
Home » analgesic
Category Archives: analgesic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nispomeben
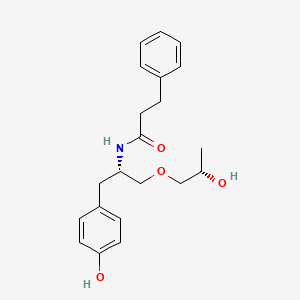
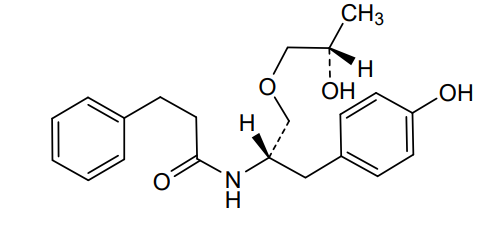
Nispomeben
CAS 1443133-41-2
MF C21H27NO4 MW357.4 g/mol
N-[(2S)-1-(4-hydroxyphenyl)-3-[(2S)-2-hydroxypropoxy]propan-2-yl]-3-phenylpropanamide
N-{(2S)-1-(4-hydroxyphenyl)-3-[(2S)-2-hydroxypropoxy]propan-2-yl}-3-phenylpropanamide
non-opioid analgesic, 470338M5XD, E1, NRD 135S E1, NRD E1, NRD.E1, NRD135S, NRD135S.E1, NRD135SE.1
Nispomeben is a small molecule drug. Nispomeben has a monoisotopic molecular weight of 357.19 Da.
- OriginatorNovaremed
- ClassAlcohols; Amides; Anti-inflammatories; Benzene derivatives; Non-opioid analgesics; Phenols; Small molecules
- Mechanism of ActionLyn protein-tyrosine kinase modulators
- Phase IINeuropathic pain
- 02 Sep 2025Updated adverse events data from a phase II trial in Neuropathic pain released by Novaremed
- 07 May 2025Novaremed completes enrolment in a phase-II clinical trial in Neuropathic pain in USA (PO) (NCT05480228)
- 16 Sep 2022Phase-II clinical trials in Neuropathic pain in USA (PO) (NCT05480228)
PAT
WO 2013/084238
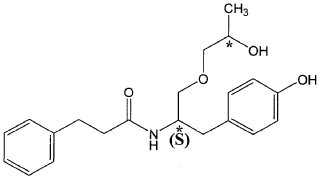
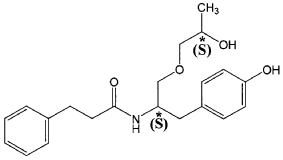
The present invention is based in part on the surprising discovery that the substantially pure enantiomers (S)2-N(3-0-((S)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide (also known as the (S,S) enantiomer or El) and (S)2-N(3-0-((R)propan 2-ol)-l -propyl -4-hydroxybenzene)-3-phenylpropyl amide (also known as the (S,R) enantiomer or E2) modulate the activity of specific tyrosine kinases in an opposite manner. It was unexpectedly found that while the (S,S) enantiomer activated protein tyrosine kinases LynA and BLK, the (S,R) enantiomer inhibited their activity. It was further unexpectedly shown that the (S,S) enantiomer was effective as a pain analgesic in animal models of pain, while the (S,R) enantiomer was shown to be ineffective or less effective in these models. Furthermore, the analgesic effect of the (S,S) enantiomer was long acting as it was efficacious for more than 24 hours post administration, in comparison to the commonly used analgesic agent gabapentin which was effective for no longer than 5 hours post administration.
The isolated enantiomers according to some embodiments of the invention may be synthesized as a racemate by known in the art methods described for example in US 7,754,771, US 7,642,290, US 7,674,829 or US 2011/0086910. The racemate may be further separated by known in the art methods for the separation of chiral compounds. According to an exemplary embodiment, the enantiomers may be synthesized as a racemate (comprising (S)2-N(3-0-((S)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide and (S)2-N(3-0-((R)propan 2-ol)-l-propyl-4-hydroxybenzene)-3-phenylpropylamide and be further separated by a supercritical fluid chromatography (SFC) in combination with chiral stationary phases. Specifically, the (S,S) and (S,R) compounds may be separated on RegisPack™ column a polysaccharide coated chiral column (with a tris-(3,5-dimethylphenyl) carbamoyl cellulose selector) generally used for enantiomeric separations of a wide range of racemate classes (Figure 7A-C).
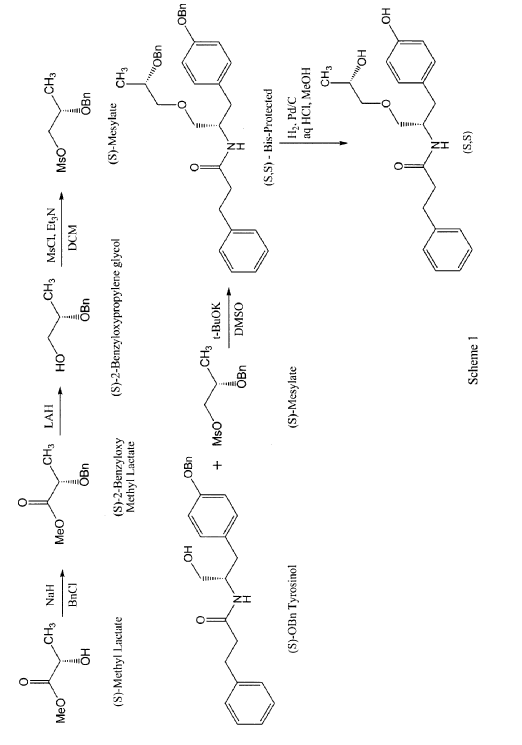
According to some embodiments, the enantiomers may be synthesized directly using for example, the process described in scheme 1 for the preparation of the (S,S) enantiomer.
PAT
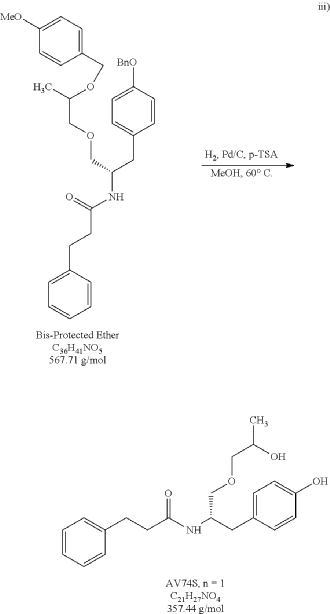
The bis-protected ether (15.7 g) was exposed to one-pot hydrogenation-debenzylation conditions (10% loading of 10% Pd/C and 0.25 eq of p-toluenesulfonic acid) in methanol. After 2 hours at 60° C. under a hydrogen atmosphere, HPLC analysis indicated that the hydrogenation of the benzyl and the debenzylation of PMB ring was complete. The reaction mixture was filtered over Celite and concentrated under reduced pressure. The residue was dissolve in ethyl acetate and a saturated aqueous sodium bicarbonate treatment was conducted to effectively remove p-toluenesulfonic acid, then DURP to provide 12.13 g of an oil (PR030-120-4). Desired product was isolated from an EA/Heptane recrystallization to provide 8.83 g of a white solid (PR030-120-6, 89.4% yield). The purity of PR030-120-6 was 99.3% via HPLC analysis. 1H NMR and Mass spec analysis supported the assigned structure for desired product.
PAT
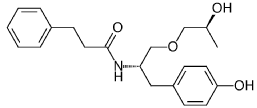
((S,S)-2-N(3-0-(propan-2-ol)-1 -propyl-4-hydroxybenzene)-3-phenylpropylamide), including its enantiomers and diastereomers may be prepared as described in WO 2013/084238,
Example 1 – Preparation of -2-N(3-Q-(propan-2-ol)-1-propyl-4-hvdroxybenzene)-3-
phenylpropylamide
(S,S)-2-N(3-0-(propan-2-ol)-1 -propyl-4-hydroxybenzene)-3-phenylpropylamide was prepared as described in WO 2013/084238 and US 201 1/0086910.
In a first step, 2 g of methyl lactate was reacted with excess of benzyl bromide to get 880 mg of (S)-benzyloxymethyl lactate. The reaction was performed by slurring sodium hydride in THF and cooling down to approximately -15°C. The reaction mixture was then allowed to warm slowly to room temperature and stirred for approximately 1 to 2 hours. The reaction was quenched with saturated ammonium chloride solution and extracted with MTBE twice followed by the removal of solvent on a rotary evaporator to obtain a crude oil. The crude product was purified by column chromatography to yield pure (S)-2-benzyloxymethyl lactate. The (R)-2-benzyloxymethyl lactate isomer was present at 0.93% only. The yield of this step may be increased by avoiding the presence of moisture in the reaction solution.
In a second step, 880 mg (S)-2-benzyloxymethyl lactate obtained in step 1 were reduced using lithium aluminum hydride to obtain (S)-2-benzyloxypropylene glycol in 83.8% yield with 98.7% purity. A solution of pure (S)-2-benzyloxymethyl lactate in methylene chloride was stirred and a solution of lithium aluminum hydride was slowly added thereto at approximately 5°C. The reaction was monitored by TLC and quenched by USP-PW water very carefully. No racemization occurred in this step.
In a third step, the (S)-2-benzyloxypropylene glycol was then reacted with methane sulfonyl chloride in methylene chloride in the presence of triethyl amine to yield the mesylate in 88% yield. A solution of step 2 was stirred in methylene chloride and methane sulfonyl chloride was added to it dropwise at <5°C. After the addition was complete, the progress of the reaction was monitored by TLC. The reaction was quenched with USP-PW water. After the layers were separated, the aqueous layer was back extracted with methylene chloride. The methylene chloride layers were then combined and washed with USP-PW water 3 times to remove most of the methane sulfonic acid. No racemization occurred in this step.
In a fourth step, the mesylate (of step 3) was coupled with S-O-benzyl tyrosinol to form the bis-protected product in 22.7% yield, with a purity of 97.4%. The reaction was carried out at room temperature using a combination of DMF as the solvent and sodium hydride as the base. The reaction went to completion after stirring for at least 12 hours at room temperature.
In a fifth step, 340 mg of the product of step 4 were reduced by hydrogenation in the presence of 10% palladium on carbon catalyst and hydrochloric acid using methylene chloride as a solvent at 50°C. The reaction went to completion in approximately 4 hours with no racemization to yield the desired product in 84.3% yield and 98.9% purity. More specifically, the catalyst was removed by filtration and the filtrate was then concentrated at 33°C. The resulting mixture of solid and oil was mixed with ethyl acetate. The resulting slurry was filtered and the solids washed with ethyl acetate and dried under vacuum at 40 to 45°C to obtain the desired product.
PAT
Example 1—Preparation of (S,S)-2-N(3-O-(propan-2-ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamide
| (S,S)-2-N(3-O-(propan-2-ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamide was prepared as described in WO 2013/084238 and US 2011/0086910. |
PAT
- Method of treating or preventing painPublication Number: US-2016317479-A1Priority Date: 2009-09-09
- Method of treating or preventing painPublication Number: US-8802734-B2Priority Date: 2009-09-09Grant Date: 2014-08-12
- Method of Treating or Preventing PainPublication Number: US-2014350099-A1Priority Date: 2009-09-09
- N-substituted benzenepropanamide or benzenepropenamide for use in the treatment of pain and inflammationPublication Number: WO-2011030205-A1Priority Date: 2009-09-09
- Method of treating or preventing painPublication Number: US-2011086910-A1Priority Date: 2009-09-09
- Isolated stereoisomeric forms of (S)2-N(3-O-(propan 2-Ol)-1-propyl-4-hydroxybenzene)-3-phenylpropylamidePublication Number: US-9381173-B2Priority Date: 2011-12-08Grant Date: 2016-07-05
- Isolated Stereoisomeric Forms Of (S)2-N(3-O-(Propan 2-Ol)-1-Propyl-4-Hydroxybenzene)-3-PhenylpropylamidePublication Number: US-2014275270-A1Priority Date: 2011-12-08
- N-substituted benzenepropanamide and benzenepropenamide for use in the prevention or the treatment of affective disordersPublication Number: US-9133103-B2Priority Date: 2011-09-21Grant Date: 2015-09-15
- N-Substituted Benzenepropanamide and Benzenepropenamide For Use in the Prevention or the Treatment of Affective DisordersPublication Number: US-2014275273-A1Priority Date: 2011-09-21
- N-substituted benzenepropanamide and benzenepropenamide for use in the prevention or the treatment of affective disordersPublication Number: EP-2758046-B1Priority Date: 2011-09-21Grant Date: 2015-10-21
- Compounds for treatment or prevention of an infection resulting from a coronavirus and/or a coronavirus-induced diseasePublication Number: EP-3939578-A1Priority Date: 2020-07-13
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: EP-3860582-B1Priority Date: 2019-01-23Grant Date: 2022-05-04
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: WO-2020152226-A1Priority Date: 2019-01-23
- Compounds for use in the treatment or prophylaxis of pain, inflammation and/or autoimmunityPublication Number: US-2022002228-A1Priority Date: 2019-01-23
- Pharmaceutical composition comprising stereoisomers of n-(1-(4-hydroxyphenyl)-3-(2-hydroxypropoxy)propan-2-yl)-3-phenylpropanamide for the prevention and treatment of type ii diabetesPublication Number: WO-2015173813-A1Priority Date: 2014-05-14



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

////////nispomeben, non-opioid analgesic, 470338M5XD, E1, NRD 135S E1, NRD E1, NRD.E1, NRD135S, NRD135S.E1, NRD135SE.1, Neuropathic pain
Methotripremazine
Methotripremazine
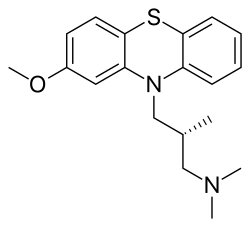
- CL 36467
- CL 39743
- N05AA02
- RP 7044
- RP-7044
- SK&F 5116
- XP-03
- XP03
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Methotrimeprazine hydrochloride | 42BB1Y2586 | 1236-99-3 | ODLGFPIWRAEFAN-PFEQFJNWSA-N |
| Methotrimeprazine maleate | 5KN5Y9V01K | 7104-38-3 | IFLZPECPTYCEBR-VIEYUMQNSA-N |
Methotrimeprazine
CAS Registry Number: 60-99-1
CAS Name: (bR)-2-Methoxy-N,N,b-trimethyl-10H-phenothiazine-10-propanamine
Additional Names: (-)-10-(3-dimethylamino-2-methylpropyl)-2-methoxyphenothiazine; levomepromazine; 2-methoxytrimeprazine; levomeprazine
Manufacturers’ Codes: RP-7044
Trademarks: Sinogan-Debil; Tisercin (EGYT); Neozine (Rh>e-Poulenc); Nirvan; Nozinan (Rh>e-Poulenc); Levoprome (Lederle)
Molecular Formula: C19H24N2OS
Molecular Weight: 328.47
Percent Composition: C 69.47%, H 7.36%, N 8.53%, O 4.87%, S 9.76%
Literature References: Prepn: Courvoisier et al.,C.R. Seances Soc. Biol. Ses Fil.151, 1378 (1957); Jacob, Robert, US2837518 (1958 to Rhône-Poulenc).Optical Rotatory Power, -17, Conc: 5 g/100mL; Solv: chloroform; Wavlen: 589.3 nm; Temp: 20 °C
Derivative Type: Maleate
CAS Registry Number: 7104-38-3
Trademarks: Minozinan; Milezin (Spofa); Neuractil; Neurocil (Bayer); Sofmin (Dainippon); Veractil
Molecular Formula: C19H24N2OS.C4H4O4
Molecular Weight: 444.54
Percent Composition: C 62.14%, H 6.35%, N 6.30%, O 18.00%, S 7.21%
Properties: Crystals, darkened by light. Dec about 190°. Sparingly sol in water (0.3% at 20°) and in ethanol (0.4%). pH of a 0.3% aq soln is 4.3. The free base is levorotatory: [a]D20 -17° (c = 5 in chloroform).
Optical Rotation: [a]D20 -17° (c = 5 in chloroform)
Therap-Cat: Analgesic.
Keywords: Analgesic (Non-Narcotic).
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Methotrimeprazine is a phenothiazine used in the management of psychosis, particular those of schizophrenia, and manic phases of bipolar disorder.
A phenothiazine with pharmacological activity similar to that of both chlorpromazine and promethazine. It has the histamine-antagonist properties of the antihistamines together with central nervous system effects resembling those of chlorpromazine. (From Martindale, The Extra Pharmacopoeia, 30th ed, p604)
Levomepromazine, also known as methotrimeprazine, is a phenothiazine neuroleptic drug. Brand names include Nozinan, Levoprome, Detenler, Hirnamin, Levotomin and Neurocil. It is a low-potency antipsychotic (approximately half as potent as chlorpromazine) with strong analgesic, hypnotic and antiemetic properties that are primarily used in palliative care.[1][2]
Serious side effects include tardive dyskinesia, akathisia, abnormalities in the electrical cycle of the heart, low blood pressure and the potentially fatal neuroleptic malignant syndrome.[1][2]
As is typical of phenothiazine antipsychotics, levomepromazine is a “dirty drug“, that is, it exerts its effects by blocking a variety of receptors, including adrenergic receptors, dopamine receptors, histamine receptors, muscarinic acetylcholine receptors and serotonin receptors.[1][2]
Medical uses
It can be used as an analgesic for moderate to severe pain in non-ambulant patients (the latter being because of its strong sedative effects).[3]
Levomepromazine is also used at lower doses for the treatment of nausea and insomnia.[1]
Levomepromazine is frequently prescribed and valued worldwide in palliative care medicine for its multimodal action, to treat intractable nausea or vomiting, and for severe delirium/agitation in the last days of life. Palliative care physicians will commonly prescribe it orally or via subcutaneous syringe drivers in combination with opioid analgesics such as hydromorphone.[1][2]
Levomepromazine is used for the treatment of psychosis, particularly those of schizophrenia, and manic phases of bipolar disorder. It should only be used with caution in the treatment of agitated depressions, as it can cause akathisia as a side effect, which could worsen the agitation.[1][2] A 2010 systematic review compared the efficacy of levomepromazine with atypical antipsychotic drugs:
Adverse effects
The most common side effect is akathisia.[2] Levomepromazine has prominent sedative and anticholinergic/sympatholytic effects (dry mouth, hypotension, sinus tachycardia, night sweats) and may cause weight gain.[2] These side effects normally preclude prescribing the drug in doses needed for full remission of schizophrenia, so it has to be combined with a more potent antipsychotic.[2] In any case, blood pressure and EKG should be monitored regularly.[2]
A rare but life-threatening side effect is neuroleptic malignant syndrome (NMS).[2] The symptoms of NMS include muscle stiffness, convulsions and fever.[2]
PAPER
Bulletin de la Societe de Pharmacie de Bordeaux (1964), 103(4), 224-30.
The authors define an extn. equil. const., pKe. When a basic mol., A, in an org. solvent (immiscible with water) is shaken with an aq. acid, part of A passes into the aq. phase in the equil. A + H+ .rdblhar. AH+, and Ke and pKe are defined by the equations Ke = [A]org[H+]H2O/[AH+]H2O and pKe = pKa -log ([A]org/[A]H2O), resp. Values of pKe are reported for levomepromazine, properidiazine, thioridazine, chlorpromazine, alimenazine, propiomazine, promethazine, and aminopromazine. Where 2 C atoms sep. the 2 N chain atoms, pKe is of the order of 5, and if 3, the value is near 4.3.
PATENT
JP 40009030
A soln. of 10.5 g. l-3-dimethylamino-2-methylpropanol in xylene is added a suspension of 2.5 g. Na in xylene and a soln. of 18 g. p-tosyl chloride in xylene is dropped in to give l-3-dimethylamino-2-methylpropanol tosylate (I), hydrochloride m. 98-100%. I is treated with 18 g. 2-methoxyphenothiazine and NaNH2 (prepd. from 1.85 g. Na) to give 80% l-3-(2-methoxy-10-phenothiazinyl)-2-methyl-1-dimethylaminopropane, m. 125-6° (hexane). Similarly are prepd. l-3-(3-ethyl-10-phenothiazinyl)-2-methyl-1-dimethylaminopropane (maleate m. 136°) and l-3-(10-phenothiazinyl)-2-methyl-1-dimethylaminopropane (maleate m. 174-5°). The products are tranquilizers.
PATENT
HU 152208
HU 157158
PL 66636
PAPER
Bulletin de la Societe Chimique de France (1968), (8), 3220-2.
Folia medica (1970), 12(1), 88-9
Journal of pharmaceutical sciences (1987), 76(7), 541-4.
SYN
| IN201203390 |
Deprotonation of 2-methoxyphenothiazine by means of KOH in refluxing touene/DMSO, followed by condensation of resulting pottasium salt with N-(3-chloro-2-methylpropyl)-N,N-dimethylamine in refluxing toluene leads to racemic levomepromazine , which upon finally resolution using (-)-dibenzoyl-L-tartaric acid in acetone or using di-p-toluoyl-L-tartaric acid and, optionally, HCOOH in EtOH at 60 °C affords the target levomepromazine

SYN

References
- ^ Jump up to:a b c d e f Brayfield A, ed. (13 December 2013). “Levomepromazine”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 12 May 2014.
- ^ Jump up to:a b c d e f g h i j k Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ “Levomepromazine”. Farmacotherapeutisch Kompas (in Dutch). Retrieved 5 October 2016.
- ^ Jump up to:a b Sivaraman P, Rattehalli RD, Jayaram MB (October 2010). “Levomepromazine for schizophrenia”. The Cochrane Database of Systematic Reviews. 10 (10): CD007779. doi:10.1002/14651858.CD007779.pub2. PMC 3283151. PMID 20927765.
External links
- “Levomepromazine”. PubChem. National Center for Biotechnology Information.
- NOZINAN – Lévomépromazine Doctissimo Guides des Medicaments
- “Levomepromazine” (PDF). Grampians Palliative Care Team Publication. Victoria, Australia. May 2010. Archived from the original (PDF) on 2011-02-26.
- “Levomepromazine in Palliative Care” (PDF). Scotland, UK. August 2013. Archived from the original (PDF) on 2013-05-22.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Pregnancy category | Only if clearly needed |
| Routes of administration | Oral, seldom IM |
| Drug class | Typical antipsychotic |
| ATC code | N05AA02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | ~50–60% |
| Metabolism | Hepatic |
| Elimination half-life | ~20 hours |
| Excretion | In feces and urine (metabolites), unchanged drug only 1% |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 60-99-1 7104-38-3 (maleate), 1236-99-3 HCl) |
| PubChem CID | 72287 |
| IUPHAR/BPS | 7603 |
| DrugBank | DB01403 |
| ChemSpider | 65239 |
| UNII | 9G0LAW7ATQ |
| KEGG | D00403 |
| ChEBI | CHEBI:6838 |
| ChEMBL | ChEMBL1764 |
| CompTox Dashboard (EPA) | DTXSID1023289 |
| ECHA InfoCard | 100.000.450 |
| Chemical and physical data | |
| Formula | C19H24N2OS |
| Molar mass | 328.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////methotripremazine, L 36467, CL 39743, N05AA02, RP 7044, RP-7044, SK&F 5116, XP-03, XP03
O(c2cc1N(c3c(Sc1cc2)cccc3)C[C@H](C)CN(C)C)C

NEW DRUG APPROVALS
one time
$10.00
LORNOXICAM
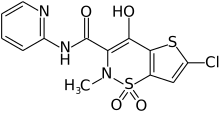
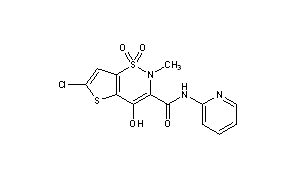
LORNOXICAM
chlortenoxicam
- Molecular FormulaC13H10ClN3O4S2
- Average mass371.819 Da
70374-39-9[RN], Chlortenoxicam, CTX, ER09126G7A
2H-thieno[2,3-e]-1,2-thiazine-3-carboxamide, 6-chloro-4-hydroxy-2-methyl-N-2-pyridinyl-, 1,1-dioxide
6233
6-Chlor-4-hydroxy-2-methyl-N-(pyridin-2-yl)-2H-thieno[2,3-e][1,2]thiazin-3-carboxamid-1,1-dioxid
6-Chloro-4-hydroxy-2-methyl-N-(2-pyridinyl)-2H-thieno[2,3-e][1,2]thiazine-3-carboxamide 1,1-dioxide
- Chlortenoxicam, Ro-13-9297
- ATC:M01AC05
- CCRIS 8589
- Ro 13-9297
Lorcam (Taisho Pharmaceutical Co.) / Xafon (Nycomed)LornoxicamCAS Registry Number: 70374-39-9
CAS Name: 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e]-1,2-thiazine-3-carboxamide 1,1-dioxide
Additional Names: 6-chloro-4-hydroxy-2-methyl-3-(2-pyridylcarbamoyl)-2H-thieno[2,3-e]-1,2-thiazine-1,1-dioxide; chlortenoxicam
Manufacturers’ Codes: Ro-13-9297; TS-110
Trademarks: Xefo (Nycomed)
Molecular Formula: C13H10ClN3O4S2
Molecular Weight: 371.82
Percent Composition: C 41.99%, H 2.71%, Cl 9.53%, N 11.30%, O 17.21%, S 17.25%
Literature References: Cyclooxygenase inhibitor; structurally similar to tenoxicam, q.v.
Prepn: R. Pfister et al.,DE2838851; eidem,US4180662 (both 1979 to Hoffmann-La Roche).Clinical pharmacokinetics: S. I. Ankier et al.,Postgrad. Med. J.64, 752 (1988). Symposium on pharmacology and clinical experience: ibid.66, Suppl. 4, S1-S50 (1990). Overview of pharmacology and safety assessment: T. P. Pruss et al.,ibid. S18.
Properties: Orange to yellow crystals, mp 225-230° (dec). pKa2 4.7. uv max: 371 nm. Partition coefficient (n-octanol/pH 7.4 buffer): 1.8. LD50 orally in mice, rats, rabbits, dogs, monkeys: >10 mg/kg (Pruss).
Melting point: mp 225-230° (dec)
pKa: pKa2 4.7
Log P: Partition coefficient (n-octanol/pH 7.4 buffer): 1.8
Absorption maximum: uv max: 371 nm
Toxicity data: LD50 orally in mice, rats, rabbits, dogs, monkeys: >10 mg/kg (Pruss)
Therap-Cat: Anti-inflammatory; analgesic.
Keywords: Analgesic (Non-Narcotic); Anti-inflammatory (Nonsteroidal); Thiazinecarboxamides.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 504-29-0 | C5H6N2 | 2-aminopyridine | 2-Pyridinamine |
| 7790-94-5 | ClHO3S | chlorosulfonic acid | Chlorosulfuric acid |
| 56946-84-0 | C5H5Cl2NO2S2 | 2,5-dichloro-N-methyl-3-thiophenesulfonamide | 3-Thiophenesulfonamide, 2,5-dichloro-N-methyl- |
| 3172-52-9 | C4H2Cl2S | 2,5-dichlorothiophene | Thiophene, 2,5-dichloro- |
SYN
Synthesis of lornoxicam (DE2838851)

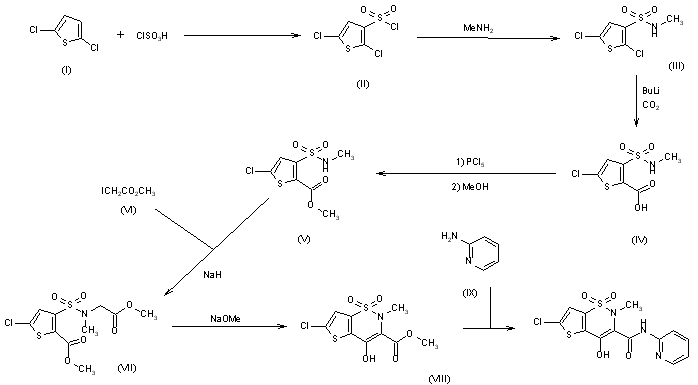
The sulfonation of 2,5-dichlorothiophene (I) with ClSO3H -SOCl2 gives 2,5-dichlorothiophene-3-sulfonic acid chloride (II), which by reaction with methylamine in CHCl3 yields the corresponding methylamide (III). The carboxylation of (III) with butyllithium and CO2 in ether affords 5-chloro-3-(N-methylsulfamoyl)thiophene-2-carboxylic acid (IV), which is esterified with PCl5 and methanol to the methyl ester (V). The condensation of (V) with methyl iodoacetate (VI) by means of NaH in DMF gives 5-chloro-3-[N-(methoxycarbonylmethyl)-N-methylsulfamoyl]thiophene-2-carboxylic acid methyl ester (VII), which is cyclized with sodium methoxide in methanol yielding 6-chloro-4-hydroxy-2-methyl-2H-thieno[2,3-e]-1,2-thiazine-3-carboxylic acid methyl ester 1,1-dioxide (VIII). Finally, this compound is treated with 2-aminopyridine (IX) in refluxing xylene.
Lornoxicam is an NSAID indicated in the treatment of mild to moderate pain, as well as rheumatoid arthritis and osteoarthritis.
Lornoxicam, also known as chlortenoxicam, is a nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class with analgesic (pain relieving), anti-inflammatory and antipyretic (fever reducing) properties. It is available in oral and parenteral formulations.
It was patented in 1977 and approved for medical use in 1997.[1] Brand names include Xefo and Xefocam among others.
Lornoxicam (chlortenoxicam) is a new nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class with analgesic, anti-inflammatory and antipyretic properties. Lornoxicam differs from other oxicam compounds in its potent inhibition of prostaglandin biosynthesis, a property that explains the particularly pronounced efficacy of the drug. Lornoxicam is approved for use in Japan.
Medical uses
Lornoxicam is used for the treatment of various types of pain, especially resulting from inflammatory diseases of the joints, osteoarthritis, surgery, sciatica, and other inflammations.[2]

Contraindications
The drug is contraindicated in patients who must not take other NSAIDs, possible reasons including salicylate sensitivity, gastrointestinal bleeding and bleeding disorders, and severe impairment of heart, liver or kidney function. Lornoxicam is not recommended during pregnancy and breastfeeding and is contraindicated during the last third of pregnancy.[2]
Adverse effects
Lornoxicam has side effects similar to other NSAIDs, most commonly mild ones like gastrointestinal disorders (nausea and diarrhea) and headache. Severe but seldom side effects include bleeding, bronchospasms and the extremely rare Stevens–Johnson syndrome.[2]
Interactions
Interactions with other drugs are typical of NSAIDs. Combination with vitamin K antagonists like warfarin increases the risk of bleeding. Combination with ciclosporin can lead to reduced kidney function, and to acute kidney injury in rare cases. Lornoxicam can also increase the adverse effects of lithium, methotrexate and digoxin and its derivatives. The effect of diuretics, ACE inhibitors and angiotensin II receptor antagonists can be reduced, but this is only relevant in patients with special risks like heart failure. As with piroxicam, cimetidine can increase plasma levels but is unlikely to cause relevant interactions.[3]
PAPER
https://www.mdpi.com/2218-0532/71/4/303

PATENT
CN 113480561
The present invention relates to the prepn. of high purity loroxicam. In particular, the prepn. method comprises a step of taking 6-chloro-4-hydroxy-2-methyl-2H-thieno[2,3-e]-1,2-Me thiazinecarboxylate-1,1-dioxide and 2-amino pyridine is used as the raw material and xylene is used as the solvent undergoes distn. reaction with solid acid catalyst, mixed gas obtained by the distn. reaction is condensed to obtain a condensate and solid acid catalyst is used to adsorb methanol in the condensate and the adsorbed condensate is recycled, filtering and refining to obtain loroxicam. The present inventive method distills out the methanol produced by the reaction to promote the pos. progress of the reaction and then catalyzes the absorption of methanol by H2SO4/MxOy solid super acid, so that the xylene returned to the reaction system does not contain methanol, which reduces the coking of the reaction, thereby improving product quality and yield. The prepd. lornoxicam has high purity, which can reach more than 99.9%, reduces the amt. of solvent and also suitable for industrial prodn.
PATENT
CN 112592356
The present invention relates to the prepn. of lornoxicam. In particular, the prepn. method comprises a step of taking 6-chloro-4-hydroxy-2-methyl-2-H-thieno[2,3-e]-1,2-thiazidecarboxylic acid Me ester-1,1-dioxide and 2-aminopyridine as raw materials, xylene is used as solvent, adding stabilizer, and carrying out aminolysis reaction, the solvent was removed by concn. under reduced pressure, adding org. solvent to make the slurry, filtering and refining to obtain lornoxicam. The inventive method uses p-toluene sulfonic acid as a stabilizer, while lowering the reaction temp., it promotes the reaction to proceed forward, and improve the product quality and yield; at the same time reduce the amt. of industrial solvents, the post-treatment process is optimized and the cost of the three wastes treatment is reduced.
PATENT
IN 2014CH02116
Example: 1Preparation of 6-chloro-4-hydroxy-l,l-dioxo-l,2-dihydro-lX6-thieno [2,3-e][l,2] thiazine-3-carboxylic acid methyl ester To the mixture of methanol ( 1000 ml) and 5-chloro-3-(methoxy carbonyl methyl sulfamoyl)-thiophene-2-carboxylicacid methyl ester ( 100 g ,0.305 moles), added sodium methoxide solution (200 ml ) at 25-30°C over a period of 30-45 min. The resulting mixture was stirred for 60 min at same temperature; allowed to heat at 65-75°C and stirred for 10-12 hrs. After completion of reaction, methanol was distilled out under reduced pressure to obtained titled residual product which is directly used to next step
(Example-2). Example: – 2:Preparation of 6-chloro-4-hydroxy-2-methyl-l,l-dioxo-l,2-dihydro-U6- thieno[2,3-e][l,2] thiazine-3-carboxylic acid methyl ester 6-chloro-4-hydroxy-1,1 -dioxo-1,2-dihydro-1 X,6-thieno [2,3-e][ 1,2] thiazine-3-carboxylic acid methyl ester was suspended in DM water (500 ml) and cooled to 10-15° C, dimethyl sulphate ( 70 g) was slowly added to the mixture at 10-15°C in 30 min. The reaction mixture was raised to 25-30°C and maintained for 2-3 hours at same temperature. After completion of reaction, mixture was cooled to 10-15°C, methylene dichloride (1600 ml) was added, reaction mixture pH was adjust to 1.0 -2.0 with hydrochloric acid at 10-15° C, stir reaction mixture to separate the layers. The methylene dichloride layer was distilled out completely at below 30°C to get an residue, followed by addition of methanol (60 ml) and distilled out methanol completely under vacuum at below 50°C to get an residue; further it was crystallized by addition of methanol 190 ml and stirred for 30 min at 50-55°C; cooled the reaction mixture at 25-30°C and stirred for 60 min at same temperature. The resultant solid was filtered, washed with methanol (40 ml) and dried at 50-55°C for 4 – 6 hrs to obtain the titled product
Example: 3Preparation of 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e]-l,2-thiazine-3-carboxamide 1,1-dioxide (Lornoxicam) 6-chloro-4-hydroxy-2-methyl-l, 1 -dioxo-1,2-dihydro-l X.6-thieno[2,3-e][l ,2] thiazine-3-carboxylic acid methyl ester ( 50 g 0.161 moles) was suspended in O-xylene (500 ml) and allow to stirred at 70-75°C to obtained clear solution. To this clear solution slowly added the mixture of THF ( 50 ml) solution of 2-Amino pyridine ( 14 g ) and ethyl magnesium bromide 2 molar solution (100 ml) at 70-75°C and allow to stirred for 3-4 hrs at same temperature. After completion of reaction, the dilute hydrochloric acid was added to the mixture at 10-15°C and stirred for 60 min. The resultant solid was filtered, washed with water (100 ml) to obtain crude Lornoxicam.
Example: 4Preparation of 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e)-l,2-thiazine-3-carboxamide 1,1-dioxide (Lornoxicam) 6-chloro-4-hydroxy-2-methyl-l,l-dioxo-l,2-dihydro-R6-thieno[2,3-e][l,2] thiazine-3-carboxylic acid methyl ester ( 50 g 0.161 moles) was suspended in O-xylene (500 ml) and allow to stirred at 70-75°C to obtained clear solution. To this clear solution slowly added the mixture of THF ( 50 ml) solution of 2-Amino pyridine ( 14 g ) and isopropyl magnesium bromide 2 molar solution (100 ml) at 70-75°C and allow to stirred for 3-4 hrs at same temperature. After completion of reaction, the dilute hydrochloric acid was added to the mixture at 10-15°C and stirred for 60 min. The resultant solid was filtered, washed with water (100 ml) to obtain crude Lornoxicam.
Example: 5Purification of Lornoxicam.The crude Lornoxicam was suspended in methanol (500 ml) and cooled to 5-10°C, resulting suspension was basified to pH 11-13 by using sodium hydroxide solution to get clear solution; followed by filtration through hyflo bed; the obtain filtrate was acidified to pH 4.5 – 5.0 with dil. HC1 (1:1) at 5-10°C; stirred the slurry for 30 min. at 5-10°C. The resultant solid was filtered, washed with DM water (100 ml) and dried at 50-55°C to obtained pure Lornoxicam.
PATENT
.EXAMPLES:Preparation of Lornoxicam crudeExample ITo 1200ml o-xylene, 20gm Methyl-6-chloro-4-hydroxy-2-methyl-2//-thieno [2, 3-e] [1, 2] thiazine-3- carboxyate 1,1-dioxide and 6.44gm 2-aminopyridine was added. The reaction mass was stirred under nitrogen atmosphere. Temperature was raised to 140-145°C and maintained for 6hrs. The reaction mass was cooled to 30-35°C and nitrogen was removed. Reaction mass was further stirred for 3hrs- Filtered and washed twice with 50ml of o-xylene. 19.8gm of crude Lornoxicam was obtained. Purification of Lornoxicam crude
Example 219.8gm of crude Lornoxicam was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around Ihr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, til! the reaction mass reached pH of 2-3, and then stirred for around I hi*. The reaction mass was cooled to room temperature, filtered, and then washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 19.1 gm of pure Lornoxicam was obtained. (HPLC purity- 99.95%)
Example 3!7.9gm of crude Lornoxicam (prepared as per example 1) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around Ihr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C till the reaction mass reached pH of 2-3, and then stirred for around Ihr. The reaction mass was cooled to room temperature, filtered and then washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 17.2 gm of pure Lornoxicam was obtained. (HPLC purity- 99.9%) clear solution and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around lhr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, till the reaction mass reached pH of 2-3, and then stirred for around lhr. The reaction mass was cooled to 30-35°C, filtered and then washed with 1:1 mixture of isopropyl alcohol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 4.85 gm of pure Lornoxicam was obtained. (HPLC purity- 99.8%)
Example 55 gm of crude Lornoxicam (prepared as per example 1) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and ethanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around lhr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, til! the reaction mass reached pH of 2-3 and then stirred for around lhr. The reaction mass was cooled to 30-35°C and filtered, washed with 1:1 mixture of ethanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 4.8 gm of pure Lornoxicam was obtained. (HPLC purity- 99.8%)
Example 619.4 gm of crude Lornoxicam (prepared as per example I) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 20% activated charcoal was further added. The reaction mass was stirred for around lhr at room temperature followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added till the reaction mass reached pH of 2-3 and then stirred for around 1 hr. The reaction mass was * filtered and washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 18.9 gm of pure Lornoxicam was obtained. (HPLC purity- 99.3%).
PATENT
https://www.sciencedirect.com/science/article/abs/pii/S0968089603007624?via%
PATENT
https://patents.google.com/patent/WO2002000167A2/en
References
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 519. ISBN 9783527607495.
- ^ Jump up to:a b c Haberfeld H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. Xefo Filmtabletten. ISBN 978-3-85200-196-8.
- ^ Klopp T, ed. (2010). Arzneimittel-Interaktionen (in German) (2010/2011 ed.). Arbeitsgemeinschaft für Pharmazeutische Information. ISBN 978-3-85200-207-1.
| Clinical data | |
|---|---|
| Trade names | Xefo, Xefocam others |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category | Not recommended; contraindicated in months 7–9 |
| Routes of administration | By mouth, parenteral |
| ATC code | M01AC05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 90–100% |
| Protein binding | 99% |
| Metabolism | CYP2C9 |
| Elimination half-life | 3–4 hours |
| Excretion | 2/3 liver, 1/3 kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 70374-39-9 |
| PubChem CID | 5282204 |
| DrugBank | DB06725 |
| ChemSpider | 10442760 |
| UNII | ER09126G7A |
| KEGG | D01866 |
| ChEBI | CHEBI:31783 |
| CompTox Dashboard (EPA) | DTXSID6046133 |
| ECHA InfoCard | 100.158.646 |
| Chemical and physical data | |
| Formula | C13H10ClN3O4S2 |
| Molar mass | 371.81 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
//////////LORNOXICAM, Ro-13-9297, TS-110, Anti-inflammatory, analgesic, chlortenoxicam, CCRIS 8589
CN1C(C(=O)NC2=CC=CC=N2)=C(O)C2=C(C=C(Cl)S2)S1(=O)=O
General References
- Balfour JA, Fitton A, Barradell LB: Lornoxicam. A review of its pharmacology and therapeutic potential in the management of painful and inflammatory conditions. Drugs. 1996 Apr;51(4):639-57. [Article]
- Vane JR: Introduction: mechanism of action of NSAIDs. Br J Rheumatol. 1996 Apr;35 Suppl 1:1-3. [Article]
- Radhofer-Welte S, Rabasseda X: Lornoxicam, a new potent NSAID with an improved tolerability profile. Drugs Today (Barc). 2000 Jan;36(1):55-76. [Article]
- Skjodt NM, Davies NM: Clinical pharmacokinetics of lornoxicam. A short half-life oxicam. Clin Pharmacokinet. 1998 Jun;34(6):421-8. [Article]
- Olkkola KT, Brunetto AV, Mattila MJ: Pharmacokinetics of oxicam nonsteroidal anti-inflammatory agents. Clin Pharmacokinet. 1994 Feb;26(2):107-20. [Article]
- Hitzenberger G, Radhofer-Welte S, Takacs F, Rosenow D: Pharmacokinetics of lornoxicam in man. Postgrad Med J. 1990;66 Suppl 4:S22-7. [Article]
- Pruss TP, Stroissnig H, Radhofer-Welte S, Wendtlandt W, Mehdi N, Takacs F, Fellier H: Overview of the pharmacological properties, pharmacokinetics and animal safety assessment of lornoxicam. Postgrad Med J. 1990;66 Suppl 4:S18-21. [Article]
- Bonnabry P, Leemann T, Dayer P: Role of human liver microsomal CYP2C9 in the biotransformation of lornoxicam. Eur J Clin Pharmacol. 1996;49(4):305-8. [Article]

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}