
Home » ANTI INFLAMATORY
Category Archives: ANTI INFLAMATORY
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Rivasterat
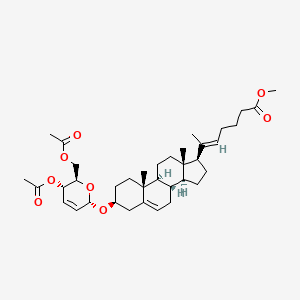
Rivasterat
CAS 2446590-96-9
MF C37H54O8 MW626.8 g/mol
methyl (E)-6-[(3S,8S,9S,10R,13S,14S,17R)-3-[[(2R,3S,6S)-3-acetyloxy-2-(acetyloxymethyl)-3,6-dihydro-2H-pyran-6-yl]oxy]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]hept-5-enoate
methyl (20E)-3β-[(4,6-di-O-acetyl-2,3-dideoxy-α-Derythro-hex-2-enopyranosyl)oxy]-27-norcholesta5,20(22)-dien-26-oate
cholesterol-derived steroid, anti-inflammatory, CURACLE, CU-06, CU-06-RE, CU06-1004, CU06-CERE/CV, CU06-EYE, CU06-HAE, CU06-IBD, CU06-ONCO, Sac-1004, 2X23JA5AKW
- OriginatorCURACLE
- ClassAnti-inflammatories; Anti-ischaemics; Antineoplastics; Eye disorder therapies; Ischaemic heart disorder therapies; Small molecules; Vascular disorder therapies
- Mechanism of ActionActin modulators; Chemokine CCL2 inhibitors; Histamine release inhibitors; Interleukin 1 beta inhibitors; Thrombin inhibitors; Vascular endothelial growth factors inhibitors
- Phase IIDiabetic macular oedema
- No development reportedAge-related macular degeneration; Cancer; Crohn’s disease; Diabetic retinopathy; Hereditary angioedema; Lung disorders; Macular degeneration; Myocardial infarction; Retinal oedema; Stroke; Ulcerative colitis; Unstable angina pectoris; Wet macular degeneration
- 28 Aug 2025No recent reports of development identified for research development in Unstable-angina-pectoris in South Korea (PO)
- 28 Jul 2025No recent reports of development identified for phase-I development in Wet macular degeneration in USA (PO)
- 28 May 2025No recent reports of development identified for phase-I development in Age-related-macular-degeneration in South Korea (PO)
Developer and Code Name
- Original code name: CU06-1004
- Developer: Curacle Co., Ltd. (South Korea)
- Drug class: Endothelial dysfunction blocker (EDB)
This class of drugs aims to restore endothelial barrier integrity rather than directly blocking VEGF like most retinal drugs.
The molecule contains:
- Steroid (cyclopenta[a]phenanthrene) core
- Unsaturated heptenoate side chain
- Acetylated sugar moiety (pyranose)
This glycosylated steroid structure is unusual for vascular-protective drugs.
Clinical Development
Phase I (Healthy Volunteers)
Key findings:
- Dose tested: 100–1200 mg
- Exposure increased more than dose proportional
- Food greatly increased absorption
- No significant drug accumulation after repeated dosing
- Minimal renal excretion detected
Phase II
Early clinical trials investigated oral therapy for diabetic macular edema with improvements in:
- Best-corrected visual acuity
- Inflammatory biomarkers
Summary
| Item | Details |
|---|---|
| Drug | Rivasterat (CU06-1004) |
| Originator | Curacle |
| Core patent | WO2013011939 |
| Chemistry | steroid glycoside |
| Key step | steroid glycosylation |
| Priority | ~2011 |
| Expiry | ~2031–2033 |
SYNTHESIS
WO2013011939
US20140148474
EP2741074
KR20130007373
SYN
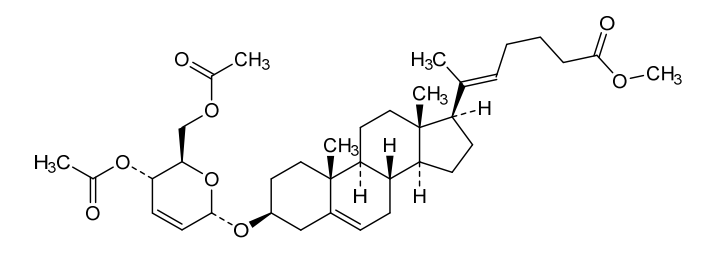
Manufacturing Example 1> Manufacturing of compound 1
[170]Compound 1 represented by the following chemical formula 1 can be manufactured using the manufacturing method described in Korean unpublished patent application number 10-2019-0166864. Specifically, it can be manufactured using the method according to the following reaction scheme 1 or reaction scheme 2.
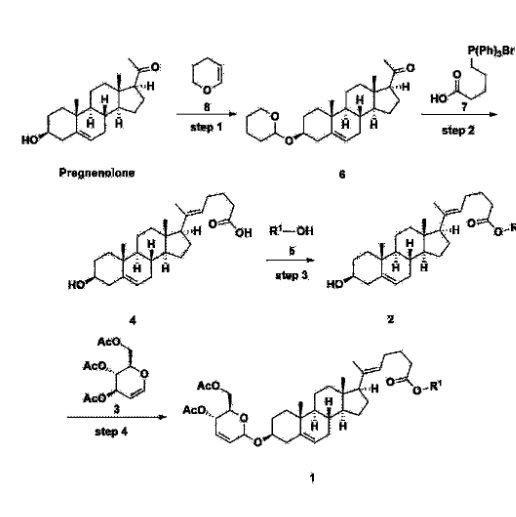
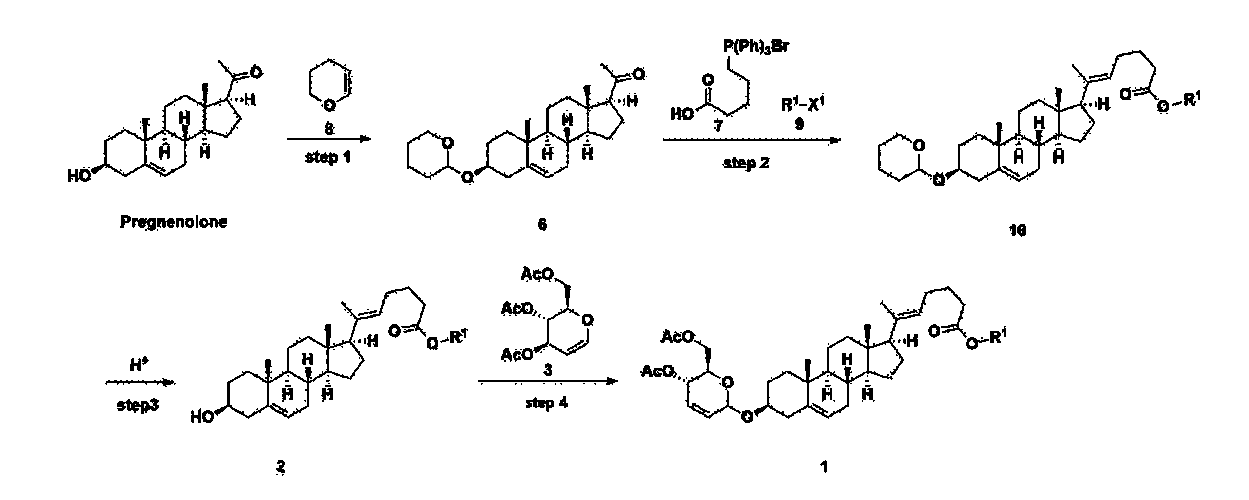
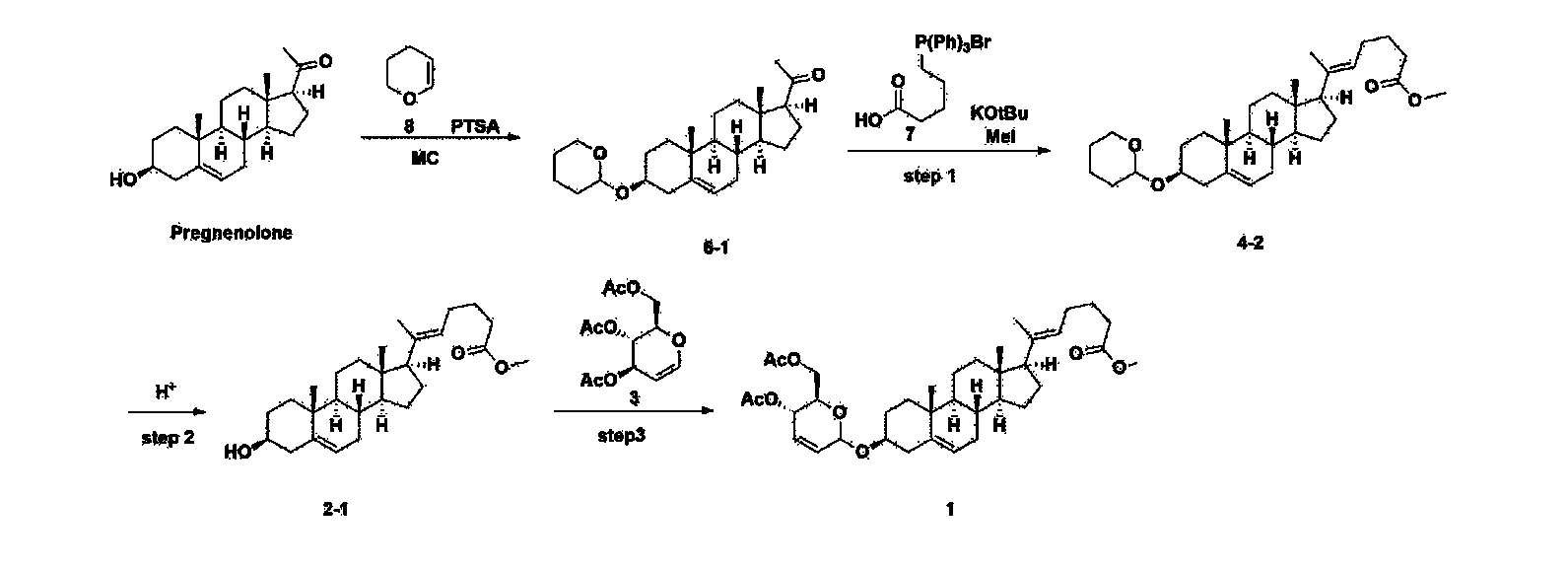
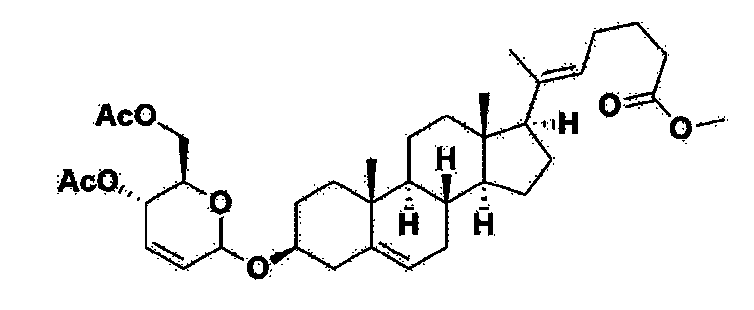
Step 1: Preparation of 6-1
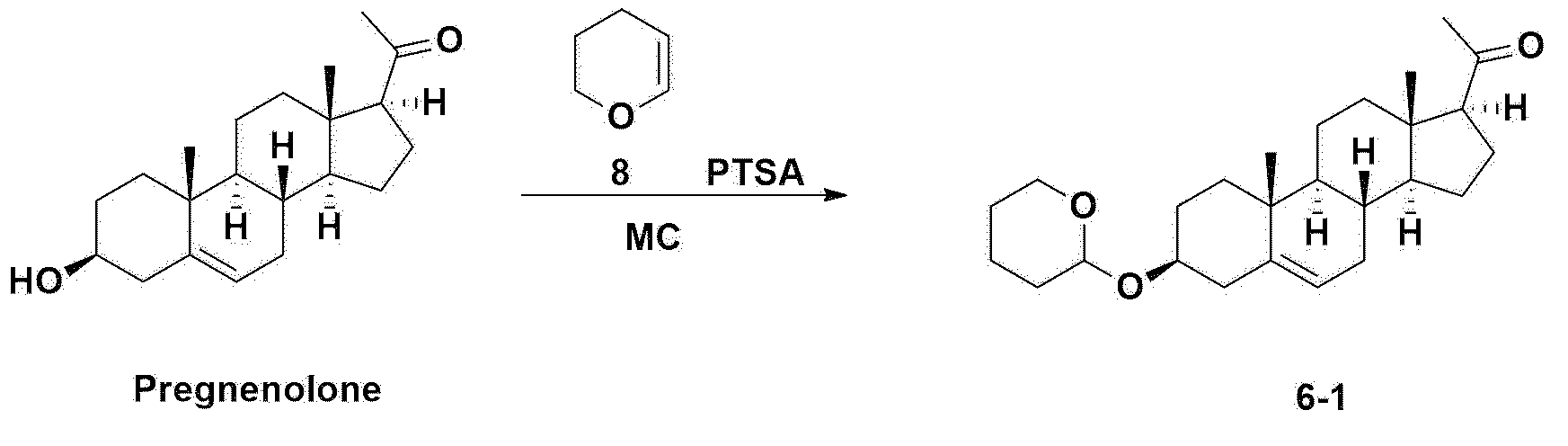
A thermometer was installed in a 5 L flask, and 200 g (0.632 mol) of pregnenolone was added to 2000 mL of dichloromethane, and 173 mL (1.896 mol) of 3,4-dihydro-2H-pyran was added. After lowering the temperature to 0-5 ℃, 3.0 g (15.8 mmol) of p-toluenesulfonic acid monohydrate dissolved in 50 mL of tetrahydrofuran (THF) was added dropwise and stirred at 0 ℃ for 1.5 hours. At 0 ℃, 800 mL of saturated sodium bicarbonate aqueous solution and 10 mL of triethylamine (TEA) were added to the reaction mixture and stirred. After separating the layers, the organic layer was washed with 800 mL of brine, and the aqueous layers were extracted again with 200 mL of dichloromethane, combined into the organic layers, dried over 200 g of anhydrous sodium sulfate, filtered, and distilled under reduced pressure. 1000 mL of MeOH and 5 mL of TEA were added to the obtained residue, heated to completely dissolve, and the temperature was lowered and stirred at -5 °C for 1 hour. The resulting solid was filtered and washed with 200 mL of MeOH to obtain 232.0 g (0.579 mol) of 6-1 (THP-Pregnenolone) as a pure white solid in a yield of 91.6%.
[204]1H-NMR (400 MHz, CDCl 3): δ 5.33-5.36 (m, 1H), 4.71-4.72 (m, 1H), 3.85-3.94 (m, 1H), 3.46-3.56 (m, 2H), 1.00-2.55 (m, 32H), 0.62 (s, 3H).
Step 2: Preparation of 4-2
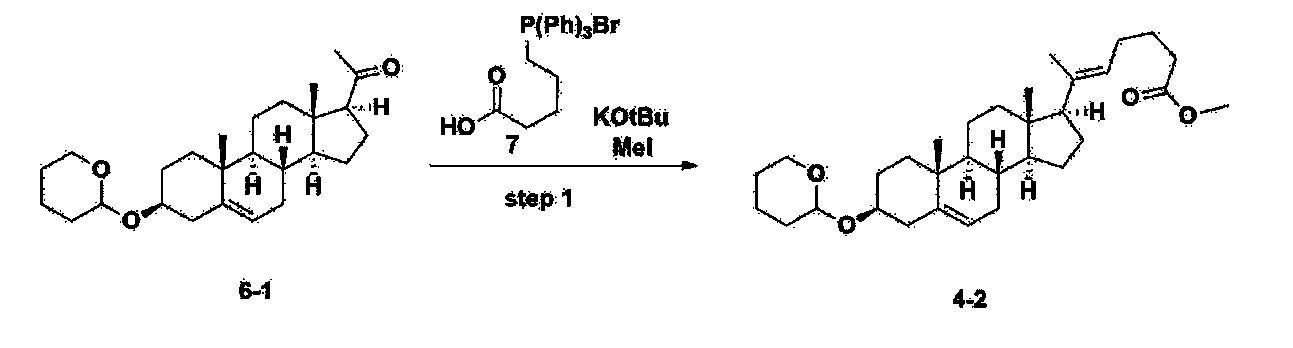
After installing a condenser, heating mantle, and mechanical stirrer in a 5L reactor, the reactor was heated to 119℃ (external temperature), cooled to room temperature while flowing nitrogen for 5 minutes, dried, and 332.5 g (0.75 mol) of 4-(carboxybutyl)triphenylphosphonium bromide and 168.1 g (1.50 mol) of potassium t-butoxide were added. Then, 2000 mL of anhydrous toluene and 750 mL of anhydrous tetrahydrofuran were added, and the reactor was heated to 119℃ (external temperature, internal mild reflux) and stirred for about 2 hours.
[209]6-1 100.0 g (0.250 mol) was dissolved in 500 mL of anhydrous toluene, added to the reaction solution, and reacted for about 20 hours. After the reaction was completed, the reaction mixture was cooled to room temperature, 320 mL (5.14 mol) of methyl iodide and 1000 mL of acetone were added, and stirred at room temperature for 15 hours. Most of the organic solvent was removed from the reaction mixture by distillation under reduced pressure, 1500 mL of ethyl acetate was added to dissolve, and the mixture was washed with 1000 mL of saturated ammonium chloride aqueous solution. The organic layer was washed twice with 1000 mL of water and 1000 mL of brine, dried with 100 g of sodium sulfate, filtered using 80 g of Celite, and concentrated.
[210]The obtained residue was dissolved in 2000 mL of methanol, stirred at 10°C for 13 hours and at 4-5°C for 1 hour, and the resulting solid was filtered, washed with 200 mL of methanol, and dried in vacuum to obtain 66.2 g of 4-2 as a white solid with a yield of 53.2%.
[211]
1H NMR(400MHz, CDCl 3): δ 5.36(t, J=5.80 Hz, 1H), 5.16(t, J=7.00 Hz, 1H), 4.71(m, 1H), 3.93(m, 1H), 3.66(s, 3H), 3.56(m, 2H), 2.37-0.88(m, 38H), 0.54(s, 3H).
Step 3: Preparation of 2-1
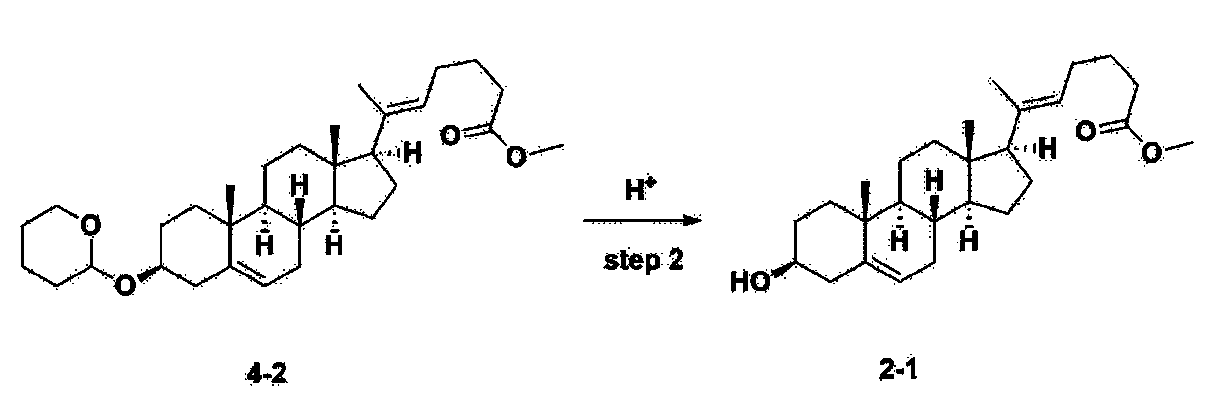
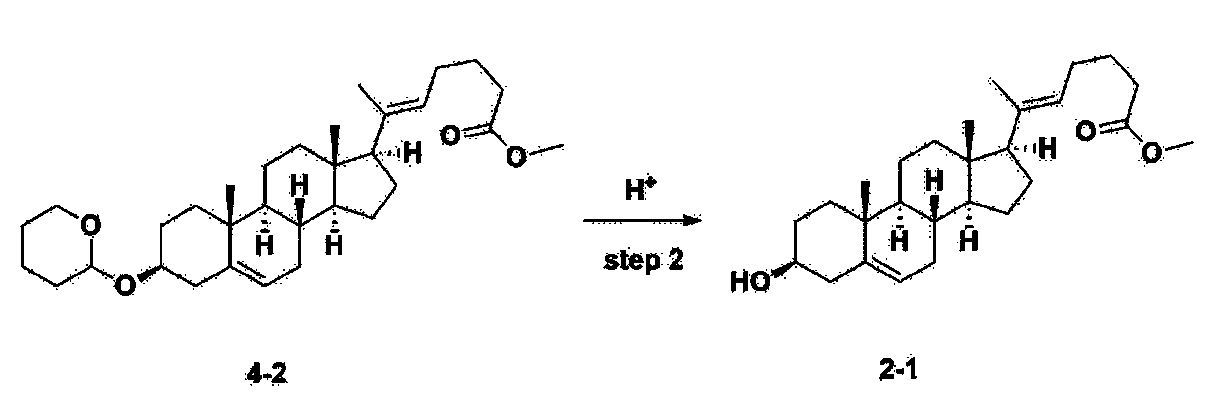
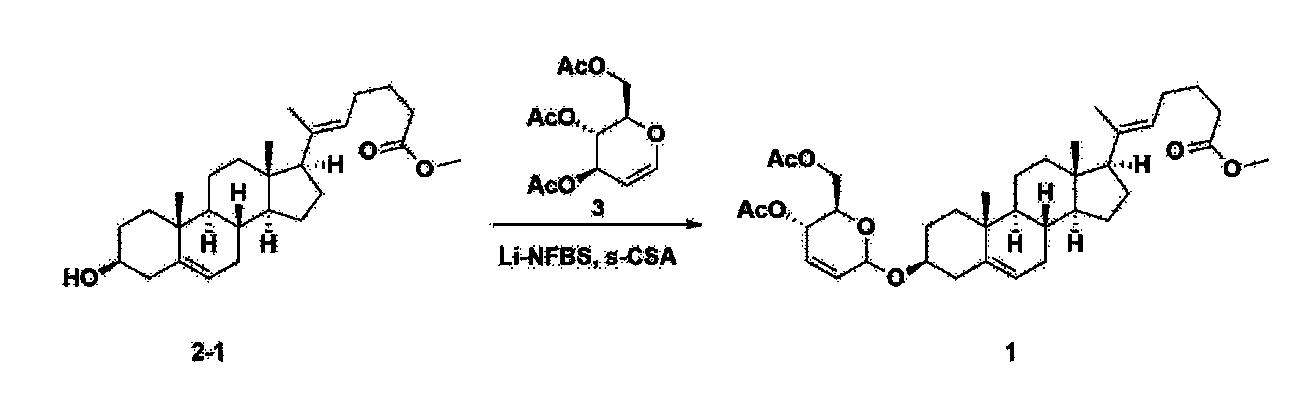
After installing a thermometer and a water bath in a 1 L flask, 42.0 g (0.101 mol) of compound 2-1 and 34.5 g (0.126 mol) of triiO-acetyl D-glucal were dissolved in 126 mL of anhydrous toluene and 252 mL of acetonitrile, and while maintaining the temperature at 30-35 ℃, 3.87 g (0.0130 mol) of lithium nonafluoro-1-butylsulfonate and 0.117 g (0.0005 mol) of (s)-camphor sulfonic acid were added and stirred for 2 hours. After completion of the reaction, the reaction was quenched with 504 mL of saturated sodium bicarbonate aqueous solution and extracted with 630 mL of heptane. The organic layer was washed twice with 504 mL of saturated sodium bicarbonate aqueous solution and then with 504 mL of brine. The organic layer was stirred with 42 g of anhydrous sodium sulfate and 34 g of charcoal, filtered with 34 g of celite, washed with 210 mL of methylene chloride, combined with the filtrate, concentrated, and dried under vacuum.
[223]
1H-NMR (400 MHz, CDCl3) : δ 5.79-5.88 (m, 2H), 5.35-5.36 (m, 1H), 5.27-5.29 (m, 1H), 5.12-5.16 (m, 2H), 4.15-4.24 (m, 3H), 3.66 (s, 3H), 3.54-3.57 (m, 1H), 0.91-2.32 (m, 38H), 0.54 (s, 3H).
PAT
- Crystalline form of vascular leakage blocker compoundPublication Number: US-12103945-B2Priority Date: 2020-05-04Grant Date: 2024-10-01
- New crystalline form of vascular leakage blocker compoundPublication Number: US-2022259256-A1Priority Date: 2020-05-04
- Preparation Method of Vascular Leakage Blockers With a High YieldPublication Number: US-2021395297-A1Priority Date: 2019-12-13
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

////////rivasterat, cholesterol-derived steroid, anti-inflammatory, CURACLE, CU-06, CU-06-RE, CU06-1004, CU06-CERE/CV, CU06-EYE, CU06-HAE, CU06-IBD, CU06-ONCO, Sac-1004, 2X23JA5AKW
Navepdekinra
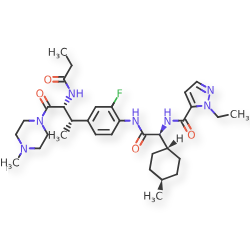
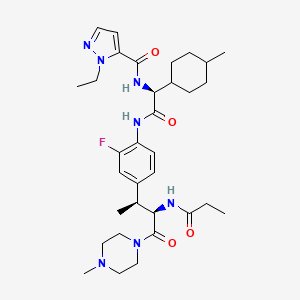
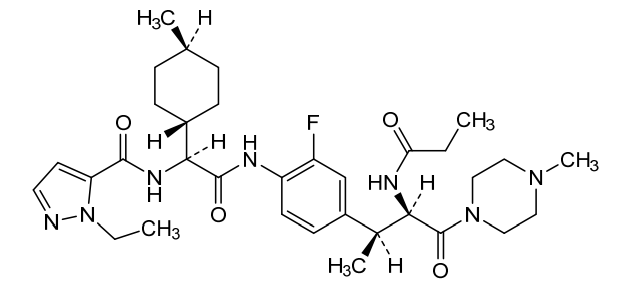
Navepdekinra
CAS 2467732-66-5
MF C33H48FN7O4 MW625.78
1H-Pyrazole-5-carboxamide, 1-ethyl-N-[(1S)-2-[[2-fluoro-4-[(1S,2R)-1-methyl-3-(4-methyl-1-piperazinyl)-3-oxo-2-[(1-oxopropyl)amino]propyl]phenyl]amino]-1-(trans-4-methylcyclohexyl)-2-oxoethyl]-
1-ethyl-N-{(1S)-2-{2-fluoro-4-[(2S,3R)-4-(4-methylpiperazin-1-yl)-4-oxo-3-propanamidobutan-2-yl]anilino}-1-[(1r,4S)-4-methylcyclohexyl]-2-oxoethyl}-1H-pyrazole-5-carboxamide
1-ethyl-N-{(1S)-2-{2-fluoro-4-[(2S,3R)-4-(4-methylpiperazin-1-yl)-4-oxo-3-propanamidobutan-2-
yl]anilino}-1-[(1r,4S)-4-methylcyclohexyl]-2-oxoethyl}-1H-pyrazole-5-carboxamide
interleukin-17A (IL-17A) inhibitor, anti-inflammatory, DC-806, LY4100504, DC 806, LY 4100504, Y64F9MC2QM
Navepdekinra (also known as DC-806 or LY4100504) is an experimental, orally active small-molecule inhibitor of interleukin-17A (IL-17A). It was primarily developed to treat autoimmune and inflammatory conditions, such as psoriasis, by disrupting the interaction between IL-17A and its receptor.
Key Properties and Development
- Mechanism: It is a potent inhibitor with an IC50 of 10.81 nM, designed to provide an oral alternative to existing injectable IL-17 biologic therapies.
- Acquisition: The drug was originally developed by DICE Therapeutics, which was acquired by Eli Lilly and Company in 2023 for approximately $2.4 billion to bolster their immunology pipeline.
Navepdekinra (DC-806) is an orally active, potent interleukin-17A (IL-17A) inhibitor (IC50 = 10.81 nM). Navepdekinra disrupts the IL-17A protein-receptor interaction, suppressing the downstream pro-inflammatory signaling pathway. Navepdekinra inhibits arthritis in a collage-induced arthritis (CIA) rat model. Navepdekinra can be used for psoriasis, psoriatic arthritis, and ankylosing spondylitis
SYN
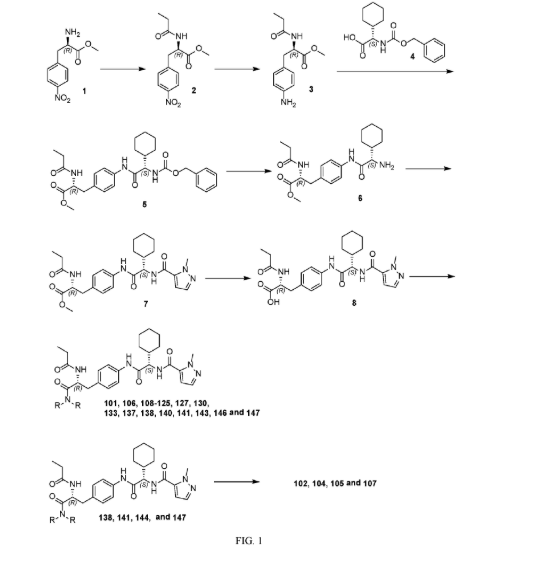
Example 210: N-[(2R,3S)-3-{4-[(2S)-2-[(1-ethyl-1H-pyrazol-5-yl)formamido]-2-[(1r,4S)-4-methylcyclo hexyl]acetamido]-3-fluorophenyl}-1-(4-methylpiperazin-1-yl)-1-oxobutan-2-yl]propanamide) (234)
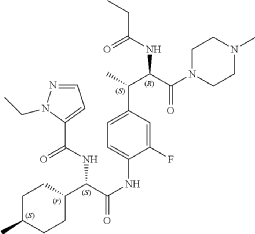
Following General Procedure R, 0.227 g, 0.310 mmol, 1.0 eq) of 82d in DMF (1 mL) were added 1-ethyl-1H-pyrazole-5-carboxylic acid (0.052 g, 0.372 mmol, 1.2 eq), DIPEA (0.43 mL, 2.482 mmol, 8.0 eq) and then HATU (0.177 g, 0.465 mmol, 1.5 eq.) and the resulting mixture was stirred at RT for 1 h. The mixture was concentrated to dryness and the residue was purified via reverse phase column chromatography on a 120 g C18 cartridge eluting with a 5-95% H 2O:MeCN eluent (0.1% ammonia) to afford 234 (0.025 g) as a white solid. 1H NMR (400 MHz, DMSO-d 6) δ 9.86 (s, 1H), 8.46 (d, J=8.3 Hz, 1H), 8.26 (d, J=8.7 Hz, 1H), 7.75 (t, J=8.3 Hz, 1H), 7.47 (d, J=2.1 Hz, 1H), 7.15-7.07 (m, 1H), 7.05-6.97 (m, 2H), 4.86 (t, J=9.4 Hz, 1H), 4.53 (t, J=8.4 Hz, 1H), 4.47 (q, J=7.2 Hz, 2H), 3.46-3.38 (m, 2H), 3.29-3.14 (m, 2H), 3.12-2.99 (m, 2H), 2.25-2.03 (m, 5H), 1.98 (s, 3H), 1.81 (ddt, J=15.0, 9.9, 5.6 Hz, 2H), 1.74-1.60 (m, 4H), 1.58-1.47 (m, 1H), 1.28 (t, J=7.1 Hz, 4H), 1.20 (d, J=7.0 Hz, 3H), 1.14-1.02 (m, 1H), 0.99 (t, J=7.6 Hz, 3H), 0.93-0.87 (m, 1H), 0.86 (d, J=6.5 Hz, 3H). UPLC-MS (basic 4 min): rt=1.76 min; m/z=626.4 for [M+H] +.
PAT
Example 1: Exemplary Scheme—Synthesis of Intermediate Compounds 62a-62d
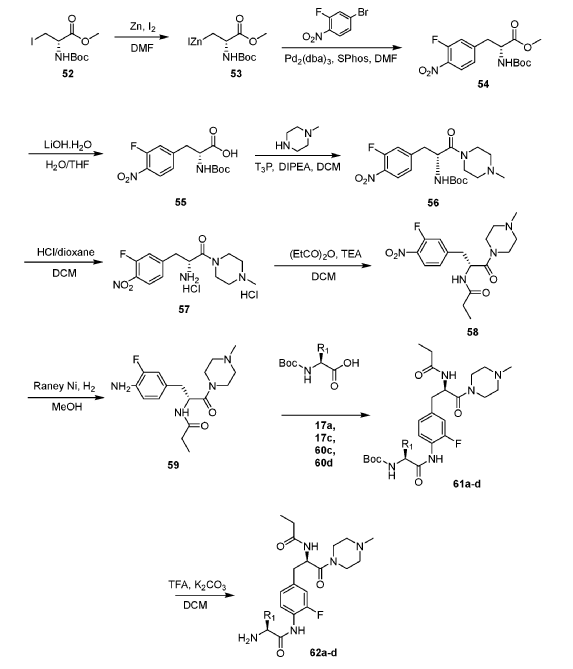
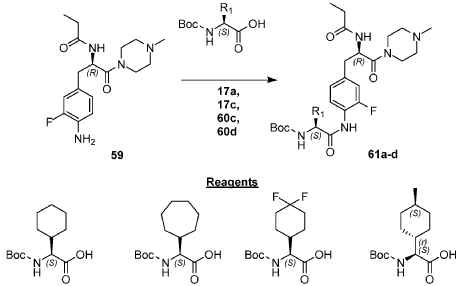
PAT
IL-17 Ligands And Uses Thereof
Publication Number: US-2020247785-A1
Priority Date: 2019-02-06
- Substituted benzenecarboxamides as IL-17A modulatorsPublication Number: US-11274094-B2Priority Date: 2019-09-16Grant Date: 2022-03-15
- Il-17a modulators and uses thereofPublication Number: US-2021101886-A1Priority Date: 2019-09-16
- IL-17 ligands and uses thereofPublication Number: US-11447468-B2Priority Date: 2019-02-06Grant Date: 2022-09-20
- Il-17 ligands and uses thereofPublication Number: US-2023053746-A1Priority Date: 2019-02-06
- IL-17 ligands and uses thereofPublication Number: US-12234225-B2Priority Date: 2019-02-06Grant Date: 2025-02-25
- Mannose 6-phosphate or asgpr receptor binding compounds for the degradation of extracellular proteinsPublication Number: WO-2023028338-A2Priority Date: 2021-08-27
- Potent asgpr-binding compounds for the degradation of immunoglobulins and other proteinsPublication Number: WO-2022235699-A2Priority Date: 2021-05-03
- Il-17a modulators and uses thereofPublication Number: WO-2021055376-A1Priority Date: 2019-09-16
- Substituted benzenecarboxamides as il-17a modulatorsPublication Number: US-2023141212-A1Priority Date: 2019-09-16
- Substituted benzenecarboxamides as il-17a modulatorsPublication Number: US-2023145481-A1Priority Date: 2019-09-16

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
- [1]. Paul R. Fatheree, et al. IL-17 Ligands And Uses Thereof. US20200247785A1.[2]. Kim D, et al. Next-Generation Anti-IL-17 Agents for Psoriatic Disease: A Pipeline Review. Am J Clin Dermatol. 2025 May;26(3):307-320. [Content Brief][3]. Xiaobing Deng, et al. The Critical and Unexpected Role of a Methyl Group in Interleukin-17A Inhibitors. bioRxiv 2025.10.02.680113
//////////navepdekinra, interleukin-17A (IL-17A) inhibitor, anti-inflammatory, DC-806, LY4100504, DC 806, LY 4100504, Y64F9MC2QM
Idrebormilast
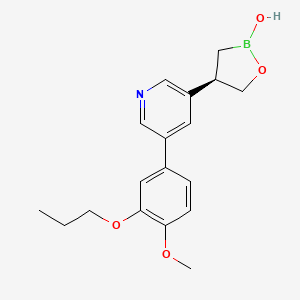
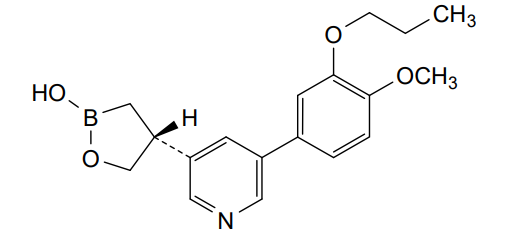
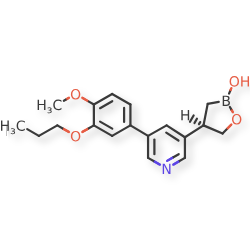
Idrebormilast
CAS 2415085-44-6
MF C18H22BNO4, MW 327.18
Pyridine, 3-[(4R)-2-hydroxy-1,2-oxaborolan-4-yl]-5-(4-methoxy-3-propoxyphenyl)-
(4R)-4-[5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl]-1,2-oxaborolan-2-ol
phosphodiesterase 4 (PDE4) inhibitor, non-steroidal anti-inflammatory, M6ZU548FWD, PF07038124, PF 07038124
PF-07038124 is under investigation in clinical trial NCT05298033 (Study of Efficacy, Safety and Tolerability of Crisaborole and PF-07038124 With and Without NBUVB in Vitiligo).
IDREBORMILAST is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 2 investigational indications.
- Study of Efficacy, Safety and Tolerability of Crisaborole and PF-07038124 With and Without NBUVB in VitiligoCTID: NCT05298033Phase: Phase 2Status: CompletedDate: 2024-06-12
- PDE4 Inhibition in Seborrheic Dermatitis and Papulopustular RosaceaCTID: NCT06013371Phase: Phase 2Status: TerminatedDate: 2025-04-24
- A Study To Determine The Safety, Tolerability, Skin Irritation Potential, And PK Following Topical Application Of PF-07038124 In Healthy ParticipantsCTID: NCT04135560Phase: Phase 1Status: CompletedDate: 2020-05-14
- Study to Evaluate the Safety, Local and Systemic Tolerability, and Pharmacokinetics of Multiple-Dose Topical Administration of PF-07038124 in Japanese Healthy ParticipantsCTID: NCT04863417Phase: Phase 1Status: CompletedDate: 2024-01-25
- Study To Assess Efficacy, Safety, Tolerability And Pharmacokinetics Of PF-07038124 Ointment In Participants With Atopic Dermatitis Or Plaque PsoriasisCTID: NCT04664153Phase: Phase 2Status: CompletedDate: 2022-08-26
SYN
PAT
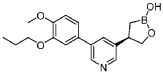
Example 4: (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol
Method A:
To a mixture of (R)-(3-((tert-butyldimethylsilyl)oxy)-2-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)propyl)boronic acid (Preparation 6, 55 g, 120 mmol) in IPA (247 mL) was added 5 M hydrogen chloride in IPA (37 mL, 185 mmol) at about 20 °C. The mixture was stirred for about 3 h and concentrated. The residue was diluted with EtOAc (500 mL) and 1 N
HCI (500 mL) was added. The layers were separated and the EtOAc layer was extracted with 0.5 N HCI (2 x 200 mL). The aqueous extracts were combined with the separated acidic aqueous layer and washed with EtOAc (3 x 250 mL). The combined acidic aqueous layers were treated with K3PO4 to pH 5-6. The mixture was extracted with EtOAc (1 x 500 mL, 2 x 200 mL). The combined EtOAc extracts were washed with brine, dried over Na2S04, filtered and concentrated to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol (34.5 g, 88%). This was further purified by preparative SFC (Prep SFC Method C) to afford 29 g as a crude product. The crude product was dissolved in methanol (250 mL) and water (50 mL) and stirred at 20 °C for about 30 min before concentrating. The concentrated solution was partitioned between brine and EtOAc. The aqueous layer was separated and extracted with EtOAc. The EtOAc extracts were combined with the separate EtOAc layer and were washed with brine, dried over Na2S04 and concentrated. The residue was dissolved in degassed EtOAc (200 mL) and degassed heptane (100 mL) was added slowly. Heptane was added until a precipitate was observed and and the resulting mixture was stirred overnight under N2. The solid was filtered to afford 8.08 g of product. The filtrate was concentrated and the residue dissolved in EtOAc (50 mL). Heptane (25 mL) was slowly added and the mixture stirred overnight open to air. The solid was filtered to afford a second batch (6.16 g). This was repeated a second time to afford 3.0 g. The filtrate was stirred overnight to afford additional batches (2.09 g and 3.1 g) respectively. The solid batches were combined to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol (22.3 g, 57%) as a crystalline solid. Ή NMR (DMSO-cfe, 400MHz): d 8.70 (d, J = 2.3 Hz, 1 H), 8.68 (s, 1 H), 8.42 (d, J = 2.0 Hz, 1 H), 7.93 (s, 1 H), 7.27 (d, J = 2.0 Hz, 1 H), 7.23-7.26 (m, 1 H), 7.06 (d, J = 8.2 Hz, 1 H), 4.28 (t, J = 8.2 Hz, 1 H), 4.03 (t, J = 6.4 Hz, 2H), 3.86 (t, J = 9.0 Hz, 1 H), 3.81 (s, 3H), 3.46-3.54 (m, 1 H), 1 .71 -1 .80 (m, 2H), 1 .28-1 .35 (m, 1 H), 1 .15 (dd, J = 10.5, 16.4 Hz, 1 H), 1 .00 (t, J = 7.4 Hz, 3H). LCMS m/z = 328 [MH]+; RT [Analytical SFC Method B] = 7.30 min. [a]20D -23.7 (c = 0.9, EtOH).
Elemental analysis calculated (%) for Ci8H22BN04: C 66.08, H 6.78, N 4.28. Found: C 65.86, H 6.59, N 4.18.
Method B:
Step 1 : To THF (18.0 mL) was added 3-(3-((tert-butyldimethylsilyl)oxy)prop-1 -en-2-yl)-5-(4-methoxy-3-propoxyphenyl)pyridine (Preparation 50, 3.0 g, 7.25 mmol), [lr(COD)CI]2 (CAS 121 12-67-3, 36.9 mg, 0.054 mmol) and (S)[(Sp)-2-(diphenylphosphino)ferrocenyl]-4-isopropyloxazoline (CAS 163169-29-7, 52.4 mg, 0.109 mmol). Additional THF (6.0 mL) was added to the mixture which was warmed to about 50 °C for about 5 min. Catecholborane (10.9 mL, 1 0M in THF) was added to the mixture and stirred at about 50 °C for about 1 h. The mixture was cooled to about 20 °C and treated with HCI (12.2 M, 1 .51 mL) over 1 min. The mixture was held at about 20 °C for about 1 h, afterwhich a precipitate had formed. The mixture was cooled to about 10 °C and filtered. The filtered solid was washed with THF (6.0 mL) and
dried overnight at 35°C under vacuum to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridine-3-yl)-1 ,2-oxaborolan-2-ol hydrochloride monohydrate (3.98 g, 91 %) as a crystalline solid. 1H NMR (CD3OD, 400MHz): d 8.98 (d, J = 1 .5 Hz, 1 H), 8.75 (s, 1 H), 8.67 (d, J = 1 .3 Hz, 1 H), 7.37-7.43 (m, 2H), 7.15 (d, J = 8.3 Hz, 1 H), 4.09 (t, J = 6.5 Hz, 2H), 3.89-3.92 (m, 1 H), 3.86-3.95 (m, 5H), 3.46 (br s, 1 H), 1 .85 (m, 2H), 1 .31 -1 .42 (m, 2H), 1 .08 (t, J = 7.4 Hz, 3H). LCMS m/z = 328 [MH]+.
Step 2: To a solution of (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridine-3-yl)-1 ,2-oxaborolan-2-ol hydrochloride monohydrate (2.0 g, 5.24 mmol) in water (60 ml_) was added EtOAc (20 ml_). To the stirred mixture was added NaOH (1 N) dropwise to adjust the pH of the aqeous layer to 7-8. The mixture was stirred at about 20 °C for about 5 min. The layers were separated and the aqueous layer was extracted with EtOAc (2 x 10 ml_). The combined EtOAc extracts were concentrated. The residue was dissolved in THF/MTBE (1 :3, 22 ml_) and stirred at about 20 °C overnight. The precipitate was filtered and dried under vacuum to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridine-3-yl)-1 ,2-oxaborolan-2-ol (1 .17 g, 68%) as a crystalline solid. Ή NMR (DMSO-cfe, 400MHz): d 8.70 (d, J = 2.3 Hz, 1 H), 8.68 (s, 1 H), 8.42 (d, J = 2.0 Hz, 1 H), 7.93 (s, 1 H), 7.27 (d, J = 2.0 Hz, 1 H), 7.23-7.26 (m, 1 H), 7.06 (d, J = 8.2 Hz, 1 H), 4.28 (t, J = 8.2 Hz, 1 H), 4.03 (t, J = 6.4 Hz, 2H), 3.86 (t, J = 9.0 Hz, 1 H), 3.81 (s, 3H), 3.46-3.54 (m, 1 H), 1 .71 -1 .80 (m, 2H), 1 .28-1 .35 (m, 1 H), 1 .15 (dd, J = 10.5, 16.4 Hz, 1 H), 1 .00 (t, J = 7.4 Hz, 3H). LCMS m/z = 328 [MH]+.
Method C:
To a solution of (R)-(3-((tert-butyldimethylsilyl)oxy)-2-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)propyl)boronic acid (Preparation 6, 29.0 g, 63.1 mmol) in THF (66 mL) was added aqueous HCI (84.2 mL, 252 mmol, 3.0 M) and stirred at 20 °C for about 1 .5 h. The mixture was concentrated. The mixture was diluted with 1 M HCI and extracted with EtOAc (3 x 100 mL). The combined EtOAc extracts were washed with 1 M HCI (3 x 50 mL). The combined aqueous extracts were neutralized with K3PO4 to pH 7-8 and extracted with EtOAc (3 x 100 mL). The combined EtOAc extracts were dried over Na2S04, filtered and concentrated to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol (19.0 g, 92%).
This was further purified by preparative SFC (Prep SFC Method C) to afford 18 g of the crude product. The crude product was dissolved in MeOH (100 mL) and water (50 mL). The mixture was partitioned between brine and EtOAc. The layers were separated and the aqueous layer was extracted with EtOAc. The combined EtOAc extracts were washed with brine, dried over Na2S04 and concentrated to afford 15 g of product. The residue was dissolved in EtOAc (60 mL) and heptane (30 mL) was slowly added over about 3 h. The mixture was stirred at about 20 °C overnight. The precipitate was filtered and dried to afford (8.08 g). This process was repeated 2 more times to afford additional batches (2.01 g and 1 .03 g), respectively. The three batches were combined in heptane (100 mL), chilled to about -78 °C for about 10 min, filtered
and dried to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol (10.4 g, 51 %) as a crystalline solid. Ή NMR (DMSO -d6, 400MHz): d 8.70 (d, J = 2.3 Hz, 1 H), 8.68 (s,
1 H), 8.42 (d, J = 2.0 Hz, 1 H), 7.93 (s, 1 H), 7.27 (d, J = 2.0 Hz, 1 H), 7.23-7.26 (m, 1 H), 7.06 (d, J = 8.2 Hz, 1 H), 4.28 (t, J = 8.2 Hz, 1 H), 4.03 (t, J = 6.4 Hz, 2H), 3.86 (t, J = 9.0 Hz, 1 H), 3.81 (s, 3H), 3.46-3.54 (m, 1 H), 1 .71 -1 .80 (m, 2H), 1 .28-1 .35 (m, 1 H), 1 .15 (dd, J = 10.5, 16.4 Hz, 1 H),
1 .00 (t, J = 7.4 Hz, 3H). LCMS m/z = 328 [MH]+.
PAT
- Substituted 1,2-oxaborolan-2-ols as PDE4 inhibitorsPublication Number: US-11559538-B2Priority Date: 2018-10-05Grant Date: 2023-01-24
- Boron-containing PDE4 inhibitorsPublication Number: CN-113166177-BPriority Date: 2018-10-05Grant Date: 2024-09-03
- Boron Containing PDE4 InhibitorsPublication Number: US-2021069219-A1Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: CA-3114702-CPriority Date: 2018-10-05Grant Date: 2023-08-08
- PDE4 INHIBITORS CONTAINING BORONPublication Number: BR-112021005870-B1Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: WO-2020070651-A1Priority Date: 2018-10-05
- Boron-containing PDE4 inhibitorsPublication Number: CN-113166177-APriority Date: 2018-10-05
- Boron Containing PDE4 InhibitorsPublication Number: US-2023148402-A1Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: EP-3861001-A1Priority Date: 2018-10-05
- Boron-containing PDE4 inhibitorsPublication Number: ES-2974208-T3Priority Date: 2018-10-05Grant Date: 2024-06-26
- PDE4 inhibitor (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: US-10946031-B2Priority Date: 2018-10-05Grant Date: 2021-03-16
- Boron-containing PDE4 inhibitorsPublication Number: KR-102576125-B1Priority Date: 2018-10-05Grant Date: 2023-09-08
- Boron-containing PDE4 inhibitorsPublication Number: KR-20210068532-APriority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: EP-3861001-B1Priority Date: 2018-10-05Grant Date: 2023-12-13
- Boron Containing PDE4 InhibitorsPublication Number: US-2020108083-A1Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: CA-3114702-A1Priority Date: 2018-10-05
- boron containing pde4 inhibitorsPublication Number: BR-112021005870-A2Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: TW-202027755-APriority Date: 2018-10-05
- Stable topical formulations of 1(r)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: CA-3191886-A1Priority Date: 2020-08-20
- STABLE TOPICAL FORMULATIONS OF 1(R)-4-(5-(4-METHOXY-3-PROPOXYPHEN IL)PYRIDIN-3-IL)-1,2-OXABOROLAN-2-OL.Publication Number: MX-2023002086-APriority Date: 2020-08-20
- Stable topical formulations of 1(r)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: WO-2022038485-A1Priority Date: 2020-08-20
- 1(R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-ol is a stable partial preparationPublication Number: CN-115916260-APriority Date: 2020-08-20
- Stable Topical Formulations of 1(R)-4-(5-(4-Methoxy-3-Propoxphenyl)Pyridin-3-YL)-1,2-OX-Aborolan-2-OLPublication Number: US-2023310472-A1Priority Date: 2020-08-20
- Combination therapyPublication Number: WO-2024231312-A2Priority Date: 2023-05-05
- Borate derivative and uses thereofPublication Number: EP-4269418-A1Priority Date: 2020-12-25
- Stable topical formulations of 1(r)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: EP-4199902-A1Priority Date: 2020-08-20
- Stable topical formulation of 1(R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: JP-2023538362-APriority Date: 2020-08-20
- STABLE TOPICAL FORMULATIONS OF 1(R)-4-(5-(4-METHOXY-3-PROPOXYPHENYL)PYRIDIN-3-IL)-1,2-OXABOROLAN-2-OLPublication Number: BR-112023002533-A2Priority Date: 2020-08-20
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////idrebormilast, phosphodiesterase 4 (PDE4) inhibitor, non-steroidal anti-inflammatory, M6ZU548FWD, PF07038124, PF 07038124
Glasmacinal



Glasmacinal
CAS 2097822-02-9
MF C37H62N2O10 MW694.90
[(2S,3R,4S,6R)-4-(dimethylamino)-2-[[(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-15-oxo-1-oxa-6-azacyclopentadec-11-yl]oxy]-6-methyloxan-3-yl] benzoate
- (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-[[2-O-Benzoyl-3,4,6-trideoxy-3-(dimethylamino)-beta-D-xylo-hexopyranosyl]oxy]-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-azacyclopentadecan-15-one
- 1-Oxa-6-azacyclopentadecan-15-one, 11-[[2-O-benzoyl-3,4,6-trideoxy-3-(dimethylamino)-beta-D-xylo-hexopyranosyl]oxy]-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-, (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-
(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-{[2-O-benzoyl-3,4,6-trideoxy-3-(dimethylamino) -β-D-xylo-hexopyranosyl]oxy}-2-ethyl3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-
azacyclopentadecan-15-one
non-antibacterial macrolide, anti-inflammatory, EP 395, M3T8D3P634
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US234729681&_cid=P12-MKVZ26-57135-1
Example 2: (2S,3R,4S,6R)-4-(dimethylamino)-2-[[(2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-15-oxo-1-oxa-6-azacyclopentadec-11-yl]oxy]-6-methyl-tetrahydropyran-3-yl] benzoate)

To a mixture of (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-11-[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyl-tetrahydropyran-2-yl]oxy-2-ethyl-3,4,10,13-tetrahydroxy-3,5,6,8,10,12,14-heptamethyl-1-oxa-6-azacyclopentadecan-15-one (Example 1) (0.5 g, 0.8500 mmol) and Triethylamine (428.2 mg, 4.23 mmol) in DCM (5 ml), cooled on ice, was added Benzoyl chloride (356.9 mg, 2.54 mmol). The reaction mixture was allowed to reach room temperature. After 3 days good conversion to the desired benzoylated product was obtained and the mixture was portioned between DCM and saturated sodium hydrogen carbonate solution. The organic phase was dried over magnesium sulphate and concentrated to a white foam. The product was purified using reversed phase chromatography (see general information)
PAT
- Azithromycin Derivatives With Epithelial Barrier Enhancement PropertiesPublication Number: US-2018354981-A1Priority Date: 2015-11-19
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-10723752-B2Priority Date: 2015-11-19Grant Date: 2020-07-28
- Azithromycin Derivatives With Epithelial Barrier Enhancement PropertiesPublication Number: US-2020317710-A1Priority Date: 2015-11-19
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-12049477-B2Priority Date: 2015-11-19Grant Date: 2024-07-30
- Azithromycin derivatives with epithelial barrier enhancement propertiesPublication Number: US-11236120-B2Priority Date: 2015-11-19Grant Date: 2022-02-01
- Compounds
- Publication Number: US-2022106349-A1
- Priority Date: 2015-11-19
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com

ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
///////glasmacinal, ANAX, ADVECT, non-antibacterial macrolide, anti-inflammatory, EP 395, M3T8D3P634
Cenacitinib



Cenacitinib
CAS 2641636-52-2
MF C19H19F2N7O3 MW431.4
Urea, N-[(1R,2S)-2-fluorocyclopropyl]-N′-[5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl]-
N-{5-[(7-fluoro-2,3-dihydro-1,4-benzodioxin-5-yl)amino]-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl}-N′-[(1R,2S)-2-fluorocyclopropyl]urea
Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
VTX958 for the Treatment of Moderately to Severely Active Crohn’s Disease
CTID: NCT05688852
Phase: Phase 2
Status: Terminated
Date: 2025-07-03
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750705&_cid=P22-MKEUDK-45432-1
Example 4: Synthesis of 1-(5-((7-fluoro-2,3-dihydrobenzo[b][1,4]dioxin-5-yl)amino)-7-(methylamino)pyrazolo[1,5-a]pyrimidin-3-yl)-3-((1R,2S)-2-fluorocyclopropyl)urea (5)


| Step 1: To a solution of 1E (100 g, 288 mmol) and 2E (57 g, 345 mmol) in dry 1,4-dioxane (3000 mL) under N 2 atmosphere was added Cs 2CO 3 (141 g, 432 mmol), Pd(OAc) 2 (5.2 g, 23.3 mmol) and BINAP (28.6 g, 46.6 mmol). After stirring at 115° C. overnight, the reaction mixture was cooled to rt. and diluted with hexane (3000 mL). The solid was collected by filtration and washed with 2×1500 mL (50% hexane in DCM). The solid was suspended into 5000 mL water and stirred for 1 h. The solid was collected by filtration and dried under vacuum to afford compound 2 (90 g, 65%) as a brown solid. |
PAT
Publication Number: US-2021139486-A1
Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: EP-4054581-A1Priority Date: 2019-11-08
- Tyk2 pseudokinase ligandsPublication Number: US-2023348478-A1Priority Date: 2019-11-08
- Substituted pyrazolo[1,5-a]pyrimidines as TYK2 pseudokinase ligandsPublication Number: US-11753411-B2Priority Date: 2019-11-08Grant Date: 2023-09-12
- TYK2 pseudokinase ligandsPublication Number: CN-114929226-BPriority Date: 2019-11-08Grant Date: 2024-09-27
- TYK2 pseudokinase ligandPublication Number: CN-114929226-APriority Date: 2019-11-08
- Preparation of a tyk2 inhibitorPublication Number: WO-2024151992-A1Priority Date: 2023-01-13
- Crystalline forms of a tyk2 inhibitorPublication Number: US-2024010654-A1Priority Date: 2022-07-06
- Crystalline forms of TYK2 inhibitorsPublication Number: CN-119816502-APriority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: EP-4551576-A1Priority Date: 2022-07-06
- Crystalline forms of a tyk2 inhibitorPublication Number: WO-2024011136-A1Priority Date: 2022-07-06
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions,Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////cenacitinib, cenacitinib, Janus kinase inhibitor, anti-inflammatory, VTX958, VTX 958, SB88R8KGL3
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
Nibrozetone
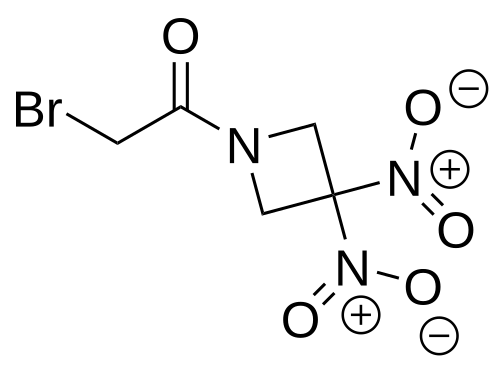
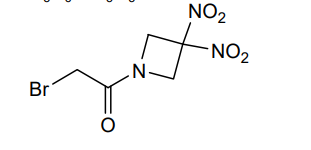
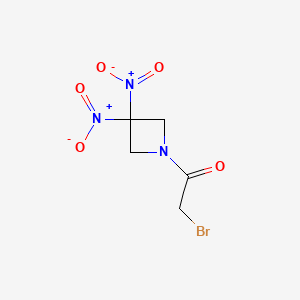
Nibrozetone
CAS 925206-65-1
MF C5H6BrN3O5 MW268.02 g/mol
2-bromo-1-(3,3-dinitroazetidin-1-yl)ethan-1-one
2-Bromo-1-(3,3-dinitroazetidin-1-yl)ethanone
2-BROMO-1-(3,3-DINITROAZETIDIN-1-YL)ETHAN-1-ONE
anti-inflammatory, RRx-001, RRx 001, ABDNAZ
Nibrozetone is an investigational new drug that is being evaluated by EpicentRx for the treatment of oral mucositis in head and neck cancer patients. It is a small molecule that combines direct inhibition of the NLRP3 inflammasome, induction of NRF2, and release of nitric oxide under hypoxic conditions.[1][2] It has received Fast Track designation from the FDA for severe oral mucositis in head and neck cancer patients.[3]
Nibrozetone (RRx-001) is an investigational, multi-action small molecule drug that is being developed by EpicentRx for a range of conditions, including head and neck cancers, small cell lung cancer, and neurodegenerative diseases like Parkinson’s and ALS. Its mechanism involves inhibiting the NLRP3 inflammasome, activating the Nrf2 pathway, and releasing nitric oxide in hypoxic tumor environments, while also protecting healthy tissues. It is being evaluated for its potential to reduce the side effects of cancer treatments and as a disease-modifying therapy itself.
How it works
- Anti-inflammatory: Nibrozetone inhibits the NLRP3 inflammasome, which is a key driver of inflammation in several diseases.
- Antioxidant: It activates the Nrf2 pathway, a cellular defense mechanism that protects against oxidative stress.
- Tumor-specific delivery: It acts as a “hypoxia-activated” drug, releasing a nitric oxide-releasing radical only in the low-oxygen environment of tumors, which can be toxic to cancer cells.
- Protective to normal tissue: The drug’s protective mechanisms are thought to keep it from causing harm to healthy tissues outside of the tumor environment.
Current and potential uses
- Oral mucositis: It is being studied to prevent and treat severe mouth sores that can be a side effect of head and neck cancer radiation therapy.
- Small cell lung cancer (SCLC): It is being investigated in a Phase 3 trial for the treatment of SCLC.
- Neurodegenerative diseases: Animal studies have shown promising neuroprotective effects in models of Parkinson’s and ALS.
- Other potential applications: Research is ongoing for its use as a treatment for other conditions, including endometriosis, toxic exposures, and various types of cancers.
- RRx-001 in Lung Cancer, Ovarian Cancer and Neuroendocrine Tumors Prior to Re-administration of Platinum Based Doublet Regimens (QUADRUPLE THREAT)CTID: NCT02489903Phase: Phase 2Status: CompletedDate: 2025-03-17
- RRx-001 for Reducing Oral Mucositis in Patients Receiving Chemotherapy and Radiation for Head and Neck CancerCTID: NCT05966194Phase: Phase 2Status: RecruitingDate: 2024-11-15
- Safety and Efficacy of RRx-001 in the Attenuation of Oral Mucositis in Patients Receiving Chemoradiation for the Treatment of Oral CancersCTID: NCT03515538Phase: Phase 2Status: CompletedDate: 2024-11-04
- Safety and Pharmacokinetic Study of RRx-001 in Cancer SubjectsCTID: NCT01359982Phase: Phase 1Status: CompletedDate: 2024-11-01
- RRx-001 Given With Irinotecan and Temozolomide for Pediatric Patients With Recurrent or Progressive Malignant Solid and Central Nervous System TumorsCTID: NCT04525014Phase: Phase 1Status: TerminatedDate: 2024-10-31
REF
- Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic MaterialsPublication Name: Cancer ResearchPublication Date: 2012-05-14PMID: 22589277DOI: 10.1158/0008-5472.can-11-2303
- Properties of delta5-3beta-hydroxysteroid oxidoreductase isolated from Streptomyces griseocarneusPublication Name: Acta microbiologica Academiae Scientiarum HungaricaePublication Date: 1975PMID: 5856
PAT
- Dinitroazetidines Are a Novel Class of Anticancer Agents and Hypoxia-Activated Radiation Sensitizers Developed from Highly Energetic MaterialsPublication Name: Cancer ResearchPublication Date: 2012-05-14PMID: 22589277DOI: 10.1158/0008-5472.can-11-2303
- Properties of delta5-3beta-hydroxysteroid oxidoreductase isolated from Streptomyces griseocarneusPublication Name: Acta microbiologica Academiae Scientiarum HungaricaePublication Date: 1975PMID: 5856
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-8927527-B2Priority Date: 2005-08-12Grant Date: 2015-01-06
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-9226915-B2Priority Date: 2005-08-12Grant Date: 2016-01-05
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: WO-2007022225-A2Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-2022016077-A1Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: US-11925617-B2Priority Date: 2005-08-12Grant Date: 2024-03-12
- Methods of synthesizing and isolating N-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: US-8471041-B2Priority Date: 2010-02-09Grant Date: 2013-06-25
- Methods of synthesizing and isolating n-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: WO-2011100090-A1Priority Date: 2010-02-09
- Methods of synthesizing and isolating n-(bromoacetyl)-3,3-dinitroazetidine and a composition including the samePublication Number: IL-221141-A0Priority Date: 2010-02-09
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: EP-1924253-A2Priority Date: 2005-08-12
- Cyclic nitro compounds, pharmaceutical compositions thereof and uses thereofPublication Number: EP-1924253-B1Priority Date: 2005-08-12Grant Date: 2014-12-10
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011100090&_cid=P11-MHTYGA-61308-1
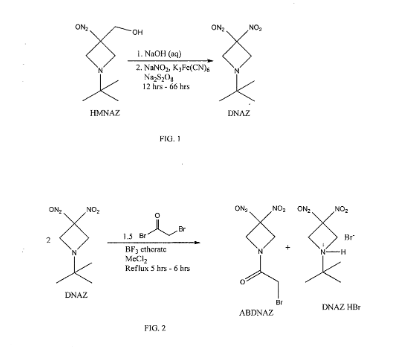
Cyclic nitro compounds, such as ABDNAZ, are being investigated for their potential use in treating cancer. Methods of synthesizing ABDNAZ have been described, such as in United States Patent No. 7,507,842 to Bednarski et al.
(“Bednarski”). In Bednarski, ABDNAZ is synthesized by reacting
l-½rt-butyl-3,3-dinitroazetidine (DNAZ) with bromoacetyl bromide and boron trifluoride etherate. For every mole of ABDNAZ produced, a mole of a hydrogen bromide salt of DNAZ (DNAZ HBr) is also produced as a coproduct. The ABDNAZ is isolated from the DNAZ HBr by cooling the reaction mixture, adding
dichloromethane, and filtering the DNAZ HBr. Solid DNAZ HBr is sensitive to impact, friction, and other external stimuli and, therefore, must be handled carefully. The dichloromethane filtrate is washed with water, dried, and then the dichloromethane is evaporated, producing a crude ABDNAZ mixture. The product is washed sequentially with diethyl ether and dried under vacuum, yielding ABDNAZ that is approximately 98% pure and at a yield of approximately 75% (based on bromoacetyl bromide). The 2% of impurities remaining in the ABDNAZ are believed to include
bromoacetic acid, unreacted DNAZ, and DNAZ HBr. This method of producing ABDNAZ is referred to herein as the Bednarski process. While the Bednarski process provides ABDNAZ at a reasonable purity and yield, the purity is not sufficient for pharmaceutical uses. In addition, solid DNAZ HBr produced during the Bednarski process is an explosive compound, which adds to the complexity of producing
Example 2
Synthesis of ABDNAZ from DNAZ
A three neck round bottom flask (3 L) equipped with a magnetic stir bar and a water jacketed reflux condenser was charged with the dichloromethane solution of DNAZ (produced as described in Example 1). A nitrogen gas purge of the apparatus was initiated and, after ten minutes, boron trifluoride diethyletherate (6.37 mL, 52 mmol) was added, followed by bromoacetyl bromide (33.77 mL, 388 mmol). The flask was sealed, except for a small vent at the top of the condenser, and the solution was heated to a mild reflux. After six hours (± 0.5 hour), heating was stopped and dichloromethane (1000 mL) and distilled water (800 mL) were added, in that order, to the heterogeneous mixture. The two-phase system was stirred vigorously for sixteen hours, until all solids (DNAZ HBr) were dissolved. The two-phase system was then transferred to a separatory funnel. The aqueous phase was removed and the organic phase was washed with additional distilled water (4 x 500 mL). The organic phase was dried with sodium sulfate (100 g – 150 g) and then transferred to a single neck, round bottom flask. The solution was concentrated on a rotary evaporator to approximately half of its initial volume and then ethanol (250 mL) was added. The remaining dichloromethane was removed by a rotary evaporator, causing precipitation of clear, colorless crystals. The flask was chilled in an ice bath for thirty minutes. The precipitate was isolated by vacuum filtration, rinsed with additional cold ethanol (5 x 150 mL), and dried to afford pure ABDNAZ (56.04 g, 81% yield): Ή NMR
(d6-acetone) δ 4.02 (s, 2H, -CH2Br ), 4.96 (br s, 2H, ring -CH2), 5.36 (br s, 2H, ring -CH2); 13C NMR (d6-acetone) δ 25.58, 58.58, 60.53, 107.69, 167.48.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007022225&_cid=P11-MHTYDP-59218-1
Example 5: Synthesis of ABDNAZ
[00139] A 25 ml, three-neck, round bottom flask was charged with 7 ml of methylene chloride and 2.50 g (12.3 mmol) of t-BuDNAZ prepared as described in Archibald et at, Journal of Organic Chemistry, 1990, 2920. Under nitrogen, 0.16 ml (1.23 mmol) of boron trifluoride etherate was added. After stirring 5 min. at ambient temperature, 0.54 ml (6.15 mol) of bromoacetyl bromide was added. The solution was heated between 50-600C for 2 h. The darkened reaction mixture was cooled to ambient temperature, diluted with 50 ml methylene chloride, and filtered. The solid was identified as the HBr salt of t-BuDNAZ. The methylene chloride filtrate was washed with two 20 ml portions of water, dried over sodium sulfate, filtered, and evaporated under reduced pressure. The resultant solid was washed with three 20 ml portions of ethyl ether and dried under vacuum to yield 1.24 g (75.2% based on bromoacetyl bromide) of BrADNAZ as a white solid (mp = 124-1250C). 1H NMR (CDCl3): δ 3.76 (s, 2H), 4.88 (br s, 2H), 5.14 (br s, 2H); 13C NMR (CDCl3): δ 165.2, 105.0, 59.72, 57.79, 23.90. CaIc. for C5H6BrN3O5: %C 22.41, %H 2.26, %N 15.68; Found: %C 22.61, %H 2.36, %N 15.58.
HPLC/MS C-8 reverse phase column with acetonitrile/water mobile phase – m/e 266.95 (100%), 268.95 (98.3%). FT-IR 3014.24 (weak), 1677.66, 1586.30, 1567.65, 1445.55 (NO2), 1367.80, 1338.00, 1251.27 cm‘1.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Oronsky B, Takahashi L, Gordon R, Cabrales P, Caroen S, Reid T (2023). “RRx-001: a chimeric triple action NLRP3 inhibitor, Nrf2 inducer, and nitric oxide superagonist”. Frontiers in Oncology. 13 1204143. doi:10.3389/fonc.2023.1204143. PMC 10258348. PMID 37313460.
- Jayabalan N, Oronsky B, Cabrales P, Reid T, Caroen S, Johnson AM, et al. (April 2023). “A Review of RRx-001: A Late-Stage Multi-Indication Inhibitor of NLRP3 Activation and Chronic Inflammation”. Drugs. 83 (5): 389–402. doi:10.1007/s40265-023-01838-z. PMC 10015535. PMID 36920652.
- Ryan C (30 March 2023). “FDA Grants Fast Track Designation to RRx-001 for Severe Oral Mucositis in Head and Neck Cancer”. OncLive.
| Clinical data | |
|---|---|
| Other names | Rrx-001 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 925206-65-1 |
| PubChem CID | 15950826 |
| DrugBank | DB12060 |
| ChemSpider | 13092644 |
| UNII | 7RPW6SU9SC |
| KEGG | D12720 |
| ChEMBL | ChEMBL3526802 |
| Chemical and physical data | |
| Formula | C5H6BrN3O5 |
| Molar mass | 268.023 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Nibrozetone, anti-inflammatory, RRx-001, RRx 001, ABDNAZ
Girocitinib
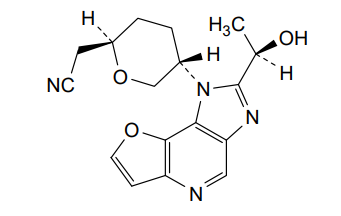
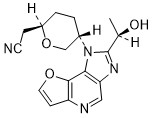
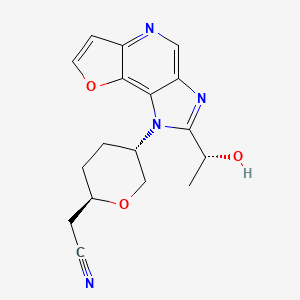
Girocitinib
CAS 2222137-79-1
MFC17H18N4O3 MW 326.36
2-[(2R,5S)-5-[4-[(1R)-1-hydroxyethyl]-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]oxan-2-yl]acetonitrile
[(2R,5S)-5-{2-[(1R)-1-hydroxyethyl]-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl}oxan-2-yl]acetonitrile
2-((2R,5S)-5-(2-((R)-1-hydroxyethyl)-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl)tetrahydro-2H-pyran-2-yl)acetonitrile
Janus kinase inhibitor, anti-inflammatory, A0IES9T8GO
In an era where targeted therapies are redefining the landscape of medical treatment, Girocitinib emerges as a beacon of hope for many. This innovative drug, developed by leading pharmaceutical research institutions, primarily targets specific proteins involved in disease progression. Classified as a tyrosine kinase inhibitor (TKI), Girocitinib has shown significant promise in the treatment of various cancers, particularly non-small cell lung cancer (NSCLC). The drug is currently in the advanced stages of clinical trials, with researchers optimistic about its potential to provide a more effective and less toxic treatment option compared to conventional therapies.
Girocitinib is designed to interfere with the signaling pathways that promote cancer cell growth and survival. It does this by inhibiting the activity of tyrosine kinases, enzymes that play a key role in the activation of many proteins by signaling pathways within the cell. Tyrosine kinases are often overactive in cancer cells, leading to unchecked proliferation and survival. By targeting these enzymes, Girocitinib effectively disrupts these malign processes, thereby slowing down or even halting the progression of the disease.
The primary indication for Girocitinib is non-small cell lung cancer (NSCLC), which accounts for approximately 85% of all lung cancer cases. NSCLC is notoriously difficult to treat, especially in its advanced stages, and current treatments often come with significant side effects. Clinical trials have shown that Girocitinib can significantly improve progression-free survival in patients with specific genetic mutations that make them more responsive to TKI therapy. These mutations can be identified through genetic testing, allowing for a more personalized treatment approach that increases the likelihood of success.
In addition to NSCLC, researchers are exploring the potential of Girocitinib to treat other types of cancer, including colorectal cancer and certain forms of leukemia. Early-stage trials have shown encouraging results, suggesting that Girocitinib could become a versatile tool in the oncology arsenal. Its ability to target specific molecular pathways makes it a promising candidate for combination therapies, which aim to enhance treatment efficacy while minimizing resistance and adverse effects.
The development of Girocitinib is a testament to the power of modern science and technology in addressing some of the most challenging health issues of our time. The drug’s journey from the laboratory to clinical trials has been marked by rigorous research and collaboration among scientists, healthcare professionals, and patients. As we await the results of ongoing studies, there is a palpable sense of anticipation in the medical community, as Girocitinib holds the promise of transforming cancer treatment for many patients.
In conclusion, Girocitinib represents a significant advancement in the field of targeted cancer therapy. Its mechanism of action, which involves the inhibition of tyrosine kinases, offers a more precise and potentially less harmful treatment option for patients with NSCLC and possibly other cancers. As research progresses, Girocitinib may well become a cornerstone in the fight against cancer, providing hope and improved outcomes for countless individuals around the world.
PDT PAT
WO2018067422
SYN
https://patents.google.com/patent/US10738060B2/en?oq=US10738060
Example 4: Synthesis of 2-[(2R,5S)-5-[2-[(R)-1-Hydroxyethyl]furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]tetrahydropyran-2-yl] acetonitrile (4)
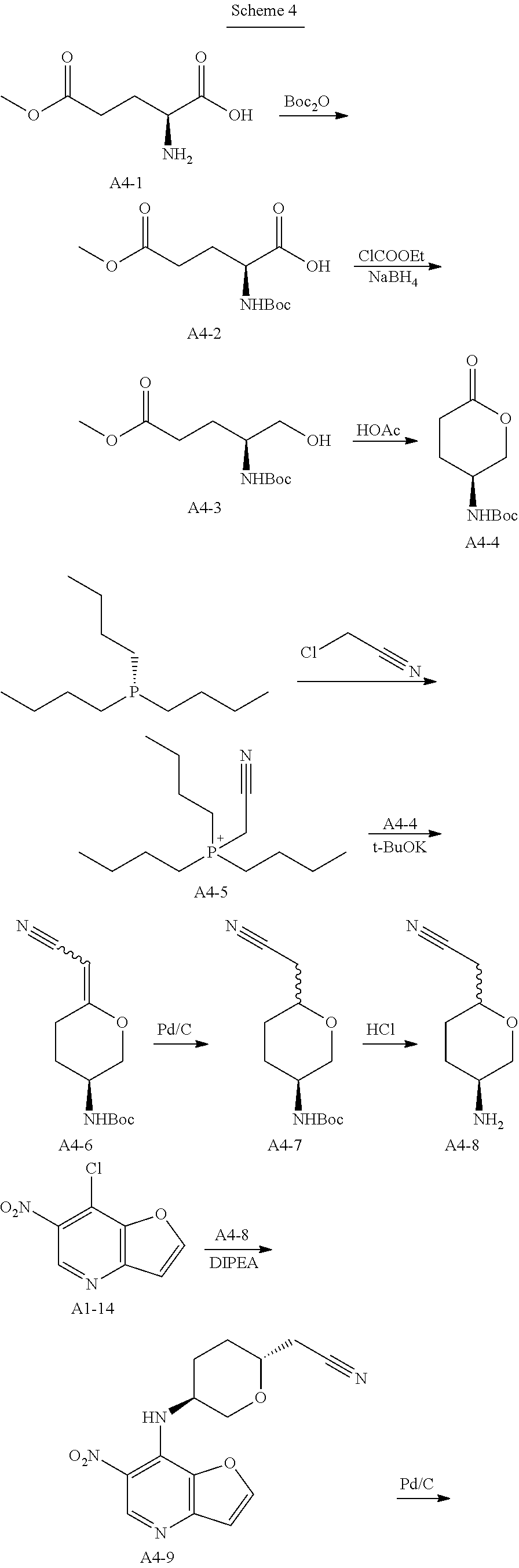
Step 1. In a round bottom flask, triethylamine (188 g, 1.86 mol, 1.0 eq) was added dropwise to a stirred solution of di-tert-butyl dicarbonate (162 g, 0.744 mol, 1.2 eq) and compound A4-1 (100 g, 0.62 mol, 1.0 eq) in water (500 mL) and 1,4-dioxane (500 mL). After stirring for 18 hrs at room temperature, the solution was extracted with MTBE (500 mL*2) and the aqueous phase was cooled on ice and carefully acidified to pH 3 by slow addition of 10% citric acid solution. The urethane was then extracted twice with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, and concentrated to give compound A4-2 as clear viscous oil (180 g, yield 100%). MS-ESI:[M+1]+: 262.1
Step 2. A solution of compound A4-2 (40 g, 0.153 mmol, 1.0 eq) in THF (600 mL) was treated with 4-methylmorpholine (17 g, 0.168, 1.1 eq) at room temperature. The resulting mixture was cooled to 0° C. before being treated with isobutyl chloroformate (22.7 g, 0.166 mmol, 1.08 eq) dropwise. The resulting reaction mixture was stirred at 0° C. for an addition 20 mins before being filtered and washed with THF. Then the clear filtrate solution was cooed to 0° C., and treated with a solution of NaBH4 (11.2 g, 0.295 mol, 1.93 eq) in water (100 mL). The resulting mixture was stirred overnight at room temperature, and then quenched with an aqueous HCl solution (1.0 mol/L,200 mL) dropwise, The mixture was extracted with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated to give compound A4-3 as a yellow oil (25 g, yield 66%). MS-ESI:[M+1]+: 248.1
Step 3. A solution of compound of A4-3 (25 g, 0.1 mol, 1.0 eq) in toluene (300 mL) and acetic acid (150 mL) was heated to reflux for 5 hrs and then cooled, concentrated under vacuum. The residual was added saturated sodium bicarbonate solution to pH 7-8 in ice-bath. Then the mixture was extracted three times with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated and recrystallized by ethyl acetate and PE to give compound A4-4 as a white powder (8.0 g, yield 37.2%). GC-MS: 215
Step 4. A solution of tributyl phosphine (72.9 g, 0.36 mol, 1.0 eq) in nitromethane (500 mL), was added dropwise chloroacetonitrile (27.2 g, 0.36 mol, 1.0 eq) in nitrogen atmosphere. The resulting reaction mixture was stirred for 16 hrs at room temperature, then concentrated. The residual oil solidified when a small amount of ethyl acetate was added. The solid was recrystallized by ethyl acetate and DCM to afford compound A4-5 as a white powder (95 g, yield 95%).
Step 5. To a solution of dry compound A4-5 (8.3 g, 30 mmol, 3.0 eq) in N,N-dimethylacetamide (30 mL) in nitrogen atmosphere, was added solid Potassium tert-butoxide (3.1 g, 28 mmol, 2.8 eq) in portions at 0° C. The resulting mixture was gradually warmed to 30° C. and stirred for 2 hrs. The resulting ylide solution was then treated with compound A4-4 (2.15 g, 10 mmol, 1.0 eq), and stirred overnight at 70° C. After cooled to room temperature, the resulting slurry was poured into the mixture of ice-water (100 mL) and saturated sodium bicarbonate solution (100 mL). The mixture was extracted twice with ethyl acetate, and the combined extracts was washed three times with brine, dried over anhydrous sodium sulfate, concentrated to give compound A4-6 as yellow oil without purification (7.5 g, yield 100%). MS-ESI:[M+1]+: 239.1
Step 6. To a solution of compound A4-6 (7.5 g, 10 mmol, 1.0 eq) in methanol (200 mL), was added 10% Pd/C (0.5 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and purified by silica gel column chromatography to give compound A4-7 as off-white powder (1.6 g, yield 66.7%). MS-ESI:[M+1]+: 241.1
Step 7. To a solution of compound A4-7 (1.6 g, 6.67 mmol, 1.0 eq) in DCM (20 mL), was added TFA (10 g, 88.5 mmol, 13.2 eq). The reaction mixture was stirred for 2 hrs at room temperature until TLC showed the reaction was complete, then concentrated under vacuum. Water (20 mL) was added and the solution was treated with aqueous sodium hydroxide solution (4 mol/L) to pH 10. Then the aqueous phase was extracted six times with DCM/methanol (10/1). The combined extracts was dried over anhydrous sodium sulfate, concentrated to give compound A4-8 as light-brown oil (950 mg, yield 100%). MS-ESI:[M+1]+: 141.1
Step 8. To a solution of compound A1-14 (prepared as step 4 to 12 in example 1) (600 mg, 3.0 mmol, 1.0 eq) in n-butanol (15 mL), was added compound A4-8 (950 mg, 6.7 mmol, 2.26 eq) and DIPEA (1.36 g, 10.5 mmol, 3.5 eq). The reaction mixture was stirred for 1 hr at 135° C., concentrated and purified by silica gel column chromatography to give compound A4-9 (2R,5S) as light-yellow powder (254 mg, yield 28.0%).MS-ESI: [M+1]+: 303.1.
1H NMR (300 MHz, d6-DMSO): 9.063 (s, 1H), 8.503 (d, 1H), 9.326 (d, 1H), 7.176 (d, 1H), 4.431-4.513 (m, 1H), 4.128-4.156 (m, 1H), 3.633-3.659 (m, 1H), 3.448-3.518 (m, 1H), 2.775-2.841 (m, 2H), 2.205-2.312 (m, 1H), 1.829-1.859 (m, 2H), 1.501-1.521 (m, 1H).
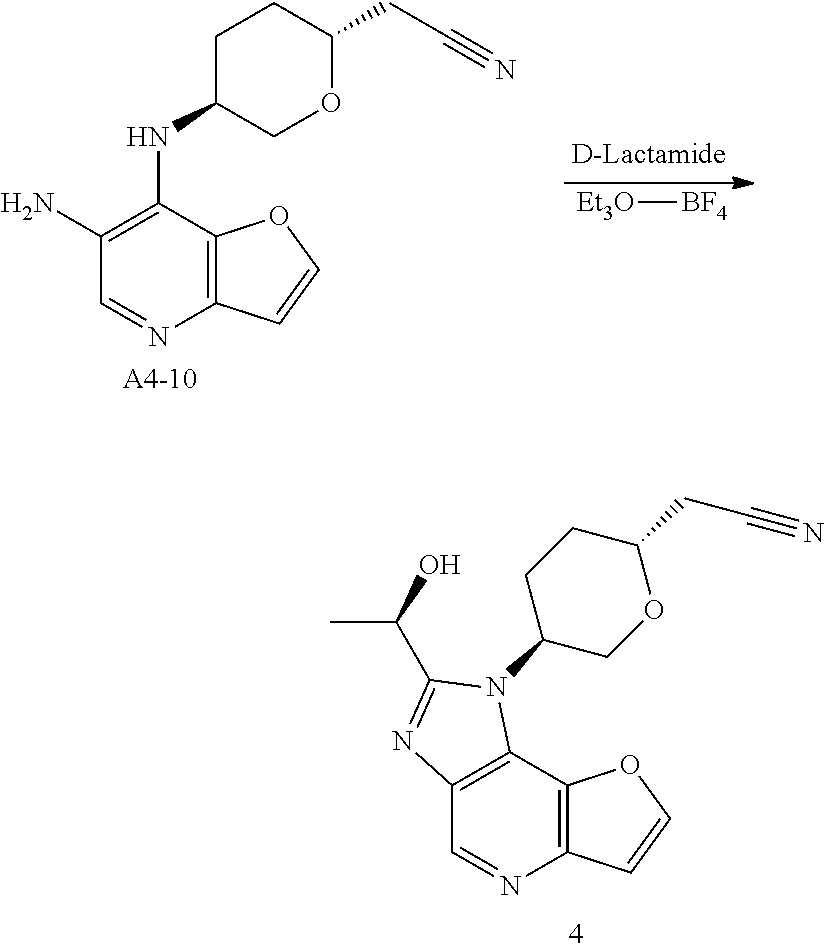
Step 9. To a solution of compound A4-9 (254 g, 0.84 mmol, 1.0 eq) in methanol (20 mL), was added 10% Pd/C (0.15 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and compound A4-10 was obtained as yellow oil (230 mg, yield 100%). MS-ESI:[M+1]+: 273.1
Step 10. A solution of D-Lactamide (388 mg, 4.2 mmol, 5.0 eq) and Et3O—BF4 (1.3 g, 6.72 mmol, 8.0 eq) in THF (10 mL) was stirred for 30 mins at room temperature in nitrogen atmosphere. Then the above solution was added to the mixture of compound A4-10 (230 mg, 0.84 mmol, 1.0 eq) in ethanol (10 mL). After stirring for 3 hrs at 85° C. until HPLC showed the reaction was complete, the mixture was concentrated, added water and extracted four times with ethyl acetate. The organic phases was discarded and the aqueous phase was treated with saturated sodium bicarbonate solution to pH 8, extracted twice with ethyl acetate. The second organic phases was dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give the title compound as light-yellow powder (120 mg, yield 43.8%). MS-ESI: [M+1]+: 327.6,
1H NMR (300 MHz, CDCl3): 9.039 (s, 1H), 7.939 (d, 1H), 7.196 (d, 1H), 5.235-5.336 (m, 1H), 4.806-4.973 (m, 1H), 4.403-4.483 (t, 1H), 4.096-6.116 (m, 2H), 2.700-2.807 (m, 4H), 2.105-2.312 (m, 2H), 1.830-1.852 (d, 3H).
SYN
US2022227777
https://patents.google.com/patent/US20220227777A1
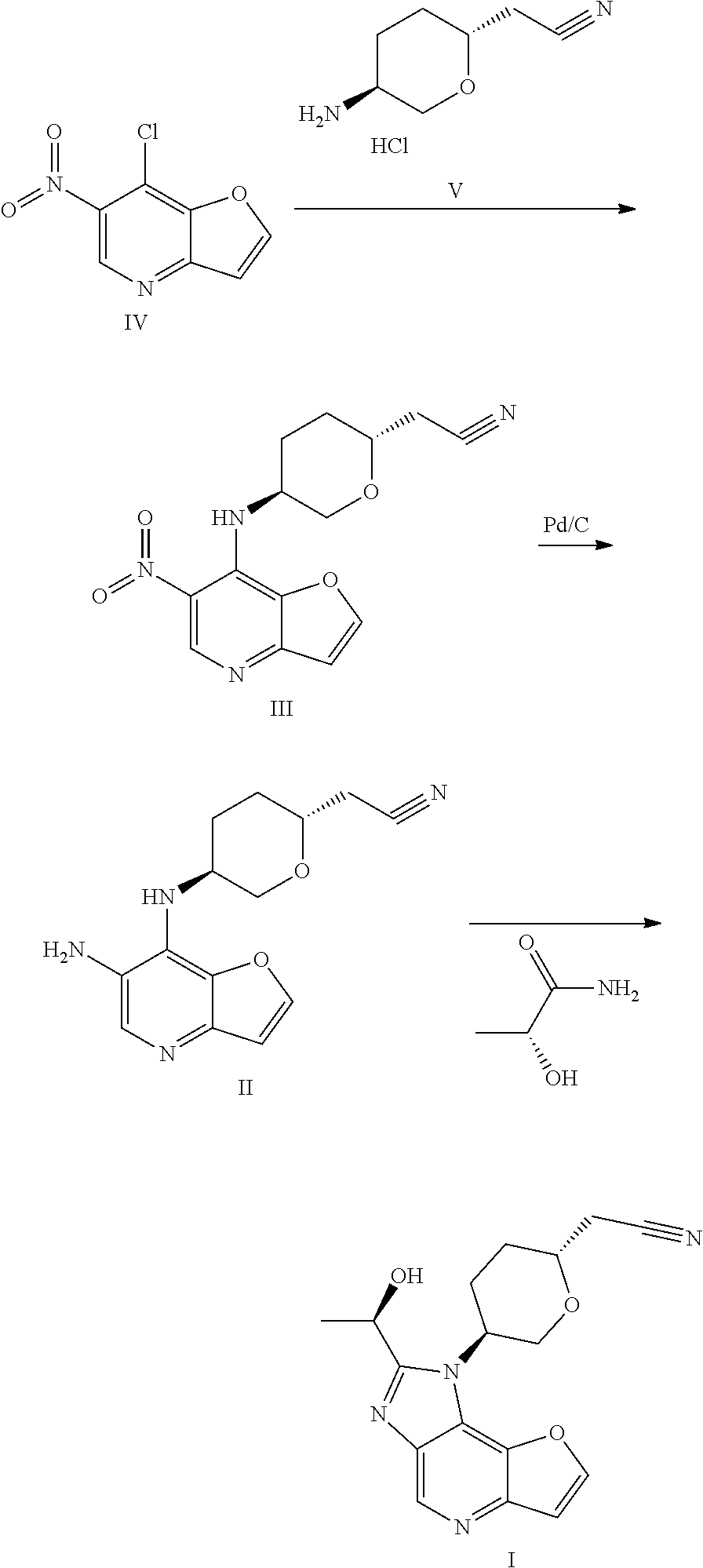
International patent application WO2018067422A1 discloses 1H-furo[3,2-b]imidazo[4,5-d]pyridine derivatives as selective JAK1 kinase inhibitors and preparation methods thereof, wherein compound I and its preparation method is disclosed.
Preparation of a Compound of Formula I
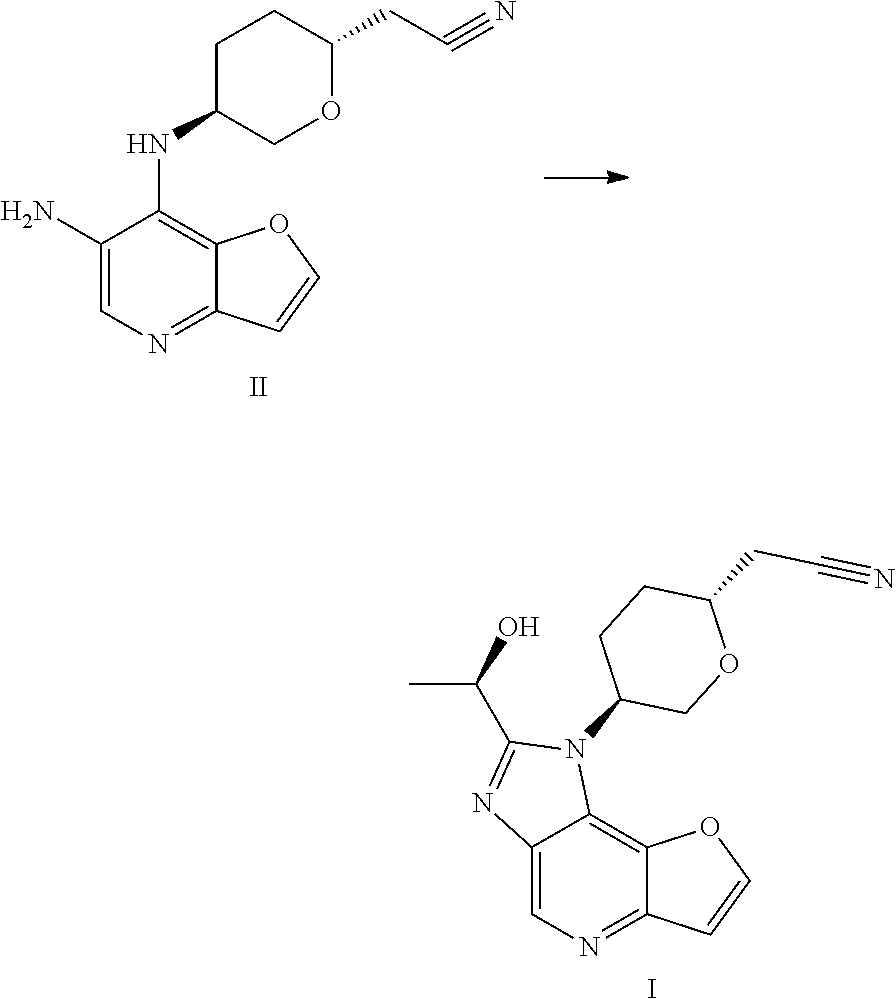
- [0204]THF (60 mL, 12 V), (R)-lactamide (6.6 g, 4.0 eq) and Et3O—BF4 (13.9 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (5.0 g, 1.0 eq) and ethanol (80 mL, 16 V) were added to another 250 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 with a syringe dropwise within 10-20 minutes; the system was heated to 85±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 2 hours; the system was cooled to room temperature; the reaction liquid was concentrated with a rotary evaporator until there was basically no fraction flowing out; 1M HCl (80 mL) was added to the residual concentrated liquid, the pH was about 1 (determined with a pH test paper); the system was extracted four times with DCM (50 mL×4); the pH of the aqueous phase was adjusted to 7-8 with saturated sodium bicarbonate solution; the system was stirred at room temperature for 0.5 hour, then was filtered, the filter cake was washed with water (60 mL) and EA (10 mL), respectively; the filter cake was dried under vacuum at 50° C. for 16 hours; 4.3 g of faint yellow solid was obtained, with a purity of 95.0%; the solid was dissolved with methanol (30 mL); 4.1 g of silicon based metal eliminator and 1.0 g of activated carbon were added, the system was heated to 50° C. and stirred for 1 hour, then was cooled, filtered, washed with methanol (30 mL); the filtrate was concentrated with rotary evaporator until there was basically no fraction flowing out; methanol (10 mL) and MTBE (25 mL) were added to the residue, the system was heated to 50° C., and was stirred for 0.5 hour, then was cooled, the system was cooled to 10±5° C. and stirred for 0.5 hour; filtered, the filter cake was washed with MTBE (25 mL); the filter cake was dried under vacuum at 50° C. for 16 hours, 3.2 g of faint yellow solid was obtained, with a purity of 97.9%.
- [0205]MS-ESI: [M+1]+: 327.6
- [0206]1H NMR (400 MHz, CDCl3): 8.988 (s, 1H), 7.922 (d, 1H), 7.175 (d, 1H), 5.200-5.265 (m, 1H), 4.859-4.942 (m, 1H), 4.350-4.406 (t, 1H), 4.020-4.108 (m, 2H), 3.067 (d, 1H), 2.619-2.779 (m, 3H), 2.108-2.269 (m, 2H), 1.790-1.895 (m, 3H).
- [0207]THF (650 mL, 12 V), (R)-lactamide (70.6 g, 4.0 eq) and Et3O—BF4 (150.6 g, 4.0 eq) were added to a 1000 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (54 g, 1.0 eq) and ethanol (860 mL, 16 V) were added to another 2000 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were slowly added to three-necked flask #2 dropwise within 1 hour; the system was heated to 85±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 2 hours; the system was cooled to room temperature; the reaction liquid was concentrated with a rotary evaporator until there was basically no fraction flowing out; 1M HCl (450 mL) was added to the residual concentrated liquid, the pH was about 1 (determined with a pH test paper); the system was extracted four times with DCM (270 mL×4); the pH of the aqueous phase was adjusted to 7-8 with saturated sodium bicarbonate solution; the system was stirred at room temperature for 0.5 hour, then was filtered, the filter cake was washed with water (540 mL); MTBE (270 mL) was added to the filter cake, the system was stirred at room temperature for 0.5 hour, filtered, the filter cake was washed with MTBE (108 mL); the filter cake was dried under vacuum at 50° C. for 16 hours; 49.2 g of light yellow solid was obtained, with an HPLC purity of 94.2%; the solid was dissolved with methanol (380 mL); silicon based metal eliminator (44 g) and activated carbon (5.4 g) were added, the system was heated to 50° C. and stirred for 1 hour, then was cooled, filtered, washed with methanol (430 mL); the filtrate was concentrated with a rotary evaporator to (80-110 mL, 1.5 V-2 V); MTBE (540 mL) was added to the residue, the system was heated to 50° C., and was stirred for 1 hour, then was cooled to 10±5° C. and stirred for 0.5 hour; filtered, the filter cake was washed with MTBE (270 mL); 42.4 g of filter cake was obtained, with an HPLC purity of 96.9%; the filter cake was dried under vacuum at 50° C. for 16 hours, 41.0 g of light yellow solid was obtained, with an HPLC purity of 96.7%, a yield of 63.3%.
- [0208]Purification of a Compound of Formula I:
- [0209]A compound of formula I (41 g) was dissolved with methanol; silica gel (50 g) was added to the solution, the system was concentrated to dryness for later use; silica gel (200 g) was added to the chromatographic column, the column was compacted with an air pump; a compound of formula I mixed with silica gel was added to the chromatographic column, the column was compacted with an air pump; the chromatographic column was eluted with an eluent (VMeOH:VDCM=1:100-1:30); qualified components were collected, concentrated to dryness; the product was dried under vacuum at 50° C. for 16 hours; 36 g of off-white solid was obtained, with an HPLC purity of 98.5%.
- [0210]The MS-ESI and 1H NMR data are consistent with example 21.
- [0211]THF (60 mL, 6 V), (R)-lactamide (13.2 g, 4.0 eq) and Et3O—BF4 (27.9 g, 4.0 eq) were added to a 100 mL three-necked flask #1, the system was stirred; the materials in #1 were stirred under nitrogen protection for later use; a compound of formula II (10 g, 1.0 eq) and ethanol (100 mL, 10 V) were added to another 250 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were slowly added to three-necked flask #2 dropwise within 20 minutes; the system was heated to 80±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 0.5 hour; the system was cooled to room temperature 20-30° C.; the reaction liquid was concentrated to about 50-80 mL with a rotary evaporator between 30-40° C.; water (100 mL, 10 V) was added to the system, then the system was concentrated with a rotary evaporator between 30-40° C. until there was basically no fraction flowing out; the system was cooled to 20-30° C.; the temperature of the system was controlled at 20-30° C., 12M HCl (5.5 g) was used to adjust the pH of the system to 2-3, the system was extracted with ethyl acetate (50 mL×2, 5V×2); the organic phase was discarded, and the aqueous phase was transferred to a flask; the temperature of the system was controlled at 20-30° C., the pH of the system was adjusted to 8-9 with saturated potassium carbonate solution (23 g); the temperature of the system was controlled at 20-25° C., the system was stirred for 2 hours, then was filtered, the filter cake was washed with water (50 mL) and MTBE (50 mL); the filter cake was dried with an air blower at 50° C. for 24 hours, 18 g of earth yellow solid was obtained, with an HPLC purity of 93.5%.
- [0212]The MS-ESI and 1H NMR data are consistent with example 21.
- [0213]THF (120 mL, 12 V), (R)-lactamide (13.2 g, 4.0 eq) and Et3O—BF4 (27.8 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in #1 were stirred under nitrogen protection for later use; a compound of formula II (10 g, 1.0 eq) and ethanol (140 mL, 14 V) were added to another 500 mL three-necked flask #2; the system was heated to 40-45° C. (internal temperature) under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 dropwise within 1 hour; the system was maintained at 40-45° C. (internal temperature) under nitrogen protection for reacting for 4.5 hours; the system was cooled to room temperature, and water (20 mL, 2V) was added; the system was concentrated with a rotary evaporator at 30-40° C. until there was basically no fraction flowing out; the system was cooled to 20-30° C.; the temperature of the system was controlled at 20-30° C., 12M HCl (3 mL) was used to adjust the pH of the system to 2-3, the system was extracted with ethyl acetate (50 mL×2, 5V×2); the organic phase was discarded, and the aqueous phase was transferred to a flask; the temperature of the system was controlled at 20-30° C., the pH of the system was adjusted to 8-9 with 50% potassium carbonate solution (15 mL); the temperature of the system was controlled at 20-25° C., the system was stirred for 2 hours, then was filtered, the filter cake was washed with water (50 mL) and acetone (50 mL); the crude product was triturated and stirred with water (50 mL) at 20-25° C. for 1 hour; the system was filtered, the filter cake was washed with water (50 mL) and acetone (50 mL); the filter cake was dried with an air blower at 50° C. for 24 hours, 17.8 g of khaki solid was obtained, with an HPLC purity of 95.3%.
- [0214]The MS-ESI and 1H NMR data are consistent with example 21.
- [0215]THF (60 mL, 12 V), (R)-lactamide (6.6 g, 4.0 eq) and Et3O—BF4 (13.9 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (5 g, 1.0 eq) and ethanol (70 mL, 14 V) were added to another 250 mL three-necked flask #2; the system was heated to 40-45° C. (internal temperature) under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 dropwise within 20 minutes; the system was maintained at 40-45° C. (internal temperature) under nitrogen protection for reacting for 3 hours; the system was cooled to room temperature and was filtered, the filter cake was washed with THF (10 mL); water (10 mL, 2V) was added to the filtrate; the filtrate was concentrated with a rotary evaporator to 10-20 mL (2V-4V), the concentrated residue was exchanged with ethyl acetate (25 mL×2) and concentrated to 10-20 mL (2V-4V); water (50 mL, 10V) was added to the concentrated residue; the internal temperature was controlled at 20-25° C., 12M HCl (4.1 g) was used to adjust the pH of the system to 1-2; activated carbon (0.5 g) was added to the system, and the system was stirred at room temperature for 2 hours, and was filtered, the filter cake was washed with water (10 mL) and 1M HCl (10 mL); the combined filtrate was extracted with ethyl acetate (25 mL×2), the organic phase was discarded; the internal temperature was controlled at 20-25° C., the pH of the system was adjusted to 9-10 with saturated potassium carbonate solution (15 g); the internal temperature was controlled at 15-20° C., the system was stirred for 1 hour, and was filtered, the filter cake was washed with water (10 mL); the filter cake was triturated with acetone aqueous solution (50 mL, V/V=1:1) for 1 hour; the system was filtered, the filter cake was washed with acetone aqueous solution (10 mL, V/V=1:1); the filter cake was dried with an air blower at 50° C. for 24 hours; 5.0 g of pale gray solid was obtained, with an HPLC purity of 95.6%, and a yield of 83.5%;
- [0216]Purification of a Compound of Formula I:
- [0217]5.0 g of the obtained solid and methanol (40 mL) were added to a flask, and were stirred for 10 minutes at room temperature, the materials were basically dissolved and the solution was clear; activated carbon (0.5 g) and silica gel (4.0 g) were added to the system; the system was heated to 50-55° C., the temperature was maintained and the system was stirred for 2 hours, then was filtered with silica gel (5 g), the filter cake was washed with methanol (50 mL); the filtrate was concentrated with a rotary evaporator to 5-10 mL; MTBE (50 mL) was added to the concentrated residue; the system was heated to reflux, and was allowed for reflux for 1 hour; the system was cooled to 5-10° C., the temperature was maintained and the system was stirred for 1 hour and was filtered, the filter cake was washed with MTBE; the filter cake was dried with a drying oven under vacuum at 50° C. for 16 hours; 3.0 g of off-white solid was obtained, with a yield of 60% and a purity of 97.9%; the filtrate was concentrated to dryness to obtain 1.4 g of yellow solid.
- [0218]The MS-ESI and 1H NMR data are consistent with example 21.
PAT
- NEW SELECTIVE JAK1 INHIBITORS AND THEIR USEPublication Number: HR-P20211965-T1Priority Date: 2016-10-03
- Novel JAK1 selective inhibitors and uses thereofPublication Number: KR-102399848-B1Priority Date: 2016-10-03Grant Date: 2022-05-19
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-113214278-BPriority Date: 2016-10-03Grant Date: 2022-10-28
- Jak1 selective inhibitors and uses thereofPublication Number: US-RE49834-EPriority Date: 2016-10-03Grant Date: 2024-02-13
- Novel jak1 selective inhibitors and uses thereofPublication Number: US-2019256523-A1Priority Date: 2016-10-03
- JAK1 selective inhibitors and uses thereofPublication Number: US-10738060-B2Priority Date: 2016-10-03Grant Date: 2020-08-11
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-108366994-BPriority Date: 2016-10-03Grant Date: 2021-10-01
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-113214278-APriority Date: 2016-10-03
- Novel jak1 selective inhibitors and uses thereofPublication Number: EP-3509591-B1Priority Date: 2016-10-03Grant Date: 2021-11-17
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereof.Publication Number: MX-2024006688-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: US-12195476-B2Priority Date: 2019-06-06Grant Date: 2025-01-14
- Novel jak1 selective inhibitors and uses thereofPublication Number: CA-3039178-A1Priority Date: 2016-10-03
- Novel jak1 selective inhibitors and uses thereofPublication Number: EP-3509591-A1Priority Date: 2016-10-03
- Novel JAK1 selective inhibitors and uses thereofPublication Number: JP-2019537559-APriority Date: 2016-10-03
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248967-A3Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: JP-2023089169-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compounds, crystal forms of furoimidazopyridine compounds and crystal forms of their saltsPublication Number: CN-113906035-BPriority Date: 2019-06-06Grant Date: 2023-11-10
- Synthesis method of furoimidazopyridine compounds, crystal forms of furoimidazopyridine compounds and crystal forms of their saltsPublication Number: CN-117327083-APriority Date: 2019-06-06
- METHOD OF SYNTHESIS OF FUROIMIDAZOPYRIDINE COMPOUND, CRYSTAL FORM OF FUROIMIDAZOPYRIDINE COMPOUND, AND CRYSTAL FORM OF ITS SALT.Publication Number: MX-2024004146-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: US-2022227777-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: AU-2020289149-B2Priority Date: 2019-06-06Grant Date: 2023-05-11
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248966-A2Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248966-A3Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248967-A2Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: WO-2020244348-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: AU-2020289149-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound and crystal form of salt thereofPublication Number: CN-113906035-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-3981771-A1Priority Date: 2019-06-06

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Girocitinib, Janus kinase inhibitor, anti-inflammatory, A0IES9T8GO
Tezepelumab-ekko

(Heavy chain)
QMQLVESGGG VVQPGRSLRL SCAASGFTFR TYGMHWVRQA PGKGLEWVAV IWYDGSNKHY
ADSVKGRFTI TRDNSKNTLN LQMNSLRAED TAVYYCARAP QWELVHEAFD IWGQGTMVTV
SSASTKGPSV FPLAPCSRST SESTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSNFGT QTYTCNVDHK PSNTKVDKTV ERKCCVECPP CPAPPVAGPS
VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVQFNWYV DGVEVHNAKT KPREEQFNST
FRVVSVLTVV HQDWLNGKEY KCKVSNKGLP APIEKTISKT KGQPREPQVY TLPPSREEMT
KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPMLD SDGSFFLYSK LTVDKSRWQQ
GNVFSCSVMH EALHNHYTQK SLSLSPGK
(Light chain)
SYVLTQPPSV SVAPGQTARI TCGGNNLGSK SVHWYQQKPG QAPVLVVYDD SDRPSWIPER
FSGSNSGNTA TLTISRGEAG DEADYYCQVW DSSSDHVVFG GGTKLTVLGQ PKAAPSVTLF
PPSSEELQAN KATLVCLISD FYPGAVTVAW KADSSPVKAG VETTTPSKQS NNKYAASSYL
SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS
(Disulfide bridge: H22-H96, H136-L213, H149-H205, H224-H’224, H225-H’225, H228-H’228, H231-H’231, H262-H322, H368-H426, H’22-H’96, H’136-L’213, H’149-H’205, H’262-H’322, H’368-H’426, L22-L87, L136-L195, L’22-L’87, L’136-L’195)
Tezepelumab-ekko
テゼペルマブ (遺伝子組換え)
| Formula | C6400H9844N1732O1992S52 |
|---|---|
| CAS | 1572943-04-4 |
| Mol weight | 144588.4306 |
PEPTIDE
UD FDA APPROVED, 12/17/2021, To treat severe asthma as an add-on maintenance therapy , Tezspire
Monoclonal antibody
Treatment of asthma and atopic dermatitis
Tezepelumab, sold under the brand name Tezspire, is a human monoclonal antibody used for the treatment of asthma.[4][5]
It blocks thymic stromal lymphopoietin (TSLP),[2] an epithelial cytokine that has been suggested to be critical in the initiation and persistence of airway inflammation.[6]
It was approved for medical use in the United States in December 2021.[2][3]
Medical uses
Tezepelumab is indicated for the add-on maintenance treatment of people aged twelve years and older with severe asthma.[2]
Research
In Phase III trials, tezepelumab demonstrated efficacy compared to placebo for patients with severe, uncontrolled asthma.[7][8]
Structural studies by X-ray crystallography showed that Tezepelumab competes against a critical part of the TSLPR binding site on TSLP.[1]
It is being studied for the treatment of chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE).[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b Verstraete K, Peelman F, Braun H, Lopez J, Van Rompaey D, Dansercoer A, et al. (April 2017). “Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma”. Nature Communications. 8 (1): 14937. Bibcode:2017NatCo…814937V. doi:10.1038/ncomms14937. PMC 5382266. PMID 28368013.
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761224s000lbl.pdf
- ^ Jump up to:a b c “Tezspire (tezepelumab) approved in the US for severe asthma”. AstraZeneca (Press release). 17 December 2021. Retrieved 17 December 2021.
- ^ Marone G, Spadaro G, Braile M, Poto R, Criscuolo G, Pahima H, et al. (November 2019). “Tezepelumab: a novel biological therapy for the treatment of severe uncontrolled asthma”. Expert Opinion on Investigational Drugs. 28 (11): 931–940. doi:10.1080/13543784.2019.1672657. PMID 31549891. S2CID 202746054.
- ^ Matera MG, Rogliani P, Calzetta L, Cazzola M (February 2020). “TSLP Inhibitors for Asthma: Current Status and Future Prospects”. Drugs. 80 (5): 449–458. doi:10.1007/s40265-020-01273-4. PMID 32078149. S2CID 211194472.
- ^ “Tezepelumab granted Breakthrough Therapy Designation by US FDA”. AstraZeneca (Press release). 7 September 2018.
- ^ “Studies found for: Tezepelumab”. ClinicalTrials.Gov. National Library of Medicine, National Institutes of Health, U.S. Department of Health and Human Services.
- ^ Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. (May 2021). “Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma”. New England Journal of Medicine. 384 (19): 1800–09. doi:10.1056/NEJMoa2034975. PMID 33979488. S2CID 234484931.
External links
- “Tezepelumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02054130 for “Study to Evaluate the Efficacy and Safety of MEDI9929 (AMG 157) in Adult Subjects With Inadequately Controlled, Severe Asthma” at ClinicalTrials.gov
- Clinical trial number NCT03347279 for “Study to Evaluate Tezepelumab in Adults & Adolescents With Severe Uncontrolled Asthma (NAVIGATOR)” at ClinicalTrials.gov
| Structural basis for inhibition of TSLP-signaling by Tezepelumab (PDB 5J13)[1] | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | thymic stromal lymphopoietin (TSLP) |
| Clinical data | |
| Trade names | Tezspire |
| Other names | MEDI9929, AMG 157, tezepelumab-ekko |
| License data | US DailyMed: Tezepelumab |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2][3] |
| Identifiers | |
| CAS Number | 1572943-04-4 |
| DrugBank | DB15090 |
| ChemSpider | None |
| UNII | RJ1IW3B4QX |
| KEGG | D11771 |
| Chemical and physical data | |
| Formula | C6400H9844N1732O1992S52 |
| Molar mass | 144590.40 g·mol−1 |
////////////Tezepelumab-ekko, Tezspire, PEPTIDE, APPROVALS 2021, FDA 2021, Monoclonal antibody
, asthma, atopic dermatitis, ANTI INFLAMATORY, テゼペルマブ (遺伝子組換え)

NEW DRUG APPROVALS
ONE TIME
$10.00
LORNOXICAM
LORNOXICAM
chlortenoxicam
- Molecular FormulaC13H10ClN3O4S2
- Average mass371.819 Da
70374-39-9[RN], Chlortenoxicam, CTX, ER09126G7A
2H-thieno[2,3-e]-1,2-thiazine-3-carboxamide, 6-chloro-4-hydroxy-2-methyl-N-2-pyridinyl-, 1,1-dioxide
6233
6-Chlor-4-hydroxy-2-methyl-N-(pyridin-2-yl)-2H-thieno[2,3-e][1,2]thiazin-3-carboxamid-1,1-dioxid
6-Chloro-4-hydroxy-2-methyl-N-(2-pyridinyl)-2H-thieno[2,3-e][1,2]thiazine-3-carboxamide 1,1-dioxide
- Chlortenoxicam, Ro-13-9297
- ATC:M01AC05
- CCRIS 8589
- Ro 13-9297
Lorcam (Taisho Pharmaceutical Co.) / Xafon (Nycomed)LornoxicamCAS Registry Number: 70374-39-9
CAS Name: 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e]-1,2-thiazine-3-carboxamide 1,1-dioxide
Additional Names: 6-chloro-4-hydroxy-2-methyl-3-(2-pyridylcarbamoyl)-2H-thieno[2,3-e]-1,2-thiazine-1,1-dioxide; chlortenoxicam
Manufacturers’ Codes: Ro-13-9297; TS-110
Trademarks: Xefo (Nycomed)
Molecular Formula: C13H10ClN3O4S2
Molecular Weight: 371.82
Percent Composition: C 41.99%, H 2.71%, Cl 9.53%, N 11.30%, O 17.21%, S 17.25%
Literature References: Cyclooxygenase inhibitor; structurally similar to tenoxicam, q.v.
Prepn: R. Pfister et al.,DE2838851; eidem,US4180662 (both 1979 to Hoffmann-La Roche).Clinical pharmacokinetics: S. I. Ankier et al.,Postgrad. Med. J.64, 752 (1988). Symposium on pharmacology and clinical experience: ibid.66, Suppl. 4, S1-S50 (1990). Overview of pharmacology and safety assessment: T. P. Pruss et al.,ibid. S18.
Properties: Orange to yellow crystals, mp 225-230° (dec). pKa2 4.7. uv max: 371 nm. Partition coefficient (n-octanol/pH 7.4 buffer): 1.8. LD50 orally in mice, rats, rabbits, dogs, monkeys: >10 mg/kg (Pruss).
Melting point: mp 225-230° (dec)
pKa: pKa2 4.7
Log P: Partition coefficient (n-octanol/pH 7.4 buffer): 1.8
Absorption maximum: uv max: 371 nm
Toxicity data: LD50 orally in mice, rats, rabbits, dogs, monkeys: >10 mg/kg (Pruss)
Therap-Cat: Anti-inflammatory; analgesic.
Keywords: Analgesic (Non-Narcotic); Anti-inflammatory (Nonsteroidal); Thiazinecarboxamides.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 504-29-0 | C5H6N2 | 2-aminopyridine | 2-Pyridinamine |
| 7790-94-5 | ClHO3S | chlorosulfonic acid | Chlorosulfuric acid |
| 56946-84-0 | C5H5Cl2NO2S2 | 2,5-dichloro-N-methyl-3-thiophenesulfonamide | 3-Thiophenesulfonamide, 2,5-dichloro-N-methyl- |
| 3172-52-9 | C4H2Cl2S | 2,5-dichlorothiophene | Thiophene, 2,5-dichloro- |
SYN
Synthesis of lornoxicam (DE2838851)

The sulfonation of 2,5-dichlorothiophene (I) with ClSO3H -SOCl2 gives 2,5-dichlorothiophene-3-sulfonic acid chloride (II), which by reaction with methylamine in CHCl3 yields the corresponding methylamide (III). The carboxylation of (III) with butyllithium and CO2 in ether affords 5-chloro-3-(N-methylsulfamoyl)thiophene-2-carboxylic acid (IV), which is esterified with PCl5 and methanol to the methyl ester (V). The condensation of (V) with methyl iodoacetate (VI) by means of NaH in DMF gives 5-chloro-3-[N-(methoxycarbonylmethyl)-N-methylsulfamoyl]thiophene-2-carboxylic acid methyl ester (VII), which is cyclized with sodium methoxide in methanol yielding 6-chloro-4-hydroxy-2-methyl-2H-thieno[2,3-e]-1,2-thiazine-3-carboxylic acid methyl ester 1,1-dioxide (VIII). Finally, this compound is treated with 2-aminopyridine (IX) in refluxing xylene.
Lornoxicam is an NSAID indicated in the treatment of mild to moderate pain, as well as rheumatoid arthritis and osteoarthritis.
Lornoxicam, also known as chlortenoxicam, is a nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class with analgesic (pain relieving), anti-inflammatory and antipyretic (fever reducing) properties. It is available in oral and parenteral formulations.
It was patented in 1977 and approved for medical use in 1997.[1] Brand names include Xefo and Xefocam among others.
Lornoxicam (chlortenoxicam) is a new nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class with analgesic, anti-inflammatory and antipyretic properties. Lornoxicam differs from other oxicam compounds in its potent inhibition of prostaglandin biosynthesis, a property that explains the particularly pronounced efficacy of the drug. Lornoxicam is approved for use in Japan.
Medical uses
Lornoxicam is used for the treatment of various types of pain, especially resulting from inflammatory diseases of the joints, osteoarthritis, surgery, sciatica, and other inflammations.[2]

Contraindications
The drug is contraindicated in patients who must not take other NSAIDs, possible reasons including salicylate sensitivity, gastrointestinal bleeding and bleeding disorders, and severe impairment of heart, liver or kidney function. Lornoxicam is not recommended during pregnancy and breastfeeding and is contraindicated during the last third of pregnancy.[2]
Adverse effects
Lornoxicam has side effects similar to other NSAIDs, most commonly mild ones like gastrointestinal disorders (nausea and diarrhea) and headache. Severe but seldom side effects include bleeding, bronchospasms and the extremely rare Stevens–Johnson syndrome.[2]
Interactions
Interactions with other drugs are typical of NSAIDs. Combination with vitamin K antagonists like warfarin increases the risk of bleeding. Combination with ciclosporin can lead to reduced kidney function, and to acute kidney injury in rare cases. Lornoxicam can also increase the adverse effects of lithium, methotrexate and digoxin and its derivatives. The effect of diuretics, ACE inhibitors and angiotensin II receptor antagonists can be reduced, but this is only relevant in patients with special risks like heart failure. As with piroxicam, cimetidine can increase plasma levels but is unlikely to cause relevant interactions.[3]
PAPER
https://www.mdpi.com/2218-0532/71/4/303

PATENT
CN 113480561
The present invention relates to the prepn. of high purity loroxicam. In particular, the prepn. method comprises a step of taking 6-chloro-4-hydroxy-2-methyl-2H-thieno[2,3-e]-1,2-Me thiazinecarboxylate-1,1-dioxide and 2-amino pyridine is used as the raw material and xylene is used as the solvent undergoes distn. reaction with solid acid catalyst, mixed gas obtained by the distn. reaction is condensed to obtain a condensate and solid acid catalyst is used to adsorb methanol in the condensate and the adsorbed condensate is recycled, filtering and refining to obtain loroxicam. The present inventive method distills out the methanol produced by the reaction to promote the pos. progress of the reaction and then catalyzes the absorption of methanol by H2SO4/MxOy solid super acid, so that the xylene returned to the reaction system does not contain methanol, which reduces the coking of the reaction, thereby improving product quality and yield. The prepd. lornoxicam has high purity, which can reach more than 99.9%, reduces the amt. of solvent and also suitable for industrial prodn.
PATENT
CN 112592356
The present invention relates to the prepn. of lornoxicam. In particular, the prepn. method comprises a step of taking 6-chloro-4-hydroxy-2-methyl-2-H-thieno[2,3-e]-1,2-thiazidecarboxylic acid Me ester-1,1-dioxide and 2-aminopyridine as raw materials, xylene is used as solvent, adding stabilizer, and carrying out aminolysis reaction, the solvent was removed by concn. under reduced pressure, adding org. solvent to make the slurry, filtering and refining to obtain lornoxicam. The inventive method uses p-toluene sulfonic acid as a stabilizer, while lowering the reaction temp., it promotes the reaction to proceed forward, and improve the product quality and yield; at the same time reduce the amt. of industrial solvents, the post-treatment process is optimized and the cost of the three wastes treatment is reduced.
PATENT
IN 2014CH02116
Example: 1Preparation of 6-chloro-4-hydroxy-l,l-dioxo-l,2-dihydro-lX6-thieno [2,3-e][l,2] thiazine-3-carboxylic acid methyl ester To the mixture of methanol ( 1000 ml) and 5-chloro-3-(methoxy carbonyl methyl sulfamoyl)-thiophene-2-carboxylicacid methyl ester ( 100 g ,0.305 moles), added sodium methoxide solution (200 ml ) at 25-30°C over a period of 30-45 min. The resulting mixture was stirred for 60 min at same temperature; allowed to heat at 65-75°C and stirred for 10-12 hrs. After completion of reaction, methanol was distilled out under reduced pressure to obtained titled residual product which is directly used to next step
(Example-2). Example: – 2:Preparation of 6-chloro-4-hydroxy-2-methyl-l,l-dioxo-l,2-dihydro-U6- thieno[2,3-e][l,2] thiazine-3-carboxylic acid methyl ester 6-chloro-4-hydroxy-1,1 -dioxo-1,2-dihydro-1 X,6-thieno [2,3-e][ 1,2] thiazine-3-carboxylic acid methyl ester was suspended in DM water (500 ml) and cooled to 10-15° C, dimethyl sulphate ( 70 g) was slowly added to the mixture at 10-15°C in 30 min. The reaction mixture was raised to 25-30°C and maintained for 2-3 hours at same temperature. After completion of reaction, mixture was cooled to 10-15°C, methylene dichloride (1600 ml) was added, reaction mixture pH was adjust to 1.0 -2.0 with hydrochloric acid at 10-15° C, stir reaction mixture to separate the layers. The methylene dichloride layer was distilled out completely at below 30°C to get an residue, followed by addition of methanol (60 ml) and distilled out methanol completely under vacuum at below 50°C to get an residue; further it was crystallized by addition of methanol 190 ml and stirred for 30 min at 50-55°C; cooled the reaction mixture at 25-30°C and stirred for 60 min at same temperature. The resultant solid was filtered, washed with methanol (40 ml) and dried at 50-55°C for 4 – 6 hrs to obtain the titled product
Example: 3Preparation of 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e]-l,2-thiazine-3-carboxamide 1,1-dioxide (Lornoxicam) 6-chloro-4-hydroxy-2-methyl-l, 1 -dioxo-1,2-dihydro-l X.6-thieno[2,3-e][l ,2] thiazine-3-carboxylic acid methyl ester ( 50 g 0.161 moles) was suspended in O-xylene (500 ml) and allow to stirred at 70-75°C to obtained clear solution. To this clear solution slowly added the mixture of THF ( 50 ml) solution of 2-Amino pyridine ( 14 g ) and ethyl magnesium bromide 2 molar solution (100 ml) at 70-75°C and allow to stirred for 3-4 hrs at same temperature. After completion of reaction, the dilute hydrochloric acid was added to the mixture at 10-15°C and stirred for 60 min. The resultant solid was filtered, washed with water (100 ml) to obtain crude Lornoxicam.
Example: 4Preparation of 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e)-l,2-thiazine-3-carboxamide 1,1-dioxide (Lornoxicam) 6-chloro-4-hydroxy-2-methyl-l,l-dioxo-l,2-dihydro-R6-thieno[2,3-e][l,2] thiazine-3-carboxylic acid methyl ester ( 50 g 0.161 moles) was suspended in O-xylene (500 ml) and allow to stirred at 70-75°C to obtained clear solution. To this clear solution slowly added the mixture of THF ( 50 ml) solution of 2-Amino pyridine ( 14 g ) and isopropyl magnesium bromide 2 molar solution (100 ml) at 70-75°C and allow to stirred for 3-4 hrs at same temperature. After completion of reaction, the dilute hydrochloric acid was added to the mixture at 10-15°C and stirred for 60 min. The resultant solid was filtered, washed with water (100 ml) to obtain crude Lornoxicam.
Example: 5Purification of Lornoxicam.The crude Lornoxicam was suspended in methanol (500 ml) and cooled to 5-10°C, resulting suspension was basified to pH 11-13 by using sodium hydroxide solution to get clear solution; followed by filtration through hyflo bed; the obtain filtrate was acidified to pH 4.5 – 5.0 with dil. HC1 (1:1) at 5-10°C; stirred the slurry for 30 min. at 5-10°C. The resultant solid was filtered, washed with DM water (100 ml) and dried at 50-55°C to obtained pure Lornoxicam.
PATENT
.EXAMPLES:Preparation of Lornoxicam crudeExample ITo 1200ml o-xylene, 20gm Methyl-6-chloro-4-hydroxy-2-methyl-2//-thieno [2, 3-e] [1, 2] thiazine-3- carboxyate 1,1-dioxide and 6.44gm 2-aminopyridine was added. The reaction mass was stirred under nitrogen atmosphere. Temperature was raised to 140-145°C and maintained for 6hrs. The reaction mass was cooled to 30-35°C and nitrogen was removed. Reaction mass was further stirred for 3hrs- Filtered and washed twice with 50ml of o-xylene. 19.8gm of crude Lornoxicam was obtained. Purification of Lornoxicam crude
Example 219.8gm of crude Lornoxicam was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around Ihr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, til! the reaction mass reached pH of 2-3, and then stirred for around I hi*. The reaction mass was cooled to room temperature, filtered, and then washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 19.1 gm of pure Lornoxicam was obtained. (HPLC purity- 99.95%)
Example 3!7.9gm of crude Lornoxicam (prepared as per example 1) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around Ihr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C till the reaction mass reached pH of 2-3, and then stirred for around Ihr. The reaction mass was cooled to room temperature, filtered and then washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 17.2 gm of pure Lornoxicam was obtained. (HPLC purity- 99.9%) clear solution and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around lhr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, till the reaction mass reached pH of 2-3, and then stirred for around lhr. The reaction mass was cooled to 30-35°C, filtered and then washed with 1:1 mixture of isopropyl alcohol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 4.85 gm of pure Lornoxicam was obtained. (HPLC purity- 99.8%)
Example 55 gm of crude Lornoxicam (prepared as per example 1) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and ethanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around lhr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, til! the reaction mass reached pH of 2-3 and then stirred for around lhr. The reaction mass was cooled to 30-35°C and filtered, washed with 1:1 mixture of ethanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 4.8 gm of pure Lornoxicam was obtained. (HPLC purity- 99.8%)
Example 619.4 gm of crude Lornoxicam (prepared as per example I) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 20% activated charcoal was further added. The reaction mass was stirred for around lhr at room temperature followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added till the reaction mass reached pH of 2-3 and then stirred for around 1 hr. The reaction mass was * filtered and washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 18.9 gm of pure Lornoxicam was obtained. (HPLC purity- 99.3%).
PATENT
https://www.sciencedirect.com/science/article/abs/pii/S0968089603007624?via%
PATENT
https://patents.google.com/patent/WO2002000167A2/en
References
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 519. ISBN 9783527607495.
- ^ Jump up to:a b c Haberfeld H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. Xefo Filmtabletten. ISBN 978-3-85200-196-8.
- ^ Klopp T, ed. (2010). Arzneimittel-Interaktionen (in German) (2010/2011 ed.). Arbeitsgemeinschaft für Pharmazeutische Information. ISBN 978-3-85200-207-1.
| Clinical data | |
|---|---|
| Trade names | Xefo, Xefocam others |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category | Not recommended; contraindicated in months 7–9 |
| Routes of administration | By mouth, parenteral |
| ATC code | M01AC05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 90–100% |
| Protein binding | 99% |
| Metabolism | CYP2C9 |
| Elimination half-life | 3–4 hours |
| Excretion | 2/3 liver, 1/3 kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 70374-39-9 |
| PubChem CID | 5282204 |
| DrugBank | DB06725 |
| ChemSpider | 10442760 |
| UNII | ER09126G7A |
| KEGG | D01866 |
| ChEBI | CHEBI:31783 |
| CompTox Dashboard (EPA) | DTXSID6046133 |
| ECHA InfoCard | 100.158.646 |
| Chemical and physical data | |
| Formula | C13H10ClN3O4S2 |
| Molar mass | 371.81 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
//////////LORNOXICAM, Ro-13-9297, TS-110, Anti-inflammatory, analgesic, chlortenoxicam, CCRIS 8589
CN1C(C(=O)NC2=CC=CC=N2)=C(O)C2=C(C=C(Cl)S2)S1(=O)=O
General References
- Balfour JA, Fitton A, Barradell LB: Lornoxicam. A review of its pharmacology and therapeutic potential in the management of painful and inflammatory conditions. Drugs. 1996 Apr;51(4):639-57. [Article]
- Vane JR: Introduction: mechanism of action of NSAIDs. Br J Rheumatol. 1996 Apr;35 Suppl 1:1-3. [Article]
- Radhofer-Welte S, Rabasseda X: Lornoxicam, a new potent NSAID with an improved tolerability profile. Drugs Today (Barc). 2000 Jan;36(1):55-76. [Article]
- Skjodt NM, Davies NM: Clinical pharmacokinetics of lornoxicam. A short half-life oxicam. Clin Pharmacokinet. 1998 Jun;34(6):421-8. [Article]
- Olkkola KT, Brunetto AV, Mattila MJ: Pharmacokinetics of oxicam nonsteroidal anti-inflammatory agents. Clin Pharmacokinet. 1994 Feb;26(2):107-20. [Article]
- Hitzenberger G, Radhofer-Welte S, Takacs F, Rosenow D: Pharmacokinetics of lornoxicam in man. Postgrad Med J. 1990;66 Suppl 4:S22-7. [Article]
- Pruss TP, Stroissnig H, Radhofer-Welte S, Wendtlandt W, Mehdi N, Takacs F, Fellier H: Overview of the pharmacological properties, pharmacokinetics and animal safety assessment of lornoxicam. Postgrad Med J. 1990;66 Suppl 4:S18-21. [Article]
- Bonnabry P, Leemann T, Dayer P: Role of human liver microsomal CYP2C9 in the biotransformation of lornoxicam. Eur J Clin Pharmacol. 1996;49(4):305-8. [Article]

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}