
Home » 0rphan drug status (Page 2)
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
HEXASODIUM PHYTATE





HEXASODIUM PHYTATE
cas 34367-89-0
myo-Inositol, 1,2,3,4,5,6-hexakis(dihydrogen phosphate) sodium salt (1:6)
hexasodium;[2,3,4,5,6-pentakis[[hydroxy(oxido)phosphoryl]oxy]cyclohexyl] hydrogen phosphate
free form RN: 83-86-3
C6H12Na6O24P6, 791.93
- Inositol, hexakis(dihydrogen phosphate) hexasodium salt, myo– (8CI)
- myo-Inositol, hexakis(dihydrogen phosphate), hexasodium salt (9CI)
- Hexasodium fytate
- Hexasodium phytate
- SNF 472
- UNII-ZBX50UG81V
- CSL-525; Hexasodium phytate; Myo-inositol hexaphosphate; SNF-472
x Na salt
14306-25-3
C6H18O24P6.xNa
free form : 83-86-3
myo-Inositol, 1,2,3,4,5,6-hexakis(dihydrogen phosphate), sodium salt
| (1R,2R,3S,4S,5R,6S)-CYCLOHEXANE-1,2,3,4,5,6-HEXAYL-HEXAKIS(DIHYDROGEN PHOSPHATE) |
- Inositol, hexakis(dihydrogen phosphate) sodium salt, myo– (8CI)
- myo-Inositol, hexakis(dihydrogen phosphate), sodium salt (9CI)
- Inositol hexaphosphate sodium salt
- Phytic acid sodium salt
- Sodium inositol hexaphosphate
- Sodium phytate
- OriginatorLaboratoris Sanifit
- DeveloperCSL Vifor; Laboratoris Sanifit
- ClassAntineoplastics; Calcium regulators; Cardiovascular therapies; Phosphates; Small molecules; Sodium compounds; Sugar alcohols
- Mechanism of ActionUndefined mechanism
- Orphan Drug StatusYes – Peripheral arterial disorders; Calciphylaxis
- Phase IIICalciphylaxis; Peripheral arterial disorders
- 09 Nov 2022Phase-III clinical trials in Peripheral arterial disorders in USA (IV) (CSL Behring pipeline, November 2022)
- 24 Oct 2022Sanifit Therapeutics completes a phase III trial in Calciphylaxis in Belgium, Poland, United Kingdom, Germany, Spain, USA (IV) (NCT04195906)
- 28 Sep 2022Hexasodium fytate is still in phase III trials for Calciphylaxis in USA (IV) (NCT04195906)
- You need to be a logged in or subscribed to view this
Hexasodium phytate (also known as SNF472) is a compound being developed as a potential treatment for calciphylaxis, a condition causing skin damage and tissue death in patients with end-stage renal disease. It works by inhibiting the formation and growth of hydroxyapatite crystals, which are implicated in calciphylaxis.
What it is:Hexasodium phytate is the hexasodium salt of myo-inositol hexaphosphate (IP6), a naturally occurring substance found in foods like beans and grains.
- How it works:It binds to hydroxyapatite crystals, the main component of vascular calcification, and prevents their growth, potentially disrupting the calciphylaxis process.
- Why it’s used:Hexasodium phytate is being investigated as a treatment for calciphylaxis, a serious complication of end-stage renal disease characterized by skin and tissue damage due to calcification of small blood vessels.
- Mechanism:It is believed to work by inhibiting the formation and growth of calcium-phosphate crystals (hydroxyapatite) in the blood vessels, thus preventing the calcification that leads to calciphylaxis.
- Clinical trials:Clinical trials have demonstrated the safety and potential efficacy of hexasodium phytate in reducing hydroxyapatite crystallization in patients undergoing hemodialysis, providing a basis for its use in treating calciphylaxis.
- Intravenous administration:It is administered intravenously during dialysis sessions to achieve supra-physiological plasma concentrations, which are thought to be necessary for its therapeutic effect.
- Benefits:It has shown promise in preclinical studies and clinical trials, potentially improving wound healing, pain, and health-related quality of life in patients with calciphylaxis.
- Active development:It is currently in active development as a novel experimental drug for the treatment of calciphylaxis and other related conditions.
Phytic acid is a major phosphorus storage compound of most seeds and cereal grains. It has the strong ability to chelate multivalent metal ions, especially zinc, calcium, and iron. Phytic acid is also considered to be a natural antioxidant and is suggested to have potential functions of reducing lipid peroxidation and as a preservative in foods. Clathrin-associated adaprot complex AP-2 has it been suggested may act as one of the receptor sites for Phytic acid. Both in vivo and in vitro experiments have demonstrated striking anticancer (preventive as well as therapeutic) effects of Phytic acid.
SCHEME

contd………

References
WO2022129148
EP4015494
CN114874473
iScience (2022), 25(3), 103950
CN111718463
CN110483240
Uzbekskii Khimicheskii Zhurnal (1995), (5-6), 72-75 JP61056142
////////HEXASODIUM PHYTATE, Hexasodium fytate, Hexasodium phytate, SNF 472, calciphylaxis, UNII-ZBX50UG81V, CSL-525, Hexasodium phytate, Myo-inositol hexaphosphate, SNF-472, ORPHAN DRUG
Omaveloxolone



Omaveloxolone
CAS
1474034-05-3
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-octadecahydropicen-4a-yl]-2,2-difluoropropanamide
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,3,4,5,6,7,8,8a,14a,14b-decahydropicen-4a-yl]-2,2-difluoropropanamide
FDA 2023, 2/28/2023, To treat Friedrich’s ataxia
Drug Trials Snapshot
WeightAverage: 554.723
Monoisotopic: 554.331999611
Chemical FormulaC33H44F2N2O3
- RTA 408
- RTA-408
- OriginatorDartmouth College; University of Texas M. D. Anderson Cancer Center
- DeveloperBiogen
- ClassAnalgesics; Anti-inflammatories; Antineoplastics; Eye disorder therapies; Neuroprotectants; Small molecules; Triterpenes
- Mechanism of ActionNF-E2-related factor 2 stimulants
- Orphan Drug StatusYes – Friedreich’s ataxia; Malignant melanoma
- MarketedFriedreich’s ataxia
- Phase IIMitochondrial disorders; Ocular inflammation; Ocular pain
- Phase I/IIMalignant melanoma
- PreclinicalBrain disorders
- DiscontinuedDuchenne muscular dystrophy; Non-small cell lung cancer; Radiation-induced skin damage
- 08 Apr 2025Biogen completes a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
- 17 Mar 2025Registered for Friedreich’s ataxia (In adolescents, In adults) in Canada (PO)
- 18 Oct 2024Biogen initiates enrolment in a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
Omaveloxolone, sold under the brand name Skyclarys, is a medication used for the treatment of Friedreich’s ataxia.[2][5] It is taken by mouth.[2]
The most common side effects include an increase in alanine transaminase and an increase of aspartate aminotransferase, which can be signs of liver damage, headache, nausea, abdominal pain, fatigue, diarrhea and musculoskeletal pain.[5]
Omaveloxolone was approved for medical use in the United States in February 2023,[2][5][6][7][8] and in the European Union in February 2024.[3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[9]
SYNTHESIS

PATENT
Sheikh, AY et al. (2018). Bardoxolonmethyl-2,2-difluoropropionamide derivatives, polymorphe forms and procedures for use thereof. DK/EP 2989114 T3. Danish Patent and Trademark Office. Available at https://patentimages.storage.googleapis.com/51/87/43/97d0fb3e69ee73/DK2989114T3.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP159939262&_cid=P21-MAKI10-93498-1

[0164] Reagents and conditions: (a) (PhO) 2PON 3 (DPPA), triethylamine, toluene, 0 °C for 5 minutes, then ambient temperature overnight, ∼94%; (b) benzene, 80 °C for 2 hours; (c) HCl, CH 3CN, ambient temperature for 1 hour; (d) CH 3CF 2CO 2H, dicyclohexylcarbodiimide, 4-(dimethylamino)pyridine, CH 2Cl 2, ambient temperature overnight, 73% from RTA 401 (4 steps).
[0165]Compound 1: RTA 401 (20.0 g, 40.6 mmol), triethylamine (17.0 mL, 122.0 mmol), and toluene (400 mL) were added into a reactor and cooled to 0 °C with stirring. Diphenyl phosphoryl azide (DPPA) (13.2 mL, 61.0 mmol) was added with stirring at 0 °C over 5 minutes, and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no RTA 401 left). The reaction mixture was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 5% ethyl acetate in CH 2Cl 2) to give compound 1 (19.7 g, ∼94%, partially converted into compound 2) as a white foam.
[0166]Compound 2: Compound 1 (19.7 g, ∼38.1 mmol) and benzene (250 mL) were added into a reactor and heated to 80 °C with stirring for 2 hours (HPLC-MS check shows no compound 1 left). The reaction mixture was concentrated at reduced pressure to afford crude compound 2 as a solid residue, which was used for the next step without purification.
[0167]Compound 3: Crude compound 2 (≤38.1 mmol) and CH 3CN (200 mL) were added into a reactor and cooled to 0 °C with stirring. HCl (12 N, 90 mL) was added at 0 °C over 1 minute, and the mixture was continually stirred at room temperature for 1 hour (HPLC-MS check shows no compound 2 left). The reaction mixture was cooled to 0 °C and 10% NaOH (∼500 mL) was added with stirring. Then, saturated NaHCO 3 (1 L) was added with stirring. The aqueous phase was extracted by ethyl acetate (2×500 mL). The combined organic phase was washed by H 2O (200 mL), saturated NaCl (200 mL), dried over Na 2SO 4, and concentrated to afford crude compound 3 (16.62 g) as a light yellow foam, which was used for the next step without purification.
[0168]RTA 408: Crude amine 3 (16.62 g, 35.9 mmol), CH 3CF 2CO 2H (4.7388 g, 43.1 mmol), and CH 2Cl 2 (360 mL) were added into a reactor with stirring at room temperature. Then, dicyclohexylcarbodiimide (DCC) (11.129 g, 53.9 mmol) and 4-(dimethylamino)pyridine (DMAP) (1.65 g, 13.64 mmol) were added and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no compound 3 left). The reaction mixture was filtered to remove solid by-products, and the filtrate was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 20% ethyl acetate in hexanes) twice to give compound RTA 408 (16.347 g, 73% from RTA 401 over 4 steps) as a white foam: 1H NMR (400 MHz, CD 3Cl) δ ppm 8.04 (s, 1H), 6.00 (s, 1H), 5.94 (s, br, 1H), 3.01 (d, 1H, J = 4.8 Hz), 2.75-2.82 (m, 1H), 1.92-2.18 (m, 4H), 1.69-1.85 (m, 7H), 1.53-1.64 (m, 1H), 1.60 (s, 3H), 1.50 (s, 3H), 1.42 (s, 3H), 1.11-1.38 (m, 3H), 1.27 (s, 3H), 1.18 (s, 3H), 1.06 (s, 3H), 1.04 (s, 3H), 0.92 (s, 3H); m/z 555 (M+1).
SYNTHESIS
J. Med. Chem. 2025, 68, 2147−2182
Omaveloxolone (Skyclarys). Omaveloxolone (6) was approved in February 2023 for the treatment of Friedreich’s Ataxia (FRDA), a genetic, neurodegenerative disease. Patients with FRDA have lowered activity of the frataxin gene (FXN), attributed to an expansion of a guanine-adenine-adenine (GAA)
triplet. The resulting decrease in frataxin limits the production of iron−sulfur clusters, leading to accumulation of iron in the mitochondria and oxidative stress which in turn leads to cell damageanddeath.49
Omaveloxoloneactivates the nuclear factor erythroid 2-related factor 2 (Nrf2), an important pathway in
oxidative stress. It acts by preventing ubiquitination and subsequent degradation of Nrf2, keeping levels high enough to counteract the oxidative stress associated with FRDA. 50
Omaveloxolone was developed by Reata Pharmaceuticals (which was acquired by Biogen in September 2023) and was granted orphan drug, fast track, priority review, and rare pediatric disease designations. 51Omaveloxolone (6) is a semisynthetic triterpenoid based on the oleanolic acid scaffold.52
advanced intermediate 6.1,The synthesis started from the53also known as CDDO orbardoxolone, which has individually been investigated fortherapeutic benefits from Nrf2 activation (Scheme 10).
Treatment of acid 6.1 with DPPA produced the azide, and subsequent heating in benzene generated isocyanate 6.2 via aCurtius rearrangement. Hydrolysis with aqueous acid generated amine 6.3, and an amidation with 2,2-difluoropropanoic acid produced omaveloxolone (6). A yield of 73% over the sequence was reported, and intermediates were used crude with no purification between steps.
(49) Ghanekar, S. D.; Miller, W. W.; Meyer, C. J.; Fenelon, K. J.;
Lacdao, A.; Zesiewicz, T. A. Orphan drugs in development for the
treatment of Friedreich’s ataxia: focus on omaveloxolone. Degener.
Neurol. Neuromuscular Dis. 2019, 9, 103−107.
(50) Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-inducer
prevents mitochondrial defects and oxidative stress in Friedreich’s
ataxia models. Front. Cell. Neurosci. 2018, 12, 188.
(51) Lee,A.Omaveloxolone:first approval. Drugs 2023, 83, 725−729.
(52) Anderson, E.; Decker, A.; Liu, X. Synthesis, pharmaceutical use,
and characterization of crystalline forms of 2,2-difluoropropionamide
derivatives of bardoxolone methyl. WO 2013163344, 2013.
(53) Honda, T.; Rounds, B. V.; Gribble, G. W.; Suh, N.; Wang, Y.;
Sporn, M. B. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9
dien-28-oic acid, a novel and highly active inhibitor of nitric oxide
production in mouse macrophages. Bioorg. Med. Chem. Lett. 1998, 8,
2711−2714.

SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Omaveloxolone (Skyclarys)
Omaveloxolone was granted FDA approval on February 28, 2023, to treat Friedrich’s ataxia in individuals aged 16 and older [2]. Omaveloxolone possesses antioxidant and anti-inflammatory properties, making it a semi-synthetic triterpenoid compound. It has the ability to function as a stimulator of nuclear factor-erythroid 2 related factor 2(Nrf2), a transcription factor that reduces oxidative stress. In individuals
suffering from FA, a genetic disorder characterized by mitochondrial dysfunction, the Nrf2 pathway is compromised, leading to a decrease in Nrf2 activity. Hence, Omaveloxolone, an Nrf2 activator, can be
employed as a therapeutic option for the management of these in dividuals [23].The process route of Omaveloxolone is described below in Scheme 724]. The substitution reaction of carboxylic acid OMAV-001 with diphenylphosphoryl azide (DPPA) gave the acyl azide OMAV-002,which underwent Curtius-rearrangement under heating conditions to produce isocyanate OMAV-003. The amine OMAV-004 was obtained under acidic conditions. OMAV-004 was condensed with 2,2-difluoro propionic acid to obtain the final product Omaveloxolone.
[23] B.L. Probst, I. Trevino, L. McCauley, R. Bumeister, I. Dulubova, W.C. Wigley, D.
A. Ferguson, RTA 408, A novel synthetic triterpenoid with broad anticancer and
anti-inflammatory activity, PLoS One 10 (2015) e0122942.
[24] E. Anderson, A. Decker, X. Liu Synthesis, Pharmaceutical Use, and
Characterization of Crystalline Forms of 2,2-difluoropropionamide Derivatives of
Bardoxolone Methyl, 2013. WO2013163344.

.
Medical uses
Omaveloxolone is indicated for the treatment of Friedreich’s ataxia.[2][5]
Friedreich’s ataxia causes progressive damage to the spinal cord, peripheral nerves, and the brain, resulting in uncoordinated muscle movement, poor balance, difficulty walking, changes in speech and swallowing, and a shortened lifespan.[5] The condition can also cause heart disease.[5] This disease tends to develop in children and teenagers and gradually worsens over time.[5]
Although rare, Friedreich’s ataxia is the most common form of hereditary ataxia in the United States, affecting about one in every 50,000 people.[5]
Mechanism of action
The mechanism of action of omaveloxolone and its related compounds has been demonstrated to be through a combination of activation of the antioxidative transcription factor Nrf2 and inhibition of the pro-inflammatory transcription factor NF-κB.[10]
Nrf2 transcriptionally regulates multiple genes that play both direct and indirect roles in producing antioxidative potential and the production of cellular energy (i.e., adenosine triphosphate or ATP) within the mitochondria. Consequently, unlike exogenously administered antioxidants (e.g., vitamin E or Coenzyme Q10), which provide a specific and finite antioxidative potential, omaveloxolone, through Nrf2, broadly activates intracellular and mitochondrial antioxidative pathways, in addition to pathways that may directly increase mitochondrial biogenesis (such as PGC1α) and bioenergetics.[11]
History
Omaveloxolone is a second generation member of the synthetic oleanane triterpenoid compounds and in clinical development by Reata Pharmaceuticals. Preclinical studies have demonstrated that omaveloxolone possesses antioxidative and anti-inflammatory activities[10][12] and the ability to improve mitochondrial bioenergetics.[11] Omaveloxolone is under clinical investigation for a variety of indications, including Friedreich’s ataxia, mitochondrial myopathies, immunooncology, and prevention of corneal endothelial cell loss following cataract surgery.
The efficacy and safety of omaveloxolone was evaluated in a 48-week randomized, placebo-controlled, and double-blind study [Study 1 (NCT02255435)] and an open-label extension.[5] Study 1 enrolled 103 individuals with Friedreich’s ataxia who received placebo (52 individuals) or omaveloxolone 150 mg (51 individuals) for 48 weeks.[5] Of the research participants, 53% were male, 97% were white, and the mean age was 24 years at study entry.[5] Nine (18%) patients were younger than age 18.[5] The primary objective was to evaluate the change in the modified Friedreich’s Ataxia Rating Scale (mFARS) score compared to placebo at week 48.[5] The mFARS is a clinical assessment that measures disease progression, namely swallowing and speech (bulbar), upper limb coordination, lower limb coordination, and upright stability.[5] Individuals receiving omaveloxolone performed better on the mFARS than people receiving placebo.[5]
The US Food and Drug Administration (FDA) granted the application for omaveloxolone orphan drug, fast track, priority review, and rare pediatric disease designations.[5][9]
Society and culture
Legal status
Omaveloxolone was approved for medical use in the United States in February 2023.[2][5]
In December 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Skyclarys, intended for the treatment of Friedreich’s ataxia.[3] The applicant for this medicinal product is Reata Ireland Limited.[3] Omaveloxolone was approved for medical use in the European Union in February 2024.[3][4]
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ Jump up to:a b c d e f “Skyclarys- omaveloxolone capsule”. DailyMed. 12 May 2023. Archived from the original on 1 July 2023. Retrieved 16 December 2023.
- ^ Jump up to:a b c d e “Skyclarys EPAR”. European Medicines Agency (EMA). 14 December 2023. Archived from the original on 15 December 2023. Retrieved 16 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Skyclarys product information”. Union Register of medicinal products. 12 February 2024. Retrieved 19 February 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA approves first treatment for Friedreich’s ataxia”. U.S. Food and Drug Administration (FDA). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Reata Pharmaceuticals Announces FDA Approval of Skyclarys (Omavaloxolone), the First and Only Drug Indicated for Patients with Friedreich’s Ataxia”. Reata Pharmaceuticals Inc. (Press release). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
- ^ Lee A (June 2023). “Omaveloxolone: First Approval”. Drugs. 83 (8): 725–729. doi:10.1007/s40265-023-01874-9. PMID 37155124. S2CID 258567442. Archived from the original on 9 December 2023. Retrieved 16 December 2023.
- ^ Subramony SH, Lynch DL (May 2023). “A Milestone in the Treatment of Ataxias: Approval of Omaveloxolone for Friedreich Ataxia”. Cerebellum. 23 (2): 775–777. doi:10.1007/s12311-023-01568-8. PMID 37219716. S2CID 258843532.
- ^ Jump up to:a b New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Jump up to:a b Reisman SA, Lee CY, Meyer CJ, Proksch JW, Ward KW (July 2014). “Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin”. Archives of Dermatological Research. 306 (5): 447–454. doi:10.1007/s00403-013-1433-7. PMID 24362512. S2CID 25733020.
- ^ Jump up to:a b Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, et al. (July 2011). “Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis”. Free Radical Biology & Medicine. 51 (1): 88–96. doi:10.1016/j.freeradbiomed.2011.03.027. PMC 3109235. PMID 21457778.
- ^ Reisman SA, Lee CY, Meyer CJ, Proksch JW, Sonis ST, Ward KW (May 2014). “Topical application of the synthetic triterpenoid RTA 408 protects mice from radiation-induced dermatitis”. Radiation Research. 181 (5): 512–520. Bibcode:2014RadR..181..512R. doi:10.1667/RR13578.1. PMID 24720753. S2CID 23906747.
External links
Clinical trial number NCT02255435 for “RTA 408 Capsules in Patients With Friedreich’s Ataxia – MOXIe” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Skyclarys |
| Other names | RTA 408 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Omaveloxolone |
| Routes of administration | By mouth |
| ATC code | N07XX25 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3][4] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1474034-05-3 |
| PubChem CID | 71811910 |
| IUPHAR/BPS | 7573 |
| DrugBank | DB12513 |
| ChemSpider | 34980948 |
| UNII | G69Z98951Q |
| KEGG | D10964 |
| ChEBI | CHEBI:229661 |
| CompTox Dashboard (EPA) | DTXSID101138251 |
| Chemical and physical data | |
| Formula | C33H44F2N2O3 |
| Molar mass | 554.723 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Zesiewicz TA, Hancock J, Ghanekar SD, Kuo SH, Dohse CA, Vega J: Emerging therapies in Friedreich’s Ataxia. Expert Rev Neurother. 2020 Dec;20(12):1215-1228. doi: 10.1080/14737175.2020.1821654. Epub 2020 Sep 21. [Article]
- Jiang Z, Qi G, Lu W, Wang H, Li D, Chen W, Ding L, Yang X, Yuan H, Zeng Q: Omaveloxolone inhibits IL-1beta-induced chondrocyte apoptosis through the Nrf2/ARE and NF-kappaB signalling pathways in vitro and attenuates osteoarthritis in vivo. Front Pharmacol. 2022 Sep 27;13:952950. doi: 10.3389/fphar.2022.952950. eCollection 2022. [Article]
- Shekh-Ahmad T, Eckel R, Dayalan Naidu S, Higgins M, Yamamoto M, Dinkova-Kostova AT, Kovac S, Abramov AY, Walker MC: KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain. 2018 May 1;141(5):1390-1403. doi: 10.1093/brain/awy071. [Article]
- Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, Ferguson DA: RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PLoS One. 2015 Apr 21;10(4):e0122942. doi: 10.1371/journal.pone.0122942. eCollection 2015. [Article]
- Lynch DR, Farmer J, Hauser L, Blair IA, Wang QQ, Mesaros C, Snyder N, Boesch S, Chin M, Delatycki MB, Giunti P, Goldsberry A, Hoyle C, McBride MG, Nachbauer W, O’Grady M, Perlman S, Subramony SH, Wilmot GR, Zesiewicz T, Meyer C: Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann Clin Transl Neurol. 2018 Nov 10;6(1):15-26. doi: 10.1002/acn3.660. eCollection 2019 Jan. [Article]
- Zighan M, Arkadir D, Douiev L, Keller G, Miller C, Saada A: Variable effects of omaveloxolone (RTA408) on primary fibroblasts with mitochondrial defects. Front Mol Biosci. 2022 Aug 12;9:890653. doi: 10.3389/fmolb.2022.890653. eCollection 2022. [Article]
- FDA Approved Drug Products: SKYCLARYS (omaveloxolone) capsules for oral use (February 2023) [Link]
- EMA Approved Drug Products: Skyclarys (omaveloxolone) Oral Capsules [Link]
- Health Canada Approved Drug Products: SKYCLARYS (Omaveloxolone) Capsules For Oral Use [Link]
///////////Omaveloxolone, Skyclarys, Friedrich’s ataxia, FDA 2023, APPROVALS 2023, RTA 408, RTA-408, omaveloxolona, RTA 408, 63415, PP415, orphan drug, fast track, priority review, rare pediatric disease



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Clofutriben



Clofutriben
Cas 1204178-50-6
HCL 1203941-88-1
- ASP 3662
- 4-(5-(2-(4-Chloro-2,6-difluorophenoxy)propan-2-yl)-4-methyl-4H-1,2,4-triazol-3-yl)-3-fluorobenzamide
- 4-{5-[2-(4-Chloro-2,6-difluorophenoxy)propan-2-yl]-4-methyl-4H-1,2,4-triazol-3-yl}-3-fluorobenzamide
- 4-[5-[2-(4-chloro-2,6-difluorophenoxy)propan-2-yl]-4-methyl-1,2,4-triazol-3-yl]-3-fluorobenzamide
- 4L1TY1U5VC
| Molecular Weight | 424.80 |
|---|---|
| Formula | C19H16ClF3N4O2 |
Clofutriben (ASP3662) is a 11β-hydroxysteroid dehydrogenase type 1 inhibitor.
Clofutriben is an orally bioavailable selective inhibitor of the enzyme 11beta-hydroxysteroid dehydrogenase type 1 (11b-HSD1; 11bHSD1; HSD11B1; HSD1; HSD-1), with potential protective activity for disorders of corticosteroid excess. Upon oral administration, clofutriben selectively binds to and inhibits the activity of HSD-1. This prevents the conversion of cortisone to the active hormone cortisol and thereby preventing the activation of the glucocorticoid receptors (GRs). By blocking cortisol production in metabolic tissues, clofutriben may inhibit the adverse metabolic effects that are caused by exogenous administration of glucocorticoids or in disorders in which cortisol is secreted in excess. HSD-1 is highly expressed in metabolic tissues, such as liver, skeletal muscle, and adipose tissue. It plays a crucial role in regulating the production of cortisol to activate the GRs.
SCHEME

PATENTS
Clinical and Translational Science (2019), 12(3), 291-301
British Journal of Pharmacology (2018), 175(19), 3784-3796
Sparrow Pharmaceuticals, Inc. WO2020106337
WO2019075394
WO2018117063
WO2010001946
PATENT
PDT PAT FOR HCL SALT, WO2012033070
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012033070
PATENT
PDT PAT FOR BASE, WO2018117063
PATENT
WO2010001946
[1]. Kiso T, et al. Analgesic effects of ASP3662, a novel 11尾-hydroxysteroid dehydrogenase 1 inhibitor, in rat models of neuropathic and dysfunctional pain. Br J Pharmacol. 2018 Oct;175(19):3784-3796. [Content Brief]
////////////Clofutriben, ASP 3662, orphan drug, 4L1TY1U5VC, Sparrow Pharmaceuticals,
Levacetylleucine

Levacetylleucine
WeightAverage: 173.212
Monoisotopic: 173.105193347
Chemical FormulaC8H15NO3
- N-Acetyl-L-leucine
- CAS 1188-21-2
- acetyl-L-leucine
- Ac-Leu-OH
- N-Acetylleucine
- NSC 206316
- UNII-E915HL7K2O
NSC-206316
(2S)-2-acetamido-4-methylpentanoic acid
FDA APPROVED 9/24/2024, To treat Niemann-Pick disease type C
Press Release
Drug Trials Snapshot
- Originator University of Munich; University of Oxford
- Developer IntraBio
- Class Acetamides; Amino acids; Esters; Neuroprotectants; Pentanoic acids; Small molecules; Vestibular disorder therapies
- Mechanism of Action Calcium channel modulators
- Orphan Drug StatusYes – Tay-Sachs disease; Niemann-Pick disease type C; Ataxia telangiectasia
Registered Niemann-Pick disease type C
- Phase IIIAtaxia telangiectasia
- Phase IISandhoff disease; Tay-Sachs disease
18 Mar 2025Phase-III clinical trials in Ataxia telangiectasia (In adolescents, In children, In the elderly, In adults) in Switzerland, Slovakia, Spain, Germany, USA, United Kingdom (PO) (NCT06673056)
- 04 Nov 2024IntraBio plans a phase III trial for Ataxia telangiectasia (In children, In adolescents, In adults, In elderly) in the US, Germany, Slovakia, Spain and Switzerland (PO, Suspension) in March 2025 (NCT06673056)
- 24 Sep 2024Registered for Niemann-Pick disease type C (In adolescents, In children, In adults) in USA (PO)
Levacetylleucine (N-acetyl-L-leucine), sold under the brand name Aqneursa, is a medication used for the treatment of neurological manifestations of Niemann-Pick disease type C.[1][2] Levacetylleucine is a modified version of the amino acid leucine.[1] It is the L-form of acetylleucine. It is taken by mouth.[1]
The most common side effects include abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting.[1][2]
Levacetylleucine was approved for medical use in the United States in September 2024.[1][2][3] Levacetylleucine is the second medication approved by the US Food and Drug Administration (FDA) for the treatment of Niemann-Pick disease type C.[2] The FDA considers it to be a first-in-class medication.[4]
DATA
N-acetyl-D, L-leucine is the active ingredient of Tanganil ® which helps treat vertigo attacks.

N-Acetyl-D, L-leucine
Unlike the majority of chemical syntheses of active principles where it is desirable to separate the enanti omers and / or to retain the selective stereo information during the synthesis steps, the synthesis of N-acetyl-D, L-leucine is carried out from L-leucine and therefore involves a racemization step. This racemization takes place before the acetylation step, via a Schiff base formed in situ with salicylic aldehyde (Yamada et al., J. Org. Chem., 1983 48, 843- 846).

Two competitive reactions are then involved: the acetylation of leucine, the main reaction, where acetic anhydride reacts with the amine function of leucinate of sodium to give N-acetyleucinate and the hydrolysis of acetic anhydride to acetic acid, a side reaction described below.

This synthesis has a molar yield of 70%. The limiting steps are essentially the secondary reaction of hydrolysis of acetic anhydride and the step of isolation of the racemized leucine before the acetylation reaction. Indeed, on an industrial scale, the quantities of products brought into play for isolations prove to be very restrictive.
There is therefore a real need to develop a new process for the preparation of N-actéyl-D, L-leucine which is faster and more economical.
The inventors thus discovered that the racemization step could be carried out after the L-leucine acetylation step making it possible to avoid a step of isolating the intermediate product and that this process could be carried out in continuous flow. Du Vigneaud & Meyer (J. Biol Chem, 1932, 98, 295-308) had already shown that it was possible to racemize different acetylated amino acids by bringing them into the presence of acetic anhydride for several hours. However, no examples had been made with acetyl leucine. By attempting to reproduce this process with acetyl-leucine, the inventors have thus found that this racemization reaction did not give satisfactory results with acetyl-leucine because of a competitive hydrolysis reaction of acetic anhydride. used. The inventors have also surprisingly discovered that the racemization reaction of N-acetyl-L-leucine could be improved by producing it in a continuous flow. It seems indeed that the realization of this continuous flow process allows better control of the mixing of the reagents and therefore to better control the reaction. The inventors have also shown that the racemization of N-acetyl-L-Leucine in continuous flow was obtained in a very short time of the order of a few minutes.
Furthermore, there is also a need to develop a new method of acetylation of leucine for the preparation of N-actyle-leucine which is faster and more economical. The inventors have discovered that the acetylation reaction of leucine can be improved by making it in a continuous flow. The process according to the invention gives good yields, in a very short time and using fewer reagents compared to the method known hitherto.
Indeed, DeWitt et al. (J Am Chem Soc (1951) 73 (7) 3359-60) described the preparation of N-acetyl-L-Leucine by reacting L-Leucine with 3 molar equivalents of acetic anhydride and sodium hydroxide for 2 hours 20 minutes. . N-acetyl-L-leucine is then obtained in a yield of only 70-80%. In addition, the authors of this publication clearly indicated that a molar ratio between L-Leucine and acetic anhydride below 2 resulted in much lower yields.
SYNTHESIS
H. D. DeWitt and A. W. Ingersoll. The Preparation of Pure N-Acetyl-L-leucine and L-Leucine. Journal of the American Chemical Society 1951 73 (7), 3359-3360. DOI: 10.1021/ja01151a108
PATENT
https://patents.google.com/patent/WO2012038515A1/en
EXAMPLES
A. Acetylation of L-Leucine in Continuous Flow

A. L. Study of the molar ratio of acetic anhydride to leucine
The objective of this study is to define the necessary molar ratio of acetic anhydride so that the acetylation reaction with acetic anhydride is complete and is not disadvantageous by competition with the acetic anhydride hydrolysis reaction. In this study, the residence time in the reactor / exchanger (1 process plate) was set at 9 seconds, for a temperature of the reaction medium of between 25 and 30 ° C.
The ratio range studied is between 0.9 and 2.0 molar equivalents. The optimum is obtained for a ratio between 1.20 and 2.00, more particularly between 1.30 and 1.60. Below this ratio, the acetylation reaction is disadvantageous compared to the acetic hydrolysis reaction. Beyond this, the drop in pH (acid instead of base) also disadvantages the acetylation reaction.
EXAMPLES 1-10:
A solution of sodium L-leucinate, for passage in continuous flow reactor, is prepared in the following manner: 700 g of L-leucine are dissolved in a solution of 576 g of sodium hydroxide and 3.5 liters of Demineralized Water. This solution is the main fluid process. The reaction between this solution and the acetic anhydride is carried out in a continuous flow in a Boostec® reactor, made of silicon carbide. The reactor / exchanger is configured with an injection-type process plate comprised between two utility plates. The volume of the process plate is 10 mL. The temperature in the reactor is maintained by the circulation of a coolant heated by a thermostatic bath. The transformation of L-leucine to N-acetyl-L-leucine is monitored online by quantitative Raman spectroscopy. This method of analysis is calibrated beforehand with solutions of known concentration prepared with pure L-leucine and N-acetyl-L-leucine.
Example 1
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 4.06 kg.h -1 and 0.42 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 0.91 equivalents. The total flow rate is therefore 4.48 kg.h -1 , which corresponds to a residence time (equivalent to the reaction time) of 8.7 s The yield of acetyl-L-leucinate determined by Raman spectroscopy online at the outlet of the reactor is 40% Example 2:
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.95 kg · h -1 and 0.45 kg · h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.01 equivalents. The total flow rate is therefore 4.40 kg.h -1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 52.degree. %.
Example 3
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.89 kg · h -1 and 0.52 kg · h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.18 equivalents. The total flow rate is therefore 4.41 kg.h -1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 57.degree. %. Example 4
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.82 kg. h -1 and 0.57 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.32 equivalents. The total flow is therefore 4.39 kg. h “1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 83%.
Example 5
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective rates set at 3.64 kg. h -1 and 0.55 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.34 equivalents. The total flow is therefore 4, 19 kg. h “1 , which corresponds to a residence time of 9.4 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 98%.
Example 6
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective rates set at 3.66 kg. h 1 and 0.62 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.50 equivalents. The total flow is therefore 4.28 kg. h “1 , which corresponds to a residence time of 9.2 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 96%.
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates fixed at 3.67 kg. h -1 and 0.64 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.54 equivalents. The total flow is therefore 4.31 kg. h “1 , which corresponds to a residence time of 9.1 sec The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 100%. Example 8
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.63 kg. h -1 and 0.73 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.78 equivalents. The total flow is therefore 4.36 kg. h “1 , which corresponds to a residence time of 9.0 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 90%.
PATENT
https://patents.google.com/patent/CN104592052A/en
Example 1:
100gL-leucine adds 1000ML2NNaOH rising temperature for dissolving, adds 1ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add 160ML HCl and adjust PH 2.5, be cooled to 4 degree, suction filtration, the 118g. of oven dry
Example 2:
100gL-leucine adds 1200ML 2NNaOH rising temperature for dissolving, adds 3ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add the 3.0. that 180ML HCl adjusts PH, be cooled to 4 degree, suction filtration, the 110g. of oven dry
Example 3:
100gL-leucine adds 1000ML 2NNaOH rising temperature for dissolving, adds 2ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add 180ML HCl and adjust PH 3.0, be cooled to 4 degree, suction filtration, the 120g. of oven dry
Medical uses
Levacetylleucine is indicated for the treatment of neurological manifestations of Niemann-Pick disease type C in people weighing at least 15 kilograms (33 lb).[1][2]
Adverse effects
The most common side effects include abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting.[2]
Levacetylleucine may cause embryo-fetal harm if used during pregnancy.[1][2]
History
The safety and efficacy of levacetylleucine for the treatment of Niemann-Pick disease type C were evaluated in a randomized, double-blind, placebo-controlled, two-period, 24-week crossover study.[2] The duration was twelve weeks for each treatment period.[2] The study enrolled 60 participants.[2] To be eligible for the study participants had to be four years of age or older with a confirmed diagnosis of Niemann-Pick disease type C and at least mild disease-related neurological symptoms.[2] Participants could receive miglustat, an enzyme inhibitor, as background treatment in the study.[2]
The US Food and Drug Administration (FDA) granted the application for levacetylleucine priority review, fast track, orphan drug, and rare pediatric disease designations.[2] The FDA granted approval of Aqneursa to IntraBio Inc.[2]
Society and culture
Legal status
Levacetylleucine was approved for medical use in the United States in September 2024.[1][2][5]
Names
Levacetylleucine is the international nonproprietary name.[6]
Research
Levacetylleucine is being studied for the treatment of GM2 gangliosidoses (Tay-Sachs and Sandhoff diseases),[7] ataxia-telangiectasia,[8] Lewy body dementia,[9] amyotrophic lateral sclerosis, restless legs syndrome, multiple sclerosis, and migraine.[10]
References
- ^ Jump up to:a b c d e f g h i “Aqneursa- levacetylleucine granule, for suspension”. DailyMed. 24 September 2024. Retrieved 5 October 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves New Drug to Treat Niemann-Pick Disease, Type C”. U.S. Food and Drug Administration (Press release). 24 September 2024. Retrieved 25 September 2024. This article incorporates text from this source, which is in the public domain.
- ^ “IntraBio Announces U.S. FDA Approval of Aqneursa for the Treatment of Niemann-Pick Disease Type C”. IntraBio (Press release). 25 September 2024. Retrieved 26 September 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ World Health Organization (2024). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 131”. WHO Drug Information. 38 (2). hdl:10665/378367. ISBN 9789240098558.
- ^ Martakis K, Claassen J, Gascon-Bayari J, Goldschagg N, Hahn A, Hassan A, et al. (March 2023). “Efficacy and Safety of N-Acetyl-l-Leucine in Children and Adults With GM2 Gangliosidoses”. Neurology. 100 (10): e1072 – e1083. doi:10.1212/WNL.0000000000201660. PMC 9990862. PMID 36456200.
- ^ Fields T, Patterson M, Bremova-Ertl T, Belcher G, Billington I, Churchill GC, et al. (January 2021). “A master protocol to investigate a novel therapy acetyl-L-leucine for three ultra-rare neurodegenerative diseases: Niemann-Pick type C, the GM2 gangliosidoses, and ataxia telangiectasia”. Trials. 22 (1): 84. doi:10.1186/s13063-020-05009-3. PMC 7821839. PMID 33482890.
- ^ Passmore P (15 April 2014). A clinical trial to test amlodipine as a new treatment for vascular dementia. ISRCTN registry (Report). doi:10.1186/isrctn31208535.
- ^ Strupp M, Bayer O, Feil K, Straube A (February 2019). “Prophylactic treatment of migraine with and without aura with acetyl-DL-leucine: a case series”. Journal of Neurology. 266 (2): 525–529. doi:10.1007/s00415-018-9155-6. PMID 30547273. S2CID 56148131.
Further reading
- Churchill GC, Strupp M, Factor C, Bremova-Ertl T, Factor M, Patterson MC, et al. (August 2021). “Acetylation turns leucine into a drug by membrane transporter switching”. Scientific Reports. 11 (1): 15812. Bibcode:2021NatSR..1115812C. doi:10.1038/s41598-021-95255-5. PMC 8338929. PMID 34349180.
- Bremova-Ertl T, Ramaswami U, Brands M, Foltan T, Gautschi M, Gissen P, et al. (February 2024). “Trial of N-Acetyl-l-Leucine in Niemann-Pick Disease Type C”. The New England Journal of Medicine. 390 (5): 421–431. doi:10.1056/NEJMoa2310151. PMID 38294974.
- Tifft CJ (February 2024). “N-Acetyl-l-Leucine and Neurodegenerative Disease”. The New England Journal of Medicine. 390 (5): 467–470. doi:10.1056/NEJMe2313791. PMID 38294981.
External links
- Clinical trial number NCT05163288 for “A Pivotal Study of N-Acetyl-L-Leucine on Niemann-Pick Disease Type C” at ClinicalTrials.gov
- Bremova-Ertl T, Ramaswami U, Brands M, Foltan T, Gautschi M, Gissen P, Gowing F, Hahn A, Jones S, Kay R, Kolnikova M, Arash-Kaps L, Marquardt T, Mengel E, Park JH, Reichmannova S, Schneider SA, Sivananthan S, Walterfang M, Wibawa P, Strupp M, Martakis K: Trial of N-Acetyl-l-Leucine in Niemann-Pick Disease Type C. N Engl J Med. 2024 Feb 1;390(5):421-431. doi: 10.1056/NEJMoa2310151. [Article]
- Fields T, M Bremova T, Billington I, Churchill GC, Evans W, Fields C, Galione A, Kay R, Mathieson T, Martakis K, Patterson M, Platt F, Factor M, Strupp M: N-acetyl-L-leucine for Niemann-Pick type C: a multinational double-blind randomized placebo-controlled crossover study. Trials. 2023 May 29;24(1):361. doi: 10.1186/s13063-023-07399-6. [Article]
- FDA Approved Drug Products: Aqneursa (levacetylleucine) for oral suspension (September 2024) [Link]
- FDA News Release: FDA Approves New Drug to Treat Niemann-Pick Disease, Type C [Link]
| Clinical data | |
|---|---|
| Trade names | Aqneursa |
| Other names | IB1001 |
| AHFS/Drugs.com | Aqneursa |
| License data | US DailyMed: Levacetylleucine |
| Pregnancy category | Not recommended |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1188-21-2 |
| PubChem CID | 70912 |
| DrugBank | DB16956 |
| ChemSpider | 1918 |
| UNII | E915HL7K2O |
| KEGG | D12967 |
| ChEBI | CHEBI:17786 |
| ChEMBL | ChEMBL56021 |
| PDB ligand | LAY (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID6045870 |
| ECHA InfoCard | 100.013.370 |
| Chemical and physical data | |
| Formula | C8H15NO3 |
| Molar mass | 173.212 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Levacetylleucine, Aqneursa, Niemann-Pick disease type C, FDA 2024, APPROVALS 2024, N-Acetyl-L-leucine, 1188-21-2, acetyl-L-leucine, Ac-Leu-OH, N-Acetylleucine, NSC 206316, UNII-E915HL7K2O, ORPHAN DRUG, NSC-206316, NSC 206316
Bevemipretide




Bevemipretide,
CAS 2356106-71-1 FREE BASE
CAS SBT-272 Trihydrochloride, 2589640-11-7
607.7 g/mol, C31H45N9O4 F553HAL9V8
- SBT-272
- (2R)-2-amino-N-[(2S)-1-[[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]amino]-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl]-5-(diaminomethylideneamino)pentanamide
- L-Tyrosinamide, D-arginyl-N-[(1S)-5-amino-1-[3-(phenylmethyl)-1,2,4-oxadiazol-5-yl]pentyl]-2,6-dimethyl-
- (2R)-2-amino-N-[(2S)-1-[[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]amino]-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl]-5-(diaminomethylideneamino)pentanamide
- L-Tyrosinamide, D-arginyl-N-[(1S)-5-amino-1-[3-(phenylmethyl)-1,2,4-oxadiazol-5-yl]pentyl]-2,6-dimethyl-
- (2R)-2-amino-N-[(1S)-1-{[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]carbamoyl}-2-(4-hydroxy-2,6-dimethylphenyl)ethyl]-5-carbamimidamidopentanamide
- Originator Stealth BioTherapeutics
- ClassAntidementias; Antiparkinsonians; Neuroprotectants; Peptidomimetics
- Mechanism of Action Adenosine triphosphatase stimulants; Cardiolipin modulators; Reactive oxygen species inhibitors
- Orphan Drug Status Yes – Amyotrophic lateral sclerosis
Phase IAmyotrophic lateral sclerosis
- Preclinical Dry age-related macular degeneration; Frontotemporal dementia; Parkinson’s disease
- No development reported Multiple system atrophy
18 Sep 2024Pharmacodynamics data from a preclinical trial in dry age related macular degenration released by Stealth BioTherapeutics
- 18 Sep 2024Preclinical trials in Dry age-related macular degeneration in USA (Opthalmic)
- 18 Sep 2024Stealth Biotherapeutics plans clinical trial for Dry age related macular degeneration (Topical)
The present technology relates generally to compounds (i.e. peptidomimetics), compositions (e.g. medicaments) and methods for treating, preventing, inhibiting, amelioration or delaying the onset of ophthalmic diseases, disorders or conditions in a mammalian subject. In some embodiments, the ophthalmic disease, disorder or condition is associated with deterioration of the integrity of the ellipsoid zone of one or more eyes of the mammalian subject. For example, the present technology may relate to administering one or more mitochondrial-targeting peptidomimetics (alone, as formulated and/or in combination with other active pharmaceutical ingredients) in effective amounts to treat, prevent, inhibit, ameliorate or delay the onset of ophthalmic diseases, disorders or conditions (e.g., macular degeneration (including (wet or dry) age-related macular degeneration), dry eye, diabetic retinopathy, diabetic macular edema, cataracts, autosomal dominant optic atrophy (DOA), Leber hereditary optic neuropathy (LHON), pigmentary retinopathy, retinitis pigmentosa, glaucoma, ocular hypertension, uveitis, chronic progressive external ophthalmoplegia (often referred to as CPEO or just PEO, e.g., Kearns-Sayre syndrome), and/or Leber congenital amaurosis (LCA)), in mammalian subjects
[0003] The following introduction is provided to assist the understanding of the reader. None of the information provided, or references cited, is admitted as being prior art to the present technology.
[0004] Diseases, disorders and degenerative conditions of the optic nerve and retina are the leading causes of blindness in the world. Many ophthalmic diseases disorders or conditions result from, or are associated with, mitochondrial dysfunction.
[0005] A significant degenerative condition of the retina is age-related macular degeneration (AMD). AMD is the most common cause of blindness in people over the age of 50 in the United States and its prevalence increases with age. AMD is classified as either wet (neovascular) or dry (non-neovascular). The dry form of the disease is more common.
Macular degeneration occurs when the central retina has become distorted and thinned. This change is usually associated with age but also characterized by intra-ocular inflammation and angiogenesis (wet AMD only) and/or intra-ocular infection. The subsequent generation of free radicals, resulting in oxidative tissue damage, local inflammation and production of growth factors (such as VEGF and FGF) and inflammatory mediators, can lead to inappropriate neovascularization in common with the wet form of AMD. Mitochondrial dysfunction is believed to play a role in age-related disorders such as AMD. (Liu et al., Appl. Sci. (2021) 11: 7385). Pieramici & Ehlers have reported that: “RPE mitochondria in AMD eyes undergo more pronounced degenerative changes, with lower mitochondrial density, organelle area and cristae number.” (Pieramici & Ehlers, Presentation at 54th Annual Retina Society Meeting, Sept.30, 2021, slide 3).
[0006] Retinopathy is a leading cause of blindness in type I diabetes and is also common in type II diabetes. The degree of retinopathy depends on the duration of diabetes, and generally begins to occur ten or more years after onset of diabetes. Diabetic retinopathy may be classified as non-proliferative, where the retinopathy is characterized by increased capillary permeability, edema and exudates, or proliferative, where the retinopathy is characterized by neovascularization extending from the retina to the vitreous, scarring, deposit of fibrous tissue and the potential for retinal detachment. Diabetic retinopathy is believed to be caused by the development of glycosylated proteins due to high blood glucose and leads to damage in small blood vessels in the eye. Diabetic retinopathy (often if left untreated) can progress to diabetic macular edema. Diabetic macular edema involves damage to the blood vessels in the retina that progress to a point where they leak fluid into the macula thereby causing the macula to swell and this results in blurred vision. Mitochondrial dysfunction has been linked to the pathogenesis of diabetic retinopathy. (Wu et al. Hindawi Oxidative Medicine and Cellular Longevity, Volume 2018, Article 3420187)
[0007] Glaucoma is made up of a collection of eye diseases that cause vision loss by damage to the optic nerve and retinal ganglion cells (RGCs). An intraocular pressure (IOP) of over 21 mmHg without optic nerve damage is known as ocular hypertension. Elevated IOP due to inadequate ocular drainage is the primary cause of glaucoma. Lowering IOP reduces the risk of progressive RGC loss in glaucoma; however, no currently available treatments directly prevent RGC damage. Glaucoma often develops as the eye ages, or it can occur as the result of an eye injury, inflammation, tumor or in advanced cases of cataract or diabetes. It can also be caused by the increase in IOP caused by treatment with steroids. Drug therapies that are proven to be effective in glaucoma reduce IOP either by decreasing vitreous humor production or by facilitating ocular draining. Such agents are often vasodilators and as such act on the sympathetic nervous system and include adrenergic antagonists. It has been stated that: “… mitochondrial dysfunction plays an important role in the pathogenesis of neurodegenerative diseases…” and “… mitochondrial damage may provide potential strategies for the treatment of glaucoma….” (Liu et al., Appl. Sci. (2021) 11: 7385).
[0008] Autosomal dominant optic atrophy (DOA) is a genetic X-linked neuro-ophthalmic condition characterized by bilateral degeneration of optic nerves. It affects approximately 1 in 10,000 (Denmark) to 1 in 30,000 (worldwide) persons. The nerve damage causes visual loss. It generally begins to manifest itself during the first decade of life and progresses thereafter. The disease itself affects primarily the retinal ganglion nerves. Mutations in the genes known as OPA1 and OPA3, which encode inner mitochondrial membrane proteins (resulting in mitochondrial dysfunction), are generally associated with DOA.
[0009] Leber Hereditary Optic Neuropathy (LHON) is a genetically-based inherited disease that generally starts to manifest itself between the ages of 15 and 35. In LHON, mitochondrial mutations affect complex I subunit genes in the respiratory chain leading to selective degeneration of retinal ganglion cells (RGCs) and optic atrophy generally within a year of disease onset. LHON is caused by mutations in the MT-NDI1, MT-ND4, MT-ND4L and MT-ND6 genes; all of which are associated with mitochondrial genome coding. LHOH affects approximately 1 in 50,000 people worldwide. It generally starts in one eye and progresses quickly to the other eye. Subjects with LHON may eventually become legally or totally blind, often before they turn 50. LHON affects vision needed for tasks such as reading, driving and recognizing others.
[0010] Retinitis pigmentosa (RP) is a group of hereditary retinal degenerative disorders characterized by progressive vision loss. RP is a leading cause of inherited blindness in the developed world. Clinically, RP is manifested by night vision difficulties due to the death of rod photoreceptors followed by the progressive loss of peripheral vision eventually leading to central vision impairment from the secondary loss of cone photoreceptors. RP is caused by mutations of at least 87 genes. The pathogenesis of RP is not well understood. However, mitochondrial dysfunction and oxidative damage are believed to play a key role in the pathogenesis of photoreceptor cell death in RP. (Gopalakrishnan et al., Scientific Reports (2020) 10: 20382)
[0011] Pigmentary retinopathy (PR) is a frequent feature of retinitis pigmentosa.
Pigmentary retinopathy is a non-specific finding that may be found in several mitochondrial diseases, such as Neurogenic weakness, Ataxia, and Retinitis Pigmentosa (NARP). PR is an inherited degenerative disorder of the retina, characterized by progressive photoreceptor damage. The damage leads to atrophy and cell death of the photoreceptors. Patients with PR can follow an autosomal-dominate, autosomal recessive or X-linked recessive pattern. The prevalence is about one in about three to four thousand individuals. Symptoms of the disease include nyctalopia (night blindness), peripheral visual field constriction, and sometimes loss of the central visual acuity or visual field.
[0012] Uveitis is array of intraocular inflammatory diseases of the eye that often results in irreversible visual loss. Uveitis is responsible for an estimated 30,000 new cases of legal blindness annually in the USA. It is believed that this disease is at least in part due to retinal tissue damage caused excessive mitochondrial oxidative stress that triggers a damaging immune response.
[0013] Chronic progressive external ophthalmoplegia (CPEO) is a condition characterized mainly by a loss of the muscle functions including in eye and eyelid movement. The condition typically appears in adults between ages 18 and 40 and slowly worsens over time. CPEO can be caused by genetic changes in any of several genes, which may be located in mitochondrial DNA or nuclear DNA. CPEO can occur as part of other underlying conditions, such as ataxia neuropathy spectrum and Kearns-Sayre syndrome. These conditions may not only involve CPEO, but various additional features that are not shared by most individuals with CPEO.
[0014] Kearns-Sayre syndrome is a condition that affects many parts of the body, especially the eyes. The features of Kearns-Sayre syndrome usually appear before age 20, and the condition is diagnosed by a few characteristic signs and symptoms. People with Kearns-Sayre syndrome have progressive external ophthalmoplegia. Affected individuals also have an eye condition called pigmentary retinopathy, which results from breakdown (degeneration) of the retina that gives it a speckled and streaked appearance.
[0015] Leber congenital amaurosis (LCA) is a rare genetic eye disorder that affects infants. The infants are often blind at birth. LCA can be associated with mitochondrial dysfunction. (Castro-Gago et al., J. Child Neurol. (1996) 11(2):108-11) Children born with LCA have light-gathering cells (rods and cones) of the retina that do not function properly. LCA has been estimated to be 1-2/100,000 births. This disorder affects males and females in equal numbers.
[0016] Drusen are small yellow or white spots between the retinal pigment epithelium and Bruch’s membrane in the retina that can be detected by an ophthalmologist during a dilated eye exam or with retinal photography. Drusen can also be imaged and monitored by optical coherence tomography (OCT). Drusen are made up of lipids and proteins. Drusen are a defining feature of macular degeneration. Drusen can be hard or soft. Larger numbers of drusen, as well as drusen of larger size, indicate higher risk for some vision loss in the future. “Hard” drusen are small and indicate lower risk of future vision loss than “soft” drusen. “Soft” drusen are larger, cluster together, and have edges that are not as clearly defined. Soft drusen are more likely to lead to vision loss.
[0017] Geometric Atrophy (GA) is generally considered part of the later stage of age-related macular degeneration (AMD) and refers to progression of the disease to a point where in regions of the retina, cells begin to waste away and die (i.e. atrophy).
[0018] Best corrected visual acuity (BCVA) is a measure of the best possible vision an eye can achieve with the use of glasses or corrective lenses. It is typically measured using Snellen lines on an eye chart. Repeated testing of the BCVA over time can be used to determine if a subject’s vision is stable, improving or deteriorating.
[0019] Low luminance visual acuity (LLVA) involves standard visual acuity testing under low-light conditions. This is often achieved by adding a neutral density filter in front of the testing eye. It is a useful visual function marker in those with geographic atrophy (GA) and neovascular age-related macular degeneration. Repeated testing of the LLVA over time can be used to determine if a subject’s vision, under low light conditions, is stable, improving or deteriorating.
[0020] Optical coherence tomography (OCT) is a non-invasive imaging method used to generate a picture of the back of the eye (i.e. the retina). OCT uses a low-powered laser to create pictures of the layers of the retina and optic nerve. The cross-sectional images are three-dimensional and color-coded. OCT can measure the thickness of the retina and optic nerve. OCT can be used to diagnose and manage Glaucoma, AMD, diabetes-related retinopathy, cystoid macular edema, macula pucker and macular hole.
[0021] Spectral domain optical coherence tomography (SDOCT) is an interferometric technique that provides depth-resolved tissue structure information encoded in the magnitude and delay of the back-scattered light by spectral analysis of the interference fringe pattern. SDOCT increases axial resolution 2- to 3-fold and scan speed 60- to 110-fold vs conventional (TD) OCT.
[0022] The ellipsoid zone can be mapped using SCOCT and the integrity of (or changes in) the ellipsoid zone can be determined from such mapping/scanning activity. (Itoh et al., Br J Ophthalmol. (2016) 100(3): 295-299). The technology is capable of evaluating the structures of the external limiting membrane (ELM), ellipsoid zone (EZ), interdigitation zone (IZ) and the retinal pigment epithelium (RPE). Id. Use of this technology is capable of accessing EZ integrity and EZ-RPE alterations. Id. The EZ and ELM, in particular, have been linked to visual outcomes and prognosis in numerous macular conditions, such as age-related macular degeneration (AMD) Id. Itoh et al. suggest that the utility of SDOCT as an assessment tool for EZ integrity for clinical trials and disease prognostication/management may prove particularly useful.
[0023] Swept source OCT (SS-OCT) and OCT angiography (OCTA) are relatively new techniques that are capable of better resolution of the retinal pigment epithelium (RPE), Bruch’s membrane (BM) and choriocapillaris (CC) structures. (Zhou et al. Biomedical Optics Express (2020) 11(4): 1834-1850) Using this technology it is possible to generate relative distance and thickness maps of the RPE-BM-CC complex. Id. Use of these techniques may provide a better understanding of the CC in three dimensions, and further
investigate potential functional relationships between RPE, BM and CC, and their involvement in age-related ocular diseases. Id.
[0024] The ellipsoid zone (EZ) of the eye is a mitochondrial rich tissue (Ball et al., Sci. Adv.8, eabn2070 (2022)). The ellipsoid zone can be imaged using optical coherence tomography (Fujita et al., Scientific Reports (2019) 9:12433). The integrity of the EZ can be quantified. (Fugita et al.). There is a clear relationship between the integrity of the ellipsoid zone and visual function. (Fugita et al., Figure.3). Ball et al. suggest that tightly packed mitochondria in the ellipsoid “focus” light for entry into the outer segment and that healthy mitochondria structure (including cristae structure) might be important for producing a Stiles-Crawford effect (SCE) and maintaining visual resolution in mammals. Pieramici & Ehlers describe mapping the ellipsoid zone to thereby observe the ellipsoid zone and possibly monitor changes in the integrity of the ellipsoid zone. (Pieramici & Ehlers, Presentation at 54th Annual Retina Society Meeting, Sept.30, 2021). Pieramici & Ehlers further described the use of Sub-RPE compartment maps as a means to find and monitor drusen formation and RPE atrophy in a subject. In the study being described (which described results from a P2 clinical trial involving treatments with elamipretide), Pieramici & Ehlers concluded, inter alia, that: (i) “Average BCVA and LLVA in NCGA and HRD patients improved significantly at 24 weeks [of treatment with elamipretide]” and (ii) “Baseline higher order OCT parameters, such as EZ integrity, correlated with improved LLVA in Elamipretide-treated eyes” (Pieramici & Ehlers at slide 15).
[0025] In brief, there are many ophthalmic diseases for which there remains a need for treatments/therapies or improved treatments/therapies. For example, there remains a need for treatments/therapies, or improved treatments/therapies, to address ophthalmic diseases, disorders or conditions such as macular degeneration (including (wet or dry) age-related macular degeneration), dry eye, diabetic retinopathy, diabetic macular edema, cataracts, autosomal dominant optic atrophy (DOA), Leber hereditary optic neuropathy (LHON), pigmentary retinopathy, retinitis pigmentosa, glaucoma, ocular hypertension, uveitis, chronic progressive external ophthalmoplegia (e.g., Kearns-Sayre syndrome), and/or Leber congenital amaurosis (LCA). This forgoing discussion addresses these needs.
SCHEME

MAIN

PATENT
WO2023069255
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023069255&_cid=P22-M93M8P-34013-1

Synthesis of (R)-2-amino-N-((S)-1-(((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5- yl)pentyl)amino)-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl)-5- guanidinopentanamide (D-Arg-DMT-NH((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5- yl)pent-1-yl), 7a (a.k.a. ((Formula IIa)):
[0134] In some embodiments, Compound 7a (a.k.a. Formula IIa) may be synthesized as illustrated in Scheme 5, below (Also see WO2019/118878, incorporated herein by reference), wherein compound 12a can be prepared as illustrated in Scheme 6, below
[0135] Step a: Synthesis of benzyl (S)-2-((R)-2-((tert-butoxycarbonyl)amino)-5- guanidinopentanamido)-3-(4-hydroxy-2,6-dimethylphenyl)propanoate (3a). To a suspension of 2,6-Dmt-OBn.HCl (2a, 45.0 g, 134 mmol) in ACN (800 mL), NMM (32.7 mL, 298 mmol) was added at 00C. The reaction mixture was stirred until the reaction mixture became transparent. Then Boc-D-Arg-OH.HCl (1a, 46.3 g, 149 mmol) and HOBt.H2O (9.11 g, 59.5 mmol) were added to reaction mixture and stirred for 15 min. Finally, EDC.HCl (38.5 g, 201 mmol) was added and mixture was stirred at 00C for 4 h. Then EtOAc (450 mL), 1N HCl in brine (300 mL) were added. The combined organic extracts were washed with 1N HCl in brine (7×150 mL), NaHCO3/brine (300 mL and until pH of aqueous layer is about pH=6 to 7), dried over Na2SO4, filtered and concentrated to afford 86.0 g (97%) of Boc-D-Arg-DMT- OBn (3a) that was used without further purification.1H-NMR (400 MHz, Methanol-d4) δ 7.33 – 7.18 (m, 5H), 6.43 (s, 2H), 5.06 (s, 2H) 4.71 (t, J=7.8Hz, 1H), 4.07 (t, J=6.7Hz,1H), 3.19 – 3.09 (m, 3H), 3.03-2.97 (m, 1H), 2.23 (s, 6H), 1.72 – 1.65 (m, 1H), 1.54 – 1.43 (m, 3H), 1.45 (s, 9H).
[0136] Step b: Synthesis of (S)-2-((R)-2-((tert-butoxycarbonyl)amino)-5-guanidinopentanamido)-3-(4-hydroxy-2,6-dimethylphenyl)propanoic acid (4a). To a solution of Boc-D-Arg-DM-Tyr-OBn (3a, 84.0 g, 142 mmol) in MeOH (1000 mL) Pd/C (10% w/w, 14.0 g) was added. The hydrogen was purged in reaction mixture at room temperature for 4h. Then reaction mixture was filtrated through filter paper and washed with MeOH (150 mL). The solvent was removed by evaporation. White foam product 4a was obtained (74.0 g, 93%) and used without further purification.1H-NMR (400 MHz, Methanol-d4) δ 6.44 (s, 2H), 4.68 (t, J = 7.2 Hz, 1H), 4.04 (t, J = 6.8 Hz, 1H), 3.15 – 3.09 (m, 3H), 3.02 – 2.94 (m, 1H), 2.29 (s, 6H), 1.74 – 1.59 (m, 1H), 1.54 – 1.43 (m, 1H), 1.45 (s, 9H).
[0137] Step c: Synthesis of tert-butyl ((6R,9S,12S)-1-amino-12-(3-benzyl-1,2,4-oxadiazol-5-yl)-9-(4-hydroxy-2,6-dimethylbenzyl)-1-imino-20,20-dimethyl-7,10,18-trioxo-19-oxa-2,8,11,17-tetraazahenicosan-6-yl)carbamate (6a). DMF (200 mL) was added to 4a (11.17 g, 24 mmol) and stirred at r.t. for 15 min. To the resulting suspension, 12a (10.65 g, 20 mmol) was added and stirred at r.t. for 20 min. After addition of HOBt (612 mg, 4.00 mmol), the suspension was cooled in ice bath. EDC . HCl (5.38 g, 28 mmol) was added in one portion, and the reaction mixture was stirred while cooled in ice bath for 2.5 h and then, for 4.5 h at r.t. The nearly homogeneous reaction mixture was quenched with EtOAc (1500 mL) and the resulting solution was washed for 10 times with brine/aq.0.5 M HCl (1:1; 400 mL). During the 6th and 9th washings, gel in the aqueous phase was formed. After addition of iPrOH (40 mL in each case) and repeated shaking the layers went clear again. Afterwards, the organic phase was washed for 6 times with brine/sat. aq. NaHCO3 (9:1; 400 mL). During the 4th washing, gel in the aqueous phase was formed. After addition of iPrOH (40 mL) and repeated shaking the layers were separated easily. The organic phase was washed with brine (200 mL) and water (100 mL) and the solvent was removed under reduced pressure. No vigorous shaking was performed upon washing with water to avoid difficulties in phase separation. As a result, 16.8 g of the crude product were obtained (6a, 97.0 % purity by HPLC, white amorphous solid).1H-NMR (300 MHz, Methanol-d4) ppm: δ = 7.33–7.16 (m, 5H), 6.38 (s, 2H), 5.18-5.07 (m, 1H), 4.64-4.55 (m, 1H), 4.10 – 3.92 (m, 3H), 3.18-2.77 (m, 6H), 2.20 (s, 6H), 1.97-1.76 (m, 2H), 1.75-1.14 (m, 8H), 1.43 (s, 9H), 1.41 (s, 9H).
[0138] Step d: Synthesis of (R)-2-amino-N-((S)-1-(((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)amino)-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl)-5-guanidinopentanamide (7a, but also referred to as (IIa – the tri-hydrochloride salt of Compound I) herein). After 6a (16.8 g) was dissolved in DCM (100 mL) and cooled to 0°C, TFA (20 mL) was added dropwise and the solution was allowed to stir at 0 °C for 10 min, and then at r.t. for 3 h (LC/MS shows no starting material). Then reaction mixture was evaporated (at 0–5 °C) and additionally re-evaporated from DCM (100 mL, at 0–5 °C). The purification by flash chromatography on reverse phase (cartridge C-18, 120G) was performed on crude material divided in 4 parts. Then all solvents were evaporated at reduced pressure at <40oC. White foam was dissolved in isopropanol (100 mL) and 5 mL of HCl in isopropanol (5-6M) was added at 0 oC and evaporated under reduced pressure. This step was repeated 3 times. Additionally, 100 mL of ACN was added and suspension was evaporated one more time. As a result, white powder of 7a was obtained as the tri-hydrochloride salt.1H-NMR (300 MHz, Methanol-d4) δ 7.36 – 7.14 (m, 5H), 6.40 (s, 2H), 5.15 (dd, J = 8.5, 6.3 Hz, 1H), 4.68 (dd, J = 8.7, 7.5 Hz, 1H), 4.07 (s, 2H), 3.97 (t, J = 6.3 Hz, 1H), 3.18 (t, J = 6.9 Hz, 2H), 3.11 (dd, J = 14.2, 8.8 Hz, 1H), 2.95 – 2.84 (m, 3H), 2.22 (s, 6H), 2.02 – 1.59 (m, 6H), 1.57 – 1.28 (m, 4H). MS: EI-MS: m/z 608.4 [M+1].


Synthesis of (S)-1-(3-Benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-Butoxycarbonyl)amino)pentan-1-Aminium 4-Methylbenzenesulfonate (12a)
step a: NH2OH; step b: T3P, NaHCO3; step c: TEA; step d: PTSA
[0139] Step a: Synthesis of N-hydroxy-2-phenylacetimidamide (9a). To a solution of nitrile 8a (1.0 mol) in EtOH (1.2 L) was added NH2OH (50% aqueous solution, 130 g, 2.0 mol).
The solution was heated to reflux and stirred for 12 hours (hrs.). After completion, the reaction mixture was concentrated under reduced pressure. The resulting residue was re-dissolved in EtOH (350 mL) and concentrated under reduced pressure again (this procedure was repeated three times). The resulting solid was triturated in hexane (350 mL), filtered, washed with hexane (100 mL), and then dried to give the desired product 9a as white solid. (10.5 kg; KF = 1295) with good results (purity by HPLC, > 98.9 A%; Assay = 22.2 w%, yield = 91%).1H NMR (300 MHz, DMSO-d6): δ 8.90 (s, 1H), 7.28-7.18 (m, 5H), 5.40 (s, 2H), 3.25 (s, 2H) ppm. MS: (M+H)+: m/z = 151.1
[0140] Step b: Synthesis of (9H-Fluoren-9-yl)methyl tert-Butyl (1-(3-Benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-Dicarbamate (11a). To a solution of protected enantiomerically pure N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-N6-(tert-butoxycarbonyl)-L-lysine (10a, 4.31 kg, 9.2 mol) and hydroxyimidamide 9a (1.1 equivalents “equiv.” or “eq.”) in ethyl acetate was added NaHCO3 (3.0 equiv.). The mixture was stirred at 25 oC for 20 minutes (min.). Then, propane phosphonic acid anhydride (T3P, 50% solution in ethyl acetate, 3.0 equivalents (equiv.)) was added and the reaction mixture was heated to 80 oC and stirred for 4 hrs. (about 60% conversion of compound 10a based on HPLC). Then compound 9a (1.1 equiv.) was added and the reaction mixture was stirred at 80oC for another 20 hr. (about 10% compound 10a remained). The reaction mixture was cooled to room temperature, saturated aqueous NaHCO3 (2.0 L) was added, the mixture was then extracted with ethyl acetate (3x 1.0 L). The combined organic layers were then washed with brine (1 L), dried over anhydrous Na2SO4, filtered and concentrated to give a crude residue, which was generally purified by silica gel column chromatography (Petroleum ether (PE):EtOAc = 5: 1) to give crude product, (9H-fluoren-9-yl)methyl tert-butyl (1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-dicarbamate (11a), solution in ACN (19.7 kg, assay = 20%, chiral HPLC purity = 99.12 A%,yield = 73%).1H-NMR (300 MHz, CDCl3): δ 7.78 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 6.3 Hz, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.35-7.30 (m, 7H), 5.52 (br, 1H), 5.09-5.05 (m, 1H), 4.56-4.37 (m, 3H), 4.22 (t, J = 6.6 Hz, 1H), 4.08 (s, 2H), 1.95-1.86 (m, 2H), 1.48-1.42 (m, 11H) ppm. MS: (M-100+H)+: m/z = 483.2.
[0141] Step c: Synthesis of tert-Butyl (S)-(5-Amino-5-(3-Benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a). To a solution of compound (9H-fluoren-9-yl)methyl tert-butyl (1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-dicarbamate (11a) was added TEA (2.5 eq.). The mixture was kept stirring with mechanical stirrer at 20~ 25 °C for 15 h. The reaction mixture was diluted by tap water and MTBE. Separated, aqueous layer was extracted by MTBE for one time. Both MTBE layers were combined, and then washed by NH4Cl. Then anhydrous Na2SO4 was added and that solution stirred for least 2 h, then filtered and washed with MTBE to afford tert-butyl (S)-(5-amino-5-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a) solution in MTBE (32.9 kg, assay = 6.5%, yield = 88%).1H-NMR (300 MHz, DMSO-d6): δ 7.33-7.25 (m, 5H), 6.78 (br, 1H), 5.09-5.05 (m, 1H), 4.56-4.37 (m, 3H), 4.06 (s, 2H), 3.98 (t, J = 6.6 Hz, 1H), 2.87-2.84 (m, 2H), 2.10 (s, 2H), 1.38-1.34 (m, 2H), 1.24 (s, 9H), 1.20-1.15 (m, 2H) ppm. MS: (M+H)+: m/z = 361.1.
[0142] Step d: Synthesis of (S)-1-(3-Benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-Butoxycarbonyl)-amino)pentan-1-Aminium 4-Methylbenzenesulfonate (12a). p-toluenesulfonic acid (PTSA) was added to solution of crude tert-butyl (S)-(5-amino-5-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a) in MTBE to afford (S)-1-(3-benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-butoxycarbonyl)amino)pentan-1-aminium 4-methylbenzenesulfonate (12a) (2.7 kg, yield = 85 %, HPLC purity > 99%, ee > 99%) as white solid.1H-NMR (400 MHz, DMSO-d6): δ 8.74 (br, 3H), 7.48 (d, J = 8.0 Hz, 2H), 7.37-7.26 (m, 5H), 7.11 (d, J = 8.0 Hz, 2H), 6.77 (t, J = 5.2 Hz, 1H), 4.82 (t, J = 6.8 Hz, 1H), 4,17 (s, 2H), 2.90-2.86 (m, 2H), 2.29 (s, 3H), 1.39-1.36 (m, 11H), 1.35-1.28 (m, 2H) ppm. MS: (M-172+H)+: m/z = 361.1.
PATENT WO2021016462
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021016462&_cid=P22-M93MJV-41323-1

WO2019118878
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019118878&_cid=P22-M93MNH-43976-1
/////////Bevemipretide, SBT-272 Trihydrochloride, SBT 272, ORHAN DRUG, Stealth BioTherapeutics
MILADEMETAN
Milademetan
| Molecular Weight | 618.53 |
|---|---|
| Formula | C30H34Cl2FN5O4 |
| CAS No. | 1398568-47-2 |
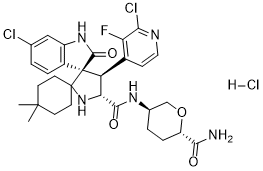
Milademetan. hcl
Chemical Formula: C30H35Cl3FN5O4
Exact Mass: 617.1972
Molecular Weight: 654.99
Elemental Analysis: C, 55.01; H, 5.39; Cl, 16.24; F, 2.90; N, 10.69; O, 9.77
1398568-47-2 (free base) 1398569-75-9 (tosylate) 2095625-97-9 (tosylate hydrate) Milademetan HCl
DS3032b; DS-3032b; DS 3032b; DS3032; DS-3032; DS 3032; DS-3032b tosylate; Milademetan tosylate; Milademetan HCl
(3’R,4’S,5’R)-N-[(3R,6S)-6-carbamoyloxan-3-yl]-6”-chloro-4′-(2-chloro-3-fluoropyridin-4-yl)-4,4-dimethyl-2”-oxo-1”,2”-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3”-indole]-5′-carboxamide hydrochloride
orphan drug, UNII:R3I80TLN7S, миладеметан , ميلاديميتان , 米拉美坦
(3’R,4’S,5’R)-N-((3R,6S)-6-Carbamoyltetrahydro-2H-pyran-3-yl)-6”-chloro-4′-(2-chloro-3-fluoro-4-pyridinyl)-4,4-dimethyl-2”-oxo-1”,2”-dihydrodispiro(cyclohexane-1,2′-pyrrolidine-3′,3”-indole)-5′-carboxamide
milademetan
rolontis
SPI-2012
Milademetan, also known as DS-3032b or DS-3032, is a potent and selective MDM2 inhibitor with potential antineoplastic activity. Upon oral administration, MDM2 inhibitor DS-3032b binds to, and prevents the binding of MDM2 protein to the transcriptional activation domain of the tumor suppressor protein p53. By preventing this MDM2-p53 interaction, the proteosome-mediated enzymatic degradation of p53 is inhibited and the transcriptional activity of p53 is restored. This results in the restoration of p53 signaling and leads to the p53-mediated induction of tumor cell apoptosis.
DS-3032 (Milademetan) is an orally available, potent and selective inhibitor of the p53-MDM2 (murine double minute 2) interaction. Milademetan binds to, and prevents the binding of MDM2 protein to the transcriptional activation domain of the tumor suppressor protein p53. Milademetan is 10-fold more potent than the first-generation inhibitor nutlin-3a. By preventing this MDM2-p53 interaction, the proteasome-mediated enzymatic degradation of p53 is inhibited and the transcriptional activity of p53 is restored. This results in the restoration of p53 signaling and leads to the p53-mediated induction of tumor cell apoptosis. DS-3032 is currently being evaluated in three phase 1 clinical trials for solid and hematological malignancies, including acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), chronic myeloid leukemia (CML) in blast phase, lymphoma and myelodysplastic syndrome (MDS).
- OriginatorRigel Pharmaceuticals
- DeveloperDaiichi Sankyo Inc; National Cancer Center Hospital East; Rain Therapeutics; University of Texas M. D. Anderson Cancer Center
- ClassAntineoplastics; Cyclohexanes; Indoles; Pyrrolidines; Small molecules
- Mechanism of ActionProto-oncogene protein c mdm2 inhibitors
- Orphan Drug StatusYes – Liposarcoma
- Phase IIILiposarcoma
- Phase IISarcoma; Solid tumours
- Phase I/IIAcute myeloid leukaemia
- Phase ILymphoma; Myelodysplastic syndromes
- PreclinicalMesothelioma
- No development reportedMultiple myeloma
- 10 Aug 2022Rain Therapeutics completes enrolment in phase-III clinical trials in Liposarcoma in (Inoperable/Unresectable, Metastatic disease, Second-line therapy or greater) in United Kingdom, Taiwan, Spain, Poland, South Korea, Italy, Hong Kong, Germany, Georgia, France, Canada, Belgium, Austria (PO) (NCT04979442)
- 09 Jun 2022Efficacy, adverse events and pharmacodynamics data from phase I/II trial in Acute myeloid leukemia presented at the 27th Congress of the European Haematology Association(EHA-2022)
- 04 May 2022Rain Therapeutics plans a phase I/II MANTRA-4 trial in Solid tumours (Combination therapy, Late-stage disease) in Second half of 2022
PATENT
WO2015033974

[Example 2]
Ethyl (3’R,4’S,5’R)-6”-chloro-4′-(3-chloro-2-fluorophenyl)-4,4-dimethyl-2”-oxo 1″,2″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indole]-5′-carboxylate
[0202]
[Chem. 58]
[0203]
(3E/Z)-6-chloro-3-(3-chloro-2-fluorobenzylidene)-1,3-dihydro-2H-indol-2-one ( WO 2006/091646) (98.7) under nitrogen atmosphere mg), (R)-BINAP (12.1 mg, 0.019 mmol), CuOAc (2.0 mg, 0.016 mmol), 4,4-dimethylcyclohexanone (61.4 mg, 0.48 mmol), glycine ethyl ester. (39.5 μL, 0.39 mmol) and a solution of triethylamine (6.8 μL, 0.049 mmol) in N,N-dimethylacetamide (2.0 mL) were added and stirred at room temperature for 22 hours. Ethyl acetate (2 mL), water (1 mL), and 20% aqueous ammonium chloride solution (1 mL) were added to the reaction mixture, and the mixture was vigorously stirred to separate the organic layer. The aqueous layer was extracted twice with ethyl acetate (2 mL each) and all the organic layers were combined and then washed with water three times (5 mL each). The obtained organic layer was concentrated under reduced pressure, ethyl acetate (6 mL) and silica gel (500 mg) were added to the residue, and the silica gel was separated by filtration. The filtrate was concentrated under reduced pressure, ethanol (1.0 mL) was added to the residue, water (1 mL) was added dropwise, and the mixture was stirred overnight at room temperature. The precipitated solid was filtered and dried under reduced pressure at 40° C. to obtain the title compound (137 mg, yield 82%, 94% ee) as a solid.
1H NMR (500 MHz, CDCl3): δ = 0.67 (s, 3H), 0.91 (s, 3H), 1.10-1.19 (m, 2H), 1.17 (t, J=7.3 Hz, 3H), 1.25-1.33 (m, 1H), 1.44- 1.72 (m, 3H), 1.87-2.01 (m, 1H), 3.16 (s, 1H), 4.07-4.21 (m, 2H), 4.52 (d, J = 8.5 Hz, 1H), 4.83 (d, J = 8.5 Hz, 1H), 6.74 (d, J = 1.5Hz, 1H), 6.81-6.86 (m, 1H), 7.06 (dd, J = 8.3, 2.8 Hz, 1H), 7.10-7.16 (m, 1H), 7.37 (dd, J = 8.3, 1.8 Hz, 1H), 7.48-7.54 (m, 1H), 7.81 (s, 1H).
(HPLC conditions for optical purity determination)
カラム: CHIRALPAK OD-3R 4.6 × 150 mm, 3μm
Moving layer: 10mM Rinic acid buffer: MeCN = 40:60
Flow rate: 1.0 min/min
カラム Temperature: 40°C
Exhaust wavelength: 254 nm
Injection volume: 5 μL
Hold time: Labeling compound = 13.8 min, エナンチオマー= 12.9 min
[Example 11]
11-1) Effects of various asymmetric catalysts
[0230]
[Chem. 67]
[0231]
(3E/Z)-6-chloro-3-[(2-chloro-3-fluoropyridin-4-yl)methylene]-1,3-dihydro-2H-indol-2-one ( WO 2012 / 121361), 4,4-dimethylcyclohexanone (1.5 eq.), glycine ethyl ester (1.2 eq.), triethylamine (15 mol%) in THF solution (10 times the volume), separately, Lewis acid (5 mol%) , an asymmetric ligand (6 mol %) and THF (10 times the amount) were stirred for 1 hour under a nitrogen atmosphere, a catalyst solution prepared was added, and the mixture was stirred at room temperature for 12 to 16 hours. After that, the resulting trans1 compound ((ethyl (3′S,4′R,5′S)-6″-chloro-4′-(2-chloro-3-fluoropyridin-4-yl) -4,4-dimethyl-2”-oxo-1”,2”-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3”-indole]-5′-carboxylate) Optical purity and HPLC yield were measured.
(HPLC conditions for measuring optical purity)
Column: CHIRALPAK OD-3R 4.6 × 150 mm, 3 µm
Mobile phase: 10 mM phosphoric acid buffer: MeCN = 40:60
Flow rate: 1.0 min/min
column Temperature: 40°C
Detection wavelength: 254 nm
Injection volume: 5 µL
Retention time: Title compound = 13.8 min, enantiomer = 12.9 min
Main results are shown in Table 1.
[0232]
[Table 1-1]
[Table 1-2]
[0233]
11-2) Effects of various solvents
[0234]
[Chem. 68]
[0235]
(3E/Z)-6-chloro-3-[(2-chloro-3-fluoropyridin-4-yl)methylene]-1,3-dihydro-2H-indol-2-one ( WO 2012 / 121361), 4,4-dimethylcyclohexanone (1.5 eq.), glycine ethyl ester (1.2 eq.), triethylamine (15 mol%), a solvent (10 times the amount), CuOAc (5 mol%), ( A catalyst solution prepared by stirring S)-BINAP (6 mol %) and a solvent (10 times the amount) under a nitrogen atmosphere for 1 hour was added, followed by stirring at room temperature for 21.5 hours. After that, by HPLC, the resulting trans2 compound (ethyl (3’S,4’R,5’S)-6”-chloro-4′-(2-chloro-3-fluoropyridin-4-yl)- HPLC of 4,4-dimethyl-2″-oxo-1″,2″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indole]-5′-carboxylate) Yields and optical purities were determined.
Table 2 shows the main results.
[0236]
[Table 2]
11-3) Examination of Cu(I) Lewis acid
PATENT
WO2014038606
WO2014038606 CLICK HERE
Example 1
[0062]
[Chem.3]
[0063]
(3′R,4′S,5′R)-N-[(3R,6S)-6-carbamoyltetrahydro-2H-pyran-3-yl]-6″-chloro-4′-(2-chloro- 3-fluoropyridin-4-yl)-4,4-dimethyl-2″-oxo-1″,2″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indole]-5 ‘
-Carboxamide The compound (35 mg, 0.24 mmol) obtained in Reference Example 2, Step 3 was added to a solution of the compound (100 mg, 0.20 mmol) obtained in Step 3 of Reference Example 1 in N,N-dimethylformamide (4 ml). , triethylamine (0.04 ml, 0.30 mmol), 1-hydroxybenzotriazole (27 mg, 0.20 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (46 mg, 0.24 mmol) were added. , and stirred for 1 hour at 50° C. After allowing to cool, the reaction solution was diluted with ethyl acetate, washed successively with water, saturated aqueous sodium hydrogencarbonate solution and saturated brine, and dried over anhydrous sodium sulfate. After evaporating the solvent under reduced pressure, the residue was purified by NH-silica gel column chromatography [chloroform:methanol=50:1 (v/v)]. After stirring for 24 hours at rt, the solvent was distilled off under reduced pressure to obtain 94 mg (76%) of the title compound as a solid.1H
– NMR (400 MHz, CDCl3 ) .) δ: 0.68 (3H, s), 0.95 (3H, s), 1.11-1.27 (2H, m), 1.35-1.81 (8H, m), 2.10-2.17 (1H, m), 2.25-2.32 (1H, m), 3.15(1H,t,J=10.5Hz), 3.27(1H,br s), 3.80(1H,dd,J=11.0,2.3Hz), 3.85-3.95(1H,m), 4.13(1H, ddd,J=10.8,4.5,1.3Hz),4.44(1H,d,J=9.2Hz),4.64(1H,d,J=9.2Hz),5.46(1H,d,J=3.7Hz),6.49( 1H,d,J=3.7Hz), 6.74(1H,d,J=1.8Hz), 7.07(1H,dd,J=8.2,1.8Hz), 7.31(1H,dd,J=8.2,2.3Hz), 7.48-7.52(2H,m),7.62(1H,s),8.05(1H,d,J=5.5Hz).MS
(ESI)m/z:618(M+H) +
Reference example 1
[0087]
[Chem.4]
[0088]
[Step 1] (3E/Z)-6-chloro-3-[(2-chloro-3-fluoropyridin-4-yl)methylene]-1,3-dihydro-2H-indol-2-one
6-chloro -1,3-dihydro-2H-indol-2-one (2.20 g, 13.11 mmol) and 2-chloro-3-fluoroisonicotinaldehyde (2.20 g, 13.8 mmol) in methanol (130 ml). , N,N-diisopropylethylamine (0.46 ml, 2.63 mmol) was added, and the mixture was heated under reflux for 16 hours. After standing to cool, the precipitate was collected by filtration, washed with cold methanol and dried to obtain 3.37 g (83%) of the title compound as a solid.
MS(APCI) m/z: 309(M+H) + .
[0089]
[Step 2] (3′S,4′R,7′S,8′S,8a′R)-6″-chloro-8′-(2-chloro-3-fluoropyridin-4-yl)-4 ,4-dimethyl-3′,4′-diphenyl-3′,4′,8′,8a′-tetrahydro-1′H-dispiro[cyclohexane-1,6′-pyrrolo[2,1-c][1 ,4]oxazine-7′,3″-indole]-1′,2″(1″H)
-dione Under a nitrogen atmosphere, the compound obtained in Step 1 (1.86 g, 6.00 mmol), (5R,6S )-5,6-diphenylmorpholin-2-one (1.67 g, 6.60 mmol) and 4,4-dimethylcyclohexanone (0.83 g, 6.60 mmol) in tetrahydrofuran (30 ml) was added with diethyl boron trifluoride. An ether complex (0.15 ml, 1.20 mmol) and molecular sieve 4A (powder) (3 g) were added, and the mixture was heated and stirred at 70° C. for 7 days. After allowing to cool, insoluble matter was removed by filtration through celite, and the filtrate was washed with saturated brine and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure and purified by silica gel column chromatography [n-hexane:ethyl acetate=4:1→1:1 (v/v)] to obtain 3.39 g (84%) of the title compound as a solid. rice field.
1 H-NMR (400 MHz, CDCl3) δ: 0.21 (3H, s), 0.53 (3H, s), 0.89-1.08 (3H, m), 1.28-1.43 (3H, m), 1.73-1.81 (1H, m), 2.23-2.33 (1H, m), 4.58 (1H, d, J = 11.0Hz), 4.86 (1H, d, J = 3.2Hz), 5.31 (1H, d, J = 11.0Hz), 6.25 (1H, d, J = 8.3Hz) ,6.67(1H,dd,J=8.3,1.8Hz),6.72-6.77(2H,m),6.93(1H,d,J=1.8Hz),7.04-7.17(6H,m),7.18-7.25(3H ,m),7.79(1H,t,J=4.6Hz),7.99(1H,s),8.29(1H,d,J=5.0Hz).MS
(APCI)m/z:670(M+H) + .
[0090]
[Step 3] (4′S,5′R)-6″-chloro-4′-(2-chloro-3-fluoropyridin-4-yl)-4,4-dimethyl-2″-oxo-1″ ,2″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indole]-5′-carboxylic acid
The compound obtained in step 2 (630 mg, 0.94 mmol) was treated with acetonitrile (10 ml). Dissolve in water (4 ml), add potassium carbonate (130 mg, 0.94 mmol) and heat under reflux for 16 hours at 85° C. After allowing to cool, add anhydrous magnesium sulfate (113 mg, 0.94 mmol) and stir at room temperature for 15 minutes. After extraction with ethyl acetate, the organic layer was washed with saturated brine and dried over anhydrous magnesium sulfate. (2-chloro-3-fluoropyridin-4-yl)-1′-[(1R,2S)-2-hydroxy-1,2-diphenylethyl]-4,4-dimethyl-2″-oxo-1″ ,2″-dihydrodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indole]-5′-carboxylic acid (650 mg, 100%) was obtained as a solid [MS (ESI) m/z :688(M+H) +]. The resulting carboxylic acid (650 mg, 0.94 mmol) was dissolved in methanol (30 ml) and water (8 ml), and diammonium cerium (IV) nitrate (1.55 g, 2.82 mmol) was added under ice-cooling. Stir at room temperature for 30 minutes. Potassium carbonate (780 mg, 5.64 mmol) was added under ice-cooling, and the mixture was stirred at the same temperature for 1 hour. After removing the insoluble matter by filtration through celite, the filtrate was concentrated under reduced pressure, water was added to the resulting residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography [chloroform:methanol=20:1→4:1 (v/v)] to obtain 152 mg (33%) of the title compound as a solid. .
1 H-NMR (500 MHz, CD 3 OD) δ: 0.74 (3H, s), 0.9 (3H, s), 1.29-1.44 (2H, m), 1.48-1.58 (2H, m), 1.64-1.76 (1H ,m),1.94-2.02(1H,m),2.11(1H,ddd,J=14.0,14.0,4.0Hz),2.43-2.53(1H,m),5.07(1H,d,J=10.3Hz), 5.32(1H,d,J=10.3Hz),6.84(1H,d,J=1.7Hz),7.16(1H,dd,J=8.3,2.0Hz),7.63(1H,dd,J=8.0,2.3Hz) ),7.75(1H,t,J=5.2Hz),8.15(1H,d,J=5.2Hz).
MS(ESI)m/z:492(M+H) + .
[0091]
Reference example 2
[0092]
[Chem.5]
[0093]
[Step 1] Methyl 2,6-anhydro-3,4,5-trideoxy-5-(dibenzylamino)-L-erythro
-hexonate 2,6-anhydro-3,4,5-trideoxy-5-( dibenzylamino)-L-erythro-hexonate methyl 2,6-anhydro-3,4,5-trideoxy-5-(dibenzylamino)-L-erythro-hexonate (1.60 g, 4.70 mmol) was The mixture was dissolved in methanol (30 ml), 1N aqueous sodium hydroxide solution (10 ml) was gradually added under ice-cooling, and the mixture was stirred at room temperature for 3 hours. Dowex 50W-X8 was added to the reaction mixture to adjust the pH to 5 to 6, insoluble materials were removed by filtration, and the filtrate was concentrated under reduced pressure to obtain 1.7 g (100%) of the title compound as a solid.
1 H-NMR (400 MHz, CDCl 3 ) δ: 1.18-1.26(1H,m), 1.36-1.48(1H,m), 1.79-1.97(2H,m), 2.62(1H,t,J=11.0Hz) ,3.18(1H,t,J=10.4Hz),3.40(1H,d,J=11.5Hz),3.51-3.61(4H,m),3.90-3.99(1H,m),7.12-7.38(10H,m ).
MS(ESI)m/z:326(M+H) + .
[0094]
[Step 2] (2S,5R)-5-(dibenzylamino)tetrahydro-2H-pyran-2-carboxamide
The compound (870 mg, 2.67 mmol) obtained in Step 1 above was dissolved in N,N-dimethylformamide (30 ml). 1-hydroxybenzotriazole (361 mg, 2.67 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (614 mg, 3.20 mmol) were added and stirred at room temperature for 15 minutes. Ammonium chloride (285 mg, 5.44 mmol) and N,N-diisopropylethylamine (1.86 ml, 10.7 mmol) were added and stirred at room temperature for 8 hours. After diluting with ethyl acetate, the organic layer was washed with saturated aqueous sodium hydrogencarbonate solution and saturated brine in that order, and dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure to give 495 mg (57%) of the title compound as a solid.
1 H-NMR (400 MHz, CDCl 3 ) δ: 1.35-1.45 (1H, m), 1.60-1.70 (1 H, m), 2.10-2.18 (1 H, m), 2.21-2.28 (1 H, m), 2.76 ( 1H,tt,J=11.4,4.0Hz),3.44(1H,t,J=10.9Hz),3.67(4H,q,J=14.2Hz),3.71-3.73(1H,m),4.04(1H,dq ,J=11.0,2.1Hz),5.35(1H,s),6.40(1H,s),7.21-7.36(10H,m).MS
(ESI)m/z:325(M+H) + .
[0095]
[Step 3] (2S,5R)-5-aminotetrahydro-2H-pyran-2-carboxamide
The compound (490 mg, 1.51 mmol) obtained in Step 2 above was dissolved in ethanol (10 ml) and treated with 20% palladium hydroxide. (100 mg) was added, and the mixture was stirred at room temperature for 16 hours under a hydrogen atmosphere. After removing the catalyst by filtration through celite, the filtrate was distilled off under reduced pressure and dried to obtain 215 mg (99%) of the title compound as a solid.
1 H-NMR (400 MHz, DMSO-d 6 ) δ: 1.11-1.22(1H,m), 1.25-1.35(1H,m), 1.83-1.91(2H,m), 2.51-2.60(1H,m), 2.90(1H,t,J=10.5Hz),3.52(1H,d,J=11.9Hz),
3.78-3.84 (1H,m),6.99(1H,br s),7.09(1H,br s). (ESI) m/z: 145(M+H) + .
PATENT
WO2012121361
PATENT
WO2015033974
PAPER
https://pubs.acs.org/doi/10.1021/acs.oprd.2c00192
Abstract

Herein, we report the structure and synthesis of the potent MDM2-p53 inhibitor BI-0282. The complex spirooxindole scaffold bearing four stereocenters embedded in a rigid polycyclic ring-system was effectively prepared on a multi-gram scale in only five synthesis steps employing a three-component 1,3-dipolar cycloaddition and a late-stage Davis–Beirut reaction as key steps.
Compound 1
Intermediate 10 (28.8 g, 44.8 mmol) is dissolved in isopropanol (300 mL) and a solution of potassium hydroxide (39.0 g, 694.9 mmol) in water (95 mL) is slowly added. After stirring for 16 h at ambient temperature, the solvents are partially removed under reduced pressure. The residue is diluted with ethyl acetate and treated with a diluted aqueous solution of citric acid. After extraction of the aqueous layer with ethyl acetate, the organic layers are combined, dried with sodium sulfate, and the solvent is removed under reduced pressure. Purification by normal phase column chromatography using dichloromethane and methanol as solvents yields rac-1 (25.8 g, 43.5 mmol) in 70% yield as an amorphous white solid.
Chiral SFC and subsequent purification by reversed phase column chromatography using acetonitrile and methanol as solvents furnishes 1 (BI-0282).
Rac-1 (60 g, 93,3 mmol) was separated by chiral SFC and reversed phase column chromatography to obtain 1 (24.4 g, 40,0 mmol, 43%) as an amorphous white solid.
Chiral HPLC (CHIRALPAK, heptane/isopropanol/trifluoroacetic acid = 70/30/0.1, flow rate 1.0 mL/min, I = 240 mM) tR = 7.8 min (1), and 11.1 min (ent-1). Preparative SFC (CHIRALPAK, carbon dioxide/(isopropanol + 1% diethylamine) = 70/30, flow rate 300 g/min, I = 290 nM).
1H NMR (500 MHz, DMSO-d6): δ 12.64 (br s, 1H), 10.29 (s, 1H), 7.67 (s, 1H), 7.47 (d, J = 8.83 Hz, 2H), 7.29–7.36 (m, 1H), 7.26 (d, J = 7.88 Hz, 1H), 7.21 (dd, J = 1.26, 8.83 Hz, 1H), 7.12 (t, J = 8.04 Hz, 1H), 6.92 (dd, J = 1.89, 7.88 Hz, 1H), 6.48 (d, J = 1.89 Hz, 1H), 5.86 (t, J = 9.14 Hz, 1H), 4.59–4.68 (m, 1H), 4.52 (dd, J = 7.88, 11.35 Hz, 1H), 4.23–4.32 (m, 1H), 4.20 (d, J = 10.09 Hz, 1H), 2.27 (dd, J = 7.57, 13.08 Hz, 1H), 2.13 (dd, J = 5.83, 13.08 Hz, 1H), 0.47–0.62 (m, 1H), 0.26–0.37 (m, 1H), 0.11–0.20 (m, 1H), −0.04 to 0.04 (m, 1H), −0.25 (s, 1H).
13C{1H} NMR (125 MHz, DMSO-d6): δ 177.5, 168.1, 156.1 (d, 1JC,F = 248.7 Hz), 146.3, 145.3, 144.0, 134.1, 130.3, 129.7, 129.5, 126.8, 126.7, 125.4 (d, 3JC,F = 4.4 Hz), 123.5 (d, 2JC,F = 13.2 Hz), 122.5, 120.0, 119.9, 119.7 (d, 2JC,F = 18.3 Hz), 118.7, 110.0, 107.3, 76.4, 69.2, 57.5, 56.8, 54.2, 51.2, 11.6, 5.5, 4.1.
HRMS (ESI) m/z: [M + H]+ calcd for C30H24Cl2FN4O4, 593.1153; found, 593.1165.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Milademetan is under investigation in clinical trial NCT02319369 (Safety, Tolerability and Pharmacokinetics of Milademetan Alone and With 5-Azacitidine (AZA) in Acute Myelogenous Leukemia (AML) or High-Risk Myelodysplastic Syndrome (MDS)).
- [1]. ARYL SULFONOHYDRAZIDES. WO 2017069289 A1.[2]. M.M. Gounder, et al. Milademetan, an oral MDM2 inhibitor, in well-differentiated/dedifferentiated liposarcoma: results from a phase 1 study in patients with solid tumors or lymphomas. European Journal of Cancer 138S2 (2020) S1–S62.[3]. Li, Yangbing, et al. Development of novel PROTAC Small-Molecule Degraders of MDM2 Protein and Peptidomimetic Inhibitors Targeting WDR5-MLL1 Protein-Protein Interaction.[4]. Viktor Arnhold, et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. ncotarget. 2018 Jan 5; 9(2): 2304–2319.
/////////Milademetan, DS3032b, DS-3032b, DS 3032b, DS3032, DS-3032, DS 3032, DS-3032b tosylate, Milademetan tosylate, Milademetan HCl, orphan drug, UNII:R3I80TLN7S, миладеметан , ميلاديميتان , 米拉美坦
CC1(C)CCC2(CC1)N[C@H]([C@H](C1=C(F)C(Cl)=NC=C1)[C@]21C(=O)NC2=CC(Cl)=CC=C12)C(=O)N[C@@H]1CC[C@H](OC1)C(N)=O

NEW DRUG APPROVALS
ONE TIME
$10.00
Daxibotulinumtoxin A

Daxibotulinumtoxin A
FDA APPROVED 2022 2022/9/7, Daxxify
| Formula | C6708H10359N1729O1995S32 |
|---|---|
| CAS | 93384-43-1 |
| Mol weight | 148171.4934 |
Daxibotulinumtoxin A-lanm
Treatment of galbellar lines, cervical dystonia, lateral canthal lines, migraine headaches and hyperhidrosis
- DeveloperRevance Therapeutics; Shanghai Fosun Pharmaceutical
- ClassAnalgesics; Anti-inflammatories; Antiarrhythmics; Antidepressants; Antimigraines; Antipruritics; Antispasmodics; Bacterial proteins; Bacterial toxins; Botulinum toxins; Eye disorder therapies; Foot disorder therapies; Muscle relaxants; Skin disorder therapies; Urologics; Vascular disorder therapies
- Mechanism of ActionAcetylcholine inhibitors; Glutamate antagonists; Membrane transport protein modulators; Neuromuscular blocking agents
- Orphan Drug StatusYes – Torticollis
- RegisteredGlabellar lines
- Phase IIITorticollis
- Phase IIMuscle spasticity
- No development reportedSkin disorders
- DiscontinuedPlantar fasciitis
- 19 Sep 2022Efficacy data from a phase IIa FHL trials in Glabellar-lines (crow’s feet) released by Revance
- 19 Sep 2022Updated efficacy and safety data from the phase III SAKURA 1, SAKURA 2 and SAKURA 3 trials in Glabellar lines released by Revance Therapeutics
- 18 Sep 2022Updated efficacy and safety data from the phase III SAKURA 1, SAKURA 2 and SAKURA 3 trials in Glabellar lines released by Revance Therapeutics
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
DAXI Impresses; Approaches FDA Approval
March 2, 2021
Dermatology Times, Dermatology Times, February 2021 (Vol. 42, No. 2), Volume 42, Issue 2
The investigational neuromodulator, evaluated in clinical trials as a treatment for glabellar lines and as a combined therapy for glabellar, dynamic forehead, and lateral canthal lines, is quickly nearing an approval by the FDA.
The long-awaited, longer-lasting neuromodulator drug candidate DaxibotulinumtoxinA for Injection (DAXI), a botulinum toxin type A formulated with a novel peptide excipient, may be nearing FDA approval.
In mid-December 2020, Revance Therapeutics shared results from its phase 2 upper facial lines study (NCT04259086),1 in which investigators looked at DAXI for combined treatment of glabellar, dynamic forehead, and lateral canthal lines.
The authors reported on a multicenter study of 48 patients enrolled to receive 40, 32, and 48 U of DAXI for injection in the glabellar complex, forehead, and lateral canthal areas, respectively. At week 4, nearly 92% of patients achieved an Investigator Global Assessment (IGA) score indicating no or mild wrinkle severity with maximum contraction on their lateral canthal lines. Nearly 96% achieved similar results on their forehead and glabellar lines at week 4.
Wrinkle severity returned to baseline at a median of 7.6 months post treatment, according to the phase 2 study findings.
The treatment was well tolerated in all upper facial regions. The most common adverse event (AE) was injection site erythema, which occurred in 6.3% of patients. The authors reported no eyelid or brow ptosis.
This was Revance’s first DAXI study on not just glabellar lines but also on forehead and periocular lines, or crow’s-feet, according to Jeffrey S. Dover, MD, FRCPC, a phase 2 study investigator and a dermatologist at SkinCare Physicians in Chestnut Hill, Massachusetts. “I think this is yet more evidence that the Revance neuromodulator produces an impressive effect on lines of negative facial expression and lasts longer than any of the other neuromodulators approved by the FDA thus far,” said Dover.
Dermatologic Surgery published 2 papers on the investigational neuromodulator in January 2021. In one study,2 investigators evaluated the use of up to 3 DAXI treatments for moderate or severe glabellar lines. They focused on data from SAKURA 1 and 2 (NCT03014622 and NCT03014635), two identical phase 3, open label, multicenter studies in which investigators evaluated single and repeat treatment of the glabellar lines with 40 U of DAXI.
The authors reported on safety results for nearly 2700 patients, including 882 who received a second treatment and 568 who got DAXI a third time. Treatment-related AEs, which were generally mild and resolved, occurred in 17.8% of patients. Eyelid ptosis occurred in 0.9% of treatments.
Investigators of 2 other studies3,4 focused on DAXI efficacy among nearly 2700 subjects enrolled in Revance’s preceding pivotal trials. Participants received repeat treatments when they returned to baseline on the IGA–Frown Wrinkle Severity (FWS) and IGA–Patient Frown Winkle Severity (PFWS) scales at 12 weeks and up to 36 weeks after treatment.
More than 96% of patients achieved no or mild severity in glabellar wrinkles on the IGA- FWS scale after each of the 3 treatments, with peak responses between weeks 2 to 4, and about one-third or more saw no or mild severity at week 24. Response rates reached highs of 92% or more at weeks 2 to 4 on the IGA-PFWS scale.
“The median duration for return to moderate or severe severity was 24 weeks,” the authors said. “If approved, I believe daxibotulinumtoxinA will change the landscape of neuromodulators significantly. The approved ones all last 3 months. They all give nice results and have few adverse effects,” Dover said.
He and other investigators have seen no rise in AEs, and those that did occur lasted no longer than those of Botox, he said.
Revance appears to be preparing for approval. The company announced on December 22, 2020, that it has a strategic commercial manufacturing agreement with Ajinomoto Bio- Pharma Services for the supply of DAXI.5
As of November 24, 2020, the FDA had deferred a decision on the neuromodulator because the required factory inspection could not be conducted due to travel restrictions related to coronavirus disease 2019.6 The FDA did not indicate any other issues.
References:
- Green JB, Mariwalla K, Coleman K, et al. A large, open-label, phase 3 safety study of DaxibotulinumtoxinA for Injection in glabellar lines: a focus on safety from the SAKURA 3 study. Derm Surg. 2021;47(1):42-46. doi:10.1097/DSS.0000000000002463
- Carruthers JD, Jean D, Fagien S, et al; SAKURA 1 and SAKURA 2 Investigator Group. DaxibotulinumtoxinA for Injection for the treatment of glabellar lines: results from each of two multicenter, randomized, double-blind, placebo-controlled, phase 3 studies (SAKURA 1 and SAKURA 2).Plast Reconstr Surg. 2020;1(145):45-58.doi: 10.1097/PRS.0000000000006327
- Fabi SG, Cohen JL, Green LJ, et al. DaxibotulinumtoxinA for Injection for the treatment of glabellar lines: efficacy results from SAKURA 3, a large, open-label, phase 3 safety study. Derm Surg. 2021;47(1):48-54. doi:10.1097/DSS.0000000000002531
- https://investors.revance.com/news-releases/news-release-details/ajinomoto-bio-pharma-services-and-revance-therapeutics-announce. December 22, 2020. Accessed January 15, 2021.
- FDA defers approval of DaxibotulinumtoxinA for Injection in glabellar lines due to COVID-19 related travel restrictions impacting manufacturing site inspection. News release. Revance Therapeutics, Inc. November 25, 2020. Accessed January 13, 2021. https://www.businesswire.com/news/home/20201125005462/en/FDA-Defers-Approval-DaxibotulinumtoxinA-Injection-Glabellar-Lines
///////////Daxibotulinumtoxin A, FDA 2022, APPROVALS 2022, DAXI, Daxibotulinumtoxin-A, DaxibotulinumtoxinA for Injection, daxibotulinumtoxinA-lanm, DAXXIFY, RT-002, Orphan Drug

NEW DRUG APPROVALS
ONE TIME
$10.00
ELRAGLUSIB
![3-(5-fluorobenzofuran-3-yl)-4-(5-methyl-5H-[1,3]dioxolo[4,5-f]indol-7-yl)-1H-pyrrole-2,5-dione.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=44582816&t=l)
ELRAGLUSIB
RN: 1034895-42-5
UNII: ND1SOF0DLU, WHO 11553, 9-ING-41
- 1H-Pyrrole-2,5-dione, 3-(5-fluoro-3-benzofuranyl)-4-(5-methyl-5H-1,3-dioxolo(4,5-F)indol-7-yl)-
- 3-(5-Fluoro-benzofuran-3-yl)-4-(5-methyl-5H-(1,3)dioxolo(4,5-F)indol-7-yl)-pyrrole-2,5-dioneAntineoplastic
Molecular Formula
- C22-H13-F-N2-O5
Molecular Weight
- 404.3517
- OriginatorNorthwestern University; University of Illinois at Chicago
- DeveloperActuate Therapeutics; Incyte Corporation; Levine Cancer Institute; University of Kansas Medical Center
- ClassAntineoplastics; Benzofurans; Dioxolanes; Indoles; Pyrroles; Small molecules
- Mechanism of ActionGlycogen synthase kinase 3 beta inhibitors
- Orphan Drug StatusYes – Glioblastoma; Neuroblastoma
- Phase IIAdenoid cystic carcinoma; Myelofibrosis; Neuroblastoma; Pancreatic cancer; Salivary gland cancer
- Phase I/IICancer
- PreclinicalBrain cancer; Chronic lymphocytic leukaemia; Colorectal cancer
- 20 Sep 2022Elraglusib – Actuate Therapeutics receives Fast Track designation for Pancreatic cancer [IV] (Combination therapy, First-line therapy, Late-stage disease, Metastatic disease, Recurrent) in USA
- 03 Jun 2022Efficacy and safety data from a phase I trial in cancer presented at the 58th Annual Meeting of the American Society of Clinical Oncology (ASCO-2022)
- 08 Apr 2022Preclinical trials in Brain cancer in USA (unspecified route)
9-ING-41 is under investigation in clinical trial NCT04218071 (Actuate 1901: 9-ING-41 in Myelofibrosis).

SYN
WO2019079299
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019079299
3-(5-Fluorobenzofuran-3-yl)-4-(5-methyl-5H-[l,3]dioxolo[4,5-f]indol-7-yl)pyrrole-2,5-dione (“9-ING-41”) has the following chemical structure:
[0004] 9-ING-41 has been reported as being useful for the treatment of cancers, including brain, lung, breast, ovarian, bladder, neuroblastoma, renal, and pancreatic cancers, as well as for treatment of traumatic brain injury.
[0005] The structure, properties, and/or biological activity of 9-ING-41 are set forth in U.S. Patent Number 8,207,216; Gaisina et al., From a Natural Product Lead to the Identification of Potent and Selective Benzofuran-3-yl-(indol-3-yl)maleimides as Glycogen Synthase Kinase 3β Inhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells, J. Med. Chem. 2009, 52, 1853-1863; and Hilliard, et al., Glycogen synthase kinase 3β inhibitors induce apoptosis in ovarian cancer cells and inhibit in-vivo tumor growth, Anti-Cancer Drugs 2011, 22:978-985.
Example 1: Preparation of 9-ING-41
[0056] Crude 9-ING-41 can be obtained by the general methods described in U.S. Patent Number 8,207,216, and in Gaisina et al., From a Natural Product Lead to the
Identification of Potent and Selective Benzofuran-3-yl-(indol-3-yl)maleimides as Glycogen Synthase Kinase 3β Inhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells, J. Med. Chem. 2009, 52, 1853-1863.
Example 2: Preparation of 9-ING-41 Crystalline Form I
[0057] Crystalline Form I of 9-ING-41 may also be prepared as follows.
Synthesis of Intermediate 1
[0058] Into a 3-L 4-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed 6-nitro-2H-l,3-benzodioxole-5-carbaldehyde (200 g, 1.02 mol, 1.00 equiv), ammonium acetate (200 g, 2.59 mol, 2.53 equiv), acetic acid (2 L), and nitromethane (313 g, 5.13 mol, 5.00 equiv). The solution was stirred for 12 h at lOOoC. The reaction repeated three times. The solutions were combined and diluted with 20 L of water. The resulting solution was extracted with 3×10 L of ethyl acetate and the organic layers were combined. The mixture was washed with 3×10 L of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in 450 g (crude) of 5-nitro-6-[(E)-2-nitroethenyl]-2H-l,3-benzodioxole (1) as a dark green solid.
Synthesis of Intermediate 2
[0059] Fe (120 g, 2.14 mol, 17.01 equiv) was slowly added in portions into a suspension of 5-nitro-6-[(Z)-2-nitroethenyl]-2H-l,3-benzodioxole (30 g, 125.97 mmol, 1.00 equiv), silica gel (120 g) in acetic acid (300 mL), toluene (200 mL), and cyclohexane (400 mL) at 80oC under nitrogen. The resulting black mixture was stirred for 8h at 80oC.The reaction repeated ten times. The reaction mixtures were combined. The solids were filtrated out. The filtrate was concentrated under vacuum and the residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1/5). The collected fractions were combined and concentrated under vacuum to give 67.3 g (33%) of 2H, 5H-[1, 3] dioxolo [4, 5-f] indole (2) as an off-white solid.
Synthesis of Intermediate 3
[0060] Sodium hydride (19.9 g, 497.50 mmol, 1.18 equiv, 60%) was added in portions into a solution of 2H,3H,5H-furo[2,3-f]indole (67.3 g, 422.78 mmol, 1.00 equiv) in N,N-
dimethylformamide (1.3 L) at 0°C under nitrogen. The mixture was stirred for lh at 0°C and CH3I (70.9 g, 499.51 mmol, 1.18 equiv) was added dropwise. The resulting solution was stirred for 3 h at room temperature. The solution was quenched by added 1 L of ice water. The resulting solution was extracted with 3×1 L of ethyl acetate and the organic layers were combined. The mixture was washed with 3×1 L of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1/10). The collected fractions were combined and concentrated under vacuum to give 71 g (97%) of 5-methyl-2H,3H,5H-furo[2,3-f]indole (3) as a light yellow solid.
Synthesis of Int rmediate 4
[0061] Ethyl 2-chloro-2-oxoacetate (220 g, 1.61 mol, 3.96 equiv) was added dropwise into a solution of 5-methyl-2H,3H,5H-furo[2,3-f]indole (70.4 g, 406.44 mmol, 1.00 equiv) in ethyl ether (1.6 L) at OoC under nitrogen. The resulting solution was warmed to room temperature and stirred for 4 h. The reaction was quenched slowly by the addition of 2 L of ice water and the pH value of the resulting solution was adjusted to 9 by Na2C03. The resulted mixture was extracted with 3×1.5 L of ethyl acetate. The organic layers were combined and dried over anhydrous sodium sulfate and concentrated under vacuum to give 92.8 g (84%) of ethyl 2-[5-methyl-2H,3H,5H-furo[2,3-f]indol-7-yl]-2-oxoacetate (4) as a light yellow solid.
[0062] 1H MR (300 MHz, DMSO-d6): δ 8.28 (s, 4H), 7.56 (s, 4H), 7.27 (s, 4H), 6.17 (s, 1H), 6.08 (s, 8H), 4.35 (q, J = 7.1 Hz, 7H), 3.85 (s, 11H), 3.35 (s, 2H), 1.35 (t, J = 7.1 Hz, 11H), 1.25 (s, 2H).
Synthesis of Intermediate 5
5
[0063] Into a 10-L 4-necked round-bottom flask was placed 2-bromo-4-fluorophenol (500 g, 2.62 mol, 1.00 equiv), N,N-dimethylformamide (5 L), potassium carbonate (1253 g, 9.07 mol, 3.46 equiv), and ethyl (2E)-4-bromobut-2-enoate (1010 g, 5.23 mol, 2.00 equiv). The resulting solution was stirred for 12 h at room temperature. The solids were collected by filtration. The reaction was then quenched by the addition of 15 L of water and extracted with 3×10 L of ethyl acetate. The organic layers were combined and washed with 4×20 L of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1/20). The collected fractions were combined and concentrated under vacuum to give 500 g (63%) of ethyl (2E)-4-(2-bromo-4-fluorophenoxy)but-2-enoate (5) as a white solid.
Synthesis of Intermediate 6
[0064] Into a 2-L 3 -necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed ethyl (2E)-4-(2-bromo-4-fluorophenoxy)but-2-enoate (125 g, 412.37 mmol, 1.00 equiv), benzyltri ethyl azanium chloride (99 g, 434.64 mmol, 1.05 equiv), sodium formate dihydrate (45.1 g), Pd(OAc)2 (2.9 g, 12.92 mmol, 0.03 equiv), sodium carbonate (92 g, 868.01 mmol, 2.10 equiv), and N,N-dimethylformamide (1.25 L). The resulting solution was stirred for 12 h at 80°C. The reaction repeated four times. The reaction mixtures were combined and the solids were filtrated out. The filtrate was diluted with 10 L of brine and extracted with 3×5 L of ethyl acetate. The organic layers were combined and washed with 4×6 L of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1/20). The collected fractions were combined and concentrated under vacuum. This resulted in 258 g (crude) of ethyl 2-(5-fluoro-l-benzofuran-3-yl)acetate (6) as light yellow oil.
Synthesis of Intermediate 7
7
[0065] Into a 5-L round-bottom flask was placed ethyl 2-(5-fluoro-l-benzofuran-3-yl)acetate (147 g, 661.53 mmol, 1.00 equiv), methanol (1 L), tetrahydrofuran (1 L), water (1 L), and Li OH (47.7 g, 1.99 mol, 3.01 equiv). The resulting solution was stirred for 3 h at room temperature. The reaction repeated twice. The mixture was concentrated under vacuum and then extracted with 1 L of dichloromethane. The aqueous layer was collected and the pH of the layer was adjust to 1-3 by hydrogen chloride (1 mol/L). The resulting solution was extracted with 3×1 L of ethyl acetate and the combined organic layers were dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in 160 g (62%) of 2-(5-fluoro-l-benzofuran-3-yl)acetic acid (7) as a white solid.
Synthesis of Intermediate 8
[0066] Into a 10-L round-bottom flask was placed 2-(5-fluoro-l-benzofuran-3-yl) acetic acid (160 g, 824.1 mmol, 1.00 equiv), H4C1 (436 g, 8.16 mol, 9.89 equiv), N,N-dimethylformamide (6L), DIEA (1064 g, 8.24 mol, 9.99 equiv), and HATU (376 g, 988.88 mmol, 1.20 equiv). The resulting solution was stirred for 12 h at room temperature. The resulting solution was diluted with 10 L of water. The solids were collected by filtration to give in 126 g (78%) of 2-(5-fluoro-l-benzofuran-3-yl) acetamide (8) as a white solid.
Synthesis of 9-ING-41 in cr stalline Form I
8 9-ING-41
[0067] t-BuOK (1200 mL, 1 mol/L in THF) was added dropwise into a solution of ethyl 2-[5-methyl-2H,3H,5H-furo[2,3-f]indol-7-yl]-2-oxoacetate (100 g, 365.9 mmol, 1.00 equiv), 2-(5-fluoro-l-benzofuran-3-yl)acetamide (72 g, 372.7 mmol, 1.02 equiv) in tetrahydrofuran (3 L) at 0°C under nitrogen. The reaction was stirred for 2h at room temperature. The reaction was cooled to 0°C and poured into of 2 L of H4C1 (saturated solution in water) and extracted with 4×2 L of dichloromethane. The organic layers were combined, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/dichloromethane/petroleum ether (1/1/5). The collected fractions were combined and concentrated under vacuum to give 107.9 g (74%) of 3-(5-fluoro-l-benzofuran-3-yl)-4-[5-methyl-2H,5H-[l,3]dioxolo[4,5-f]indol-7-yl]-2,5-dihydro-lH-pyrrole-2,5-dione as a red solid. This red solid is 9-ING-41 crystalline Form I. MS-ESI: [M+H]+ = 405.
PATENT
WO2019032958
PATENT
US20100004308
REF
Journal of Medicinal Chemistry (2009), 52(7), 1853-1863
PATENT
WO2008077138
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Elraglusib is a maleimide-based, small molecule inhibitor of glycogen synthase kinase-3 (GSK-3; serine/threonine-protein kinase GSK3) with potential antineoplastic activity. Upon intravenous administration, elraglusib binds to and competitively inhibits GSK-3, which may lead to downregulation of nuclear factor kappa B (NF-kappaB) and decreased expression of NF-kappaB target genes including cyclin D1, B-cell lymphoma 2 (Bcl-2), anti-apoptotic protein XIAP, and B-cell lymphoma extra-large (Bcl-XL). This may inhibit NF-kappaB-mediated survival and chemoresistance in certain tumor types. GSK-3, a constitutively active serine/threonine kinase that plays a role in numerous pathways involved in protein synthesis, cellular proliferation, differentiation, and metabolism, is aberrantly overexpressed in certain tumor types and may promote tumor cell survival and resistance to chemotherapy and radiotherapy.
Actuate Therapeutics Announces Initiation of a Multicenter Randomized Trial of Elraglusib Plus FOLFIRINOX As First Line Therapy for Advanced Pancreatic Cancer
Published: Feb 07, 2022
CHICAGO and FORT WORTH, Texas, Feb. 07, 2022 (GLOBE NEWSWIRE) — Actuate Therapeutics (Actuate), a clinical stage biopharmaceutical company, today announced the opening of a randomized study of elraglusib (9-ING-41) plus FOLFIRINOX alone or with Losartan for patients with advanced pancreatic cancer in the first-line setting (NCT05077800). Elraglusib is Actuate’s proprietary small molecule glycogen synthase kinase-3 beta (GSK-3β) inhibitor which is being developed for adults and children with advanced refractory cancers. This multicenter investigator-initiated study, which is receiving substantial support from the Lustgarten Foundation for Pancreatic Cancer Research, is being led by Colin D. Weekes MD at the Massachusetts General Hospital and will also enroll patients at the University of Washington, University of Colorado Denver, and Johns Hopkins University.
“Novel approaches for patients with advanced pancreatic cancer are urgently required,” said Dr Weekes. “The pre-clinical and clinical data being generated with elraglusib in a spectrum of cancers, including pancreatic cancer, is extremely encouraging and we are delighted to have initiated this study with elraglusib. Elraglusib is the first clinically relevant specific GSK-3β inhibitor that we can thoroughly investigate. In preclinical models, elraglusib has multiple biologic effects relevant to targeting pancreatic cancer including direct cytotoxicity, reversal of chemoresistance, reversal of pathologic fibrosis, and there is increasing evidence of its immune-modulatory activity. In our study, we are particularly focused on elraglusib’s potential to synergize with TGF-β suppression mediated by Losartan. This study builds on the work of our investigative teams demonstrating the roles of TGF-β and GSK-3β in acquired chemotherapy resistance. This study uniquely attempts to harness the mechanisms that pancreatic cancer utilizes to combat the effects of chemotherapy as an Achilles heel for therapeutic intent. We believe that a multi-pronged attack as represented by elraglusib plus Losartan is a potentially sophisticated approach to a complex, often lethal, situation. It is an honour to lead this multicenter collaboration with my clinical and pre-clinical colleagues across the US and Europe. We are very grateful for the critical support of this program by the Lustgarten Foundation.”
“At the Lustgarten Foundation, we understand time is everything for patients and their families,” said Andrew Rakeman, PhD, VP of Research. “Dr. Weekes’ study will help us understand and address a critical issue in pancreatic cancer treatment—acquired chemotherapy resistance. This trial builds on exciting observations from previous preclinical and clinical research. The Foundation established the Clinical Accelerator Initiative for projects like this; bringing more trials based on the best science to the clinic and expanding our understanding of pancreatic cancer biology and treatment. We believe Dr. Weekes’ trial and others like it have the potential to change the way we think about treating pancreatic cancer, ultimately transforming it into a curable disease.”
“We are honored and excited to collaborate with Dr. Weekes, his colleagues at world-leading cancer research centers, and the Lustgarten Foundation on this important trial, which will advance the development of elraglusib for treating patients with one of the most challenging types of cancer,” said Daniel Schmitt, Actuate’s President & CEO. “The results we have seen to date with elraglusib combined with chemotherapy in pancreatic cancer are very promising, and this Phase 2 trial in combination with FOLFIRINOX leverages significant positive preclinical and clinical experience for potentially better outcomes for patients.”
Based on positive data from a prior Phase 2 open-label single arm study of elraglusib plus gemcitabine/nab-paclitaxel, Actuate has also recently initiated an international randomized controlled study of elraglusib in combination with gemcitabine/nab-paclitaxel, in patients with advanced pancreatic cancer in the first-line setting (NCT03678883, EudraCT#:2018-003739-32). Actuate is also conducting studies in pediatric patients with refractory tumors in preparation for a neuroblastoma-specific clinical program (NCT04239092). Actuate is also collaborating with investigators at the Dana-Farber Cancer Institute and Brigham and Women’s Hospital on a Phase 2 study focused on elraglusib combined with cytotoxic therapy for patients with advanced salivary gland carcinomas (NCT05010629).
About Actuate Therapeutics, Inc.
Actuate is a clinical stage pharmaceutical company focused on the development and commercialization of novel therapeutics for cancers and inflammatory diseases. For additional information, please visit the Company’s website at http://www.actuatetherapeutics.com.
///////////ELRAGLUSIB, WHO 11553, 9-ING-41, Orphan Drug
Cn1cc(C2=C(C(=O)NC2=O)c3coc4ccc(F)cc34)c5cc6OCOc6cc15

NEW DRUG APPROVALS
one time
$10.00
Valemetostat tosilate
Valemetostat tosilate
バレメトスタットトシル酸塩
| Formula | C26H34ClN3O4. C7H8O3S |
|---|---|
| CAS | 1809336-93-3 |
| Mol weight | 660.2205 |
PMDA JAPAN approved 2022/9/26, Ezharmia
- 1,3-Benzodioxole-5-carboxamide, 7-chloro-N-((1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl)-2-(trans-4-(dimethylamino)cyclohexyl)-2,4-dimethyl-, (2R)-, compd. with 4-methylbenzenesulfonate (1:1)
Antineoplastic, histone methyltransferase inhibitor
1809336-39-7 (free base). 1809336-93-3 (tosylate) 1809336-92-2 (mesylate) 1809336-94-4 (fumarate) 1809336-95-5 (tarate)
Synonym: Valemetostat; DS-3201; DS 3201; DS3201; DS-3201b
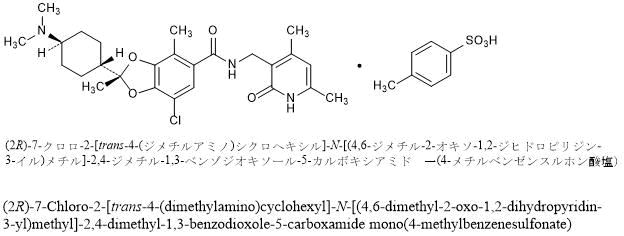
(2R)-7-Chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide mono(4-methylbenzenesulfonate)
C26H34ClN3O4▪C7H8O3S : 660.22
[1809336-93-3]
1809336-39-7 (free base)
Chemical Formula: C26H34ClN3O4
Exact Mass: 487.2238
Molecular Weight: 488.02
(2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide
Valemetostat, also known as DS-3201 is a potent, selective and orally active EZH1/2 inhibitor. DS-3201 selectively inhibits the activity of both wild-type and mutated forms of EZH1 and EZH2. Inhibition of EZH1/2 specifically prevents the methylation of lysine 27 on histone H3 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways, enhances transcription of certain target genes, and results in decreased proliferation of EZH1/2-expressing cancer cells.
- OriginatorDaiichi Sankyo Inc
- DeveloperCALYM Carnot Institute; Daiichi Sankyo Inc; Lymphoma Academic Research Organisation; Lymphoma Study Association; University of Texas M. D. Anderson Cancer Center
- ClassAmides; Amines; Antineoplastics; Benzodioxoles; Chlorinated hydrocarbons; Cyclohexanes; Pyridones; Small molecules
- Mechanism of ActionEnhancer of zeste homolog 1 protein inhibitors; Enhancer of zeste homolog 2 protein inhibitors
- Orphan Drug StatusYes – Adult T-cell leukaemia-lymphoma; Peripheral T-cell lymphoma
- New Molecular EntityYes
- RegisteredAdult T-cell leukaemia-lymphoma
- Phase IIB-cell lymphoma; Peripheral T-cell lymphoma
- Phase I/IISmall cell lung cancer
- Phase INon-Hodgkin’s lymphoma; Prostate cancer; Renal cell carcinoma; Urogenital cancer
- PreclinicalDiffuse large B cell lymphoma
- No development reportedAcute myeloid leukaemia; Precursor cell lymphoblastic leukaemia-lymphoma
- 26 Sep 2022First global approval – Registered for Adult T-cell leukaemia-lymphoma (Monotherapy, Second-line therapy or greater) in Japan (PO)
- 26 Sep 2022Updated efficacy and adverse events data from a phase II trial in Adult T-cell leukaemia-lymphoma released by Daiichi Sankyo
- 28 Dec 2021Preregistration for Adult T-cell leukaemia-lymphoma (Monotherapy, Second-line therapy or greater) in Japan (PO

PATENT
WO 2015141616
Watson, W. D. J. Org. Chem. 1985, 50, 2145.
Lengyel, I. ; Cesare, V. ; Stephani, R. Synth. Common. 1998, 28, 1891.
PATENT
WO2022009911
The equipment and measurement conditions for the powder X-ray diffraction measurement in the examples are as follows.
Model: Rigaku Rint TTR-III
Specimen: Appropriate
X-ray generation conditions: 50 kV, 300 mA
Wavelength: 1.54 Å (Copper Kα ray)
Measurement temperature: Room temperature
Scanning speed: 20°/min
Scanning range: 2 to 40°
Sampling width: 0.02°
[0043]
(Reference Example 1) Production of ethyl trans-4-[(tert-butoxycarbonyl)amino]cyclohexanecarboxylate
[0044]
[hua 6]
[0045]
Under a nitrogen atmosphere, ethanol (624 L) and ethyl trans-4-aminocyclohexanecarboxylate monohydrochloride (138.7 kg, 667.8 mol) were added to a reaction vessel and cooled. Triethylamine (151.2 kg, 1495 .5 mol) and di-tert-butyl dicarbonate (160.9 kg, 737.2 mol) were added dropwise while maintaining the temperature below 20°C. After stirring at 20-25°C for 4 hours, water (1526 kg) was added dropwise at 25°C or lower, and the mixture was further stirred for 2 hours. The precipitated solid was collected by filtration, washed with a mixture of ethanol:water 1:4 (500 L), and dried under reduced pressure at 40°C to obtain 169.2 kg of the title compound (yield 93.4%). .
1 H NMR (500 MHz, CDCl 3 ): δ 4.37 (br, 1H), 4.11 (q, J = 2.8 Hz, 2H), 3.41 (br, 1H), 2.20 (tt, J = 4.8, 1.4 Hz, 1H),2.07(m,2H),2.00(m,2H),1.52(dq,J=4.6,1.4Hz,2H),1.44(s,9H),1.24(t,J=2.8Hz,3H), 1.11(dq,J=4.6,1.4Hz,2H)
[0046]
(Reference Example 2) Production of tert-butyl = [trans-4-(hydroxymethyl)cyclohexyl]carbamate
[0047]
[hua 7]
[0048]
Under a nitrogen atmosphere, tetrahydrofuran (968 kg), ethyl = trans-4-[(tert-butoxycarbonyl)amino]cyclohexanecarboxylate (110 kg, 405.4 mol), lithium chloride (27.5 kg, 648 kg) were placed in a reaction vessel. .6 mol), potassium borohydride (32.8 kg, 608.1 mol), and water (2.9 L, 162.2 mol) were added, the temperature was slowly raised to 50°C, and the mixture was further stirred for 6 hours. Cooled to 0-5°C. Acetone (66 L) and 9 wt % ammonium chloride aqueous solution (1210 kg) were added dropwise while maintaining the temperature at 20° C. or lower, and the mixture was stirred at 20-25° C. for 1 hour. Additional ethyl acetate (550 L) was added, the aqueous layer was discarded and the organic layer was concentrated to 550 L. Ethyl acetate (1650 L) and 9 wt% aqueous ammonium chloride solution (605 kg) were added to the residue, and the aqueous layer was discarded after stirring. Washed sequentially with water (550 L). The organic layer was concentrated to 880 L, ethyl acetate (660 L) was added to the residue, and the mixture was concentrated to 880 L while maintaining the internal temperature at 40-50°C. The residue was cooled to 0-5° C. and stirred for 1 hour, petroleum ether (1760 L) was added dropwise over 30 minutes, and the mixture was stirred at the same temperature for 2 hours. The precipitated solid was collected by filtration, washed with a petroleum ether:ethyl acetate 3:1 mixture (220 L) cooled to 0-5°C, and dried at 40°C under reduced pressure to give 86.0 kg of the title compound (yield: obtained at a rate of 92.3%).
1 H NMR (500 MHz, CDCl 3 ): δ 4.37 (br, 1H), 3.45 (d, J = 2.2 Hz, 2H), 3.38 (br, 1H), 2.04 (m, 2H),
1.84(m,2H),1.44(m,10H),1.28-1.31(m,1H),1.00-1.13(m,4H)
[0049]
(Reference Example 3) Production of tert-butyl = [trans-4-(2,2-dibromoethenyl)cyclohexyl]carbamate
[0050]
[hua 8]
[0051]
(Step 1)
Under a nitrogen atmosphere, ethyl acetate (50 L), tert-butyl = [trans-4-(hydroxymethyl)cyclohexyl]carbamate (2.5 kg, 10.90 mol), potassium bromide ( 39.3 g, 0.33 mol), 2,2,6,6-tetramethylpiperidine 1-oxyl (51.1 g, 0.33 mol), 4.8% aqueous sodium hydrogen carbonate solution (26.25 kg ) was added and cooled to 0-5°C, 9.9% sodium hypochlorite (8.62 kg, 11.45 mol) was added at 5°C or lower, and the mixture was further stirred at 0°C for 4 hours. Sodium sulfite (250 g) was added to the mixture and stirred at 0-5°C for 30 minutes before warming to 20-25°C. Thereafter, the aqueous layer was discarded and washed with a 20% aqueous sodium chloride solution (12.5 kg), then the organic layer was dried over sodium sulfate and concentrated to 7.5 L. Ethyl acetate (12.5 L) was added to the residue, the mixture was concentrated again to 7.5 L, and used in the next reaction as a tert-butyl=(trans-4-formylcyclohexyl)carbamate solution.
[0052]
(Step 2)
Under a nitrogen atmosphere, tetrahydrofuran (30 L) and triphenylphosphine (5.72 kg, 21.8 mol) were added to a reaction vessel, heated to 40°C, and stirred for 5 minutes. Carbon tetrabromide (3.61 kg, 10.9 mol) was added over 30 minutes and stirred at 40-45° C. for another 30 minutes. A mixture of tert-butyl (trans-4-formylcyclohexyl)carbamate solution and triethylamine (2.54 kg, 25.1 mol) was added below 45°C over 20 minutes and stirred at 40°C for an additional 15 hours. After cooling the reaction solution to 0° C., water (0.2 L) was added at 10° C. or lower, and water (25 L) was added. After heating to 20-25° C., the aqueous layer was discarded, ethyl acetate (4.5 kg) and 10% aqueous sodium chloride solution (25 kg) were added, and after stirring, the aqueous layer was discarded again. After the obtained organic layer was concentrated to 15 L, 2-propanol (19.65 kg) was added and concentrated to 17.5 L. 2-Propanol (11.78 kg) and 5 mol/L hydrochloric acid (151.6 g) were added to the residue, and the mixture was stirred at 25-35°C for 2.5 hours. Water (16.8 L) was added dropwise to the resulting solution, and the mixture was stirred at 20-25°C for 30 minutes and then stirred at 0°C for 2 hours. The precipitated solid was collected by filtration, washed with a mixture (11 kg) of acetonitrile:water 60:40 cooled to 0-5°C, and dried at 40°C under reduced pressure to give 3.05 kg of the title compound (yield 73%). .0%).
1 H NMR (500 MHz, CDCl3):δ6.20(d,J=3.6Hz,1H),4.37(br,1H),3.38(br,1H),2.21(dtt,J=3.6,4.6,1.4Hz,1H),2.05-2.00(m,2H),1.80-1.83(m,2H),1.44(s,9H),1.23(ddd,J=9.9,5.3,1.2 Hz,2H), 1.13(ddt,J=4.6,1.4,5.2 Hz,2H)
[0053]
(Reference Example 4) Production of tert-butyl = (trans-4-ethynylcyclohexyl) carbamate
[0054]
[Chemical 9]
[0055]
Under a nitrogen atmosphere, toluene (1436 kg), tert-butyl = [trans-4-(2,2-dibromoethenyl)cyclohexyl]carbamate (110 kg, 287.1 mol), and N,N,N ‘,N’-Tetramethylethane-1,2-diamine (106.7 kg, 918.8 mol) was added and cooled to -10°C. An isopropylmagnesium chloride-tetrahydrofuran solution (2.0 mol/L, 418 kg, 863 mol) was added dropwise at -5°C or lower, and stirred at -10°C for 30 minutes. After the reaction, 5 mol/L hydrochloric acid (465 kg) was added at 5°C or lower, heated to 20-25°C, and further 5 mol/L hydrochloric acid (41.8 kg) was used to adjust the pH to 5.0-. adjusted to 6.0. After discarding the aqueous layer, the organic layer was washed twice with water (550 L) and concentrated to 550 L. 2-Propanol (1296 kg) was added to the concentrate and concentrated to 550 L again. Further, 2-propanol (1296 kg) was added to the residue, and after concentrating to 550 L, water (770 L) was added dropwise in 4 portions. At that time, it was stirred for 30 minutes after each addition. After the addition, the mixture was stirred for 1 hour and further stirred at 0° C. for 1 hour. The precipitated solid was collected by filtration, washed with a 5:7 mixture of 2-propanol:water (550 L) cooled to 0-5°C, and dried at 40°C under reduced pressure to yield 57.8 kg of the title compound. obtained at a rate of 90.2%).
1 H NMR (500 MHz, CDCl 3 ): δ 4.36 (br, 1H), 3.43 (br, 1H), 2.18-2.23 (m, 1H), 1.97-2.04 (m, 5H), 1.44-1.56 (m, 11H ),1.06-1.14(m,2H)
[0056]
(Reference Example 5) Production of 4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile
[0057]
[Chemical 10]
[0058]
Under a nitrogen atmosphere, water (300 L), 2-cyanoacetamide (20 kg, 238 mol), 1-pentane-2-4-dione (26.2 kg, 262 mol), potassium carbonate (3.29 mol) were added to a reaction vessel. kg, 23.8 mol) was added and stirred at room temperature for 6 hours or longer. After the reaction, the precipitated solid was collected by filtration, washed with water (60 L), further washed with a mixture of methanol (40 L) and water (40 L), and dried under reduced pressure at 40°C to give the title compound as 34 Obtained in .3 kg (97.3% yield).
1 H NMR (500 MHz, DMSO-d 6 ): δ 2.22 (s, 3H), 2.30 (s, 3H), 6.16 (s, 1H), 12.3 (brs, 1H)
[0059]
(Reference Example 6) Production of 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one monohydrochloride
[0060]
[Chemical 11]
[0061]
Under a nitrogen atmosphere, water (171 L), methanol (171 L), 4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (17.1 kg, 116 mol), concentrated After adding hydrochloric acid (15.8 kg, 152 mol) and 5% palladium carbon (55% wet) (3.82 kg), the inside of the reaction vessel was replaced with hydrogen. Then, the mixture was pressurized with hydrogen and stirred overnight at 30°C. After the reaction, the reaction vessel was purged with nitrogen, the palladium on carbon was removed by filtration, and the palladium on carbon was washed with a 70% aqueous solution of 2-propanol (51 L). Activated carbon (0.86 kg) was added to the filtrate and stirred for 30 minutes. Activated carbon was removed by filtration and washed with 70% aqueous 2-propanol solution (51 L). The filtrate was concentrated under reduced pressure until the liquid volume became 103 L, and 2-propanol (171 L) was added. The mixture was again concentrated under reduced pressure until the liquid volume reached 103 L, then 2-propanol (171 L) was added, and the mixture was stirred for 1 hour or longer. Precipitation of a solid was confirmed, and the solution was concentrated to a volume of 103 L. Further, 2-propanol (51 L) was added, and after concentration under reduced pressure until the liquid volume reached 103 L, the mixture was stirred at 50° C. for 30 minutes. After adding acetone (171 L) over 1 hour while keeping the internal temperature at 40° C. or higher, the mixture was stirred at 40 to 45° C. for 30 minutes. The solution was cooled to 25°C and stirred for 2 hours or longer, and the precipitated solid was collected by filtration, washed with acetone (86 L) and dried under reduced pressure at 40°C to give 19.7 kg of the title compound (yield 90.4%). ).
1 H NMR (500 MHz, methanol-d 4 ): δ 2.27 (s, 3H), 2.30 (s, 3H), 4.02 (s, 2H), 6.16 (s, 1H)
[0062]
(Example 1-1) Production of methyl 5-chloro-3,4-dihydroxy-2-methylbenzoate
[0063]
[Chemical 12]
[0064]
Under a nitrogen atmosphere, water (420 L), toluene (420 L), acetonitrile (420 L), and methyl 3,4-dihydroxy-2-methylbenzoate (1) (60 kg, 329 mol) were added to the reactor and cooled. After that, sulfuryl chloride (133.4 kg, 988 mol) was added dropwise while maintaining the temperature at 20°C or lower. After the reaction, the mixture was separated into an organic layer 1 and an aqueous layer, acetonitrile (60 L) and toluene (120 L) were added to the aqueous layer, and the mixture was stirred. Water (420 L) and acetonitrile (210 L) were added to the organic layer 1, and after cooling, sulfuryl chloride (88.9 kg, 659 mol) was added dropwise at 20°C or lower, and sulfuryl chloride (53.2 kg, 394 mol) was added. ) was added in portions. After the reaction, the mixture was separated into an organic layer 3 and an aqueous layer, and the organic layer 2 was added to the aqueous layer and stirred. Water (420 L), acetonitrile (210 L) were added to the combined organic layer, sulfuryl chloride (44.5 kg, 329 mol) was added dropwise below 20°C, and sulfuryl chloride (106.4 kg, 788 mol) was added. ) was added in portions. After the reaction, the organic layer 4 and the aqueous layer were separated, acetonitrile (60 L) and toluene (120 L) were added to the aqueous layer, and the mixture was stirred. The combined organic layers were washed three times with 20 wt % aqueous sodium chloride solution (300 L) and then concentrated under reduced pressure to 600 L. After repeating the operation of adding toluene (300 L) and concentrating under reduced pressure to 600 L again twice, the mixture was heated and stirred at 60° C. for 1 hour. After cooling to room temperature, the precipitated solid was collected by filtration, washed with toluene (120 L), and dried under reduced pressure at 40°C to give 52.1 kg of the crude title compound (2) (yield: 73.0%). ).
[0065]
Under a nitrogen atmosphere, toluene (782 L) and crude title compound (52.1 kg, 241 mol) were added to a reactor and heated to 80°C. After confirming that the crystals were completely dissolved, they were filtered and washed with heated toluene (261 L). The mixture was cooled to 60° C. and stirred for 0.5 hours after crystallization. After cooling to 10°C, the precipitated solid was collected by filtration, washed with toluene (156 L), and dried under reduced pressure at 40°C to give 47.9 kg of the title compound (2) (yield 91.9%). Acquired.
1 H NMR (500 MHz, methanol-d 4 ): δ 2.41 (s, 3H), 3.82 (s, 3H), 7.41 (s, 1H)
[0066]
(Example 1-2) Examination of chlorination conditions 1 Since
it is difficult to remove compound (1), which is the starting material, and compound (4), which is a by-product of the reaction, even in subsequent steps, need to control. Therefore, chlorination was investigated in the same manner as in Example 1-1 using compound (1) as a starting material. Table 1 shows the results.
[0067]
[Chemical 13]
[0068]
[Table 1]
[0069]
HPLC condition
detection: 220 nm
column: ACQUITY UPLC BEH C18 (2.1 mm ID x 50 mm, 1.7 μm, Waters)
column temperature: 40 ° C
mobile phase: A: 0.1 vol% trifluoroacetic acid aqueous solution, B: acetonitrile
Gradient conditions:
[0070]
[Table 2]
[0071]
Flow rate: 1.0 mL/min
Injection volume: 1 μL
Sample solution: acetonitrile/water (1:1)
wash solution: acetonitrile/water (1:1)
purge solution: acetonitrile/water (1:1)
seal wash solution : Acetonitrile/water (1:1)
Sample cooler temperature: None
Measurement time: 5 minutes
Area measurement time: about 0.5 minutes – 4.0 minutes
Comp. 1: 1.11 min, Comp. 2: 1.55 min,
Comp. 3: 1.44 min, Comp. 4: 1.70 min
[0072]
(Example 1-3) Examination of chlorination conditions 2
Compound (1) was used as a starting material, sulfuryl chloride was used as a chlorination reagent, and chlorination in various solvents was examined. Table 3 shows the results.
[0073]
[table 3]
[0074]
(Example 2) Methyl (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5- Manufacture of carboxylates
[0075]
[Chemical 14]
[0076]
Toluene (9.0 L), tert-butyl = (trans-4-ethynylcyclohexyl) carbamate (2.23 kg, 9.99 mol), methyl = 5-chloro-3,4- were added to a reaction vessel under a nitrogen atmosphere. Dihydroxy-2-methylbenzoate (1.80 kg, 8.31 mol), tri(o-tolyl)phosphine (76.0 g, 250 mmol), triruthenium dodecacarbonyl (53.0 g, 82.9 mmol) ) was added, and the mixture was heated and stirred at 80 to 90° C. for 7 hours under an oxygen-containing nitrogen stream. The reaction solution was cooled to room temperature to obtain a toluene solution of the title compound.
[0077]
(Example 3) (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carvone acid production
[0078]
[Chemical 15]
[0079]
Methyl = (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole obtained in Example 2 -5-carboxylate toluene solution (13 L, equivalent to 7.83 mol), methanol (9.0 L), 1,2-dimethoxyethane (3.6 L), 5 mol / L sodium hydroxide aqueous solution ( 2.50 L, 12.5 mol) was added and stirred at 55-65° C. for 3 hours. After adding water (5.4 L), the mixture was allowed to stand and separated into an organic layer and an aqueous layer. After cooling to room temperature, 1,2-dimethoxyethane (16.2 L) was added to the aqueous layer, and after adjusting the pH to 4.0 to 4.5 with 3 mol/L hydrochloric acid, toluene (5.4 L) was added. added. After heating to 50-60° C., the organic layer and aqueous layer were separated, and the organic layer was washed with a 20 wt % sodium chloride aqueous solution (7.2 L). Then, 1,2-dimethoxyethane (21.6 L) was added to the organic layer, and after concentration under reduced pressure to 9 L, 1,2-dimethoxyethane (21.6 L) was added and heated to 50-60°C. After that, filtration was performed to remove inorganic substances. Then, after washing with 1,2-dimethoxyethane (1.8 L), the 1,2-dimethoxyethane solution of the title compound (quantitative value 89.6% (Example 2 total yield from ), corresponding to 7.45 mol).
[0080]
(Example 4) (1S)-1-phenylethanaminium (2R)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1, Preparation of 3-benzodioxole-5-carboxylate
[0081]
[Chemical 16]
[0082]
(2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5 obtained in Example 3 – A solution of carboxylic acid in dimethoxyethane (21.6 L, corresponding to 7.45 mol) was heated to 75-80°C, and then (1S)-1-phenylethanamine (1.02 kg, 8.42 mmol). was added and stirred for 4 hours. A mixture of 1,2-dimethoxyethane (9.2 L) and water (3.4 L) heated to 50-60° C. was added, stirred, and then cooled to room temperature. The precipitated solid was collected by filtration and washed with 1,2-dimethoxyethane (9 L) to give a crude title compound (1.75 kg (converted to dry matter), yield 38.5% (Example 2 total yield from ) and an optical purity of 93.8% ee).
[0083]
Under a nitrogen atmosphere, a 1,2-dimethoxyethane aqueous solution (13.6 L) was placed in a reaction vessel, and (1S)-1-phenylethanaminium obtained in step 1 (2R)-2-{trans-4-[(tert -Butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate crude (1.70 kg equivalent, 3.11 mol) was added. After that, 5 mol/L hydrochloric acid (0.56 L, 2.8 mol) was added dropwise. After stirring at room temperature for 10 minutes or longer, the mixture was heated to 75° C. or higher, and (1S)-1-phenylethanamine (360 g, 2.97 mmol) was dissolved in 1,2-dimethoxyethane (2.6 L). The solution was added dropwise over 1 hour. It was then washed with 1,2-dimethoxyethane (0.9 L), stirred for 2 hours and cooled to 0-5°C. The slurry was collected by filtration and washed with 1,2-dimethoxyethane (5.1 L) cooled to 0-5° C. to give the title compound (1.56 kg, yield 91.9%, obtained with an optical purity of 99.5% ee).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.15-1.23(m,2H), 1.28-1.35(m,2H), 1.42(s,9H),
1.59(s,3H), 1.60-1.61(d ,3H,J=7.0Hz,3H),1.80-1.86(dt,J=12.0,3.0Hz,1H),1.95-1.96(m,4H),2.27(s,3H),3.24-3.28(m,1H ),4.39-4.43(q,J=7.0Hz,1H),7.07(s,1H),7.37-7.45(m,5H)
[0084]
(Example 5) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride Manufacturing A
[0085]
[Chemical 17]
[0086]
(Step 1)
Under a nitrogen atmosphere, 1,2-dimethoxyethane (200 L) and (1S)-1-phenylethanaminium (2R)-2-{trans-4-[(tert-butoxycarbonyl) were placed in a reaction vessel. Amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate (equivalent to 87.64 kg, 160 mol), 35% hydrochloric acid (16.7 kg, 160 mol) was added and heated to 45-55° C., 35% hydrochloric acid (36.7 kg, 352 mol) was added dropwise in 7 portions and stirred for 3 hours after dropping. After cooling to room temperature, the reaction solution was added to a mixture of water (982 L) and 5 mol/L sodium hydroxide (166.34 kg, 702 mol). 3 mol/L hydrochloric acid (22.4 kg) was added dropwise to the resulting solution at 30°C, crystal precipitation was confirmed, and the mixture was stirred for 30 minutes or more, cooled to 10°C, and further stirred for 2 hours. After stirring, 3 mol/L hydrochloric acid (95.1 kg) was added dropwise at 10°C to adjust the pH to 7.0. The slurry liquid was collected by filtration, washed with water (293 L) cooled to 10° C., and (2R)-2-(trans-4-aminocyclohexyl)-7-chloro-2,4-dimethyl-1,3- Benzodioxol-5-carboxylic acid trihydrate was obtained (57.63 kg (converted to dry matter), yield 94.7%).
1 H NMR (500 MHz, methanol- d4 + D2O): 1.32-1.44 ( m, 4H), 1.61 (s, 3H), 1.89-1.94 (m, 1H), 2.01-2.13 (m, 4H) ,2.27(s,3H),2.99-3.07(m,1H),7.06(s,3H)
[0087]
(Step 2)
Under nitrogen atmosphere, 1,2-dimethoxyethane (115 L), (2R)-2-(trans-4-aminocyclohexyl)-7-chloro-2,4-dimethyl-1,3 -benzodioxole-5-carboxylic acid trihydrate (57.63 kg equivalent, 152 mmol), formic acid (34.92 kg, 759 mol), 37% formaldehyde aqueous solution (93.59 kg, 1153 mol) was added and stirred at 55-65°C for 2 hours. Cool to room temperature, add 2-propanol (864 L) and concentrate to 576 L under reduced pressure. 2-Propanol (231 L) was added thereto and concentrated under reduced pressure to 576 L. Further, 2-propanol (231 L) was added and concentrated under reduced pressure to 576 L. After concentration, 35% hydrochloric acid (20.40 kg, 196 mol) was added dropwise over 2 hours and stirred at room temperature for 30 minutes. Ethyl acetate (576 L) was added to the resulting slurry over 30 minutes and concentrated to 692 L. Ethyl acetate (461 L) was added followed by further concentration to 519 L. Ethyl acetate (634 L) was added to the residue and the mixture was stirred at room temperature for 2 hours. The precipitated solid was collected by filtration, washed with ethyl acetate (491 L) and dried under reduced pressure at 40°C to give the title compound (51. 56 kg, 87.1% yield).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.38-1.47 (m, 2H), 1.53-1.61 (m, 2H), 1.67 (s, 3H), 1.99-2.05 (m, 1H), 2.13 -2.18(m,4H),2.38(s,3H),2.84(s,6H),3.19-3.25(dt,J=12.5,3.5Hz,1H),
7.53(s,1H)
[0088]
(Example 6) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride Manufacturing B
[0089]
[Chemical 18]
[0090]
Under a nitrogen atmosphere, formic acid (20 mL), 37% formaldehyde aqueous solution (15 mL), dimethoxyethane (10 mL), (1S)-1-phenylethanaminium (2R)-2-{trans-4- [(tert-Butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate (10 g, 18.3 mmol) was added and Stirred for 10 hours. After cooling to room temperature and filtering the insolubles, 2-propanol (100 mL) was added and the mixture was concentrated under reduced pressure until the liquid volume became 30 mL. While stirring at room temperature, ethyl acetate (120 mL) and concentrated hydrochloric acid (6.1 mL) were added to form a slurry. This was concentrated under reduced pressure to 30 mL, ethyl acetate (120 mL) was added, and then concentrated under reduced pressure to 30 mL again. After adding ethyl acetate (120 mL), the precipitated solid was collected by filtration, washed with ethyl acetate (50 mL) and dried under reduced pressure at 40°C to give 6.56 g of the title compound (yield 92.0%). Acquired.
[0091]
(Example 7) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl ) Preparation of methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide p-toluenesulfonate
[0092]
[Chemical 19]
[0093]
Under nitrogen atmosphere, acetone (6.5 L), purified water (1.3 L), (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4- Dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride (650.4 g, 1.67 mol), 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one Monohydrochloride (330.1 g, 1.75 mol) and triethylamine (337 g, 3.33 mol) were added and stirred at room temperature for 30 minutes. After that, 1-hydroxybenzotriazole monohydrate (255 g, 1.67 mol), 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (383 g, 2.00 mmol) were added, and the mixture was stirred overnight at room temperature. Stirred. After adjusting the pH to 11 with 5 mol/L sodium hydroxide, toluene (9.8 L) was added, and after stirring, the mixture was separated into an organic layer 1 and an aqueous layer. Toluene (3.3 L) was added to the aqueous layer, and after stirring, the aqueous layer was discarded, and the obtained organic layer was combined with the previous organic layer 1. The combined organic layers were concentrated under reduced pressure to 9.75 L, toluene (6.5 L) was added and washed twice with purified water (3.25 L). The resulting organic layer was concentrated under reduced pressure to 4.875 L and 2-propanol (1.625 L) was added. A solution of p-toluenesulfonic acid monohydrate (0.12 kg, 0.631 mol) dissolved in 4-methyl-2-pentanone (1.14 L) was added to the organic layer heated to 68°C. The mixture was added dropwise over 5 hours and stirred at 68°C for 30 minutes. Furthermore, a solution of p-toluenesulfonic acid monohydrate (0.215 kg, 1.13 mol) dissolved in 4-methyl-2-pentanone (2.11 L) was added dropwise over 3.5 hours, Stirred at 68° C. for 30 minutes. After that, 4-methyl-2-pentanone (6.5 L) was added dropwise over 1 hour. After cooling to room temperature, the precipitated solid was collected by filtration, washed with 4-methyl-2-pentanone (3.25 L) and dried under reduced pressure at 40°C to give 1.035 kg of the crude title compound (yield 94%). .2%).
[0094]
Under a nitrogen atmosphere, 2-propanol (6.65 L) and the obtained crude title compound (950 g) were added to the reactor and stirred. Purified water (0.23 L) was added to completely dissolve the solid at 68° C., filtered, and washed with warm 2-propanol (0.95 L). After confirming that the solid was completely dissolved at an internal temperature of 68°C, the solution was cooled to 50°C. After cooling, seed crystals* (9.5 g, 0.01 wt) were added and stirred at 50° C. overnight. tert-Butyl methyl ether (11.4 L) was added dropwise thereto in 4 portions over 30 minutes each. At that time, it was stirred for 30 minutes after each addition. After cooling to room temperature, the precipitated solid was collected by filtration, washed with a mixture of 2-propanol (0.38 L) and tert-butyl methyl ether (3.42 L), and further treated with tert-butyl methyl ether (4.75 L). ) and dried under reduced pressure at 40° C. to obtain the title compound (915.6 g, yield 96.4%).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.35-1.43 (m, 2H), 1.49-1.57 (m, 2H), 1.62 (s, 3H),
1.94-2.00 (dt, J = 12.5, 3.0Hz ,1H),2.09-2.13(m,4H),2.17(s,3H),2.24(s,3H),2.35(s,3H),2.36(s,3H),2.82(s,6H),3.16- 3.22(dt,J=12.0,3.5Hz,1H),4.42(s,2H),
6.10(s,1H),6.89(s,1H),7.22-7.24(d,J=8.0Hz,2H),7.69 -7.71(dt,J=8.0,1.5 Hz,2H)
*Seed crystal preparation method
Under a nitrogen atmosphere, 2-propanol (79.0 L) and the obtained crude title compound (7.90 kg) were added to a reactor and stirred. Purified water (7.9 L) was added to completely dissolve the solid, and activated carbon (0.40 kg) was added and stirred. After filtering the activated carbon, it was washed with 2-propanol (79.0 L) and concentrated to 58 L. 2-Propanol (5 L) was added to the residue, and after heating to 64° C., tert-butyl methyl ether (19.8 L) was added, and after crystal precipitation was confirmed, tert-butyl methyl ether (75. 1 L) was added in three portions. At that time, it was stirred for 30 minutes after each addition. After cooling to room temperature, the precipitated solid was collected by filtration, washed with a mixture of 2-propanol (7.9 L) and tert-butyl methyl ether (15.8 L), and dried under reduced pressure at 40°C to obtain seed crystals. The title compound was obtained (7.08 kg, 89.6% yield).
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
///////Valemetostat tosilate, japan 2022, approvals 2022, Ezharmia, バレメトスタットトシル酸塩 , DS-3201, DS 3201, DS3201, DS-3201b, Orphan Drug
CN(C)[C@@H]1CC[C@H](CC1)[C@]2(C)Oc3c(C)c(cc(Cl)c3O2)C(=O)NCC4=C(C)C=C(C)NC4=O

NEW DRUG APPROVALS
ONE TIME
$10.00
Danavorexton, TAK 925


Danavorexton, TAK 925
2114324-48-8
- Molecular FormulaC21H32N2O5S
- Average mass424.554 Da
1-Piperidinecarboxylic acid, 3-[(methylsulfonyl)amino]-2-[[(cis-4-phenylcyclohexyl)oxy]methyl]-, methyl ester, (2R,3S)-
Methyl (2R,3S)-3-[(methylsulfonyl)amino]-2-[[(cis-4-phenylcyclohexyl)oxy]methyl]-1-piperidinecarboxylate
- OriginatorTakeda
- ClassCyclohexanes; Esters; Ethers; Piperidines; Sleep disorder therapies; Small molecules; Sulfonamides
- Mechanism of ActionOrexin receptor type 2 agonists
- Orphan Drug StatusYes – Narcolepsy
- Phase IHypersomnia; Narcolepsy; Respiration disorders; Sleep apnoea syndrome
- 01 Jun 2022Takeda Pharmaceuticals completes a phase I clinical trials in Respiratory disorder (In adults) in Netherlands (IV) (ISRCTN63027076)
- 02 Apr 2022Efficacy and safety data from phase a Ib trial in Hypersomnia presented at the 74th Annual Meeting of the American Academy of Neurology 2022 (AAN-2022)
- 10 Mar 2022Phase-I clinical trials in Sleep apnoea syndrome in Australia (IV) (NCT05180890)
Danavorexton (developmental code name TAK-925) is a selective orexin 2 receptor agonist.[1] It is a small-molecule compound and is administered intravenously.[1][2] The compound was found to dose-dependently produce wakefulness to a similar degree as modafinil in a phase 1 clinical trial.[1][3] As of March 2021, danavorexton is under development for the treatment of narcolepsy, idiopathic hypersomnia, and sleep apnea.[2][1][4] It is related to another orexin receptor agonist known as TAK-994, the development of which was discontinued for safety reasons in October 2021.[1][5]
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00626
TAK-925, a potent, selective, and brain-penetrant orexin 2 receptor (OX2R) agonist, [methyl (2R,3S)-3-((methylsulfonyl)amino)-2-(((cis-4-phenylcyclohexyl)oxy)methyl)piperidine-1-carboxylate, 16], was identified through the optimization of compound 2, which was discovered by a high throughput screening (HTS) campaign. Subcutaneous administration of compound 16 produced wake-promoting effects in mice during the sleep phase. Compound 16 (TAK-925) is being developed for the treatment of narcolepsy and other related disorders.


aReagents and conditions: (a) chiral column separation; (b) RCOCl, Et3N, THF, rt (for 15 and 16); (c) ethyl chlorocarbonate, DIEA, THF, rt (for 17); (d) isocyanatoethane, Et3N, THF, 0 °C−rt (for 18).
Methyl (2R,3S)-3-((methylsulfonyl)amino)-2-(((cis-4- phenylcyclohexyl)oxy)methyl)piperidine-1-carboxylate (16) To a mixture of 14 (58 mg, 0.16 mmol) and Et3N (0.044 mL, 0.32 mmol) in THF (3 mL) was added methyl chlorocarbonate (0.024 mL, 0.32 mmol) at rt. The mixture was stirred at rt overnight. The mixture was quenched with water and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane/EtOAc, 1:1 to 0:100) to give 16 (64 mg, 0.15 mmol, 95%) as a colorless oil. Crystallization of 16 (1.8 g, 4.1 mmol) from EtOH-H2O gave 16 (1.7 g, 3.9 mmol, 95%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 1.40−1.55 (5H, m), 1.56−1.73 (5H, m), 1.87 (1H, brd, J = 13.2 Hz), 1.96 (1H, brd, J = 13.6 Hz), 2.44−2.57 (1H, m), 2.83 (1H, brs), 2.95 (3H, s), 3.40 (1H, brs), 3.53−3.62 (5H, m), 3.73 (1H, brt, J = 9.7 Hz), 3.84 (1H, brs), 4.47 (1H, brs), 7.15 (1H, brt, J = 7.2 Hz), 7.18 (1H, brs), 7.19 (2H, brd, J = 8.1 Hz), 7.27 (2H, brt, J = 7.4 Hz). 13C NMR (151 MHz, DMSO-d6, the minor rotamer’s signals are marked with an asterisk) δ24.05, 24.39*, 26.00, 26.17*, 27.60*, 27.79, 28.68, 30.15*, 37.54, 38.13*, 39.91, 42.99, 51.01, 52.07, 53.90*, 54.49, 61.48, 61.89*, 71.68, 125.68, 126.51, 128.14, 147.34, 155.27*, 156.08. MS (ESI/APCI) mass calculated for [M + H]+ (C21H33N2O5S) requires m/z 424.6, found m/z 425.2. mp 113 °C. Anal. Calcd for C21H32N2O5S: C, 59.41; H, 7.60; N, 6.60. Found: C, 59.45; H, 7.59; N, 6.55. [α] 20 D +16.3 (c 0.1, CHCl3
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Other names | TAK-925 |
| Routes of administration | Intravenous[1][2] |
| Drug class | Orexin receptor agonist |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2114324-48-8 |
| PubChem CID | 130310079 |
| ChemSpider | 68011464 |
| UNII | 1QMD83K4YN |
| ChEMBL | ChEMBL4650341 |
| Chemical and physical data | |
| Formula | C21H32N2O5S |
| Molar mass | 424.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
References
- ^ Jump up to:a b c d e f Jacobson LH, Hoyer D, de Lecea L (January 2022). “Hypocretins (orexins): The ultimate translational neuropeptides”. J Intern Med. doi:10.1111/joim.13406. PMID 35043499.
- ^ Jump up to:a b c “Danavorexton – Takeda”. Adis Insight. Springer Nature Switzerland AG. Retrieved 7 March 2021.
- ^ Evans, R., Hazel, J., Faessel, H., Wu, J., Hang, Y., Alexander, R., … & Hartman, D. (2019). Results of a phase 1, 4-period crossover, placebo-controlled, randomized, single dose study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of TAK-925, a novel orexin 2 receptor agonist, in sleep-deprived healthy adults, utilizing modafinil as an active comparator. Sleep Medicine, 64, S106. https://scholar.google.com/scholar?cluster=10933819770107034612
- ^ Evans R, Tanaka S, Tanaka S, Touno S, Shimizu K, Sakui S, et al. (December 2019). “A Phase 1 single ascending dose study of a novel orexin 2 receptor agonist, TAK-925, in healthy volunteers (HV) and subjects with narcolepsy type 1 (NT1) to assess safety, tolerability, pharmacokinetics, and pharmacodynamic outcomes”. Sleep Medicine. 64: S105–S106. doi:10.1016/j.sleep.2019.11.290.
- ^ Tong A (6 October 2021). “Takeda flashes red light on ‘breakthrough’ narcolepsy drug after PhII trials turned up mysterious safety signal”. Endpoints News.
External links
///////////////Danavorexton, TAK 925, ORPHAN DRUG, PHASE 1

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}