







| Novel Tricyclic Compounds [US2011311474] | 2011-12-22 |
Home » 0rphan drug status (Page 15)
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Reslizumab


Reslizumab
(Cinqair®) Approved Active, FDA 2016-03-23
An interleukin-5 (IL-5) antagonist used to treat severe asthma.
CAS 241473-69-8
![]()
Research Code CDP-835; CEP-38072; CTx-55700; SCH-5570; SCH-55700; TRFK-5,

Anti-interleukin-5 monoclonal antibody – Celltech/Schering-Plough
Reslizumab was approved by the U.S. Food and Drug Administration (FDA) on March 23, 2016. It was developed and marketed as Cinqair® by Teva.
Reslizumab is an interleukin-5 antagonist, which binds to human IL-5 and prevents it from binding to the IL-5 receptor, thereby reducing eosinophilic inflammation. It is indicated for the maintenance treatment of patients with severe asthma in patients aged 18 years and older.
Cinqair® is available as injection for intravenous infusion, containing 100 mg of reslizumab in 10 mL solution in single-use vials. The recommended dose is 3 mg/kg once every four weeks.
- Originator Celltech R&D; Schering-Plough
- Developer Celltech R&D; Teva Pharmaceutical Industries
- Class Antiasthmatics; Monoclonal antibodies
- Mechanism of Action Interleukin 5 receptor antagonists
- Orphan Drug Status Yes – Oesophagitis
- 23 Mar 2016 Registered for Asthma in USA (IV) – First global approval
- 04 Mar 2016 Pooled efficacy data from two phase III trials in Asthma presented at the 2016 Annual Meeting of the American Academy of Allergy, Asthma and Immunology (AAAAI-2016)
- 10 Dec 2015 Preregistration for Asthma in Canada (IV)
Reslizumab (trade name Cinqair) is a humanized monoclonal antibody intended for the treatment of eosinophil-meditated inflammations of the airways, skin and gastrointestinal tract.[1] The FDA approved reslizumab for use with other asthma medicines for the maintenance treatment of severe asthma in patients aged 18 years and older on March 23, 2016. Cinqair is approved for patients who have a history of severe asthma attacks (exacerbations) despite receiving their current asthma medicines.[2]

Teva Announces FDA Acceptance of the Biologics License Application for Reslizumab
Investigational Biologic for the Treatment of Inadequately Controlled Asthma in Patients with Elevated Blood Eosinophils Accepted for Review
JERUSALEM–(BUSINESS WIRE)–Jun. 15, 2015– Teva Pharmaceutical Industries Ltd., (NYSE: TEVA) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the Biologics License Application (BLA) for reslizumab, the company’s investigational humanized monoclonal antibody (mAb) which targets interleukin-5 (IL-5), for the treatment of inadequately controlled asthma in adult and adolescent patients with elevated blood eosinophils, despite an inhaled corticosteroid (ICS)-based regimen.
“Despite currently available medicines, uncontrolled asthma remains a serious problem for patients, physicians and healthcare systems, highlighting the need for targeted new treatment options,” said Dr. Michael Hayden, President of Global R&D and Chief Scientific Officer at Teva Pharmaceutical Industries Ltd. “The reslizumab BLA filing acceptance represents a significant milestone for Teva as we work toward serving a specific asthma patient population that is defined by elevated blood eosinophil levels and inadequately controlled symptoms despite standard of care therapy. In clinical trials, patients treated with reslizumab showed significant reductions in the rate of asthma exacerbations and significant improvement in lung function. If approved, we believe reslizumab will serve as an important new targeted treatment option to achieve better asthma control for patients with eosinophil-mediated disease.”
The BLA for reslizumab includes data from Teva’s Phase III BREATH clinical trial program. The program consisted of four separate placebo-controlled Phase III trials involving more than 1,700 adult and adolescent asthma patients with elevated blood eosinophils, whose symptoms were inadequately controlled with inhaled corticosteroid-based therapies. Results from these studies demonstrated that reslizumab, in comparison to placebo, reduced asthma exacerbation rates by at least half and provided significant improvement in lung function and other secondary measures of asthma control when added to an existing ICS-based therapy. Common adverse events in the reslizumab treatment group were comparable to placebo and included worsening of asthma, nasopharyngitis, upper respiratory infections, sinusitis, influenza and headache. Two anaphylactic reactions were reported and resolved following medical treatment at the study site.
Results from the reslizumab BREATH program were recently presented at the American Thoracic Society 2015 Annual Meeting and the American Academy of Allergy, Asthma and Immunology 2015 Annual Meeting, in addition to being published in The Lancet Respiratory Medicine. The BLA for reslizumab has been accepted for filing by the FDA for standard review, with FDA Regulatory Action expected in March 2016.
About Reslizumab
Reslizumab is an investigational humanized monoclonal antibody which targets interleukin-5 (IL-5). IL-5 is a key cytokine involved in the maturation, recruitment, and activation of eosinophils, which are inflammatory white blood cells implicated in a number of diseases, such as asthma. Elevated levels of blood eosinophils are a risk factor for future asthma exacerbations. Reslizumab binds circulating IL-5 thereby preventing IL-5 from binding to its receptor.
About Asthma
Asthma is a chronic (long term) disease usually characterized by airway inflammation and narrowing of the airways, which can vary over time. Asthma may cause recurring periods of wheezing (a whistling sound when you breathe), chest tightness, shortness of breath and coughing that often occurs at night or early in the morning. Without appropriate treatment, asthma symptoms may become more severe and result in an asthma attack, which can lead to hospitalization and even death.
About Eosinophils
Eosinophils are a type of white blood cell that are present at elevated levels in the lungs and blood of many asthmatics. Evidence shows that eosinophils play an active role in the pathogenesis of the disease. IL-5 has been shown to play a crucial role in maturation, growth and activation of eosinophils. Increased levels of eosinophils in the sputum and blood have been shown to correlate with severity and frequency of asthma exacerbations.
About Teva
Teva Pharmaceutical Industries Ltd. (NYSE and TASE: TEVA) is a leading global pharmaceutical company that delivers high-quality, patient-centric healthcare solutions to millions of patients every day. Headquartered in Israel, Teva is the world’s largest generic medicines producer, leveraging its portfolio of more than 1,000 molecules to produce a wide range of generic products in nearly every therapeutic area. In specialty medicines, Teva has a world-leading position in innovative treatments for disorders of the central nervous system, including pain, as well as a strong portfolio of respiratory products. Teva integrates its generics and specialty capabilities in its global research and development division to create new ways of addressing unmet patient needs by combining drug development capabilities with devices, services and technologies. Teva’s net revenues in 2014 amounted to $20.3 billion. For more information, visit www.tevapharm.com.
The U.S. Food and Drug Administration today approved Cinqair (reslizumab) for use with other asthma medicines for the maintenance treatment of severe asthma in patients aged 18 years and older. Cinqair is approved for patients who have a history of severe asthma attacks (exacerbations) despite receiving their current asthma medicines.
Asthma is a chronic disease that causes inflammation in the airways of the lungs. During an asthma attack, airways become narrow making it hard to breathe. Severe asthma attacks can lead to asthma-related hospitalizations because these attacks can be serious and even life-threatening. According to the Centers for Disease Control and Prevention, as of 2013, more than 22 million people in the U.S. have asthma, and there are more than 400,000 asthma-related hospitalizations each year.
“Health care providers and their patients with severe asthma now have another treatment option to consider when the disease is not well controlled by their current asthma therapies,” said Badrul Chowdhury, M.D., Ph.D., director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Cinqair is administered once every four weeks via intravenous infusion by a health care professional in a clinical setting prepared to manage anaphylaxis. Cinqair is a humanized interleukin-5 antagonist monoclonal antibody produced by recombinant DNA technology in murine myeloma non-secreting 0 (NS0) cells. Cinqair reduces severe asthma attacks by reducing the levels of blood eosinophils, a type of white blood cell that contributes to the development of asthma.
The safety and efficacy of Cinqair were established in four double-blind, randomized, placebo‑controlled trials in patients with severe asthma on currently available therapies. Cinqair or a placebo was administered to patients every four weeks as an add-on asthma treatment. Compared with placebo, patients with severe asthma receiving Cinqair had fewer asthma attacks, and a longer time to the first attack. In addition, treatment with Cinqair resulted in a significant improvement in lung function, as measured by the volume of air exhaled by patients in one second.
Cinqair can cause serious side effects including allergic (hypersensitivity) reactions. These reactions can be life-threatening. The most common side effects in clinical trials for Cinqair included anaphylaxis, cancer, and muscle pain.
Cinqair is made by Teva Pharmaceuticals in Frazer, Pennsylvania.

References
- 1Walsh, GM (2009). “Reslizumab, a humanized anti-IL-5 mAb for the treatment of eosinophil-mediated inflammatory conditions”. Current opinion in molecular therapeutics 11 (3): 329–36. PMID 19479666.
- 2http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491980.htm
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm491980.htm
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from rat) |
| Target | IL-5 |
| Clinical data | |
| Trade names | Cinquil |
| Identifiers | |
| ATC code | R03DX08 (WHO) |
| ChemSpider | none |
/////////CDP-835, CEP-38072, CTx-55700, SCH-5570, SCH-55700, TRFK-5, Reslizumab, Cinqair®, teva, interleukin-5 (IL-5) antagonist, severe asthma, FDA 2016, Orphan Drug StatuS
AVORALSTAT
Avoralstat, BCX4161,
CAS 918407-35-9
UNII: UX17773O15
513.5513, C28-H27-N5-O5
2-Pyridinecarboxylic acid, 3-(2-(((4-(aminoiminomethyl)phenyl)amino)carbonyl)-4-ethenyl-5-methoxyphenyl)-6-(((cyclopropylmethyl)amino)carbonyl)-
3-(2-((4-Carbamimidoylphenyl)carbamoyl)-4-ethenyl-5-methoxyphenyl)-6-((cyclopropylmethyl)carbamoyl)pyridine-2-carboxylic acid
Hereditary angioedema (HAE)
Kallikrein inhibitor
BioCryst Pharmaceuticals
![]()
BioCryst is also investigating second-generation plasma kallikrein inhibitors to avoralstat, for treating HAE (in February 2016, this program was listed as being in preclinical development).
Prevent acute attacks in patients with hereditary angioedema (HAE); Treat hereditary angioedema (HAE)
U.S. – Fast Track (Treat hereditary angioedema (HAE));
U.S. – Orphan Drug (Prevent acute attacks in patients with hereditary angioedema (HAE))
26 Feb 2016Clinical trials in Hereditary angioedema (Prevention) in USA (PO, Hard-gelatin capsule) before February 2016
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in France (PO, Soft-gelatin capsule)
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in Germany (PO, Soft-gelatin capsule)

| Conditions | Interventions | Phases | Recruitment | Sponsor/Collaborators |
|---|---|---|---|---|
| Hereditary Angioedema|HAE | Drug: BCX4161|Drug: Placebo | Phase 2|Phase 3 | Recruiting | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161|Drug: Placebo | Phase 2 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
Avoralstat, also known as BCX-4161, is a potent and orally active Kallikrein inhibitor and Bradykinin inhibitor. Avoralstat may be potentially useful for treatment for Hereditary angioedema. Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.

Selective inhibitor of plasma kallikrein that subsequently suppresses bradykinin production
Hereditary angioedema (HAE) is a serious and potentially life-threatening rare genetic illness, caused by mutations in the C1-esterase inhibitor (C1 INH) gene, located on chromosome 11q. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by C1 INH gene mutations which produce low levels of C1 -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective C1 protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; 100(Suppl 2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herin by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that C1 INH is the most important inhibitor of kallikrein in human plasma and that C1 INH deficiency leads to unopposed activation of the kallikrein-bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack.
Approved therapies include purified plasma-derived C1 INH (Cinryze®, Berinert), the recombinant peptide kallikrein inhibitor ecallantide (Kalbitor®), and the bradykinin receptor B2 inhibitor iticabant (Firazyr®). All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
There are many delivery routes for active pharmaceutical ingredients (APIs). Generally, the oral route of administration is favored. Oral administration provides a number of advantages, such as, but not limited to, patient convenience, flexibility of timing of administration, location of administration and non-invasiveness. Oral administration also provides more prolonged drug exposure compared with intermittent intravenous infusion, which may be important for drugs with schedule-dependent efficacy. For example, a drug with a short half-life can achieve a greater exposure time by either continuous infusion or by continuous oral dosing. The use of oral therapy further has the potential to reduce the cost of healthcare resources for inpatient and ambulatory patient care services.
In the pharmaceutical arts, it is known that a number of APIs cannot be administered effectively by the oral route. The main reasons why these compounds cannot be administered by the oral route are: a) rapid enzymatic and metabolic degradation; b) chemical and/or biological instability; c) low solubility in aqueous medium; and/or d) limited permeability in the gastrointestinal tract. For such compounds, non-oral routes of delivery, such as parenteral administration, mainly via intramuscular or subcutaneous injections, may be developed. However, non-oral administration poses a disadvantage for the patient as well as healthcare providers, and for this reason, it is important to develop alternative routes of administration for such compounds, such as oral routes of administration.
While the oral route of administration is the most convenient for the patient and the most economical, designing formulations for administration by the oral route involves many complications. Several methods are available to predict the ease by which an API may be formulated into a formulation suitable for administration by the oral route. Such methods include, but are not limited to, and Lipinski rule (also referred to as the Rule of Five) and the Biopharmaceutical Drug Disposition Classification System (BDDCS).
The BDDCS divides APIs into four classifications, depending on their solubility and permeability. Class I APIs have high solubility and high permeability; Class II APIs have low solubility and high permeability; Class III APIs have high solubility and low permeability; and Class IV APIs have low solubility and low permeability. APIs in higher classes in the BDDCS face greater challenges in formulating into an effective, pharmaceutically acceptable product than those in lower classes. Of the four classes, APIs falling into Class IV are the most difficult to formulate into a formulation for administration by the oral route that is capable of delivering an effective amount of the API as problems of both solubility and permeability must be addressed (note the BDDCS does not inherently address chemical stability). The role of BDDCS in drug development is described generally in L.Z. Benet J Pharm Sci. 2013, 102(1), 34-42.
Lipinski’s rule (described in Lipinski et al. Adv. Drug Deliv. Rev. 46 (1-3): 3-26) states, in general, that in order to develop a successful formulation for administration by the oral route, an API can have no more than one violation of the following criteria:
i) not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms)
ii) not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms) iii) a molecular mass less than 500 daltons
iv) an octanol-water partition coefficient log P not greater than 5.
J. Zhang et al. Medicinal Chemistry, 2006, 2, 545-553, describes a number of small molecule amidine compounds which have activity as inhibitors of kallikrein. The molecules described in this document fall into Class IV of the BDDCS as described above. The compounds are poorly soluble in aqueous and physiological fluids, and are poorly permeable as demonstrated by oral dosing in rats and in vitro experiments with Caco-2 cells.
Furthermore, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, one of the compounds described in Zhang et al., is a Class IV API and violates criteria iii) and iv) as set forth in the Lipinski Rule.
Furthermore, the compounds described in Zhang et al., including 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, exhibit poor stability with respect to oxidation in air, to light
(photodegradation) and in aqueous and physiological fluids, as well as to elevated temperatures.
Therefore, the compounds described by Zhang et al. including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, not only exhibit poor solubility and permeability characteristics, but also poor stability characteristics. As a result, such compounds are predicted to be especially difficult to formulate into an effective, orally deliverable
pharmaceutical composition that is capable of delivering an effective amount of the compound to a subject.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties, such as, but not limited to, melting point, thermal behaviors (e.g. measured by thermogravimetric analysis (TGA), or differential scanning calorimetry (DSC), x-ray diffraction pattern, infrared absorption fingerprint, and solid state NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms and solvates of a pharmaceutical product can provide alternate forms of the compound that display a number of desirable and advantageous properties, such as, but not limited to, ease of handling, ease of processing, ease of formulation, storage stability, and/or ease of purification. Further, new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof may further provide for improved pharmaceutical products, by providing compounds that are more soluble in a set of pharmaceutical excipients. Still further, the provision of new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof enlarges the repertoire of compounds that a formulation scientist has available for formulation optimization, for example by providing a pharmaceutical product with different properties, such as, but not limited to, improved processing characteristics, improved handling characteristics, improved solubility profiles, improved dissolution profile and/or improved shelf-life. Therefore, there is a need for additional polymorphs of pharmaceutically useful compounds, such as, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid and the compounds disclosed herein.
In one aspect, the present invention provides an oral formulation that is capable of delivering an effective amount of the amidine compounds described by Zhang et al. to a subject. In particular, the present invention provides an oral formulation that is capable of delivering an effective amount of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid to a subject. In one specific aspect, the 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid is present in a particular crystal form designated Form A. In light of the art suggesting the difficulties in formulating such an oral formulation, this result was unexpected.
As described herein, the amidine compounds described in Zhang et al., including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (specifically including particular crystal Form A), may now be conveniently used in oral administration and further used in oral administration for the treatment of a number of diseases and conditions in a subject, such as, but not limited to, HAE as described herein.
Avoralstat & next generation kallikrein inhibitors for HAE
Avoralstat
![]()
May 16 is HAE awareness day
See BioCryst’s video regarding HAE to learn more
Avoralstat is being developed as an oral prophylactic treatment for patients suffering from Hereditary Angioedema (HAE). Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
In May 2014 BioCryst, announced that the OPuS-1 (OralProphylaxiS-1) Phase 2a proof of concept clinical trial met its primary efficacy endpoint, several secondary endpoints and all other objectives established for the trial. OpuS-1 enrolled 24 HAE patients with a history of HAE attack frequency of at least 1 per week. Treatment with avoralstat demonstrated a statistically significant mean attack rate reduction of 0.45 attacks per week versus placebo, p<0.001. The mean attack rate per week was 0.82 on BCX4161 treatment, compared to 1.27 on placebo.
In December 2014, BioCryst initiated enrollment in OPuS-2 (Oral ProphylaxiS-2). OPuS-2 is a blinded, randomized, 12-week, three-arm, parallel cohort design trial evaluating the efficacy and safety of two different dose regimens of avoralstat administered three-times daily, 300 mg and 500 mg, compared with placebo. The primary efficacy endpoint for the trial will be the mean angioedema attack rate, which will be reported for each avoralstat dose group compared to placebo. The trial is being conducted in the U.S., Canada and Europe. On October 8, 2015, announced that it has completed enrollment of approximately 100 HAE patients with a history of moderately frequent to very frequent attacks in OPuS-2. BioCryst expects to report the OPuS-2 trial results in early 2016.
PATENT
WO200234711
http://www.google.com/patents/WO2002034711A1?cl=en
PATENT
PATENT
Examples
Example 1 – Synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl- phenyll-6-(cvclopropylmethyl-carbarnoyl)-pyridine-2-carboxylic acid
The synthesis of the above compound and intermediates is described below. In this section, the following abbreviations are used:

The synthesis of starting material, (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) is described in Scheme 1.
f 0HCY ° ΒΓΥΥ°

Preparation of 6-bromobenzofdl[1,3ldioxole-5-carbaldehvde (1b)

1a 1b
To a mixture of piperonal (1a) (498 g, 3.32 mol) in glacial acetic acid (1000 mL) was added a solution of bromine (200 mL, 3.89 mol) in glacial acetic acid (500 mL) over a period of 30 min and stirred at room temperature for 24h. The reaction mixture was poured into water (2000 mL) and the solid that separated was collected by filtration. The solid was dissolved in boiling ethanol (4000 mL) and cooled to room temperature. The solid obtained on cooling was collected by filtration to furnish 6-bromobenzo[d][1 ,3]dioxole-5-carbaldehyde (lb) (365 g, 48 %) as a white solid, MP 126 °C; HNMR (300 MHz, DMSO-d6): δ 10.06 (s, 1 H), 7.42 (s,1 H), 7.29 (s, 1 H), 6.20 (d, J=12.3, 2H); IR (KBr) 3434, 2866, 1673,1489, 1413, 259, 1112, 1031 , 925 cm“1; Analysis calculated for CeH5BrO3.O 25H C, 41.15; H, 2.37; Found: C, 41.07; H, 2.11.
Preparation of 2-bromo-5-hvdroxy-4-methoxybenzaldehyde (1c)

1c
A solution of potassium tert-butoxide (397 g, 3.36 mol) in DMSO (1.5 L) was heated at 50 °C for 30 min. Methanol (1.5 L) was added to it and continued heating at 50 °C for additional 30 min. To the hot reaction mixture was added 6-bromo-benzo[d][1,3]dioxole-5-carbaldehyde (1 b) (350g, 1.53 mol) and continued heating at 50 °C for 30 min. The reaction mixture was cooled to room temperature and quenched with water (2.3 L) and sodium hydroxide (61.2 g, 1.53 mol). The reaction mixture was washed with ether (2 x 1.5 L), acidified to pH 2 using cone. HCI and extracted with ethyl acetate ( 1 L). The ethyl acetate layers were combined and concentrated under vacuum to dryness. The residue obtained was treated with water (1.5 L) and ethyl acetate (1 L). The solid obtained was collected by filtration to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (97 g, 27.5% as a first crop). The layers from the filtrate were separated and aqueous layer was extracted with ethyl acetate (200 ml_). The ethyl acetate layers were combined dried over MgS04 and concentrated under vacuum to dryness to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (192 g, 54.4%, second crop) as an orange solid, MP 108 °C; ‘HNMR (300MHz, DMSO-cfe): S 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H); IR (KBr) 3477, 2967, 2917,
2837, 2767, 2740, 1657, 1595, 1428, 1270, 1210, 1164, 1022 cm“‘; Analysis calculated for C8H7Br03.H20: C, 38.58; H, 3.64: Found: C, 38.60; H, 3.60.
Preparation of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehvde ( d)

To a solution 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (120 g, 520 mmol) in DMF (1000 mL) was added potassium carbonate (79 g, 572 mmol) and benzyl bromide (68 mL, 572 mmol). The reaction mixture was stirred at room temperature overnight and quenched with water (3000 mL). The solid obtained was collected by filtration, washed with ether and dried under vacuum to furnish 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (113.19 g, 67.9%) as a white solid, MP 144 °C;1HNMR (300 MHz, DMSO-c/6): δ 10.06 (s, 1H), 7.47-7.34 (m, 7H), 5.17 (s, 2H), 3.92 (s, 3H); IR (KBr) 2898, 2851 , 1673, 1592, 1502, 1437, 1402, 1264, 1210, 1158, 1017, 754 cm“1; Analysis calculated for C 5H13Br03: C, 56.10; H, 4.08; Found: C, 55.44; H, 4.08.
Preparation of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e)
15 046578
146

1d 1e
To a solution of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (100 g, 311 mmol) in
ethanol (1500 mL) was added triethyl orthoformate (103 mL, 622 mmol), ammonium nitrate
(7.5 g, 93.3 mmol) and stirred at room temperature overnight. The reaction mixture was
treated with ether (1200 mL) and stirred for 15 min before filtration. The filtrate was
concentrated under vacuum to dryness to give 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (134 g) as a brown syrup; The product was used in the next step
without further purification; 1H N R (300 MHz, DMSO-cf6) δ 7.45 – 7.37 (m, 4H), 7.36 – 7.33
(m, 1 H), 7.17 – 7.14 (m, 1 H), 7.10 (s, 1 H), 5.10 (s, 2H), 3.80 (s, 3H), 3.58 – 3.33 (m, 5H),
1.13 – 1.07 (m, 6H); IR (KBr) 2974, 2879, 1601 , 1503, 1377, 1260, 1163, 1060 cm“1;
Analysis calculated for C19H23Br04: C, 57.73; H, 5.86; Found: C, 57.21 ; H, 5.94.
acid (1fi

To a solution of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (120 g,
300 mmol) in dry ether (1000 mL) at -78 °C was added n-butyllithium (1.6 M solution in
hexanes, 244 mL, 390 mmol) over a period of 30 min and further stirred at -78 °C for 30 min.
A solution of tri-n-butylborate (110 mL, 405 mmol) in dry ether (300 mL) was added to this
solution at -78 °C over a period of 30 min. The reaction mixture was further stirred for 2 h at -78 °C and warmed to 0 °C. The reaction mixture was quenched with 3N HCI (300 mL) at 0
°C and heated at reflux for 1 h. After cooling to room temperature, the solid obtained was
collected by filtration washed with water (250 mL) dried in vaccum to afford (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (30.85 gm, 37.6% as a white solid. The organic
layer from above filtrate was extracted with 1.5 N NaOH (3 x 200 mL). The combined basic
extracts were acidified with cone. HCI (pH about 4). The solid obtained was collected by
filtration, washed with water and dried under vacuum to furnish a second crop of (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (22.3 g, 26%) as a light orange solid
MP 158 °C; 1H NMR (300 MHz, DMSO-cfe) δ 10.08 (s, 1 H), 7.52 (s, 1 H), 7.48 – 7.33 (m, 5H),
7.24 (s, 1H), 5.18 (s, 2H), 3.89 (s, 3H); 1H NMR (300 MHz, DMSO-d6/D20) δ 10.06 (s, 1H),
7.52 (s, 1H), 7.49 – 7.32 (m, 5H), 7.23 (s, 1 H), 5.18 (s, 2H), 3.89 (s, 3H); MS (ES+) 309.1 (M+Na); IR (KBr) 3335, 2937, 1647, 1545, 1388, 1348, 1268, 1146, 1095 cm-1; Analysis calculated for C15H15BO5.0.25H2O: C, 62.00; H, 5.38; Found: C, 61.77; H, 5.19.
Synthesis of methyl-6-(cvclopropylmethylcarbamoyl¾-3-ftrifluoromethylsulfonyloxyVpicolinate
The synthesis of the intermediate methyl 6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethyl sulfonyloxy)picolinate (2h) is described in Scheme 2.

Preparation of 2-bromo-3-hvdroxy-6-methylpyridine (2b)

H3C N Br
2a 2b
To a solution of 3-hydroxy-6-methylpyridine (2a) (3000 g, 27.5 mol) in pyridine (24 L) cooled to 15 °C was added a solution of bromine (4.83 kg, 1.55 L, 30.2 mol) in pyridine (3 L) over a period of 50 min maintaining the internal temperature between 20 to 25 DC. After stirring for 19 h at room temperature the solvent was removed under vacuum and the residue was triturated with water. The solid separated was collected by filtration, washed with water and dried under vacuum to give 2-bromo-3-hydroxy-6-methylpyridine (2b) (3502 g, 67.7 %) as a light brown solid which was used as such without further purification; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.18 (d, J = 8.0 Hz, 1 H), 7.08 (d, J
MS (ES+) 188.35, 186.36 (M+1).
(2c)

2b 2c
A mixture of 2-bromo-3-hydroxy-6-methylpyridine (2b) (3000 g, 15.96 mol), anhydrous potassium carbonate (3308 g, 23.94 mol), and iodomethane (2.491 kg, 1.09 L, 17.556 mol) in 30 L of acetone was heated at 40 °C overnight. The reaction mixture was cooled to room temperature and filtered through Celite. Evaporation of the solvent followed by silica gel chromatography (Hexane: ethyl acetate = 7:3) afforded the desired compound, 2-bromo-3-methoxy-6-methylpyridine (2c) which was used as such for the next step; 1H NMR (300 MHz, DMSO-cfe) δ 7.42 (dd, J = 8.3, 1.5 Hz, 1H), 7.29 – 7.19 (m, 1H), 3.84 (d, J = 1.6 Hz, 3H), 2.37 (d, J = 1.7 Hz, 3H).

2c
2d
To a solution of 2-bromo-3-methoxy-6-methylpyridine (2c) (310 g, 1.53 mol) in 6000 mL of water at 60 °C was added KMnO, (725 g, 4.59 mol) in small portions over a 90 min period with vigorous mechanical stirring. A dark purple solution resulted. This solution was kept at 90 °C for a further 3 h and filtered through Celite while still hot to give a colourless filtrate.
After cooling, the aqueous solution was acidified to pH 1-2 by adding 6 N HCI. The white solid obtained was collected by filtration to give on drying 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (302g, 85%) of product, which was used as such in the next reaction without further purification. An analytical sample was obtained by recrystallization from methanol to give 6-bromo-5-methoxy-2-pyridinecarboxylic acid; 1H NMR (300 MHz, DMSO-tfe) δ 7.40 – 7.28 (m, 1H), 7.17 (d, J = 8.3 Hz, 1 H), 3.83 (d, J = 1.7 Hz, 3H).
Preparation of 6-bromo-N-(cvclopropylmethyl)-5-methoxypicolinamide (2e)

To a solution of 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (12 g, 52 mol) in pyridine (70 mL) was added EDCI (11.5 g, 59 mmol) and cyclopropylmethylamine (3.6 g, 52 mmol). The reaction mixture was stirred at room temperature overnight and then concentrated under vacuum. The reaction mixture was diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and the water layer was extracted with ethyl acetate (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), brine (500 mL), dried over magnesium sulphate, filtered and concentrated under vacuum to furnish 10.43g of crude product. The crude product was converted into a slurry (silica gel 20 g) and purified by flash column chromatography (silica gel 230 g, eluting with 0-100% ethyl acetate in hexane) to yield compound 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (8.02 g, 54%) as off white solid, mp 67-70 °C; 1HNMR (300 MHz, DMSO-d6) δ 8.51 (t, J = 5.8, 1 H), 8.02 (d, J = 8.4, 1 H), 7.65 (d, J = 8.5, 1 H), 3.96 (s, 3H), 3.14 (t, J = 6.5, 2H), 1.11 -0.99 (m, 1 H), 0.47 – 0.36 (m, 2H), 0.27 – 0.20 (m, 2H); MS (ES+) 307.0, 309.0 (100%
M+Na)
Preparation of methyl 6-(cvclopropylmethylcarbamoyl)-3-methoxypicolinate (2f)

To a solution of 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (7.5 g, 27.6 mol) in methanol (300 mL) in a 2-L stainless steel bomb was added Pd(OAc)2(750 mg), 1 ,1-bis(diphenylphosphino)-ferrocene (750 mg), and triethylamine (3.9 mL, 27.6 mmol). The reaction mixture was vacuum flushed and charged with CO gas to 150 psi. The reaction mixture was and heated with stirring at 150°C overnight and cooled to room temperature. The catalyst was filtered through a pad of celite, and concentrated to dryness to furnish crude product. The crude was purified by flash column chromatography (silica gel 150 g,
eluting with, 0%, 5%, 10%, 20%, 30%, 50% ethyl acetate/hexanes (250 mL each) as eluents to give methyl 6-(cyclopropylmethyl-carbamoyl)-3-methoxypicolinate (2f) (6.29 g, 86.1 %) as a salmon coloured solid, MP 107 °C; 1HNMR (300 MHz, DMSO-cfe) δ 8.28 (t, J = 6.0, 1H), 7.91 (d, J = 8.8, 1H), 7.55 (d, J = 8.8, 1 H), 3.68 (s, 3H), 3.64 (s, 3H), 2.90 (t, J = 6.5, 2H), 0.89 – 0.68 (m, 1 H), 0.26 – 0.09 (m, 2H), 0.08 – 0.00 (m, 2H); MS (ES+) 287.1 (M+Na); IR (KBr) 3316, 2921 , 1730, 1659, 1534, 1472, 1432, 1315, 1272, 1228, 1189, 1099, 1003, 929, 846, 680 cm“1; Analysis calculated for C13H16 204: C, 59.08; H, 6.10; N, 10.60; Found: C, 58.70; H, 5.97; N, 10.23.
Preparation of 6-(cvclopropylmethylcarbamoyl 3-hvdroxypicolinic acid (2q)

2f 2g
Aluminium chloride method:
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-methoxypicolinate (2f) (0.16 mmol) in dichloromethane (840 mL) was added AICI3 (193 g, 1.5 mol). The reaction mixture was heated at reflux for 12 h under nitrogen. After slowly adding ~2L of 1 N HCI, the organic layer was separated. The aqueous layer was re-extracted several times with ethyl acetate/DME. The combined organic layer was washed with brine, dried (MgSO.4), and evaporated in vacuo to furnish crude 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid. To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid was added a solution of acetyl chloride (1 10 mL) in methanol (1.1 L). The reaction mixture was stirred for 12 h at room temperature and then concentrated to dryness in vacuo. After co-evaporating once with methanol, the compound was purified by flash-column chromatography (silica gel, 500 g, eluted with chloroform and 3% methanol in chloroform) to furnish 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g).
Boron tribromide method:
To a stirring solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-ethoxypicolinate (2f) (58.0 g, 208 mmol) was added BBr3 (79 mL, 834 mmol) in CH2CI2 (1.3 L) at 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 18h. The reaction mixture was evaporated to dryness and anhydrous methanol (1 L) was added to the light yellowish solid residue. Insoluble solid was collected by filtration (36 g). Mother liquor was evaporated and co-evaporated with MeOH (2 x 200 mL). The insoluble solid (36 g) was treated with MeOH (500 mL) and acetyl chloride (50 mL) and stirred at room temperature for 18 h (at this point reaction mixture was clear). The mixture was evaporated to dryness and diluted with water and extracted with EtOAc. White solid that separated out from EtOAc layer was collected by filtration, washed with water (2 x 20 mL), dried in vacuo at 50 °C to afford 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (5.36 g, 10 %) as a white solid, MP 92-95 °C. 1HNMR (DMSO-cfe) δ 11.04 (s, 1 H, exchangeable with D20), 8.37 (t, J = 6.0, 1 H, exchangeable with D20), 8.12 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 3.90 (m, 3 H), 3.15 (m, 2 H), 1.04 ( m, 1 H), 0.41 (m, 2 H), 0.24 (m, 2 H). IR (KBr): 3346, 3205, 1684 cm“1; MS (ES+): 251.1 (M+1); Analysis calculated for C12H14N2O4.0.1 H2O: C, 57.18; H, 5.67; N, 11.14; Found: C, 57.11 ; H, 5.61; N, 11.09.
Preparation of methyl-6-(cvclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy) picolinate (2h

To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (28 mmol) in DMF (200 mL) were added triethylamine (12 mL, 84 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (12 g, 34 mmol). The reaction mixture was stirred for 1.5 h at room temperature and then poured into ice. After diluting with water and extracting with ethyl acetate, the aqueous phase was re-extracted, and then the combined organic layer was washed with water and concentrated under vacuum to give methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)picolinate (2h), which was used in the next step without purification.
1H NMR (300 MHz, CDCI3) δ 8.50 (d, J = 8.6, 1 H), 8.07 (s, 1 H), 7.88 (d, J = 8.6, 1 H), 4.09 (d, J = 12.6, 3H), 3.48 – 3.24 (m, 2H), 1.18 – 1.01 (m, 1 H), 0.69 – 0.44 (m, 2H), 0.42 – 0.20 (m, 2H). MS (ES*): 405.17, 100%, M+Na.
Synthesis of 3-f2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid:
The synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i) is described as shown in Scheme 3.

3-f4-Benzyloxy-2-formyl-5-methoxy-phenylV6-(cvcloDroDvlmethvl-carbarnovn-pyridine-2-carboxylic acid methyl ester (3a)
5 046578
153

3a
To a solution of methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)
picolinate (2h) (24.3g, 63 mmol) in DME (225 mL) were added water (25 mL), (4- (benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (27.3 g, 95 mmol), NaHC03(15.9 g,
5 189 mmol), and bis(triphenylphosphine)palladium(ll) chloride (0.885 g). The reaction
mixture was stirred at 70°C overnight under nitrogen. After extracting with ethyl acetate, the organic layer was washed with water and brine and dried (MgSO^), and then concentrated
under vacuum. The compound was purified by flash-column chromatography (silica gel, 300 g, eluting with 10%, 20%, 30% and 40% ethyl acetate in hexane) to furnish 3-(4-benzyloxy- 10 2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
methyl ester (3a) (25 g, 83%) as off white solid, MP 48-50°C: 1H NMR (300 MHz, DMSO-cfe) δ 9.61(s, 1 H), 8.40 (d, J= 7.9 Hz, 1H), 8.14 (t, J= 5.0 Hz, 1H), 7.87 (d, J= 8.1 Hz, 1 H), 7.58
(s, 1H), 7.54-7.30 (m, 5H), 6.71 (s, 1 H), 5.24 (s, 2H), 3.93 (s, 3H), 3.70 (s, 3H), 3.45-3.34 (m,
2H), 1.19-1.05 (m, 1 H), 0.64-0.54 (m, 2H), 0.37-0.30 (m, 2H); IR ( Br) 1735, 1678, 1594,
15 1513, 1437, 1283, 1217, 1141, 1092 cm“1; MS (ES+) 497.29 (M+Na); Analysis calculated for
C27H2eN206: C, 68.34; H, 5.52; N, 5.90; Found; C, 68.16; H, 5.62; N, 5.80.
2-(6-(Cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-vn-4-methoxy-5- vinylbenzoic acid (3b)

To a solution of 3-(4-benzyloxy-2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl- carbamoyl)-pyridine-2-carboxylic acid methyl ester (3a) (24g, 50.6 mmol) in acetonitrile (50
mL), 2-methyl-2-propanol (350 mL), and water (125 mL) were added sodium dihydrogen
phosphate (12.5 g) and 2-methyl-2-butene (55 mL, 519 mmol). The reaction mixture was cooled in an ice bath and then sodium chlorite (28 g) was added. After stirring for 1 h, the reaction mixture was extracted with ethyl acetate and washed with water. The aqueous layer was re-extracted and then the combined organic layers were dried (MgS04). The solvent was evaporated in vacuo to furnish 5-(benzyloxy)-2-(6- ((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (29 g) which was used for the next step. MS (ES+): 513.24, (M+Na(; (ES ): 489.26, M-1.
Methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxytoarbonyltohenyl)-6-(cvclopropylmethylcarbamovnpicolinate (3c)

To a mixture of 5-(benzyloxy)-2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxy-carbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (31 g, 63.2 mmol), and triethylamine (17.7 mL, 126.4 mmol) in dichloromethane (300 mL), was added MEM-chloride (9.03 mL, 79 mmol), and stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 40 g) to furnish methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 89%) as a thick gum; H NMR (300 MHz, CDCI3) δ 8.35 (d, J = 8.0 Hz, 1 H), 8.15 (t, J = 5.7 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.49 (d, J = 6.8 Hz, 2H), 7.36 (ddd, J = 7.5, 14.8, 22.4 Hz, 3H), 6.66 (s, 1 H), 5.37-5.13 (m, 4H), 3.90 (s, 3H), 3.69 (s, 3H), 3.60-3.49 (m, 2H), 3.49 (s, 2H), 3.39 (dd, J = 4.4, 8.4 Hz, 2H), 3.34 (s, 3H), 1.19-1.00 (m, 1H), 0.57 (q, J = 5.8 Hz, 2H), 0.38-0.25 (m, 2H). MS (ES+): 601.24 (M+Na); (ES“): 577.27 (M-1);1H NMR (300 MHz, DMSO-cfe) δ 8.69 (t, 7 = 6.1 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.63 (s, 1H), 7.41 (m, 5H), 6.92 (s, 1 H), 5.20 (m, 4H), 3.83 (s, 3H), 3.57 (s, 3H), 3.44 (m, 2H), 3:33 (m, 2H), 3.21 (m, 5H), 1.14 (m, 1H), 0.44 (m, 2H), 0.27 (m, 2H). IR (KBr):
1732, 1671 cm“1. MS (ES+): 601.1(M+Na); Analysis calculated for C31H 2Oe: C, 64.35; H, 5.92; N, 4.84; Found: C, 64.27; H, 6.04; N, 4.79.
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(4-hvdroxy-5-methoxy-2-(((2-methoxyethoxy¾methoxy)carbonyl)phenyl)picolinate (3d)

3c 3d
To a solution of methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)-carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 56.68 mmol) in ethanol (650 mL) was added 10% Pd/C (4 g) and hydrogenated at 45 psi for 5 h. The catalyst was removed by filtration through Celite and the filtrate was concentrated under vacuum to yield methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)picolinate (3d) (31.87 g, 86%), which was pure enough to be used as such for the next step. An analytical sample of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) was obtained by purification of 350 mg of above crude using flash column chromatography (silica gel, eluting with ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethyl-carbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-phenyl)picolinate (3d) as a clear gum; 1HNMR (300 MHz, DMSO-d6) δ 9.74 (s, 1 H), 8.68 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1 H), 7.95 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 6.83 (s, 1H), 5.19 (s, 2H), 3.77 (m, 3H), 3.58 (s, 3H), 3.44 (m, 2H), 3.34 (m, 2H), 3.21 (m, 5H), 1.04 (m, 1 H), 0.44 (m, 2H), 0.27 (m, 2H); IR (KBr): 1731 , 1664 cm‘1. MS (ES*): 489.0 (M+1); Analysis calculated for C^e^O,,: C, 59.01; H, 5.78; N, 5.73; Found: C, 58.92; H, 6.15; N, 5.29.
6-(Cvclopropylmethylcarbamovn-3-(5-methoxy-2-(((2-methoxyethoxy^methoxy)-carbonyl)-4- (trifluoromethylsulfonyloxy)phenyl)picolinate (3e)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2- methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) (14.3 g, 29.3 mmol) in dichloromethane (150 mL) were added pyridine (12 mL, 146 mmol) and triflic anhydride (7.5 mL g, 44 mmol). After stirring overnight at room temperature under N2. the reaction mixture was poured into ice water and then extracted twice with dichloromethane. After washing the combined organic extracts with water and drying (MgS0 ), the solvent was evaporated in vacuo. The compound was purified by flash chromatography over silica gel column using ethyl acetate: hexane to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)-carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (1 g, 93%); H NMR (300 MHz, CDCy a 8.41 (d, J = 8.0, 1H), 8.17 (s, 1H), 8.03 (s, 1H), 7.79 (d, J = 8.0, 1 H), 6.82 (s, 1H), 5.32 (q, J = 6.1, 2H), 3.97 (s, 3H), 3.74 (s, 3H), 3.67 – 3.57 (m, 2H), 3.55 – 3.45 (m, 2H), 3.41 (dd, J = 8.2, 14.5, 2H), 3.34 (s, 3H), 1.36 – 1.17 (m, 1H), 0.58 (d, J = 7.1 , 2H), 0.33 (d, J = 5.1 , 2H).
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(5-methoxy-2-f((2-methoxyethoxy)- methoxy)carbonvn-4-vinylphenyl)picolinate (3f)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (37.4
g, 60.30 mmol) and potassium vinyltrifluoroborate (16.87 g, 120.6 mmol) in DMF (450 mL) and water (45 mL) was bubbled N2 for 5 min. To this mixture was added NaHC03 (20.26 g, 241.2 mmol) and dichloro-bis(triphenylphosphine)palladium (II) (6.34 g, 9.0 mmol). The reaction mixture was stirred at 70 °C for 20 h under N2(reaction progress was checked by 1H N R because product and starting material had same Rf in TLC). The reaction mixture was cooled down to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water, brine, dried ( gS04) and filtered. The filtrate was concentrated under vacuum to yield crude methyl 6-(cyclopropylmethyl-carbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-4-vinylphenyl)-picolinate (3f). The crude product was purified by flash column chromatography (silica gel, 1 kg, eluting with 0-100% ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate [31) (26.54 g, 88%) as an amber gum; H NMR (300 MHz, DMSO-c¾ δ 8.70 (t, J = 6.1 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1 H), 8.12 (s, 1 H), 8.00 (d, J = 8.0 Hz, 1 H), 6.98 (m, 2H), 5.94 (dd, J = 1.2, 17.8 Hz, 1H), 5.43 (d, J = 12.5 Hz, 1 H), 5.21 (d, J = 6.5 Hz, 2H), 3.88 (s, 3H), 3.64 (s, 3H), 3.48 (d, J = 3.1 Hz, 2H), 3.35 (m, 5H), 3.22 (m, 2H), 1.11 (s, 1H), 0.44 (dt, J = 4.9, 5.5 Hz, 2H), 0.28 (q, J = 4.8 Hz, 2H). IR (KBr); 1732, 1670 cm“1. MS (ES+) 499.1 (M+1).
2-(6-(cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzolc acid (3g)

A mixture of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate (3f) (27.4 mmol) in DME (160 mL) and 6N HCI (40 mL) was stirred at room temperature for 6 h or till TLC showed complete conversion. The solvent was removed under vacuum. The residue obtained was suspended in water, the solid separated out was collected by filtration, washed with water and dried under vacuum to give 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (7.0 g, 63%) as a white
solid MP 40 – 42 °C; H NMR (300 MHz, DMSO-de) δ 8.69 (t, J= 6.0 Hz, 1H, NH), 8.20 (d, J= 7.9 Hz, 1H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1H), 6.97 (dd, J= 18.0, 11.3 Hz, 1H), 6.88 (s, 1H), 5.92 (d, J= 7.9 Hz, 1H), 5.38 (d, J= 11.1 Hz, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); IR (KBr): 3084, 1728, 1650, 1533, 1212, 1143 cm-1; MS (ES+) 433.26 (M+Na); (ES-): 409.28 (M-1); Analysis calculated for θ22Η22Ν2Ο6.0.25Η2Ο; C, 63.68; H, 5.47; N, 6.75; Found C, 63.75; H, 5.56; N, 6.65
Methyl-3-(2-(4-carbamimidoylprienylcarbamoyl)-5-metrioxy-4-vinylphenyl)-6- (cvclopropylmethylcarbamoyl)picolinate (3h)

To a solution of 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (2.35 g, 5.7 mmol) and 4-aminobenzimidamide dihydrochloride (3j) (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 mL) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 in CMA 50) yielding methyl-3-(2-(4-carbamimidoylphenyl-carbamoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (2.2 g, 65%) as a white solid MP 266 °C; 1H NMR (300 MHz, DMSO-c/6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 – 7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for 
C, 58.93; H, 5.63; N,11.85; Found: C, 58.75; H, 5.65; N, 11.92.
46578
159
3-r2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy -vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i)

3h 3i
To a solution of methyl-3-(2-(4-carbamirriidoylphenylcarbarnoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (1 g, 1.9 mmol) in methanol (10 mL) and THF
(10 mL) was added 2 N NaOH (10 mL). The reaction mixture was stirred at room
temperature for 3 h, and concentrated in vacuo to remove methanol and THF. The aqueous layer was acidified with 6N HCI to pH 6-7 and the solid obtained was collected by filtration
washed with water and ether to furnish on drying 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
(3i)(0.775 g, 80%) as the hydrochloride salt as an off white solid.
1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1 H), 9.11 (s, 2H), 8.97 (s, 2H), 8.74 (s, 1 H), 7.90
(d, J = 7.8, 1 H), 7.80 (s, 1 H), 7.72 – 7.58 (m, 4H), 6.99 (dd, J = 11.3, 17.7, 1 H), 6.78 (s, 1H),
5.95 (d, J = 17.2, 1H), 5.38 (d, J = 11.9, 1H), 3.82 (s, 3H), 3.18 (s, 2H), 1.06 (s, 1 H), 0.43 (d,
J = 7.9, 2H), 0.25 (d, J = 4.7, 2H); MS (ES+) 514.0 (M+1 ); Analysis calculated for
C2eH27N5O5.HCI.H2O: C, 59.21; H, 5.32; N, 12.33; Found: C, 59.43; H, 5.21; N, 12.06.
Example 1A- Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride in Form
C

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 to -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1.6 kg) and water (0.57 kg). The mixture was heated to 46 °C. Smopex-234 (21 g) and Acticarbone 2SW (10 g) were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 “C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The
suspension was stirred at 0-3.0 “for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjackel= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 11 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C.
Example-1 B: Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbartiovQpyridine-2-carboxylic acid hydrochloride in Form A
The procedure was carried out in an identical manner to Example 1 A, with the exception that after the final filtration the filter cake was rinsed with 2.87 kg methyl ierf-butyl ether instead of 2.87 kg water, and pulled dry. The product was dried at 40-43 °C and 50 mbar to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) as Form A.
PATENT
Methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (compound 6a) is (I) (pages 85 and 86). Avoralstat hydrochloride (compound of formula XVIII) is (II) (claim 40, page 109). A Markush structures is presented (claim 1, page 99).
The synthesis of (II) via intermediate (I) is described (example 1, pages 80-93).
A synthesis of the compound 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (Compound 3i) is described in Schemes A-C.
O y OHCk n Br^ ^OCH3
B Brr22,, AAccOOHH Y^ V”“ \ \ tt–BBuuOOKK
OHC^^^O ” Br^\^0 MeOH ” OHC
1a 1b 66%

1d 95% 1 e

1f
Scheme A


3h 31
Scheme C
Examples. In this section, the following abbreviations are used:



Example-1 : Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b)

7b
Step (1): Preparation of 6-Bromobenzo 1 ,3]dioxole-5-carbaldehyde (1 b):

1b
A solution of bromine (33.0 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to a solution of piperonal (1a) (29.9 kg, 199.16 mol) in acetic acid (105 L) at room
temperature over a period of 50 min and the reaction mixture was stirred at room temperature for 14.2 h. Additional solution of bromine (33 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to the reaction mixture over a period of 2 h and the reaction mixture was stirred for 22 h. The reaction mixture was quenched by addition of ice water (500 L) with stirring over a period of 6 h and continued stirring for additional 1.25 h. The mixture was allowed to settle and most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (600 L) was added to the solid, stirred, mixture was allowed to settle and then most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (100 L) was added to the decanted mixture, stirred for 15 min and the solid obtained was collected by filtration using a centrifuge. The solid was washed with water (2 x 100 L) and air-dried in a tray drier for 3.75 h to afford the crude product 1 b (52 kg). The crude product (51.2 kg) was stirred in n-hexane (178 L) for 3 h, collected by filtration, washed with n-hexane (25 L) and dried to afford 6-bromobenzo[1 ,3]dioxole-5-carbaldehyde (1b) (40.1 1 kg, 87.9%) as a light brown solid. MP: 109-112°C. 1H NMR (300 MHz, CDCI3) δ 10.21 (s, 1 H), 7.37 (s, 1 H), 7.07 (s, 1 H), 6.10 (s, 2H); HNMR (DMSO-cf6): δ 10.06 (s, 1 H), 7.42 (s, 1 H), 7.29 (s, 1 H), 6.20 (d, J =12.3 Hz, 2H)
The process is also illustrated in Fig. 1.
Average yield of isolated 1 b from step-1 is 78 – 88%.
Step (2): Preparation of 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c)

A solution of potassium terf-butoxide (10.7 kg, 95.36 mol) in DMSO (49 L) was stirred at 50 °C for 30 min. Methanol (49 L) was added slowly over a period of 4.25 h and stirred at 50 °C for 30 min. 6-Bromobenzo[1 ,3]dioxole-5-carbaldehyde (1 b) (9.91 kg, 43.27 mol) was added to the reaction mixture in small portions over a period of 45 min and stirred at 50 °C for 1 h. The reaction mixture was cooled to room temperature and split into two equal portions. Each portion was quenched with water (50.9 L) and basified with 50% aqueous NaOH solution (2.4 L). Each portion was extracted with MTBE (4 x 36 L) to remove impurities. The aqueous layer was acidified with cone. HCI to pH ~ 3 to obtain
product as a yellow solid. The solid was collected by filtration using a centrifuge, washed with water (2 x 35 L) and air-dried to afford 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) (4.37 kg, 40.7%, contains 7 % water); Mp: 100-102°C; 1HNMR (300MHz, DMSO-d6): δ 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H).
The process is also illustrated in Fig. 2.
Average yield of isolated product 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) from step-2 is 40-50%.
Step (3): 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-y benzaldehyde (4a)

2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) [1.3 kg (93%, 7% water content), 5.25 mol] was dissolved in toluene (13 L) in a reaction flask equipped with a Dean Stark apparatus. The solution was heated at reflux with stirring to distil off about 25% of the toluene along with water (90 ml_). The solution was cooled to 90 °C then
bis(pinacolato)diboron (1.5 kg, 5.82 mol), KOAc (772.6 g, 7.87 mol) and Pd(PPh3) (24.3 g, 0.02 mol) were added and the reaction mixture was heated at reflux for 10h. After confirming the completion of reaction by TLC (mobile phase: 100% DCM), the reaction mixture was cooled to room temperature and was kept standing overnight. The reaction mixture was filtered through celite and the celite cake was washed with toluene (4 L). The filtrate of this batch was mixed with the filtrate of another batch (batch size 1.3 kg obtained from an identical reaction). The mixed filtrate was washed with water (17.5 L), brine (17.5 L), dried over Na2S04, filtered and the solution was passed through a pad of silica gel (2 kg, mesh size 230-400). The silica gel pad was washed with toluene. The combined filtrate and washing was concentrated under reduced pressure and the residual crude product was stirred with n-hexane (23 L) for 1 h to obtain a solid product. The solid was collected by filtration, washed with n-hexane (5 L) and dried to afford 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)benzaldehyde (4a) (2.47 kg, 84.6%). H NMR (300 MHz, CDCI3) δ 10.54 (s, 1 H), 7.57 (s, 1 H), 7.33 (s, 1 H), 5.89 (s, 1 H), 4.01 (s, 3H), 1.37 (s, 12H); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1 H), 9.95 (s, 1 H), 7.33 (s, 1 H), 7.23 (s, 1 H), 3.87 (s, 3H), 1.33 (s, 12H); MS (ES+) 301.1 (M+Na); 579.1 (2M+Na); Analysis calculated for C14H19B05: C, 60.46; H, 6.89; Found: C, 60.60; H, 6.87
The average yield of 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) from step (3) is 78 – 90%.
The process is also illustrated in Fig. 3.
Step (4): Preparation of 3-Bromo-2,6-dimethylpyridine (5b)

2,6-lutidine (5a) (115 kg, 1073.3 mol) was added into pre-chilled oleum (20-23%, 1015 kg, 2276.7 mol) at 0 °C over a period of 4.5 h (temperature r6ached 14 °C during the addition). Bromine (88.18 kg, 1103.6 mol) was then added at 5-10 °C over a period of 1 h. The reaction mixture was slowly heated to 150 °C over a period of 12h. TLC analysis indicated about 40-50% conversion to product and the formation of a dimer by-product (5%). The reaction mixture was cooled to room temperature and then additional bromine (88.18 kg, 1103.6 mol) was added slowly. The reaction mixture was slowly heated to maintain a temperature of 65-75 °C over a period of 15h. TLC analysis indicated a 65-70 % conversion to product and the formation of 5% dimer by product. The reaction mixture was quenched by addition of water (500L) while maintaining the reaction temperature below 20 °C. The mixture was basified with 6.6 M NaOH (3800 L) while maintain the temperature at < 40 °C. EtOAc (220 L) was added and the mixture was stirred for 1 h then allowed to settle over a period of 2 h. The layers were separated and the aqueous layer was treated with NaOH (10 kg) in water (10 L) and extracted with EtOAc (160 L). The organic extracts were combined washed with brine (100 L), dried over Na2S04 (50.0 kg), filtered and the solvent was evaporated under atmospheric pressure. The residue was vacuum distilled and the desired product 3-bromo-2,6-dimethylpyridine (5b) was collected at 58-60 °C, 2 mmHg (98.45 kg, 49.2 %) as a colorless liquid.
The process is also illustrated in Fig. 4.
Step (5): Preparation of 3-Bromopyridine-2,6-dicarboxylic acid (5c)

5b 5c
To a stirred solution of 3-bromo-2,6-dimethylpyridine (5b) (98 kg, 5326 mol) in water (1310 L) was added KMn0 (225 kg, 1423.6 mol) in 5 equal portions in 1 h intervals at 70 °C. After stirring for 1 h at 70 °C, additional KMn04 (225 Kg, 1423.6 mol) was added in 5 equal portion in 1 h intervals at 90 °C. The reaction mixture was stirred for 12 h at 90 °C. The suspension was filtered hot through celite to obtain a clear solution. The solvent was distilled off to remove about 30% of the total volume. The remaining concentrated solution was chilled to 0 °C and made acidic (to pH 3-4) by the addition of cone. HCI (120 L). The white precipitate obtained was collected by filtration and dried at 70 °C to afford 3-bromopyridine-2,6-dicarboxylic acid (5c) as a white solid (109 kg, 84%).
The process is also illustrated in Fig. 5.
Step (6): Preparation of Dimethyl 3-Bromopyridine-2,6-dicarboxylate (5d)

To a stirred solution of 3-bromopyridine-2,6-dicarboxylic acid (5c) (20.0 kg, 81.29 mol) in methanol (100 L) was added cone. H2S04 (4.4 L) over a period of 30 min. The reaction mixture was heated to 65 °C and maintained at that temperature for 5 h (the reaction was monitored by TLC analysis to determine completion of reaction). The reaction mixture was cooled to room temperature basified by careful addition of aqueous NaHC03 solution (prepared from 10 kg NaHC03 in 120 L of water) and further diluted with water (120 L). The white solid obtained was collected by filtration, washed with plenty of water and then oven-dried at 40 °C to obtain dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (9.2 kg, 41.3%) as a white solid; 1HNMR (300 MHz, DMSO-cf6) δ 8.47 (d, J = 8.4, 1 H), 8.08 (dd, J = 4.5, 8.4, 1 H), 3.95 (s, 3H), 3.91 (s, 3H); MS (ES+) 570.6 (2M+Na); Analysis calculated for C9H8BrN04: C, 39.44; H, 2.94; Br, 29.15 N, 5. 1 ;
Found: C, 39.52; H, 2.92; Br, 29.28; N, 5.03.
The process is also illustrated in Fig. 6.
6582
Step (7): Preparation of Methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (

To a stirred solution of dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (27 kg, 98.52 mol) in ierf-butanol (135 L) was added at room temperature cyclopropylmethanamine (7.83 kg, 110.1 mol). The reaction mixture was heated at 65 °C for 17 h. The progress of reaction was monitored by TLC and HPLC (HPLC analysis showed the formation of 74% of the product 5e after 17 h. The reaction mixture was cooled to room temperature and then cone. HCI (2.7 L) was added slowly and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in hot /-PrOH (54 L) filtered through a celite pad. The filtrate was cooled with stirring to 10 °C to obtain a white precipitate. The solid obtained was collected by filtration, washed with cold
i-PrOH (13 kg), n-hexane (15 L) and dried to provide pure methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e) (15.7 kg, 50.9%). The filtrate was concentrated under reduced pressure and the crude product can be purified by silica gel column chromatography eluting with tert-butanol in hexanes to furnish additional 10% methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e). HNMR (300 MHz, DMSO-cf6) δ 8.83 (t, J = 5.9, 1 H), 8.47 – 8.41 (m, 1 H), 8.06 (d, J = 8.4, 1 H), 3.96 (s, 3H), 3.16 (t, J = 6.5, 2H), 1.14 – 0.99 (m, 1 H), 0.42 (m, 2H), 0.30 -0.19 (m, 2H); MS (ES+) 337.0 (M+23), 650.8 (2M+23); Analysis calculated for
C12H13BrN203: C, 46.03; H, 4.18; N, 8.95; Br, 25.52; Found: C, 46.15; H, 4.17; N, 8.72; Br, 25.26.
The average isolated yield for step (7) is 50% to 60%.
The process is also illustrated in Fig. 7.
Step (8): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a)
2

6a
THF (37.5 L) was charged to a 100 L reactor followed by ethyl 3-bromo-6- (cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate (5e) (2.5 kg, 7.98 mol) under a nitrogen atmosphere. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) (2.88 kg, 10.36 mol) was added, followed by the addition of PPh3 (53.13 g, 0.20 mol), PdCI2(PPh3)2 (120.4 g, 0.17 mol) and a solution of Na2C03(2.12 kg, 20.00 mol) in demineralized water (10.0 L) under nitrogen atmosphere. The reaction mixture was degassed again two times by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 6.5 h, cooled to room temperature and filtered through a Celite bed. Water (75 L) was added to the filtrate and the product was extracted with ethyl acetate (75 L). The aqueous layer was back extracted with ethyl acetate (2 χ 60 L). The combined ethyl acetate extract was divided into two equal portions and each portion was washed with brine (37 L), dried over Na2S04, filtered and concentrated under reduced pressure to give crude methyl 6- ((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) as a reddish viscous material (-4.5 Kg) which was used as such for the next step without further purification. An analytical sample was prepared by purification of a small sample by flash column chromatography (silica gel, eluting with 0-100% ethyl acetate in hexane) to furnish methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)-picolinate (6a) as an off-white solid; HNMR (300 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.52 (s, 1 H), 8.79 (t, J = 6.1 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 6.90 (s, 1 H), 3.85 (s, 3H), 3.62 (s, 3H), 3.22 (m, 2H), 1.16 -1.02 (m, 1 H), 0.49 – 0.38 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 791.0 (2M+Na), (ES-) 382.7 (M-1), 767.3 (2M-1); Analysis calculated for C20H20N2O6.0.25 H20: C, 61.77; H, 5.31 ; N, 7.20; Found: C, 61.54; H, 5.13; N, 7.05.
The process is also illustrated in Fig. 8.
46582
Step (9): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b)

6a 6b
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) (2.11 kg, estimated about 3.83 mol from step-8) in dichloromethane (16.0 L) and pyridine (1.4 L, 17.4 mol) cooled to -10°C and maintained at that temperature for 1 h was added a solution of triflic anhydride (980.0 ml_, 5.8 mol) in dichloromethane (6.0 L) drop wise over a period of 3 h at -10 °C. The reaction mixture was stirred at -5°C for 1.3 h, quenched with saturated aqueous NaHCO3(10.4 L) and stirred for 30 mins. The organic layer was separated, washed successively with saturated aqueous NaHC03 (10.4 L), 1 HCI (2 x 16.6 L), water (13.2 L), brine (13.2 L), dried over MgS04, filtered and concentrated under reduced pressure to give the crude product. The crude product was stirred with 15% ethyl acetate in n-hexane (7.0 L) for 1 h. The solid obtained was collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (7.0 L) for 1 h, was collected by filtration and washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (8.0 L) for 1 h, collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was dried to afford methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)-oxy)phenyl)picolinate (6b) as a light brown solid (1.7 kg, 86% yield, for combined steps 8 & 9). Average isolated yield for combined steps 8 and 9 was 70% to 86%; Ή NMR (300 MHz, DMSO-cf6): δ 9.64 (s, 1 H), 8.78 (t, J = 6.1 , 1 H), 8.29 (d, J = 8.0, 1 H), 8.16 (d, J = 8.0, 1 H), 8.03 (s, 1H), 7.39 (s, 1 H), 4.00 (s, 3H), 3.63 (s, 3H), 3.22 (m, 2H), 1.11 (m, 1 H), 0.52 – 0.39 (m, 2H), 0.28 (m, 2H); MS (ES+) 538.9 (M+Na). The process is also illustrated in Fig. 9.
Step (10): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c)

A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4- (((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b) (12 kg, 23.24 mol) in DME (106 L) was charged into reactor under nitrogen. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. Potassium trifluoro(vinyl)borate (3.9 kg, 29.1 1 mol), PdCI2(PPh3)2 (815 g, 1.13 mol), KHC03 (4.65 g, 46.44 mol) and demineralized water (12 L) was then added under a N2 atmosphere. The reaction mixture was degassed by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 5 h. The reaction mixture was cooled to room temperature and then filtered through a Celite bed. Demineralized water (118 L) was added to the filtrate followed by ethyl acetate (124 L). The mixture was stirred for 20 min and then the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 95 L). The combined organic extract was washed with brine (95 L), dried over Na2S04, and filtered. The solvent was evaporated under reduced pressure to give the crude product. The crude product was purified by column chromatography (silica gel, 120 kg, 230-400 mesh size, eluting with ethyl acetate in n-hexane) to obtain methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (6 kg, 72%). 1H NMR (300 MHz, CDCI3): δ (ppm) 9.64 (s, 1 H), 8.35 (d, J = 7.8 Hz, 1 H), 8.06-8.03 (m, 2H), 7.78(d, J = 7.8 Hz, 1 H), 7.02-6.92 (m, 1 H), 6.61 (s, 1 H), 5.86 (d, J = 17.7 Hz, 1 H), 5.38 (d, J = 1 1.4 Hz, 1 H), 3.84 (s, 3H), 3.67 (s, 3H), 3.35-3.29 (m, 2H),1.08-1.03 (m, 1H), 0.55-0.49 (m, 2H), 0.29-0.2 4(m, 2H). 1HNMR (300 MHz, DMSO-d6) 6 9.68 (s, 1 H), 8.77 (t, J = 6.1 , 1 H), 8.35 – 8.21 (m, 1 H), 8.16 – 8.01 (m, 2H), 7.14 -6.87 (m, 2H), 6.01 (dd, J = 1.2, 17.8, 1 H), 5.45 (dd, J = 1.1 , 1 1.3, 1 H), 3.91 (s, 3H), 3.64 (s, 3H), 3.23 (m, 2H), 1.21 – 1.01 (m, 1H), 0.51 – 0.40 (m, 2H), 0.34 – 0.20 (m, 2H). MS
(ES+) 417.0 (M+Na); Analysis calculated for C22H22N205: C, 66.99; H, 5.62; N, 7.10;
Found: C, 66.75; H, 5.52; N, 7.06.
The process is also illustrated in Fig. 10.
Step (1 1): Preparation of 2-(6-((cyclopropylmethyl)carbamoyl)-2- (methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d)

To a stirred solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (1.57 kg, 3.80 mol) in acetonitrile (15.4 L) was added ferf-butyl alcohol (22.2 L), demineralized water (3.2 L) and sodium dihydrogen phosphate monohydrate (323.74 g, 2.346 mol). The reaction mixture was cooled to 0 °C and added 2-methyl-2-butene (5.3 L, 50.0 mol) and stirred at 0 °C for 30 min. A solution of 80% sodium chlorite (1.36 kg, 12.0 mol) in demineralized water (5.2 L) was added to the reaction mixture over a period of 2.5 h at 0 °C [temperature rises to 7 °C during the addition]. The reaction mixture was stirred at 0 °C for 2 h, diluted with water (40 L) and ethyl acetate (24 L). After stirring the mixture, it was allowed to settle and the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 20 L) then acidified with 5.9 % aqueous acetic acid (2 L) and extracted once with ethyl acetate (10 L). The organic extracts were combined washed with water (2 x 20 L), a solution of acetic acid (125 mL) in water (20.0 L), brine (2 χ 20 L), dried over Na2S04, filtered and concentrated under reduced pressure (vapor temperature below 40 °C). The residue obtained was dissolved in acetone (7 L) (residue didn’t dissolve completely). The solution was poured slowly into a reactor containing stirred n-hexane (70.0 L) to precipitate the solid product and the mixture was stirred for 2 h. The solid obtained was collected by filtration, washed with 10% acetone in n-hexane (6.3 L), AJ-hexane (6.3 L), dried to afford 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4- methoxy-5-vinylbenzoic acid (6d) as an off-white solid (1.29 Kg, yield: 79.0%). Average isolated yield for step 1 1 is 74% to 84%. 1H NMR (300 MHz, DMSO-d6): δ (ppm) 12.50 (brs, 1 H), 8.69(t, J= 6.0 Hz, 1 H, NH), 8.20 (d, J= 7.9 Hz, 1 H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1 H), 6.97 (dd, J= 18.0, 1 1.3 Hz, 1 H), 6.88 (s, 1 H), 5.92 (d, J= 7.9 Hz, 1 H), 5.38 (d, J= 1 1.1 Hz, 1 H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); MS (ES+) 433.26, (M+Na); (ES-) 409.28 (M-1). The process is also illustrated in Fig. 1 1.
Step (12): Preparation of Methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate methanesulfonate (7a

Pyridine (3.8 L, 47.17 mol) and EDCI (5.31 kg, 27.66 mol) were sequentially added to a cooled solution (0 °C) of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (9 kg, 21.92 mol) and 4-aminobenzamidine dihydrochloride (5.13 kg, 24.65 mol) in /-PrOH (90 L). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. TLC analysis indicated incomplete reaction. Additional EDCI (1.08 kg, 5.6 mol) was added and the reaction mixture was stirred for 8 h. The reaction was still incomplete as indicated by TLC analysis, additional EDCI (0.54 kg, 2.8 mol) was added and the reaction mixture was stirred for 5 h. TLC analysis indicated there was trace amount of unreacted starting material remaining. The reaction mixture was cooled to 0 °C and a solution of
methanesulfonic acid (MSA) (9.13 kg, 95 mol) in MeOH (38.7 L) was added to the cooled mixture over a period of 4 h. The reaction mixture was allowed to warm to room temperature and stirred for 15 h. The product was collected by filtration, washed with a mixture of /‘-PrOH and MeOH (4:1 , 45 L). The wet cake was slurried in a mixture of /-PrOH and MeOH (2:1 , 135 L) stirred for 1 h and the product was collected by filtration and washed with a mixture of /‘-PrOH and MeOH (4:1 , 46.8 L). The product was dried in
2015/046582
a vacuum oven at 45 °C to afford methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) as a pink-colored solid (12.71 kg, 93%). Average isolated yield for this step: >90%.
1H NMR (300 MHz, DMSO-c/6) δ 10.71 (s, 1 H), 9.16 (s, 2H), 8.80 (s, 2H), 8.68 (t, J = 6.1 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (s, 1H), 7.84 – 7.72 (m, 4H), 7.12 – 6.97 (m, 2H), 6.04 (dd, J = 17.8, 1.3 Hz, 1 H), 5.45 (d, J = 12.6 Hz, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.25 – 3.16 (m, 2H), 2.32 (s, 3H), 1.10 – 1.01 (m, 1 H), 0.48 – 0.37 (m, 2H), 0.30 – 0.22 (m, 2H); MS (ES+) 528.0 (M+1); Analysis calculated for
C29H29N5O5.CH3SO3H.2H2O. C, 54.62; H, 5.65; N, 10.62; S, 4.86; Found: C, 54.95; H, 5.55; N, 10.61 ; S, 4.87.
The process is also illustrated in Fig. 12.
Step (13): Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-rnethoxy-4- vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate

(3i) ,a 3i
A pre-cooled (0-5 °C) aq. NaOH solution [prepared from solid NaOH (4 kg, 100 mol) in water (86 L)] was added to a suspension of methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) (28.7 kg, 46 mol) in acetonitrile (86 L) cooled to 0 to 5 °C over a period of 25 mins. The reaction mixture was stirred at 0 to 5 °C for 2.5 h (TLC analysis showed the reaction was complete). The reaction mixture was filtered through a sparkler filter, washed with a mixture of 1 :1 CH3CN / H20 ( 57.4 L). Acetic acid (3.2 L, 55.9 mol) in water (56 L) was added to the filtrate at room temperature over a period of 25 mins and the resulting mixture was stirred at room temperature for 2.5 h. The solid product obtained was collected by filtration, washed with a 1 :4 mixture of CH3CN / H20 (57.5 L). The solid was dried at 45°C in a vacuum oven to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate (3i) as an off-white solid (12,77 kg, 54.1%). Average yield for this step is 50% to 75%. Mp: >200°C; H NMR (300 MHz, DMSO-d6): δ 13.49 (s, 1 H), 8.94 (bs, 4H), 8.56 (t, 1 H), 7.82 – 7.71 (m, 2H), 7.67 -7.56 (m, 4H), 7.51 (d, J = 7.8, 1 H), 6.98 (dd, J = 11.3, 17.8, 1 H), 6.68 (s, 1 H), 5.92 (d, J = 16.6, 1 H), 5.36 (d, J = 12.4, 1 H), 3.80 (s, 3H), 3.16 (m, 2H), 1.05 (m, 1 H), 0.43 (m, 2H), 0.24 (m, 2H); MS (ES+) 514.1 (M+1), 536.1 (M+Na), (ES-) 512.1 ; Analysis calculated for C28H27N5O5.3H2O: C, 59.25; H, 5.86; N, 12.34; Found C, 59.50; H,
5.75; N, 12.05. If needed this material can be crystallized from a mixture of acetone and water.
The process is also illustrated in Fig. 13.
Step 14: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b

A pre-cooled (5-8 °C) aqueous NaOH solution (prepared from solid NaOH (1.97 kg, 49.25 mol) in demineralized water (41 L) was added to a pre-cooled (0-5 °C) suspension of (3i) (13.8 kg, 26.9 mol) in acetonitrile (41 L). The reaction mixture was stirred at 0-5 °C for 30 min (until the reaction mixture becomes homogeneous). The reaction mixture was filtered through a sparkler filter washed with 50% acetonitrile in demineralized water (4.4 L). The filtrate was charged into a reactor and cooled to 0-5 °C. Aqueous HCI [prepared from cone. HCI (9.3 L) in demineralized water (36 L)] was added slowly with stirring to keep the reaction temperature at or below 15 °C, the resulting mixture was stirred at 10-15 °C for 13 h. The reaction mixture was cooled to 0-5 °C and stirred for 1 h. The solid obtained was collected by filtration and washed with demineralized water (36 L). The solid product was suspended in water (69 L) stirred for 30 mins and collected by filtration washed twice with water (20 L each). The solid product was dried in a vacuum oven at 45°C to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethyl carbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (1 1.21 Kg, 75.77%). Mp: >200°C; 1H NMR (300 MHz, DMSO-ci6): δ 12.98 (br s, 1 H), 10.86 (s, 1 H), 9.24 (s, 3H), 9.04 (s, 2H), 8.22 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 5.7 Hz, 2H), 7.78 (s, 4H), 7.09-6.99 (m, 2H), 6.07 (d, J = 17.7 Hz, 1 H), 5.45(d, J = 11.4 Hz, 1 H), 3.88 (s, 3H), 3.26-3.24 (m, 2H), 1.09 (m, 1 H), 0.47 (m, 2H), 0.28 (m, 2H).
Average isolated yield for this step varies from 63% to 80%.
The process is also illustrated in Fig. 14.
Example-2: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

6d 8a
To a solution of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (2.35 g, 5.7 mmol) and 4-aminobenzamidine dihydrochloride (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 ml_) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The
reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and concentrated in vacuum. The residue obtained was purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 and 0-100% chloroform in CMA 50) to furnish methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)-carbamoyl)picolinate hydrochloride (8a) (2.2 g, 65%) as a white solid; MP 266 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 -7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1 H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1 H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for C29H29N505 (H20)1 5 (HCI): C, 58.93; H, 5.63; N, 1 1.85; Found: C, 58.75; H, 5.65; N, 1 1.92.
Step-2: preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

8a 8b j0 a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml), was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with a solution of sulfuric acid (0.483 ml, 9.00 mmol) in water (5 mL) and stirred for 10 min at room temperature. To this cold water (5 ml) was added and stirred at room temperature until product crystallized out. Cold water (5 mL) was added to the slurry and stir for additional 20 min, additional cold water (5 mL) was added prior to filtration of solid. The solid obtained was collected by filtration washed with water (5 mL and 2.5 mL), dried under vacuum overnight to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b) (1.103 g, 90 % yield) as a white solid; MP 221.7 °C; H NMR (300 MHz, DMSO-d6) δ 12.30 – 10.91 (bs, 1 H, D20 exchangeable), 10.69 (bs, 1 H, D20 exchangeable), 9.24 (t, J = 6.0 Hz, 1 H), 9.16 (s, 2H, D2O exchangeable), 8.78 (s, 2H, D2O exchangeable), 8.24 (d, J = 8.0 Hz, 1 H), 8.04 – 7.91 (m, 2H), 7.84 – 7.67 (m, 4H), 7.13 – 6.94 (m, 2H), 6.03 (dd, J = 17.8, 1 .4 Hz, 1 H), 5.51 – 5.37 (m, 1 H), 3.88 (s, 3H), 3.24 (t, J = 6.4 Hz, 2H), 1.16 – 1.01 (m, 1 H), 0.52 – 0.41 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for: C28H27N605 1.0H2SO4 1.5H20: C, 52.66; H, 5.05; N, 10.97; S, 5.02; Found: C, 52.81 ; H, 4.95; N, 10.94; S, 4.64.
Example-3: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane s

To a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml) was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with methanesulfonic acid (0.584 ml, 9.00 mmol) and stirred for 1 h at room temperature. Cold water (5.00 ml) was added to the reaction mixture and stirred at room temperature until product crystallized out. To the slurry was added water (5 ml) of water stirred for additional 20 min, followed by the addition of water (5 ml) prior to filtration. The solid obtained was collected by filtration washed with water (5 ml and 2.5 ml), dried under vacuum to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane sulfonate salt (8c)
(1 .138 g, 1.867 mmol, 93 % yield) as a white solid; MP 221.2 °C; 1 H NMR (300 MHz,
DMSO-d6) δ 12.89 (s, 1 H, D2O exchangeable), 10.69 (s, 1 H, D2O exchangeable), 9.24
(t, J = 6.0 Hz, 1 H), 9.16 (s, 2H,), 8.85 (s, 2H), 8.24 (d, J = 8.0 Hz, 1 H), 8.06 – 7.91 (m, 2H), 7.86 – 7.70 (m, 4H), 7.15 – 6.96 (m, 2H), 6.03 (dd, J = 17.8, 1.4 Hz, 1 H), 5.52 – 5.35 (m, 1 H), 3.88 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.34 (s, 3H), 1.17 – 1.01 (m, 1 H), 0.53 -0.43 (m, 2H), 0.32 – 0.23 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for:
CzeH^NsOsCHsSOsH 1.5H20: C, 54.71 ; H, 5.38; N, 11.00; S, 5.04; Found: C, 54.80; H, 5.14; N, 10.94; S, 4.90.
Example-4: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) in Form C (Compound XX)

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled
46582
to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 – -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1 .6 kg) and 0.57 kg water. The mixture was heated to 46 °C. 21 g of Smopex-234 and 10 g Acticarbone 2SW were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 °C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The suspension was stirred at 0-3.0 °for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjacke,= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 1 17 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C (Compound XX).
/////avoralstat, BCX4161, Fast Track, Treat hereditary angioedema (HAE), Orphan Drug, PRECLINICAL
COc1cc(c(cc1C=C)C(=O)Nc2ccc(cc2)C(=N)N)c3cc(ncc3C(=O)O)C(=O)NCC4CC4
Avoralstat

Avoralstat, BCX4161,
CAS 918407-35-9
UNII: UX17773O15
513.5513, C28-H27-N5-O5
2-Pyridinecarboxylic acid, 3-(2-(((4-(aminoiminomethyl)phenyl)amino)carbonyl)-4-ethenyl-5-methoxyphenyl)-6-(((cyclopropylmethyl)amino)carbonyl)-
3-(2-((4-Carbamimidoylphenyl)carbamoyl)-4-ethenyl-5-methoxyphenyl)-6-((cyclopropylmethyl)carbamoyl)pyridine-2-carboxylic acid
Hereditary angioedema (HAE)
Kallikrein inhibitor
BioCryst Pharmaceuticals
![]()
BioCryst is also investigating second-generation plasma kallikrein inhibitors to avoralstat, for treating HAE (in February 2016, this program was listed as being in preclinical development).
Prevent acute attacks in patients with hereditary angioedema (HAE); Treat hereditary angioedema (HAE)
U.S. – Fast Track (Treat hereditary angioedema (HAE));
U.S. – Orphan Drug (Prevent acute attacks in patients with hereditary angioedema (HAE))
26 Feb 2016Clinical trials in Hereditary angioedema (Prevention) in USA (PO, Hard-gelatin capsule) before February 2016
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in France (PO, Soft-gelatin capsule)
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in Germany (PO, Soft-gelatin capsule)
| Conditions | Interventions | Phases | Recruitment | Sponsor/Collaborators |
|---|---|---|---|---|
| Hereditary Angioedema|HAE | Drug: BCX4161|Drug: Placebo | Phase 2|Phase 3 | Recruiting | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161|Drug: Placebo | Phase 2 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
Avoralstat, also known as BCX-4161, is a potent and orally active Kallikrein inhibitor and Bradykinin inhibitor. Avoralstat may be potentially useful for treatment for Hereditary angioedema. Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
Selective inhibitor of plasma kallikrein that subsequently suppresses bradykinin production
Hereditary angioedema (HAE) is a serious and potentially life-threatening rare genetic illness, caused by mutations in the C1-esterase inhibitor (C1 INH) gene, located on chromosome 11q. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by C1 INH gene mutations which produce low levels of C1 -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective C1 protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; 100(Suppl 2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herin by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that C1 INH is the most important inhibitor of kallikrein in human plasma and that C1 INH deficiency leads to unopposed activation of the kallikrein-bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack.
Approved therapies include purified plasma-derived C1 INH (Cinryze®, Berinert), the recombinant peptide kallikrein inhibitor ecallantide (Kalbitor®), and the bradykinin receptor B2 inhibitor iticabant (Firazyr®). All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
There are many delivery routes for active pharmaceutical ingredients (APIs). Generally, the oral route of administration is favored. Oral administration provides a number of advantages, such as, but not limited to, patient convenience, flexibility of timing of administration, location of administration and non-invasiveness. Oral administration also provides more prolonged drug exposure compared with intermittent intravenous infusion, which may be important for drugs with schedule-dependent efficacy. For example, a drug with a short half-life can achieve a greater exposure time by either continuous infusion or by continuous oral dosing. The use of oral therapy further has the potential to reduce the cost of healthcare resources for inpatient and ambulatory patient care services.
In the pharmaceutical arts, it is known that a number of APIs cannot be administered effectively by the oral route. The main reasons why these compounds cannot be administered by the oral route are: a) rapid enzymatic and metabolic degradation; b) chemical and/or biological instability; c) low solubility in aqueous medium; and/or d) limited permeability in the gastrointestinal tract. For such compounds, non-oral routes of delivery, such as parenteral administration, mainly via intramuscular or subcutaneous injections, may be developed. However, non-oral administration poses a disadvantage for the patient as well as healthcare providers, and for this reason, it is important to develop alternative routes of administration for such compounds, such as oral routes of administration.
While the oral route of administration is the most convenient for the patient and the most economical, designing formulations for administration by the oral route involves many complications. Several methods are available to predict the ease by which an API may be formulated into a formulation suitable for administration by the oral route. Such methods include, but are not limited to, and Lipinski rule (also referred to as the Rule of Five) and the Biopharmaceutical Drug Disposition Classification System (BDDCS).
The BDDCS divides APIs into four classifications, depending on their solubility and permeability. Class I APIs have high solubility and high permeability; Class II APIs have low solubility and high permeability; Class III APIs have high solubility and low permeability; and Class IV APIs have low solubility and low permeability. APIs in higher classes in the BDDCS face greater challenges in formulating into an effective, pharmaceutically acceptable product than those in lower classes. Of the four classes, APIs falling into Class IV are the most difficult to formulate into a formulation for administration by the oral route that is capable of delivering an effective amount of the API as problems of both solubility and permeability must be addressed (note the BDDCS does not inherently address chemical stability). The role of BDDCS in drug development is described generally in L.Z. Benet J Pharm Sci. 2013, 102(1), 34-42.
Lipinski’s rule (described in Lipinski et al. Adv. Drug Deliv. Rev. 46 (1-3): 3-26) states, in general, that in order to develop a successful formulation for administration by the oral route, an API can have no more than one violation of the following criteria:
i) not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms)
ii) not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms) iii) a molecular mass less than 500 daltons
iv) an octanol-water partition coefficient log P not greater than 5.
J. Zhang et al. Medicinal Chemistry, 2006, 2, 545-553, describes a number of small molecule amidine compounds which have activity as inhibitors of kallikrein. The molecules described in this document fall into Class IV of the BDDCS as described above. The compounds are poorly soluble in aqueous and physiological fluids, and are poorly permeable as demonstrated by oral dosing in rats and in vitro experiments with Caco-2 cells.
Furthermore, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, one of the compounds described in Zhang et al., is a Class IV API and violates criteria iii) and iv) as set forth in the Lipinski Rule.
Furthermore, the compounds described in Zhang et al., including 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, exhibit poor stability with respect to oxidation in air, to light
(photodegradation) and in aqueous and physiological fluids, as well as to elevated temperatures.
Therefore, the compounds described by Zhang et al. including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, not only exhibit poor solubility and permeability characteristics, but also poor stability characteristics. As a result, such compounds are predicted to be especially difficult to formulate into an effective, orally deliverable
pharmaceutical composition that is capable of delivering an effective amount of the compound to a subject.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties, such as, but not limited to, melting point, thermal behaviors (e.g. measured by thermogravimetric analysis (TGA), or differential scanning calorimetry (DSC), x-ray diffraction pattern, infrared absorption fingerprint, and solid state NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms and solvates of a pharmaceutical product can provide alternate forms of the compound that display a number of desirable and advantageous properties, such as, but not limited to, ease of handling, ease of processing, ease of formulation, storage stability, and/or ease of purification. Further, new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof may further provide for improved pharmaceutical products, by providing compounds that are more soluble in a set of pharmaceutical excipients. Still further, the provision of new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof enlarges the repertoire of compounds that a formulation scientist has available for formulation optimization, for example by providing a pharmaceutical product with different properties, such as, but not limited to, improved processing characteristics, improved handling characteristics, improved solubility profiles, improved dissolution profile and/or improved shelf-life. Therefore, there is a need for additional polymorphs of pharmaceutically useful compounds, such as, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid and the compounds disclosed herein.
In one aspect, the present invention provides an oral formulation that is capable of delivering an effective amount of the amidine compounds described by Zhang et al. to a subject. In particular, the present invention provides an oral formulation that is capable of delivering an effective amount of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid to a subject. In one specific aspect, the 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid is present in a particular crystal form designated Form A. In light of the art suggesting the difficulties in formulating such an oral formulation, this result was unexpected.
As described herein, the amidine compounds described in Zhang et al., including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (specifically including particular crystal Form A), may now be conveniently used in oral administration and further used in oral administration for the treatment of a number of diseases and conditions in a subject, such as, but not limited to, HAE as described herein.
Avoralstat & next generation kallikrein inhibitors for HAE
Avoralstat
![]()
May 16 is HAE awareness day
See BioCryst’s video regarding HAE to learn more
Avoralstat is being developed as an oral prophylactic treatment for patients suffering from Hereditary Angioedema (HAE). Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
In May 2014 BioCryst, announced that the OPuS-1 (OralProphylaxiS-1) Phase 2a proof of concept clinical trial met its primary efficacy endpoint, several secondary endpoints and all other objectives established for the trial. OpuS-1 enrolled 24 HAE patients with a history of HAE attack frequency of at least 1 per week. Treatment with avoralstat demonstrated a statistically significant mean attack rate reduction of 0.45 attacks per week versus placebo, p<0.001. The mean attack rate per week was 0.82 on BCX4161 treatment, compared to 1.27 on placebo.
In December 2014, BioCryst initiated enrollment in OPuS-2 (Oral ProphylaxiS-2). OPuS-2 is a blinded, randomized, 12-week, three-arm, parallel cohort design trial evaluating the efficacy and safety of two different dose regimens of avoralstat administered three-times daily, 300 mg and 500 mg, compared with placebo. The primary efficacy endpoint for the trial will be the mean angioedema attack rate, which will be reported for each avoralstat dose group compared to placebo. The trial is being conducted in the U.S., Canada and Europe. On October 8, 2015, announced that it has completed enrollment of approximately 100 HAE patients with a history of moderately frequent to very frequent attacks in OPuS-2. BioCryst expects to report the OPuS-2 trial results in early 2016.
PATENT
WO200234711
http://www.google.com/patents/WO2002034711A1?cl=en
PATENT
PATENT
Examples
Example 1 – Synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl- phenyll-6-(cvclopropylmethyl-carbarnoyl)-pyridine-2-carboxylic acid
The synthesis of the above compound and intermediates is described below. In this section, the following abbreviations are used:
The synthesis of starting material, (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) is described in Scheme 1.
f 0HCY ° ΒΓΥΥ°
Preparation of 6-bromobenzofdl[1,3ldioxole-5-carbaldehvde (1b)
1a 1b
To a mixture of piperonal (1a) (498 g, 3.32 mol) in glacial acetic acid (1000 mL) was added a solution of bromine (200 mL, 3.89 mol) in glacial acetic acid (500 mL) over a period of 30 min and stirred at room temperature for 24h. The reaction mixture was poured into water (2000 mL) and the solid that separated was collected by filtration. The solid was dissolved in boiling ethanol (4000 mL) and cooled to room temperature. The solid obtained on cooling was collected by filtration to furnish 6-bromobenzo[d][1 ,3]dioxole-5-carbaldehyde (lb) (365 g, 48 %) as a white solid, MP 126 °C; HNMR (300 MHz, DMSO-d6): δ 10.06 (s, 1 H), 7.42 (s,1 H), 7.29 (s, 1 H), 6.20 (d, J=12.3, 2H); IR (KBr) 3434, 2866, 1673,1489, 1413, 259, 1112, 1031 , 925 cm“1; Analysis calculated for CeH5BrO3.O 25H C, 41.15; H, 2.37; Found: C, 41.07; H, 2.11.
Preparation of 2-bromo-5-hvdroxy-4-methoxybenzaldehyde (1c)
1c
A solution of potassium tert-butoxide (397 g, 3.36 mol) in DMSO (1.5 L) was heated at 50 °C for 30 min. Methanol (1.5 L) was added to it and continued heating at 50 °C for additional 30 min. To the hot reaction mixture was added 6-bromo-benzo[d][1,3]dioxole-5-carbaldehyde (1 b) (350g, 1.53 mol) and continued heating at 50 °C for 30 min. The reaction mixture was cooled to room temperature and quenched with water (2.3 L) and sodium hydroxide (61.2 g, 1.53 mol). The reaction mixture was washed with ether (2 x 1.5 L), acidified to pH 2 using cone. HCI and extracted with ethyl acetate ( 1 L). The ethyl acetate layers were combined and concentrated under vacuum to dryness. The residue obtained was treated with water (1.5 L) and ethyl acetate (1 L). The solid obtained was collected by filtration to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (97 g, 27.5% as a first crop). The layers from the filtrate were separated and aqueous layer was extracted with ethyl acetate (200 ml_). The ethyl acetate layers were combined dried over MgS04 and concentrated under vacuum to dryness to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (192 g, 54.4%, second crop) as an orange solid, MP 108 °C; ‘HNMR (300MHz, DMSO-cfe): S 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H); IR (KBr) 3477, 2967, 2917,
2837, 2767, 2740, 1657, 1595, 1428, 1270, 1210, 1164, 1022 cm“‘; Analysis calculated for C8H7Br03.H20: C, 38.58; H, 3.64: Found: C, 38.60; H, 3.60.
Preparation of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehvde ( d)
To a solution 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (120 g, 520 mmol) in DMF (1000 mL) was added potassium carbonate (79 g, 572 mmol) and benzyl bromide (68 mL, 572 mmol). The reaction mixture was stirred at room temperature overnight and quenched with water (3000 mL). The solid obtained was collected by filtration, washed with ether and dried under vacuum to furnish 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (113.19 g, 67.9%) as a white solid, MP 144 °C;1HNMR (300 MHz, DMSO-c/6): δ 10.06 (s, 1H), 7.47-7.34 (m, 7H), 5.17 (s, 2H), 3.92 (s, 3H); IR (KBr) 2898, 2851 , 1673, 1592, 1502, 1437, 1402, 1264, 1210, 1158, 1017, 754 cm“1; Analysis calculated for C 5H13Br03: C, 56.10; H, 4.08; Found: C, 55.44; H, 4.08.
Preparation of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e)
15 046578
146
1d 1e
To a solution of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (100 g, 311 mmol) in
ethanol (1500 mL) was added triethyl orthoformate (103 mL, 622 mmol), ammonium nitrate
(7.5 g, 93.3 mmol) and stirred at room temperature overnight. The reaction mixture was
treated with ether (1200 mL) and stirred for 15 min before filtration. The filtrate was
concentrated under vacuum to dryness to give 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (134 g) as a brown syrup; The product was used in the next step
without further purification; 1H N R (300 MHz, DMSO-cf6) δ 7.45 – 7.37 (m, 4H), 7.36 – 7.33
(m, 1 H), 7.17 – 7.14 (m, 1 H), 7.10 (s, 1 H), 5.10 (s, 2H), 3.80 (s, 3H), 3.58 – 3.33 (m, 5H),
1.13 – 1.07 (m, 6H); IR (KBr) 2974, 2879, 1601 , 1503, 1377, 1260, 1163, 1060 cm“1;
Analysis calculated for C19H23Br04: C, 57.73; H, 5.86; Found: C, 57.21 ; H, 5.94.
acid (1fi
To a solution of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (120 g,
300 mmol) in dry ether (1000 mL) at -78 °C was added n-butyllithium (1.6 M solution in
hexanes, 244 mL, 390 mmol) over a period of 30 min and further stirred at -78 °C for 30 min.
A solution of tri-n-butylborate (110 mL, 405 mmol) in dry ether (300 mL) was added to this
solution at -78 °C over a period of 30 min. The reaction mixture was further stirred for 2 h at -78 °C and warmed to 0 °C. The reaction mixture was quenched with 3N HCI (300 mL) at 0
°C and heated at reflux for 1 h. After cooling to room temperature, the solid obtained was
collected by filtration washed with water (250 mL) dried in vaccum to afford (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (30.85 gm, 37.6% as a white solid. The organic
layer from above filtrate was extracted with 1.5 N NaOH (3 x 200 mL). The combined basic
extracts were acidified with cone. HCI (pH about 4). The solid obtained was collected by
filtration, washed with water and dried under vacuum to furnish a second crop of (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (22.3 g, 26%) as a light orange solid
MP 158 °C; 1H NMR (300 MHz, DMSO-cfe) δ 10.08 (s, 1 H), 7.52 (s, 1 H), 7.48 – 7.33 (m, 5H),
7.24 (s, 1H), 5.18 (s, 2H), 3.89 (s, 3H); 1H NMR (300 MHz, DMSO-d6/D20) δ 10.06 (s, 1H),
7.52 (s, 1H), 7.49 – 7.32 (m, 5H), 7.23 (s, 1 H), 5.18 (s, 2H), 3.89 (s, 3H); MS (ES+) 309.1 (M+Na); IR (KBr) 3335, 2937, 1647, 1545, 1388, 1348, 1268, 1146, 1095 cm-1; Analysis calculated for C15H15BO5.0.25H2O: C, 62.00; H, 5.38; Found: C, 61.77; H, 5.19.
Synthesis of methyl-6-(cvclopropylmethylcarbamoyl¾-3-ftrifluoromethylsulfonyloxyVpicolinate
The synthesis of the intermediate methyl 6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethyl sulfonyloxy)picolinate (2h) is described in Scheme 2.
Preparation of 2-bromo-3-hvdroxy-6-methylpyridine (2b)
![]()
H3C N Br
2a 2b
To a solution of 3-hydroxy-6-methylpyridine (2a) (3000 g, 27.5 mol) in pyridine (24 L) cooled to 15 °C was added a solution of bromine (4.83 kg, 1.55 L, 30.2 mol) in pyridine (3 L) over a period of 50 min maintaining the internal temperature between 20 to 25 DC. After stirring for 19 h at room temperature the solvent was removed under vacuum and the residue was triturated with water. The solid separated was collected by filtration, washed with water and dried under vacuum to give 2-bromo-3-hydroxy-6-methylpyridine (2b) (3502 g, 67.7 %) as a light brown solid which was used as such without further purification; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.18 (d, J = 8.0 Hz, 1 H), 7.08 (d, J
MS (ES+) 188.35, 186.36 (M+1).
(2c)
![]()
2b 2c
A mixture of 2-bromo-3-hydroxy-6-methylpyridine (2b) (3000 g, 15.96 mol), anhydrous potassium carbonate (3308 g, 23.94 mol), and iodomethane (2.491 kg, 1.09 L, 17.556 mol) in 30 L of acetone was heated at 40 °C overnight. The reaction mixture was cooled to room temperature and filtered through Celite. Evaporation of the solvent followed by silica gel chromatography (Hexane: ethyl acetate = 7:3) afforded the desired compound, 2-bromo-3-methoxy-6-methylpyridine (2c) which was used as such for the next step; 1H NMR (300 MHz, DMSO-cfe) δ 7.42 (dd, J = 8.3, 1.5 Hz, 1H), 7.29 – 7.19 (m, 1H), 3.84 (d, J = 1.6 Hz, 3H), 2.37 (d, J = 1.7 Hz, 3H).
2c
2d
To a solution of 2-bromo-3-methoxy-6-methylpyridine (2c) (310 g, 1.53 mol) in 6000 mL of water at 60 °C was added KMnO, (725 g, 4.59 mol) in small portions over a 90 min period with vigorous mechanical stirring. A dark purple solution resulted. This solution was kept at 90 °C for a further 3 h and filtered through Celite while still hot to give a colourless filtrate.
After cooling, the aqueous solution was acidified to pH 1-2 by adding 6 N HCI. The white solid obtained was collected by filtration to give on drying 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (302g, 85%) of product, which was used as such in the next reaction without further purification. An analytical sample was obtained by recrystallization from methanol to give 6-bromo-5-methoxy-2-pyridinecarboxylic acid; 1H NMR (300 MHz, DMSO-tfe) δ 7.40 – 7.28 (m, 1H), 7.17 (d, J = 8.3 Hz, 1 H), 3.83 (d, J = 1.7 Hz, 3H).
Preparation of 6-bromo-N-(cvclopropylmethyl)-5-methoxypicolinamide (2e)
To a solution of 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (12 g, 52 mol) in pyridine (70 mL) was added EDCI (11.5 g, 59 mmol) and cyclopropylmethylamine (3.6 g, 52 mmol). The reaction mixture was stirred at room temperature overnight and then concentrated under vacuum. The reaction mixture was diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and the water layer was extracted with ethyl acetate (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), brine (500 mL), dried over magnesium sulphate, filtered and concentrated under vacuum to furnish 10.43g of crude product. The crude product was converted into a slurry (silica gel 20 g) and purified by flash column chromatography (silica gel 230 g, eluting with 0-100% ethyl acetate in hexane) to yield compound 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (8.02 g, 54%) as off white solid, mp 67-70 °C; 1HNMR (300 MHz, DMSO-d6) δ 8.51 (t, J = 5.8, 1 H), 8.02 (d, J = 8.4, 1 H), 7.65 (d, J = 8.5, 1 H), 3.96 (s, 3H), 3.14 (t, J = 6.5, 2H), 1.11 -0.99 (m, 1 H), 0.47 – 0.36 (m, 2H), 0.27 – 0.20 (m, 2H); MS (ES+) 307.0, 309.0 (100%
M+Na)
Preparation of methyl 6-(cvclopropylmethylcarbamoyl)-3-methoxypicolinate (2f)
To a solution of 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (7.5 g, 27.6 mol) in methanol (300 mL) in a 2-L stainless steel bomb was added Pd(OAc)2(750 mg), 1 ,1-bis(diphenylphosphino)-ferrocene (750 mg), and triethylamine (3.9 mL, 27.6 mmol). The reaction mixture was vacuum flushed and charged with CO gas to 150 psi. The reaction mixture was and heated with stirring at 150°C overnight and cooled to room temperature. The catalyst was filtered through a pad of celite, and concentrated to dryness to furnish crude product. The crude was purified by flash column chromatography (silica gel 150 g,
eluting with, 0%, 5%, 10%, 20%, 30%, 50% ethyl acetate/hexanes (250 mL each) as eluents to give methyl 6-(cyclopropylmethyl-carbamoyl)-3-methoxypicolinate (2f) (6.29 g, 86.1 %) as a salmon coloured solid, MP 107 °C; 1HNMR (300 MHz, DMSO-cfe) δ 8.28 (t, J = 6.0, 1H), 7.91 (d, J = 8.8, 1H), 7.55 (d, J = 8.8, 1 H), 3.68 (s, 3H), 3.64 (s, 3H), 2.90 (t, J = 6.5, 2H), 0.89 – 0.68 (m, 1 H), 0.26 – 0.09 (m, 2H), 0.08 – 0.00 (m, 2H); MS (ES+) 287.1 (M+Na); IR (KBr) 3316, 2921 , 1730, 1659, 1534, 1472, 1432, 1315, 1272, 1228, 1189, 1099, 1003, 929, 846, 680 cm“1; Analysis calculated for C13H16 204: C, 59.08; H, 6.10; N, 10.60; Found: C, 58.70; H, 5.97; N, 10.23.
Preparation of 6-(cvclopropylmethylcarbamoyl 3-hvdroxypicolinic acid (2q)
2f 2g
Aluminium chloride method:
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-methoxypicolinate (2f) (0.16 mmol) in dichloromethane (840 mL) was added AICI3 (193 g, 1.5 mol). The reaction mixture was heated at reflux for 12 h under nitrogen. After slowly adding ~2L of 1 N HCI, the organic layer was separated. The aqueous layer was re-extracted several times with ethyl acetate/DME. The combined organic layer was washed with brine, dried (MgSO.4), and evaporated in vacuo to furnish crude 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid. To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid was added a solution of acetyl chloride (1 10 mL) in methanol (1.1 L). The reaction mixture was stirred for 12 h at room temperature and then concentrated to dryness in vacuo. After co-evaporating once with methanol, the compound was purified by flash-column chromatography (silica gel, 500 g, eluted with chloroform and 3% methanol in chloroform) to furnish 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g).
Boron tribromide method:
To a stirring solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-ethoxypicolinate (2f) (58.0 g, 208 mmol) was added BBr3 (79 mL, 834 mmol) in CH2CI2 (1.3 L) at 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 18h. The reaction mixture was evaporated to dryness and anhydrous methanol (1 L) was added to the light yellowish solid residue. Insoluble solid was collected by filtration (36 g). Mother liquor was evaporated and co-evaporated with MeOH (2 x 200 mL). The insoluble solid (36 g) was treated with MeOH (500 mL) and acetyl chloride (50 mL) and stirred at room temperature for 18 h (at this point reaction mixture was clear). The mixture was evaporated to dryness and diluted with water and extracted with EtOAc. White solid that separated out from EtOAc layer was collected by filtration, washed with water (2 x 20 mL), dried in vacuo at 50 °C to afford 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (5.36 g, 10 %) as a white solid, MP 92-95 °C. 1HNMR (DMSO-cfe) δ 11.04 (s, 1 H, exchangeable with D20), 8.37 (t, J = 6.0, 1 H, exchangeable with D20), 8.12 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 3.90 (m, 3 H), 3.15 (m, 2 H), 1.04 ( m, 1 H), 0.41 (m, 2 H), 0.24 (m, 2 H). IR (KBr): 3346, 3205, 1684 cm“1; MS (ES+): 251.1 (M+1); Analysis calculated for C12H14N2O4.0.1 H2O: C, 57.18; H, 5.67; N, 11.14; Found: C, 57.11 ; H, 5.61; N, 11.09.
Preparation of methyl-6-(cvclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy) picolinate (2h
To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (28 mmol) in DMF (200 mL) were added triethylamine (12 mL, 84 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (12 g, 34 mmol). The reaction mixture was stirred for 1.5 h at room temperature and then poured into ice. After diluting with water and extracting with ethyl acetate, the aqueous phase was re-extracted, and then the combined organic layer was washed with water and concentrated under vacuum to give methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)picolinate (2h), which was used in the next step without purification.
1H NMR (300 MHz, CDCI3) δ 8.50 (d, J = 8.6, 1 H), 8.07 (s, 1 H), 7.88 (d, J = 8.6, 1 H), 4.09 (d, J = 12.6, 3H), 3.48 – 3.24 (m, 2H), 1.18 – 1.01 (m, 1 H), 0.69 – 0.44 (m, 2H), 0.42 – 0.20 (m, 2H). MS (ES*): 405.17, 100%, M+Na.
Synthesis of 3-f2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid:
The synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i) is described as shown in Scheme 3.
3-f4-Benzyloxy-2-formyl-5-methoxy-phenylV6-(cvcloDroDvlmethvl-carbarnovn-pyridine-2-carboxylic acid methyl ester (3a)
5 046578
153
3a
To a solution of methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)
picolinate (2h) (24.3g, 63 mmol) in DME (225 mL) were added water (25 mL), (4- (benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (27.3 g, 95 mmol), NaHC03(15.9 g,
5 189 mmol), and bis(triphenylphosphine)palladium(ll) chloride (0.885 g). The reaction
mixture was stirred at 70°C overnight under nitrogen. After extracting with ethyl acetate, the organic layer was washed with water and brine and dried (MgSO^), and then concentrated
under vacuum. The compound was purified by flash-column chromatography (silica gel, 300 g, eluting with 10%, 20%, 30% and 40% ethyl acetate in hexane) to furnish 3-(4-benzyloxy- 10 2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
methyl ester (3a) (25 g, 83%) as off white solid, MP 48-50°C: 1H NMR (300 MHz, DMSO-cfe) δ 9.61(s, 1 H), 8.40 (d, J= 7.9 Hz, 1H), 8.14 (t, J= 5.0 Hz, 1H), 7.87 (d, J= 8.1 Hz, 1 H), 7.58
(s, 1H), 7.54-7.30 (m, 5H), 6.71 (s, 1 H), 5.24 (s, 2H), 3.93 (s, 3H), 3.70 (s, 3H), 3.45-3.34 (m,
2H), 1.19-1.05 (m, 1 H), 0.64-0.54 (m, 2H), 0.37-0.30 (m, 2H); IR ( Br) 1735, 1678, 1594,
15 1513, 1437, 1283, 1217, 1141, 1092 cm“1; MS (ES+) 497.29 (M+Na); Analysis calculated for
C27H2eN206: C, 68.34; H, 5.52; N, 5.90; Found; C, 68.16; H, 5.62; N, 5.80.
2-(6-(Cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-vn-4-methoxy-5- vinylbenzoic acid (3b)
To a solution of 3-(4-benzyloxy-2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl- carbamoyl)-pyridine-2-carboxylic acid methyl ester (3a) (24g, 50.6 mmol) in acetonitrile (50
mL), 2-methyl-2-propanol (350 mL), and water (125 mL) were added sodium dihydrogen
phosphate (12.5 g) and 2-methyl-2-butene (55 mL, 519 mmol). The reaction mixture was cooled in an ice bath and then sodium chlorite (28 g) was added. After stirring for 1 h, the reaction mixture was extracted with ethyl acetate and washed with water. The aqueous layer was re-extracted and then the combined organic layers were dried (MgS04). The solvent was evaporated in vacuo to furnish 5-(benzyloxy)-2-(6- ((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (29 g) which was used for the next step. MS (ES+): 513.24, (M+Na(; (ES ): 489.26, M-1.
Methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxytoarbonyltohenyl)-6-(cvclopropylmethylcarbamovnpicolinate (3c)
To a mixture of 5-(benzyloxy)-2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxy-carbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (31 g, 63.2 mmol), and triethylamine (17.7 mL, 126.4 mmol) in dichloromethane (300 mL), was added MEM-chloride (9.03 mL, 79 mmol), and stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 40 g) to furnish methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 89%) as a thick gum; H NMR (300 MHz, CDCI3) δ 8.35 (d, J = 8.0 Hz, 1 H), 8.15 (t, J = 5.7 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.49 (d, J = 6.8 Hz, 2H), 7.36 (ddd, J = 7.5, 14.8, 22.4 Hz, 3H), 6.66 (s, 1 H), 5.37-5.13 (m, 4H), 3.90 (s, 3H), 3.69 (s, 3H), 3.60-3.49 (m, 2H), 3.49 (s, 2H), 3.39 (dd, J = 4.4, 8.4 Hz, 2H), 3.34 (s, 3H), 1.19-1.00 (m, 1H), 0.57 (q, J = 5.8 Hz, 2H), 0.38-0.25 (m, 2H). MS (ES+): 601.24 (M+Na); (ES“): 577.27 (M-1);1H NMR (300 MHz, DMSO-cfe) δ 8.69 (t, 7 = 6.1 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.63 (s, 1H), 7.41 (m, 5H), 6.92 (s, 1 H), 5.20 (m, 4H), 3.83 (s, 3H), 3.57 (s, 3H), 3.44 (m, 2H), 3:33 (m, 2H), 3.21 (m, 5H), 1.14 (m, 1H), 0.44 (m, 2H), 0.27 (m, 2H). IR (KBr):
1732, 1671 cm“1. MS (ES+): 601.1(M+Na); Analysis calculated for C31H 2Oe: C, 64.35; H, 5.92; N, 4.84; Found: C, 64.27; H, 6.04; N, 4.79.
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(4-hvdroxy-5-methoxy-2-(((2-methoxyethoxy¾methoxy)carbonyl)phenyl)picolinate (3d)
3c 3d
To a solution of methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)-carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 56.68 mmol) in ethanol (650 mL) was added 10% Pd/C (4 g) and hydrogenated at 45 psi for 5 h. The catalyst was removed by filtration through Celite and the filtrate was concentrated under vacuum to yield methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)picolinate (3d) (31.87 g, 86%), which was pure enough to be used as such for the next step. An analytical sample of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) was obtained by purification of 350 mg of above crude using flash column chromatography (silica gel, eluting with ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethyl-carbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-phenyl)picolinate (3d) as a clear gum; 1HNMR (300 MHz, DMSO-d6) δ 9.74 (s, 1 H), 8.68 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1 H), 7.95 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 6.83 (s, 1H), 5.19 (s, 2H), 3.77 (m, 3H), 3.58 (s, 3H), 3.44 (m, 2H), 3.34 (m, 2H), 3.21 (m, 5H), 1.04 (m, 1 H), 0.44 (m, 2H), 0.27 (m, 2H); IR (KBr): 1731 , 1664 cm‘1. MS (ES*): 489.0 (M+1); Analysis calculated for C^e^O,,: C, 59.01; H, 5.78; N, 5.73; Found: C, 58.92; H, 6.15; N, 5.29.
6-(Cvclopropylmethylcarbamovn-3-(5-methoxy-2-(((2-methoxyethoxy^methoxy)-carbonyl)-4- (trifluoromethylsulfonyloxy)phenyl)picolinate (3e)
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2- methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) (14.3 g, 29.3 mmol) in dichloromethane (150 mL) were added pyridine (12 mL, 146 mmol) and triflic anhydride (7.5 mL g, 44 mmol). After stirring overnight at room temperature under N2. the reaction mixture was poured into ice water and then extracted twice with dichloromethane. After washing the combined organic extracts with water and drying (MgS0 ), the solvent was evaporated in vacuo. The compound was purified by flash chromatography over silica gel column using ethyl acetate: hexane to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)-carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (1 g, 93%); H NMR (300 MHz, CDCy a 8.41 (d, J = 8.0, 1H), 8.17 (s, 1H), 8.03 (s, 1H), 7.79 (d, J = 8.0, 1 H), 6.82 (s, 1H), 5.32 (q, J = 6.1, 2H), 3.97 (s, 3H), 3.74 (s, 3H), 3.67 – 3.57 (m, 2H), 3.55 – 3.45 (m, 2H), 3.41 (dd, J = 8.2, 14.5, 2H), 3.34 (s, 3H), 1.36 – 1.17 (m, 1H), 0.58 (d, J = 7.1 , 2H), 0.33 (d, J = 5.1 , 2H).
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(5-methoxy-2-f((2-methoxyethoxy)- methoxy)carbonvn-4-vinylphenyl)picolinate (3f)
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (37.4
g, 60.30 mmol) and potassium vinyltrifluoroborate (16.87 g, 120.6 mmol) in DMF (450 mL) and water (45 mL) was bubbled N2 for 5 min. To this mixture was added NaHC03 (20.26 g, 241.2 mmol) and dichloro-bis(triphenylphosphine)palladium (II) (6.34 g, 9.0 mmol). The reaction mixture was stirred at 70 °C for 20 h under N2(reaction progress was checked by 1H N R because product and starting material had same Rf in TLC). The reaction mixture was cooled down to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water, brine, dried ( gS04) and filtered. The filtrate was concentrated under vacuum to yield crude methyl 6-(cyclopropylmethyl-carbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-4-vinylphenyl)-picolinate (3f). The crude product was purified by flash column chromatography (silica gel, 1 kg, eluting with 0-100% ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate [31) (26.54 g, 88%) as an amber gum; H NMR (300 MHz, DMSO-c¾ δ 8.70 (t, J = 6.1 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1 H), 8.12 (s, 1 H), 8.00 (d, J = 8.0 Hz, 1 H), 6.98 (m, 2H), 5.94 (dd, J = 1.2, 17.8 Hz, 1H), 5.43 (d, J = 12.5 Hz, 1 H), 5.21 (d, J = 6.5 Hz, 2H), 3.88 (s, 3H), 3.64 (s, 3H), 3.48 (d, J = 3.1 Hz, 2H), 3.35 (m, 5H), 3.22 (m, 2H), 1.11 (s, 1H), 0.44 (dt, J = 4.9, 5.5 Hz, 2H), 0.28 (q, J = 4.8 Hz, 2H). IR (KBr); 1732, 1670 cm“1. MS (ES+) 499.1 (M+1).
2-(6-(cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzolc acid (3g)
A mixture of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate (3f) (27.4 mmol) in DME (160 mL) and 6N HCI (40 mL) was stirred at room temperature for 6 h or till TLC showed complete conversion. The solvent was removed under vacuum. The residue obtained was suspended in water, the solid separated out was collected by filtration, washed with water and dried under vacuum to give 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (7.0 g, 63%) as a white
solid MP 40 – 42 °C; H NMR (300 MHz, DMSO-de) δ 8.69 (t, J= 6.0 Hz, 1H, NH), 8.20 (d, J= 7.9 Hz, 1H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1H), 6.97 (dd, J= 18.0, 11.3 Hz, 1H), 6.88 (s, 1H), 5.92 (d, J= 7.9 Hz, 1H), 5.38 (d, J= 11.1 Hz, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); IR (KBr): 3084, 1728, 1650, 1533, 1212, 1143 cm-1; MS (ES+) 433.26 (M+Na); (ES-): 409.28 (M-1); Analysis calculated for θ22Η22Ν2Ο6.0.25Η2Ο; C, 63.68; H, 5.47; N, 6.75; Found C, 63.75; H, 5.56; N, 6.65
Methyl-3-(2-(4-carbamimidoylprienylcarbamoyl)-5-metrioxy-4-vinylphenyl)-6- (cvclopropylmethylcarbamoyl)picolinate (3h)
To a solution of 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (2.35 g, 5.7 mmol) and 4-aminobenzimidamide dihydrochloride (3j) (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 mL) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 in CMA 50) yielding methyl-3-(2-(4-carbamimidoylphenyl-carbamoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (2.2 g, 65%) as a white solid MP 266 °C; 1H NMR (300 MHz, DMSO-c/6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 – 7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for ![]()
C, 58.93; H, 5.63; N,11.85; Found: C, 58.75; H, 5.65; N, 11.92.
46578
159
3-r2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy -vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i)
3h 3i
To a solution of methyl-3-(2-(4-carbamirriidoylphenylcarbarnoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (1 g, 1.9 mmol) in methanol (10 mL) and THF
(10 mL) was added 2 N NaOH (10 mL). The reaction mixture was stirred at room
temperature for 3 h, and concentrated in vacuo to remove methanol and THF. The aqueous layer was acidified with 6N HCI to pH 6-7 and the solid obtained was collected by filtration
washed with water and ether to furnish on drying 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
(3i)(0.775 g, 80%) as the hydrochloride salt as an off white solid.
1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1 H), 9.11 (s, 2H), 8.97 (s, 2H), 8.74 (s, 1 H), 7.90
(d, J = 7.8, 1 H), 7.80 (s, 1 H), 7.72 – 7.58 (m, 4H), 6.99 (dd, J = 11.3, 17.7, 1 H), 6.78 (s, 1H),
5.95 (d, J = 17.2, 1H), 5.38 (d, J = 11.9, 1H), 3.82 (s, 3H), 3.18 (s, 2H), 1.06 (s, 1 H), 0.43 (d,
J = 7.9, 2H), 0.25 (d, J = 4.7, 2H); MS (ES+) 514.0 (M+1 ); Analysis calculated for
C2eH27N5O5.HCI.H2O: C, 59.21; H, 5.32; N, 12.33; Found: C, 59.43; H, 5.21; N, 12.06.
Example 1A- Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride in Form
C
The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 to -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1.6 kg) and water (0.57 kg). The mixture was heated to 46 °C. Smopex-234 (21 g) and Acticarbone 2SW (10 g) were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 “C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The
suspension was stirred at 0-3.0 “for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjackel= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 11 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C.
Example-1 B: Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbartiovQpyridine-2-carboxylic acid hydrochloride in Form A
The procedure was carried out in an identical manner to Example 1 A, with the exception that after the final filtration the filter cake was rinsed with 2.87 kg methyl ierf-butyl ether instead of 2.87 kg water, and pulled dry. The product was dried at 40-43 °C and 50 mbar to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) as Form A.
PATENT
Methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (compound 6a) is (I) (pages 85 and 86). Avoralstat hydrochloride (compound of formula XVIII) is (II) (claim 40, page 109). A Markush structures is presented (claim 1, page 99).
The synthesis of (II) via intermediate (I) is described (example 1, pages 80-93).
A synthesis of the compound 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (Compound 3i) is described in Schemes A-C.
O y OHCk n Br^ ^OCH3
B Brr22,, AAccOOHH Y^ V”“ \ \ tt–BBuuOOKK
OHC^^^O ” Br^\^0 MeOH ” OHC
1a 1b 66%
1d 95% 1 e
![]()
1f
Scheme A
3h 31
Scheme C
Examples. In this section, the following abbreviations are used:
Example-1 : Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b)
7b
Step (1): Preparation of 6-Bromobenzo 1 ,3]dioxole-5-carbaldehyde (1 b):
1b
A solution of bromine (33.0 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to a solution of piperonal (1a) (29.9 kg, 199.16 mol) in acetic acid (105 L) at room
temperature over a period of 50 min and the reaction mixture was stirred at room temperature for 14.2 h. Additional solution of bromine (33 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to the reaction mixture over a period of 2 h and the reaction mixture was stirred for 22 h. The reaction mixture was quenched by addition of ice water (500 L) with stirring over a period of 6 h and continued stirring for additional 1.25 h. The mixture was allowed to settle and most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (600 L) was added to the solid, stirred, mixture was allowed to settle and then most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (100 L) was added to the decanted mixture, stirred for 15 min and the solid obtained was collected by filtration using a centrifuge. The solid was washed with water (2 x 100 L) and air-dried in a tray drier for 3.75 h to afford the crude product 1 b (52 kg). The crude product (51.2 kg) was stirred in n-hexane (178 L) for 3 h, collected by filtration, washed with n-hexane (25 L) and dried to afford 6-bromobenzo[1 ,3]dioxole-5-carbaldehyde (1b) (40.1 1 kg, 87.9%) as a light brown solid. MP: 109-112°C. 1H NMR (300 MHz, CDCI3) δ 10.21 (s, 1 H), 7.37 (s, 1 H), 7.07 (s, 1 H), 6.10 (s, 2H); HNMR (DMSO-cf6): δ 10.06 (s, 1 H), 7.42 (s, 1 H), 7.29 (s, 1 H), 6.20 (d, J =12.3 Hz, 2H)
The process is also illustrated in Fig. 1.
Average yield of isolated 1 b from step-1 is 78 – 88%.
Step (2): Preparation of 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c)
A solution of potassium terf-butoxide (10.7 kg, 95.36 mol) in DMSO (49 L) was stirred at 50 °C for 30 min. Methanol (49 L) was added slowly over a period of 4.25 h and stirred at 50 °C for 30 min. 6-Bromobenzo[1 ,3]dioxole-5-carbaldehyde (1 b) (9.91 kg, 43.27 mol) was added to the reaction mixture in small portions over a period of 45 min and stirred at 50 °C for 1 h. The reaction mixture was cooled to room temperature and split into two equal portions. Each portion was quenched with water (50.9 L) and basified with 50% aqueous NaOH solution (2.4 L). Each portion was extracted with MTBE (4 x 36 L) to remove impurities. The aqueous layer was acidified with cone. HCI to pH ~ 3 to obtain
product as a yellow solid. The solid was collected by filtration using a centrifuge, washed with water (2 x 35 L) and air-dried to afford 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) (4.37 kg, 40.7%, contains 7 % water); Mp: 100-102°C; 1HNMR (300MHz, DMSO-d6): δ 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H).
The process is also illustrated in Fig. 2.
Average yield of isolated product 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) from step-2 is 40-50%.
Step (3): 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-y benzaldehyde (4a)
2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) [1.3 kg (93%, 7% water content), 5.25 mol] was dissolved in toluene (13 L) in a reaction flask equipped with a Dean Stark apparatus. The solution was heated at reflux with stirring to distil off about 25% of the toluene along with water (90 ml_). The solution was cooled to 90 °C then
bis(pinacolato)diboron (1.5 kg, 5.82 mol), KOAc (772.6 g, 7.87 mol) and Pd(PPh3) (24.3 g, 0.02 mol) were added and the reaction mixture was heated at reflux for 10h. After confirming the completion of reaction by TLC (mobile phase: 100% DCM), the reaction mixture was cooled to room temperature and was kept standing overnight. The reaction mixture was filtered through celite and the celite cake was washed with toluene (4 L). The filtrate of this batch was mixed with the filtrate of another batch (batch size 1.3 kg obtained from an identical reaction). The mixed filtrate was washed with water (17.5 L), brine (17.5 L), dried over Na2S04, filtered and the solution was passed through a pad of silica gel (2 kg, mesh size 230-400). The silica gel pad was washed with toluene. The combined filtrate and washing was concentrated under reduced pressure and the residual crude product was stirred with n-hexane (23 L) for 1 h to obtain a solid product. The solid was collected by filtration, washed with n-hexane (5 L) and dried to afford 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)benzaldehyde (4a) (2.47 kg, 84.6%). H NMR (300 MHz, CDCI3) δ 10.54 (s, 1 H), 7.57 (s, 1 H), 7.33 (s, 1 H), 5.89 (s, 1 H), 4.01 (s, 3H), 1.37 (s, 12H); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1 H), 9.95 (s, 1 H), 7.33 (s, 1 H), 7.23 (s, 1 H), 3.87 (s, 3H), 1.33 (s, 12H); MS (ES+) 301.1 (M+Na); 579.1 (2M+Na); Analysis calculated for C14H19B05: C, 60.46; H, 6.89; Found: C, 60.60; H, 6.87
The average yield of 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) from step (3) is 78 – 90%.
The process is also illustrated in Fig. 3.
Step (4): Preparation of 3-Bromo-2,6-dimethylpyridine (5b)
2,6-lutidine (5a) (115 kg, 1073.3 mol) was added into pre-chilled oleum (20-23%, 1015 kg, 2276.7 mol) at 0 °C over a period of 4.5 h (temperature r6ached 14 °C during the addition). Bromine (88.18 kg, 1103.6 mol) was then added at 5-10 °C over a period of 1 h. The reaction mixture was slowly heated to 150 °C over a period of 12h. TLC analysis indicated about 40-50% conversion to product and the formation of a dimer by-product (5%). The reaction mixture was cooled to room temperature and then additional bromine (88.18 kg, 1103.6 mol) was added slowly. The reaction mixture was slowly heated to maintain a temperature of 65-75 °C over a period of 15h. TLC analysis indicated a 65-70 % conversion to product and the formation of 5% dimer by product. The reaction mixture was quenched by addition of water (500L) while maintaining the reaction temperature below 20 °C. The mixture was basified with 6.6 M NaOH (3800 L) while maintain the temperature at < 40 °C. EtOAc (220 L) was added and the mixture was stirred for 1 h then allowed to settle over a period of 2 h. The layers were separated and the aqueous layer was treated with NaOH (10 kg) in water (10 L) and extracted with EtOAc (160 L). The organic extracts were combined washed with brine (100 L), dried over Na2S04 (50.0 kg), filtered and the solvent was evaporated under atmospheric pressure. The residue was vacuum distilled and the desired product 3-bromo-2,6-dimethylpyridine (5b) was collected at 58-60 °C, 2 mmHg (98.45 kg, 49.2 %) as a colorless liquid.
The process is also illustrated in Fig. 4.
Step (5): Preparation of 3-Bromopyridine-2,6-dicarboxylic acid (5c)
![]()
5b 5c
To a stirred solution of 3-bromo-2,6-dimethylpyridine (5b) (98 kg, 5326 mol) in water (1310 L) was added KMn0 (225 kg, 1423.6 mol) in 5 equal portions in 1 h intervals at 70 °C. After stirring for 1 h at 70 °C, additional KMn04 (225 Kg, 1423.6 mol) was added in 5 equal portion in 1 h intervals at 90 °C. The reaction mixture was stirred for 12 h at 90 °C. The suspension was filtered hot through celite to obtain a clear solution. The solvent was distilled off to remove about 30% of the total volume. The remaining concentrated solution was chilled to 0 °C and made acidic (to pH 3-4) by the addition of cone. HCI (120 L). The white precipitate obtained was collected by filtration and dried at 70 °C to afford 3-bromopyridine-2,6-dicarboxylic acid (5c) as a white solid (109 kg, 84%).
The process is also illustrated in Fig. 5.
Step (6): Preparation of Dimethyl 3-Bromopyridine-2,6-dicarboxylate (5d)
To a stirred solution of 3-bromopyridine-2,6-dicarboxylic acid (5c) (20.0 kg, 81.29 mol) in methanol (100 L) was added cone. H2S04 (4.4 L) over a period of 30 min. The reaction mixture was heated to 65 °C and maintained at that temperature for 5 h (the reaction was monitored by TLC analysis to determine completion of reaction). The reaction mixture was cooled to room temperature basified by careful addition of aqueous NaHC03 solution (prepared from 10 kg NaHC03 in 120 L of water) and further diluted with water (120 L). The white solid obtained was collected by filtration, washed with plenty of water and then oven-dried at 40 °C to obtain dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (9.2 kg, 41.3%) as a white solid; 1HNMR (300 MHz, DMSO-cf6) δ 8.47 (d, J = 8.4, 1 H), 8.08 (dd, J = 4.5, 8.4, 1 H), 3.95 (s, 3H), 3.91 (s, 3H); MS (ES+) 570.6 (2M+Na); Analysis calculated for C9H8BrN04: C, 39.44; H, 2.94; Br, 29.15 N, 5. 1 ;
Found: C, 39.52; H, 2.92; Br, 29.28; N, 5.03.
The process is also illustrated in Fig. 6.
6582
Step (7): Preparation of Methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (
To a stirred solution of dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (27 kg, 98.52 mol) in ierf-butanol (135 L) was added at room temperature cyclopropylmethanamine (7.83 kg, 110.1 mol). The reaction mixture was heated at 65 °C for 17 h. The progress of reaction was monitored by TLC and HPLC (HPLC analysis showed the formation of 74% of the product 5e after 17 h. The reaction mixture was cooled to room temperature and then cone. HCI (2.7 L) was added slowly and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in hot /-PrOH (54 L) filtered through a celite pad. The filtrate was cooled with stirring to 10 °C to obtain a white precipitate. The solid obtained was collected by filtration, washed with cold
i-PrOH (13 kg), n-hexane (15 L) and dried to provide pure methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e) (15.7 kg, 50.9%). The filtrate was concentrated under reduced pressure and the crude product can be purified by silica gel column chromatography eluting with tert-butanol in hexanes to furnish additional 10% methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e). HNMR (300 MHz, DMSO-cf6) δ 8.83 (t, J = 5.9, 1 H), 8.47 – 8.41 (m, 1 H), 8.06 (d, J = 8.4, 1 H), 3.96 (s, 3H), 3.16 (t, J = 6.5, 2H), 1.14 – 0.99 (m, 1 H), 0.42 (m, 2H), 0.30 -0.19 (m, 2H); MS (ES+) 337.0 (M+23), 650.8 (2M+23); Analysis calculated for
C12H13BrN203: C, 46.03; H, 4.18; N, 8.95; Br, 25.52; Found: C, 46.15; H, 4.17; N, 8.72; Br, 25.26.
The average isolated yield for step (7) is 50% to 60%.
The process is also illustrated in Fig. 7.
Step (8): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a)
2
6a
THF (37.5 L) was charged to a 100 L reactor followed by ethyl 3-bromo-6- (cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate (5e) (2.5 kg, 7.98 mol) under a nitrogen atmosphere. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) (2.88 kg, 10.36 mol) was added, followed by the addition of PPh3 (53.13 g, 0.20 mol), PdCI2(PPh3)2 (120.4 g, 0.17 mol) and a solution of Na2C03(2.12 kg, 20.00 mol) in demineralized water (10.0 L) under nitrogen atmosphere. The reaction mixture was degassed again two times by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 6.5 h, cooled to room temperature and filtered through a Celite bed. Water (75 L) was added to the filtrate and the product was extracted with ethyl acetate (75 L). The aqueous layer was back extracted with ethyl acetate (2 χ 60 L). The combined ethyl acetate extract was divided into two equal portions and each portion was washed with brine (37 L), dried over Na2S04, filtered and concentrated under reduced pressure to give crude methyl 6- ((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) as a reddish viscous material (-4.5 Kg) which was used as such for the next step without further purification. An analytical sample was prepared by purification of a small sample by flash column chromatography (silica gel, eluting with 0-100% ethyl acetate in hexane) to furnish methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)-picolinate (6a) as an off-white solid; HNMR (300 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.52 (s, 1 H), 8.79 (t, J = 6.1 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 6.90 (s, 1 H), 3.85 (s, 3H), 3.62 (s, 3H), 3.22 (m, 2H), 1.16 -1.02 (m, 1 H), 0.49 – 0.38 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 791.0 (2M+Na), (ES-) 382.7 (M-1), 767.3 (2M-1); Analysis calculated for C20H20N2O6.0.25 H20: C, 61.77; H, 5.31 ; N, 7.20; Found: C, 61.54; H, 5.13; N, 7.05.
The process is also illustrated in Fig. 8.
46582
Step (9): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b)
6a 6b
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) (2.11 kg, estimated about 3.83 mol from step-8) in dichloromethane (16.0 L) and pyridine (1.4 L, 17.4 mol) cooled to -10°C and maintained at that temperature for 1 h was added a solution of triflic anhydride (980.0 ml_, 5.8 mol) in dichloromethane (6.0 L) drop wise over a period of 3 h at -10 °C. The reaction mixture was stirred at -5°C for 1.3 h, quenched with saturated aqueous NaHCO3(10.4 L) and stirred for 30 mins. The organic layer was separated, washed successively with saturated aqueous NaHC03 (10.4 L), 1 HCI (2 x 16.6 L), water (13.2 L), brine (13.2 L), dried over MgS04, filtered and concentrated under reduced pressure to give the crude product. The crude product was stirred with 15% ethyl acetate in n-hexane (7.0 L) for 1 h. The solid obtained was collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (7.0 L) for 1 h, was collected by filtration and washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (8.0 L) for 1 h, collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was dried to afford methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)-oxy)phenyl)picolinate (6b) as a light brown solid (1.7 kg, 86% yield, for combined steps 8 & 9). Average isolated yield for combined steps 8 and 9 was 70% to 86%; Ή NMR (300 MHz, DMSO-cf6): δ 9.64 (s, 1 H), 8.78 (t, J = 6.1 , 1 H), 8.29 (d, J = 8.0, 1 H), 8.16 (d, J = 8.0, 1 H), 8.03 (s, 1H), 7.39 (s, 1 H), 4.00 (s, 3H), 3.63 (s, 3H), 3.22 (m, 2H), 1.11 (m, 1 H), 0.52 – 0.39 (m, 2H), 0.28 (m, 2H); MS (ES+) 538.9 (M+Na). The process is also illustrated in Fig. 9.
Step (10): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c)
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4- (((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b) (12 kg, 23.24 mol) in DME (106 L) was charged into reactor under nitrogen. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. Potassium trifluoro(vinyl)borate (3.9 kg, 29.1 1 mol), PdCI2(PPh3)2 (815 g, 1.13 mol), KHC03 (4.65 g, 46.44 mol) and demineralized water (12 L) was then added under a N2 atmosphere. The reaction mixture was degassed by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 5 h. The reaction mixture was cooled to room temperature and then filtered through a Celite bed. Demineralized water (118 L) was added to the filtrate followed by ethyl acetate (124 L). The mixture was stirred for 20 min and then the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 95 L). The combined organic extract was washed with brine (95 L), dried over Na2S04, and filtered. The solvent was evaporated under reduced pressure to give the crude product. The crude product was purified by column chromatography (silica gel, 120 kg, 230-400 mesh size, eluting with ethyl acetate in n-hexane) to obtain methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (6 kg, 72%). 1H NMR (300 MHz, CDCI3): δ (ppm) 9.64 (s, 1 H), 8.35 (d, J = 7.8 Hz, 1 H), 8.06-8.03 (m, 2H), 7.78(d, J = 7.8 Hz, 1 H), 7.02-6.92 (m, 1 H), 6.61 (s, 1 H), 5.86 (d, J = 17.7 Hz, 1 H), 5.38 (d, J = 1 1.4 Hz, 1 H), 3.84 (s, 3H), 3.67 (s, 3H), 3.35-3.29 (m, 2H),1.08-1.03 (m, 1H), 0.55-0.49 (m, 2H), 0.29-0.2 4(m, 2H). 1HNMR (300 MHz, DMSO-d6) 6 9.68 (s, 1 H), 8.77 (t, J = 6.1 , 1 H), 8.35 – 8.21 (m, 1 H), 8.16 – 8.01 (m, 2H), 7.14 -6.87 (m, 2H), 6.01 (dd, J = 1.2, 17.8, 1 H), 5.45 (dd, J = 1.1 , 1 1.3, 1 H), 3.91 (s, 3H), 3.64 (s, 3H), 3.23 (m, 2H), 1.21 – 1.01 (m, 1H), 0.51 – 0.40 (m, 2H), 0.34 – 0.20 (m, 2H). MS
(ES+) 417.0 (M+Na); Analysis calculated for C22H22N205: C, 66.99; H, 5.62; N, 7.10;
Found: C, 66.75; H, 5.52; N, 7.06.
The process is also illustrated in Fig. 10.
Step (1 1): Preparation of 2-(6-((cyclopropylmethyl)carbamoyl)-2- (methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d)
To a stirred solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (1.57 kg, 3.80 mol) in acetonitrile (15.4 L) was added ferf-butyl alcohol (22.2 L), demineralized water (3.2 L) and sodium dihydrogen phosphate monohydrate (323.74 g, 2.346 mol). The reaction mixture was cooled to 0 °C and added 2-methyl-2-butene (5.3 L, 50.0 mol) and stirred at 0 °C for 30 min. A solution of 80% sodium chlorite (1.36 kg, 12.0 mol) in demineralized water (5.2 L) was added to the reaction mixture over a period of 2.5 h at 0 °C [temperature rises to 7 °C during the addition]. The reaction mixture was stirred at 0 °C for 2 h, diluted with water (40 L) and ethyl acetate (24 L). After stirring the mixture, it was allowed to settle and the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 20 L) then acidified with 5.9 % aqueous acetic acid (2 L) and extracted once with ethyl acetate (10 L). The organic extracts were combined washed with water (2 x 20 L), a solution of acetic acid (125 mL) in water (20.0 L), brine (2 χ 20 L), dried over Na2S04, filtered and concentrated under reduced pressure (vapor temperature below 40 °C). The residue obtained was dissolved in acetone (7 L) (residue didn’t dissolve completely). The solution was poured slowly into a reactor containing stirred n-hexane (70.0 L) to precipitate the solid product and the mixture was stirred for 2 h. The solid obtained was collected by filtration, washed with 10% acetone in n-hexane (6.3 L), AJ-hexane (6.3 L), dried to afford 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4- methoxy-5-vinylbenzoic acid (6d) as an off-white solid (1.29 Kg, yield: 79.0%). Average isolated yield for step 1 1 is 74% to 84%. 1H NMR (300 MHz, DMSO-d6): δ (ppm) 12.50 (brs, 1 H), 8.69(t, J= 6.0 Hz, 1 H, NH), 8.20 (d, J= 7.9 Hz, 1 H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1 H), 6.97 (dd, J= 18.0, 1 1.3 Hz, 1 H), 6.88 (s, 1 H), 5.92 (d, J= 7.9 Hz, 1 H), 5.38 (d, J= 1 1.1 Hz, 1 H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); MS (ES+) 433.26, (M+Na); (ES-) 409.28 (M-1). The process is also illustrated in Fig. 1 1.
Step (12): Preparation of Methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate methanesulfonate (7a
Pyridine (3.8 L, 47.17 mol) and EDCI (5.31 kg, 27.66 mol) were sequentially added to a cooled solution (0 °C) of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (9 kg, 21.92 mol) and 4-aminobenzamidine dihydrochloride (5.13 kg, 24.65 mol) in /-PrOH (90 L). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. TLC analysis indicated incomplete reaction. Additional EDCI (1.08 kg, 5.6 mol) was added and the reaction mixture was stirred for 8 h. The reaction was still incomplete as indicated by TLC analysis, additional EDCI (0.54 kg, 2.8 mol) was added and the reaction mixture was stirred for 5 h. TLC analysis indicated there was trace amount of unreacted starting material remaining. The reaction mixture was cooled to 0 °C and a solution of
methanesulfonic acid (MSA) (9.13 kg, 95 mol) in MeOH (38.7 L) was added to the cooled mixture over a period of 4 h. The reaction mixture was allowed to warm to room temperature and stirred for 15 h. The product was collected by filtration, washed with a mixture of /‘-PrOH and MeOH (4:1 , 45 L). The wet cake was slurried in a mixture of /-PrOH and MeOH (2:1 , 135 L) stirred for 1 h and the product was collected by filtration and washed with a mixture of /‘-PrOH and MeOH (4:1 , 46.8 L). The product was dried in
2015/046582
a vacuum oven at 45 °C to afford methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) as a pink-colored solid (12.71 kg, 93%). Average isolated yield for this step: >90%.
1H NMR (300 MHz, DMSO-c/6) δ 10.71 (s, 1 H), 9.16 (s, 2H), 8.80 (s, 2H), 8.68 (t, J = 6.1 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (s, 1H), 7.84 – 7.72 (m, 4H), 7.12 – 6.97 (m, 2H), 6.04 (dd, J = 17.8, 1.3 Hz, 1 H), 5.45 (d, J = 12.6 Hz, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.25 – 3.16 (m, 2H), 2.32 (s, 3H), 1.10 – 1.01 (m, 1 H), 0.48 – 0.37 (m, 2H), 0.30 – 0.22 (m, 2H); MS (ES+) 528.0 (M+1); Analysis calculated for
C29H29N5O5.CH3SO3H.2H2O. C, 54.62; H, 5.65; N, 10.62; S, 4.86; Found: C, 54.95; H, 5.55; N, 10.61 ; S, 4.87.
The process is also illustrated in Fig. 12.
Step (13): Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-rnethoxy-4- vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate
(3i) ,a 3i
A pre-cooled (0-5 °C) aq. NaOH solution [prepared from solid NaOH (4 kg, 100 mol) in water (86 L)] was added to a suspension of methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) (28.7 kg, 46 mol) in acetonitrile (86 L) cooled to 0 to 5 °C over a period of 25 mins. The reaction mixture was stirred at 0 to 5 °C for 2.5 h (TLC analysis showed the reaction was complete). The reaction mixture was filtered through a sparkler filter, washed with a mixture of 1 :1 CH3CN / H20 ( 57.4 L). Acetic acid (3.2 L, 55.9 mol) in water (56 L) was added to the filtrate at room temperature over a period of 25 mins and the resulting mixture was stirred at room temperature for 2.5 h. The solid product obtained was collected by filtration, washed with a 1 :4 mixture of CH3CN / H20 (57.5 L). The solid was dried at 45°C in a vacuum oven to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate (3i) as an off-white solid (12,77 kg, 54.1%). Average yield for this step is 50% to 75%. Mp: >200°C; H NMR (300 MHz, DMSO-d6): δ 13.49 (s, 1 H), 8.94 (bs, 4H), 8.56 (t, 1 H), 7.82 – 7.71 (m, 2H), 7.67 -7.56 (m, 4H), 7.51 (d, J = 7.8, 1 H), 6.98 (dd, J = 11.3, 17.8, 1 H), 6.68 (s, 1 H), 5.92 (d, J = 16.6, 1 H), 5.36 (d, J = 12.4, 1 H), 3.80 (s, 3H), 3.16 (m, 2H), 1.05 (m, 1 H), 0.43 (m, 2H), 0.24 (m, 2H); MS (ES+) 514.1 (M+1), 536.1 (M+Na), (ES-) 512.1 ; Analysis calculated for C28H27N5O5.3H2O: C, 59.25; H, 5.86; N, 12.34; Found C, 59.50; H,
5.75; N, 12.05. If needed this material can be crystallized from a mixture of acetone and water.
The process is also illustrated in Fig. 13.
Step 14: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b
A pre-cooled (5-8 °C) aqueous NaOH solution (prepared from solid NaOH (1.97 kg, 49.25 mol) in demineralized water (41 L) was added to a pre-cooled (0-5 °C) suspension of (3i) (13.8 kg, 26.9 mol) in acetonitrile (41 L). The reaction mixture was stirred at 0-5 °C for 30 min (until the reaction mixture becomes homogeneous). The reaction mixture was filtered through a sparkler filter washed with 50% acetonitrile in demineralized water (4.4 L). The filtrate was charged into a reactor and cooled to 0-5 °C. Aqueous HCI [prepared from cone. HCI (9.3 L) in demineralized water (36 L)] was added slowly with stirring to keep the reaction temperature at or below 15 °C, the resulting mixture was stirred at 10-15 °C for 13 h. The reaction mixture was cooled to 0-5 °C and stirred for 1 h. The solid obtained was collected by filtration and washed with demineralized water (36 L). The solid product was suspended in water (69 L) stirred for 30 mins and collected by filtration washed twice with water (20 L each). The solid product was dried in a vacuum oven at 45°C to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethyl carbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (1 1.21 Kg, 75.77%). Mp: >200°C; 1H NMR (300 MHz, DMSO-ci6): δ 12.98 (br s, 1 H), 10.86 (s, 1 H), 9.24 (s, 3H), 9.04 (s, 2H), 8.22 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 5.7 Hz, 2H), 7.78 (s, 4H), 7.09-6.99 (m, 2H), 6.07 (d, J = 17.7 Hz, 1 H), 5.45(d, J = 11.4 Hz, 1 H), 3.88 (s, 3H), 3.26-3.24 (m, 2H), 1.09 (m, 1 H), 0.47 (m, 2H), 0.28 (m, 2H).
Average isolated yield for this step varies from 63% to 80%.
The process is also illustrated in Fig. 14.
Example-2: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)
6d 8a
To a solution of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (2.35 g, 5.7 mmol) and 4-aminobenzamidine dihydrochloride (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 ml_) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The
reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and concentrated in vacuum. The residue obtained was purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 and 0-100% chloroform in CMA 50) to furnish methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)-carbamoyl)picolinate hydrochloride (8a) (2.2 g, 65%) as a white solid; MP 266 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 -7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1 H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1 H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for C29H29N505 (H20)1 5 (HCI): C, 58.93; H, 5.63; N, 1 1.85; Found: C, 58.75; H, 5.65; N, 1 1.92.
Step-2: preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)
8a 8b j0 a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml), was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with a solution of sulfuric acid (0.483 ml, 9.00 mmol) in water (5 mL) and stirred for 10 min at room temperature. To this cold water (5 ml) was added and stirred at room temperature until product crystallized out. Cold water (5 mL) was added to the slurry and stir for additional 20 min, additional cold water (5 mL) was added prior to filtration of solid. The solid obtained was collected by filtration washed with water (5 mL and 2.5 mL), dried under vacuum overnight to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b) (1.103 g, 90 % yield) as a white solid; MP 221.7 °C; H NMR (300 MHz, DMSO-d6) δ 12.30 – 10.91 (bs, 1 H, D20 exchangeable), 10.69 (bs, 1 H, D20 exchangeable), 9.24 (t, J = 6.0 Hz, 1 H), 9.16 (s, 2H, D2O exchangeable), 8.78 (s, 2H, D2O exchangeable), 8.24 (d, J = 8.0 Hz, 1 H), 8.04 – 7.91 (m, 2H), 7.84 – 7.67 (m, 4H), 7.13 – 6.94 (m, 2H), 6.03 (dd, J = 17.8, 1 .4 Hz, 1 H), 5.51 – 5.37 (m, 1 H), 3.88 (s, 3H), 3.24 (t, J = 6.4 Hz, 2H), 1.16 – 1.01 (m, 1 H), 0.52 – 0.41 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for: C28H27N605 1.0H2SO4 1.5H20: C, 52.66; H, 5.05; N, 10.97; S, 5.02; Found: C, 52.81 ; H, 4.95; N, 10.94; S, 4.64.
Example-3: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane s
To a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml) was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with methanesulfonic acid (0.584 ml, 9.00 mmol) and stirred for 1 h at room temperature. Cold water (5.00 ml) was added to the reaction mixture and stirred at room temperature until product crystallized out. To the slurry was added water (5 ml) of water stirred for additional 20 min, followed by the addition of water (5 ml) prior to filtration. The solid obtained was collected by filtration washed with water (5 ml and 2.5 ml), dried under vacuum to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane sulfonate salt (8c)
(1 .138 g, 1.867 mmol, 93 % yield) as a white solid; MP 221.2 °C; 1 H NMR (300 MHz,
DMSO-d6) δ 12.89 (s, 1 H, D2O exchangeable), 10.69 (s, 1 H, D2O exchangeable), 9.24
(t, J = 6.0 Hz, 1 H), 9.16 (s, 2H,), 8.85 (s, 2H), 8.24 (d, J = 8.0 Hz, 1 H), 8.06 – 7.91 (m, 2H), 7.86 – 7.70 (m, 4H), 7.15 – 6.96 (m, 2H), 6.03 (dd, J = 17.8, 1.4 Hz, 1 H), 5.52 – 5.35 (m, 1 H), 3.88 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.34 (s, 3H), 1.17 – 1.01 (m, 1 H), 0.53 -0.43 (m, 2H), 0.32 – 0.23 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for:
CzeH^NsOsCHsSOsH 1.5H20: C, 54.71 ; H, 5.38; N, 11.00; S, 5.04; Found: C, 54.80; H, 5.14; N, 10.94; S, 4.90.
Example-4: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) in Form C (Compound XX)
The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled
46582
to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 – -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1 .6 kg) and 0.57 kg water. The mixture was heated to 46 °C. 21 g of Smopex-234 and 10 g Acticarbone 2SW were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 °C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The suspension was stirred at 0-3.0 °for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjacke,= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 1 17 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C (Compound XX).
/////avoralstat, BCX4161, Fast Track, Treat hereditary angioedema (HAE), Orphan Drug, PRECLINICAL
COc1cc(c(cc1C=C)C(=O)Nc2ccc(cc2)C(=N)N)c3cc(ncc3C(=O)O)C(=O)NCC4CC4
ONL 1204 a small molecule peptide

OR

ONL 1204
CAS 1349038-53-4
(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[2-[(3R)-3-[[(2S)-2-[[(2S)-2-[[2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]-3-phenylpropanoyl]amino]-3-methylbutanoyl]amino]-3-hydroxybutanoyl]amino]acetyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-3-phenylpropanoyl]amino]-2-oxopiperidin-1-yl]acetyl]amino]-4-methylpentanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]pyrrolidine-2-carbonyl]amino]propanoic acid
His-His- Ile-Tyr-Leu-Gly-Ala-Val-Asn-Tyr-Ile-Tyr-NH2
ONL Therapeutics Inc.
Fas receptor (CD95)
Peptide, Retinal detachment, OPTHALMIC DRUGS
C71 H100 N18 O16, 1461.66
L-Histidyl-L-histidyl-L-isoleucyl-L-tyrosyl-L-leucylglycyl-L-alanyl-L-valyl-L-asparaginyl-L-tyrosyl-L-isoleucyl-L-tyrosinamide
RFVTGHFXGL YPA
ORPHAN DRUG DESIGNATION DATA
His-His- Ile-Tyr-Leu-Gly-Ala-Val-Asn-Tyr-Ile-Tyr-NH2
01/13/2016
Treatment of retinal detachment
ONL Therapeutics, Inc
1600 Huron Parkway
Second Floor
Ann Arbor, Michigan 48109…….http://www.accessdata.fda.gov/scripts/opdlisting/oopd/OOPD_Results_2.cfm?Index_Number=501215
![]()
ONL1204, ONL’s lead therapeutic candidate, is a first-in-class small molecule peptide designed to protect key retinal cells, including photoreceptors, against the apoptosis (programmed cell death) that occurs in a range of retinal diseases and conditions. It is this death of these retinal cells that is the root cause of vision loss and the leading cause of blindness.
Researchers have shown that ONL1204 effectively inhibits the Fas pathway; one of the body’s primary mechanisms for inducing programmed cell death (apoptosis). Specifically, the compound’s activity inhibits the Fas receptor, blocks the activation of the Fas pathway, and prevents the apoptosis cascade which results in the death of key retinal cells, including photoreceptor.
While initial development efforts for ONL1204 are focused on retinal detachment, preclinicalin vivo data, along with a growing body of literature, support potential application in age-related macular degeneration (AMD) and other chronic retinal diseases. Combined, the estimated market for the initial indications that ONL plans to target is >$12 billion globally.
ONL Therapeutics, Inc., a biopharmaceutical company developing novel therapies for preserving sight in a range of retinal diseases, today announced that the United States Food and Drug Administration (FDA) has granted orphan drug designation to ONL1204 for the treatment of retinal detachment. ONL1204 is a novel, first-in-class small molecule peptide designed to protect key retinal cells, including photoreceptors, from cell death that occurs in a range of retinal diseases and conditions. Death of these retinal cells is the root cause of vision loss and the leading cause of blindness. ONL expects to advance ONL1204 into clinical trials for retinal detachment patients in 2016.
Retinal detachment occurs when the retina is separated from the underlying layer of cells called the retinal pigment epithelium (RPE). The RPE provides nutritional support to the highly-active photoreceptors in the retina. When there is a detachment, the photoreceptors no longer receive these nutrients and undergo cell death processes that dramatically impact a patient’s vision. Retinal detachments occur in approximately 50,000 people each year in the United States and affect people of all ages, although risk increases as people reach fifty years of age.
Patients experiencing a retinal detachment are normally treated by surgical reattachment of the retina to reconnect the photoreceptors with the RPE and prevent additional loss of vision. However, these procedures do not address the photoreceptor death and vision loss, which can be significant, that occurs prior to surgery. ONL1204 will be delivered to patients upon diagnosis and is intended to block photoreceptor cells from dying until surgery can be completed.
“When retinal detachments involve the center of vision called the macula, more than a third of patients have final best corrected vision of 20/60 or worse after successful surgery,” said David Zacks, M.D., Ph.D., co-founder and chief science officer of ONL Therapeutics. “Those are truly poor outcomes from successful surgeries. We are very pleased the FDA has recognized this need and that ONL is the only company to have received an orphan designation for this disease. It reinforces our belief that ONL1204 can play a key role in preventing vision loss in these patients by protecting their photoreceptors.”
The FDA’s Orphan Drug Designation program provides certain incentives for companies developing therapeutics to treat rare diseases or conditions that affect less than 200,000 individuals in the US. A drug candidate and its developer must meet several key criteria in order to qualify for, and obtain, orphan drug status. Once a drug has received orphan drug designation, the developer qualifies for a range of benefits, including federal grants, tax credits, reduction in certain regulatory fees, and the potential for seven years of market exclusivity for the drug following FDA marketing approval.
About ONL Therapeutics
ONL Therapeutics (ONL) is a biopharmaceutical company committed to protecting and improving the vision of patients with retinal disease. By advancing a novel breakthrough technology designed to protect key retinal cells from Fas-mediated cell death, ONL is pioneering an entirely new approach to preserving sight. The death of key retinal cells is the root cause of vision loss and leading cause of blindness, and is implicated in a wide range of retinal diseases, including retinal detachment and both the wet and dry forms of age related macular degeneration (AMD).
read
FDA grants orphan status for ONL Therapeutics’ ONL1204 to treat retinal detachment
The US Food and Drug Administration (FDA) has granted orphan drug designation for ONL Therapeutics’ first-in-class small molecule peptide, ONL1204, for the treatment of retinal detachment.
/////
Use smiles
N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)O)C(=O)NCC(=O)N[C@@H](Cc2cncn2)C(=O)N[C@@H](Cc3ccccc3)C(=O)N[C@@H]6CCCN(CC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](Cc4ccc(O)cc4)C(=O)N5CCC[C@H]5C(=O)N[C@@H](C)C(=O)O)C6=O
OR
CC(C)CC(C(=O)NC(CC1=CC=C(C=C1)O)C(=O)N2CCCC2C(=O)NC(C)C(=O)O)NC(=O)CN3CCCC(C3=O)NC(=O)C(CC4=CC=CC=C4)NC(=O)C(CC5=CN=CN5)NC(=O)CNC(=O)C(C(C)O)NC(=O)C(C(C)C)NC(=O)C(CC6=CC=CC=C6)NC(=O)C(CCCN=C(N)N)N
OR
C[C@@H](CC)[C@H](NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@H](CC(=O)N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc2ccc(O)cc2)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](Cc3cncn3)N)Cc4cncn4)[C@@H](C)CC)C(C)C)C(=O)N[C@@H](Cc5ccc(O)cc5)C(N)=O
Elotuzumab
Elotuzumab
Approved nov 30 2012
A SLAMF7-directed immunostimulatory antibody used to treat multiple myeloma.
(Empliciti®)
HuLuc-63;BMS-901608
cas 915296-00-3
![]()


Elotuzumab (brand name Empliciti, previously known as HuLuc63) is a humanized monoclonal antibody used in relapsed multiple myeloma.[1] The package insert denotes its mechanism as a SLAMF7-directed (also known as CD 319) immunostimulatory antibody.[2]
Approvals and indications
In May 2014, it was granted “Breakthrough Therapy” designation by the FDA. [3] On November 30, 2015, FDA approved elotuzumab as a treatment for patients with multiple myeloma who have received one to three prior medications.[1] Elotuzumab was labeled for use with lenalidomide and dexamethasone. Each intravenous injection of elotuzumab should be premedicated with dexamethasone, diphenhydramine, ranitidine and acetaminophen.[2]
Elotuzumab is APPROVED for safety and efficacy in combination with lenalidomide and dexamethasone.
Monoclonal antibody therapy for multiple myeloma, a malignancy of plasma cells, was not very clinically efficacious until the development of cell surface glycoprotein CS1 targeting humanized immunoglobulin G1 monoclonal antibody – Elotuzumab. Elotuzumab is currently APPROVED in relapsed multiple myeloma.
Elotuzumab (HuLuc63) binds to CS1 antigens, highly expressed by multiple myeloma cells but minimally present on normal cells. The binding of elotuzumab to CS1 triggers antibody dependent cellular cytotoxicity in tumor cells expressing CS1. CS1 is a cell surface glycoprotein that belongs to the CD2 subset of immunoglobulin superfamily (IgSF). Preclinical studies showed that elotuzumab initiates cell lysis at high rates. The action of elotuzumab was found to be enhanced when multiple myeloma cells were pretreated with sub-therapeutic doses of lenalidomide and bortezomib. The impressive preclinical findings prompted investigation and analysis of elotuzumab in phase I and phase II studies in combination with lenalidomide and bortezomib.
Elotuzumab As Part of Combination Therapy: Clinical Trial Results
Elotuzumab showed manageable side effect profile and was well tolerated in a population of relapsed/refractory multiple myeloma patients, when treated with intravenous elotuzumab as single agent therapy. Lets’ take a look at how elotuzumab fared in combination therapy trials,
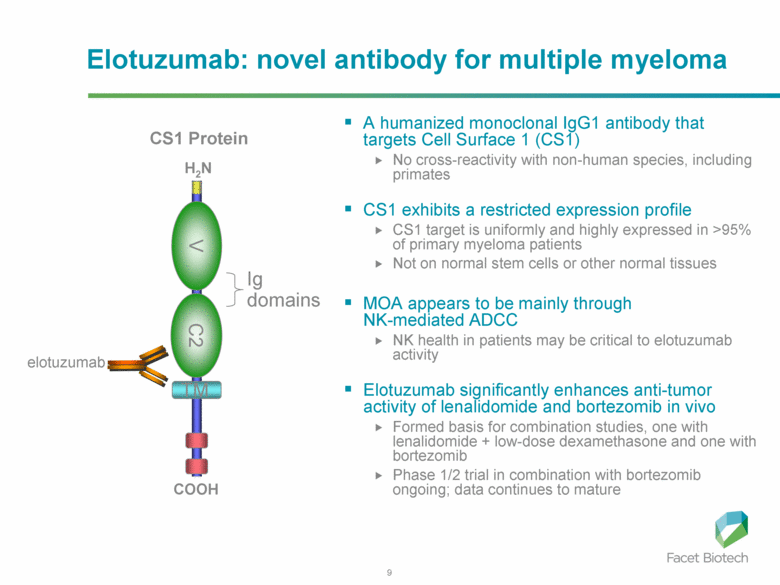
In phase I trial of elotuzumab in combination with Velcade/bortezomib in patients with relapsed/refractory myeloma, the overall response rate was 48% and activity was observed in patients whose disease had stopped responding to Velcade previously. The trial results found that elotuzumab enhanced Velcade activity.
A phase I/II trial in combination with lenalidomide and dexamethasone in refractory/relapsed multiple myeloma patients showed that 82% of patients responded to treatment with a partial response or better and 12% of patients showed complete response. Patients who had received only one prior therapy showed 91% response rate with elotuzumab in combination with lenalidomide and dexamethasone.
Phase I/II trials of the antibody drug has been very impressive and the drug is currently into Phase III trials. Two phase III trials are investigating whether addition of elotuzumab with Revlimid and low dose dexamethasone would increase the time to disease progression. Another phase III trial (ELOQUENT 2) is investigating and comparing safety and efficacy of lenalidomide plus low dose dexamethasone with or without 10mg/kg of elotuzumab in patients with relapsed/refractory multiple myeloma.
Elotuzumab is being investigated in many other trials too. It is being evaluated in combination with Revlimid and low-dose dexamethasone in multiple myeloma patients with various levels of kidney functions, while another phase II study is investigating elotuzumab’s efficacy in patients with high-risk smoldering myeloma.
The main target of multiple myeloma drug development is to satisfy the unmet need for drugs that would improve survival rates. Elotuzumab is an example that mandates much interest in this area and should be followed with diligence.

On November 30, 2015, the U. S. Food and Drug Administration approved elotuzumab (EMPLICITI, Bristol-Myers Squibb Company) in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received one to three prior therapies.
Elotuzumab is a monoclonal antibody directed against Signaling Lymphocyte Activation Molecule Family 7 (SLAMF7). SLAMF7 is present on myeloma cells and is also present on natural killer cells.
The approval was based on a multicenter, randomized, open-label, controlled trial evaluating progression-free survival (PFS) and overall response rate (ORR) in patients with relapsed or refractory multiple myeloma who had received 1 to 3 prior lines of therapy. A total of 646 patients were randomized (1:1) to receive elotuzumab in combination with lenalidomide and dexamethasone (n=321) or lenalidomide plus dexamethasone alone (n=325). Patients continued treatment until disease progression or the development of unacceptable toxicity.
The trial demonstrated a statistically significant improvement in both PFS and ORR, the trial’s co-primary endpoints. The median PFS in the elotuzumab-containing arm was 19.4 months and 14.9 months in the lenalidomide plus dexamethasone alone arm (hazard ratio 0.70, 95% CI: 0.57, 0.85; p = 0.0004). The ORR in the elotuzumab-containing arm was 78.5% (95% CI: 73.6, 82.9) compared to 65.5% (95% CI: 60.1, 70.7) in the lenalidomide plus dexamethasone alone arm (p=0.0002).
The safety data reflect exposure in 318 patients to elotuzumab in combination with lenalidomide and dexamethasone and 317 patients to lenalidomide plus dexamethasone. The most common adverse reactions (greater than or equal to 20%), with an increased rate in the elotuzumab arm compared to the control arm, were fatigue, diarrhea, pyrexia, constipation, cough, peripheral neuropathy, nasopharyngitis, upper respiratory tract infection, decreased appetite, and pneumonia.
Other important adverse reactions include infusion reactions, infections, second primary malignancies, hepatotoxicity, and interference with determination of complete response. As elotuzumab is an IgG kappa monoclonal antibody, it can be detected in the serum protein electrophoresis and immunofixation assays used to assess response.
Serious adverse events occurred in 65.4% of patients in the elotuzumab-containing arm compared to 56.5% in the lenalidomide plus dexamethasone alone arm. The most common serious adverse reactions were pneumonia, pyrexia, respiratory tract infection, anemia, pulmonary embolism, and acute renal failure.
The recommended dose and schedule for elotuzumab is 10 mg/kg intravenously every week for the first two cycles and every 2 weeks, thereafter, until disease progression or unacceptable toxicity with lenalidomide 25 mg daily orally on days 1 through 21. Dexamethasone is administered as follows: In weeks with elotuzumab infusion, dexamethasone is to be administered in divided doses, 8 mg intravenously prior to infusion and 28 mg orally; in weeks without elotuzumab infusion, dexamethasone is to be administered 40 mg orally. Pre-medication with an H1 blocker, H2 blocker, and acetaminophen should be administered prior to elotuzumab infusion.
Elotuzumab is being approved prior to the Prescription Drug User Fee Act (PDUFA) goal date of February 29, 2016. This application was granted priority review and had breakthrough therapy designation. A description of these expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics, available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358301.pdf
Full prescribing information is available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/761035s000lbl.pdf
Empliciti’s Cost
Empliciti will be sold in the U.S. in two vials sizes: A smaller vial that contains 300 mg of the drug, and a larger vial that contains 400 mg.
Bristol-Myers Squibb has informed The Beacon that the wholesale price per vial of Empliciti will be $1,776 for the 300 mg vial and $2,368 for the 400 mg vial.
Using these prices and an assumed patient weight of between 154 and 176 pounds, Empliciti will cost $18,944 per four-week cycle for each of the first two cycles of treatment, and $9,472 per cycle thereafter. This means, in turn, that Empliciti’s cost per year will be $142,080 in the first year and $123,136 in subsequent years.
In comparison, Velcade costs between $4,800 and $8,500 per four-week cycle, depending on how often it is dosed. Ninlaro costs $8,670 per four-week cycle. And Kyprolis costs $10,500 per four-week cycle at the standard (20 – 27 mg/m2) dose.
Additional details about the FDA approval of Empliciti can be found in this press release from the FDA, a related press release from Bristol-Myers Squibb and AbbVie, and the full Empliciti prescribing information.
The results of the ELOQUENT-2 trial were published in Lonial, S. et al., “Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma,” The New England Journal of Medicine, June 2, 2015 (abstract). Slides from the ASCO presentation summarizing the ELOQUENT-2 results can be viewed here (PDF, courtesy of Dr. Lonial). This Beacon news article provides an in-depth look at the trial results.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | SLAMF7 (CD319) |
| Clinical data | |
| Trade names | Empliciti |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
IV |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Identifiers | |
| CAS Number | 915296-00-3 |
| ATC code | None |
| IUPHAR/BPS | 8361 |
| UNII | 1351PE5UGS |
| Chemical data | |
| Formula | C6476H9982N1714O2016S42 |
| Molecular mass | 145.5 kDa |
References
1 “Press Announcement—FDA approves Empliciti, a new immune-stimulating therapy to treat multiple myeloma”. U.S. Food and Drug Administration. Retrieved 3 December 2015.
2“Empliciti (elotuzumab) for Injection, for Intravenous Use. Full Prescribing Information” (PDF). Empliciti (elotuzumab) for US Healthcare Professionals. Bristol-Myers Squibb Company, Princeton, NJ 08543 USA.
3 “Bristol-Myers Squibb and AbbVie Receive U.S. FDA Breakthrough Therapy Designation for Elotuzumab, an Investigational Humanized Monoclonal Antibody for Multiple Myeloma” (Press release). Princeton, NJ & North Chicago, IL: Bristol-Myers Squibb. 2014-05-19. Retrieved 2015-02-05.
///////
Pacritinib

Pacritinib
パクリチニブ;
| Formula |
C28H32N4O3
|
|---|---|
| CAS |
937272-79-2
|
| Mol weight |
472.5787
|
UPDATE FDA APPROVED 2/28/2022, Vonjo
To treat intermediate or high-risk primary or secondary myelofibrosis in adults with low platelets
A Jak2 inhibitor potentially for the treatment of acute myeloid Leukemia and myelofibrosis.
UNII-G22N65IL3O
пакритиниб
باكريتينيب
帕瑞替尼
ONX-0803; SB-1518
CAS No. 937272-79-2
472.57868 g/mol, C28H32N4O3
S*Bio Pte Ltd. and concert innovator
11-(2-pyrrolidin-1-ylethoxy)-14,19-dioxa-5,7,26-triazatetracyclo(19.3.1.1(2,6).1(8,12))heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
| Pacritinib (SB1518) is a potent and selective inhibitor of Janus Kinase 2 (JAK2) and Fms-Like Tyrosine Kinase-3 (FLT3) with IC50s of 23 and 22 nM, respectively. | ||||||
UPDATED
Pacritinib, sold under the brand name Vonjo, is an anti-cancer medication used to treat myelofibrosis.[1][2] It is a macrocyclic Janus kinase inhibitor. It mainly inhibits Janus kinase 2 (JAK2) and Fms-like tyrosine kinase 3 (FLT3).
Common side effects include diarrhea, low platelet counts, nausea, anemia, and swelling in legs.[2]
Medical uses
Pacritinib in indicated to treat adults who have a rare form of a bone marrow disorder known as intermediate or high-risk primary or secondary myelofibrosis and who have platelet (blood clotting cells) levels below 50,000/µL.[1][2]
History
The effectiveness and safety of pacritinib were demonstrated in a study that included 63 participants with intermediate or high-risk primary or secondary myelofibrosis and low platelets who received pacritinib 200 mg twice daily or standard treatment.[2] Effectiveness was determined based upon the proportion of participants who had a 35% or greater spleen volume reduction from baseline to week 24.[2] Nine participants (29%) in the pacritinib treatment group had a 35% or greater spleen volume reduction, compared to one participant (3%) in the standard treatment group.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pacritinib priority review, fast track, and orphan drug designations.[2]
Society and culture
Names
Pacritinib is the International nonproprietary name (INN).[3][4]
References
- ^ Jump up to:a b c “Enforcement Reports”. Accessdata.fda.gov. Retrieved 5 March 2022.
- ^ Jump up to:a b c d e f g h “FDA approves drug for adults with rare form of bone marrow disorder”. U.S. Food and Drug Administration. 1 March 2022. Retrieved 3 March 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ World Health Organization (2010). “International nonproprietary names for pharmaceutical substances (INN). proposed INN: list 104” (PDF). WHO Drug Information. 24 (4): 386. hdl:10665/74579.
- ^ World Health Organization (2011). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 66”. WHO Drug Information. 25 (3). hdl:10665/74683.
External links
- “Pacritinib”. Drug Information Portal. U.S. National Library of Medicine.
OLD—
Pacritinib (INN[1]) is a macrocyclic Janus kinase inhibitor that is being developed for the treatment of myelofibrosis. It mainly inhibits Janus kinase 2 (JAK2). The drug is in Phase III clinical trials as of 2013.[2] The drug was discovered in Singapore at the labs of S*BIO Pte Ltd. It is a potent JAK2 inhibitor with activity of IC50 = 23 nM for the JAK2WT variant and 19 nM for JAK2V617F with very good selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively).[3][4] The drug is acquired by Cell Therapeutics, Inc. (CTI) and Baxter international and could effectively address an unmet medical need for patients living with myelofibrosis who face treatment-emergent thrombocytopenia on marketed JAK inhibitors.[5]
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.
Synthesis Reference
A245943 — William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, Goh KC, Ong WC, Goh SK, Hart S, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW: Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011 Jul 14;54(13):4638-58. doi: 10.1021/jm200326p. Epub 2011 Jun 15.
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.

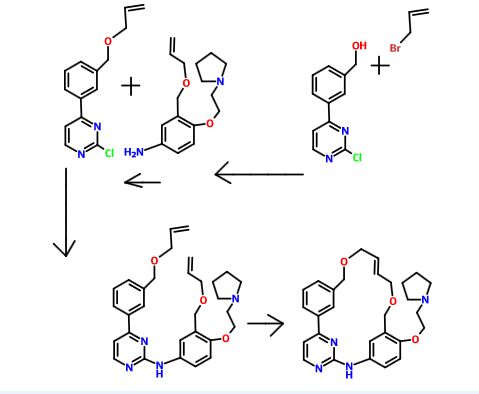
The compound 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) was first described in PCT/SG2006/000352 and shows significant promise as a pharmaceutically active agent for the treatment of a number of medical conditions and clinical development of this compound is underway based on the activity profiles demonstrated by the compound.
-
In the development of a drug suitable for mass production and ultimately commercial use acceptable levels of drug activity against the target of interest is only one of the important variables that must be considered. For example, in the formulation of pharmaceutical compositions it is imperative that the pharmaceutically active substance be in a form that can be reliably reproduced in a commercial manufacturing process and which is robust enough to withstand the conditions to which the pharmaceutically active substance is exposed.
-
In a manufacturing sense it is important that during commercial manufacture the manufacturing process of the pharmaceutically active substance be such that the same material is reproduced when the same manufacturing conditions are used. In addition it is desirable that the pharmaceutically active substance exists in a solid form where minor changes to the manufacturing conditions do not lead to major changes in the solid form of the pharmaceutically active substance produced. For example it is important that the manufacturing process produce material having the same crystalline properties on a reliable basis and also produce material having the same level of hydration.
-
In addition it is important that the pharmaceutically active substance be stable both to degradation, hygroscopicity and subsequent changes to its solid form. This is important to facilitate the incorporation of the pharmaceutically active substance into pharmaceutical formulations. If the pharmaceutically active substance is hygroscopic (“sticky”) in the sense that it absorbs water (either slowly or over time) it is almost impossible to reliably formulate the pharmaceutically active substance into a drug as the amount of substance to be added to provide the same dosage will vary greatly depending upon the degree of hydration. Furthermore variations in hydration or solid form (“polymorphism”) can lead to changes in physico-chemical properties, such as solubility or dissolution rate, which can in turn lead to inconsistent oral absorption in a patient.
-
Accordingly, chemical stability, solid state stability, and “shelf life” of the pharmaceutically active substance are very important factors. In an ideal situation the pharmaceutically active substance and any compositions containing it, should be capable of being effectively stored over appreciable periods of time, without exhibiting a significant change in the physico-chemical characteristics of the active substance such as its activity, moisture content, solubility characteristics, solid form and the like.
-
In relation to 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene initial studies were carried out on the hydrochloride salt and indicated that polymorphism was prevalent with the compound being found to adopt more than one crystalline form depending upon the manufacturing conditions. In addition it was observed that the moisture content and ratio of the polymorphs varied from batch to batch even when the manufacturing conditions remained constant. These batch-to-batch inconsistencies and the exhibited hygroscopicity made the hydrochloride salt less desirable from a commercial viewpoint.
-
Accordingly it would be desirable to develop one or more salts of 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene which overcome or ameliorate one or more of the above identified problems.
PATENT
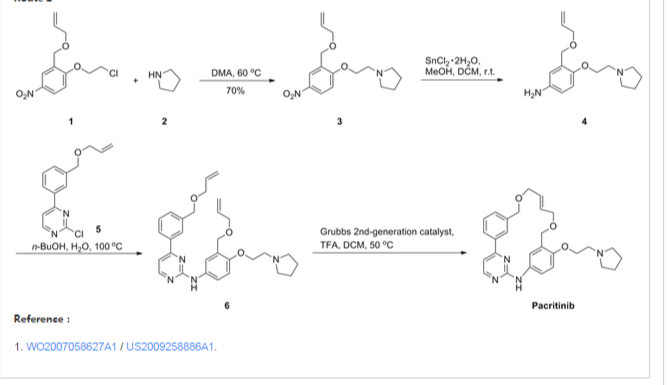
US 2011263616
http://www.google.com/patents/US20110263616
11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26triaza-tetra-cyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) which have been found to have improved properties. In particular the present invention relates to the maleate salt of this compound. The invention also relates to pharmaceutical compositions containing this salt and methods of use of the salt in the treatment of certain medical conditions.

PATENT
http://www.google.com/patents/US8415338
Representative Procedure for the Synthesis of Compounds Type (XVIIId) [3-(2-Chloro-pyrimidin-4-yl)-phenyl]-methanol (XIIIa2)

Compound (XIIIa2) was obtained using the same procedure described for compound (XIIIa1); LC-MS (ESI positive mode) m/z 221 ([M+H]+).
4-(3-Allyloxymethyl-phenyl)-2-chloro-pyrimidine (XVa2)

Compound (XVa2) was obtained using the same procedure described for compound (XVa1); LC-MS (ESI positive mode) m/z 271 ([M+H]+).
[4-(3-Allyloxymethyl-phenyl)-pyrimidin-2-yl]-[3-allyloxymethyl-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amine (XVIId1)

Compound (XVIId1) was obtained using the same procedure described for compound (XVIIb1); LC-MS (ESI positive mode) m/z 501.
Macrocycle Example 3 Compound 13

Compound (13) was obtained using the same procedure described for compound (1) HPLC purity at 254 nm: 99%; LC-MS (ESI positive mode) m/z 473 ([M+H]+); 1H NMR (MeOD-d4) δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34-8.31 (m, 1H), 7.98-7.96 (m, 1H), 7.62-7.49 (m, 2H), 7.35 (d, 1H), 7.15-7.10 (m, 1H), 7.07-7.02 (m, 1H), 5.98-5.75 (m, 2H, 2×=CH), 4.67 (s, 2H), 4.67 (s, 2H), 4.39-4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88-3.82 (m, 2H), 3.70 (t, 2H), 2.23-2.21 (m, 2H), 2.10-2.07 (m, 2H).
PAPER
J MC 2011, 54 4638
http://pubs.acs.org/doi/abs/10.1021/jm200326p

Discovery of the activating mutation V617F in Janus Kinase 2 (JAK2V617F), a tyrosine kinase critically involved in receptor signaling, recently ignited interest in JAK2 inhibitor therapy as a treatment for myelofibrosis (MF). Herein, we describe the design and synthesis of a series of small molecule 4-aryl-2-aminopyrimidine macrocycles and their biological evaluation against the JAK family of kinase enzymes and FLT3. The most promising leads were assessed for their in vitro ADME properties culminating in the discovery of 21c, a potent JAK2 (IC50 = 23 and 19 nM for JAK2WT and JAK2V617F, respectively) and FLT3 (IC50 = 22 nM) inhibitor with selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively). Further profiling of 21c in preclinical species and mouse xenograft and allograft models is described. Compound 21c(SB1518) was selected as a development candidate and progressed into clinical trials where it is currently in phase 2 for MF and lymphoma.
 Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
S*BIO Pte. Ltd., 1 Science Park Road, #05-09, The Capricorn, Singapore Science Park II, Singapore 117528
J. Med. Chem., 2011, 54 (13), pp 4638–4658
DOI: 10.1021/jm200326p
Publication Date (Web): May 23, 2011
Copyright © 2011 American Chemical Society
(21c)
The title compound was synthesized from 21a and pyrrolidine (yield, 83%; mixture of trans/cis85:15 by NMR). LC-MS (ESI positive mode) m/z 473 ([M + H]+). HRMS: theoretical C28H32N4O3MW, 472.2474; found, 473.2547. 1H NMR (MeOD-d4): δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34–8.31 (m, 1H, CH), 7.98–7.96 (m, 1H), 7.62–7.49 (m, 2H), 7.35 (d, 1H), 7.15–7.10 (m, 1H), 7.07–7.02 (m, 1H), 5.98–5.75 (m, 2H), 4.67 (s, 2H), 4.67 (s, 2H), 4.39–4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88–3.82 (m, 2H), 3.70 (t, 2H), 2.23–2.21 (m, 2H), 2.10–2.07 (m, 2H); chloride content (titration) 7.7% (1.18 equivs); water content (Karl Fischer) 6.1% (1.85 equivs); Anal. Calcd. for C28H32N4O3·1.18HCl·1.85H2O: C, 61.46; H, 6.46; N, 10.24; Cl, 7.65. Found: C, 61.99; H, 6.91; N, 10.25; Cl, 7.45.
References
1 “International Nonproprietary Names for Pharmaceutical Substances (INN) List 104” (PDF). WHO Drug Information 24 (4): 386. 2010.
2“JAK-Inhibitoren: Neue Wirkstoffe für viele Indikationen”. Pharmazeutische Zeitung (in German) (21). 2013.
3William, A. D.; Lee, A. C. -H.; Blanchard, S. P.; Poulsen, A.; Teo, E. L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E. T.; Goh, K. C.; Ong, W. C.; Goh, S. K.; Hart, S.; Jayaraman, R.; Pasha, M. K.; Ethirajulu, K.; Wood, J. M.; Dymock, B. W. (2011). “Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma”. Journal of Medicinal Chemistry 54 (13): 4638–58. doi:10.1021/jm200326p. PMID 21604762.
4Poulsen, A.; William, A.; Blanchard, S. P.; Lee, A.; Nagaraj, H.; Wang, H.; Teo, E.; Tan, E.; Goh, K. C.; Dymock, B. (2012). “Structure-based design of oxygen-linked macrocyclic kinase inhibitors: Discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3)”. Journal of Computer-Aided Molecular Design 26 (4): 437–50. doi:10.1007/s10822-012-9572-z. PMID 22527961.
5http://www.pmlive.com/pharma_news/baxter_licenses_cancer_drug_from_cti_in_$172m_deal_519143
| US8153632 * | Nov 15, 2006 | Apr 10, 2012 | S*Bio Pte Ltd. | Oxygen linked pyrimidine derivatives |
| US8415338 * | Apr 4, 2012 | Apr 9, 2013 | Cell Therapeutics, Inc. | Oxygen linked pyrimidine derivatives |
| US20110294831 * | Dec 9, 2009 | Dec 1, 2011 | S*Bio Pte Ltd. | 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene citrate salt |
| Patent | Submitted | Granted |
|---|---|---|
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US8153632] | 2009-03-19 | 2012-04-10 |
| ANTIVIRAL JAK INHIBITORS USEFUL IN TREATING OR PREVENTING RETROVIRAL AND OTHER VIRAL INFECTIONS [US2014328793] | 2012-11-30 | 2014-11-06 |
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US2013172338] | 2013-02-20 | 2013-07-04 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE MALEATE SALT [US2011263616] | 2011-10-27 | |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE CITRATE SALT [US2011294831] | 2011-12-01 | |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| Oxygen linked pyrimidine derivatives [US8415338] | 2012-04-04 | 2013-04-09 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(16E)-11-[2-(1-Pyrrolidinyl)ethoxy]-14,19-dioxa-5,7,26-triazatetracyclo[19.3.1.12,6.18,12]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 46216796 |
| ChemSpider | 28518965 |
| ChEMBL | CHEMBL2035187 |
| Synonyms | SB1518 |
| Chemical data | |
| Formula | C28H32N4O3 |
| Molecular mass | 472.58 g/mol |


S*Bio Pte Ltd
Address: 1 Science Park Rd, Singapore 117528
Phone:+65 6827 5000

S*BIO Pte Ltd. provides research and clinical development services for small molecule drugs for the treatment of cancer in Singapore. The company’s products include JAK2 inhibitors, such as SB1518 for leukemia/myelofibrosis, lymphoma, and polycythemia; and SB1578 for RA/psoriasis. The company also offers SB939, a histone deacetylases for MDS/AML+combo, prostate cancer, sarcoma, pediatric tumor, and myelofibrosis; SB2602, a mTOR inhibitor; SB2343, a mTOR/PI3K inhibitor; and SB1317, a CDK/Flt3 inhibitor. The company was founded in 2000 and is based in Singapore. S*BIO Pte Ltd. operates as a subsidiary of Chiron Corporation Limited.
SEE……..http://apisynthesisint.blogspot.in/2016/01/pacritinib.html

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
|
|
| Clinical data | |
|---|---|
| Trade names | Vonjo |
| Other names | SB1518 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C28H32N4O3 |
| Molar mass | 472.589 g·mol−1 |
| 3D model (JSmol) | |
///////Vonjo, FDA APPTOVESD 2022, APPROVALS 2022, PACRITINIB, パクリチニブ, priority review, fast track, orphan drug, UNII-G22N65IL3O, пакритиниб , باكريتينيب , 帕瑞替尼 , SB 1518
c1cc2cc(c1)-c3ccnc(n3)Nc4ccc(c(c4)COC/C=C/COC2)OCCN5CCCC5
C1CCN(C1)CCOC2=C3COCC=CCOCC4=CC=CC(=C4)C5=NC(=NC=C5)NC(=C3)C=C2
VAL-083

VAL-083
(1R,2S)-1-((R)-oxiran-2-yl)-2-((S)-oxiran-2-yl)ethane-1,2-diol
Galactitol, 1,2:5,6-dianhydro-
- 1,2:5,6-Dianhydrodulcitol
- 1,2:5,6-Dianhydrogalactitol
- 1,2:5,6-Diepoxydulcitol
Dianhydrodulcitol; Dianhydrogalactitol; VAL083; VAL 083, Dulcitol diepoxide, NSC 132313
CAS 23261-20-3
MF C6H10O4, MW 146.14
VAL-083 is a bi-functional alkylating agent; inhibit U251 and SF188 cell growth in monolayer better than TMZ and caused apoptosis
VAL-083 is a bi-functional alkylating agent, with potential antineoplastic activity. Upon administration, VAL-083 crosses the blood brain barrier (BBB) and appears to be selective for tumor cells. This agent alkylates and crosslinks DNA which ultimately leads to a reduction in cancer cell proliferation. In addition, VAL-083 does not show cross-resistance to other conventional chemotherapeutic agents and has a long half-life in the brain. Check for active clinical trials or closed clinical trials using this agent
Currently, VAL-083 is approved in China to treat chronic myelogenous leukemia and lung cancer, while the drug has also secured orphan drug designation in Europe and the US to treat malignant gliomas.
LAUNCHED CHINA FOR Cancer, lung
Del Mar Pharmaceuticals Inc……..Glioblastoma…………..PHASE2
DelMar and MD Anderson to accelerate development of anti-cancer drug VAL-083
DelMar Pharmaceuticals has collaborated with the University of Texas MD Anderson Cancer Center (MD Anderson) to speed up the clinical development of its VAL-083 anti-cancer drug.
VAL-083 is a BI-Functional alkylating agent; INHIBIT U251 and SF188 Cell Growth in monolayer Better than TMZ and Caused apoptosis. IC50 Value : 5 uM (INHIBIT U251, SF188, T98G Cell Growth in monolayer after 72h) [1]. in vitro :.. VAL-083 INHIBITED U251 and SF188 Cell Growth in monolayer and as neurospheres Better than TMZ and Caused apoptosis after 72 hr Formation Assay In the colony, VAL-083 (5 uM) SF188 Growth suppressed by about 95% are T98G cells classically TMZ-resistant and express MGMT, but VAL-083 inhibited their growth in monolayer after 72 hr in a dose-dependent manner (IC50, 5 uM). VAL-083 also inhibited the growth of CSCs (BT74, GBM4, and GBM8) . by 80-100% in neurosphere self-Renewal assays Conversely, there was minimal normal Effect on Human Neural stem cells [1]. in Vivo : Clinical Trial : Safety Study of VAL-083 in Patients With Recurrent Malignant glioma or Secondary Progressive Brain Tumor. Phase 1 / Phase 2

VAL-083 has demonstrated activity in cyclophosphamide, BCNU and phenylanine mustard resistant cell lines and no evidence of cross-resistance has been encountered in published clinical studies. Based on the presumed alkylating functionality of VAL-083, published literature suggests that DNA repair mechanisms associated with Temodar and nitrosourea resistance, such as 06-methylguanine methyltransferace (MGMT), may not confer resistance to VAL-083. VAL-083 readily crosses the blood brain barrier where it maintains a long half-life in comparison to the plasma. Published preclinical and clinical research demonstrates that VAL-083 is selective for brain tumor tissue. VAL-083 has been assessed in multiple studies as chemotherapy in the treatment of newly diagnosed and recurrent brain tumors. In published clinical studies, VAL-083 has previously been shown to have a statistically significant impact on median survival in high grade gliomas when combined with radiation vs. radiation alone. The main dose-limiting toxicity related to the administration of VAL-083 in previous clinical studies was myelosuppression

Glioblastoma is the most common form of primary brain cancer
DelMar Pharmaceuticals has collaborated with the University of Texas MD Anderson Cancer Center (MD Anderson) to speed up the clinical development of its VAL-083 anti-cancer drug.
VAL-083 is a small-molecule chemotherapeutic designed to treat glioblastoma multiforme (GBM), the most common and deadly cancer that starts within the brain.
Under the deal, MD Anderson will begin a new Phase II clinical trial with VAL-083 in patients with GBM at first recurrence / progression, prior to Avastin (bevacizumab) exposure.
During the trial, eligible patients will have recurrent GBM characterised by a high expression of MGMT, the DNA repair enzyme implicated in drug-resistance, and poor patient outcomes following current front-line chemotherapy.
” … Our research shows that VAL-083 may offer advantages over currently available chemotherapies in a number of tumour types.”
The company noted that MGMT promoter methylation status will be used as a validated biomarker for enrollment and tumours must exhibit an unmethylated MGMT promoter for patients to be eligible for the trial.
DelMar chairman and CEO Jeffrey Bacha said: “The progress we continue to make with our research shows that VAL-083 may offer advantages over currently available chemotherapies in a number of tumour types.
“This collaboration will allow us to leverage world-class clinical and research expertise and a large patient population from MD Anderson as we extend and accelerate our clinical focus to include GBM patients, following first recurrence of their disease.
“We believe that VAL-083’s unique cytotoxic mechanism offers promise for GBM patients across the continuum of care as a potential superior alternative to currently available cytotoxic chemotherapies, especially for patients whose tumours exhibit a high-expression of MGMT.”
The deal will see DelMar work with the scientists and clinicians at MD Anderson to accelerate its research in order to transform the treatment of patients whose cancers fail or are unlikely to respond to existing treatments.
In more than 40 clinical trials, VAL-083 showed clinical activity against several cancers including lung, brain, cervical, ovarian tumours and leukemia both as a single-agent and in combination with other treatments.
PATENT
WO 2012024368
https://www.google.com/patents/WO2012024368A3?cl=en
Dianhydrogalactitol (DAG or dianhydrodulcitol) can be synthesized from dulcitol which can be produced from natural sources (such as Maytenus confertiflora) or commercial sources.The structure of DAG is given below as Formula (I).

One method for the preparation of dulcitol from Maytenus confertiflora is as follows: (1) The Maytenus confertiflora plant is soaked in diluted ethanol (50-80%) for about 24 hours, and the soaking solution is collected. (2) The soaking step is repeated, and all soaking solutions are combined. (3) The solvent is removed by heating under reduced pressure. (4) The concentrated solution is allowed to settle overnight and the clear supernatant is collected. (5) Chloroform is used to extract the supernatant. The chloroform is then removed under heat and reduced pressure. (6) The residue is then dissolved in hot methanol and cooled to allow crystallization. (7) The collected crystals of dulcitol are filtered and dried under reduced pressure. The purified material is dulcitol, contained in the original Maytenus confertiflora plant at a concentration of about 0.1% (1/1000).
DAG can be prepared by two general synthetic routes as described below:
Route 1 :
Dulcitol DAG
Route 2. Dulcitol

In Route 1 , “Ts” represents the tosyl group, or p-toluenesulfonyl group. PATENT
However, the intermediate of Route 1, 1,6-ditosy)dulcitol, was prepared with low yield (~36%), and the synthesis of 1,6-ditosyldulcitol was poorly reproducible. Therefore, the second route process was developed, involving two major steps: (1) preparation of dibromodulcitol from dulcitol; and (2) preparation of dianhydrodulcitol from dibromodulcitol.
Dibromodulcitol is prepared from dulcitol as follows: (1) With an aqueous HBr solution of approximately 45% HBr concentration, increase the HBr concentration to about 70% by reacting phosphorus with bromine in concentrated HBr in an autoclave. Cool the solution to 0° C. The reaction is:
2P+3Br2→2PBr3+H20→HBr†+H3P04. (2) Add the dulcitol to the concentrated HBr solution and reflux at 80° C to complete the reaction. (3) Cool the solution and pour the mixture onto ice water. Dibromodulcitol is purified through recrystallization.
The results for the preparation of dibromodulcitol (DBD) are shown in Table 1, below.
TABLE 1

For the preparation of DAG from DBD, DBD was poorly dissolved in methanol and ethanol at 40° C (different from what was described in United States PATENT
Patent No. 3,993,781 to Horvath nee Lengyel et al., incorporated herein by this reference). At refluxing, DBD was dissolved but TLC showed that new impurities formed that were difficult to remove from DBD.
The DBD was reacted with potassium carbonate to convert the DBD to dianhydrogalactitol.
The results are shown in Table 2, below.
TABLE 2

In the scale-up development, it was found the crude yield dropped significantly. It is unclear if DAG could be azeotropic with BuOH. It was confirmed that t-BuOH is essential to the reaction. Using MeOH as solvent would result in many impurities as shown spots on TLC. However, an improved purification method was developed by using a slurry with ethyl ether, which could provide DAG with good purity. This was developed after a number of failed attempts at recrystallization of DAG.

Bromination of dulcitol with HBr at 80°C gives dibromodulcitol , which upon epoxidation in the presence of K2CO3 in t-BuOH or NaOH in H2O or in the presence of ion exchange resin Varion AD (OH) (4) affords the target dianhydrogalactitol .
PATENT



SCHEME 5



PATENT
CN 103923039
http://www.google.com/patents/CN103923039A?cl=en
The resulting Dulcitol 9g and 18ml mass percent concentration of 65% hydrobromic acid at 78 ° C under reflux for 8 hours to give 1,6-dibromo dulcitol, and the product is poured into ice crystals washed anhydrous tert-butyl alcohol, and dried to give 1,6-dibromo dulcitol crystal, then 10.0gl, 6- dibromo dulcitol sample is dissolved in t-butanol, adding solid to liquid 2 % obtained through refining process 1,6_ dibromo dulcitol seed stirred and cooled to 0 ° C, allowed to stand for seven days to give 1,6_ dibromo dulcitol crystal, anhydrous t-butanol, dried to give 1,6-dibromo dulcitol. 5g of the resulting 1,6_ dibromo Euonymus dissolved in 50ml tert-butanol containing 5g of potassium carbonate, the elimination reaction, at 80 ° C under reflux time was 2 hours, the resulting product was dissolved in t-butanol, Join I% stock solution to the water quality of 1,2,4,5_ two Dulcitol including through a purification step to get less than 1% of 1,2,5,6_ two to water Dulcitol seeded stirring, cooling to 0 ° C, allowed to stand for I-day, two to go get 1,2,5,6_ water Dulcitol crystals washed anhydrous tert-butyl alcohol, and dried to give 1,2,5,6 two to crystalline water Dulcitol and lyophilized to give two to water Dulcitol lyophilized powder, containing I, 2,4,5- two to water Dulcitol less than 0.3%.
PATENT
PATENT
-
DAG can be prepared by two general synthetic routes as described below:
-

-
In Route 1, “Ts” represents the tosyl group, or p-toluenesulfonyl group.
-
However, the intermediate of Route 1, 1,6-ditosyldulcitol, was prepared with low yield (˜36%), and the synthesis of 1,6-ditosyldulcitol was poorly reproducible. Therefore, the second route process was developed, involving two major steps: (1) preparation of dibromodulcitol from dulcitol; and (2) preparation of dianhydrodulcitol from dibromodulcitol.
-
Dibromodulcitol is prepared from dulcitol as follows: (1) With an aqueous HBr solution of approximately 45% HBr concentration, increase the HBr concentration to about 70% by reacting phosphorus with bromine in concentrated HBr in an autoclave. Cool the solution to 0° C. The reaction is: 2P+3Br2→2PBr3+H2O→HBr↑+H3PO4. (2) Add the dulcitol to the concentrated HBr solution and reflux at 80° C. to complete the reaction. (3) Cool the solution and pour the mixture onto ice water. Dibromodulcitol is purified through recrystallization.
PATENT
US 20150329511
PAPER
Molecules 2015, 20(9), 17093-17108; doi:10.3390/molecules200917093
Article
Antibacterial and Anti-Quorum Sensing Molecular Composition Derived from Quercus cortex (Oak bark) Extract
Microbiological Department, Orenburg State University, 13 Pobedy Avenue, Orenburg 460018, Russia
* Author to whom correspondence should be addressed.
1,2: 5,6-dianhydrogalactitol ** in table 1
Paper
Takano, Seiichi; Iwabuchi, Yoshiharu; Ogasawara, Kunio
Journal of the American Chemical Society, 1991 , vol. 113, 7 pg. 2786 – 2787
Journal of the American Chemical Society, 1991 , vol. 113, 7 pg. 2786 – 2787

REFERENCES
Currently, VAL-083 is approved in China to treat chronic myelogenous leukemia and lung cancer, while the drug has also secured orphan drug designation in Europe and the US to treat malignant gliomas.
[1]. Fotovati A, Hu KJ, Wakimoto H, VAL-083, A NOVEL N7 ALKYLATING AGENT, SURPASSES TEMOZOLOMIDE ACTIVITY AND INHIBITS CANCER STEM CELLS, PROVIDING A NEW POTENTIAL TREATMENT OPTION FOR GLIOBLASTOMA MULTIFORME. Neuro-oncology, 2012, 14, AbsET-37, Suppl. 6
1: Szende B, Jeney A, Institoris L. The diverse modification of N-butyl-N-(4-hydroxybutyl) nitrosamine induced carcinogenesis in urinary bladder by dibromodulcitol and dianhydrodulcitol. Acta Morphol Hung. 1992;40(1-4):187-93. PubMed PMID: 1365762.
2: Anderlik P, Szeri I, Bános Z. Bacterial translocation in dianhydrodulcitol-treated mice. Acta Microbiol Hung. 1988;35(1):49-54. PubMed PMID: 3293340.
3: Huang ZG. [Clinical observation of 15 cases of chronic myelogenous leukemia treated with 1,2,5,6-dianhydrodulcitol]. Zhonghua Nei Ke Za Zhi. 1982 Jun;21(6):356-8. Chinese. PubMed PMID: 6957285.
4: Anderlik P, Szeri I, Bános Z, Wessely M, Radnai B. Higher resistance of germfree mice to dianhydrodulcitol, a lymphotropic cytostatic agent. Acta Microbiol Acad Sci Hung. 1982;29(1):33-40. PubMed PMID: 6211912.
5: Bános Z, Szeri I, Anderlik P. Effect of Bordetella pertussis vaccine on the course of lymphocytic choriomeningitis (LCM) virus infection in suckling mice pretreated with dianhydrodulcitol (DAD). Acta Microbiol Acad Sci Hung. 1979;26(2):121-5. PubMed PMID: 539467.
6: Bános Z, Szeri I, Anderlik P. Dianhydrodulcitol treatment of lymphocytic choriomeningitis virus infection in suckling mice. Acta Microbiol Acad Sci Hung. 1979;26(1):29-34. PubMed PMID: 484266.
7: Gerö-Ferencz E, Tóth K, Somfai-Relle S, Gál F. Effect of dianhydrodulcitol (DAD) on the primary immune response of normal and tumor bearing rats. Oncology. 1977;34(4):150-2. PubMed PMID: 335301.
8: Kopper L, Lapis K, Institóris L. Incorporation of 3H-dibromodulcitol and 3H-dianhydrodulcitol into ascites tumor cells. Autoradiographic study. Neoplasma. 1976;23(1):47-52. PubMed PMID: 1272473.
9: Bános S, Szeri I, Anderlik P. Combined phytohaemagglutinin and dianhydrodulcitol treatment of lymphocytic choriomeningitis virus infection in mice. Acta Microbiol Acad Sci Hung. 1975;22(3):237-40. PubMed PMID: 1155228.
Carbohydrate Research, 1982 , vol. 108, p. 173 – 180
Deryabin, Dmitry G.; Tolmacheva, Anna A.
Molecules, 2015 , vol. 20, 9 pg. 17093 – 17108
Gati; Somfai-Relle
Arzneimittel-Forschung/Drug Research, 1982 , vol. 32, 2 pg. 149 – 151
| WO2013128285A2 * | Feb 26, 2013 | Sep 6, 2013 | Del Mar Pharmaceuticals | Improved analytical methods for analyzing and determining impurities in dianhydrogalactitol |
| WO2013128285A3 * | Feb 26, 2013 | Dec 27, 2013 | Del Mar Pharmaceuticals | Improved analytical methods for analyzing and determining impurities in dianhydrogalactitol |
| US9029164 | Nov 18, 2013 | May 12, 2015 | Del Mar Pharmaceuticals | Analytical methods for analyzing and determining impurities in dianhydrogalactitol |
| US3470179 * | Jun 14, 1966 | Sep 30, 1969 | Sandoz Ag | 4-substituted-3,4-dihydroquinazolines |
| US20020032230 * | May 21, 2001 | Mar 14, 2002 | Dr. Reddy’s Laboratories Ltd. | Novel compounds having antiinflamatory activity: process for their preparation and pharmaceutical compositions containing them |
| US20020037328 * | May 31, 2001 | Mar 28, 2002 | Brown Dennis M. | Hexitol compositions and uses thereof |
| CN101045542A * | Apr 6, 2007 | Oct 3, 2007 | 中国科学院过程工程研究所 | Method for preparing water softening aluminium stone of sodium aluminate solution carbonation resolving |
| CN101654270A * | Sep 10, 2009 | Feb 24, 2010 | 沈阳工业大学 | Method for eliminating periodic thinning of granularity of seed product |
| CN101775413A * | Mar 23, 2010 | Jul 14, 2010 | 禹城绿健生物技术有限公司 | Technique for producing xylitol and dulcitol simultaneously |
| CN103270035A * | Aug 17, 2011 | Aug 28, 2013 | 德玛医药 | Method of synthesis of substituted hexitols such as dianhydrogalactitol |
/////////////////
C1C(O1)C(C(C2CO2)O)O
O[C@H]([C@H]1OC1)[C@@H](O)[C@H]2CO2
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin
Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
Upadacitinib, ABT-494, упадацитиниб , أوباداسيتينيب , 乌帕替尼 ,
ABT 494
(-)-(3S,4R) cis form
CAS 1310726-60-3 FREE FORM
| MF | C17H19F3N6O |
|---|---|
| MW | 380.36757 g/mol |
Tartrate form
C17 H19 F3 N6 O . C4 H6 O6 . 4 H2 O ………….CAS 1607431-21-9
1-Pyrrolidinecarboxamide, 3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)-, (3S,4R)-, (2R,3R)-2,3-dihydroxybutanedioate, hydrate (1:1:4)
FREE FORM
(3s,,4R)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide.
(35,,4R)-3-ethyl-4-(3H- imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide,
(cis,)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide
1-Pyrrolidinecarboxamide, 3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)-, (3S,4R)-
rel-(-)-(3S,4R)-3-Ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide
A Jak1 inhibitor potentially for the treatment of rheumatoid arthritis.
pharmaceutically acceptable salts thereof, stereoisomers thereof, and isomers thereof, is provided in U.S. Patent No. 8,426,411,
Abbott Laboratories ABBOTT ……INNOVATOR
(3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide
1310726-60-3 Cas
1-Pyrrolidinecarboxamide, 3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)-, (3S,4R)-
ABT-494
UNII:4RA0KN46E0
упадацитиниб [Russian] [INN]
أوباداسيتينيب [Arabic] [INN]
乌帕替尼 [Chinese] [INN]
Arthritis, psoriatic PHASE 3 ABBVIE
Upadacitinib (code name ABT-494) is a drug which is currently under investigation for the treatment of rheumatoid arthritis, Crohn’s disease, ulcerative colitis, and psoriatic arthritis. It was developed by the biotech company AbbVie.
Upadacitinib tartrate, a selective Jak1 inhibitor, is in phase III clinical trials at AbbVie (previously Abbott) for the treatment of patients with moderate to severe rheumatoid arthritis or active psoriatic arthritis with inadequate responses to conventional or biologic disease-modifying antirheumatic drugs (DMARDs). Phase III clinical trials are also ongoing for the treatment of moderately to severely active Crohn’s disease, ulcerative colitis, moderate to severe atopic dermatitis, and active ankylosing spondylitis.
In 2015, orphan drug designation was assigned to the compound for the treatment of pediatric juvenile idiopathic arthritis (JIA) categories excluding systemic JIA. In 2017, additional orphan drug designation was assigned in the U.S. for the treatment of pediatric systemic JIA.
In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie.
Upadacitinib tartrate [USAN]
1607431-21-9
Mechanism of action
The Janus kinases (JAKs) are a family of cytoplasmic tyrosine kinases whose function is to transduce cytokine-mediated signals via the JAK-STAT pathway. There are four JAK subtypes, each of which has overlapping receptor responsibilities. Inhibitors of this enzyme family (jakinibs) have shown efficacy in treating certain inflammatory and autoimmune diseases such as rheumatoid arthritis and Crohn’s disease. However, the first generation of these drugs, tofacitinib and ruxolitinib, lacked subtype selectivity, affecting JAK1/JAK3 and JAK1/JAK2 respectively. This has led to dose-limiting side effects in this otherwise promising class of drugs.[2][3] Upadacitinib is a second generation Janus kinase inhibitor that is selective for the JAK1 subtype of this enzyme over the JAK2 (74-fold), JAK3 (58-fold) and TYK2 subtypes.[4]
Clinical trials
Phase I studies
A phase I study revealed that upadacitinib followed a bi-exponential disposition with a terminal half-life of 6–16 hours.[1] There was no significant accumulation over the dose range of 3–36 mg per day. No interaction was found in rheumatoid arthritis patients taking methotrexate. The most common adverse event was headache but its incidence was similar to that when taking placebo (15.6% for upadacitinib vs. 16.7% for placebo). An investigation into absorption and metabolism found that dosing after a high-fat meal had no effect on upadacitinib total drug exposure over time (area under the curve or AUC).[5] Inhibition of CYP3A by ketoconazole increased total AUC, indicating the importance of this metabolic route.
Phase II studies
Two phase IIb studies were initiated to study the efficacy and safety of upadacitinib in patients with rheumatoid arthritis and one phase II study was initiated in patients with Crohn’s disease.
BALANCE I
In the first study, 276 rheumatoid arthritis patients were recruited who had previously experienced inadequate response to anti–tumor necrosis factor (TNF) therapy and were currently on a stable dose of methotrexate.[6] Patients were randomized to receive 3, 6, 12, or 18 mg twice daily or placebo. The primary endpoint was a 20% improvement in symptoms according to the American College of Rheumatology improvement criteria (ACR20). At the completion of the study it was found that response rates were significantly higher in those receiving upadacitinib versus in those receiving placebo alone (36–42% and 22– 26%, respectively). Adverse events included headache, nausea, and infection but no infections were serious.
BALANCE II
In the second phase IIb study, 300 rheumatoid arthritis patients were recruited who have had an inadequate response to methotrexate.[7] Patients were randomized to receive 3, 6, 12, or 18 mg twice daily or placebo. The primary endpoint was a 20% improvement in symptoms according to the American College of Rheumatology improvement criteria (ACR20). At the completion of the study it was found that response rates were significantly higher in those receiving upadacitinib versus in those receiving placebo alone. (62%, 68%, 80%, 64%, and 76% for the 3, 6, 12, 18, and 24 mg doses, respectively) than with placebo (46%). Improvement in symptoms was rapid, with significant changes in disease scores by week 2. Adverse events were mild with infection being the most serious. One case of community-acquired pneumonia occurred at 12 mg.
CELEST
In this 16-week study, 220 patients were recruited with moderately to severely active Crohn’s disease. Participants must have also experienced an inadequate response to or intolerance to Immunotherapy or TNF inhibitors.[8][9] Patients were randomized to therapy with upadacitinib at 3, 6, 12, 24 mg twice daily or 24 mg once daily for 16 weeks or placebo, followed by blinded extension therapy for 36 weeks. The co-primary endpoints were the proportion of patients who achieved clinical remission (soft stool frequency or daily abdominal pain score) at week 16 and endoscopic remission at week 12 or 16. Secondary endpoints included significant clinical response (≥30% reduction in symptoms) at week 16 and endoscopic response (≥25% decrease in symptoms) at week 12 or 16. At 16 weeks 22% of patients taking the 24 mg twice daily dose achieved endoscopic remission with upadacitinib compared to 0% of patients taking placebo. 27% of patients taking the 6 mg twice daily dose achieved clinical remission compared to 11% of patients taking placebo. Adverse events did not appear to be dose-related. A single case of non-melanoma skin cancer was reported in the 24 mg twice daily group.
Phase III studies
Abbvie has planned a total of six phase III trials that will evaluate over 4,000 patients with moderate to severe rheumatoid arthritis.[10] Two phase III trials are planned studying patients with psoriatic arthritis and one in patients with ulcerative colitis.
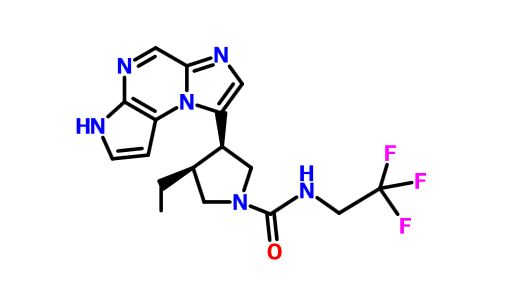
AbbVie, a global biopharmaceutical company, today announced the start of a large Phase 3 clinical trial program to study the use of ABT-494, an investigational, once-daily, oral selective JAK1 inhibitor for the treatment of rheumatoid arthritis (RA). This program will include adult patients with inadequate responses (IR) to conventional or biologic disease-modifying antirheumatic drugs (DMARDs), as well as methotrexate-naive patients.
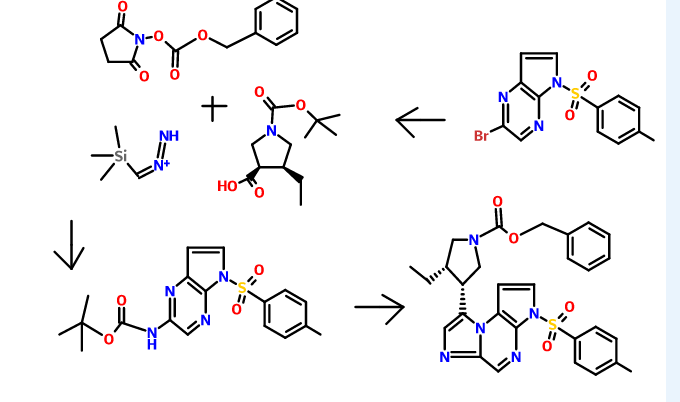

PATENT
WO2015061665
The synthesis of the compounds of the invention, including (35,,4R)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine- l-carboxamide, pharmaceutically acceptable salts thereof, stereoisomers thereof, and isomers thereof, is provided in U.S. Patent No. 8,426,411, the entire content of which is incorporated herein by reference.
For example, (3lS,,4R)-3-ethyl-4-(3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide can be synthesized according to the following scheme:
N-Alkylation using alkyl halide, a-haloketone or oc-haloamide
A round bottom flask is charged with a base such as NaH (60% dispersion in mineral oil), K2CO3, or CS2CO3 (preferably NaH (60% dispersion in mineral oil), 0.9-1.5 equiv., preferably 0.95 equiv.) and an organic solvent (such as N, N-dimethylformamide (DMF), dichloromethane (DCM), 1,4-dioxane, or N-methyl-2-pyrrolidone (NMP), preferably DMF). The mixture is cooled to about -10 °C to ambient temperature (preferably about 0°C) and a solution of an appropriately substituted amine (preferably 1 equiv.) in an organic solvent (such as DMF) is added. Alternatively, the base may be added portionwise to a solution of the amine and an organic solvent at about 0°C to ambient temperature. The reaction mixture is stirred for about 5-90 min (preferably about 15-30 min) at about -10°C to ambient temperature (preferably about 0°C) followed by the addition of an alkyl halide, a-haloketone, or cc-haloamide (1-2 equiv., preferably 1.2 equiv.). Alternatively, a solution of an amine and a base in an organic solvent may be added to a solution of an alkyl halide, α-haloketone, or a-haloamide in an organic solvent at about 0°C. The reaction mixture is stirred at about -10°C to ambient temperature (preferably ambient temperature) for about 0.5-24 h (preferably about 1 h). Optionally, the organic solvent may be removed under reduced pressure.
Optionally, the reaction mixture or residue may be diluted with water, aqueous NH4CI, or aqueous NaHC03. If a precipitate forms the solid may be optionally collected via vacuum filtration to give the target compound. Alternatively, an organic solvent (such as ethyl acetate (EtOAc) or DCM) is added to the aqueous mixture and the layers are separated. The aqueous layer may optionally be extracted further with an organic solvent (such as EtOAc and/or DCM). The combined organic layers are optionally washed with additional aqueous solutions such as brine, dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure.
The procedure above is illustrated below in the preparation of ie/t-butyl 2-amino-2-oxoethyl(5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-yl)carbamate from ie/t-butyl (5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)carbamate.
To a solution of iert-butyl 5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-ylcarbamate (1.00 g, 2.57 mmol, Example #3 Step E) and DMF (13 mL) under nitrogen at about 0 °C was added NaH (60% dispersion in mineral oil, 0.113 g, 2.83 mmol) in one portion. After about 30 min, 2-bromoacetamide (0.391 g, 2.83 mmol) was added in one portion. After about 30 min, the ice bath was removed and the solution was stirred at ambient temperature for about 2 h. Saturated aqueous NH4Cl/water (1: 1, 100 mL) was added. After stirring for about 10 min, the mixture was filtered using water to wash the filter cake. The aqueous phase was extracted with EtOAc (50 mL). The filter cake was dissolved in EtOAc and added to the organic layer. The organic layer was dried over Na2S04, filtered, and concentrated under reduced pressure. The material was purified by silica gel chromatography eluting with a gradient of 20-100% EtOAc/heptane to give tert-butyl 2-amino-2-oxoethyl(5-tosyl-5H-pyrrolo[3,2-b]pyrazin-2-yljcarbamate (0.980 g, 82%): LC/MS (Table 1, Method n) Rt = 0.70 min; MS m/z 446 (M+H)+.
Similar reaction condition can also be used to synthesize benzyl 3-ethyl-4-(2-((5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)amino)acetyl)pyrrolidine-l-carboxylate from iert-butyl (5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-yl)carbamate and benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine- 1 -carboxylate.
Cyclization of a ketone using a dithiaphosphetane reagent (e.g., synthesizing (3S,4R)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate from benzyl 3-ethyl-4-(2-((5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-yl)amino)acetyl)pyrrolidine-l-carboxylate)
To a solution of a ketone (preferably 1 equiv.) in an organic solvent such as tetrahydrofuran (THF) or 1,4-dioxane (preferably 1,4-dioxane) is added a thiolating reagent such as Lawesson’s reagent or Belleau’s reagent (2,4-bis(4-phenoxyphenyl)-l,3-dithia-2,4-diphosphetane-2,4-disulfide) (0.5-2.0 equiv., preferably Lawesson’s reagent, 0.5-0.6 equiv.). The reaction is heated at about 30°C to 120°C (preferably about 60-70°C) for about 0.5-10 h (preferably about 1-2 h). Optionally, additional thiolating reagent (0.5-2.0 equiv., preferably 0.5-0.6 equiv.) can be added to the reaction mixture and heating can be continued for about 0.5-10 h (preferably about 1-2 h). The reaction mixture is concentrated under reduced pressure.
Preparation of 8-((ds)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine from (3S,4R)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate
To a solution of (cis)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-l-carboxylate (0.838 g, 1.541 mmol) is added a solution of HBr (2.50 mL, 15.19 mmol, 33% in acetic acid). The reaction mixture is stirred at ambient temperature for about 1 h. The reaction is diluted with diethyl ether or Et20 (50 mL) and water (20 mL). The layers are stirred for about 3 min and the organic layer is decanted then the procedure is repeated 5 times. The aqueous layer is cooled to about 0°C and is basified with saturated aqueous NaHC03 solution (10 mL) to about pH 7. The aqueous layer is extracted with EtOAc (3 x 50 mL), combined, and dried over anhydrous Na2S04, filtered and concentrated to give a brown solid. The solid is dissolved in DCM (50 mL) and washed with water (3 x 20 mL), dried over anhydrous Na2S04, filtered and concentrated to afford 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine (0.453, 61%) as a brown residue: LC/MS (Table 1, Method a) Rt = 1.73 min; MS m/r. 410 (M+H)+.
Hydrolysis of a sulfonamide (e.g., 8-((3R,4S)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine to 8-((3R,4S)-4-ethylpyrrolidin-3-yl)-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine)
To a flask containing a sulfonamide, for example, a sulfonyl-protected pyrrole, (preferably 1 equiv.) in an organic solvent (such as 1,4-dioxane, methanol (MeOH), or THF/MeOH, preferably 1,4-dioxane) is added an aqueous base (such as aqueous Na2C03 or aqueous NaOH, 1-30 equiv., preferably 2-3 equiv. for aqueous NaOH, preferably 15-20 equiv. for aqueous Na2C03). The mixture is stirred at about 25-100 °C (preferably about 60 °C) for about 1-72 h (preferably about 1-16 h). In cases where the reaction does not proceed to completion as monitored by TLC, LC/MS, or HPLC, additional aqueous base (such as aqueous Na2C03, 10-20 equiv., preferably 10 equiv. or aqueous NaOH, 1-5 equiv., preferably 1-2 equiv.) and/or a cosolvent (such as ethanol (EtOH)) is added. The reaction is continued at about 25-100°C (preferably about 60°C) for about 0.25-3 h (preferably about 1-2 h). In any case where an additional base labile group is present (for example, an ester a
trifluoromethyl, or a cyano group), this group may also be hydrolyzed. The reaction is worked up using one of the following methods. Method 1. The organic solvent is optionally removed under reduced pressure and the aqueous solution is neutralized with the addition of a suitable aqueous acid (such as aqueous HC1). A suitable organic solvent (such as EtOAc or DCM) and water are added, the layers are separated, and the organic solution is dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure to give the target compound. Method 2. The organic solvent is optionally removed under reduced pressure, a suitable organic solvent (such as EtOAc or DCM) and water are added, the layers are separated, and the organic solution is dried over anhydrous Na2S04 or MgS04, filtered, and concentrated to dryness under reduced pressure to give the target compound. Method 3. The reaction mixture is concentrated under reduced pressure and directly purified by one of the subsequent methods.
Formation of a urea using CDI or thiocarbonyldiimidazole, respectively (e.g., from 8-((3R,45)-4-ethylpyrrolidin-3-yl)-3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazine to (35,4R)-3-ethyl-4-(3H-imidazo[l,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide)
To a solution or slurry of an amine or amine salt (1-3 equiv., preferably 1-2 equiv.) in an organic solvent such as DCM, THF, or DMF (preferably DMF) at about 20 – 80 °C (preferably about 65 °C) is optionally added an organic base, such as triethylamine (TEA), N,N-diisopropylethylamine (DIEA), pyridine (preferably TEA) (1-10 equiv., preferably 1-5 equiv.) followed by CDI or 1,1 ‘-thiocarbonyldiimidazole (0.5-2 equiv., preferably 1 equiv.). After about 0.5-24 h (preferably about 1-3 h), a second amine or amine salt (1-10 equiv., preferably 1-3 equiv.) is added neat or as a solution or slurry in an organic solvent such as DCM, THF, or DMF (preferably DMF). The reaction is held at about 20 – 80 °C (preferably about 65 °C ) for about 2 – 24 h (preferably about 3 h). If the reaction mixture is heated, it is cooled to ambient temperature. The reaction mixture is partitioned between an organic solvent (such as EtOAc, DCM or 1,4-dioxane) and an aqueous base (such as saturated aqueous NaHC03 or saturated aqueous Na2C03, preferably saturated aqueous NaHC03). Optionally, the reaction mixture is concentrated under reduced pressure and the residue is partitioned as above. In either case, the aqueous layer is then optionally extracted with additional organic solvent such as EtOAc or DCM. The combined organic layers may optionally be washed with brine and concentrated in vacuo or dried over anhydrous Na2S04 or MgS04 and then decanted or filtered prior to concentrating under reduced pressure to give the target compound. Optionally, the reaction mixture is concentrated under reduced pressure and the residue is directly purified.
Chiral preparative HPLC purification
Chiral purification is performed using Varian 218 LC pumps, a Varian CVM 500 with
switching valves and heaters for automatic solvent, column and temperature control and a Varian 701 Fraction collector. Detection methods include a Varian 210 variable wavelength detector, an in-line polarimeter (PDR-chiral advanced laser polarimeter, model ALP2002) used to measure qualitative optical rotation (+/-) and an evaporative light scattering detector (ELSD) (a PS-ELS 2100 (Polymer Laboratories)) using a 100: 1 split flow. ELSD settings are as follows: evaporator: 46 °C, nebulizer: 24 °C and gas flow: 1.1 SLM. The absolute stereochemistry of the purified compounds was assigned arbitrarily and is drawn as such. Compounds of the invention where the absolute stereochemistry has been determined by the use of a commercially available enantiomerically pure starting material, or a stereochemically defined intermediate, or X-ray diffraction are denoted by an asterisk after the example number.
(ci5,)-3-ethyl-4-(3H-imidazo[l,2-fl]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-l-carboxamide isolated using the above method has an Rt min of 1.52, and m/z ESI+ (M+H)+ of 381.
The starting materials and intermediates of the above synthesis scheme may be obtained using the following schemes:
Preparation of starting material of l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid
Step A: ethyl pent-2-ynoate to (Z)-ethyl pent-2-enoate
To a slurry of Lindlar catalyst (0.844 g, 0.396 mmol) in THF (100 mL) and pyridine (10.00 mL) is added ethyl pent-2-ynoate (5.22 mL, 39.6 mmol). The reaction mixture is sparged with hydrogen for about 10 min and an atmosphere of hydrogen is maintained via balloon. After about 15 h the reaction mixture is filtered through a pad of Celite®, diluted with Et20 (30 mL) and washed with saturated aqueous CuS04 (40 mL), followed by water (40 mL). The organic layer is separated, dried over anhydrous MgS04, filtered, and concentrated in vacuo to provide crude (Z)-ethyl pent-2-enoate (5 g, 98%). 1H NMR (DMSO-d6) δ 1.05 (t, 3H), 1.28 (t, 3H), 2.65 (m, 2H), 4.18 (q, 2 H), 5.72 (m, 1H), 6.21 (m, 1H).
Step B: (ds)-ethyl l-benzyl-4-ethylpyrrolidine-3-carboxylate (from (Z)-ethyl pent-2-enoate and N-benzyl-l-methoxy-N-((trimethylsilyl)methyl)methanamine)
To a solution of N-benzyl-l-methoxy-N-((trimethylsilyl)methyl)methanamine (9.98 mL, 39.0 mmol) and (Z)-ethyl pent-2-enoate (5 g, 39.0 mmol) in DCM (50 mL) is added trifluoroacetic acid (TFA) (0.030 mL, 0.390 mmol) at RT. After about 2 days, the reaction mixture is concentrated in vacuo to provide crude (cis)-ethyl 1 -benzyl-4-ethylpyrrolidine-3- carboxylate (9.8 g, 96%) as an oil. LC/MS (Table 1, Method a) Rt = 1.62 min; MS m/z: 262 (M+H)+.
Step C: ethyl l-benzyl-4-ethylpyrrolidine-3-carboxylate to (ds)-ethyl 4-ethylpyrrolidine-3-carboxylate
A Parr shaker is charged with PdOH2 on carbon (2.243 g, 3.19 mmol) and (cis)-et yl l-benzyl-4-ethylpyrrolidine-3-carboxylate (16.7 g, 63.9 mmol) followed by EtOH (100 mL). The reaction mixture is degassed and purged with hydrogen gas and shaken on the parr shaker at 60 psi for about 4 days at ambient temperature. The reaction mixture is degassed and purged with nitrogen. The suspension is filtered through a pad of Celite® washing with EtOH (~ 900 mL). The solvent is removed under reduced pressure to afford (cis)-ethyl 4-ethylpyrrolidine-3 -carboxylate (8.69 g, 79%) as an oil: LC/MS (Table 1, Method a) Rt = 1.11 min; MS m/z: 172 (M+H)+.
Step D: (ds)-ethyl 4-ethylpyrrolidine-3-carboxylate to (ds)-l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid
To a flask charged with (cis)-et yl 4-ethylpyrrolidine-3-carboxylate (8.69g, 50.7 mmol) is added aqueous HCl (6N, 130 mL, 782 mmol). The solution is heated at about 75°C for about 12 h. aqueous HCl (6N, 100 mL, 599 mmol) is added and stirred at about 80 °C for about 20 h. Aqueous HCl (6N, 100 mL, 599 mmol) is added and continued stirring at about 80 °C for about 20 h. The reaction mixture is cooled to ambient temperature and the solvent is removed under reduced pressure. 1,4-Dioxane (275 mL) and water (50 mL) are added followed by portionwise addition of Na2C03 (13.5 g, 127 mmol). Di-ie/t-butyl dicarbonate (13.3 g, 60.9 mmol) is added and the reaction mixture is stirred at ambient temperature for about 16 h. The solid is filtered and washed with EtOAc (250 mL). The aqueous layer is acidified with aqueous HCl (IN) to about pH 3-4. The layers are partitioned and the aqueous layer is extracted with EtOAc (3 x 100 mL). The combined organic layers are dried over anhydrous Na2S04, filtered and removed under reduced pressure. As the organic layer is almost fully concentrated (~ 10 mL remaining), a solid precipitated. Heptane (30 mL) is added and the solid is filtered washing with heptane to afford (cis)-l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid (3.9 g, 32%) as an off white solid as product: LC/MS (Table 1, Method c) Rt = 0.57 min; MS m/z: 242 (M-H)~.
Synthesis of Intermediate benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine-l-carboxylate
Acidic cleavage of a Boc-protected amine (e.g., l-(tert-butoxycarbonyl)-4-ethylpyrrolidine-3-carboxylic acid to 4-ethylpyrrolidine-3-carboxylic acid
hydrochloride)
To a solution of a Boc-protected amine (preferably 1 equiv.) in an organic solvent (such as DCM, 1,4-dioxane, or MeOH) is added TFA or HC1 (preferably 4 N HC1 in 1,4-dioxane, 2-35 equiv., preferably 2-15 equiv.). The reaction is stirred at about 20-100 °C (preferably ambient temperature to about 60 °C) for about 1-24 h (preferably about 1-6 h). In any case where an additional acid labile group is present (for example, a t-butyl ester), this group may also be cleaved during the reaction. Optionally, additional TFA or HC1
(preferably 4 N HC1 in 1,4-dioxane solution, 2-35 equiv., preferably 2-15 equiv.) may be added to the reaction mixture in cases where the reaction does not proceed to completion as monitored by TLC, LC/MS, or HPLC. Once the reaction has proceeded to an acceptable level, the reaction mixture can be concentrated in vacuo to provide the amine as a salt.
Alternatively, the reaction may be partitioned between an organic solvent (such as EtOAc, DCM or 1,4-dioxane) and an aqueous base (such as saturated aqueous NaHC03 or saturated aqueous Na2C03, preferably saturated aqueous NaHC03). The aqueous layer can be optionally extracted with additional organic solvent such as EtOAc or DCM. The combined organic layers may optionally be washed with brine, dried over anhydrous Na2S04 or MgS04, then decanted or filtered, prior to concentrating under reduced pressure to give the target compound.
Cbz-protection of an amine (e.g., 4-ethylpyrrolidine-3-carboxylic acid hydrochloride to l-((benzyloxy)carbonyl)-4-ethylpyrrolidine-3-carboxylic acid)
A solution of an amine or an amine salt (preferably 1 equiv.) and a base (for example, Na2C03 or NaOH, 1-3 equiv., preferably Na2C03, 1.6 equiv.) in water or aqueous organic solvent (for example, water / 1,4-dioxane or water / acetonitrile (MeCN), preferably water/ 1,4-dioxane) is stirred at ambient temperature for about 1-10 min (preferably 5 min). A solution of benzyl 2,5-dioxopyrrolidin-l-yl carbonate (1-2 equiv., preferably 1.0 equiv.) in an organic solvent such as 1,4-dioxane or MeCN is added to the reaction. The reaction is stirred at ambient temperature for about 8-144 h (preferably about 72 h). Optionally, the reaction mixture is concentrated under reduced pressure. The resulting aqueous solution is diluted with an organic solvent (such as EtOAc or DCM). The organic extracts are optionally washed with water and/or brine, dried over anhydrous Na2S04 or MgS04, filtered or decanted, and concentrated under reduced pressure. Alternatively, the resulting aqueous solution is acidified by adding an acid such as aqueous NH4C1 or HC1 and is then extracted with an organic solvent (such as EtOAc or DCM).
Formation of a bromomethyl ketone from an acid (e.g., l-((benzyloxy)carbonyl)-4-ethylpyrrolidine-3-carboxylic acid to benzyl 3-(2-bromoacetyl)-4-ethylpyrrolidine-l-carboxylate)
To a solution of a carboxylic acid (preferably 1 equiv.) in an organic solvent (DCM or 1,2-dichloroethane (DCE), preferably DCM) is slowly added oxalyl chloride (1.2-3.0 equiv., preferably 2.2 equiv.) followed by dropwise addition of DMF (0.01-0.20 equiv., preferably about 0.15 equiv.). The reaction is stirred at about 0-40 °C (preferably ambient temperature) for about 3-24 h (preferably about 14 h) before it is concentrated under reduced pressure to a constant weight to give the crude acid chloride. A solution of a crude acid chloride
(preferably 1 equiv.) in an organic solvent (such as THF, MeCN, Et20, or THF/MeCN, preferably THF/MeCN) is added to trimethylsilyldiazomethane (2.0 M in Et20) or diazomethane solution in Et20 (prepared from DIAZALD® according to Aldrich protocol or J. Chromatogr. Sci. 1991, 29:8) (2-10 equiv., preferably 3.5 equiv. of
trimethylsilyldiazomethane) at about -20-20 °C (preferably about 0 °C) in a suitable organic solvent such as THF, MeCN, Et20, or THF/MeCN (preferably THF/MeCN). The reaction mixture is stirred for about 0.5-5 h (preferably about 3 h) at about -20-20 °C (preferably about 0 °C) before the dropwise addition of 48% aqueous HBr (5-40 equiv., preferably about 10 equiv.). After about 0-30 min, (preferably about 5 min) the reaction mixture can be concentrated to dryness to give the desired product, neutralized by a dropwise addition of saturated aqueous NaHC03 or is optionally washed with brine after optional addition of an organic solvent (such as EtOAc or DCM, preferably EtOAc). In cases where the reaction mixture is subjected to an aqueous work-up, the organic layer is dried over anhydrous Na2S04 or MgS04 (preferably MgS04), filtered, and concentrated under reduced pressure.
Synthesis of Intermediate tert-butyl (5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-yl)carbamate
Step A: 3,5-dibromopyrazin-2-amine to 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine
To a solution of 3,5-dibromopyrazin-2-amine (125 g, 494 mmol), TEA (207.0 mL, 1483 mmol), and copper (I) iodide (0.941 g, 4.94 mmol) in THF (1255 mL) is added
PdCl2(PPh3)2 (3.47 g, 4.94 mmol). The reaction mixture is cooled at about -5-0°C and a solution of (trimethylsilyl)acetylene (65.0 mL, 470 mmol) in THF (157 mL) is added dropwise over about 15 min. The reaction mixture is stirred at about -5-0°C for about 1.5 h and then allowed to warm to room temperature (RT) overnight. The reaction mixture is then filtered through a CELITE® pad and washed with THF until no further product eluted. The filtrate is concentrated under reduced pressure to give a brown-orange solid. The solid is triturated and sonicated with warm petroleum ether (b.p. 30-60°C, 400 mL), cooled to RT, collected, washed with petroleum ether (b.p. 30-60°C; 2 x 60 mL), and dried to give 5-bmmo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine (124 g, 93%, 93% purity) as a brown solid: LC/MS (Table 1, Method b) Rt = 2.51 min; MS m/z: 270, 272 (M+H)+.
Step B: 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine to 2-bromo-5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine
To a solution of 5-bromo-3-((trimethylsilyl)ethynyl)pyrazin-2-amine (3.00g, 11.1 mmol) in DMF (60 mL) at about 0 °C is added NaH (60% dispersion in mineral oil, 0.577g, 14.4 mmol) in three portions. After about 15 min, p-toluenesulfonyl chloride (2.75g, 14.4 mmol) is added and the reaction is allowed to warm slowly to ambient temperature. After about 16 h, the reaction mixture is poured onto ice-cold water (120 mL) and the precipitate is collected by vacuum filtration. The crude solid is dissolved in DCM (15 mL) and purified by silica gel chromatography eluting with DCM to give 2-bromo-5-tosyl-5H-pyrrolo[2,3-bjpyrazine (2.16 g, 52%): LC/MS (Table 1, Method c) Rt = 1.58 min; MS m/z: 352, 354 (M+H)+.
Step C: 2-bromo-5-tosyl-5H-pyrrolo[2,3-b]pyrazine to methyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine-2-carboxylate
CO is bubbled into an orange solution of 2-bromo-5-tosyl-5H-pyrrolo[2,3-b]pyrazine (50. Og, 142 mmol) in DMF (2.50 L) within a 5 L round bottom flask for about 2 min.
Bis(triphenylphosphine)-palladium(II) dichloride (9.96g, 14.2 mmol), TEA (59 mL, 423 mmol) and MeOH (173.0 mL, 4259 mmol) are added and the flask is fitted with a balloon of CO. The mixture is heated at about 95°C under an atmosphere of CO (1 atmosphere). After stirring overnight, the reaction mixture is cooled to ambient temperature overnight and poured into ice water (3.2 L). The mixture is stirred for about 10 min and the precipitate is collected by filtration, while washing with water, and dried for 1 h. The crude material is dissolved in DCM, separated from residual water, dried over anhydrous MgS04, filtered, added silica gel, and concentrated under reduced pressure to prepare for chromatography. The crude material is purified by silica gel column chromatography eluting with 0-5% MeOH in DCM to yield methyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylate with 5 mol% DCM as an excipient (40.7 g, 86%, 93% purity): LC/MS (Table 1, Method a) Rt = 2.35 min;
MS m/z 332 (M+H)+.
Step D: methyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazine-2-carboxylate to 5-tosyl-5H-pyrrolo[2,3-/>]pyrazine-2-carboxylic acid
HC1 (6 N aqueous, 714 mL) is added to a yellow solution of methyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylate (17.8g, 53.6 mmol) in 1,4-dioxane (715 mL) within a 2 L round bottom flask, and the mixture is heated at about 60°C for about 16 h. The reaction mixture is cooled to ambient temperature. The organic solvent is removed under reduced pressure and the precipitate is collected, washed with water, and dried to yield 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid (14.4 g, 85%) as a yellow solid: LC/MS (Table 1, Method a) Rt = 1.63 min; MS m/z 316 (Μ-Η)“.
Step E: 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid to tert-butyl 5-tosyl-5H-pyrrolo[2,3-Z>]pyrazin-2-ylcarbamate
In a 500 mL round bottom flask, 5-tosyl-5H-pyrrolo[2,3-b]pyrazine-2-carboxylic acid (14.4 g, 45.3 mmol), diphenylphosphoryl azide (9.78 mL, 45.3 mmol) and TEA (13.9 mL, 100 mmol) in ie/t-butanol (i-BuOH) (200 mL) are added to give an orange suspension. The mixture is heated at about 70°C for about 16 h, cooled to ambient temperature and the insoluble material is removed by filtration. The solvent is removed under reduced pressure and the crude material is purified by silica gel column chromatography eluting with 25-60% EtOAc in heptane to yield tert-butyl 5-tosyl-5H-pyrrolo[2,3-b]pyrazin-2-ylcarbamate (9.75 g, 54%) as an off-white solid: LC/MS (Table 1, Method a) Rt = 2.79 min; MS m/z 389 (M+H)+.
PATENT
WO2011068881


PATENT
http://www.google.com/patents/US20110311474
Preparation #F.1.1: 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazine
-

-
To a solution of (cis)-benzyl 3-ethyl-4-(3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)pyrrolidine-1-carboxylate (0.838 g, 1.541 mmol, prepared using E from Example #36 Step D with TFA, N, R, S.1 with Example #3 Step E, and T with Lawesson’s reagent) was added a solution of HBr (2.50 mL, 15.19 mmol, 33% in acetic acid). The reaction mixture was stirred at ambient temperature for about 1 h. The reaction was diluted with Et2O (50 mL) and water (20 mL). The layers were stirred for about 3 min and the organic layer was decanted then the procedure was repeated 5 times. The aqueous layer was cooled to about 0° C. was basified with saturated aqueous NaHCO3solution (10 mL) to about pH 7. The aqueous layer was extracted with EtOAc (3×50 mL), combined, and dried over anhydrous Na2SO4, filtered and concd to give a brown solid. The solid was dissolved in DCM (50 mL) and washed with water (3×20 mL), dried over anhydrous Na2SO4, filtered and coned to afford 8-((cis)-4-ethylpyrrolidin-3-yl)-3-tosyl-3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazine (0.453, 61%) as a brown residue: LC/MS (Table 1, Method a) Rt=1.73 min; MS m/z: 410 (M+H)+.

SEE…………..1-((cis)-4-ethylpyrrolidin-3-yl)-6-tosyl-6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine (0.250 g, 0.609 mmol, Example #36, step F)
PATENTS
WO-2017066775
WO-2015061665
WO-2011068881
References
- ^ Jump up to:a b c Mohamed, Mohamed-Eslam F.; Camp, Heidi S.; Jiang, Ping; Padley, Robert J.; Asatryan, Armen; Othman, Ahmed A. (December 2016). “Pharmacokinetics, Safety and Tolerability of ABT-494, a Novel Selective JAK 1 Inhibitor, in Healthy Volunteers and Subjects with Rheumatoid Arthritis”. Clinical Pharmacokinetics. 55 (12): 1547–1558. doi:10.1007/s40262-016-0419-y. ISSN 1179-1926. PMID 27272171.
- Jump up^ Fleischmann, Roy (May 2012). “Novel small-molecular therapeutics for rheumatoid arthritis”. Current Opinion in Rheumatology. 24 (3): 335–341. doi:10.1097/BOR.0b013e32835190ef. ISSN 1531-6963. PMID 22357358.
- Jump up^ Riese, Richard J.; Krishnaswami, Sriram; Kremer, Joel (August 2010). “Inhibition of JAK kinases in patients with rheumatoid arthritis: scientific rationale and clinical outcomes”. Best Practice & Research. Clinical Rheumatology. 24 (4): 513–526. doi:10.1016/j.berh.2010.02.003. ISSN 1532-1770. PMID 20732649.
- Jump up^ “Characterization of ABT-494, a Second Generation Jak1 Selective Inhibitor”. ACR Meeting Abstracts. Retrieved 2017-05-21.
- Jump up^ Mohamed, Mohamed-Eslam F.; Jungerwirth, Steven; Asatryan, Armen; Jiang, Ping; Othman, Ahmed A. (2017-05-14). “Assessment of effect of CYP3A Inhibition, CYP Induction, OATP1B Inhibition, and High-Fat Meal on Pharmacokinetics of the JAK1 inhibitor Upadacitinib”. British Journal of Clinical Pharmacology. doi:10.1111/bcp.13329. ISSN 1365-2125. PMID 28503781.
- Jump up^ Kremer, Joel M.; Emery, Paul; Camp, Heidi S.; Friedman, Alan; Wang, Li; Othman, Ahmed A.; Khan, Nasser; Pangan, Aileen L.; Jungerwirth, Steven; Keystone, Edward C. (December 2016). “A Phase IIb Study of ABT‐494, a Selective JAK‐1 Inhibitor, in Patients With Rheumatoid Arthritis and an Inadequate Response to Anti–Tumor Necrosis Factor Therapy”. Arthritis & Rheumatology (Hoboken, N.j.). 68 (12): 2867–2877. doi:10.1002/art.39801. ISSN 2326-5191. PMC 5132116
 . PMID 27389975.
. PMID 27389975. - Jump up^ Genovese, Mark C.; Smolen, Josef S.; Weinblatt, Michael E.; Burmester, Gerd R.; Meerwein, Sebastian; Camp, Heidi S.; Wang, Li; Othman, Ahmed A.; Khan, Nasser; Pangan, Aileen L.; Jungerwirth, Steven (December 2016). “Efficacy and Safety of ABT‐494, a Selective JAK‐1 Inhibitor, in a Phase IIb Study in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate”. Arthritis & Rheumatology (Hoboken, N.j.). 68 (12): 2857–2866. doi:10.1002/art.39808. ISSN 2326-5191. PMC 5132065 . PMID 27390150.
- Jump up^ “A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study of ABT-494 for the Induction of Symptomatic and Endoscopic Remission in Subjects With Moderately to Severely Active Crohn’s Disease Who Have Inadequately Responded to or Are Intolerant to Immunomodulators or Anti-TNF Therapy – Full Text View – ClinicalTrials.gov”. Retrieved 2017-05-22.
- Jump up^ “AbbVie Announces Positive Phase 2 Study Results for Upadacitinib (ABT-494), an Investigational JAK1-Selective Inhibitor, in Crohn’s Disease | AbbVie Newsroom”. Retrieved 2017-05-22.
- Jump up^ Phase 3 upadacitinib trials
-
Upadacitinib 
Clinical data Synonyms ABT-494 Routes of
administrationOral Pharmacokinetic data Metabolism Hepatic (CYP3A major, CYP2D6 minor) [1] Biological half-life 6-15 hours[1] Identifiers CAS Number PubChem CID IUPHAR/BPS ChemSpider UNII ChEMBL Chemical and physical data Formula C17H19F3N6O Molar mass 380.38 g·mol−1 3D model (JSmol)

/////Upadacitinib, ABT 494, упадацитиниб , أوباداسيتينيب , 乌帕替尼 , ORPHAN DRUG, PHASE 3
c21cnc4c(n1c(cn2)[C@@H]3[C@@H](CN(C3)C(=O)NCC(F)(F)F)CC)ccn4
OR
CC[C@@H]1CN(C[C@@H]1c4cnc3cnc2nccc2n34)C(=O)NCC(F)(F)F
FDA approves new orphan drug Uptravi (selexipag) to treat pulmonary arterial hypertension

KEEPING WATCHING THIS POSTS FOR SYNTHESIS UPDATES
12/22/2015
On December 21, the U.S. Food and Drug Administration approved Uptravi (selexipag) tablets to treat adults with pulmonary arterial hypertension (PAH), a chronic, progressive, and debilitating rare lung disease that can lead to death or the need for transplantation.
December 22, 2015
On December 21, the U.S. Food and Drug Administration approved Uptravi (selexipag) tablets to treat adults with pulmonary arterial hypertension (PAH), a chronic, progressive, and debilitating rare lung disease that can lead to death or the need for transplantation.
“Uptravi offers an additional treatment option for patients with pulmonary arterial hypertension,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “The FDA supports continued efforts to provide new treatment options for rare diseases.”
PAH is high blood pressure that occurs in the arteries that connect the heart to the lungs. It causes the right side of the heart to work harder than normal, which can lead to limitations on exercise ability and shortness of breath, among other more serious complications.
Uptravi belongs to a class of drugs called oral IP prostacyclin receptor agonists. The drug acts by relaxing muscles in the walls of blood vessels to dilate (open) blood vessels and decrease the elevated pressure in the vessels supplying blood to the lungs.
Uptravi’s safety and efficacy were established in a long-term clinical trial of 1,156 participants with PAH. Uptravi was shown to be effective in reducing hospitalization for PAH and reducing the risks of disease progression compared to placebo. Participants were exposed to Uptravi in this trial for a median duration of 1.4 years.
Common side effects observed in those treated with Uptravi in the trial include headache, diarrhea, jaw pain, nausea, muscle pain (myalgia), vomiting, pain in an extremity, and flushing.
Uptravi was granted orphan drug designation. Orphan drug designation provides incentives such as tax credits, user fee waivers, and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Uptravi is marketed by San Francisco-based Actelion Pharmaceuticals US, Inc.
Selexipag, Uptravi
475086-01-2 CAS
(C26H32N4O4S, Mr = 496.6 g/mol)
A prostacyclin receptor (PGI2) agonist used to treat pulmonary arterial hypertension (PAH).
NIPPON SHINYAKU….INNOVATOR
Selexipag (brand name Uptravi) is a drug developed by Actelion for the treatment of pulmonary arterial hypertension (PAH). Selexipag and its active metabolite, ACT-333679 (MRE-269) (the free carboxylic acid), are agonists of the prostacyclin receptor, which leads to vasodilation in the pulmonary circulation.[1]
The US FDA granted it Orphan Drug status[2] (for PAH). It was approved by the U.S. FDA on 22 December 2015.[2]

ACT-333679 or MRE-269, the active metabolite of selexipag




PATENT
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer’s disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
PATENT
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a isthe drug
2a isthe drug
N-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614
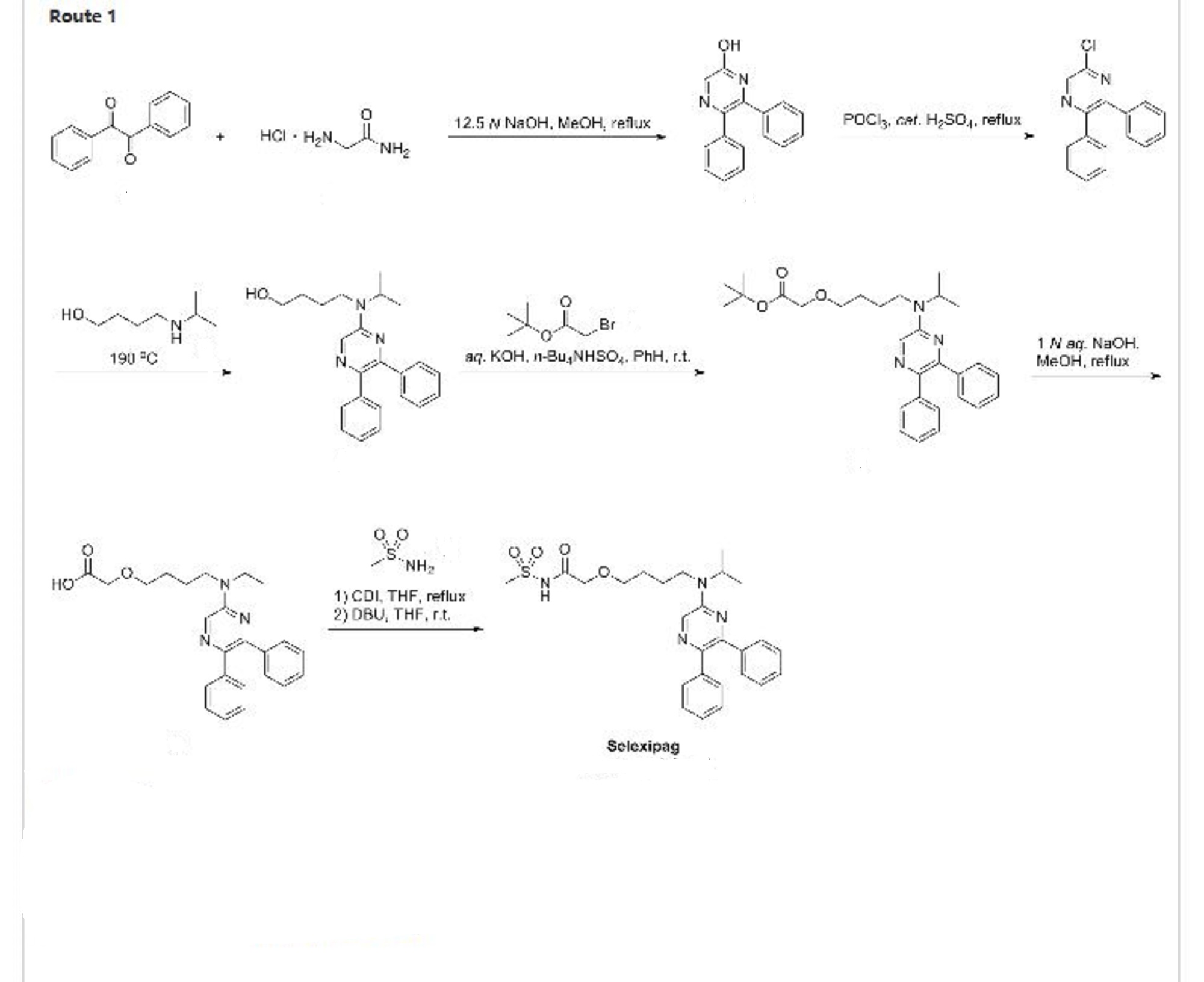
PATENT
Example 1 t- butylamine Form I crystal of the salt
Compound A (40 mg) with 0.5mL dimethoxyethane (hereinafter, referred to as. “DME”) was dissolved in, and t- butylamine (1.1 eq) were added, 25 1 ° C. at 8 it was stirred for hours. Thereafter, the reaction solution was added t- butyl methyl ether (1mL), at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, I-form crystals of t- butylamine salt ( 3 to afford 9.9mg). B Powder X-ray diffraction spectrum of type I crystal obtained t- butylamine salt using the apparatus shown in Figure 1.
Melting point: 152.5 ℃
elemental analysis (C 3 0 H 4 3 N 5 O 4 S + 0.0 3 H 2 as O)
calculated value (%) C: 6 3 .1 8 H: 7 . 6 1 N: 12 .2 8 measured value (%) C: 6 2. 8 5 H: 7 . 6 4 N: 12.52 1 H-NMR (DMSO-D 6 ): delta 8 .15 (s, 1H), 7 .55 – 7 . 8 0 (M, 2H), 7 .10- 7 . .45 (M, 10H), 4 7 . 0-4 8 5 (M, 1H), 3 . 6 6 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 3 (s, 3 H), 1.50-1. 7 5 (M, 4H), 1.2 3 (s, 9H), 1.22 (D, 6 H)
Example 2 I-form crystal of the potassium salt
Compound A tetrahydrofuran with (40mg) 12mL (hereinafter, referred to as. “THF”) was dissolved in, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added, 40 ℃ It was heated and stirred in for 15 minutes. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. After repeated two more times this step, at -20 ° C. 3 and held hours. The resulting precipitated crystals were collected by filtration under reduced pressure, and dried to obtain Form I crystal of the potassium salt. B Powder X-ray diffraction spectrum of type I crystal of the obtained potassium salt using the apparatus shown in Fig. 1 H-NMR (DMSO-D 6 ): delta 8 .14 (s, 1H), 7 .1 8 – 7 . 3 8 . (M, 10H), 4 7 . 2-4 8 4 (M, 1H) , 3 . 6 5 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 2 (s, 3 H), 1.55-1. 7 0 ( M, 4H), 1.2 3 (D, 6 H)
Example 3 II-form crystals of the potassium salt
Compound A with (40mg) was dissolved in THF and 12mL, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added and heated with stirring for 15 min at 40 ℃. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. This operation was repeated two more times, at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, after drying, 40 ℃, relative humidity 7 while 5% of thermo-hygrostat 7 left for days to give crystalline Form II of the potassium salt. B Powder X-ray diffraction spectrum of crystalline Form II of the resulting potassium salt using the apparatus Fig 3 is shown in.
Example 4 III type crystal of the potassium salt
Compound A , in addition to (100mg) acetonitrile (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (1.1 eq) was added and stirred for 200 minutes at 20 ℃. While stirring the mixture 7 after a heated stirring for 1 hour to 0 ° C., and then cooled to 10 ℃ over 10 hours. Further heated while the mixture 6 is heated to 0 ℃, t- butyl methyl ether (0. 3 after adding mL), cooled to 20 ℃ over 10 hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, III type crystal of the potassium salt ( 7 to afford 5mg). The powder X-ray diffraction spectrum of the type III crystal of the obtained potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, of about 7 endothermic peak was observed at around 4 ° C..
Elemental analysis (C 2 6 H 3 1 N 4 O 4 . SK + 0 7 8 H 2 as O)
calculated value (%) C: 5 6 .91 H: 5.9 8 N: 10.21
measured value (%) C: 5 6 . 6 1 H: 5.55 N:. 10 3 6
EXAMPLE 5 IV-type crystal of the potassium salt
Compound A , in addition to (50mg) and ethyl acetate (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (2.2 eq) was added and 2 at 20 ° C. 3 and stirred for hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried to obtain Form IV crystal of the potassium salt (41mg). The powder X-ray diffraction spectrum of crystalline Form IV of the resulting potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, an endothermic peak was observed at around approximately 91 ℃.

Selexipag (C26H32N4O4S, Mr = 496.6 g/mol) ist ein Diphenylpyrazin-Derivat. Es wird in der Leber zum aktiven Metaboliten ACT-333679 (MRE-269) biotransformiert. Selexipag unterscheidet sich strukturell von Prostazyklin und anderen Prostazylin-Rezeptor-Agonisten.

References
- 1 Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
- 2 New Drug Approved for Rare Lung Disorder. PPN. 23 Dec 2015 Has link to GRIPHON study results
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
| Patent | Submitted | Granted |
|---|---|---|
| Methods of identifying critically ill patients at increased risk of development of organ failure and compounds for the treatment hereof [US8877710] | 2009-12-30 | 2014-11-04 |
| Form-I crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide and method for producing the same [US8791122] | 2010-06-25 | 2014-07-29 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2015057325] | 2013-03-26 | 2015-02-26 |
| INHIBITION OF NEOVASCULARIZATION BY SIMULTANEOUS INHIBITION OF PROSTANOID IP AND EP4 RECEPTORS [US2014275200] | 2014-03-05 | 2014-09-18 |
| INHIBITION OF NEOVASCULARIZATION BY INHIBITION OF PROSTANOID IP RECEPTORS [US2014275238] | 2014-03-05 | 2014-09-18 |
| Fibrosis inhibitor [US8889693] | 2014-04-10 | 20 |
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound derivatives and medicines [US7205302] | 2004-05-27 | 2007-04-17 |
| METHODS OF IDENTIFYING CRITICALLY ILL PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ORGAN FAILURE AND COMPOUNDS FOR THE TREATMENT HEREOF [US2014322207] | 2014-07-11 | 2014-10-30 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2014329824] | 2014-07-18 | 2014-11-06 |
| Sustained Release Composition of Prostacyclin [US2014303245] | 2012-08-10 | 2014-10-09 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2013261177] | 2011-09-30 | 2013-10-03 |
| METHODS OF TREATMENT OF PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ISCHEMIC EVENTS AND COMPOUNDS HEREOF [US2013040898] | 2011-04-29 | 2013-02-14 |
| Substituted Diphenylpyrazine Derivatives [US2013005742] | 2010-08-06 | 2013-01-03 |
| USE OF FORM-I CRYSTAL OF 2–N-(METHYLSULFONYL)ACETAMIDE [US2014148469] | 2014-01-22 | 2014-05-29 |
| CRYSTALS OF 2- {4- [N- (5,6-DIPHENYLPYRAZIN-2-YL) -N-ISOPROPYLAMINO]BUTYLOXY}-N- (METHYLSULFONYL) ACETAMIDE [US2014155414] | 2014-01-22 | 2014-06-05 |
| PROSTACYCLIN AND ANALOGS THEREOF ADMINISTERED DURING SURGERY FOR PREVENTION AND TREATMENT OF CAPILLARY LEAKAGE [US2014044797] | 2012-03-30 | 2014-02-13 |
 |
|
| Names | |
|---|---|
| IUPAC name
2-{4-[(5,6-diphenylpyrazin-2-yl)(propan-2-yl)amino]butoxy}-N-(methanesulfonyl)acetamide
|
|
| Other names
ACT-293987, NS-304
|
|
| Identifiers | |
| 475086-01-2 |
|
| ChEMBL | ChEMBL238804 |
| ChemSpider | 8089417 |
| 7552 | |
| Jmol interactive 3D | Image |
| KEGG | D09994 |
| PubChem | 9913767 |
| UNII | P7T269PR6S |
| Properties | |
| C26H32N4O4S | |
| Molar mass | 496.6 g·mol−1 |
SEE……….http://apisynthesisint.blogspot.in/2015/12/fda-approves-new-orphan-drug-uptravi.html
//////////
CC(C)N(CCCCOCC(=O)NS(=O)(=O)C)C1=CN=C(C(=N1)C2=CC=CC=C2)C3=CC=CC=C3
Uridine triacetate, ウリジントリアセタート FDA approves first emergency treatment for overdose of certain types of chemotherapy

12/11/2015 12:05 PM EST
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
UPDATED IN SEPT 2016…………..
Uridine triacetate
Uridine, 5-hydroxy-, 2′,3′,5′-triacetate
Orphan drug
FAST TRACK
MF C15H18N2O9, MW 370.314
|
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
|
Vistogard [Trade name]
Xuriden [Trade name]
(2R,3R,4R,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl diacetate
Wellstat (Originator)
PN-401; RG-2133; TAU
MOA:Pyrimidine analog
Indication:Hereditary orotic aciduria; Chemotherapy induced poisoning
To treat patients with hereditary orotic aciduria
| Drug Name(s) | XURIDEN |
| FDA Application No. | (NDA) 208169 |
| Active Ingredient(s) | URIDINE TRIACETATE |
| Company | WELLSTAT THERAP |
| Original Approval or Tentative Approval Date | September 4, 2015 |
FDA APPROVAL SUMMARY
Chemotherapy induced poisoning, VISTOGARD, FDA 2015-12-11
Hereditary orotic aciduria, Xuriden, FIRST APPROVAL, 2015-09-04
|
|||||||
| External Identifiers |
|
|---|
Uridine triacetate is a drug used in the treatment of hereditary orotic aciduria[1] and to treat patients following an overdose ofchemotherapy drugs 5-fluorouracil or capecitabine, or in patients exhibiting early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of 5-fluorouracil or capecitabine administration.[2][3]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics and it is marketed in USA by BTG. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine triacetate is a prodrug of uridine.[4]
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTP incorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [3]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidine metabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
Marketed as the product Xuriden (FDA), uridine triacetate is indicated for the treatment of hereditary orotic aciduria. Marketed as the product Vistogard (FDA), uridine triacetate is indicated for the emergency treatment of adult and pediatric patients in the following situations: following a fluorouracil or capecitabine overdose regardless of the presence of symptoms; or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration.

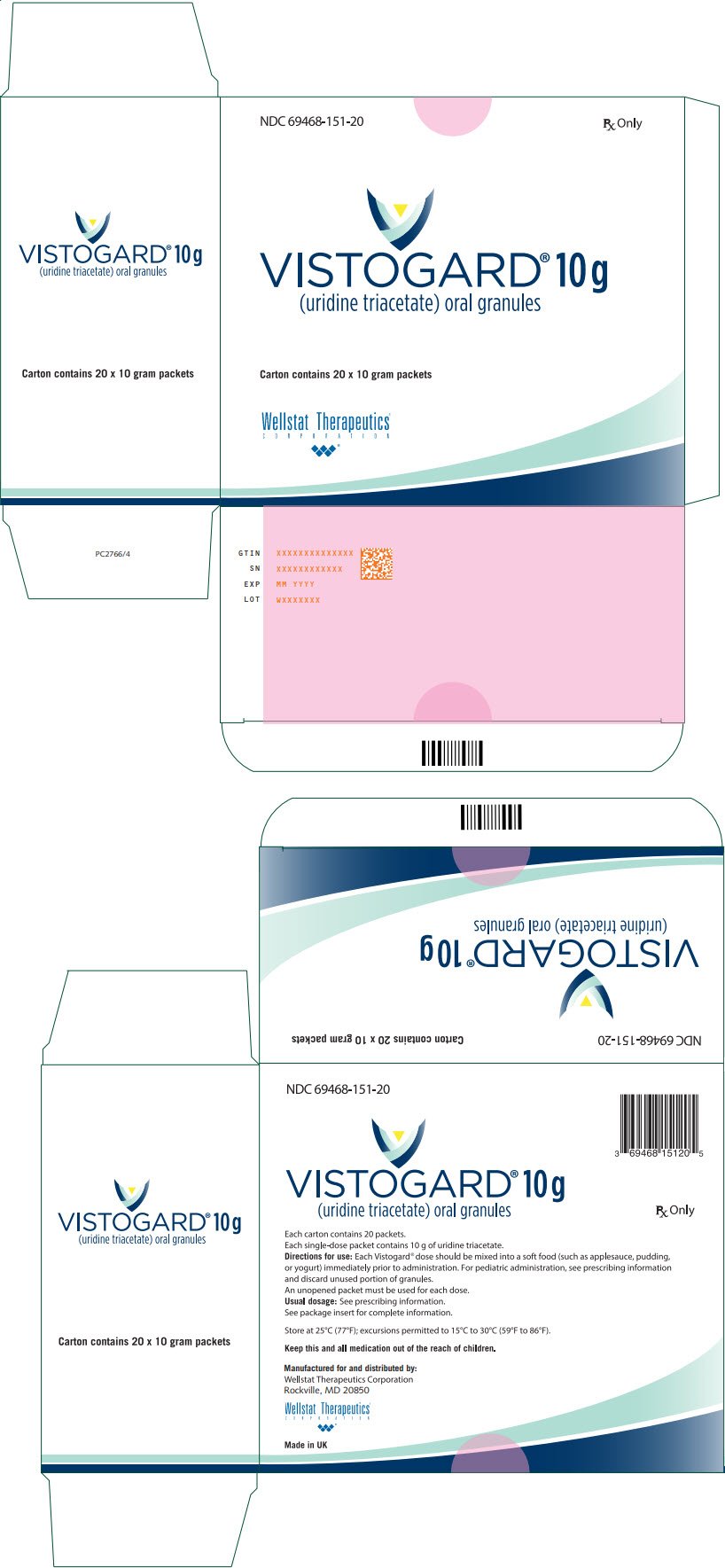
Uridine Triacetate was approved by the U.S. Food and Drug Administration (FDA) on Sep 4, 2015. It was developed by Wellstat Therapeutics, then marketed as Xuriden® by Wellstat Therapeutics in US. Then it was also approved by FDA for overdose of certain types of chemotherapy on Dec 11, 2015 and marketed as Vistogard®.
Uridine Triacetate is a prodrug of the nucleoside uridine used to treat hereditary orotic aciduria. Hereditary orotic aciduria is inherited from a recessive gene. The disease is due to a defective or deficient enzyme, which results in the body being unable to normally synthesize uridine, a necessary component of ribonucleic acid (RNA). Signs and symptoms of the disease include blood abnormalities (anemia, decreased white blood cell count, decreased neutrophil count), urinary tract obstruction due to the formation of orotic acid crystals in the urinary tract, failure to thrive, and developmental delays.
Xuriden® is approved as oral granules that can be mixed with food or in milk or infant formula, and is administered once daily. The starting dosage is 60 mg/kg once daily; the dose may be increased to 120 mg/kg (not to exceed 8 grams) once daily for insufficient efficacy.
Mechanism Of Action
Uridine triacetate is an acetylated form of uridine. Following oral administration, uridine triacetate is deacetylated by nonspecific esterases present throughout the body, yielding uridine in the circulation (Figure 1).
Figure 1: Uridine Triacetate Conversion to Uridine

URIDEN provides uridine in the systemic circulation of patients with hereditary orotic aciduria who cannot synthesize adequate quantities of uridine due to a genetic defect in uridine nucleotide synthesis.
Uridine triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578] such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Uridine triacetate is also used for replacement therapy in the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase (UMPS) deficiency. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria.
Route 1


Reference:1. J. Am. Chem. Soc. 1953, 75, 2017-2019.
2. Angew. Chem. internat. Edit. 1971, 10, 75.
3. US3116282.
PATENT
Production Example 1

5.6 g of uracil and 0.1 g of ammonium sulfate were dissolved in 22.4 ml of 1,1,1,3,3,3-hexamethyldisilazane and reacted at 120° C. for 2.5 hours. After the completion of the reaction, the reaction mixture was distilled to give 11.8 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine. 1H-NMR (400 MHz, in C2D6CO): δ=0.29 (s, 9H), 0.31 (s, 9H), 6.35 (d, J=5.6 Hz, 1H), 8.19 (d, J=5.5Hz, 1H)
Referential Example 11.21 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 1.15 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.8 ml of acetonitrile and cooled to 5° C. Next, 0.94 g of SnCl4 was added dropwise thereinto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted for 3 hours. The reaction mixture was analyzed by HPLC. Thus, β-uridine triacetate was obtained with a reaction yield of 83%.
Example 1

0.93 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 0.92 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.7 ml of acetonitrile and cooled to 4° C. Then 0.49 g of FeCl3 was added thereto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted. The reaction was monitored by HPLC. After the completion of the reaction, the reaction mixture was added dropwise at 4° C. into a cold aqueous solution of sodium hydrogencarbonate which had been preliminarily prepared. After filtering off the catalyst residue, the filtrate was separated and the aqueous layer was extracted with 20 ml portions of ethyl acetate thrice. The organic layers were combined, washed with a saturated aqueous solution of sodium chloride and dried over sodium sulfate. After distilling off the solvent, 1.2 g (purity 80%) of the target compound was obtained as a viscous white solid.
Namely, the target compound could be obtained at a yield comparable to REFERNTIAL EXAMPLE 1 wherein SnCl4 was employed as the catalyst. 1H-NMR (400 MHz, in CDCl3): δ=2.11 (s, 3H), 2.14 (s, 3H), 2.15 (s, 3H), 4.35 (m, 3H), 5.33 (m, 2H), 5.79 (d, J=8.2 Hz, 1H), 6.04 (d, J=4.9 Hz, 1H), 7.39 (d, J=8.2 Hz, 1H)

CLIP
12/11/2015 12:05 PM EST
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
CLIP
With support from Almac, Wellstat delivers for a rare disease.
Proximity of API and finished drug development helps uridine triacetate to market for two indications
By Rick Mullin
“The initial contact was a cold call by Almac in 2010 or 2011,” recalls Mike Bamat, senior vice president of R&D at Wellstat Therapeutics, a small drug company in Gaithersburg, Md. “There were probably a couple of calls. It was one of those things where timing is everything.”
Almac, a Craigavon, Northern Ireland-based pharmaceutical services company, was looking to get in on Wellstat’s development of uridine triacetate, a synthetic pyrimidine analog, as an antidote for fluorouracil and capecitabine toxicity and overdose in cancer patients receiving those chemotherapies. And the calls, which Almac records indicate followed some communication between the companies, happened to come just when Wellstat was looking to change service partners as it moved toward commercial development of the drug.

Uridine triacetate
Discovery: Wellstat Therapeutic’s research on the therapeutic potential of exogenous uridine leads to a determination that uridine triacetate is a safe means of delivering the agent
Applications: Treatment of hereditary orotic aciduria (HOA), an extremely rare disease in which the body does not produce uridine, causing overproduction of orotic acid; emergency treatment of toxic reaction to or overdose of the cancer treatments fluorouracil and capecitabine
Methods of action: Treating HOA, uridine triacetate restores intracellular nucleotide concentrations, normalizing orotic acid production; as a chemotherapy antidote, it increases intracellular levels of uridine to dilute fluorouracil and capecitabine
Years in development: Since 2008 for chemotherapy antidote, and 2013 for HOA
Approved: Xuriden for HOA, Sept. 4, 2015; Vistogard for chemotherapy antidote, Dec. 11, 2015
The job went to Almac, as did work that sprang up as the result of another phone call to Wellstat—this one from the U.S. Food & Drug Administration.
As Bamat explains, uridine triacetate caught FDA’s attention regarding another potential indication—an extremely rare and life-threatening disease called hereditary orotic aciduria, or HOA. A consequence of the body’s inability to produce uridine, a necessary component of ribonucleic acid, HOA can manifest in a range of symptoms including blood abnormalities, developmental delays, and urinary tract obstruction caused by overproduction of orotic acid. There have been 20 reported cases of HOA since the 1950s. Only four cases are currently known in the U.S., Bamat says, and likely fewer than 20 in the world.
Wellstat landed approvals for Xuriden, the HOA treatment, in September of last year and Vistogard, the chemotherapy antidote, in December.
The story of Xuriden centers on a raft of FDA incentives for super-rare diseases that enabled Wellstat to move forward on an expedited application for a drug that will never be made in any great volume. But bringing Xuriden and Vistogard to market may also be viewed as the story of a drug discovery firm becoming a commercial enterprise thanks to its partnership with a service provider.
As Wellstat began late-stage development of the chemotherapy antidote, its research partner at the time, QS Pharma, was acquired by the service firm WIL Research. The look and feel of the partnership changed, according to Bamat.
“We kind of lost the small, easy-to-work-with relationship we had with them,” he says. Wellstat also needed support on development and manufacturing of a finished drug product composed of granules delivered in packets or sachets. The drug is administered orally, usually sprinkled on food such as applesauce or yogurt.
Almac was deemed a good fit because of its experience with developing drugs in granule form for “sachet presentation,” a packaging method more common in Europe than in the U.S. The Northern Ireland firm’s ability to develop and manufacture the active pharmaceutical ingredient (API) and the drug product in one location—at its headquarters—would also prove to be a significant advantage.
The distance between Gaithersburg and Craigavon, however, was a concern, according to Bamat. “We debated it. Especially those of us who knew we would be going there,” he says. “We couldn’t just jump in a car and go. But we looked at a variety of things, including cost and value, and it was all very positive at Almac.”
According to David Downey, vice president of commercial operations at Almac, bringing Wellstat’s work on uridine triacetate to commercial production posed several challenges, the first being to secure supply of uridine starting material, which is extracted from sugar beets by Euticals, an Italian firm. Next was developing a method to control particle size in both the API and the finished product. Almac also had to validate process equipment as it scaled up production.
“Uridine triacetate is Wellstat’s first commercial product,” Downey says. “So we were provided with a process more fit for development than for commercial production.”
The basic formulation of a granule drug product is simple, according to Downey: The API and excipient are mixed in a dry blender. The challenge is developing an analytical regimen to assure the granules are blended uniformly. Meeting the challenge required a high level of coordination between API and drug product process development.
“Wellstat needed a partner that could support them from the API to the drug product,” Downey says. The physical proximity between the Almac facilities in Craigavon conducting API and drug product work was a key advantage, he claims.

Uridine triacetate is formulated into granules presented in packets and sprinkled on food.
Credit: Wellstat Therapeutics
“If you listen to our business development people, you’ll hear them use the term, ‘crossing car parks as opposed to crossing oceans,’ ” Downey says, explaining that many competitors who offer API and finished drug services run these operations thousands of kilometers apart from each other, sometimes on different continents.
Before it signed on with Almac, Wellstat had been working with uridine triacetate for about 10 years. Its focus on developing the antidote drug started in 2008. Branching into the HOA treatment, however, upped the stakes.
Clinical study development for an HOA therapy was expedited via a full house of regulatory incentives from FDA, according to Bamat. “We had orphan drug designation, rare pediatric designation, breakthrough therapy designation, and priority review,” he says. “So they really went all out in helping us develop this.”
Although Wellstat was interested in developing a life saving drug for children, it was concerned about paying for it, given the tiny market. “At that time, the rare pediatric disease priority review voucher program was just on the radar,” Bamat says. “FDA said, ‘Consider this new program. Maybe it’s a way that at some risk you could recoup some of your costs.’ We looked at it and were willing to take the risk.”
It paid off. Wellstat was able to sell its priority review voucher—which entitles a company that brings a rare pediatric drug to market to receive expedited review of a subsequent drug—to AstraZeneca last year for an undisclosed amount. Other vouchers sold in 2015 brought high sums, including $350 million for one that AbbVie bought from United Therapeutics in August.
Bamat says Wellstat is not likely to change focus after its success with uridine triacetate. It continues to investigate new indications for the compound and will likely work with Almac on anything going into commercial development.
He emphasizes the importance of maintaining an effective working relationship with an outsourcing partner. “My main consideration is that these are people we can really work with on a day-to-day, week-to-week basis,” Bamat says. “Will the communication be good? Will they be honest and transparent with us, and will we be the same for them? That was a key factor, and we felt it was a plus with Almac.”
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2-3 hours |
| Biological half-life | 2-2.5 hours |
| Excretion | Renal |
| Identifiers | |
| DrugBank | DB09144 |
| Chemical data | |
| Formula | C15H18Cl0N2O9S0 |
| Molar mass | 370.31 g·mol−1 |
References
- HIGHLIGHTS OF PRESCRIBING INFORMATION OF XURIDEN
- Jump up^ BTG Announces FDA Approval of VISTOGARD® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine
- Jump up^ “FDA Approved Drugs:Uridine Triacetate”. FDA. 2015-12-11. Retrieved 2016-04-29.
- “Uridine triacetate”. DrugBank.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7807654 | 2010-10-05 | Compositions and methods for treatment of mitochondrial diseases |
| US2010222296 | 2010-09-02 | PYRIMIDINES, SUCH AS URIDINE, IN TREATMENTS FOR PATIENTS WITH BIPOLAR DISORDER |
| US7737128 | 2010-06-15 | Pyrimidines, such as uridine, in treatments for patients with bipolar disorder |
| US2010098678 | 2010-04-22 | Methods of Treatment of Mitochondrial Disorders |
| US2010041620 | 2010-02-18 | METHODS FOR IMPROVING FRONTAL BRAIN BIOENERGETIC METABOLISM |
| US2010041621 | 2010-02-18 | METHODS AND COMPOSITIONS FOR IMPROVING COGNITIVE PERFORMANCE |
| US7582619 | 2009-09-01 | Compositions and methods for treatment of mitochondrial diseases |
| US2008226684 | 2008-09-18 | METHOD AND PROCESS FOR THE PRODUCTION OF MULTI-COATED RECOGNITIVE AND RELEASING SYSTEMS |
| US7105498 | 2006-09-12 | Acylated uridine and cytidine and uses thereof |
| US6956028 | 2005-10-18 | Compositions and methods for treatment of mitochondrial diseases |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015307542 | 2015-10-29 | MODIFIED NUCLEIC ACID MOLECULES AND USES THEREOF |
| US2015167017 | 2015-06-18 | ALTERNATIVE NUCLEIC ACID MOLECULES AND USES THEREOF |
| US8821899 | 2014-09-02 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8771713 | 2014-07-08 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8741316 | 2014-06-03 | Highly porous, recognitive polymer systems |
| US2012294869 | 2012-11-22 | Methods for Treating Fatty Liver Disease |
| US2012078529 | 2012-03-29 | DETERMINING THE SEVERITY OF 5-FLUOROURACIL OVERDOSE |
| US8067392 | 2011-11-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7915233 | 2011-03-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7884202 | 2011-02-08 | Nucleobase Having Perfluoroalkyl Group and Process for Producing the Same |
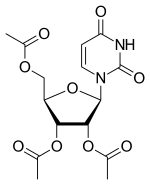
Uridine triacetate
- Molecular FormulaC15H18N2O9
- Average mass370.311 Da
ウリジントリアセタート
[(2R,3R,4R,5R)-3,4-diacetyloxy-5-(2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl acetate
Uridine, 2′,3′,5′-triacetate
uridini triacetas
Vistogard [Trade name]
Xuriden [Trade name]
(2R,3R,4R,5R)-2-(acetoxymethyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl diacetate
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
FDA APPROVED2015/9/4 . AS Xuriden
Uridine triacetate (INN),[1] formerly known as vistonuridine, is an orally active tri-acetylated prodrug of uridine[2] used:
- in the treatment of hereditary orotic aciduria (brand name Xuriden /ˈzʊərədɛn/ ZOOR-ə-den);[3]
- to treat patients following an overdose of chemotherapy drugs 5-fluorouracil (5-FU) or capecitabine regardless of the presence of symptoms, or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration (brand name Vistogard).[4][5][6]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine Triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. Uridine, a pyrimidine nucleotide, has been used in a variety of diseases including depressive disorders and inherited myopathies. (NCI04)
Pharmacology from NCIt
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTPincorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidinemetabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
from DrugBank
Uridine triacetate is an acetate ester that is uracil in which the three hydroxy hydrogens are replaced by acetate group. A prodrug for uridine, it is used for the treatment of hereditary orotic aciduria and for management of fluorouracil toxicity. It has a role as a prodrug, a neuroprotective agent and an orphan drug. It is a member of uridines and an acetate ester.
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 65” (PDF). World Health Organization. p. 92. Retrieved 12 March 2017.
- ^ “Uridine triacetate — DrugBank Page”. 12 March 2017.
- ^ “Xuriden (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “Vistogard (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “BTG Announces FDA Approval of Vistogard® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine”. BTG International Ltd. 11 December 2015. Retrieved 12 March 2017.
- ^ “Approved Drugs — Uridine Triacetate”. U.S. Food and Drug Administration. Retrieved 12 March 2017.
External links
Patents
- US7776838
- US5968914
- US6258795
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7776838 |
| Expiration | Aug 17, 2027 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
from FDA Orange Book
| FDA Orange Book Patents: 2 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6258795 |
| Expiration | Jul 10, 2019 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
from FDA Orange Book
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2–3 hours |
| Elimination half-life | 2–2.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.021.710 |
| Chemical and physical data | |
| Formula | C15H18N2O9 |
| Molar mass | 370.31 g·mol−1 |
| 3D model (JSmol) | |
////////////Uridine triacetate, ウリジントリアセタート , FDA 2015, breakthrough therapy designation ,
//////////174105-38-8, Priority review drug , Orphan drug, FDA 2015, Vistogard, uridine triacetate, fast track designations, PN-401, RG-2133, TAU, XURIDEN
|
CC(=O)OC[C@H]1O[C@H]([C@H](OC(C)=O)[C@@H]1OC(C)=O)N1C=CC(=O)NC1=O
|

{kind=link}