocytopenic purpura (TTP) or chronic idiopathic thrombocytopenic purpura (ITP). It is used in combination with prednisone to treat childhood leukemia.
The main side-effects of vincristine are peripheral neuropathy, hyponatremia, constipation, and hair loss.
Peripheral neuropathy can be severe, and hence a reason to avoid, reduce, or stop the use of vincristine. One of the first symptoms of peripheral neuropathy is foot drop: A person with a family history of foot drop and/or Charcot-Marie-Tooth disease (CMT) should avoid the taking of vincristine.[2]
Accidental injection of vinca alkaloids into the spinal canal (intrathecal administration) is highly dangerous, with a mortality rate approaching 100 percent. The medical literature documents cases of ascending paralysis due to massive encephalopathy and spinal nerve demyelination, accompanied by intractable pain, almost uniformly leading to death; a handful of survivors were left with devastating neurological damage with no hope of recovery. Rescue treatments consist of washout of the cerebrospinal fluid and administration of protective medications.[3] A significant series of inadvertent intrathecal vincristine administration occurred in China in 2007 when batches of cytarabine andmethotrexate (both often used intrathecally) manufactured by the company Shanghai Hualian were found to be contaminated with vincristine.[4]

Having been used as a folk remedy for centuries, studies in the 1950s revealed that C. roseus contained 70 alkaloids, many of which are biologically active. While initial studies for its use in diabetes mellitus were disappointing, the discovery that it caused myelosuppression (decreased activity of the bone marrow) led to its study in mice withleukemia, whose lifespan was prolonged by the use of a vinca preparation. Treatment of the ground plant with Skelly-B defatting agent and an acid benzene extract led to a fraction termed “fraction A”. This fraction was further treated withaluminium oxide, chromatography, trichloromethane, benz-dichloromethane, and separation by pH to yield vincristine.[5]
Vincristine was approved by the United States Food and Drug Administration (FDA) in July 1963 as Oncovin. The drug was initially discovered by a team led by Dr. J.G. Armstrong, then marketed by Eli Lilly and Company.
Like LSD, the microtubule toxin vincristine allegedly causes not-unpleasant visual hallucinations in humans. Other side-effects of vincristine include depression, agitation, and insomnia. Very small doses are needed for the effects of LSD or vincristine, for example, these drugs are active at concentrations of 4.3E-7 M-1 vincristine and 1.0E-8 M-1 LSD.
Many researchers have favored the drug-receptor theory to explain drug-induced hallucinations, usually at the 5-HT2A receptor. In the drug-receptor theory, signal amplification takes place when one molecule of drug binds to a receptor, which activates G-proteins, which affects more proteins, thus signaling cascades explain how a small amount of LSD can lead to widespread changes in the cell.
Van Woerkom suggests instead that LSD binds an element of the cytoskeleton, in a fashion similar to colchicine or vinblastine, which directly bind tubulin. The amount of LSD needed to produce hallucinations is so vanishly small, that it seems hard to believe that a submicromolar dosage of LSD could act on a substrate as vast as the cytoskeleton. However, some microtubule inhibitors such as vincristine are effective at very low dosages. The potency of vincristine may partly explain the success of this drug as a chemotherapeutic drug.
Three generic drug makers supply vincristine in the United States – APP, Mayne, and Sicor (Teva).
- ^ “Pharmacognosy of Vinca Alkaloids”.
- Graf, W. D.; Chance, P. F.; Lensch, M. W.; Eng, L. J.; Lipe, H. P.; Bird, T. D. (1996). “Severe Vincristine Neuropathy in Charcot-Marie-Tooth Disease Type 1A”. Cancer 77 (7): 1356–1362. doi:10.1002/(SICI)1097-0142(19960401)77:7<1356::AID-CNCR20>3.0.CO;2-#. PMID 8608515.
- Qweider, M.; Gilsbach, J. M.; Rohde, V. (2007). “Inadvertent Intrathecal Vincristine Administration: A Neurosurgical Emergency. Case Report”. Journal of Neurosurgery: Spine 6 (3): 280–283. doi:10.3171/spi.2007.6.3.280. PMID 17355029.
- Jake Hooker and Walt Bogdanich (January 31, 2008). “Tainted Drugs Tied to Maker of Abortion Pill”. New York Times.
- Johnson, I. S.; Armstrong, J. G.; Gorman, M.; Burnett, J. P. (1963). “The Vinca Alkaloids: A New Class of Oncolytic Agents” (pdf). Cancer Research 23 (8 Part 1): 1390–1427.PMID 14070392.
External links
- Vincristine chemotherapy
- Vincristine and vinblastine
- Description and Natural History of the Periwinkle
- The Boger Route to (-)-Vindoline
- U.S. National Library of Medicine: Drug Information Portal – Vincristine
-
Cytostatic Vinca alkaloids rosea L. Catharanthus roseus G.Don) are now well known anticancer and particularly useful. Given the small amount of vincristine in Catharanthus present, quite a number of ways of preparation have been proposed by chemists. Thus FR-A-2296418 describes the synthesis of vincristine by coupling Catha-ranthine and vindoline. Other laboratories have achieved the transformation of vinblastine vincristine oxidation under controlled conditions, very strict.
-
FR-A-2210393 and US-A-3899493 perform the oxidation by chromic acid at -30, -90 ° C in a mixture of acetic acid-acetone or chloroform-acetic acid at -55 ° C.
-
In U.S. 4,375,432, chromic compound is also used in acid medium at -65 ° C, -50 ° C in a medium based solvent THF. In addition, EP-A-37289 boasts an oxidation mixture ferrous salt, hydrogen peroxide, perchlorate in acetonitrile. ZA-A-82 08939 discloses a method with chromic acid and an ether-chloroform.
-
HU-A-23638 offers diterbutylchromate in pelargonic acid, and finally EP-A-117861 gets vinblastinel transformation vincristine oxidant potassium permanganate in acetic acid medium. It is clear that these dimeric alkaloids are a valuable material because of their low levels in vegetable raw materials, and therefore the processes of synthesis or semi-synthesis performance are of extreme interest.
-
Vincristine is used in cancer chemotherapy, particularly for the treatment of certain acute leukemias.
-
This alkaloid is obtained mainly by extraction from leaves of Catharanthus Ro-seus (U.S. Patent No. 3,205,220) where it is accompanied by other alkaloids bis-Indo-holic, especially vinblastine.Vinblastine (I, R = CH 3), however, is present at a concentration much higher than that of vincristine and is therefore a precursor of choice for the semisynthesis of the latter.
-
Several processes of vincristine from vinblastine were disclosed. We note in particular patents or patent applications include:
- a) Belgian Patent 739,337 (Gedeon Richter) which describes a method for the oxidation of vinblastine vincristine in a mixture chromic acid, acetic acid and acetone.
- b) Belgian Patent 823560 (Gedeon Richter) the oxidation is performed with oxygen in the presence of formic acid and of a catalyst based on platinum at room temperature.
- c) European Patent Application 18231 (Gedeon Richter): is carried out by oxidation with chromic acid or an alkali metal dichromate in the presence of acetic anhydride and, optionally, of ethanol and an organic solvent immis target with water.
- d) European Patent Application 37289 (Eli Lil-ly): the oxidation is effected by the perchlorate of iron (II) in the presence of hydrogen peroxide and acetonitrile.
-
In addition, the European patent application 37. 290 discloses a process for the oxidation of vinblastine base with Na 2 Cr 2 O 7 in the presence of sulfuric acid in tetrahydrofuran. This reaction led to -50 ° C, is achieved with a yield of 80-92% calculated for each estimation.
-
Observed yields or purity of the products obtained characterizing the processes described above are, however, significant disadvantages.
-
Frequently a secondary product formed is N-demethyl vinblastine need then reformulate for vincristine.
Thus Potier and Kutney obtained products with the C18’S-C2’R absolute configuration, which is critical for anti-tumor activity, by a coupling reaction of the N.sup.b -oxide of catharanthine, or its derivatives, with vindoline, in the presence of trifluoroacetic anhydride, followed by a reduction reaction. [See Potier et. al. J. Am. Chem. Soc. 98. 7017 (1976) and Kutney et. al. Helv. Chim. Acta, 59, 2858 (1976)].
The Potier and Kutney coupling process has disadvantages. The yields are not satisfactory except for the coupling of catharanthine N-oxide with vindoline and even there the preparative yield is low. While vindoline is the most abundant alkaloid of Vinca rosea and is thus readily available, the other possible components of the Potier-Kutney coupling process (catharanthine, allocatharanthine, voacangine,) are relatively inaccessible, costly, and they do not allow a wide range of structural variation of that component of the coupling process.
- …………………………………………………………………………………………………………………………………………………………………………………………………………..
-
EP 0117861 B1
-
clips
-
The process of the present invention produces a simple vincristine, in quantity and purity requiring little or no additional purification by recrystallization or chromatography.
-
[0009]The reagent used is oxidation permanganate ion dissolved in toluene or dichloromethane as solvent. An alternative consists in immobilizing the resin on a permanganate anion, for example a polymer such as polystyrene comprising ammonium groups. Solubilization can be achieved by the action of a complexing agent crown ether (“crown-ether”) of potassium permanganate.
-
[0010]The permanganate anion can also be solubilized by preparing an ammonium salt or quaternary phosphonium corresponding which is soluble in methylene chloride or toluene. For this purpose, it is preferable to use potassium permanganate benzyltriethylammonium.
-
[0011]Obtaining from vincristine vinblastine using a permanganate salt is unexpected since the potassium permanganate used in some acetone oxide derivatives of vinblastine at the portion of the molecule velbanamine (Kutney, Balsevich and Worth, Heterocycles, 11, 69, 1978). The N-methyl group of the vindoline part intact.
-
[0012]The formation of N-CHO indoline skeleton on a bis-indole group vinblastine using a permanganate salt has never been reported.
-
[0013]According to one embodiment of the method of the present invention, vinblastine, preferably in the form of sulphate, is treated in the presence of an organic acid such as acetic acid, with an excess of potassium permanganate dissolved in dichloromethane or toluene in the presence of “18-crown-6” or ether derivatives dibenzo-or di-cyclohexylcorrespondants. The reaction is conducted at a temperature between -40 ° C and -75 ° C and is preferably followed by thin layer chromatography. The reaction time generally ranges from 5 minutes to 3 hours.
-
[0014]Potassium permanganate is preferably dissolved in dichloromethane and the oxidation reaction is then carried out at -70 ° C.
-
[0015]The solubility of potassium permanganate is indeed substantially increased in the presence of a macrocyclic polyether as the “18-crown-6” ether (1, 4, 7, 10, 13, 16-hexaoxacy-clooctadécane) or derivative dibenzo – or corresponding dicyclohexyl-hexyl.
-
[0016]The reaction mixture is then treated simultaneously by a mild reducing and alkaline. For this purpose, use is preferably an aqueous solution of bisulfite, disulfite or sodium metabisulfite and ammonia.
-
[0017]The organic phase was separated and the aqueous phase is extracted several times with methylene chloride. The combined organic phases were concentrated in vacuo to give a residue containing 80-85% of base vincristine, a 90-95% yield.
-
[0018]Alternatively, you can proceed with the extraction of the reaction mixture after reduction without conducting a simultaneous alkalinization. The acidic aqueous solution was then extracted with dichloromethane. This route is a novel process for purification of vincristine formed in the reaction medium.
-
[0019]According to another embodiment of the present invention, vincristine is obtained by oxidation of vinblastine by reacting a quaternary ammonium permanganate. The ammonium cation is preferably benzyltriethylammonium group or benzyl trimethyl ammonium (see eg Angew. Chem., Intern. Ed. 13, 170, 1974). The reaction is carried out in 2 to 6 hours at -60 ° C in an inert solvent wherein the ammonium salt is soluble, and an acid, preferably an organic acid of low molecular weight. A mixture of dichloromethane and glacial acetic acid can be used. After treatment with a mild reducing agent in aqueous medium, the resulting acidic solution is extracted with dichloromethane, and the organic phase is made alkaline by washing with a basic aqueous solution and concentrated. Vincristine solvate is isolated with a yield higher than 90%.
-
[0020]The latest variant of the method of the invention is particularly advantageous in terms of economic and technical.
-
[0021]Purification or separation may be effected by crystallization and chromatography using techniques well known this from the crude product of the reaction. The product can also be lyophilized.
-
[0022]In most cases, vincristine thus obtained can be converted directly into an addition salt with an organic or inorganic acid, preferably pharmaceutically acceptable. This salt is preferably a sulfate that may arise in a more or less solvated or hydrated.
-
[0023]We can also prepare vincristine dissolved in a physiologically acceptable solvent and ready to be injected.
-
[0024]In particular, vincristine sulfate is obtained by addition of H 2 S0 4 to a solution of vincristine gross or recrystallized from ethanol, dissolved in a mixture of methylene chloride and anhydrous ethanol, partial removal in vacuo chloride methylene and crystallization.
-
[0025]Vincristine sulfate thus obtained has a purity sufficient for use as a medicament, particularly in the form of injectable solutions.

Madagascar Periwinkle: Public Domain Illustration by Sydenham Edwards
The Madagascar periwinkle, an attractive flowering plant, contains the powerful anti-cancer chemicals vinblastine and vincristine. Velvet beans, which are named from the covering of soft hairs on the young plant, contain L-dopa, a very helpful chemical in the treatment of Parkinson’s disease. The Madagascar periwinkle and the velvet bean are just two of the large number of plants that have been found to contain medicinal chemicals. There are almost certainly many more plants that have undiscovered health benefits.
The Madagascar Periwinkle
The Madagascar periwinkle is native to Madagascar and India, but is now grown in many countries as a garden plant. It has also escaped from gardens and grows as a weed. The red, purple, pink or white flowers often have a center which is a different color from the rest of the flower. Madagascar periwinkles may grow up to one meter tall and have glossy green leaves.
The sap of the Madagascar periwinkle, which has a milky appearance and is poisonous, contains vinblastine, vincristine and many other alkaloids. Researchers are discovering that many of these alkaloids are biologically active inside the human body.
Vinblastine and Vincristine
Vinblastine and vincristine have very similar chemical structures, but their effects on the body are not the same. Vinblastine is used to treat specific types of cancer, such as Hodgkin’s disease, breast cancer, testicular cancer and non-small cell lung cancer. Vincristine is used in the treatment of acute lymphoblastic leukemia (ALL) and has provided a great breakthrough in successful treatment of this disease in children. When vincristine is added to the treatment regimen for children suffering from ALL, the survival rate reaches eighty percent. Vincristine is not so impressive in the treatment of ALL in adults.
Cells contain a supporting network of protein tubules, which are known as microtubules. Microtubules also play a vital role in the process of cell division. Before a cell divides, each chromosome in the cell is replicated. The replicated chromosomes are separated from their partners and pulled to opposite ends of the cell by microtubules during a process called mitosis. The cell then divides down the middle.
Vinblastine and vincristine stop microtubule formation during mitosis and therefore prevent cells from reproducing. This effect is strongest in cells that have a high rate of division, such as cancer cells. However, vinblastine and vincristine also affect cells lining the intestine, the cells in the bone marrow that produce blood cells, and the cells in the hair follicles, since these too have a high rate of cell division.
Possible vinblastine or vincristine side effects include constipation, hair loss, a low platelet count, which can cause increased bleeding, a low white blood cell count, which can lead to increased infections, or a low red blood cell count, resulting in anemia. There may occasionally be nerve damage, possibly due to the effect of the medicines on the microctubules in the nerve cells. Vincristine is more likely to cause nerve damage than vinblastine.

,,,,,,,,,,,,,


Total synthesis of (+)-vincristine (2). TFA, trifluoroacetic acid or trifluoroacetyl; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene.
Stereocontrolled total synthesis of (+)-vincristine

………………….
see docstoc presentation
click below
Vincristine
var docstoc_docid=”51697405″;var docstoc_title=”Vincristine”;var docstoc_urltitle=”Vincristine”;
…………………….
isolation
Kumar A, Patil D, Rajamohanan PR, Ahmad A (2013)
Isolation, Purification and Characterization of Vinblastine and Vincristine from Endophytic Fungus Fusarium oxysporumIsolated from Catharanthus roseus. PLoS ONE 8(9): e71805. doi:10.1371/journal.pone.0071805
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0071805
Isolation, purification and characterization of vinblastine and vincristine from the endophytic fungus Fusarium oxysporum
A two stage fermentation procedure was employed for the isolation of vinblastine and vincristine by Fusarium oxysporum. In the first stage, 500 ml Erlenmeyer flasks containing 100 ml medium (MGYP, (0.3%) malt extract, (1.0%) glucose, (0.3%) yeast extract and (0.5%) peptone) were inoculated with 7 days old culture and incubated at 28°C on a rotary shaker (240 rpm) for 4–5 days, which was used as seed culture (I stage). Later, 10 ml seed culture was transferred to 500 ml Erlenmeyer flask containing 100 ml production medium called as vinca medium-1 (Glucose: 3%, Succinic acid: 1%, Sodium benzoate: 100 mg, Peptone: 1%, Magnesium sulphate: 3.6 mg, Biotin: 1 mg, Thiamine: 1 mg, Pyridoxal: 1 mg, Calcium pentothenate: 1 mg, Phosphate buffer: 1 ml (pH 6.8), L-Tryptophan: 0.1%, Geranium oil: 0.05%.) which were incubated at 28°C for 20 days as shake culture (II stage), after which it was harvested and used for further study. Culture filtrates and mycelia were separated with the help of muslin cloth and then lyophilized. Lyophilized culture filtrate was extracted using ethyl acetate as a solvent system. The organic layer was separated from the aqueous layer using separating funnel. The extraction was repeated thrice and the solvent was dried using anhydrous sodium sulphate and concentrated under vacuum using rotavapour at 40°C in order to get crude extract. A small amount of crude extract was dissolved in ethyl acetate and subjected to thin layer chromatography (TLC) on silica gel-G (0.5 mm thickness) using chloroform:methanol (8:2) as a solvent system. The TLC plates were sprayed with ceric ammonium sulphate reagent. Vinca alkaloids spots produced brilliant violet color as well as purple color with above spraying reagent. Purification of fungal vinblastine and vincristine were done by silica gel column chromatography. The crude extract was loaded on silica gel column (60–120 mesh size, 40 cm×2 cm length width) pre-equilibrated with chloroform and eluted with a gradient of chloroform:methanol (100% chloroform, 9:1, 8:2, 7:3, 1:1 and 3:7 and 100% methanol). Fractions containing compounds with Rf values similar to that of the standard vinblastine and vincristine were pooled and subjected to preparative TLC on a 0.5 mm thick (20 cm×20 cm) silica plate and developed in chloroform:methanol (8:2) solvent system. The putative bands of fungal vinblastine and vincristine were scraped and eluted out with methanol. Purity of the isolated compounds was checked on TLC in the solvent systems such as (a) chloroform:methanol (8:2) (b) chloroform:methanol (9:1) and (c) ethyl acetate: acetonitrile (8:2).
http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0071805
see also
http://www.ncbi.nlm.nih.gov/pubmed/20209002
ALSO
large-scale isolation of native catharantine, vindoline and 3′,4′-anhydrovinblastine whereby the isolation of vincristine, vinblastine, leurosine and the corresponding desacetoxy, desacetyl and N-desmethyl derivatives in a manner known per se can also be accomplished.
For the isolation of the two monoindole alkaloids: vindoline and catharantine from the dried plant Vinca rosea L. Svoboda [J. Am. Pharm. Assoc. 48, (11), 659 (1959)] described a method, which can be accomplished only with a very modest yield. From 1 kg. of the dried plant–subjecting the whole plant to a suitable treatment–approximately 0.6 g. of vindoline and 0.05 g. of catharantine were obtained.
3′,4′-ANHYDROVINBLASTINE UNTIL NOW HAS NEITHER BEEN ISOLATED FROM THE PLANT Vinca rosea L. nor identified in it.
For the preparation of the diindole alkaloid components starting from the leaves of Vinca rosea L. there are more methods known in the art (U.S. Pat. nos. 3,097,137; 3,205,220; 3,225,030 and Hungarian Pat. Nos. 153,200; 154,715; 160,967 and 164,958 as well as Austrian Pat. Nos. 313,435, 313,485, Australian pat. No. 458,629 and Swiss Pat. No. 572,488 and British Pat Nos. 1,412,932, 1,382,460 corresponding to the preceding two patents). According to these known processes from 1 kg. of the dried leaves of Vinca rosea L. about 0.1 to 0.2 g. of leurosine can be obtained and vinblastine, vincristine and optionally the corresponding N-desmethyl, desacetyl and desacetoxy derivatives are also simultaneously isolated.
Further on it is well known that the synthetic catharantine and vindoline may be coupled by the Polonovszky reaction to give 3′,4′-anhydrovinblastine which can thereafter be epoxidized to leurosine [Potier et al. Tetrahedron Letters 3945 (1976); DT-OS 25 58,124; Helv. Chim. Acta 59, 2858 (1976); Heterocycles 4, 997 (1976), Belgian patent specification No. 842,200 equivalent to U.S. patent application Ser. No. 582,372]. Leurosine itself has a valuable tumour growth inhibiting activity and the N-desmethyl-N-formyl derivative thereof is the most promising substance against leukemia (Hungarian Pat. No. 165,986 equivalent to U.S. patent application Ser. No. 422,100, and Austrian Pat. No. 332,566 which has issued as British Pat. No. 1,412,932).
The first generic version of the oral chemotherapy drug Xeloda (capecitabine) has been approved by the U.S. Food and Drug Administration to treat cancers of the colon/rectum or breast,

capecitabine
- R-340, Ro-09-1978, Xeloda
pentyl [1-(3,4-dihydroxy-5-methyltetrahydrofuran-2-yl)-5-fluoro-2-oxo-1H-pyrimidin-4-yl]carbamate

MONDAY Sept. 16, 2013 — The first generic version of the oral chemotherapy drug Xeloda (capecitabine) has been approved by the U.S. Food and Drug Administration to treat cancers of the colon/rectum or breast, the agency said Monday in a news release.

This year, an estimated 142,820 people will be diagnosed with cancer of the colon/rectum, and 50,830 are predicted to die from the disease, the FDA said, citing the U.S. National Cancer Institute. An estimated 232,340 women will be diagnosed with cancer of the breast this year, and some 39,620 will die from it.
The most common side effects of the drug are diarrhea, vomiting; pain, redness, swelling or sores in the mouth; fever and infection, the FDA said.
The agency stressed that approved generics have the same high quality and strength as their brand-name counterparts.
License to produce the generic drug was given to Israel-based Teva Pharmaceuticals. The brand name drug is produced by the Swiss pharma firm Roche.
Capecitabine (INN) /keɪpˈsaɪtəbiːn/ (Xeloda, Roche) is an orally-administered chemotherapeutic agent used in the treatment of metastatic breast and colorectal cancers. Capecitabine is a prodrug, that is enzymatically converted to 5-fluorouracil in the tumor, where it inhibits DNA synthesis and slows growth of tumor tissue. The activation of capecitabine follows a pathway with three enzymatic steps and two intermediary metabolites, 5′-deoxy-5-fluorocytidine (5′-DFCR) and 5′-deoxy-5-fluorouridine (5′-DFUR), to form 5-fluorouracil

Indications
Capecitabine is FDA-approved for:
- Adjuvant in colorectal cancer Stage III Dukes’ C – used as first-line monotherapy.
- Metastatic colorectal cancer – used as first-line monotherapy, if appropriate.
- Metastatic breast cancer – used in combination with docetaxel, after failure of anthracycline-based treatment. Also as monotherapy, if the patient has failed paclitaxel-based treatment, and if anthracycline-based treatment has either failed or cannot be continued for other reasons (i.e., the patient has already received the maximum lifetime dose of an anthracycline).
In the UK, capecitabine is approved by the National Institute for Health and Clinical Excellence (NICE) for colon and colorectal cancer, and locally advanced or metastatic breast cancer.[1] On March 29, 2007, the European Commission approved Capecitabine, in combination with platinum-based therapy (with or without epirubicin), for the first-line treatment of advanced stomach cancer.
Capecitabine is a cancer chemotherapeutic agent that interferes with the growth of cancer cells and slows their distribution in the body. Capecitabine is used to treat breast cancer and colon or rectum cancer that has spread to other parts of the body.
Formulation
Capecitabine (as brand-name Xeloda) is available in light peach 150 mg tablets and peach 500 mg tablets.
- Lacy, Charles F; Armstrong, Lora L; Goldman, Morton P; Lance, Leonard L (2004). Lexi-Comp’s Drug Information Handbook (12th Edition). Lexi-Comp Inc. ISBN 1-59195-083-X
- Fischer, David S; Knobf, M Tish; Durivage, Henry J; Beaulieu, Nancy J (2003). The Cancer Chemotherapy Handbook (6th Edition). Mosby. ISBN 0-323-01890-4
- Thomson Centerwatch: Drugs Approved by the FDA (Xeloda) Retrieved 6/05
- Mercier C, Ciccolini J (2007). “Severe or lethal toxicities upon capecitabine intake: is DPYD genetic polymorphism the ideal culprit?”. Trends in pharmacological sciences 28 (12): 597–598. doi:10.1016/j.tips.2007.09.009. PMID 18001850.
- “Subtopics”. Nice.org.uk. Retrieved 2012-08-15.
- Fingerprints May Vanish With Cancer Drug – US News and World Report
- Cancer Drug Erases Man’s Fingerprints – CNN
- “Stritch School of Medicine”. Stritch.luc.edu. Retrieved 2012-08-15.
- Xeloda.com (patient information, tools, and resources)
- OralChemo Advisor (patient information)

Capecitabine is an orally-administered anticancer agent widely used in the treatment of metastatic breast and colorectal cancers. Capecitabine is a ribofuranose-based nucleoside, and has the sterochemical structure of a ribofuranose having an β-oriented 5-fluorocytosine moiety at C-I position.
US Patent Nos. 5,472,949 and 5,453,497 disclose a method for preparing capecitabine by glycosylating tri-O-acetyl-5-deoxy-β-D-ribofuranose of formula I using 5-fluorocytosine to obtain cytidine of formula II; and carbamoylating and hydrolyzing the resulting compound, as shown in Reaction Scheme 1 :
Reaction Scheme 1

1
The compound of formula I employed as an intermediate in Reaction
Scheme 1 is the isomer having a β-oriented acetyl group at the 1 -position, for the reason that 5-fluorocytosine is more reactive toward the β-isomer than the α-isomer in the glycosylation reaction due to the occurrence of a significant neighboring group participation effect which takes place when the protecting group of the 2-hydroxy group is acyl.
Accordingly, β-oriented tri-O-acetyl-5-deoxy-β-D-ribofuranose (formula
I) has been regarded in the conventional art to the essential intermediate for the preparation of capecitabine. However, such a reaction gives a mixture of β- and α-isomers from which cytidine (formula II) must be isolated by an uneconomical step.
Meanwhile, US Patent No. 4,340,729 teaches a method for obtaining capecitabine by the procedure shown in Reaction Scheme 2, which comprises hydrolyzing 1-methyl-acetonide of formula III to obtain a triol of formula IV; acetylating the compound of formula IV using anhydrous acetic anhydride in pyridine to obtain a β-/α-anomeric mixture of tri-O-acetyl-5-deoxy-D-ribofuranose of formula V; conducting vacuum distillation to purify the β-/α-anomeric mixture; and isolating the β-anomer of formula I therefrom:
Reaction Scheme 2
III IV
However, the above method is also hampered by the requirement to perform an uneconomical and complicated recrystallization steps for isolating the β-anomer from the mixture of β-/α-anomers of formula V, which leads to a low yield of only about 35% to 40% (Guangyi Wang et al., J. Med. Chem., 2000, vol. 43, 2566-2574; Pothukuchi Sairam et al., Carbohydrate Research, 2003, vol. 338, 303-306; Xiangshu Fei et al., Nuclear Medicine and Biology, 2004, vol. 31, 1033-1041; and Henry M. Kissman et al., J. Am. Chem. Soc, 1957, vol. 79, 5534-5540).
Further, US Patent No. 5,476,932 discloses a method for preparing capecitabine by subjecting 5′-deoxy-5-fluorocytidine of formula VI to a reaction with pentylchloroformate to obtain the compound of formula VII having the amino group and the 2-,3-hydroxy groups protected with C5Hi1CO2 groups; and removing the hydroxy-protecting groups from the resulting compound, as shown in Reaction Scheme 3 :
Reaction Scheme 3

Vl VII 1
However, this method suffers from a high manufacturing cost and also requires several complicated steps for preparing the 5′-deoxy-5-fluorocytidine of formula VI: protecting the 2-,3-hydroxy groups; conducting a reaction thereof with 5-fluorocytosine; and deprotecting the 2-,3-hydroxy groups.
Accordingly, the present inventors have endeavored to develop an efficient method for preparing capecitabine, and have unexpectedly found an efficient, novel method for preparing highly pure capecitabine using a trialkyl carbonate intermediate, which does not require the uneconomical β-anomer isolation steps.
synthesis

more info and description
Aspects of the present invention relate to capecitabine and processes for the preparation thereof.
The drug compound having the adopted name “capecitabine” has a chemical name 5′-deoxy-5-fluoro-N-[(pentyloxy) carbonyl] cytidine and has structural formula I.
H

OH OH I
This compound is a fluoropyrimidine carbamate with antineoplastic activity. The commercial product XELODA™ tablets from Roche Pharmaceuticals contains either 150 or 500 mg of capecitabine as the active ingredient.
U.S. Patent No. 4,966,891 describes capecitabine generically and a process for the preparation thereof. It also describes pharmaceutical compositions, and methods of treating of sarcoma and fibrosarcoma. This patent also discloses the use of ethyl acetate for recrystallization of capecitabine. The overall process is summarized in Scheme I.

Scheme I
U.S. Patent No. 5,453,497 discloses a process for producing capecitabine that comprises: coupling of th-O-acetyl-5-deoxy-β-D-hbofuranose with 5- fluorocytosine to obtain 2′,3′-di-O-acetyl-5′-deoxy-5-fluorocytidine; acylating a 2′, 3′- di-O-acetyl-5′-deoxy-5-fluorocytidine with n-pentyl chloroformate to form 5′-deoxy- 2′,3′-di-O-alkylcarbonyl-5-fluoro-N-alkyloxycarbonyl cytidine, and deacylating the 2′ and 3′ positions of the carbohydrate moiety to form capecitabine. The overall process is summarized in Scheme II.

Capecitabine
Scheme Il
The preparation of capecitabine is also disclosed by N. Shimma et al., “The Design and Synthesis of a New Tumor-Selective Fluoropyrimidine Carbamate, Capecitabine,” Bioorganic & Medicinal Chemistry, Vol. 8, pp. 1697-1706 (2000). U.S. Patent No. 7,365,188 discloses a process for the production of capecitabine, comprising reacting 5-fluorocytosine with a first silylating agent in the presence of an acid catalyst under conditions sufficient to produce a first silylated compound; reacting the first silylated compound with 2,3-diprotected-5- deoxy-furanoside to produce a coupled product; reacting the coupled product with a second silylating agent to produce a second silylated product; acylating the second silylated product to produce an acylated product; and selectively removing the silyl moiety and hydroxyl protecting groups to produce capecitabine. The overall process is summarized in Scheme III. te

R: hydrocarbyl

Scheme III
Further, this patent discloses crystallization of capecitabine, using a solvent mixture of ethyl acetate and n-heptane. International Application Publication No. WO 2005/080351 A1 describes a process for the preparation of capecitabine that involves the refluxing N4– pentyloxycarbonyl-5-fluorocytosine with trimethylsiloxane, hexamethyl disilazanyl, or sodium iodide with trimethyl chlorosilane in anhydrous acetonitrile, dichloromethane, or toluene, and 5-deoxy-1 ,2,3-tri-O-acetyl-D-ribofuranose, followed by hydrolysis using ammonia/methanol to give capecitabine. The overall process is summarized in Scheme IV.

Scheme IV
International Application Publication No. WO 2007/009303 A1 discloses a method of synthesis for capecitabine, comprising reacting 5′-deoxy-5- fluorocytidine using double (trichloromethyl) carbonate in an inert organic solvent and organic alkali to introduce a protective lactone ring to the hydroxyl of the saccharide moiety; reacting the obtained compound with chloroformate in organic alkali; followed by selective hydrolysis of the sugar component hydrolytic group using an inorganic base to give capecitabine. The overall process is summarized in Scheme V.

Scheme V
Even though all the above documents collectively disclose various processes for the preparation of capecitabine, removal of process-related impurities in the final product has not been adequately addressed. Impurities in any active pharmaceutical ingredient (API) are undesirable, and, in extreme cases, might even be harmful to a patient. Furthermore, the existence of undesired as well as unknown impurities reduces the bioavailability of the API in pharmaceutical products and often decreases the stability and shelf life of a pharmaceutical dosage form.
nmr
1H NMR(CD3OD) δ 0.91(3H5 t), 1.36~1.40(4H, m), 1.41(3H, d), 1.68~1.73(2H, m), 3.72(1H, dd), 4.08(1H, dd), 4.13~4.21(3H, m), 5.7O(1H, s), 7.96(1H, d)

- The acetylation of 5′-deoxy-5-fluorocytidine (I) with acetic anhydride in dry pyridine gives 2′,3′-di-O-acetyl-5′-deoxy-5-fluorocytidine (II), which is condensed with pentyl chloroformate (III) by means of pyridine in dichromethane yielding 2′,3′-di-O-acetyl-5′-deoxy-5-fluoro-N4-(pentyloxycarbonyl)cytidine (IV). Finally, this compound is deacetylated with NaOH in dichloromethane/water. The diacetylated cytidine (II) can also be obtained by condensation of 5-fluorocytosine (V) with 1,2,3-tri-O-acetyl-5-deoxy-beta-D-ribofuranose (VI) by means of trimethylchlorosilane in acetonitrile or HMDS and SnCl4 in dichloromethane..
-
- EP 602454, JP 94211891, US 5472949.
- Capecitabine. Drugs Fut 1996, 21, 4, 358,
- Bioorg Med Chem Lett2000,8,(7):1697,
- Capecitabine. Drugs Fut 1996, 21, 4, 358,
- EP 602454, JP 94211891, US 5472949.
Paclitaxel Against Cancer: A Short Review
| Priyadarshini K1* and 2Department of Biotechnology, Loyola Academy Degree & PG College, Secunderabad, IndiaKeerthi Aparajitha U2 | ||||||
| http://www.omicsonline.org/paclitaxel-against-cancer-a-short-review-2161-0444.1000130.php?aid=9996 | ||||||
| Corresponding Author : | Priyadarshini K Department of Biotechnology JSS College for Arts Commerce & Science Mysore, India E-mail: prits_bhargav88@yahoo.com |
|||||
| Received November 16, 2012; Accepted November 28, 2012; Published November 30, 2012 | ||||||
| Citation: Priyadarshini K, Keerthi Aparajitha U (2012) Paclitaxel Against Cancer: A Short Review. Med chem 2:139-141. doi:10.4172/2161-0444.1000130 | ||||||
| Copyright: © 2012 Priyadarshini K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. | ||||||
|
||||||
