
Home » 2020 APPROVALS (Page 3)
Category Archives: 2020 APPROVALS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tirabrutinib
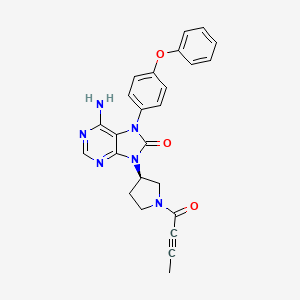
Tirabrutinib
チラブルチニブ塩酸塩
GS-4059
ONO-4059
6-amino-9-[(3R)-1-but-2-ynoylpyrrolidin-3-yl]-7-(4-phenoxyphenyl)purin-8-one
| Formula | C25H22N6O3. HCl |
|---|---|
| CAS | 1439901-97-9 HCL1351636-18-4FREE FORM |
| Mol weight | 490.9415 |
JAPAN APPROVED 2020/3/25 Velexbru
Antineoplastic, Bruton’s tyrosine kinase inhibitor
8H-Purin-8-one,6-amino-7,9-dihydro-9-((3R)-1-(1-oxo-2-butyn-1-yl)-3-pyrrolidinyl)-7-(4-phenoxyphenyl)
6-Amino-9-((3R)-1-(2-butynoyl)-3-pyrrolidinyl)-7-(4-phenoxyphenyl)-7,9-dihydro-8H-purin-8-one
Tirabrutinib (Velexbru®) is an orally administered, small molecule, Bruton’s tyrosine kinase (BTK) inhibitor being developed by Ono Pharmaceutical and its licensee Gilead Sciences for the treatment of autoimmune disorders and haematological malignancies. Tirabrutinib irreversibly and covalently binds to BTK in B cells and inhibits aberrant B cell receptor signalling in B cell-related cancers and autoimmune diseases. In March 2020, oral tirabrutinib was approved in Japan for the treatment of recurrent or refractory primary central nervous system lymphoma. Tirabrutinib is also under regulatory review in Japan for the treatment of Waldenström’s macroglobulinemia and lymphoplasmacytic lymphoma. Clinical development is underway in the USA, Europe and Japan for autoimmune disorders, chronic lymphocytic leukaemia, B cell lymphoma, Sjogren’s syndrome, pemphigus and rheumatoid arthritis. This article summarizes the milestones in the development of tirabrutinib leading to the first approval of tirabrutinib for the treatment of recurrent or refractory primary central nervous system lymphoma in Japan.

PATENT
WO 2011152351
https://patents.google.com/patent/WO2011152351A1/en
Example 19 (2) : 6-amino-9-[(3R) -1- (2-butinoyl) -3-pyrrolidinyl] -7- (4-phenoxyphenyl) -7,9-dihydro-8H-purine- 8-on

TLC: Rf 0.68 (ethyl acetate: methanol = 9: 1);
1 H-NMR (CDCl 3 ): δ 1.94-2.03, 2.23-2.39, 2.80-3.01, 3.50-3.63, 3.67-3.80, 3.86-4.02, 4.03-4.18, 4.23-4.33, 4.42-4.51, 5.11-5.25, 7.04-7.23, 7.34-7.45, 8.20-8.23.
PATENT
WO 2013081016
WO 2015193740
WO 2015181633
WO 2015185998
WO 2016024228
WO 2016024231
WO 2016163531
WO 2016024227
WO 2017033113
PATENT
US 20170035881
https://patents.google.com/patent/US20170035881A1/en
PATENT WO 2017033113
https://patents.google.com/patent/WO2017033113A1/en

///////Tirabrutinib, japan 2020, 2020 approvals, Velexbru , チラブルチニブ塩酸塩 , GS 4059, ONO 4059,
CC#CC(=O)N1CCC(C1)N2C3=NC=NC(=C3N(C2=O)C4=CC=C(C=C4)OC5=CC=CC=C5)N
Berotralstat
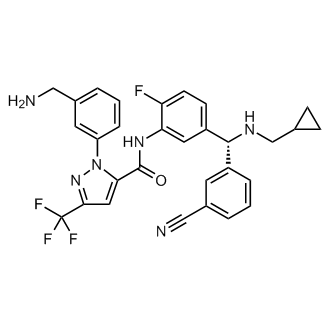

Berotralstat
CAS 1809010-50-1
DIHCl 1809010-52-3
Molecular Formula, C30-H26-F4-N6-O, Molecular Weight, 562.5684
1-(3-(Aminomethyl)phenyl)-N-(5-((R)-(3-cyanophenyl)((cyclopropylmethyl)amino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide
1H-Pyrazole-5-carboxamide, 1-(3-(aminomethyl)phenyl)-N-(5-((R)-(3-cyanophenyl)((cyclopropylmethyl)amino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-
To treat patients with hereditary angioedema
FDA APPROVED 12/4/2020, Orladeyo, 110MG CAPSULE 0RAL
New Drug Application (NDA): 214094
Company: BIOCRYST PHARMACEUTICALS INC
New Drug Application (NDA): 214094
Company: BIOCRYST PHARMACEUTICALS INC
Berotralstat Hydrochloride
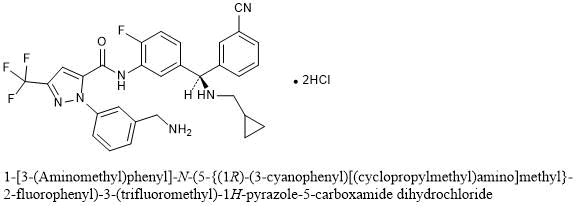
1-[3-(Aminomethyl)phenyl]-N-(5-{(1R)-(3-cyanophenyl)[(cyclopropylmethyl)amino]methyl}-2-fluorophenyl)-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide dihydrochloride
C30H26F4N6O▪2HCl : 635.48
[1809010-52-3]
Berotralstat, also known as BCX-7353, is a kallikrein inhibitor. BCX7353 is a synthetic, once-daily, small molecule drug that can be taken as an oral capsule to treat HAE attacks and for prophylaxis.
Hereditary angioedema (HAE) is rare disorder caused by a SERPING1 gene mutation that triggers severe swelling of the skin and upper airway. Treatment options for HAE with deficient and dysfunctional C1-inhibitor are expanding to include small-molecule drugs that inhibit protein interactions in the kallikrein-kinin system
Serine proteases make up the largest and most extensively studied group of proteolytic enzymes. Their critical roles in physiological processes extend over such diverse areas as blood coagulation, fibrinolysis, complement activation, reproduction, digestion, and the release of physiologically active peptides. Many of these vital processes begin with cleavage of a single peptide bond or a few peptide bonds in precursor protein or peptides. Sequential limited proteolytic reactions or cascades are involved in blood clotting, fibrinolysis, and complement activation. The biological signals to start these cascades can be controlled and amplified as well. Similarly, controlled proteolysis can shut down or inactivate proteins or peptides through single bond cleavages.
Kallikreins are a subgroup of serine proteases. In humans, plasma kallikrein (KLKB1) has no known homologue, while tissue kallikrein-related peptidases (KLKs) encode a family of fifteen closely related serine proteases. Plasma kallikrein participates in a number of pathways relating to the intrinsic pathway of coagulation, inflammation, and the complement system.
Coagulation is the process by which blood forms clots, for example to stop bleeding. The physiology of coagulation is somewhat complex insofar as it includes two separate initial pathways, which converge into a final common pathway leading to clot formation. In the final common pathway, prothrombin is converted into thrombin, which in turn converts fibrinogen into fibrin, the latter being the principal building block of cross- linked fibrin polymers which form a hemostatic plug. Of the two initial pathways upstream of the final common pathway, one is known as the contact activation or intrinsic pathway, and the other is known as the tissue factor or extrinsic pathway.
The intrinsic pathway begins with formation of a primary complex on collagen by high-molecular- weight kininogen (HMWK), prekallikrein, and FXII (Factor XII; Hageman factor). Prekallikrein is converted to kallikrein, and FXII is activated to become FXIIa. FXIIa then converts Factor XI (FXI) into FXIa, and FXIa in turn activates Factor IX (FIX), which with its co-factor F Villa form the“tenase” complex, which activates Factor X (FX) to FXa. It is FXa which is responsible for the conversion of prothrombin into thrombin within the final common pathway.
Prekallikrein, the inactive precursor of plasma kallikrein, is synthesized in the liver and circulates in the plasma bound to FDVTWK or as a free zymogen. Prekallikrein is cleaved by activated factor XII(FXIIa) to release activated plasma kallikrein (PK). Activated plasma kallikrein displays endopeptidase activity towards peptide bonds after arginine (preferred) and lysine. PK then generates additional FXIIa in a feedback loop which in turn activates factor XI (FXI) to FXIa to connect to the common pathway. Although the initial activation of the intrinsic pathway is through a small amount of FXIIa activating a small amount of PK, it is the subsequent feedback activation of FXII by PK that controls the extent of activation of the intrinsic pathway and hence downstream coagulation. Hathaway, W. E., et al. (1965) Blood 26:521-32.
Activated plasma kallikrein also cleaves HMWK to release the potent vasodilator peptide bradykinin. It is also able to cleave a number of inactive precursor proteins to generate active products, such as plasmin (from plasminogen) and urokinase (from prourokinase). Plasmin, a regulator of coagulation, proteolytically cleaves fibrin into fibrin degradation products that inhibit excessive fibrin formation.
Patients who have suffered acute myocardial infarction (MI) show clinical evidence of being in a hypercoagulable (clot-promoting) state. This hypercoagulability is
paradoxically additionally aggravated in those receiving fibrinolytic therapy. Increased generation of thrombin, as measured by thrombin-antithrombin III (TAT) levels, is observed in patients undergoing such treatment compared to the already high levels observed in those receiving heparin alone. Hoffmeister, H. M. et al. (1998) Circulation 98:2527-33. The increase in thrombin has been proposed to result from plasmin-mediated activation of the intrinsic pathway by direct activation of FXII by plasmin.
Not only does the fibrinolysis-induced hypercoagulability lead to increased rates of reocclusion, but it is also probably responsible, at least in part, for failure to achieve complete fibrinolysis of the clot (thrombus), a major shortcoming of fibrinolytic therapy (Keeley, E. C. et al. (2003) Lancet 361 : 13-20). Another problem in fibrinolytic therapy is the accompanying elevated risk of intracranial hemorrhage. Menon, V. et al. (2004) (Chest l26:549S-575S; Fibrinolytic Therapy Trialists’ Collaborative Group (1994) Lancet 343 :311-22. Hence, an adjunctive anti -coagulant therapy that does not increase the risk of bleeding, but inhibits the formation of new thrombin, would be greatly beneficial. Plasma kallikrein inhibitors also have therapeutic potential for treating hereditary angioedema (HAE). HAE is is a serious and potentially life-threatening rare genetic illness, caused by mutations in the Cl -esterase inhibitor (C1INH) gene, located on chromosome 1 lq. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by CllNH gene mutations which produce low levels of Cl -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective Cl protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; l00(Suppl2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herein by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that CllNH is the most important inhibitor of kallikrein in human plasma and that CllNH deficiency leads to unopposed activation of the kallikrein- bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack. All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
Therefore, a need exists to develop inhibitors of PK that can tip the balance of fibrinolysis/thrombosis at the occluding thrombus toward dissolution, thereby promoting reperfusion and also attenuating the hypercoagulable state, thus preventing thrombus from reforming and reoccluding the vessel. In particular, the creation of plasma kallikrein inhibitors that are specific and capable of being formulated for in vivo use could lead to a new class of therapeutics. Thus, what is needed are improved compositions and methods for preparing and formulating plasma kallikrein inhibitors.
For example, in patients with angioedema conditions, small polypeptide PK inhibitor DX-88 (ecallantide) alleviates edema in patients with hereditary angioedema (HAE). Williams, A. et al. (2003) Transfus. Apher. Sci. 29:255-8; Schneider, L. et al.
(2007) J Allergy Clin Immunol. 120:416-22; and Levy, J. H. et al. (2006) Expert Opin. Invest. Drugs 15: 1077-90. A bradykinin B2 receptor antagonist, Icatibant, is also effective in treating HAE. Bork, K. et al. (2007) J. Allergy Clin. Immunol. 119:1497-1503. Because plasma kallikrein generates bradykinin, inhibition of plasma kallikrein is expected to inhibit bradykinin production.
For example, in coagulation resulting from fibrinolytic treatment (e.g., treatment with tissue plasminogen activator or streptokinase), higher levels of plasma kallikrein are found in patients undergoing fibrinolysis. Hoffmeister, H. M. et al. (1998) J. Cardiovasc. Pharmacol. 31 :764-72. Plasmin-mediated activation of the intrinsic pathway has been shown to occur in plasma and blood and was markedly attenuated in plasma from individuals deficient in any of the intrinsic pathway components. Ewald, G. A. et al. (1995) Circulation 91 :28-36. Individuals who have had an acute MI were found to have elevated levels of activated plasma kallikrein and thrombin. Hoffmeister, H. M., et al. (1998) Circulation 98:2527-33.
DX-88 reduced brain edema, infarct volume, and neurological deficits in an animal model of ischemic stroke. Storini, C. et al. (2006) J Pharm. Exp. Ther. 318:849-854. Cl- inhibitor reduced infarct size in a mouse model of middle cerebral artery occlusion
(MCAO). De Simoni, M. G. et al. (2004) Am. J. Pathol. 164: 1857-1863; and Akita, N. et al. (2003) Neurosurgery 52:395-400). B2 receptor antagonists were found to reduce the infarct volume, brain swelling, and neutrophil accumulation and were neuroprotective in an MCAO animal model. Zausinger, S. et al. (2003 ) Acta Neurochir. Suppl. 86:205-7;
Lumenta, D. B. et al. (2006) Brain Res. 1069:227-34; Ding-Zhou, L. et al. (2003) Br. J Pharmacol. 139: 1539-47.
Regarding blood loss during cardiopulmonary bypass (CPB), it has been found that the kallikrein-kinin (i.e., contact) system is activated during CABG. Wachtfogel, Y. T. (1989) Blood 73:468. Activation of the contact system during CPB results in up to a 20- fold increase in plasma bradykinin. Cugno, M. et al. (2006) Chest 120:1776-82; and Campbell, D. J. et al. (2001 ) Am. J. Physiol. Reg. Integr. Comp. Physiol. 281 : 1059-70.
Plasma kallikrein inhibitors P8720 and PKSI-527 have also been found to reduce joint swelling in rat models of arthritis. De La Cadena, R. A. et al. (1995) FASEB J. 9:446- 52; Fujimori, Y. (1993) Agents Action 39:42-8. It has also been found that inflammation in animal models of arthritis was accompanied by activation of the contact system. Blais, C. Jr. et al. (1997) Arthritis Rheum. 40: 1327-33.
Additionally, plasma kallikrein inhibitor P8720 has been found to reduce inflammation in an acute and chronic rat model of inflammatory bowel disease (IBD). Stadnicki, A. et al. (1998) FASEB J. 12:325-33; Stadnicki, A. et al. (1996) Dig. Dis. Sci.
41 :9l2-20; and De La Cadena, R. A., et al. (1995) FASEB J. 9:446-52. The contact system is activated during acute and chronic intestinal inflammation. Sartor, R. B. et al. (1996) Gastroenterology 110: 1467-81. It has been found that B2 receptor antagonist, an antibody to high molecular weight kininogen, or reduction in levels of kininogen reduced clinicopathology in animal models of IBD. Ibid !; Arai, Y. et al. (1999) Dig. Dis. Sci.
44:845-51; and Keith, J. C. et al. (2005) Arthritis Res. Therapy 7 :R769-76.
H-D-Pro-Phe-Arg-chloromethylketone (CMK), an inhibitor of PK and FXII and a physiological inhibitor (Cl -inhibitor), has been found to reduce vascular permeability in multiple organs and reduce lesions in lipopolysaccharide (LPS)- or bacterial-induced sepsis in animals. Liu, D. et al. (2005) Blood 105:2350-5; Persson, K. et al. (2000) J. Exp. Med. 192: 1415-24. Clinical improvement was observed in sepsis patients treated with Cl- inhibitor. Zeerleder, S. et al. (2003) Clin. Diagnost. Lab. Immunol. 10:529-35; Caliezi, C., et al. (2002) Crit. Care Med. 30:1722-8; and Marx, G. et al. (1999) Intensive Care Med.
25: 1017-20. Fatal cases of septicemia are found to have a higher degree of contact activation. Martinez-Brotons, F. et al. (1987) Thromb. Haemost. 58:709-713; and Kalter, E. S. et al. (1985) J. Infect. Dis. 151 : 1019-27.
It has also been found that prePK levels are higher in diabetics, especially those with proliferative retinopathy, and correlate with fructosamine levels. Gao, B.-B., et al. (2007) Nature Med. 13: 181-8; and Kedzierska, K. et al. (2005) Archives Med. Res. 36:539- 43. PrePK is also found to be highest in those with a sensorimotor neuropathy. Christie,
M. et al. (1984) Thromb. Haemostas. (Stuttgart) 52:221-3. PrePK levels are elevated in diabetics and are associated with increased blood pressure. PrePK levels independently correlate with the albumin excretion rate and are elevated in diabetics with
macroalbuminuria, suggesting prePK may be a marker for progressive nephropathy. Jaffa, A. A. et al. (2003) Diabetes 52: 1215-21. Bl receptor antagonists have been found to decrease plasma leakage in rats treated with streptozotocin. Lawson, S. R. et al. (2005)
Eur. J. Pharmacol. 514:69-78. Bl receptor antagonists can also prevent streptozotocin- treated mice from developing hyperglycemia and renal dysfunction. Zuccollo, A. et al. (1996) Can. J. Physiol. Pharmacol. 74:586-9.
PATENT
WO 2015134998
https://patents.google.com/patent/WO2015134998A1/en
PATENT
WO 2020092898
https://patents.google.com/patent/WO2020092898A1/en
Example 1 : Synthetic protocol for racemic compound 54e
Reproduced from WO 2015/134998 and U.S. Patent Application Publication No. 2017/0073314 A1 (both incorporated by reference)

Preparation of 1 -(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide
(54e)
Step-l : Preparation of 3-((3-amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b)
To a solution of 3-formylbenzonitrile (54a) (29 g, 217 mmol) in tetrahydrofuran (200 mL) cooled to 0 °C was added freshly prepared Grignard reagent (52c) (245 mL, 221 mmol, ~ 0.9 M in THF) stirred at 0 °C for 1 h, and room temperature for 18 h. The reaction mixture was quenched with 1 N HC1 (aq. 440 mL), stirred for 3 h, neutralized with NaOH (2 N, aq.) to pH = ~ 8. The reaction mixture was extracted with ethyl acetate (600, 300 mL). The combined extracts were washed with brine (120 mL), dried over MgS04, filtered and concentrated in vacuum. The crude product was purified by flash column
chromatography [silica gel, eluting with hexanes/ethyl acetate (1 :0 to 1 : 1) to give 3-((3- amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b) (36.28 g) as a brown gum which was used as such for next step; MS (ES+) 265.3 (M+23).
Step-2: Preparation of tert-butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2- fluorophenylcarbamoyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c)
To a solution of 3-((3-amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b) (24.682 g, 102 mmol) in DMF (480 mL) was added l-(3-((tert- butoxycarbonylamino)methyl)phenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxylic acid (lOd) (35.0 g, 91 mmol), N-ethyl-N-isopropylpropan-2-amine (132 mL, 758 mmol), bromotripyrrolidin-l-ylphosphonium hexafluorophosphate(V) (PyBrOP, 42.8 g, 91 mmol) and stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (1000 mL), washed with water (500, 400 mL), brine (400 mL), dried over MgS04, filtered and concentrated in vacuum. The crude product was purified by flash column chromatography [silica gel, eluting with hexanes/ethyl acetate (1 :0 to 1 : 1)] to afford tert- butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c) (4.583 g, 5% for two steps) as a yellow solid; ¾ NMR (300 MHz, DMSO-i¾) d 10.57 (s, 1H), 7.81 (t, J= 1.7 Hz, 1H), 7.73 – 7.66 (m, 2H), 7.64 – 7.19 (m, 10H), 6.25 (d, J= 4.0 Hz, 1H), 5.78 (d, J= 4.0 Hz, 1H), 4.19 (d, J= 6.1 Hz, 2H), 1.37 (s, 9H); 19F NMR (282 MHz, DMSO-i¾) d -60.81 , -123.09; MS (ES+) 632.3 (M+23).
Step-3: Preparation of tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d)
To a solution of tert-butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2- fluorophenylcarbamoyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c) (1.333 g, 2.187 mmol) in dichloromethane (40 mL) at 0°C was added thionyl chloride (0.340 mL, 4.59 mmol) and warmed to room temperature over 2 h. The reaction mixture was quenched with triethyl amine (2.0 mL, 14.35 mmol) stirred at room temperature for 1 h. It was then treated with cyclopropylmethanamine (4.30 mL, 48.0 mmol), concentrated to remove most of dichloromethane followed by addition of acetonitrile (30 mL), stirring at 70 °C for 14 h, and concentration in vacuum to dryness. The residue was treated with chlorofrom (200 mL), washed with water (100 mL), dried over MgS04 followed by filtration and
concentration. The crude product was purified by flash column chromatography [silica gel eluting with hexanes/ethyl acetate (1 :0 to 2: 1)] to afford tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d) (184 mg, 13%) as colorless gum; ¾ NMR (300 MHz, DMSO-ά) d 10.56 (s, 1H), 7.89 (t, J= 1.7 Hz, 1H), 7.77 – 7.71 (m, 1H), 7.70 – 7.30 (m, 10H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 4.19 (d, J= 6.2 Hz, 2H), 2.26 (d, J= 6.6 Hz, 2H), 1.37 (s, 9H), 1.00 – 0.80 (m, 1H), 0.45 – 0.28 (m, 2H), 0.12 – -0.01 (m, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.80 , -123.20; MS (ES+) 663.4 (M+l). Step-4: Preparation of l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e)
To a solution of tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d) (161 mg, 0.243 mmol) in 1,4- Dioxane (18 mL) was added hydrogen chloride (2.60 mL, 10.40 mmol, 4 M in l,4-dioxane) and stirred at room temperature for 16 h. the reaction mixture was treated with hexanes, decanted, washed with hexanes, and decanted again. The insoluble crude product was purified by flash column chromatography [silica gel, eluting with chloroform/CMA80 (1 :0 to 2:1)] to afford l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e). The pure product was dissolved in methanol (10 mL) and added 4 N HC1 (aq. 0.14 mL) followed by concentration in vacuum to dryness to give HC1 salt of l-(3- (aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl-methylamino)methyl)-2- fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e) (74 mg, 48%) white solid; ¾ NMR (300 MHz, DMSO- d, D20 ex NMR) d 8.13 (t, J = 1.7 Hz, 1H), 7.98 – 7.84 (m, 3H), 7.73 – 7.64 (m, 3H), 7.63 – 7.48 (m, 4H), 7.44 (dd, J = 10.2, 8.6 Hz, 1H),
5.75 (s, 1H), 4.12 (s, 2H), 2.76 (d, J = 7.2 Hz, 2H), 1.17 – 0.94 (m, 1H), 0.68 – 0.47 (m, 2H), 0.34-0.24 (m, 2H); 19F NMR (282 MHz, DMSO- d) d -60.82, -120.02; MS (ES+): 563.3 (M+l); Analysis calculated for C30H26F4N6O2.O HCT3.0 H2O: C, 52.26; H, 4.97; N, 12.19; Found: C, 52.26; H, 5.00; N, 11.72.
Example 2: Separation of enantiomers of racemic compound 54e
Reproduced from WO 2015/134998 and U.S. Patent Application Publication No. 2017/0073314 A1 (both incorporated by reference)

Compound I (free base) Separation of (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lFl-pyrazole-5-carboxamide (Compound I), and (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lFl-pyrazole-5-carboxamide ((-
)-enantiomer)
Isomers of Racemic l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lF[- pyrazole-5-carboxamide (54e) (0.4 g) were separated by using preparative SFC method using the following conditions to furnish:
Preparative SFC Method used:
Column 20mm x 25.0 cm ChromegaChiral CCS from
Regis Technologies (Morton Grove, IL)
CO2 Co-solvent (Solvent B) Methanol: Isopropanol (1 : 1) with 1%
Isopropylamine
Isocratic Method 20 % Co-solvent at 80 mL/min
System Pressure 200 bar
Column Temperature 25 °C
Sample Diluent Methanol: Isopropanol
Chiral Purity of peaks was determined by following Analytical SFC Method:
Column 4.6 x 100 mm ChiralPak AS from Chiral
Technologies (West Chester, PA)
CO2 Co-solvent (Solvent B) Methanol: Isopropanol (1 : 1) with 0.1%
Isopropylamine
Isocratic Method 5-65 % Co-solvent Gradient at 4 mL/min System Pressure 100 bar
Column Temperature 25 °C
Sample Diluent Methanol
Peak-l (Compound I) 2.1 min 144 mg >95% ee (UV 254)
98.6 % purity (UV 254)
Peak-2 ((-)-enantiomer) 2.4 min 172 mg 95.5 % ee (UV 254)
96.5 % purity (UV 254) 1. Peak-l assigned as (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3- (trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (144 mg, >95%ee) free base as white solid; Optical rotation: [O]D = (+) 6.83 [CH3OH, 1.2]; ‘H NMR (300 MHz, DMSO-£¾) d 10.53 (s, 1H, D2O exchangeable), 7.88 (t, J= 1.7 Hz, 1H), 7.77 – 7.71 (m, 1H), 7.67 (dt, 7= 7.7, 1.4 Hz, 1H), 7.63 (dd, J= 7.5, 2.1 Hz, 1H), 7.56 (s, 1H), 7.54 – 7.47 (m, 2H), 7.47 – 7.38 (m, 2H), 7.34 (ddt, J= 8.6, 5.9, 2.8 Hz, 2H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 3.77 (s, 2H), 2.31 – 2.21 (m, 2H), 0.97 – 0.80 (m, 1H), 0.42 – 0.33 (m, 2H), 0.10 – -0.02 (m, 2H); 19F NMR (282 MHz, DMSO-Ts) d -60.73 , -123.20; MS (ES+) 563.3 (M+l), 561.3 (M-l). To a solution of free base mixture of (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3- (trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (120 mg) in methanol (15 mL) was added hydrogen chloride (0.969 mL, 1.938 mmol), stirred at room temperature for 10 min, evaporated to dryness to afford (+)-l-(3- (aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl-methylamino)methyl)-2- fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (100 mg) hydrochloride salt as white solid; ¾ NMR (300 MHz, DMSO-Ts) d 10.84 (s, 1H, D2O exchangeable), 10.44 (s, 2H, D2O exchangeable), 8.44 (s, 3H, D2O exchangeable), 8.30 (s, 1H, D2O exchangeable), 8.09 (d, J= 7.9 Hz, 1H), 7.99 (d, J = 6.8 Hz, 1H), 7.91 – 7.83 (m, 1H), 7.80 – 7.50 (m, 7H), 7.42 (dd, J= 10.3, 8.6 Hz, 1H), 5.78 (d, J= 6.9 Hz, 1H), 4.13 (d, J= 5.7 Hz, 2H), 2.88 – 2.62 (m, 2H), 1.42 – 0.99 (m, 1H), 0.73 – 0.46 (m, 2H), 0.32 (d, J= 4.4 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.81 , -119.99; MS (ES+): MS (ES+) 563.3 (M+l), MS (ES-) 561.3 (M-l), 597.3 (M+Cl); Analysis calculated for C30H26F4N6O 2HC1 l.75H20: C, 54.02; H, 4.76; Cl, 10.63; N, 12.60; Found: C, 54.12; H, 4.83; Cl, 10.10; N, 11.97. Peak-2 assigned as (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (172 mg, 95.5 % ee) as free base was repurified by flash column chromatography (silica gel 12 g, eluting 0-30% MeOH in chloroform for 15 min) to afford (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) free base as an off-white solid; Optical rotation: [O]D = (-) 5.44
[CH3OH, 1.25]; ¾ NMR (300 MHz, DMSO-i¾) d 7.88 (t, J= 1.6 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.70 – 7.61 (m, 2H), 7.57 (s, 1H), 7.54 – 7.47 (m, 2H), 7.45 – 7.41 (m,
2H), 7.34 (ddq, J= 8.7, 6.1, 3.5, 2.8 Hz, 2H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 3.78 (s, 2H), 2.25 (d, J= 6.9 Hz, 2H), 0.90 (ddd, J= 9.8, 8.0, 5.2 Hz, 1H), 0.47 – 0.29 (m, 2H), 0.04 (dd, J= 5.0, 1.5 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.73 , -123.19; MS (ES+) 563.3 (M+l), MS (ES-), 561.3 (M-l). To a solution of free base of (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (0.124 g, 0.220 mmol) in methanol (15 mL) was added hydrogen chloride (1.102 mL, 2.204 mmol), stirred at room temperature for 10 min, evaporated to dryness to afford (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (0.121 g) hydrochloride salt as an off-white solid; Ή NMEE ¾ NMR (300 MHz, DMSO-i¾) d 10.82 (s, 1H, D20 exchangeable), 10.36 (s, 2H, D2O exchangeable), 8.38 (s, 3H, D2O exchangeable), 8.27 (s, 1H), 8.06 (d, J= 7.9 Hz, 1H), 7.98 (d, J= 6.7 Hz, 1H), 7.87 (d, J= 7.7 Hz, 1H), 7.78 – 7.49 (m, 7H), 7.48 – 7.37 (m, 1H), 5.78 (s, 1H), 4.13 (d, j= 5.7 Hz, 2H), 2.72 (s, 2H), 1.14 (s, 1H), 0.56 (d, j= 7.7 Hz, 2H), 0.31 (d, J= 5.0 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.82 , -120.03; MS (ES+): MS (ES+) 563.3 (M+l), MS (ES-), 561.3 (M-l), 597.2 (M+Cl); Analysis calculated for C30H26F4N6O.2HCI. I .75H2O: C, 54.02; H, 4.76; Cl, 10.63; N, 12.60; Found: C, 54.12; H, 4.83; Cl, 10.10; N, 11.97.
Example 3 : Preparation of a Seed Crystal of Compound I*2

A solution of Compound I ( see Example 2) in methyl tert-butyl ether (MTBE) (1 equiv) is added to a solution of HC1 (aq) (2 equiv) in methanol (cold), followed by heating to about 30°C, and keeping it at about 30°C for not longer than 5 hours while stirring at about 115 rpm. Compound I bis(HCl) is collected by filtration and dried. The crystalline material obtained can be used as a seed for the crystallization protocol described in
Example 4. Example 4: Large-Scale Synthetic & Crystallization Protocol for Compound I*2(HC1 )

Compound I (free base) Compound I bis(HCI)
37% Aqueous hydrochloric acid (38.1 kg, 32.3 L, 2.14 equiv.) was charged to a clean and empty crystallization vessel, methanol (228.9 kg, 39.5 equiv.) was added, and the contents were cooled to -7 ± 3°C. A solution of Compound I free base (approx. 101.8 kg; 180.9 moles) in MTBE (approx. 1,300 L) was filtered through a polish filter into the crystallization vessel at temperature -5 ± 5°C. After rinse with MTBE, pre-weighed Compound I»2(HCl) seed crystals (1.39 kg, 0.012 equiv.; Example 3) were charged to the crystallization vessel via the manhole. The vessel content was heated to 30-33°C, and the agitation speed was set to 25-50 rpm. After confirmed crystallization, the slurry was agitated for another three to four hours. The product slurry was transferred to centrifuge and isolated by centrifugation. The product was washed with MTBE (585 L). After dry spinning the wet product, Compound I*2(HC1), it was discharged from the centrifuge, and the product was dried at < 40°C under vacuum in a cone drier. Product Compound I»2(HCl) yield: 100 kg; 157.4 mol; approx. 85%.
‘H NMR (300 MHz, DMSO-c/i,) data is shown in the following table:

19F NMR (282 MHz, DMSO- is) data is shown in the following table:

Compound I has two basic sites. The conjugate acid of the primary amine was calculated to have a pKa value of 8.89, and the conjugate acid of the secondary amine was calculated to have a pKa value of 7.86.
The XRPD pattern of Compound I»2(HCl) is shown in Fig. 1. Compound I»2(HCl) has characteristic peaks in its XRPD pattern at values of two theta (°2Q) of 5.28, 8.96, 14.27, 16.18, 19.79, 21.16, 22.01, 23.31, 24.64, and 30.31. TG-IR analysis indicated two, distinct weight loss regions: the first was completed by 125 °C while the second began at approximately 208 °C. IR analysis of the off gasses from this experiment detected only trace amounts of water at the initial weight loss while HC1 gas was detected at the 208°C event. No other solvents were detected in the sample. Thus, it was determined that Compound I*2(HC1) initially loses water when heated and, when heated to above 200°C, the salt begins to break apart and HC1 gas is evolved. The IR signal for all these events is very weak indicating that they are occurring over a range and not at a specified temperature. An exemplary TG-IR spectrum is shown in Fig. 2.
REFERENCES
1: Sohtome Y, Sodeoka M. Development of Chaetocin and S-Adenosylmethionine Analogues as Tools for Studying Protein Methylation. Chem Rec. 2018 Dec;18(12):1660-1671. doi: 10.1002/tcr.201800118. Epub 2018 Oct 16. Review. PubMed PMID: 30324709.
2: Bensussen A, Torres-Sosa C, Gonzalez RA, Díaz J. Dynamics of the Gene Regulatory Network of HIV-1 and the Role of Viral Non-coding RNAs on Latency Reversion. Front Physiol. 2018 Sep 28;9:1364. doi: 10.3389/fphys.2018.01364. eCollection 2018. PubMed PMID: 30323768; PubMed Central PMCID: PMC6172855.
////////berotralstat, Orladeyo, BIOCRYST, APPROVALS 2020, FDA 2020, ORPHAN DRUG, CX-7353, CX 7353,
NCc1cccc(c1)n2nc(cc2C(=O)Nc3cc(ccc3F)[C@H](NCC4CC4)c5cccc(c5)C#N)C(F)(F)F
Viltolarsen
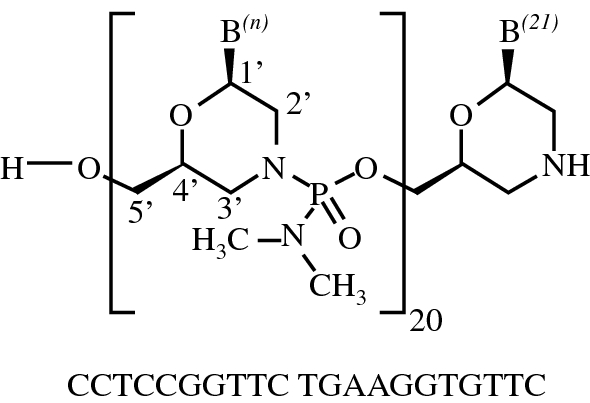
Viltolarsen
维托拉生
ビルトラルセン
| Formula | C244H381N113O88P20 |
|---|---|
| CAS | 2055732-84-6 |
| Mol weight | 6924.8155 |
APPROVED FDA 2020/8/12, Viltepso
APPROVED JAPAN PMDA 2020/3/25, VILTEPSO
- NCNP-01
- NS-065
- NS-065/NCNP-01
- WHO 10771
- WHO-10771
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Viltepso | Injection, solution | 250 mg/1 | Intravenous | Ns Pharma, Inc. | 2020-08-13 | Not applicable |  |
SYNWatanabe N, Nagata T, Satou Y, Masuda S, Saito T, Kitagawa H, Komaki H, Takagaki K, Takeda S: NS-065/NCNP-01: An Antisense Oligonucleotide for Potential Treatment of Exon 53 Skipping in Duchenne Muscular Dystrophy. Mol Ther Nucleic Acids. 2018 Dec 7;13:442-449. doi: 10.1016/j.omtn.2018.09.017.
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US9079934 | No | 2011-08-31 | 2031-08-31 | |
Viltolarsen
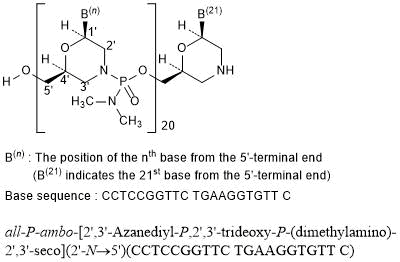
all-P-ambo-[2′,3′-Azanediyl-P,2′,3′-trideoxy-P-(dimethylamino)-2′,3′-seco](2′-N→5′)(CCTCCGGTTC TGAAGGTGTT C)
C244H381N113O88P20 : 6924.82
[2055732-84-6]
Viltolarsen, sold under the brand name Viltepso, is a medication used for the treatment of Duchenne muscular dystrophy (DMD).[3][4][2] Viltolarsen is an antisense oligonucleotide.[3][2]
The most common side effects include upper respiratory tract infection, injection site reaction, cough, and pyrexia (fever).[3][4][2]
Viltolarsen was approved for medical use in the United States in August 2020.[3][4] After golodirsen was approved in December 2019, viltolarsen is the second approved targeted treatment for people with this type of mutation in the United States.[3][5] Approximately 8% of people with DMD have a mutation that is amenable to exon 53 skipping.[3]
Medical uses
Viltolarsen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping.[3][2]
DMD is a rare genetic disorder characterized by progressive muscle deterioration and weakness.[3] It is the most common type of muscular dystrophy.[3] DMD is caused by mutations in the DMD gene that results in an absence of dystrophin, a protein that helps keep muscle cells intact.[3] The first symptoms are usually seen between three and five years of age and worsen over time.[3] DMD occurs in approximately one out of every 3,600 male infants worldwide; in rare cases, it can affect females.[3]
Adverse effects
The most common side effects include upper respiratory tract infection, injection site reaction, cough, and pyrexia (fever).[3][4][2]
Although kidney toxicity was not observed in the clinical studies, the clinical experience is limited, and kidney toxicity, including potentially fatal glomerulonephritis, has been observed after administration of some antisense oligonucleotides.[3]
History
Viltolarsen was evaluated in two clinical studies with a total of 32 participants, all of whom were male and had genetically confirmed DMD.[3] The increase in dystrophin production was established in one of those two studies, a study that included sixteen DMD participants, with eight participants receiving viltolarsen at the recommended dose.[3] In the study, dystrophin levels increased, on average, from 0.6% of normal at baseline to 5.9% of normal at week 25.[3] Trial 1 provided data for evaluation of the benefits of viltolarsen.[4] The combined populations from both trials provided data for evaluation of the side effects of viltolarsen.[4] Trial 1 was conducted at six sites in the United States and Canada and Trial 2 was conducted at five sites in Japan.[4] All participants in both trials were on a stable dose of corticosteroids for at least three months before entering the trials.[4]
The U.S. Food and Drug Administration (FDA) concluded that the applicant’s data demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit in people with DMD who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping.[3] A clinical benefit of the drug has not been established.[3] In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of available therapies.[3]
The application for viltolarsen was granted priority review designation and the FDA granted the approval to NS Pharma, Inc.[3]
References
- ^ https://www.drugs.com/pregnancy/viltolarsen.html
- ^ Jump up to:a b c d e f “Viltepso- viltolarsen injection, solution”. DailyMed. 12 August 2020. Retrieved 18 August 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u “FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation”. U.S. Food and Drug Administration (FDA) (Press release). 12 August 2020. Retrieved 12 August 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h “Drug Trials Snapshots: Viltepso”. U.S. Food and Drug Administration. 12 August 2020. Retrieved 18 August 2020. This article incorporates text from this source, which is in the public domain.
- ^ Anwar S, Yokota T (August 2020). “Golodirsen for Duchenne muscular dystrophy”. Drugs of Today. 56 (8): 491–504. doi:10.1358/dot.2020.56.8.3159186. PMID 33025945.
Further reading
- Dhillon S (July 2020). “Viltolarsen: First Approval”. Drugs. 80 (10): 1027–1031. doi:10.1007/s40265-020-01339-3. PMID 32519222. S2CID 219542850.
- Dzierlega K, Yokota T (June 2020). “Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy”. Gene Ther. 27 (9): 407–416. doi:10.1038/s41434-020-0156-6. PMID 32483212. S2CID 219157034.
- Hwang J, Yokota T (October 2019). “Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies”. Expert Rev Mol Med. 21: e5. doi:10.1017/erm.2019.5. PMID 31576784.
- Roshmi RR, Yokota T (October 2019). “Viltolarsen for the treatment of Duchenne muscular dystrophy”. Drugs Today. 55 (10): 627–639. doi:10.1358/dot.2019.55.10.3045038. PMID 31720560.
External links
- “Viltolarsen”. Drug Information Portal. U.S. National Library of Medicine (NLM).
- Clinical trial number NCT02740972 for “Safety and Dose Finding Study of NS-065/NCNP-01 in Boys With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Viltepso |
| Other names | NS-065/NCNP-01 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Viltolarsen |
| Pregnancy category | US: N (Not classified yet)[1] |
| Routes of administration | Intravenous |
| Drug class | Antisense oligonucleotide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 2055732-84-6 |
| DrugBank | DB15005 |
| ChemSpider | 71115970 |
| UNII | SXA7YP6EKX |
| KEGG | D11528 |
| ChEMBL | ChEMBL4298062 |
| Chemical and physical data | |
| Formula | C244H381N113O88P20 |
| Molar mass | 6924.910 g·mol−1 |
//////////Viltolarsen, Viltepso, 维托拉生 , FDA 2020, EU 2020, APPROVALS 2020, NCNP-01, NS-065, NS-065/NCNP-01, WHO 10771, WHO-10771, ビルトラルセン
Pemigatinib


Pemigatinib
INCB054828
| Formula | C24H27F2N5O4 |
|---|---|
| CAS | 1513857-77-62379919-96-5 HCL |
| Mol weight | 487.4991 |
2020/4/17FDA APPROVED, PEMAZYRE
佩米替尼 [Chinese] [INN]
3-(2,6-Difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholinomethyl)-1,3,4,6-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
2H-Pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one, 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-8-(4-morpholinylmethyl)-
3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
- Originator Incyte Corporation
- Developer Incyte Corporation; Innovent Biologics
- ClassAntineoplastics; Ethers; Fluorobenzenes; Morpholines; Pyridines; Pyrimidinones; Pyrroles; Small molecules
- Mechanism of Action Type 1 fibroblast growth factor receptor antagonists; Type 3 fibroblast growth factor receptor antagonists; Type 4 fibroblast growth factor receptor antagonists; Type-2 fibroblast growth factor receptor antagonists
- Orphan Drug Status Yes – Myeloproliferative disorders; Lymphoma; Cholangiocarcinoma
- MarketedCholangiocarcinoma
- Phase IIBladder cancer; Lymphoma; Myeloproliferative disorders; Solid tumours; Urogenital cancer
- Phase I/IICancer
- 05 Nov 2020Preregistration for Cholangiocarcinoma (Late-stage disease, Metastatic disease, First line therapy, Inoperable/Unresectable) in Japan (PO) in November 2020
- 05 Nov 2020Incyte Corporation stops enrolment in the FIGHT-205 trial for Bladder cancer due to regulatory feedback
- 26 Oct 2020Preregistration for Cholangiocarcinoma (Second-line therapy or greater, Inoperable/Unresectable, Late-stage disease, Metastatic disease) in Canada (PO)
Pemigatinib, also known as INCB054828, is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) types 1, 2, and 3 (FGFR1/2/3), with potential antineoplastic activity. FGFR inhibitor INCB054828 binds to and inhibits FGFR1/2/3, which may result in the inhibition of FGFR1/2/3-related signal transduction pathways. This inhibits proliferation in FGFR1/2/3-overexpressing tumor cells.
Pemigatinib (INN),[2] sold under the brand name Pemazyre, is a medication for the treatment of adults with previously treated, unresectable locally advanced or metastatic bile duct cancer (cholangiocarcinoma) with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test.[3][4] Pemigatinib works by blocking FGFR2 in tumor cells to prevent them from growing and spreading.[3]
Pemigatinib belongs to a group of medicines called protein kinase inhibitors.[5] It works by blocking enzymes known as protein kinases, particularly those that are part of receptors (targets) called fibroblast growth factor receptors (FGFRs).[5] FGFRs are found on the surface of cancer cells and are involved in the growth and spread of the cancer cells.[5] By blocking the tyrosine kinases in FGFRs, pemigatinib is expected to reduce the growth and spread of the cancer.[5]
The most common adverse reactions are hyperphosphatemia and hypophosphatemia (electrolyte disorders), alopecia (spot baldness), diarrhea, nail toxicity, fatigue, dysgeusia (taste distortion), nausea, constipation, stomatitis (sore or inflammation inside the mouth), dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain and dry skin.[3][4] Ocular (eye) toxicity is also a risk of pemigatinib.[3][4]
Medical uses
Cholangiocarcinoma is a rare form of cancer that forms in bile ducts, which are slender tubes that carry the digestive fluid bile from the liver to gallbladder and small intestine.[3] Pemigatinib is indicated for the treatment of adults with bile duct cancer (cholangiocarcinoma) that is locally advanced (when cancer has grown outside the organ it started in, but has not yet spread to distant parts of the body) or metastatic (when cancer cells spread to other parts of the body) and who have tumors that have a fusion or other rearrangement of a gene called fibroblast growth factor receptor 2 (FGFR2).[3] It should be used in patients who have been previously treated with chemotherapy and whose cancer has a certain type of abnormality in the FGFR2 gene.[6]
History
Pemigatinib was approved for use in the United States in April 2020 along with the FoundationOne CDX (Foundation Medicine, Inc.) as a companion diagnostic for patient selection.[3][4][7]
The approval of pemigatinib in the United States was based on the results the FIGHT-202 (NCT02924376) multicenter open-label single-arm trial that enrolled 107 participants with locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or rearrangement who had received prior treatment.[3][4][6] The trial was conducted at 67 sites in the United States, Europe, and Asia.[6] During the clinical trial, participants received pemigatinib once a day for 14 consecutive days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects.[3][4][6] To assess how well pemigatinib was working during the trial, participants were scanned every eight weeks.[3] The trial used established criteria to measure how many participants experienced a complete or partial shrinkage of their tumors during treatment (overall response rate).[3] The overall response rate was 36% (95% CI: 27%, 45%), with 2.8% of participants having a complete response and 33% having a partial response.[3] Among the 38 participants who had a response, 24 participants (63%) had a response lasting six months or longer and seven participants (18%) had a response lasting 12 months or longer.[3][4]
The U.S. Food and Drug Administration (FDA) granted the application for pemigatinib priority review, breakthrough therapy and orphan drug designations.[3][4][8][9] The FDA granted approval of Pemazyre to Incyte Corporation.[3]
On 24 August 2018, orphan designation (EU/3/18/2066) was granted by the European Commission to Incyte Biosciences Distribution B.V., the Netherlands, for pemigatinib for the treatment of biliary tract cancer.[5] On 17 October 2019, orphan designation EU/3/19/2216 was granted by the European Commission to Incyte Biosciences Distribution B.V., the Netherlands, for pemigatinib for the treatment of myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB, or FGFR1, or with PCM1-JAK2.[10]
PATENT
US 20200281907
| The present disclosure is directed to, inter alia, methods of treating cancer in a patient in need thereof, comprising administering pemigatinib, which is 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one, having the structure shown below: |
Pemigatinib is described in U.S. Pat. No. 9,611,267, the entirety of which is incorporated herein by reference. Pemigatinib is further described in US Publication Nos.: 2019/0337948 and 2020/0002338, the entireties of which are incorporated herein by reference.
| Provided herein is a method of treating cancer comprising administering a therapy to a patient in need thereof, wherein the therapy comprises administering a therapeutically effective amount of pemigatinib to the patient while avoiding the concomitant administration of a CYP3A4 perpetrator. |
Example 1. Synthesis of Pemigatinib
Step 1: 4-(ethylamino)-1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde
| A mixture of 4-chloro-1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde (CAS #958230-19-8, Lakestar Tech, Lot: 124-132-29: 3.0 g, 17 mmol) and ethylamine (10M in water, 8.3 mL, 83 mmol) in 2-methoxyethanol (20 mL, 200 mmol) was heated to 130° C. and stirred overnight. The mixture was cooled to room temperature then concentrated under reduced pressure. The residue was treated with 1N HCl (30 mL) and stirred at room temperature for 1 h then neutralized with saturated NaHCO 3 aqueous solution. The precipitate was collected via filtration then washed with water and dried to provide the desired product (2.9 g, 92%). LC-MS calculated for C 10H 12N 3O [M+H] + m/z: 190.1; found: 190.1. |
Step 2: 5-{[(2,6-difluoro-3,5-dimethoxyphenyl)amino]methyl}-N-ethyl-1H-pyrrolo[2,3-b]pyridin-4-amine
| A mixture of 4-(ethylamino)-1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde (7.0 g, 37 mmol), 2,6-difluoro-3,5-dimethoxyaniline (9.1 g, 48 mmol) and [(1S)-7,7-dimethyl-2-oxobicyclo[2.2.1]hept-1-yl]methanesulfonic acid (Aldrich, cat #21360: 2 g, 7 mmol) in xylenes (250 mL) was heated to reflux with azeotropic removal of water using Dean-Stark for 2 days at which time LC-MS showed the reaction was complete. The mixture was cooled to room temperature and the solvent was removed under reduced pressure. The residue was dissolved in tetrahydrofuran (500 mL) and then 2.0 M lithium tetrahydroaluminate in THF (37 mL, 74 mmol) was added slowly and the resulting mixture was stirred at 50° C. for 3 h then cooled to room temperature. The reaction was quenched by addition of water, 15% aqueous NaOH and water. The mixture was filtered and washed with THF. The filtrate was concentrated and the residue was washed with CH 2Cl 2 and then filtered to get the pure product (11 g, 82%). LC-MS calculated for C 18H 21F 2N 4O 2[M+H] + m/z: 363.2; found: 363.1. |
Step 3: 3-(2,6-Difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
| A solution of triphosgene (5.5 g, 18 mmol) in tetrahydrofuran (30 mL) was added slowly to a mixture of 5-{[(2,6-difluoro-3,5-dimethoxyphenyl)amino]methyl}-N-ethyl-1H-pyrrolo[2,3-b]pyridin-4-amine (5.6 g, 15 mmol) in tetrahydrofuran (100 mL) at 0° C. and then the mixture was stirred at room temperature for 6 h. The mixture was cooled to 0° C. and then 1.0 M sodium hydroxide in water (100 mL, 100 mmol) was added slowly. The reaction mixture was stirred at room temperature overnight and the formed precipitate was collected via filtration, washed with water, and then dried to provide the first batch of the purified desired product. The organic layer in the filtrate was separated and the aqueous layer was extracted with methylene chloride. The combined organic layer was concentrated and the residue was triturated with methylene chloride then filtered and dried to provide another batch of the product (total 5.5 g, 92%). LC-MS calculated for C 19H 19F 2N 4O 3[M+H] + m/z: 389.1; found: 389.1. |
Step 4: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
| To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (900 mg, 2.32 mmol) in N,N-dimethylformamide (20 mL) cooled to 0° C. was added sodium hydride (185 mg, 4.63 mmol, 60 wt % in mineral oil). The resulting mixture was stirred at 0° C. for 30 min then benzenesulfonyl chloride (0.444 mL, 3.48 mmol) was added. The reaction mixture was stirred at 0° C. for 1.5 h at which time LC-MS showed the reaction completed to the desired product. The reaction was quenched with saturated NH 4Cl solution and diluted with water. The white precipitate was collected via filtration then washed with water and hexanes, dried to afford the desired product (1.2 g, 98%) as a white solid which was used in the next step without further purification. LC-MS calculated for C 25H 23F 2N 4O 5S [M+H] + m/z: 529.1; found: 529.1. |
Step 5: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde
| To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one (1.75 g, 3.31 mmol) in tetrahydrofuran (80 mL) at −78° C. was added freshly prepared lithium diisopropylamide (1M in tetrahydrofuran (THF), 3.48 mL, 3.48 mmol). The resulting mixture was stirred at −78° C. for 30 min then N,N-dimethylformamide (1.4 mL, 18 mmol) was added slowly. The reaction mixture was stirred at −78° C. for 30 min then quenched with water and extracted with EtOAc. The organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 20% EtOAc in DCM to give the desired product as a white solid (1.68 g, 91%). LC-MS calculated for C 26H 23F 2N 4O 6S (M+H) + m/z: 557.1; found: 556.9. |
Step 6: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
| To a solution 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde (1.73 g, 3.11 mmol) in dichloromethane (50 mL) was added morpholine (0.95 mL, 11 mmol), followed by acetic acid (2 mL, 30 mmol). The resulting yellow solution was stirred at room temperature overnight then sodium triacetoxyborohydride (2.3 g, 11 mmol) was added. The mixture was stirred at room temperature for 3 h at which time LC-MS showed the reaction went to completion to the desired product. The reaction was quenched with saturated NaHCO 3 then extracted with ethyl acetate (EtOAc). The organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 40% EtOAc in DCM to give the desired product as a yellow solid (1.85 g, 95%). LC-MS calculated for C 30H 32F 2N 5O 6S (M+H) + m/z: 628.2; found: 628.0. |
Step 7: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one (pemigatinib)
| To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one (1.5 g, 2.4 mmol) in tetrahydrofuran (40 mL) was added tetra-n-butylammonium fluoride (1M in THF, 7.2 mL, 7.2 mmol). The resulting solution was stirred at 50° C. for 1.5 h then cooled to room temperature and quenched with water. The mixture was extracted with dichloromethane (DCM) and the organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 10% MeOH in DCM to give the desired product as a white solid, which was further purified by prep HPLC (pH=2, acetonitrile/H 2O). LC-MS calculated for C 24H 28F 2N 5O 4 (M+H) + m/z: 488.2; found: 488.0. 1H NMR (500 MHz, DMSO) δ 12.09 (s, 1H), 8.06 (s, 1H), 7.05 (t, J=8.1 Hz, 1H), 6.87 (s, 1H), 4.78 (s, 2H), 4.50 (s, 2H), 4.17 (q, J=6.8 Hz, 2H), 3.97 (br, 2H), 3.89 (s, 6H), 3.65 (br, 2H), 3.37 (br, 2H), 3.15 (br, 2H), 1.37 (t, J=6.8 Hz, 3H). |
PATENT
WO 2019213506
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019213506
PATENT
WO 2019213544
The present disclosure is directed to, inter alia, solid forms, including crystalline forms and amorphous forms, of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)- 1 ,3,4,7 -tetrahydro-2H-pyrrolo [3 ‘,2’ : 5 ,6]pyrido [4,3 -d]pyrimidin-2-one
(Compound 1), and processes and intermediates for preparing the compound. The structure of Compound 1 is shown below.
Compound 1
Compound 1 is described in US Patent No. 9,611,267, the entirety of which is incorporated herein by reference.
Example 1
Synthesis of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-l^, 4,7-tetrahydro-2H-pyrrolo[3f,2f:5,6]pyrido[4r3-d]pyrimidin-2-one (Compound 1) Scheme 1.
Step 1: Synthesis of 4-((4-chloro-5-(l, 3-dioxolan-2-yl)-l-(phenylsulfonyl)-lH-pyrrolo[2, 3-b ] pyridin-2-yl) methyl) morpholine
To a l-L flask was added 4-chloro-5-(l,3-dioxolan-2-yl)-l-(phenylsulfonyl)-lH-pyrrolo [2,3-b] pyridine (50.0 g, 137 mmol) (see, e.g., Example 2) and tetrahydrofuran (THF, 266 g, 300 mL) under N2. To this mixture at -70 °C was added 2.0 M lithium
diisopropylamide in THF/heptane/ethyl benzene (77.4 g, 95 mL, 190 mmol, 1.4 eq.). The mixture was stirred at -70 °C for 1 h. To the mixture was added /V- formyl morpholine (29.7 g, 258 mmol, 1.9 eq.) in THF (22. 2 g, 25 mL) dropwise. The reaction was done in 30 min after addition. LC/MS showed that the desired product, 4-chloro-5-(l, 3-dioxolan-2-yl)-l-(phenylsulfonyl)- 1 //-pyrrolo [2, 3-61 pyridine-2-carbaldehyde, was formed cleanly. The reaction was quenched with acetic acid (16.4 g, 15.6 mL, 274 mmol, 2.0 eq.) and the dry ice cooling was removed. To the mixture was added morpholine (33.7 g, 33.5 mL, 387 mmol, 2.83 eq.) followed by acetic acid (74.0 g, 70 mL, 1231 mmol, and 9.0 eq.) at 0 °C (internal temperature rose from 0 °C to 18 °C) and stirred overnight. Sodium triacetoxyborohydride (52.50 g, 247.7 mmol, 1.8 eq.) was added and the reaction mixture temperature rose from 20 °C to 32 °C. The mixture was stirred at room temperature for 30 min. HPLC & LC/MS indicated the reaction was complete. Water (100 g, 100 mL) was added followed by 2.0 M sodium carbonate (Na2C03) in water (236 g, 200 mL, 400 mmol, 2.9 eq.) slowly (off gas!). The mixture was stirred for about 30 min. The organic layer was separated and water (250 g, 250 mL) and heptane (308 g, 450 mL) were added. The resulting slurry was stirred for 1 h and the solid was collected by filtration. The wet cake was washed with heptane twice (75.00 mL x 2, 51.3 g x 2) before being dried in oven at 50 °C overnight to give the desired product, 4-((4-chloro-5-( 1 3-dioxolan-2-yl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridin-2-yl)methyl)morpholine as a light brown solid (52.00 g, 81.8 % yield): LCMS calculated for C21H23CIN2O5S [M+H]+: 464.00; Found: 464.0; ftf NMR ^OO MHz, DMSO-de) d 8.48 (s, 1 H), 8.38 (m, 2H), 7.72 (m, 1H), 7.64 (m, 2H), 6.83 (s, 1H), 6.13 (s, 1H), 4.12 (m, 2H), 4.00 (m, 2H), 3.92 (s, 2H), 3.55 (m, 4H), 2.47 (m, 4H).
Step 2: Synthesis of 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-lH-pyrrolo[2, 3-b] pyridine-5 -carbaldehyde
To a 2 L reactor with a thermocouple, an addition funnel, and a mechanical stirrer was charged 4-((4-chloro-5 -(1 ,3 -dioxolan-2-yl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo [2,3 -6]pyridin-2-yl)methyl)morpholine (20.00 g, 43.1 mmol) and dichloromethane (265 g, 200 mL) at room temperature. The resulting mixture was stirred at room temperature (internal temperature
was 19.5 °C) to achieve a solution. To the resulting solution was added an aqueous hydrochloric acid solution (0.5 M, 240 g, 200.0 ml, 100 mmol, 2.32 eq.) at room temperature in 7 min. After over 23 h agitations at room temperature, the bilayer reaction mixture turned into a thick colorless suspension. When HPLC showed the reaction was complete, the slurry was cooled to 0-5 °C and aqueous sodium hydroxide solution (1 N, 104 g, 100 mL, 100 mmol, and 2.32 eq.) was added in about 10 min to adjust the pH of the reaction mixture to 10-11. «-Heptane (164 g, 240 mL) was added and the reaction mixture and the mixture were stirred at room temperature for 1 h. The solid was collected by filtration and the wet cake was washed with water (2 x 40 mL), heptane (2 x 40 ml) before being dried in oven at 50 °C under vacuum to afford the desired product, 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-/i |pyridine-5-carbaldehyde as a light brown solid (16.9 g, 93% yield): LCMS calculated for C19H19CIN3O4S [M+H]+: 420.00; Found: 420.0; ¾ NMR (400 MHz, DMSO-de) d 10.33 (s, 1H), 8.76 (s, 1 H), 8.42 (m, 2H), 7.74 (m, 1H), 7.65 (m, 2H), 6.98 (s, 1H), 3.96 (m, 2H), 3.564 (m, 4H), 2.51 (m, 4H).
Step 3: Synthesis ofN-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-lH-pyrrolo [2, 3-h] pyridin-5-yl) methyl) -2, 6-difluoro-3,5-dimethoxyaniline
To a 2-L reactor equipped with a thermocouple, a nitrogen inlet and mechanical stirrer were charged AOV-dimethyl formamide (450 mL, 425 g), 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridine-5-carbaldehyde (30.0 g, 71.45 mmol) and 2,6-difluoro-3,5-dimethoxyanihne (14.2 g, 75.0 mmol). To this suspension (internal temperature 20 °C) was added chlorotrimethylsilane (19.4 g, 22. 7 mL, 179 mmol) dropwise in 10 min at room temperature (internal temperature 20-23 °C). The suspension changed into a solution in 5 min after the chlorotrimethylsilane addition. The solution was stirred at room temperature for 1.5 h before cooled to 0-5 °C with ice-bath. Borane-THF complex in THF (1.0 M, 71.4 mL, 71.4 mmol, 64.2 g, 1.0 eq.) was added dropwise via additional funnel over 30 min while maintaining temperature at 0-5 °C. After addition, the mixture was stirred for 4 h. Water (150 g, 150 mL) was added under ice-bath cooling in 20 min, followed by slow addition of ammonium hydroxide solution (28% N¾, 15.3 g, 17 ml, 252 mmol, 3.53 eq.) to pH 9-10 while maintaining the temperature below 10 °C. More water (250 mL, 250 g) was added through the additional funnel. The slurry was stirred for 30 min and the solids were collected by filtration. The wet cake was washed with water (90 g x 2, 90 ml x 2) and heptane (61.6 g x2, 90 ml x 2). The product w as suction dried overnight to give the desired product LG-((4-chloro-2-(morphohnomethyl)-l-(phenylsulfonyl)-li/-pyrrolo[2,3-Z>]pyridin-5-yl)methyl)-2,6- difluoro-3,5-dimethoxyaniline (41.6 g, 96% yield): LCMS calculated for C27H28ClF2N405S[M+H]+: 593.10; Found: 593.1 ; ¾ NMR (400 MHz, DMSO-d6) 5 8.36 (m, 2H), 8.28 (s, 1H), 7.72 (m, 1H), 7.63 (m, 2H), 6.78 (s, 1H), 6.29 (m, 1H), 5.82 (m, 1H), 4.58 (m, 2H), 3.91 (s, 2H), 3.76 (s, 6H), 3.56 (m, 4H), 2.47 (m, 4H).
Step 4: Synthesis of l-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-lH-pyrrolo [2, 3-b ] pyridin-5-yl) methyl)-! -(2, 6-difluoro-3, 5-dimethoxyphenyl)-3-ethylurea
To a 2-L, 3-neck round bottom flask fitted with a thermocouple, a nitrogen bubbler inlet, and a magnetic stir were charged /V-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-li/-pyrrolo[2,3-b]pyridin-5-yl)methyl)-2,6-difluoro-3,5-dimethoxyaniline (67.0 g, 113 mmol) and acetonitrile (670 ml, 527 g). The suspension was cooled to 0-5 °C.
To the mixture was charged ethyl isocyanate (17.7 mL, 15.9 g, 224 mmol, 1.98 eq.) over 30 sec. The temperature stayed unchanged at 0.7 °C after the charge. Methanesulfonic acid (16.1 mL, 23.9 g, 248 mmol, 2.2 eq.) was charged dropwise over 35 min while maintaining the temperature below 2 °C. The mixture was warmed to room temperature and stirred overnight. At 24 h after addition showed that the product was 93.7%, unreacted SM was 0.73% and the major impurity (bis-isocyanate adduct) was 1.3%. The mixture was cooled with an ice-bath and quenched with sodium hydroxide (NaOH) solution (1.0M, 235 mL, 244 g, 235 mmol, 2.08 eq.) over 20 min and then saturated aqueous sodium bicarbonate
(NaHCCh) solution (1.07 M, 85 mL, 91 g, 0.091 mol, 0.80 eq.) over 10 min. Water (550 mL, 550 g) was added and the liquid became one phase. The mixture was stirred for 2 h and the solids were collected by filtration, washed with water (165 mL, 165 g) to give l-((4-chloro-2-(morpholinomethyl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo| 2.3-6 |p\ ri din-5 -y l (methy l )- 1 -(2,6-difluoro-3,5-dimethoxyphenyl)-3-ethylurea ( 70.3 g, 93.7% yield).
The crude l-((4-chloro-2-(morpholinomethyl)-l -(phenylsulfonyl)- li/-pyrrolo [2, 3-61 pyridin-5-yl) methyl)- 1 -(2, 6-difluoro-3, 5-dimethoxyphenyl)-3-ethylurea (68.5 g, 103 mmol) was added in to acetonitrile (616 mL, 485 g). The mixture was heated 60-65 °C and an amber colored thin suspension was obtained. The solid was filtered off with celite and the celite was washed with acetonitrile (68.5 mL, 53.8 g). To the pale yellow filtrate was added water (685 g, 685 ml) to form a slurry. The slurry was stirred overnight at room temperature and filtered. The solid was added to water (685 mL, 685 g) and stirred at 60 °C for 2 h. The solid was filtered and re-slurred in heptane (685 mL, 469 g) overnight. The product was dried in an oven at 50 °C under vacuum for 48 h to afford l-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridin-5-yl)methyl)- 1 -(2.6-difluoro-3.5-
dimethoxyphenyl)-3-ethylurea as a colorless solid (62.2 g, 90.8% yield, 99.9% purity by HPLC area%). KF was 0.028%. Acetonitrile (by ‘H NMR) was about 1.56%, DCM (by ‘H NMR) 2.0%: LCMS calculated for C30H33CIF2N5O6S [M+H]+: EM: 664.17; Found: 664.2; ¾ NMR (400 MHz, DMSO-de) d 8.33 (m, 2H), 8.31 (s, 1H), 7.72 (m, 1H), 7.64 (m, 1H), 6.96 (m, 2H), 6.73 (s, 1H), 6.43 (m, 1H), 4.87 (s, 2H), 3.90 (s, 2H), 3.77 (s, 6H), 3.54 (m, 4H),
3.03 (m, 2H), 2.46 (m, 4H), 0.95 (m, 3H).
Step 5: Synthesis of 3-(2, 6-difluoro-3, 5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-l, 3, 4, 7-tetrahydro-2H-pyrrolo[ 3 2’:5, 6 ]pyrido[ 4, 3-d]pyrimidin-2-one
To a 2000 mL flask equipped with a thermal couple, a nitrogen inlet, and a mechanical stirrer were charged dry l-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-1 //-pyrrolo| 2.3-6 |pyridin-5-yl)methyl)- 1 -(2.6-dinuoro-3.5-dimetho\yphenyl)-3-ethylurea (30.0 g, 45.2 mmol, KF=0. l l%) and tetrahydrofuran (1200 mL, 1063 g). To this suspension at room temperature was charged 1.0 M lithium hexamethyldisilazide in THF (62.3 mL, 55.5 g, 62.3 mmol, 1.38 eq). The mixture turned into a solution after the base addition. The reaction mixture was stirred for 2 h and HPLC shows the starting material was not detectable. To this mixture was added 1.0 M hydrochloric acid (18.1 mL, -18.1 g. 18.1 mmol, 0.4 eq.). The solution was concentrated to 600 mL and water (1200 mL, 1200 g) was added. Slurry was formed after water addition. The slurry was stirred for 30 min at room temperature and the solid was collected by filtration. The wet cake was washed with water twice (60 mLx2,
60 gx2) and dried at 50 °C overnight to give 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-l,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4, 3-d]pyrimidin-2-one as a light brown solid (26.58 g, as-is yield 93.7%): THF by ‘H NMR 0.32%, KF 5.26%, adjusted yield was 88.5%: LCMS calculated for C30H32F2N5O6S [M+H]+: EM: 628.20; Found: 628.2; ¾ NMR (400 MHz, DMSO-de) d 8.41 (m, 2H), 8.07 (s, 1H), 7.70 (m, 1H), 7.63 (m, 2H), 7.05 (m, 1H), 6.89 (s, 1H), 4.76 (s, 2H), 4.09 (m, 2H), 3.93 (s, 2H), 3.89 (s, 6H), 3.60 (m, 4H), 2.50 (m, 4H), 1.28 (m, 3H).
Step 6: Synthesis of 3-( 2, 6-difluoro-3, 5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-1,3, 4, 7 -tetrahydro-2H-pyrrolo [ 3 ‘, 2 5, 6 ]pyrido[ 4, 3-dJpyrimidin-2-one
To a stirring suspension of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholinomethyl)-7-(phenylsulfonyl)-l,3,4,7-tetrahydro-2i/-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (10.0 g, 15.93 mmol) in l,4-dioxane (100 ml, 103 g) in a 500 mL flask equipped with a nitrogen inlet, a condenser, a thermocouple and a heating mantle was added 1 M aqueous sodium hydroxide (63.7 ml, 66.3 g, 63.7 mmol). The reaction mixture was heated at 75 °C for 18 h. LCMS showed the reaction was complete. Water (100 mL, 100 g) was added to give a thick suspension. This slurry was stirred at room temperature for 1 h and filtered. The cake was washed with water (3 x 10 mL, 3 x 10 g) and heptane (2 x 10 mL, 2 x 6.84 g). The cake was dried overnight by pulling a vacuum through the filter cake and then dried in an oven at 50 °C under vacuum overnight to give 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-l,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5, 6]pyrido[4,3-d]pyrimidin-2-one (6.8 g, 87.6% yield): LCMS calculated for C24H28F2N5O4 [M+H]+: 488.20; Found: 488.2.
PATENT
US 20130338134
https://patents.google.com/patent/US20130338134A1/en
Step 1: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
- [0832]
- [0833]
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (Example 49, Step 3: 900 mg, 2.32 mmol) in N,N-dimethylformamide (20 mL) cooled to 0° C. was added sodium hydride (185 mg, 4.63 mmol, 60 wt % in mineral oil). The resulting mixture was stirred at 0° C. for 30 min then benzenesulfonyl chloride (0.444 mL, 3.48 mmol) was added. The reaction mixture was stirred at 0° C. for 1.5 h at which time LC-MS showed the reaction completed to the desired product. The reaction was quenched with saturated NH4Cl solution and diluted with water. The white precipitate was collected via filtration then washed with water and hexanes, dried to afford the desired product (1.2 g, 98%) as a white solid which was used in the next step without further purification. LC-MS calculated for C25H23F2N4O5S [M+H]+ m/z: 529.1; found: 529.1.
Step 2: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde
- [0834]
- [0835]
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (1.75 g, 3.31 mmol) in tetrahydrofuran (80 mL) at −78° C. was added freshly prepared lithium diisopropylamide (1M in tetrahydrofuran (THF), 3.48 mL, 3.48 mmol). The resulting mixture was stirred at −78° C. for 30 min then N,N-dimethylformamide (1.4 mL, 18 mmol) was added slowly. The reaction mixture was stirred at −78° C. for 30 min then quenched with water and extracted with EtOAc. The organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 20% EtOAc in DCM to give the desired product as a white solid (1.68 g, 91%). LC-MS calculated for C26H23F2N4O6S (M+H)+ m/z: 557.1; found: 556.9.
Step 3: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
- [0836]
- [0837]
To a solution 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde (1.73 g, 3.11 mmol) in dichloromethane (50 mL) was added morpholine (0.95 mL, 11 mmol), followed by acetic acid (2 mL, 30 mmol). The resulting yellow solution was stirred at room temperature overnight then sodium triacetoxyborohydride (2.3 g, 11 mmol) was added. The mixture was stirred at room temperature for 3 h at which time LC-MS showed the reaction went to completion to the desired product. The reaction was quenched with saturated NaHCO3 then extracted with ethyl acetate (EtOAc). The organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 40% EtOAc in DCM to give the desired product as a yellow solid (1.85 g, 95%). LC-MS calculated for C30H32F2N5O6S (M+H)+ m/z: 628.2; found: 628.0.
Step 4: 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one
- [0838]
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (1.5 g, 2.4 mmol) in tetrahydrofuran (40 mL) was added tetra-n-butylammonium fluoride (1M in THF, 7.2 mL, 7.2 mmol). The resulting solution was stirred at 50° C. for 1.5 h then cooled to room temperature and quenched with water. The mixture was extracted with dichloromethane (DCM) and the organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 10% MeOH in DCM to give the desired product as a white solid, which was further purified by prep HPLC (pH=2, acetonitrile/H2O). LC-MS calculated for C24H28F2N5O4 (M+H)+ m/z: 488.2; found: 488.0. 1H NMR (500 MHz, DMSO) δ 12.09 (s, 1H), 8.06 (s, 1H), 7.05 (t, J=8.1 Hz, 1H), 6.87 (s, 1H), 4.78 (s, 2H), 4.50 (s, 2H), 4.17 (q, J=6.8 Hz, 2H), 3.97 (br, 2H), 3.89 (s, 6H), 3.65 (br, 2H), 3.37 (br, 2H), 3.15 (br, 2H), 1.37 (t, J=6.8 Hz, 3H).
PATENTS
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2013338134-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| US-2017137424-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| US-2019127376-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| US-9611267-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2017-04-04 |
| WO-2014007951-A2 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| JP-6336665-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2018-06-06 |
| JP-6545863-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2019-07-17 |
| JP-6711946-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2020-06-17 |
| TW-201402574-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| US-10131667-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2018-11-20 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| JP-2015521600-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| JP-2017222709-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| JP-2018135377-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| JP-2019178156-A | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| JP-6301321-B2 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2018-03-28 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3176170-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| EP-3176170-B1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | 2018-11-14 |
| EP-3495367-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| ES-2704744-T3 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2019-03-19 |
| HU-E031916-T2 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| DK-2861595-T5 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2018-01-15 |
| DK-3176170-T3 | Substituted tricyclic relations as fgfr inhibitors | 2012-06-13 | 2019-01-28 |
| EP-2861595-A2 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| EP-2861595-B1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | 2016-12-21 |
| EP-2861595-B9 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | 2017-06-21 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019191707-A1 | Heterocyclic compounds as immunomodulators | 2018-03-30 | |
| AU-2013287176-A1 | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | |
| CA-2876689-A1 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | |
| CN-107383009-B | Substituted tricyclic compounds as FGFR inhibitors | 2012-06-13 | 2020-06-09 |
| DK-2861595-T3 | Substituted tricyclic compounds as fgfr inhibitors | 2012-06-13 | 2017-02-13 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019213544-A2 | Solid forms of an fgfr inhibitor and processes for preparing the same | 2018-05-04 | |
| WO-2019213544-A3 | Solid forms of an fgfr inhibitor and processes for preparing the same | 2018-05-04 | |
| TW-202003511-A | Heterocyclic compounds as immunomodulators | 2018-03-30 | |
| US-10669271-B2 | Heterocyclic compounds as immunomodulators | 2018-03-30 | 2020-06-02 |
| US-2019300524-A1 | Heterocyclic compounds as immunomodulators | 2018-03-30 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| TW-201946630-A | Salts of an FGFR inhibitor | 2018-05-04 | |
| TW-202003516-A | Solid forms of an FGFR inhibitor and processes for preparing the same | 2018-05-04 | |
| US-2019337948-A1 | Solid forms of an fgfr inhibitor and processes for preparing the same | 2018-05-04 | |
| US-2020002338-A1 | Salts of an fgfr inhibitor | 2018-05-04 | |
| WO-2019213506-A1 | Salts of an fgfr inhibitor | 2018-05-04 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2019227007-A1 | Tricyclic heterocyclic compounds as sting activators | 2018-05-25 | |
| TW-201946626-A | Heterocyclic compounds as immunomodulators | 2018-05-11 | |
| US-10618916-B2 | Heterocyclic compounds as immunomodulators | 2018-05-11 | 2020-04-14 |
| US-2019345170-A1 | Heterocyclic compounds as immunomodulators | 2018-05-11 | |
| WO-2019217821-A1 | Tetrahydro-imidazo[4,5-c]pyridine derivatives as pd-l1 immunomodulators | 2018-05-11 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020040009-A1 | Tricyclic heteraryl compounds as sting activators | 2018-07-31 | |
| WO-2020028565-A1 | Tricyclic heteraryl compounds as sting activators | 2018-07-31 | |
| WO-2020028566-A1 | Heteroaryl amide compounds as sting activators | 2018-07-31 | |
| WO-2019238873-A1 | A method of precision cancer therapy | 2018-06-13 | |
| US-2019359608-A1 | Tricyclic heterocyclic compounds as sting activators | 2018-05-25 |
| Title | Priority Date | Grant Date | |
|---|---|---|---|
| WO-2020131627-A1 | Substituted pyrazolo[1,5-a]pyridine compounds as inhibitors of fgfr tyrosine kinases | 2018-12-19 | |
| WO-2020131674-A1 | 7-((3,5-dimethoxyphenyl)amino)quinoxaline derivatives as fgfr inhibitors for treating cancer | 2018-12-19 | |
| WO-2020081898-A1 | Non-invasive urinary biomarkers for the detection of urothelial carcinoma of the bladder | 2018-10-20 | |
| US-2020115378-A1 | Dihydropyrido[2,3-d]pyrimidinone compounds as cdk2 inhibitors | 2018-10-11 | |
| US-2020039994-A1 | Heteroaryl amide compounds as sting activators | 2018-07-31 |
References
- ^ “Pemigatinib (Pemazyre) Use During Pregnancy”. Drugs.com. 11 August 2020. Retrieved 24 September 2020.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3): 479. hdl:10665/330907.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves First Targeted Treatment for Patients with Cholangiocarcinoma, a Cancer of Bile Ducts”. U.S. Food and Drug Administration (FDA) (Press release). 17 April 2020. Retrieved 17 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to pemigatinib for cholangiocarcinoma”. U.S. Food and Drug Administration (FDA). 17 April 2020. Retrieved 20 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e “EU/3/18/2066”. European Medicines Agency (EMA). 19 December 2018. Retrieved 20 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d “Drug Trials Snapshot: Pemazyre”. U.S. Food and Drug Administration (FDA). 17 April 2020. Retrieved 5 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Pemazyre: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 21 April 2020.
- ^ “Pemigatinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). Retrieved 19 April 2020.
- ^ “Pemigatinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). Retrieved 19 April 2020.
- ^ “EU/3/19/2216”. European Medicines Agency (EMA). 23 January 2020. Retrieved 19 April 2020. This article incorporates text from this source, which is in the public domain.
Further reading
- Roskoski R (January 2020). “The role of fibroblast growth factor receptor (FGFR) protein-tyrosine kinase inhibitors in the treatment of cancers including those of the urinary bladder”. Pharmacol. Res. 151: 104567. doi:10.1016/j.phrs.2019.104567. PMID 31770593.
External links
- “Pemigatinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Pemigatinib”. National Cancer Institute.
- Clinical trial number NCT02924376 for “Efficacy and Safety of Pemigatinib in Subjects With Advanced/Metastatic or Surgically Unresectable Cholangiocarcinoma Who Failed Previous Therapy – (FIGHT-202)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Pemazyre |
| Other names | INCB054828 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620028 |
| License data | US DailyMed: Pemigatinib |
| Pregnancy category | US: N (Not classified yet)[1] |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 1513857-77-6 |
| PubChem CID | 86705695 |
| DrugBank | DB15102 |
| ChemSpider | 68007304 |
| UNII | Y6BX7BL23K |
| KEGG | D11417 |
| ChEMBL | ChEMBL4297522 |
| Chemical and physical data | |
| Formula | C24H27F2N5O4 |
| Molar mass | 487.508 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CCN1C2=C3C=C(NC3=NC=C2CN(C1=O)C4=C(C(=CC(=C4F)OC)OC)F)CN5CCOCC5 | |
| InChI[hide]InChI=1S/C24H27F2N5O4/c1-4-30-21-14(11-27-23-16(21)9-15(28-23)13-29-5-7-35-8-6-29)12-31(24(30)32)22-19(25)17(33-2)10-18(34-3)20(22)26/h9-11H,4-8,12-13H2,1-3H3,(H,27,28)Key:HCDMJFOHIXMBOV-UHFFFAOYSA-N |
/////////Pemigatinib, 佩米替尼 , PEMAZYRE, FDA 2020, 2020 APPROVALS, INCB054828, INCB 054828, Orphan Drug Status, Myeloproliferative disorders, Lymphoma, Cholangiocarcinoma, INCYTE
O=C1N(CC)C2=C3C(NC(CN4CCOCC4)=C3)=NC=C2CN1C5=C(F)C(OC)=CC(OC)=C5F.[H]Cl
TUCATINIB

Tucatinib
ツカチニブ;
N6-(4,4-dimethyl-4,5-dihydro-1,3-oxazol-2-yl)-N4-(3-methyl-4-{[1,2,4]triazolo[1,5-a]pyridin-7-yloxy}phenyl)quinazoline-4,6-diamine
| Formula | C26H24N8O2 |
|---|---|
| CAS | 937263-43-9 |
| Mol weight | 480.5212 |
To treat advanced unresectable or metastatic HER2-positive breast cancer
Drug Trials Snapshot
FDA APPROVED 4/17/2020 Tukysa
- ARRY 380
- ARRY-380
- ONT 380
- ONT-380
Tucatinib (INN),[1] sold under the brand name Tukysa, is a small molecule inhibitor of HER2 for the treatment of HER2-positive breast cancer.[2][3] It was developed by Array BioPharma and licensed to Cascadian Therapeutics (formerly Oncothyreon, subsequently part of Seattle Genetics).[4]
Common side effects are diarrhea, palmar-plantar erythrodysesthesia (burning or tingling discomfort in the hands and feet), nausea, fatigue, hepatotoxicity (liver damage), vomiting, stomatitis (inflammation of the mouth and lips), decreased appetite, abdominal pain, headache, anemia and rash.[5][6] Pregnant or breastfeeding women should not take Tucatinib because it may cause harm to a developing fetus or newborn baby.[5]
Tucatinib was approved for medical use in Australia in August 2020.[7]
Medical uses
Tucatinib is a kinase inhibitor indicated in combination with trastuzumab and capecitabine for treatment of adults with advanced unresectable or metastatic HER2-positive breast cancer, including those with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting.[8]
Clinical trials
Two early stage clinical trials have reported encouraging results, both of which had options to enroll subjects with central nervous system (CNS) metastases.[2][9][10][11][12][10] HER2CLIMB is a Phase 2 randomized, double-blinded, placebo-controlled study of tucatinib in combination with trastuzumab and capecitabine in patients with pretreated, unresectable locally advanced or metastatic HER2-positive breast cancer.[13]
History
In April 2020, the U.S. Food and Drug Administration (FDA) approved tucatinib in combination with chemotherapy (trastuzumab and capecitabine) for the treatment of adults with advanced forms of HER2-positive breast cancer that can’t be removed with surgery, or has spread to other parts of the body, including the brain, and who have received one or more prior treatments.[5][6][14]
The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, Health Sciences Authority (HSA, Singapore) and Swissmedic (SMC, Switzerland) on the review.[5] This was the first Project Orbis partnership between the FDA, HSA and Swissmedic.[5] As of 17 April 2020, the application is still under review at the other agencies.[5]
Tucatinib is a kinase inhibitor meaning it blocks a type of enzyme (kinase) and helps prevent the cancer cells from growing.[5] Tucatinib is approved for treatment after adults have taken one or more anti-HER2-based regimens in the metastatic setting.[5] The FDA approved tucatinib based on the results of the HER2CLIMB trial (NCT02614794) enrolling 612 subjects who had HER2-positive advanced unresectable or metastatic breast cancer and had prior treatment with trastuzumab, pertuzumab and ado-trastuzumab emtansine (T-DM1).[5][6] Subjects with previously treated and stable brain metastases, as well as those with previously treated and growing or untreated brain metastases, were eligible for the clinical trial, and 48% of enrolled subjects had brain metastases at the start of the trial.[5]
Subjects received either tucatinib 300 mg twice daily plus trastuzumab and capecitabine (tucatinib arm, n=410) or placebo plus trastuzumab and capecitabine (control arm, n=202).[6] The primary endpoint was progression-free survival (PFS), or the amount of time when there was no growth of the tumor, assessed by a blinded independent central review, evaluated in the initial 480 randomized patients.[5][6] The median PFS in subjects who received tucatinib, trastuzumab, and capecitabine was 7.8 months (95% CI: 7.5, 9.6) compared to 5.6 months (95% CI: 4.2, 7.1) in those subjects who received placebo, trastuzumab, and capecitabine (HR 0.54; 95% CI: 0.42, 0.71; p<0.00001).[5][6] Overall survival and PFS in subjects with brain metastases at baseline were key secondary endpoints.[5] The median overall survival in subjects who received tucatinib, trastuzumab, and capecitabine was 21.9 months (95% CI: 18.3, 31.0) compared to 17.4 months (95% CI: 13.6, 19.9) in subjects who received placebo, trastuzumab, and capecitabine (HR: 0.66; 95% CI: 0.50, 0.87; p=0.00480).[5][6] The median PFS in subjects with brain metastases at baseline who received tucatinib, trastuzumab and capecitabine was 7.6 months (95% CI: 6.2, 9.5) compared to 5.4 months (95% CI: 4.1, 5.7) in subjects who received placebo, trastuzumab and capecitabine (HR: 0.48; 0.34, 0.69; p<0.00001).[5][6]
The FDA granted the application for tucatinib priority review, breakthrough therapy, fast track, and orphan drug designations.[5][6][15] The FDA granted approval of Tukysa to Seattle Genetics, Inc.[5]
SYN
Recently, the Mao team reported a new route for the efficient synthesis of Tucatinib.
The results were published on Synthesis (DOI: 10.1055/s-0037-1610706).
Previously, the synthesis report route of Tucatinib was published by Array BioPharma in a patent document (WO 2007059257, 2007). The synthetic route reported in the patent is shown in the figure below:

Using 4-nitro-2-cyanoaniline as the raw material, the first step is to condense with DMF-DMA to prepare imine 3 (yield 87%); subsequent catalytic hydrogenation of palladium on carbon to reduce the nitro group to obtain the amine 4 (90% yield); followed by 1,1&39;-thiocarbonyldiimidazole (TCDI) and The amino alcohol undergoes condensation to prepare the thiourea derivative 5 (yield is only 34%); further with the intermediate 6 to undergo ring-closure reaction to obtain the key intermediate 7 (yield 62%) ; Finally, under the action of p-toluenesulfonic acid, intramolecular dehydration and ring closure to form oxazoline, complete the synthesis of the target compound tucatinib.
Reverse synthesis analysis
The author broke the bond of Tucatinib from two points a and b and split them into three fragments. : Thioether oxazoline 17, nitrobenzene 3 and the key fragment of the original research route 6.
Preparation of key fragment 6
4-nitro-3-methylphenol 8 as a starting point The material, with pyridine derivative 9, undergoes aromatic affinity substitution reaction to prepare aryl ether 10 (yield 64%); then it is condensed with DMF-DMA, and then treated with hydroxylamine hydrochloride. The step yield was 81% to obtain the oxime derivative 12; subsequently, the ring was closed under the treatment of trifluoroacetic anhydride, the mostAfter palladium-catalyzed hydrogenation to reduce the nitro group, the key aniline triazole 6 was successfully prepared, with a total yield of 32.8%.
aromatic ring skeleton construction
fragment 3 was synthesized according to the method reported in the literature. The estimated aromatic ring fragment was then constructed with the aniline triazole 6 prepared above:
Compound 6 and fragment 3 were cyclized in acetic acid , 14 was successfully prepared, and finally the nitro group was reduced by palladium-catalyzed hydrogenation to obtain the key arylamine 15 with a two-step yield of 76.4%.
Fragment 17 and Tucatinib synthesis
amino alcohol and 1,1&39;-thiocarbonyl diimidazole (TCDI) The ring is closed to obtain 16, which is then treated with methyl trifluoromethanesulfonate to obtain oxazoline 17, with a total yield of 67.23% in the two steps.
oxazoline17 and arylamine 15 in the presence of cesium carbonate, heated in DMF for 20 hours, and finally completed the synthesis of Tucatinib with a yield of 76%.
Comparison of the new route and the patent route
The yield of the last step of the patent is unknown, starting with key intermediates 3 and 6, total income The rate is less than 19%.
The overview of the new route is as follows:
Correspondingly, starting from the intermediate 3 and 6, the total yield of the new route There is a significant improvement to 39%. Moreover, the purity of the product and other aspects also meet the requirements of API.
Comment
Tucatinib (Tukysa) Tucatinib/Tucatinib as a small-molecule oral tyrosine kinase (TKI) inhibitor for HER2 Positive breast cancer has highly specific targeting selectivity. The study of the new synthetic route
effectively improves the production efficiency in terms of ensuring the purity of the compound, and the raw materials used are relatively simple and easy to obtain.
Medicinal chemists have completed the research and development and synthesis of compounds (from 0 to 1), while process chemists have optimized the synthetic routes and processes, so that the compounds can be prepared more simply, efficiently, economically and environmentally.
SYN PATENT
CN 111825604
PAPER
Synthesis (2019), 51(13), 2660-2664
Abstract
A new and improved synthetic route to tucatinib is described that involves three key intermediates. The first of these, 4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylaniline, was prepared on a 100 g scale in 33% yield over five steps and 99% purity. Next, N 4-(4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylphenyl)quinazoline-4,6-diamine was isolated in 67% yield over three steps and >99% purity. Then, 4,4-dimethyl-2-(methylthio)-4,5-dihydrooxazole trifluoromethanesulfonate was prepared under mild conditions in 67% yield over two steps. Finally, tucatinib was obtained in 17% yield over nine steps and in >99% purity (HPLC). Purification methods used to isolate the product and the intermediates involved in the route are also reported.

References
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 161. hdl:10665/331046.
- ^ Jump up to:a b “ONT-380 Active Against CNS Mets in HER2-Positive Breast Cancer”. Cancer Network. 15 December 2015. Retrieved 17 April 2020.
- ^ Martin M, López-Tarruella S (October 2018). “Emerging Therapeutic Options for HER2-Positive Breast Cancer”. American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting. 35 (36): e64–70. doi:10.1200/EDBK_159167. PMID 27249772.
- ^ “Tucatinib” (PDF). Statement on a Nonproprietary Name Adopted by the USAN Council.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves First New Drug Under International Collaboration, A Treatment Option for Patients with HER2-Positive Metastatic Breast Cancer”. U.S. Food and Drug Administration (FDA) (Press release). 17 April 2020. Retrieved 17 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g h i “FDA approves tucatinib for patients with HER2-positive metastatic brea”. U.S. Food and Drug Administration (FDA). 17 April 2020. Retrieved 20 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Tukysa”. Therapeutic Goods Administration (TGA). 21 August 2020. Retrieved 22 September 2020.
- ^ “Tukysa (tucatinib) tablets, for oral use” (PDF). Seattle Genetics. Retrieved 17 April2020.
- ^ “Oncothyreon Inc. Announces Data For ONT-380 In HER2-Positive Breast Cancer Patients With And Without Brain Metastases At The San Antonio Breast Cancer Symposium”. BioSpace (Press release). 9 December 2015. Retrieved 18 April 2020.
- ^ Jump up to:a b Borges VF, Ferrario C, Aucoin N, Falkson CI, Khan QJ, Krop IE, et al. “Efficacy results of a phase 1b study of ONT-380, a CNS-penetrant TKI, in combination with T-DM1 in HER2+ metastatic breast cancer (MBC), including patients (pts) with brain metastases”. Journal of Clinical Oncology. 2016 ASCO Annual Meeting.
- ^ “SABCS15: Promising phase 1 results lead to phase 2 for ONT-380 in HER2+ breast cancer”. Colorado Cancer Blogs. Retrieved 10 June 2016.
- ^ “A Study of Tucatinib (ONT-380) Combined With Capecitabine and/or Trastuzumab in Patients With HER2+ Metastatic Breast Cancer”. ClinicalTrials.gov. 31 December 2013. Retrieved 18 April 2020.
- ^ “A Study of Tucatinib vs. Placebo in Combination With Capecitabine & Trastuzumab in Patients With Advanced HER2+ Breast Cancer (HER2CLIMB)”. ClinicalTrials.gov. Retrieved 18 April 2020.
- ^ “Tukysa: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 20 April 2020.
- ^ “Tucatinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration(FDA). 24 December 1999. Retrieved 20 April 2020.
External links
- “Tucatinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Tucatinib”. National Cancer Institute.
- Clinical trial number NCT02614794 for “A Study of Tucatinib vs. Placebo in Combination With Capecitabine & Trastuzumab in Patients With Advanced HER2+ Breast Cancer (HER2CLIMB)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Tukysa |
| Other names | ONT-380, ARRY-380 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620032 |
| License data | US DailyMed: Tucatinib |
| Pregnancy category | AU: DUS: N (Not classified yet) |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-only |
| Identifiers | |
| CAS Number | 937263-43-9 |
| PubChem CID | 51039094 |
| DrugBank | DB11652 |
| ChemSpider | 34995558 |
| UNII | 234248D0HH |
| KEGG | D11141 |
| ChEMBL | ChEMBL3989868 |
| Chemical and physical data | |
| Formula | C26H24N8O2 |
| Molar mass | 480.532 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CC1=C(C=CC(=C1)NC2=NC=NC3=C2C=C(C=C3)NC4=NC(CO4)(C)C)OC5=CC6=NC=NN6C=C5 | |
| InChI[hide]InChI=1S/C26H24N8O2/c1-16-10-17(5-7-22(16)36-19-8-9-34-23(12-19)28-15-30-34)31-24-20-11-18(4-6-21(20)27-14-29-24)32-25-33-26(2,3)13-35-25/h4-12,14-15H,13H2,1-3H3,(H,32,33)(H,27,29,31)Key:SDEAXTCZPQIFQM-UHFFFAOYSA-N |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Tukysa | Tablet | 150 mg/1 | Oral | Seattle Genetics, Inc. | 2020-04-17 | Not applicable | | |
| Tukysa | Tablet | 150 mg | Oral | Seattle Genetics, Inc. | 2020-08-27 | Not applicable |  | |
| Tukysa | Tablet | 50 mg/1 | Oral | Seattle Genetics, Inc. | 2020-04-17 | Not applicable | | |
| Tukysa | Tablet | 50 mg | Oral | Seattle Genetics, Inc. | 2020-10-08 | Not applicable | |
Showing 1 to 4 of 4 entries
///////tucatinib, FDA 2020, TUKSYA, 2020 APROVALS, ARRY 380, ONT 380, ツカチニブ ,
Ripretinib

Ripretinib
リプレチニブ;
| Formula | C24H21BrFN5O2 |
|---|---|
| CAS | 1442472-39-0 |
| Mol weight | 510.3582 |
Antineoplastic, Receptor tyrosine kinase inhibitor
US FDA APPROVED 2020/5/15 QUINLOCK
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Qinlock | Tablet | 50 mg | Oral | Deciphera Pharmaceuticals. Llc | Not applicable | Not applicable | | |
| Qinlock | Tablet | 50 mg/1 | Oral | Deciphera Pharmaceuticals, LLC | 2020-05-15 | Not applicable | |
SYN

Ripretinib, sold under the brand name Qinlock, is a medication for the treatment of adults with advanced gastrointestinal stromal tumor (GIST), a type of tumor that originates in the gastrointestinal tract.[3] It is taken by mouth.[3] Ripretinib is a kinase inhibitor, meaning it works by blocking a type of enzyme called a kinase, which helps keep the cancer cells from growing.[3]
The most common side effects include alopecia (hair loss), fatigue, nausea, abdominal pain, constipation, myalgia (muscle pain), diarrhea, decreased appetite, palmar-plantar erythrodysesthesia syndrome (a skin reaction in the palms and soles) and vomiting.[3][4] Alopecia is a unique side effect to ripretinib, which is not seen with other tyrosine kinase inhibitors used to treat GISTs.
Ripretinib was approved for medical use in the United States in May 2020,[3] and in Australia in July 2020.[1] Ripretinib is the first new drug specifically approved in the United States as a fourth-line treatment for advanced gastrointestinal stromal tumor (GIST).
Medical uses
Ripretinib is indicated for the treatment of adults with advanced gastrointestinal stromal tumor (GIST), a type of tumor that originates in the gastrointestinal tract, who have received prior treatment with three or more kinase inhibitor therapies, including imatinib.[3] GIST is type of stomach, bowel, or esophagus tumor.[4]
Adverse effects
The most common side effects include alopecia (hair loss), fatigue, nausea, abdominal pain, constipation, myalgia (muscle pain), diarrhea, decreased appetite, palmar-plantar erythrodysesthesia syndrome (a skin reaction in the palms and soles) and vomiting.[3][4]
Ripretinib can also cause serious side effects including skin cancer, hypertension (high blood pressure) and cardiac dysfunction manifested as ejection fraction decrease (when the muscle of the left ventricle of the heart is not pumping as well as normal).[3][4]
Ripretinib may cause harm to a developing fetus or a newborn baby.[3][4]
History
Ripretinib was approved for medical use in the United States in May 2020.[3][5][6][4]
The approval of ripretinib was based on the results of an international, multi-center, randomized, double-blind, placebo-controlled clinical trial (INVICTUS/NCT03353753) that enrolled 129 participants with advanced gastrointestinal stromal tumor (GIST) who had received prior treatment with imatinib, sunitinib, and regorafenib.[3][7] The trial compared participants who were randomized to receive ripretinib to participants who were randomized to receive placebo, to determine whether progression free survival (PFS) – the time from initial treatment in the clinical trial to growth of the cancer or death – was longer in the ripretinib group compared to the placebo group.[3] During treatment in the trial, participants received ripretinib 150 mg or placebo once a day in 28-day cycles, repeated until tumor growth was found (disease progression), or the participant experienced intolerable side effects.[3][7] After disease progression, participants who were randomized to placebo were given the option of switching to ripretinib.[3][7] The trial was conducted at 29 sites in the United States, Australia, Belgium, Canada, France, Germany, Italy, the Netherlands, Poland, Singapore, Spain, and the United Kingdom.[4]
The major efficacy outcome measure was progression-free survival (PFS) based on assessment by blinded independent central review (BICR) using modified RECIST 1.1 in which lymph nodes and bone lesions were not target lesions and a progressively growing new tumor nodule within a pre-existing tumor mass must meet specific criteria to be considered unequivocal evidence of progression.[7] Additional efficacy outcome measures included overall response rate (ORR) by BICR and overall survival (OS).[7] The trial demonstrated a statistically significant improvement in PFS for participants in the ripretinib arm compared with those in the placebo arm (HR 0.15; 95% CI: 0.09, 0.25; p<0.0001).[7]
The U.S. Food and Drug Administration (FDA) granted the application for ripretinib priority review and fast track designations, as well as breakthrough therapy designation and orphan drug designation.[3][8] The FDA granted approval of Qinlock to Deciphera Pharmaceuticals, Inc.[3]
The FDA collaborated with the Australian Therapeutic Goods Administration (TGA) and Health Canada on the review of the application as part of Project Orbis.[3][7] The FDA approved ripretinib three months ahead of schedule.[3][7] As of May 2020, the review of the applications was ongoing for the Australian TGA and for Health Canada.[3][7]
Names
Ripretinib is the International nonproprietary name (INN) and the United States Adopted Name (USAN).[9][10]
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US8940756 | No | 2012-06-07 | 2032-06-07 | |
| US8461179 | No | 2012-06-07 | 2032-06-07 | |
| US8188113 | No | 2010-07-27 | 2030-07-27 | |
PATENT
US 8461179
PATENT
WO 2013184119
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013184119
[0125] Example A13: A mixture of Example C5 (2.191 g, 7.94 mmol), Example Bl (1.538 g, 8.33 mmol) and KF on alumina (40 wt%) (9.22 g, 63.5 mmol) in DMA (40 mL) was sonicated for 2 h. The mixture was filtered through a shallow bed of silica gel and rinsed well with EtOAc. The filtrate was washed with satd. NaHC03 (lx), 5% LiCl (2x), then brine (lx), dried (MgS04), and concentrated to dryness to afford 3-(5-amino-2-bromo-4-fluorophenyl)-7-chloro-l -ethyl- l,6-naphthyridin-2(lH)-one (2.793 g, 89% yield) as a brown solid. 1H NMR (400 MHz, DMSO-<¾): δ 8.77 (s, 1 H), 8.00 (s, 1 H), 7.74 (s, 1 H), 7.37 (d, 1 H), 6.77 (d, 1 H), 5.45 (s, 2 H), 4.27 (q, 2 H), 1.20 (t, 3 H); MS (ESI) m z: 398.0 [M+H]+.
[0126] Example A14: A suspension of Example A13 (1.50 g, 3.78 mmol) in dioxane (15 mL) was treated with methylamine (40% in water) (26.4 mL, 303 mmol) in a pressure tube and heated to 100°C overnight. The mixture was cooled to RT, treated with a large amount of brine, then diluted with EtOAc until all of the solids dissolved. The layers were separated, the aqueous layer extracted with additional EtOAc (lx) and the combined organics were washed with satd. NaHC03 (lx), dried (MgS04) and concentrated to dryness. The resulting solid was suspended in MeCN/H20, frozen and lyophilized to afford 3-(5-amino-2-bromo-4-fluorophenyl)-l-ethyl-7-(methylamino)-l,6-naphthyridin-2(lH)-one (1.32g, 89% yield) as a light brown solid. 1H NMR (400 MHz, DMSO-<¾): δ 8.37 (s, 1 H), 7.62 (s, 1 H), 7.30 (d, 1 H), 6.99 (q, 1 H), 6.73 (d, 1 H), 6.21 (s, 1 H), 5.33 (s, 2 H), 4.11 (q, 2 H), 2.84 (d, 3 H), 1.19 (t, 3 H); MS (ESI) m/z: 393.0 [M+H]+.
[0263] Example 31: A mixture of Example A14 (0.120 g, 0.307 mmol) and TEA (0.043 mL, 0.307 mmol) in THF (3.0 mL) was treated with phenyl isocyanate (0.040 g, 0.337 mmol) and stirred at RT for 4 h. Over the course of the next 4 days the mixture was treated with additional phenyl isocyanate (0.056 mL) and stirred at RT. The resulting solid was filtered, rinsed with THF, then triturated with MeOH to afford l-(4-bromo-5-(l-ethyl-7-(methylamino)-2-oxo- 1 ,2-dihydro- 1 ,6-naphthyridin-3 -yl)-2-fluorophenyl)-3 -phenylurea (101 mg, 64.5% yield) as a bright white solid. 1H NMR (400 MHz, DMSO-<¾): δ 9.09 (s, 1 H), 8.68 (s, 1 H), 8.41 (s, 1 H), 8.17 (d, 1 H), 7.70 (s, 1 H), 7.65 (d, 1 H), 7.41 (d, 2 H), 7.27 (m, 2 H), 7.03 (m, 1 H), 6.96 (t, 1 H), 6.23 (s, 1 H), 4.13 (q, 2 H), 2.86 (d, 3 H), 1.20 (t, 3 H); MS (ESI) m/z: 510.1 [M+H]+.
References
- ^ Jump up to:a b c “Qinlock Australian Prescription Medicine Decision Summary”. Therapeutic Goods Administration (TGA). 21 July 2020. Retrieved 17 August 2020.
- ^ “Ripretinib (Qinlock) Use During Pregnancy”. Drugs.com. 10 August 2020. Retrieved 17 August 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t “FDA Approves First Drug for Fourth-Line Treatment of Advanced Gastrointestinal Stromal Tumors”. U.S. Food and Drug Administration (FDA) (Press release). 15 May 2020. Retrieved 15 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g “Drug Trial Snapshot: Qinlock”. U.S. Food and Drug Administration (FDA). 15 May 2020. Retrieved 2 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “FDA Grants Full Approval of Deciphera Pharmaceuticals’ Qinlock (ripretinib) for the Treatment of Fourth-Line Gastrointestinal Stromal Tumor”. Deciphera Pharmaceuticals, Inc. (Press release). 15 May 2020. Retrieved 15 May 2020.
- ^ “Qinlock: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 May 2020.
- ^ Jump up to:a b c d e f g h i “FDA approves ripretinib for advanced gastrointestinal stromal tumor”. U.S. Food and Drug Administration (FDA). 15 May 2020. Retrieved 18 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Ripretinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 2 October 2014. Retrieved 15 May 2020.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1): 106. hdl:10665/330896. License: CC BY-NC-SA 3.0 IGO.
- ^ “Ripretinib” (PDF). United States Adopted Name (USAN) Drug Finder. Retrieved 17 May 2020.
Further reading
- Schneeweiss M, Peter B, Bibi S, et al. (May 2018). “The KIT and PDGFRA switch-control inhibitor DCC-2618 blocks growth and survival of multiple neoplastic cell types in advanced mastocytosis”. Haematologica. 103 (5): 799–809. doi:10.3324/haematol.2017.179895. PMC 5927976. PMID 29439183.
External links
- “Ripretinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Ripretinib”. NCI Drug Dictionary. National Cancer Institute.
- “Ripretinib”. National Cancer Institute.
- Clinical trial number NCT03353753 for “Phase 3 Study of DCC-2618 vs Placebo in Advanced GIST Patients Who Have Been Treated With Prior Anticancer Therapies (invictus)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Pronunciation | rip re’ ti nib |
| Trade names | Qinlock |
| Other names | DCC-2618 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620035 |
| License data | US DailyMed: Ripretinib |
| Pregnancy category | AU: D[1]US: N (Not classified yet)[2]Use should be avoided |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-only [3] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 1442472-39-0 |
| PubChem CID | 71584930 |
| DrugBank | DB14840 |
| ChemSpider | 67886378 |
| UNII | 9XW757O13D |
| KEGG | D11353 |
| ChEMBL | ChEMBL4216467 |
| Chemical and physical data | |
| Formula | C24H21BrFN5O2 |
| Molar mass | 510.367 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CCN1C(=O)C(=CC2=C1C=C(NC)N=C2)C1=C(Br)C=C(F)C(NC(=O)NC2=CC=CC=C2)=C1 | |
| InChI[hide]InChI=1S/C24H21BrFN5O2/c1-3-31-21-12-22(27-2)28-13-14(21)9-17(23(31)32)16-10-20(19(26)11-18(16)25)30-24(33)29-15-7-5-4-6-8-15/h4-13H,3H2,1-2H3,(H,27,28)(H2,29,30,33)Key:CEFJVGZHQAGLHS-UHFFFAOYSA-N |
////////////Ripretinib, QINLOCK, リプレチニブ , 2020 APPROVALS, FDA 2020
Triheptanoin

Triheptanoin
Approved US FDA 30/6/2020 Dojolvi UX 007
Triheptanoin is a source of heptanoate fatty acids, which can be metabolized without the enzymes of long chain fatty acid oxidation.4 In clinical trials, patients with long chain fatty acid oxidation disorders (lc-FAODs) treated with triheptanoin are less likely to develop hypoglycemia, cardiomyopathy, rhabdomyolysis, and hepatomegaly.1,2 Complications in lc-FAOD patients are reduced from approximately 60% to approximately 10% with the addition of triheptanoin.2
Triheptanoin was granted FDA approval on 30 June 2020.4
Triheptanoin, sold under the brand name Dojolvi, is a medication for the treatment of children and adults with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD).[1][2][3]
The most common adverse reactions include abdominal pain, diarrhea, vomiting, and nausea.[1][2][3]
Triheptanoin was approved for medical use in the United States in June 2020.[4][2][3]
Triheptanoin is a triglyceride that is composed of three seven-carbon (C7:0) fatty acids. These odd-carbon fatty acids are able to provide anaplerotic substrates for the TCA cycle. Triheptanoin is used clinically in humans to treat inherited metabolic diseases, such as pyruvate carboxylase deficiency and carnitine palmitoyltransferase II deficiency. It also appears to increase the efficacy of the ketogenic diet as a treatment for epilepsy.
Since triheptanoin is composed of odd-carbon fatty acids, it can produce ketone bodies with five carbon atoms, as opposed to even-carbon fatty acids which are metabolized to ketone bodies with four carbon atoms. The five-carbon ketones produced from triheptanoin are beta-ketopentanoate and beta-hydroxypentanoate. Each of these ketone bodies easily crosses the blood–brain barrier and enters the brain.
Medical uses
Dojolvi is indicated as a source of calories and fatty acids for the treatment of children and adults with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD).[1][2]
History
Triheptanoin was designated an orphan drug by the U.S. Food and Drug Administration (FDA) in 2006, 2008, 2014, and 2015.[5][6][7][8] Triheptanoin was also designated an orphan drug by the European Medicines Agency (EMA).[9][10][11][12][13][14][15][16]
Triheptanoin was approved for medical use in the United States in June 2020.[4][2]
The FDA approved triheptanoin based on evidence from three clinical trials (Trial 1/NCT018863, Trial 2/NCT022141 and Trial 3/NCT01379625).[3] The trials enrolled children and adults with LC-FAOD.[3] Trials 1 and 2 were conducted at 11 sites in the United States and the United Kingdom, and Trial 3 was conducted at two sites in the United States.[3]
Trial 1 and Trial 2 were used to evaluate the side effects of triheptanoin.[3] Both trials enrolled children and adults diagnosed with LC-FAOD.[3] In Trial 1, participants received triheptanoin for 78 weeks.[3] Trial 2 enrolled participants from other trials who were already treated with triheptanoin (including those from Trial 1) as well as participants who were never treated with triheptanoin before.[3] Trial 2 is still ongoing and is planned to last up to five years.[3]
The benefit of triheptanoin was evaluated in Trial 3 which enrolled enrolled children and adults with LC-FAOD.[3] Half of the participants received triheptanoin and half received trioctanoin for four months.[3] Neither the participants nor the investigators knew which treatment was given until the end of the trial.[3] The benefit of triheptanoin in comparison to trioctanoin was assessed by measuring the changes in heart and muscle function.[3]
Names
Triheptanoin is the international nonproprietary name.[17]
SYN
https://onlinelibrary.wiley.com/doi/abs/10.1002/ejlt.201100425




References
- ^ Jump up to:a b c d “Dojolvi- triheptanoin liquid”. DailyMed. 30 June 2020. Retrieved 24 September2020.
- ^ Jump up to:a b c d e “Ultragenyx Announces U.S. FDA Approval of Dojolvi (UX007/triheptanoin), the First FDA-Approved Therapy for the Treatment of Long-chain Fatty Acid Oxidation Disorders”. Ultragenyx Pharmaceutical. 30 June 2020. Retrieved 30 June 2020 – via GlobeNewswire.
- ^ Jump up to:a b c d e f g h i j k l m n o “Drug Trials Snapshots: Dojolvi”. U.S. Food and Drug Administration. 30 June 2020. Retrieved 16 July 2020.
- ^ Jump up to:a b “Dojolvi: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 30 June 2020.
- ^ “Triheptanoin Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 26 May 2006. Retrieved 30 June 2020.
- ^ “Triheptanoin Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 February 2008. Retrieved 30 June 2020.
- ^ “Triheptanoin Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 21 October 2014. Retrieved 30 June 2020.
- ^ “Triheptanoin Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 15 April 2015. Retrieved 30 June 2020.
- ^ “EU/3/12/1081”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/12/1082”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1495”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1508”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1524”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1525”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1526”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/16/1710”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3): 694. hdl:10665/330879. License: CC BY-NC-SA 3.0 IGO.
Further reading
- de Almeida Rabello Oliveira M, da Rocha Ataíde T, de Oliveira SL, de Melo Lucena AL, de Lira CE, Soares AA, et al. (March 2008). “Effects of short-term and long-term treatment with medium- and long-chain triglycerides ketogenic diet on cortical spreading depression in young rats”. Neurosci. Lett. 434 (1): 66–70. doi:10.1016/j.neulet.2008.01.032. PMID 18281154. S2CID 7754768.
- Mochel F, DeLonlay P, Touati G, Brunengraber H, Kinman RP, Rabier D, et al. (April 2005). “Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy”. Mol. Genet. Metab. 84 (4): 305–12. doi:10.1016/j.ymgme.2004.09.007. PMID 15781190.
- Borges K, Sonnewald U (July 2012). “Triheptanoin–a medium chain triglyceride with odd chain fatty acids: a new anaplerotic anticonvulsant treatment?”. Epilepsy Res. 100 (3): 239–44. doi:10.1016/j.eplepsyres.2011.05.023. PMC 3422680. PMID 21855298.
External links
- “Triheptanoin”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01379625 for “Study of Triheptanoin for Treatment of Long-Chain Fatty Acid Oxidation Disorder (Triheptanoin)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Dojolvi |
| Other names | UX007 |
| AHFS/Drugs.com | Professional Drug Facts |
| License data | US DailyMed: Triheptanoin |
| Pregnancy category | US: N (Not classified yet) |
| Routes of administration | By mouth |
| Drug class | Glycerolipids |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 620-67-7 |
| PubChem CID | 69286 |
| DrugBank | DB11677 |
| ChemSpider | 62497 |
| UNII | 2P6O7CFW5K |
| KEGG | D11465 |
| ChEMBL | ChEMBL4297585 |
| CompTox Dashboard (EPA) | DTXSID40862306 |
| ECHA InfoCard | 100.009.681 |
| Chemical and physical data | |
| Formula | C24H44O6 |
| Molar mass | 428.610 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CCCCCCC(=O)OCC(COC(=O)CCCCCC)OC(=O)CCCCCC | |
| InChI[hide]InChI=1S/C24H44O6/c1-4-7-10-13-16-22(25)28-19-21(30-24(27)18-15-12-9-6-3)20-29-23(26)17-14-11-8-5-2/h21H,4-20H2,1-3H3 Key:PJHKBYALYHRYSK-UHFFFAOYSA-N |
//////////Triheptanoin, Dojolvi, UX 007, FDA 2020, 2020 APPROVALS
Prescription Products
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Dojolvi | Liquid | 0.96 g/1mL | Oral | Ultragenyx Pharmaceutical Inc. | 2020-07-01 | Not applicable | |
Cetuximab sarotalocan sodium
Cetuximab Sarotalocan Sodium (Genetical Recombination)
.png)
Cetuximab Sarotalocan Sodium is an antibody-drug-conjugate (molecular weight: 156,000-158,000) consisting of tetrasodium salt of Sarotalocan (6-({[3-({(OC-6-13)-bis({3-[bis(3-sulfopropyl)(3-sulfonatopropyl)azaniumyl]propyl}dimethylsilanolato-κO,κO‘)[(phtalocyaninato(2-)κN29,κN30,κN31,κN32)-1-yl]silicon}oxy)propoxy]carbonyl}amino)hexanoyl (C70H96N11O24S6Si3; molecular weight: 1,752.22)) attached to an average of 2-3 Lys residues of Cetuximab.
[2166339-33-7 , Cetuximab sarotalocan]
Cetuximab sarotalocan sodium
Enarodustat
Enarodustat
エナロデュスタット
JTZ 951
| Formula | C17H16N4O4 |
|---|---|
| CAS | 1262132-81-9 |
| Mol weight | 340.3333 |
PMDA 2020/9/25 APPROVED ENAROY
Anti-anemic, Hypoxia inducible factor-prolyl hydroxylase (HIF-PH) inhibitor
Originator Japan Tobacco
Developer Japan Tobacco; JW Pharmaceutical
Class Acetic acids; Amides; Antianaemics; Pyridones; Small molecules; Triazoles
Mechanism of Action Hypoxia-inducible factor-proline dioxygenase inhibitors
Preregistration Anaemia
27 Dec 2019 Japan Tobacco and SalubrisBio enter into a development and marketing agreement for enarodustat (JTZ 951) in China, Hong Kong, Macau and Taiwan for Anaemia
29 Nov 2019 Preregistration for Anaemia in Japan (PO)
31 Oct 2019 Phase I development in Anaemia is ongoing in USA
Enarodustat is a potent and orally active factor prolyl hydroxylase inhibitor, with an EC50 of 0.22 μM. Enarodustat has the potential for renal anemia treatment
PATENT
WO 2011007856
PAPER
ACS Medicinal Chemistry Letters (2017), 8(12), 1320-1325
https://pubs.acs.org/doi/10.1021/acsmedchemlett.7b00404
Abstract

Inhibition of hypoxia inducible factor prolyl hydroxylase (PHD) represents a promising strategy for the discovery of a next generation treatment for renal anemia. We identified several 5,6-fused ring systems as novel scaffolds of the PHD inhibitor on the basis of pharmacophore analysis. In particular, triazolopyridine derivatives showed potent PHD2 inhibitory activities. Examination of the predominance of the triazolopyridines in potency by electrostatic calculations suggested favorable π–π stacking interactions with Tyr310. Lead optimization to improve the efficacy of erythropoietin release in cells and in vivo by improving cell permeability led to the discovery of JTZ-951 (compound 14), with a 5-phenethyl substituent on the triazolopyridine group, which increased hemoglobin levels with daily oral dosing in rats. Compound 14 was rapidly absorbed after oral administration and disappeared shortly thereafter, which could be advantageous in terms of safety. Compound 14 was selected as a clinical candidate.

(7-Hydroxy-5-phenethyl-[1,2,4]triazolo[1,5-a]pyridine-8-carbonyl)glycine (14)
To a solution of SI-5 (2.28 g, 6.19 mmol) in EtOH (9.1 mL) was added 2N NaOH aq. (12.4 mL, 24.8 mmol) at room temperature. After stirring at 90 °C for 2 h, 6N HCl aq. (4.1 mL, 24.6 mmol). This was allowed to gradually cool with stirring and crystals were precipitated. The crystals were collected by filtration to give the title compound 14 (2.16 g, 103% yield). 1H NMR (400 MHz, DMSO-D6) δ: 14.22 (s, 1H), 12.98 (br s, 1H), 9.84 (t, J = 5.6 Hz, 1H), 8.58 (s, 1H), 7.33– 7.18 (m, 5H), 6.80 (s, 1H), 4.22 (d, J = 5.6 Hz, 2H), 3.40 (t, J = 7.7 Hz, 2H), 3.12 (t, J = 7.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ: 170.28, 167.70, 165.32, 152.95, 148.53, 146.49, 140.05, 128.33, 128.20, 126.17, 106.72, 95.56, 41.00, 31.95, 31.72. HRMS m/z: [M+H]+ calcd for C17H17N4O4, 341.1244; found, 341.1243. Anal. (C17H16N4O4) calcd C 59.99%, H 4.74%, N 16.46%; found C 60.02%, H, 4.78%, N, 16.42%. Melting point: 186 °C Purity: 100.0%.
PATENT
WO 2018097254
PATENT
US 20200017492
/////////////Enarodustat, 2020 APPROVALS, JAPAN 2020, エナロデュスタット , JTZ 951, ENAROY, 2020 APPROVALS,
Abametapir アバメタピル , абаметапир , أباميتابير , 阿巴甲吡 ,

Abametapir
アバアバメタピル , абаметапир , أباميتابير , 阿巴甲吡 ,
5,5′-dimethyl-2,2′-bipyridine, 6,6′-Bi-3-picoline
- BRN 0123183
- HA 44
- HA-44
- HA44
| Formula |
C12H12N2
|
|---|---|
| CAS |
1762-34-1
|
| Mol weight |
184.2371
|
Xeglyze, FD APPROVED 24/7/2020
|
Pediculicide, Metalloproteinase inhibitor
|
|
| Disease |
Head lice infestation
|
|---|
- Originator Hatchtech
- DeveloperDr Reddys Laboratories; Hatchtech
- ClassAntiparasitics; Heterocyclic compounds; Pyridines; Small molecules
- Mechanism of ActionChelating agents; Metalloprotease inhibitors
- Registered Pediculosis
- 27 Jul 2020Registered for Pediculosis (In adolescents, In children, In infants, In adults) in USA (Topical)
- 18 Jun 2020FDA assigns PDUFA action date of 12/08/2020 for Abametapir for Pediculosis (Dr Reddy’s Laboratories website, June 2020)
- 31 Mar 2019Abametapir is still in preregistration phase for Pediculosis in USA
Abametapir is a novel pediculicidal metalloproteinase inhibitor used to treat infestations of head lice.4 The life cycle of head lice (Pediculus capitis) is approximately 30 days, seven to twelve of which are spent as eggs laid on hair shafts near the scalp.2 Topical pediculicides generally lack adequate ovicidal activity,2 including standard-of-care treatments such as permethrin, and many require a second administration 7-10 days following the first to kill newly hatched lice that resisted the initial treatment. The necessity for follow-up treatment may lead to challenges with patient adherence, and resistance to agents like permethrin and pyrethrins/piperonyl butoxide may be significant in some areas.3
Investigations into novel ovicidal treatments revealed that several metalloproteinase enzymes were critical to the egg hatching and survival of head lice, and these enzymes were therefore identified as a potential therapeutic target.1 Abemetapir is an inhibitor of these metalloproteinase enzymes, and the first topical pediculicide to take advantage of this novel target. The improved ovicidal activity (90-100% in vitro) of abemetapir allows for a single administration, in contrast to many other topical treatments, and its novel and relatively non-specific mechanism may help to curb the development of resistance to this agent.1
Abametapir was first approved for use in the United States under the brand name Xeglyze on July 27, 2020.6
Abametapir, sold under the brand name Xeglyze, is a medication used for the treatment of head lice infestation in people six months of age and older.[1][2]
The most common side effects include skin redness, rash, skin burning sensation, skin inflammation, vomiting, eye irritation, skin itching, and hair color changes.[2]
Abametapir is a metalloproteinase inhibitor.[1] Abametapir was approved for medical use in the United States in July 2020.[1][3]
Abametapir, a metalloproteinase inhibitor, demonstrated potent pediculicidal activity in preclinical studies. In vitro assays showed abametapir lotion (0.74–1% w/v) achieved >95% mortality of Pediculus humanus capitis adults and eggs within 10 minutes of exposure. Ex vivo human hair assays confirmed ovicidal efficacy, with >90% inhibition of egg hatching compared to vehicle controls. Mechanistic studies indicated abametapir disrupted metalloproteinase-dependent processes essential for louse development and egg viability. Toxicology studies in rodents and rabbits showed no significant systemic toxicity at topical doses up to 3% formulation, supporting its advancement into clinical evaluation as a single-application pediculicide.
Medical uses
Abametapir is indicated for the topical treatment of head lice infestation in people six months of age and older.[1][2]
History
The U.S. Food and Drug Administration (FDA) approved abametapir based on evidence from two identical clinical trials of 699 participants with head lice.[2] The trials were conducted at fourteen sites in the United States.[2]
The benefit and side effects of abametapir were evaluated in two clinical trials that enrolled participants with head lice who were at least six months old.[2]
About half of all enrolled participants was randomly assigned to abametapir and the other half to placebo.[2] Abametapir lotion or placebo lotion were applied once as a ten-minute treatment to infested hair.[2] The benefit of abametapir in comparison to placebo was assessed after 1, 7 and 14 days by comparing the counts of participants in each group who were free of live lice.[2]
SYN
Ronald Harding, Lewis David Schulz, Vernon Morrison Bowles, “Pediculicidal composition.” WIPO Patent WO2015107384A2, published July, 2015.


SYN
References
- ^ Jump up to:a b c d e “Xeglyze (abametapir) lotion, for topical use” (PDF). U.S. Food and Drug Administration (FDA). Dr. Reddy’s Laboratories. Inc. Retrieved 25 July 2020.
- ^ Jump up to:a b c d e f g h i “Drug Trial Snapshot: Xeglyze”. U.S. Food and Drug Administration (FDA). 24 July 2020. Retrieved 6 August 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Abametapir: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 25 July 2020.
Further reading
- Bowles VM, VanLuvanee LJ, Alsop H, Hazan L, Shepherd K, Sidgiddi S, et al. (September 2018). “Clinical studies evaluating abametapir lotion, 0.74%, for the treatment of head louse infestation”. Pediatr Dermatol. 35 (5): 616–621. doi:10.1111/pde.13612. PMC 6175393. PMID 29999197.
External links
- “Abametapir”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Xeglyze |
| Other names | Ha44 |
| AHFS/Drugs.com | Professional Drug Facts |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Topical |
| Drug class | Pediculicide, Metalloproteinase inhibitor |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.157.434 |
| Chemical and physical data | |
| Formula | C12H12N2 |
| Molar mass | 184.242 g·mol−1 |
| 3D model (JSmol) | |
///////Abametapir, 2020 APPROVALS, FDA 2020, Xeglyze, アバメタピル , абаметапир , أباميتابير , 阿巴甲吡 , BRN 0123183, HA 44, head lice
CC1=CC=C(N=C1)C1=CC=C(C)C=N1
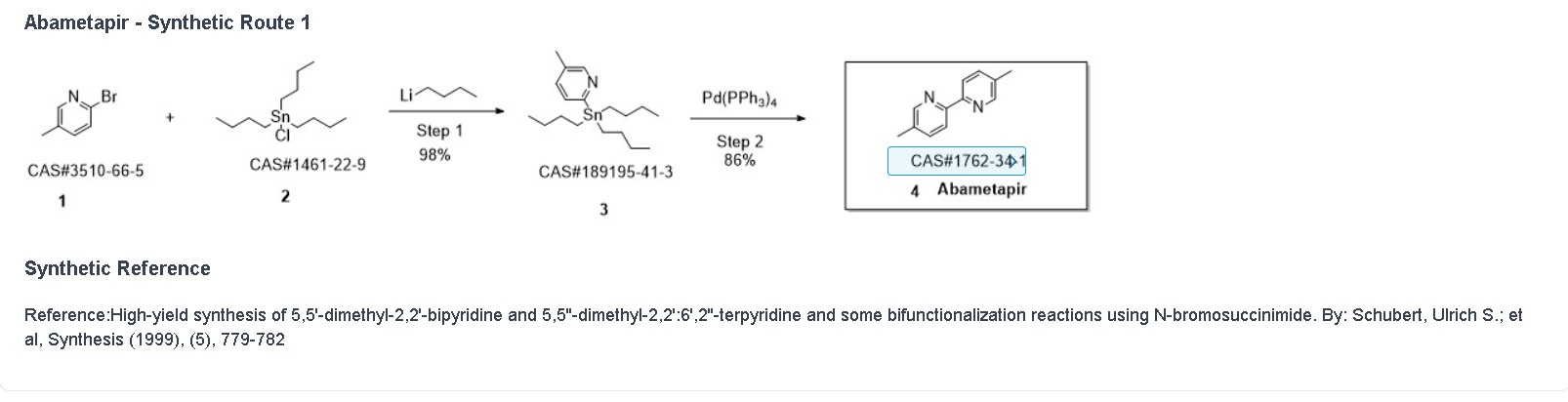

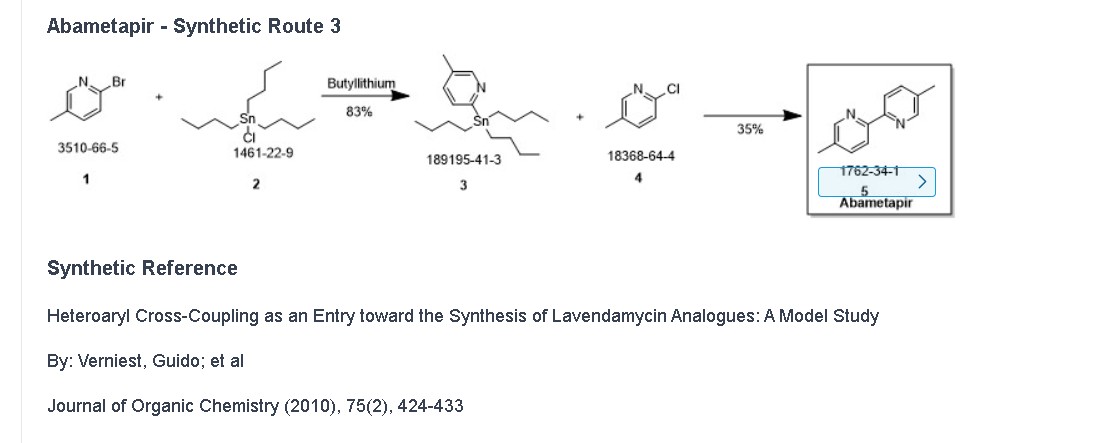
.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}