Buparlisib
Novartis Ag
5-[2,6-Di(4-morpholinyl)-4-pyrimidinyl]-4-(trifluoromethyl)-2-pyridinamine.
5-[2,6-Di(morpholin-4-yl)pyrimidin-4-yl]-4-(trifluoromethyl)pyridin-2-amine
5-(2,6-Di-4-morpholinyl-4-pyrimidinyl)-4- trifluoromethylpyridin-2-amine
944396-07-0
Chemical Formula: C18H21F3N6O2
Mass: 410.16781
NVP-BKM-120, BKM-120;
Novartis AG phase 3 for breast cancer
Phosphoinositide 3-kinase inhibitor
Buparlisib, also known as BKM120, is an orally bioavailable specific oral inhibitor of the pan-class I phosphatidylinositol 3-kinase (PI3K) family of lipid kinases with potential antineoplastic activity. PI3K inhibitor BKM120 specifically inhibits class I PIK3 in the PI3K/AKT kinase (or protein kinase B) signaling pathway in an ATP-competitive manner, thereby inhibiting the production of the secondary messenger phosphatidylinositol-3,4,5-trisphosphate and activation of the PI3K signaling pathway. This may result in inhibition of tumor cell growth and survival in susceptible tumor cell populations. Activation of the PI3K signaling pathway is frequently associated with tumorigenesis. Dysregulated PI3K signaling may contribute to tumor resistance to a variety of antineoplastic agents.
NVP-BKM-120 is an oral selective phosphatidylinositol 3-kinase (PI3K) inhibitor in phase III clinical development at Novartis for the treatment of breast cancer in combination with fulvestrant in postmenopausal women with hormone receptor-positive HER2-negative locally advanced or metastatic breast cancer which progressed on or after aromatase inhibitor treatment.
Early clinical development at Novartis Oncology, a division of Novartis, is also ongoing for the treatment of solid tumors, advanced endometrial carcinoma, non-small cell lung cancer (NSCLC), bladder cancer, gastrointestinal stromal cancer and for the treatment of metastatic castration-resistant prostate cancer.
Novartis is conducting phase II clinical trials for the treatment of follicular lymphoma, diffuse large B-cell lymphoma, mantle cell lymphoma and squamous cell carcinoma of head and neck.
The University of Kansas is evaluating the compound in phase I clinical trials for the treatment of advanced colorectal cancer in combination with irinotecan, while additional phase I trials are ongoing at the Dana-Farber Cancer Institute for the treatment of renal cell carcinoma. The Dana-Farber Cancer Institute is also conducting phase II clinical trials for the oral treatment of recurrent glioblastoma and preclinical studies for the treatment of ovarian cancer. Novartis is also conducting early clinical studies for the treatment of metastatic melanoma
pyrimidine derivative 5-(2,6-Di- 4-morpholinyl-4-pyrimidinyl)-4-trifluoromethylpyridin-2-amine (Compound A, see below), its hydrates, its salts and hydrates and solvates of its salts, to said specific solid forms thereof, to pharmaceutical compositions containing said solid forms, to processes for the preparation of pharmaceutical compositions containing said solid forms, to methods of using said solid forms and to pharmaceutical compositions for the therapeutic treatment of warm-blooded animals, especially humans. Background of the invention
WO 2007/084786 (priority date: January 20, 2006) describes certain pyrimidine derivatives having PI3 inhibiting properties, their use as pharmaceuticals and manufacturing processes thereof. One pyrimidine derivative disclosed in WO 2007/084786 is the selective
phosphatidylinositol 3-kinase inhibitor compound 5-(2,6-Di-4-morpholinyl-4-pyrimidinyl)-4- trifluoromethylpyridin-2-amine, hereinafter referred to as “Compound A” or “the compound of formula A”.
Compound A is described in WO 2007/084786 in free form and as the hydrochloric acid salt. The manufacturing process for preparing Compound A is described in Example 10 of this document. The manufacturing processes described therein are, although suitable, regarded as disadvantageous for commercial production.
Due to the high potency of pyrimidine derivatives, in particular PI3K inhibitors, there is a need for improved manufacturing methods of such compounds. In particular there is a need to provide processes that fulfill one or more of the following criteria: scalable, safer; simpler; higher yielding and more economical when compared to known.
…………………………………….
WO 2007084786
http://www.google.com/patents/WO2007084786A1?cl=en
Example 10
Preparation of 4-(“trifluoromethyπ-5-(2,6-dimorpholmoρyrirnidin-4-yπpyridin-2- amine
c
[0388] To a slurry of 2-moφholino-4,6-dichloropyrimidine (prepared as in
Method 22, 2.0 g, 8.54 mmol) in NMP (14 mL), triethylamine (1.43 mL, 10.25 mmol) was added. The heterogeneous mixture was stirred for 15 minutes, then treated with morpholine (0.75 mL, 8.54 mmol). Upon refluxing at 85 0C under argon for 2 hours, the solution was cooled, then added to EtOAc (160 mL). The organic solution was washed with 25 mL of NaHCO3(sat.) (2 x), water (2 x) and brine, dried over Na2SO4, filtered and concentrated. The crude material was dissolved in 200 mL EtOAc and filtered through a SiO2 pad, further eluting with EtOAc, yielding 2.2 g (93%) of 2,4-dimorpholino-6- chloropyrimidine as an off-white solid. LCMS (m/z): 285.0 (MH+), 1H NMR (CDCl3): δ 5.86 (s, IH), 3.71-3.76(m, 12H), 3.52-3.56(m, 4H).
[0389] 4-(trifluoromethyl)-5-(2,6-dimoφholmopyrimidin-4-yl)pyridin-2-amine 8
[0390] Argon gas was bubbled through a heterogeneous mixture of 2,4- dimoφholino-6-chloropyrimidine (4.1 g, 14.3 mmol) and 4-(trifluoromethyl)-5-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)pyridm-2-amine (16.5 g, 57.3 mmol) in 1,2- dimethoxyethane and 2M Na2Cθ3 (3:1) for 20 minutes. 1,1′-
Bis(diphenylphosphino)ferrocene palladium (IT) chloride (292 mg, 0.36 mmol) was added and the high pressure glass vessel containing the mixture was sealed. The reaction mixture was then heated at 900C for 15 hours, cooled and diluted with EtOAc (300 mL). The organic solution was washed with 300 mL of a mixture of water: Na2Cθ3(sat.):NH4θH(conc.) = 5:4:1, then NH4Cl(sat), and brine (2x), dried over Na2SO4, filtered and concentrated. The crude material was purified by SiO2 chromatography (50- 90% EtOAc/hexanes with 0.1% TEA) resulting in 5.62 g (95%) of 4-(trifluoromethyl)-5- (2,6-dimorpholinopyrimidin-4-yl)pyridin-2-amine as an off-white solid.
LCMS (m/z): 411.3 (MH+);
1H NMR (CDCl3): δ 8.27 (s, IH), 6.78 (s, IH), 5.97 (s, IH), 4.77 (bs, 2H), 3.59-3.80(m, 12H), 3.58-3.61(m, 4H).
…………….
WO2012044727 or equi as below
http://www.google.com/patents/EP2621908A2?cl=en
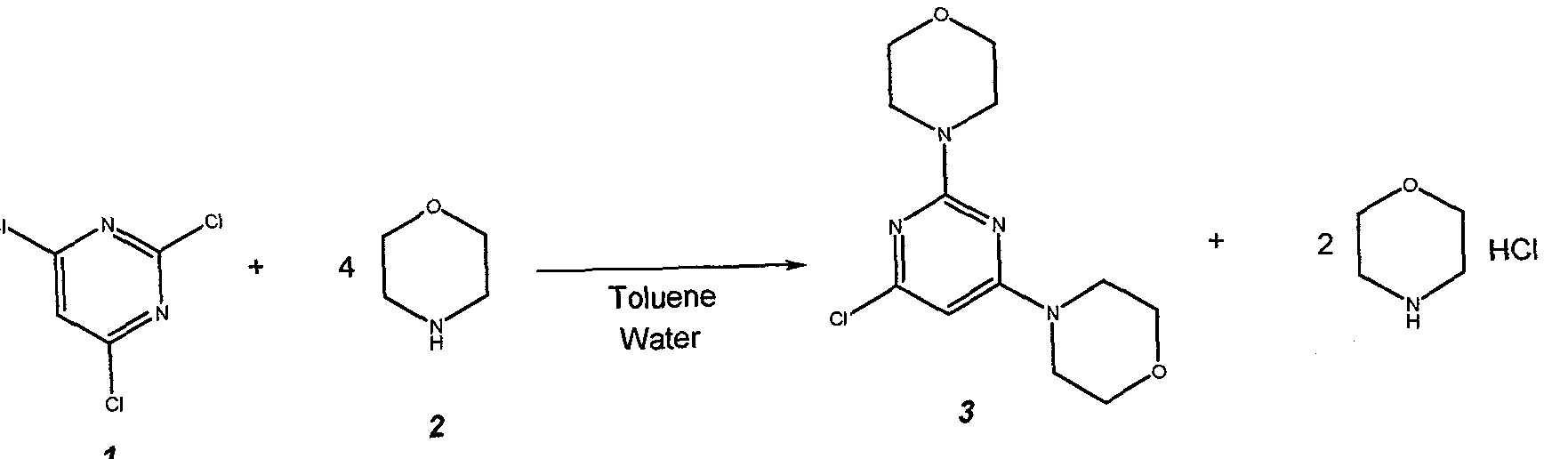
Example 1: 4,4′-(6-Chloropyrimidine-2,4-diyl)di[morpholine] (3) U 2011/053808
63
Prepare a solution of 22 g (0.12 mol) of 2,4,6-trichloropyrimidine 1 , in 95.2 g (110 mL) of toluene and charge it to the 25 mL addition funnel. Charge a nitrogen-flushed 500 mL round bottom 4- neck flask that equipped with a condenser, heating mantle, thermocouple, 125 mL addition funnel, mechanical stirrer and nitrogen inlet / outlet with 62.7 g (63 mL, 0.72 mol) of morpholine 2, 95.2 g (110 mL) of toluene and 44 g (44 mL) of water. Add the toluene solution of 1 over 10 minutes. Heat the reaction mixture to 83 ± 3 °C. Stir at 83 ± 3 °C for 2 h. Check the progress of the reaction. Cool to 30 + 3 °C. Transfer the 2-phase mixture to a 1L separatory funnel.
Separate the phases. Wash the organic phase (top) twice with 200 mL (2 x 100 mL) of warm (30 °C) water. Separate the phases after each wash. Transfer the organic (top) phase back to the 500 mL reaction flask that equipped with a condenser, heating mantle, thermocouple, 125 mL addition funnel, mechanical stirrer and nitrogen inlet / outlet. Stir and add 50.0 mL of 10.0 N aqueous hydrochloric acid solution. Heat the solution to 53 ± 3 °C and stir for 12 – 18 h. Check the progress of the reaction. Cool to 22 + 3 °C. Transfer the 2-phase mixture to a 1 L separatory funnel. Separate the phases. Transfer the aqueous (bottom) phase to a 500 mL round bottom 4-neck flask equipped with a cooling bath, thermocouple, addition funnel, pH probe, mechanical stirrer and nitrogen inlet / outlet. Stir and cool to 0 ± 3 °C. Add 85.0 g of 25% aqueous sodium hydroxide solution by drops over 30 minutes, maintaining a batch temperature of 10 ± 10 °C throughout the addition. Warm to 20 ± 3 °C and stir for 30 minutes. Isolate the solids by vacuum filtration. Wash the cake with 3 x 100 mL of water. Dry the solids (55°C, 30 mbar) for 24 hours to afford 30.9 g (91.9% yield) of 3 as a white crystalline solid.
Example 2:
4,4′-[6-(4>4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine-2,4-diyl]di[morpholine] (4)

Charge a nitrogen-flushed 2 L round bottom 4-neck flask that equipped with a condenser, heating mantle, thermocouple, rubber septum, mechanical stirrer and nitrogen inlet / outlet with 100.0 g (0.351 mol) of 4,4′-(6-chloropyrimidine -2,4-diyl)di[morpholine] 3 and 943 g (1200 mL) of acetonitrile. Stir and heat to 60 + 3 °C. Hold this solution at 60 + 3 °C for charge to batch. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, nitrogen inlet/outlet and rubber septum with 115.9 g (0.457 mol) of bis(pinacolato)- diboron, 51.7 g (0.527 mol) of potassium acetate, 12.9 g (0.014 mol) of tris(dibenzylideneacetone) – dipalladium(O), 7.9 g (0.029 mol) of tricyclohexylphosphine and 393 g (500 mL) of acetonitrile. Stir and heat the slurry to 84 ± 3 °C (reflux). Collect 00 mL of distillate. Transfer the warm 3 acetonitrile solution via peristaltic pump to the 3 L reactor containing the reaction mixture over 30 minutes and continue collecting distillate. Wash the 2 L flask and transfer lines with 79 g (100 mL) of acetonitrile and transfer the wash to the batch. Maintain distillation at 84 ± 3 °C and collect an additional 900 mL of distillate (batch volume ~ 1100 mL). Check the progress of the reaction 2 h from the start of the addition of 3. Cool the reaction mixture to 70 ± 3 °C and charge 693 g (800 mL) of toluene over 1-2 min. The batch will cool upon the addition of the toluene. Further cool the reaction mixture to 50 ± 3 °C. Charge to a clean 1 L flask, 347 g (400 mL) of toluene and warm it to 50 °C. This will be used as the cake wash. Filter the reaction mixture through a 15 g pad of Celite 545. Wash the filter cake with the warm (50 °C) toluene (400 mL) and collect this wash separately from the batch. This wash will be charged to the distillation residue later in the process. Transfer the filtrate back to the 3 L reactor. Concentrate the batch (25 °C to 40 °C internal temperature, 50 mbar) until a batch volume of 250 mL is reached.
Charge toluene cake wash held in reserve (~400 mL) and continue to concentrate the batch (37 °C to 43 °C internal temperature, 50 mbar) until a batch volume of 250 mL is reached. Check for complete removal of acetonitrile using the described Process Steering Control. Warm to 50 °C and stir for 15 min. Add 164 g (240 mL) of heptane over 30 minutes maintaining 50 °C throughout the addition. Stir the resulting suspension for 1 h. Cool the slurry to 23 ± 3 °C over 1 h and hold at this temperature for at least 1 h. Blanket the filtering funnel used for isolation of the product with nitrogen (to avoid moisture) and quickly filter the solids. Wash the filter cake twice with a mixture of 22 g (25 mL) of toluene and 51 g (75 mL) of heptane. Dry the solids at 50 °C, 35 mbar for 16 h to afford 4.4 g (72.7% corrected yield) of 4 as a sandy, beige solid. Example 3: 5-Bromo-4-(trifluoromethyl)pyridin-2-amine (4a)
4b 4a
Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, nitrogen inlet/outlet and rubber septum with 112.14 g (0.63 mol) of N-bromosuccinimide (NBS) and 645 g (725 mL) of tetrahydrofuran. Stir and cool the slurry to -5 ± 3 °C. Charge a nitrogen- flushed 1 L round bottom 4-neck flask that equipped with a thermocouple, mechanical stirrer and nitrogen inlet / outlet with 97.26 g (0.6 mol) of 2-amino-4-(trifluoromethyl)pyridine, 4b and 511 g (575 mL) of tetrahydrofuran. Stir to dissolve the 4b. Transfer the 4b solution to the addition funnel on the reactor and add the solution to the NBS slurry over 2 h maintaining an internal temperature of 0 ± 3 °C throughout the addition. Rinse the 1 L flask and addition funnel with 44 g (50 mL) of tetrahydrofuran and add the wash to the reaction mixture. Warm the solution to 20 + 3 °C over 30 minutes. Check for completeness of the reaction. Quench by charging a solution of 24.6 g of sodium thiosulfate pentahydrate dissolved in 475 mL of water over 10 minutes, maintaining a batch temperature of 20 ± 3 °C throughout the addition. Stir for 1 h after the quench. Concentrate (internal temp = 25 °C, 50 mbar) to remove tetrahydrofuran. Add 379 g (500 mL) of fert-butyl methyl ether. Stir and warm the resulting solution/suspension to 30 ± 3 °C and stir for 15 minutes. Separate the phases. Wash the extract four times with a solution of 32 g of sodium chloride dissolved in 768 g (768 mL) of water (4 x 200 mL per wash), separating the phases after each wash. Finally, wash the extract with 150 g (150 mL) of water. Separate the phases. Charge 152 g (200 mL) of terf-butyl methyl ether. Partially concentrate (57 ± 3 °C) to a volume of 350 mL. Cool to 50 °C and add 265 g (350 mL) of ferf-butyl methyl ether. Resume the concentration (57 ± 3 °C) until a batch volume of 350 mL is reached. Cool to 50 °C and add 265 g (350 mL) of fe/f-butyl methyl ether. Again, resume the concentration (57 ± 3 °C) until a batch volume of 350 mL is reached. Cool to 50 °C and add 103 g (150 mL) of terf-butyl methyl ether to raise the batch volume to 500 mL. Charge 1026 g (1500 mL) of heptane over 15 minutes maintaining 45 ± 3 °C throughout the addition. Slowly increase the vacuum and concentrate (internal temp = 40 °C to 50 °C) to a batch volume of 1000 mL. Release the vacuum and seed the batch. Resume the distillation, further increase the vacuum (slowly) and concentrate (internal temp = 25 °C to 40 °C) to a batch volume of 500 mL. Stir the resulting suspension at 0 °C for 30 min. Filter the solids. Wash the filter cake with 68 g (100 mL) of cold (0 °C) heptane (containing 30 ppm Octastat). Dry the solids (40 °C, 50 mbar) for 16 h to afford 109.8 g (78.0% yield) 4a as an orange solid.
Example 4: 5-(2,6-Di-4-morpholinyl^^yrimidinyl)-^trifluoromethylpyridin-2-ami^ (5)

Charge a 500 mL round bottom 3-neck flask that equipped with a thermocouple, mechanical stirrer, nitrogen inlet/outlet and cooling bath with 202.8 g (0.622 mol) of cesium carbonate and 260 g (260 mL) of water. Stir and cool the resulting solution to 22 ± 3 °C. Transfer the solution to the addition funnel. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, pH probe, nitrogen inlet/outlet and 500 mL addition funnel with 50.0 g (0.207 mol) of 5-bromo-4-(trifluoromethyl) pyridin-2-amine 4a, 190.9 g (0.456 mol) of 4,4′-[6-(4,4,5,5- tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyrimidine-2,4-diyl]di[morpholine] 4, 6.75 g (0.0103 mol) of 1,1′-bis(di-ferf-butylphosphino) ferrocene palladium dichloride and 556 g (625 mL) of thf. Stir the slurry at 22 ± 3 °C. Add the aqueous cesium carbonate solution via the addition funnel to the slurry over 1 – 2 min. Stir rapidly (to ensure good mixing), heat to 45 ± 3 °C over 15 min and hold at this temperature for at least 30 minutes. Check for completeness of the reaction. Cool to 22 ± 3 °C. Separate the phases. Partially concentrate the THF (25 °C, 90 mbar) to a volume of 400 mL. Add 654 g (750 mL) of isopropyl acetate, resume the vacuum distillation and concentrate to a volume of 400 mL. Add 610 g (700 mL) of isopropyl acetate, stir and filter the hazy solution through a 25 g pad of Celite. Wash the reactor and filter cake with 87 g (100 mL) of isopropyl acetate and add the wash to the batch. Add 1 L of 0. 25N aqueous N-acetyl-L- cysteine solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Again, add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Charge 34.5 g of Si-Thiol functionalized silica gel and stir the suspension at 60 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Add 1 L of 1 N aqueous hydrochloric acid solution and stir for 15 minutes. Separate the phases and retain the aqueous phase which now contains product. Extract the organic phase again by adding 500 mL of 1N aqueous HCI solution and stirring for 15 minutes. Separate the phases and combine the aqueous extracts. Adjust the pH to 2.3 ± 0.2 by the addition of ~280 mL of 4N aqueous sodium hydroxide solution. Charge 17.2 g of Si-Thiol functionalized silica gel and stir the suspension at 50 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Adjust the pH to 5.0 ± 0.2 by the slow addition of ~75 mL of 4N aqueous sodium hydroxide solution maintaining a batch temperature of 15 ± 3 °C. Stir the slurry for at least 16 h at 22 ± 3 °C to allow the product to completely solidify. Filter the solids and wash the filter cake once with 250 g (250 mL) of water. Dry the solids (50 °C, 35 mbar) for 16 h to obtain 75 g (89% yield) of 5 as a tan solid. Following this procedure, Compound 5 is the hemihydrate polymorph form HA of the Compound of Formula A.
Alternative procedure:
Charge a 500 mL round bottom 3-neck flask that equipped with a thermocouple, mechanical stirrer, nitrogen inlet/outlet and cooling bath with 202.8 g (0.622 mol) of cesium carbonate and 260 g (260 mL) of water. Stir and cool the resulting solution to 22 ± 3 °C. Transfer the solution to the addition funnel. Charge a nitrogen-flushed 3 L reactor that equipped with an overhead stirrer, condenser, pH probe, nitrogen inlet/outlet and 500 mL addition funnel with 50.0 g (0.207 mol) of 5-bromo-4-(trifluoromethyl) pyridin-2-amine 4a, 90.9 g (0.456 mol) of
4,4′[6(4,4,5,5tetramethyl1 ,3,2 dioxaborolan2yl)pyrimidine2,4diyl]di[morpholine] 4, 6.75 g (0.0103 mol) of 1 ,1′-bis(di-fert-butylphosphino) ferrocene palladium dichloride and 556 g (625 mL) of tetrahydrofuran. Stir the slurry at 22 ± 3 °C. Add the aqueous cesium carbonate solution via the addition funnel to the slurry over 1-2 min. Stir rapidly (to ensure good mixing), heat to 45 ± 3 °C over 15 min and hold at this temperature for at least 30 minutes. Check for completeness of the reaction . Cool to 22 + 3 °C. Separate the phases. Partially concentrate the THF (25 C, 90 mbar) to a volume of 400 mL. Add 654 g (750 mL) of isopropyl acetate, resume the vacuum distillation and concentrate to a volume of 400 mL. Add 610 g (700 mL) of isopropyl acetate, stir and filter the hazy solution through a 25 g pad of Celite. Wash the reactor and filter cake with 87 g (100 mL) of isopropyl acetate and add the wash to the batch. Add 1 L of 0.125N aqueous N- acetyl-L-cysteine solution and stir at 60 ± 3 °C for 1 h. Cool to 22 + 3 °C C and drain the aqueous wash. Add 1 L of 0.25N aqueous N-acetyl-L-cysteine pH = 7 solution and stir at 60 + 3 °C for 1 h. Cool to 22 + 3 °C and drain the aqueous wash. Again, add 1 L of 0.25N aqueous N- acetyl-L-cysteine pH = 7 solution and stir at 60 + 3 °C for 1 h. Cool to 22 ± 3 °C and drain the aqueous wash. Charge 34.5 g of Si-Thiol functionalized silica gel and stir the suspension at 60 + 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Add 1 L of N aqueous hydrochloric acid solution and stir for 15 minutes. Separate the phases and retain the aqueous phase which now contains product. Extract the organic phase again by adding 500 mL of 1N aqueous hydrochloric acid solution and stirring for 15 minutes. Separate the phases and combine the aqueous extracts. Adjust the pH to 2.3 + 0.2 by the addition of ~280 mL of 4N aqueous sodium hydroxide solution. Charge 17.2 g of Si-Thiol functionalized silica gel and stir the suspension at 50 ± 3 °C for 1 h. Cool to 22 ± 3 °C and filter to remove the silica gel. Adjust the pH to 5.0 ± 0.2 by the slow addition of ~75 mL of 4N aqueous sodium hydroxide solution maintaining a batch temperature of 15 ± 3 °C. Stir the slurry for at least 16 h at 22 ± 3 °C to allow the product to completely solidify. Filter the solids and wash the filter cake once with 250 g (250 mL) of water. Dry the solids (50 °C, 35 mbar) for 16 h to obtain 75 g (89% yield) of 5 as a tan solid. Following this procedure, Compound 5 is the hemihydrate polymorph form HA of the Compound of Formula A.
…………..
WO-2014064058
Improved process for manufacturing 5-(2,6-di-4-morpholinyl-4-pyrimidinyl)-4-trifluoromethylpyridin-2-amine
Improved process for the preparation of buparlisib, an oral PI3K inhibitor Novartis is developing for the treatment of solid tumors, including breast cancer and hematological tumors. In January 2014, a phase III development was ongoing and Novartis expected to file for regulatory approval for breast cancer in 2015. Buparlisib was originally claimed in WO2007084786, protection for which expires in both the US and Europe in January 2027. Also see WO2012044727 for a more recent process case.
Burger, M.T.; Pecchi, S.; Wagman, A.; et al.
Discovery of BKM120, a pan class I PI3 kinase inhibitor in phase I/II clinical trials
240th ACS Natl Meet (August 22-26, Boston) 2010, Abst MEDI 489
Vu, A.T.; Morris, J.; Malhotra, S.V.
Efficient and improved synthesis of a PI3K inhibitor anticancer agent
241st ACS Natl Meet (March 27-30, Anaheim) 2011, Abst ORGN 115

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











 GADOBUTROL
GADOBUTROL




 WORLDCUP FOOTBALL WEEK 2014 BRAZIL
WORLDCUP FOOTBALL WEEK 2014 BRAZIL