
Home » Antineoplastic (Page 5)
Category Archives: Antineoplastic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
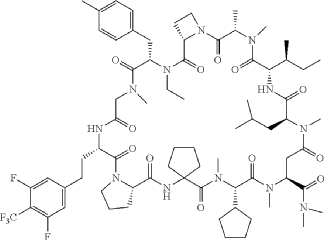
Paluratide
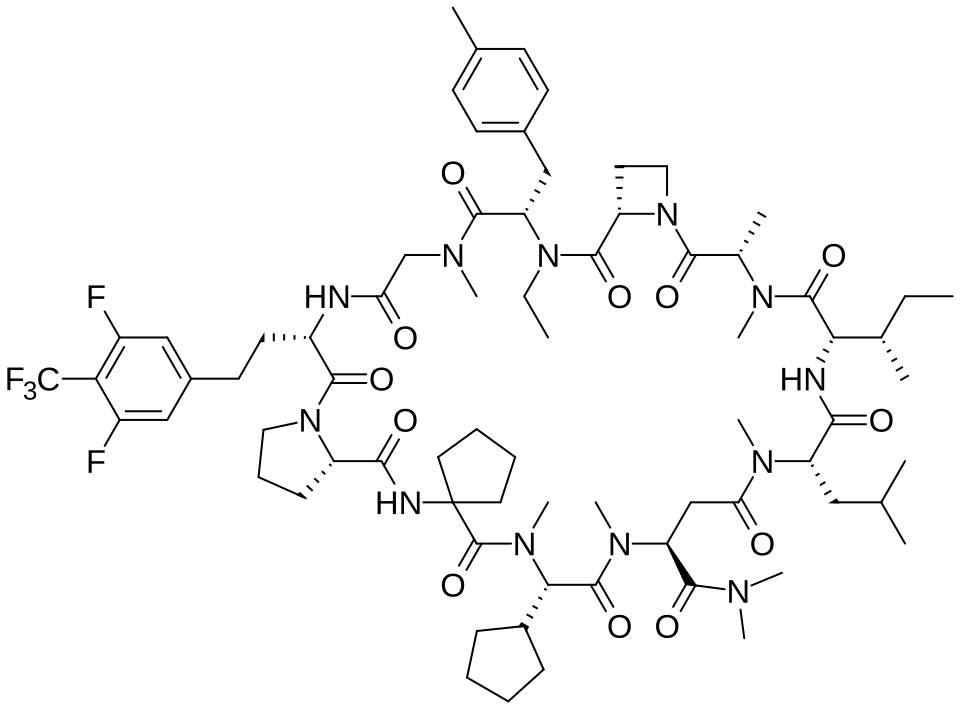


Paluratide
CAS 2676177-63-0
MFC73H105F5N12O12 MW 1437.7 g/mol
1,11-anhydro[N-methyl-L-alanyl-(2S)-azetidine-2-carbonyl-N-ethyl-4-methyl-L-phenylalanyl-N-methylglycyl-3-{[3,5-difluoro-4-(trifluoromethyl)phenyl]methyl}-L-alanyl-L-prolyl-2-
aminocyclopentane-1-carbonyl-(2S)-N-methyl-3-cyclopentylglycyl-1-
(dimethylamino)-N-methyl-L-aspart-4-yl-N-methyl-L-leucyl-Lisoleucine]
(3S,9S,12S,17S,20S,23S,27S,30S,36S)-20-[(2S)-butan-2-yl]-30-cyclopentyl-3-[2-[3,5-difluoro-4-(trifluoromethyl)phenyl]ethyl]-10-ethyl-N,N,7,17,18,24,28,31-octamethyl-9-[(4-methylphenyl)methyl]-23-(2-methylpropyl)-2,5,8,11,16,19,22,25,29,32,35-undecaoxospiro[1,4,7,10,15,18,21,24,28,31,34-undecazatricyclo[34.3.0.012,15]nonatriacontane-33,1′-cyclopentane]-27-carboxamide
G-protein Ras (rat sarcoma virus) inhibitor, antineoplastic, LUNA 18, CHUGAI, AW3YP3CD9X
Paluratide (development code LUNA18) was an investigational cyclic peptide KRAS inhibitor developed by Chugai Pharmaceutical, a member of the Roche Group, for the treatment of cancers with KRAS mutations.[1] The compound was notable as an orally bioavailable macrocyclic peptide that could target intracellular protein-protein interactions, a class of targets traditionally considered “undruggable.”[2]
Development was discontinued in July 2025 due to a narrow therapeutic window compared to competing KRAS inhibitors.[3]
Ras Inhibitor LUNA18 is an orally bioavailable cyclic peptide and Ras inhibitor, with potential antineoplastic activity. Upon oral administration, Ras inhibitor LUNA18 selectively targets, binds to and inhibits Ras, thereby inhibiting Ras-dependent signaling and inhibits proliferation of tumor cells in which Ras is overexpressed and/or mutated. Ras serves an important role in cell signaling, division and differentiation. Mutations of Ras may induce constitutive signal transduction leading to tumor cell growth, proliferation, invasion, and metastasis.
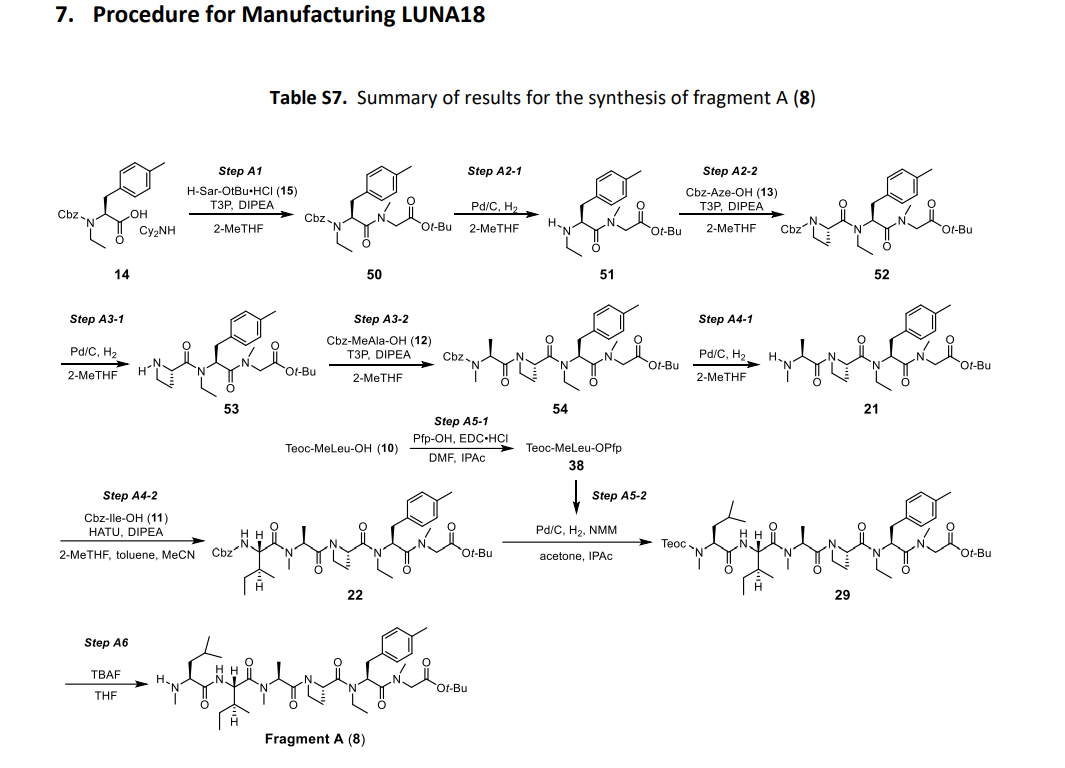
Paluratide (LUNA18 is synthesized using a novel liquid-phase peptide synthesis (LPPS) method, not traditional solid-phase methods, to overcome challenges with N-alkylated cyclic peptides. This process involves a convergent route of 24 telescoped chemical transformations, a final crystallization step, and a focus on specific strategies to manage side reactions like diketopiperazine formation and low reactivity of sterically hindered amino acids.
Key aspects of the synthesis
- Liquid-phase synthesis: A novel, high-yielding LPPS process was developed to enable the large-scale production of paluratide. This is a departure from traditional solid-phase methods, which have limitations with solubility and waste.
- Convergent synthetic route: The synthesis uses a convergent approach, meaning smaller fragments of the peptide are synthesized separately and then joined together. The overall process includes 24 telescoped chemical transformations followed by a final crystallization step.
- Addressing synthesis challenges: Specific strategies were employed to overcome key difficulties:
- Low reactivity: Amino acids with N-alkylation are sterically hindered, so more reactive and stable protecting groups were used to ensure efficient coupling.
- Side reactions: The method was designed to prevent side reactions like diketopiperazine formation in intermediates and incomplete hydrolysis of active esters.
- Instability: The peptide backbone is sensitive to acidic conditions, so a mildly acidic aqueous medium was chosen for workup and purification to maintain stability.
- Protecting group selection: Cbz-protected amino acid active esters were preferred over Boc-protected ones because they are less prone to forming N-carboxyanhydrides (NCA) under activating conditions, which can reduce yield and purity.
- Purification: A final crystallization step is used for purification.
PAT
- Method for producing eutectic of cyclic peptidePublication Number: WO-2024195801-A1Priority Date: 2023-03-20
- Method for producing cyclic peptide crystalsPublication Number: WO-2024085235-A1Priority Date: 2022-10-20
- Composition containing peptide, surfactant, and polymerPublication Number: WO-2024080308-A1Priority Date: 2022-10-12
- Methods for producing cyclic compounds comprising n-substituted amino acid residuesPublication Number: EP-4086272-A1Priority Date: 2021-05-07
- Methods for producing cyclic compounds comprising n-substituted amino acid residuesPublication Number: US-2022411462-A1Priority Date: 2021-05-07
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.5c00260?ref=PDF



PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US383248369&_cid=P20-MI3YXS-80609-1



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Mechanism of action
Paluratide functions as a pan-RAS inhibitor, targeting multiple RAS isoforms including KRAS, NRAS, and HRAS.[1] The compound binds with high affinity to KRASG12D, with a dissociation constant (Kd) of 0.043 nM, and blocks the interaction between KRASG12D and the guanine nucleotide exchange factor SOS1 with an IC50 of less than 2.2 nM.[4]
Unlike covalent KRAS inhibitors that target specific mutations (such as sotorasib for KRASG12C), paluratide was designed to inhibit RAS proteins through disruption of protein-protein interactions with guanine nucleotide exchange factors (GEFs).[1] This mechanism allows the drug to affect RAS signalling regardless of the specific mutation, theoretically providing broader applicability across different KRAS-mutant cancers. The compound also demonstrates activity against downstream signalling pathways, affecting ERK and AKT phosphorylation.[4]
Medical uses
Paluratide was being developed for the treatment of locally advanced or metastatic solid tumors harbouring RAS gene alterations.[5] The drug demonstrated significant cellular activity against multiple cancer types with KRAS mutations in preclinical studies, including colorectal cancer, gastric cancer, non-small cell lung cancer, and pancreatic cancer.[1]
Chemistry
Paluratide is an 11-member (11-mer) cyclic peptide with a molecular weight in the range of 1000–2000 g/mol, classified as a “middle-size” cyclic peptide.[1] The compound features extensive N-alkylation, a modification that reduces hydrogen bond donors and improves oral absorption while maintaining cellular permeability.[2] Its structure allows it to navigate the challenging boundary between small molecules and biologics, achieving properties of both classes. The compound demonstrated oral bioavailability ranging from 21% to 47% in preclinical animal studies without requiring special formulations.[1]
Discovery
Paluratide was discovered through Chugai Pharmaceutical’s cyclic peptide platform using an mRNA display library screening approach.[1] The initial hit compound, designated AP8747, was identified from the mRNA display library and subsequently underwent extensive chemical optimization without scaffold hopping (maintaining the basic cyclic peptide structure).[1] The optimization focused on increasing plasma stability, improving absorption, reducing clearance, and reducing hydrogen bond donors to achieve oral bioavailability.
The final clinical compound, LUNA18, emerged after modifications to four amino acid positions (positions 5, 7, 10, and 11) from an intermediate compound (compound 40). Key structure-activity relationship findings included: the side chain at position 5 preferring aromatic over aliphatic groups; physicochemical properties being adjustable at position 11; and biological activity enhancement through modifications at positions 7 and 10.[1]
Chugai also developed a novel synthetic methodology that enabled the broadly applicable synthesis of highly N-alkylated cyclic peptide-like drugs.[6] This method overcame three major technical challenges: formation of diketopiperazine, insufficient reactivity of amidation due to steric hindrance, and instability of cyclic peptides under acidic conditions. Using this approach, more than 4,000 cyclic peptides were synthesized with a process yield of 31% and final product purity of 97%.[6]
Clinical trials
A Phase 1 dose-escalation and cohort expansion study (NCT05012618) was initiated in August 2021 to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary activity of paluratide administered as a single agent or in combination with other anti-cancer drugs.[5] The study, in the United States and Japan, was designed to enrol approximately 195 patients with locally advanced or metastatic solid tumors positive for documented RAS alterations.[5]
Paluratide was administered orally as capsules.[5] The study also evaluated combination therapy with cetuximab, an EGFR inhibitor.[5]
References
- Tanada M, Tamiya M, Matsuo A, Chiyoda A, Takano K, Ito T, et al. (August 2023). “Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor”. Journal of the American Chemical Society. 145 (30): 16610–16620. Bibcode:2023JAChS.14516610T. doi:10.1021/jacs.3c03886. PMID 37463267.
- Ohta A, Tanada M, Shinohara S, Morita Y, Nakano K, Yamagishi Y, et al. (November 2023). “Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets”. Journal of the American Chemical Society. 145 (44): 24035–24051. Bibcode:2023JAChS.14524035O. doi:10.1021/jacs.3c07145. PMID 37874670.
- Taylor NP (24 October 2025). “Roche axes 4 Chugai solid tumor assets in early-phase clear-out”. Fierce Biotech.
- “LUNA18 (Paluratide) – KRAS Inhibitor, ERK Inhibitor, RAS Inhibitor”. MedChemExpress.
- “A Dose-escalation Study of LUNA18 in Patients With Locally Advanced or Metastatic Solid Tumors (With Expansion)”. ClinicalTrials.gov. 29 July 2025. NCT05012618.
- Nomura K, Hashimoto S, Takeyama R, Tamiya M, Kato T, Muraoka T, et al. (October 2022). “Broadly Applicable and Comprehensive Synthetic Method for N-Alkyl-Rich Drug-like Cyclic Peptides”. Journal of Medicinal Chemistry. 65 (19): 13401–13412. doi:10.1021/acs.jmedchem.2c01296. PMID 36109865.
- “Chugai Announces 2025 2nd Quarter Results” (Press release). Chugai Pharmaceutical. 24 July 2025.
External links
- Phase 1 Clinical Trial Information at ClinicalTrials.gov
- Development of LUNA18 at Journal of the American Chemical Society
| Clinical data | |
|---|---|
| Other names | LUNA18 |
| Routes of administration | Oral administration |
| Legal status | |
| Legal status | Development discontinued |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2676177-63-0 |
| PubChem CID | 166509683 |
| ChemSpider | 129321315 |
| UNII | AW3YP3CD9X |
| Chemical and physical data | |
| Formula | C73H105F5N12O12 |
| Molar mass | 1437.707 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////Paluratide, antineoplastic, LUNA 18, CHUGAI, AW3YP3CD9X
Neladalkib
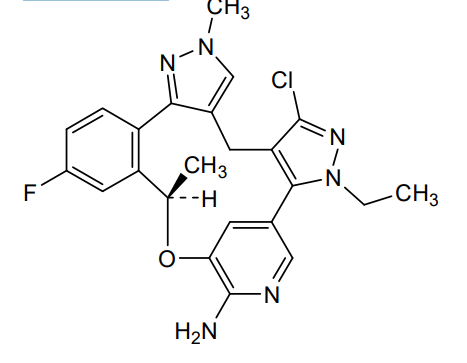
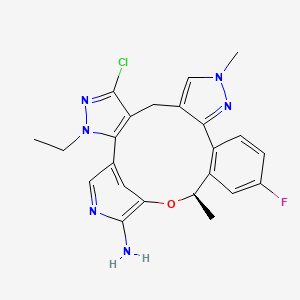
Neladalkib
CAS 2739866-40-9
MF C23H22ClFN6O MW 452.9 g/mol
(19R)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(25),2(6),4,8,11,13(18),14,16,21,23-decaen-22-amine

anaplastic lymphoma kinase (ALK) inhibitor, antineoplastic, NVL-655, NVL 655, J32P26A6BC, ALK-IN-27
Neladalkib is a small molecule drug. The usage of the INN stem ‘-alkib’ in the name indicates that Neladalkib is a ALK (anaplastic lymphoma kinase) inhibitor. Neladalkib is under investigation in clinical trial NCT06765109 (Neladalkib (NVL-655) for TKI-naive Patients With Advanced ALK-Positive NSCLC). Neladalkib has a monoisotopic molecular weight of 452.15 Da.
ALK Inhibitor NVL-655 is an orally bioavailable, brain-penetrant, selective small molecule inhibitor of the receptor tyrosine kinase (RTK) anaplastic lymphoma kinase (ALK), with potential antineoplastic activity. Upon oral administration, ALK inhibitor NVL-655 specifically targets, binds to and inhibits ALK fusion proteins and activating mutations, including the acquired resistance mutations solvent front mutation (SFM) G1202R and the compound mutations G1202R/L1196M and G1202R/G1269A. The inhibition of ALK leads to the disruption of ALK-mediated signaling and the inhibition of cell growth in ALK-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a variety of tumor cell types. NVL-655 is able to penetrate the blood-brain-barrier (BBB) and may therefore exert its activity against EGFR-driven central nervous system (CNS) primary tumors and CNS metastases.
- Expanded Access Program of Neladalkib (NVL-655) for Patients With Advanced ALK+ NSCLC or Other ALK+ Solid TumorsCTID: NCT06834074Status: AvailableDate: 2025-09-22
- Neladalkib (NVL-655) for TKI-naive Patients With Advanced ALK-Positive NSCLCCTID: NCT06765109Phase: Phase 3Status: RecruitingDate: 2025-08-29
- A Study of Neladalkib (NVL-655) in Patients With Advanced NSCLC and Other Solid Tumors Harboring ALK Rearrangement or Activating ALK Mutation (ALKOVE-1)CTID: NCT05384626Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-07-24
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023196910&_cid=P20-MHSIQF-58684-1
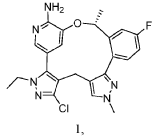
SYN
PAT
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2022098212-A1Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2022340586-A9Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2023076627-A1Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-11667649-B2Priority Date: 2020-05-05Grant Date: 2023-06-06
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: US-2023322797-A1Priority Date: 2022-04-07
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: WO-2023196900-A1Priority Date: 2022-04-07
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: WO-2023196900-A9Priority Date: 2022-04-07
- Methods of treating solid tumor using (19r)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentaazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(24),2(6),4,8,11,13,15,17,21(25),22-decaen-22-aminePublication Number: WO-2023196910-A1Priority Date: 2022-04-07
- Methods of treating solid tumor using (19r)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentaazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(24),2(6),4,8,11,13,15,17,21(25),22-decaen-22-aminePublication Number: EP-4504189-A1Priority Date: 2022-04-07
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////neladalkib, antineoplastic, NVL-655, NVL 655, J32P26A6BC, ALK-IN-27
Nefextinib
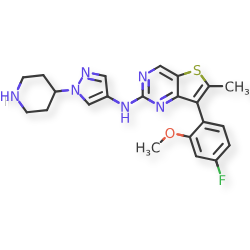
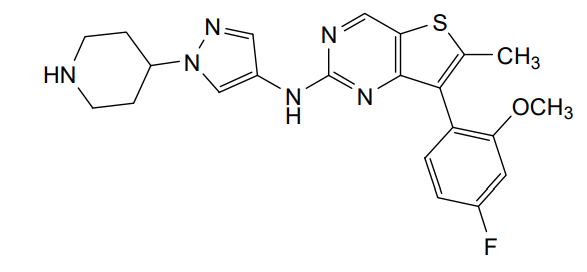
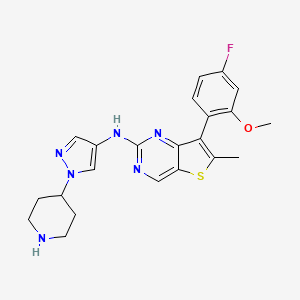
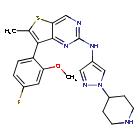
Nefextinib
CAS 2070931-57-4
MF C22H23FN6OS MW 438.52
7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-[1-(piperidin4-yl)-1H-pyrazol-4-yl]thieno[3,2-d]pyrimidin-2-amine
7-(4-FLUORO-2-METHOXYPHENYL)-6-METHYL-N-(1-(PIPERIDIN-4-YL)-1H-PYRAZOL-4-YL) THIENO (3,2-D)PYRIMIDIN-2-AMINE
tyrosine kinase inhibitor, antineoplastic, DL772G3NN7, MAX-40279, MAX 40279
Nefextinib is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) and FMS-like tyrosine kinase 3 (FLT3; CD135; STK1; FLK2), with potential antineoplastic activity. Upon oral administration, nefextinib binds to and inhibits both FGFR and FLT3, including FLT3 mutant forms, which results in the inhibition of FGFR/FLT3-mediated signal transduction pathways. This inhibits proliferation in FGFR/FLT3-overexpressing tumor cells. FGFR, a family of receptor tyrosine kinases, is upregulated in many tumor cell types. FLT3, a class III receptor tyrosine kinase (RTK), is overexpressed or mutated in most B-lineage neoplasms and in acute myeloid leukemias. They both play key roles in cellular proliferation and survival.
- A Phase 2 Study to Evaluate the Safety and Efficacy of Max-40279-01 in Patients With Advanced Gastric Cancer or Gastroesophageal Junction CancerCTID: NCT05395780Phase: Phase 2Status: Unknown statusDate: 2022-06-02
- MAX-40279 in Subjects With Acute Myelogenous Leukemia (AML)CTID: NCT03412292Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- MAX-40279-01 in Patients With Advanced Solid TumorsCTID: NCT04183764Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- Study of MAX-40279 in Patients With Relapsed or Refractory Acute Myelogenous Leukemia (AML)CTID: NCT04187495Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- A Clincal Study of Max-40279-01 in Patients With Advanced Colorectal CancerCTID: NCT05130021Phase: Phase 2Status: Unknown statusDate: 2021-12-06
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017012559&_cid=P22-MHRG1L-67142-1
[0488]N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidin-4-yl)-1H-pyrazol-4-amine (compound 31)
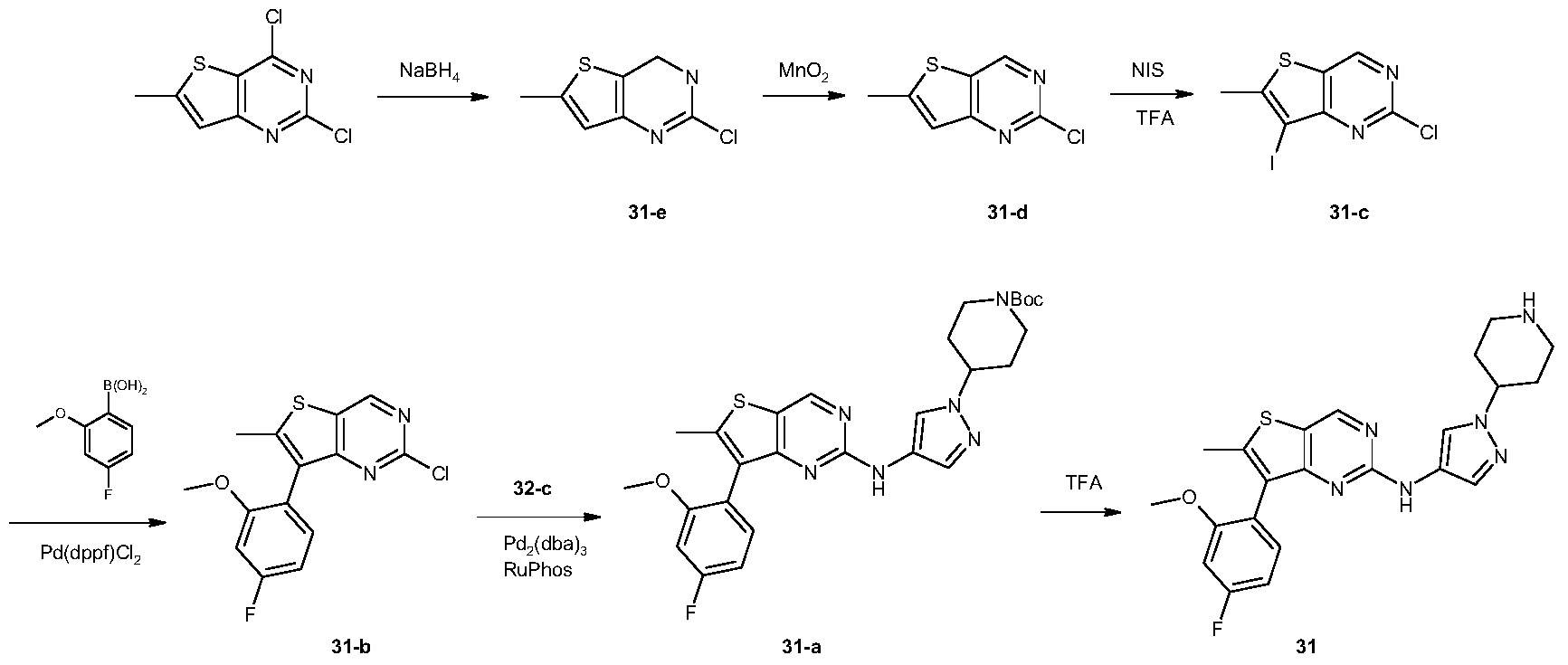
[0491]2,4-Dichloro-6-methylthiophene[3,2-d]pyrimidine (10 g, 45.6 mmol) was dissolved in tetrahydrofuran (100 mL) and ethanol (100 mL). The reaction mixture was cooled to 0 °C, and sodium borohydride (12.5 g, 198 mmol) was added in portions. The reaction mixture was brought to room temperature and stirred for 16 hours. It was then diluted with water (500 mL) and adjusted to pH 7 with 1 N hydrochloric acid solution. The aqueous phase was extracted with ethyl acetate (150 mL × 3). The organic phase was washed successively with water (100 mL × 3) and saturated brine (100 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a white solid 31-e (7.5 g, yield: 88%). This product required no further purification. LC-MS (ESI): m/z = 187 [M+H] + .
[0492]Synthesis of compound 31-d
[0493]Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0 °C, and activated manganese dioxide (35 g, 400 mmol) was added. The reaction mixture was brought to room temperature and stirred for 16 hours. The reaction mixture was filtered through diatomaceous earth, and the filter cake was washed with chloroform (100 mL × 3). The combined filtrates were concentrated under reduced pressure to give a white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS (ESI): m/z = 185 [M + H]+.
[0494]Synthesis of compound 31-c
[0495]Compound 31-d (3.1 g, 16.8 mmol) was dissolved in trifluoroacetic acid (30 mL) at 0 °C. N-iodosuccinimide (5.7 g, 25.3 mmol) was added in portions. The reaction mixture was brought to room temperature and stirred for 1 hour. The reaction was quenched with water (50 mL) and extracted with dichloromethane (50 mL × 3). The organic phase was washed successively with water (50 mL × 3) and saturated brine (50 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a white solid 31-c (4.9 g, yield: 94%). This product required no further purification. LC-MS (ESI): m/z = 311 [M + H] + .
[0496]Synthesis of compound 31-b
[0497]Compound 31-c (615 mg, 1.98 mmol), 2-methoxy-4-fluorophenylboronic acid (405 mg, 2.38 mmol), and sodium carbonate (630 mg, 5.94 mmol) were suspended in dioxane (5 mL) and water (5 mL). A [1,1′-bis(diphenylphosphine)ferrocene]palladium dichloride dichloromethane complex (163 mg, 0.2 mmol) was added. The mixture was purged three times with nitrogen and heated to 80 °C for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was separated into layers by dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated and purified by silica gel column chromatography (petroleum ether:dichloromethane = 1:1) to give a white solid 31-b (240 mg, yield: 39%). LC-MS (ESI): m/z = 309 [M+H] + .
[0498]Synthesis of compound 31-a
[0499]Compound 31-b (240 mg, 0.78 mmol) and compound 32-c (208 mg, 0.78 mmol) were dissolved in N,N-dimethylformamide (3 mL), and potassium carbonate (323 mg, 2.34 mmol), 2-dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol), and tris(dibenzylacetone)palladium (134 mg, 0.24 mmol) were added. The reaction was carried out under nitrogen protection at 110 °C for 16 hours. After cooling to room temperature, the reaction mixture was separated into layers by dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin-layer chromatography (petroleum ether: ethyl acetate = 1:1) to give a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z = 539[M+H] + .
[0500]Synthesis of Compound 31
[0501]31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), and trifluoroacetic acid (3 mL) was added. The mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure, and the residue was separated into layers by ethyl acetate (50 mL) and 1N hydrochloric acid aqueous solution (50 mL). The aqueous phase was adjusted to pH = 10 with saturated potassium carbonate aqueous solution, and a solid precipitated. The solid was filtered, and the filter cake was washed with water (20 mL × 3). The solid was dried under vacuum to give a light yellow solid 31 (22 mg, yield: 14%). LC-MS (ESI): m/z = 439 [M+H] + .
[0502]
1H-NMR(400MHz,MeOD)δ:8.78(d,J=5Hz,1H),7.87(s,1H),7.48(s,1H),7.35(m,1H),7.05(dd,J=11Hz,J=2Hz,1H),6.91(m,1H),4.10(m,1H),3.79(s,3H),3.22(m,2H),2.77(m,2H),2.47(s,3H),2.03(m,2H),1.73(m,2H)ppm
PAT
- Condensed ring pyrimidine compound, intermediate, its preparation method, composition and applicationPublication Number: CN-106366093-BPriority Date: 2015-07-21Grant Date: 2020-08-18
- Condensation ring pyrimidine compounds, intermediates, methods for producing them, compositions and applicationsPublication Number: JP-6875372-B2Priority Date: 2015-07-21Grant Date: 2021-05-26
- Condensed ring pyrimidine compounds, intermediates, preparation methods, compositions and applications thereofPublication Number: KR-102591886-B1Priority Date: 2015-07-21Grant Date: 2023-10-20
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: EP-3354653-B1Priority Date: 2015-07-21Grant Date: 2019-09-04
- Fused ring pyrimidine compounds, intermediates, production methods, compositions and applications thereofPublication Number: JP-2018520202-APriority Date: 2015-07-21
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: US-10494378-B2Priority Date: 2015-07-21Grant Date: 2019-12-03
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: US-2018208604-A1Priority Date: 2015-07-21
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: WO-2017012559-A1Priority Date: 2015-07-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////nefextinib, tyrosine kinase inhibitor, antineoplastic, DL772G3NN7, MAX-40279, MAX 40279
Lunbotinib
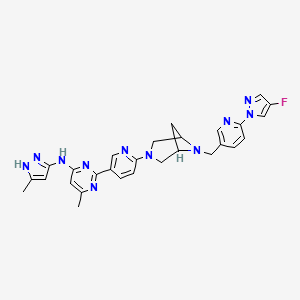
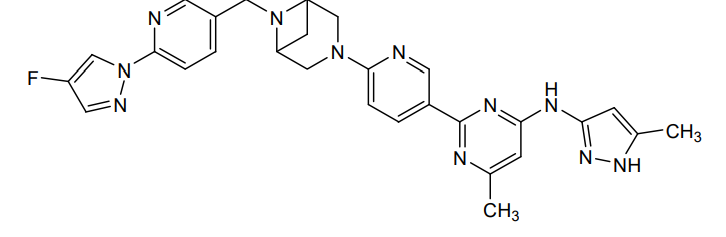
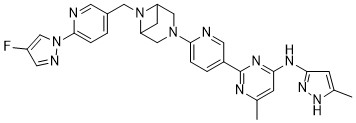
Lunbotinib
CAS 2479961-46-9
MF C28H28FN11 MW537.6 g/mol
2-[6-(6-{[6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl]methyl}-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl]-6-methyl-N-(5-methyl1H-pyrazol-3-yl)pyrimidin-4-amine
tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
- 2-(6-(6-((6-(4-fluoropyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo(3.1.1)heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
- 2-[6-[6-[[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
Lunbotinib is an orally bioavailable selective inhibitor of the proto-oncogene receptor tyrosine kinase rearranged during transfection (RET), with potential antineoplastic activity. Upon oral administration, lunbotinib selectively binds to various RET fusions and mutations, including solvent front resistance mutations, and inhibits the activity of RET. This results in an inhibition of cell growth of tumors that exhibit increased RET activity due to these fusions and mutations. RET overexpression, activating mutations, and fusions result in the upregulation and/or overactivation of RET tyrosine kinase activity in various cancer cell types. Dysregulated RET activity plays a key role in the development and progression of certain cancers. Lunbotinib is able to penetrate the blood-brain barrier (BBB) and may also be able to overcome resistance mechanisms to first generation selective RET inhibitors (SRIs).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020168939&_cid=P12-MHKH7H-14851-1
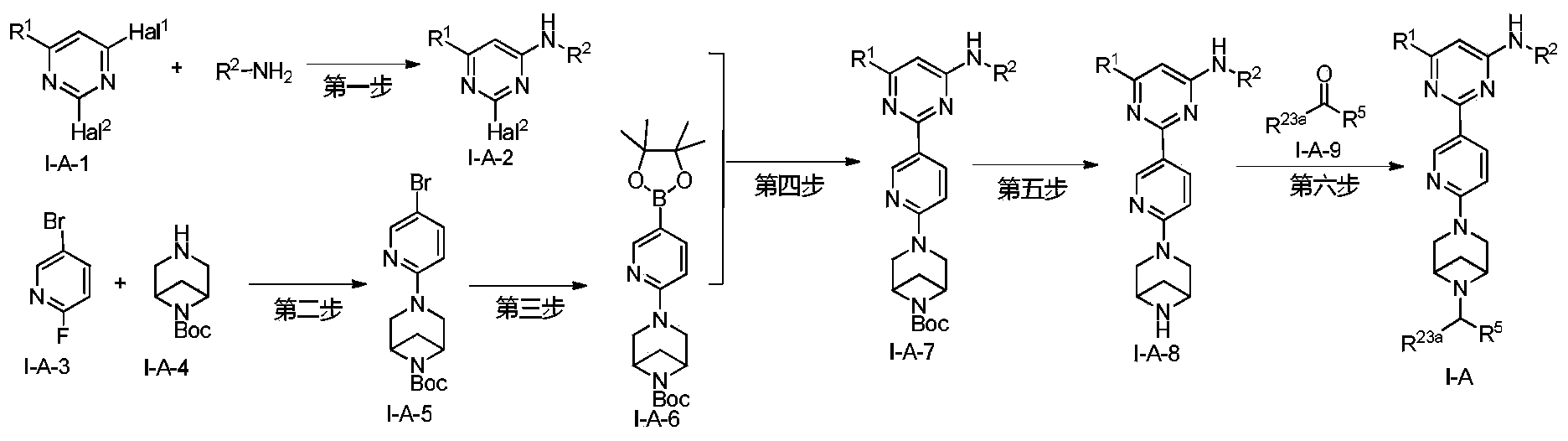
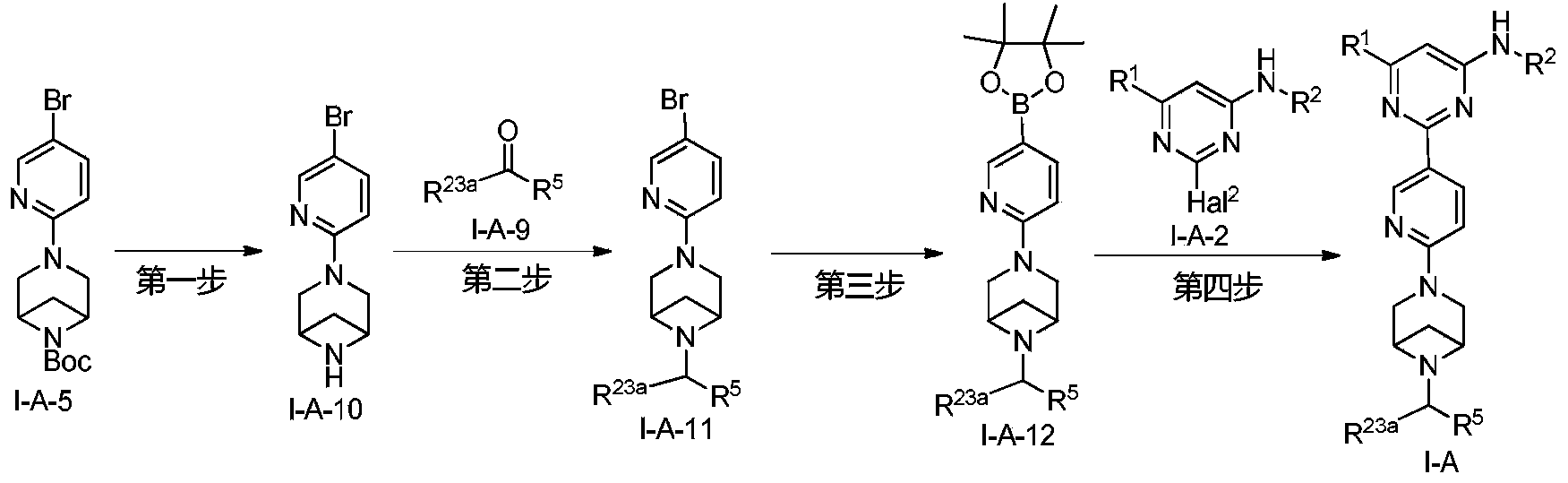
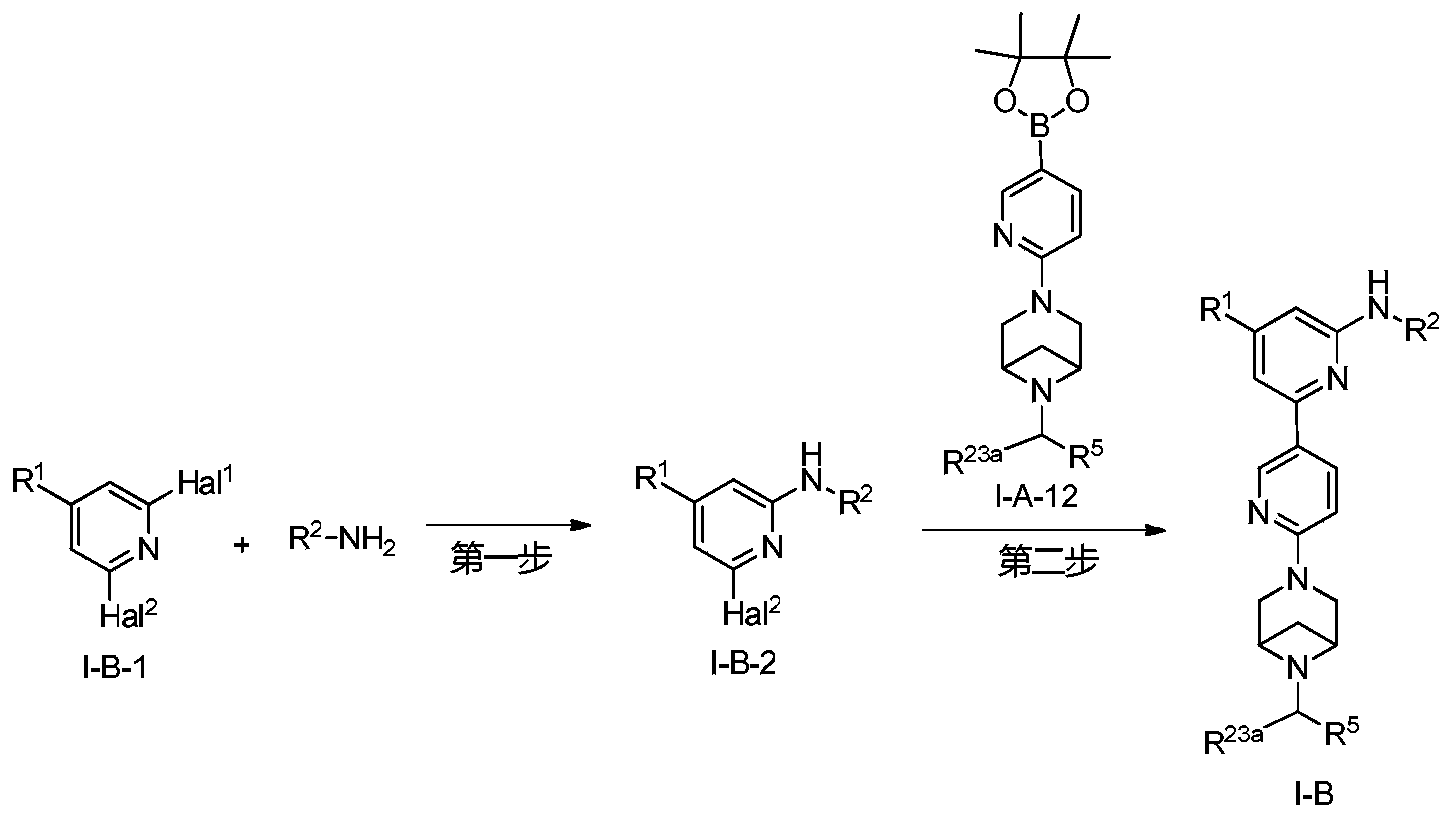
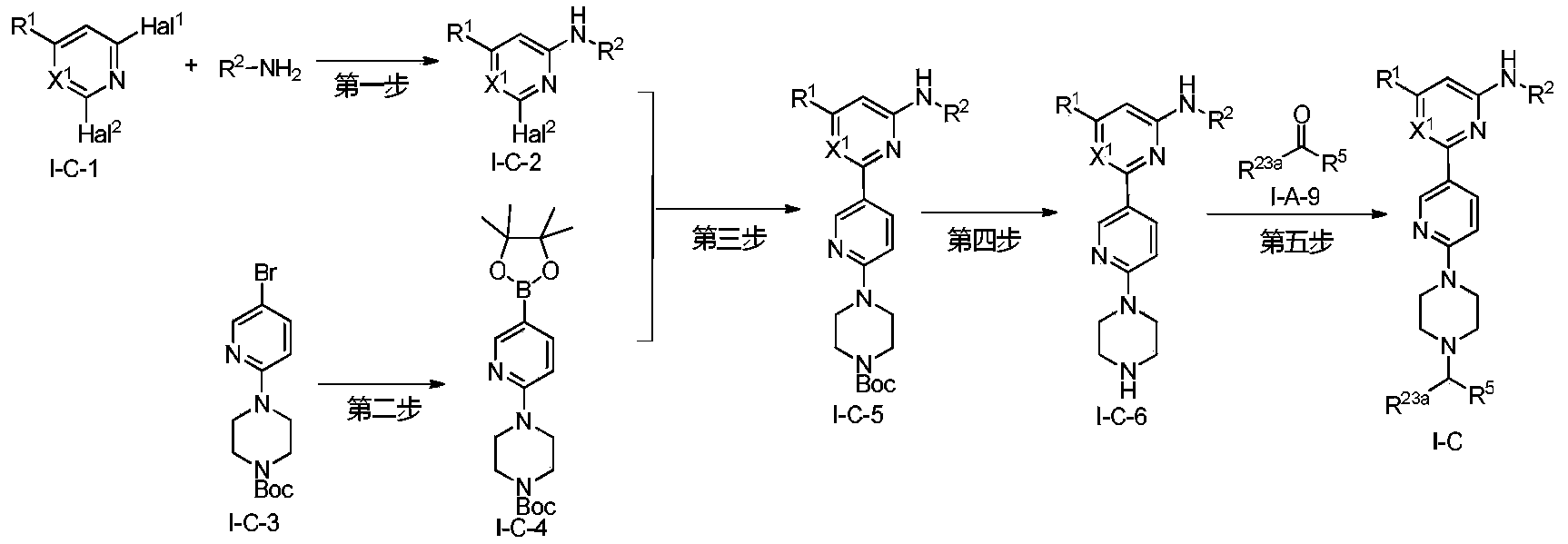
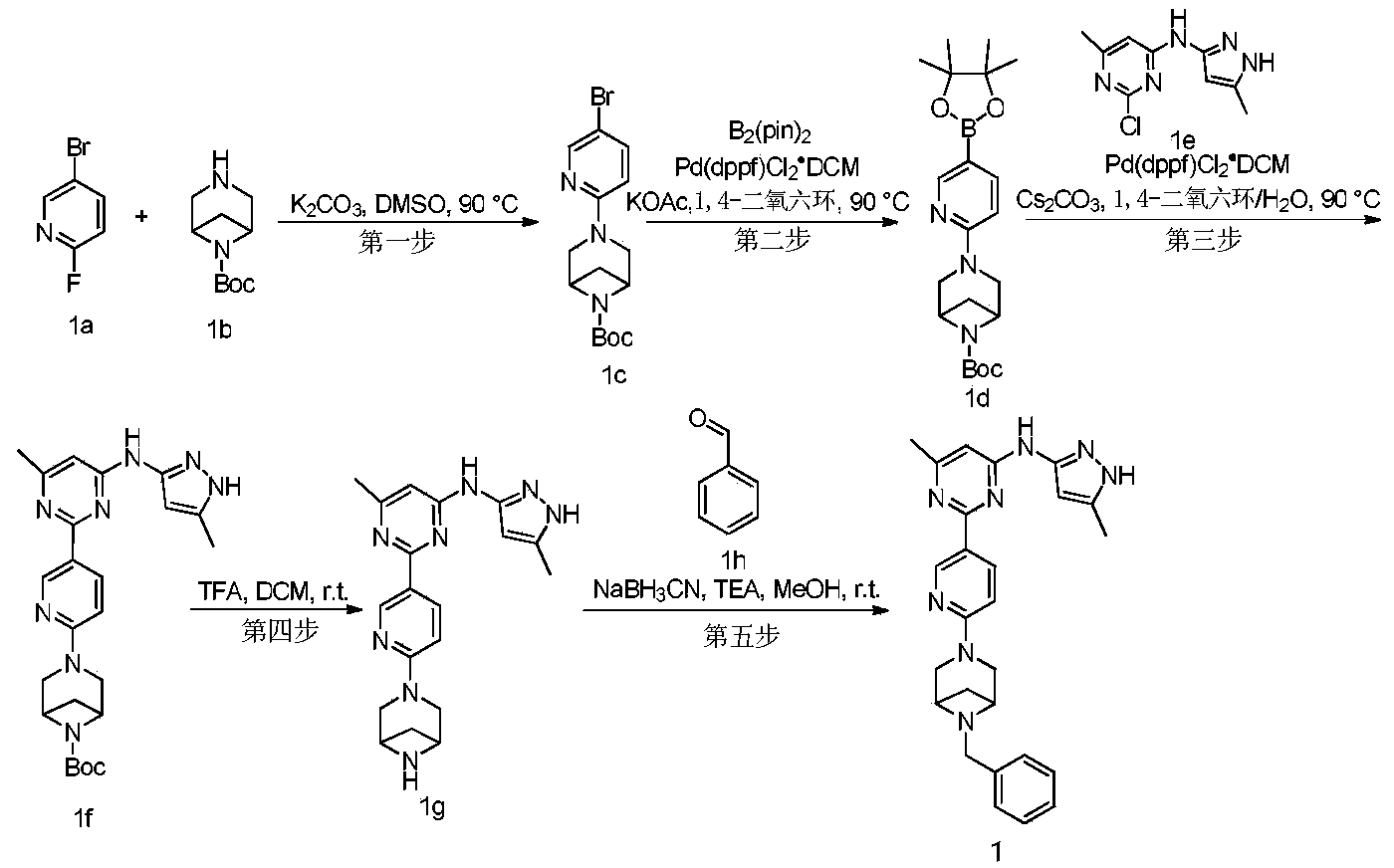
Example 6: 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 17)
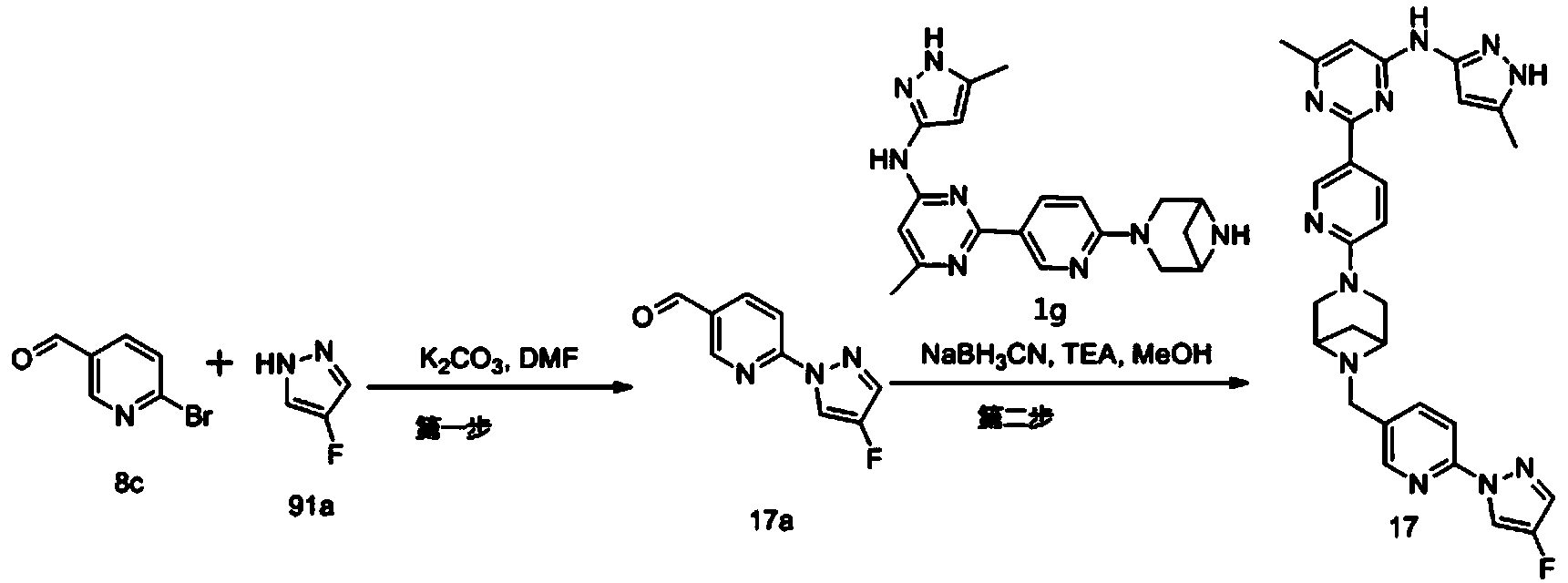
Step 1: Preparation of 6-(4-fluoro-1H-pyrazol-1-yl)nicotinaldehyde (compound 17a)
[0396]Compound 8c (2.0 g), 91a hydrochloride (1.58 g), and potassium carbonate (4.45 g) were sequentially added to DMF (15 mL), and the mixture was heated to 80 °C and stirred for 14 h. The reaction mixture was cooled to room temperature, diluted with water (100 mL), and extracted with DCM (50 mL x 2). The organic phases were combined, washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography (PE:EA = 10:1) to give compound 17a (0.81 g). MS m/z (ESI): 192.1 [M+H]
[0397]Step 2: Preparation of 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 17)
[0398]1 g of trifluoroacetate (22.82 mg) and compound 17a (27.47 mg) were added to methanol (1.0 mL), followed by the sequential addition of triethylamine (4.45 mg) and sodium cyanoborohydride (13.86 mg), and the reaction was carried out at room temperature for 14 h. After the reaction was completed, the reaction solution was concentrated to dryness under reduced pressure and purified by Prep-HPLC to obtain compound 17 (7.0 mg). MS m/z (ESI): 538.3 [M+H]
[0399]
1H NMR(400MHz,DMSO-d 6)δ11.98(s,1H),9.66(s,1H),9.12(d,J=2.16Hz,1H),8.67(dd,J=4.54,0.64Hz,1H),8.43(dd,J=8.94,2.28Hz,1H),8.41(d,J=1.68,1H),7.98(dd,J=8.48Hz,2.12 1H),7.92(d,J=4.28,1H),7.87(d,J=8.4,1H),6.78(d,J=9.0Hz,2H),6.31(br,1H),3.78-3.71(m,4H),3.68-3.52(m,4H),2.59-2.52(m,1H),2.33(s,3H),2.25(s,3H),1.60(d,J=8.36Hz,1H).
PAT
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: US-2022144847-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-113316578-BPriority Date: 2019-02-19Grant Date: 2023-10-31
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117263945-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117327078-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing same, methods for their preparation and usePublication Number: JP-7615056-B2Priority Date: 2019-02-19Grant Date: 2025-01-16
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: US-2023295174-A1Priority Date: 2020-07-28
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: WO-2020168939-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same, and preparation methods and uses thereofPublication Number: CN-113316578-APriority Date: 2019-02-19
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: EP-3929198-A1Priority Date: 2019-02-19
- Heterocyclic compounds, drug compositions containing them, methods of their manufacture and usePublication Number: JP-2022521859-APriority Date: 2019-02-19
- Use of heterocyclic compound for treating diseases related to ret genetic change and method thereforPublication Number: WO-2024240017-A1Priority Date: 2023-05-19
- Uses and methods of heterocyclic compounds for treating diseases associated with kinase resistance mutationsPublication Number: CN-116801882-APriority Date: 2021-03-24
- Use of heterocyclic compound in treating diseases related to kinase drug-resistant mutation and method thereforPublication Number: EP-4316490-A1Priority Date: 2021-03-24
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: EP-4190781-A1Priority Date: 2020-07-28
- Salts, crystal forms of pyrimidine compounds and methods for their preparationPublication Number: JP-2023535361-APriority Date: 2020-07-28
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Lunbotinib, tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
Lirodegimod
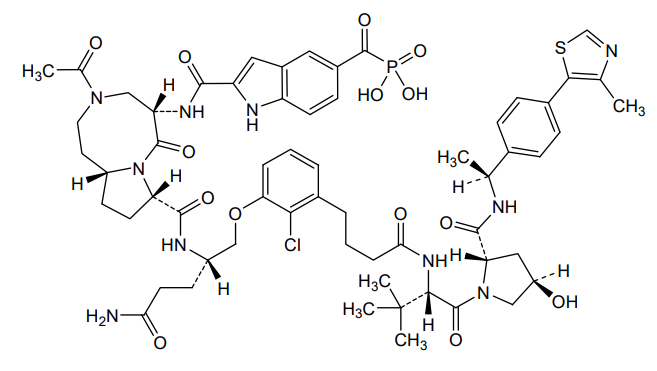
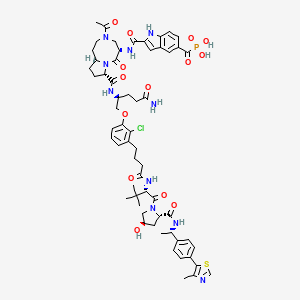
Lirodegimod
CAS 2502186-79-8
MF C60H74ClN10O14PS, MW 1257.79

[2-[[(5S,8S,10aR)-3-acetyl-8-[[(2S)-5-amino-1-[2-chloro-3-[4-[[(2S)-1-[(2S,4R)-4-hydroxy-2-[[(1S)-1-[4-(4-methyl-1,3-thiazol-5-yl)phenyl]ethyl]carbamoyl]pyrrolidin-1-yl]-3,3-dimethyl-1-oxobutan-2-yl]amino]-4-oxobutyl]phenoxy]-5-oxopentan-2-yl]carbamoyl]-6-oxo-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocin-5-yl]carbamoyl]-1H-indole-5-carbonyl]phosphonic acid
KT 333, KT333, ANTINEOPLASTIC, Fast Track (United States), Orphan Drug (United States), 4Q6ZHJ2MNA
Lirodegimod is a small molecule drug. The usage of the INN stem ‘-imod’ in the name indicates that Lirodegimod is a immunomodulator, both stimulant/suppressive and stimulant. Lirodegimod has a monoisotopic molecular weight of 1256.45 Da.
Safety, PK, PD, Clinical Activity of KT-333 in Adult Patients With Refractory Lymphoma, Large Granular Lymphocytic Leukemia, Solid Tumors
CTID: NCT05225584
Phase: Phase 1
Status: Completed
Date: 2025-03-19
PAT
- Stat3 degraders and uses thereofPublication Number: US-2023212201-A1Priority Date: 2021-12-11
- Stat3 degraders and uses thereofPublication Number: US-2025019388-A1Priority Date: 2021-12-11
- Stat degraders and uses thereofPublication Number: US-2024016942-A1Priority Date: 2020-03-17
- Stat degraders and uses thereofPublication Number: WO-2020206424-A1Priority Date: 2019-04-05
- Stat degraders and uses thereofPublication Number: US-11746120-B2Priority Date: 2019-04-05Grant Date: 2023-09-05
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Lirodegimod, KT 333, KT333, ANTINEOPLASTIC, Fast Track, Orphan Drug, 4Q6ZHJ2MNA
Inlexisertib
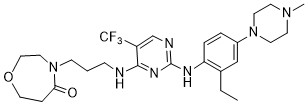
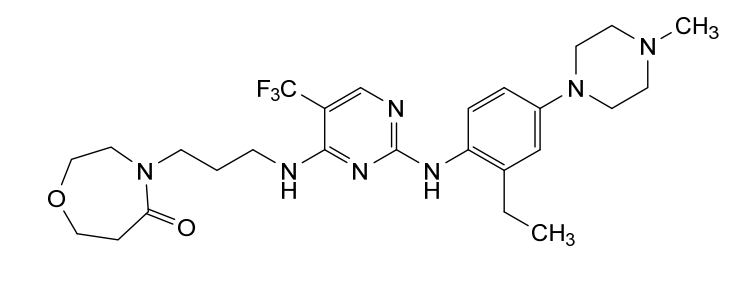
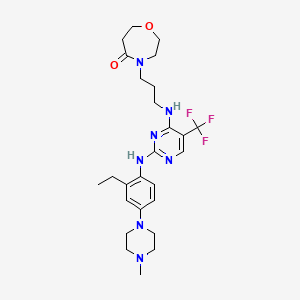
Inlexisertib
CAS 2543673-19-2
MF C26H36F3N7O2, 535.62
4-(3-((2-((2-ethyl-4-(4-methylpiperazin-1-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4-yl)amino)propyl)-1,4-oxazepan-5-one
4-[3-[[2-[2-ethyl-4-(4-methylpiperazin-1-yl)anilino]-5-(trifluoromethyl)pyrimidin-4-yl]amino]propyl]-1,4-oxazepan-5-one

serine/ threonine kinase inhibitor, antineoplastic, DCC 3116, JM2ZTM8S7S
Inlexisertib is an orally bioavailable inhibitor of the serine/threonine-protein kinase ULK 1 and 2, with potential antineoplastic activity. Upon oral administration, inlexisertib targets and binds to ULK1/2. This inhibits cancer autophagy, which mutant RAS cancer cells use for their survival, and results in tumor cell death. ULK1/2 mediates the autophagocytotic process and is often upregulated in cancers, especially in mutant RAS cancers. Autophagy plays a key role in a tumor cell proliferation and survival, and mediates tumor cell resistance.
- A Study of Inlexisertib (DCC-3116) in Combination With Anticancer Therapies in Participants With Advanced MalignanciesCTID: NCT05957367Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-06-05
- A Phase 1/2 Study of Inlexisertib (DCC-3116) in Patients With RAS/MAPK Pathway Mutant Solid TumorsCTID: NCT04892017Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-05-06
SYN
https://patents.google.com/patent/US11530206B2/en
PAT
Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereof
Publication Number: JP-7593947-B2
Priority Date: 2019-05-10
Grant Date: 2024-12-03
- PHENYLAMINOPYRIMIDINE AMIDE INHIBITORS OF AUTOPHAGY AND METHODS OF THEIR APPLICATIONPublication Number: HR-P20231730-T1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-12071432-B2Priority Date: 2019-05-10Grant Date: 2024-08-27
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118878519-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118878520-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118930524-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: AU-2020275392-B2Priority Date: 2019-05-10Grant Date: 2023-09-14
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-114127057-BPriority Date: 2019-05-10Grant Date: 2024-07-12
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-3966207-B1Priority Date: 2019-05-10Grant Date: 2023-11-01
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-4342469-A2Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: ES-2966807-T3Priority Date: 2019-05-10Grant Date: 2024-04-24
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-3966207-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: KR-20220008873-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitor and how to use itPublication Number: JP-2022531801-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-11530206-B2Priority Date: 2019-05-10Grant Date: 2022-12-20
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-2023039712-A1Priority Date: 2019-05-10
- Combination of dcc-3116 and mapkap pathway inhibitors for use in the treatment of cancerPublication Number: WO-2024050351-A1Priority Date: 2022-09-02
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-2020354352-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: WO-2020231806-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: AU-2020275392-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitor and method of usePublication Number: CN-114127057-APriority Date: 2019-05-10
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020231806&_cid=P12-MHCSWS-98394-1

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Inlexisertib, serine/ threonine kinase inhibitor, antineoplastic, DCC 3116, JM2ZTM8S7S
Icovamenib
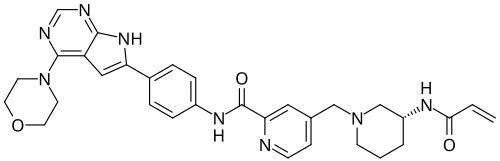
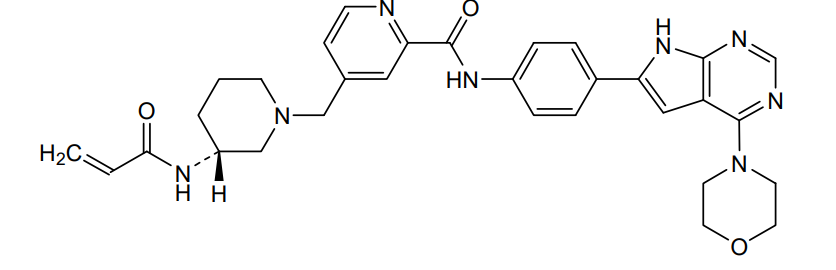
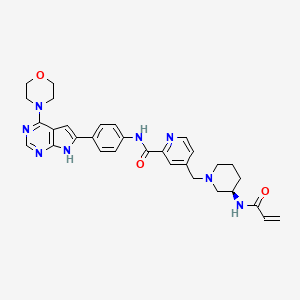
Icovamenib
CAS 2448172-22-1
MF C31H34N8O3 MW 566.7 g/mol
N-{4-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl}-4-{[(3R)-3-(prop-2-enamido) piperidin-1-yl]methyl}pyridine-2-carboxamide
N-[4-(4-morpholin-4-yl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)phenyl]-4-[[(3R)-3-(prop-2-enoylamino)piperidin-1-yl]methyl]pyridine-2-carboxamide
menin-MLL (mixed-lineage leukemia) protein interaction inhibitor,
antineoplastic, BMF-219, BMF 219, 2Z737MY35A, Menin-MLL inhibitor 21
Icovamenib is an investigational irreversible covalent inhibitor of menin. It is developed by Biomea Fusion for diabetes, lymphoma, leukemia, and multiple myeloma.[1][2][3]
Icovamenib is an orally bioavailable, irriversible inhibitor of menin, an essential co-factor of oncogenic menin-mixed lineage leukemia (MLL; myeloid/lymphoid leukemia; KMT2A) fusion proteins, with potential antineoplastic activity. Upon oral administration, icovamenib specifically targets and binds to menin, thereby preventing the interaction between the two proteins menin and MLL and the formation of the menin-MLL complex. This reduces the expression of downstream target genes, such as MYC and Bcl2, and results in an inhibition of the proliferation of MLL-rearranged tumor cells. Menin, an essential transcriptional regulator, plays a key role in oncogenic signaling in cancers driven by oncogenic MLL-fusions.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US299042443&_cid=P20-MH9YDY-31032-1
Example 9
Synthesis of Compound 10
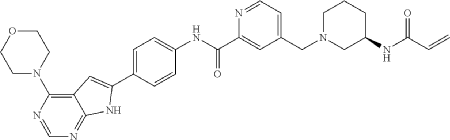
Compound 10
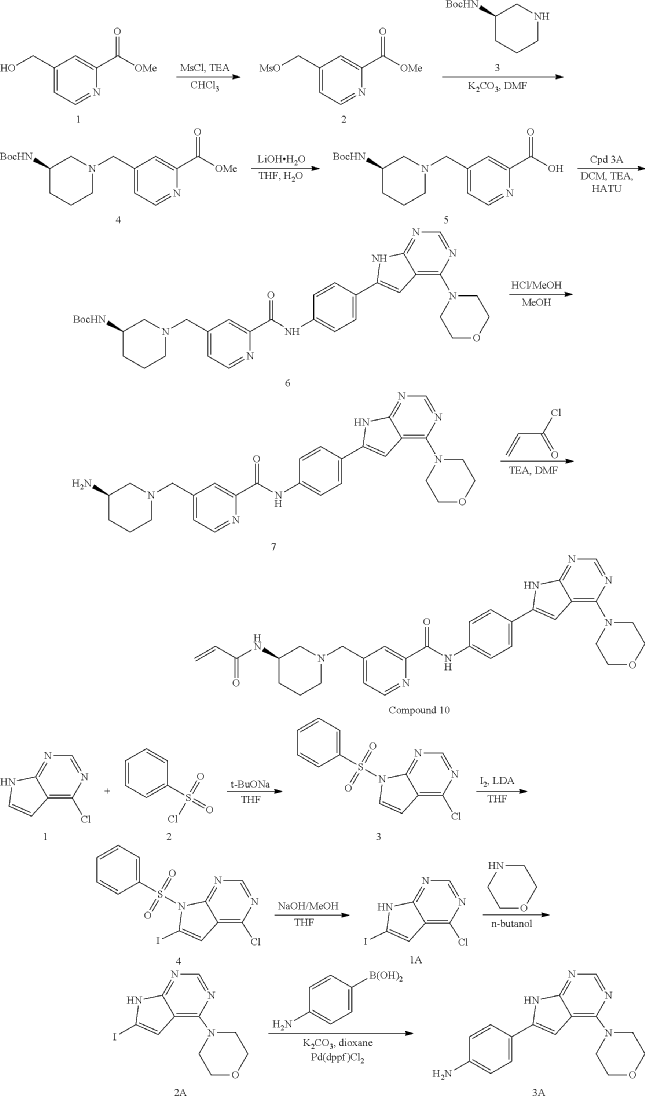
General Procedure for Preparation of Intermediate 2
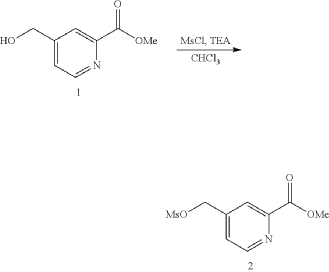
| 1H NMR: CDCl 3 400 MHz 8.80 (d, J=4.85 Hz, 1H), 8.15 (d, J=0.66 Hz, 1H), 7.53 (dt, J=4.91, 0.85 Hz, 1H), 5.27-5.34 (m, 2H), 4.00-4.08 (m, 3H), 3.11 (s, 3H) |
General Procedure for Preparation of Intermediate 5—
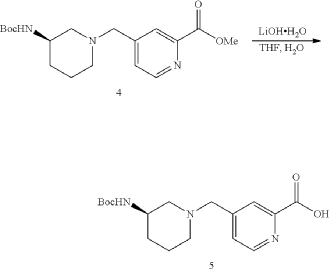
| To a solution of Intermediate 4 (1.50 g, 4.29 mmol, 1 eq) in THF (7.00 mL) was added LiOH.H 2O (540.3 mg, 12.8 mmol, 3 eq) in H 2O (7.00 mL). The mixture was stirred at 25° C. for 3 h. TLC (Dichloromethane:Methanol=10:1, R f=0) showed the reaction was complete. The mixture was poured into H 2O (20.0 mL) and extracted with DCM (10.0 mL×3). Then the organic phases dried over Na 2SO 4, filtered and concentrated under vacuum. The crude without purification. Give the Intermediate 5 (1.20 g, crude) as a yellow solid. |
General Procedure for Preparation of Intermediate 6—
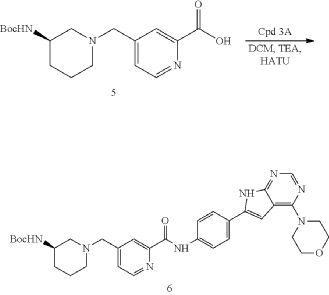
| To a solution of Intermediate 5 (0.80 g, 2.39 mmol, 1 eq), Intermediate 3A (704.4 mg, 2.39 mmol, 1 eq), TEA (1.69 g, 16.7 mmol, 2.32 mL, 7 eq) in DCM (10.0 mL) was added HATU (1.36 g, 3.58 mmol, 1.5 eq). The mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was poured into H 2O (40.0 mL) and extracted with DCM (20.0 mL×3). Then the organic phases were washed with brine (50.0 mL) dried over Na 2SO 4, filtered and concentrated under vacuum. The crude for next step without purification. Give the Intermediate 6 (0.60 g, crude) as a yellow solid. |
General Procedure for Preparation of Intermediate 7—
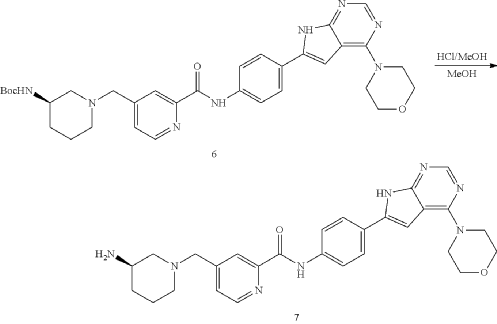
| To a solution of Intermediate 6 (0.50 g, 816.0 umol, 1 eq) in MeOH (5.00 mL) was added HCl/MeOH (4 M, 5.00 mL, 24.51 eq). The mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was concentrated under vacuum. The crude for next step without purification. Give the Intermediate 7 (0.50 g, crude, HCl) as a yellow solid. |
General Procedure for Preparation of Compound 10—
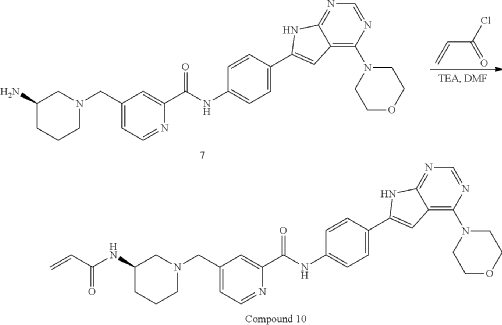
| To a solution of Intermediate 3 (0.50 g, 910.6 umol, 1 eq, HCl) in DMF (10.0 mL) was added TEA (645.0 mg, 6.37 mmol, 887.2 uL, 7 eq) and prop-2-enoyl chloride (82.4 mg, 910.6 umol, 74.2 uL, 1 eq). Then the mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was poured into H 2O (50.0 mL), then was filtered and filter cake was concentrated in vacuum. The crude product was purified by reversed-phase HPLC (column: Phenomenex Luna C18 200*40 mm*10 um; mobile phase: [water(0.05% HCl)-ACN]; B %: 10%-30%, 10 min) and (column: Xtimate C18 150*25 mm*5 um; mobile phase: [water(10 mM NH 4HCO 3)-ACN]; B %: 30%-60%, 10 min). Give the Intermediate Compound 10 (20.0 mg, 35.0 umol, 3.85% yield, 99.3% purity) as a yellow solid. |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024172911&_cid=P20-MH9YNT-37455-1
PAT
- Substituted pyridines as irreversible inhibitors of menin-MLL interactionPublication Number: US-11702421-B2Priority Date: 2018-12-31Grant Date: 2023-07-18
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2023227458-A1Priority Date: 2018-12-31
- N-[4-[4-(4-MORPHOLINYL)-7H-PYRROLO[2,3-d]PYRIMIDIN-6-YL]PHENYL]-4-[[3(R)-[(1-OXO-2-PROPEN-1-YL)AMINO]-1-PIPERIDINYL]METHYL]-2-PYRIDINE CARBOXAMIDE AND USES THEREOFPublication Number: US-2024376112-A1Priority Date: 2018-12-31
- Covalent inhibitors of menin-mll interaction for diabetes mellitusPublication Number: WO-2023018825-A1Priority Date: 2021-08-11
- Covalent inhibitors of Mennin-MLL interaction for diabetesPublication Number: CN-118076357-APriority Date: 2021-08-11
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2020223853-A1Priority Date: 2018-12-31
- Substituted pyridines as irreversible inhibitors of menin-MLL interactionPublication Number: US-11084825-B2Priority Date: 2018-12-31Grant Date: 2021-08-10
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2022169627-A1Priority Date: 2018-12-31
- Crystalline forms of an irreversible inhibitor of menin-mll interactionPublication Number: US-2023086137-A1Priority Date: 2021-08-20
- Crystalline form of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo -2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide, an irreversible menin-mll inhibitor for the treatment of cancerPublication Number: EP-4387972-A1Priority Date: 2021-08-20
- Crystalline forms of N-[4-[4-(4-morpholinyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo-2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide as an irreversible inhibitor of menin-MLL interactionPublication Number: US-12018032-B2Priority Date: 2021-08-20Grant Date: 2024-06-25
- Crystalline form of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo -2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide, an irreversible menin-mll inhibitor for the treatment of cancerPublication Number: WO-2023022912-A1Priority Date: 2021-08-20
- Crystalline forms of an irreversible inhibitor of menin-mll interactionPublication Number: US-2024343731-A1Priority Date: 2021-08-20
- Synthetic methods for preparing a pyridinecarboxamide compoundPublication Number: WO-2024011450-A1Priority Date: 2022-07-13
- Menin-mll inhibitors and compositions for proliferation of beta cellsPublication Number: WO-2024006391-A1Priority Date: 2022-06-28
- Flt3 combination therapy for cancer and compositions thereforPublication Number: WO-2023225005-A1Priority Date: 2022-05-17
- Treatment of cancer with menin inhibitors and immuno-oncology agentsPublication Number: WO-2023172925-A1Priority Date: 2022-03-08
- Treatment of hematological malignancies with menin inhibitors and p-glycoprotein inhibitorsPublication Number: WO-2023150635-A1Priority Date: 2022-02-04
- Crystalline forms of N[4[4-(4-Morpholinyl)-7H-Pyrrolo[2-3-D]Pyrimidin-6-yl]Phenyl]-4-[[3(R)-[(1-Oxo-2-Protein-1-yl)Amino]-1-Piperidinyl]Methyl]2-Pyridinecarboxamide]Publication Number: US-12215113-B2Priority Date: 2023-01-18Grant Date: 2025-02-04
- CRYSTALLINE FORMS OF N-[4-[4-(4-MORPHOLINYL)-7H-PYRROLO[2,3-d]PYRIMIDIN-6-YL]PHENYL]-4-[[3(R)-[(1-OXO-2-PROPEN-1-YL)AMINO]-1-PIPERIDINYL]METHYL]-2-PYRIDINECARBOXAMIDE AS IRREVERSIBLE INHIBITORS OF MENIN-MLL INTERACTIONPublication Number: US-2024417404-A1Priority Date: 2023-01-18
- Crystalline forms of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6- yl]phenyl]-4-[[3(r)-[(l-oxo-2-propen-l-yl)amino]-l-piperidinyl]methyl]-2-pyridinecarboxamide as a covalent inhibitor of menin-mll interactionPublication Number: WO-2024155710-A1Priority Date: 2023-01-18
- Crystalline forms of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(l-oxo-2-propen-l-yl)amino]-l-piperidinyl]methyl]-2- pyridinecarboxamide as a covalentinhibitor of menin-mll interactionPublication Number: WO-2024155719-A1Priority Date: 2023-01-18
- Combinations of lsd1 inhibitors and menin inhibitors for treating cancerPublication Number: WO-2024110649-A1Priority Date: 2022-11-24
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Rodriguez, Jose E.; Abitbol, Alexander; Abuzgaya, Fathi; Perez, Cesar; Mourya, Sanchita; Munneke, Brian; Morris, Stephan W.; Butler, Thomas (20 June 2023). “91-LB: COVALENT-111, a Phase 1/2 Trial of BMF-219, a Covalent Menin Inhibitor, in Patients with Type 2 Diabetes Mellitus—Preliminary Results”. Diabetes. 72 (Supplement_1) 91-LB. doi:10.2337/db23-91-LB. S2CID 259444592.
- Ravandi-Kashani, F.; Kishtagari, A.; Carraway, H.; Schiller, G.; Curran, E.; Yadav, B.; Cacovean, A.; Morris, S.; Butler, T.; Lancet, J. (23 June 2022). “P587: Covalent-101: A Phase 1 Study of BMF-219, A Novel Oral Irreversible Menin Inhibitor, in Patients with Relapsed/Refractory Acute Leukemia, Diffuse Large B-Cell Lymphoma, and Multiple Myeloma”. HemaSphere. 6: 486–487. doi:10.1097/01.HS9.0000845236.32931.83.
- Somanath, Priyanka; Lu, Daniel; Law, Brian; Archer, Tenley C.; Cacovean, Alexandru; Palmer, James T.; Kinoshita, Taisei; Butler, Thomas (5 November 2021). “Novel Irreversible Menin Inhibitor, BMF-219, Shows Potent Single Agent Activity in Clinically Relevant DLBCL Cells”. Blood. 138 (Supplement 1): 4318. doi:10.1182/blood-2021-148045.
| Clinical data | |
|---|---|
| Other names | BMF-219 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2448172-22-1 |
| PubChem CID | 154988914 |
| ChemSpider | 115037287 |
| UNII | 2Z737MY35A |
| Chemical and physical data | |
| Formula | C31H34N8O3 |
| Molar mass | 566.666 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Icovamenib, antineoplastic, BMF-219, BMF 219, 2Z737MY35A, Menin-MLL inhibitor 21
Foselutoclax
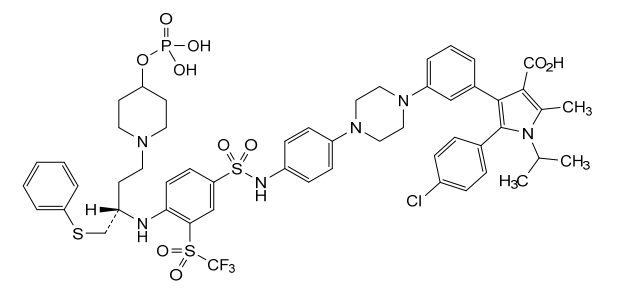
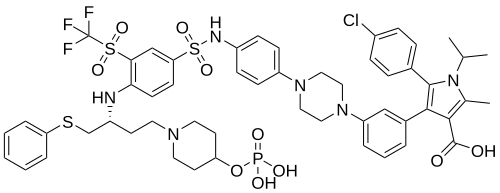
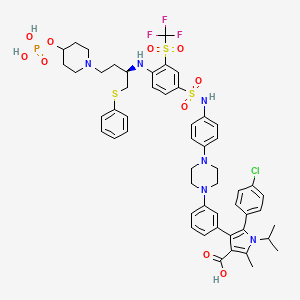
Foselutoclax
CAS 2271269-01-1
MF C53H59ClF3N6O10PS3 MW 1159.7 g/mol
(10R)-14-chloro-25-methyl-7,7-dioxo-10-[(phenylsulfanyl)methyl]-134-(phosphonooxy)-21-(propan-2-yl)-83-(trifluoromethanesulfonyl)-21H-7λ6-thia-6,9-diaza-4(1,4)-piperazina-13(1)-piperidina-2(2,3)-pyrrola-1(1),3(1,3),5,8(1,4)-tetrabenzenatridecaphane-24-carboxylic acid
5-(4-chlorophenyl)-2-methyl-4-[3-[4-[4-[[4-[[(2R)-1-phenylsulfanyl-4-(4-phosphonooxypiperidin-1-yl)butan-2-yl]amino]-3-(trifluoromethylsulfonyl)phenyl]sulfonylamino]phenyl]piperazin-1-yl]phenyl]-1-propan-2-ylpyrrole-3-carboxylic acid
B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, VT53CL5GES, UBX 1325
Foselutoclax is an investigational new drug that is being evaluated for the treatment of age-related eye diseases, particularly diabetic macular edema (DME) and wet age-related macular degeneration (AMD). Developed by Unity Biotechnology, this senolytic compound acts as a potent inhibitor of Bcl-xL, a protein that senescent cells rely on for survival.[1] Foselutoclax is designed to selectively eliminate senescent cells in the retina, potentially addressing the underlying causes of vision loss in these conditions.[2]
- Assess the Efficacy and Safety of Repeat Intravitreal Injections of Foselutoclax (UBX1325) in Patients With DME (ASPIRE)CTID: NCT06011798Phase: Phase 2Status: CompletedDate: 2025-08-05
- Safety, Tolerability and Evidence of Activity Study of UBX1325 in Patients With Diabetic Macular Edema (BEHOLD)CTID: NCT04857996Phase: Phase 2Status: CompletedDate: 2024-05-16
- Safety and Tolerability Study of UBX1325 in Patients With Diabetic Macular Edema or Neovascular Age-Related Macular DegenerationCTID: NCT04537884Phase: Phase 1Status: CompletedDate: 2022-03-10
REF
- Therapeutic targeting of cellular senescence in diabetic macular edema: preclinical and phase 1 trial resultsPublication Name: Nature MedicinePublication Date: 2024-02PMID: 38321220DOI: 10.1038/s41591-024-02802-4
- Senolytics in the treatment of diabetic retinopathyPublication Name: Frontiers in PharmacologyPublication Date: 2022-08-26PMCID: PMC9462063PMID: 36091769DOI: 10.3389/fphar.2022.896907
- Senolytic drugs: from discovery to translationPublication Name: Journal of Internal MedicinePublication Date: 2020-08-04PMCID: PMC7405395PMID: 32686219DOI: 10.1111/joim.13141
- bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell deathPublication Name: CellPublication Date: 1993-08-27PMID: 8358789DOI: 10.1016/0092-8674(93)90508-n
PAT
Treatment of Lung Diseases Using Pharmaceutical Agents that Eliminate Senescent Cells
Publication Number: US-2020354336-A9
Priority Date: 2017-08-11
- Senescent Cells and for Treating CancerPublication Number: US-2022017485-A1Priority Date: 2018-06-13
- Acyl sulfonamides that are bcl family antagonists for use in clinical management of conditions caused or mediated by senescent cells and for treating cancerPublication Number: EP-4335516-A2Priority Date: 2018-06-13
- Methods of Inhibiting Pathological AngiogenesisPublication Number: US-2020253991-A1Priority Date: 2017-10-31
- Methods of inhibiting pathological angiogenesisPublication Number: US-11129838-B2Priority Date: 2017-10-31Grant Date: 2021-09-28
- Treatment of Lung Diseases Using Pharmaceutical Agents that Eliminate Senescent CellsPublication Number: US-2020199103-A1Priority Date: 2017-08-11
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US279621490&_cid=P21-MGPXU3-15237-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US421382898&_cid=P21-MGPXWE-19244-1
A crystalline solid meglumine salt of of (R)-5-(4-chlorophenyl)-1-isopropyl-2-methyl-4-(3-(4-(4-((4-((1-(phenylthio)-4-(4-((phosphonooxy)methyl)piperidin-1-yl)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonamido)phenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylic acid, the compound of Formula I:
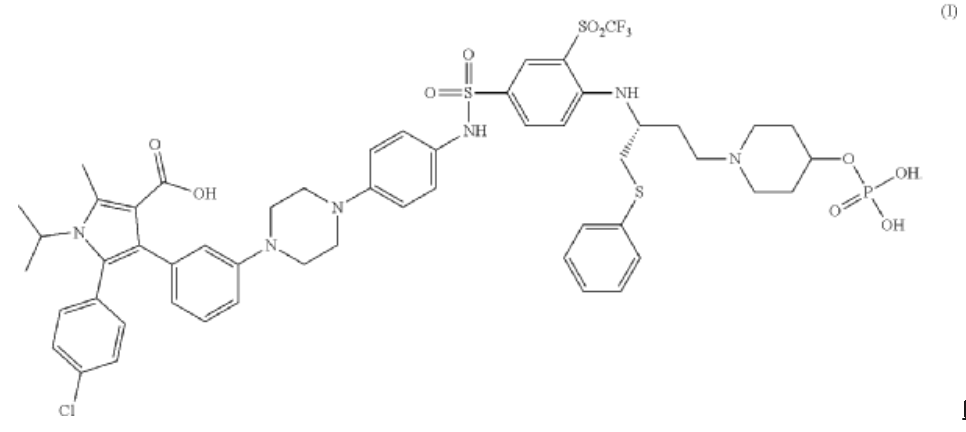
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US348024244&_cid=P21-MGPXWE-19244-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | UBX1325 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2271269-01-1 |
| PubChem CID | 147562879 |
| IUPHAR/BPS | 13366 |
| ChemSpider | 115277082 |
| UNII | VT53CL5GES |
| Chemical and physical data | |
| Formula | C53H59ClF3N6O10PS3 |
| Molar mass | 1159.69 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Crago SM (22 June 2023). “Design for Phase 2B ASPIRE Study of UBX1325 for DME announced by UNITY”. Modern Retina. Archived from the original on 13 August 2024.
- Macha N, Yu M, Sapieha P, Klier S, Ghosh A, White L, et al. (September 2024). “Multifocal Electroretinography Changes after UBX1325 (Foselutoclax) Treatment in Neovascular Age-Related Macular Degeneration”. Journal of Clinical Medicine. 13 (18): 5540. doi:10.3390/jcm13185540. PMC 11433175. PMID 39337030.
//////////foselutoclax, antineoplastic, VT53CL5GES, UBX 1325
Ezobresib
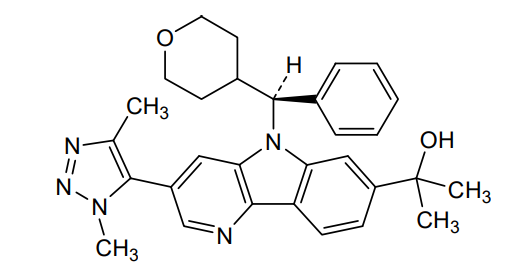
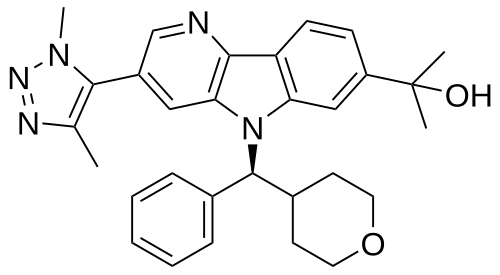
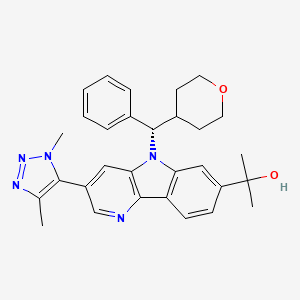
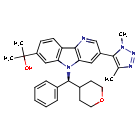
Ezobresib
CAS 1800340-40-2
MF C30H33N5O2 MW 495.6 g/mol
2-{3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol
bromodomain and extra-terminal motif (BET) inhibitor,
antineoplastic, BMS-986158, BMS 986158, Bristol Myers Squibb, antineoplastic, UNII-X8BW0MQ5PI
2-[3-(3,5-dimethyltriazol-4-yl)-5-[(S)-oxan-4-yl(phenyl)methyl]pyrido[3,2-b]indol-7-yl]propan-2-ol
Ezobresib is an investigational new drug that has been evaluated for the treatment of cancer. It inhibits Bromodomain and Extra-Terminal domain (BET) proteins, with potential antineoplastic activity.[1] Developed by Bristol Myers Squibb, this therapeutic agent has been studied for its efficacy in treating various cancers, including solid tumors and hematological malignancies.[2] Despite showing promise in early-phase clinical trials, recent developments suggest that Bristol Myers Squibb has decided to discontinue further development of ezobresib.[3]
BMS-986158 is under investigation in clinical trial NCT02419417 (Study of BMS-986158 in Subjects With Select Advanced Cancers).
Ezobresib is an inhibitor of the Bromodomain (BRD) and Extra-Terminal domain (BET) family of proteins, with potential antineoplastic activity. Upon administration, ezobresib binds to the acetyl-lysine binding site in the BRD of BET proteins, thereby preventing the interaction between BET proteins and acetylated histones. This disrupts chromatin remodeling and prevents the expression of certain growth-promoting genes, resulting in an inhibition of tumor cell growth. BET proteins (BRD2, BRD3, BRD4 and BRDT) are transcriptional regulators that bind to acetylated lysines on the tails of histones H3 and H4, and regulate chromatin structure and function; they play an important role in the modulation of gene expression during development and cellular growth
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US206490064&_cid=P21-MGLNPO-16484-1
Examples 54 & 55
2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
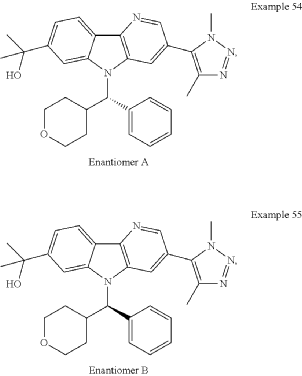
Step 1: 2-Chloro-5-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)pyridin-3-amine
Step 2: Methyl 3-((2-chloro-5-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)pyridin-3-yl)amino)benzoate
Step 3: Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate
Alternate synthesis of Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 4: Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 5: 2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Alternate Synthesis of Examples 54
2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Step 1: 2-Chloro-5-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)pyridin-3-amine
Step 2: Methyl 3-((2-chloro-5-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)pyridin-3-yl)amino)benzoate
Step 3: Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate
Alternate synthesis of Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 4: Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 5: 2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Alternate Synthesis of Examples 54
2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Step 1: (S)-methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 2. (S)-2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Step 1: (S)-methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 2. (S)-2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
PATENT
LIT
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- Recent updates on 1,2,3-triazole-containing hybrids with in vivo therapeutic potential against cancers: A mini-reviewPublication Name: European Journal of Medicinal ChemistryPublication Date: 2023-05-05PMID: 36893627DOI: 10.1016/j.ejmech.2023.115254
- Development of BET Inhibitors as Potential Treatments for Cancer: Optimization of Pharmacokinetic PropertiesPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2022-07-05PMCID: PMC9290009PMID: 35859878DOI: 10.1021/acsmedchemlett.2c00219
- Synthesis of BMS-986158Publication Name: SynfactsPublication Date: 2021-11-17DOI: 10.1055/s-0041-1737090
- Development of BET inhibitors as potential treatments for cancer: A new carboline chemotypePublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2021-11-01PMID: 34560263DOI: 10.1016/j.bmcl.2021.128376
- Discovery and Preclinical Pharmacology of an Oral Bromodomain and Extra-Terminal (BET) Inhibitor Using Scaffold-Hopping and Structure-Guided Drug DesignPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-09-20PMID: 34543572DOI: 10.1021/acs.jmedchem.1c00625
- Retrospective assessment of rat liver microsomal stability at NCATS: data and QSAR modelsPublication Name: Scientific ReportsPublication Date: 2020-11-26PMCID: PMC7693334PMID: 33244000DOI: 10.1038/s41598-020-77327-0
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- Predictive models of aqueous solubility of organic compounds built on A large dataset of high integrityPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2019-07-15PMCID: PMC8274818PMID: 31176566DOI: 10.1016/j.bmc.2019.05.037
- Highly predictive and interpretable models for PAMPA permeabilityPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2017-02-01PMCID: PMC5291813PMID: 28082071DOI: 10.1016/j.bmc.2016.12.049
- BET inhibitor resistance emerges from leukaemia stem cellsPublication Name: NaturePublication Date: 2015-09-14PMCID: PMC6069604PMID: 26367796DOI: 10.1038/nature14888
- Efficacy of BET Bromodomain Inhibition in Kras-Mutant Non–Small Cell Lung CancerPublication Name: Clinical cancer research : an official journal of the American Association for Cancer ResearchPublication Date: 2013-11-14PMCID: PMC3838895PMID: 24045185DOI: 10.1158/1078-0432.ccr-12-3904
- Discovery and Preclinical Evaluation of [4-[[1-(3-fluorophenyl)methyl]-1H-indazol-5-ylamino]-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yl]carbamic Acid, (3S)-3-Morpholinylmethyl Ester (BMS-599626), a Selective and Orally Efficacious Inhibitor of Human Epidermal Growth Factor Receptor 1 and 2 KinasesPublication Name: Journal of Medicinal ChemistryPublication Date: 2009-10-12PMID: 19821562DOI: 10.1021/jm9010065
- [Statistical analysis of cerebrospinal fluid acid-base equilibrium and cerebrospinal fluid lactate concentration in cases of brain tumors, cerebrocranial injuries and meningoencephalitis]Publication Name: Neurologia i neurochirurgia polskaPublication Date: 1976-07PMID: 8740
PAT
- Tricyclic compounds as anticancer agentsPublication Number: WO-2015100282-A1Priority Date: 2013-12-24
- Tricyclic compound as anticancer agentsPublication Number: EP-3466949-B1Priority Date: 2013-12-24Grant Date: 2020-12-23
- Novel tricyclic compounds as anticancer agentsPublication Number: TW-202028203-APriority Date: 2013-12-24
- Novel tricyclic compounds as anticancer agentsPublication Number: TW-I726544-BPriority Date: 2013-12-24Grant Date: 2021-05-01
- Tricyclic compounds as anticancer agentsPublication Number: CN-108558871-BPriority Date: 2013-12-24Grant Date: 2022-02-18
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | BMS-986158 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1800340-40-2 |
| PubChem CID | 118196485 |
| DrugBank | DB15435 |
| ChemSpider | 58828664 |
| UNII | X8BW0MQ5PI |
| KEGG | D12710 |
| ChEMBL | ChEMBL4297458 |
| Chemical and physical data | |
| Formula | C30H33N5O2 |
| Molar mass | 495.627 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Ma Z, Zhang C, Bolinger AA, Zhou J (October 2024). “An updated patent review of BRD4 degraders”. Expert Opinion on Therapeutic Patents. 34 (10): 929–951. doi:10.1080/13543776.2024.2400166. PMC 11427152. PMID 39219068.
- “Clinical Trials Using Ezobresib”. National Cancer Institute.
- Brown A. “Bristol backs out of BET inhibition”. ApexOnco.
////////////Ezobresib, antineoplastic, BMS-986158, BMS 986158, Bristol Myers Squibb, antineoplastic, UNII-X8BW0MQ5PI
Dapolsertib
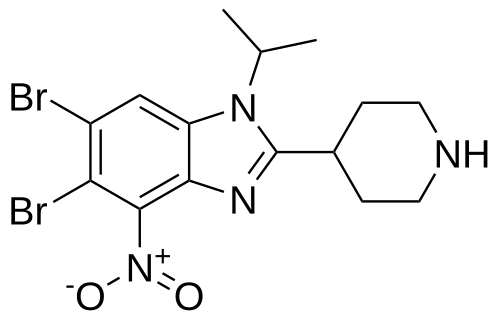
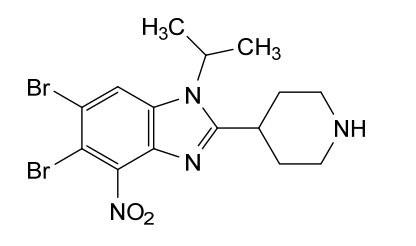
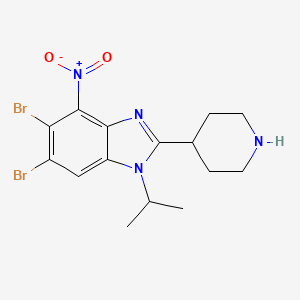
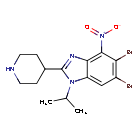
Dapolsertib
CAS 1616359-00-2
MF C15H18Br2N4O MW 446.14 g/mol
5,6-dibromo-4-nitro-2-piperidin-4-yl-1-propan-2-ylbenzimidazole
5,6-dibromo-4-nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzimidazole
serine/ threonine kinase inhibitor, antineoplastic
Ryvu Therapeutics SA, MEN1703, SEL24-B489
- SEL24-B489
- SEL-24 free base
- 9M7X64VTLI
- SEL-24
Dapolsertib is an investigational new drug that is being evaluated for the treatment of cancer. It is dual inhibitor of PIM family of serine/threonine protein kinases and mutant forms of FMS-related tyrosine kinase 3 (FLT3) that is being developed by Ryvu Therapeutics SA.[1]
Dapolsertib is an orally available inhibitor of PIM family serine/threonine protein kinases and mutant forms of FMS-related tyrosine kinase 3 (FLT3; STK1) with potential antineoplastic activity. Upon oral administration, dapolsertib binds to and inhibits the kinase activities of PIM-1, -2 and -3, and mutant forms of FLT3, which may result in the interruption of the G1/S phase cell cycle transition, an inhibition of cell proliferation, and an induction of apoptosis in tumor cells that overexpress PIMs or express mutant forms of FLT3. FLT3, a tyrosine kinase receptor that is overexpressed or mutated in various cancers, plays a role in signaling pathways that regulate hematopoietic progenitor cell proliferation, and in leukemic cell proliferation and survival. PIM kinases, downstream effectors of many cytokine and growth factor signaling pathways, including the FLT3 signaling pathway, play key roles in cell cycle progression and apoptosis inhibition and may be overexpressed in various malignancies.
- MEN1703 (SEL24) in Participants With Acute Myeloid LeukemiaCTID: NCT03008187Phase: Phase 1/Phase 2Status: CompletedDate: 2025-04-29
- MEN1703 (SEL24) to Treat Relapsed or Refractory Aggressive B-cell Non-Hodgkin Lymphoma (JASPIS-01)CTID: NCT06534437Phase: Phase 2Status: RecruitingDate: 2025-04-11
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014096388&_cid=P12-MG5YKY-59978-1
3.9. Compounds of Example 26:
3.9. Compounds of Example 26:
5,6-dibromo-4-nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzodiazole (Example 26A):
4,5-dibromo-1-N-(propan-2-yl)benzene-1,2-diamine (2,8g, 9,lmmol) and
isonipeconic acid (1,17g, 9,lmmol) were taken up in phosphoric acid (17,82g, 0,18mol). The resulting mixture was stirred at 180°C for 3,5 hours. The mixture was allowed to cool to RT and diluted with water to 200ml. The solution was basified to pH 14.0 using solid NaOH. The resulting precipitate was then filtered off and washed repeatedly with MeOH. The filtrate was concentrated in-vacuo. The product was purified on Al2O3 (basic) using DCM/MeOH/NH3 sat. in MEOH (25: 15: 1). The obtained product (8,7mmol, 3,9g) was dissolved in cone. H2SO4 (30ml). Next KNO3 (8,7mmol, 0,89g) was added in one portion at 0° C. The resulting mixture was stirred at 0°C for 3h and at RT overnight. Then the mixture was poured onto ice. The product was filtered and washed with water.The product was purified on on Al2O3 (basic) using DCM/MeOH/NH3 sat. in MEOH (25: 15: 1) to afford 5,6-dibromo-4- nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzodiazole (1,9g). 1H NMR (600 MHz, DMSO) δ 8.74 (bs, 1H), 8.48 (s, 1H), 8.35 (bs, 1H), 4.94 (hept, J = 6.8 Hz, 1H), 3.52 – 3.46 (m, 1H), 3.42 – 3.37 (m, 2H), 3.08 (bs, 2H), 2.07 – 1.96 (m, 4H), 1.60 (d, J = 6.9 Hz, 6H). m/z 446,8; rt 2,7min.
5,6-dibromo-4-nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzodiazole (Example 26A):
4,5-dibromo-1-N-(propan-2-yl)benzene-1,2-diamine (2,8g, 9,lmmol) and
isonipeconic acid (1,17g, 9,lmmol) were taken up in phosphoric acid (17,82g, 0,18mol). The resulting mixture was stirred at 180°C for 3,5 hours. The mixture was allowed to cool to RT and diluted with water to 200ml. The solution was basified to pH 14.0 using solid NaOH. The resulting precipitate was then filtered off and washed repeatedly with MeOH. The filtrate was concentrated in-vacuo. The product was purified on Al2O3 (basic) using DCM/MeOH/NH3 sat. in MEOH (25: 15: 1). The obtained product (8,7mmol, 3,9g) was dissolved in cone. H2SO4 (30ml). Next KNO3 (8,7mmol, 0,89g) was added in one portion at 0° C. The resulting mixture was stirred at 0°C for 3h and at RT overnight. Then the mixture was poured onto ice. The product was filtered and washed with water.The product was purified on on Al2O3 (basic) using DCM/MeOH/NH3 sat. in MEOH (25: 15: 1) to afford 5,6-dibromo-4- nitro-2-(piperidin-4-yl)-1-(propan-2-yl)-1H-1,3-benzodiazole (1,9g). 1H NMR (600 MHz, DMSO) δ 8.74 (bs, 1H), 8.48 (s, 1H), 8.35 (bs, 1H), 4.94 (hept, J = 6.8 Hz, 1H), 3.52 – 3.46 (m, 1H), 3.42 – 3.37 (m, 2H), 3.08 (bs, 2H), 2.07 – 1.96 (m, 4H), 1.60 (d, J = 6.9 Hz, 6H). m/z 446,8; rt 2,7min.
PAT
Novel benzimidazole derivatives as kinase inhibitors
Publication Number: WO-2014096388-A2
Priority Date: 2012-12-21
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: KR-20150095908-APriority Date: 2012-12-21
- Benzimidazole derivatives as kinase inhibitorsPublication Number: US-10174013-B2Priority Date: 2012-12-21Grant Date: 2019-01-08
- Novel Benzimidazole Derivatives as Kinase InhibitorsPublication Number: US-2015336967-A1Priority Date: 2012-12-21
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: US-2017152249-A1Priority Date: 2012-12-21
- Benzimidazole derivatives as Kinase InhibitorsPublication Number: US-9388192-B2Priority Date: 2012-12-21Grant Date: 2016-07-12
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: EP-2935244-B1Priority Date: 2012-12-21Grant Date: 2018-06-27
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: ES-2688395-T3Priority Date: 2012-12-21Grant Date: 2018-11-02
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: JP-2016503779-APriority Date: 2012-12-21
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: JP-6169185-B2Priority Date: 2012-12-21Grant Date: 2017-07-26
- Novel benzimidazole derivatives as kinase inhibitorsPublication Number: KR-101779272-B1Priority Date: 2012-12-21Grant Date: 2017-09-18
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | MEN1703, SEL24-B489 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1616359-00-2 |
| PubChem CID | 76286825 |
| IUPHAR/BPS | 13204 |
| ChemSpider | 81367232 |
| UNII | 9M7X64VTLI |
| ChEMBL | ChEMBL4467168 |
| Chemical and physical data | |
| Formula | C15H18Br2N4O2 |
| Molar mass | 446.143 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Wu M, Li C, Zhu X (December 2018). “FLT3 inhibitors in acute myeloid leukemia”. Journal of Hematology & Oncology. 11 (1) 133. doi:10.1186/s13045-018-0675-4. PMC 6280371. PMID 30514344.
//////////Dapolsertib, antineoplastic, MEN1703, SEL24-B489, MEN 1703, SEL24 B489, Ryvu Therapeutics SA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}